Способ получения f-18 меченых aβ лигандов
Номер патента: 22897
Опубликовано: 31.03.2016
Авторы: Самсон Фабрис, Смуда Кристоф, Фрибе Маттиас, Браун Райнер, Гарке Гуннар, Берндт Матиас, Шильдан Андреас, Патт Марианне



















Формула / Реферат
1. Способ получения соединения формулы I

который включает следующие стадии.
Стадия 1. Введение радиоактивной метки в соединение формулы II с помощью F-18 фторирующего агента, при этом получаем соединение формулы I, если R=H, или соединение формулы III, если R=PG

.
Стадия 2. Отщепление защитной группы PG, при этом получаем соединение формулы I, если R=PG.
Стадия 3. Очистка соединения формулы I, где
n=1-6;
X выбирают из группы, включающей:
a) СН,
b) N;
R выбирают из группы, включающей:
a) Н,
b) PG;
PG представляет собой аминозащитную группу;
LG представляет собой уходящую группу.
На стадии 3 используют метод ВЭЖХ, где ВЭЖХ растворитель или смесь растворителей представляют собой часть препарата для инъекций соединения I, пригодного для инъекций людям, где ВЭЖХ растворитель выбирают из группы, включающей этанол, водный буфер или смесь этанол/водный буфер.
2. Способ в соответствии с п.1, где PG выбирают из группы, включающей:
a) Boc,
b) тритил и
c) 4-метокситритил.
3. Способ в соответствии с п.1 или 2, где
LG выбирают из группы, включающей:
a) галоген и
b) сульфонилокси;
галоген представляет собой хлор, бром или йод.
4. Способ в соответствии с п.3, где сульфонилокси выбирают из группы, включающей:
a) метансульфонилокси,
b) п-толуолсульфонилокси,
c) (4-нитрофенил)сульфонилокси,
d) (4-бромфенил)сульфонилокси.
5. Способ в соответствии с п.1, где n=3 и X=СН.
6. Способ в соответствии с п.1, где n=3, X=СН, R=Вос и LG=метансульфонилокси.
7. Способ в соответствии с пп.1-6, где водный буфер выбирают из группы растворов: хлорид натрия, натрий-фосфатный буфер, аскорбиновая кислота, аскорбатный буфер или их смесей.
8. Способ в соответствии с пп.1-7, где ВЭЖХ растворитель представляет собой смесь этанола и водного буфера, включающего аскорбиновую кислоту или соли аскорбиновой кислоты.
9. Способ в соответствии с пп.1-8, где используют 10-50 мкмоль соединения формулы II.
10. Способ в соответствии с пп.1-9, где способ осуществляют в виде полностью автоматизированного процесса.
Текст
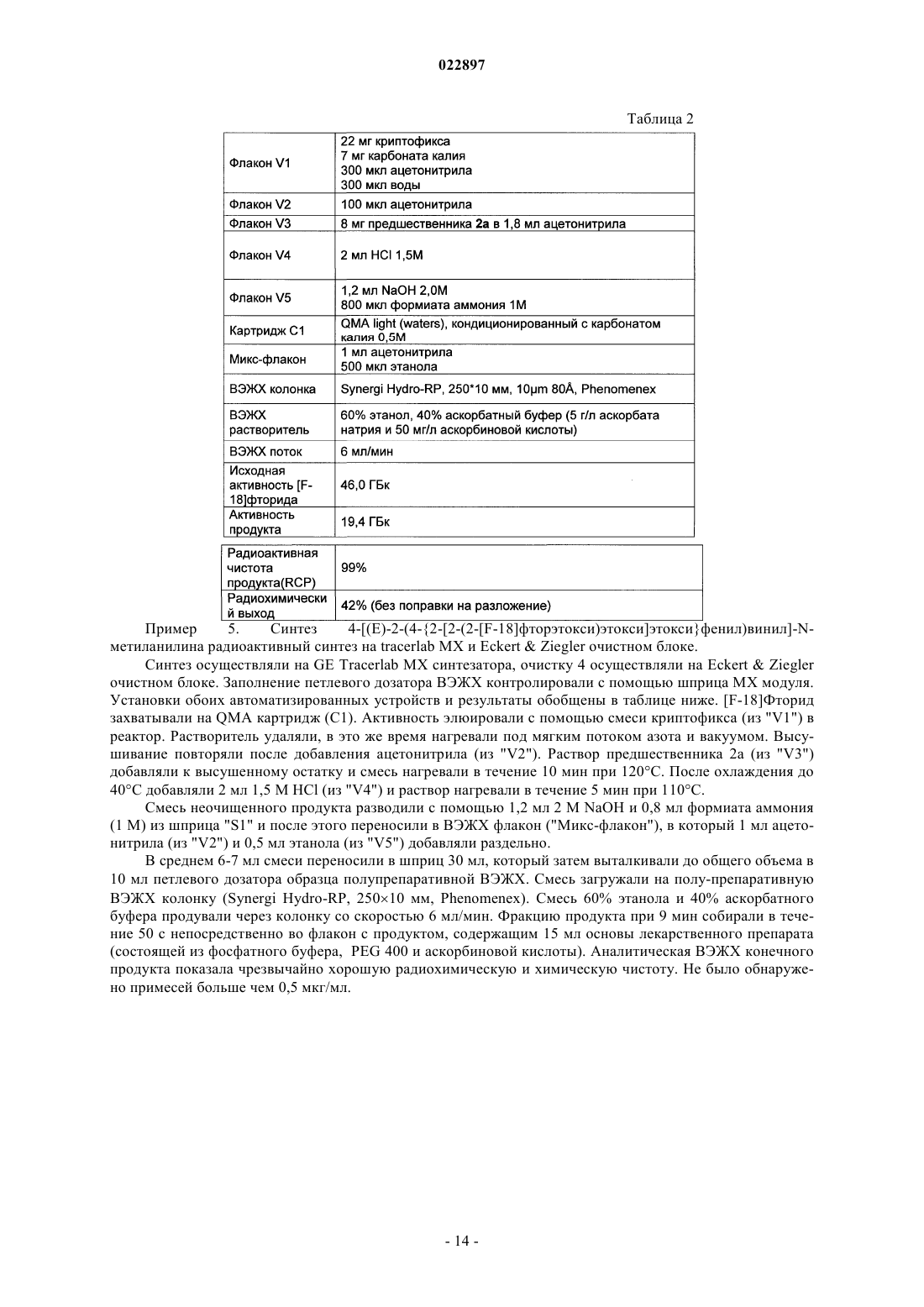
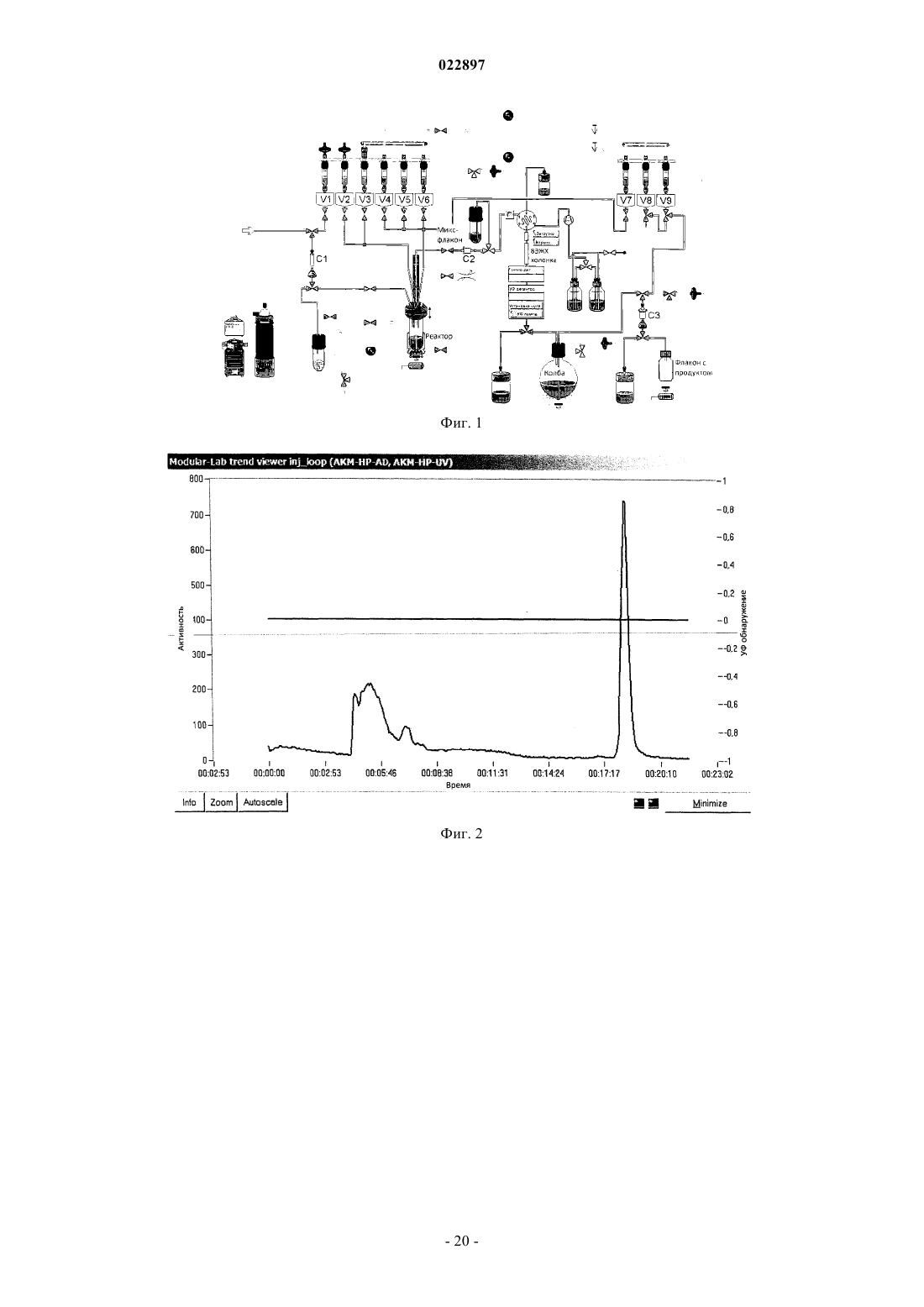
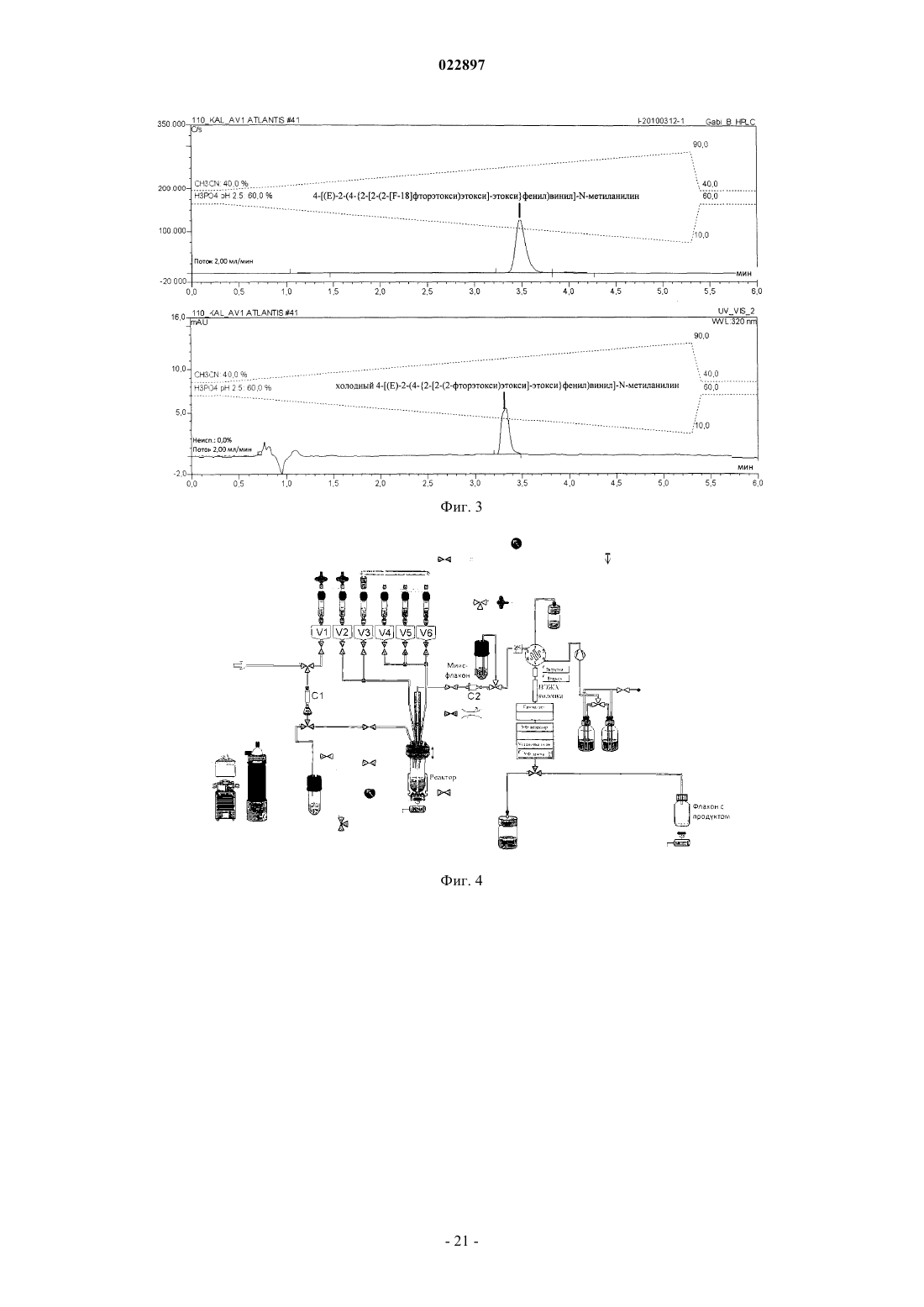
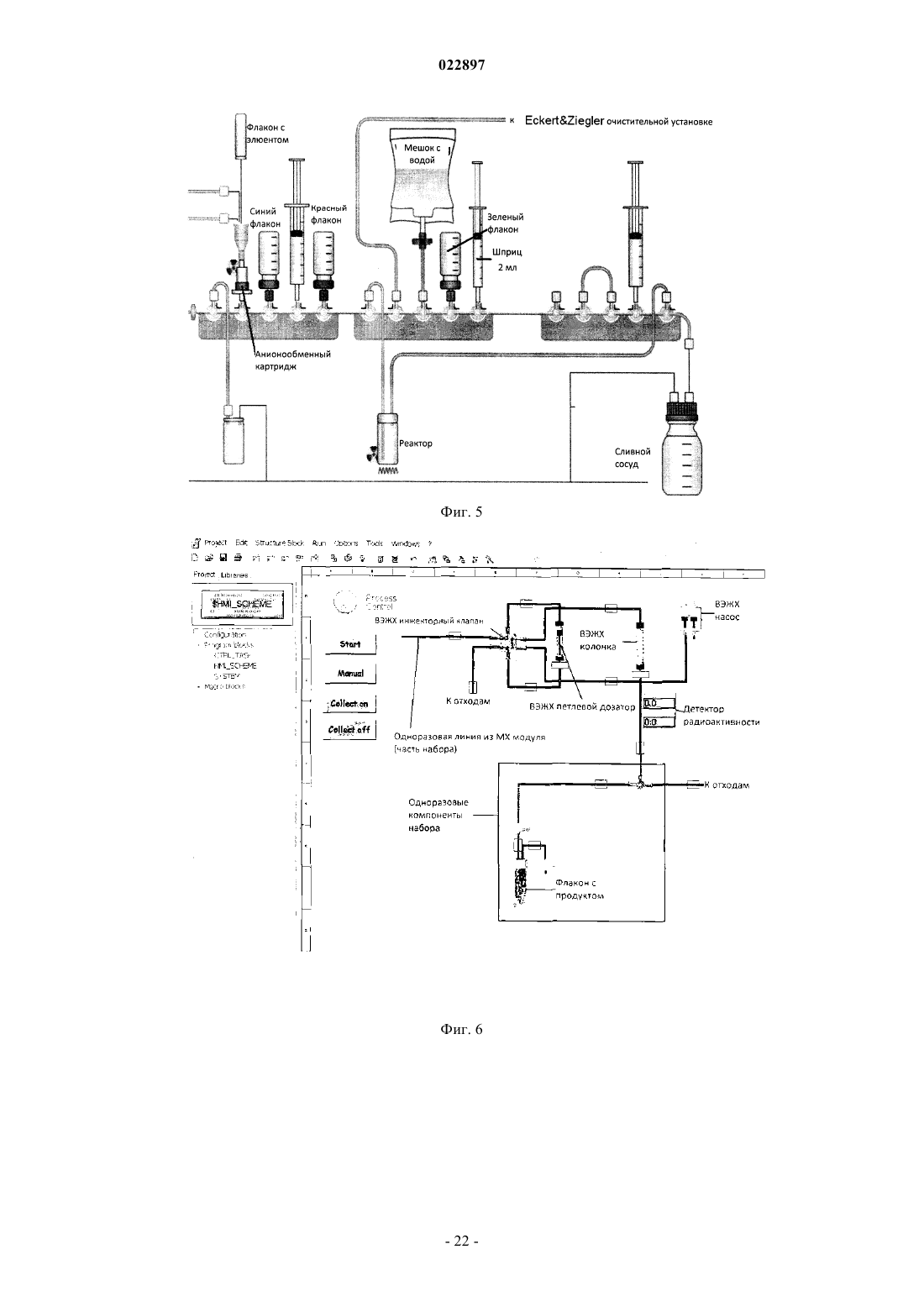
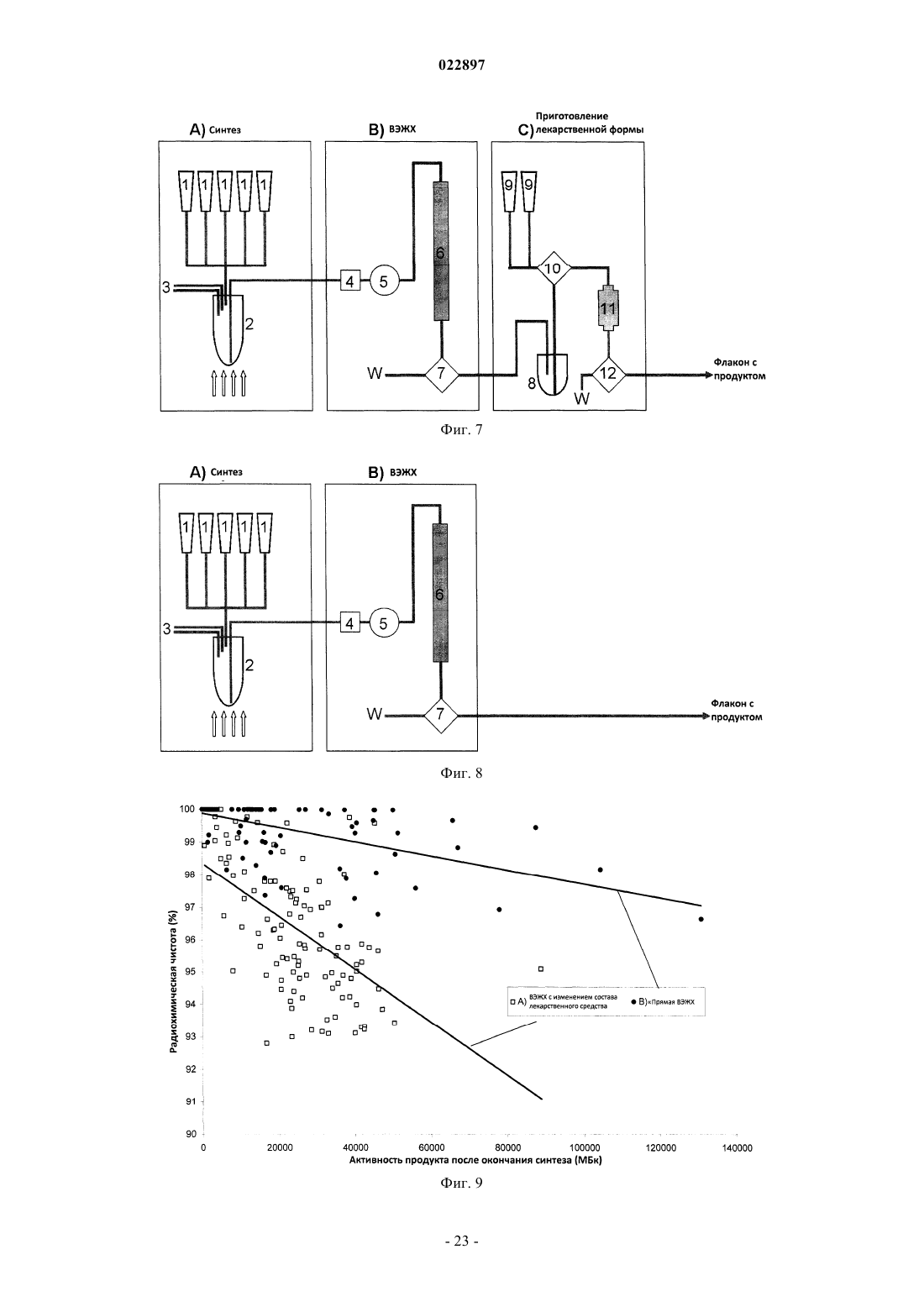
Изобретение относится к способу получения соединения формулы I, который обеспечивает введение F-18:(71)(73) Заявитель и патентовладелец: ПИРАМАЛЬ ИМЭДЖИНГ СА (CH) Область техники, к которой относится изобретение Настоящее изобретение относится к способам, которые обеспечивают [F-18]фторпегилированные производные (арил/гетероарил-винил)фенил метиламина. Предпосылки создания изобретения Болезнь Альцгеймера (AD) представляет собой прогрессирующее нейродегенеративное нарушение,характеризующееся потерей памяти, познавательных способностей и стабильности поведения. AD определяется патологически с помощью внеклеточных сенильных бляшек, состоящих из фибриллярных отложений бета-амилоидного пептида (А) и нейрофибриллярных клубков, состоящих из спаренных спиральных филаментов гиперфосфорилированного тау. 39-43 аминокислоты, содержащиеся в А пептидах,имеют происхождение из более крупного предшественника амилоидного белка (АРР). В амилоидогенном метаболическом пути А пептиды отщепляются от АРР путем последовательного протеолиза с помощью бета- и гамма-секретаз. А пептиды высвобождаются в виде растворимых белков и обнаруживаются в незначительных концентрациях в цереброспинальной жидкости (CSF) в нормальном стареющем головном мозге. При прогрессировании AD А пептиды агрегируют и образуют амилоидные отложения в паренхиме и сосудистой сети головного мозга, которые могут быть обнаружены посмертно в виде диффузных и сенильных бляшек и сосудистого амилоида при осуществлении гистологического анализа(недавний обзор см.: Blennow et al. Lancet. 2006 Jul. 29; 368(9533):387-403). Болезнь Альцгеймера (AD) становится большой медицинской и социально-экономической проблемой во всем мире. Большие усилия направлены на разработку техник и методов для раннего обнаружения и эффективного лечения заболевания. В настоящее время диагноз AD по академическим клиническим параметрам нарушений памяти составляет приблизительно 85-90% точности (Petrella J.R. et al.Radiology. 2003, 226:315-36). Он основывается на исключении различных заболеваний, вызывающих сходные симптомы, и тщательном неврологическом и психиатрическом исследовании, а также на нейропсихологическом тестировании. Молекулярная визуализация является эффективной для более раннего обнаружения прогрессирования заболевания или терапевтической эффективности по сравнению с другими наиболее распространенными методами в области неврологии, онкологии и кардиологии. Среди нескольких перспективных технологий молекулярной визуализации, таких как оптическая визуализация, MRI, SPECT и PET, PET представляет чрезвычайный интерес относительно разработки лекарственных средств в связи с их высокой чувствительностью и способностью обеспечивать количественные и кинетические данные. Например, позитронно-активные изотопы включают, например, углерод, йод, азот и кислород. Эти изотопы могут заменять их нерадиоактивные аналоги в целевых соединениях с получением PET индикаторов, которые обладают аналогичными биологическими свойствами. Из этих изотопов F-18 является предпочтительным изотопом для мечения в связи с его периодом полураспада 110 мин, который предоставляет возможность приготовления диагностических индикаторов и последующего изучения биохимических процессов. Дополнительно, его низкая + энергия (634 кэВ) также является благоприятной. Посмертное гистологическое исследование головного мозга все еще является единственным точным диагнозом болезни Альцгеймера. Таким образом, полагают, что in vivo обнаружение одной патологической характерной особенности заболевания - отложения амилоидных агрегатов в головном мозге будет оказывать сильное влияние на ранее выявление AD и дифференциацию ее от других форм деменции. Дополнительно, большинство терапий, модифицирующих заболевание, которые разрабатываются,нацелены на снижение амилоидной нагрузки в головном мозге. Таким образом, визуализация амилоидной нагрузки в головном мозге может обеспечивать существенный инструмент для стратификации пациентов и мониторинга лечения (недавний обзор см.: Nordberg. Eur. J. Nucl. Med. Mol. Imaging. 2008 Mar.; 35 Suppl. 1:S46-50). Дополнительно, также известно, что амилоидные отложения принимают участие в амилоидозах, при которых амилоидные белки (например, тау) атипично отложены в различных органах и/или тканях, вызывая заболевание. Недавний обзор см. Chiti et al. Annu Rev. Biochem. 2006; 75:333-66. Фторпегилированные (арил/гетероарил-винил)фенил метиламины, такие как 4-[(Е)-2-(4-2-[2-(2-фторэтокси)этокси]этоксифенил)винил]-N-метиланилин и 4-[(Е)-2-(6-2-[2-(2-фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилин,метили с помощью F-18 фторида, и они описаны в патентных заявках WO 2006/066104, WO 2007/126733 и представителях соответствующих патентных семейств. Пригодность этих радиоактивных индикаторов для обнаружения А бляшек описана в литературе(2008), 1-7; S.R. Choi et al., The Journal of Nuclear Medicine, 50 (2009), 1887-1894). Чтобы не ограничивать использования таких F-18 меченых диагностических средств, необходимы процессы, которые предоставляют возможность хорошо отлаженного и безопасного приготовления F-18 меченых индикаторов. Дополнительно, такие процессы должны обеспечивать высокий выход полного синтеза для предоставления возможности получения количеств диагностического средства для доставки радиоактивного индикатора, несмотря на период полураспада 110 мин, к оборудованию без циклотрона или оборудованию для получения радиофармацевтического препарата. Синтез F-18 меченых фторпегилированных (арил/гетероарил-винил)фенил метиламинов был описан ранее. 4-[(Е)-2-(4-2-[2-(2-[F-18]Фторэтокси)этокси]этоксифенил)винил]-N-метиланилинa) W. Zhang et al., Nuclear Medicine and Biology, 32 (2005), 799-809. 4 мг предшественника 2 а (2-[2-(2-4-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]феноксиэтокси)этокси]этил метансульфонат) в 0,2 мл ДМСО подвергали реакции с комплексом[F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощьюHCl и нейтрализовали с помощью NaOH. Смесь экстрагировали с помощью этилацетата. Растворитель высушивали и упаривали. Остаток растворяли в ацетонитриле и очищали путем полупрепаративной ВЭЖХ (ацетонитрил/5 мМ диметилглутаратный буфер рН 7 9/1). 20% (с поправкой на разложение), 11%(без поправки на разложение) 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина получали в течение 90 мин. Дополнительное изменение состава лекарственного препарата,-2 022897 необходимое для получения раствора, пригодного для инъекций людям, не описано.b) WO 2006/066104. 4 мг предшественника 2 а (2-[2-(2-4-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]феноксиэтокси)этокси]этил метансульфонат) в 0,2 мл ДМСО подвергали реакции с комплексом[F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощьюHCl и нейтрализовали с помощью NaOH. Смесь экстрагировали с помощью этил ацетата. Растворитель высушивали и упаривали, остаток растворяли в ацетонитриле и очищали путем полупрепаративной ВЭЖХ. 30% (с поправкой на разложение), 17% (без поправки на разложение) 4-[(E)-2-(4-2-[2-(2-[F18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина получали через 90 мин. Дополнительное изменение состава лекарственного препарата, необходимое для получения раствора, пригодного для инъекций людям, не описано.c) С.С. Rowe et al., Lancet Neurology, 7 (2008), 129-135. После введения радиоактивной метки, кислотного гидролиза и очистки путем полупрепаративной ВЭЖХ 4-[(E)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилин приготавливали в виде лекарственного средства с помощью твердофазной экстракции (SPE).d) H. Wang et al., Nuclear Medicine and Biology, 38 (2011), 121-127. 5 мг (9,33 мкмоль) предшественника 2 а (2-[2-(2-4-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]феноксиэтокси)этокси]этил метансульфонат) в 0,5 мл ДМСО подвергали реакции с комплексом [F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощью HCl и нейтрализовали с помощью NaOH. Неочищенный продукт разводили с помощью ацетонитрила/0,1 M формиата аммония (6/4) и очищали путем полупрепаративной ВЭЖХ. Фракцию продукта собирали, разводили водой, пропускали через С 18 картридж и элюировали с помощью этанола, получая 17% (без поправки на разложение) 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина в течение 50 мин. В том же источнике описано превращение незащищенного мезилатного предшественника. 5 мг (11,48 мкмоль) незащищенного мезилатного предшественника (2-2-[2-(4-(Е)-2-[4(метиламино)фенил]винилфенокси)этокси]этоксиэтил 4-метансульфонат) в 0,5 мл ДМСО подвергали реакции с комплексом [F-18]фторид/криптофикс/карбонат калия. Неочищенный продукт разводили с помощью ацетонитрила/0,1 M формиата аммония (6/4) и очищали путем полупрепаративной ВЭЖХ. Фракцию продукта собирали, разводили водой, пропускали через С 18 картридж и элюировали с помощью этанола,получая 23%(без поправки на разложение) 4-[(E)-2-(4-2-[2-(2-[F18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина в течение 30 мин. Было обнаружено, что процесс, в котором радиоактивный индикатор очищают только с помощьюSPE (без ВЭЖХ), обеспечивает получение продукта с приемлемой радиохимической чистотой (95%),тем не менее, химическая чистота чрезвычайно низкая, например, побочные продукты вследствие избытка предшественника не могут быть удалены. е) US 20100113763. 2 а(2-[2-(2-4-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]феноксиэтокси)этокси]этил метансульфонат) подвергали реакции с [F-18]фторидным реагентом в смеси трет-спирта и ацетонитрила. После фторирования растворитель упаривали и добавляли смесь HCl и ацетонитрила. После снятия защиты (нагревание при 100-120 С) смесь неочищенного продукта очищали путем ВЭЖХ(С 18, 60% ацетонитрил, 40% 0,1 M формиат аммония). Дополнительное изменение состава лекарственного препарата, необходимое для получения раствора, пригодного для инъекций людям, не описано.a) S.R. Choi et al., The Journal of Nuclear Medicine 50 (2009) 1887-1894. 1 мг предшественника 2b (2-2-[2-(5-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]пиридин-2-илокси)этокси]этоксиэтил 4-метилбензолсульфонат) в 1 мл ДМСО подвергали реакции с комплексом [F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощью HCl и нейтрализовали с помощью NaOH. ДМСО и неорганические компоненты удаляли путем твердофазной экстракции на картридже SepPak light C18 (Waters). Неочищенный продукт очищали путем полупрепаративной ВЭЖХ (55% ацетонитрил, 45% 20 мМ NH4OAc + 0,5%(мас./об.) аскорбат натрия). Фракцию продукта разводили водой и пропускали черезSepPak light C18 картридж. Радиоактивный индикатор элюировали с помощью этанола. Выход для 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина составил 10-30% (с поправкой на разложение).b) WO 2010/078370. 1,5 мг (2,45 мкмоль) предшественника 2b (2-2-[2-(5-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]пиридин-2-илокси)этокси]этоксиэтил 4-метилбензолсульфонат) в 2 мл ДМСО подвергали реакции с комплексом [F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощью HCl и разводили с помощью 1% раствора NaOH для нейтрализации. Смесь загружали на картридж с обращенной фазой. Картридж промывали водой (содержащей 5% (мас./об.) аскорбат натрия). Неочищенный продукт элюировали с помощью ацетонитрила в резервуар, содержащий воду + 5% (мас./об.) аскорбат натрия и ВЭЖХ растворитель. После очистки путем полупрепаративной ВЭЖХ фракцию продукта собирали в резервуар, содержащий воду + 0,5% (мас./об.) аскорбат натрия. Раствор пропускали через С 18 картридж, картридж промывали водой (содержащей 0,5%(мас./об.) аскорбат натрия и конечный продукт элюировали с помощью этанола во флакон, содержащий 0,9% раствор хлорида натрия с 0,5% (мас./об.) аскорбатом натрия.c) Y. Liu et al., Nuclear Medicine and Biology, 37 (2010), 917-925. 1 мг (1,63 мкмоль) предшественника 2b (2-2-[2-(5-[(Е)-2-4-[(трет-бутоксикарбонил)(метил)амино]фенилвинил]пиридин-2-илокси)этокси]этоксиэтил 4-метилбензолсульфонат) в 1 мл ДМСО подвергали реакции с комплексом [F-18]фторид/криптофикс/карбонат калия. С промежуточного соединения снимали защиту с помощью HCl и разводили с помощью 1% раствора NaOH. Смесь загружали на картридж Oasis HLB. Картридж промывали водой, высушивали в потоке аргона и продукт элюировали с помощью этанола во флакон, содержащий солевой раствор. Несмотря на то что радиохимические примеси удаляли посредством этой процедуры, нерадиоактивные побочные продукты, образовавшие вследствие гидролиза избытка предшественника, оставались в растворе конечного продукта. Выход для 4-[(E)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-Nметиланилина составил 34% (без поправки на разложение) в течение 50 мин на уровне радиоактивности от 10-100 мКи (370-3700 МБк) [F-18]фторида.Synthera адаптировали для синтеза 4-[(E)-2-(6-2-[2-(2-[F18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина. Дополнительно, интегрировали полупрепаративную ВЭЖХ систему и дополнительный модуль Synthera для изменения состава лекарственного препарата.WFNMB, Cape Town, South Africa, 18-23 September 2010). Приготовление 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-Nметиланилина сопровождалось использованием модуля синтеза IBA Synthera, соединенного с ВЭЖХ полупрепаративной системой очистки и дополнительным модулем для приготовления лекарственного средства (разведение ВЭЖХ фракции, захват на С 18 картридж, промывка и элюирование с помощью этанола). Несмотря на то что процессы очистки с помощью картриджей исследованы, оптимальное качество продукта относительно радиохимической чистоты и отделения от нерадиоактивных побочных продуктов показано и обосновано только для ВЭЖХ очистки. Таким образом, F-18 меченые фторпегилированные(арил/гетероарил-винил)фенил метиламины были очищены путем ВЭЖХ, используя системы растворителей, состоящие из ацетонитрила и водного буфера. Очевидно, собранные фракции продукта не могут непосредственно использоваться для введения пациенту. Ацетонитрил и другие компоненты системы растворителей, которые являются недопустимыми для инъекций людям, должны быть удалены. Это может сопровождаться упариванием или с помощью твердофазной экстракции (например, захват на С 18 твердофазный экстрагирующий картридж и элюирование этанолом, см. фиг. 1: картридж для окончательной твердофазной экстракции С 3, элюирование этанолом из V8; см. также фиг. 7: картридж для окончательной твердофазной экстракции 11, элюирование этанолом одного из флаконов 9). Тем не менее, в особенности при более высоких уровнях радиоактивности может происходить разложение радиоактивного индикатора вследствие процессов радиолиза. Эта проблема хорошо известна,для предотвращения радиолиза в процессе очистки 4-[(E)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина аскорбат натрия (в качестве акцептора радикалов) добавляли к ВЭЖХ растворителю и к промывочным растворам (S.R. Choi et al., WO 2010/078370). Тем не менее, концентрация радиоактивного индикатора после ВЭЖХ путем упаривания или путем твердофазной экстракции является решающей стадией приготовления. В обобщенных экспериментах более высокие радиохимические чистоты F-18 меченых фторпегилированных (арил/гетероарил-винил)фенил метиламинов могут быть обнаружены после ВЭЖХ, перед твердофазной экстракцией по сравнению с композицией после твердофазной экстракции. Общая схема процесса приготовления для F-18 меченых фторпегилированных (арил/гетероарилвинил)фенил метиламинов, как было описано ранее, проиллюстрирована на фиг. 7. Процесс приготовления может быть разделен на три основные части:C) приготовление лекарственного средства. Стадии приготовления безводного [F-18]фторида, введение радиоактивной метки в молекулу предшественника и снятие защиты осуществляют в части А устройства для синтеза (фиг. 7). Смесь неочищенного продукта переносят во вторую часть В для очистки путем ВЭЖХ (на силикагеле с обращенной фазой, используя элюент ацетонитрил/буфер). Для получения радиоактивного индикатора в лекарственном препарате, пригодном для инъекций людям, растворитель (ацетонитрил), присутствующий во фракции продукта, должен быть удален и поменян на композицию, которая является пригодной для приготовления лекарственного средства. Обычно (и это описано в вышеприведенных ссылках) фракцию продукта разводили водой (сосуд "8", фиг. 7, часть С) и затем пропускали через картридж с обращенной фазой ("11", фиг. 7, часть С). Картридж промывали водным раствором из одного из резервуаров 9 (фиг. 7,часть С) и в завершение элюировали из картриджа с помощью этанольного раствора (или этанола) из другого резервуара 9 во флакон с продуктом, который необязательно содержит дополнительные части и наполнители конечного лекарственного препарата. Это является очевидным для квалифицированного специалиста в данной области техники, что иллюстрации на фиг. 7 представляют собой упрощение процесса и оборудования и что другие компоненты, такие как клапаны, флаконы, трубки и др. могут быть частью такого процесса или оборудования."GMP соответствующий" процесс приготовления для 4-[(E)-2-(6-2-[2-(2-[F18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилин описан в WO 2010/078370 и С.-Н. Yao et al., Applied Radiation and Isotopes, 68 (2010), 2293-2297. Для предотвращения разложения 4-[(E)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина аскорбат натрия добавляли к ВЭЖХ растворителю (45% ацетонитрил, 55% 20 мМ ацетат аммония, содержащий 0,5% (мас./об.) аскорбат натрия) и конечный лекарственный препарат (0,5% (мас./об.) аскорбат натрия). Процесс позволяет получить вплоть до 18,5 ГБк (25,47,7%, с поправкой на разложение) 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина. Радиохимическая чистота составила 95,32,2%. Несмотря на то что аскорбат/аскорбиновую кислоту добавляли к растворителям, задействованным в очистку, радиохимическая чистота составила только приблизительно 95,32,2% на уровнях активности продукта вплоть до 18,5 ГБк (Yao et al.), возможно, вследствие разложения путем радиолиза. При более высоких уровнях активности продукта была обнаружена еще более высокая изменчивость радиохимической чистоты для приготовления 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина (пример 7, фиг. 9, метод А). Наряду с изменчивостью радиохимической чистоты, для изменения состава лекарственного препарата при действующем способе (превращение радиоактивного индикатора из ВЭЖХ среды в инъецируемый раствор) необходима дополнительная продолжительность процесса и требуется более сложное оборудование. Например, для процесса для синтеза 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилина, описанного Silva et al. и Casale et al., требуется три модуля для суммарного процесса производства. Синтез неочищенного продукта (схематически проиллюстрирован на фиг. 7, часть А) осуществляют на модуле IBA Synthera, полупрепаративную ВЭЖХ систему используют для очистки (схематически проиллюстрирован на фиг. 7, часть В) и дополнительный модуль синтеза IBA Synthera используют для изменения состава лекарственного препарата (схематически проиллюстрирован на фиг. 7, часть С). Задачей, на решение которой направлено настоящее изобретение, является обеспечение улучшенного способа ВЭЖХ очистки для F-18 меченых фторпегилированных (арил/гетероарил-винил)фенил метиламинов, который обеспечивает высокие химические и радиохимические чистоты радиоактивного индикатора, избегая концентрации меченого продукта после очистки для предотвращения радиолиза, в особенности при более высоких уровнях радиоактивности. Этот процесс должен быть пригодным для приготовления больших количеств (радиоактивности) радиоактивного индикатора для предоставления возможности распределения визуализирующих способностей за пределами собственной фармацевтической продукции. До настоящего времени максимальная описанная активность для F-18 меченого фторпегилированного (арил/гетероарил-винил)фенил метиламина составляла 18,5 ГБк (Yao et al.). Тем не менее,еще более высокие выходы могут быть поддержаны для широкого использования и доступности радиоактивного индикатора. Предпосылкой нового способа приготовления должна быть высокая радиохимическая чистота (например, 95%) в пределах широкого спектра радиоактивности. Более точно, такой способ должен быть пригодным для приготовления более высоких уровней активности радиоактивного индикатора по сравнению с описанными ранее (например, 20 ГБк, или даже 50 ГБк, или даже 100 ГБк) с подтвержденными радиохимическими чистотами 95%. В качестве дополнительной характерной особенности такой способ должен быть менее сложным по сравнению с ранее описанными способами. Проблемы, описанные выше, решают путем модифицированной процедуры очистки. Для упрощения общей схемы производства модифицируют состав растворителей для ВЭЖХ очистки. Вместо смеси ацетонитрил/буфер используют смесь этанол/буфер. Преимуществом новой смеси растворителей ВЭЖХ является то, что все составные компоненты ВЭЖХ растворителя - в отличие от ранее описанных композиций - являются хорошо переносимыми в виде части лекарственного препарата, следовательно, пригодны для инъекций людям. Следовательно, изменение состава лекарственного препарата для удаления составных компонентов ВЭЖХ растворителя (как проиллюстрировано на фиг. 7, часть С) уже больше не требуется. Это дополнительное преимущество нового способа - упрощенная схема - схематически проиллюстрировано на фиг. 8. (Очевидно, эта иллюстрация является упрощением, которое показывает общую схему нового способа, описанного в настоящем изобретении) Согласно изображению на фиг. 8 фракцию продукта собирают непосредственно (с помощью переключающего клапана "7") во флакон с продуктом (который может содержать другие части конечного лекарственного препарата). Вследствие уменьшенной сложности общее время приготовления при использовании нового способа, описанного в настоящем документе, сокращается, что непосредственно способствует более высоким выходам без поправки на разложение по сравнению с ранее использованным способом, где используют ВЭЖХ очистку с дополнительным (требующим затраты времени) изменением состава лекарственного препарата на твердофазном картридже (SPE). Основным преимуществом нового способа, описанного в настоящем документе, является достоверная высокая радиохимическая чистота F-18 меченых фторпегилированных (арил/гетероарилвинил)фенил метиламинов, синтезированных с помощью нового способа. В примере 7 и на фиг. 9 показана радиохимическая чистота в зависимости от способа очистки и количества (радиоактивности) радиоактивно меченого продукта при окончании синтеза. Точки/квадраты (каждая представляет отдельный эксперимент) и прямые на графике, отображающие тенденции, на фиг. 9 убедительно свидетельствуют о том, что радиохимическая чистота, полученная после ВЭЖХ с изменением состава лекарственного препарата путем SPE, изменяется существенно (фиг. 9, незарисованные квадраты). В особенности, при более высоких уровнях радиоактивности (20 ГБк) радиохимическая чистота часто составляет даже 95%. В отличие от этого, изменчивость радиохимических чистот, полученных с помощью нового способа согласно настоящему изобретению, значительно более низкая и достигаются высокие радиохимические чистоты 95%, даже при уровнях радиоактивности продукта больше чем 50 ГБк или даже больше чем 100 ГБк (фиг. 9, зарисованные точки). Сущность изобретения Настоящее изобретение обеспечивает способ получения радиоактивно меченого соединения формулы I и его подходящих солей неорганической или органической кислоты, его гидратов, комплексов,сложных эфиров, амидов, сольватов и пролекарств и, необязательно, фармацевтически приемлемого носителя, разбавителя, адъюванта или наполнителя. Способ включает следующие стадии: радиоактивное фторирование соединения формулы II; необязательно, отщепление защитной группы; очистка соединения формулы I путем ВЭЖХ с использованием системы растворителей, которая может являться частью инъецируемого препарата. Способ, обеспечиваемый настоящим изобретением, схематически проиллюстрирован на фиг. 8. Радиоактивное фторирование соединения формулы II и, необязательно, отщепление защитной группы осуществляют на находящейся слева части оборудования (фиг. 8, часть А). Очистку соединения формулы I осуществляют таким образом, что фракция продукта, полученная путем ВЭЖХ (фиг. 8, часть В),может быть непосредственно перенесена во флакон с продуктом, где флакон с продуктом необязательно содержит дополнительные фармацевтически приемлемые носители, разбавители, адъювант или наполнители. Дальнейшая часть процесса и оборудования, как проиллюстрировано на фиг. 7 (часть С), уже больше не является необходимой согласно способу в соответствии с настоящим изобретением. Описание изобретения В первом аспекте настоящее изобретение относится к способу получения соединения формулы I который включает следующие стадии. Стадия 1. Введение радиоактивной метки в соединение формулы II с помощью F-18 фторирующего агента, получая соединение формулы I, если R=H, или получая соединение формулы III, если R=PG Стадия 2. Необязательно, если R=PG, отщепление защитной группы PG, получая соединение формулы I. Стадия 3. Очистка соединения формулы I (причем на этой стадии используют метод ВЭЖХ, где ВЭЖХ растворитель или смесь растворителей представляют собой часть препарата для инъекций соединения I, пригодного для инъекций людям, где ВЭЖХ растворитель выбирают из группы, включающей этанол, водный буфер или смесь этанол/водный буфер), гдеa) СН,b) N. В одном предпочтительном варианте осуществления X=СН. В другом предпочтительном варианте осуществления X=N.a) Н,b) PG,PG представляет собой аминозащитную группу. В предпочтительном варианте осуществления PG выбирают из группы, включающей:c) 4-метокситритил. В более предпочтительном варианте осуществления R представляет собой Н. В другом более предпочтительном варианте осуществления R представляет собой Boc.LG представляют собой уходящую группу. В предпочтительном варианте осуществления LG выбирают из группы, включающей:b) сульфонилокси. Галоген представляет собой хлор, бром или йод. Предпочтительно галоген представляет собой бром или хлор. В предпочтительном варианте осуществления сульфонилокси выбирают из группы, включающей метансульфонилокси, п-толуолсульфонилокси, трифторметилсульфонилокси, 4-цианофенилсульфонилокси,4-бромфенилсульфонилокси,4-нитрофенилсульфонилокси,2-нитрофенилсульфонилокси,4-изопропил-фенилсульфонилокси, 2,4,6-триизопропил-фенилсульфонилокси, 2,4,6-триметилфенилсульфонилокси,4-трет-бутил-фенилсульфонилокси,4-адамантилфенилсульфонилокси и 4-метоксифенилсульфонилокси. В более предпочтительном варианте осуществления сульфонилокси выбирают из группы, включающей:a) метансульфонилокси,b) п-толуолсульфонилокси,c) (4-нитрофенил)сульфонилокси,d) (4-бромфенил)сульфонилокси. В еще более предпочтительном варианте осуществления LG представляет собой метансульфонилокси. В другом еще более предпочтительном варианте осуществления LG представляет собой п-толуолсульфонилокси. Предпочтительное соединение формулы I представляет собой: Другое предпочтительное соединение формулы I представляет собой: Предпочтительное соединение формулы II представляет собой: Другое предпочтительное соединение формулы II представляет собой: Другое предпочтительное соединение формулы II представляет собой: Другое предпочтительное соединение формулы II представляет собой: Другое предпочтительное соединение формулы II представляет собой: Стадия 1 включает прямую реакцию введения [F-18]фтор метки в соединения формулы II для получения соединения формулы I (если R=Н) или соединения формулы III (если R=PG). Способ введения радиоактивной метки включает стадию взаимодействия соединения формулы II сF-18 фторирующим агентом для получения соединения формулы III или соединения формулы I. В предпочтительном варианте осуществления, производное [DF-18]фторида представляет собой 4,7,13,16,21,24 гексаокса-1,10-диазабицикло[8,8,8]гексакозан K[F-18]F (криптофикс K[F-18]F), K[F-18]F, H[F-18]F,KH[F-18]F2, Cs[F-18]F, Na[F-18]F или тетраалкиламмониевую соль [F-18]F (например, фторид [F18]тетрабутиламмония). Более предпочтительно фторирующий агент представляет собой K[F-18]F,H[F-18]F, фторид [F-18]тетрабутиламмония, Cs[F-18]F или KH[F-18]F2, более предпочтительно K[F-18],Cs[F-18]F или фторид [F-18]тетрабутиламмония. Еще более предпочтительный F-18 фторирующий агент представляет собой криптофикс/калий[F18]фторид, предпочтительно полученный из [F-18]фторида, криптофикса и карбоната калия. Реакции радиоактивного фторирования осуществляют в ацетонитриле, диметилсульфоксиде или диметилформамиде или их смеси. Но также можно использовать другие растворители, которые хорошо известны специалисту в данной области техники. Вода и/или спирты могут быть задействованы в реакцию в качестве сорастворителя. Реакции радиоактивного фторирования осуществляют в течение менее чем 60 мин. Предпочтительное время реакций составляет менее чем 30 мин. Дальнейшее предпочтительное время реакций составляет менее чем 15 мин. Эти и другие условия для такого радиоактивного фторирования известны экспертам (Coenen, Fluorine-18 Labeling Methods: Features and Possibilities of Basic В одном варианте осуществления 7,5-75 мкмоль, предпочтительно 10-50 мкмоль, более предпочтительно 10-30 мкмоль и еще более предпочтительно 12-25 мкмоль и еще более предпочтительно 13-25 мкмоль соединения формулы II используют на стадии 1. В другом варианте осуществления более чем 7,5 мкмоль, предпочтительно более чем 10 мкмоль,более предпочтительно более чем 12 мкмоль и еще более предпочтительно более чем 13 мкмоль соединения формулы II используют на стадии 1. В другом варианте осуществления более чем 5 мг, предпочтительно более чем 6 мг и более предпочтительно более чем 7 мг соединения формулы II используют на стадии 1. В другом варианте осуществления 7 мг соединения формулы II используют на стадии 1. В другом варианте осуществления 8 мг соединения формулы II используют на стадии 1. В одном предпочтительном варианте осуществления радиоактивное фторирование соединения формулы II осуществляют в ацетонитриле или в смеси ацетонитрила и сорастворителей, где процент ацетонитрила составляет по меньшей мере 50%, более предпочтительно по меньшей мере 70%, еще более предпочтительно по меньшей мере 90%. Необязательно, если R=PG, стадия 2 включает снятие защиты с соединения формулы III, получая соединение формулы I. Условия реакции являются известными или очевидными для специалиста в данной области техники, которые выбирают, но не ограничиваясь только ими, из условий, описанных в пособии Greene and Wuts, Protecting groups in Organic Synthesis, 3-е изд., p. 494-653, включенном в настоящее описание в качестве ссылки. Предпочтительные условия реакции включают добавление кислоты и перемешивание при 0-180 С; добавление основания и нагревание при 0-180 С или их комбинацию. Предпочтительно стадию 1 и стадию 2 осуществляют в одном реакционном сосуде. Стадия 3 включает очистку соединения формулы I, используя систему ВЭЖХ разделения, где,ВЭЖХ растворитель элюент, а именно этанол, водный буфер или смесь этанол/водный буфер может быть частью инъецируемого лекарственного препарата соединения формулы I. Собранная фракция продукта может быть разведена или смешана с другими частями лекарственного препарата. Смесь ВЭЖХ растворителей состоит из этанола или водного буфера или смеси этанол/водный буфер, где водный буфер состоит из компонентов или наполнителя, которые могут быть инъецированы человеку. Примерами такого водного буфера являются растворы хлорида натрия, натрий-фосфатный буфер, аскорбиновая кислота, аскорбатный буфер или их смеси. В предпочтительном варианте осуществления способ приготовления соединения формулы I осуществляют путем использования модуля (обзор: Krasikowa, Synthesis Modules and Automation in F-18Molecular Imaging. Springer, Berlin Heidelberg, p. 289-316), который предоставляет возможность автоматизированного синтеза. Более предпочтительно способ осуществляют путем использования модуля в одном сосуде. Еще более предпочтительно способ осуществляют на общеизвестных модулях некассетного типа (например, EckerZiegler Modular-Lab, GE Tracerlab FX, Raytest SynChrom) и модулях кассетного типа (например, GE Tracerlab MX, GE Fastlab, IBA Synthera, EckertZiegler Modular-LabPharmTracer), необязательно, дополнительное оборудование, такое как ВЭЖХ или диспергирующие устройства, присоединены к указанным модулям. В предпочтительном аспекте настоящее изобретение относится к полностью автоматизированному и/или дистанционно управляемому способу получения соединения формулы I, где соединения формулыI, II и III и стадии 1, 2 и 3 описаны выше. В предпочтительном варианте осуществления этот способ представляет собой полностью автоматизированный процесс, соответствующий GMP принципам, который обеспечивает лекарственный препарат формулы I для применения для введения (инъекции) человеку. Определения В контексте настоящего изобретения предпочтительные соли представляют собой фармацевтически приемлемые соли соединений в соответствии с изобретением. Изобретение также включает соли, которые в связи с их частью не пригодны для фармацевтических применений, но которые могут применяться,например для выделения или очистки соединений в соответствии с изобретением. Фармацевтически приемлемые соли соединений в соответствии с изобретением включают соли присоединения кислот минеральных кислот, карбоновых кислот и сульфоновых кислот, например соли соляной кислоты, бромисто-водородной кислоты, серной кислоты, фосфорной кислоты, метансульфоновой кислоты, этансульфоновой кислоты, толуолсульфоновой кислоты, бензолсульфоновой кислоты, нафталин дисульфоновой кислоты, уксусной кислоты, трифторуксусной кислоты, пропионовой кислоты,молочной кислоты, винной кислоты, яблочной кислоты, лимонной кислоты, фумаровой кислоты, малеиновой кислоты и бензойной кислоты. Фармацевтически приемлемые соли соединений в соответствии с изобретением также включают соли общепринятых оснований, такие как, в качестве примера и предпочтения, соли щелочных металлов(например, соли натрия и соли калия), соли щелочно-земельных металлов (например, соли кальция и соли магния) и соли аммония, имеющие происхождение из аммиака или органических аминов, имеющих от 1 до 16 атомов углерода, такие как, в качестве примера и предпочтения, этиламин, диэтиламин, три- 10022897N-метилпиперидин. Термин "галоген", или "гало" относится к Cl, Br, F или I. Термин "аминозащитная группа", как используется в настоящем описании самостоятельно или в качестве части другой группы, известен или очевиден для специалиста в данной области техники, который выбирают, но не ограничиваясь только ими, из класса защитных групп, а именно карбаматы, амиды,имиды, N-алкиламины, N-ариламины, имины, енамины, бораты, N-P-защитные группы, N-сульфенил,N-сульфонил и N-силил, и которые выбирают, но не ограничиваясь только ими, из групп, которые описаны в пособии Greene and Wuts, Protecting groups in Organic Synthesis, 3-е изд., с. 494-653, включенном в настоящее описание в качестве ссылки. Аминозащитная группа предпочтительно представляет собой карбобензилокси (Cbz), п-метоксибензил карбонил (Moz или MeOZ), трет-бутилоксикарбонил (Boc),9-флуоренилметилоксикарбонил (FMOC), бензил (Bn), п-метоксибензил (РМВ), 3,4-диметоксибензил(DMPM), п-метоксифенил (РМР) или защищенная аминогруппа представляет собой 1,3-диоксо-1,3 дигидро-2 Н-изоиндол-2-ил (фталимидо) или азидогруппу. Термин "уходящая группа", как используется в настоящем описании самостоятельно или в качестве части другой группы, известен или очевиден для специалиста в данной области техники и обозначает,что атом или группу атомов отсоединяют от химического вещества с помощью нуклеофильного агента. Примеры представлены, например, в Synthesis (1982), p. 85-125, табл. 2 (с. 86 (последнюю запись этой табл. 2 необходимо откорректировать: "n-C4F9S(O)2-О-нонафлат" вместо "n-C4H9S(O)2-O-нонафлат"),Carey and Sundberg, Organische Synthese, (1995), p. 279-281, табл. 5.8; или Netscher, Recent Res. Dev. Org.Driving Force in Molecular Imaging. Springer, Berlin Heidelberg, p. 15-50, а именно: схема 4 c. 25, схема 5 с. 28, табл. 4 с. 30, фиг. 7 c. 33). Термин "сульфонилокси" относится к -O-S(O)2-Q, где Q представляет собой необязательно замещенный арил или необязательно замещенный алкил. Термин "алкил", как используется в настоящем описании самостоятельно или в качестве части другой группы, относится к C1-С 10 неразветвленной или разветвленной алкильной группе, такой как, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гептил,гексил, децил или адамантил. Предпочтительно алкил представляет собой C1-С 6 неразветвленный или разветвленный алкил или С 7-С 10 неразветвленный или разветвленный алкил. Низший алкил представляет собой C1-С 6 неразветвленный или разветвленный алкил. Термин "арил", как используется в настоящем описании самостоятельно или в качестве части другой группы, относится к моноциклическим или бициклическим ароматическим группам, содержащим от 6 до 10 углеродов в кольцевой части, таким как фенил, нафтил или тетрагидронафтил. Когда используется термин "замещенный", это обозначает, что указание о том, что один или несколько водородов на атоме, указанном в выражении с использованием "замещенный", заменен/заменены одним или несколькими компонентами из группы, включающей галоген, нитро, циано,трифторметил, алкил и О-алкил, при условии, что не превышается нормальная валентность соответствующего атома, и что замещение приводит к химически стабильному соединению, т.е. соединению, которое достаточно устойчиво для перенесения выделения до пригодной степени чистоты из реакционной смеси. Если специально не указано иначе, то при ссылке на соединения формулы согласно настоящему изобретению per se, a также на любую их фармацевтическую композицию, настоящее изобретение включает все гидраты, соли и комплексы. Термин "F-18" обозначает изотоп фтора 18F. Термин "F-19" обозначает изотоп фтора 19F. Примеры Определение радиохимической и химической чистоты. Радиохимические и химические чистоты 4-[(E)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина и 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина определяли с помощью аналитической ВЭЖХ (колонка: Atlantis Т 3; 1504,6 мм, 3 мкс,Waters; растворитель А: 5 мМ K2HPO4 pH 2,2; растворитель В: ацетонитрил; поток: 2 мл/мин, градиент: 0:00 мин 40% В, 0:00-05:50 мин 40-90% В, 05:50-05:60 мин 90-40% В, 05:60-09:00 мин 40% В). Время удерживания 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина: 3,5-3,9 мин в зависимости от ВЭЖХ системы, используемой для контроля качества. В связи с различным оборудованием (например, разливания по пробиркам) наблюдали отличия по времени удерживания между различными ВЭЖХ системами. Идентичность 4-[(Е)-2-(4-2-[2-(2-[F18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина доказывали путем совместной инъекции с нерадиоактивным эталоном 4-[(Е)-2-(4-2-[2-(2-[F-19]фторэтокси)этокси]этоксифенил)винил]-Nметиланилин. Время удерживания 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-Nметиланилина: 3,47 мин. Идентичность 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3 ил)винил]-N-метиланилина путем совместной элюции с нерадиоактивным эталоном -[(E)-2-(6-2-[2-(2- 11022897[F-19]фторэтокси)этокси]этоксипиридин-3-ил)винил]-N-метиланилин. Пример 1. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на EckertZiegler модульной лаборатории(60362 МБк) захватывали на QMA картридж. Активность элюировали с помощью смеси мезилат калия/криптофикс/н-Bu4MHCO3/метанол в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила. Раствор 4 мг 2 а в 1 мл трет-амиловый спирт/ацетонитрил (9:1) добавляли к высушенному остатку и смесь нагревали в течение 20 мин при 120 С. При осуществлении нагревания выпускную трубу реактора открывали для предоставления возможности испарения растворителя. Добавляли смесь 2,2 мл 1,5 М HCl, 1,1 мл ацетонитрила и 30 мг аскорбата натрия и реактор нагревали при 100 С в течение 10 мин. Неочищенный продукт нейтрализовали (1,5 мл 2 М NaOH + 0,3 мл буфера) и переносили на полупрепаративную ВЭЖХ колонку (Synergy Hydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера (рН 7,0) продували через колонку со скоростью 3 мл/мин. Фракцию продукта при 18 мин (фиг. 2) непосредственно собирали во флакон с продуктом, содержащим 8,5 мл основы лекарственного препарата (фосфатный буфер, аскорбиновая кислота, PEG 400). Аналитическая ВЭЖХ конечного продукта (фиг. 3) показала чрезвычайно хорошую радиохимическую и химическую чистоту. Только холодный 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилин обнаруживали на УФ-хроматограмме (фиг. 3, нижняя часть), все нерадиоактивные примеси были удалены. Определенная радиохимическая чистота составила 99,6%. Пример 2. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на Tracerlab FXN.Tracerlab FXN синтезатор адаптировали для "ВЭЖХ подхода с прямым срезом" (фиг. 4).[F-18]Фторид (3700 МБк) захватывали на QMA картридж. Активность элюировали с помощью смеси карбонат калия/криптофикс/ацетонитрил/вода в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила. Раствор 7 мг 2 а в 1 мл ацетонитрила добавляли к высушенному остатку и смесь нагревали в течение 8 мин при 120 С. После охлаждения до 60 С добавляли смесь 0,5 мл 2 М HCl и 0,5 мл ацетонитрила и реактор нагревали при 110 С в течение 4 мин. Неочищенный продукт нейтрализовали (1 мл 1 М NaOH + 2 мл буфера) и переносили на полупрепаративную ВЭЖХ колонку (Synergy Hydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера (рН 7,0) продували через колонку со скоростью 3 мл/мин. Фракцию продукта при 16 мин (фиг. 2) непосредственно собирали во флакон с продуктом, содержащим 8,5 основы лекарственного препарата (фосфатный буфер, аскорбиновая кислота,PEG 400). Определенная радиохимическая чистота составила 99%. Пример 3. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на Tracerlab MX и EckertZiegler очистном блоке. Набор собирали для синтеза 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина (табл. 1). Таблица 1 Состав набора для производства 4-[(E)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина на tracerlab MX и EckertZiegler очистном блоке Конструкцию Tracerlab MX кассеты адаптировали (фиг. 5). [F-18]Фторид захватывали на QMA картридж. Активность элюировали с помощью смеси карбонат калия/криптофикс/ацетонитрил/вода (из"флакона элюента") в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила. Раствор 8 мг 2 а в 1,8 мл ацетонитрила (ацетонитрил из"синего закрытого колпачком флакона" добавляли к твердому 2 а в "красный закрытый колпачком флакон" при осуществлении последовательности) добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. Добавляли 1,5 М HCl (из "зеленого закрытого колпачком флакона") и реактор нагревали при 110 С в течение 5 мин. Неочищенный продукт нейтрализовали (1 мл 1 М NaOH + 0,3 мл буфера, из "шприца 2 мл") и переносили в инжекторный клапан EckertZiegler ВЭЖХ (фиг. 6) с помощью левого шприцевого насоса MX модуля. Неочищенный продукт очищали на Synergy Hydro-RP,25010 мм, Phenomenex ВЭЖХ колонку, используя смесь 60% этанола и 40% аскорбатного буфера (рН 7,0). Фракцию продукта при 17,5 мин (фиг. 2) непосредственно собирали во флакон с продуктом, содержащим 8,5 основы лекарственного препарата (фосфатный буфер, аскорбиновая кислота, PEG 400). Пример 4. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на EckertZiegler модульной лаборатории. Синтез осуществляли на EckertZiegler ModularLab синтезаторе, используя ацетонитрил в качестве растворителя для фторирования. Установки синтезатора и результаты обобщены в табл. 2.[F-18]Фторид захватывали на QMA картридж (С 1). Активность элюировали с помощью смеси криптофикса (из "V1") в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления 100 мкл ацетонитрила (из "V2"). Раствор предшественника 2 а (из "V3") добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. После охлаждения до 40 С добавляли 2 мл 1,5 М HCl (из "V4") и раствор нагревали в течение 5 мин при 110 С. Смесь неочищенного продукта разводили с помощью 1,2 мл 2 М NaOH и 0,8 мл формиата аммония(1 М) из флакона "V5" и после этого переносили в ВЭЖХ флакон ("Микс-флакон"), содержащий предварительно 1 мл ацетонитрила и 0,5 мл этанола. Смесь переносили в 10 мл петлевого дозатора образца полупрепаративной ВЭЖХ, используя избыточное давление азота, в ВЭЖХ сосуд ("Микс-флакон") и с помощью жидкостного датчика, который контролирует окончание загрузки. Смесь загружали на полупрепаративную ВЭЖХ колонку (SynergiHydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера продували через колонку со скоростью 6 мл/мин. Фракцию продукта при 7 мин собирали непосредственно во флакон с продуктом, содержащим 15 мл основы лекарственного препарата (состоящей из фосфатного буфера,PEG 400 и аскорбиновой кислоты). Аналитическая ВЭЖХ конечного продукта показала чрезвычайно хорошую радиохимическую и химическую чистоту. Не было обнаружено примесей больше чем 0,3 мкг/мл. Пример 5. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на tracerlab MX и EckertZiegler очистном блоке. Синтез осуществляли на GE Tracerlab MX синтезатора, очистку 4 осуществляли на EckertZiegler очистном блоке. Заполнение петлевого дозатора ВЭЖХ контролировали с помощью шприца MX модуля. Установки обоих автоматизированных устройств и результаты обобщены в таблице ниже. [F-18]Фторид захватывали на QMA картридж (С 1). Активность элюировали с помощью смеси криптофикса (из "V1") в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила (из "V2"). Раствор предшественника 2 а (из "V3") добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. После охлаждения до 40 С добавляли 2 мл 1,5 М HCl (из "V4") и раствор нагревали в течение 5 мин при 110 С. Смесь неочищенного продукта разводили с помощью 1,2 мл 2 М NaOH и 0,8 мл формиата аммония(1 М) из шприца "S1" и после этого переносили в ВЭЖХ флакон ("Микс-флакон"), в который 1 мл ацетонитрила (из "V2") и 0,5 мл этанола (из "V5") добавляли раздельно. В среднем 6-7 мл смеси переносили в шприц 30 мл, который затем выталкивали до общего объема в 10 мл петлевого дозатора образца полупрепаративной ВЭЖХ. Смесь загружали на полу-препаративную ВЭЖХ колонку (Synergi Hydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера продували через колонку со скоростью 6 мл/мин. Фракцию продукта при 9 мин собирали в течение 50 с непосредственно во флакон с продуктом, содержащим 15 мл основы лекарственного препарата(состоящей из фосфатного буфера, PEG 400 и аскорбиновой кислоты). Аналитическая ВЭЖХ конечного продукта показала чрезвычайно хорошую радиохимическую и химическую чистоту. Не было обнаружено примесей больше чем 0,5 мкг/мл. Пример 6. Синтез 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-Nметиланилина радиоактивный синтез на tracerlab MX и EckertZiegler очистном блоке. Синтез осуществляли на GE Tracerlab MX синтезаторе, очистку 4 осуществляли на EckertZiegler очистном блоке. Заполнение петлевого дозатора ВЭЖХ контролировали с помощью детектора жидкостиEckertZiegler Очистного блока. Установки обоих автоматизированных устройств и результаты обобщены в таблице ниже. [F-18]Фторид захватывали на QMA картридж (С 1). Активность элюировали с помощью смеси криптофикса (из "V1") в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила (из "V2"). Раствор предшественника (из "V3") добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. После охлаждения до 40 С добавляли 2 мл 1,5 М HCl (из "V4") и раствор нагревали в течение 5 мин при 110 С. Смесь неочищенного продукта разводили с помощью 1,2 мл 2 М NaOH и 0,8 мл формиата аммония (1 М) из шприца "S1". 1 мл ацетонитрила (из "V2") и 0,5 мл этанола (из "V5") добавляли раздельно к смеси и после этого переносили в правый шприц автоматизированного устройства GETracerlab MX. Смесь переносили в 10 мл петлевого дозатора образца полупрепаративной ВЭЖХ, используя правый шприц GE Tracerlab MX автоматизированного устройства с помощью жидкостного датчика,который контролирует окончание загрузки. Смесь загружали на полупрепаративную ВЭЖХ колонку(Synergi Hydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера продували через колонку со скоростью 6 мл/мин. Фракцию продукта при 9 мин собирали непосредственно в течение 50 секунд во флакон с продуктом, содержащим 15 мл основы лекарственного препарата (состоящей из фосфатного буфера, PEG 400 и аскорбиновой кислоты). Аналитическая ВЭЖХ конечного продукта показала чрезвычайно хорошую радиохимическую и химическую чистоту. Не было обнаружено примесей больше чем 0,7 мкг/мл. Пример 7. Влияние способа очистки на радиохимическую чистоту. Серии синтезов 4-[(Е)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина осуществляли на двух различных синтезаторах (EckertZiegler модульной лаборатории и GE tracerlabMX), как в целом описано в примерах 1, 3-6. Введение радиоактивных меток осуществляли с использованием 4-10 мг предшественника 2 а в ацетонитриле, а также в смеси трет-амиловый спирт/ацетонитрил при 100-120 С в течение 10-20 мин (в случае введения радиоактивных меток в растворитель третамиловый спирт упаривали перед снятием защиты). N-Boc защитную группу удаляли путем нагревания сHCl (1,5-2 М). Смеси неочищенных продуктов индивидуально очищали с помощью одного из двух методов А) или В). Метод А). Смесь неочищенного продукта, полученную после снятия защиты, нейтрализовали с помощью смеси 2 М NaOH и 0,1 M формиата аммония и инъецировали в полупрепаративную ВЭЖХ (например, колонка: Gemini C18, 10250 мм, 5 мкм, Phenomenex; растворитель: 70% ацетонитрил, 30% буфер формиат аммония 0,1 M с 5 мг/мл аскорбата натрия, скорость потока 3 мл/мин). Фракцию продукта собирали в колбу, содержащую около 160 мл воды с 10 мг/мл аскорбата натрия. Смесь пропускали через С 18 картридж (tC18 SepPak environmental, Waters). Картридж промывали приблизительно 8-10 мл 20% EtOH в воде (содержащей 10 мг/мл аскорбата натрия). В завершение, продукт элюировали с 1,5-3 мл этанола во флакон, содержащий 8,5-17 мл "основы лекарственного препарата" (содержащей PEG 400, фосфатный буфер и аскорбиновую кислоту). Метод В). Смесь неочищенного продукта, полученную после снятия защиты, нейтрализовали с помощью смеси 2 М NaOH и 0,1 M формиата аммония и инъецировали в полупрепаративную ВЭЖХ (колонка: например: Gemini C18, 10250 мм, 5 мкм, Phenomenex или Synergi Hydro-RP, 25010 мм, 10 мкм 80 ,Phenomenex или Synergi Hydro-RP, 25010 мм, 4 мкм 80 , Phenomenex; растворитель: 60-70% этанол,40-30% аскорбатный буфер = 5 мг/мл аскорбата; поток 3, или 4, или 6 мл/мин). Фракцию продукта непосредственно собирали во флакон, содержащий основу лекарственного препарата (содержащую PEG 400,фосфатный буфер и аскорбиновую кислоту), получая 10-24 мл конечного лекарственного препарата. Время снижения пикового значения корректировали с помощью программного обеспечения для получения лекарственного препарата, содержащего 15% EtOH. Каждый незарисованный квадрат (каждый один результат для синтеза, включающего очистку с помощью метода А, 110 экспериментов) и каждая зарисованная точка (каждый один результат для синтеза,включающего очистку с помощью метода В, 105 экспериментов) на фиг. 9 представляет собой отдельный эксперимент относительно приготовления 4-[(E)-2-(4-2-[2-(2-[F-18]фторэтокси)этокси]этоксифенил)винил]-N-метиланилина. Тенденцию радиохимической чистоты в корреляции с радиоактивностью конечного продукта иллюстрировали с помощью прямых на графике, отображающих основные тенденции. Радиохимическая чистота, полученная после ВЭЖХ с изменением состава лекарственного препарата с помощью SPE (метод А), изменяется существенно (фиг. 9, незарисованные квадраты). В особенности при более высоких уровнях радиоактивности (20 ГБк) радиохимическая чистота часто даже 95%. В отличие от этого, изменчивость является значительно ниже для метода В). Соответственно, высокие радиохимические чистоты 95% достигали при уровнях активности продукта больше чем 50 ГБк и даже больше чем 100 ГБк (фиг. 9, зарисованные точки). Пример 8. Синтез 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3-ил)винил]-Nметиланилина на Tracerlab FXNTracerlab FXN синтезатор адаптировали для "ВЭЖХ подхода с прямым срезом" (фиг. 4).[F-18]Фторид (10 ГБк) захватывали на QMA картридж. Активность элюировали с помощью смеси карбонат калия/криптофикс/ацетонитрил/вода в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила. Раствор 8 мг 2b в 1,5 мл ацетонитрила добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. После охлаждения до 60 С добавляли 1 мл 1,5 М HCl и реактор нагревали при 110 С в течение 5 мин. Неочищенный продукт нейтрализовали (1 мл 1 М NaOH /формиат аммония), разводили (с помощью 0,5 мл EtOH и 1,5 мл MeCN) и переносили на полупрепаративную ВЭЖХ колонку (SynergyHydro-RP, 25010 мм, Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера (5 г/л аскорбата натрия и 50 мг/л аскорбиновой кислоты, рН 7,0) продували через колонку со скоростью 3 мл/мин. Фракцию продукта при 10 мин (см. фиг. 10) непосредственно собирали в течение 100 с и смешивали с 15 мл основы лекарственного препарата (фосфатный буфер, аскорбиновая кислота, PEG 400). 4,2 ГБк (42% без поправки на разложение) получали через 61 мин суммарного времени синтеза. Радиохимическая чистота Пример 9. Синтез метиланилина на Tracerlab FXNTracerlab FXN синтезатор адаптировали для "ВЭЖХ подхода с прямым срезом" (фиг. 4).[F-18]Фторид (6,85 ГБк) захватывали на QMA картридж. Активность элюировали с помощью смеси карбонат калия/криптофикс/ацетонитрил/вода в реактор. Растворитель удаляли, в это же время нагревали под мягким потоком азота и вакуумом. Высушивание повторяли после добавления ацетонитрила. Раствор 8 мг 2 с в 1,5 мл ацетонитрила добавляли к высушенному остатку и смесь нагревали в течение 10 мин при 120 С. После охлаждения до 60 С неочищенный продукт разводили с помощью 4 мл ВЭЖХ элюент и переносили на полупрепаративную ВЭЖХ колонку (Synergy Hydro-RP, 25010 мм,Phenomenex). Смесь 60% этанола и 40% аскорбатного буфера (5 г/л аскорбата натрия и 50 мг/л аскорбиновой кислоты, рН 7,0) продували через колонку со скоростью 3 мл/мин. Фракцию продукта при 12 мин непосредственно собирали в течение 100 с и смешивали с 15 мл основы лекарственного препарата (фосфатный буфер, аскорбиновая кислота, PEG 400). 2,54 ГБк (37% без поправки на разложение) получали через 53 мин суммарного времени синтеза. Радиохимическая чистота (определенная с помощью ВЭЖХ, tR=3,78 мин) составляла 99%. Описание чертежей Фиг. 1. Установки Tracerlab FXN для очистки с изменением состава лекарственного препарата(адаптированные с помощью программного обеспечения tracerlab). Фиг. 2. Хроматограмма очистки с использованием Synergy колонки на EckertZiegler модульной лаборатории (канал радиоактивности). Фиг. 3. Аналитическая ВЭЖХ радиоактивно меченого продукта (вверху канал радиоактивности,внизу УФ-канал). Фиг. 4. Установки Tracerlab FXN для очистки с изменением состава лекарственного препарата(адаптированные с помощью программного обеспечения tracerlab). Фиг. 5. Установки Tracerlab MX (адаптированные с помощью программного обеспеченияCoincidence FDG). Фиг. 6. Установки EckertZiegler очистного блока (адаптированные с помощью программного обеспечения Modual-Lab). Фиг. 7. Схематическая иллюстрация процесса и оборудования для приготовления F-18 меченых фторпегилированных (арил/гетероарил-винил)фенил метиламинов, включающая три части: А) синтез,В) ВЭЖХ, С) приготовление лекарственного препарата; включая (1) флаконы для реагентов и растворителей, (2) реакционный сосуд, (3) целевую линию для F-18, необязательно газовые линии, вакуум и др.,(4) необязательно детектор жидкости или фильтр и др., (5) инжекторный клапан, (6) ВЭЖХ колонку,(7) клапан для снижения максимального значения, (W) спускную(ые) линию(и), (8) сосуд для сбора/разведения ВЭЖХ фракции, (9) флаконы растворителей для промывания и элюции, (10) клапан,(11) картридж, например С 18 картридж для улавливания продукта, (12) клапан. Фиг. 8. Схематическая иллюстрация процесса и оборудования для приготовления F-18 меченых фторпегилированных (арил/гетероарил-винил)фенил метиламинов, включающая две части: А) синтез,В) ВЭЖХ; включая (1) флаконы для реагентов и растворителей, (2) реакционный сосуд, (3) целевую линию для F-18, необязательно газовые линии, вакуум и др., (4) необязательно, детектор жидкости или фильтр и др., (5) инжекторный клапан, (6) ВЭЖХ колонку, (7) клапан для снижения максимального зна- 18022897 чения. Фиг. 9. Влияние способа очистки на радиохимическую чистоту. Фиг. 10. Хроматограмма очистки 4-[(Е)-2-(6-2-[2-(2-[F-18]фторэтокси)этокси]этоксипиридин-3 ил)винил]-N-метиланилина на EckertZiegler модульной лаборатории (канал радиоактивности) ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы I который включает следующие стадии. Стадия 1. Введение радиоактивной метки в соединение формулы II с помощью F-18 фторирующего агента, при этом получаем соединение формулы I, если R=H, или соединение формулы III, если R=PG: Стадия 2. Отщепление защитной группы PG, при этом получаем соединение формулы I, если R=PG. Стадия 3. Очистка соединения формулы I, гдеPG представляет собой аминозащитную группу;LG представляет собой уходящую группу. На стадии 3 используют метод ВЭЖХ, где ВЭЖХ растворитель или смесь растворителей представляют собой часть препарата для инъекций соединения I, пригодного для инъекций людям, где ВЭЖХ растворитель выбирают из группы, включающей этанол, водный буфер или смесь этанол/водный буфер. 2. Способ в соответствии с п.1, где PG выбирают из группы, включающей:b) сульфонилокси; галоген представляет собой хлор, бром или йод. 4. Способ в соответствии с п.3, где сульфонилокси выбирают из группы, включающей:a) метансульфонилокси,b) п-толуолсульфонилокси,c) (4-нитрофенил)сульфонилокси,d) (4-бромфенил)сульфонилокси. 5. Способ в соответствии с п.1, где n=3 и X=СН. 6. Способ в соответствии с п.1, где n=3, X=СН, R=Вос и LG=метансульфонилокси. 7. Способ в соответствии с пп.1-6, где водный буфер выбирают из группы растворов: хлорид натрия, натрий-фосфатный буфер, аскорбиновая кислота, аскорбатный буфер или их смесей. 8. Способ в соответствии с пп.1-7, где ВЭЖХ растворитель представляет собой смесь этанола и водного буфера, включающего аскорбиновую кислоту или соли аскорбиновой кислоты. 9. Способ в соответствии с пп.1-8, где используют 10-50 мкмоль соединения формулы II. 10. Способ в соответствии с пп.1-9, где способ осуществляют в виде полностью автоматизированного процесса.
МПК / Метки
МПК: A61K 51/04, C07B 59/00
Метки: меченых, получения, лигандов, способ, а&beta
Код ссылки
<a href="https://eas.patents.su/25-22897-sposob-polucheniya-f-18-mechenyh-abeta-ligandov.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения f-18 меченых aβ лигандов</a>
Способ получения f-18 меченых aβ лигандов

Номер патента: 22896
Опубликовано: 31.03.2016
Авторы: Берндт Матиас, Смуда Кристоф, Патт Марианне, Хульч Кристина, Самсон Фабрис, Шильдан Андреас, Фрибе Маттиас
МПК: A61K 51/04, C07B 59/00
Метки: меченых, а&beta, способ, лигандов, получения
Формула / Реферат:
1. Способ получения соединения формулы Iкоторый включает стадии:стадия 1 - введение радиоактивной метки в соединение формулы II с помощью F-18 фторирующего агента, получая соединение формулы I, если R=Н, или получая соединение формулы III, если R=PGв которой реакцию радиоактивного фторирования осуществляют в ацетонитриле или в смеси ацетонитрила и сорастворителя, где процент ацетонитрила составляет по меньшей мере 50%,стадия 2 - если R=PG,...
Применение мелатонинергических лигандов для получения фармацевтических композиций, предназначенных для профилактики или лечения патологий желудочно-кишечной системы

Номер патента: 2823
Опубликовано: 31.10.2002
Авторы: Ренар Пьер, Делагранж Филипп, Беннежан Каролин, Мерль Анн, Пеллиссье Сонья
МПК: A61P 1/00, A61K 31/4045
Метки: системы, получения, профилактики, композиций, лигандов, предназначенных, фармацевтических, лечения, патологий, мелатонинергических, применение, желудочно-кишечной
Формула / Реферат:
1. Применение мелатонина и мелатонинергических лигандов для получения фармацевтических композиций для профилактики и/или лечения патологий желудочно-кишечной системы. 2. Применение по п.1 мелатонина для получения фармацевтических композиций для профилактики и/или лечения патологий желудочно-кишечной системы. 3. Применение по п.1 мелатонинергических лигандов для получения фармацевтических композиций для профилактики и/или лечения патологий...
Способ стабилизации органофосфитных лигандов в процессах, катализируемых комплексными катализаторами

Номер патента: 1483
Опубликовано: 23.04.2001
Авторы: Брайант Дэвид Роберт, Леунг Так Вай, Шоу Бернард Лесли
МПК: C07C 45/50, C07F 9/02, B01J 31/18...
Метки: процессах, катализаторами, катализируемых, органофосфитных, стабилизации, способ, комплексными, лигандов
Формула / Реферат:
1. Способ стабилизации комплексного катализатора металл-органополифосфитный лиганд против деактивации в процессах гидроформилирования, включающих реакцию одного или нескольких реагентов в присутствии комплексного катализатора металл-органополифосфитный лиганд и необязательно свободного органополифосфитного лиганда для получения потока продуктов реакции, содержащего один или несколько продуктов, отличающийся тем, что указанный процесс проводят в...
Производные 1-арилсульфонилбензазола в качестве лигандов 5-гидрокситриптамина-6

Номер патента: 6057
Опубликовано: 25.08.2005
Авторы: Келли Майкл Джерард, Зоу Пинг
МПК: A61K 31/4045, C07D 209/08, A61P 25/28...
Метки: лигандов, производные, 1-арилсульфонилбензазола, качестве, 5-гидрокситриптамина-6
Формула / Реферат:
1. Соединение формулы I где W означает SO2; X означает CR7; Y означает CR8 или N; Z означает O; каждый из R1 и R2 означает H; n означает целое число 2, 3 или 4; каждый из R3 и R4 независимо означает H, CNR10NR11R12 или группу: C1-C6алкил, C2-C6алкенил, C2-C6алкинил, C3-C6циклоалкил, циклогетероалкил, арил или гетероарил, каждую необязательно замещенную, либо R3 и R4 вместе с атомом, к которому они присоединены, могут образовывать необязательно...
8-сульфонил-1,3,4,8-тетрагидро-2н-[1,4]оксазепино[6,7-e]индольные производные и их использование в качестве 5-нт6 рецепторных лигандов

Номер патента: 16456
Опубликовано: 30.05.2012
Авторы: Рингберг Эрик, Хаммер Кристин, Рингом Руне, Бертс Веи, Хенрикссон Софиа, Линдквист Бенгт, Брандт Петер
МПК: C07D 498/18, C07D 498/04, A61K 31/553...
Метки: 8-сульфонил-1,3,4,8-тетрагидро-2н-[1,4]оксазепино[6,7-e]индольные, использование, рецепторных, лигандов, 5-нт6, производные, качестве
Формула / Реферат:
1. Соединение формулы (I)в которой R1 выбирают из фенила, нафтила, пиридинила, изоксазолила, имидазолила, 1,4-бензодиоксинила, бензофуранила, фуранила, 1,3-бензотиазолила, хроманила, тиенила и бензотиенила, каждый из которых необязательно независимо замещен в одном или двух положениях заместителем, выбранным из:(a) галогена,(b) C1-3алкила,(c) фтор-С1-3алкила,(d) C1-3алкокси,(e) фтор-С1-3алкокси,(f) C1-3алкилсульфонила,(g) -CN и(h) фенила;R2...
Предыдущий патент: Способ получения f-18 меченых aβ лигандов
Следующий патент: Стабильные полипептиды, вариабельные домены антитела и антагонисты против tnfr1
Случайный патент: Коммутаторный узел