Способ получения марбофлоксацина и его промежуточного соединения
Номер патента: 21239
Опубликовано: 29.05.2015
Авторы: Коленц Иванка, Искра Ернеи, Рузиц Милош, Зупет Рок, Пецавар Аница, Пуцель Йоже, Плапер Игор


















Формула / Реферат
1. Способ получения соединения формулы (I)

где R выбран из Н, C1-6 алкила, катиона щелочного металла, X представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил,
отличающийся тем, что осуществляют взаимодействие соединения формулы (II)

где R и X являются такими, как указано выше, и R' выбран из Н или формила,
с соединением формулы (III)

где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила.
2. Способ по п.1, где соединение формулы (I), где R и R' представляет собой Н, а X представляет собой 4-метилпиперазинил, получают путем взаимодействия соединения формулы (II), где R представляет собой Н или этил, R' представляет собой Н, а X представляет собой 4-метилпиперазинил, с гидроксидом тетраметиламмония.
3. Способ по п.1 или 2, где способ осуществляют в отсутствие растворителей.
4. Способ по любому из пп.1-3, где соотношение соединения формулы (II) и соединения формулы (III) составляет от 1:3 до 1:6.
5. Способ однореакторного получения марбофлоксацина формулы (IV)

где R представляет собой Н, а X представляет собой 4-метилпиперазинил,
отличающийся тем, что включает следующие этапы:
а) взаимодействие соединения формулы (II)

где R выбран из группы Н, C1-6 алкила, катиона щелочного металла,
X представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил и
R' выбран из Н или формила,
с соединением формулы (III)

где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила,
b) взаимодействие полученного промежуточного соединения без выделения с муравьиной кислотой и формальдегидом и, необязательно,
с) в случае, когда X представляет собой галоген, его замещение соответствующим пиперазином традиционными способами с получением после выделения соединения формулы (IV).
6. Способ по п.5, в котором используют соединение формулы (II), где R выбран из Н, метила, этила; X представляет собой 4-метилпиперазин и R' представляет собой Н или формил.
7. Способ по п.5 или 6, в котором используют соединение формулы (II), где R представляет собой Н; X представляет собой 4-метилпиперазин и R' представляет собой Н (формула (IIa)).
8. Способ по любому из пп.5-7, где этап (а) проводят в отсутствие растворителя.
9. Способ по любому из пп.5-8, где соотношение соединения формулы (II) и соединения формулы (III) составляет от 1:3 до 1:6.
10. Способ по любому из пп.7-9, в котором соединение формулы (IIa) получают путем гидролиза соединения формулы (II), где R представляет собой C1-C6 алкил, R' представляет собой формил и X представляет собой N-метилпиперазин (формула (IIc)), с кислотой или с основанием или путем взаимодействия соединения формулы (II), где R представляет собой Н, R' представляет собой Н, а X представляет собой F (формула (IId)), с N-метилпиперазином.
11. Способ по п.10, в котором соединение формулы (IIc) получают путем осуществления взаимодействия соединения формулы (II), где R представляет C1-C6 алкил, R' представляет собой формил и X представляет собой F (формула (IIb)), с N-метилпиперазином.
12. Способ по п.10, в котором соединение формулы (IId) получают путем гидролиза соединения формулы (II), где R представляет C1-C6 алкил, R' представляет собой формил и X представляет собой F (формула (IIb)), с кислотой или с основанием.
13. Способ по любому из пп.5-12, дополнительно включающий этапы:
a') растворения марбофлоксацина, его промежуточного соединения или предшественника, содержащего группу пиперазина, в воде в присутствии кислоты,
b') необязательно добавления активированного угля к смеси этапа (a'),
c') фильтрации смеси этапа (a') или (b') через фильтр, предпочтительно фильтр с активированным углем,
d') осаждения продукта этапа (с') путем добавления основного реагента.
Текст
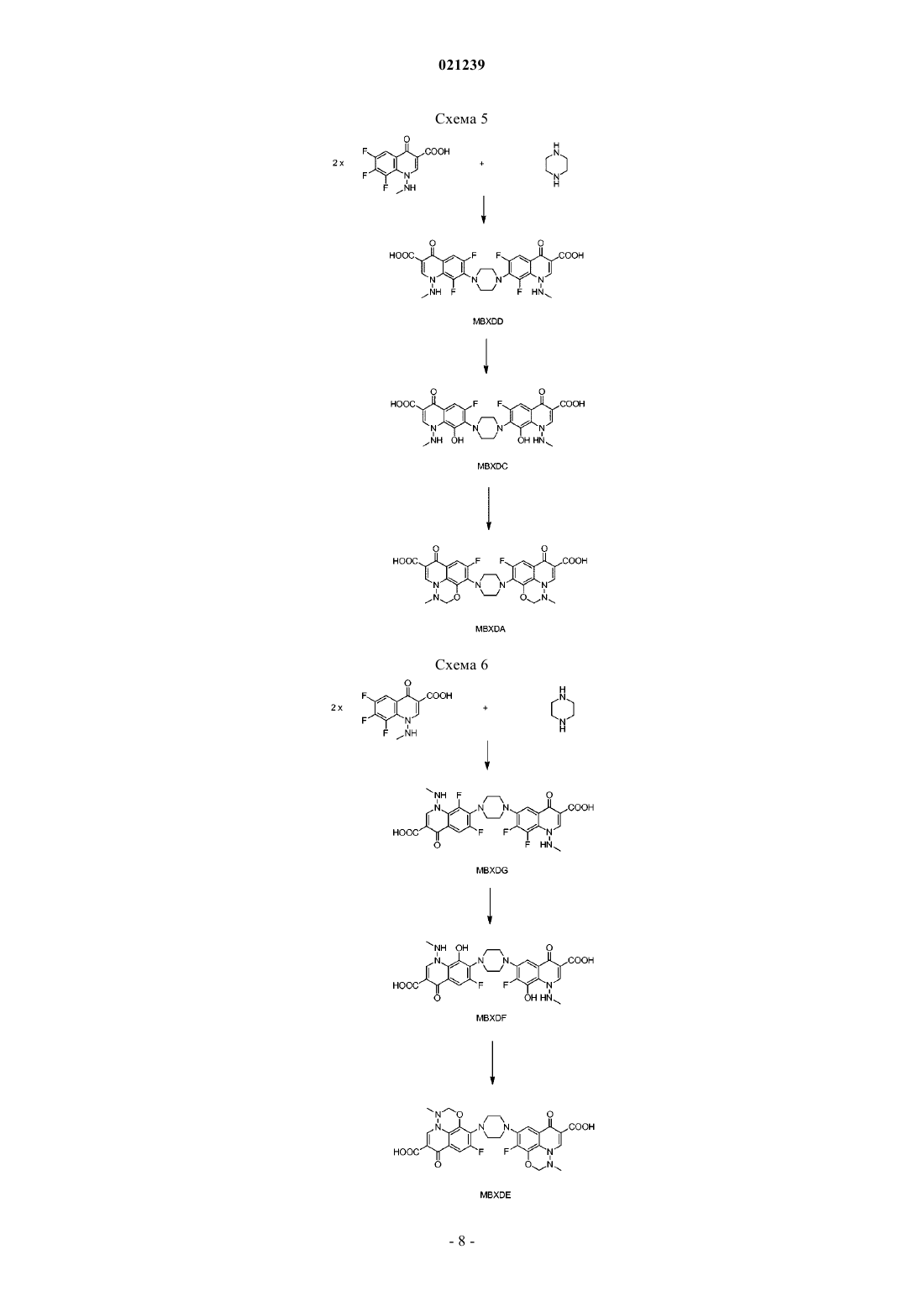
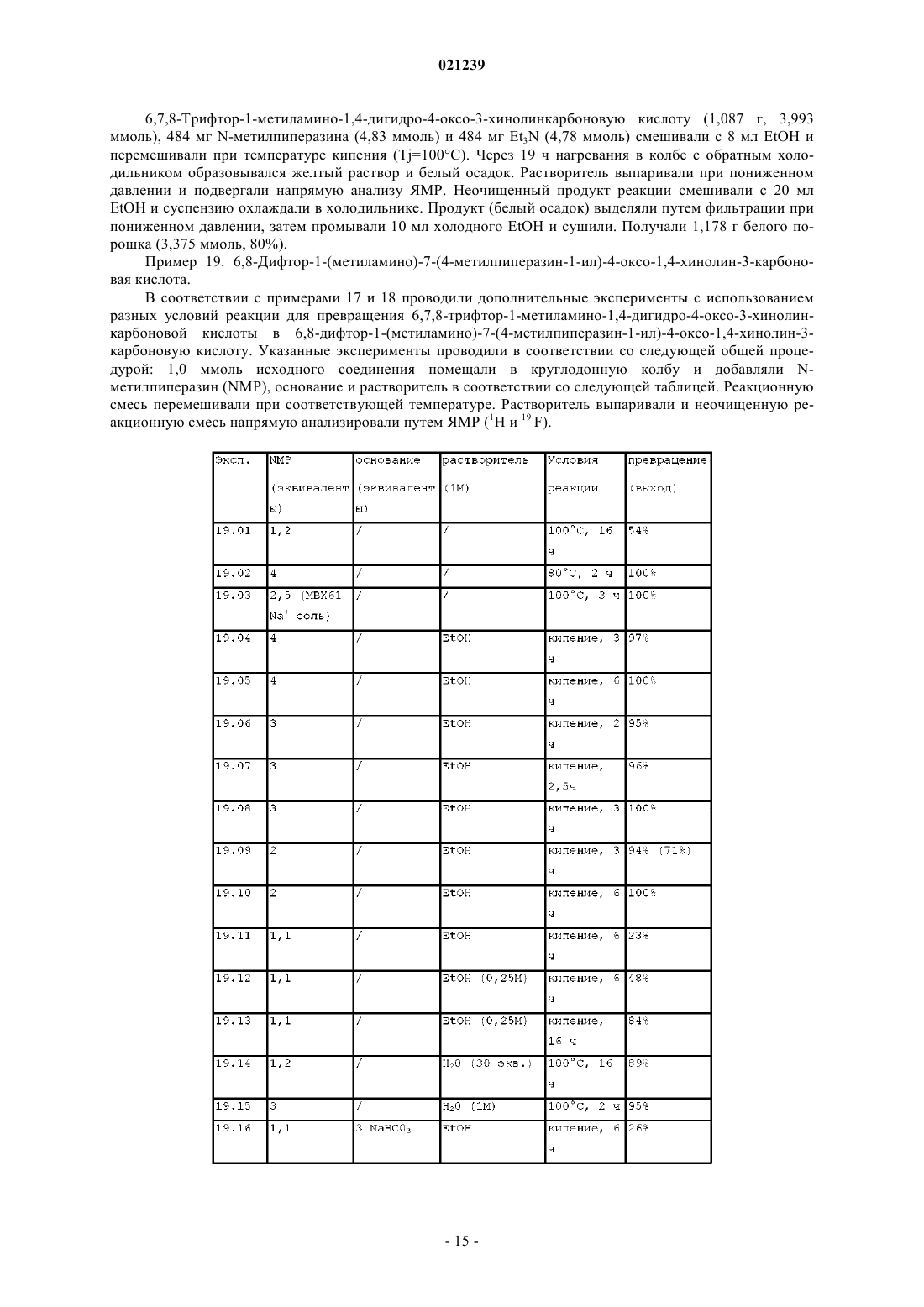
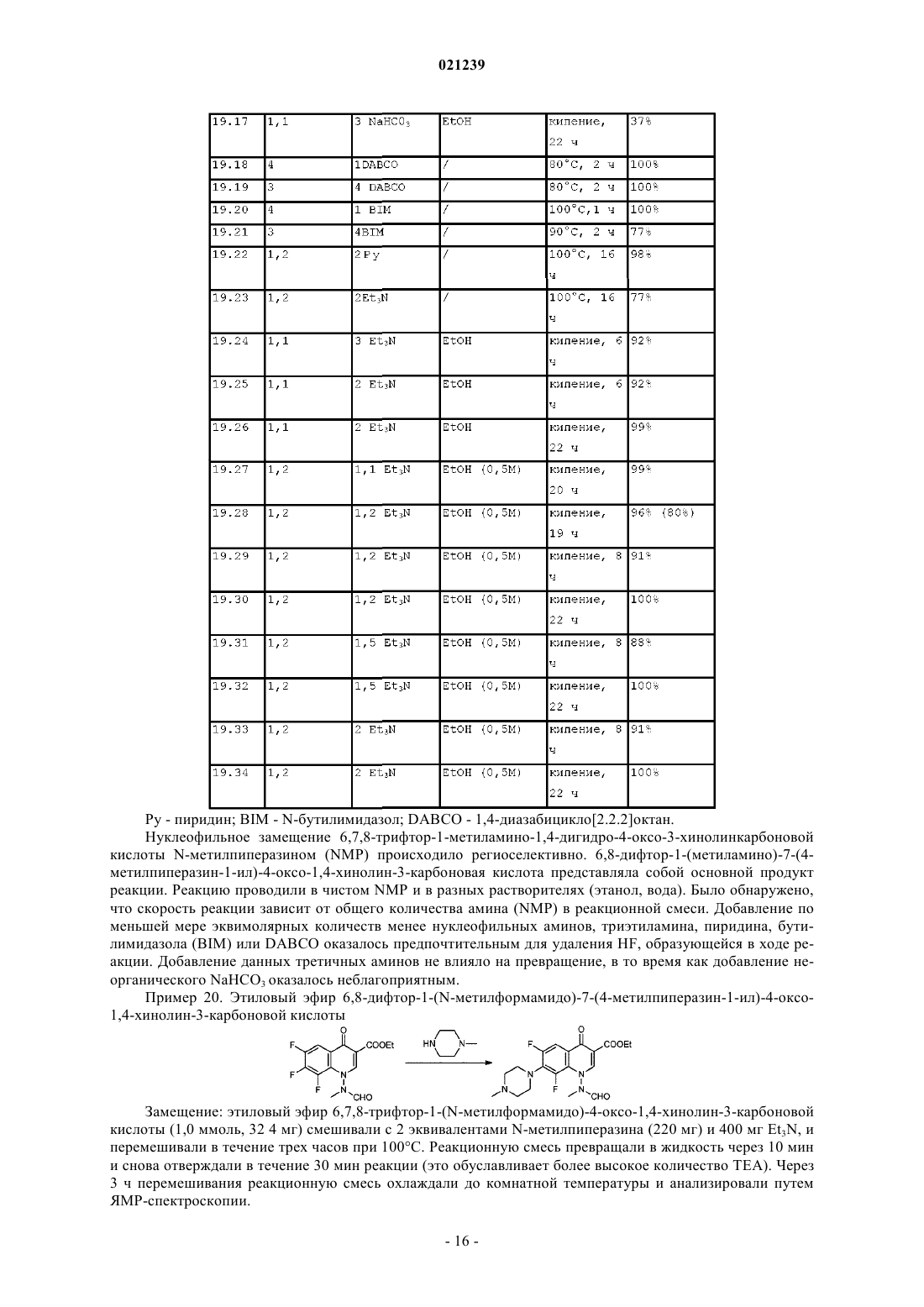
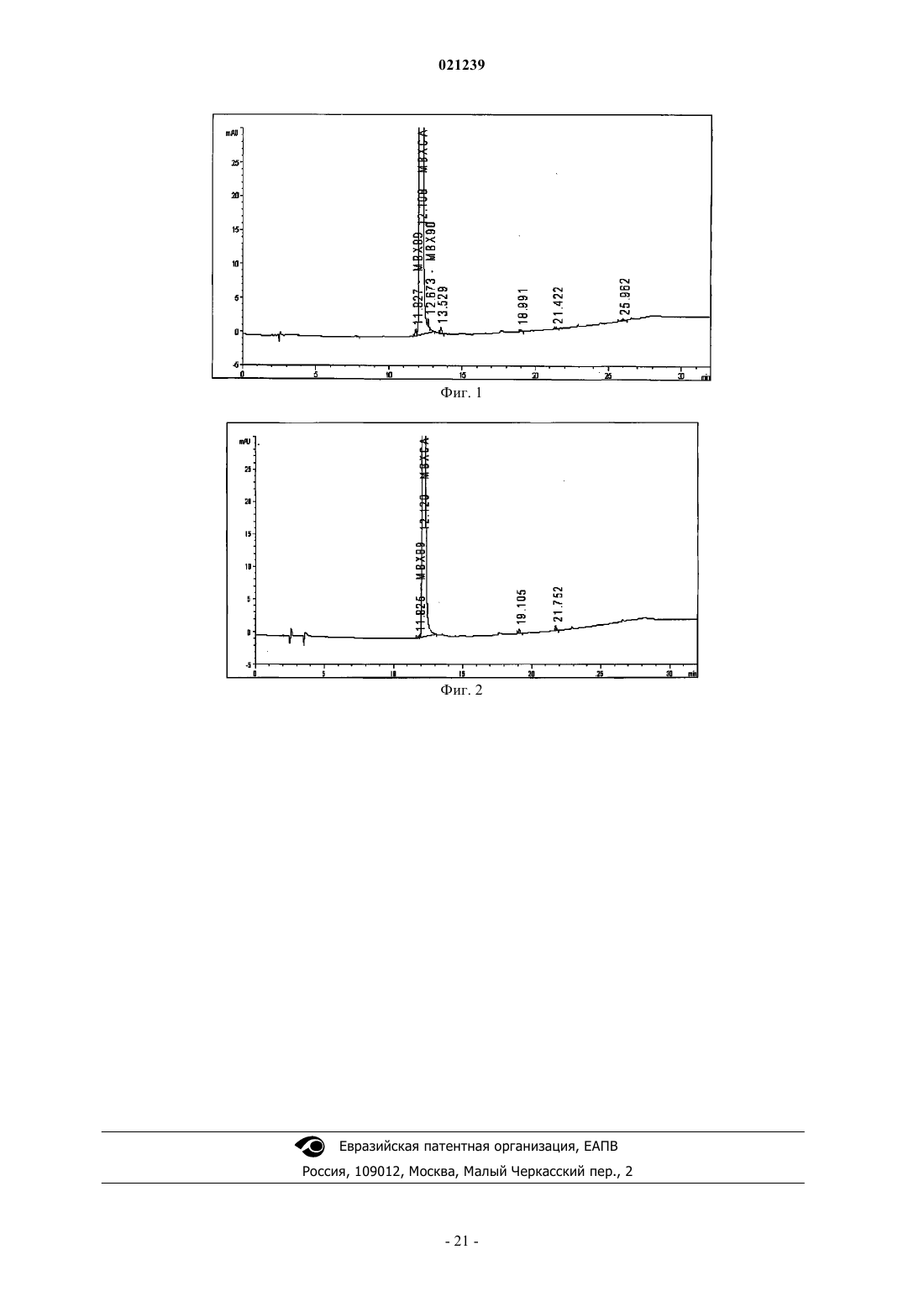
СПОСОБ ПОЛУЧЕНИЯ МАРБОФЛОКСАЦИНА И ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ Изобретение раскрывает новый способ получения марбофлоксацина и его промежуточного соединения, включающий реакцию гидроксида аммония формулы (III), NR1R2R3R4, где R1, R2, R3 иR4 независимо выбраны из группы Н, алкила, алкиларила, арила и/или гетероарила, с соединением формулы (II), где R выбран из Н, алкила, арилалкила, катиона щелочного металла, катиона NH4,катиона NR1R2R3R4; X представляет собой галоген, такой как хлор, бром, фтор; пиперазинил,который может быть замещен или незамещен, и R' выбран из Н, формила или COO-алкила. Область техники Изобретение относится к способу получения промежуточного соединения марбофлоксацина (marbofloxacin) и его производных, а также к способу получения марбофлоксацина и фармацевтических препаратов, содержащих указанное вещество. Предшествующий уровень техники Марбофлоксацин представляет собой общее название 9-фтор-2,3-дигидро-3-метил-10-(4-метил-1 пиперазинил)-7-оксо-7 Н-пиридол(3,2,1-ij)(4,2,1)бензоксадиазин-6-карбоновой кислоты формулы Марбофлоксацин представляет собой сильный антибиотик группы фторхинолонов. В EP 259804 описан марбофлоксацин, а также синтез для его получения многоэтапным способом,который непригоден для крупномасштабного производства, поскольку для него необходимы высокие температуры и реагенты, не подходящие для крупномасштабного производства, что приводит к низким общим выходам. Указанный способ получения показан на схеме 1. В EP 680482 описан альтернативный подход к получению марбофлоксацина, при котором гидроксигруппу вводят в молекулу посредством реакции промежуточного соединения с гидроксидом щелочного металла в водной среде. Используемым исходным веществом является 2,3,4,5-тетрафторбензойная кислота. Недостатками данного способа являются относительно большой избыток гидроксида щелочного металла и очень длительная процедура. Способ синтеза в соответствии с данным патентом показан на схеме 2. Схема 1 В Research Disclosure No. 291,1988, стр. 548-551 описан альтернативный путь синтеза, в котором также исходным веществом является 2,3,4,5-тетрафторбензойная кислота. Дальнейшие этапы способа показаны на схеме 3.IT 1313683 относится к способу получения марбофлоксацина посредством процесса с использованием бензилового эфира. Эфир дебензилируют в водном растворе путем гидрирования с помощью 5%Pd/BaSO4 и полученный продукт циклизуют с использованием HCOOH/НСОН. Принимая во внимание известный уровень техники, до сих пор существует необходимость в улучшенном способе получения марбофлоксацина и его промежуточных соединений, подходящем для крупномасштабного производства. Краткое описание изобретения Цель настоящего изобретения заключается в предложении нового способа получения промежуточного соединения марбофлоксацина и его применения для получения марбофлоксацина и его производных. В соответствии с первым воплощением настоящего изобретения предложен способ получения соединения формулы I где R представляет собой водород, C1-6 алкил, катион щелочного металла иX представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил, при котором осуществляют взаимодействие соединения формулы II где R и X являются такими, как указано выше, и R' выбран из Н или формила, с соединением формулы III где R1, R2, R3 и R4 являются такими, как указано выше. В соответствии со вторым воплощением настоящего изобретения предложен способ однореакторного получения марбофлоксацина формулы IV где R представляет собой Н, и X представляет собой 4-метилпиперазинил, включающий этапы: а) взаимодействия соединения формулы II где R выбран из Н, C1-6 алкила, катиона щелочного металла, X выбран из галогена, такого как хлор,бром или фтор; и 4-метилпиперазинила,R' выбран из Н или формила,с соединением формулы III где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила, иb) взаимодействия полученного промежуточного соединения в том же реакторе с муравьиной кислотой и формальдегидом, и необязательноc) в случае, когда X представляет собой галоген, его замещение соответствующим пиперазином традиционными способами с получением после выделения соединения формулы IV. Подробное описание изобретения В соответствии с первым воплощением настоящего изобретения предложен способ получения соединения формулы IX представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил, при котором осуществляют взаимодействие соединения формулы II где R и X являются такими, как указано выше, и R' выбран из Н или формила, с соединением формулы III где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила. Если специально не ограничен иным образом, термин "алкил", а также производные термины, употребляемые в настоящем описании, содержат в своем объеме прямую цепь, разветвленную цепь и циклические группы. Если специально не указано иное, каждый может быть незамещен или замещен одним или более заместителями, выбранными из, но не ограничиваясь ими, галогена, гидрокси, алкокси или алкилтио, при условии, что указанные заместители стерически совместимы и выполняются правила образования химической связи и энергии напряжения. Предпочтительно, термин "алкил" относится к насыщенному алифатическому углеводородному радикалу с прямой и разветвленной цепью, содержащему от 1 до 6 атомов углерода, такому как метил,этил, пропил, изопропил, трет-бутил, или к арилалкилу с прямой или разветвленной цепью, такому как бензил, трифенилметил. Предпочтительно, соединение формулы I и/или II согласно настоящему изобретению относится к соединению, где R выбран из группы, состоящей из Н, метила, этила, катиона щелочного металла или; X предпочтительно выбран из 4-метилпиперазина и R' предпочтительно выбран из Н. Предпочтительное соединение формулы III в соответствии с первым аспектом настоящего изобретения относится к соединению, где R1, R2, R3 и R4 независимо выбраны из группы метила, этила, н- и изо-пропила, н-бутила, более предпочтительные гидроксиды представляют собой соединения, где R1, R2,R3 и R4 являются одинаковыми, или где R1, R2 и R3 являются одинаковыми и R4 представляет собой (C2C16) алкил, наиболее предпочтительные соединения представляют собой гидроксид тетраметиламмония и гидроксид тетраэтиламмония. В соответствии с первым воплощением настоящего изобретения способ получения соединения формулы I является предпочтительным, где R и R' представляет собой Н, и X представляет собой 4 метилпиперазинил, в котором соединение для формулы II, где R представляет собой Н или этил, R' представляет собой Н, и X представляет собой 4-пиперазинил, подвергают взаимодействию с гидроксидом тетраметиламмония. Реакцию можно проводить в отсутствие растворителя или в присутствии растворителя, или смеси растворителей. В случае, когда реакцию проводят в отсутствие растворителя, гидроксид аммония формулы III предпочтительно выбран из гидратированной формы, такой как пентагидрат, 7,5 гидрат, декагидрат. Было обнаружено, что жидкая фаза, получаемая с использованием гидратированной формы соединения III,более легка в обращении, поскольку она менее вязкая. Но реакцию также можно осуществлять с чистым соединением III без какого-либо растворителя. В качестве альтернативы можно использовать водный раствор гидроксида аммония формулы III. В данном случае реакционную смесь нагревают до 100C и воду оставляют испаряться до образования масляной жидкой фазы, а затем реакцию продолжают дальше. Если используют водный раствор соединения III, предпочтительно удалять избыточную воду путем выпаривания, например путем перегонки, предпочтительно путем вакуумной (пониженное давление) перегонки. Если перегонку осуществляют при нормальном давлении, было обнаружено, что время реакции, необходимое для завершения реакции, составляет более 20 ч, в случае использования вакуумной перегонки время реакции может быть уменьшено до 3-5 ч. Вакуумную перегонку предпочтительно осуществляют при температуре в диапазоне от 60 до 100C, более предпочтительно при примерно от 80 до 100C. Воду предпочтительно удаляют до такой степени, что оставшееся количество воды соответствует от 3 до 5 моль воды на моль гидроксида аммония формулы III. После выпаривания избыточной воды ре-3 021239 акцию предпочтительно продолжают при повышенной температуре, более предпочтительно при температуре примерно 100C. В случае использования растворителя его выбирают из воды или органического растворителя, такого как полярный или неполярный органический растворитель. Указанный органический растворитель может быть выбран из ДМСО, ТГФ, метилтетрагидрофурана, этиленгликоля, полиэтиленгликолей, таких как ПЭГ 300, ПЭГ 400, ПЭГ 500. Предпочтительными растворителями являются ДМСО, ТГФ, метилтетрагидрофуран, этиленгликоль и полиэтиленгликоль. В случае использования органического растворителя присутствие воды нежелательно и соединениеIII предпочтительно используют в негидратированной форме. Более того, органические растворители и, в частности, гигроскопичные растворители, такие как ДМФ, ДМСО, предпочтительно сушат перед использованием, например, путем удаления воды, растворенной в растворителе, путем перегонки. Реакцию можно проводить в отсутствие или в присутствии катализатора фазового переноса, выбранного из краун-эфиров или солей четвертичного аммония. Катализатор фазового переноса может быть выбран из группы хлорида тетрабутиламмония, цианида тетрабутиламмония, фторида тетрабутиламмония, йодида тетрабутиламмония, гидроксида тетрабутиламмония, хлорида тетрабутилфосфония, хлорида трикаприлилметиламмония, хлорида тетраэтиламмония, бромида тетраметиламмония, бромида триоктилэтилфосфония, хлорида триоктилметиламмония,хлорида триоктилпропиламмония, бромида тетрапропиламмония, хлорида тетрафениларсония, бромида тетрафенилфосфония, хлорида тетрафенилфосфония, гидроксида бензилтриметиламмония, 18-краун-6,дибензо-18-краун-6, дициклогексил-18-краун-6 или их смесей. Предпочтительно используют бромид тетра-н-бутиламмония или йодид тетра-н-бутиламмония. В качестве альтернативы органический растворитель может быть выбран из группы ионных жидкостей. Ионные жидкости могут быть выбраны из группы 1-R-3-R1-имидазолия Н(+) х А(-), где R и R1 представляют собой низший алкил и А(-) представляет собой одно- или многоосновный анион кислоты. Термин "низший алкил" относится к C1-C10 углеводородам, которые могут содержать линейные и/или разветвленные цепи, насыщенные и/или ненасыщенные атомы и гетероатомы. Примерами подходящих ионных жидкостей являются метилсульфат 1,2,3-триметилимидазолия,хлорид 1-бутил-2,3-диметилимидазолия, ацетат 1-бутил-3-метилимидазолия, бромид 1-бутил-3 метилимидазолия, гексафторфосфат 1-бутил-3-метилимидазолия, гидросульфат 1-бутил-3-метилими дазолия, метансульфонат 1-бутил-3-метилимидазолия, метилсульфат 1-бутил-3-метилимидазолия, тетрахлоралюминат 1-бутил-3-метилимидазолия, тетрафторборат 1-бутил-3-метилимидазолия, тиоцианат 1 бутил-3-метилимидазолия, этилсульфат 1-этил-2,3-диметилимидазолия, ацетат 1-этил-3-метилимидазолия, хлорид 1-этил-3-метилимидазолия, этилсульфат 1-этил-3-метилимидазолия, гидросульфат 1 этил-3-метилимидазолия, метансульфонат 1-этил-3-метилимидазолия, метилсульфат 1-этил-3 метилимидазолия,тетрахлоралюминат 1-этил-3-метилимидазолия,тетрафторборат 1-этил-3 метилимидазолия, тиоцианат 1-этил-3-метилимидазолия, тетрафторборат 1-гексил-3-метилимидазолия,тетрафторборат 1-гексил-3-метилимидазолия, трифторметансульфонат 1-гексил-3-метилимидазолия,гексафторфосфат 1-метил-3-октилимидазолия, трифторметансульфонат 1-метил-3-октилимидазолия,хлорид метилимидазолия, гидросульфат метилимидазолия, метилсульфат метил-три-н-бутиламмония. Предпочтительно, используют этилсульфат 1-этил-3-метилимидазолия или метилсульфат 1-этил-3 метилимидазолия. Ионные жидкости могут быть получены, например, от компании Merck, Германия. Также могут быть использованы смеси одной или более ионных жидкостей с водой. Соотношение соединения формулы II и гидроксида аммония формулы III составляет от 1:1 до 1:10,предпочтительно от 1:3 до 1:7, наиболее предпочтительно от 1:4 до 1:6. Реакцию можно осуществлять в присутствии минерального основания, более предпочтительно в присутствии карбоната или гидрокарбоната щелочного металла, такого как Na2CO3, NaHCO3, K2CO3,KHCO3. Время реакции и температура реакции могут быть выбраны в соответствии с общими знаниями. Предпочтительно, реакцию проводят в течение 1-20 ч при температурах в диапазоне 60-120C. Более предпочтительно, реакцию проводят при температуре 80-110C в течение 4-8 ч. После проведения реакции полученную смесь охлаждают, предпочтительно до комнатной температуры, и избыточный гидроксид нейтрализуют путем добавления кислоты. Примеры подходящих кислот,которые могут быть использованы, включают неорганические кислоты, такие как соляная кислота, фосфорная кислота и серная кислота; и органические кислоты, такие как муравьиная кислота, уксусная кислота, п-толуолсульфоновая кислота, метансульфоновая кислота и трифторуксусная кислота. Продукт выделяют в виде свободного основания или в форме соли путем добавления воды или органического растворителя или их смесей. Согласно предпочтительному воплощению настоящего изобретения органические растворители выбраны из С 1-С 5 спирта, ацетонитрила или тетрагидрофурана, метилтетрагидрофурана, уксусной кислоты, муравьиной кислоты, ацетона и/или простых эфиров. Более предпочтительно, спирты выбраны из метанола, этанола и изопропанола, и простые эфиры выбраны из тетрагидрофурана, метилтетрагидрофурана, метил-трет-бутилового эфира, диизопропилового эфира. Второе воплощение настоящего изобретения представляет собой однореакторный способ получения марбофлоксацина формулы IV где R представляет собой Н, и X представляет собой 4-метилпиперазинил, включающий этапы а) взаимодействия соединения формулы II где R выбран из Н, C1-6 алкила, катиона щелочного металла, и X выбран из галогена, такого как хлор, бром или фтор; 4-метилпиперазинила R' выбран из Н или формила, с соединением формулы III где R1, R2, R3 и R4 являются такими, как указано выше,b) взаимодействия полученного промежуточного соединения с муравьиной кислотой и формальдегидом в том же реакторе, и необязательно с) в случае, когда X представляет собой галоген, его замещают соответствующим пиперазином традиционными способами с получением после выделения соединения формулы IV. Предпочтительно реакцию осуществляют путем взаимодействия соединения формулы II, в которомR представляет собой Н, метил или этил, X представляет собой 4-метилпиперазинил; R' представляет собой Н и R1, R2, R3, R4 являются одинаковыми и представляют собой метильную или этильную группу. Этап а) осуществляют в тех же условиях реакции, что и способ получения соединения формулы (I) в соответствии с первым аспектом настоящего изобретения. Предпочтительно, реакцию осуществляют в отсутствие растворителей. Реакция может протекать при температуре от 60 до 120C, необходимое время реакции составляет от 1 до 20 ч. Предпочтительно реакцию проводят в течение 4-8 ч при температуре от 80 до 110C. Соотношение соединения формулы II и соединения формулы III составляет от 1:1 до 1:10,предпочтительно от 1:3 до 1:7, наиболее предпочтительно от 1:4 до 1:6. Этап b) осуществляют напрямую из реакционной смеси, полученной после этапа а), без выделения продукта путем добавления муравьиной кислоты и водного раствора формальдегида. Молярное соотношение соединения формулы II: HCOOH:HCHO составляет предпочтительно 1:5-30:1-6, более предпочтительно 1:20-25:2-3. Реакция может протекать при температуре от комнатной температуры до 100C,предпочтительно при температурах 40-80C. Необходимое время реакции обычно составляет от 0,1 ч до 3 ч, предпочтительно от 0,25 до 0,5 ч. Соединение, полученное таким образом, может быть выделено из реакционной смеси традиционными способами в виде свободного основания или в форме соли с кислотой или основанием. Указанное соединение может быть выделено путем кристаллизации, осаждения или экстракции. Замещение согласно необязательному этапу с) может быть осуществлено в соответствии со способами, известными в данной области техники. Данную реакцию предпочтительно проводят при температуре кипения в органическом растворителе, таком как толуол, в присутствии основания, такого как низший триалкиламин, например триэтиламин, или карбонат щелочного металла, например карбонат натрия. В качестве альтернативы, реакция может быть проведена в основном органическом растворителе,таком как пиридин, 1,4-диазабицикло[2.2.2]октан (DABCO) или триэтиламин. Реакция также может быть проведена в чистом пиперазине, предпочтительно в чистом NMP (N-метилпирролидон). Было обнаружено, что скорость реакции зависит от общего количества амина (например, NMP) в реакционной смеси. Для того чтобы поддерживать как можно более низкое количество нуклеофильного пиперазина, может быть добавлен другой тип аминов, предпочтительно пиридин или третичный амин, такой как триэтиламин, бутилимидазол или DABCO. Добавление по меньшей мере эквимолярных количеств данных дополнительных менее нуклеофильных аминов является предпочтительным. Продукт выделяют в виде свободного основания или в форме соли путем добавления воды или органического растворителя или их смесей. Согласно предпочтительному воплощению настоящего изобретения используют воду и/или органические растворители, выбранные из С 1-С 5 спирта, ацетонитрила или тетрагидрофурана, метилтетрагидрофурана, уксусной кислоты, муравьиной кислоты, ацетона и/или простых эфиров. Более предпочтительно используют воду и/или спирты, выбранные из метанола, этанола и изопропанола; и простые эфиры, выбранные из тетрагидрофурана, метилтетрагидрофурана, метил-третбутилового эфира, диизопропилового эфира. Наиболее предпочтительно продукт выделяют путем добавления воды с осаждением продукта, тем самым побочные продукты остаются растворенными в водной фазе. Полученный продукт может быть дополнительно очищен традиционными способами, такими как кристаллизация, мацерация, осаждение, и другими традиционными способами. Продукт можно кристаллизовать из спиртов, таких как метанол, этанол, изопропанол, н-пропанол; воды; простых эфиров, таких как диэтилэфир, толуола и их смесей. Продукт можно кристаллизовать в соответствии со способами,описанными в EP 259804, ЕР 680482, IPCOM 000186605D. Полученная кристаллическая форма марбофлоксацина может быть такой же, что описана в IPCOM 000173285D. Исходные соединения формулы II могут быть получены любым известным способом, таким как описанный в EP 155587, ЕР 530360, US 4604401, RD 291097, EP 680482. Согласно наиболее предпочтительному воплощению настоящего изобретения соединение, которое подвергают взаимодействию с гидроксидом аммония (III) с получением соединения (I), представляет собой соединение формулы (II), где как R, так и R' представляют собой водород, и X представляет собой 4-метилпиперазинил (формула IIa). Соединение (IIa) может быть получено в соответствии с EP 0155587B1. Соединения согласно формуле (II), имеющие структуру, отличную от (IIa), сначала предпочтительно превращают в соединение (IIa). Предпочтительное исходное вещество для получения соединения (IIa) представляет собой соединение согласно формуле (II), где R представляет собой C1-C6 алкил, предпочтительно этил, R' представляет собой формил, и X представляет собой F (формула, IIb). Соединение IIb также может быть получено в соответствии с EP 0155587 В 1. Вышеуказанные реакции приведены на схеме 4. Схема 4 Соединение (IIb) подвергают взаимодействию с N-метилпиперазином с получением соответствующего продукта замещения, где X представляет собой 4-метилпиперазинил (соединение IIc). Реакция замещения может быть проведена в соответствии с EP 0155587 В 1 или EP 0680482 В 1. Данную реакцию предпочтительно проводят при температуре кипения в органическом растворителе, таком как толуол, в присутствии основания, такого как низший триалкиламин, например триэтиламин, или карбонат щелочного металла, например карбонат натрия. В качестве альтернативы, реакция может быть проведена в основном органическом растворителе, таком как пиридин, 1,4-диазабицикло[2.2.2]октан (DABCO) или триэтиламин. Поскольку основные условия способствуют гидролизу сложноэфирной группы и формильной группы, часто получают смесь (IIc) и (IIa). Затем соединение (IIc) гидролизуют с получением соединения (IIa). Гидролиз осуществляют с помощью подходящего основания или предпочтительно с помощью разбавленной кислоты, более предпочтительно с помощью водной HCl или H2SO4, такой как 10% H2SO4. Поскольку соединение (IIc) легко растворимо в воде, соединение (IIc) легко превращают в (IIa). Соединение (IIb) может быть превращено в(IIa) в однореакторной реакции без выделения (IIc). В соответствии с альтернативным способом соединение (IIb) сначала гидролизуют с получением соединения (IId) с последующим замещением атома фтора в положении 7 с получением соединения (IIa). Гидролиз (IIb) можно осуществлять с помощью разбавленной кислоты, предпочтительно водной HCl илиH2SO4, например, 10% H2SO4, или с помощью основания, такого как нижний триалкиламин, например,триэтиламин. Типичная реакционная среда для гидролиза представляет собой смесь триэтиламина (предпочтительно от примерно 2 до 4 об.%) в растворителе, таком как этанол, содержащая достаточное количество (предпочтительно примерно 0,5-1 об.%) воды. Соединение (IIb) является гидрофобным, и оно лишь плохо смачивается водными средами. Следовательно, для облегчения смешивания соединения (IIb) с водными средами его сначала предпочтительно смешивают с маленьким количеством полярного растворителя, предпочтительно EtOH, MeCN или АсОН, до реакции гидролиза. Данные растворители образуют пленку вокруг кристаллов (IIb) и улучшают смачивание данного соединения водными растворителями. Таким образом, реакция гидролиза значительно ускоряется. На соединение (IIb) можно действовать полярным растворителем путем простого смешивания указанного растворителя с соединением (IIb), предпочтительно в соотношении, составляющем по меньшей мере 0,1 мл растворителя на ммоль соединения (IIb). Согласно альтернативному воплощению настоящего изобретения соединение (IIb) растворяют в подходящем растворителе, предпочтительно в этаноле или другом низшем алифатическом спирте, предпочтительно C1-C8 спирте и более предпочтительно C1-C3 спирте, и затем данный раствор смешивают с кислотой. Гидролиз (IIb) с получением (IIc) предпочтительно проводят при повышенной температуре,предпочтительно при температуре в диапазоне температуры кипения от 95 до 102C и более предпочтительно от 98 до 100C. В ходе реакции растворитель, используемый для растворения (IIb), предпочтительно удаляют путем выпаривания. Для реакции замещения можно использовать те же условия, определенные выше. Нуклеофильное замещение (IId) пиперазином, таким как N-метилпиперазин (NMP) происходит региоселективно. (IIa) представляет собой основной продукт реакции. Реакция может быть проведена в чистом пиперазине,предпочтительно в чистом NMP, или в растворителе, таком как этанол, вода или их смесь, предпочтительно этаноле. Было обнаружено, что скорость реакции зависит от общего количества амина (например,NMP) в реакционной смеси. Для того чтобы поддерживать как можно более низкое количество нуклеофильного пиперазина, может быть добавлен другой тип аминов, предпочтительно пиридин или третичный амин, такой как триэтиламин, бутилимидазол или DABCO. Добавление, по меньшей мере, эквимолярных количеств данных дополнительных менее нуклеофильных аминов является предпочтительным. Предполагается, что данные амины удаляют HF, образующуюся в ходе реакции. HF может протонировать пиперазин и, таким образом, уменьшает его нуклеофильность. Добавление указанных аминов не влияет на реакцию замещения. Согласно предпочтительному воплощению настоящего изобретения пиперазин, в частности, NMP используют в количестве 1-5 эквивалентов, более предпочтительно 1-2 эквивалента и наиболее предпочтительно 1,2 эквивалента. Необязательный дополнительный амин, такой как Et3N, предпочтительно используют в том же количестве, что и пиперазин. Растворители, такие как EtOH, можно использовать в любом соответствующем количестве, например, в количестве, равном 0-100 мл/ммоль, предпочтительно 0,5-10 мл/ммоль или более предпочтительно 1 мл/ммоль исходного вещества. Реакцию замещения (IId) и (IIb) предпочтительно проводят при повышенной температуре, предпочтительно при температуре в диапазоне от 70 до 83C, наиболее предпочтительно при температуре кипения, например, при 83C. Затем соединение (IIa) подвергают взаимодействию с гидроксидом аммония (III), предпочтительно с гидроксидом тетраметиламмония, с получением соответствующего соединения формулы (I), которое затем превращают в марбофлоксацин (IV), как описано выше. Таким образом, в предпочтительном способе согласно настоящему изобретению в качестве исходного вещества используют соединение (IIb), и он протекает через (IIc), (IIa), (I) с получением марбофлоксацина. В качестве альтернативы, в указанном способе в качестве исходного вещества используют соединение (IIb), и он протекает через (IId), (IIa), (I) с получением марбофлоксацина. Все этапы реакции предпочтительно проводят в инертной атмосфере,такой как атмосфера азота (для избежания окисления кислородом). Димерные примеси могут образовываться, например, по реакциям, приведенным на схемах 5 и 6. Причина заключается в том, что N-метилпиперазин содержит некоторое количество пиперазина в виде примеси (или может превращаться в пиперазин в ходе реакции), и реакция может протекать по обоим центрам NH с образованием, таким образом, димерной примеси. Димерные примеси связывали с проблемой осадка при приготовлении растворов, таких как растворы для инъекций или инфузий. Авторы настоящего изобретения обнаружили, что образования осадка из раствора можно избежать и что фильтруемость растворов фторхинолонов может быть улучшена путем уменьшения содержания димерных примесей до менее 0,01% (площадь ВЭЖХ), предпочтительно менее 0,005% (площадь ВЭЖХ), более предпочтительно до 0,003% (площадь ВЭЖХ). Чистоту продукта оценивают путем высокоэффективной жидкостной хроматографии (ВЭЖХ) с колонкой пентафторфенилпропила (PFP) (тип Luna PFP, 1504,6 мм, 3 мкм, Phenomenex, США); детектор: УФ 315 нм; скорость потока: 0,8 мл/мин; вводимый объем: 5 мкл; подвижная фаза: А: 0,02 МNaH2PO4H2O+0,1% TEA, pH 2,5; В: ацетонитрил:метанол =5:95 (об./об.); градиент: 0'=10 В, 25'=100 В,30'=100 В, 32'=10 В. Данный метод ВЭЖХ, как правило, применим для анализа марбофлоксацина, производных марбофлоксацина, его промежуточных соединений и предшественников, содержащих группу пиперазина, в частности соединений, содержащих группу пиперазина, указанных в настоящем описании. Было обнаружено, что в вышеуказанных условиях время удерживания димерных примесей по меньшей мере в два раза больше, чем время удерживания марбофлоксацина. Настоящее изобретение также относится к способу очистки марбофлоксацина, производных марбофлоксацина, его промежуточных соединений и предшественников, содержащих группу пиперазина. Способ очистки согласно настоящему изобретению включает следующие этапы: a') растворение марбофлоксацина или производного марбофлоксацина, его промежуточного соединения или предшественника,содержащего группу пиперазина, в воде в присутствии кислоты, b') необязательно добавление активированного угля к смеси этапа (a'), c') фильтрация смеси этапа (a') или (b') через фильтр, предпочтительно фильтр с активированным углем, d') осаждение продукта путем добавления основного реагента. Марбофлоксацин или соединение, содержащее группу пиперазина, используемое на этапе (a'),предпочтительно представляет собой соединение, синтезированное согласно настоящему изобретению. Таким образом, согласно третьему воплощению настоящее изобретение относится к способу, включающему этапы синтеза согласно первому воплощению или второму воплощению настоящего изобретения в комбинации с этапами очистки (a') - (d'). Раствор, полученный на этапе (а') или (b'), перемешивают в течение от 0,5 до 24 ч, предпочтительно 2-12 ч при температурах от комнатной температуры до 60C. рН смеси этапа (а') предпочтительно доводят до значения в диапазоне от 2,0 до 6,0, предпочтительно в диапазоне от 5,0 до 5,3. Кислота, используемая на этапе (a'), предпочтительно выбрана из органических кислот, в частности,монокарбоновых кислот и поликарбоновых кислот, содержащих от 2 до 25, предпочтительно от 2 до 10 атомов углерода; моноциклических и полициклических арилкислот, таких как бензойная кислота. Предпочтительными примерами данных кислот являются уксусная кислота, муравьиная кислота,пропионовая кислота, метансульфоновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, адипиновая кислота, молочная кислота, левулиновая кислота, сорбиновая кислота и фруктовые кислоты, такие как винная кислота, яблочная кислота, аскорбиновая кислота или лимонная кислота. Наиболее предпочтительными органическими кислотами являются уксусная кислота,молочная кислота и глюконовая кислота. В качестве альтернативы, на этапе (а) могут быть использованы неорганические кислоты, такие как HCl. рН смеси этапа (b') предпочтительно доводят до значения в диапазоне от 6,4 до 8,0, предпочтительно от 6,8 до 7,4. Основный реагент этапа (d') предпочтительно представляет собой гидроксид щелочного металла,такой как гидроксид натрия или калия, гидроксиды щелочноземельных металлов, аммиак или амин,предпочтительно гидроксид щелочного металла. Способ очистки согласно настоящему изобретению наиболее подходит для удаления примесей, которые, как правило, вызывают осадки во время производства и хранения растворов для парентерального введения, в частности, димерных примесей, наиболее предпочтительно димерных примесей, показанных на схемах реакции 5 и 6. Димерные примеси могут быть удалены на любой стадии синтеза, когда группа пиперазина уже присутствует в молекуле. Очистку проводят или до замещения атома фтора в положении 8 гидроксильной группой и/или на стадии конечного продукта. Очищенный марбофлоксацин согласно настоящему изобретению обладает чистотой по меньшей мере 99,5%, предпочтительно 99,7%, более предпочтительно 99,8%, наиболее предпочтительно 99,9%,определенной путем ВЭЖХ. В частности, марбофлоксацин согласно настоящему изобретению содержит менее 0,01% (площадь ВЭЖХ), предпочтительно менее 0,005% (площадь ВЭЖХ), более предпочтительно менее 0,003% (площадь ВЭЖХ) димерных примесей. Согласно другому воплощению настоящего изобретения марбофлоксацин очищают путем препаративной ВЭЖХ. Препаративную ВЭЖХ можно осуществлять с колонками с нормальной фазой и предпочтительно с обращенной фазой. Предпочтительные колонки с обращенной фазой представляют собой колонки С 12, такие как Synergi MAX-RP, производимые Phenomenex 25021,2 мм, размер частиц 10 мкм, колонки с обращенной фазой С 18, такие как XBridge C18 и колонки Symmetry С 18 (Waters) и колонки Zorbax Eclipse-XDB (Agilent), и колонки пентафторфенилпропила (PFP), такие как Luna PFP (2), производимые Phenomenex,15030 мм, размер частиц 5 мкм. Предпочтительная подвижная фаза для обращенно-фазовой хроматографии содержит (1) буфер:NH4COOCH3 или NaH2PO4, 10-20 мМ, область рН 2-5; для улучшения формы пика может быть добавлен 0,1-0,2% триэтиламин с последующей (2) органической фазой: ацетонитрил или смеси ацетонитрила с метанолом (ацетонитрил : метанол = от 25:75 (об.%) до 5:95 (об.%). Предпочтительные колонки с нормальной фазой для ВЭЖХ представляют собой колонки с полярным модифицированным аминопропилом диоксидом кремния, такие как Nucleosil 100-10NH2 25016 мм, производимые Macherey-Nagel, размер частиц 10 мкм. Предпочтительные подвижные фазы для ВЭЖХ с нормальными фазами представляют собой смеси гексанов и этанола в соотношении от 50:50 до 30:70 (об.%). Димерные примеси, показанные на схемах 5 и 6, подходят в качестве эталонных стандартов и также являются объектом настоящего изобретения. Соединения формулы (IV), полученные в соответствии со способом согласно настоящему изобретению, могут быть использованы для приготовления фармацевтической композиции. Подходящие фармацевтические композиции включают композиции для перорального и парентерального введения, такие как описанные в WO 02/058669, WO 2008/030469, WO 2007/085760, WO 2007/101560, DE 102006010643,IPCOM000186605D. Фармацевтические композиции согласно настоящему изобретению предпочтительно помещают в упаковку, которая соответствует стандартам европейской фармакопеи. На фиг. 1 показана хроматограмма ВЭЖХ марбофлоксацина до очистки в соответствии со способом согласно настоящему изобретению, и На фиг. 2 показана хроматограмма ВЭЖХ марбофлоксацина после очистки в соответствии со способом согласно настоящему изобретению. Следующие примеры иллюстрируют настоящее изобретение, не ограничивая его объем. Примеры Метод ВЭЖХ высокого разрешения применяют для определения количества и чистоты соединений формулы I, II и IV. Тесты проводят в колонке X-Bridge C18, 1504,6 мм, 3,5 мкм. Подвижная фаза представляет собой градиент А) 5 мМ NH4COOCH3 рН 7,0 В) ацетонитрила. Градиент: 0'=10%В, 10'=20%В,25'-30'=90%В, 32'=10%В. Хроматограф оснащен УФ-детектором, установленным на 250 нм и 315 нм, скорость потока составляет 1,0 мл/мин при 30C. Пример 1. а) 6,8-Дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота и натриевая соль 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4 дигидрохинолин-3-карбоновой кислоты 4,137 г этил-6,8-дифтор-1-(N-метилформамидо)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (10,14 ммоль) помещали в 40 мл 10% H2SO4 и перемешивали при 100C в течение 7 ч. Реакционную смесь охлаждали и образовывались кристаллы. Смесь охлаждали до 4C и фильтровали с отсасыванием. Осадок на фильтре промывали смесью H2O/EtOH/ТГФ (1/1/5) и сушили. Получали 3,260 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты в виде желтых кристаллов (91%). В случае, когда необходима натриевая соль, продукт, полученный на предыдущем этапе, помещали в 5 мл EtOH и 10 мл CH2Cl2, и добавляли 1,20 г NaOH, растворенного в 2 мл воды. Раствор перемешивали при комнатной температуре в течение 1 ч, сушили Na2SO4 и выпаривали. Выделяли 2,90 г чистого продукта, указанного в названии (желтый порошок, 7,71 ммоль, 76%). 400 мг этил-6,8-дифтор-1-(N-метилформамидо)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (0,979 ммоль) помещали в 2 мл 10% H2SO4 и перемешивали при 100C в течение 2 ч. Реакционную смесь охлаждали и образовывались кристаллы. К данной смеси медленно добавляли 1,7 мл 25% водн. NH3. Сначала образовывалась очень плотная суспензия, которая растворялась при последующем добавлении раствора аммиака. В конце образовывался прозрачный раствор с рН, равным 9. Сульфат аммония осаждали путем добавления 10 мл EtOH, отфильтровывали и промывали 5 млH2O/EtOH (1/2). Маточный раствор сушили на роторном испарителе и добавляли 10 мл смеси EtOH/H2O(7/3) с осаждением остаточной неорганической соли, которую снова отфильтровывали. Оставшийся желтый раствор сушили на роторном испарителе с получением 321 мг желтого порошка (0,912 ммоль, 93%). Пример 2. а) 6-Фтор-8-гидрокси-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3 карбоновая кислота 178 мг натриевой соли 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4 дигидрохинолин-3-карбоновой кислоты (0,47 0 ммоль) смешивали с 360 мг Me4NOH5H2O (2,00 ммоль) и перемешивали при 100C в течение 4 ч. В ходе реакции соль аммония плавится и образуется темнокоричневое масло. Реакционную смесь охлаждали до комнатной температуры и добавляли 0,10 мл вали с отсасыванием, и осадок на фильтре промывали 2 мл холодного EtOH. Получали 90 мг продукта. Пример 3. Формиатная соль 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино[6,5,4-ij]хинолин-6-карбоновой кислоты 180 мг натриевой соли 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (0,481 ммоль) смешивали с 360 мг Me4NOH5H2O (2,00 ммоль) и перемешивали при 100C в течение 3 ч. В ходе реакции соль аммония плавится, и образуется темнокоричневое масло. Реакционную смесь охлаждали до комнатной температуры и добавляли 1 мл HCOOH с последующим добавлением 0,4 мл 37% водного раствора HCHO, и перемешивали при 70C в течение еще одного часа. Реакционную смесь охлаждали до комнатной температуры, и добавляли 5 мл EtOH с осаждением продукта, который фильтровали с отсасыванием, и осадок на фильтре промывали 2 мл холодного EtOH. Получали 111 мг серого порошка. Пример 4. Формиатная соль 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино[6,5,4-ij]хинолин-6-карбоновой кислоты 1,14 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,00 ммоль) смешивали с 3,06 г Me4NOH5H2O (16,96 ммоль) и перемешивали при 100C в течение 5 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры и добавляли 1,44 мл HCOOH (85% водный раствор) с последующим добавлением 0,5 мл 37% водного раствора HCHO, и колбу охлаждали на водяной бане при 22C. Добавляли еще 1,44 мл 85% HCOOH и реакционную смесь нагревали до 70C в течение 30 мин, и после охлаждения к реакционной смеси добавляли 20 мл EtOH и оставляли в холодильнике на 16 ч. Осадок фильтровали при пониженном давлении и промывали холодным этанолом (10 мл). После сушки получали 1,23 г сероватого порошка (90%). Пример 5. 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино[6,5,4-ij]хинолин-6-карбоновая кислота 1,145 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,01 ммоль) смешивали с 2,72 г Me4NOH5H2O (15,00 ммоль) и перемешивали при 100C в течение 8 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры, и добавляли 3,00 мл HCOOH с последующим добавлением 0,5 мл 37% водного раствора HCHO (6,0 ммоль), и колбу охлаждали на водяной бане при 22C. Сразу же образовывался осадок. Колбу нагревали до 70C, в ходе чего осадок растворялся. После перемешивания при 70C в течение 30 мин (осадок снова образовывался через 5 мин) реакционную колбу охлаждали до комнатной температуры, и к реакционной смеси добавляли 20 мл EtOH, и оставляли в холодильнике на 16 ч. Осадок фильтровали при пониженном давлении и промывали холодным этанолом(10 мл). После сушки получали 1,165 г сероватого порошка (85%) с чистотой 97,11% (ВЭЖХ). Неочищенный продукт реакции смешивали с 0,9 мл 25% водного раствора NH3 и кристаллизовали в смеси 26 мл EtOH и 14 мл Н 2 О. Получали 0,673 г порошка (61%) с чистотой 98,75% (ВЭЖХ). Пример 6. 9-Фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино 1,140 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,00 ммоль) смешивали с 2,72 г Me4NOH5H2O (15,01 ммоль) и перемешивали при 100C в течение 8 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры, и добавляли 3,0 мл HCOOH с последующим до- 11021239 бавлением 0,5 мл 37% водного раствора HCHO (6,0 ммоль), и колбу охлаждали на водяной бане при 22C. Сразу же образовывался осадок. Колбу нагревали до 70C, в ходе чего осадок растворялся, и перемешивали в течение 30 мин (после перемешивания при 70C в течение 5 мин осадок снова образовывался). Реакционную колбу охлаждали до комнатной температуры, и к реакционной смеси добавляли 20 мл Н 2 О, и оставляли в холодильнике на 16 ч. Осадок фильтровали при пониженном давлении и промывали холодным этанолом (10 мл). После сушки получали 1,022 г сероватого порошка (75%) с чистотой 97,11%(ВЭЖХ). Неочищенный продукт реакции смешивали с 0,9 мл 25% водного раствора NH3 и кристаллизовали в смеси 20 мл EtOH и 6 мл CHCl3. Получали 0,771 г желтого порошка (71%) с чистотой 99,50%, определенной путем ВЭЖХ. Пример 7. 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино 1,142 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,01 ммоль) смешивали с 3,26 г Me4NOH5H2O (18,01 ммоль) и перемешивали при 100C в течение 4 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры, и добавляли 3,0 мл HCOOH с последующим добавлением 0,5 мл 37% водного раствора HCHO (6,0 ммоль), и колбу охлаждали на водяной бане при 22C. Сразу же образовывался осадок. Колбу нагревали до 70C, в ходе чего осадок растворялся, и перемешивали в течение 30 мин (после перемешивания при 70C в течение 5 мин осадок снова образовывался). Реакционную колбу охлаждали до комнатной температуры и сушили на роторном испарителе. К реакционной смеси добавляли 20 мл Н 2 О и охлаждали в холодильнике. Осадок фильтровали при пониженном давлении. После сушки получали 1,147 г белого порошка (84%). Неочищенный продукт реакции смешивали с 5 мл воды и 2 мл 25% водного раствора NH3 и получали прозрачный раствор. К данному раствору добавляли 7 мл EtOH и сушили при пониженном давлении. Продукт кристаллизовали в смеси 15 мл EtOH и 10 мл CHCl3 с получением 0,4321 г белого порошка 1,136 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (2,98 ммоль) смешивали с 2,7 3 г Me4NOH5H2O (15,00 ммоль) и перемешивали при 100C в течение 7 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры, и добавляли 3,0 мл HCOOH с последующим добавлением 0,5 мл 37% водного раствора HCHO (6,0 ммоль), и колбу охлаждали на водяной бане при 22C. Сразу же образовывался осадок. Колбу нагревали до 70C, в ходе чего осадок растворялся, и перемешивали в течение 30 мин (после перемешивания при 70C в течение 5 мин осадок снова образовывался). Реакционную колбу охлаждали до комнатной температуры и сушили на роторном испарителе. К реакционной смеси добавляли 20 мл Н 2 О и охлаждали в холодильнике. Осадок фильтровали при пониженном давлении. После сушки получали 1,039 г серого порошка (77%). Неочищенный продукт реакции нейтрализовали 2 мл 25% водного раствора NH3 и прозрачный раствор разбавляли 15 мл EtOH и 9 мл Н 2 О. Раствор частично сушили при пониженном давлении до образования осадка. В данный момент смесь охлаждали в холодильнике и осадок выделяли путем фильтрации при пониженном давлении с получением 0,675 г порошка (65%) с чистотой 98,84%, определенной путем ВЭЖХ. Пример 9. 9-Фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино 1,140 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (3,01 ммоль) смешивали с 3,30 г Me4NOH5H2O (18,20 ммоль) и перемешивали при 100C в течение 4 ч. В ходе реакции соль аммония плавится и образуется темно-коричневое масло. Реакционную смесь охлаждали до комнатной температуры, и добавляли 3,0 мл HCOOH с последующим до- 12021239 бавлением 0,5 мл 37% водного раствора HCHO (6,0 ммоль), и колбу охлаждали на водяной бане при 22C. Сразу же образовывался осадок. Колбу нагревали до 70C, в ходе чего осадок растворялся, и перемешивали в течение 30 мин (после перемешивания при 70C в течение 5 мин осадок снова образовывался). Реакционную колбу охлаждали до комнатной температуры и сушили на роторном испарителе. К реакционной смеси добавляли 20 мл Н 2 О и охлаждали в холодильнике. Осадок фильтровали при пониженном давлении с получением 0,847 г твердого вещества, тогда как маточный раствор разбавляли EtOH и концентрировали при пониженном давлении до образования осадка, который снова фильтровали с получением дополнительных 0,208 г твердого вещества. Выход смешанного твердого вещества составлял 1,055 г, 77%. Неочищенный продукт реакции (формиатная соль) кристаллизовали в H2O/EtOH (25 мл/10 мл) с получением 0,722 г (53%) желтого порошка. Формиатную соль помещали в 20 мл смесиEtOH/CH2Cl2 (1/1) и добавляли 0,5 мл 2 5% водн. NH3 с получением прозрачного раствора. Раствор сушили Na2SO4 и растворитель выпаривали при пониженном давлении с получением 0,580 г желтого порошка В 100 мл реактор с роторной мешалкой загружали 10,16 г 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты (28,83 ммоль) и 26,50 гMe4NOH5H2O (146,25 ммоль), которые предварительно смешивали. Температуру нагревательной рубашки устанавливали на 100C и перемешивание на 100 с-1, тогда как воду оставляли испаряться из реактора во время реакции. Реакционную смесь перемешивали при указанной температуре в течение 5 ч и получали гомогенное темно-коричневое масло. Температуру реактора охлаждали до 20C, добавляли 30 мл HCOOH и хорошо перемешивали таким образом, что все масло превращалось в коричневую суспензию. По каплям добавляли 4,5 мл 37% водн. HCHO и нагревали при 70C в течение 30 мин. Реакционную смесь охлаждали до 20C и добавляли 20 мл воды с осаждением продукта в форме формиатного комплекса. Суспензию охлаждали до 0C и фильтровали при пониженном давлении, и осадок на фильтре промывали дополнительными 10 мл холодной воды с получением 8,38 г белого порошка. Маточный раствор частично выпаривали при пониженном давлении и, когда твердое вещество начинало осаждаться,его снова фильтровали с получением дополнительных 0,80 г порошка. К маточному раствору добавляли 50 мл EtOH с осаждением продукта и после фильтрации при пониженном давлении получали дополнительные 0,80 г белого порошка. Продукт собирали и 9,98 г белого порошка суспендировали в смеси 50 мл EtOH и 50 мл CH2Cl2. К суспензии добавляли 25% водн. NH3 с нейтрализацией формиатного комплекса, и после добавления 12 мл NH3 весь продукт растворяли и образовывалось маленькое количество твердого вещества. К сухому органическому раствору добавляли 5 г безводного Na2SO4 и его отфильтровывали, и растворитель выпаривали при пониженном давлении. Получали 8,99 г слегка желтого порошка с выходом 86%. Пример 11. Кристаллизация из смеси этанол/толуол/вода 2:1:1. 8,4 г неочищенного марбофлоксацина суспендировали в смеси 83 мл этанола, 41 мл толуола и 41 мл воды, и нагревали в колбе с обратным холодильником. Из образованного прозрачного желтого раствора отгоняли 83 мл смеси растворителей, в результате чего температура повышалась от 74 до примерно 79C,и образовывался желтый осадок. Суспензию охлаждали до 20-25C, перемешивали в течение 1 ч, фильтровали и осадок на фильтре промывали 3 порциями 6 мл этанола с получением после сушки в вакуумной сушилке продукта с выходом более 95%. Пример 12. Кристаллизация марбофлоксацина из формиата марбофлоксацина. 26 г формиата марбофлоксацина суспендировали в смеси 65 мл этанола и 27 мл воды. При перемешивании медленно (примерно 30 мин) по каплям добавляли раствор 25% аммиака в этаноле (20 мл 25%NH3/10 мл EtOH) до растворения вещества и достижения значения рН, равного 7-9. Реакционную смесь перемешивали в течение примерно 15 мин и фильтровали. Фильтрат выпаривали при 110C до отгонки примерно 60 мл растворителя и начала осаждения марбофлоксацина. После перегонки суспензию охлаждали и перемешивали в течение от 0,5 до 1 ч при 0-5C, фильтровали с получением после сушки при 40C/50 мбар в течение от 3 до 5 ч продукта с выходом 100%. Пример 13. Кристаллизация из этанола. 1 г марбофлоксацина растворяли при нагревании в колбе с обратным холодильником в 160 мл этанола, после фильтрации раствор охлаждали и кристаллизованный продукт выделяли с выходом более 90%. Пример 14. 6,7,8-Трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота 10 ммоль этилового эфира 6,7,8-трифтор-1-(N-метилформамидо)-1,4-дигидро-4-оксо-3 хинолинкарбоновой кислоты помещали в круглодонную колбу. Добавляли 20 мл 10% H2SO4 и перемешивали при температуре песчаной бани 100C в течение периодов времени, указанных в следующей таблице. Реакционную смесь охлаждали до 4C, фильтровали, осадок промывали водой и определяли превращение и выход. Повторяли указанный эксперимент, но исходное соединение смешивали с 1,0 мл растворителя(EtOH, AcOH или MeCN, как указано в следующей таблице) перед добавлением 10% H2SO4. Исходное соединение нерастворимо в водной фазе. Путем смешивания исходного соединения с маленьким количеством полярного растворителя (EtOH, MeCN, AcOH) образовывалась пленка вокруг кристаллов, улучшающая смачивание кристаллов водным раствором кислоты. Без добавления полярного растворителя перед добавлением водного раствора кислоты смачивание кристаллов нарушается, и реакция протекает медленнее. Пример 15. 6,7,8-трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота. 3,30 г этилового эфира 6,7,8-трифтор-1-(N-метилформамидо)-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты (10,054 ммоль) помещали в круглодонную колбу, оснащенную магнитной мешалкой. Добавляли 1 мл MeCN и перемешивали в течение одной минуты. Добавляли 20 мл 10% H2SO4 и перемешивали. Колбу помещали на песчаную баню (Т=100C) и перемешивали в течение 21 ч. Суспензию охлаждали до 4C и фильтровали под вакуумом. Желтый порошок дважды промывали холодной водой и сушили. Получали 2,646 г желтого порошка (9,721 ммоль, 96,7%) и идентифицировали путем ЯМРспектроскопии, что он представлял собой соединение, указанное в названии. Пример 16. 6,7,8-Трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота. Этиловый эфир 6,7,8-трифтор-1-(N-метилформамидо)-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты (6,868 г, 20,92 ммоль) смешивали с 1 мл EtOH (с уменьшением гидрофобности субстрата). Затем добавляли 40 мл 10% водного раствора H2SO4 и смесь перемешивали при температуре бани, равной 100C, в течение 12 ч. Образовывалась белая суспензия, которую охлаждали до 0C и фильтровали при пониженном давлении. Белый порошок промывали холодной водой и холодным EtOH и сушили. Получали 5, 135 г желтого порошка и идентифицировали, что он представлял собой соединение, указанное в названии, путем 19F и 1 Н ЯМР-спектроскопии. Выход гидролиза составлял 90%. Пример 17. 6,8-Дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-хинолин-3-карбоновая кислота 6,7,8-Трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту (272 мг, 1,0 ммоль), полученную, как описано в примере 16, и 400 мг N-метилпиперазина (4,0 ммоль) смешивали с 1 мл EtOH и перемешивали при температуре кипения (температура рубашки Tj=100C). Через два часа реакции образовывался прозрачный раствор, затем продукт осаждался и образовывалась очень плотная суспензия. Реакцию прекращали через три часа перемешивания при Tj = 100C. Образец подвергали напрямую анализу ЯМР и наблюдали только два сигнала, показывающие, что реакция была количественной. Неочищенный продукт реакции разбавляли EtOH и нейтрализовали путем добавления водного раствора NH3 до достижения рН, равного 8. Суспензию охлаждали до 0C и продукт выделяли путем фильтрации при пониженном давлении, затем промывали 10 мл холодного EtOH и сушили. Получали 138 мг 6,7,8-Трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновую кислоту (1,087 г, 3,993 ммоль), 484 мг N-метилпиперазина (4,83 ммоль) и 484 мг Et3N (4,78 ммоль) смешивали с 8 мл EtOH и перемешивали при температуре кипения (Tj=100C). Через 19 ч нагревания в колбе с обратным холодильником образовывался желтый раствор и белый осадок. Растворитель выпаривали при пониженном давлении и подвергали напрямую анализу ЯМР. Неочищенный продукт реакции смешивали с 20 млEtOH и суспензию охлаждали в холодильнике. Продукт (белый осадок) выделяли путем фильтрации при пониженном давлении, затем промывали 10 мл холодного EtOH и сушили. Получали 1,178 г белого порошка (3,375 ммоль, 80%). Пример 19. 6,8-Дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-хинолин-3-карбоновая кислота. В соответствии с примерами 17 и 18 проводили дополнительные эксперименты с использованием разных условий реакции для превращения 6,7,8-трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты в 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-хинолин-3 карбоновую кислоту. Указанные эксперименты проводили в соответствии со следующей общей процедурой: 1,0 ммоль исходного соединения помещали в круглодонную колбу и добавляли Nметилпиперазин (NMP), основание и растворитель в соответствии со следующей таблицей. Реакционную смесь перемешивали при соответствующей температуре. Растворитель выпаривали и неочищенную реакционную смесь напрямую анализировали путем ЯМР (1 Н и 19 F).Py - пиридин; BIM - N-бутилимидазол; DABCO - 1,4-диазабицикло[2.2.2]октан. Нуклеофильное замещение 6,7,8-трифтор-1-метиламино-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты N-метилпиперазином (NMP) происходило региоселективно. 6,8-дифтор-1-(метиламино)-7-(4 метилпиперазин-1-ил)-4-оксо-1,4-хинолин-3-карбоновая кислота представляла собой основной продукт реакции. Реакцию проводили в чистом NMP и в разных растворителях (этанол, вода). Было обнаружено,что скорость реакции зависит от общего количества амина (NMP) в реакционной смеси. Добавление по меньшей мере эквимолярных количеств менее нуклеофильных аминов, триэтиламина, пиридина, бутилимидазола (BIM) или DABCO оказалось предпочтительным для удаления HF, образующейся в ходе реакции. Добавление данных третичных аминов не влияло на превращение, в то время как добавление неорганического NaHCO3 оказалось неблагоприятным. Пример 20. Этиловый эфир 6,8-дифтор-1-(N-метилформамидо)-7-(4-метилпиперазин-1-ил)-4-оксо 1,4-хинолин-3-карбоновой кислоты Замещение: этиловый эфир 6,7,8-трифтор-1-(N-метилформамидо)-4-оксо-1,4-хинолин-3-карбоновой кислоты (1,0 ммоль, 32 4 мг) смешивали с 2 эквивалентами N-метилпиперазина (220 мг) и 400 мг Et3N, и перемешивали в течение трех часов при 100C. Реакционную смесь превращали в жидкость через 10 мин и снова отверждали в течение 30 мин реакции (это обуславливает более высокое количество TEA). Через 3 ч перемешивания реакционную смесь охлаждали до комнатной температуры и анализировали путем ЯМР-спектроскопии. Замещение: повторяли вышеуказанную реакцию, но Et3N заменяли на 1 эквивалент DABCO. В обоих случаях замещение было количественным, и анализ неочищенных реакционных смесей демонстрировал, что в некоторой степени происходил гидролиз этилового эфира (ЕЕ) с образованием группы свободной карбоновой кислоты (СА), приводя к смеси продуктов. Результаты суммированы в следующей таблице. Этиловый эфир легко растворим в воде.(1,0 ммоль, 324 мг) смешивали с 2 эквивалентами N-метилпиперазина (220 мг) и перемешивали в течение одного часа при 100C. Реакционную смесь превращали в жидкость через 10 мин и снова отверждали в течение 30 мин реакции. Через 1 ч реакции реакционную смесь охлаждали до комнатной температуры и добавляли 10% водный раствор H2SO4 (5 мл), и снова перемешивали при 100C в течение 2 ч. Желтый раствор охлаждали до 0C так, что продукт осаждался. Его выделяли путем фильтрации при пониженном давлении. Получали чистую 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-хинолин 3-карбоновую кислоту в форме сульфатной соли (как определено путем ЯМР) в виде слегка желтого порошка (279 мг, 58%). Пример 22. Синтез 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4]оксадиазино[6,5,4-ij]хинолин-6-карбоновой кислоты (марбофлоксацина, МВХ). 13,5 г гидрохлорида 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты и приблизительно 63 г 25% водного раствора гидроксида тетраметиламмония загружали в реактор и медленно нагревали до 100C. После достижения данной температуры воду удаляли путем перегонки при пониженном давлении (от 0,8 до 0,3 бар) таким образом, что удаляли приблизительно от 25 до 32 мл воды через 3 ч. Реакционную смесь перемешивали в течение еще 3 ч после завершения превращения, реакционную смесь охлаждали до 0-10C и медленно добавляли приблизительно 40,5 мл муравьиной кислоты при энергичном перемешивании. Температуру поддерживали ниже 20C, предпочтительно от 0 до 10C. Затем медленно добавляли приблизительно 6,1 мл формальдегида. После добавления реакционную смесь нагревали до 70C и выдерживали при данной температуре в течение примерно 30 мин. Реакционную смесь охлаждали до комнатной температуры (20-30C), добавляли приблизительно 27 мл очищенной воды и смесь перемешивали в течение 30 мин. Затем реакционную смесь охлаждали до 05C и перемешивали при данной температуре в течение по меньшей мере 2 ч. Продукт формиат марбофлоксацина (MBXBZ) центрифугировали и промывали 10-15 г охлажденной (0-5C) очищенной воды. Продукт сушили в центробежной сушилке и собирали. Влажный продукт MBXBZ добавляли к смеси 67 мл этанола, 67 мл метиленхлорида и 16,2 мл раствора аммиака (приблизительно 25%). Если фазы не отделялись, добавляли еще 63 мл метиленхлорида и 33 мл очищенной воды. рН водной фазы доводили до 7-9,5, предпочтительно от 7,5 до 8,5. Смесь перемешивали в течение приблизительно от 15 мин до 1 ч, а затем слои разделяли и обе фазы подвергали анализу технологического контроля (IPC). Если результаты IPC показывали, что экстракция не была полной, добавляли приблизительно 63 мл метиленхлорида к водному слою и экстракцию повторяли до соответствия спецификации IPC. Смешивали органические фазы и добавляли приблизительно 6,8 мг безводного сульфата натрия, и необязательно 0,4 мг активированного угля. Смесь перемешивали в течение по меньшей мере 30 мин и фильтровали, затем органический растворитель отгоняли с получением неочищенного марбофлоксацина. Очистка неочищенного марбофлоксацина. В инертной атмосфере 5 г очищенной воды, 12 г 96% этанола и 4,3 г толуола (соотношение между указанными растворителями находилось в пределах следующих диапазонов: этанол : толуол : вода: 1,82,8 : 1 : 1,1-1,2) загружали в реактор и добавляли влажный неочищенный марбофлоксацин (МВХСА),полученный на предыдущем этапе, в атмосфере азота. Смесь медленно нагревали в колбе с обратным холодильником (70-80C) до получения прозрачного раствора. Указанный раствор перемешивали в течение 0,5 ч при данной температуре, а затем выпаривали одну половину азеотропной смеси растворителей(толуол : вода : этанол = 51% : 6% : 43%). Затем оставшуюся смесь медленно охлаждали до 5C (допустимый интервал составляет от 0 и 25C) при перемешивании (необязательно можно добавить 1 масс.% продукта динатрий-EDTA). Смесь перемешивали в течение от 1 до 3 ч и затем продукт выделяли путем центрифугирования, промывали 13 г этанола, сушили в центробежной сушилке и собирали. Продукт сушили при температуре 40-45C, р 100 мбар в течение 8 ч. Пример 23. Очистка марбофлоксацина. Марбофлоксацин растворяли в 20 мас.ч. воды путем добавления уксусной кислоты. Марбофлоксацин полностью растворялся при рН 5,3. Добавляли активированный уголь и смесь перемешивали в течение ночи. Затем смесь фильтровали с использованием фильтра с активированным углем. рН фильтрата доводили до 7,2 путем использования KOH, полученную суспензию перемешивали в течение 1 ч при комнатной температуре, а затем выделяли осажденный продукт. Получали марбофлоксацин с чистотой 99,9% (площадь ВЭЖХ). Анализ ВЭЖХ проводили с использованием колонки пентафторфенилпропила (PFP) (тип Luna=5:95 (об./об.); градиент: 0'=10 В, 25'=100 В, 30'= 100 В, 32'=10 В. Хроматограмма ВЭЖХ марбофлоксацина до очистки показана на фиг. 1, хроматограмма ВЭЖХ после очистки показана на фиг. 2. Как видно из указанных хроматограмм, все продукты со временем удерживания более 24 мин были успешно удалены. Пример 24. 6,7,8-Трифтор-1-(метиламино)-4-оксо-1,4-дигидрохинолин-3-карбоновая кислота 180 л очищенной воды и 86 л 30% серной кислоты загружали в 400 л реактор в инертной атмосфере. Полученный раствор добавляли к 36 кг этил-6,7,8-трифтор-1-(N-метилформамидо)-4-оксо-1,4 дигидрохинолин-3-карбоксилата, растворенного в 40 л 96% этанола, и перемешивали в течение 30 мин. Суспензию нагревали при перемешивании до 99-100C, в то же время отгоняли этанол. За реакцией наблюдали посредством технологического анализа и ее прекращали, когда содержание исходного вещества составляло менее 2% площади. Смесь охлаждали до 0-5C и перемешивали в течение по меньшей мере 2 ч. Продукт отфильтровывали, последовательно промывали холодной водой и 96% этанолом и тщательно отсасывали. Получали 28 кг продукта, указанного в названии, обладающего чистотой, составляющей 98% площади. Пример 25. 6,8-Дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3 карбоновая кислота В 400 л реактор загружали 22,9 кг 6,7,8-трифтор-1-(метиламино)-4-оксо-1,4-дигидрохинолин-3 карбоновой кислоты, полученной, как описано в примере 24, 25,5 л 1-метилпиперазина и 196 л абсолютного этанола и перемешивали в течение 15 мин в инертной атмосфере. Реакционную смесь нагревали до температуры кипения (83C) и перемешивали при данной температуре в течение по меньшей мере 5 ч. После завершения реакции смесь охлаждали до 25C и перемешивали в течение от 2 до 24 ч. Осадок фильтровали, промывали абсолютным этанолом и сушили. Получали 28,9 кг 6,8-дифтор-1-(метиламино)7-(4-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты. Пример 26. Синтез 9-фтор-3-метил-10-(4-метилпиперазин-1-ил)-7-оксо-3,7-дигидро-2 Н-[1,3,4] оксадиазино[6,5,4-ij]хинолин-6-карбоновой кислоты (марбофлоксацина, МВХ) В 150 л реактор загружали 92 кг 25% водного раствора гидроксида тетраметиламмония и 30,7 кг 6,8-дифтор-1-(метиламино)-7-(4-метилпиперазин-1-ил)-4-оксо-1, 4-дигидрохинолин-3-карбоновой кислоты, полученной, как описано в примере 25, и нагревали до минимум 95C (температура рубашки Tj=100C). После достижения данной температуры воду удаляли путем отгонки при пониженном давлении(от 0,6 до 0,3 бар) для ускорения превращения. После завершения превращения, за которым наблюдали посредством технологического анализа, к реакционной смеси добавляли 55 л воды, смесь охлаждали до 0-10C и добавляли 51 кг муравьиной кислоты. Суспензию нагревали до комнатной температуры и добавляли 13,4 кг 37% формальдегида. После добавления реакционную смесь нагревали до приблизительно 70C и выдерживали при данной температуре до завершения реакции, наблюдаемой посредством технологического анализа. Реакционную смесь охлаждали до 0-5C, перемешивали при данной температуре в течение 2 ч и фильтровали. Продукт, формиат марбофлоксацина, промывали водой и тщательно отсасывали. Формиат марбофлоксацина, полученный на предыдущем этапе, загружали в 1000 л реактор совместно с 450 л воды. рН раствора доводили до 5,0-5,3 с использованием аммиака (воды.), затем добавляли активированный уголь и суспензию перемешивали в течение по меньшей мере 30 мин, предпочтительно 16-20 ч. Суспензию фильтровали через фильтр, в реакторе собирали фильтрат, к которому добавляли 260 л метиленхлорида, и рН доводили до 7,5-8,0 с использованием водного раствора аммиака. Смесь перемешивали, а затем слои разделяли, продукт собирали в органической фазе. К водной фазе добавляли метиленхлорид для дополнительной экстракции и экстракцию повторяли до достижения концентрации продукта в водной фазе менее 2 г/л. Органические фазы смешивали, необязательно сушили, затем органический растворитель отгоняли с получением 24,2 кг влажного неочищенного марбофлоксацина. Для дополнительной очистки продукта использовали стандартные способы кристаллизации. Пример 27. Очистка марбофлоксацина с использованием препаративной обращенно-фазовой хроматографии ВЭЖХ. Марбофлоксацин растворяли в метаноле путем добавления нескольких капель 0,1 Н метанольного раствора NaOH. Концентрация образца составляла с = 5-10 мг/мл. Образец содержал примерно от 0,03 до 0,1% димерных примесей. Образец загружали в колонку. При каждом вводе загружали примерно от 5 до 50 мг образца. После завершения хроматографических циклов фракции собирали, отгоняли органическую фазу с получением фракции, содержащей марбофлоксацин в концентрации примерно с = 0,5 мг/мл. Содержание димерной примеси анализировали методом ВЭЖХ, описанным в примере 23. Содержание димерных примесей составляло менее 5 м.д. Для ВЭЖХ использовали следующие условия: подвижная фаза: 1) буфер: NH4COOCH3 или NaH2PO4, 10-20 мМ, диапазон рН 2-5; для улучшения формы пика добавляли 0,1-0,2% триэтиламина; 2) органическая фаза: ацетонитрил или смеси ацетонитрила с метанолом (ацетонитрил : метанол = от 25:75 (об.%) до 5:95 (об.%. Детектирование: УФ-детектор, длина волны 315 нм. Хроматографическая колонка: колонка с обращенной фазой С 12 (Synergi MAX-RP) или С 18 (LunaPFP). Количество димерной примеси определяли путем ВЭЖХ с использованием колонки YMC PackODS-AQ 1504,6 мм, размер частиц 5 мкм; детектор: УФ 306 нм; скорость потока: 1,2 мл/мин; вводимый объем: 20 мкл; Т: 25C; подвижная фаза: А: 0,1 М Н 3 РО 4; В: ацетонитрил; градиент: 0-15'=20%В, 1520'=80%В, 21-25'= 20%В; после прохождения: 3 мин. Данный способ более чувствительный (предел детектирования примерно 5 м.д.), чем способ из примера 23. Пример 28. Очистка марбофлоксацина с использованием препаративной хроматографии ВЭЖХ с нормальными фазами. Марбофлоксацин растворяли в метаноле путем добавления нескольких капель 0,1 Н метанольного раствора NaOH. Концентрация образца составляла с = 5-10 мг/мл. Образец содержал примерно от 0,03 до 0,1% димерных примесей. Образец загружали в колонку. При каждом вводе загружали примерно от 5 до 20 мг образца. После завершения препаративных хроматографических циклов фракции собирали, отгоняли органическую фазу с получением сухого остатка. Сухой остаток растворяли в метаноле с получением раствора марбофлоксацина в концентрации примерно с = 1-2 мг/мл. Содержание димерной примеси анализировали методом ВЭЖХ, описанным в примере 23. Содержание димерных примесей составляло менее 5 м.д. Для ВЭЖХ использовали следующие условия. Подвижная фаза: смесь гексанов (30 об.%) и этанола (70 об.%). Детектирование: УФ-детектор, длина волны 315 нм. Хроматографическая колонка: колонка Nucleosil 100-10NH2 25016 мм, производимаяMacherey-Nagel, размер частиц 10 мкм. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (I) где R выбран из Н, C1-6 алкила, катиона щелочного металла,X представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил,отличающийся тем, что осуществляют взаимодействие соединения формулы (II) с соединением формулы (III) где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила. 2. Способ по п.1, где соединение формулы (I), где R и R' представляет собой Н, а X представляет собой 4-метилпиперазинил, получают путем взаимодействия соединения формулы (II), где R представляет собой Н или этил, R' представляет собой Н, а X представляет собой 4-метилпиперазинил, с гидроксидом тетраметиламмония. 3. Способ по п.1 или 2, где способ осуществляют в отсутствие растворителей. 4. Способ по любому из пп.1-3, где соотношение соединения формулы (II) и соединения формулы(III) составляет от 1:3 до 1:6. 5. Способ однореакторного получения марбофлоксацина формулы (IV) где R представляет собой Н, а X представляет собой 4-метилпиперазинил,отличающийся тем, что включает следующие этапы: а) взаимодействие соединения формулы (II) где R выбран из группы Н, C1-6 алкила, катиона щелочного металла,X представляет собой галоген, такой как хлор, бром, фтор; 4-метилпиперазинил иR' выбран из Н или формила,с соединением формулы (III) где R1, R2, R3 и R4 независимо выбраны из C1-6 алкила,b) взаимодействие полученного промежуточного соединения без выделения с муравьиной кислотой и формальдегидом и, необязательно,с) в случае, когда X представляет собой галоген, его замещение соответствующим пиперазином традиционными способами с получением после выделения соединения формулы (IV). 6. Способ по п.5, в котором используют соединение формулы (II), где R выбран из Н, метила, этила;X представляет собой 4-метилпиперазин и R' представляет собой Н или формил. 7. Способ по п.5 или 6, в котором используют соединение формулы (II), где R представляет собой Н; X представляет собой 4-метилпиперазин и R' представляет собой Н (формула (IIa. 8. Способ по любому из пп.5-7, где этап (а) проводят в отсутствие растворителя. 9. Способ по любому из пп.5-8, где соотношение соединения формулы (II) и соединения формулы(III) составляет от 1:3 до 1:6. 10. Способ по любому из пп.7-9, в котором соединение формулы (IIa) получают путем гидролиза соединения формулы (II), где R представляет собой C1-C6 алкил, R' представляет собой формил и X представляет собой N-метилпиперазин (формула (IIc, с кислотой или с основанием или путем взаимодействия соединения формулы (II), где R представляет собой Н, R' представляет собой Н, а X представляет собой F (формула (IId, с N-метилпиперазином. 11. Способ по п.10, в котором соединение формулы (IIc) получают путем осуществления взаимодействия соединения формулы (II), где R представляет C1-C6 алкил, R' представляет собой формил и X представляет собой F (формула (IIb, с N-метилпиперазином. 12. Способ по п.10, в котором соединение формулы (IId) получают путем гидролиза соединения формулы (II), где R представляет C1-C6 алкил, R' представляет собой формил и X представляет собой F(формула (IIb, с кислотой или с основанием. 13. Способ по любому из пп.5-12, дополнительно включающий этапы:a') растворения марбофлоксацина, его промежуточного соединения или предшественника, содержащего группу пиперазина, в воде в присутствии кислоты,b') необязательно добавления активированного угля к смеси этапа (a'),c') фильтрации смеси этапа (a') или (b') через фильтр, предпочтительно фильтр с активированным углем,d') осаждения продукта этапа (с') путем добавления основного реагента.
МПК / Метки
МПК: C07D 215/58
Метки: промежуточного, способ, получения, марбофлоксацина, соединения
Код ссылки
<a href="https://eas.patents.su/22-21239-sposob-polucheniya-marbofloksacina-i-ego-promezhutochnogo-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения марбофлоксацина и его промежуточного соединения</a>
Способ получения иминного промежуточного соединения

Номер патента: 9659
Опубликовано: 28.02.2008
Авторы: Краснаи Дьёрдь, Надь Калман, Верецкеине Донат Дьёрдьи, Грегорне Борош Ливия, Котаи Надь Петер, Баркоци Йожеф, Якфальви Элемер, Шимиг Дьюла, Мемет Норберт
МПК: C07C 249/02, C07C 209/26, C07C 211/42...
Метки: промежуточного, соединения, способ, получения, иминного
Формула / Реферат:
1. Способ получения [4(S,R)-(3,4-дихлорфенил)-3,4-дигидро-1(2Н)-нафталин-1-илиден]метиламина формулы осуществлением реакции 4-(3,4-дихлорфенил)-3,4-дигидро-1(2Н)-нафталин-1-она формулы с монометиламином, при котором указанную реакцию проводят в присутствии тионилхлорида в растворителе типа простого эфира. 2. Способ по п.1, где в качестве растворителя типа простого эфира используют тетрагидрофуран, диоксан, диэтиловый эфир, диизопропиловый эфир...
Способ получения глимепирида и соответствующего промежуточного соединения

Номер патента: 8193
Опубликовано: 27.04.2007
Авторы: Ярраг Камаль, Радль Станислав
МПК: C07D 207/38, C07C 211/35
Метки: промежуточного, способ, соответствующего, глимепирида, получения, соединения
Формула / Реферат:
1. Способ получения глимепирида формулы I отличающийся тем, что пивалат транс-4-метилциклогексиламина формулы VII подвергают взаимодействию с алкил[4-(2-{[(3-этил-4-метил-2-оксо-2,5-дигидро-1H-пиррол-1-ил)карбонил]амино}этил)фенил]сульфонилкарбаматом общей формулы IV где R представляет собой С1-С5алкил, с получением глимепирида формулы I. 2. Способ по п.1, где пивалат транс-4-метилциклогексиламина формулы VII получен путем осуществления...
Промежуточное соединение для получения паклитаксела и способ получения промежуточного соединения

Номер патента: 678
Опубликовано: 28.02.2000
Авторы: Чандер Мадхави С., Систи Николас Дж., Свинделл Чарльз С.
МПК: C07D 305/14
Метки: соединение, соединения, получения, промежуточного, паклитаксела, способ, промежуточное
Формула / Реферат:
1. Промежуточное соединение для получения паклитаксела, где промежуточное соединение имеет общую формулу где P1 представляет гидрирующуюся бензильную защитную группу. 2. Промежуточное соединение паклитаксела по п.1, где гидрирующуюся бензильную защитную группу выбирают из группы, включающей бензилоксиметил и бензил. 3. Способ получения промежуточного соединения для получения паклитаксела, где промежуточное соединение имеет общую формулу ...
Способ получения фармацевтического промежуточного соединения

Номер патента: 14530
Опубликовано: 30.12.2010
Авторы: Реитер Йожеф, Тринка Петер, Барта Ференц, Катона Зольтан, Надь Калман, Мезеи Тибор, Верецкеине Донат Дьёрдьи, Понго Ласло
МПК: C07D 295/088
Метки: получения, способ, фармацевтического, промежуточного, соединения
Формула / Реферат:
1. Способ получения {2-[4-(a-фенил-n-хлорбензил)пиперазин-1-ил]этокси}уксусной кислоты N,N-диметиламида формулы (I)и его энантиомеров, который включает взаимодействие 1-(a-фенил-n-хлорбензил)пиперазина формулы (II)или его энантиомера с b-хлорэтокси-уксусной кислоты N,N-диметиламидом формулы (III)в инертном растворителе в присутствии катализатора и вещества, связывающего кислоту.2. Способ по п.1, отличающийся тем, что в качестве растворителя...
Полиморфные формы производного 1-пиррола, промежуточного соединения для получения аторвастатина

Номер патента: 7104
Опубликовано: 30.06.2006
Авторы: Баркоци Йожеф, Грефф Зольтан, Надь Калман, Верецкейне Донат Дьёрди, Котаи Надь Петер, Шимиг Дьюла, Барта Ференц, Сент Кирайи Жужа
МПК: C07D 405/06
Метки: соединения, производного, промежуточного, формы, 1-пиррола, аторвастатина, полиморфные, получения
Формула / Реферат:
1. Кристаллическая форма I трет-бутилового эфира (4R-циc)-6-[2-[3-фенил-4-фенилкарбамоил-2-(4-фторфенил)-5-(1-метилэтил)пиррол-1-ил]этил]-2,2-диметил-[1,3]даоксан-4-ил-уксусной кислоты формулы характеризующаяся тем, что имеет порошковую рентгенограмму, представленную в табл. 1 и на фиг. 1, полученную с помощью СuКa -излучения: Таблица 1. Положение дифракционных полос и относительная интенсивность (> 10% полиморфной формы I) 2. Способ...
Предыдущий патент: Способ добычи нефти
Следующий патент: Производные 5,6-дигидро-2h-[1,4]оксазин-3-иламина в качестве ингибиторов бета-секретазы (bace)
Случайный патент: Устройство для измерения концентрации ионов в атмосферном воздухе (его варианты)