Дейтерированные морфинановые соединения





















Формула / Реферат
1. Соединение формулы (I)
или его фармацевтически приемлемая соль,
в которой R1 выбирают из СН3, CD3, -O-CD2CD3 или -O-CD(CD3)2; a
R2 выбирают из СН3 или CD3;
при условии, что по меньшей мере один из R1 и R2 является дейтерированным.
2. Соединение по п.1, в котором R1 представляет собой -O-CD2CD3 или -O-CD(CD3)2.
3. Соединение по п.2, в котором R1 представляет собой -O-CD(CD3)2.
4. Соединение по п.1, в котором соединение выбирают из любого соединения, приведенного в представленной ниже таблице
или его фармацевтически приемлемая соль.
5. Соединение по п.1, в котором R1 представляет собой -CD3.
6. Соединение по п.1, выбранное из любого из
или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности.
7. Соединение по любому из пп.1-6, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности.
8. Фармацевтическая композиция, не содержащая пирогенов, включающая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
9. Способ лечения субъекта, страдающего от или склонного к заболеванию или состоянию, выбранному из эмоциональной неустойчивости; псевдобульбарного аффекта; аутизма; неврологических нарушений и нейродегенеративных заболеваний; повреждений мозга; нарушений, связанных с расстройством сознания; сердечно-сосудистых заболеваний; глаукомы; поздней дискинезии; рака; ревматоидного артрита; диабетической невропатии; ретинопатических заболеваний; заболеваний или нарушений, вызванных апоптозом, индуцированным гомоцистеином; заболеваний или нарушений, вызванных повышенными уровнями гомоцистеина; хронической боли; неустранимой боли; невропатической боли, симпатически опосредуемой боли; боли, связанной с дисфункцией желудочно-кишечного тракта; боли в ротовой полости; боли спины; центрального болевого синдрома; комплексного регионального болевого синдрома; эпилептических припадков; эпилептической гемиплегии; приобретенной эпилептиформной афазии (синдром Ландау-Клеффнера); тяжелой миоклонической эпилепсии раннего детского возраста (SMEI); эпилептической энцефалопатии раннего детского возраста; постинсультных судорог; фебрильных судорог; посттравматических судорог; звона в ушах; сексуальной дисфункции; трудноизлечимого кашля; дерматита; нарушений, связанных с аддикциями; синдрома Ретта (RTT); нарушений в работе голосовых связок вследствие неконтролируемых спазмов гортанных мышц; нейротоксичности метотрексата и усталости, вызванной раком, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8.
10. Способ по п.9, в котором субъект страдает от или склонен к диабетической невропатической боли.
11. Способ по п.9, в котором субъект страдает от или склонен к эпилептическим припадкам.
12. Способ лечения субъекта, страдающего от или подверженного условиям, относящимся к воздействию химических веществ, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8.
13. Способ лечения субъекта, страдающего от или склонного к боли, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8.
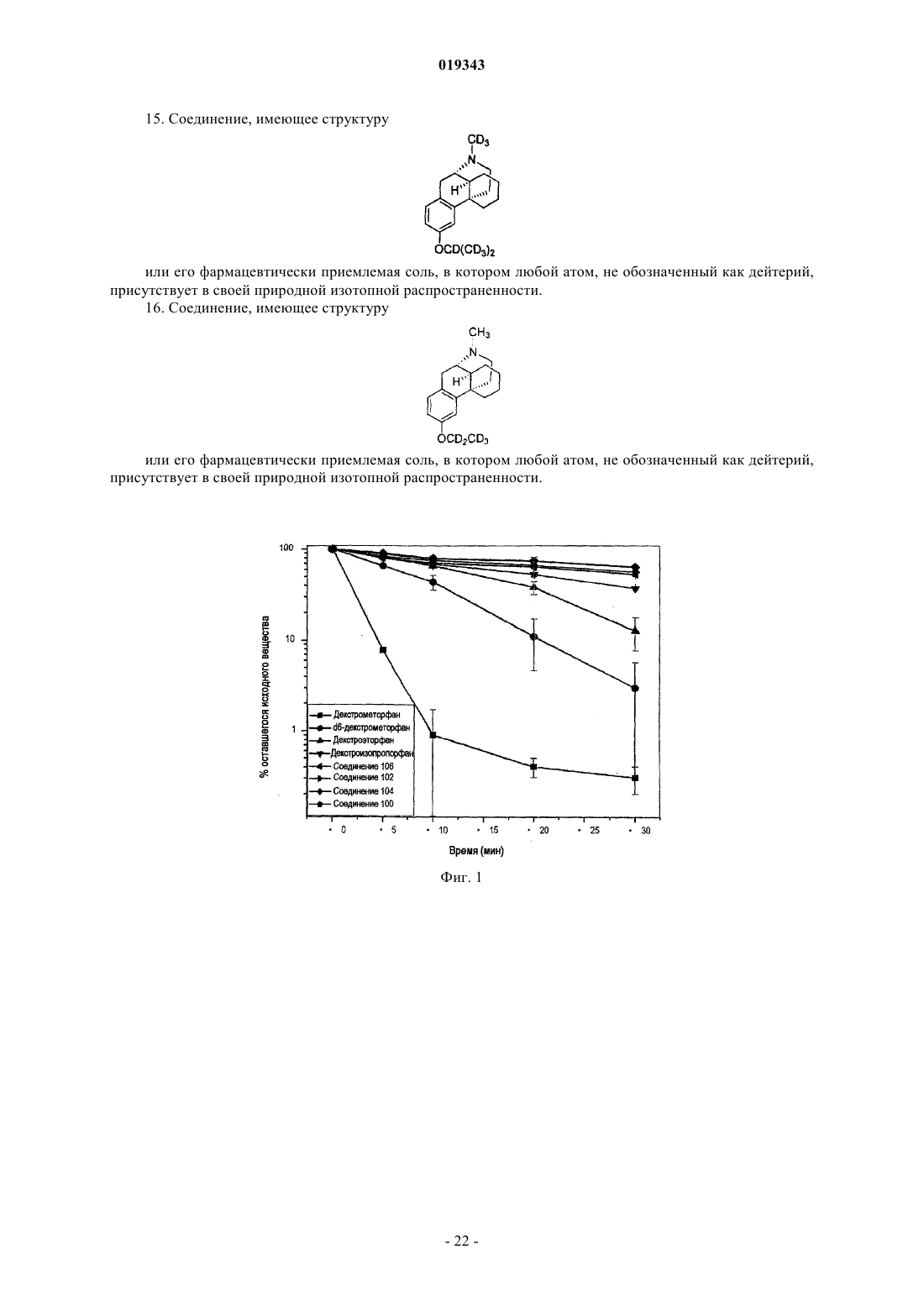
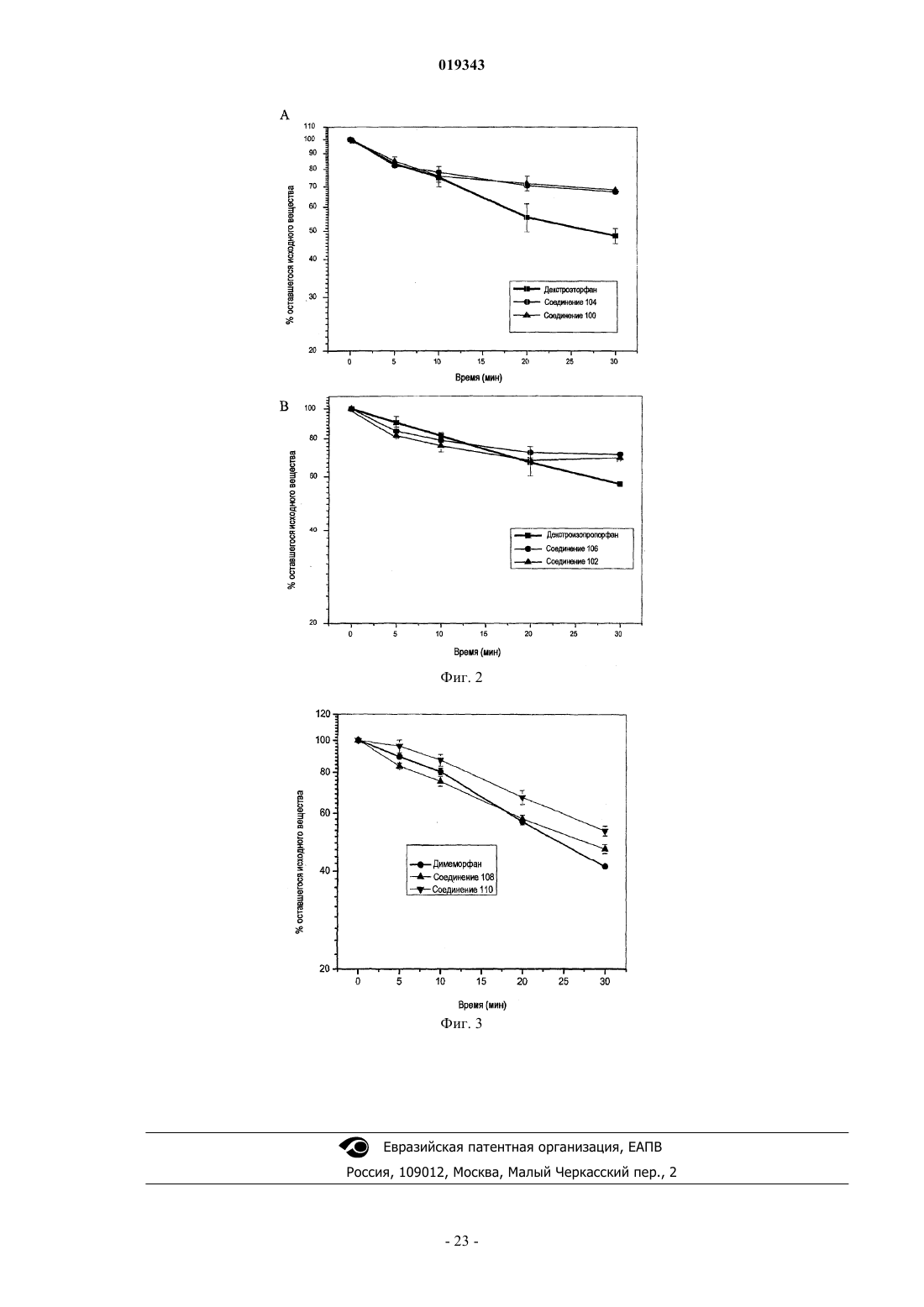
14. Соединение, имеющее структуру
или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности.
15. Соединение, имеющее структуру
или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности.
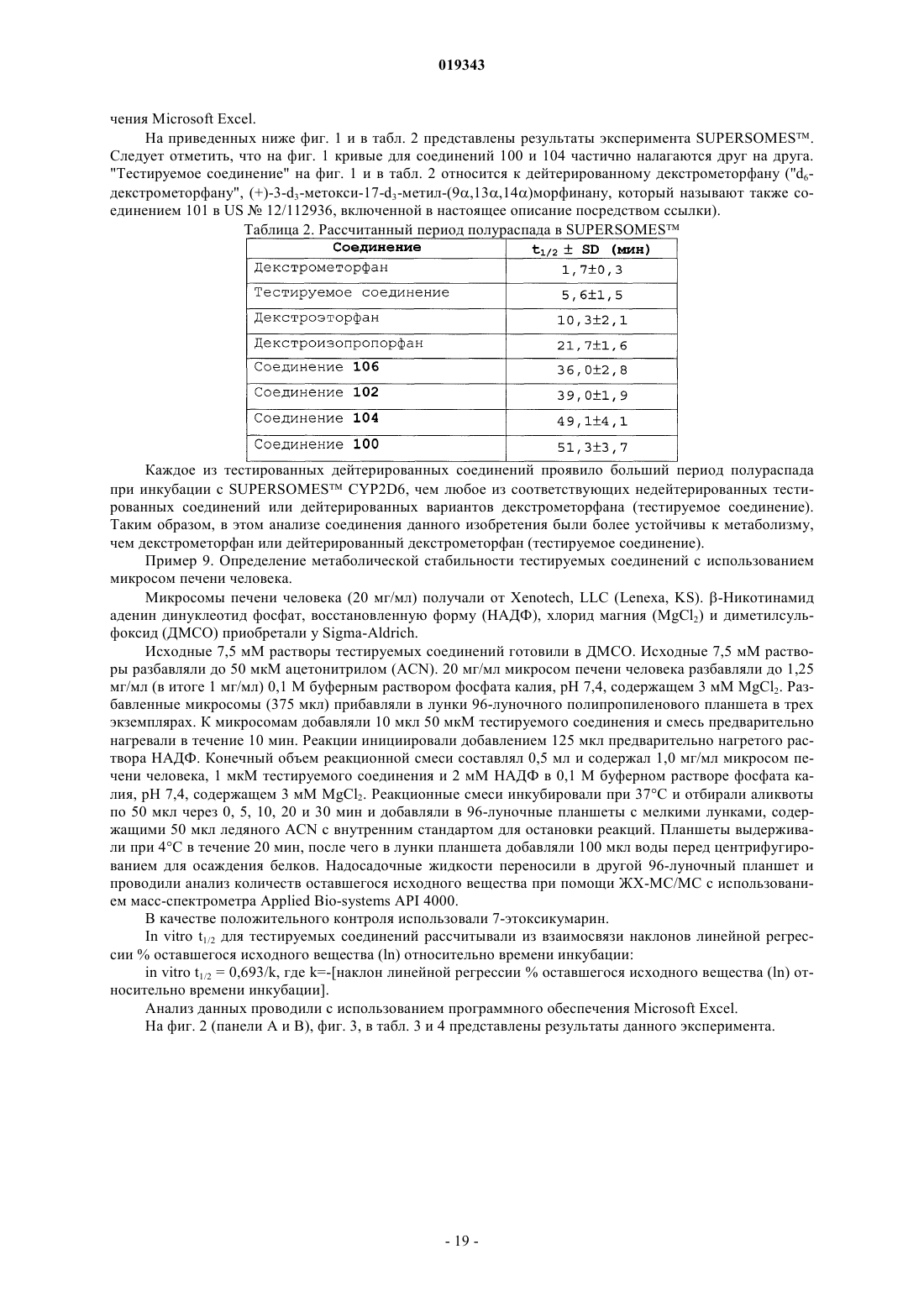
16. Соединение, имеющее структуру
или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности.
Текст
Данное изобретение относится к дейтерированным морфинановым соединениям и их фармацевтически приемлемым солям. В данном изобретении также предоставлены композиции, содержащие соединение данного изобретения, и использование подобных композиций в способах лечения заболеваний и состояний,которые преимущественно лечатся введением агониста рецептора 1, который обладает также антагонистической активностью относительно NDMA. По данной заявке испрашивается приоритет на основании предварительной заявки США 61/098511, поданной 19 сентября 2008 г., которая включена в настоящее описание в виде ссылки во всей полноте. Область техники, к которой относится изобретение Данное изобретение относится к новым морфинановым соединениям и их фармацевтически приемлемым солям. В данном изобретении представлены также композиции, включающие соединение данного изобретения, и применение подобных композиций в способах лечения заболеваний и состояний, которые преимущественно лечат введением агониста сигма-1 рецептора, который обладает также антагонистической активностью в отношении NMDA. Уровень техники Декстрометорфан, известный также под своим химическим названием (+)-3-метокси-17-метил(9,13,14)морфинан, в настоящее время является одним из наиболее широко используемых противокашлевых средств. Помимо отмеченной выше физиологической активности декстрометорфан является также агонистом 2 рецептора, антагонистом N-метил-D-аспартата (NMDA) и антагонистом 34 никотинового рецептора. Декстрометорфан ингибирует нейротрансмиттеры, такие как глутамат, от активации рецепторов в мозге. Кроме того, ингибируется поглощение допамина и серотонина. Декстрометорфан одобрен для применения в ряду отпускаемых без рецепта противокашлевых препаратов. В настоящее время он находится в I фазе клинических испытаний для лечения субъектов со спазмами голосовых связок и в III фазе клинических испытаний для лечения синдрома Ретта(http://www.clinicaltrials.gov). Декстрометорфан исследуют с другими лекарственными средствами во II фазе клинических испытаний, характеризующих механизмы развития боли у субъектов с синдромом раздраженного кишечника (http://www.clinicaltrials.gov/). Декстрометорфан также находится в I фазе клинических испытаний для лечения гипералгезии у субъектов, находящихся на метадоновой поддержке(http://www.clinicaltrials.gov/). Кроме того, комбинация гидробромида декстрометорфана и сульфата хинидина в настоящее время находится в III фазе клинических испытаний для лечения диабетической невропатической боли(http://www.clinicaltrials.gov). Эта лекарственная комбинация, известная также как Zenvia, находится вIII фазе клинических испытаний для лечения нарушения, связанного с непроизвольным выражением эмоций (IEED), известного также как псевдобульбарный аффект, у субъектов, страдающих болезнью Альцгеймера,инсультом,болезнью Паркинсона и черепно-мозговой травмой(http://www.clinicaltrials.gov). Декстрометорфан метаболизируется в печени. Разложение начинается с О- и N-деметилирования с образованием первичных метаболитов декстрорфана и 3-метоксиморфинана, оба из которых в дальнейшем N- и О-деметилируются соответственно в 3-гидроксиморфинан. Предполагается, что три данных метаболита являются терапевтически активными. Основным метаболическим катализатором является фермент 2D6 цитохрома Р 450 (CYP2D6), который отвечает за реакции О-деметилирования декстрометорфана и 3-метоксиморфинана. N-деметилирование декстрометорфана и декстрофана катализируется ферментами в родственном семействе CYP3A. Конъюгаты декстрорфана и 3-гидроксиморфинана могут быть обнаружены в плазме крови и моче человека в течение часов его переваривания. Злоупотребление декстрометорфаном связано с его активным метаболитом, декстрорфаном. РСРподобные эффекты, приписываемые декстрометорфану, с большей достоверностью дает декстрорфан и,таким образом, потенциал злоупотребления у людей можно отнести за счет метаболизма декстрометорфана в декстрорфан (Miller S. C. et al., Addict Biol., 2005, 10(4): 325-7, Nicholson K. L. et al., Psychopharmacology (Berl), 199 Sep 1, 146(1): 49-59, Pender E.S. et al., Pediart Emerg Care, 1991, 7: 163-7). В одном из исследований психотропных воздействий декстрометорфана было найдено, что для людей, являющихся активными метаболизаторами (ЕМ), сообщалось о большем потенциале злоупотребления по сравнению со слабыми метаболизаторами (РМ), что свидетельствует о том, что декстрорфан вносит вклад в потенциал злоупотребления декстрометорфаном (Zawertailo L. A, et al., J. Clin. Psychopharmacol, 1998, Aug.,18(4): 332-7). У значительной части населения имеется функциональная нехватка фермента CYP2D6. Таким образом, поскольку для основного пути метаболизма декстрометорфана требуется CYP2D6, сниженная активность приводит к намного более длительной продолжительности действия и большим эффектам лекарственного средства у субъектов с дефицитом CYP2D6. Помимо врожденной функциональной недостаточности некоторые лекарственные средства, такие как антидепрессанты, являются эффективными ингибиторами фермента CYP2D6. Помимо присущей им функциональной недостаточности некоторые препараты, такие как антидепрессанты, являются эффективными ингибиторами фермента CYP2D6. Со своим более медленным метаболизмом у некоторых людей декстрометорфан, особенно в сочетании с другим лекарственным средством(ами), может привести к серьезным отрицательным событиям. Более длительное по сравнению с рекомендованным время действия лекарственного средства в организме может оказать непрерывное благоприятное влияние, но оно может также вызвать или продлить нежелательные побочные эффекты. Нежелательные побочные эффекты при рекомендованных дозах терапевтического лечения декстрометорфаном включают тошноту, потерю аппетита, диарею, сонливость,головокружение и импотенцию. Димеморфан, аналог декстрометорфана, известен также под своим химическим названием как (+)(9,13,14)-3,17-диметилморфинан, является ненаркотическим противокашлевым средством. Предполагается, что противокашлевая активность димеморфана происходит от прямого воздействия на кашлевый центр в продолговатом мозге (Ida, H., Clin Ther., 1997, Mar-Apr; 19(2): 215-31). Помимо его противокашлевых свойств было показано, что димеморфан обладает противосудорожным и нейропротекторным действием, возможно, происходящим вследствие антагонизма N-метил-Dаспартата (NDMA) декстрометорфана (DM) и/или высокой афинности DM -рецепторов (Chou, Y-C. etal., Brain Res., 1999, Mar. 13; 821(2): 516-9). Было найдено, что активация -1 рецептора оказывает противосудорожное действие на крыс и мышей, по типу DM, но без побочных поведенческих эффектов, вызванных DM и его метаболитом, декстрорфаном (Shin, E.J. et al., Br. J. Pharmacol., 2005, Apr.; 144(7): 90818 и Shin, E.J. et al., Behavioural Brain Research, 2004, 151: 267-276) . Известно, что метаболизм димеморфана у людей протекает через катализируемое цитохромом Р 450N-деметилирование, а также окисление 3-метила. У здоровых взрослых мужчин метаболизируется свыше 98% дозы димеморфана и было показано, что ни один из метаболитов не обладает противокашлевыми свойствами (Chou Y.-С, et al., Life Sci., 2005, Jul. 1; 77(7): 735-45 и Chou Y.-C., et al., J. Pharm. Sci.,2009, Jul.: 1-15). Кроме того, было показано, что два эфирных аналога декстрометорфана, [(+)-3-этокси-17 метилморфинан], называемый также "декстроэторфан", и [(+)-3-(2-пропокси)-17-метилморфинан], называемый также "декстроизопропорфан", обладают противосудорожной активностью (Newman, A et al., J.Med. Chem., 1992, 35(22): 4135-42 и Tortella, F. et al., J. Pharmacol. and Exp. Therap., 1994, 268(2): 727733), также нейропротекторным действием на крыс (Tortella, F. et al., Neurosci. Lett., 1995, 198(2): 79-82). Соответственно, желательно предоставить новые соединения, которые обладают благоприятной активностью декстрометорфана, димеморфана, декстроэторфана и декстроизопропорфана и могут обладать также другими преимуществами, например сниженными неблагоприятными побочными действиями, с пониженной склонностью к метаболизму, чтобы дополнительно продлить свое фармакологически эффективное существование, повысить приверженность субъекта лечению и, возможно, понизить фармакологическую изменчивость популяции и/или уменьшить ее потенциальные возможности в отношении опасных межлекарственных взаимодействий, или понизить вероятность злоупотребления декстрометорфаном вследствие образования тяжелых метаболитов, таких как декстрорфан. Сущность изобретения Настоящее изобретение относится к соединению формулы I или его фармацевтически приемлемой соли, в которойR1 выбирают из СН 3, CD3, -O-CD2CD3 или -O-CD(CD3)2; aR2 выбирают из СН 3 или CD3; при условии, что по меньшей мере один из R1 и R2 является дейтерированным. В предпочтительном варианте в соединении формулы I R1 представляет собой -O-CD2CD3 или -OCD(CD3)2. Ещ более предпочтительно, когда R1 представляет собой -O-CD(CD3)2. В предпочтительном варианте соединение формулы I выбирают из любого соединения, приведенного в представленной ниже таблице: или его фармацевтически приемлемой соли. В предпочтительном варианте в соединении формулы I R1 представляет собой -CD3. Предпочтительно соединение формулы I представляет собой любое из или его фармацевтически приемлемую соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. В предпочтительном варианте любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности. Другим объектом изобретения является фармацевтическая композиция, не содержащая пирогенов,включающая соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Ещ одним объектом изобретения является способ лечения субъекта, страдающего от или склонного к заболеванию или состоянию, выбранному из эмоциональной неустойчивости; псевдобульбарного аффекта; аутизма; неврологических нарушений и нейродегенеративных заболеваний; повреждений мозга; нарушений, связанных с расстройством сознания; сердечно-сосудистых заболеваний; глаукомы; поздней дискинезии; рака; ревматоидного артрита; диабетической невропатии; ретинопатических заболеваний; заболеваний или нарушений, вызванных апоптозом, индуцированным гомоцистеином; заболеваний или нарушений, вызванных повышенными уровнями гомоцистеина; хронической боли; неустранимой боли; невропатической боли, симпатически опосредуемой боли; боли, связанной с дисфункцией желудочно-кишечного тракта; боли в ротовой полости; боли спины; центрального болевого синдрома; комплексного регионального болевого синдрома; эпилептических припадков; эпилептической гемиплегии; приобретенной эпилептиформной афазии (синдром Ландау-Клеффнера); тяжелой миоклонической эпилепсии раннего детского возраста (SMEI); эпилептической энцефалопатии раннего детского возраста; постинсультных судорог; фебрильных судорог; посттравматических судорог; звона в ушах; сексуальной дисфункции; трудноизлечимого кашля; дерматита; нарушений, связанных с аддикциями; синдрома Ретта(RTT); нарушений в работе голосовых связок вследствие неконтролируемых спазмов гортанных мышц; нейротоксичности метотрексата, и усталости, вызванной раком, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества указанной выше фармацевтической композиции. Предпочтительно применять указанный выше способ, когда субъект страдает от или склонен к диабетической невропатической боли или к эпилептическим припадкам. Другим объектом настоящего изобретения является способ лечения субъекта, страдающего от или подверженного условиям, относящимся к воздействию химических веществ, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8. Ещ одним объектом изобретения является способ лечения субъекта, страдающего от или склонного к боли, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества вышеуказанной фармацевтической композиции. Предложенное изобретение также относится к соединению, имеющему структуру или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. В другом варианте предложенное изобретение относится к соединению, имеющему структуру или его фармацевтически приемлемой соли, где любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности. В ещ одном другом варианте предложенное изобретение относится к соединению, имеющему структуру или его фармацевтически приемлемой соли, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. Краткое описание чертежей На фиг. 1 изображена метаболическая устойчивость соединений данного изобретения в CYP2D6SUPERSOMES. На фиг. 2 на панелях А и В изображена метаболическая устойчивость декстроэторфана (панель А),декстроизопропорфана (панель В) и соединений данного изобретения в микросомах печени человека. На фиг. 1 изображена метаболическая устойчивость димеморфана и соединений данного изобретения в микросомах печени человека. Подробное описание Определения Термины "улучшать" и "лечить" используются взаимозаменяемо и включают как терапевтическое лечение, так и/или профилактическое лечение (уменьшение вероятности развития). Оба термина означают снижение, подавление, ослабление, уменьшение, прекращение или стабилизацию развития или прогрессирования заболевания (например, описанного здесь заболевания или нарушения), снижение тяжести заболевания или улучшения симптомов, связанных с данным заболеванием."Заболевание" означает любое состояние или нарушение, которое нарушает или препятствует нормальному функционированию клетки, ткани или органа. Очевидно, что в синтезированном соединении имеет место некоторое различие в природной распространенности изотопов в зависимости от происхождения химических веществ, используемых в синтезе. Таким образом, препарат декстрометорфана или аналогов декстрометорфана будет, по существу,содержать небольшие количества дейтерированных изотопологов. Концентрация распространенных в природе стабильных изотопов водорода и углерода, несмотря на это изменение, невелика и несущественна по сравнению со степенью замещения стабильными изотопами соединений данного изобретения. См., например, Wada E. et al., Seikagaku 1994, 66:15; Gannes L. Z. et al., Comp. Biochem. Physiol. Mol. Integr. Physiol. 1998, 119:725. Если не утверждается иначе, в случае, когда положение конкретно обозначено как "Н" или "водород", подразумевается, что в данном положении имеется водород со своим природным составом изотопной распространенности. Также, если не утверждается иначе, в случае, когда положение конкретно обозначено как "D", или "дейтерий", подразумевается, что в данном положении имеется дейтерий со своей распространенностью, которая по меньшей мере в 3340 раз превышает природную распространенность дейтерия, составляющую 0,015% (то есть термин "D" или "дейтерий" указывает по меньшей мере на 50,1% включение дейтерия). Использованный в настоящем описании термин "фактор изотопного обогащения" означает соотношение между изотопной распространенностью D в определенном положении соединения данного изобретения и существующей в природе распространенностью этого изотопа. Природная распространенность дейтерия составляет 0,015%. В других вариантах осуществления фактор изотопного обогащения в соединении данного изобретения для каждого атома дейтерия, находящегося в положении, обозначенном как потенциальное положение дейтерирования данного соединения, составляет по меньшей мере 3500 (52,5% включения дейтерия), по меньшей мере 4000 (60% включения дейтерия), по меньшей мере 4500 (67,5% включения дейтерия), по меньшей мере 5000 (75% включения дейтерия), по меньшей мере 5500 (82,5% включения дейтерия), по меньшей мере 6000 (90% включения дейтерия), по меньшей мере 6333,3 (95% включения дейтерия), по меньшей мере 6466,7 (97% включения дейтерия), по меньшей мере 6600 (99% включения дейтерия) или по меньшей мере 6633,3 (99,5% включения дейтерия). Понятно, что фактор изотопного обога-4 019343 щения для каждого атома дейтерия, находящегося в положении, обозначенном как положение дейтерирования, не зависит от других дейтерированных положений. Например, при наличии в соединении двух положений дейтерирования одно положение может быть дейтерировано на 52,5%, тогда как другое положение может быть дейтерировано на 75%. Полученное соединение рассматривалось бы как соединение, в котором фактор изотопного обогащения составляет по меньшей мере 3500 (52,5%). Термин "изотополог" относится к веществам, имеющим ту же химическую структуру и формулу,что и конкретное соединение данного изобретения, за исключением положений изотопного замещения и/или уровня изотопного обогащения в одном или более положениях, например Н относительно D. Использованный здесь термин "соединение" относится к совокупности молекул, имеющих идентичную химическую структуру, за исключением того, что среди атомов, составляющих данные молекулы, может иметь место варьирование изотопов. Таким образом, специалистам в данной области будет ясно, что соединение, представленное конкретной химической структурой, содержащей указанные атомы дейтерия, будет также содержать меньшие количества изотопологов, содержащих атомы водорода в одном или более из указанных положений дейтерия в данной структуре. Относительное количество подобных изотопологов в соединении данного изобретения будет зависеть от ряда факторов, включая изотопную чистоту дейтерированных реагентов, использованных для получения соединения, и эффективность включения дейтерия на различных стадиях синтеза, используемого для получения соединения. Однако, как указано выше, относительное количество подобных изотопологов будет составлять менее 49,9% соединения. Соль соединения данного изобретения образуется при взаимодействии кислоты и основной группы соединения, такой как функциональная аминогруппа, или основания и кислотной группы соединения,такой как функциональная карбоксильная группа. Согласно следующему варианту осуществления соединение представляет собой фармацевтически приемлемую соль присоединения кислоты. Использованный здесь термин "фармацевтически приемлемый" относится к компоненту, который в рамках вынесенного медицинского заключения подходит для использования в контакте с тканями людей и других млекопитающих без чрезмерной токсичности, раздражения, аллергической реакции и так далее и соизмерим с допустимым соотношением польза/риск."Фармацевтически приемлемая соль" означает любую подходящую соль, которая при введении реципиенту способна привести к образованию либо прямым, либо косвенным образом соединения данного изобретения. "Фармацевтически приемлемый противоион" означает ионную часть соли, которая не является токсичной при высвобождении из соли при введении реципиенту. Кислоты, обычно используемые для получения фармацевтически приемлемых солей, включают неорганические кислоты, такие как сероводородная кислота, хлористо-водородная кислота, бромистоводородная кислота, йодисто-водородная кислота, серная кислота и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфокислота, салициловая кислота, винная кислота, дивинная кислота, аскорбиновая кислота, малеиновая кислота, бензолсульфоновая кислота, фумаровая кислота,глюконовая кислота, глюкуроновая кислота, муравьиная кислота, глутаминовая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, молочная кислота, щавелевая кислота, пара-бромфенилсульфоновая кислота, угольная кислота, янтарная кислота, лимонная кислота,бензойная кислота и уксусная кислота, а также родственные и органические кислоты. Таким образом,подобные фармацевтически приемлемые соли включают сульфат, пиросульфат, гидросульфат, сульфит,гидросульфит, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид,йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себацат, фумарат, малеат, бутин-1,4-диоат, гексин-1,4-диоат,бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, сульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гидроксибутират, гликолят, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат,нафталин-2-сульфонат, манделат и другие соли. В одном из вариантов осуществления фармацевтически приемлемые соли присоединения кислоты включают соли, образованные минеральными кислотами, такими как хлористо-водородная кислота и бромисто-водородная кислота, а особенно соли, образованные органическими кислотами, такими как малеиновая кислота. Использованный здесь термин "устойчивые соединения" относится к соединениям, которые обладают достаточной устойчивостью, чтобы допустить их получение, и которые сохраняют целостность соединения в течение периода времени, достаточного для того, чтобы быть применимыми в описанных здесь целях (например, составление из них терапевтических продуктов, промежуточных соединений для использования при получении терапевтических соединений, выделяемых или хранящихся промежуточных соединений, лечение заболевания или состояния, поддающихся действию терапевтических агентов)."Стереоизомер" представляет собой как энантиомеры, так и диастереомеры. "D" представляет собой дейтерий. Каждый из "трет-", "т-" и "т-" обозначает собой третичный. США - Соединенные Штаты Америки. "FDA" обозначает Управление по контролю за пищевыми продуктами и медикаментами. "NDA" обозначает применение новых лекарственных средств, "кт" и "КТ" - комнатная температура; "ч" - часы. В данной спецификации на переменную величину могут быть общие ссылки (например, "каждыйR") или могут быть конкретные ссылки (например, R1 или R2). Если не указано иначе, при общей ссылке на переменную величину, подразумевается, что она включает все конкретные варианты осуществления этой конкретной переменной величины. Терапевтические соединения В настоящем изобретении предоставлено соединение формулы I или его фармацевтически приемлемой соли, в которойR1 выбирают из СН 3, CD3, -O-CD2CD3 или -O-CD(CD3)2; аR2 выбирают из СН 3 или CD3 ; при условии, что по меньшей мере один из R1 и R2 является дейтерированным. В предпочтительном варианте в соединении формулы I R1 представляет собой -O-CD2CD3 или -OCD(CD3)2. Ещ более предпочтительно, когда R1 представляет собой -O-CD(CD3)2. В предпочтительном варианте соединение формулы I выбирают из любого соединения, приведенного в представленной ниже таблице: или его фармацевтически приемлемой соли. В предпочтительном варианте в соединении формулы I R1 представляет собой -CD3. Предпочтительно соединение формулы I представляет собой любое из или его фармацевтически приемлемую соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. В предпочтительном варианте любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности. Синтез соединения формулы I можно легко осуществить, обратившись к описанным здесь иллюстративным синтезам и примерам, и при использовании методик и промежуточных соединений, аналогичных промежуточным соединениям, описанным, например, у Schnider О.Grussner A., Helv. Chim. Acta.,1951, 34: 2211; Grussner A.Schnider O.; GB 713146 (1954); Toyo Pharma K. K., Japan JP 60089474 A(1983); Newman A. H. et al., J. Med. Chem., 1992, 35: 4135. Подобные способы можно осуществить при использовании соответствующих дейтерированных и, необязательно, других изотопсодержащих реагентов и/или промежуточных соединений, для синтеза описанных здесь соединений, или применяя стан-6 019343 дартные методы синтеза, известные в данной области, для введения изотопных атомов в химическую структуру. Иллюстративные синтезы Следующие дейтерированные реагенты и блок-реагенты, которые можно использовать для получения соединений формулы I, являются коммерчески доступными: этил-d5 йодэтан, этил-2,2,2-d3 йодид,этил-1,1-d2 йодид, изопропил-d7 йодид, изопропил-d7 бромид, изопропил-1,1,1,3,3,3-d6 йодид и изопропил-1,1,1,3,3,3-d6 бромид. Подходящий способ синтеза соединений формулы I, в которых R1 представляет собой -О-(С 2 С 4)алкил, приведен на схеме 1. Схема 1. Синтез соединения формулы I (R1 представляет собой -О-(С 2-С 4)алкил) В результате обработки известного 17-этоксикарбонил-3-метоксиморфинана (10) (по поводу его получения см. Murdter Т. Е. et al., Journal of Labelled Compounds and Radiopharmaceuticals 2002, 45: 11531158) трибромидом бора согласно методике, описанной Newman A. H. et al. , Journal of Medicinal Chemistry 1992, 35: 4135-4142, получают 17-этоксикарбонил-3-гидроксиморфинан (11). В результате обработки 3-гидроксиморфинана 11 соответствующим образом дейтерированным алкилйодидом в присутствии карбоната калия по методике, которая аналогична методике в описанной выше статье, образуются дейтерированные 17-этоксикарбонил-3-алкоксиморфинаны (12). Восстановление карбамата морфинана 12 либо алюмогидридом лития, либо алюмодейтеридом лития в ТГФ (THF) по методике, аналогичной методике Newman, приводит к дейтерированному 3-алкокси 17-метилморфинановому (13) или 3-алкокси-17-тридейтерометилморфинановому (14) соединениям формулы I соответственно. Подходящий способ синтеза соединений формулы I, в которых R1 представляет собой -(С 1 С 4)алкил, приведен на схеме 2. Схема 2. Синтез соединения формулы I (R1 представляет собой -(C1-С 4)алкил)N-фенилтрифторметансульфонимидом согласно методике, описанной Kim С.-Н. в патенте США 2005/0256147 А 1, образуется соответствующий трифлат фенола (15). В результате катализируемого палладием кросс-сочетания 15 с соответствующим образом дейтерированной (C1-C4)алкилбороновой кислотой (16) по аналогии с методикой из упомянутого выше патента, получают дейтерированные 17-этоксикарбонил-3-(C1C4)алкилморфинаны (17). Восстановление карбамата морфинана 17 либо алюмогидридом лития, либо алюмодейтеридом лития в ТГФ по аналогии с методикой, описанной Newman et al. , Journal of MedicinalChemistry 1992, 35: 4135-4142, приводит к дейтерированным 3-(C1-C4)алкил-17-метилморфинановым или 3-(C1-C4)алкил-17-дейтерометилморфинановым соединениям формулы I соответственно. Реагент алкилбороновой кислоты 16, использованный в схеме 2, получают, как описано далее в схеме 3. Схема 3. Синтез эфира алкилбороновой кислоты 16 В результате обработки соответствующим образом дейтерированного (C1-C4)алкилгалогенида (20) элементарным литием в пентане по аналогии с методикой, описанной Dawildowski D. et al., в WO 2005/082911 A1, получают соответствующий анион (С 1-С 4)алкиллития, который можно немедленно обработать триизопропилборатом с последующим гидролизом водным хлористым водородом по аналогии с методикой, описанной Brown H. C. et al., Organometallics 1985, 4: 816-821, получая соответствующим образом дейтерированные (С 1-С 4)алкилбороновые кислоты (16). Приведенные выше конкретные способы и соединения не подразумеваются как ограничивающие. Химические структуры в приведенных здесь схемах изображают переменные величины, которые таким образом соответственно определены при помощи определений химических групп (фрагменты, атомы и так далее) в соответствующем положении в формулах соединений настоящего описания, независимо от того, идентифицированы они названием той же переменной величины (то есть R1 или R2) или нет. Вопрос о пригодности химической группы в структуре соединения для использования в синтезе другого соединения находится в пределах эрудиции специалиста в данной области. Другие способы синтеза соединений формулы I и их синтетических предшественников, включая те из них, подходы к которым не показаны в подробностях на приведенных здесь схемах, находятся в пределах возможностей химиков стандартной квалификации в данной области. Превращения синтетической химии и методология защитных групп (введение и снятие защиты), применимые при синтезе используемых соединений, известны в данной области и включают, например, превращения и методологии, описанные в Larock R., Comprehensive Organic Transformations, VCH Publishers (1989); Greene T. W. et al., Protective Groups in Organic Synthesis, 3rd Ed., John Wiley and Sons (1999); Fieser L. et al., Fieser and Fieser's Reagents for Organic Synthesis,John Wiley and Sons (1994); and Paquette L., ed., Encyclopedia of Reagents for Organic Synthesis, John Wileyand Sons (1995) и его последующих изданиях. Комбинации заместителей и переменных величин, представленных данным изобретением, являются только комбинациями, которые приводят к образованию стабильных соединений. Композиции В изобретении предоставлены также композиции, не содержащие пирогенных веществ, включающие соединение формулы I (например, включая любую из приведенных в настоящем описании формул) или фармацевтически приемлемую соль указанного соединения, и приемлемый носитель. В одном из вариантов осуществления композиция включает эффективное количество данного соединения или его фармацевтически приемлемой соли. Предпочтительно композиция данного изобретения составлена для фармацевтического использования ("фармацевтическая композиция"), где носитель представляет собой фармацевтически приемлемый носитель. Носитель(и) являются "приемлемыми" в смысле совместимости с другими ингредиентами состава и, в случае фармацевтически приемлемого носителя, не опасными для его реципиента в количестве, которое используется в лекарственном препарате. Фармацевтически приемлемые носители, адъюванты и наполнители, которые можно использовать в фармацевтических композициях данного изобретения, включают, без ограничения перечисленным, иониты, оксид алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновая кислота, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, вода, соли или электролиты, такие как сульфат протамина, динатрия гидрофосфат, гидрофосфат калия, хлорид натрия, соли цинка, коллоидный оксид кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы,полиэтиленгликоль,натрийкарбоксиметилцеллюлозу,полиакрилаты,воски,полиэтиленполиоксипропиленовые блок-сополимеры, полиэтиленгликоль и ланолин. В случае необходимости, растворимость и биодоступность соединений настоящего изобретения в фармацевтических композициях можно повысить при помощи методов, хорошо известных в данной области. Один из методов включает использование в препарате липидных эксципиентов. См. "Oral LipidBased Formulations: Enhancing the Bioavailability of Poorly Water-Soluble Drugs (Drugs and the Pharmaceutical Sciences)", David J. Hauss, ed. Informa Healthcare, 2007; и "Role of Lipid Excipients in Modifying Oral andParenteral Drug Delivery: Basic Principles and Biological Examples", Kishor M. Wasan, ed. Wiley-Interscience,2006. Другой известный метод повышения биодоступности заключается в использовании аморфной формы соединения данного соединения, необязательно составленной с полоксамером, таким как LUTROL и PLURONIC (корпорация BASF) или блок-сополимеров этиленоксида и пропиленоксида. См. патент Соединенных Штатов 7014866 и патентные публикации Соединенных Штатов 20060094744 и 20060079502. Фармацевтические композиции данного изобретения включают композиции, подходящие для перо-8 019343 рального, ректального, назального, местного (включая буккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и интрадермальное) введения. В некоторых вариантах осуществления соединение приведенных здесь формул вводят чрескожно (например, при использовании чрескожного пластыря или методик ионтофореза). Другие составы можно подходящим образом представить в виде разовой дозы, например таблеток, капсул с продолжительным высвобождением и в виде липосом, и можно изготовить любым способом, известным в области фармации. См., например, Remington: The Science and Practice of Pharmacy, Lippincott WilliamsWilkins, Baltimore,MD (20th ed. 2000) . Подобные препаративные способы включают стадию соединения с молекулой, подлежащей введению, ингредиентов, таких как носитель, составляющий один или более дополнительных ингредиентов. В общем, композиции получают путем равномерного и близкого соединения активных ингредиентов и жидких носителей, липосом или тонкоизмельченных твердых носителей или и тех, и других, а затем, при необходимости, формования продукта. В некоторых вариантах осуществления соединение вводят перорально. Композиции настоящего изобретения, подходящие для перорального введения, можно представить в виде дискретных единиц,таких как капсулы, пакетики или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента, порошка или гранул, раствора или суспензии в водной жидкости или неводной жидкости, или жидкой эмульсии масло-в-воде, жидкой эмульсии вода-в-масле, упакованных в липосомы, или в виде болюса и так далее. Для содержания подобных суспензий могут оказаться применимыми мягкие желатиновые капсулы, которые могут выгодно повысить скорость абсорбции соединения. В случае таблеток для перорального использования, обычно используемые носители включают лактозу и кукурузный крахмал. Кроме того, обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в форме капсулы применимые разбавители включают лактозу и высушенный кукурузный крахмал. При пероральном введении водных суспензий активный ингредиент соединяют с эмульгирующими и суспендирующими агентами. При желании можно добавить некоторые подслащивающие и/или вкусовые, и/или окрашивающие агенты. Композиции, подходящие для перорального введения, включают леденцы, содержащие ингредиенты во вкусовой основе, обычно сахарозе, или аравийской или трагакантовой камеди, и пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахарозе, или аравийской камеди. Композиции, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные вещества, бактериостаты и растворенные вещества, придающие данному составу изотоничность с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загущающие агенты. Данные составы можно представить в виде контейнеров с разовой дозой или многократными дозами, например запаянных ампул и сосудов, и можно хранить в высушенном лиофильной сушкой (лиофилизированном) состоянии, с необходимостью лишь добавить стерильный жидкий носитель, например воду для инъекций, непосредственно перед использованием. Приготовленные для немедленного приема растворы для инъекций и суспензии можно получить из стерильных порошков, гранул и таблеток. Подобные растворы для инъекций могут иметь вид, например, стерильной водной или масляной суспензии для инъекций. Данную суспензию можно составить в соответствии с известными в данной области техники методиками с использованием подходящих диспергирующих или увлажняющих агентов (например, таких как Tween 80) и суспендирующих агентов. Стерильный препарат для инъекций может также представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально-приемлемом разбавителе или растворителе, например, раствор в 1,3-бутандиоле. В число приемлемых носителей и растворителей, которые можно использовать, входят маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды удобно использовать стерильные нелетучие масла. Для этой цели можно использовать любое мягкое нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, применимы для получения препаратов для инъекций, как и природные фармацевтически приемлемые масла, такие как оливковое или касторовое масло, в особенности в их полиоксиэтилированном варианте. Данные растворы или суспензии в масле могут также включать разбавитель или диспергатор на основе длинноцепочечного спирта. Фармацевтические композиции данного изобретения можно вводить в виде суппозиториев для ректального введения. Данные композиции можно получить, смешивая соединение данного изобретения с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре, и, следовательно, будет плавиться в прямой кишке, высвобождая активные компоненты. Подобные вещества включают, без ограничения перечисленным, масло какао,пчелиный воск и полиэтиленгликоли. Фармацевтические композиции данного изобретения можно вводить при помощи назального аэрозоля или ингаляции. Подобные композиции получают в соответствии с методиками, которые хорошо известны в области фармацевтических препаратов, и могут быть получены в виде растворов в физиологическим растворе с использованием бензилового спирта или других подходящих консервантов, усилителей абсорбции для повышения биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих агентов, известных в данной области техники. См., например: Raboniwitz J.D. и Zaffaroni А. С, патент США 6803031, Alexza Molecular Delivery Corporation. Местное введение фармацевтических композиций данного изобретения особенно полезно в том случае, когда желательная обработка включает области или органы, легкодоступные для местного применения. Для местного применения, локально для кожи, фармацевтическая композиция должна быть составлена с подходящей мазью, содержащей активные компоненты, суспендированные или растворенные в носителе. Носители для местного введения соединений данного изобретения включают, но без ограничения указанным, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтиленовое полиоксипропиленовое соединение, эмульгирующий воск и воду. Альтернативным образом,фармацевтическую композицию можно составить с подходящим лосьоном или кремом, содержащим активное соединение, суспендированное или растворенное в носителе. Подходящие носители включают, но без ограничения указанным, минеральное масло, сорбитанмоностеарат, полисорбат 60, воск на основе цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол,бензиловый спирт и воду. Фармацевтическую композицию данного изобретения можно также применять местно к нижнему отделу кишечного тракта при помощи препарата ректального суппозитория или в виде подходящего клизменного препарата. В данное изобретение также включены местно-чрескожные пластыри и ионтофоретическое введение. Применение рассматриваемых терапевтических средств может быть локальным для того, чтобы вводить их в интересующую область. Для предоставления обсуждаемых композиций в интересующую область можно использовать различные методики, такие как инъекцию, использование катетеров, троакаров, прожектилей, геля Pluronic, стентов, полимеров с пролонгированным высвобождением лекарственного вещества или другого устройства, предусмотренного для внутреннего доступа. Таким образом, соединения данного изобретения могут быть включены в композиции для покрытия имплантируемого медицинского устройства, такого как протезы, искусственные клапаны, сосудистые протезы, стенты или катетеры. Подходящие покрытия и общее получение имплантируемых устройств с покрытием известны в данной области техники и их примеры приведены в патентах США 6099562,5886026 и 5304121. Данные покрытия представляют собой обычно биосовместимые полимерные вещества, такие как гидрогелевый полимер, полиметилдисилоксан, поликапролактон, полиэтиленгликоль, полимолочная кислота, этиленвинилацетат и их смеси. На покрытия необязательно может быть дополнительно нанесено верхнее покрытие из слоя фторсиликона, полисахаридов, полиэтиленгликоля, фосфолипидов или их смесей для придания композиции характеристик регулируемого высвобождения. Покрытия для инвазивных устройств должны быть включены в определение фармацевтически приемлемого носителя, адъюванта или среды, как эти термины используются в настоящем описании. В настоящем описании представлен способ нанесения покрытия на имплантируемое медицинское устройство, включающий стадию контакта указанного устройства с описанной выше композицией покрытия. Специалистам в данной области техники будет очевидно, что нанесение покрытия на данное устройство будет происходить перед имплантацией млекопитающему. В настоящем описании представлен способ пропитки имплантируемого устройства, высвобождающего лекарственное вещество, включающий стадию контакта указанного устройства, высвобождающего лекарственное вещество, с соединением или композицией данного изобретения. Имплантируемые устройства, высвобождающие лекарственное вещество, включают, но не ограничиваются указанным, капсулы или шары из биоразлагаемого полимера, неразлагаемые, диффундирующие полимерные капсулы и биоразлагаемые полимерные пластины. В настоящем описании представлено имплантируемое медицинское устройство с покрытием из соединения или композиции, содержащей соединение данного изобретения, такое, что указанное соединение является терапевтически активным. В настоящем описании представлено имплантируемое устройство, высвобождающее лекарственное вещество, пропитанное или содержащее соединение или композицию, включающую соединение данного изобретения, так что указанное соединение высвобождается из указанного устройства и является терапевтически активным. В случае, когда орган или ткань доступны по причине извлечения из субъекта, подобный орган или ткань можно погрузить в среду, содержащую композицию данного изобретения, композицией данного изобретения можно намазать данный орган или композицию данного изобретения можно применить любым другим удобным способом. Соединение настоящего изобретения присутствует в составе фармацевтической композиции в эффективном количестве. Использованный здесь термин "эффективное количество" относится к такому количеству, которого при введении в надлежащем режиме дозирования достаточно для снижения или уменьшения тяжести,продолжительности или развития нарушения, которое лечат, предупреждения развития нарушения, ко- 10019343 торое лечат, вызывания регресса нарушения, которое лечат, или усиления, или улучшения профилактического или терапевтического эффекта(ов) другого вида терапии. Взаимосвязь дозировок для животных и людей (на основе миллиграмм на квадратный метр поверхности тела) описана Freireich et al., (1966) Cancer Chemother. Rep. 50:219. Площадь поверхности тела можно приблизительно определить, исходя из высоты и массы субъекта. См., например, Scientific Tables,Geigy Pharmaceuticals, Ardsley N. Y., 1970, 537. В одном из вариантов осуществления эффективное количество соединения данного изобретения может изменяться в интервале от 0,4 до 400 мг, от 4,0 до 350 мг, от 10 до 90 мг или от 30 до 45 мг, включительно, которое может быть дано один, два или три раза в день в зависимости от различных факторов,принимаемых во внимание специалистами в данной области. Кроме того, эффективные дозы будут изменяться, как признают специалисты в данной области, в зависимости от заболеваний, которые лечат, тяжести заболевания, способа введения, пола, возраста и общего состояния здоровья субъекта, использования эксципиентов, возможности совместного использования с другими видами терапевтического лечения, такими как использование других агентов, и заключения лечащего врача. Например, руководство для выбора эффективной дозы можно определить, обратившись к предписанной информации для декстрометорфана. Способы лечения Согласно варианту осуществления в изобретении предоставлен способ лечения субъекта, страдающего или склонного к заболеванию или состоянию, которое преимущественно лечится декстрометорфаном, включающий стадию введения указанному субъекту эффективного количества соединения формулы I, или его фармацевтически приемлемой соли, или композиции, содержащей указанное соединение. Такие заболевания или состояния хорошо известны в данной области и описаны, но не ограничиваются заболеваниями или состояниями, описанными в патентах США 4316888, 4446140, 4694010, 4898860,5166207, 5336980, 5350756, 5366980, 5863927, RE38115, 6197830, 6207164, 6583152 и 7114547, а также в патентных публикациях США 2001/0044446, 2002/0103109, 2004/0087479, 2005/0129783, 2005/0203125 и 2007/0191411. Такие заболевания или состояния включают, но не ограничиваются перечисленным, эмоциональную неустойчивость; псевдобульбарный аффект; аутизм; неврологические нарушения и нейродегенеративные заболевания, например, такие как слабоумие, боковой амиотрофический склероз (ALS, известный также как болезнь Лу Герига), болезнь Альцгеймера и рассеянный склероз; нарушения, связанные с расстройством сознания; сердечно-сосудистые заболевания, например, такие как болезнь периферических сосудов, удар, инфаркты миокарда и атеросклероз; глаукому, позднюю дискинезию; диабетическую невропатию; ретинопатические заболевания; заболевания или нарушения, вызванные апоптозом, индуцированным гомоцистеином; заболевания или нарушения, вызванные повышенными уровнями гомоцистеина; боль, включая, но не ограничиваясь перечисленным, хроническую боль; неустранимую боль; невропатическую боль, симпатически опосредуемую боль, например, такую как аллодиния, гиперпатия,гипералгезия, дизестезия, парестезия, деафферентационная боль и боль при болезненной анестезии; боль,связанную с желудочно-кишечной дисфункцией, включая, например, синдром раздраженного кишечника; и боль в ротовой полости; эпилептические припадки; звон в ушах; сексуальную дисфункцию; трудноизлечимый кашель; дерматит; нарушения, связанные с аддикциями, например, такие как аддикция или зависимость от стимуляторов, никотина, морфина, героина, других опиатов, амфетаминов, кокаина и алкоголя; синдром Ретта (RTT); нарушения в работе голосовых связок вследствие неконтролируемых спазмов гортанных мышц, включая, например, абдукторную спастическую дисфонию, аддукторную спастическую дисфонию, дисфонию при мышечном напряжении и тремор голосовых связок; нейротоксичность метотрексата; усталость, вызванную раком; и состояния, вызванные воздействием химических агентов. Предпочтительно применять указанный выше способ, когда субъект страдает от или склонен к диабетической невропатической боли или к эпилептическим припадкам. Другим объектом настоящего изобретения является способ лечения субъекта, страдающего от или подверженного условиям, относящимся к воздействию химических веществ, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции согласно настоящему изобретению. Ещ одним объектом изобретения является способ лечения субъекта, страдающего от или склонного к боли, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества вышеуказанной фармацевтической композиции. Диагностические методы и наборы Соединения и композиции данного изобретения также применимы в качестве реагентов в способах определения концентрации декстрометорфана в растворе или биологическом образце, таком как плазма крови, при исследовании метаболизма декстрометорфана и других аналитических исследованиях. В настоящем описании представлен способ определения концентрации недейтерированного аналога соединения формулы I в растворе или биологическом образце, который включает стадии:a) прибавления соединения формулы I в известной концентрации к раствору биологического образ- 11019343b) подвергания раствора или биологического образца действию измерительного устройства, отличающего соответствующий недейтерированный аналог от соединения формулы I;c) калибровки данного измерительного устройства для корреляции определенного количества соединения формулы I с известной концентрацией соединения формулы I, добавленной к биологическому образцу или раствору, иd) определения количества соответствующего недейтерированного аналога в биологическом образце при помощи указанного измерительного устройства иe) определения концентрации соответствующего недейтерированного аналога в растворе или образце при помощи корреляции между количеством и концентрацией, полученной для соединения формулы I. Измерительные устройства, которые способны отличить соответствующий недейтерированный аналог от соединения формулы I, включают любое измерительное устройство, которое может различить два соединения, отличающиеся друг от друга изотопной распространенностью. Примеры измерительных устройств включают масс-спектрометр, ЯМР-спектрометр или ИК-спектрометр. В другом варианте осуществления предоставлен способ определения количества недейтерированного аналога соединения формулы I в растворе или биологическом образце, который включает:a) добавление известного количества соединения формулы I к раствору или биологическому образцу;b) детектирование по меньшей мере одного сигнала соединения формулы I и по меньшей мере одного сигнала соответствующего недейтерированного аналога при помощи измерительного устройства,способного различать два этих соединения;c) корреляцию по меньшей мере одного сигнала, детектированного для соединения формулы I, с известным количеством соединения формулы I, добавленного к раствору или биологическому образцу; иd) определение количества соответствующего недейтерированного аналога в растворе или биологическом образце с использованием корреляции между по меньшей мере одним сигналом, детектированным для соединения формулы I, и количеством соединения формулы I, добавленным к раствору или биологическому образцу. В настоящем описании представлен способ оценки метаболической стабильности соединения формулы I, включающий стадии контакта соединения формулы I с источником метаболизирующего фермента в течение некоторого промежутка времени, и сравнение количества соединения формулы I с продуктами метаболизма соединения формулы I после данного промежутка времени. В родственном варианте представлен способ оценки метаболической стабильности соединения формулы I в субъекте после введения соединения формулы I. Данный способ включает стадии получения образцов сыворотки, крови, ткани, мочи или кала субъекта в течение некоторого промежутка времени после введения субъекту соединения формулы I, и сравнение количества соединения формулы I с продуктами метаболизма соединения формулы I в образце сыворотки, крови, ткани, мочи или кала. В настоящем описании также представлены наборы реагентов для использования при лечении псевдобульбарного нарушения, диабетической невропатии, синдрома Ретта (RTT), нарушений голосовых связок вследствие неконтролируемых спазмов гортанных мышц, включая, например, абдукторную спастическую дисфонию, аддукторную спастическую дисфонию, дисфонию при мышечном напряжении и тремор голосовых связок; нейротоксичности метотрексата и усталости, вызванной раком. Данные наборы включают: а) фармацевтическую композицию, содержащую соединение формулы I, или его соль, где указанная композиция находится в контейнере, и b) инструкции, описывающие способ использования данной фармацевтической композиции для лечения псевдобульбарного аффекта, диабетической невропатии, синдрома Ретта (RTT), нарушений голосовых связок вследствие неконтролируемых спазмов гортанных мышц, включая, например, абдукторную спастическую дисфонию, аддукторную спастическую дисфонию, дисфонию при мышечном напряжении и тремор голосовых связок; нейротоксичности метотрексата и усталости, вызванной раком. Контейнер может представлять собой любой сосуд или другое герметизированное или герметизируемое приспособление, которое может вмещать указанную фармацевтическую композицию. Примеры включают бутылки, ампулы, бутылки с делениями или содержащие много отсеков, где в каждом отделении или отсеке содержится разовая доза указанной композиции, пакет из фольги с отделениями, в котором в каждом отделении содержится разовая доза указанной композиции или дозатор, который дозирует разовые дозы указанной композиции. Данный контейнер может иметь любой удобный вид или форму,известные в данной области, изготовленную из фармацевтически приемлемого материала, например бумажной или картонной коробки, стеклянной или пластиковой бутылки, или банки, или повторно герметизируемого мешка (например, для вмещения таблеток "повторного заполнения" для помещения в другой контейнер), или блистерной упаковки с индивидуальными дозами для выдавливания из данной упаковки в соответствии с терапевтическим режимом. Используемый контейнер может зависеть от конкретной содержащейся дозированной формы, например, обычная картонная коробка, как правило, не будет использоваться для помещения в нее жидкой суспензии. Для продажи разовой дозы допускается совме- 12019343 стное использование более чем одного контейнера в одной упаковке. Например, таблетки могут содержаться в бутылке, которая, в свою очередь, помещена в коробку. В одном из вариантов осуществления контейнер представляет собой блистерную упаковку. Наборы могут также включать устройство для введения или для отмеривания разовой дозы фармацевтической композиции. Подобное устройство может включать ингалятор, если указанная композиция представляет собой композицию для ингаляции, шприц и иглу, если указанная композиция представляет собой композицию для инъекции, шприц, ложку, насос или сосуд, имеющий или не имеющий маркировки объема, если указанная композиция представляет собой пероральную жидкую композицию или любое другое устройство для измерения или доставки, подходящее для находящегося в наборе дозированного препарата композиции. В варианте осуществления наборов композиция, включающая второй активный агент, может находиться в сосуде или контейнере, отделенном от сосуда, в котором содержится композиция, включающая соединение формулы I. Примеры Пример 1. Синтез гидрохлорида (+)-3-(этокси-d5)-17-(метил-d3)-(9,13,14)морфинана (100). Соединение получают, как схематично показано далее. Подробности синтеза приведены ниже. Синтез (+)-3-метокси-17-метил-(9,13,14)морфинана (свободное основание, 8). В реакционную колбу добавляли соль HBr (+)-3-метокси-17-метил-(9,13,14)морфинана (7; 3,00 г, 8,5 ммоль), NH3 в CH3OH (2,0 М, 8,5 мл, 17,0 ммоль) и мешалку. Реакционную смесь перемешивали при кт в течение 1 ч. Полученное вещество концентрировали на роторном испарителе, затем разбавлялиCHCl3 (50 мл) и H2O (50 мл). Слои разделяли и экстрагировали водный слой CHCl3 (50 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали на роторном испарителе, получая 2,88 г 8 в виде пушистого твердого вещества белого цвета. 1(дд, J1=8,2, J2=2,7, 1H), 6,80 (д, J2=2,7, 1H), 7,02 (д, J=8,8, 1H). Синтез (+)-3-метокси-(9,13,14)морфинана (9). Твердый (+)-3-метокси-17-метил-(9,13,14)морфинан (8; 6,79 г, 25,1 ммоль) помещали в реакционную колбу с CHCl3 и мешалкой. Добавляли K2CO3 (13,85 г, 100,2 ммоль) и перемешивали смесь при комнатной температуре в атмосфере N2 в течение 10 мин перед добавлением хлористого ацетила (7,866 г,100,2 ммоль). Полученную реакционную смесь, все еще в атмосфере N2, перемешивали в условиях кипения в течение 7 ч, затем фильтровали через слой целита. Органический фильтрат концентрировали на роторном испарителе, а полученное сырое вещество растворяли в СН 3 ОН, затем перемешивали в условиях кипения с обратным холодильником в течение 1 ч. Раствор концентрировали на роторном испарителе,затем сушили в вакууме, получая 6,78 г 9 в виде твердого вещества почти белого цвета. 1CHCl3 (100 мл). Прибавляли диизопропилэтиламин (DIEA; 16,32 г, 126,3 ммоль) и перемешивали смесь в течение 10 мин при комнатной температуре в атмосфере азота перед прибавлением этилхлорформиата(13,094 г, 76,8 ммоль). Реакционную смесь перемешивали в условиях кипения в атмосфере азота в течение 3 ч, в это время методом ТСХ (20% этилацетат/гексан) было показано полное расходование исходного вещества. Органический слой отделяли и промывали сначала 1 М HCl, а затем насыщенным NaHCO3. Водные слои от каждого промывания объединяли и снова экстрагировали 50 мл CHCl3. Органический слой от повторной экстракции объединяли с органическим слоем от промываний и сушили объединенные органические слои над Na2SO4. Затем органический раствор фильтровали, концентрировали на роторном испарителе, потом очищали при помощи автоматической колоночной флэш-хроматографии (030% этилацетат/гексан), получая 5,37 г 10 в виде прозрачного масла светло-желтого цвета. 1(20 мл) и охлаждали полученный раствор до 0 С. Прибавляли BBr3 (9,24 г, 36,9 ммоль) и перемешивали реакционную смесь в атмосфере N2 при 0 С в течение 20 мин (в это время методом ТСХ в 20% смеси этилацетат/гексан было показано завершение реакции). В лабораторный стакан с мешалкой помещали раствор 27% NH4OH и медленно прибавляли реакционную смесь при перемешивании. Полученную смесь перемешивали в течение 20 мин, затем экстрагировали смесью 4:1 CHCl3/СН 3 ОН. Органический слой сушили над Na2SO4, фильтровали, затем концентрировали на роторном испарителе. Сырое вещество очищали при помощи автоматической колоночной флэш-хроматографии (СН 3 ОН с 1% NH4OH/CHCl3, 010%). Чистые фракции концентрировали на роторном испарителе, получая 1,48 г 11 в виде твердого вещества белого цвета. 1H-ЯМР (300 МГц, CDCl3):1,04-1,12 (м, 1 Н), 1,22-1,36 (м, 7 Н), 1,45-1,59 (м, 3 Н), 1,63-1,67 (м, 2 Н),2,30-2,33 (м, 1 Н), 2,52-2,66 (м, 2 Н), 3,06 (дт, J1=18,4, J2=5,9, 1H), 3,84 (ддд, J1=35,8, J2=13,8, J3=6,1, 1H),4,10-4,18 (м, 2 Н), 4,31 (дт, J1=53,9, J2=3,l, 1H), 6,64 (M, 1H), 6,78 (с, 1 Н), 6,93 (кажущийся т, J=7,8, 1H). Синтез (+)-3-(этокси-d5)-17-этоксикарбонил-(9,13,14)морфинана (20). К раствору спирта 11 (1,50 г, 4,8 ммоль) в ДМФА (25 мл) прибавляли при перемешивании K2CO3(2,00 г, 14,5 ммоль, 3,05 экв.) и йодистый этил-d5 (1,15 г, 7,1 ммоль, 1,50 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре (кт) в атмосфере N2, гасили добавлением H2O и экстрагировали Et2O (330 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме, получая масло желтого цвета. В результате очистки автоматизированной колоночной флэш-хроматографией (0-40% EtOAc/гексаны) получали промежуточное соединение 20(1,53 г, выход 91%). Синтез гидрохлорида (+)-3-(этокси-d5)-17-(метил-d3)-(9,13,14)морфинана (100). К суспензии LiAlD4 (0,184 г, 4,4 ммоль, 2,0 экв.) в ТГФ (10 мл), перемешиваемой при -78 С, прибавляли раствор карбамата 20 (0,763 г, 2,2 ммоль) в ТГФ (5 мл). После 1 ч перемешивания при кт методом ТСХ реакции не фиксировали и добавляли еще 2,0 экв. LiAlD4 (0,184 г, 4,4 ммоль, 2,0 экв.). Реакционную смесь перемешивали в течение ночи при кт, затем гасили добавлением гептагидрата сульфата магния до прекращения выделения газа. Смесь фильтровали, концентрировали в вакууме и очищали полученное сырое вещество автоматизированной колоночной флэш-хроматографией(CHCl3/CH3OH/NH3OH - 90/10/1), получая свободный амин 100. Данное вещество растворяли в 1,25 МHCl в СН 3 ОН, затем концентрировали при пониженном давлении и сушили в высоком вакууме, получая 14,3 мг продукта 100 в виде соли HCl. 1 Н ЯМР (300 МГц, ДМСО-d6):0,94-1,63 (м, 8 Н), 1,72-1,80 (м, 1 Н), 1,94 (д, J=11,9, 1 Н), 2,43-2,47 (м,1 Н), 2,96 (дд, J1=19,2, J2=6,1, 2 Н , 3,09-3,17 (м, 2 Н), 3,57-3,61 (м, 1 Н), 6,79-6,82 (м, 2 Н), 7,11 (д, J=8,8, 1 Н),9,58 (ушир.с, 1 Н). ВЭЖХ (метод: колонка 150 мм C18-RP - метод градиента 5-95% ACN; длина волны: 280 нм): время удерживания: 3,08 мин, чистота: 95%. МС (М+Н): 294,2. Пример 2. Синтез гидрохлорида (+)-3-(этокси-d5)-17-метил-(9,13,14)морфинана (104). Соединение 104 получали, как описано в приведенном выше примере 1, за исключением того, что для восстановления карбамата 20 в 104 вместо LiAlD4 использовали LiAlH4. Синтез гидрохлорида (+)-3-(этокси-d5)-17-метил-(9,13,14)морфинана (104). К суспензии LiAlH4 (0,166 г, 4,4 ммоль, 2,0 экв.) в ТГФ (10 мл), перемешиваемой при -78 С, прибавляли раствор карбамата 20 (0,763 г, 2,2 ммоль) в ТГФ (5 мл). Спустя 1 ч добавляли еще 2,0 экв. LiAlH4(0,184 г, 4,4 ммоль, 2,0 экв.). Реакционную смесь перемешивали в течение ночи при кт, затем гасили добавлением гептагидрата сульфата магния до прекращения выделения газа. Смесь фильтровали, концентрировали в вакууме и очищали полученное сырое вещество автоматизированной колоночной флэшхроматографией (CHCl3/CH3OH/NH3OH - 90/10/1), получая свободный амин 104. Данное вещество растворяли в 1,25 М HCl в СН 3 ОН, затем концентрировали при пониженном давлении и сушили в высоком вакууме, получая 31 мг продукта 104 в виде HCl соли. 1 Н ЯМР (300 МГц, ДМСО-d6)0,94-1,64 (м, 8 Н), 1,74-1,82 (м, 1 Н), 1,97 (д, J=12,4, 1 Н), 2,44-2,47 (м,1 Н), 2,81 (с, 3 Н), 2,96 (дд, J1=20,0, J2=5,8, 2 Н), 3,09-3,18 (м, 2 Н), 3,55-3,62 (м, 1 Н), 6,79-6,82 (м, 2 Н), 7,12(д, J=9,1, 1 Н), 9,68 (ушир.с, 1 Н). ВЭЖХ (метод: колонка 150 мм C18-RP - метод градиента 5-95% ACN; длина волны: 280 нм): время удерживания: 3,00 мин, чистота: 95%. МС (М+Н): 291,2. Пример 3. Синтез (+)-3-(изопропокси-d7)-17-(метил-d3)-(9,13,14)морфинана (102). Соединение 102 получали, как описано ниже. Подробности синтеза приведены далее. Синтез (+)-3-(изопропокси-d7)-17-этоксикарбонил-(9,13,14)морфинана (21). К раствору спирта 11 (1,50 г, 4,8 ммоль, полученного согласно примеру 1) в ДМФА (25 мл) прибавляли при перемешивании K2CO3 (2,0 г, 14,5 ммоль, 3,05 экв.) и 2-йодпропан-d7 (0,71 мл, 7,1 ммоль, 1,50 экв.). Реакционную смесь перемешивали в течение ночи при комнатной температуре (кт) в атмосфере N2,гасили добавлением H2O и экстрагировали Et2O (330 мл). Объединенные органические слои сушили надNa2SO4, фильтровали и концентрировали в вакууме, получая бесцветное масло. В результате очистки автоматизированной колоночной флэш-хроматографией (0-40% EtOAc/гексан) получали промежуточное соединение 21 (1,48 г, выход 85%). Синтез (+)-3-(изопропокси-d7)-17-(метил-d3)-(9,13,14)морфинана (102). К суспензии LiAlD4 (0,340 г, 8,1 ммоль, 4,0 экв.) в ТГФ (10 мл), перемешиваемой при -78 С, прибавляли раствор карбамата 21 (0,739 г, 2,0 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивали в течение ночи при кт, затем гасили добавлением гептагидрата сульфата магния до прекращения выделения газа. Смесь фильтровали, фильтрат концентрировали в вакууме и растворяли полученное вещество в СН 3 ОН. Полученный раствор подкисляли фумаровой кислотой до рН 4, что приводило к выпадению соли. Смесь перемешивали в течение 5 мин и добавляли Et2O, чтобы высадить из раствора оставшуюся соль. Соль выделяли фильтрованием и сушили, получая 660 мг конечного продукта 102 в виде соли фумаровой кислоты. 1(дд, J1=8,4, J2=2,4, 1H), 6,79 (д, J1=2,5, 1H), 6,82 (c, 1H), 7,03 (д, J1=8,3, 1H). ВЭЖХ (метод: колонка 150 мм C18-RP - метод градиента 5-95% ACN; длина волны: 280 нм): время удерживания: 3,11 мин, чистота: 95%. МС (М+Н): 310,3. Пример 4. Синтез (+)-3-(изопропокси-d7)-17-метил-(9,13,14)морфинана (106). Соединение 106 получали, как описано в приведенном выше примере 3, за исключением того, что для восстановления карбамата 21 в 106 вместо LiAlD4 использовали LiAlH4. Синтез (+)-3-(изопропокси-d7)-17-метил-(9,13,14)морфинана (106). К суспензии LiAlH4 (0,308 г, 8,1 ммоль, 4,0 экв.) в ТГФ (10 мл), перемешиваемой при -78 С, прибавляли раствор карбамата 21 (0,739 г, 2,0 ммоль) в ТГФ (5 мл). Реакционную смесь перемешивали в течение ночи при кт, затем гасили добавлением гептагидрата сульфата магния до прекращения выделения газа. Смесь фильтровали, фильтрат концентрировали в вакууме и растворяли полученное вещество в СН 3 ОН. Полученный раствор подкисляли фумаровой кислотой до рН 4, что приводило к выпадению соли. Смесь перемешивали в течение 5 мин и добавляли Et2O, чтобы высадить из раствора оставшуюся соль. Соль выделяли фильтрованием и сушили, получая 330 мг конечного продукта 106 в виде соли фумаровой кислоты. 1H-ЯМР (300 МГц, CDCl3):1,09 (квд, J1=12,6, J2=3,8, 1H), 1,22-1,58 (м, 6 Н), 1,65 (д, J=12,6, 1 Н),2,06 (тд, J1=13,5, J2=4,3, 1 Н), 2,20 (д, J=12,4, 1 Н), 2,35 (д, J=13,3, 1H), 2,46-2,53 (м, 1H), 2,78 (с, 3 Н), 2,963,12 (м, 2 Н), 3,25-3,30 (м, 1H), 3,62-3,64 (м, 1H), 6,73 (дд, J1=8,3, J2=2,5, 1H), 6,80 (д, J=2,5, 1H), 6,86 (с,2 Н), 7,05 (д, J=8,3, 1H). ВЭЖХ (метод: колонка 150 мм C18-RP - метод градиента 5-95% ACN; длина волны: 280 нм): время удерживания: 3,18 мин, чистота: 95%. МС (М+Н): 307,4. Пример 5. Синтез (+)-3-(метил-d3)-17-метил-(9,13,14)морфинана (108). Соединение 108 получали, как описано ниже. Подробности синтеза приведены далее. Синтез (+)-17-этилкарбамат-3-трифторметилсульфонилокси-(9,13,14)морфинана (22). К раствору 11 (9 г, 28,6 ммоль, см. пример 1) и триэтиламина (16 мл, 114 ммоль) в СН 2 Сl2 (400 мл) прибавляли N-фенилтрифторметансульфонимид "PhNTf2" (20,7 г, 57,2 ммоль) при охлаждении на бане со льдом. Реакционную смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Смесь разбавляли CH2Cl2 (500 мл) и промывали раствор насыщенным раствором гидрокарбоната натрия, водой и насыщенным раствором соли, затем сушили над сульфатом натрия. После фильтрования и концентрирования при пониженном давлении сырой продукт очищали колоночной хроматографией на силикагеле (этилацетат/гептаны, 0-10%), получая 12 г (94%) 22 в виде прозрачного масла. Синтез (+)-17-этилкарбамат-3-(метил-d3)-(9,13,14)морфинана (23). К раствору 22 (22 г, 43,8 ммоль) в ТГФ (500 мл) прибавляли N-метил-2-пирролидон ("NMP") (26,2 мл, 153,1 ммоль) при температуре окружающей среды. Реакционную смесь дегазировали продувкой N2 в течение 10 мин. Прибавляли ацетилацетонат железа (III) "Fe(acac)3" (1,65 г, 4,4 ммоль) и CD3MgI (1M вEt2O, 53 мл, 47,6 ммоль, Sigma Aldrich, 99% атомн. D) и нагревали реакционную смесь при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали и добавляли воду (500 мл). Слои разделяли и экстрагировали водный слой CH2Cl2 (3100 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (этилацетат/гептаны,0-10%), получая 4 г (94%, из расчета на регенерированное исходное вещество) 23 и 16 г регенерированного 22. Синтез (+)-3-(метил-d3)-17-метил-(9,13,14)морфинана (108). Смесь 23 (1,5 г, 4,8 ммоль) в ТГФ (70 мл) обрабатывали LiAlH4 (1 M B ТГФ, 19,2 мл) при 0 С. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Для гашения реакционной смеси добавляли воду (1 мл), затем NaOH (24%, 10 мл). Смесь перемешивали в течение 30 мин, в течение которых выпадал осадок белого цвета. Данный осадок отфильтровывали и концентрировали фильтрат при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ (см. описанные ниже условия), получая 108. Свободный амин растворяли в МТВЕ (30 мл) и нагревали до кипения с обратным холодильником. Прибавляли по каплям H3PO4 (в изопропаноле), что приводило к образованию осадка белого цвета. Добавление H3PO4 продолжали до тех пор, пока выпадение осадка белого цвета не прекращалось. Осадок отфильтровывали и промывали МТВЕ (100 мл) с получением 1,2 г твердого вещества. Данное вещество перекристаллизовывали из МеОН/МТВЕ с получением 108 в виде фосфатной соли (0,75 г, 47%) . 1 Пример 6. Синтез (+)-3-метил-17-(метил-d3)-(9,13,14)морфинана (109). Соединение 109 получали, как описано ниже. Подробности синтеза приведены далее."NMP" (4,3 мл, 25,1 ммоль) при температуре окружающей среды. Реакционную смесь дегазировали продувкой N2 в течение 10 мин. Прибавляли ацетилацетонат железа (III) "Fe(acac)3" (270 мг, 0,72 ммоль) иMeMgBr (3M в Et2O, 2,9 мл, 7,8 ммоль) и нагревали реакционную смесь при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали и добавляли воду (50 мл). Слои разделяли и экстрагировали водный слой CH2Cl2 (3100 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле (этилацетат/гептаны, 0-10%) с получением 0,84 г 24 (75% из расчета на регенерированное исходное вещество) и 2 г регенерированного 22. Синтез (+)-3-(метил)-17-(метил-d3)-(9,13,14)морфинана (109). Смесь 24 (1 г, 6,2 ммоль) в ТГФ(30 мл) обрабатывали LiAlD4 (0,9 г, 24,8 ммоль, Cambridge Isotopes, 98% атомн. D) при 0 С и оставляли реакционную смесь нагреваться до температуры окружающей среды и перемешивали в течение ночи. Для гашения реакционной смеси добавляли воду (1 мл), затем NaOH (24%, 5 мл). Реакционную смесь перемешивали в течение 30 мин, в течение которых выпадал осадок белого цвета. Твердое вещество отфильтровывали и концентрировали фильтрат при пониженном давлении. Сырой продукт растворяли вEtOAc (30 мл) и экстрагировали 10% HCl (330 мл). Объединенный водный слой промывали CH2Cl2 (30 мл) и нейтрализовывали 10% NaOH. Затем водный слой экстрагировали CH2Cl2 (330 мл), объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 109. Свободный амин растворяли в МТВЕ (30 мл) и нагревали до кипения с об- 17019343 ратным холодильником. Прибавляли по каплям H3PO4 (в изопропаноле), что приводило к образованию осадка белого цвета. Добавление H3PO4 продолжали до тех пор, пока выпадение осадка белого цвета не прекращалось. Твердое вещество отфильтровывали и промывали МТВЕ (100 мл). Продукт перекристаллизовывали из МеОН/МТВЕ с получением 109 в виде фосфатной соли (0,4 г, 36%). 1 Н-ЯМР (300 МГц, CD3OD):1,09-1,60 (м, 7 Н), 1,68-1,71 (м, 1 Н), 1,98-2,02 (м, 1 Н), 2,04-2,15 (м,1 Н), 2,31 (с, 3 Н), 2,50-2,55 (м, 1 Н), 2,64-2,65 (м, 1 Н), 3,06-3,07 (м, 1 Н), 3,16 (ушир.с, 2 Н), 3,54-3,55 (м,1 Н), 7,02-7,17 (м, 3 Н). 13 С-ЯМР (75 МГц, CD3OD):20,2, 21,7, 25,8, 35,0, 35,8, 60,3, 125,8, 127,4, 128,0, 130,8, 137,2, 137,4. ВЭЖХ (метод: колонка 20 мм C18-RP - метод градиента 2-95% ACN/вода/0,1% муравьиная кислота; длина волны: 210 нм): время удерживания: 2,51 мин, чистота: 97,7%. МС (М+Н): 259,2. Пример 7. Синтез (+)-3-(метил-d3)-17-(метил-d3)-(9,13,14)морфинана (110). Соединение 110 получали, как описано ниже. Подробности синтеза приведены далее. Синтез (+)-3-(метил-d3)-17-(метил-d3)-(9,13,14) морфинана (110). Смесь 23 (2,5 г, 8 ммоль, см. пример 5) в ТГФ (70 мл) обрабатывали LiAlD4 (1,7 г, 32 ммоль, Cambridge Isotopes, 98% атомн. D) при 0 С. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение ночи. Для гашения реакционной смеси добавляли воду (1 мл), затем NaOH(24%, 10 мл). Реакционную смесь перемешивали в течение 30 мин, в течение которых выпадал осадок белого цвета. Осадок отфильтровывали и концентрировали фильтрат при пониженном давлении. Сырой продукт очищали препаративной ВЭЖХ (см. условия, описанные в примере 5) с получением 110. Свободный амин растворяли в МТВЕ (50 мл) и нагревали до кипения с обратным холодильником. Прибавляли по каплям H3PO4 (в изопропаноле), что приводило к образованию осадка белого цвета. ДобавлениеH3PO4 продолжали до тех пор, пока выпадение осадка белого цвета не прекращалось. Твердое вещество отфильтровывали и промывали МТВЕ (100 мл) с получением 1,2 г твердого вещества. Продукт перекристаллизовывали из МеОН/МТВЕ с получением 110 в виде фосфатной соли (1 г, 36%). 1H-ЯМР (300 МГц, CD3OD):1,09-1,13 (м, 1 Н), 1,24-1,33 (м, 1 Н), 1,39-1,72 (м, 6 Н), 1,95-2,04 (м,1 Н), 2,16-2,18 (м, 1 Н), 2,50-2,54 (м, 1 Н), 2,60-2,68 (м, 1 Н), 3,07-3,16 (м и с, 3 Н), 3,54-3,55 (м, 1 Н), 7,027,17 (м, 3 Н). 13 С-ЯМР (75 МГц, D2O):21,4, 23,2, 25,5, 25,6, 34,7, 39,3, 43,0, 47,8, 60,4, 126,4, 127,5, 128,3, 131,2,138,0. ВЭЖХ (метод: колонка 20 мм C18-RP -метод градиента 2-95% ACN/вода/0,1% муравьиная кислота; длина волны: 210 нм): время удерживания: 2,61 мин, чистота 99,9%. МС (М+Н): 262,2. Пример 8. Оценка метаболической стабильности в CYP2D6 SUPERSOMESCYP2D6 SUPERSOMES человека приобретали у GenTest (Woburn, M.A, США). Исходные 7,5 мМ растворы тестируемых соединений (соединения 100, 102, 104, 106, декстрометорфан, дейтерированный аналог декстрометорфана, в котором каждую метильную группу заменяли CD3 (d6-декстрометорфан",химическое название (+)-3-d3-метокси-17-d3-метил-(9,13,14) морфинан, называемый также соединением 101 в US12/112936, и "тестируемым соединением" на приведенных далее фиг. 1 и в табл. 2), или изопропильный эфирный аналог декстрометорфана ("декстроизопропорфан") готовили в ДМСО. Исходные 7,5 М растворы разбавляли до 50 мкМ ацетонитрилом (ACN). Суперсомы CYP2D6 в концентрации 1000 пмоль/мл разбавляли до 62,5 пмоль/мл 0,1 М буферным раствором фосфата калия, рН 7,4, содержащим 3 мМ MgCl2. Разбавленные SUPERSOMES добавляли в лунки 96-луночного полипропиленового планшета с глубокими лунками в трех экземплярах. К суперсомам добавляли 10 мкл 50 мкМ тестируемого соединения и предварительно нагревали смесь в течение 10 мин. Реакции инициировали добавлением предварительно нагретого раствора НАДФ. Конечный объем реакционной смеси составлял 0,5 мл и содержал 50 пмоль/мл SUPERSOMES CYP2D6, 1 мкМ тестируемого соединения и 2 мМ НАДФ в 0,1 М буферном растворе фосфата калия, рН 7,4 и 3 мМ MgCl2. Реакционные смеси инкубировали при 37 С и отбирали аликвоты по 50 мкл через 0, 5, 10, 2 0 и 30 мин, и добавляли в 96-луночные планшеты с мелкими лунками, содержащими 50 мкл ледяного CAN с внутренним стандартом для остановки реакции. Планшеты выдерживали при 4 С в течение 20 мин, после чего в лунки планшета добавляли 100 мкл воды перед центрифугированием для осаждения белков центрифугированием. Надосадочные жидкости переносили в другой 96-луночный планшет и проводили анализ количеств оставшегося исходного вещества методом ЖХ-МС/МС с использованием масс-спектрометра Applied Bio-systems API 4000. Период полураспада in vitro (t1/2) для каждого из тестируемых соединений рассчитывали из взаимосвязи наклонов линейной регрессии % оставшегося исходного вещества (ln) относительно времени инкубации: in vitro t1/2=0,693/k, где k=-[наклон линейной регрессии % оставшегося исходного вещества (ln) относительно времени инкубации]. Анализ данных проводили с использованием программного обеспе- 18019343 чения Microsoft Excel. На приведенных ниже фиг. 1 и в табл. 2 представлены результаты эксперимента SUPERSOMES. Следует отметить, что на фиг. 1 кривые для соединений 100 и 104 частично налагаются друг на друга."Тестируемое соединение" на фиг. 1 и в табл. 2 относится к дейтерированному декстрометорфану (d6 декстрометорфану", (+)-3-d3-метокси-17-d3-метил-(9,13,14)морфинану, который называют также соединением 101 в US12/112936, включенной в настоящее описание посредством ссылки). Таблица 2. Рассчитанный период полураспада в SUPERSOMES Каждое из тестированных дейтерированных соединений проявило больший период полураспада при инкубации с SUPERSOMES CYP2D6, чем любое из соответствующих недейтерированных тестированных соединений или дейтерированных вариантов декстрометорфана (тестируемое соединение). Таким образом, в этом анализе соединения данного изобретения были более устойчивы к метаболизму,чем декстрометорфан или дейтерированный декстрометорфан (тестируемое соединение). Пример 9. Определение метаболической стабильности тестируемых соединений с использованием микросом печени человека. Микросомы печени человека (20 мг/мл) получали от Xenotech, LLC (Lenexa, KS). -Никотинамид аденин динуклеотид фосфат, восстановленную форму (НАДФ), хлорид магния (MgCl2) и диметилсульфоксид (ДМСО) приобретали у Sigma-Aldrich. Исходные 7,5 мМ растворы тестируемых соединений готовили в ДМСО. Исходные 7,5 мМ растворы разбавляли до 50 мкМ ацетонитрилом (ACN). 20 мг/мл микросом печени человека разбавляли до 1,25 мг/мл (в итоге 1 мг/мл) 0,1 М буферным раствором фосфата калия, рН 7,4, содержащем 3 мМ MgCl2. Разбавленные микросомы (375 мкл) прибавляли в лунки 96-луночного полипропиленового планшета в трех экземплярах. К микросомам добавляли 10 мкл 50 мкМ тестируемого соединения и смесь предварительно нагревали в течение 10 мин. Реакции инициировали добавлением 125 мкл предварительно нагретого раствора НАДФ. Конечный объем реакционной смеси составлял 0,5 мл и содержал 1,0 мг/мл микросом печени человека, 1 мкМ тестируемого соединения и 2 мМ НАДФ в 0,1 М буферном растворе фосфата калия, рН 7,4, содержащем 3 мМ MgCl2. Реакционные смеси инкубировали при 37 С и отбирали аликвоты по 50 мкл через 0, 5, 10, 20 и 30 мин и добавляли в 96-луночные планшеты с мелкими лунками, содержащими 50 мкл ледяного ACN с внутренним стандартом для остановки реакций. Планшеты выдерживали при 4 С в течение 20 мин, после чего в лунки планшета добавляли 100 мкл воды перед центрифугированием для осаждения белков. Надосадочные жидкости переносили в другой 96-луночный планшет и проводили анализ количеств оставшегося исходного вещества при помощи ЖХ-МС/МС с использованием масс-спектрометра Applied Bio-systems API 4000. В качестве положительного контроля использовали 7-этоксикумарин.In vitro t1/2 для тестируемых соединений рассчитывали из взаимосвязи наклонов линейной регрессии % оставшегося исходного вещества (ln) относительно времени инкубации:in vitro t1/2 = 0,693/k, где k=-[наклон линейной регрессии % оставшегося исходного вещества (ln) относительно времени инкубации]. Анализ данных проводили с использованием программного обеспечения Microsoft Excel. На фиг. 2 (панели А и В), фиг. 3, в табл. 3 и 4 представлены результаты данного эксперимента. Таблица 3. Рассчитанный период полураспада в микросомах печени человека В случае как декстроэторфана, так и декстроизопропорфана дейтерирование алкилового простого эфира (R1) приводило к существенному повышению периода полураспада (t1/2) в микросомах печени человека по сравнению с недейтерированным аналогом. Таблица 4. Рассчитанный период полураспада в микросомах печени человека В случае димеморфана дейтерирование R1 приводило к существенному повышению периода полураспада (t1/2) в микросомах печени человека по сравнению с недейтерированным аналогом. Дейтерирование N-метильного фрагмента (R2) приводило к дополнительному существенному повышению t1/2. Без дальнейшего описания предполагается, что специалист в данной области сможет, используя предшествующее описание и иллюстративные примеры, получить и использовать соединения данного изобретения и осуществить на практике заявленные способы. Следует понять, что предшествующее описание и примеры представляют собой лишь подробное описание некоторых предпочтительных вариантов осуществления. Специалисту в данной области будет ясно, что можно произвести различные модификации и эквиваленты, не выходя из духа и рамок изобретения. Все патенты, статьи из журналов и другие документы, обсуждаемые или цитированные выше, включены в настоящее описание посредством ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) или его фармацевтически приемлемая соль,в которой R1 выбирают из СН 3, CD3, -O-CD2CD3 или -O-CD(CD3)2; aR2 выбирают из СН 3 или CD3; при условии, что по меньшей мере один из R1 и R2 является дейтерированным. 2. Соединение по п.1, в котором R1 представляет собой -O-CD2CD3 или -O-CD(CD3)2. 3. Соединение по п.2, в котором R1 представляет собой -O-CD(CD3)2. 4. Соединение по п.1, в котором соединение выбирают из любого соединения, приведенного в представленной ниже таблице или его фармацевтически приемлемая соль. 5. Соединение по п.1, в котором R1 представляет собой -CD3. 6. Соединение по п.1, выбранное из любого из или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. 7. Соединение по любому из пп.1-6, в котором любой атом, не обозначенный как дейтерий, присутствует в своей природной изотопной распространенности. 8. Фармацевтическая композиция, не содержащая пирогенов, включающая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. 9. Способ лечения субъекта, страдающего от или склонного к заболеванию или состоянию, выбранному из эмоциональной неустойчивости; псевдобульбарного аффекта; аутизма; неврологических нарушений и нейродегенеративных заболеваний; повреждений мозга; нарушений, связанных с расстройством сознания; сердечно-сосудистых заболеваний; глаукомы; поздней дискинезии; рака; ревматоидного артрита; диабетической невропатии; ретинопатических заболеваний; заболеваний или нарушений, вызванных апоптозом, индуцированным гомоцистеином; заболеваний или нарушений, вызванных повышенными уровнями гомоцистеина; хронической боли; неустранимой боли; невропатической боли, симпатически опосредуемой боли; боли, связанной с дисфункцией желудочно-кишечного тракта; боли в ротовой полости; боли спины; центрального болевого синдрома; комплексного регионального болевого синдрома; эпилептических припадков; эпилептической гемиплегии; приобретенной эпилептиформной афазии(синдром Ландау-Клеффнера); тяжелой миоклонической эпилепсии раннего детского возраста (SMEI); эпилептической энцефалопатии раннего детского возраста; постинсультных судорог; фебрильных судорог; посттравматических судорог; звона в ушах; сексуальной дисфункции; трудноизлечимого кашля; дерматита; нарушений, связанных с аддикциями; синдрома Ретта (RTT); нарушений в работе голосовых связок вследствие неконтролируемых спазмов гортанных мышц; нейротоксичности метотрексата и усталости, вызванной раком, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8. 10. Способ по п.9, в котором субъект страдает от или склонен к диабетической невропатической боли. 11. Способ по п.9, в котором субъект страдает от или склонен к эпилептическим припадкам. 12. Способ лечения субъекта, страдающего от или подверженного условиям, относящимся к воздействию химических веществ, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8. 13. Способ лечения субъекта, страдающего от или склонного к боли, включающий стадию введения нуждающемуся в этом субъекту терапевтически эффективного количества фармацевтической композиции по п.8. 14. Соединение, имеющее структуру или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности. 16. Соединение, имеющее структуру или его фармацевтически приемлемая соль, в котором любой атом, не обозначенный как дейтерий,присутствует в своей природной изотопной распространенности.
МПК / Метки
МПК: C07D 471/08, A61K 31/439
Метки: соединения, дейтерированные, морфинановые
Код ссылки
<a href="https://eas.patents.su/24-19343-dejjterirovannye-morfinanovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Дейтерированные морфинановые соединения</a>
Дейтерированные производные катехоламина, а также лекарственные средства, содержащие эти соединения

Номер патента: 9258
Опубликовано: 28.12.2007
Автор: Алкен Рудольф-Гизберт
МПК: A61P 25/00, C07B 59/00, A61K 31/198...
Метки: производные, соединения, содержащие, также, лекарственные, катехоламина, дейтерированные, средства, эти
Формула / Реферат:
1. Дейтерированные производные катехоламина общей формулы I причем R1 обозначает H или D, R2 обозначает H или D, R3 обозначает H, D, C1-С6алкил или С5-С6циклоалкил, дейтерированный C1-С6алкил или C5-C6циклоалкил, R4 обозначает H или D, R5 обозначает H или D. 2. Дейтерированные производные катехоламина по п.1, причем R1 обозначает H или D, R2 обозначает H или D, R3 обозначает H, D, C1-С6алкил или С5-С6циклоалкил, дейтерированный C1-С6алкил или...
Дейтерированные производные катехоламина и лекарственные средства, содержащие указанные соединения

Номер патента: 17983
Опубликовано: 30.04.2013
Авторы: Шнайдер Франк, Алкен Рудольф-Гизберт
МПК: C07C 229/26, C07B 59/00
Метки: производные, соединения, содержащие, дейтерированные, лекарственные, катехоламина, указанные, средства
Формула / Реферат:
1. Дейтерированные производные катехоламина общей формулы Iгде R1 представляет собой H;R2 представляет собой D;R3 представляет собой H, метил или этил;R4 представляет собой H;R5 представляет собой Н или D;R6 представляет собой Н или D, где оба остатка R6 не являются одновременно D и где оба остатка R6 не являются одновременно H,их стереоизомеры, энантиомеры или диастереомеры в оптически чистой форме, а также их физиологически приемлемые соли.2....
Дейтерированные производные 1,3-бензодиоксола

Номер патента: 14432
Опубликовано: 30.12.2010
Автор: Танг Роджер
МПК: C07D 295/00
Метки: производные, 1,3-бензодиоксола, дейтерированные
Формула / Реферат:
1. Выделенное соединение формулы (I)или его соль; или пролекарство или соль его пролекарства; или его гидрат, сольват или полиморф; в которомD-является дейтерием;каждый Y независимо выбран из дейтерия или водорода;каждый водород независимо необязательно замещен дейтерием икаждый углерод независимо необязательно замещен 13С.2. Соединение или его пролекарство по п.1, в котором Y1является дейтерием.3. Соединение по п.2, в котором до 4 атомов...
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Перроне Марк Х., Кюродо Алан Х., Лидли Роберт Дж., Данвидди Кристофер Т., Юзан Андре
МПК: A61K 31/715, A61P 7/02
Метки: профилактики, агрегации, лечения, тромбоцитов, активностью, заболеваний, способ, набор, эти, фармкомпозиция, содержащий, содержащая, антагониста, анти-ха, тромбообразованию, сопутствующих, соединения, применение
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения

Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Вийенов Николь, Пеглион Жан-Луи, Дессинже Эмея, Шименти Стефано, Толлон Катрин, Вилен Жан-Поль, Кеньяр Паскаль, Гумен Бертран
МПК: A61K 31/55, C07D 223/16, A61P 9/00...
Метки: способ, 1,2,4,5-тетрагидро-3н-бензазепина, эти, композиции, фармацевтические, соединения, содержащие, получения
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или...