(1-фенил-2-пиридин-4-ил)этиловые эфиры бензойной кислоты в качестве ингибиторов фосфодиэстеразы 4 (pde4)
Номер патента: 19113
Опубликовано: 30.01.2014
Авторы: Армани Элизабетта, Амари Габриеле, Дельканале Маурицио


















Формула / Реферат
1. Соединение общей формулы (I) в виде (-)-энантиомера

где n равно 0 или 1;
R1 и R2 могут быть одинаковыми или разными и выбраны из группы, состоящей из
линейного или разветвленного C1-С6алкила, возможно замещенного одним или более чем одним атомом галогена;
OR3, где R3 представляет собой линейный или разветвленный C1-С6алкил, возможно замещенный одним или более чем одним атомом галогена либо одной или более чем одной С3-С7циклоалкильной группой; и
HNSO2R4, где R4 представляет собой линейный или разветвленный С1-С4алкил, возможно замещенный одним или более чем одним атомом галогена,
где по меньшей мере один из R1 и R2 представляет собой HNSO2R4;
или его фармацевтически приемлемая неорганическая или органическая соль, гидрат или сольват.
2. Соединение по п.1, где R1 представляет собой HNSO2R4, где R4 представляет собой метил, R2 представляет собой OR3, где R3 представляет собой циклопропилметил, и n равно 0.
3. Соединение по п.1, где R1 представляет собой HNSO2R4, где R4 представляет собой метил, R2 представляет собой OR3, где R3 представляет собой циклопропилметил, и n равно 1.
4. Соединение по п.1, где R1 представляет собой OR3, R2 представляет собой HNSO2R4, где R4 представляет собой метил, и n равно 1.
5. Соединение по п.1, где R1 представляет собой метил, R2 представляет собой HNSO2R4, где R4 представляет собой метил, и n равно 1.
6. Соединение по п.1, где оба R1 и R2 представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 0.
7. Соединение по п.1, где оба R1 и R2 представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 1.
8. Способ получения соединения по любому из пп.1-7, включающий стадию взаимодействия альдегида (1)

с метилдихлорпиридином (2)

с получением рацемического спирта (3)

который возможно окисляют до соответствующего N-оксидного производного (4)

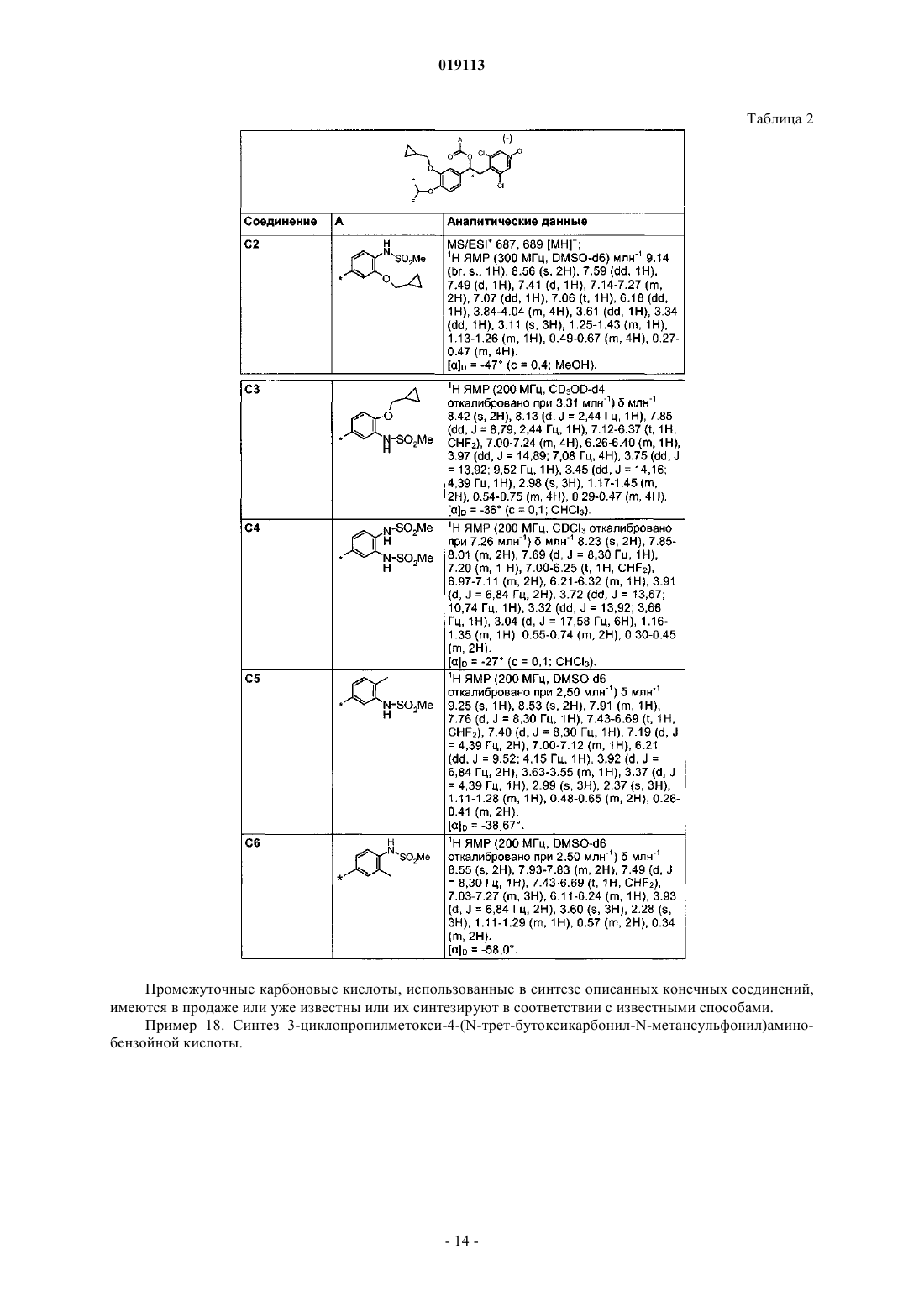
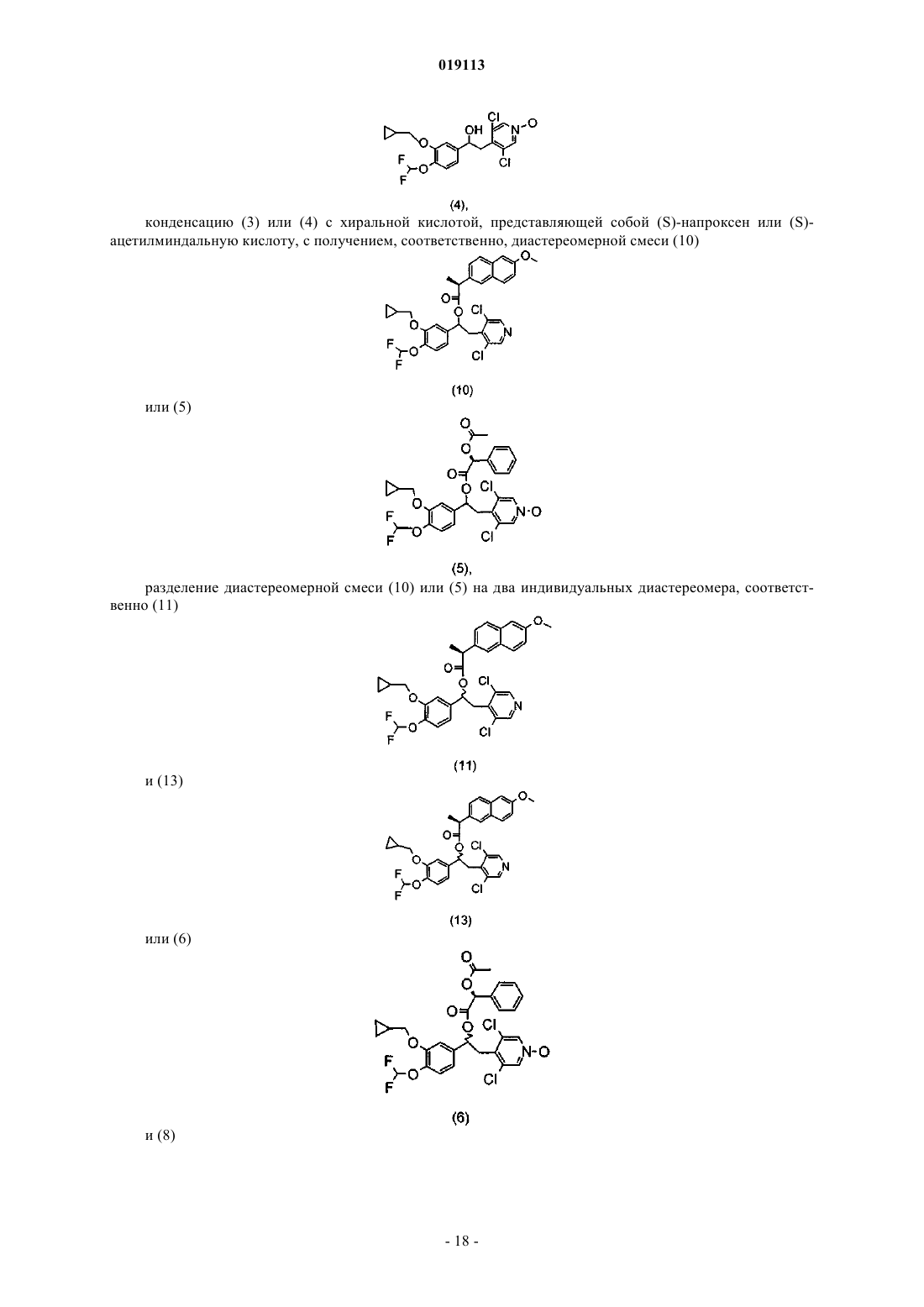
конденсацию (3) или (4) с хиральной кислотой, представляющей собой (S)-напроксен или (S)-ацетилминдальную кислоту, с получением, соответственно, диастереомерной смеси (10)

или (5)

разделение диастереомерной смеси (10) или (5) на два индивидуальных диастереомера, соответственно (11)

и (13)

или (6)

и (8)

путем хроматографии или кристаллизации с получением, после расщепления (+)-спирта (14)

или (+) (7)

и затем приведение во взаимодействие соединения (+) (14) или (+) (7) с подходящей бензойной кислотой (15)

с получением соединений общей формулы (I), где R1 и R2 такие, как определено в п.1.
9. Соединение общей формулы (II)

где n равно 0 или 1, а хиральный атом углерода, отмеченный (*), имеет (S)-конфигурацию.
10. Применение соединения формулы (I) по любому из пп.1-7 в комбинации со вторым фармацевтическим активным компонентом, выбранным из классов β2-агонистов, М3-антагонистов или кортикостероидов.
11. Применение по п.10, где второй активный компонент представляет собой формотерол или кармотерол.
12. Фармацевтическая композиция, обладающая активностью ингибитора фосфодиэстеразы 4 (PDE4), содержащая соединение формулы (I) по любому из пп.1-7 и один или более чем один фармацевтически приемлемый носитель и/или эксципиент.
13. Фармацевтическая композиция по п.12, дополнительно содержащая второй фармацевтически активный компонент, выбранный из классов β2-агонистов, М3-антагонистов или кортикостероидов.
14. Применение соединения формулы (I) по любому из пп.1-7 в качестве лекарственного средства, обладающего активностью ингибитора PDE4.
15. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения заболевания дыхательных путей, характеризующегося обструкцией дыхательных путей.
16. Применение по п.15, где указанное заболевание представляет собой астму или COPD (хроническую обструктивную болезнь легких).
17. Устройство, представляющее собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist), содержащее фармацевтическую композицию по п.12 или 13.
18. Набор, содержащий фармацевтическую композицию по п.12 или 13 и устройство, которое представляет собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist).
19. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения аллергического ринита.
20. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения атопического дерматита.
Текст
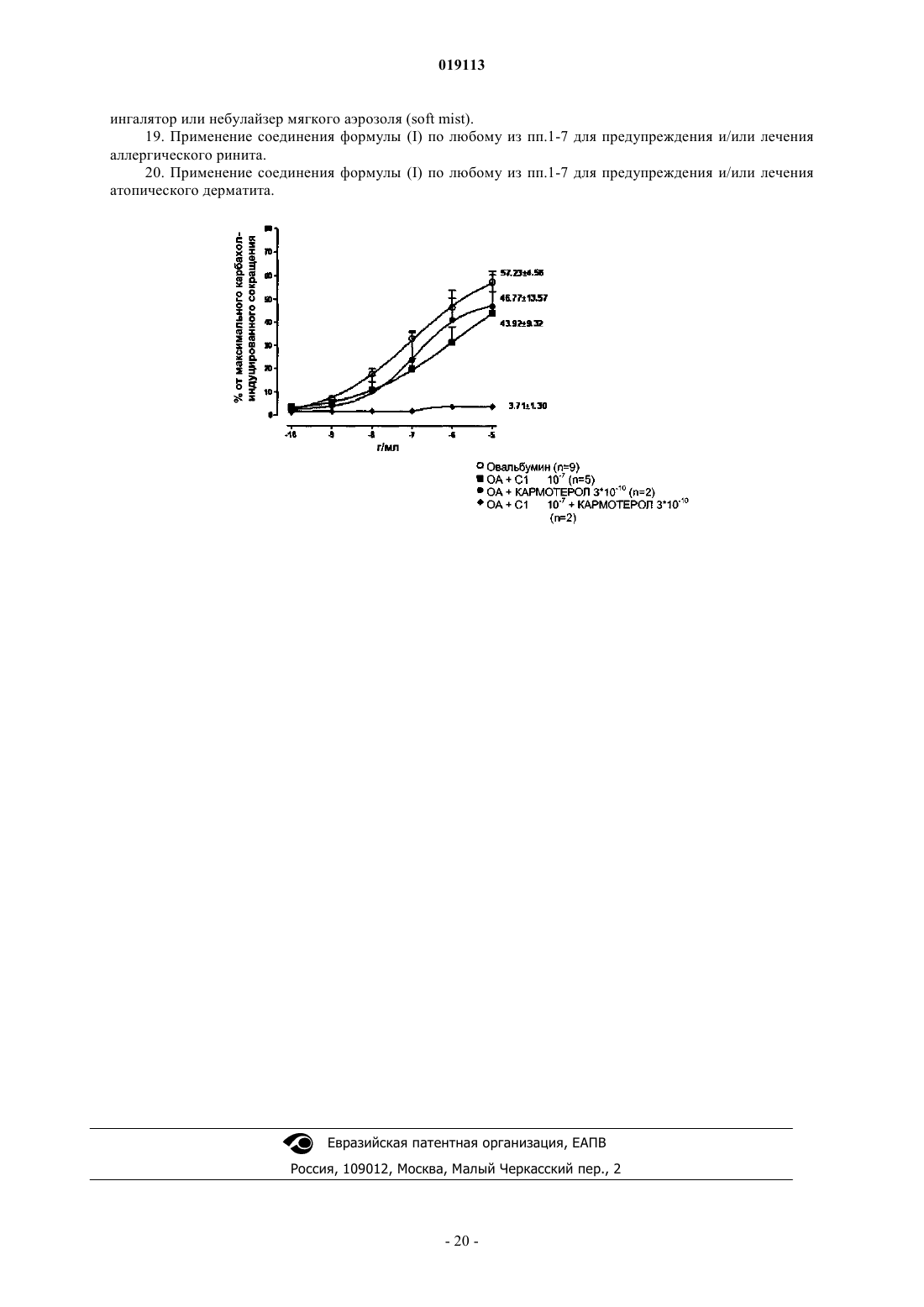
(1-ФЕНИЛ-2-ПИРИДИН-4-ИЛ)ЭТИЛОВЫЕ ЭФИРЫ БЕНЗОЙНОЙ КИСЛОТЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗЫ 4 (PDE4) Настоящее изобретение относится к ингибиторам фермента фосфодиэстеразы 4 (PDE4). Более конкретно изобретение относится к соединениям, которые являются производным 1 фенил-2-пиридин-4-ил-этиловых спиртов, способам получения таких соединений, композициям,содержащим их, комбинациям и терапевтическим применениям таких соединений формулы (I), гдеn равно 0 или 1; R1 и R2 могут быть одинаковыми или разными и выбраны из группы, состоящей из линейного или разветвленного C1-С 6 алкила; OR3, где R3 представляет собой C1-С 6 алкил, возможно замещенный одной или более С 3-С 7 циклоалкильными группами; и HNSO2R4, где R4 представляет собой С 1-С 4 алкил, возможно замещенный одним или более атомами галогена либо С 1-С 4-группой,где по меньшей мере один из R1 и R2 представляет собой HNSO2R4. Другие переменные описаны в формуле изобретения. Область изобретения Настоящее изобретение относится к ингибиторам фермента фосфодиэстеразы 4 (PDE4). Более конкретно изобретение относится к производным 1-фенил-2-пиридинилалкиловых спиртов, к способам их получения, композициям, содержащим их, комбинациям и их терапевтическим применениям. Предшествующий уровень техники Обструкция дыхательных путей является характерной чертой ряда тяжелых респираторных заболеваний, включая астму и хроническую обструктивную болезнь легких (COPD). События, приводящие к обструкции дыхательных путей, включают отек стенок воздухоносных путей, увеличение образования слизи и воспаление. Лекарственные средства для лечения респираторных заболеваний, таких как астма и COPD, в настоящее время вводят путем ингаляции. Одним из преимуществ ингаляционного способа над системным способом является возможность доставки лекарственного средства непосредственно к месту действия,что позволяет избежать любых системных побочных эффектов, обеспечивая, таким образом, более быстрый клинический ответ и более высокий терапевтический индекс. В настоящее время ингалируемые кортикостероиды представляют собой предпочтительную поддерживающую терапию для лечения астмы, и вместе с бронхолитическими 2-агонистами для ослабления острых симптомов они образуют основу современной терапии данного заболевания. Существующее сегодня лечение COPD в большой степени является симптоматическим и осуществляется посредством применения бронходилятационной терапии ингалируемыми антихолинергическими средствами и ингалируемыми агонистами 2-адренорецепторов. Однако кортикостероиды не снижают воспалительный ответ при COPD, как это они делают при астме. Другой класс терапевтических агентов, которые рассматриваются ввиду их противовоспалительного действия в отношении лечения воспалительных респираторных заболеваний, таких как астма и COPD,представлен ингибиторами ферментов фосфодиэстераз (PDE), в частности фосфодиэстеразы 4 типа (далее обозначаемой как PDE4). Описаны различные соединения, действующие в качестве ингибиторов PDE4. Однако применимость некоторых ингибиторов PDE4 первого поколения, таких как ролипрам и пикламиласт, ограничена их нежелательными побочными эффектами, такими как тошнота, секреция кислот желудочного сока и рвота, вследствие их действия на PDE4 в центральной нервной системе и вследствие их действия наPDE4 в париетальных клетках в кишечнике. Причина указанных побочных эффектов всесторонне исследована. Обнаружено, что PDE4 существует в двух различных формах, представляющих собой разные конформации, которые были обозначены как PDE4 с высокоаффинным сайтом связывания с ролипрамом или HPDE4, главным образом представленная в центральной нервной системе и в париетальных клетках,и PDE4 с низкоаффинным сайтом связывания с ролипрамом или LPDE4 (Jacobitz, S. et al. Mol. Pharmacol,1996, 50, 891-899), которая обнаружена в иммунных и воспалительных клетках. Хотя обе формы повидимому проявляют каталитическую активность, они различаются по своей чувствительности к ингибиторам. В частности, соединения с высокой аффинностью к LPDE4 в меньшей степени склонны к индуцированию побочных эффектов, таких как тошнота, рвота и повышенная желудочная секреция. Попытка направленного воздействия на LPDE4 привела к небольшому улучшению в селективности у ингибиторов PDE4 второго поколения, таких как циломиласт и рофлумиласт. Однако даже эти соединения не обеспечивают хорошую селективность в отношении LPDE4. Соединения, селективно ингибирующие активность LPDE4, описаны в WO 2009/018909. 1-Фенил-2-пиридинилалкиленовые спирты и их применение в качестве ингибиторов PDE4 также описано в WO 2008/006509. Согласно настоящему изобретению предложен ряд эффективных новых ингибиторов PDE4, обладающих превосходной селективностью к LPDE4. Неожиданно обнаружено, что наличие заместителей сульфонамидо на остатке бензоата существенно улучшает такую эффективность. Более того, неожиданно обнаружено, что сульфониламидопроизводные по изобретению, которые представляют собой (-)-энантиомеры (см. атом углерода, отмеченный снизу звездочкой), обладают более сильным действием, чем соответствующие (+)-энантиомеры и рацематы. Теперь обнаружено, что в случае использования соединений по изобретению в комбинации с 2 агонистом длительного действия получается неожиданно полезный терапевтический эффект, в частности синергический эффект, в лечении воспалительных или обструктивных заболеваний дыхательных путей. Сущность изобретения Изобретение относится к соединениям общей формулы (I) в виде (-)-энантиомеров, действующих в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4), к способам их получения, композициям,содержащим их, и их терапевтическим применениямR1 и R2 могут быть одинаковыми или разными и выбраны из группы состоящей из линейного или разветвленного С 1-С 6 алкила, возможно замещенного одним или более чем одним атомом галогена;OR3, где R3 представляет собой линейный или разветвленный С 1-С 6 алкил, возможно замещенный одним или более чем одним атомом галогена либо одной или более чем одной C3-С 7 циклоалкильной группой; иHNSO2R4, где R4 представляет собой линейный или разветвленный С 1-С 4 алкил, возможно замещенный одним или более чем одним атомом галогена,где по меньшей мере один из R1 и R2 представляет собой HNSO2R4. Изобретение также охватывает фармацевтически приемлемые гидраты, сольваты, комплексы присоединения, их неорганические или органические соли, например соли натрия, калия и лизина. Настоящее изобретение также относится к способу получения соединений формулы (I), как приведено на схеме 1, который включает приведение во взаимодействие альдегида (1) с метилдихлорпиридином (2) с получением рацемического спирта (3). Этот последний затем конденсируют с хиральной кислотой, такой как (S)-напроксен или (S)-ацетилминдальная кислота, с получением диастереомерной смеси(10) или (5), соответственно, согласно путям 1 или 2 схемы 1. Разделение на индивидуальные диастереомеры, соответственно (11) и (13) или (6) и (8), осуществляют с использованием хроматографии, кристаллизации или других общеизвестных способов, получая после расщепления, соответственно, энантиомерные спирты (-) (12) и (+) (14) или (+) (7) и (-) (9). И наконец, путем взаимодействия энантиомеров (+) (14) или (+) (7) с подходящей бензойной кислотой (15) получают соединения общей формулы (I). Настоящее изобретение также относится к способу получения соединений формулы (I), где n равно 0, как представлено на схеме 1, который включает приведение во взаимодействие любого энантиомерного спирта, например (+) (14), с бензойной кислотой (15). Настоящее изобретение также относится к способу получения соединений формулы (I), где n равно 1, как представлено на схеме 1, который включает окисление (+)-энантиомерного спирта (14) посредством окисляющего агента, такого как 3-хлорпербензойная кислота, перуксусная кислота или перекись водорода, с получением (+)-энантиомера спирта (7), который путем взаимодействия с бензойной кислотой формулы (15) дает соединения формулы (I), где n равно 1. Настоящее изобретение также относится к способу получения соединений формулы (I), где n равно 1, как приведено на схеме 1, который включает окисление сложных эфиров формулы (I), где n равно 0,посредством окисляющего агента, такого как 3-хлорпербензойная кислота, перуксусная кислота или перекись водорода. Настоящее изобретение также относится к промежуточным соединениям общей формулы (II) где n такой, как определено выше, а атом углерода, отмеченный снизу звездочкой, демонстрирует (S)конфигурацию. Согласно настоящему изобретению также предложены фармацевтические композиции, содержащие соединение формулы (I) и один или более чем один фармацевтически приемлемый носитель и/или эксципиент. В частности, согласно настоящему изобретению предложены фармацевтические композиции, подходящие для введения путем ингаляции. Согласно настоящему изобретению также предложены комбинации соединения формулы (I) со вторым компонентом, выбранным из классов длительно действующих 2-агонистов, М 3-антагонистов и кортикостероидов. Согласно настоящему изобретению также предложены комбинации соединения формулы (I) с 2 агонистом длительного действия, выбранным из группы, состоящей из кармотерола, GSK-642444, индакатерола, милветерола, арформотерола, формотерола, сальбутамола, формотерола, левальбутерола, тер-2 019113 буталина, AZD-3199, BI-1744-CL, LAS-100977, бамбутерола, изопротеренола, прокатерола, кленбутерола, репротерола, фенотерола и ASF-1020. Согласно настоящему изобретению также предложены комбинации соединения формулы (I) с М 3 антагонистом, выбранным из группы, состоящей из аклидиния, тиотропия, ипратропия и окситропия. Согласно настоящему изобретению также предложены комбинации соединения формулы (I) с кортикостероидом, выбранным из группы, состоящей из дексаметазона, флутиказона, флутиказона фуроата,преднизолона, бетаметазона, будесонида, мометазона, мометазона фуроата, триамцинолона ацетонида,циклесонида, TPI-1020, беклометазона, беклометазона дипропионата, преднизона, дефлазакорта, гидрокортизона, QAE-397 и флунизолида. В предпочтительном воплощении согласно настоящему изобретению предложены комбинации соединения формулы (I) с формотеролом или кармотеролом. Согласно настоящему изобретению также предложены соединения формулы (I) для применения в качестве лекарственного средства. Также предложено применение соединений формулы (I) в изготовлении лекарственного средства для предупреждения или лечения любого заболевания, в которое вовлечена активность рецепторов PDE4 и при котором желательно ингибирование PDE4-рецепторной активности. Согласно настоящему изобретению также предложен способ предупреждения или лечения любого заболевания, в которое вовлечена активность рецепторов PDE4 и при котором желательно ингибирование активности рецепторнов PDE4, включающий введение пациенту, нуждающемуся в таком введении,терапевтически эффективного количества соединения формулы (I). В вышеупомянутых применениях или способах используют соединение формулы (I) либо само по себе, либо в комбинации с другими активными ингредиентами из тех, о которых сообщалось ранее. Вышеупомянутые заболевания, в которые вовлечены активность рецепторов PDE4 и ингибирование рецепторов PDE4, включают заболевания дыхательных путей, характеризующиеся обструкцией дыхательных путей, такие как астма и COPD. Кроме того, изобретение также относится к применению соединений формулы (I) для ингибирования PDE4 in vitro. Изобретение также относится к устройству, которое может представлять собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist), содержащий соединение формулы (I). Изобретение также относится к набору, содержащему фармацевтические композиции соединений формулы (I), как таковых или в комбинации с дополнительным фармацевтическим ингредиентом, в смеси с одним или более чем одним фармацевтически приемлемым носителем и/или эксципиентом, и устройство, которое может представлять собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist). Определения Термин "атомы галогена", как он использован здесь, включает фтор, хлор, бром и йод. Использованное здесь выражение "линейный или разветвленный C1-Схалкил", где х представляет собой целое число больше 1, как, например, C1-C6- или С 1-С 4 алкил, относится к алкильным группам с прямой или разветвленной цепью, где количество атомов углерода лежит в диапазоне от 1 до х (например, от 1 до 6 или от 1 до 4). Так, примеры алкильных групп могут включать метил, этил, н-пропил, изопропил, трет-бутил, пентил, гексил и тому подобное. Возможно, в указанных группах один или более чем один атом водорода может быть заменен на атом галогена, предпочтительно хлора или фтора. Как оно используется здесь, выражение "C3-С 7 циклоалкил" относится к циклическим неароматическим углеводородным группам, содержащим от 3 до 7 атомов углерода в кольце. Таким образом, примеры таких групп могут включать циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Если не указано иное, то в том случае, когда дается ссылка на хиральные соединения, степень чистоты "по существу чистый" здесь означает по меньшей мере более чем приблизительно на 97% хирально чистый, предпочтительно более чем на 99% и наиболее предпочтительно более чем на 99,9%. Графические материалы На чертеже продемонстрировано наличие синергического действия для предпочтительного воплощения настоящего изобретения. ОА = овальбумин. С 1 = 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир 3-циклопропилметокси-4-метансульфониламинобензойной кислоты.CARM = кармотерол. Подробное описание изобретения Изобретение относится к соединениям, действующим в качестве ингибиторов фермента фосфодиэстеразы 4 (PDE4). Указанные соединения ингибируют превращение циклических нуклеотидов, в частности циклического аденозинмонофосфата (цАМФ), в их неактивные 5'-мононуклеотидные формы. В дыхательных путях физиологические ответы на повышенные внутриклеточные уровни циклических нуклеотидов, в частности цАМФ, приводят к подавлению активности иммунных и провоспалительных клеток, таких как тучные клетки, макрофаги, Т-лимфоциты, эозинофилы и нейтрофилы, в результате чего снижается высвобождение воспалительных медиаторов, включая цитокины, такие какIL(интерлейкин)-1, IL-3 и фактор некроза опухоли - альфа (TNF-). Это также приводит к релаксации гладкой мускулатуры дыхательных путей и уменьшению отека. Каталитический сайт PDE4 был идентифицирован ранее. Главным образом, он содержит гидрофобный участок, в котором присутствуют два "субкармана", например S0 и S1, и гидрофильный участок, содержащий ионы металлов Zn2+ и Mg2+, который, в свою очередь, содержит "субкарман" S2, располагающийся вокруг ионов металлов, и "субкарман" S3, который отклоняется приблизительно на 90 от среднего гидрофобного "кармана". Большинство известных соединений содержат группировку, способную взаимодействовать с "субкарманами" S0 и S1 гидрофобного участка, такую как замещенная катехольная группа, и другую группировку, способную опосредованно взаимодействовать с ионами металлов "субкармана" S2, например гетероцикл, такой как пиридин или пирролидон. Настоящее изобретение относится к соединениям, которые могут поддерживать взаимодействия с"субкарманами" S0 и S1 посредством замещенной группировки катехола и взаимодействие с участком ионов металлов посредством пиридинового кольца подобно другим известным ингибиторам PDE4, но отличаются от них наличием группы сульфониламинобензойной кислоты, которая позволяет им устанавливать дополнительное взаимодействие с "субкарманом" S3. В частности, настоящее изобретение относится к соединениям общей формулы (I), как определено ранее, в том числе их фармацевтически приемлемым неорганическим и органическим солям, гидратам,сольватам или комплексам присоединения. Предпочтительными группами соединений формулы (I) являются соединения, гдеR1 представляет собой линейный или разветвленный С 1-С 6 алкил, R2 представляет собой HNSO2R4 и n равно 0;R1 представляет собой линейный или разветвленный С 1-С 6 алкил, R2 представляет собой HNSO2R4 и n равно 1;R2 представляет собой линейный или разветвленный C1-С 6 алкил, R1 представляет собой HNSO2R4 и n равно 0;R2 представляет собой линейный или разветвленный С 1-С 6 алкил, R1 представляет собой HNSO2R4 и n равно 1; Оба, R1 и R2, представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 0; Оба, R1 и R2, представляют собой HNSO2R4, и n равно 1; Оба, R1 и R2, представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 1. Специалистам в данной области очевидно, что соединения общей формулы (I) содержат по меньшей мере один асимметрический центр, изображенный в настоящем описании в виде атома углерода с расположенной под ним звездочкой, и, следовательно, существуют в виде оптических стереоизомеров. Настоящее изобретение относится к соединениям формулы (I), которые являются (-)энантиомерами с (S)-конфигурацией у атома углерода, обозначенного снизу звездочкой. Настоящее изобретение также относится к промежуточным соединениям формулы (II), где атом углерода, обозначенный снизу звездочкой, демонстрирует (S)-конфигурацию. Соединения формулы (I) в наномолярном диапазоне концентраций демонстрируют ингибирующую активность in vitro в отношении фермента PDE4, и они обеспечивают значительную активность в легких при внутритрахеальном введении в животной модели COPD. Кроме того, они могут демонстрировать устойчивые уровни в легких при отсутствии детектирования их в плазме крови, что является показателем краткосрочного системного действия. В соответствии с предпочтительными воплощениями согласно настоящему изобретению предложены соединения формулы (I), представленные ниже Вышеупомянутые соединения соответственно идентифицированы как (-)-энантиомеры, которые,тем не менее, имеют (S)-конфигурацию при атоме углерода, отмеченном звездочкой. По существу, эти же соединения также могут быть идентифицированы так, как представлено в следующей таблице. Предпочтительно, соединения по изобретению характеризуются более высокой селективностью кLPDE4, чем к HPDE4, по результатам определения их величин IC50. В случае LPDE4, IC50 представляет собой молярную концентрацию тестируемого соединения, вызывающую 50%-ное ингибирование исчезновения цАМФ, оцененное, как описано в Cortijo J. et al. Br. J.Pharmacol. 1993, 108: 562-568. В случае HPDE4, вместо этого, IC50 представляет собой молярную концентрацию тестируемого соединения, вызывающую 50%-ное ингибирование связывания [Н 3]-ролипрама,оцененное, как описано в Duplantier A.J. et al. J. Med. Chem. 1996, 39: 120-125. Предпочтительно, чтобы отношение IC50 HPDE4/LPDE4 для соединений по изобретению составляло более 5, более предпочтительно более 10, еще более предпочтительно более 20 и наиболее предпочтительно более 100. Соединения формулы (I) могут быть получены традиционно в соответствии с известными способами. Некоторые способы, которые могут быть использованы, описаны ниже и приведены на схеме 1. Методика получения соединений формулы (I). В соответствии с конкретным воплощением настоящего изобретения соединения формулы (I) могут быть получены, например, следуя путям синтеза, описанным на схеме 1. Рацемический спирт (3) можно получить путем приведения во взаимодействие альдегида (1) с метилдихлорпиридином (2). Путь 1 - рацемический спирт (3) можно разделить на (-)-энантиомер (12) и (+)-энантиомер (14) известными способами, например путем приведения во взаимодействие рацемической смеси с подходящим хиральным вспомогательным агентом с получением таким образом смеси диастереомеров. Такие диастереомеры могут быть разделены кристаллизацией или хроматографией или посредством использования ферментов в соответствии с известными способами. Впоследствии хиральное вспомогательное средство может быть удалено из диастереомеров с получением желаемого хирального спирта в качестве единственного энантиомера. Альтернативно, рацемическая смесь спиртов может быть разделена с помощью хроматографии с использованием хиральной неподвижной фазы в соответствии с известными методамиStereochemistry: Analytical Methods and Pharmacology", Irving W. Wainer, CRC Press, 1993). В частности, можно провести реакцию конценсации рацемического спирта (3) с хиральной кислотой, такой как (S)-напроксен, и полученную диастереомерную смесь (10) можно разделить на два индивидуальных диастереомера (11) и (13) с помощью хроматографии. После расщепления индивидуальных диастереомерных сложных эфиров путем гидролиза в водном растворителе или путем алкоголиза в спиртовом растворителе с использованием кислотных или основных условий можно получить энантиомерно чистые промежуточный (-)-спирт (12) и (+)-спирт (14). Путь 2 - рацемат (4), полученный окислением рацемата (3), осуществляемым согласно традиционным методам, может быть приведен во взаимодействие с хиральной кислотой, такой как (S)ацетилминдальная кислота, с получением таким образом смеси двух диастереомеров (5). Путем растирания в порошок с диэтиловым эфиром и кристаллизации в растворителе, таком как изопропанол, этанол или метанол, или путем хроматографического разделения можно получить индивидуальные диастереомерные сложные эфиры (6) и (8). После расщепления индивидуальных диастереомерных сложных эфиров путем гидролиза в водном растворителе или путем алкоголиза в спиртовом растворителе с использованием кислотных или основных условий, можно получить энантиомерно чистый промежуточный (+)-спирт (7) и (-)-спирт (9). Соединения общей формулы (I), где n равно 0, могут быть получены путем приведения во взаимодействие надлежащего энантиомерного (+)-спирта (14) с бензойной кислотой (15) в присутствии подходящего сильного основания, такого как диизопропиламид лития (LDA), NaH или диметиламинопиридин(DMAP), и в присутствии конденсирующего агента, такого как гидрохлорид 1-этил-3-[3-диметиламинопропил]карбодиимида (EDC) или N-гидроксибензотриазол (НОВТ), в растворителе, таком как дихлорметан. Можно использовать другие растворители, такие как диметилформамид (DMF), тетрагидрофуран(THF), хлороформ, диоксан или любой другой апротонный растворитель, известный специалистам в данной области техники. В конкретном воплощении данное взаимодействие также может быть выполнено в отсутствие растворителей. Соединения формулы (I), где n равно 1, могут быть получены путем окисления соответствующих соединений формулы (I), где n равно 0, посредством окисляющего агента, такого как 3-хлорпербензойная кислота, перуксусная кислота или перекись водорода, в таких растворителях, как хлороформ, дихлорметан или уксусная кислота (путь В). Альтернативно, соединения формулы (I), где n равно 1, также могут быть получены сначала путем окисления (+)-энантиомеров спирта (14) в указанных выше рабочих условиях с получением в результате этого (+)-энантиомеров спирта (7). Таким образом, последующая реакция между данным энантиомером спирта с бензойной кислотой формулы (15) обеспечивает получение вышеупомянутых соединений формулы (I), где n равно 0 (путь А). Отделение (+)-энантиомеров (7) и (-)-энантиомеров (9) от рацемического спирта (4), который, в свою очередь, был получен окислением рацемического спирта (3), может быть осуществлено известными способами, как описано выше для разделения энантиомеров рацемического спирта (3). Специалисту следует знать, что при получении соединений по изобретению также можно применять дополнительные вариации в стадиях синтеза, приведенных на схеме 1. Авторы изобретения особенно отмечают очередность реакций, которые могут быть проведены с целью получения желаемых соединений или их промежуточных соединений, а также выбор адаптируемых рабочих условий, включая растворители, возможные окисляющие агенты, конденсирующие агенты и тому подобное. В качестве примера, в случае наличия химически реакционноспособных заместителей в любом из исходных веществ или их промежуточных соединений, которые могли бы вызвать нежелательные побочные реакции, можно ввести подходящую защиту этих же заместителей перед проведением данного взаимодействия. Далее по аналогии можно провести последующее удаление защиты с тем, чтобы снова получить вышеупомянутый химически реакционноспособный заместитель или группу в свободной форме. Введение и удаление защиты с функциональных групп описано в "Protective Groups in OrganicKocienski, Georg Thieme Verlag (1994). В соответствии с представленным способом получения соединений по настоящему изобретению и их вариантов, исходные вещества формулы (1) и (2), а также любой дополнительный реагент [например,формулы (15)], вспомогательный агент хиральности (auxiliar of chirality), растворитель или агент, которые используются, известны или легко могут быть получены в соответствии с известными способами. Согласно настоящему изобретению также предложены фармацевтические композиции соединений формулы (I) в смеси с одним или более чем одним фармацевтически приемлемым носителем, например,описанным в Remington's Pharmaceutical Sciences Handbook, XVII Ed., Mack Pub., N.Y., U.S.A. Примеры включают разбавители (такие как сахароза, маннит, лактоза, крахмалы) и известные эксципиенты, в том числе суспендирующие агенты, солюбилизаторы, буферные вещества, связующие вещества, разрыхлители, консерванты, красители, корригенты, смазывающие вещества и тому подобное. При введении соединений по настоящему изобретению также предпочтительны капсулы, таблетки и гели с пролонгированным высвобождением. Введение соединений по настоящему изобретению может быть выполнено в соответствии с нуждами пациента, например перорально, интраназально, парентерально, например подкожно, внутривенно,внутримышечно, интрастернально и путем инфузии, путем ингаляции, ректально, вагинально, местно,локально, трансдермально и путем глазного введения. Для введения соединений по изобретению можно использовать различные твердые пероральные лекарственные формы, включая такие твердые формы, как таблетки, желатиновые капсулы (gelcaps), капсулы, каплеты, гранулы, пастилки и нерасфасованные порошки. Для введения соединений по изобретению также можно использовать различные жидкие пероральные лекарственные формы, включая водные и неводные растворы, эмульсии, суспензии, сиропы и эликсиры. Такие лекарственные формы также могут содержать известные подходящие инертные разбавители,такие как вода, и известные подходящие эксципиенты, такие как консерванты, смачивающие вещества,подсластители, корригенты, а также агенты для эмульгирования и/или суспендирования соединений по изобретению. Соединения по изобретению можно вводить, например, внутривенно в форме изотонического стерильного раствора. Также возможны другие известные препараты. Суппозитории для ректального введения указанных соединений по изобретению могут быть получены путем смешивания соединения с подходящим эксципиентом, таким как масло какао, салицилаты и полиэтиленгликоли. Композиции для вагинального введения могут быть в форме крема, геля, пасты, пены или аэрозоля,содержащих в дополнение к активному ингредиенту традиционные носители. Фармацевтические композиции для местного введения могут быть в форме кремов, мазей, лини-8 019113 ментов, лосьонов, эмульсий, суспензий, гелей, растворов, паст, порошков, спреев и капель, подходящих для применения на коже, введения в глаз, ухо или нос. Местное введение также может включать трансдермальное введение, например, посредством трансдермальных пластырей. Для лечения заболеваний дыхательных путей соединения по изобретению предпочтительно вводят путем ингаляции. Ингалируемые препараты включают ингалируемые порошки, пропеллентсодержащие дозируемые аэрозоли или не содержащие пропеллента ингалируемые композиции. Для введения в виде сухого порошка можно применять известные ингаляторы для однократного или многократного приема. В этом случае порошок может быть внесен в желатиновые, пластиковые или другие капсулы, картриджи или блистерные упаковки или находиться в резервуаре. Разбавитель или носитель, обычно химически инертный к соединениям по изобретению, например лактоза или любая другая добавка, подходящая для улучшения вдыхаемой фракции, может быть добавлена к порошкообразным соединениям по изобретению. Ингаляционные аэрозоли, содержащие газ-пропеллент, такой как гидрофторалканы, могут содержать соединения по изобретению или в растворе, или в диспергированной форме. Композиции, приводимые в движение с помощью пропеллента, также могут содержать другие ингредиенты, такие как сорастворители, стабилизаторы и возможно другие эксципиенты. Не содержащие пропеллента ингалируемые композиции, содержащие соединения по изобретению,могут быть в форме растворов или суспензий в водной, спиртовой или гидроспиртовой среде, и их можно доставлять посредством известных струйных или ультразвуковых небулайзеров или небулайзеров мягкого аэрозоля, таких как Respimat. Соединения по изобретению могут быть введены в виде единственного активного агента или в комбинации с одним или более чем одним другим фармацевтически активным ингредиентом, включая ингредиенты, используемые в настоящее время в лечении респираторных расстройств, например 2 агонисты, кортикостероиды и М 3-антагонисты. Дозировки соединений по изобретению могут зависеть от разнообразных факторов, включая конкретное подвергаемое лечению заболевание, тяжесть симптомов, путь введения, частоту интервалов дозировки, конкретное используемое соединение, эффективность, токсикологический профиль и фармакокинетический профиль соединения. Предпочтительно соединения формулы (I) могут быть введены, например, в дозировке от 0,001 до 1000 мг/сутки, предпочтительно от 0,1 до 500 мг/сутки. При введении путем ингаляции дозировка соединений формулы (I) составляет предпочтительно от 0,01 до 20 мг/сутки, предпочтительно от 0,1 до 10 мг/сутки. Предпочтительно соединения формулы (I) сами по себе или в комбинации с другими активными ингредиентами могут быть введены для предупреждения и/или лечения любого обструктивного респираторного заболевания, такого как астма, хронический бронхит и хроническая обструктивная болезнь легких (COPD). Тем не менее, соединения формулы (I) могут быть введены для предупреждения и/или лечения любого заболевания, в которое вовлечена активность рецепторов PDE4 и при котором желательно ингибирование PDE4-рецепторной активности, или болезненного состояния, которое опосредовано активностью PDE4 (например, болезненного состояния, при котором PDE4 сверхэкспрессируется или характеризуется повышенной активностью). Примеры таких заболеваний включают аллергические болезненные состояния, такие как атопический дерматит, крапивница, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильная гранулема, псориаз, воспалительный артрит, ревматоидный артрит, септический шок, неспецифический язвенный колит, болезнь Крона, реперфузионное повреждение миокарда и головного мозга, хронический гломерулонефрит, эндотоксический шок, муковисцидоз, артериальный рестеноз, атеросклероз, кератоз, ревматоидный спондилит, остеоартрит, pyresis,сахарный диабет, пневмокониоз, токсическую и аллергическую контактную экзему, атопическую экзему,себоррейную экзему, простой лишай, солнечный ожог, зуд в аногенитальной области, гнездную алопецию, гипертрофические рубцы, дискоидную красную волчанку, системную красную волчанку, фолликулярную и обширную пиодермию, эндогенные и экзогенные акне, розовые угри, болезнь Бехчета (Beghet'sdisease), анафилактическую пурпуру, нефрит, воспалительное заболевание кишечника, лейкоз, рассеянный склероз, желудочно-кишечные заболевания, аутоиммунные заболевания и им подобные. Они также включают неврологические и психиатрические расстройства, такие как болезнь Альцгеймера, рассеянный склероз, амиотрофический боковой склероз (amylolaterosclerosis, ALS), множественная системная атрофия (MSA), шизофрения, болезнь Паркинсона, болезнь Гентингтона, болезнь Пика, депрессия, удар и повреждение спинного мозга. Теперь настоящее изобретение будет описано далее посредством следующих примеров. Пример 1. Получение 1-(3-циклопропилметокси-4-дифторметокси-фенил)-2-(3,5-дихлорпиридин-4 ил)этанола (3). Раствор 3-циклопропилметокси-4-дифторметоксибензальдегида (5,00 г) и 3,5-дихлор-4-метил-9 019113 пиридина (2,57 г) в 50 мл безводного THF охлаждали до -30 С. Порциями добавляли твердый трет-бутилат калия (tBuOK; 1,96 г) при поддержании температуры от-30 до -20 С, получая, таким образом, темно-красный раствор. После завершения добавления смесь перемешивали при -30 С в течение 1 ч. Затем к реакционной смеси добавляли насыщенный водный растворNH4Cl (50 мл), поддерживая температуру от -5 до -10 С. Цвет реакционной смеси изменялся на желтый. Затем смесь экстрагировали EtOAc. Органический слой сушили над Na2SO4 и растворитель выпаривали. Остаток обрабатывали 30 мл смеси петролейный эфир/EtOAc = 8/2; осадок отфильтровывали и сушили, получая 4,83 г указанного в заголовке соединения, которое использовали на следующей стадии без дополнительной очистки.MS (масс-спектрометрия)/ESI+ 404-406 [МН]+. Пример 2. Получение 1-(3-циклопропил метокси-4-дифторметоксифенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этанола (4). Соединение (3) (13,0 г) растворяли в CH2Cl2 (250 мл), затем добавляли м-хлорпербензойную кислоту (16,5 г) и полученный раствор перемешивали при комнатной температуре в течение 2 ч. ДобавлялиNa2S2O3 (25,4 г) и смесь интенсивно перемешивали при к.т. в течение 1 ч. Твердый остаток отфильтровывали, раствор промывали 1 н. NaOH (3100 мл), затем органическую фазу сушили над Na2SO4 и растворитель выпаривали, получая 10,3 г желаемого продукта (4) в виде белого твердого вещества, которое использовали на следующих стадиях без дополнительной очистки.MS/ESI+ 420-422 [МН]+. Пример 3. Получение 1-(3-циклопропил метокси-4-дифторметоксифенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этилового эфира ацетоксифенилуксусной кислоты (5, смесь диастереомеров). Соединение (4) (19,95 г), (S)-ацетилминдальную кислоту (9,22 г), 1-этил-3-[3 диметиламинопропил]карбодиимида гидрохлорид (18 г) и 4-диметиламинопиридин (2,89 г) растворяли в безводном CH2Cl2 (300 мл) в атмосфере N2. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли 5%-ный водный раствор NaHCO3 (200 мл) и водную фазу экстрагировали CH2Cl2 (3100 мл). Объединенные органические фазы сушили над Na2SO4 и растворитель упаривали при пониженном давлении, получая указанное в заголовке соединение (5) в виде смеси двух диастереомеров (32 г); разделение этих двух диастереомеров описано в примерах 4 и 6. Пример 4. Получение 1-(3-циклопропилметокси-4-дифторметокси-фенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этилового эфира (+)-ацетоксифенилуксусной кислоты (6). Неочищенную диастереомерную смесь (5) (32 г) растирали с Et2O (100 мл), подвергали ультразвуковой обработке и фильтровали. Эту процедуру повторяли четыре раза, чтобы получить твердую смесь,обогащенную по диастереомеру (6). Это твердое вещество кристаллизовали из iPrOH (80 мл) и отфильтровывали, получая 9,65 г соединения (6) с диастереомерной чистотой более 95%. Диастереомерную чистоту определяли посредством HPLC-анализа и посредством аналитической хиральной HPLC, проводимой на колонке Chiracel OD (изократическое элюирование смесью гексан:изопропанол 40:60, скорость потока 0,45 мл/мин, время удерживания = 27,2 мин).[]D = +14 (с = 0,54; МеОН). Пример 5. Получение (+)-1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этанола (7). Соединение (6) (6,42 г) суспендировали в метаноле (350 мл), затем добавляли насыщенный растворNaHCO3 (175 мл). Белую суспензию интенсивно перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли CH2Cl2 (700 мл) и промывали 5%-ным водным раствором NaHCO3(300 мл); водную фазу экстрагировали CH2Cl2 (2300 мл), объединенные органические слои сушили надNa2SO4 и растворитель выпаривали под вакуумом. Полученное неочищенное белое твердое вещество растирали с Et2O (2100 мл) и отфильтровывали, получая 3,88 г соединения (7) с энантиомерной чистотой более 99%. Энантиомерную чистоту определяли посредством аналитической хиральной HPLC, проводимой на колонке Chiracel OD (изократическое элюирование смесью гексан:изопропанол 30:70, скорость потока 0,35 мл/мин, время удерживания = 22,3 мин).[]D = +68 (с = 0,5; МеОН). Пример 6. Получение 1-(3-циклопропил метокси-4-дифторметокси-фенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этилового эфира (+)-ацетоксифенилуксусной кислоты (8). Неочищенную диастереомерную смесь (5) растирали с Et2O (100 мл), подвергали ультразвуковой обработке и фильтровали. Эту процедуру повторяли четыре раза и фильтраты собирали и упаривали при пониженном давлении, получая твердую смесь, обогащенную по диастереомеру (8), который кристаллизовали из iPrOH (100 мл), получая 6,4 г соединения (8) в виде белого твердого вещества с диастереомерной чистотой более 99%. Диастереомерную чистоту определяли посредством HPLC-анализа и посредством аналитической хиральной HPLC, проводимой на колонке Chiracel OD (изократическое элюирование смесью гексан:изопропанол 40:60, скорость потока 0,45 мл/мин, время удерживания = 21,6 мин).[]D = +26 (с = 0,55; МеОН). Пример 7. Получение (-)-1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлор-1 оксипиридин-4-ил)этанола (9). Соединение (8) (1,18 г) суспендировали в метаноле (50 мл), затем добавляли насыщенный растворNaHCO3 (25 мл). Белую суспензию интенсивно перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь разбавляли CH2Cl2 (700 мл), затем добавляли 5%-ный водный раствор NaHCO3(300 мл) и фазы разделяли. Водную фазу экстрагировали CH2Cl2 (2100 мл), объединенные органические слои сушили над Na2SO4 и растворитель выпаривали под вакуумом. Полученное неочищенное белое твердое вещество растирали дважды, используя Et2O (50 мл), и один раз, используя CH2Cl2 (20 мл), затем отфильтровывали, получая 0,74 г соединения (7) с энантиомерной чистотой более 99%. Энантиомерную чистоту определяли посредством аналитической хиральной HPLC, проводимой на колонке Chiracel OD[]D = -61 (c = 0,5; MeOH). Пример 8. 1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир 2-(6-метоксинафталин-2-ил)пропионовой кислоты (10, смесь диастереомеров 11 и 13). Соединение (3) (12,0 г) растворяли в DMF (100 мл), затем добавляли (S)-2-(6-метоксинафталин-2 ил)пропионовую кислоту (7,5 г), 4-диметиламинопиридин (3,6 г) и гидрохлорид 1-этил-3-[3 диметиламинопропил]карбодиимида (5,7 г). После перемешивания при к.т. в течение 4 ч добавляли воду(1000 мл). Смесь экстрагировали EtOAc (500 мл 2), объединенные органические слои сушили над сульфатом натрия и растворитель выпаривали при пониженном давлении, что позволяло получить 17,0 г масла, которое кристаллизовали из EtOH, получая таким образом 11,5 г указанного в заголовке соединения в виде смеси диастереомеров (11) и (13). 1 Н ЯМР (200 МГц, CDCl3) млн-1 8.43 и 8.60 (2s, 1 Н каждый, 2 Н), 7.51-7.68 (m, 3H), 7.10-7.23 (m, 3H),6.85-6.97 (m, 2 Н), 6.51-6.68 (m, 1 Н), 6.22-6.97 (t, 1 Н, CHF2), 6.00-6.13 (m, 1 Н), 3.93-3.95 (s, 3H, ОСН 3),3.72-3.84 (m, 2 Н), 3.07-3.57 (m, 3H), 1.42-1.45 (d, 3H, СН 3), 0.94-1.25 (m, 1H), 0.51-0.67 (m, 2H), 0.12-0.36MS/ESI+ 616, 618 [MH]+. Пример 9. 1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир (+)-2-(6-метоксинафталин-2-ил)пропионовой кислоты (элюируемый вторым диастереомер) (13). Соединение выделяли из диастереомерной смеси примера 8 посредством HPLC-разделения, используя колонку Daisogel 10 мкм, 50300 мм; элюент: н-гексан/метил-трет-бутиловый эфир/изопропиловый спирт: 90/9,9/0,1; скорость потока: 80 мл/мин; загрузка: 300 мг на одно введение; время элюирования: от 11 до 20 мин. Собранные фракции упаривали и остаток кристаллизовали из смеси нгексан/изопропиловый спирт. 1 Н ЯМР (200 МГц, CDCl3) млн-1 8.60 (s, 2H), 7.68-7.75 (m, 2H), 7.58-7.59 (m, 1 Н), 7.27-7.29 (d, 1 Н),7.12-7.24 (m, 2 Н), 6.98-7.04 (m, 1 Н), 6.73-6.78 (dd, 1 Н), 6.67-6.68 (d, 1 Н), 6.60-7.35 (t, 1 Н, CHF2), 5.99-6.06[]D = +52,8 (с = 0,5; МеОН). Пример 10. 1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир (+)-2-(6-метоксинафталин-2-ил)пропионовой кислоты (элюируемый первым диастереомер) (11). Соединение выделяли из диастереомерной смеси примера 8 посредством HPLC-разделения, используя колонку Daisogel 10 мкм, 50300 мм; элюент: н-гексан/метил-трет-бутиловый эфир/изопропиловый спирт: 90/9,9/0,1; скорость потока: 80 мл/мин; загрузка: 300 мг на одно введение; время элюирования: от 7 до 10 мин. Собранные фракции упаривали и остаток кристаллизовали из смеси н- 11019113[]D = +45 (с = 0,5; МеОН). Пример 11. (+)-1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил) этанол (14). К суспензии 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этилового эфира (+)-2-(6-метоксинафталин-2-ил)пропионовой кислоты (13) (14,0 г) в метаноле (110 мл) добавляли трет-бутилат калия (5,1 г). Полученную смесь перемешивали при к.т. в течение 2 ч, получая прозрачный раствор. Медленно, при перемешивании, добавляли воду до начала выпадения осадка (мутный раствор). После перемешивания в течение следующих 60 мин выпавшее в осадок твердое вещество отфильтровывали, промывали водой и растворяли в хлороформе (100 мл). Раствор сушили над сульфатом натрия и растворитель удаляли под вакуумом. Остаток кристаллизовали в смеси хлороформ/гексан (1/2,5), получая 8,1 г белого твердого вещества. 1 Н ЯМР (200 МГц, CDCl3) млн-18.45 (s, 2H), 7.19-7.08 (d, 1H), 7.06-7.00 (d, 1H), 6.95-6.85 (dd, 1H),6.99-6.24 (t, 1H, CHF2), 5.18-5.00 (m, 1H), 3.98-3.78 (m, 2H), 3.54-3.35 (m, 1H), 3.31-3.15 (m, 1H), 2.04-1.94(-)-1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4 ил)этанол (12). Исходя из диастереомера (11), следуя процедуре примера 10, получали спирт (12).[]D = -9,15 (c = 1, CHCl3). Пример 13. Получение спирта (7) путем окисления спирта (14). Соединение (14) (3,0 г) растворяли в CH2Cl2 (100 мл). Добавляли 70%-ную м-хлор-пербензойную кислоту (5,4 г) и полученный раствор перемешивали при комнатной температуре в течение 18 ч. Затем добавляли твердый Na2S2O3 (5 г) и смесь интенсивно перемешивали при к.т. в течение 30 мин. Твердый остаток отфильтровывали; органический раствор разбавляли дополнительными 100 мл CH2Cl2 и промывали водным насыщенным раствором NaHCO3 (3100 мл). Органическую фазу сушили над Na2SO4 и растворитель выпаривали. Остаток растирали в EtOAc (20 мл), получая 1,9 г желаемого продукта 7 в виде белого твердого вещества, которое использовали на следующей стадии без дополнительной очистки. 1 Н ЯМР (200 МГц, CDCl3) млн-1 8.14 (s, 2H), 7.18-7.09 (d, 1H), 7.07-7.02 (d, 1H), 6.92-6.83 (dd, 1H),7.01-6.22 (t, 1H, CHF2), 5.10-4.96 (m, 1H), 3.96-3.84 (d, 2H), 3.45-3.29 (m, 1H), 3.23-3.07 (m, 1H), 3.24-3.17[]D = +65,0 (с = 0,5; МеОН). Пример 14. Получение спирта (9) путем окисления спирта (12). Спирт (9) можно получить, следуя процедуре, описанной в примере 13, с использованием спирта(14) (2,0 г), 4-диметиламинопиридина (0,3 г), 3-циклопропилметокси-4-(N-трет-бутоксикарбонил-Nметансульфонил)аминобензойной кислоты (2,0 г) в безводном CH2Cl2 (180 мл) при к.т. в атмосфере азота. После перемешивания при к.т. в течение ночи смесь промывали 5%-ным водным HCl (2100 мл); органическую фазу отделяли и промывали насыщенным водным раствором NaHCO3 (2100 мл), сушили над Na2SO4 и упаривали досуха. Неочищенное вещество очищали флэш-хроматографией на силикагеле при градиентном элюировании (гексан/EtOAc от 10/1 до 6/4), что позволило получить 1,4 г указанного в заголовке соединения. Стадия 2. Получение С 1. 1-(3-Циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир 3 циклопропилметокси-4-(N-трет-бутоксикарбонил-N-метансульфонил)аминобензойной кислоты (1,4 г) растворяли в CH2Cl2 (140 мл). Добавляли 4 М раствор HCl в диоксане (40 мл) и полученную смесь перемешивали при к.т. в течение 24 ч. Затем реакционную смесь упаривали досуха и остаток растирали вiPrOH (50 мл) и после этого в EtOH (50 мл), затем используя Et2O (70 мл), что позволило получить 0,880 г соединения (С 1). Аналитическая характеристика С 1 приведена в табл. 1. Таблица 1 Аналогично могут быть получены следующие соединения: 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир (-)4-циклопропилметокси-3-метансульфониламинобензойной кислоты,1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир (-)3,4-бис-метансульфониламинобензойной кислоты,1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлорпиридин-4-ил)этиловый эфир (-)3-метансульфониламино-4-метилбензойной кислоты и 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлор-1-оксипиридин-4-ил)этиловый эфир (-)-4-метансульфониламино-3-метилбензойной кислоты. Пример 16. Получение 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлор-1-оксипиридин-4-ил)этилового эфира (-)-3-циклопропилметокси-4-метансульфониламинобензойной кислоты(С 2). Соединение (С 2) получали в соответствии с той же методикой синтеза, что и в примере 15, исходя из промежуточного спирта (7). Альтернативно, соединение (С 2) может быть получено, исходя из соединения (С 1), как описано в следующем далее примере 17. Пример 17. Получение 1-(3-циклопропилметокси-4-дифторметоксифенил)-2-(3,5-дихлор-1-оксипиридин-4-ил)этилового эфира (-)-3-циклопропилметокси-4-метансульфониламинобензойной кислоты(С 2), исходя из соединения (С 1). Соединение (С 1) (0,69 г) растворяли в CH2Cl2 (20 мл). Добавляли 70%-ную м-хлор-пербензойную кислоту (0,355 г) и полученный раствор перемешивали при комнатной температуре в течение 18 ч. Затем добавляли твердый Na2S2O3 (0,244 г) и смесь интенсивно перемешивали при к.т. в течение 30 мин. Твердый остаток отфильтровывали; органический раствор разбавляли дополнительными 20 мл CH2Cl2 и промывали водным насыщенным раствором NaHCO3 (320 мл). Органическую фазу сушили над Na2SO4 и растворитель выпаривали. Остаток растирали в EtOH (20 мл), получая 0,710 г желаемого соединения(С 2) в виде белого твердого вещества. Приведенные далее соединения получали, следуя тому же пути и используя подходящие реагенты. Промежуточные карбоновые кислоты, использованные в синтезе описанных конечных соединений,имеются в продаже или уже известны или их синтезируют в соответствии с известными способами. Пример 18. Синтез 3-циклопропилметокси-4-(N-трет-бутоксикарбонил-N-метансульфонил)аминобензойной кислоты.H2SO4 (2 мл) и смесь нагревали до 60 С в течение 18 ч. Реакционную смесь концентрировали до приблизительно 200 мл, разбавляли EtOAc (200 мл) и промывали водным насыщенным раствором NaHCO3 (220 мл). Органический слой сушили над Na2SO4 и растворитель выпаривали, получая 10,5 г желаемого промежуточного соединения. Стадия 2. Метиловый эфир 3-ииклопропилметокси-4-нитробензойной кислоты. Метиловый эфир 3-гидрокси-4-нитробензойной кислоты (10,5 г) растворяли в безводном DMF (150 мл) в атмосфере N2. Добавляли K2CO3 (24,3 г), KI (2,6 г) и циклопропилметилбромид (10,3 мл) и смесь перемешивали при 50 С в течение 6 ч. Реакционную смесь разбавляли водой (300 мл) и экстрагировалиEt2O (2200 мл); объединенные органические слои сушили над Na2SO4 и растворитель выпаривали, получая 12,7 г желаемого промежуточного соединения. Стадия 3. Метиловый эфир 4-амино-3-ииклопропилметоксибензойной кислоты. Метиловый эфир 3-циклопропилметокси-4-нитробензойной кислоты (12,7 г) растворяли в МеОН(10 мл) и гидрирование продолжали в течение дополнительных 2 чн до полного превращения. Катализатор отфильтровывали через набивку целита, смесь разбавляли EtOAc (200 мл) и промывали водным насыщенным раствором NaHCO3 (2100 мл). Органический слой сушили над Na2SO4 и растворитель выпаривали, получая 10,7 г желаемого промежуточного соединения. Стадия 4. Метиловый эфир 3-циклопропилметокси-4-метансульфониламинобензойной кислоты. Метил-3-(циклопропилметокси)-4-аминобензоат (8,86 г) растворяли в пиридине (80 мл) при комнатной температуре в атмосфере N2. Добавляли метансульфонилхлорид (4,04 мл) и смесь перемешивали при к.т. в течение 18 часов. Реакционную смесь упаривали досуха, неочищенное вещество обрабатывали 1 н. HCl (500 мл) и экстрагировали CH2Cl2 (3200 мл). Органический слой сушили над Na2SO4 и растворитель выпаривали, получая 11,7 г желаемого промежуточного соединения.MS/ESI+ 300 [МН]+. Стадия 5. Метиловый эфир 3-циклопропилметокси-4-(N-трет-бутоксикарбонил-N-метансульфонил)аминобензойной кислоты. Метиловый эфир 3-циклопропилметокси-4-метансульфониламинобензойной кислоты (3,0 г) растворяли в CH2Cl2 (150 мл). Добавляли диметиламинопиридин (DMAP; 1,22 г) и Boc2O (2,18 г) и смесь перемешивали при к.т. в течение 1 ч. Реакционную смесь промывали 5%-ным водным HCl (250 мл),органический слой сушили над Na2SO4 и растворитель выпаривали. Остаток растирали в Et2O и отфильтровывали, что позволило получить 4,0 г желаемого промежуточного соединения, которое использовали на следующих стадиях без дополнительной очистки. Стадия 6. 3-Циклопропилметокси-4-(N-трет-бутоксикарбонил-N-метансульфонил)аминобензойная кислота. Метиловый эфир циклопропилметокси-4-(N-трет-бутоксикарбонил-N-метансульфонил)аминобензойной кислоты (4,0 г) растворяли в МеОН (100 мл). Добавляли 1 н. NaOH (15 мл) и полученную смесь перемешивали при к.т. в течение 1 ч, затем нагревали до 50 С в течение 2 ч. Реакционную смесь далее разбавляли EtOAc (250 мл) и промывали 1 н. HCl (2100 мл). Органический слой сушили над Na2SO4 и растворитель выпаривали, получая 3,5 г желаемого производного кислоты.t = триплет,q = квартет,dd = дублет дублетов,m = мультиплет,br = уширенный,ESI = ионизация электрораспылением. Фармакологическая активность соединений по изобретению Пример 19. Определение PDE4-ингибирующей активности in vitro в анализе с мононуклеарными клетками периферической крови (РВМС). Анализ, основанный на известной ингибирующей активности, проявляемой ингибиторами PDE4 в отношении индуцируемого липополисахаридами (LPS) высвобождения фактора некроза опухоли альфа(TNF-) мононуклеарными клетками периферической крови (РВМС, проводят в соответствии с ранее описанным способом (Hatzelmann A. et al. J. Pharmacol. Exp. Ther. 2001, 297: 267-279; Draheim R. et al. J.Pharmacol. Exp. Ther. 2004, 308: 555-563). Криоконсервированные РВМС человека (100 мкл/лунка) инкубируют в 96-луночных планшетах(105 клеток/лунка) в течение 30 мин в присутствии или в отсутствие (50 мкл) тестируемых соединений,концентрация которых находится в диапазоне от 10-12 М до 10-6 М. Затем добавляют LPS (3 нг/мл). Через 18 ч инкубации при 37 С в инкубаторе с увлажнением в атмосфере 95% воздуха и 5% СО 2 собирают культуральную среду и TNF- измеряют с использованием ELISA (иммуноферментный твердофазный анализ). Результаты, относящиеся к соединениям С 1-С 6, выраженные в виде среднего значения 95% доверительный интервал для молярной концентрации тестируемого соединения, вызывающей 50%-ное ингибирование LPS-индуцируемого высвобождения TNF- (IC50), находятся в диапазоне от 0,06 до 4,4 нМ. Эффекты тестируемых соединений рассчитывают в виде процента ингибирования высвобождения TNF, принимая LPS-индуцируемое продуцирование TNF- в отсутствие ингибирующего соединения за 100%, а базисное продуцирование TNF- РВМС в отсутствие LPS за 0%. Пример 20. Оценка способности ингибировать низкоаффинную LPDE4 в сравнении со способностью конкурировать за высокоаффинную HPDE4. Аффинность к LPDE4 и HPDE4 оценивают, как описано ранее в Cortijo J. et al. Br. J. Pharmacol. 1993, 108: 562-568 и Duplantier A.J. et al. J. Med. Chem. 1996, 39: 120-125, соответственно. Концентрация тестируемого соединения находится в диапазоне от 10-12 до 10-5 М. Значения аффинностей к LPDE4 и HPDE4, протестированные на соединениях С 1-С 6, находятся в диапазоне от 82 до 477. В случае LPDE4 IC50 представляет собой молярную концентрацию тестируемого соединения, вызывающую 50%-ное ингибирование исчезновения цАМФ, тогда как в случае HPDE4 IC50 представляет собой молярную концентрацию тестируемого соединения, вызывающую 50%-ное ингибирование связывания [Н 3]-ролипрама. Результаты указывают на то, что соединения по изобретению ингибируют LPDE4 с субнаномолярной аффинностью и значительно более селективны к LPDE4 по сравнению с HPDE4. Пример 21. Синергическая активность комбинации с фиксированной дозой кармотерола/С 1 на индуцированное карбахолом сокращение трахеи морских свинок. Зигзагообразные сегменты трахеи получают от самцов морских свинок, чувствительных к овальбумину (ОА), и из трахеи получают два препарата. Каждый препарат помещают в ванну для органов (20 мл), заполненную насыщенным кислородом (95% O2 и 5% СО 2) нормальным раствором КребсаХенселейта, и сохраняют при 37 С. Препараты трахей присоединяют к изометрическим динамометрическим датчикам при тонусе покоя 1 г. После периода уравновешивания в течение 60 мин препараты трахей предварительно обрабатывают в течение 30 мин, используя С 1 (10-7 М), кармотерол (310-10 М), совместно С 1 и кармотерол или разбавитель, соответственно, с последующим суммарным введением ОА(10-10 - 10-5 г/мл). По окончании введения ОА добавляют карбахол в максимальной концентрации (10-5 М) для получения максимального сокращения каждого препарата. Эффекты выражают в процентных величинах от индуцированного карбахолом максимального ответа (100%). 30-Минутная предварительная обработка препарата с использованием С 1 (10-7 М) вызывала ингибирование индуцированного ОА сокращения на 23%. Аналогичным образом, ингибирование, вызываемое кармотеролом (310-10 М), составляет 18%. Комбинация С 1 (10-7 М) и кармотерола (310-10 М) вызывала уменьшение индуцированного ОА сокращения на 93%. Результаты этого исследования показывают, что как кармотерол, так и С 1 эффективно антагонизируют индуцированное карбахолом сокращение дыхательных путей морской свинки. Более того, в соответствии с их взаимодополняющим молекулярным механизмом действия, в рамках функционального агонизма-антагонизма, фиксированные комбинации демострируют синергический эффект в отношении контроля холинергического сокращения мышц трахеи (trachealis) морской свинки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение общей формулы (I) в виде (-)-энантиомераR1 и R2 могут быть одинаковыми или разными и выбраны из группы, состоящей из линейного или разветвленного C1-С 6 алкила, возможно замещенного одним или более чем одним атомом галогена;OR3, где R3 представляет собой линейный или разветвленный C1-С 6 алкил, возможно замещенный одним или более чем одним атомом галогена либо одной или более чем одной С 3-С 7 циклоалкильной группой; иHNSO2R4, где R4 представляет собой линейный или разветвленный С 1-С 4 алкил, возможно замещенный одним или более чем одним атомом галогена,где по меньшей мере один из R1 и R2 представляет собой HNSO2R4; или его фармацевтически приемлемая неорганическая или органическая соль, гидрат или сольват. 2. Соединение по п.1, где R1 представляет собой HNSO2R4, где R4 представляет собой метил, R2 представляет собой OR3, где R3 представляет собой циклопропилметил, и n равно 0. 3. Соединение по п.1, где R1 представляет собой HNSO2R4, где R4 представляет собой метил, R2 представляет собой OR3, где R3 представляет собой циклопропилметил, и n равно 1. 4. Соединение по п.1, где R1 представляет собой OR3, R2 представляет собой HNSO2R4, где R4 представляет собой метил, и n равно 1. 5. Соединение по п.1, где R1 представляет собой метил, R2 представляет собой HNSO2R4, где R4 представляет собой метил, и n равно 1. 6. Соединение по п.1, где оба R1 и R2 представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 0. 7. Соединение по п.1, где оба R1 и R2 представляют собой HNSO2R4, где R4 представляет собой метил, и n равно 1. 8. Способ получения соединения по любому из пп.1-7, включающий стадию взаимодействия альдегида (1) с получением рацемического спирта (3) который возможно окисляют до соответствующего N-оксидного производного (4) разделение диастереомерной смеси (10) или (5) на два индивидуальных диастереомера, соответственно (11) путем хроматографии или кристаллизации с получением после расщепления (+)-спирта (14) и затем приведение во взаимодействие соединения (+) (14) или (+) (7) с подходящей бензойной кислотой с получением соединений общей формулы (I), где R1 и R2 такие, как определено в п.1. 9. Соединение общей формулы (II) где n равно 0 или 1, а хиральный атом углерода, отмеченный , имеет (S)-конфигурацию. 10. Применение соединения формулы (I) по любому из пп.1-7 в комбинации со вторым фармацевтическим активным компонентом, выбранным из классов 2-агонистов, М 3-антагонистов или кортикостероидов. 11. Применение по п.10, где второй активный компонент представляет собой формотерол или кармотерол. 12. Фармацевтическая композиция, обладающая активностью ингибитора фосфодиэстеразы 4(PDE4), содержащая соединение формулы (I) по любому из пп.1-7 и один или более чем один фармацевтически приемлемый носитель и/или эксципиент. 13. Фармацевтическая композиция по п.12, дополнительно содержащая второй фармацевтически активный компонент, выбранный из классов 2-агонистов, М 3-антагонистов или кортикостероидов. 14. Применение соединения формулы (I) по любому из пп.1-7 в качестве лекарственного средства,обладающего активностью ингибитора PDE4. 15. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения заболевания дыхательных путей, характеризующегося обструкцией дыхательных путей. 16. Применение по п.15, где указанное заболевание представляет собой астму или COPD (хроническую обструктивную болезнь легких). 17. Устройство, представляющее собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist), содержащее фармацевтическую композицию по п.12 или 13. 18. Набор, содержащий фармацевтическую композицию по п.12 или 13 и устройство, которое представляет собой сухой порошковый ингалятор для однократного или многократного приема, дозирующий ингалятор или небулайзер мягкого аэрозоля (soft mist). 19. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения аллергического ринита. 20. Применение соединения формулы (I) по любому из пп.1-7 для предупреждения и/или лечения атопического дерматита.
МПК / Метки
МПК: C07D 213/61, A61K 31/44, A61P 11/00, C07D 213/89
Метки: фосфодиэстеразы, 1-фенил-2-пиридин-4-ил)этиловые, эфиры, кислоты, бензойной, pde4, ингибиторов, качестве
Код ссылки
<a href="https://eas.patents.su/21-19113-1-fenil-2-piridin-4-iletilovye-efiry-benzojjnojj-kisloty-v-kachestve-ingibitorov-fosfodiesterazy-4-pde4.html" rel="bookmark" title="База патентов Евразийского Союза">(1-фенил-2-пиридин-4-ил)этиловые эфиры бензойной кислоты в качестве ингибиторов фосфодиэстеразы 4 (pde4)</a>
Соединения, эффективные в качестве агонистов &beta- 2 – адренорецептора и ингибиторов фосфодиэстеразы pde4

Номер патента: 5824
Опубликовано: 30.06.2005
Авторы: Брундель Паулус, Стерк Герт, Кристианс Йоханнес, Ван-Дер-Лан Ивонне, Эльтце Манфрид, Хатцельманн Армин, Бундшу Даниела, Тиммерманн Хендрик
МПК: C07D 237/14, A61P 11/06, A61K 31/50...
Метки: ингибиторов, соединения, адренорецептора, агонистов, beta, качестве, эффективные, фосфодиэстеразы
Формула / Реферат:
1. Соединения формулы I где Ar1означает фенильный радикал формулы (Ia), замещенный R1 и R2, или радикал дигидробензофуранил формулы (Ib), замещенный R3, R4 и R5 где R1 означает C1-C4алкокси или C1-C4алкокси, который полностью или преимущественно замещен фтором, R2 означает C1-C8алкокси или C1-C4алкокси, который полностью или преимущественно замещен фтором, R3 означает C1-C4алкокси, R4 и R5 вместе, включая два атома углерода, к которым...
Производные пирролидина в качестве ингибиторов фосфодиэстеразы, специфичной к циклическому амф

Номер патента: 5686
Опубликовано: 28.04.2005
Авторы: Одинго Джошуа, Шлахтер Стивен, Бэрджесс Лоренс И., Мартинс Тимоти Дж., Годино Джон Дж., Фаулер Керри У., Кесицки Эдвард А., Джоунс Закари С., Ньюхаус Брэдли Дж., Оливер Эйми
МПК: C07D 295/20, A61K 31/40
Метки: специфичной, качестве, фосфодиэстеразы, пирролидина, амф, ингибиторов, циклическому, производные
Формула / Реферат:
1. Соединение, имеющее формулу где R1 выбран из группы, включающей водород, C1-6алкил, мостиковый алкил, выбранный из группы, включающей норборнил, адамантил, бицикло[2.2.2]октил, бицикло[3.2.1]гептил, бицикло[3.2.1]октил, бицикло[4.1.0]гептил, бицикло[3.1.0]гексил и декагидронафтил, замещенный или незамещенный арил, выбранный из группы включающей фенил, нафтил, бифенил, тетрагидронафтил, инданил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил,...
Тетрагидротиопиранфталазиноновые производные в качестве ингибиторов pde4

Номер патента: 5856
Опубликовано: 30.06.2005
Авторы: Бундшу Данила, Клей Ханс-Петер, Стерк Ян Герт, Тиммерманн Хендрик, Ван-Дер-Лан Ивонне Йоханна, Хатцельманн Армин
МПК: C07D 409/04, A61K 31/502, A61P 11/06...
Метки: качестве, производные, тетрагидротиопиранфталазиноновые, ингибиторов
Формула / Реферат:
1. Соединения формулы I где R1 и R2 оба обозначают водород или вместе образуют дополнительную связь, A обозначает S (серу), S(O) (сульфоксид) или S(O)2 (сульфон), Ar обозначает бензольное производное формулы (a) или (b) где R3 обозначает галоген, C1-C4алкокси или C1-C4алкокси, более половины или все атомы водорода которого замещены фтором, R4 обозначает галоген, C1-C8алкокси, C3-C7циклоалкокси, C3-C7циклоалкилметокси или C1-C4алкокси, более...
Производные пиразоло[3,4-в]пиридина в качестве ингибиторов фосфодиэстеразы

Номер патента: 18670
Опубликовано: 30.09.2013
Авторы: Кайрнар Винайяк Васантрао, Рай Абиджит, Рамайя Мандадапу Рагу, Барегама Лалит Кумар, Салла Манохар, Виджайкришнан Лалита, Рудра Сонали, Балачандран Сарала, Агарвал Риту, Палле Венката П., Гупта Ниди, Дастидар Сунанда Г., Кондаскар Атул
МПК: A61P 11/00, A61K 31/437, A61P 17/00...
Метки: фосфодиэстеразы, производные, качестве, пиразоло[3,4-в]пиридина, ингибиторов
Формула / Реферат:
1. Соединение, имеющее структуру формулы Iили его фармацевтически приемлемые соли, гдеR1 и R2 независимо представляют собой водород, С6-10арил, моноциклический гетероарил, содержащий 5 или 6 кольцевых атомов, либо бициклический или трициклический гетероарил, имеющий 8-10 атомов в цикле, с гетероатомами, выбранными из N, О или S, -COR4, -S(O)mR4 (где R4 представляет собой водород, С1-6алкил, С3-8циклоалкил, С6-10арил, аралкил (где арил...
Производные фталазина в качестве ингибиторов фосфодиэстеразы 4

Номер патента: 3702
Опубликовано: 28.08.2003
Авторы: Гранчини Джанкарло, Наполетано Мауро, Пеллачини Франко, Норчини Габриеле, Мораццони Габриеле
МПК: C07D 237/30, A61K 31/502
Метки: фталазина, качестве, ингибиторов, производные, фосфодиэстеразы
Формула / Реферат:
1. Соединение формулы I где ---- представляет одинарную или двойную связь; B представляет метилен; A представляет пиридин, замещенный одним заместителем или большим количеством заместителей; R представляет два атома водорода или C = O группу, когда ---- представляет одинарную связь, или, когда ---- представляет двойную связь, R представляет водород, необязательно замещенный арил или гетероцикл, (C1-8)алкил, (C2-8)алкенил или (C2-8)алкинил,...
Предыдущий патент: Гигроскопическая прокладка
Следующий патент: Новая кристаллическая форма 17α-ацетокси-21-метокси-11β-[4-n,n-диметиламинофенил]-19-норпрегна-4,9-диен-3,20-диона и способ ее получения
Случайный патент: Изображающий гиперспектрометр