α-кристаллическая форма карбабензпирида
Номер патента: 19080
Опубликовано: 30.12.2013
Авторы: Сяркевич Олег, Костюк Григорий, Маргитич Виктор, Жебровска Филя, Ванат Михайло


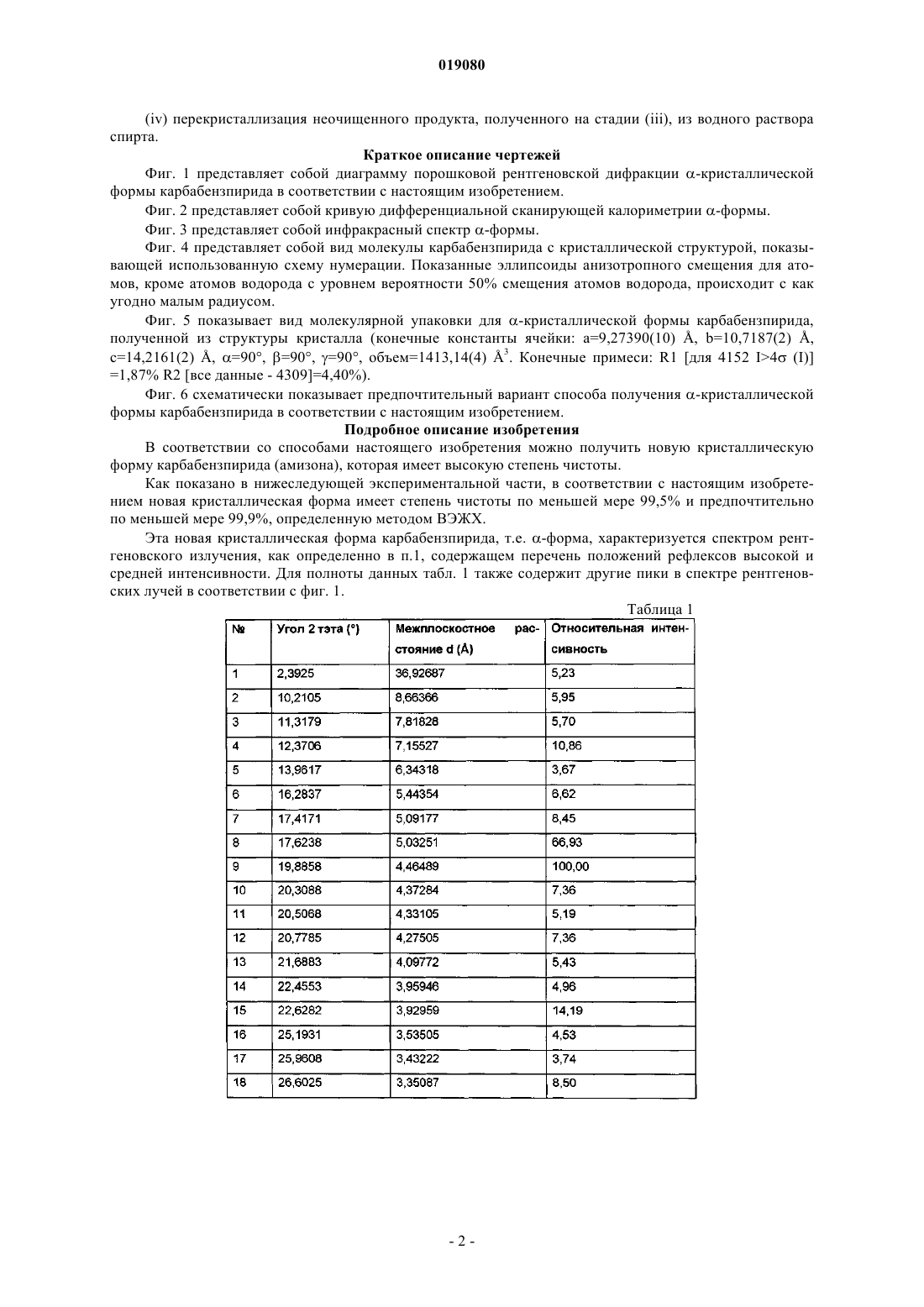






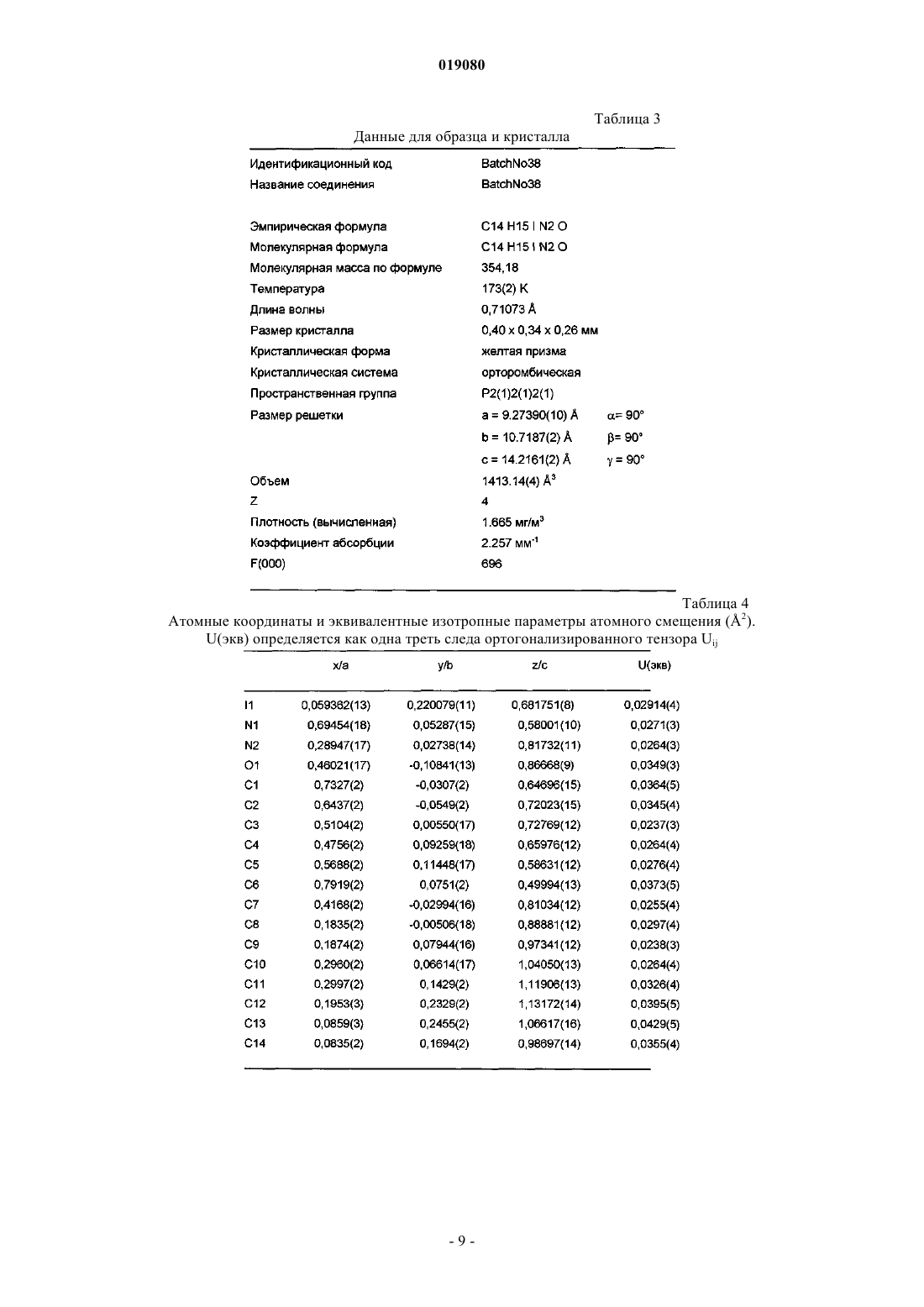
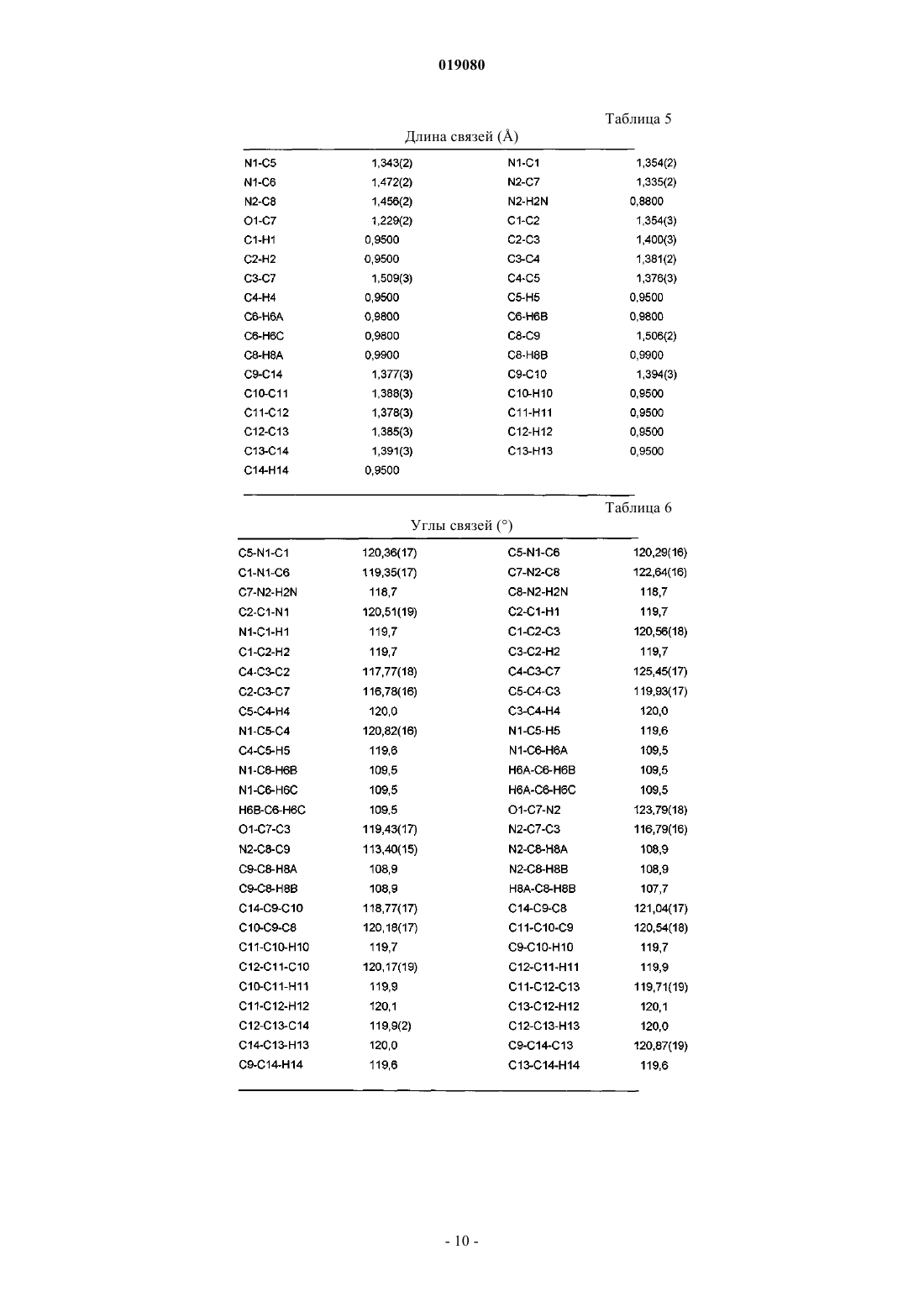



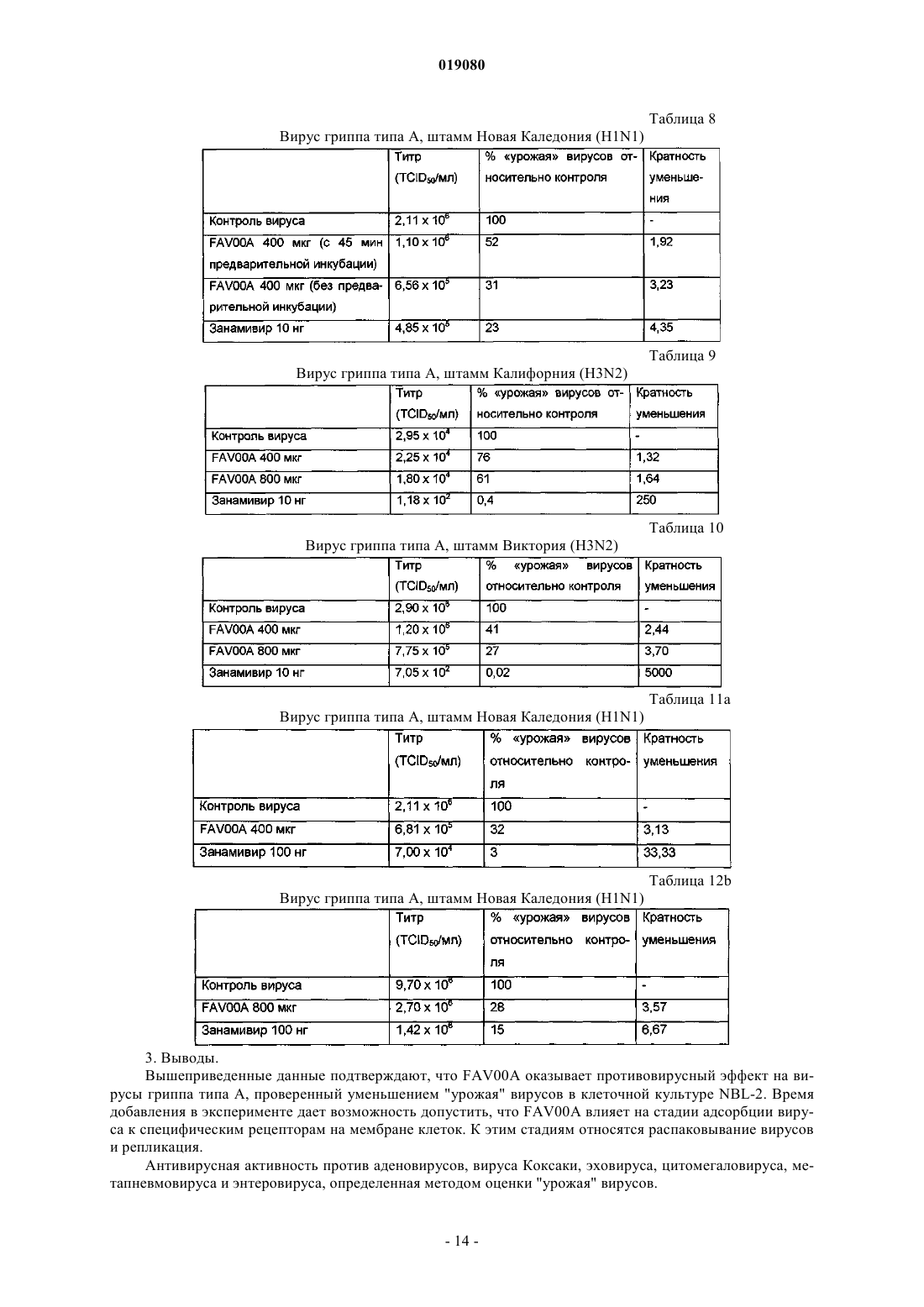


Формула / Реферат
1. α-Кристаллическая форма карбабензпирида формулы (I)

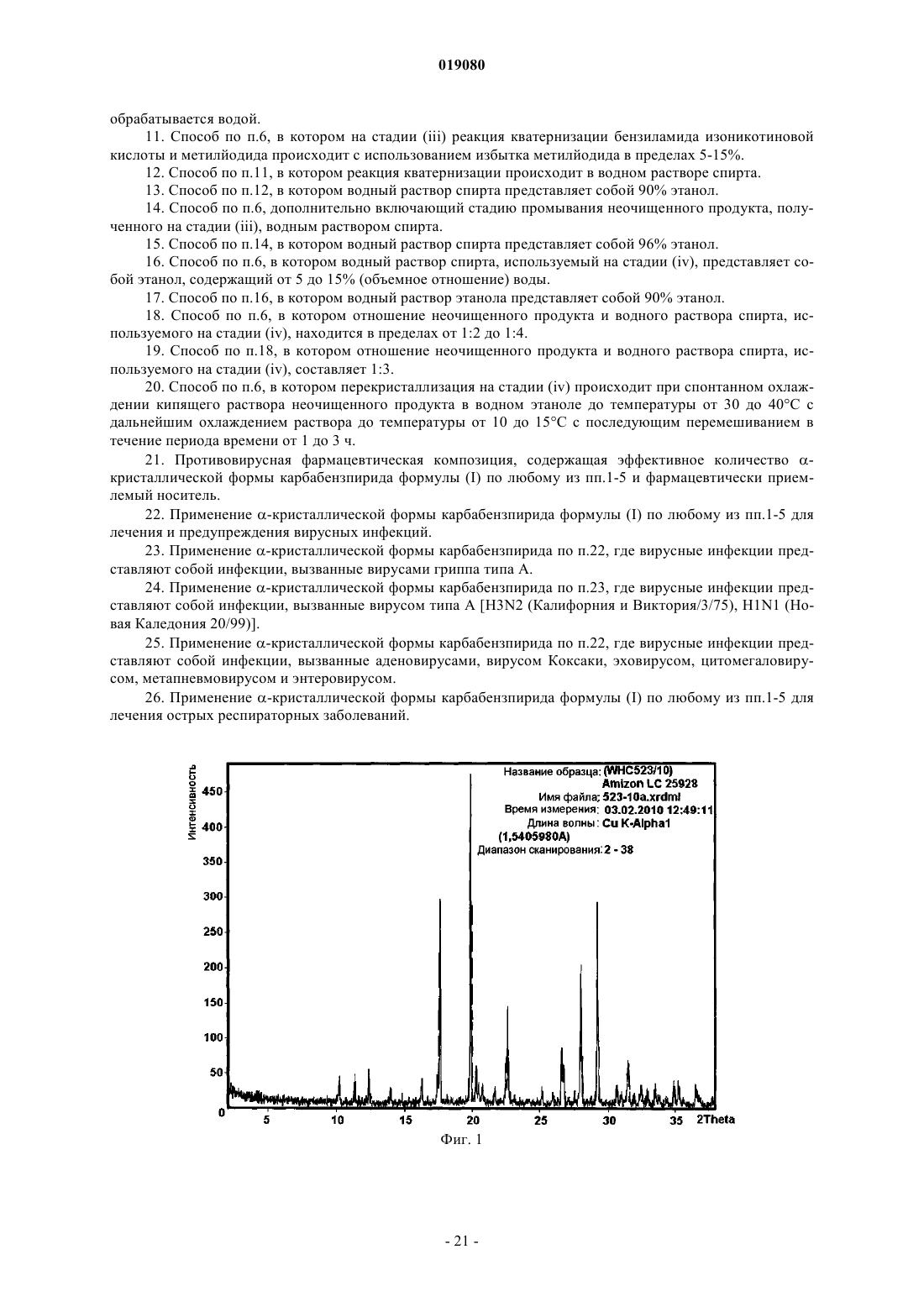
которая имеет параметры, определенные с помощью метода порошковой рентгеновской дифракции с использованием для измерения дифрактометра (медный антикатод), выраженные относительно межплоскостного расстояния d, угла Брэгга 2 тэта и относительной интенсивности (выражается в процентах к наиболее интенсивному лучу), указанные в нижеприведенной таблице с перечнем положений рефлексов высокой и средней интенсивности.

2. α-Кристаллическая форма карбабензпирида по п.1, имеющая степень чистоты по меньшей мере 99,5%, определенную методом ВЭЖХ.
3. α-Кристаллическая форма карбабензпирида по п.2, имеющая степень чистоты по меньшей мере 99,9%, определенную методом ВЭЖХ.
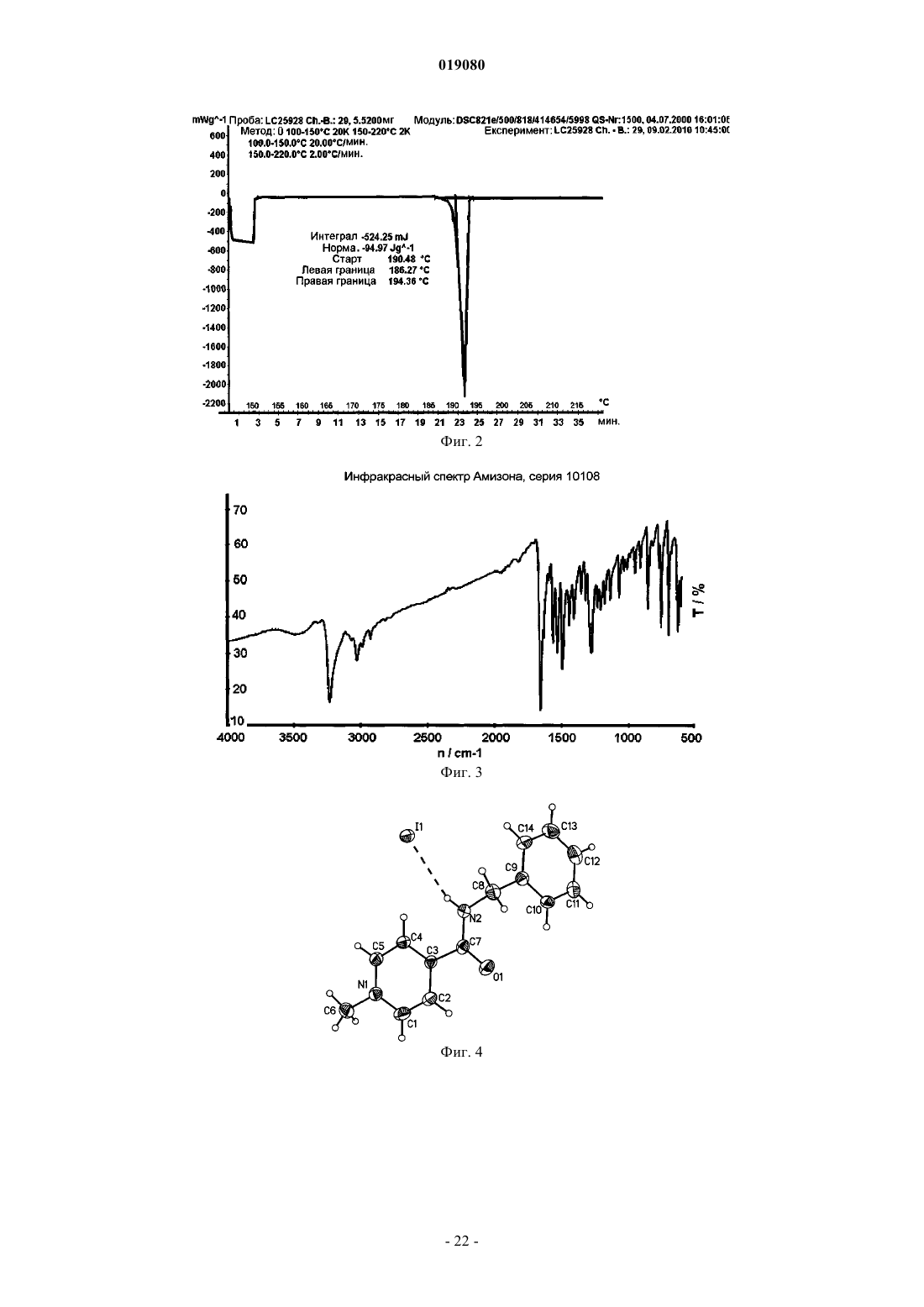
4. α-Кристаллическая форма карбабензпирида по п.1, имеющая один эндотермический максимум на ДСК-кривой от 187 до 193°C.
5. α-Кристаллическая форма карбабензпирида по п.1, имеющая ИК-спектр с характерными пиками, показанными в нижеследующей таблице.

6. Способ получения кристаллической формы карбабензпирида по п.1, включающий следующие стадии:
(i) конденсация изоникотиновой кислоты с бензиламином при повышенных температурах;
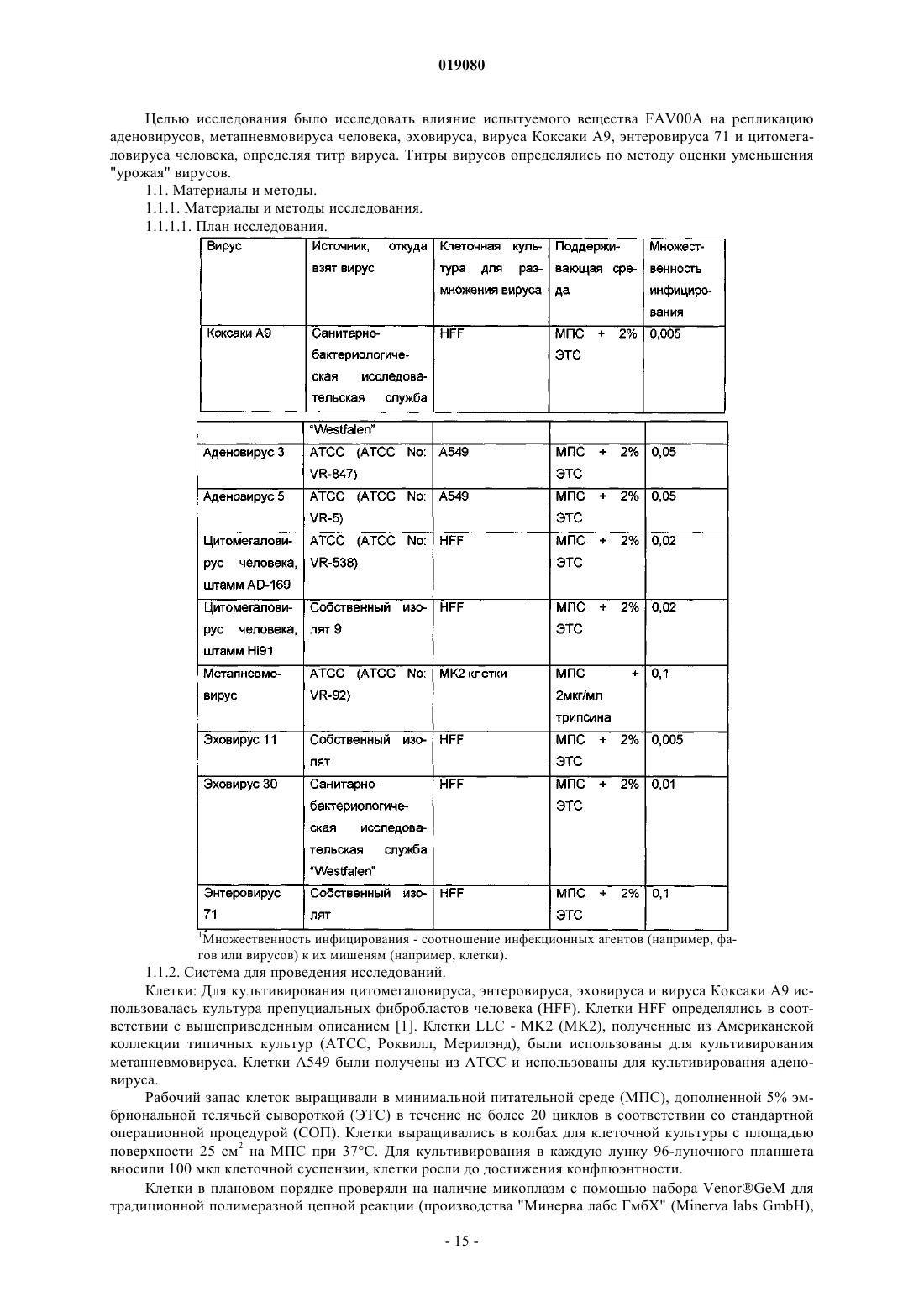
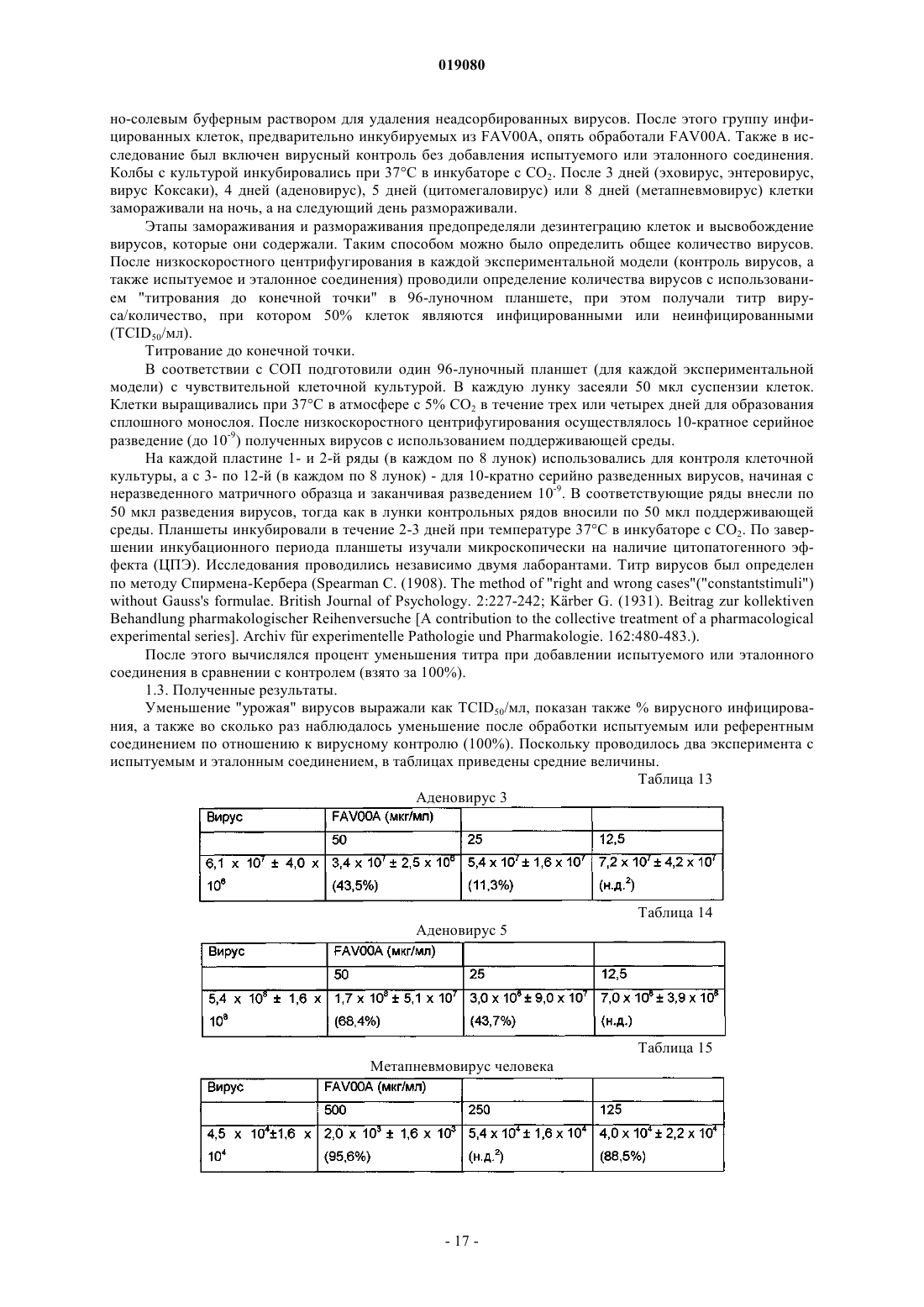
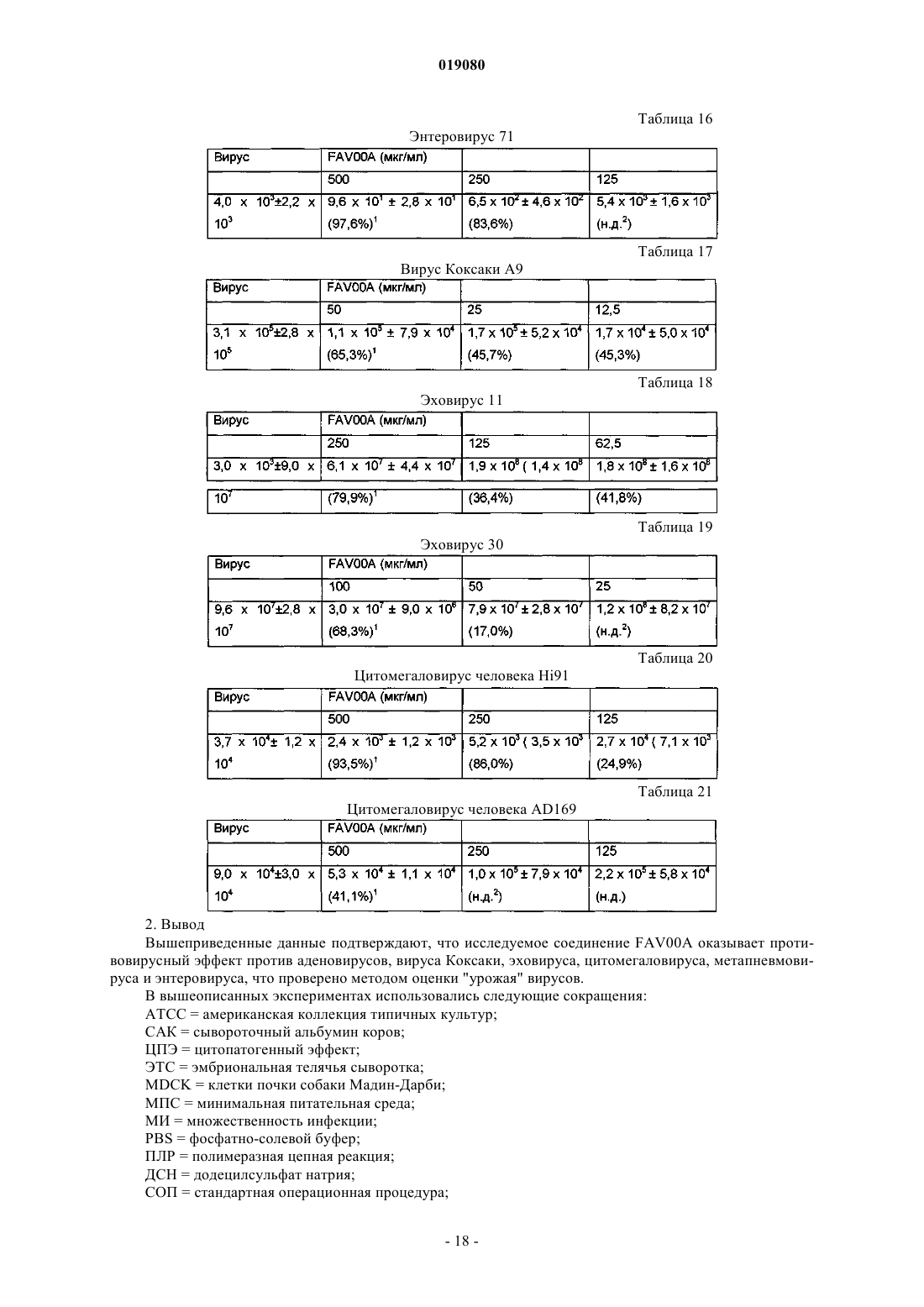
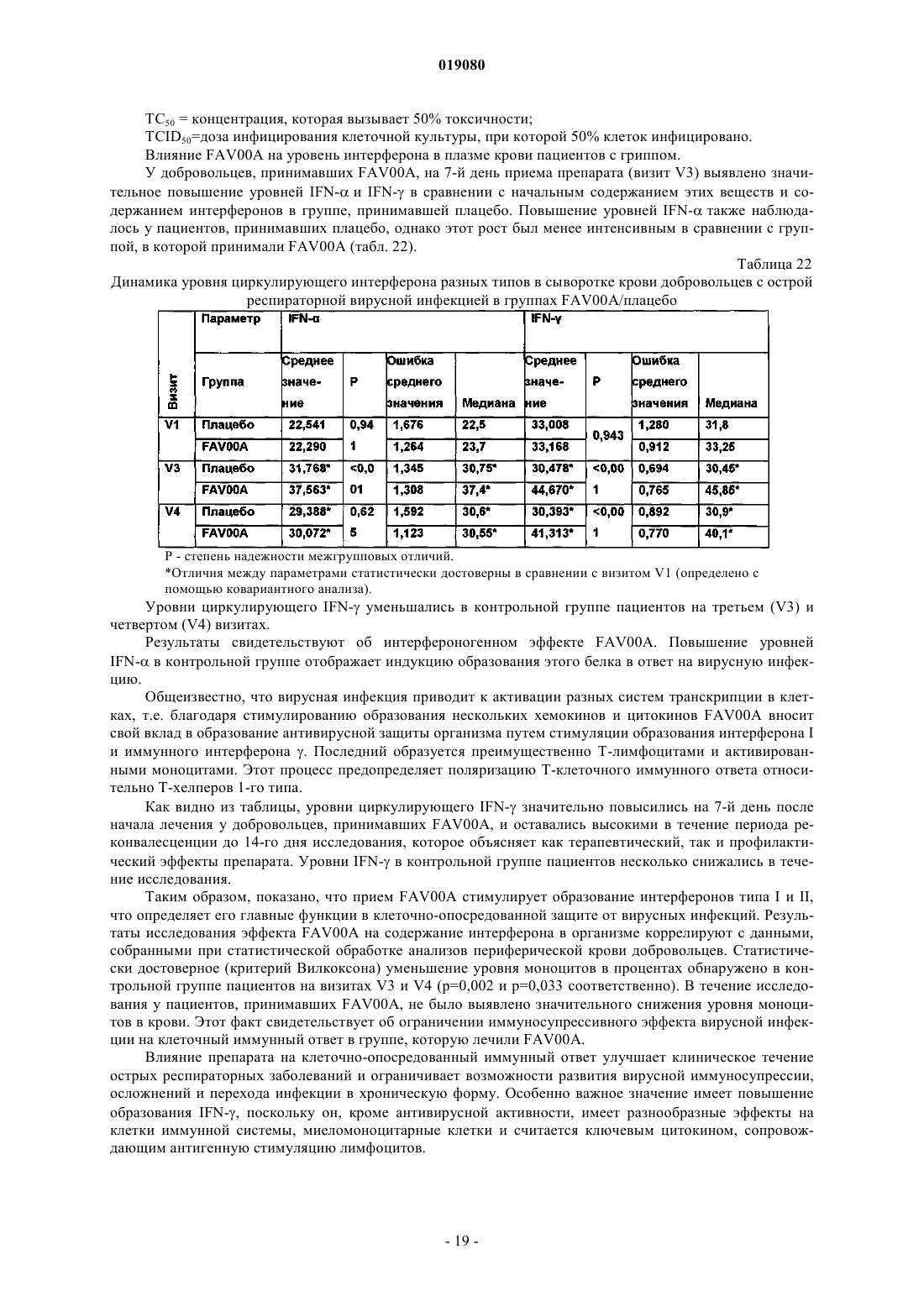
(ii) кристаллизация и выделение продукта конденсации, полученного на стадии (i);
(iii) реакция кристаллического продукта, полученного на стадии (ii), с метилйодидом и
(iv) перекристаллизация неочищенного продукта, полученного на стадии (iii), из водного раствора спирта.
7. Способ по п.6, в котором реакция конденсации между изоникотиновой кислотой и бензиламином в соответствии со стадией (i) происходит с использованием избытка бензиламина в пределах 10-25%.
8. Способ по п.6, в котором продукт реакции конденсации между изоникотиновой кислотой и бензиламином, т.е. бензиламид изоникотиновой кислоты (БАИНК), кристаллизуется из реакционной смеси с использованием растворителя, выбранного из группы, состоящей из этилацетата, ацетонитрила и изопропанола.
9. Способ по п.8, дополнительно включающий использование активированного угля.
10. Способ по любому из пп.6, 8 или 9, в котором продукт, полученный на стадии (ii), т.е. БАИНК, обрабатывается водой.
11. Способ по п.6, в котором на стадии (iii) реакция кватернизации бензиламида изоникотиновой кислоты и метилйодида происходит с использованием избытка метилйодида в пределах 5-15%.
12. Способ по п.11, в котором реакция кватернизации происходит в водном растворе спирта.
13. Способ по п.12, в котором водный раствор спирта представляет собой 90% этанол.
14. Способ по п.6, дополнительно включающий стадию промывания неочищенного продукта, полученного на стадии (iii), водным раствором спирта.
15. Способ по п.14, в котором водный раствор спирта представляет собой 96% этанол.
16. Способ по п.6, в котором водный раствор спирта, используемый на стадии (iv), представляет собой этанол, содержащий от 5 до 15% (объемное отношение) воды.
17. Способ по п.16, в котором водный раствор этанола представляет собой 90% этанол.
18. Способ по п.6, в котором отношение неочищенного продукта и водного раствора спирта, используемого на стадии (iv), находится в пределах от 1:2 до 1:4.
19. Способ по п.18, в котором отношение неочищенного продукта и водного раствора спирта, используемого на стадии (iv), составляет 1:3.
20. Способ по п.6, в котором перекристаллизация на стадии (iv) происходит при спонтанном охлаждении кипящего раствора неочищенного продукта в водном этаноле до температуры от 30 до 40°C с дальнейшим охлаждением раствора до температуры от 10 до 15°C с последующим перемешиванием в течение периода времени от 1 до 3 ч.
21. Противовирусная фармацевтическая композиция, содержащая эффективное количество кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 и фармацевтически приемлемый носитель.
22. Применение α-кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 для лечения и предупреждения вирусных инфекций.
23. Применение α-кристаллической формы карбабензпирида по п.22, где вирусные инфекции представляют собой инфекции, вызванные вирусами гриппа типа А.
24. Применение α-кристаллической формы карбабензпирида по п.23, где вирусные инфекции представляют собой инфекции, вызванные вирусом типа A [H3N2 (Калифорния и Виктория/3/75), H1N1 (Новая Каледония 20/99)].
25. Применение α-кристаллической формы карбабензпирида по п.22, где вирусные инфекции представляют собой инфекции, вызванные аденовирусами, вирусом Коксаки, эховирусом, цитомегаловирусом, метапневмовирусом и энтеровирусом.
26. Применение α-кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 для лечения острых респираторных заболеваний.
Текст
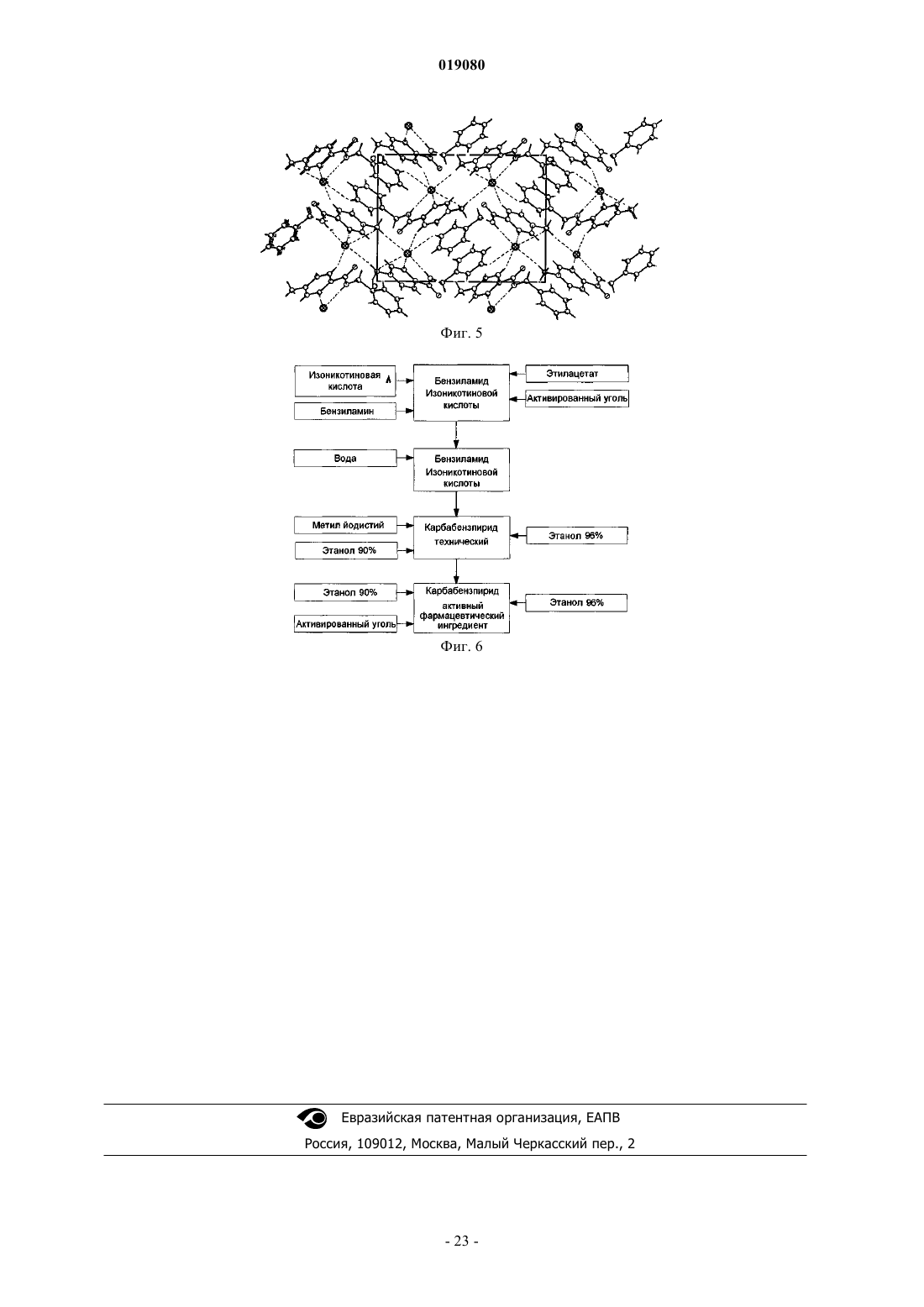
ИСПРАВЛЕННОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ Изобретение относится к новой кристаллической форме карбабензпирида формулы (I) и способу ее получения. Также изобретение касается применения новой кристаллической формы карбабензпирида для лечения и предупреждения вирусных инфекций. Примечание: библиография отражает состояние при переиздании(71)(73) Заявитель и патентовладелец: ФАРМАК ИНТЕРНЭШНЛ ХОЛДИНГ ГМБХ (AT) Область техники изобретения Настоящее изобретение касается новой кристаллической формы карбабензпирида и способа его получения. Также изобретение касается фармацевтической композиции, содержащей новую кристаллическую форму, т.е. -кристаллическую форму карбабензпирида. В завершение можно сказать, что изобретение касается применения -кристаллической формы карбабензпирида для изготовления лекарственного средства для лечения или предупреждения вирусных инфекций. Карбабензпирид имеет следующую формулу (I): и также известен как амизон. Уровень техники Фармацевтически приемлемые соли карбабензпирида имеют ценные фармакологические свойства. Их главной характеристикой является способность лечить и предупреждать вирусные инфекции, особенно вызванные вирусами гриппа А. Для фармацевтического применения особый интерес представляет вещество с высокой степенью чистоты. Кроме того, желательно использовать стабильный, хорошо настроенный и масштабируемый производственный процесс, в результате которого получается продукт соответствующего качества, пригодный для изготовления фармацевтических композиций. В SU 583612 (1975) описан синтез карбабензпирида для применения в фармацевтических целях, однако отсутствует надлежащее описание получения активного вещества воссоздаваемым способом. Следовательно, существует потребность в способе синтеза, который бы обеспечивал получение материала с высокой степенью чистоты, соответствующей требованиям для фармацевтического применения. Сущность изобретения Настоящее изобретение касается -кристаллической формы карбабензпирида формулы (I) которой присущи указанные ниже параметры, определенные с помощью метода порошковой рентгеновской дифракции с использованием для измерения дифрактометра (медный антикатод), выраженные относительно межплоскостного расстояния d, угла Брэгга 2 тэта и относительной интенсивности (выражается в процентах наиболее интенсивного луча), что приведены в таблице ниже с перечнем положений рефлексов высокой и средней интенсивности. Эта новая -кристаллическая форма карбабензпирида получена способом, который в соответствии с настоящим изобретением состоит из следующих стадий:(i) конденсация изоникотиновой кислоты с бензиламином при повышенных температурах;(ii) кристаллизация и выделение продукта конденсации, полученного на стадии (i);(iii) реакция кристаллического продукта, полученного на стадии (ii), с метилйодидом;(iv) перекристаллизация неочищенного продукта, полученного на стадии (iii), из водного раствора спирта. Краткое описание чертежей Фиг. 1 представляет собой диаграмму порошковой рентгеновской дифракции -кристаллической формы карбабензпирида в соответствии с настоящим изобретением. Фиг. 2 представляет собой кривую дифференциальной сканирующей калориметрии -формы. Фиг. 3 представляет собой инфракрасный спектр -формы. Фиг. 4 представляет собой вид молекулы карбабензпирида с кристаллической структурой, показывающей использованную схему нумерации. Показанные эллипсоиды анизотропного смещения для атомов, кроме атомов водорода с уровнем вероятности 50% смещения атомов водорода, происходит с как угодно малым радиусом. Фиг. 5 показывает вид молекулярной упаковки для -кристаллической формы карбабензпирида,полученной из структуры кристалла (конечные константы ячейки: а=9,27390(10) , b=10,7187(2) ,с=14,2161(2) , =90, =90, =90, объем=1413,14(4) 3. Конечные примеси: R1 [для 4152 I4 (I)]=1,87% R2 [все данные - 4309]=4,40%). Фиг. 6 схематически показывает предпочтительный вариант способа получения -кристаллической формы карбабензпирида в соответствии с настоящим изобретением. Подробное описание изобретения В соответствии со способами настоящего изобретения можно получить новую кристаллическую форму карбабензпирида (амизона), которая имеет высокую степень чистоты. Как показано в нижеследующей экспериментальной части, в соответствии с настоящим изобретением новая кристаллическая форма имеет степень чистоты по меньшей мере 99,5% и предпочтительно по меньшей мере 99,9%, определенную методом ВЭЖХ. Эта новая кристаллическая форма карбабензпирида, т.е. -форма, характеризуется спектром рентгеновского излучения, как определенно в п.1, содержащем перечень положений рефлексов высокой и средней интенсивности. Для полноты данных табл. 1 также содержит другие пики в спектре рентгеновских лучей в соответствии с фиг. 1. Таблица 1-Форма карбабензпирида также характеризуется ДСК-кривой, как показано на фиг. 2 (на этой фигуре также указаны условия измерения). -Кристаллическая форма имеет эндотермический максимум на ДСК-кривой в пределах от 187 до 199C и, в частности, от 189 до 191C. Кроме того, -форма карбабензпирида характеризуется ИК-спектром, как показано на фиг. 3, с характерными пиками, перечисленными в табл. 2. Таблица 2 Как было упомянуто, -кристаллическая форма имеет степень чистоты по меньшей мере 99,5% и предпочтительно по меньшей мере 99,9%, определенную методом ВЭЖХ. Такую высокую степень чистоты не удалось получить согласно предшествующему уровню техники. В частности, это стало возможным благодаря использованию в соответствии с настоящим изобретением способа для значительного уменьшения содержания в конечном продукте метилйодида, известного генотоксичного вещества. Как уже было упомянуто, в соответствии с настоящим изобретением способ состоит из четырех основных стадий, а именно:(i) конденсация изоникотиновой кислоты с бензиламином при повышенных температурах;(ii) кристаллизация и выделение продукта конденсации, полученного на вышеописанной стадии (i);(iii) реакция кристаллического продукта, полученного на вышеуказанной стадии (ii), с метилйодидом и(iv) перекристаллизация неочищенного продукта, полученного на стадии (iii), из водного раствора спирта. Этот способ проиллюстрирован на нижеследующей схеме. где изоникотиновая кислота обозначена ИНК, бензиламин - БА, а БАИНК означает бензиламид изоникотиновой кислоты. Конденсация изоникотиновой кислоты с бензиламином происходит при повышенных температурах, т.е. в пределах от 160 до 220C. Наибольшее предпочтение отдается температурному диапазону от 200 до 210C. Молярное соотношение изоникотиновой кислоты и бензиламина находится в пределах 1:(1,1-1,25). Наибольшее предпочтение отдается соотношению приблизительно 1:1,23. В результате реакции изоникотиновой кислоты и бензиламина образуется вода, которая удаляется при помощи дистилляции с избытком бензиламина. Продукт конденсации, а именно бензиламид изоникотиновой кислоты (БАИНК), выделяется из реакционной смеси при добавлении растворителя, который выбирается из группы растворителей, а именно этилацетата, ацетонитрила и изопропанола. Наибольшее предпочтение отдается этилацетату в качестве растворителя. В соответствии с предпочтительным вариантом способа согласно настоящему изобретению раствор БАИНК, содержащий вышеупомянутый растворитель, предпочтительно этилацетат, обрабатывается активированным углем. Активированный уголь используется в количестве от 0,5 до 1,5% от объема растворителя, предпочтительно приблизительно 1%. Время обработки активированным углем составляет от 20 до 40 мин, предпочтительно около 30 мин. Температура, при которой производится обработка активированным углем, изменяется от 65 до 75C, предпочтительно около 70C. Далее активированный уголь или древесный уголь фильтруется, а фильтрат спонтанно охлаждается до температуры в пределах от 25 до 35C, предпочтительно около 30C. Спонтанное охлаждение означает, что раствор просто оставляют стоять, пока он не достигнет желательной температуры. При этом не применяется никаких дополнительных средств или мер для ускорения способа охлаждения. После такого спонтанного охлаждения до вышеупомянутой температуры применяется охлаждающий агент, который снижает температуру приблизительно до уровня от 0 до 5C. После перемешивания собирают полученный неочищенный продукт. Он получается в форме пасты,которая обрабатывается водой. Массовое отношение пасты БАИНК и воды может быть от 1:2 до 1:3,предпочтительно приблизительно 1:2. Эта водная система нагревается до температуры в пределах от 30 до 40C, предпочтительно от 32 до 35C. Время перемешивания составляет от 1,5 до 2,5 ч, предпочтительно около 2 ч. После фильтрации осадок дважды промывается холодной водой, а потом сушится, например, при 25C в течение 18 ч. В результате вышеупомянутых стадий способа получают БАИНК, который является гомогенным кристаллическим порошком от желтой до желто-зеленой окраски и содержит не более чем 0,5% примесей. Такая высокая степень чистоты дает возможность точно вычислить количество метилйодида, используемого на третьей стадии способа в соответствии с настоящим изобретением. Обычно реакция кватернизации БАИНК и метилйодида проводится с использованием избытка последнего в пределах от 5 до 15%, предпочтительно от 8 до 12%. Наибольшее предпочтение отдается избытку метилйодида, который составляет приблизительно 10%. В соответствии с предпочтительным вариантом способа согласно настоящему изобретению реакция кватернизации происходит в водном растворе спирта. Содержание воды в спирте обычно составляет от 5 до 15%, предпочтительно от 8 до 12%. Наибольшее предпочтение отдается содержанию воды около 10%, а спиртом является этанол. Неочищенный продукт реакции кватернизации может выделяться с применением фильтрации и промываться предпочтительно водным раствором спирта. Предпочтительно используется 96% этанол. Чистая -форма карбабензпирида в соответствии с настоящим изобретением получается благодаря перекристаллизации неочищенного продукта, полученного на стадии (iii), из водного раствора этанола. Обычно количество воды, имеющейся в этаноле, составляет от 5 до 15%, предпочтительно от 7 до 13%. Наибольшее предпочтение для использования на стадии (iv) отдается 90% этанолу. В соответствии с предпочтительным вариантом заявленного способа отношение растворителя к неочищенному продукту, который используется на стадии (iv), составляет от 1:2 до 1:4. Наиболее предпочтительным является отношение около 1:3. После растворения неочищенного продукта в растворителе, предпочтительно в 90% этаноле, раствор спонтанно (т.е. без использования охлаждающих агентов) охлаждается от температуры растворителя до уровня от 30 до 40C. Наибольшее предпочтение отдается спонтанному охлаждению горячего раствора (т.е. без использования охлаждающих агентов) до температуры приблизительно 30C. После этого температуру снижают до уровня от 10 до 15C. После перемешивания в течение периода от 1 до 3 ч чистый продукт можно отфильтровать и промыть, например, 2 раза холодным 96% спиртом. В соответствии с предпочтительным вариантом способа перекристаллизации к раствору неочищенного продукта добавляется активированный древесный уголь. Изобретение также касается фармацевтических композиций, содержащих в качестве активного вещества -кристаллическую форму соединения (I) с одним или несколькими фармацевтически приемлемыми, инертными наполнителями. Композиция может применяться перорально, назально, ректально или парентерально в форме таблеток, таблеток, покрытых оболочкой, желатиновых капсул, пастилок, растворов для орального применения, назальных спреев, растворов для инъекций, суппозиториев, растворов для ингаляции или порошков и т.п. Эффективная доза может изменяться от 125 до 2500 мг в день в 1 или 4 отдельных приема. Фармацевтические композиции в соответствии с настоящим изобретением могут использоваться для лечения или предупреждения вирусных инфекций. В частности, их используют при инфекциях, вызванных такими вирусами гриппа (А), как вирусы штаммов типа A [H3N2 (Калифорния и Виктория/3/75), H1N1 (Новая Каледония 20/99)]. Кроме того, новая -кристаллическая форма карбабензпирида в соответствии с настоящим изобретением, а также фармацевтические композиции, которые содержат это соединение, обладают противовирусной активностью относительно аденовирусов, вирусов Коксаки, эховирусов, цитомегаловируса, метапневмовируса и энтеровируса, определенных по результатам анализа воспроизведения вирусов. И, в конечном итоге, выявлено, что новая -кристаллическая форма оказывает влияние на содержание интерферона в плазме крови пациентов с гриппом. В соответствии с настоящим изобретением влияние -кристаллической формы карбабензпирида соединения (I) на клеточно-опосредованный иммунный ответ приводит к улучшению клинического течения острых респираторных заболеваний и ограничивает возможности развития вирусной иммуносупрессии, осложнений и перехода инфекции в хроническую форму. Увеличение образования интерферона-гамма (IFN-) имеет особенно важное значение, поскольку кроме противовирусной активности он имеет различное влияние на клетки иммунной системы, миеломоноцитарные клетки и считается ключевым цитокином, сопровождающим процесс антигенной стимуляции лимфоцитов. Следующие примеры иллюстрируют изобретение, однако никоим образом не ограничивают его. Экспериментальная часть Получение БАИНК. Пример 1. Смесь 91,4 г (0,7424 М) изоникотиновой кислоты и 97,9 г (0,9136 М) бензиламина (1:1,23) нагревается до температуры 180C с использованием обратного конденсатора и перемешивается в течение 1 ч. При этом температура реакции уменьшается до 160C. Тогда реакционная масса нагревается до температуры 210C в течение 2 ч с использованием прямого конденсатора для отгонки водного бензиламина. Реакция нагревается до температуры 220C в течение 1,5 ч с использованием прямого конденсатора для отгонки избытка бензиламина. Содержимое реактора охлаждается до температуры 100C, добавляется 240 мл этилацетата, потом реакционная масса перемешивается в течение 20 мин и добавляется 2,4 г древесного угля, после чего реакционная масса перемешивается при температуре 70-75C в течение 30 мин, фильтруется через древесный уголь, а полученный раствор спонтанно охлаждается до 30C, а потом до 0-5C с использованием охлаждающего агента, в течение 1 ч перемешивается и фильтруется. Паста БАИНК растворяется в 180 мл воды, нагревается до температуры 32-35C и перемешивается в течение 2 ч. Реакционная масса фильтруется, а осадок в фильтре промывается дважды 50 мл охлажденной воды. Продукт высушивается при температуре 25C в течение 18 ч. Количество БАИНК: 136,5 г (86,6%). Аналитический контроль. Содержание: 101,44%. Содержание сопутствующих примесей: Пример 2. Этот пример отличается от примера 1 тем, что: 1) смесь 294 г (2,39 М) изоникотиновой кислоты и 316,0 г (2,95 М) бензиламина нагревается; 2) добавляется 772 мл этилацетата; 3) паста БАИНК растворяется в 500 мл воды. Количество БАИНК: 413,6 г (81,6%). Пример 3. Этот пример отличается от примера 1 тем, что: 1) смесь 45,7 г (0,3715 М) изоникотиновой кислоты и 49,1 г (0,4589 М) бензиламина нагревается; 2) добавляется 120 мл этилацетата; 3) паста БАИНК растворяется в 80 мл воды. Количество БАИНК: 65,1 г (82,6%). Получение неочищенного карбабензпирида. Пример 4. 106,1 г (0,5 М) БАИНК помещается в реактор, оборудованный мешалкой, обратным конденсатором и капельной воронкой, добавляется 230 мл 90% спирта, реакционная масса нагревается до 38-40C и перемешивается в течение 30 мин до получения раствора. Добавляется 2,3 г древесного угля, и нагревается реакционная масса до температуры 60-70C на 30 мин, после чего реакционная масса фильтруется, а древесный уголь в фильтре дважды промывается 5 мл 90% спирта. Полученный раствор нагревается до температуры 40-41C и по каплям добавляется 78,1 г (0,55 М) метилйодида. Реакционная масса перемешивается при температуре 40-41C в течение 1 ч, нагревается до кипения и кипятится в течение 1 ч. Реакционная масса спонтанно охлаждается до температуры 40C, а потом на водяной бане до температуры 1015C, при этой температуре смесь перемешивается в течение 1,5 ч (без затравки кристаллов). Реакционная масса фильтруется, а осадок на фильтре дважды промывается 55 мл охлажденного 96% спирта. Продукт высушивается при 25C в течение 18 ч и взвешивается. Количество неочищенного карбабензпирида: 164,6 г (92,9%). Аналитический контроль. Содержание: 102,05%. Содержание сопутствующих примесей: Пример 5. Этот пример отличается от примера 4 тем, что для реакции кватернизации используют 398 г БАИНК (1,88 М), 868 мл 90% спирта и 292,0 г (2,06 М) метилйодида. Количество неочищенного карбабензпирида (пасты): 586,8 г. Пример 6. Этот пример отличается от примера 4 тем, что для реакции кватернизации используют 30 г (0,14 М) БАИНК, 63,5 мл 90% спирта, 21,42 г (0,15 М) метилйодида. Количество неочищенного карбабензпирида (пасты): 42,9 г Получение очищенной -кристаллической формы карбабензпирида. Пример 7. 580 г неочищенного карбабензпирида растворяют в 1744 мл 90% спирта (1:3) (m/V) и добавляют 17 г активированного древесного угля. Реакционную массу нагревают до температуры кипения, перемешивают и кипятят в течение 30 мин и фильтруют. Полученный раствор спонтанно охлаждают до температуры 30C, а потом на охлаждающей водяной бане до температуры 10-15C, после чего при такой температуре перемешивают в течение 1 ч (без затравки кристаллов), фильтруют для получения раствора, а фильтр дважды промывают 105 мл охлажденного 96% спирта. Продукт высушивается при температуре 25C в течение 18 ч и взвешивается. Количество очищенного карбабензпирида: 502,8 г (62,5% на основе изоникотиновой кислоты). Аналитический контроль. Содержание: 100,97%. Содержание сопутствующих примесей: Пример 8. Этот пример отличается от примера 7 тем, что для реакции перекристаллизации используется 30 г неочищенного карбабензпирида, 90 мл 90% спирта и 0,1 г активированного древесного угля. Количество очищенного карбабензпирида: 25,6 г (60,3% на основе изоникотиновой кислоты). Аналитический контроль. Содержание: 100,58%. Содержание сопутствующих примесей: Пример 9. Этот пример отличается от примера 7 тем, что для перекристаллизации используется 500 г неочищенного карбабензпирида, 1500 мл 90% спирта и 15 г активированного древесного угля. Количество очищенного карбабензпирида: 425,79 г (75,4% на основе изоникотиновой кислоты). Аналитический контроль. Содержание: 99,48%. Содержание сопутствующих примесей: Пример 10. 50 г неочищенного карбабензпирида растворяют в 150 мл 90% спирта (1:3) (m/V), добавляют 1,5 г активированного древесного угля, реакционную массу нагревают до температуры кипения, помешивают и кипятят в течение 30 мин и фильтруют. Полученный раствор спонтанно охлаждается до температуры 30C, а потом на охлаждающей водяной бане до температуры 10-15C, при этой температуре перемешивается в течение 1 ч (без затравки кристаллов), фильтруется для получения раствора, а фильтр дважды промывается 20 мл охлажденного 96% спирта. Продукт высушивается при 25C в течение 18 ч и взвешивается. Количество очищенного карбабензпирида: 44 г (70,8% на основе изоникотиновой кислоты). Аналитический контроль. Температура плавления: 191,3C. Содержание: 99,81%. Содержание сопутствующих примесей: Пример 11. Этот пример отличается от примера 10 тем, что в качестве растворителя используется 70% спирт. Количество очищенного карбабензпирида: 33 г (53,1% на основе изоникотиновой кислоты). Аналитический контроль. Температура плавления: 191,5C. Содержание: 101,01%. Содержание сопутствующих примесей: Пример 12. Этот пример отличается от примера 10 тем, что в качестве растворителя используется вода. Количество очищенного карбабензпирида: 40,6 г (65,4% на основе изоникотиновой кислоты). Аналитический контроль. Температура плавления: 191,4C. Содержание: 100,19%. Пример 13. Этот пример отличается от примера 10 тем, что неочищенный карбабензпирид очищают и подвергают второй перекристаллизации с использованием в качестве растворителя воды. 40 г очищенного карбабензпирида растворяют в 460 мл воды при температуре 30-35C, перемешивают в течение 20 мин, а потом спонтанно охлаждают до температуры 22-25C, далее перемешивают в течение 1 ч, а потом охлаждают в течение 2 ч до температуры 7-10C и фильтруют. Количество очищенного карбабензпирида: 29,5 г (73,8% на основе очищенного карбабензпирида). Аналитический контроль. Температура плавления: 191,1C. Содержание: 99,11%. Содержание сопутствующих примесей: Информация о структуре кристалла (кристаллическая форма карбабензпирида). Для кристаллографического анализа с использованием рентгеновских лучей использовалась желтая призма C14H15IN2O с приблизительными размерами 0,260,340,40 мм. Данные относительно интенсивности рентгеновского излучения определялись при 173(2) K на системе Bruker SMART APEX II, оборудованной графитовым монохроматором и MoK запаянной трубкой с ультратонким фокусом(=0,71073), работающим на мощности 1250 Вт (50 кВ, 25 мА). Детектор был расположен на расстоянии 60 мм от кристалла. Было получено 458 изображений с шириной сканирования 1,5 ви 211 дополнительных изображений с шириной сканирования 1,5 в . Все изображения были получены с выдержкой 20 с/изображение. Общее время получения данных составляло 5 ч. Изображения были интегрированы с помощью пакета программного обеспечения Bruker SAINT с использованием алгоритма интеграции узких изображений. Интеграция данных выполнялась с использованием ромбической элементарной ячейки, что дало в целом 18403 отражений к максимальному углу падения =30,54 (разделение - 0,7 ),из которых 4309 были независимыми (избыточность 4,27, полнота = 99,7%, Rint=2,67%, Rsig=2,29%) и 4152 (96,4%) превышали 4(F2). Окончательные параметры элементарной ячейки (а=9,27390(10) ,b=10,7187(2) , с=14,2161(2) ,=90, =90, =90, объем=1413,14(4) 3) основываются на уточнении центров масс XYZ 9894 отражений более 20(I) с 2,38230,54. Анализ данных показал незначительный разброс при сборе данных. Данные были откорректированы с учетом эффекта поглощения с использованием численного метода (SADABS). Отношение минимального к максимальному очевидному пропусканию составляло 0,79. Вычисленные коэффициенты минимального и максимального пропускания(на основании размера кристалла) составляют 0,4625 и 0,5872. Структура была определена и уточнена с помощью пакета программного обеспечения BrukerSHELXTL (версия 6.1) с использованием пространственной группы Р 2(1) 2(1) 2(1), с Z=4 для состава элементарной ячейки C14H15IN2O. Окончательное уточнение с использованием анизотропного приближения полноматричного метода наименьших квадратов при F2 с 164 переменными сошлось приR1=1,87% для наблюдаемых данных и при wR2=4,40% - для всех данных. Степень соответствия была 1,117. Наибольший пик при окончательном дифференцированном воссоздании электронной плотности был 0,260 е-/3, а дырка - 0,752 е-/3 со среднеквадратичным отклонением 0,093 е-/3. Плотность, рассчитанная на основании окончательной модели, составила 1,665 г/см 3 и F(000), 696 е-. Результаты рентгеноструктурного графического анализа кристалла подытожены в следующих таблицах. Таблица 3 Данные для образца и кристалла Таблица 4 Атомные координаты и эквивалентные изотропные параметры атомного смещения (2).U(экв) определяется как одна треть следа ортогонализированного тензора Uij Антивирусная активность против штаммов вируса гриппа типа A [H3N2 (Калифорния и Виктория/3/75), H1N1 (Новая Каледония 20/99)] Целью нижеописанного исследования было проверить антивирусную активность кристаллической формы карбабензпирида, имеющую кодовое название "FAV00A", против разных штаммов вирусов гриппа типа А. Ингибирование вирусной инфекции в культуре клеток MDCK (NBL-2) (клетки почки собаки Мадин-Дарби) проверяли, определяя уменьшение титров вирусов с помощью метода оценки уменьшения"урожая" вирусов. В качестве контроля эффективности FAV00A использовалось вещество сравнения(Занамивир - ингибитор нейраминидазы) с известным противовирусным эффектом к вирусам гриппа. 1. Материалы и методы. Система для проведения исследований. Клетки. Линия клеток почки собаки Мадин-Дарби (MDCK, NBL-2) была получена из Американской коллекции типичных культур (АТСС), Роквилл, Мэриленд, АТСС- номер CCL-34. Клеточную культуру хранили в жидком азоте. Рабочий запас клеток NBL-2 выращивали в минимальной питательной среде, дополненной 5% эмбриональной телячьей сывороткой (ЭТС) в течение не более 20 циклов в соответствии со стандартной операционной процедурой (СОП). Клетки NBL-2 выращивались в колбах для клеточной культуры с площадью поверхности 25 см 2 на минимальной питательной среде (МПС) при 37C и переносились в разведении 1:10 или 1:20 дважды в неделю. В каждую лунку 96-луночного планшета вносили 100 мкл клеточной суспензии, которая содержала 8104 клеток. В соответствии с СОП клетки в плановом порядке проверяли на наличие микоплазм с помощью набора VenorGeM для традиционной полимеразной цепной реакции (производства "Минерва Биолабс ГмбХ" (Minerva Biolabs GmbH), Берлин, Германия). Клетки, которые использовались в исследовании, не были загрязнены. Вирусы. К вирусам гриппа, которые использовались для проверки антивирусной активности, принадлежали: вирус гриппа типа A H1N1 (штамм Новая Каледония 20/99), вирус гриппа типа A H3N2 (штаммы Виктория/3/75 и Калифорния), вирус гриппа типа В (штаммы Тайвань 2/62 и Цзянсу). Запас вирусов был получен при размножении вирусов гриппа на бессывороточной МПС с добавлением 2 мкг/мл трипсина с использованием клеток NBL-2 в качестве хозяина. Для гарантирования стабильности вируса к зараженной вирусом культуре клеток NBL-2 после 2-3 дней инкубации при 37C добавили 1% сывороточного альбумина коров (САК), перед замораживанием на ночь при температуре 80C. Из очищенного супернатанта клеточной культуры, используя низкоскоростное центрифугирование, приготовили маточные растворы с вирусами, а потом разделили на аликвоты по 500 мкл. Этот исходный материал с вирусами хранился при 80C до момента использования. Соответствующие титры инфекционности вирусов в исходных растворах определялись титрованием на монослое NBL-2 клеток в 96-луночных планшетах и оценивалась доза заражения 50% культуры тканей (TCID50). Инфекционный титр определялся методом Спирмена-Кербера (Spearman С. (1908). The Среда для клеточной культуры: МПС (MEM), номер по каталогу Т 031-10, "Биохром"(Biochrom). ЭТС (FCS), номер по каталогу F-7524, "Сигма" (Sigma). Фосфатно-солевой буферный раствор (PBS) 10: Фосфатно-солевой буферный раствор, номер по каталогу L 182-10, "Дульбекко Биохром" (DulbeccoBiochrom) Добавить до 1 л воды для инъекций, номер по каталогу 0370-3452, "Браун" (Braun). Автоклавируют и хранят при комнатной температуре, срок годности один год; два месяца в случае использования.PBS 1: 450 мл воды для инъекций 50 мл 10 PBS. Хранить при комнатной температуре, срок годности один год; два месяца в случае использования.NaHCO3, 7,5: 75 г NaHCO3, номер по каталогу S-4019, "Сигма" (Sigma). Растворяют в 1 л воды (Millipore - Н 2 О). Хранить при температуре 2-8C, срок годности один год; два месяца в случае использования.Hepes (N-2-гидроксиэтилпиперазин N'-2-этансульфоновая кислота), 1 М: 238,31 г Hepes, номер по каталогу S-4019, "Сигма" (Sigma). 33,75 мл NaOH 32. Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 2-8C, срок годности один год; четыре месяца в случае использования.L-глутамин, 0,2 М: 29,2 г L-глутамина, номер по каталогу G-5763, "Сигма" (Sigma). Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 18C в течение одного года; два месяца при температуре 2-8C в случае использования. Пенициллин/стрептомицин: 10 МЕГА Пенициллин (пенициллин), "Грюненталь"(Grnenthal). 10 г стрептомицин-сульфат, номер по каталогу S-6501, "Сигма" (Sigma). Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 18C в течение одного года; две недели при температуре 2-8C в случае использования. Трипсин/ЭДТК раствор: 2,0 г трипсина 1: 250 ВАЕЕ 1570 единицы/мл, номер по каталогу Т-4799, "Сигма" (Sigma). 0,6 г ЭДТК-Na (Tritriplex III), номер по каталогу 8418.0100, "Мерк "(Merck). Растворить в 1 л 1 PBS. Хранить при температуре 18C в течение одного года; один месяц при температуре 2-8C в случае использования. В качестве поддерживающей среды использовали бессывороточные МПС с добавлением 2% Hepes,2% NaHCO3, 1% пенициллина/стрептомицина, с содержанием 2 мкг/мл трипсина. 2. Определение антивирусной активности (метод оценки уменьшения "урожая" вирусов). Метод оценки уменьшения "урожая" вирусов - мощный метод для оценки эффективности противовирусных соединений. Этот метод дает возможность выполнить количественный анализ образования инфекционных вирионов в обработанной препаратом культуре в сравнении с "урожаем" вирусов в необработанном контрольном образце. При этой процедуре инфицированные вирусом культуры инкубируют с противовирусными соединениями в течение достаточного для репликации периода, а потом оценивают на наличие нового потомства вирусов методом титрования на отдельном монослое клеточной культурыsimplex virus. J. Virol. Methods. 28:101-106). 2.1. Принцип исследования. Исследование выполнялось в колбах для клеточной культуры в трех параллелях. Были проведены два независимых эксперимента. Концентрации испытуемого и эталонного соединений предварительно определялись при измерении цитотоксичности, как описано выше. При наивысшей использованной концентрации не должно было обнаруживаться ни одного токсического эффекта на клетки. Предварительные эксперименты были сначала проведены в двух разных вариантах с использованием 400 мкг испытуемого вещества FAV00A и 10 нг Занамивира в качестве эталонного соединения. Эксперимент проводился с использованием вируса гриппа типа В штамма Тайвань 2/62 и вируса гриппа типа А штамма Новая Каледония. При первом варианте исследования клетки NBL-2 высевали в колбы для клеточной культуры с площадью поверхности 25 см 2 на МПС при 37C в разведении 1:10. Клетки подсчитывали, а потом заражали после промывания фосфатно-солевым буферным раствором при множественности инфицирования 0,01 для обоих штаммов вируса гриппа. К одному ряду (в трех параллелях) инфицированных клеток одновременно прибавили 400 мкг FAV00A (одновременно с вирусом). Клетки инкубировали в течение 45 мин при 37C, чтобы позволить вирусу осуществить адсорбцию,а потом трижды промывали фосфатно-солевым буферным раствором для удаления неадсорбированных вирусов. После этого группу инфицированных клеток, предварительно инкубируемых из 400 мкгFAV00A, опять обработали 400 мкг FAV00A. Вторую группу инфицированных клеток обработали 10 нг Занамивира. Также в исследование был включен вирусный контроль без добавления испытуемого или эталонного соединения. Второй вариант исследования выполнялся подобно первому, но без этапа предварительной инкубации с 400 мкг FAV00A (одновременное добавление FAV00A). При этом клетки сначала инфицировали вирусами, после чего инкубировали в течение 45 мин для того, чтобы позволить вирусу адсорбироваться. После трехкратного промывания фосфатно-солевым буферным раствором культуру инфицированных клеток обрабатывали 400 мкг FAV00A или 10 нг Занамивира. Колбы с культурой инкубировали при 37C в инкубаторе с 5% CO2 в течение 24 ч, а потом замораживали на ночь, а на следующий день размораживали. Этапы замораживания и размораживания предопределяли дезинтеграцию клеток и высвобождение вирусов, которые они содержали. Таким образом, можно было определить общее количество вируса. После низкоскоростного центрифугирования в каждой экспериментальной модели (контроль вируса, а также испытуемое и эталонное соединение) определяли количество вируса, используя "титрование к конечной точке" в 96-луночном планшете, при этом получали титр вируса/количество, при котором 50% клеток являются инфицированными или неинфицированными (TCID50/мл). Все дальнейшие эксперименты со всеми испытуемыми вирусами гриппа проводили по второму варианту. Для этого выполняли подсчет клеток, которые потом инфицировали после промывания фосфатно-солевым буферным раствором при множественности инфицирования 0,01 для всех использованных типов вируса гриппа, кроме H3N2 (Калифорния), который использовали для инфицирования клеток при множественности инфицирования 0,001. Были исследованы две разные концентрации (400 и 800 мкгFAV00A; 100 и 1000 нг Занамивира). Титрование до конечной точки. В соответствии с СОП подготовили один 96-луночный планшет (для каждой экспериментальной модели) с чувствительной клеточной культурой. В каждую лунку засеяли 50 мкл суспензии клеток. Клетки выращивали при 37C в атмосфере с 5% СО 2 в течение трех или четырех дней для образования сплошного монослоя. После низкоскоростного центрифугирования осуществляли 10-кратное серийное разведение (до 10-9) полученных вирусов с использованием поддерживающей среды. На каждой пластине 1- и 2-й ряды (в каждом по 8 лунок) использовали для контроля клеточной культуры, а с 3- по 12-й - для 10-кратно серийно разведенных вирусов, начиная с неразведенного матричного образца и заканчивая разведением 10-9. В соответствующие ряды внесли по 50 мкл разведения вирусов, тогда как в лунки контрольных рядов вносили по 50 мкл поддерживающей среды. Планшеты инкубировали в течение 2-3 дней при температуре 37C в инкубаторе с СО 2. По завершении инкубационного периода планшеты изучали микроскопически на наличие цитопатогенного эффекта (ЦПЭ). Исследования проводили независимо двумя лаборантами. Титр вирусов был подсчитан по методу Спирмена-Кербера. После этого вычислялся процент уменьшения титра при добавлении испытуемого или эталонного соединения в сравнении с контролем (взято за 100%). 2.2. Полученные результаты. Уменьшение "урожая" вирусов выражали как TCID50/мл, показан также % вирусного инфицирования, а также во сколько раз наблюдалось уменьшение в случае обработки испытуемым или эталонным соединением по отношению к вирусному контролю (100%). Поскольку проводили два эксперимента с испытуемым и эталонным соединением, в таблицах приведены средние величины. Таблица 8 Вирус гриппа типа А, штамм Новая Каледония (H1N1) Таблица 9 Вирус гриппа типа А, штамм Калифорния (H3N2) Таблица 10 Вирус гриппа типа А, штамм Виктория (H3N2) Таблица 11 а Вирус гриппа типа А, штамм Новая Каледония (H1N1) Таблица 12b Вирус гриппа типа А, штамм Новая Каледония (H1N1) 3. Выводы. Вышеприведенные данные подтверждают, что FAV00A оказывает противовирусный эффект на вирусы гриппа типа А, проверенный уменьшением "урожая" вирусов в клеточной культуре NBL-2. Время добавления в эксперименте дает возможность допустить, что FAV00A влияет на стадии адсорбции вируса к специфическим рецепторам на мембране клеток. К этим стадиям относятся распаковывание вирусов и репликация. Антивирусная активность против аденовирусов, вируса Коксаки, эховируса, цитомегаловируса, метапневмовируса и энтеровируса, определенная методом оценки "урожая" вирусов. Целью исследования было исследовать влияние испытуемого вещества FAV00A на репликацию аденовирусов, метапневмовируса человека, эховируса, вируса Коксаки А 9, энтеровируса 71 и цитомегаловируса человека, определяя титр вируса. Титры вирусов определялись по методу оценки уменьшения 1 Множественность инфицирования - соотношение инфекционных агентов (например, фагов или вирусов) к их мишеням (например, клетки). 1.1.2. Система для проведения исследований. Клетки: Для культивирования цитомегаловируса, энтеровируса, эховируса и вируса Коксаки А 9 использовалась культура препуциальных фибробластов человека (HFF). Клетки HFF определялись в соответствии с вышеприведенным описанием [1]. Клетки LLC - MK2 (MK2), полученные из Американской коллекции типичных культур (АТСС, Роквилл, Мерилэнд), были использованы для культивирования метапневмовируса. Клетки А 549 были получены из АТСС и использованы для культивирования аденовируса. Рабочий запас клеток выращивали в минимальной питательной среде (МПС), дополненной 5% эмбриональной телячьей сывороткой (ЭТС) в течение не более 20 циклов в соответствии со стандартной операционной процедурой (СОП). Клетки выращивались в колбах для клеточной культуры с площадью поверхности 25 см 2 на МПС при 37C. Для культивирования в каждую лунку 96-луночного планшета вносили 100 мкл клеточной суспензии, клетки росли до достижения конфлюэнтности. Клетки в плановом порядке проверяли на наличие микоплазм с помощью набора VenorGeM для традиционной полимеразной цепной реакции (производства "Минерва лабс ГмбХ" (Minerva labs GmbH),- 15019080 Берлин, Германия). Клетки, использованные в исследовании, не были загрязненными. Среда для клеточной культуры: МПС (MEM), номер по каталогу Т 031-10, "Биохром" (Biochrom). ЭТС (FCS), номер по каталогу F-7524, "Сигма" (Sigma). Фосфатно-солевой буферный раствор (PBS) 10: Фосфатно-солевой буферный раствор, номер по каталогу L 182-10, "Дульбекко Биохром" (DulbeccoBiochrom). Добавить до 1 л воды для инъекций, номер по каталогу 0370-3452, "Браун" (Braun). Автоклавируют и хранят при комнатной температуре, срок годности один год; два месяца в случае использования.PBS 1: 450 мл воды для инъекций 50 мл 10 PBS. Хранить при комнатной температуре, срок годности один год; два месяца в случае использования.NaHCO3, 7,5: 75 г NaHCO3, номер по каталогу S-4019, "Сигма" (Sigma). Растворяют в 1 л воды (Millipore - Н 2 О). Хранить при температуре 2-8C, срок годности один год; два месяца в случае использования.Hepes, (N-2-гидроксиэтилпиперазин N-2-этансульфоновая кислота) 1 М: 238,31 г Hepes, номер по каталогу S-4019, "Сигма" (Sigma). 33,75 мл NaOH 32%. Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 2-8C, срок годности один год; четыре месяца в случае использования.L-глутамин, 0,2 М: 29,2 г L-глутамина, номер по каталогу G-5763, "Сигма" (Sigma). Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 18C в течение одного года; два месяца при температуре 2-8C в случае использования. Пенициллин/стрептомицин: 10 МЕГА Пенициллин (пенициллин), "Грюненталь" (Grnenthal). 10 г стрептомицин-сульфат, номер по каталогу S-6501, "Сигма" (Sigma). Растворить в 1 л воды (Millipore - Н 2 О). Хранить при температуре 18C в течение одного года; две недели при температуре 2-8C в случае использования. Трипсин/ЭДТК раствор: 2,0 г трипсина 1:250 ВАЕЕ 1570 единицы/мл, номер по каталогу Т-4799, "Сигма" (Sigma). 0,6 г ЭДТК-Na (Tritriplex III), номер по каталогу 8418.0100, "Мерк" (Merck). Растворить в 1 л 1 PBS. Хранить при температуре 18C в течение одного года; один месяц при температуре 2-8C в случае использования. В качестве поддерживающей среды использовалась бессывороточная МПС с добавлением 2%Hepes, 2% NaHCO3, 1% пенициллина/стрептомицина, содержащая 2 мкг/мл трипсина. 1.2. Определение антивирусной активности (метод оценки уменьшения "урожая" вирусов). Метод оценки уменьшения "урожая" вирусов - мощный метод для оценки эффективности противовирусных соединений. Этот метод дает возможность выполнить количественный анализ образования инфекционных вирионов в обработанной препаратом культуре по сравнению с "урожаем" вирусов в необработанном контрольном образце. При этой процедуре инфицированные вирусом культуры инкубируют с противовирусными соединениями в течение достаточного для репликации периода, а потом оценивают на наличие нового потомства вирусов методом титрования на отдельном монослое клеточной культуры (Shipman С. Jr., Smith S.H., Carlson R.H., Drach J.C. (1976). Antiviral activity of arabinosyladeninevirus. J. Virol. Methods. 28:101-106). 1.2.1. Принцип исследования. Исследование выполнялось в колбах для клеточной культуры в трех параллелях. Были проведены два независимых эксперимента. Клетки подсчитывали, а потом заражали после промывания фосфатносолевым буферным раствором при множественности инфицирования. К клеточной культуре одновременно добавили FAV00A (вместе с вирусом). Потом клетки инкубировали в течение 45 мин при 37C, чтобы позволить вирусу осуществить адсорбцию, а после трижды промывали фосфат- 16019080 но-солевым буферным раствором для удаления неадсорбированных вирусов. После этого группу инфицированных клеток, предварительно инкубируемых из FAV00A, опять обработали FAV00A. Также в исследование был включен вирусный контроль без добавления испытуемого или эталонного соединения. Колбы с культурой инкубировались при 37C в инкубаторе с CO2. После 3 дней (эховирус, энтеровирус,вирус Коксаки), 4 дней (аденовирус), 5 дней (цитомегаловирус) или 8 дней (метапневмовирус) клетки замораживали на ночь, а на следующий день размораживали. Этапы замораживания и размораживания предопределяли дезинтеграцию клеток и высвобождение вирусов, которые они содержали. Таким способом можно было определить общее количество вирусов. После низкоскоростного центрифугирования в каждой экспериментальной модели (контроль вирусов, а также испытуемое и эталонное соединения) проводили определение количества вирусов с использованием "титрования до конечной точки" в 96-луночном планшете, при этом получали титр вируса/количество, при котором 50% клеток являются инфицированными или неинфицированными(TCID50/мл). Титрование до конечной точки. В соответствии с СОП подготовили один 96-луночный планшет (для каждой экспериментальной модели) с чувствительной клеточной культурой. В каждую лунку засеяли 50 мкл суспензии клеток. Клетки выращивались при 37C в атмосфере с 5% СО 2 в течение трех или четырех дней для образования сплошного монослоя. После низкоскоростного центрифугирования осуществлялось 10-кратное серийное разведение (до 10-9) полученных вирусов с использованием поддерживающей среды. На каждой пластине 1- и 2-й ряды (в каждом по 8 лунок) использовались для контроля клеточной культуры, а с 3- по 12-й (в каждом по 8 лунок) - для 10-кратно серийно разведенных вирусов, начиная с неразведенного матричного образца и заканчивая разведением 10-9. В соответствующие ряды внесли по 50 мкл разведения вирусов, тогда как в лунки контрольных рядов вносили по 50 мкл поддерживающей среды. Планшеты инкубировали в течение 2-3 дней при температуре 37C в инкубаторе с СО 2. По завершении инкубационного периода планшеты изучали микроскопически на наличие цитопатогенного эффекта (ЦПЭ). Исследования проводились независимо двумя лаборантами. Титр вирусов был определен по методу Спирмена-Кербера (Spearman С. (1908). The method of "right and wrong cases"("constantstimuli")experimental series]. Archiv fr experimentelle Pathologie und Pharmakologie. 162:480-483.). После этого вычислялся процент уменьшения титра при добавлении испытуемого или эталонного соединения в сравнении с контролем (взято за 100%). 1.3. Полученные результаты. Уменьшение "урожая" вирусов выражали как TCID50/мл, показан также % вирусного инфицирования, а также во сколько раз наблюдалось уменьшение после обработки испытуемым или референтным соединением по отношению к вирусному контролю (100%). Поскольку проводилось два эксперимента с испытуемым и эталонным соединением, в таблицах приведены средние величины. Таблица 13 Аденовирус 3 2. Вывод Вышеприведенные данные подтверждают, что исследуемое соединение FAV00A оказывает противовирусный эффект против аденовирусов, вируса Коксаки, эховируса, цитомегаловируса, метапневмовируса и энтеровируса, что проверено методом оценки "урожая" вирусов. В вышеописанных экспериментах использовались следующие сокращения: АТСС = американская коллекция типичных культур; САК = сывороточный альбумин коров; ЦПЭ = цитопатогенный эффект; ЭТС = эмбриональная телячья сыворотка;MDCK = клетки почки собаки Мадин-Дарби; МПС = минимальная питательная среда; МИ = множественность инфекции;TCID50=доза инфицирования клеточной культуры, при которой 50% клеток инфицировано. Влияние FAV00A на уровень интерферона в плазме крови пациентов с гриппом. У добровольцев, принимавших FAV00A, на 7-й день приема препарата (визит V3) выявлено значительное повышение уровней IFN- и IFN- в сравнении с начальным содержанием этих веществ и содержанием интерферонов в группе, принимавшей плацебо. Повышение уровней IFN- также наблюдалось у пациентов, принимавших плацебо, однако этот рост был менее интенсивным в сравнении с группой, в которой принимали FAV00A (табл. 22). Таблица 22 Динамика уровня циркулирующего интерферона разных типов в сыворотке крови добровольцев с острой респираторной вирусной инфекцией в группах FAV00A/плацебо Р - степень надежности межгрупповых отличий. Отличия между параметрами статистически достоверны в сравнении с визитом V1 (определено с помощью ковариантного анализа). Уровни циркулирующего IFN- уменьшались в контрольной группе пациентов на третьем (V3) и четвертом (V4) визитах. Результаты свидетельствуют об интерфероногенном эффекте FAV00A. Повышение уровнейIFN- в контрольной группе отображает индукцию образования этого белка в ответ на вирусную инфекцию. Общеизвестно, что вирусная инфекция приводит к активации разных систем транскрипции в клетках, т.е. благодаря стимулированию образования нескольких хемокинов и цитокинов FAV00A вносит свой вклад в образование антивирусной защиты организма путем стимуляции образования интерферона I и иммунного интерферона . Последний образуется преимущественно Т-лимфоцитами и активированными моноцитами. Этот процесс предопределяет поляризацию Т-клеточного иммунного ответа относительно Т-хелперов 1-го типа. Как видно из таблицы, уровни циркулирующего IFN- значительно повысились на 7-й день после начала лечения у добровольцев, принимавших FAV00A, и оставались высокими в течение периода реконвалесценции до 14-го дня исследования, которое объясняет как терапевтический, так и профилактический эффекты препарата. Уровни IFN- в контрольной группе пациентов несколько снижались в течение исследования. Таким образом, показано, что прием FAV00A стимулирует образование интерферонов типа I и II,что определяет его главные функции в клеточно-опосредованной защите от вирусных инфекций. Результаты исследования эффекта FAV00A на содержание интерферона в организме коррелируют с данными,собранными при статистической обработке анализов периферической крови добровольцев. Статистически достоверное (критерий Вилкоксона) уменьшение уровня моноцитов в процентах обнаружено в контрольной группе пациентов на визитах V3 и V4 (р=0,002 и р=0,033 соответственно). В течение исследования у пациентов, принимавших FAV00A, не было выявлено значительного снижения уровня моноцитов в крови. Этот факт свидетельствует об ограничении иммуносупрессивного эффекта вирусной инфекции на клеточный иммунный ответ в группе, которую лечили FAV00A. Влияние препарата на клеточно-опосредованный иммунный ответ улучшает клиническое течение острых респираторных заболеваний и ограничивает возможности развития вирусной иммуносупрессии,осложнений и перехода инфекции в хроническую форму. Особенно важное значение имеет повышение образования IFN-, поскольку он, кроме антивирусной активности, имеет разнообразные эффекты на клетки иммунной системы, миеломоноцитарные клетки и считается ключевым цитокином, сопровождающим антигенную стимуляцию лимфоцитов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. -Кристаллическая форма карбабензпирида формулы (I) которая имеет параметры, определенные с помощью метода порошковой рентгеновской дифракции с использованием для измерения дифрактометра (медный антикатод), выраженные относительно межплоскостного расстояния d, угла Брэгга 2 тэта и относительной интенсивности (выражается в процентах к наиболее интенсивному лучу), указанные в нижеприведенной таблице с перечнем положений рефлексов высокой и средней интенсивности. 2. -Кристаллическая форма карбабензпирида по п.1, имеющая степень чистоты по меньшей мере 99,5%, определенную методом ВЭЖХ. 3. -Кристаллическая форма карбабензпирида по п.2, имеющая степень чистоты по меньшей мере 99,9%, определенную методом ВЭЖХ. 4. -Кристаллическая форма карбабензпирида по п.1, имеющая один эндотермический максимум на ДСК-кривой от 187 до 193C. 5. -Кристаллическая форма карбабензпирида по п.1, имеющая ИК-спектр с характерными пиками,показанными в нижеследующей таблице. 6. Способ получения -кристаллической формы карбабензпирида по п.1, включающий следующие стадии:(i) конденсация изоникотиновой кислоты с бензиламином при повышенных температурах;(ii) кристаллизация и выделение продукта конденсации, полученного на стадии (i);(iii) реакция кристаллического продукта, полученного на стадии (ii), с метилйодидом и(iv) перекристаллизация неочищенного продукта, полученного на стадии (iii), из водного раствора спирта. 7. Способ по п.6, в котором реакция конденсации между изоникотиновой кислотой и бензиламином в соответствии со стадией (i) происходит с использованием избытка бензиламина в пределах 10-25%. 8. Способ по п.6, в котором продукт реакции конденсации между изоникотиновой кислотой и бензиламином, т.е. бензиламид изоникотиновой кислоты (БАИНК), кристаллизуется из реакционной смеси с использованием растворителя, выбранного из группы, состоящей из этилацетата, ацетонитрила и изопропанола. 9. Способ по п.8, дополнительно включающий использование активированного угля. 10. Способ по любому из пп.6, 8 или 9, в котором продукт, полученный на стадии (ii), т.е. БАИНК,- 20019080 обрабатывается водой. 11. Способ по п.6, в котором на стадии (iii) реакция кватернизации бензиламида изоникотиновой кислоты и метилйодида происходит с использованием избытка метилйодида в пределах 5-15%. 12. Способ по п.11, в котором реакция кватернизации происходит в водном растворе спирта. 13. Способ по п.12, в котором водный раствор спирта представляет собой 90% этанол. 14. Способ по п.6, дополнительно включающий стадию промывания неочищенного продукта, полученного на стадии (iii), водным раствором спирта. 15. Способ по п.14, в котором водный раствор спирта представляет собой 96% этанол. 16. Способ по п.6, в котором водный раствор спирта, используемый на стадии (iv), представляет собой этанол, содержащий от 5 до 15% (объемное отношение) воды. 17. Способ по п.16, в котором водный раствор этанола представляет собой 90% этанол. 18. Способ по п.6, в котором отношение неочищенного продукта и водного раствора спирта, используемого на стадии (iv), находится в пределах от 1:2 до 1:4. 19. Способ по п.18, в котором отношение неочищенного продукта и водного раствора спирта, используемого на стадии (iv), составляет 1:3. 20. Способ по п.6, в котором перекристаллизация на стадии (iv) происходит при спонтанном охлаждении кипящего раствора неочищенного продукта в водном этаноле до температуры от 30 до 40C с дальнейшим охлаждением раствора до температуры от 10 до 15C с последующим перемешиванием в течение периода времени от 1 до 3 ч. 21. Противовирусная фармацевтическая композиция, содержащая эффективное количество кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 и фармацевтически приемлемый носитель. 22. Применение -кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 для лечения и предупреждения вирусных инфекций. 23. Применение -кристаллической формы карбабензпирида по п.22, где вирусные инфекции представляют собой инфекции, вызванные вирусами гриппа типа А. 24. Применение -кристаллической формы карбабензпирида по п.23, где вирусные инфекции представляют собой инфекции, вызванные вирусом типа A [H3N2 (Калифорния и Виктория/3/75), H1N1 (Новая Каледония 20/99)]. 25. Применение -кристаллической формы карбабензпирида по п.22, где вирусные инфекции представляют собой инфекции, вызванные аденовирусами, вирусом Коксаки, эховирусом, цитомегаловирусом, метапневмовирусом и энтеровирусом. 26. Применение -кристаллической формы карбабензпирида формулы (I) по любому из пп.1-5 для лечения острых респираторных заболеваний.
МПК / Метки
МПК: C07D 213/81, A61K 31/4425, A61P 31/16
Метки: alpha-кристаллическая, форма, карбабензпирида
Код ссылки
<a href="https://eas.patents.su/24-19080-alpha-kristallicheskaya-forma-karbabenzpirida.html" rel="bookmark" title="База патентов Евразийского Союза">α-кристаллическая форма карбабензпирида</a>
α-кристаллическая форма замещенных селеноксантенов

Номер патента: 18106
Опубликовано: 30.05.2013
Авторы: Хомичёнок Виктор Владимирович, Подгородниченко Владимир Константинович, Гончарова Анна Яковлевна, Цыб Анатолий Федорович, Розиев Рахимджан Ахметджанович
МПК: C07D 345/00
Метки: замещенных, alpha-кристаллическая, селеноксантенов, форма
Формула / Реферат:
Средство, представляющее собой α-кристаллическую форму 9-фенил-симм-октагидроселеноксантена, обладающее антиоксидантным, детоксицирующим, иммуномодулирующим, антиатерогенным, антисклеротическим, анаболическим, гиполипедическим действием и соответствующее структурной формулес порошковой рентгенограммой, полученной на Cu-K-источнике излучения с показателями характеристического отражения, выраженными в градусах угла дифракции 2θ: 6,0;...
&alpha-кристаллическая форма трет-бутиламиновой соли периндоприла

Номер патента: 5008
Опубликовано: 28.10.2004
Авторы: Бейе Стефан, Пфеффер Брюно, Жино Ив-Мишель, Кокерель Жерар
МПК: C07D 209/42, A61P 9/12, A61K 31/475...
Метки: соли, форма, трет-бутиламиновой, периндоприла, alpha-кристаллическая
Формула / Реферат:
1. a-Кристаллическая форма соединения формулы (I) отличающаяся следующей диаграммой рентгеновской дифракции порошка, измеряемой с применением дифрактометра Siemens D5005 (медный антикатод) и выражаемой в терминах: межплоскостное расстояние d, угол 2 тета Брага (Bragg's), интенсивность и относительная интенсивность (выражаемые в процентах наиболее интенсивного луча): 2. Способ получения a -кристаллической формы соединения формулы (I) по...
Кристаллическая форма ii линезолида

Номер патента: 4434
Опубликовано: 29.04.2004
Автор: Бергрен Майкл С.
МПК: C07D 413/10
Метки: линезолида, кристаллическая, форма
Формула / Реферат:
1. (S)-N-[[3-[3-Фтор-4-(4-морфолинил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид в виде кристаллической формы II, имеющей спектр порошковой дифракции рентгеновских лучей. d-расстояние, A Угол два-тета, ш Относительная интенсивность 12,44 7,10 2 9,26 9,54 9 6,37 13,88 6 6,22 14,23 24 5,48 16,18 3 5,28 16,79 100 5,01 17,69 2 4,57 19,41 4 4,50 19,69 2 4,45 19,93 6 4,11 21,61 15 3,97 22,39 23 ...
Кристаллическая форма целекоксиба

Номер патента: 4470
Опубликовано: 29.04.2004
Авторы: Бахар Мехмет, Гоктепе Мехмет, Гундуз А.Халит
МПК: A61K 31/415, C07D 231/12
Метки: кристаллическая, форма, целекоксиба
Формула / Реферат:
1. Кристаллический 4-[5-(4-метилфенил)-3-(трифторметил)-1H-пиразол-1-ил]бензолсульфонамид, характеризующийся, по меньшей мере, следующими данными порошковой дифрактограммы рентгеновских лучей: Угол [° 2q] Отн. инт. [%] 14.800 69.0 16.050 78.9 17.875 63.7 19.615 100.0 21.455 96.6 22.080 68.1 22.385 65.4 23.425 62.5 25.330 64.5 29.355 60.8 2. Кристаллический...
Кристаллическая форма эплеренона

Номер патента: 8449
Опубликовано: 29.06.2007
Авторы: Год Генри Т., Вечорек Джозеф Дж., Гэнсер Скотт, Йан Крис Й., Бартон Кэтлин П., Борчардт Томас Б., Мудипалли Партха С., Карлос Марлон В., Пилипоскас Дэниел Р., Ферро Леонард Дж., Стал Гленн Л., Литтл Клэй Р., Питц Марк А., Синг Йуен-Лунг Л., Десай Субхаш
МПК: C07J 71/00, A61K 31/58
Метки: форма, кристаллическая, эплеренона
Формула / Реферат:
1. Фармацевтическая композиция в оральной дозированной форме, включающая от около 10 до около 1000 мг эплеренона и по крайней мере один фармацевтически приемлемый наполнитель, причем эплеренон, присутствующий в композиции, имеет фазовую чистоту формы L кристаллического эплеренона от около 90 до около 100% и форма L кристаллического эплеренона характеризуется порошковой рентгенограммой при длине волны 1,54056 Е, включающей один или более пик,...
Предыдущий патент: Биобезопасный нанокомпозитный полимерный сорбент для селективного связывания изотопов sr и cs из жидких сред и сырьевая смесь для его изготовления
Следующий патент: Устройство для производства пресной воды
Случайный патент: Соединения и композиции как ингибиторы протеинкиназы b-raf