Ненуклеозидные ингибиторы обратной транскриптазы, композиции, их содержащие, и их применение
Номер патента: 24804
Опубликовано: 31.10.2016
Авторы: Ст-Ондж Мигель, Коте Бернар, Нгуйен Натали, Ли Чунь Синг, Говро Данни, Берч Джейсон







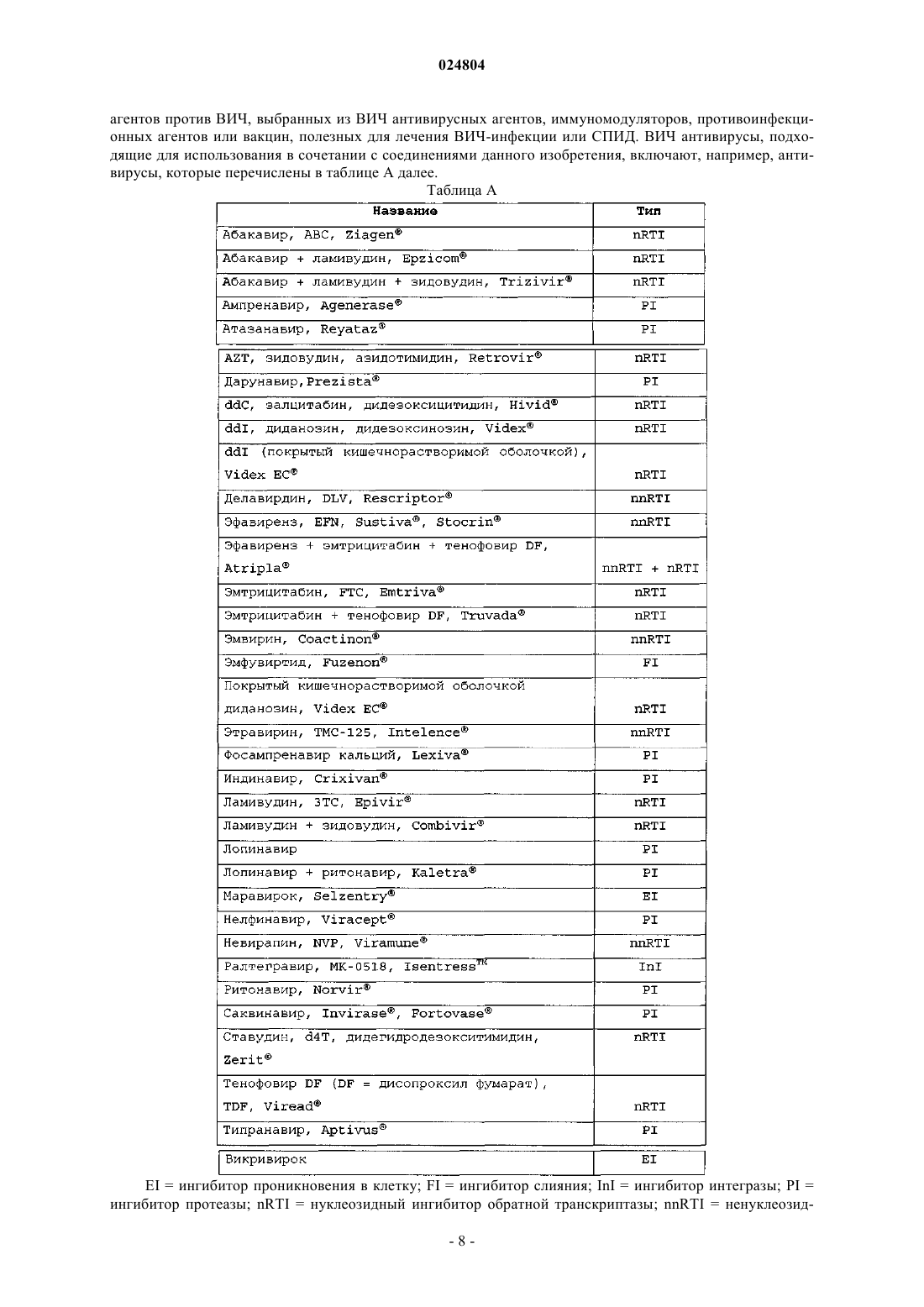












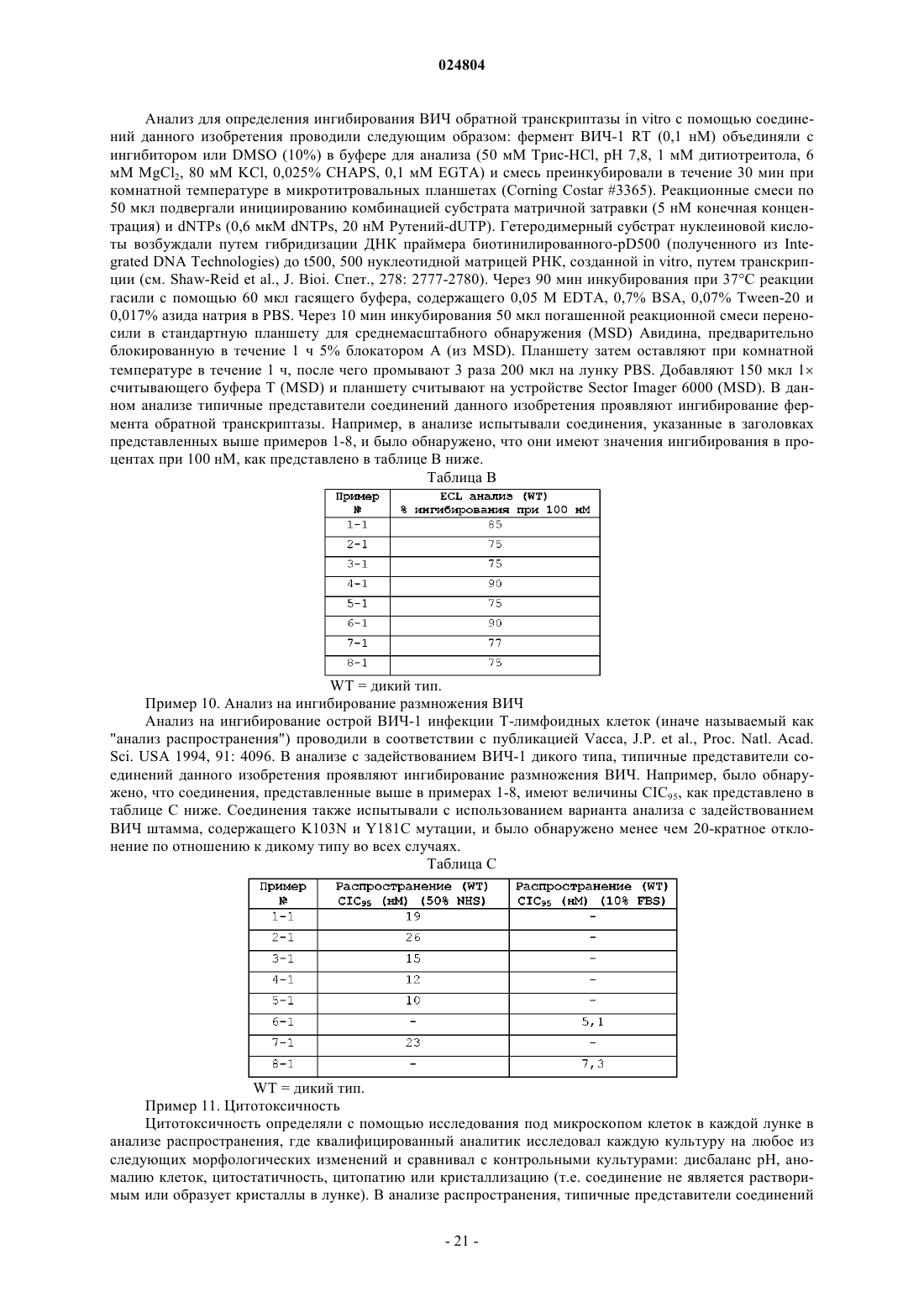


Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль,
где R1 представляет собой AryА;
где AryА представляет собой фенил, который необязательно замещен в целом 1-3 заместителями, выбранными из (2) C1-6галогеналкила, (11) галогена, (12) CN;
R2 и R3, каждый, представляют собой независимо (1) Н, (3) C1-6галогеналкил, (7) галоген;
R4 представляет собой Н;
R5 представляет собой C1-6алкил.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где фенил является необязательно замещенным 1-3 заместителями, каждый из которых представляет собой независимо (2) С1-4галогеналкил.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 представляет собой (2) С1-4галогенкалкил, (5) галоген; R3 представляет собой (1) Н, (3) С1-4галогеналкил, (6) галоген.
4. Соединение по п.1 или его фармацевтически приемлемая соль, которые представляют собой соединение формулы (II)

где X1 и X2, каждый, представляют собой независимо (3) С1-4галогеналкил, (9) галоген, (10) CN;
R2 и R3, каждый, представляют собой независимо (1) Н, (3) С1-4галогеналкил, (14) галоген;
R5 представляет собой С1-4алкил.
5. Соединение по п.4 или его фармацевтически приемлемая соль,
где X1 и X2, каждый, представляют собой независимо (2) С1-4фторалкил, (4) галоген, (5) CN,
R2 представляет собой (2) С1-4фторалкил, (5) галоген;
R3 представляет собой (1) Н, (3) С1-4галогеналкил, (6) галоген и
R5 представляет собой C1-3алкил.
6. Соединение по п.5 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (III)

7. Соединение по п.6 или его фармацевтически приемлемая соль, где R5 представляет собой СН3 или СН2СН3.
8. Соединение по п.7 или его фармацевтически приемлемая соль, где R3 представляет собой Н.
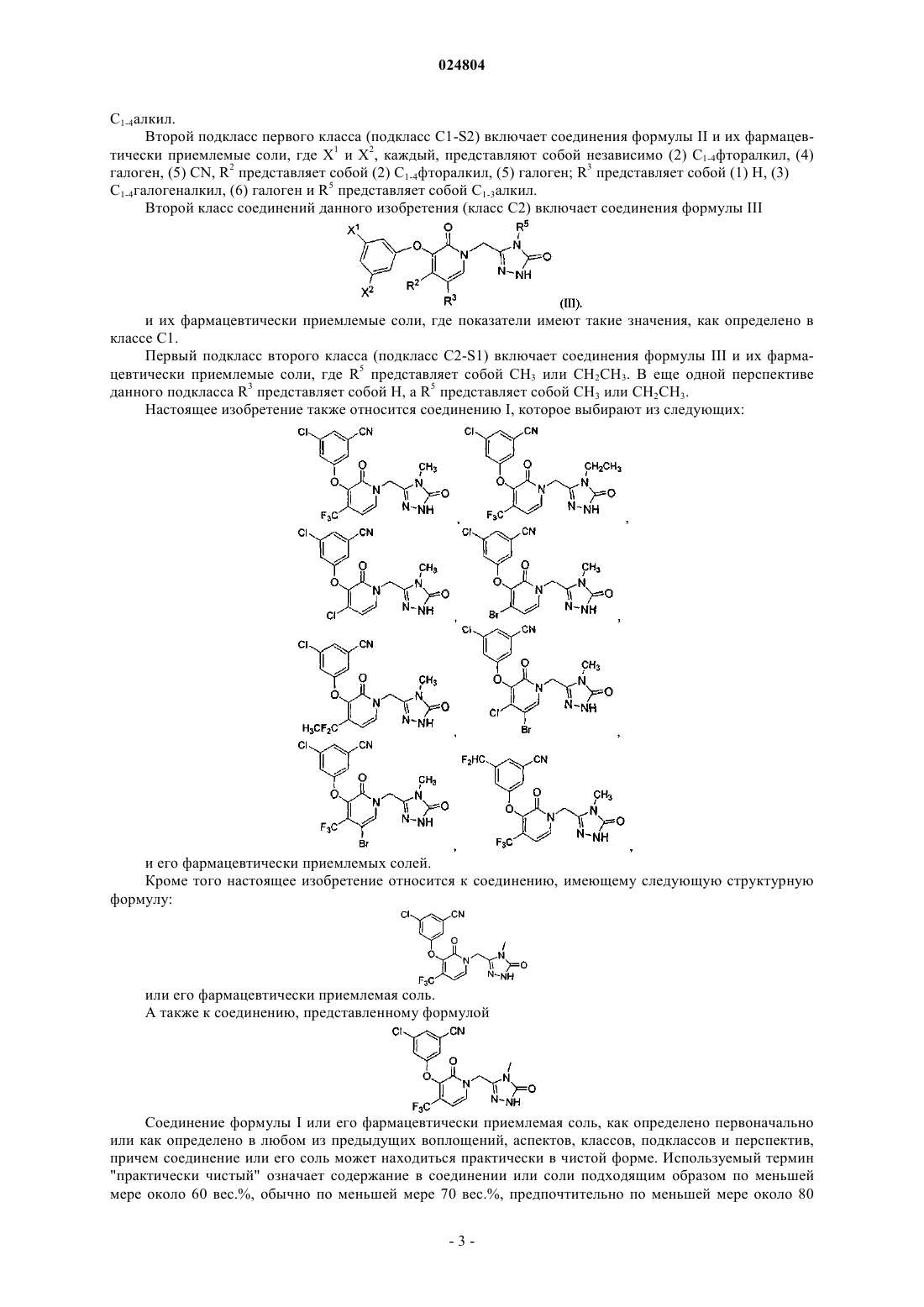
9. Соединение формулы (I) по п.1, где соединение выбрано из группы, состоящей из

и его фармацевтически приемлемые соли.
10. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения согласно любому одному из пп.1-9 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
11. Применение соединения по любому одному из пп.1-9 или его фармацевтически приемлемой соли в приготовлении лекарственных средств для профилактики или лечения инфекций, вызываемых ВИЧ, или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом.
12. Соединение, представленное формулой

или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.12 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
14. Соединение, представленное формулой

15. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.14 и фармацевтически приемлемый носитель.
16. Применение соединения по п.12 или его фармацевтически приемлемой соли в профилактике или лечении инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом.
17. Применение соединения по п.14 в приготовлении лекарственных средств для профилактики или лечения инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом.
Текст
НЕНУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ, КОМПОЗИЦИИ,ИХ СОДЕРЖАЩИЕ, И ИХ ПРИМЕНЕНИЕ Медведев В.Н. (RU) Гетероароматические соединения формулы I: (I) представляют собой ингибиторы обратной транскриптазы, где R1, R2, R3, R4 и R5 имеют значения, определенные в описании. Соединения формулы I и их фармацевтически приемлемые соли являются полезными в ингибировании ВИЧ обратной транскриптазы, профилактике и лечении инфекций, вызываемых ВИЧ, и в профилактике, препятствовании возникновению или развитию и в лечении СПИД. Соединения и их соли могут быть применены в качестве ингредиентов в фармацевтических композициях,необязательно в сочетании с другими антивирусами, иммуномодуляторами, антибиотиками или вакцинами. Область изобретения Настоящее изобретение направлено на некоторые соединения 3-(необязательно замещенный ароматический гидрокарбилокси)-1-[(5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил])пиридин-2(1 Н)-она и к их применению для профилактики ВИЧ-инфекции и ВИЧ репликации, лечения ВИЧ-инфекции и ВИЧ репликации, профилактики СПИД, лечения СПИД, и для препятствования возникновению и/или развитию СПИД. Предпосылки создания изобретения Ретровирус, называемый вирусом иммунодефицита человека (ВИЧ), особенно штаммы, известные как ВИЧ типа-1 (ВИЧ-1) и типа-2 (ВИЧ-2), этиологически связаны с иммуносуппрессивным заболеванием, известным как синдром приобретенного иммунодефицита (СПИД). ВИЧ серопозитивные индивидуумы изначально асимптоматичны, но обычно обнаруживают СПИД-ассоциированный комплекс (ARC) с последующим проявлением СПИД. Пораженные индивидуумы проявляют сильную иммуносуппрессию, которая делает их весьма восприимчивыми к изнуряющим и в конечном итоге смертельным оппортунистическим инфекциям. Размножение ВИЧ клеткой хозяина требует внедрения вирусного генома в ДНК клетки хозяина. Так как ВИЧ является ретровирусом, цикл размножения ВИЧ требует транскрипции вирусного генома РНК в ДНК через фермент, известный, как обратная транскриптаза (RT). Обратная транскриптаза имеет три известные ферментные функции: фермент действует как РНКзависимая полимераза ДНК, как рибонуклеаза и как ДНК-зависимая полимераза ДНК. В роли РНКзависимой полимеразы ДНК обратная транскриптаза транскрибирует однонитевую копию ДНК вирусной РНК. В качестве рибонуклеазы обратная транскриптаза разрушает первоначальную вирусную РНК и высвобождает ДНК, только что полученную из первоначальной РНК. И в качестве ДНК-зависимой полимеразы ДНК обратная транскриптаза делает вторую комплементарную нить ДНК с использованием первой нити ДНК в качестве матрицы. Две нити образуют двунитевую ДНК, которая внедряется в геном клетки хозяина с помощью фермента интегразы. Известно, что соединения, которые ингибируют ферментные функции обратной транскриптазы ВИЧ, будут ингибировать размножение ВИЧ в инфицированных клетках. Данные соединения полезны для профилактики или лечения ВИЧ-инфекции у человека. Среди соединений, одобренных для использования в лечении ВИЧ-инфекции и СПИД находятся ингибиторы обратной транскриптазы 3'-азидо-3'дезокситимидин (AZT), 2',3'-дидезоксиинозин (ddl), 2',3'-дидезоксицитидин (ddC), d4T, 3TC, невирапин,делавирдин, эфавиренз, абакавир, эмтрицитабин и тенофовир. Хотя каждое из предшествующих лекарств является эффективным для лечения ВИЧ-инфекции и СПИД, остается необходимость разработки дополнительных ВИЧ антивирусных лекарств, включающих дополнительные ингибиторы обратной транскриптазы. Особенную проблему представляет развитие мутантных штаммов ВИЧ, которые являются устойчивыми к известным ингибиторам. Использование ингибиторов обратной транскриптазы для лечения СПИД часто ведет к вирусам, которые менее чувствительны к ингибиторам. Данная резистентность является обычно результатом мутаций, которые появляются в сегменте обратной транскриптазы генного пула. Продолжительное использование антивирусных соединений для предотвращения ВИЧ-инфекции будет неминуемо приводить к появлению новых резистентных штаммов ВИЧ. Соответственно, существует особая потребность в новых ингибиторах обратной транскриптазы, которые эффективны против мутантных штаммов ВИЧ. Следующие ссылки представляют интерес в качестве уровня техники изобретения:Clemo et al., J. Chem. Soc. 1954, pp. 2693-2702 раскрывает некоторые производные 4-оксо-3-(2 пиридил)пиридоколиновой системы и, в частности, раскрывает 6-метил-6'-фенокси-2,2'-метилендипиридин.Sweeney et al., BioorganicMedicinal Chem. Letters 2008, vol. 18, pp. 4348-4351 раскрывает серии триазолинонов, которые, как было обнаружено, являются ненуклеозидными ингибиторами ВИЧ обратной транскриптазы.WO 2001/034578 раскрывает некоторые замещенные азолы (включая, например, некоторые имидазолы и бензимидазолы), обладающие анти-Helicobacter pylori воздействием. В частности, WO '578 описывает 1-[(3-метил-4-фенокси-2-пиридинил)метил]-1 Н-бензимидазол (см. соединение 91 на стр. 40).WO 2004/085406 и соответствующая US 7189718 раскрывают некоторые бензилпиридазиноны в качестве ингибиторов обратной транскриптазы.WO 2005/102989 и соответствующая US 7166738 раскрывают некоторые N-фенил 2 фенилацетамиды, как ненуклеозидные ингибиторы обратной транскриптазы.WO 2006/067587 раскрывают некоторые производные биарилового эфира, как модуляторы фермента обратной транскриптазы.WO 2007/045572 и WO 2007/045573 раскрывают некоторые 2-(2-феноксифенил) Nфенилацетамиды, как ненуклеозидные ингибиторы обратной транскриптазы.WO 2008/076225 раскрывает некоторые индазолы, бензотриазолы и схожие бициклические соединения в качестве ингибиторов ВИЧ обратной транскриптазы.WO 2009/067166 раскрывает некоторые арилокси-, циклоалкилокси- и гетероциклилоксипиридины и родственные соединения. Соединения являются ингибиторами ВИЧ обратной транскриптазы полезны-1 024804 ми, например, для лечения ВИЧ-инфекции. Среди раскрытых соединений находятся некоторые 3-(3,5 дизамещенный фенокси)-1-(1H-пиразоло[3,4-b]пиридин-3-илметил)-4-(замещенный)пиридин-2(1H)-оны.US 2004/0192704 раскрывает некоторые 3-(фенокси)бензилзамещенные 5-членные триазолоны, оксадиазолоны и тиадиазолоны. Соединения раскрываются как ненуклеозидные ингибиторы обратной транскриптазы, полезные для лечения или профилактики ВИЧ опосредованных заболеваний.US 2007/0021442 и WO 2007/015812 раскрывают некоторые замещенные ароматические соединения. Соединения являются ненуклеозидными ингибиторами обратной транскриптазы, полезными для лечения ВИЧ-инфекции. Краткое содержание изобретения Данное изобретение относится к некоторым 3-(необязательно замещенный фенокси)-1-[(5-оксо-4,5 дигидро-1 Н-1,2,4-триазол-3-ил)метил])пиридин-2(1H)-оновым соединениям и их использованию для ингибирования ВИЧ обратной транскриптазы, профилактики ВИЧ-инфекции, лечения ВИЧ-инфекции и профилактики, лечения и для препятствования возникновению или развитию СПИД и/или ARC. Более конкретно, данное изобретение включает соединения формулы I и их фармацевтически приемлемые соли:AryА представляет собой арил, который необязательно замещен в целом от 1 до 3 заместителями,выбранными из (2) C1-6 галогеналкила, (11) галогена, (12) CN;R2 и R3, каждый, представляют собой независимо (1) Н, (3) С 1-6 галогеналкил, (7) галоген;R5 представляет собой C1-6 алкил. Данное изобретение также включает фармацевтические композиции, содержащие соединение формулы I или его фармацевтически приемлемую соль. Соединения формулы I настоящего изобретения могут быть полезны в лечении СПИД, для препятствования возникновению или развитию СПИД, профилактики СПИД, профилактики ВИЧ-инфекции и лечения ВИЧ-инфекции. Другие воплощения, аспекты, классы и подклассы и особенности данного изобретения или далее описаны, или будут очевидными из данного описания, примеров и приложенных формул изобретения. Подробное описание изобретения Соединения формулы I, представленной выше, и их фармацевтически приемлемые соли являются ингибиторами ВИЧ обратной транскриптазы. Соединения являются полезными для ингибирования ВИЧ обратной транскриптазы и для ингибирования размножения ВИЧ in vivo и in vitro. Более конкретно, соединения формулы I ингибируют функцию полимеразы ВИЧ-1 обратной транскриптазы. Основываясь на тестировании представленных соединений изобретения в анализе, изложенном в примере 9 ниже, известно, что соединения формулы I ингибируют активность РНК-зависимой полимеразы ДНК ВИЧ-1 обратной транскриптазы. Представленные соединения данного изобретения (см. соединения примеров 1-8) также проявляют активность против устойчивых к лекарствам форм ВИЧ (например, мутантных штаммов ВИЧ-1, в которых обратная транскриптаза имеет мутацию на лизин 103 аспарагин (K103N) и/или тирозин 181 цистеин (Y181C), и таким образом, может проявлять пониженный перекрестный иммунитет против принятых в настоящее время методов антивирусной терапии. Первое воплощение данного изобретения (иначе называемое как "воплощение Е 1") представляет собой соединение формулы I (иначе или более просто называемое как "соединение I") , или его фармацевтически приемлемую соль, где AryА представляет собой фенил, причем фенил является необязательно замещенным от 1 до 3 заместителями, каждый из которых представляет собой независимо (2) С 1-4 галогеналкил, (11) галогена, (12) CN. Второе воплощение данного изобретения ("воплощение Е 2") представляет собой соединение формулы I или его фармацевтически приемлемую соль, где R2 представляет собой (2) С 1-4 галогеналкил,(5) галоген; R3 представляет собой (1) Н, (3) С 1-4 галогеналкил,(6) галоген. Первый класс соединений данного изобретения (иначе называемый как "класс С 1") включает соединения формулы II и их фармацевтически приемлемые соли, гдеX1 и X2, каждый, представляют собой независимо (3) С 1-4 галогеналкил, (9) галоген, (10) CN; R2 и R3,каждый, представляют собой независимо (1) Н, (3) С 1-4 галогеналкил, (14) галоген; R5 представляет собой С 1-4 алкил. Второй подкласс первого класса (подкласс C1-S2) включает соединения формулы II и их фармацевтически приемлемые соли, где X1 и X2, каждый, представляют собой независимо (2) С 1-4 фторалкил, (4) галоген, (5) CN, R2 представляет собой (2) С 1-4 фторалкил, (5) галоген; R3 представляет собой (1) Н, (3) С 1-4 галогеналкил, (6) галоген и R5 представляет собой C1-3 алкил. Второй класс соединений данного изобретения (класс С 2) включает соединения формулы III и их фармацевтически приемлемые соли, где показатели имеют такие значения, как определено в классе С 1. Первый подкласс второго класса (подкласс C2-S1) включает соединения формулы III и их фармацевтически приемлемые соли, где R5 представляет собой СН 3 или СН 2 СН 3. В еще одной перспективе данного подкласса R3 представляет собой Н, a R5 представляет собой СН 3 или СН 2 СН 3. Настоящее изобретение также относится соединению I, которое выбирают из следующих: и его фармацевтически приемлемых солей. Кроме того настоящее изобретение относится к соединению, имеющему следующую структурную формулу: или его фармацевтически приемлемая соль. А также к соединению, представленному формулой Соединение формулы I или его фармацевтически приемлемая соль, как определено первоначально или как определено в любом из предыдущих воплощений, аспектов, классов, подклассов и перспектив,причем соединение или его соль может находиться практически в чистой форме. Используемый термин"практически чистый" означает содержание в соединении или соли подходящим образом по меньшей мере около 60 вес.%, обычно по меньшей мере 70 вес.%, предпочтительно по меньшей мере около 80 вес.%, более предпочтительно по меньшей мере около 90 вес.% (например, от примерно 90 до примерно 99 вес.%), еще более предпочтительно по меньшей мере около 95 вес.% (например, от примерно 95 до примерно 99 вес.%, или от примерно 98 до 100 вес.%) и наиболее предпочтительно по меньшей мере около 99 вес.% (например, 100 вес.%) продукта, содержащего соединение формулы I или его соли (например, продукт выделяется из реакционной смеси, дающей соединение или соль). Уровень чистоты соединений и солей может быть определен с использованием стандартного метода анализа, такого как тонкослойная хроматография, гелевый электрофорез, жидкостная хроматография высокого разрешения и/или масс-спектрометрия. Если применяется более чем один метод анализа и методы дают экспериментально существенные различия уровней определенной чистоты, тогда руководствуются методом, дающим наивысший уровень чистоты. Соединение или соль 100% чистоты - это то, которое свободно от поддающихся обнаружению примесей, что определяется с помощью стандартного метода анализа. В отношении соединения изобретения, которое имеет один или более асимметричных центров и может встречаться в виде смесей стереоизомеров, практически чистым соединением может быть или практически чистая смесь стереоизомеров или практически чистый отдельный диастереомер или энантиомер. Соединения настоящего изобретения могут находиться в форме пролекарства. Термин "пролекарство" относится к производному соединения формулы I или его фармацевтически приемлемой соли, которое in vivo превращается в соединение I. Пролекарства соединений формулы I могут проявлять усиленную растворимость, абсорбцию и/или липофильность по сравнению с собственно соединениями, приводя таким образом к увеличению биодоступности и эффективности. Превращение продукта in vivo может быть результатом катализируемой ферментом химической реакции, метаболической химической реакции и/или спонтанной химической реакции (например, сольволиза). Когда соединение содержит, например,гидроксигруппу, продукт может быть производным гидроксигруппы, таким как сложный эфир (OC(O)R), карбонатный эфир (-ОС(О)OR), фосфатный эфир (-OP(=O)(OH)2), или простой эфир (-OR). Другие примеры включают следующие: когда соединение формулы I содержит карбоново-кислотную группу, продукт может быть сложным эфиром или амидом, а когда соединение формулы I содержит группу первичного амина или другой подходящий азот, который может быть производным, пролекарством может быть амид, карбамат, мочевина, имин или Mannich основание. Одна или более функциональных групп соединения I могут быть производными для получения их пролекарств. Обычные процедуры для отбора и получения подходящих производных пролекарств описываются, например, в Design of Prodrugs, под редакцией H.Bundgaard, Elsevier, 1985; J.J. Hale et al., J. Med. Chem. 2000, vol.43, pp.1234-1241;C.S. Larsen and J. Ostergaard, "Design and application of prodrugs" в Textbook of Drug Design and Discovery,3 е издание под редакцией C.S. Larsen, 2002, pp.410-458: и Beaumont et al., Current Drug Metabolism 2003,vol. 4, pp. 461-458; описания каждой из которых включены путем ссылки на них. Другие воплощения данного изобретения включают:(a) Фармацевтическую композицию, пригодную для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновения СПИД у субъекта, нуждающегося в этом, включающую эффективное количество соединения формулы I, как определено выше, или фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.(b) Фармацевтическую композицию, пригодную для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновения СПИД у субъекта, нуждающегося в этом, которая включает соединение, представленное формулой или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.(с) Фармацевтическую композицию, пригодную для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновения СПИД у субъекта, нуждающегося в этом, которая включает соединение, представленное формулой и фармацевтически приемлемый носитель. Данное изобретение также включает применение соединения формулы I или его фармацевтически приемлемой соли при производстве лекарственного средства для лечения или профилактики инфекции,вызываемой ВИЧ, или лечения, профилактики или препятствования возникновению или развитию СПИД. В этих использованиях соединения данного изобретения могут применяться необязательно в сочетании с одним или более агентами против ВИЧ, выбранными из ВИЧ антивирусных агентов, противо-4 024804 инфекционных агентов и иммуномодуляторов. Кроме того настоящее изобретение относится к применению соединения, представленного формулой или его фармацевтически приемлемой солью в профилактике или лечении инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом. Также изобретение относится к применению соединения, представленного формулой в приготовлении лекарственных средств для профилактики или лечения инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом. В фармацевтической композиции соединение формулы I может применяться в количестве, эффективном против ВИЧ-1, и агентом против ВИЧ является ВИЧ-1 антивирус, выбранный из группы, состоящей из ингибиторов ВИЧ-1 протеазы, ингибиторов ВИЧ-1 обратной транскриптазы, ингибиторов ВИЧ-1 интегразы, ингибиторов ВИЧ-1 слияния и ингибиторов ВИЧ-1 внедрения. Используемый в описании термин "алкил" относится к одновалентной прямой или разветвленной цепи насыщенного алифатического углеводородного радикала, имеющего число атомов углерода в определенном интервале. Таким образом, например, "C1-6 алкил" (или "С 1-С 6 алкил") относится к любому из гексилалкильных или пентилалкильных изомеров, также как и н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу. В качестве другого примера, "С 1-4 алкил" относится к н-, изо-, втор- и трет-бутилу,н- и изопропилу, этилу и метилу. Термин "галоген" относится к фтору, хлору, брому и йоду (иначе называемым как фтор, хлор, бром и йод). Термин "галогеналкил" относится к алкильной группе, определенной выше, в которой один или более водородных атомов заменены галогеном (т.е. F, Cl, Br или I). Таким образом, например, "С 1-6 галогеналкил" (или "С 1-С 6 галогеналкил") относится к C1-C6 линейной или разветвленной алкильной группе, определенной выше, с одним или более галогеновыми заместителями. Термин "фторалкил" имеет аналогичное значение, за исключением того, что галогеновые заместители ограничены фтором. Подходящие фторалкилы включают серии (CH2)0-4CF3 (т.е. трифторметил, 2,2,2-трифторметил, 3,3,3 трифтор-н-пропил и т.д.). Представляющим особенный интерес фторалкилом является CF3. Термин "арил" относится к (i) фенилу, (ii) 9- или 10-членной бициклической, сконденсированной карбоциклической кольцевой системе, в которой по меньшей мере одно кольцо является ароматическим,и (iii) 11- или 14-членной трициклической, сконденсированной карбоциклической кольцевой системе, в которой по меньшей мере одно кольцо является ароматическим. Подходящие арилы включают, например, фенил, нафтил, тетрагидронафтил (тетралинил), инденил, антраценил и флуоренил. Классом арилов,представляющих интерес в изобретении, является фенил и нафтил. Арилом, представляющим особенный интерес, является фенил. Как должно быть признано специалистом в данной области, некоторые соединения данного изобретения могут существовать в виде таутомеров. Все таутомерные формы этих соединений, либо выделенные отдельно, либо в смеси, охватываются объемом данного изобретения. Например, в случае, когда оксо (=0) заместитель допускается на гетероароматическом кольце и кето-енольный таутомеризм является возможным, понятно, что заместитель фактически может присутствовать целиком или частично в виде гидроксиформы, как показано на примере:"Стабильным" соединением является соединение, которое может быть получено и выделено и чья структура и свойства остаются или могут остаться, по существу, неизменными в течение периода времени, достаточного для возможного использования соединения для целей, описанных в настоящем описании (например, терапевтического или профилактического назначения субъекту). Соединения данного изобретения ограничиваются до стабильных соединений, охватываемых формулой I. Как результат отбора заместителей и примеров заместителей некоторые соединения данного изобретения могут иметь асимметричные центры и могут существовать в виде смесей стереоизомеров, или в виде индивидуальных диастереомеров, или энантиомеров. Все изомерные формы этих соединений, будучи или индивидуальными или в смесях, охватываются объемом данного изобретения. Атомы в соединении формулы I могут обнаруживать обилие их естественных изотопов, или один или более атомов могут быть искусственно обогащены в особенности изотопом, имеющим то же атомное число, но атомная масса или массовое число отличается от атомной массы или массового числа атома,преимущественно встречающегося в природе. Подразумевается, что данное изобретение включает все подходящие изотопные варианты соединений общей формулы I. Например, различные изотопные формы водорода (Н) включают протий (1 Н) и дейтерий (2 Н). Протий представляет собой преимущественный изотоп водорода, встречающийся в природе. Обогащение до дейтерия может приводить к некоторым терапевтическим преимуществам, таким как увеличение in vivo периода полураспада, или понижение требований к дозировке, или может дать соединение, полезное в качестве стандарта для характеристики биологических образцов. Изотопно-обогащенные соединения общей формулы I могут быть получены без излишнего экспериментирования путем общепринятой технологии, хорошо известной специалистам в данной области или с помощью процессов, аналогичных тем, что описаны в схемах и примерах настоящего описания с использованием соответствующих изотопно-обогащенных реагентов и/или промежуточных соединений. Соединения данного изобретения могут использоваться в ингибировании ВИЧ обратной транскриптазы (например, ВИЧ-1 дикого типа и/или его мутантные штаммы), профилактике или лечении инфекций, вызываемых вирусом иммунодефицита человека (ВИЧ) и для профилактики, лечения или препятствования возникновению или развитию последствий патологических состояний, таких как СПИД. Предотвращение СПИД, лечение СПИД, препятствование возникновению или развитию СПИД, или лечение или профилактика инфекций, вызываемых ВИЧ, определяются как включающие, но не ограниченные ими, лечение широкого ряда форм ВИЧ-инфекции: AIDS (СПИД), ARS (относящийся к СПИД комплекс), как симптоматические, так и бессимптомные, острые или возможные состояния от воздействия ВИЧ. Например, данное изобретение может быть применено для лечения инфекций, вызываемых ВИЧ после подвержения воздействию ВИЧ таким образом, как переливание крови, замена биологической жидкости, порезы (раны), случайный укол использованной иглой, или заражение через кровь пациента во время хирургической операции. Еще, например, данное изобретение может быть также применено для предотвращения переноса ВИЧ от беременной женщины ее еще не родившемуся ребенку или от ВИЧинфицированной женщины, которая кормит (при грудном кормлении) ребенка, самому ребенку с помощью назначения эффективного количества соединения I или его пролекарства или фармацевтически приемлемой соли. Соединения могут быть назначены в форме фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к соли, которая обладает эффективностью исходного соединения и которая не является биологически или иным образом нежелательной (например, является ни токсичной, ни иным образом вредной для ее реципиента). Подходящие соли включают кислотноаддитивные соли, которые могут, например, быть образованы путем смешивания раствора соединения данного изобретения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота,серная кислота, уксусная кислота или бензойная кислота. Когда соединения, применяемые в данном изобретении, несут кислотный фрагмент (например, -СООН или фенольную группу), их фармацевтически приемлемые соли могут включать соли щелочных металлов (например, соли натрия или калия), соли щелочноземельных металлов (например, соли кальция или магния) , и соли, образованные с подходящими органическими лигандами,такие как четвертично-аммониевые соли. Также, в случае присутствия кислотной (-СООН) или спиртовой группы, могут применяться фармацевтически приемлемые сложные эфиры для изменения характеристик растворимости или гидролиза соединения. Термин "введение" и его варианты (например, "введение" соединения) в отношении к соединению формулы I означает предоставление соединения индивидууму, нуждающемуся в лечении или профилактике. Соединение может предоставляться в сочетании с одним или более другими активными агентами(например, антивирусными агентами, полезными для лечения или профилактики ВИЧ-инфекций или СПИД), тогда "введение" и его варианты предполагают, что каждый включает предоставление соединения и других агентов в одно и то же время или в разное время. Когда агенты сочетания вводятся в одно и то же время, они могут вводиться вместе в одной композиции или они могут вводиться по отдельности. Под используемым в заявке термином "композиция" имеется в виду, что охватывается продукт,включающий определенные ингредиенты, так же как и любой продукт, который является результатом,прямо или косвенно, от объединения определенных ингредиентов. Ингредиенты, подходящие для включения в фармацевтическую композицию, являются фармацевтически приемлемыми ингредиентами, что означает, что ингредиенты должны быть совместимы друг с другом и не вредоносными для их реципиента. Термин "субъект", используемый в настоящем описании, относится к животным, предпочтительно млекопитающим, более предпочтительно к человеку, который являлся объектом лечения, наблюдения или эксперимента. Термин "эффективное количество", используемый в настоящем описании, означает количество активного соединения или фармацевтического агента, которое вызывает биологическую или лечебную реакцию в тканях, системах животного или человека, которое определяется исследователем, ветеринарным врачом, лечащим врачом или другим врачом-консультантом. В одном воплощении эффективным количеством является "терапевтически эффективное количество" для смягчения симптомов заболевания или состояния, подвергаемого лечению. В другом воплощении эффективным количеством является "профилактически эффективное количество" для профилактики симптомов заболевания или состояния для их недопущения. Термин также включает количество активного соединения, подходящее для ингибирования ВИЧ обратной транскриптазы (дикого типа и/или его мутантных штаммов) и тем самым вызывает желаемую ответную реакцию (т.е. "эффективное для ингибирования количество"). Когда активное соединение (т.е. активный ингредиент) назначают в виде соли, ссылки на количество активного ингредиента относятся к свободной форме (т.е. несолевой форме) соединения. Соединения формулы I необязательно в форме соли могут быть введены любым способом, который создает контакт активного агента с местом приложения действия агента. Они могут вводиться с помощью любого общепринятого способа, доступного для применения в отношении лекарственных средств,как отдельных терапевтических агентов, так и в сочетании терапевтических агентов. Их могут вводить одиночно, но обычно их вводят с фармацевтическим носителем, на основании выбора способа введения и стандартной фармацевтической практики. Соединение изобретения, например, может быть введено перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, интрастернальную (внутригрудинную) инъекции или технику вливания), путем спрейной ингаляции, или ректально, в форме единичной дозы фармацевтической композиции, содержащей эффективное количество соединения и общепринятого нетоксичного фармацевтически приемлемого носителя, адъюванта и средств доставки. Жидкие препараты, подходящие для перорального введения (например, суспензии,сиропы, эликсиры и аналогичные), могут быть приготовлены согласно технологии, известной в данной области, и могут использоваться любые обычные среды, такие как вода, гликоль, масла, спирты и аналогичные. Твердые препараты, подходящие для перорального введения (например, порошки, пилюли, капсулы и таблетки), могут быть приготовлены согласно технологии, известной в данной области, и могут использоваться твердые инертные наполнители, такие как крахмал, сахар, каолин, смазывающие агенты,связующие агенты, измельчающие агенты и аналогичные. Парентеральные композиции могут быть приготовлены согласно технологии, известной в данной области и обычно используются стерилизованная вода в качестве носителя и, необязательно, другие ингредиенты, такие как способствующие растворению средства. Инъецируемые растворы могут быть приготовлены согласно методам, известным в данной области, причем носитель включает физиологический раствор, раствор глюкозы или раствор, содержащий смесь физиологического раствора и глюкозы. Дальнейшее описание способов, подходящих для использования в приготовлении фармацевтических композиций, для применения в данном изобретении и ингредиентов, подходящих для использования в указанных композициях, представлено в Remington's Pharmaceutical Sciences, 18 е издание под редакцией A.R.Gennaro, Mack Publishing Co., 1990 и в Remington TheScience and Practice of Pharmacy, 21 е издание, Lippincott WilliamsWilkins, 2005. Соединения формулы I могут быть введены перорально в диапазоне доз 0,001-1000 мг/кг веса тела млекопитающего (например, человека) в день в единичной дозе или раздельными дозами. Одним предпочтительным диапазоном доз является 0,01-500 мг/кг веса в день перорально в единичной дозе или раздельными дозами. Другим предпочтительным диапазоном доз является 0,1-100 мг/кг веса в день перорально в единичной дозе или раздельными дозами. Для перорального введения композиции могут быть приготовлены в форме таблеток или капсул, содержащих 1,0-500 мг активного ингредиента, в частности 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 мг активного ингредиента для симптоматического регулирования дозы пациенту, подвергаемому лечению. Особый уровень дозы и частота дозировки для любого частного пациента может быть изменена и будет зависеть от множества факторов, включающих интенсивность действия применяемых конкретных соединений, метаболическую стабильность и продолжительность воздействия данного соединения, возраст, вес, общее состояние здоровья, пол пациента, диету и время введения, скорость выведения из организма, сочетание лекарственных препаратов,тяжесть конкретного состояния и проходящего курса терапии. Как отмечено выше, Соединения формулы I могут вводиться с одним или более агентами против ВИЧ. "Агентом против ВИЧ" является любой агент, который прямо или косвенно является эффективным в ингибировании ВИЧ обратной транскриптазы или другого фермента, необходимого для размножения ВИЧ или инфицирования, в лечении или профилактике ВИЧ-инфекции, и/или лечении, профилактике или препятствовании возникновению или развитию СПИД. Понятно, что агент против ВИЧ является эффективным при лечении, предотвращении или препятствовании возникновению или развитию ВИЧинфекции или СПИД и/или заболеваний или состояний, возникающих как следствие или ассоциируемых с ними. Например, соединения данного изобретения могут эффективно вводиться, будь то в период прединфекционный и/или постконтактный, в сочетании с эффективными количествами одного или более агентов против ВИЧ, выбранных из ВИЧ антивирусных агентов, иммуномодуляторов, противоинфекционных агентов или вакцин, полезных для лечения ВИЧ-инфекции или СПИД. ВИЧ антивирусы, подходящие для использования в сочетании с соединениями данного изобретения, включают, например, антивирусы, которые перечислены в таблице А далее. Таблица АEI = ингибитор проникновения в клетку; FI = ингибитор слияния; InI = ингибитор интегразы; PI = ингибитор протеазы; nRTI = нуклеозидный ингибитор обратной транскриптазы; nnRTI = ненуклеозид-8 024804 ный ингибитор обратной транскриптазы. Некоторые из лекарственных средств, перечисленных в таблице, используются в форме соли, например абакавир сульфат, делавирдин мезилат, индинавир сульфат,атазанавир сульфат, нелфинавир мезилат, саквинавир мезилат. Понятно, что диапазон комбинаций соединений данного изобретения с анти-ВИЧ агентами на ограничивается ВИЧ антивирусными агентами, перечисленными в таблице А, но в принципе включает любую комбинацию с любой фармацевтической композицией, полезной для лечения или профилактики СПИД. ВИЧ антивирусные агенты и другие агенты будут, как правило, применяться в этих комбинациях в общепринятых диапазонах доз и схемах приема лекарств, как описано в данной области, включая, например, дозировки, описанные в изданиях Physicians' Desk Reference, таком, как 63 е издание (2009) и более ранние издания. Диапазоны доз для соединений изобретения в этих комбинациях могут быть теми же, как вышеупомянутые. Соединения данного изобретения являются также полезными при подготовке и проведении скрининговых исследований для антивирусных соединений. Например, соединения данного изобретения также полезны для выделения мутантных ферментов, которые являются превосходным скрининговым инструментом для более сильных антивирусных соединений. Кроме того, соединения данного изобретения являются также полезными при установке или разрушении центра связывания других антивирусов с ВИЧ обратной транскриптазой, например, путем конкурентного ингибирования. Таким образом, соединения данного изобретения являются коммерческим продуктом для продажи в этих целях. Применяемые сокращения включают следующие: АсОН = уксусная кислота;LCAP = жидкостная хроматография относительной площади;PBS = забуференный фосфатом солевой раствор;TFAA = трифторуксусный ангидрид. Соединения данного изобретения могут быть легко приготовлены в соответствии со следующими реакционными схемами и примерами или их модификациями с использованием легко доступных исходных материалов, реагентов и общепринятых методов синтеза. В этих реакциях также возможно осуществлять использование вариантов, которые сами по себе известны в данной области техники, но не упоминаются подробнее. Кроме того, другие методы получения соединений изобретения будут, очевидно, выражены для специалиста в данной области техники в свете следующих схем реакций и примеров. Если не указано иное, все переменные величины имеют значения, определенные выше. Схема I изображает метод получения соединений формулы I, в которой гидроксипиридин 1-1 подвергается алкилированию хлортриазолиноном 1-2 для получения соединения 1-3, которое может быть селективно алкилировано алкилгалогенидом (например, метилйодидом, этилйодидом, и т.д.) для получения желаемого соединения 1-4. Схема II изображает альтернативный путь к соединениям данного изобретения, в котором фторгидроксипиридин II-1 может быть алкилирован хлортриазолиноном II-2 для получения алкилированного продукта II-3, который может быть превращен в желаемое соединение II-5 посредством нуклеофильного ароматического замещения (SNAr) с использованием подходящего гидроксиарена II-4. Схема II Гидроксипиридины формулы 1-1 (схема I) могут быть получены в соответствии со схемой III, в которой реакция SNAr между пиридином III-1 (таким, как коммерчески доступный 2-хлор-3-фтор-4(трифторметил)пиридин) и гидроксиареном II-4 может дать хлорпиридин III-2, который может быть подвержен гидролизу в основных условиях до гидроксипиридина 1-1. Схема III Другой способ получения гидроксипиридинов формулы 1-1 поясняется на схеме IV, где SNAr сочетание коммерчески доступного 2-хлор-3-фтор-4-нитропиридон-N-оксида IV-1 с подходящим гидроксиареном II-4 дает N-оксид IV-2, который вначале может быть превращен в дигалогениды IV-3, а затем гидролизоваться до гидроксипиридина IV-4. Дальнейшее получение производных гидроксипиридина IV-4 возможно с помощью процессов сочетания переходов, катализируемых металлом, таких как сочетанияStille или бороновой кислоты с использованием PdLn катализатора (где L представляет собой лиганд, такой как трифенилфосфин, три-трет-бутилфосфин или ксантфос) для образования гидроксипиридинов IV5, или химии аминирования для образования гидроксипиридинов IV-6, где R2 представляет собой Схема V изображает введение заместителя в пятом положении гидроксипиридинов с помощью ре- 10024804 акции бромирования и последующих катализируемых металлом преобразований химических составов,таких как сочетания Stille или бороновой кислоты с использованием PdLn, в котором L имеет значение,определенное на схеме IV, с образованием гидроксипиридинов V-3, или химии аминирования для образования гидроксипиридинов V-4, где R3 представляет собой N(RA)RB. Схема V Как показано на схеме IV, фторгидроксипиридины II-1 (схема II) достижимы из коммерчески доступных 3-фторпиридинов VI-1 с помощью образования N-оксида и перегруппировки, как описано у Следующие примеры служат только для иллюстрации изобретения и его методики. Примеры не должны расцениваться как ограничивающие объем или сущность изобретения. Термин "комнатная температура" в примерах относится к температуре окружающей среды, которая обычно была в пределах от примерно 20 до примерно 26 С. Пример 1. 3-Хлор-5-(1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо-4(трифторметил)-1,2-дигидропиридин-3-илокси)бензонитрил (1-1) Смесь 3-бром-5-хлорфенола (3,74 г; 18,0 ммоль), 2-хлор-3-фтор-4-(трифторметил)пиридина (3,00 г; 15,0 ммоль) и К 2 СО 3 (2,49 г; 18,0 ммоль) в NMP (15 мл) нагревали до 120 С в течение одного часа, затем охлаждали до комнатной температуры. Затем смесь разбавляли 250 мл EtOAc и промывали 3250 мл 1:1 Н 2 О:солевой раствор. Органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (120 г колонка, загруженная толуолом, 100:0 - 0:100 гексаны:CH2Cl2 на протяжении 40 мин) давала указанное в заголовке (целевое) соединение (1-2) в виде белого твердого вещества. Повторная очистка смешанных фракций дополнительно давала целевое соединение. 1 Н ЯМР (400 МГц, CDCl3):8,55 (д, J=5,0 Гц, 1 Н); 7,64 (д, J=5,0 Гц, 1 Н); 7,30 (с, 1 Н); 6,88 (с, 1 Н); 6,77 (с, 1 Н). Стадия 1(b): 3-(3-бром-5-хлорфенокси)-4-(трифторметил)пиридин-2-ол (1-3) К суспензии 3-(3-бром-5-хлорфенокси)-2-хлор-4-(трифторметил)пиридина (1-2; 3,48 г; 8,99 ммоль) в tBuOH (36 мл) добавляли КОН (1,51 г; 27,0 ммоль) и смесь нагревали до 75 С в течение ночи, в резуль- 11024804 тате из раствора осаждалось желтое маслянистое твердое вещество, и анализ LCMS показывал завершение превращения. Смесь охлаждали до комнатной температуры и нейтрализовали путем добавления 50 мл насыщенного водного раствора NH4Cl. Смесь разбавляли 50 мл Н 2 О, затем экстрагировали 2100 млEtOAc. Объединенные органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (120 г колонка, сухая загрузка, 100:0 -90:10 СН 2Cl2/MeOH на протяжении 40 мин) давала целевое соединение (1-3) в виде рыхлого белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО):12,69 (с, 1 Н), 7,59 (д, J=6,9 Гц, 1 Н); 7,43 (т, J=1,7 Гц, 1 Н); 7,20 (т, J=1,9 Гц, 1 Н); 7,13 (т, J=2,0 Гц, 1 Н); 6,48 (д, J=6,9 Гц, 1 Н). Стадия 1 (с): 3-хлор-5-[2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4)NMP (29 мл) добавляли CuCN (7,90 г; 88 ммоль) и смесь нагревали до 17 5 С в течение 5 ч, затем медленно охлаждали до комнатной температуры. С усиленной вентиляцией в вытяжном шкафу добавляли 100 мл ледяного АсОН, затем 100 мл EtOAc и смесь фильтровали через Целит (орошение EtOAc). Фильтрат промывали 3200 мл 1:1 Н 2 О:солевой раствор, затем органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (120 г колонка, сухая загрузка, 100:0 90:10 CH2Cl2:МеОН на протяжении 40 мин), затем растирание полученного твердого вещества с Et2O(для удаления остаточного NMP, который был элюирован совместно с продуктом) давали целевое соединение (1-4). 1 Н ЯМР (400 МГц, ДМСО):12,71 (с, 1 Н); 7,75 (с, 1 Н); 7,63-7,57 (м, 2 Н); 7,54 (с, 1 Н); 6,49 (д, J=6,9 Гц, 1 Н). Стадия 1(d): 5-(хлорметил)-2,4-дигидро-3 Н-1,2,4-триазол-3-он (1-5) Суспензию 3-хлор-5-[(2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрила (1-4; 2,00 г; 6,36 ммоль), 5-(хлорметил)-2,4-дигидро-3 Н-1,2,4-триазол-3-она (1-5; 0,849 г; 6,36 ммоль) и K2CO3 (0,878 г; 6,36 ммоль) в ДМФА (32 мл) перемешивали в течение 2 часов при комнатной температуре, в результате чего анализ LCMS показывал завершение превращения. Смесь разбавляли 200 мл Me-THF и промывали 150 мл смеси 1:1:1 H2O:солевой раствор:насыщенный водный раствор NH4Cl, и затем промывали 2150 мл 1:1 H2O:солевой раствор. Водные фракции далее экстрагировали 150 мл Me-THF, затем объединенные органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Очистка с помощьюISCO CombiFlash (80 г колонка, сухая загрузка, 100:0 - 90:10 EtOAc:EtOH на протяжении 25 мин) давала целевое соединение (1-6) в виде белого твердого вещества. 1l(f): 3-хлор-5-(1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо-4(трифторметил)-1,2-дигидропиридин-3-илокси)бензонитрил (1-1) Раствор 3-хлор-5-(2-оксо-1-[(5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-4-(трифторметил)1,2-дигидропиридин-3-илокси)бензонитрила (1-6; 2,37 г; 5,76 ммоль) и K2CO3 (0,796 г; 5,76 ммоль) в ДМФА (58 мл) охлаждали до 0 С, затем добавляли метилйодид (0,360 мл; 5,76 ммоль). Смеси давали нагреться до комнатной температуры и перемешивали в течение 90 мин, в результате чего анализ LCMS показывал 95% превращения и желаемый продукт 75% LCAP чистоты с остатком не прореагировавшего исходного материала и продуктами бис-метилирования. Смесь разбавляли 200 мл Me-THF и промывали 3200 мл смеси 1:1 Н 2 О:солевой раствор. Водные фракции далее экстрагировали 200 мл Me- 12024804THF, затем объединенные органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Полученное белое твердое вещество сначала растирали со 100 мл EtOAc, затем с 50 мл THF, что давало (после высушивания) целевое соединение (1-1)95% LCAP. Очистка до 99% LCAP возможна при использовании препаративной LCMS (Max-RP, 10030 мм колонка; 30-60% CH3CN в 0,6% водного НСООН на протяжении 8,3 мин; 25 мл/мин). 1 Смесь 3-хлор-1-йодфенола (208 г; 816,0 ммоль), 2-хлор-3-фтор-4-(трифторметил)пиридина (155 г; 777,0 ммоль) и K2CO3 (161 г; 1165,0 ммоль) в NMP (1,5 л) выдерживали при 60 С в течение 2,5 ч, а затем оставляли при комнатной температуре на 2 дня. Далее смесь снова нагревали до 60 С в течение 3 ч, затем охлаждали до комнатной температуры. Далее смесь разбавляли 4 л EtOAc и промывали 2 л воды + 1 л солевого раствора. Объединенные органические вещества промывали 2500 мл полуконцентрированного солевого раствора, затем 500 мл солевого раствора, сушили над MgSO4 и концентрировали, получая сырой неочищенный продукт 1 А-2. 1 Н ЯМР (500 МГц, ДМСО):8,67 (д, J=5,0 Гц, 1 Н), 7,98 (д, J=5,0 Гц, 1 Н), 7,63-7,62 (м, 1 Н), 7,427,40 (м, 1 Н), 7,22 (т, J=2,1 Гц, 1 Н). Стадия 1 А(b): 2-хлор-3-(3-хлор-5-йодфенокси)-4-(трифторметил)пиридин (1 А-3) К суспензии 3-(3-хлор-5-йодфенокси)-2-хлор-4-(трифторметил) пиридина (1 А-2; 421 г; 97 0 ммоль) в t-BuOH (1 л) добавляли КОН (272 г, 4850 ммоль) и смесь нагревали до 75 С в течение 1 ч, в результате чего анализ HPLC показывал 95% превращения. t-BuOH выпаривали, и смесь разбавляли водой (7 мл/г,2,4 л) и затем охлаждали до 0 С, после чего добавляли 12 н HCl (240 мл) до тех пор, пока значение рН не достигало 5. Данную смесь экстрагировали EtOAc (20 мл/г, 6,5 л), снова экстрагировали EtOAc 15 мл/г(1,5 л), промывали 1 водой:солевым раствором 1:1 (10 мл/г, 3,2 л), 1 солевым раствором (10 мл/г, 3,2 л), сушили над MgSO4, фильтровали и концентрировали, получая сырой неочищенный продукт. Сырой продукт суспендировали в МТВЕ (2,25 л, 7 мл/г), после чего к суспензии на протяжении 10 мин добавляли гексаны (1 л, 3 мл/г) и смесь выдерживали 30 мин при комнатной температуре. Продукт фильтровали на устройстве Buchner, орошали смесью МТВЕ/гексаны 1:2 (2 мл/г = 640 мл), затем гексанами (640 мл) и сушили на фритте (порошковая керамика) , получая соединение 1 А-3. 1CuCN (73,7 г; 823 ммоль), а затем смесь дегазировали дополнительно 5 мин. Смесь нагревали до 120 С в течение 17 ч, затем охлаждали до комнатной температуры и распределяли между 6 л MeTHF и 2 л аммо- 13024804 нийного буфера (4:3:1 = насыщенный раствор NH4Cl/вода/NH4 ОН 30%). Органический слой промывали 2 л буфера, затем 1 л буфера и 1 л солевого раствора, сушили над MgSO4 и концентрировали. Неочищенное твердое вещество перемешивали в 2,2 л MeCN при нагревании с обратным холодильником в течение 45 мин, затем охлаждали в водяной ванне до комнатной температуры на протяжении 1 ч, выдерживали 30 мин, затем фильтровали и орошали холодным MeCN (2400 мл). Твердое вещество сушили на фритте в атмосфере N2 в течение 60 ч, получая целевое соединение 1-4. 1 Н ЯМР (400 МГц, ДМСО):12,71 (с, 1 Н); 7,75 (с, 1 Н); 7,63-7,57 (м, 2 Н); 7,54 (с, 1 Н); 6,49 (д, J=6,9 Гц, 1 Н). Стадия 1A(d) и 1 А(е) Указанное в заголовке соединение 1-1 получали затем из соединения 1-4 с использованием процедур, аналогичных описанным в стадиях 1(d) и 1 (е), представленных выше в примере 1. Пример 2. 3-Хлор-5-(1-[(4-этил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо-4-(трифторметил)-1,2-дигидропиридин-3-илокси)бензонитрил (2-1) Указанное в заголовке соединение получали с использованием процедуры, описанной в примере,причем йодометан, применяемый в стадии l(f), заменяли йодоэтаном. 1 Н ЯМР (400 МГц, ДМСО):11,68 (с, 1 Н); 7,92 (д, J=7,3 Гц, 1 Н); 7,76 (с, 1 Н); 7,60 (с, 1 Н); 7,52 (с,1H); 6,69 (д, J=7,3 Гц, 1 Н); 5,20 (с, 2 Н); 3,65-3,56 (м, 2 Н); 1,11 (т, J=7,1 Гц, 3 Н). Пример 3. 3-Хлор-5-(4-хлор-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо 1,2-дигидропиридин-3-илокси)бензонитрил (3-1)(1,11 г; 5,78 ммоль) и K2CO3 (0,798 г; 5,78 ммоль) в 3:1 ТТФ:ДМФА (23 мл) перемешивали при комнатной температуре в течение 5 ч. ТГФ удаляли в вакууме, затем смесь разбавляли насыщенным водным раствором NaHCO3 и экстрагировали три раза этилацетатом. Объединенные органические экстракты промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме. Полученный остаток растирали с Et2O, получая целевое соединение в виде коричневого твердого вещества с чистотой, подходящей для непосредственного использования. 1 Н ЯМР (400 МГц, MeOD):8,57 (д, J=7,5 Гц, 1 Н); 8,21 (д, J=7,5 Гц, 1 Н); 7,43 (с, 1 Н); 7,26 (с, 1 Н); 7,16 (с, 1 Н). Стадия 3(b): 3-(3-бром-5-хлорфенокси)-2,4-дихлорпиридин (3-3) Суспензию 3-(3-бром-5-хлорфенокси)-2-хлор-4-нитропиридин 1-оксида (3-2; 1,41 г; 3,71 ммоль) в АсОН (37 мл) нагревали до 60 С и затем добавляли ацетилхлорид (2,64 мл; 37,1 ммоль). Через 60 мин при 60 С смесь охлаждали до комнатной температуры и АсОН удаляли в вакууме. Остаток разбавляли насыщенным водным раствором NaHCO3 и экстрагировали три раза этилацетатом. Объединенные органические экстракты промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме. Полученное оранжевое масло разбавляли CHCl3 (37 мл), охлаждали до 0 С, затем добавляли PCl3 (4,87 мл; 55,7 ммоль). Смесь нагревали до 60 С в течение ночи, затем охлаждали до комнатной температуры и гасили путем осторожного добавления насыщенного водного раствора NaHCO3 до тех пор, пока водный слой не становился щелочным. Смесь три раза экстрагировали CH2Cl2, затем объединенные экстракты промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме, получая целевое соединение в виде белого твердого вещества с чистотой, подходящей для непосредственного использования. 1 К раствору 3-(3-бром-5-хлорфенокси)-2,4-дихлорпиридина (3-3; 81 мг; 0,2 3 ммоль) в t-BuOH (1,0 мл) добавляли KOH (39 мг; 0,69 ммоль) и смесь нагревали до 75 С в течение ночи. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали EtOAc. Органическую фазу промывали солевым раствором, сушили (MgSO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (4 г колонка, сухая загрузка, 100:0 - 20:80 гексаны:EtOAc) давала соединение, указанное в заголовке. 1(106 мг; 0,092 ммоль). Смесь нагревали до 90 С в течение 2 ч, затем охлаждали до комнатной температуры. Смесь промывали EtOAc и водой, приводя к осаждению желаемого соединения, которое собирали с помощью фильтрования. Дальнейшее промывание водой, EtOAc, CH2Cl2 и МеОН давало целевое соединение в виде белого твердого вещества. 1 Н ЯМР (400 МГц, MeOD):7,52 (с, 1 Н); 7,43 (д, J=7,1 Гц, 1 Н); 7,29 (с, 1 Н); 7,27 (с, 1 Н); 6,58 (д,J=7,1 Гц, 1 Н). Стадии 3(е) и (f): 3-хлор-5-(4-хлор-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2 оксо-1, 2-дигидропиридин-3-илокси)бензонитрил (3-1) Указанное в заголовке соединение получали с использованием модификаций стадий 1(е) и (f), заменяя 3-хлор-5-[2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4) в стадии 1 (е) на 3 хлор-5-[(4-хлор-2-гидроксипиридин-3-ил)окси]бензонитрил (3-5). 1(6,39 г; 25,2 ммоль),диметоксо-бис-(1,5-циклооктадиен)дииридий(I) (0,208 г; 0,315 ммоль) и 4,4-ди-трет-бутил-2,2-дипиридил (0,169 г; 0,62 9 ммоль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Гексаны удаляли в вакууме, затем смесь повторно растворяли в ацетоне (70 мл) и добавляли Oxone (т.е. пероксимоносульфат калия) (12,9 г; 21,0 ммоль) в воде (70 мл) . После перемешивания в течение 10 мин реакцию гасили путем добавления 5% водного раствора Na2SO3 и продукт экстрагировали три раза Et2O. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (100:0 - 70:30 гексаны:EtOAc) давала целевое соединение в виде коричневого масла.(3,67; 19,1 ммоль) и K2CO3 (2,72 г; 19,7 ммоль) в ТГФ (79 мл) перемешивали при комнатной температуре в течение ночи. Смесь концентрировали в вакууме, затем разбавляли насыщенным водным растворомNaHCO3 и экстрагировали CH2Cl2. Органические экстракты сушили (MgSO4) и концентрировали в вакууме. Полученный остаток растирали с Et2O, получая целевое соединение в виде желтого твердого вещества с чистотой, подходящей для непосредственного использования. 1 Н ЯМР (400 МГц, CDCl3):8,40 (д, J=7,4 Гц, 1 Н); 8,04 (д, J=7,4 Гц, 1 Н); 7,54 (с, 1 Н); 7,12 (с, 1 Н); 6,88 (т, J=2,0 Гц, 1 Н). Стадия 4(с): 2,4-дибром-3-(3-хлор-5-йодфенокси)пиридин (4-4) Суспензию 2-хлор-3-(3-хлор-5-йодфенокси)-4-нитропиридин 1-оксида (4-3; 3,00 г; 7,03 ммоль) в АсОН (23 мл) нагревали до 80 С, затем добавляли ацетил бромид (5,19 мл; 70,3 ммоль). Через 4 ч при 60 С смесь охлаждали до комнатной температуры и АсОН удаляли в вакууме. Остаток разбавляли насыщенным водным раствором NaHCO3 и экстрагировали три раза CH2Cl2. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Полученный остаток разбавляли CHCl3 (23 мл), затем добавляли PBr3 (9,94 мл; 105 ммоль). Смесь нагревали до 80 С в течение ночи, затем охлаждали до комнатной температуры с выливанием на лед и гасили путем медленного добавления 1 М (водного) раствора NaOH. Смесь экстрагировали три раза CH2Cl2 с выходом мутного органического слоя. Растворитель удаляли в вакууме (без высушивания) и полученное твердое вещество растирали с CH2Cl2. Фильтрат концентрировали в вакууме, последующая очистка с помощью ISCO CombiFlash (100:0 - 80:20 гексаны:EtOAc) давала дополнительное твердое вещество, которое объединяли с растертым материалом,получая целевое соединение. 1 К раствору дибромида (2,75 г; 5,62 ммоль) в t-BuOH (23 мл) добавляли КОН (0,946 г; 16,9 ммоль) и смесь нагревали до 75 С в течение ночи, что показывало смесь 4:1 желаемого продукта и его 2-бром-4 гидрокси изомера. Смесь разбавляли насыщенным водным раствором NaHCO3 и экстрагировали три разаEtOAc. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Полученное твердое вещество растирали с МеОН, получая целевое соединение в виде белого твердого вещества. 1 Н ЯМР (400 МГц, MeOD):7,48 (т, J=1,5 Гц, 1 Н); 7,33 (д, J=7,1 Гц, 1 Н); 7,20 (т, J=1,7 Гц, 1 Н); 6,94 Раствор 4-бром-3-(3-хлор-5-йодфенокси)пиридин-2-ола (4-5; 1,72 г; 4,03 ммоль) в ДМФА продували с помощью N2 в течение 5 мин. Затем добавляли цианид цинка(II) (0,284 г; 2,42 ммоль) и Pd(PPh3)4 (0,466 г; 0,403 ммоль) и смесь нагревали до 40 С в течение ночи. После охлаждения до комнатной температуры смесь разбавляли насыщенным водным раствором NaHCO3 и экстрагировали три раза CH2Cl2. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Растирание полученного твердого вещества с водой, затем с МеОН, затем с Et2O давало целевое соединение в виде белого твердо- 16024804 го вещества. 1 Н ЯМР (400 МГц, ДМСО):12,33 (с, 1 Н); 7,73 (с, 1 Н); 7,51 (с, 1 Н); 7,44 (с, 1 Н); 7,36 (д, J=7,1 Гц,1 Н); 6,56 (д, J=7,0 Гц, 1 Н). Стадии 4(f) и (g): 3-(4-бром-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо 1,2-дигидропиридин-3-илокси)-5-хлорбензонитрил (4-1) Указанное в заголовке соединение получали с использованием модификаций стадий 1 (е) и (f), заменяя 3-хлор-5-[(2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4) в стадии 1(е) на 3[(4-бром-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрил (4-6). 1 Раствор 3-[(4-бром-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрила (4-6; 300 мг; 0,922 ммоль) и трибутил(1-этоксивинил) олова (832 мг; 2,30 ммоль) в ДМФА (4 мл) дегазировали с помощью N2 в течение 5 мин. Затем добавляли Pd(PPh3)4 (53 мг; 0,046 ммоль) и смесь нагревали до 80 С в течение ночи. После охлаждения до комнатной температуры добавляли водный раствор KF (2,3 М, 5 мл) и полученный осадок удаляли с помощью фильтрования и промывали EtOAc. После фазы разделения водную фазу далее экстрагировали EtOAc, затем объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (100:0 - 0:100 гексаны:EtOAc) давала целевое соединение, загрязненное остаточным трифенилфосфиноксидом, которое использовали без дальнейшей очистки. 1 Н ЯМР (400 МГц, MeOD):7,45 (с, 1 Н); 7,40 (д, J=6, 9 Гц, 1 Н); 7,19 (с, 1 Н); 7,17 (с, 1 Н); 6,58 (д,J=5,8 Гц, 1 Н); 4,66 (д, J=2,9 Гц, 1 Н); 4,52 (д, J=2,9 Гц, 1 Н); 3,78 (дд, J=14,0, 7,0 Гц, 2 Н); 1,17-1,09 (м, 3 Н). Стадия 5(b): 3-[(4-ацетил-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрил (5-3) К раствору 3-хлор-5-[4-(1-этоксиэтенил)-2-гидроксипиридин-3-ил]оксибензонитрила (5-2; 157 мг; содержит O=PPh3) в ацетоне (2,5 мл) добавляли 1,0 мл 10% водного раствора HCl и смесь перемешивали в течение 2 дней. Ацетон удаляли в вакууме, затем смесь разбавляли водой и экстрагировали три разаCH2Cl2. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Растирание с Et2O давало целевое соединение в виде белого твердого вещества. 1 Н ЯМР (400 МГц, MeOD):7,53 (с, 1 Н); 7,45 (д, J=6,9 Гц, 1 Н); 7,34 (с, 1 Н); 7,31 (с, 1 Н); 6,58 (д,J=6,9 Гц, 1 Н); 2,51 (с, 3 Н). Стадия 5(с): 3-хлор-5-[4-(1,1-дифторэтил)-2-гидроксипиридин-3-ил]оксибензонитрил (5-4) К раствору 3-[(4-ацетил-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрила (5-3; 102 мг; 0,353 ммоль) в CH2Cl2 (1,4 мл) добавляли DAST (233 мкл; 1,77 ммоль) и смесь перемешивали при комнатной температуре. Дополнительные эквиваленты DAST добавляли через 4, 24 и 32 ч и перемешивание продолжали в течение 48 ч, в результате чего анализ LCMS показывал завершение превращения. Реакционную смесь гасили путем осторожного добавления насыщенного водного раствора NaHCO3 и затем про- 17024804 дукт три раза экстрагировали CH2Cl2. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме, получая целевое соединение, которое использовалось непосредственно без дальнейшей очистки. Стадии 5(d) и (е): 3-хлор-5-(4-(1,1-дифторэтил)-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3 ил)метил]-2-оксо-1,2-дигидропиридин-3-илокси)бензонитрил (5-1) Указанное в заголовке соединение получали с использованием модификаций стадий 1(е) и (f), заменяя 3-хлор-5-[2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4) в стадии 1 (е) на 3 хлор-5-[4-(1,1-дифторэтил)-2-гидроксипиридин-3-ил]оксибензонитрил (5-4). 1 К суспензии 3-[(4-бром-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрила (4-6; 1,5 г; 5,34 ммоль) в ацетонитриле (30 мл) добавляли NBS (1,5 г, 8,4 3 ммоль). Смесь перемешивали при комнатной температуре в течение 2-3 ч до тех пор, пока анализ LCMS не показывал завершение превращения. Добавляли воду (20 мл) и твердое вещество собирали, промывали последовательно водой, МеОН и Et2O, получая бромид, в виде бледно-желтого твердого вещества с чистотой, подходящей для непосредственногоиспользования. 1H ЯМР (400 МГц, ДМСО):12,67 (с, 1 Н); 7,92 (с, 1 Н); 7,74 (с, 1 Н); 7,61 (с, 1 Н); 7,57 (с, 1 Н). Стадии 6(b) и (с): 3-(5-бром-4-хлор-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]2-оксо-1, 2-дигидропиридин-3-илокси)-5-хлорбензонитрил (6-1) Указанное в заголовке соединение получали с использованием модификаций стадий 1(е) и (f), заменяя 3-хлор-5-[2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4) в стадии 1 (е) на 3-[(5 бром-4-хлор-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрил (6-2). 1H ЯМР (400 МГц, ДМСО):11,70 (с, 1 Н) ; 8,26 (с, 1 Н) ; 7,76 (с, 1 Н) ; 7,64 (с, 1 Н) ; 7,57 (т, J=2,0 Гц, 1 Н); 5,12 (с, 2 Н); 3,13 (с, 3 Н). Пример 7. 3-(5-Бром-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо-4(трифторметил)-1,2-дигидропиридин-3-илокси)-5-хлорбензонитрил (7-1) К суспензии 3-[(4-бром-2-гидроксипиридин-3-ил)окси]-5-хлорбензонитрила (4-6; 1,30 г; 4,13 ммоль) в ацетонитриле (30 мл) добавляли NBS (0,800 г, 4,49 ммоль). Смесь перемешивали при комнатной температуре в течение 2-3 ч до тех пор, пока анализ LCMS не показывал завершение превращения. Добавляли воду (20 мл) и твердое вещество собирали, промывали последовательно водой, МеОН и Et2O, получая бромид в виде бледно-желтого твердого вещества с чистотой, подходящей для непосредственного использования.H ЯМР (400 МГц, ДМСО):12,94 (с, 1 Н); 7,95 (с, 1 Н); 7,74 (с, 1 Н); 7,67 (с, 1 Н); 7,63 (с, 1 Н). Стадии 7 (b) и (с): 3-(5-бром-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2-оксо 4-(трифторметил)-1,2-дигидропиридин-3-илокси)-5-хлорбензонитрил (7-1) Указанное в заголовке соединение получали с использованием модификаций стадий 1(е) и (f), заменяя 3-хлор-5-[2-гидрокси-4-(трифторметил)пиридин-3-ил]оксибензонитрил (1-4) в стадии 1(е) на 3-[5 бром-2-гидрокси-4-(трифторметил)пиридин-3-ил]окси-5-хлорбензонитрил (7-2). 1 К раствору N-метилгидразинкарбоксамида (3,13 г; 35,1 ммоль; Can. J. Chem. 1951, 29, 478) в ТГФ добавляли бензилоксиацетилхлорид (5,45 мл; 35,1 ммоль). Смесь охлаждали до 0 С, затем добавляли 5 М(водный раствор) (60 мл) и смесь нагревали до 95 С в течение ночи. После охлаждения до комнатной температуры смесь нейтрализовали с помощью добавления по каплям 6 М водного раствора HCl. После разбавления водой водный слой дважды экстрагировали EtOAc. Объединенные органические экстракты сушили (Na2SO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (80:20 - 0:100 гексаны:EtOAc) давала целевое соединение. 1 Н ЯМР (500 МГц, CDCl3):10,03 (с, 1 Н); 7,43-7,34 (м, 5 Н); 4,57 (с, 2 Н); 4,47 (с, 2 Н); 3,35 (с, 3 Н). Стадия 8 (b): 5-(гидроксиметил)-4-метил-2,4-дигидро-3 Н-1,2,4-триазол-3-он (8-3) К суспензии 5-[(бензилокси)метил]-4-метил-2,4-дигидро-3 Н-1,2,4-триазол-3-она (8-2; 4,20 г; 19,2 ммоль) в EtOH (84 мл) добавляли Pd(OH)2 (20 вес.%; 2,02 г; 2,87 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Фильтрование через Solka Floe и концентрирование в вакууме давали 2,33 г (18,1 ммоль; 94%) целевого соединения с чистотой, подходящей для непосредственного использования. 1 Н ЯМР (500 МГц, ДМСО):11,53 (с, 1 Н); 5,51 (т, J=5,7 Гц, 1 Н); 4,33 (д, J=5,8 Гц, 2 Н); 3,17 (с, 3 Н). Стадия 8(с): 5-(хлорметил)-4-метил-2,4-дигидро-3 Н-1,2,4-триазол-3-он (8-4) К суспензии 5-(гидроксиметил)-4-метил-2,4-дигидро-3 Н-1,2,4-триазол-3-она (8-3; 2,33 г; 18,0 ммоль) в CH3CN (47 мл) добавляли тионилхлорид (1,52 мл; 20,7 ммоль) и смесь перемешивали при комнатной температуре в течение 5 ч. После концентрирования в вакууме полученный остаток растирали с гексанами, получая целевое соединение в виде белого твердого вещества. 1 затем добавляли метилтриоксорений(VII) (0,0 62 г; 0,24 9 ммоль) и перекись водорода (30%; 6,2 мл; 61 ммоль) и смесь нагревали до комнатной температуры. После перемешивания в течение 2 ч реакцию гасили путем добавления MnO2 (5 мг) и перемешивали дополнительно 30 мин. После разбавления дополнительным количеством CH2Cl2 смесь фильтровали через Solka Floe, сушили (MgSO4) и концентрировали в вакууме, получая 3-фтор-4-(трифторметил)пиридин 1-оксид с чистотой, подходящей для непосредственного использования. N-оксид (3,70 г; 20,4 ммоль) растворяли в трифторуксусном ангидриде (21,6 мл; 153 ммоль) в гидрогенизаторе и нагревали до 85 С в течение 15 ч. После охлаждения до 0 С добавляли воду с последующим добавлением твердого K2CO3 до тех пор, пока значение рН не становилось 9. Водную фракцию экстрагировали EtOAc и Me-THF и объединенные органические экстракты сушили(MgSO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash (70:30 - 0:100 гексаны:EtOAc) давала целевое соединение в виде розового твердого вещества. 1 Н ЯМР (400 МГц, ацетон):11,48 (с, 1 Н); 7,55 (д, J=7,1 Гц, 1 Н); 6,39 (т, J=6,2 Гц, 1 Н). Стадия 8(е): 3-(дифторметил)бензонитрил (8-6) К раствору 3-цианобензальдегида (1,00 г; 7,63 ммоль) в CH2Cl2 (38 мл) добавляли DAST (1,01 мл,7,63 ммоль) и смесь перемешивали в течение 1 ч при комнатной температуре. Реакцию гасили 1 н водным раствором HCl и дважды экстрагировали CH2Cl2. Объединенные органические экстракты сушили и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash давала целевое соединение в виде желтого масла. 1 Н ЯМР (400 МГц, ДМСО):8,09 (с, 1 Н); 8,05 (д, J=7,8 Гц, 1 Н); 7,94 (д, J=7,9 Гц, 1 Н); 7,80-7,71 (м,1 Н); 7,12 (т, J=55,4 Гц, 1 Н). Стадия 8(е): 3-(дифторметил)-5-гидроксибензонитрил (8-7) Указанное в заголовке соединение получали с использованием модификации стадии 4 (а), заменяя 1-хлор-3-йодбензол на 3-(дифторметил)бензонитрил (8-6), а гексаны в качестве растворителя заменяли на МТВЕ. 1 Н ЯМР (400 МГц, ДМСО):10,69 (с, 1 Н); 7,46 (с, 1 Н); 7,32 (с, 1 Н); 7,26 (с, 1 Н); 7,02 (т, J=55,4 Гц,1 Н). Стадия 8(f): 3-фтор-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-4-(трифторметил)пиридин-2-(1 Н)-он (8-8) К раствору 3-фтор-4-(трифторметил)пиридин-2-ола (8-5; 50 мг; 0,34 ммоль) в диоксане (1,7 мл) добавляли K2CO3 (51 мг; 0,373 ммоль) и смесь перемешивали в течение 5 мин. Затем добавляли 5(хлорметил)-4-метил-2,4-дигидро-3 Н-1,2,4-триазол-3-он (8-4; 68 мг; 0,373 ммоль) и смесь перемешивали в течение 2 ч при комнатной температуре. Смесь разбавляли водой и экстрагировали CH2Cl2. Органические экстракты сушили (MgSO4) и концентрировали в вакууме. Очистка с помощью ISCO CombiFlash давала целевое соединение. 1 Н ЯМР (400 МГц, CDCl3):10,14 (с, 1 Н); 8,11 (д, J=5,2 Гц, 1 Н); 7,18 (т, J=4,7 Гц, 1 Н); 5,40 (с, 2 Н); 3,41 (с, 3 Н). Стадия 8(g): 3-(дифторметил)-5-(1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-2 оксо-4-(трифторметил)-1,2-дигидропиридин-3-илокси)бензонитрил (8-1) Смесь 3-фтор-1-[(4-метил-5-оксо-4,5-дигидро-1 Н-1,2,4-триазол-3-ил)метил]-4-(трифторметил)пиридин-2(1 Н)-она (8-8; 30 мг; 0,103 ммоль), 3-(дифторметил)-5-гидроксибензонитрила (8-7; 34,7 мг; 0,205 ммоль) и K2CO3 (28,4 мг; 0,205 ммоль) нагревали до 75 С и перемешивали в течение ночи. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали CH2Cl2. Объединенные органические экстракты сушили (MgSO4) и концентрировали в вакууме. Очистка с помощью масснаправленной HPLC, давала 2,1 мг (0,0048 ммоль; 4,6%) целевого соединения в виде белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО):11,70 (с, 1 Н); 7,90 (д, J=7,3 Гц, 1 Н); 7,80 (д, J=8,5 Гц, 2 Н); 7,57 (с, 1 Н); 7,05 (т, J=55,2 Гц, 1 Н); 6,69 (д, J=7,3 Гц, 1 Н); 5,17 (с, 2 Н); 3,10 (с, 3 Н). Пример 9. ECL анализ на ингибирование ВИЧ обратной транскриптазы Анализ для определения ингибирования ВИЧ обратной транскриптазы in vitro с помощью соединений данного изобретения проводили следующим образом: фермент ВИЧ-1 RT (0,1 нМ) объединяли с ингибитором или DMSO (10%) в буфере для анализа (50 мМ Трис-HCl, рН 7,8, 1 мМ дитиотреитола, 6 мМ MgCl2, 80 мМ KCl, 0,025% CHAPS, 0,1 мМ EGTA) и смесь преинкубировали в течение 30 мин при комнатной температуре в микротитровальных планшетах (Corning Costar 3365). Реакционные смеси по 50 мкл подвергали инициированию комбинацией субстрата матричной затравки (5 нМ конечная концентрация) и dNTPs (0,6 мкМ dNTPs, 20 нМ Рутений-dUTP). Гетеродимерный субстрат нуклеиновой кислоты возбуждали путем гибридизации ДНК праймера биотинилированного-pD500 (полученного из Integrated DNA Technologies) до t500, 500 нуклеотидной матрицей РНК, созданной in vitro, путем транскрипции (см. Shaw-Reid et al., J. Bioi. Спет., 278: 2777-2780). Через 90 мин инкубирования при 37 С реакции гасили с помощью 60 мкл гасящего буфера, содержащего 0,05 М EDTA, 0,7% BSA, 0,07% Tween-20 и 0,017% азида натрия в PBS. Через 10 мин инкубирования 50 мкл погашенной реакционной смеси переносили в стандартную планшету для среднемасштабного обнаружения (MSD) Авидина, предварительно блокированную в течение 1 ч 5% блокатором А (из MSD). Планшету затем оставляют при комнатной температуре в течение 1 ч, после чего промывают 3 раза 200 мкл на лунку PBS. Добавляют 150 мкл 1 считывающего буфера Т (MSD) и планшету считывают на устройстве Sector Imager 6000 (MSD). В данном анализе типичные представители соединений данного изобретения проявляют ингибирование фермента обратной транскриптазы. Например, в анализе испытывали соединения, указанные в заголовках представленных выше примеров 1-8, и было обнаружено, что они имеют значения ингибирования в процентах при 100 нМ, как представлено в таблице В ниже. Таблица ВWT = дикий тип. Пример 10. Анализ на ингибирование размножения ВИЧ Анализ на ингибирование острой ВИЧ-1 инфекции Т-лимфоидных клеток (иначе называемый какSci. USA 1994, 91: 4096. В анализе с задействованием ВИЧ-1 дикого типа, типичные представители соединений данного изобретения проявляют ингибирование размножения ВИЧ. Например, было обнаружено, что соединения, представленные выше в примерах 1-8, имеют величины CIC95, как представлено в таблице С ниже. Соединения также испытывали с использованием варианта анализа с задействованием ВИЧ штамма, содержащего K103N и Y181C мутации, и было обнаружено менее чем 20-кратное отклонение по отношению к дикому типу во всех случаях. Таблица СWT = дикий тип. Пример 11. Цитотоксичность Цитотоксичность определяли с помощью исследования под микроскопом клеток в каждой лунке в анализе распространения, где квалифицированный аналитик исследовал каждую культуру на любое из следующих морфологических изменений и сравнивал с контрольными культурами: дисбаланс рН, аномалию клеток, цитостатичность, цитопатию или кристаллизацию (т.е. соединение не является растворимым или образует кристаллы в лунке). В анализе распространения, типичные представители соединений данного изобретения не проявляют цитотоксичность вплоть до верхней дозы, исследуемой в данном анализе при концентрации при или выше ее величины CIС 95. Например, соединения, указанные в заголовках примеров 1-8, не проявляют цитотоксичность вплоть до 125 нМ. Кроме того, что предшествующее определение сообщает принципы данного изобретения с примерами, предоставленными для целей иллюстрации, методика изобретения охватывает все обычные разновидности, варианты и/или модификации, которые попадают в объем следующей формулы изобретения. Все указанные публикации, патенты и патентные заявки включены в данное описание путем ссылки на них. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) или его фармацевтически приемлемая соль,где R1 представляет собой AryА; где AryА представляет собой фенил, который необязательно замещен в целом 1-3 заместителями,выбранными из (2) C1-6 галогеналкила, (11) галогена, (12) CN;R2 и R3, каждый, представляют собой независимо (1) Н, (3) C1-6 галогеналкил, (7) галоген;R5 представляет собой C1-6 алкил. 2. Соединение по п.1 или его фармацевтически приемлемая соль, где фенил является необязательно замещенным 1-3 заместителями, каждый из которых представляет собой независимо (2) С 1-4 галогеналкил. 3. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 представляет собой(2) С 1-4 галогенкалкил, (5) галоген; R3 представляет собой (1) Н, (3) С 1-4 галогеналкил, (6) галоген. 4. Соединение по п.1 или его фармацевтически приемлемая соль, которые представляют собой соединение формулы (II) где X1 и X2, каждый, представляют собой независимо (3) С 1-4 галогеналкил, (9) галоген, (10) CN;R2 и R3, каждый, представляют собой независимо (1) Н, (3) С 1-4 галогеналкил, (14) галоген;R5 представляет собой С 1-4 алкил. 5. Соединение по п.4 или его фармацевтически приемлемая соль,где X1 и X2, каждый, представляют собой независимо (2) С 1-4 фторалкил, (4) галоген, (5) CN,R2 представляет собой (2) С 1-4 фторалкил, (5) галоген;R5 представляет собой C1-3 алкил. 6. Соединение по п.5 или его фармацевтически приемлемая соль, где соединение представляет собой соединение формулы (III) 7. Соединение по п.6 или его фармацевтически приемлемая соль, где R5 представляет собой СН 3 или СН 2 СН 3. 8. Соединение по п.7 или его фармацевтически приемлемая соль, где R3 представляет собой Н. 9. Соединение формулы (I) по п.1, где соединение выбрано из группы, состоящей из и его фармацевтически приемлемые соли. 10. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения согласно любому одному из пп.1-9 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. 11. Применение соединения по любому одному из пп.1-9 или его фармацевтически приемлемой соли в приготовлении лекарственных средств для профилактики или лечения инфекций, вызываемых ВИЧ,или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом. 12. Соединение, представленное формулой или его фармацевтически приемлемая соль. 13. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.12 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. 14. Соединение, представленное формулой 15. Фармацевтическая композиция, пригодная для профилактики или лечения инфекции HIV или для профилактики, лечения или препятствования возникновению СПИД у субъекта, нуждающегося в этом, содержащая эффективное количество соединения по п.14 и фармацевтически приемлемый носитель. 16. Применение соединения по п.12 или его фармацевтически приемлемой соли в профилактике или лечении инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом. 17. Применение соединения по п.14 в приготовлении лекарственных средств для профилактики или лечения инфекций, вызываемых ВИЧ, или для профилактики, лечения или для препятствования возникновению СПИД у субъекта, нуждающегося в этом.
МПК / Метки
МПК: C07D 401/06, A61P 31/18, A61K 31/4439
Метки: ненуклеозидные, содержащие, транскриптазы, композиции, применение, обратной, ингибиторы
Код ссылки
<a href="https://eas.patents.su/24-24804-nenukleozidnye-ingibitory-obratnojj-transkriptazy-kompozicii-ih-soderzhashhie-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Ненуклеозидные ингибиторы обратной транскриптазы, композиции, их содержащие, и их применение</a>
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 5598
Опубликовано: 28.04.2005
Автор: Симоно Брюно
МПК: A61K 31/55, C07D 471/14
Метки: обратной, ингибиторы, ненуклеозидные, транскриптазы
Формула / Реферат:
1. Соединение общей формулы I где R2 выбирают из группы, включающей H, F, Cl, C1-C4алкил, C3-C4циклоалкил и CF3; R4 обозначает H или Me; R5 обозначает H, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает Me, то R5 не обозначает Et; R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбирают из группы, включающей или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R11 обозначает...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 16386
Опубликовано: 30.04.2012
Авторы: Кеннеди-Смит Джошуа, Суини Захари Кевин, Ву Джеффри
МПК: A61K 31/4174, A61P 31/18, A61K 31/513...
Метки: ингибиторы, обратной, ненуклеозидные, транскриптазы
Формула / Реферат:
1. Соединение формулы Iв которой А обозначает:(a) CR3=CR2,(b) CH2R3CH2R2 или(c) CHR2;или (i) R1 обозначает (II) и R2 обозначает водород, C1-C3-алкил, C1-C3-галогеналкил, C2-C6-алкенил или CH2ORc; и, в дополнение, если А обозначает или (a), или (b), то R2 также может обозначать галоген, NRaRb, CN или ORc;или (ii) R1 обозначает C1-C6-алкил и R2 обозначает (II);X1 обозначает О или S;X2 обозначает О или, если А обозначает (а), то X2 обозначает О или...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 9700
Опубликовано: 28.02.2008
Авторы: Сунд Кристьян, Сальберг Кристер, Линдстрем Стефан, Антонов Дмитрий, Роуэ Натали, Янссон Катарина
МПК: A61K 31/426, A61K 31/44, A61K 31/425...
Метки: транскриптазы, ингибиторы, обратной, ненуклеозидные
Формула / Реферат:
1. Соединение формулы Z где А представляет собой СН или N; R1 представляет собой заместитель у атома углерода в кольце, содержащем А, выбранном из -S(=O)pRa, где Ra представляет собой -C1-С4-алкил, -ORx, -NRxRx, -NHNRxRx, -NHNHC(=O)ORx, -NRxOH; -C(=O)-Rb, где Rb представляет собой -C1-С4-алкил, ORx, -NRxRx, -NHNRxRx, -NHC1-С3-алкил-C(=O)ORx; -NRxRc, где Rc представляет собой Н, C1-С4-алкил, -NRxRx; -C(=O)Rd, -CN, S(=O)pRx, где Rd представляет...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 9478
Опубликовано: 28.02.2008
Авторы: Ким Хонгву, Жирарде Жан-Люк, Хун Чжи, Де Ла Роса Марта А., Нилар Шахул, Шоу Стефания, Хэматейк Роберт, Гунич Эсмир, Кох Йунг-Хио, Яо Наньхуа, Чжан Чжицзюн
МПК: C07D 233/84, A61K 31/4196, A61K 31/4709...
Метки: транскриптазы, ингибиторы, обратной, ненуклеозидные
Формула / Реферат:
1. Способ лечения пациента, инфицированного ВИЧ, включающий введение пациенту фармацевтической композиции, содержащей соединение в дозе, эффективной, чтобы снизить вирусную репродукцию, причем соединение имеет структуру в соответствии с формулой (А) или формулой (В) в которых R1 представляет необязательно замещенный C1-C10алкил, галоген или CF3, R2 представляет необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно...
Ненуклеозидные ингибиторы обратной транскриптазы как антагонисты пролиферации и индукторы дифференцировки клеток

Номер патента: 7004
Опубликовано: 30.06.2006
Авторы: Спадафора Коррадо, Маттеи Элизабетта, Лорендзини Родольфо Нелло, Лавия Патриция, Нерви Клара, Паломбини Гильельмо
МПК: A61K 31/55, A61K 31/535, A61K 31/495...
Метки: ингибиторы, обратной, пролиферации, ненуклеозидные, индукторы, транскриптазы, антагонисты, дифференцировки, клеток
Формула / Реферат:
1. Применение по меньшей мере одного соединения-ингибитора обратной транскриптазы для получения фармацевтической композиции для противодействия утрате дифференцировки клеток и уменьшения скорости пролиферации клеток при раковых и не раковых патологиях, где вышеуказанное соединение выбрано из класса соединений 5,11-дигидро-6Н-дипиридо[3,2-b:2',3'-е][1,4]диазепина. 2. Применение по п.1, где соединение способно стимулировать дифференцировку клеток....
Предыдущий патент: Способ предварительной очистки сточных вод и способ очистки бытовых сточных вод с использованием способа предварительной очистки
Следующий патент: Способ изготовления топливных пеллет и других продуктов из лигноцеллюлозной биомассы
Случайный патент: Устройство для получения изображения радужной оболочки глаза