Контролируемый синтез зипразидона и его композиции
Номер патента: 7866
Опубликовано: 27.02.2007
Авторы: Хауард Гарри Ральф Млад., Гроубин Адам Уорт, Лиман Кайл Роберт, Буш Фрэнк Роберт



















Формула / Реферат
- формула изобретения, действующая на территории Договаривающихся государств, для которых ниже не указана особая редакция формулы
1. Композиция, содержащая зипразидон и количество дехлорзипразидона, выбранное из количества, не превышающего приблизительно 1000 миллион-1 (млн-1), не превышающего приблизительно 500 млн-1 и не превышающего приблизительно 100 млн-1.
2. Композиция по п.1, где зипразидон представляет собой зипразидон в форме свободного основания, моногидрат гидрохлорида зипразидона, дигидрат мезилата зипразидона или тригидрат мезилата зипразидона.
3. Фармацевтическая композиция для лечения у млекопитающего расстройства или состояния, выбранного из шизофрении, тревоги, боли при мигрени, синдрома Туретта, глаукомы, ишемической ретинопатии, деменции альцгеймеровского типа, биполярного расстройства, расстройства настроения, агорафобии, социальной фобии, панического расстройства, посттравматического стрессового расстройства, острого стрессового расстройства, тревожного расстройства, вызванного веществами, неуточненных (NOS) тревожных расстройств, дискинезий, поведенческого проявления задержки психического развития, расстройства поведения и аутистического расстройства, содержащая количество композиции по п.1, эффективное для лечения указанного расстройства или состояния, и фармацевтически приемлемый носитель.
4. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, выбранное из количества, составляющего не более чем приблизительно
A) 1000 млн-1,
Б) не более чем приблизительно 500 млн-1, и
B) не более чем приблизительно 100 млн-1, при котором:
а) получают один или более чем один образец одной или более чем одной партии 6-хлор-1,3-дигидро-2H-индол-2-она;
б) определяют уровень примеси оксиндола в каждом образце со стадии (а);
в) выбирают партию 6-хлор-1,3-дигидро-2H-индол-2-она, которая содержит оксиндол на уровне
для (А) - не выше приблизительно 0,3%, на основании определения или определений, проведенных на стадии (б),
для (Б) - не выше приблизительно 0,15%, на основании определения или определений, проведенных на стадии (б), и
для (В) - не выше приблизительно 0,03%, на основании определения или определений, проведенных на стадии (б), и
г) используют партию, выбранную на стадии (в), для синтеза указанной композиции зипразидона.
5. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более чем приблизительно 1000 млн-1, при котором:
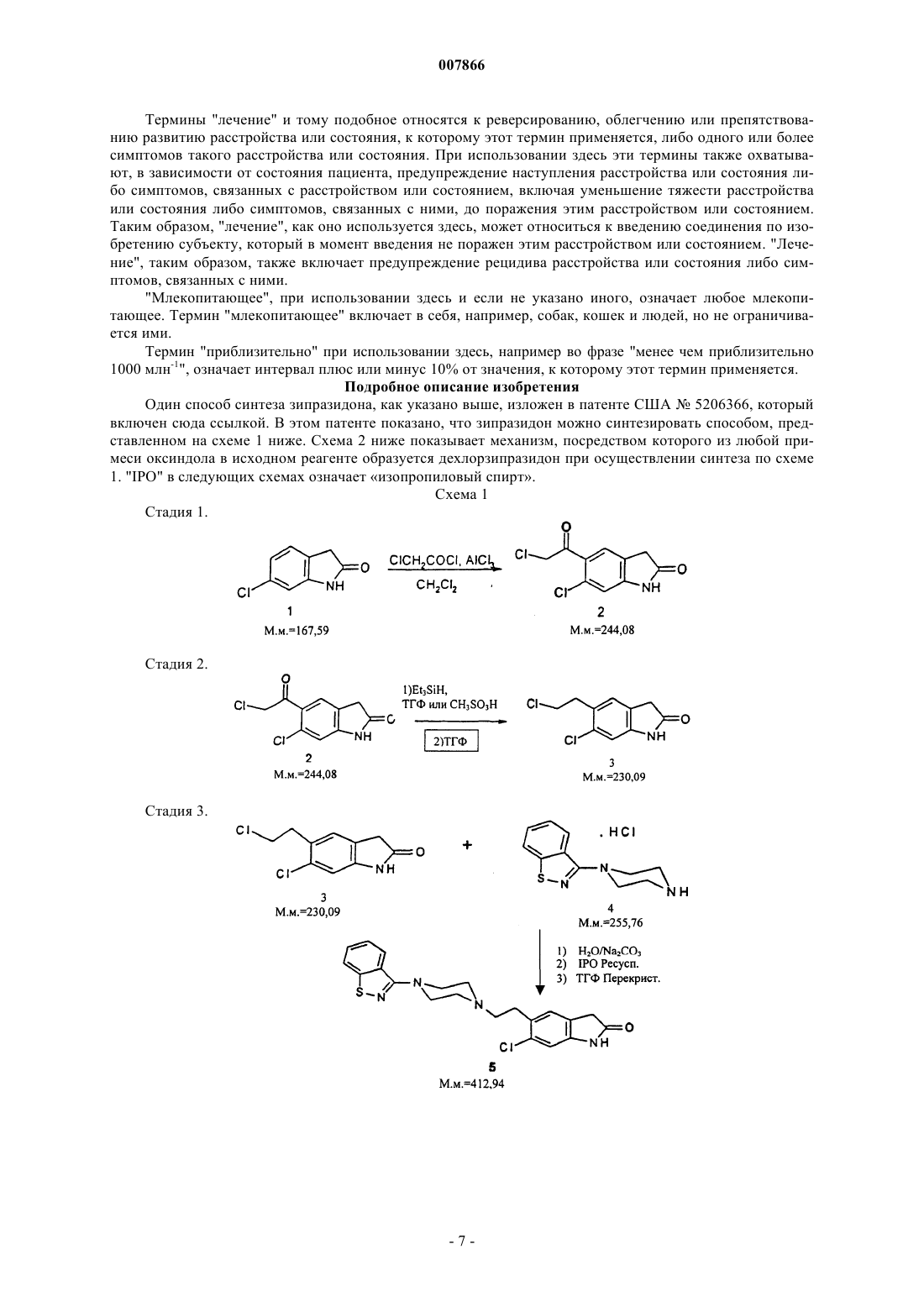
а) осуществляют ацилирование композиции, содержащей 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, хлорангидридом хлоруксусной кислоты по методу ацилирования Фриделя-Крафтса, синтезируя композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он;
б) обрабатывают композицию, полученную на стадии (а), восстанавливая в содержащейся в ней хлорацетильной группе оксо с образованием композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она;
в) выделяют образец композиции, полученной на стадии (б);
г) определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в образце, выделенном на стадии (в);
д) определяют, превышает ли или не превышает количество, определенное на стадии (г), приблизительно 0,28%; и
е) если количество, определенное на стадии (г), превышает приблизительно 0,28%, то композицию, полученную на стадии (б), очищают путем перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%, и из очищенной таким образом композиции синтезируют композицию зипразидона; или
ж) если количество, определенное на стадии (г), не превышает приблизительно 0,28%, синтезируют композицию зипразидона из композиции, полученной на стадии (б).
6. Способ, в котором применяется высокоэффективная жидкостная хроматография (ВЭЖХ) для определения количества 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, при котором:
а) готовят раствор образца из указанной композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2Н-индол-2-он, путем растворения части указанной композиции в органическом растворителе с последующим разбавлением органическим растворителем растворенной части таким образом, что получают концентрацию (мас./об.), составляющую, исходя из массы указанной части и объема растворителя, приблизительно 1 мг/мл;
б) пропускают раствор образца через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую, по существу, из смеси 0,05М KН2РО4 с рН=5,5-6,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.) при температуре колонки от приблизительно 30 до 40шС с детекцией в УФ-свете при 254 нм УФ;
в) детектируют пик, появляющийся на хроматограмме, полученной на стадии (б), между 8 и 10 мин;
г) определяют площадь пика (обозначаемую Ас), детектированного на стадии (в);
д) готовят стандарт из композиции, состоящей, по существу, из 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, путем растворения и разбавления части указанной композиции в органическом растворителе таким образом, чтобы концентрация 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она (мас./об.) приблизительно была равна, исходя из массы части и объема растворителя, выбранному значению содержания, при котором или выше которого требуется детекция 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он;
е) пропускают стандарт через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую, по существу, из смеси 0,05М KН2РО4 с рН =5,5-6,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.), при температуре колонки от приблизительно 30 до 40шС с детекцией в УФ-свете при 254 нм УФ;
ж) определяют площадь пика (обозначаемую Apur1) пика на хроматограмме, полученной на стадии (е); и
з) рассчитывают количество 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, для чего:
1) рассчитывают коэффициент отклика для 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она по следующей формуле:
Rpur1=(Apur1)(DF)/(Wpur1)(PF)
где Apur1 такое, как определено выше,
Wpur1 = масса композиции в стандарте,
PF = коэффициент эффективности 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она и
DF = коэффициент разбавления стандарта; и
2) рассчитывают % мас./мас. 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она по следующей формуле:
% мac./мac.=(Ac)(DF)(100)/(Rpur1)(WS2)
где Ас такое, как определено выше;
Rpur1 = коэффициент отклика, рассчитанный в (з)(1) выше;
WS2 = масса части композиции, используемой на стадии (а); и
DF = коэффициент разбавления раствора образца.
7. Способ синтеза композиции зипразидона, которая содержит количество дехлорзипразидона не более приблизительно 1000 млн-1, при котором:
а) восстанавливают композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он и примесь 5-(хлорацетил)-1,3-дигидро-2H-индол-2-она, путем обработки триэтилсиланом в присутствии сильной кислоты с получением композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; и
б) синтезируют композицию, содержащую зипразидон, из композиции, полученной на стадии (а).
8. Способ по п.7, при котором дополнительно:
1) выделяют до стадии (б) образец композиции, полученной на стадии (а), и в указанном образце определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2Н-индол-2-она;
2) определяют, превышает ли количество, определенное в (1), приблизительно 0,28% или не превышает; и
3) если количество, определенное в (1), превышает приблизительно 0,28%, то композицию, полученную на стадии (а), очищают путхь перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%; и затем переходят к осуществлению стадии (б), используя композицию, полученную на стадии (а), очищенную таким образом; или
4) если количество в (1) не превышает приблизительно 0,28%, тогда переходят к осуществлению стадии (б).
9. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающего количества, выбранного из
A) приблизительно 1000 млн-1,
Б) приблизительно 500 млн-1 и
B) приблизительно 100 млн-1,
при котором:
а) очищают композицию, содержащую 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, до получения композиции, содержащей количество указанной примеси оксиндола, составляющее
для (А) - приблизительно 0,3%,
для (Б) - приблизительно 0,15% и
для (В) - приблизительно 0,03%; и
б) используют композицию, полученную на стадии (а), для синтеза композиции зипразидона.
10. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающее приблизительно
A) 1000 млн-1,
Б) 500 млн-1 или
B) 100 млн-1,
при котором
а) осуществляют перекристаллизацию и/или ресуспендирование композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, до получения композиции, содержащей не более чем приблизительно
для (А) - 0,3%,
для (Б) - 0,15% и
для (В) - 0,03%,
указанной примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; и
б) используют композицию, полученную на стадии (а), для синтеза композиции зипразидона.
Текст
007866 Предшествующий уровень техники Предпринимаются усилия для получения лекарственных продуктов высокого качества и содержащих минимальное количество примесей. Контроль примесей требует изучения различных вариантов решений относительно условий реакции и протоколов проверки для гарантии того, что лекарственные средства, которые будут вводить людям, являются чистыми. В руководстве, изданном регулирующими органами, в том числе Управлением по контролю за продуктами и лекарствами США (FDA), предлагается примеси в лекарственных средствах, если они присутствуют, идентифицировать, если они присутствуют на уровне 0,1% (т.е. 1000 млн-1) или более в лекарственных веществах, принимаемых в дозировке 2 г/сут. или ниже. (Учитываем, что млн-1 означает миллионные доли, так что 1%=10000 млн-1; 0,1%=1000 млн-1; 0,01%=100 млн-1 и 0,001%=10 млн-1). Например,FDA показало, что идентификация примесей ниже различимого уровня, составляющего 0,1%, для лекарственного вещества, принимаемого в дозировке 2 г/сут., как правило, не считается необходимой (FederalRegister Vol. 65,140, pp. 45085-45090, 45086 и 45089 (July 20, 2000. Однако FDA также указывает,что для некоторых примесей, в зависимости от их специфичных свойств, может быть необходимым более строгий контроль (Id. на 45086). Кроме того, рекомендуется проводить исследования для получения информации о безопасности предполагаемого количества примеси, если это предполагаемое количество квалификационный порог, составляющий 0,05% (500 млн-1 для лекарственного вещества, принимаемого в дозировке 2 г/сут. или менее (Id. на 45087 и 45089. Зипразидон (5-(2-(4-1,2-бензизотиазол-3-ил-1-пиперазинил)этил)-6-хлор-1,3-дигидро-2-(1 Н)-индол 2-он) является сильным антипсихотическим агентом и используется для лечения различных расстройств,включая шизофрению, тревожность и боль при мигрени. Зипразидон одобрен FDA для лечения шизофрении и продается в США под торговой маркой Geodon. Также было показано, что зипразидон пригоден при лечении синдрома Туретта (патент США 6127373), глаукомы и ишемической ретинопатии (ЕР 985414 А 2), а также психиатрических состояний, включая деменцию альцгеймеровского типа, биполярные нарушения, расстройства настроения, панические расстройства, агорафобию, социальную фобию,паническое расстройство, посттравматическое стрессовое расстройство, острое стрессовое расстройство,тревожное расстройство, вызванное веществами, неуточненные тревожные расстройства, дискинезии и поведенческие проявления задержки психического развития, расстройство поведения и аутистическое расстройство (патент США 6245766). Указанные выше патенты США и заявка на европейский патент включены в данное описание изобретения ссылкой во всей полноте. В патенте США 4831031 описан класс соединений, включающий зипразидон, и синтез таких соединений. Другой способ синтеза зипразидона описан в патенте США 5206366. Способ синтеза конкретно зипразидона гидрохлорида моногидрата описан в патенте США 5312925. Способ синтеза зипразидона мезилата дигидрата описан в патенте США 6245765; и способ синтеза зипразидона мезилата тригидрата описан в патенте США 6110918. В патентах США 5338846; 5359068 и 6111105 также описаны способы синтеза зипразидона и/или промежуточных соединений для него. Указанные выше патенты США включены в данное описание изобретения ссылкой во всей полноте. Структура зипразидона может быть изображена следующим образом:(Н. Howard et al., "Ziprasidone Hydrochloride", Drugs of the Future 1994, 19(6): 560-563). Как видно из представленной выше структуры, соединение зипразидон содержит один атом хлора. Способы введения галогенов в органические соединения обобщены во многих руководствах по органической химии. Например, в J. March, Advanced Organic Chemistry, 4th Edition, pp. 587-591 и указанных в этом руководстве ссылках рассматривается химизм галогенирования. Более конкретно, образование хлорароматических соединений часто осуществляют рядом способов, также хорошо известных специалистам в данной области и также рассматриваемых в J. March, Advanced Organic Chemistry, 4th Edition.Chapter 11, "Aromatic Electrophilic Substitution". Химизм присоединения галогена или более конкретно хлора к ароматической группе, таким образом, хорошо известен специалистам в данной области. Также известно, что этот химизм часто ведет к смесям нескольких молекул, одной из которых обычно является непрореагировавшее исходное вещество, не содержащее атома хлора. Кроме того, проблемой, хорошо известной специалистам в данной области, является избыточное хлорирование; часто образуются некоторые примеси дихлорсоединений, когда требуется монохлор-, и некоторые примеси трихлорсоединений, когда требуется дихлор-. Избыточное хлорирование обычно контролируют, ограничивая количество используемого хлорирующего реагента. К сожалению, следует ожидать, что контроль количества избыточно хлорированных аналогов лекарственного вещества путем ограничения количества хлорирующего реагента, применяемого для введении хлора в качестве заместителя в ароматическое кольцо, приведет к-1 007866 большему количеству дехлорпримеси (непрореагировавшего исходного вещества, не содержащего атом хлора). Краткое изложение сущности изобретения Аналог дехлорзипразидона представляет собой 5-[2-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил] этил]-1,3-дигидро-2H-индол-2-он (ниже - дехлорзипразидон). Любая партия лекарственного вещества зипразидона, синтезированная на основе известных методов синтеза галогенсодержащих ароматических соединений, упомянутых выше, будет содержать некоторое количество примеси, представляющей собой дехлорзипразидон. Следует ожидать, что контроль количества избыточно хлорированных аналогов лекарственного вещества путем ограничения количества хлорирующего реагента, применяемого при введении хлора в качестве заместителя в ароматическое кольцо, приведет к большему количеству дехлорпримеси. Данное изобретение относится к методикам, которые авторы изобретения разработали для контроля синтеза лекарственного вещества зипразидона с целью обеспечения низких уровней дехлорзипразидона. В конкретной лекарственной субстанции авторов изобретения, предназначенной для применения в фармацевтических композициях, постоянно наблюдается уровень дехлорзипразидона, не выше чем приблизительно 100 млн-1. Однако данное изобретение относится к композициям зипразидона, содержащим уровни дехлорзипразидона, составляющие до приблизительно 1000 млн-1, но не превышающие этого значения, а также к способам контроля уровней дехлорзипразидона до приблизительно 1000 млн-1, но не выше этого значения, в композиции зипразидона. Данное изобретение, кроме того, относится к композиции зипразидона, содержащей низкие уровни дехлорзипразидона, предпочтительно не выше приблизительно 1000 млн-1 дехлорзипразидона, более предпочтительно - не выше приблизительно 500 млн-1 дехлорзипразидона и еще более предпочтительно не выше 100 млн-1 дехлорзипразидона. При использовании здесь термин "зипразидон", если не указано иного, включает в себя зипразидон в форме свободного основания и фармацевтически приемлемые соли зипразидона. Общая идея получения фармацевтически приемлемых солей класса соединений, включающего зипразидон, предложена в патенте США 4831031 (см., например, столбец 3), включенном в данное описание изобретения ссылкой. В одном воплощении зипразидон в композиции по настоящему изобретению представляет собой зипразидон в форме свободного основание. В другом воплощении зипразидон в композиции по настоящему изобретению представляет собой моногидрат гидрохлорида зипразидона. В еще одном воплощении, зипразидон представляет собой дигидрат мезилата зипразидона, и в другом воплощении зипразидон представляет собой тригидрат мезилата зипразидона. Термин "лекарственное вещество зипразидон", как он используется здесь, и если не указано иного,относится к композиции зипразидона, как определено выше, которую применяют в приготовлении фармацевтической композиции. Такая фармацевтическая композиция может содержать фармацевтические носители, эксципиенты, вкусоароматизаторы и другие ингредиенты, которые известны как используемые в фармацевтических композициях и описаны более подробно ниже. Согласно изобретению также предложена фармацевтическая композиция для лечения у млекопитающего, в том числе человека, расстройства или состояния, выбранного из шизофрении, тревоги, боли при мигрени, синдрома Туретта, глаукомы, ишемической ретинопатии, деменции альцгеймеровского типа, биполярного расстройства, расстройства настроения, агорафобии, социальной фобии, панического расстройства, посттравматического стрессового расстройства, острого стрессового расстройства, тревожного расстройства, вызванного веществами, неуточненных (NOS) тревожных расстройств, дискинезий, поведенческого проявления задержки психического развития, расстройства поведения и аутистического расстройства, содержащая количество лекарственного вещества зипразидона, представляющего собой композицию, содержащую зипразидон и количество дехлорзипразидона, не превышающее приблизительно 1000 млн-1, причем количество лекарственного вещества зипразидона эффективно при лечении указанного расстройства или состояния, и фармацевтически приемлемый носитель. В одном воплощении количество дехлорзипразидона в лекарственном веществе зипразидоне составляет не более 500 млн-1. В предпочтительном воплощении количество дехлорзипразидона в лекарственном веществе зипразидоне составляет не более 100 млн-1. Согласно изобретению также предложен способ лечения у млекопитающего, в том числе человека,нуждающегося в этом, расстройства или состояния, выбранного из шизофрении, тревоги, боли при мигрени, синдрома Туретта, глаукомы, ишемической ретинопатии, деменции альцгеймеровского типа, биполярного расстройства, расстройства настроения, агорафобии, социальной фобии, панического расстройства, посттравматического стрессового расстройства, острого стрессового расстройства, индуцированного веществами тревожного расстройства, неуточненных (NOS) тревожных расстройств, дискинезий, поведенческих проявлений задержки психического развития, расстройства поведения и аутистического расстройства, при котором указанному млекопитающему вводят количество лекарственного вещества зипразидона, представляющего собой композицию, содержащую зипразидон и количество дехлорзипразидона, не превышающее приблизительно 1000 млн-1, причем это количество лекарственного вещества зипразидона эффективно при лечении указанного расстройства или состояния.-2 007866 В одном воплощении указанное количество дехлорзипразидона в лекарственном веществе зипразидоне не превышает приблизительно 500 млн-1. В предпочтительном воплощении количество дехлорзипразидона в лекарственном веществе зипразидоне составляет не более 100 млн-1 дехлорзипразидона. Согласно изобретению также предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающее приблизительно 1000 млн-1, который начинается с композиции хлорсодержащего реагента, имеющей достаточно низкий уровень не содержащей хлор примеси,для синтеза указанной композиции зипразидона. В одном воплощении хлорсодержащий реагент представляет собой композицию 6-хлороксиндола (6-хлор-1,3-дигидро-2H-индол-2-она). В более конкретном воплощении в данном изобретении предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающее приблизительно 1000 млн-1,при котором: а) получают один или более чем один образец из одной или более чем одной партии 6-хлор-1,3 дигидро-2H-индол-2-она; б) определяют уровень примеси оксиндола в каждом образце со стадии (а); в) выбирают партию 6-хлор-1,3-дигидро-2H-индол-2-она, которая содержит оксиндол на уровне не выше приблизительно 0,3%, на основании определения или определений, проведенных на стадии (б); и г) используют партию, выбранную на стадии (в), для синтеза указанной композиции зипразидона. В одном воплощении на стадии (в) выбирают партию 6-хлор-1,3-дигидро-2H-индол-2-она, которая содержит оксиндол на уровне не выше приблизительно 0,15%. В предпочтительном воплощении, на стадии (в) выбирают партию 6-хлор-1,3-дигидро-2H-индол-2-она, которая содержит оксиндол на уровне не выше приблизительно 0,03%. Хотя существует много известных путей получения 6-хлороксиндола, исходными веществами для этого обычно являются замещенный 4-хлортолуол или 1,4-дихлорнитробензол (см. G.J. Quallich and P.M."Development of an Efficient Process to 6-Chlorooxindole", представленный на 208-м национальном собрании Американского химического общества (ACS National Meeting) в Вашингтоне на симпозиуме по техническим достижениям в органической химии (Symposium on Technical Achievements in Organic Chemistry), 1994 (лекция 126. Однако идея контроля хлорсодержащих изомеров, избыточного хлорирования или дехлор-примесей в синтезе 6-хлороксиндола не описана в предшествующем уровне техники. Документ G.J. Qualich and P.M. Morrissey, см. выше, включен сюда ссылкой во всей полноте. Другие способы синтеза 6-хлороксиндола может определить специалист в данной области, и эти способы включены в стадию получения партии 6-хлороксиндола в описанном выше способе по данному изобретению. Кроме того, партию 6-хлороксиндола можно получить путем покупки органических химических реагентов у производителей, например Plaistow, Ltd., Little Island, County Cork, Ireland of Finorga, Route de Givors,38670 Chasse-sur-Rhone, France. При использовании здесь, под "партией 6-хлороксиндола" подразумевается композиция, состоящая по существу из 6-хлороксиндола, которая может содержать низкие уровни примесей, одной из которых может быть оксиндол. Уровень примеси оксиндола в образце партии 6-хлороксиндола можно определить, используя стандартные аналитические методики, известные специалистам в данной области. Например, уровень примеси оксиндола можно определить методами нормально-фазовой ВЭЖХ (высокоэффективной жидкостной хроматографии), обращенно-фазовой ВЭЖХ или газовой хроматографии. Конкретный способ определения уровня оксиндола в образце партии 6-хлороксиндола в данном описании обозначается как "Способ детекции Б" и раскрыт в разделе данной заявки "Подробное описание изобретения" ниже. Согласно изобретению также предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более чем 1000 млн-1, при котором: а) осуществляют ацилирование композиции, содержащей 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, хлорангидридом хлоруксусной кислоты по методу ацилирования Фриделя-Крафтса,синтезируя композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он; б) обрабатывают композицию, полученную на стадии (а), восстанавливая оксо в содержащейся в ней хлорацетильной группе с образованием композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро 2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; в) выделяют образец композиции, полученной на стадии (б); г) определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в образце, выделенном на стадии (в); д) определяют, превышает ли или не превышает количество, определенное на стадии (г), приблизительно 0,28%; и е) если количество, определенное на стадии (г), превышает приблизительно 0,28%, то композицию,полученную на стадии (б), очищают путем перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%, и из очищенной таким образом композиции синтезируют композицию зипразидона; или-3 007866 ж) если количество, определенное на стадии (г), не превышает приблизительно 0,28%, синтезируют композицию зипразидона из композиции, полученной на стадии (б). В предпочтительном воплощении композиция зипразидона, полученная способом, описанным в предыдущем абзаце, содержит количество дехлорзипразидона не более чем приблизительно 500 млн-1,при этом значение 0,28%, полученное на стадиях (д) и (е), доведено соответственно до приблизительно 0,14%. В более предпочтительном воплощении композиция зипразидона, полученная способом, описанным в предыдущем абзаце, содержит количество дехлорзипразидона не выше 100 млн-1, при этом значение 0,28%, полученное на стадиях (д) и (е), доведено соответственно до приблизительно 0,028%. Определение 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, как указано для стадии (г) способа, описанного в предыдущем абзаце, можно осуществить стандартными методами аналитической химии, например, при помощи обращенно-фазовой ВЭЖХ или других подходящих хроматографических методов. Однако в предпочтительном воплощении 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он определяют на стадии (б) способом детекции А, описанным ниже. В одном воплощении очистку композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2Hиндол-2-он, на стадии (е) осуществляют путем ресуспендирования. Ресуспендирование представляет собой процесс, аналогичный перекристаллизации, но при котором все вещество полностью не растворяется. Композицию, содержащую 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, на стадии (е) можно тем не менее очищать путем перекристаллизации, ресуспендирования или их комбинацией. Предпочтительным способом очистки 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она является перекристаллизация и/или ресуспендирование в смешиваемом с водой растворителе, предпочтительно в смеси ацетонитрил/вода. Согласно изобретению также предложен способ ВЭЖХ, называемый в данном описании "Способ детекции А", для определения количества 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции,содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он. Более конкретно в данном изобретении предложен способ "Способ детекции А", в котором применяется ВЭЖХ для определения количества 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции,содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, при котором: а) готовят раствор образца из указанной композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3 дигидро-2H-индол-2-он, путем растворения части указанной композиции в органическом растворителе с последующим разбавлением органическим растворителем растворенной части таким образом, что получают концентрацию (мас./об.), составляющую, исходя из массы указанной части и объема растворителя,приблизительно 1 мг/мл; б) пропускают раствор образца через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую, по существу, из смеси 0,05 М КН 2 РО 4 с рН=5,5-6,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.), при температуре колонки от приблизительно 30 до 40 С с детекцией в УФ свете при 254 нм УФ; в) детектируют пик, появляющийся на хроматограмме, полученной на стадии (б), между 8 и 10 мин; г) определяют площадь пика (обозначаемую Ас), детектированного на стадии (в); д) готовят стандарт из композиции, состоящей по существу из 5-(2-хлорэтил)-1,3-дигидро-2Hиндол-2-она, путем растворения и разбавления части указанной композиции в органическом растворителе таким образом, чтобы концентрация 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она (мас./об.) приблизительно была равна, исходя из массы части и объема растворителя, выбранному значению содержания,при котором или выше которого требуется детекция 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он; е) пропускают стандарт через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую по существу из смеси 0,05 М КН 2 РО 4 с рН=5,56,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.), при температуре колонки от приблизительно 30 до 40 С с детекцией в УФ свете при 254 нм УФ; ж) определяют площадь пика (обозначаемую Apur1) на хроматограмме, полученной на стадии (е); и з) рассчитывают количество 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, для чего: 1) рассчитывают коэффициент отклика для 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она по следующей формуле:% мac./мac.=(Ac)(DF)(100)/(Rpur1)(WS2) где Ас является таким, как определено выше;WS2 = масса части композиции, используемой на стадии (а); иDF = коэффициент разбавления раствора образца. Органические растворители, которые используют на стадиях (а) и (д), включают, но не ограничены ими, ТГФ (тетрагидрофуран), метанол, ацетонитрил или мобильную фазу, описанную на стадии (а). Другие органические растворители также могут быть полезны в данном способе. В предпочтительном воплощении способа ВЭЖХ, описанного в предыдущем абзаце, раствор образца растворяют в ТГФ, и затем разбавляют, используя мобильную фазу. В другом предпочтительном воплощении, стандарт готовят путем разбавления композиции, состоящей по существу из 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в ТГФ. В другом предпочтительном воплощении в способе ВЭЖХ используют скорость потока 1,0 мл/мин. В другом предпочтительном воплощении используют объемы введения стандарта и раствора образца по меньшей мере приблизительно 20 мкл, более предпочтительно приблизительно 20 мкл. В другом предпочтительном воплощении описанного выше способа ВЭЖХ температура колонки составляет 35 С. В другом предпочтительном воплощении соотношение КН 2 РО 4:ацетонитрил:метанол составляет 75:15:10. В другом предпочтительном воплощении рН КН 2 РО 4 составляет 6,0. Термин "колонка, содержащая стационарную фазу с привитыми цианогруппами", используемый здесь, означает колонку ВЭЖХ, содержащую стационарную фазу, состоящую, по существу, из фазы с привитыми цианогруппами. Колонки со стационарной фазой с привитыми цианогруппами известны специалистам в данной области. Такие колонки ВЭЖХ, например колонка ВЭЖХ Zorbax (Mac-Mod Analytical, P.O. Box 2600, 127 Commons Courts, Pennsylvania 19317, USA), доступны специалистам в данной области техники из коммерческих источников."Коэффициент эффективности", используемый в расчете на стадии (з) в указанном выше способе,относится к чистоте композиции, которая содержит, по существу, 5-(2-хлорэтил)-1,3-дигидро-2H-индол 2-он, в отношении к 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-ону в ней. Коэффициент эффективности специалист в данной области может определить путем вычитания количеств веществ, если они присутствуют, которые обычно определяются специалистом в данной области при анализе чистоты органической композиции. К таким веществам относятся, например, вода, растворитель или растворители и "остаток при сжигании" (т.е. неорганическое вещество, например натрий или калий). Определение и количественную оценку такого вещества может осуществить специалист в данной области. Следовательно, с учетом таких веществ, коэффициент эффективности композиции, состоящей, по существу, из 5-(2-хлорэтил)-1,3 дигидро-2H-индол-2-она, может составлять, например, 98% 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она."Коэффициенты разбавления" (для раствора образца и для стандарта), используемые в расчете на стадии (з) в описанном выше способе, относятся к количеству, которым анализируемую композицию и композицию, состоящую, по существу, из 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, разбавляют на стадиях (а) и (д) соответственно. Следовательно, коэффициентом разбавления для раствора образца будет объем растворителя, используемый для получения раствора образца на стадии (а) данного способа. Например, если на стадии (а) используют 80 мг композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3 дигидро-2H-индол-2-он, и, соответственно, 80 мл растворителя, тогда коэффициент разбавления будет составлять 80. Коэффициент разбавления для стандарта А будет зависеть от значения, выбранного на стадии (д). Например, если выбирают детекцию 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она при приблизительно 100 млн-1 или выше, тогда концентрация 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в стандарте будет составлять приблизительно 0,0001. Если, например, на стадии (д) используют 20 мг композиции,состоящей, по существу, из 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, тогда коэффициент разбавления стандарта составляет 20/0,0001, или 2105. В качестве другого примера: если выбирают детекцию 5-(2 хлорэтил)-1,3-дигидро-2H-индол-2-она при приблизительно 500 млн-1 или выше, тогда концентрация 5(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в стандарте будет составлять приблизительно 0,0005. Если,например, на стадии (д) используют 20 мг композиции, состоящей, по существу, из 5-(2-хлорэтил)-1,3 дигидро-2H-индол-2-она, тогда коэффициент разбавления стандарта составит 20/0,0005, или 4104. В качестве другого примера, если выбирают детекцию 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она при приблизительно 1000 млн-1 или выше, тогда концентрация 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в стандарте будет составлять приблизительно 0,001. Если, например, на стадии (д) снова используют 20 мг композиции, состоящей, по существу, из 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, тогда коэффициент разбавления стандарта будет равен 20/0,001, и коэффициент разбавления стандарта составит 2104. Согласно изобретению также предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более приблизительно 1000 млн-1, при котором: а) восстанавливают композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он и примесь 5-(хлорацетил)-1,3-дигидро-2H-индол-2-она, путем обработки триэтилсиланом в присутствии сильной кислоты с получением композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2 она и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2 она; и б) синтезируют композицию, содержащую зипразидон, из композиции, полученной на стадии (а). В предпочтительном воплощении сильная кислота на стадии (а) представляет собой трифторуксусную ки-5 007866 слоту или метансульфоновую кислоту. В другом предпочтительном воплощении данный способ дополнительно включает в себя стадии, на которых: 1) выделяют до стадии (б) образец композиции, полученной на стадии (а), и в указанном образце определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; 2) определяют, превышает ли количество, определенное в (1), приблизительно 0,28% или не превышает; и 3) если количество, определенное в (1), превышает приблизительно 0,28%, то композицию, полученную на стадии (а), очищают путем перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%; и затем переходят к осуществлению стадии (б), используя композицию, полученную на стадии (а), очищенную таким образом; или 4) если количество в (1) не превышает приблизительно 0,28%, тогда переходят к осуществлению стадии (б). В другом воплощении предложен способ синтеза композиции зипразидона, содержащей не более 500 млн-1 дехлорзипразидона. Значение 0,28%, используемое для анализа образца со стадии (а) можно довести, соответственно, до 0,14%. В другом воплощении, вышеуказанный способ используют для синтеза композиции зипразидона, содержащей не более 100 млн-1. Значение 0,28%, используемое для анализа способа, можно аналогично довести соответственно до 280 млн-1. В другом воплощении данного способа, количество 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в образце композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, определяют способом, включающим в себя способ детекции А. Очистку композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, на стадии(a)(2) указанного выше способа, производят путем ресуспендирования и/или перекристаллизации. Перекристаллизацию и/или ресуспендирование проводят в подходящей смеси растворителей, предпочтительно в растворителе, смешиваемом с водой, и более предпочтительно - в смеси ацетонитрил/вода. Согласно изобретению также предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более приблизительно 1000 млн-1, при котором: а) очищают композицию, содержащую 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, до получения композиции, содержащей не более приблизительно 0,3% указанной примеси оксиндола; и б) композицию, полученную на (а), используют для синтеза композиции зипразидона. В одном воплощении композицию на стадии (а) очищают до получения композиции, содержащей не более чем приблизительно 0,15% примеси оксиндола, и синтезируют композицию, которая содержит количество дехлорзипразидона не более чем приблизительно 500 млн-1. В другом воплощении композицию на стадии (а) очищают до получения композиции, содержащей не более приблизительно 0,03% примеси оксиндола, и синтезируют композицию, которая содержит количество дехлорзипразидона не более приблизительно 100 млн-1. Способы очистки композиции, содержащей -хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола,которую можно использовать в данном изобретении, включают экстракцию и/или перекристаллизацию и/или ресуспендирование. В одном воплощении очистку проводят путем перекристаллизации и/или ресуспендирования из органических растворителей. См., например, G.J. Qualich and P.M. Morrisey, см. выше, и приведенные в данном описании ссылки; а также F.R. Busch and R.J. Shine, см. выше. Согласно изобретению также предложен способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более чем приблизительно 1000 млн-1, при котором: а) осуществляют перекристаллизацию и/или ресуспендирование композиции, содержащей 6-хлор-5(2-хлорэтил)-1,3-дигидро-2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, до получения композиции, содержащей не более чем приблизительно 0,3% указанной примеси 5-(2-хлорэтил)1,3-дигидро-2H-индол-2-она; и б) используют композицию, полученную на стадии (а), для синтеза композиции зипразидона. В одном воплощении на стадии (а) осуществляют перекристаллизацию или ресуспендирование композиции до получения композиции, содержащей не более чем приблизительно 0,15% указанной примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, и синтезируют композицию, содержащую количество дехлорзипразидона не более чем приблизительно 500 млн-1. В другом воплощении на стадии (а) осуществляют перекристаллизацию и/или ресуспендирование композиции до получения композиции, содержащей не более приблизительно 0,03% примеси 5-(2 хлорэтил)-1,3-дигидро-2H-индол-2-она, и синтезируют композицию, которая содержит количество дехлорзипразидона не более чем приблизительно 100 млн-1. В предпочтительном воплощении условия перекристаллизации/ресуспендирования на стадии (а) данного способа включают применение полярных органических растворителей и/или полярных органических растворителей, смешанных с небольшим объемом воды. Предпочтительно перекристаллизацию и/или ресуспендирование осуществляют с использованием смешивающегося с водой растворителя, например смеси ацетонитрил/вода.-6 007866 Термины "лечение" и тому подобное относятся к реверсированию, облегчению или препятствованию развитию расстройства или состояния, к которому этот термин применяется, либо одного или более симптомов такого расстройства или состояния. При использовании здесь эти термины также охватывают, в зависимости от состояния пациента, предупреждение наступления расстройства или состояния либо симптомов, связанных с расстройством или состоянием, включая уменьшение тяжести расстройства или состояния либо симптомов, связанных с ними, до поражения этим расстройством или состоянием. Таким образом, "лечение", как оно используется здесь, может относиться к введению соединения по изобретению субъекту, который в момент введения не поражен этим расстройством или состоянием. "Лечение", таким образом, также включает предупреждение рецидива расстройства или состояния либо симптомов, связанных с ними."Млекопитающее", при использовании здесь и если не указано иного, означает любое млекопитающее. Термин "млекопитающее" включает в себя, например, собак, кошек и людей, но не ограничивается ими. Термин "приблизительно" при использовании здесь, например во фразе "менее чем приблизительно 1000 млн-1", означает интервал плюс или минус 10% от значения, к которому этот термин применяется. Подробное описание изобретения Один способ синтеза зипразидона, как указано выше, изложен в патенте США 5206366, который включен сюда ссылкой. В этом патенте показано, что зипразидон можно синтезировать способом, представленном на cхеме 1 ниже. Схема 2 ниже показывает механизм, посредством которого из любой примеси оксиндола в исходном реагенте образуется дехлорзипразидон при осуществлении синтеза по cхеме 1. "IPO" в следующих схемах означает изопропиловый спирт. Схема 1 Стадия 1. Одной из методик контроля, которую установили авторы изобретения, было определение очистки дехлор-примесей в процессе химического синтеза зипразидона, и затем установление достаточных пределов качества исходного реагента, используемого для синтеза зипразидона. В процессе синтеза зипразидона одно или более чем одно промежуточное соединение претерпевает экстракцию и кристаллизацию при прохождении реакции. Хотя каждое промежуточное соединение и соответствующая ему дехлорпримесь структурно близки и обладают близкой растворимостью, существуют небольшие различия в их физико-химических свойствах. Так, авторы изобретения осуществляли эксперименты по очистке для определения количества дехлор-примеси, которая удаляется экстракцией и перекристаллизацией. Обращаясь к схемам 1 и 2: соединение 6, оксиндол, представляющий собой потенциальную примесь в партии исходного вещества, соединения 1, 6-хлороксиндола, может в условиях реакции превратиться в соединение 9, дехлорзипразидон. Неожиданно авторы изобретения обнаружили более чем трехкратную очистку от соединения 6 и его последующих аналогов в условиях взаимодействия и выделения в процессе синтеза. Следовательно, имеющееся в продаже соединение 1, содержащее до 0,03% соединения 6, даст лекарственное вещество зипразидон, содержащее менее 0,01% (100 млн-1) соединения 9. Контроль максимального предела, составляющего приблизительно 0,03% (300 млн-1), соединения 6 в покупаемом соединении 1, основывается на уровнях соединения 6, детектированных в предыдущих партиях соединения 1, на информации по очистке, полученной в ходе разработки синтеза, и измерениях,полученных в процессе обработки этих промежуточных соединений. Как указано выше, оксиндол можно детектировать стандартными аналитическими способами. Одним конкретным способом, который обнаружили авторы изобретения для определения оксиндола в композиции, содержащей 6-хлороксиндол, является описанный далее способ, и он будет упоминаться в данном описании как "способ детекции Б": Способ детекции Б. Принцип. Для отделения соединения 1 от его потенциальных примесей используют нормально-фазовую жидкостную хроматографию (ЖХ). Сравнение площадей пиков и времени удерживания образцов и рабочего стандарта соединения 1 обеспечивает количественный анализ и тест на идентичность для соединения 1. Сравнение площадей пиков конкретных примесей, если они присутствуют, с разбавленными растворами примесей обеспечивает количественное определение их содержания. Аппаратура. 1. Стандартное лабораторное оборудование. 2. Подходящий жидкостной хроматограф. а. Насос - подача постоянного потока. б. УФ детектор - 254 нм. в. Дозатор, способный осуществлять введение по 50 мкл. г. Система получения данных.-8 007866 3. Колонка. Колонка Waters Associates Nova-Pak с силикагелем, частицы 4 мкм, 1503,9 мм (внутренний диаметр) (2 колонки, помещенные последовательно, или эквивалентная длина 300 мм) или эквивалент. 4. Весы, позволяющие взвешивать 50 мг 0,1 мг - например Mettler AE240. Реагенты. 1. Гексан - ВЭЖХ качество. 2. Тетрагидрофуран (ТГФ). 3. Изопропанол (IPO) - ВЭЖХ качество. 4. 15-краун-5 (1,4,7,10,13-пентаоксациклопентадекан). 5. Рабочий стандарт соединения 6 (оксиндол). Условия хроматографии. 1. Скорость потока: 1,5 мл/мин. 2. Вводимый объем: 50 мкл. 3. Длина волны для детекции: 254 нм. 4. Мобильная фаза: гексан/изопропанол/тетрагидрофуран/15-краун-5 1000/9/9/0,5 (об./об./об./об.) 5. Температура колонки: температура окружающей среды (приблизительно 23 С). В указанных выше условиях соединение 6 элюируется между 15-19 мин. Время элюирования соединения 1 - между 17-21 мин. Приготовление мобильной фазы. В 2-литровую колбу добавляют в следующем порядке: 1000 мл гексана, 9 мл изопропанола, 9 мл тетрагидрофурана и 0,5 мл 15-краун-5. Хорошо перемешивают и дегазируют под вакуумом с ультразвуковой обработкой или перемешиванием в течение 20 с. Подготовка образца и стандартов. 1. Приготовление образца и рабочего стандарта соединения 1. Взвешивают (в двух параллелях) с точностью до 0,1 мг приблизительно 20 мг рабочего стандарта соединения 1 и образцов и вносят в отдельные колбы на 100 мл. Следует готовить по две навески для рабочего стандарта и каждой серии образца. Вносят пипеткой 10 мл ТГФ в каждую колбу, обрабатывают ультразвуком в течение приблизительно 1 мин, добавляют мобильную фазу в количестве, достаточном для заполнения каждой колбы приблизительно на 80% от ее емкости, встряхивают и отставляют уравновешиваться до комнатной температуры. Разбавляют до вместимости (сколько требуется) мобильной фазой. (См. замечание оператору 1.) Раствор рабочего стандарта обозначают как РОТ 1. Раствор образца обозначают как А 1. 2. Приготовление рабочего стандарта соединения 6. Соединение Код Масса образца Колба Соединение 6 Ох 10 мг 100 мл Вносят пипеткой 10 мл ТГФ в колбу, обрабатывают ультразвуком в течение 1 мин и добавляют мобильную фазу в количестве, достаточном для заполнения колбы приблизительно на 80% емкости, встряхивают и позволяют нагреваться до комнатной температуры. Разбавляют до вместимости дополнительным количеством мобильной фазы и обозначают PUR1. Б. Дополнительно разбавляют раствор PUR1 с получением растворов PUR2 следующим образом. Помечают колбу как раствор PUR2. Соединение Код Переносимый объем Вместимость колбы Соединение 6 Ох 2 мл PUR1 100 мл Разбавляют этот раствор (PUR2) в колбе до вместимости мобильной фазой. В. Готовят конечную концентрацию примеси оксиндола путем разбавления раствора PUR2 с получением раствора PUR3. Соединение Код Переносимый объем Вместимость колбы Соединение 6 Ох 2 мл PUR2 100 мл Вносят пипеткой указанный объем раствора PUR2 в колбу и добавляют 10 мл ТГФ. Заполняют каждую колбу до приблизительно 80% емкости мобильной фазой и оставляют нагреваться до комнатной температуры. Разбавляют до вместимости мобильной фазой. Обозначают колбу как раствор PUR3. Пригодность системы. Полную пригодность системы следует определить до первого анализа и после любого значительного изменения системы. Эти критерии смотри в разделе Пригодность системы, который следует ниже в этой методике. Пригодность системы перед каждым анализом. Перед каждым анализом, выполняемым по этой методике, следует выполнить следующие условия. 1. Рассчитать разделение соединения 1 и соединения 6, используя приготовление, подробно опи-9 007866 санное в разделе Пригодность системы. 2. Убедиться, что достигнут адекватный предел количественного определения (LOQ). Используя раствор PUR3, осуществляют 2 параллельных ввода. Площади пиков соединения 6 должны совпадать в пределах 25%. Рассчитывают % совпадения площадей следующим образом: 2(А-В) 100% совпадения площадей =,А+В где А = площадь пика оксиндола (соединение 6) для ввода 1; В = площадь пика оксиндола (соединение 6) для ввода 2. 3. Для раствора рабочего стандарта соединения 1 РОТ 1 измеряют время удерживания соединения 1. Время удерживания должно находится в интервале 17-21 мин. Методика. 1. Вводят стандарт чистоты PUR3, четыре образца (A), PUR3, четыре образца и так далее. Между стандартами можно вводить не более четырех образцов. Для стандарта чистоты (PUR3) определяют площадь пика и время удерживания каждой конкретной примеси. Для каждого введенного образца измеряют время удерживания и площадь хроматографического пика каждого наблюдаемого пика (см. замечание оператору 3). Тест на идентичность. Тест пройден удовлетворительно, если тестируемый раствор образца соединения 1 (А) имеет основной пик, время удерживания которого равно (2%) времени удерживания раствора рабочего стандарта соединения 1 (РОТ 1). Вычисления. Чистота. 1. Для каждого образца устанавливают наличие оксиндола, если он присутствует, в соответствии с относительным временем удерживания, указанным в таблице, которую можно найти в разделе Условия хроматографии, и путем сравнения с временем удерживания пиков в соответствующем стандарте чистоты (PUR3). Их идентичность считается установленной, если время удерживания отличается не более чем на 2% (пик образца по сравнению с пиком в стандарте конкретной примеси). Конкретные примеси следует количественно определять с использованием стандарта чистоты (PUR3). 2. Рассчитывают стандартный коэффициент отклика для оксиндола:SRi=(Ai)(DF)/(Wi)(PFi),где SRi = стандартный коэффициент отклика оксиндола; Аi = площадь конкретной примеси в PUR3;PFi = коэффициент эффективности рабочего стандарта оксиндола (например, 0,993);DF = коэффициенты разбавления для оксиндола: = 250000. 3. Рассчитывают процентное содержание оксиндола следующим образом:% оксиндола = (A(s(DF)(100)/SRi(avg)(Ws,где SRi(avg) = средний стандартный отклик рабочего стандарта оксиндола;Ws = масса образца оксиндола в мг;DF = коэффициент разведения = 100; 100 = перевод в %. Пригодность системы. Условия, изложенные ниже, обеспечивают условия хроматографии, которые гарантируют, что система работает в режиме, пригодном для осуществления методики. Если имеется несоответствие чеголибо, следует сделать соответствующие корректировки в системе до начала процесса. До первого анализа в этой системе или после любого существенного изменения (например, замены колонки, ремонта автоматического пробоотборника и т.д.) необходимо оценивать пригодность системы. 1. Воспроизводимость. Осуществляют пять параллельных введений раствора рабочего стандарта соединения 1. Измеряют площадь пика соединения 1. Относительное стандартное отклонение (коэффициент отклонения) площадей пиков соединения 1 не должен превышать 2,0%. Осуществляют шесть параллельных введений раствора стандарта чистоты (PUR3), содержащего соединение 6. Измеряют площадь пика соединения 6. Относительное стандартное отклонение (коэффициент отклонения) площадей пиков соединения 6 не должно превышать 15%. 2. Эффективность. Рассчитывают число теоретических тарелок (N) для хроматографической колонки, используя одно характерное введение раствора рабочего стандарта соединения 1. Число теоретических тарелок должно быть равно или превышать 6000.- 10007866 Рассчитывают асимметричность пиков (Т) для пика соединения 1 с помощью одного характерного введения раствора рабочего стандарта. Асимметричность пиков не должна превышать 2,0.T=(W0,05/2f) 4. Разделение. Рассчитывают разделение (R) соединения 1 и соединения 6, готовя образец соединения 1 с добавлением 0,1% (мас./мас.) соединения 6 следующим образом. Готовят раствор соединения 1, как указано выше на стадии 1 раздела Приготовление образца и стандартов. Перед разбавлением до вместимости добавляют 10 мл раствора PUR2, приготовленного на стадии Б в разделе, относящемся к приготовлению стандартов примесей, приведенном выше. Разделение соединения 1 и соединения 6 должно составлять 1,0.t = время удерживания, измеряемое от времени момента до момента элюирования максимума пика;W = ширина пика, измеряемая при экстраполяции относительно прямых боковых линий до линии основания. Замечания оператору. 1. Время, требуемое для уравновешивания колонок с нормальной фазой, обычно больше, чем для колонок с обращенной фазой. Новую колонку сначала следует промыть 4 л мобильной фазы. Пара последних пиков, которая должна быть разделена в процессе уравновешивания, представляет собой соединение 6 и соединение 1. Для уравновешивания делают несколько вводов стандарта. 2. Подготовку стандарта и образца (разбавление и подогрев до комнатной температуры) следует выполнять одновременно, чтобы гарантировать одинаковый объем. 3. Можно пропустить ТГФ контроль (10 мл ТГФ, разбавленные до вместимости мобильной фазой),чтобы гарантировать, что между какими-либо интересующими пиками нет влияния. Как описано выше, согласно изобретению также предложен дополнительный способ контроля низкого уровня дехлорзипразидона путем обеспечения низкого уровня дехлор-промежуточного соединения стадии 2 (соединение 8 схемы 2, 5-(2-хлорэтил)оксиндол). Это является важным моментом контроля благодаря возможности контроля процесса после восстановления карбонила. В результате восстанавливающих условий происходит гидродегалогенирование, и, таким образом, в синтезе, изображенном на схеме 1, - потеря хлора, который требуется в С-6 положении. Была разработана аналитическая методика, описанный выше метод ВЭЖХ, для оценки 6-хлор-5-(2 хлорэтил)-1,3-дигидро-2H-индол-2-она, соединения 3 cхемы 1, на количественное содержание и чистоту. Одним из важных назначений этой методики является определение уровней соединения 8, в млн-1, дехлор-примеси в выделенном материале. Методика "Способ детекции А", изложенная ниже, дает пример того, как аналитик может конкретно применить этот метод определения с помощью ВЭЖХ для количественного определения низких уровней соединения 8 без взаимного влияния с другими примесями,имеющими отношение к данному процессу. Этот пример не предназначен для того, и его не следует истолковывать так, чтобы ограничить изобретение, более полно описанное здесь и заявленное ниже. Способ детекции А. После определения пригодности системы можно применить следующий аналитический хроматографический способ с использованием ВЭЖХ для количественного определения уровней дехлор(cоединение 8) в cоединении 3. Аппаратура. 1. Подходящая система ВЭЖХ, снабженная стандартным оборудованием. 2. Нагреватель колонки, способный работать при 35 С, например BAS Temperature controller, модель LC22A. 3. Блок предварительного нагрева мобильной фазы: например Bioanalytical Systems, Inc. (BAS),по каталогу EW8146. Примечание: Это требуется для улучшения эффективности колонки. 4. Колонка - Zorbax-SB-CN (каталожный 883975.905) 15 см длина х 4,6 мм внутренний диаметр(продается Mac Mod Analytical, Chadds Ford, PA). Реагенты. 1. 0,05 M первичный кислый фосфат калия (КН 2 РО 4), буферный раствор с рН=6,0. Растворяют 6,8 г КН 2 РО 4 в 1 л очищенной воды. Доводят рН раствора до 6,00,1 с помощью 5 н. раствора гидроксида калия. Если нужно, можно приготовить большие объемы. 2. Мобильная фаза: (75:15:10 об./об./об.) 0,05 М КН 2 РО 4, рН=6,0: ацетонитрил:метанол. Фильтруют и дегазируют при пониженном давлении при перемешивании или обработке ультразвуком в течение приблизительно 5 мин. Можно приготовить большие объемы мобильной фазы, используя соответствующие количества компонентов. Условия хроматографии.- 11007866 Параметр Мобильная фаза Температура колонки Детекция Скорость потока Объем ввода Метод количественного определения Время пропускания Заданное значение Как указано выше 35 С УФ, 254 нм 1,0 мл/мин 20 мкл Площадь 60 мин В указанных выше условиях соединение 8 будет элюироваться через приблизительно 8-10 мин. Относительное время удерживания конкретных примесей приведено в таблице ниже. СоединениеRr Соединение 1 0,36 Соединение 2 0,45 Соединение 8 0,49 Соединение 3 1,00 Примечание. Относительное время удерживания (Rr) представляет собой время удерживания пика конкретной примеси относительно времени удерживания соединения 3. Приготовление стандартных эталонных растворов. Стандарт соединения 8. Стандарт соединения 8 в мерной колбе на 200 мл. Добавляют приблизительно 20 мл ТГФ и обрабатывают ультразвуком, пока образец полностью не растворится (приблизительно 1 мин). Разбавляют раствор до вместимости метанолом. Хорошо смешивают при дополнительном встряхивании и переворачивании. Обозначают его как раствор соединения 8 F1. Концентрация соединения 8 в растворе F1 составляет приблизительно 0,1 мг/мл. Разбавляют 2 мл F1 до 100 мл метанолом и хорошо перемешивают. Обозначают его как раствор соединения 8 F2. Концентрация соединения 8 в растворе F2 составляет приблизительно 0,002 мг/мл. Приготовление растворов образцов. Образец соединения 3 (для определения соединения 8). Готовят один испытываемый раствор на образец. Отвешивают приблизительно 50 мг (записывают с точностью до 0,1 мг) образца соединения 3 в 50 мл мерную колбу. Добавляют приблизительно 20 мл ТГФ и обрабатывают ультразвуком, пока образец полностью не растворится (приблизительно 2 мин). Разбавляют до вместимости мобильной фазой. Тщательно смешивают путем дополнительного встряхивания и переворачиваний. Обозначают его как раствор 1 соединения 3 (раствор образца I). Концентрация раствора I (раствора образца I) составляет приблизительно 1,0 мг/мл. Примечание. Раствор I стабилен вплоть до 24 ч в обычных лабораторных условиях. Пригодность системы. Полную пригодность системы следует определять до начала анализа и после любого существенного изменения системы. Эти условия см. в разделе Пригодность системы в конце данной методики. Проверка пригодности системы перед каждым анализом. Перед каждым анализом, выполняемым по этой методике, следует достигнуть следующих условий. 1. Рассчитывают разделения (R) соединения 2 и соединения 8, используя стандарт PUR1. Разделение этой пары пиков должно составлять 1,0. 2. Убеждаются, что достигнут адекватный предел количественного определения (LOQ). Используя раствор PUR1, осуществляют 2 параллельных ввода. Площади пиков соединения 8 должны совпадать в пределах 20%. Вычисляют % совпадения площадей следующим образом: 2(А-В) 100% совпадения площадей = ,А+В где А = площадь пика соединения 8 первого ввода. В = площадь пика соединения 8 второго ввода. 3. Для стандартного раствора соединения 3 А 1 измеряют время удерживания соединения 3. Время удерживания должно находиться в интервале 16-24 мин. Методика. Определяют пригодность системы ВЭЖХ, используя руководства и методики по определению пригодности системы, представленные ниже в этой методике испытания. Проверка LOQ, разделения и времени удерживания как описано в разделе Пригодность системы, проверяемая перед каждым анализом,следует выполнять каждый раз, когда используют систему. Оставшееся условие пригодности системы следует оценивать перед первым анализом в данной системе и после каждого существенного изменения. Оценка содержания соединения 8 в образце соединения 3 (раствор (I. Вводят аликвоты по 20 мкл раствора образца (I). Измеряют площадь хроматографического пика со- 12007866 единения 8 для каждого введения. Определяют уровни присутствия соединения 8 в испытываемом образце (I) как описано в разделе Вычисления. Вычисления. Вычисление содержания соединения 8. 1. Определяют коэффициент отклика для соединения 8 следующим образом:Rpur1 = ,Wpur1PF где Apur1 = площадь пика примеси (соединение 8) в PUR1;PF = коэффициент эффективности для соединения 8 и соединения 2 (например, 0,993);% мас./мас. = ,Rpur1WS2 где Ас = площадь пика соединения 8 в образце I;Rpur1 = коэффициент отклика для соединения 8 в стандарте;WS2 = масса соединения 3 в образце I, в мг; 50 = коэффициент разведения; 100 = перевод в процентыИспользуют средний коэффициент отклика для всех вводов PUR1, выполняемых в ходе анализа. Пригодность системы. Условия, изложенные ниже, создают условия хроматографии, которые гарантируют, что система работает способом, пригодным для осуществления процесса. Если какое-нибудь из них не выполняется,следует сделать соответствующие корректировки в системе до начала процесса. До первого анализа на этой системе или после любого существенного изменения (например, замены колонки, ремонта автоматического пробоотборника и т.д.) необходимо оценивать пригодность системы. 1. Точность ввода. Анализ. Осуществляют 5 параллельных вводов стандарта соединения 3 А 1. Измеряют площадь каждого пика соединения 3. Относительное стандартное отклонение площадей пиков не должно превышать 1,0%. Оценка чистоты. Осуществляют шесть параллельных вводов раствора стандарта PUR1. Измеряют площадь каждого пика соединения 8. Относительное стандартное отклонение площадей пиков не должно превышать 10%. 2. Эффективность. Рассчитывают число теоретических тарелок (N) для хроматографической колонки, используя пик соединения 3 в стандарте А 1. Число теоретических тарелок должно быть не менее 5000 при расчете тангенциальным методом.t = время удерживания, измеренное от момента ввода до момента элюирования максимума пика.W = ширина пика, измеренная при экстраполяции относительно прямых боковых линий до линии основания. 3. Время удерживания. Измеряют время удерживания для пика соединения 3 в стандарте А 1. Время удерживания пика соединения 3 должно находиться в интервале 16-24 мин. 4. Асимметричность пиков. Вычисляют асимметричность пиков (Т) для пика соединения 3 в стандарте А 1. Асимметричность пиков не должна превышать 2,0.W0,05 = ширина пика на 5%-ной высоте;f = расстояние от максимума пика до переднего края пика, измеренное на 5%-ной высоте пика. 4. Разделение. Рассчитывают разделение (R) соединения 2 и соединения 8 в PUR1. Разделение этой пары пиков должно быть 1,0.W = ширина пика на линии основания (измеренная путем экстраполяции до линии основания касательных к точкам перегиба, для каждого компонента). В качестве составной части разработки данного химического процесса были проанализированы многие альтернативные способы синтеза для восстановления соединения 2 в соединение 3 на приведенной выше схеме 1. Например, исследовали каталитическое гидрирование карбонила в соединении 2, используя 5%-ный палладий на угле, палладий на окиси алюминия, платину на угле или платину на оксиде алюминия. Эксперименты с палладием повторяли с загрузкой 10% катализатора, и в каждом случае дегалогенирование представляло проблему. Авторы изобретения обнаружили, что триэтилсилан (TES) в присутствии сильной кислоты обеспечивает восстановление карбонила без загрязнения гидродегалогенированием. Следовательно, этот тип восстановления является предпочтительным для получения соединения 3. Кроме того, было обнаружено, что трифторуксусная кислота или метансульфоновая кислота являются предпочтительными сильными кислотами для данного химического процесса, так как позволяют избежать образования дехлор-примесей, таких как соединение 8. Для повторной обработки серий, которые обладают, как посчитали авторы, высоким содержание дехлор-аналогов, была разработана очистка. Были разработаны методы повторной обработки соединений 1 или 3. Авторы разработали и осуществили экспериментальную программу тестирования очистки исходного материала, каждого промежуточного соединения и конечного лекарственного средства для определения в синтезе зипразидона наиболее эффективного момента для удаления дехлор-соединений, если они присутствуют. Авторы обнаружили, что наиболее эффективным является удаление примесей путем перекристаллизации и/или ресуспендирования соединений 1 или 3. Перекристаллизация соединений 2, 4 или конечного лекарственного средства была очень неэффективной, приводя к очень небольшим уменьшениям уровней дехлор-примесей. Конкретно, для очистки соединения 3 испытывали многие условия, включая ацетонитрил, смеси метиленхлорид/толуол, этилацетат/гексаны, изопропиловый спирт, толуол, ТГФ, смеси изопропиловый спирт/DMAC (диметилацетамид), метанол, смеси изопропиловый спирт/уксусная кислота и ацетонитрил/вода. Эксперименты по очистке для выяснения предпочтительных условий перекристаллизации и ресуспендирования соединения 3 обобщены в таблице ниже. В целом, большинство испытанных растворителей были неэффективными для удаления соединения 6 из соединения 3. Перекристаллизация/ресуспендирование из смеси ацетонитрил/вода имеет преимущество перед другими исследованными методиками, поскольку приводит к уменьшению уровня примеси от 1300 до 300 млн-1. Таким образом,такие перекристаллизация и/или ресуспендирование являются предпочтительными для контроля дехлорпримеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она. Очистка соединения 3 Лекарственное вещество зипразидон по данному изобретению можно вводить, как указано в данном описании, в качестве нейролептического агента, как описано в, например, патенте США 4831031, см.- 14007866 выше. Вводить ее млекопитающему, включая человека, как таковую или, предпочтительно, в комбинации с фармацевтически приемлемыми носителями или разбавителями в фармацевтической композиции,в соотвествии с традиционной фармацевтической практикой. Фармацевтические композиции можно вводить перорально или парентерально, в том числе внутривенно или внутримышечно. Подходящие фармацевтические носители включают твердые разбавители или наполнители и стерильные водные растворы и различные органические растворители. Фармацевтические композиции, далее, легко вводят во множество лекарственных форм, таких как таблетки, порошки, леденцы, сиропы и инъекционные растворы. Эти фармацевтические композиции, если требуется, могут содержать дополнительные ингредиенты, такие как вкусоароматизаторы, связывающие вещества и эксципиенты. Так, для перорального введения можно применять таблетки, содержащие различные эксципиенты, такие как цитрат натрия, карбонат кальция и фосфат кальция, вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, совместно со связывающими агентами, такими как поливинилпирролидон,сахароза, желатин и гуммиарабик. Кроме того, для таблетирования часто используют смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые вещества подобного типа также можно использовать как наполнители в мягких и жестких заполненных желатиновых капсулах. Предпочтительные для этого вещества включают лактозу, или молочный сахар, и высокомолекулярные полиэтиленгликоли. Когда для перорального введения требуются водные суспензии или эликсиры, лекарственное вещество зипразидон в них можно объединять с различными подслащивающими или ароматизирующими агентами, подкрашивающими веществами или пигментами и, если требуется, эмульгирующими или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль,глицерин и их комбинации. Для парентерального введения можно применять раствор или суспензию лекарственного вещества зипразидона в кунжутном или арахисовым масле, водном пропиленгликоле или в стерильном водном растворе. Такие водные растворы, если необходимо, следует подходящим образом забуферивать, и жидкий разбавитель следует сначала делать изотоническим при помощи достаточного количества солевого раствора или глюкозы. Эти конкретные водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. Все используемые стерильные водные среды легко доступны стандартными способами, известными специалистам в данной области. Общеизвестно, что эффективная доза зипразидона зависит от предполагаемого пути введения и других факторов, таких как показание для лечения, возраст и масса субъекта. В общем случае суточная доза будет находиться в интервале от приблизительно 0,5 мг лекарственного вещества зипразидона до приблизительно 500 мг, в одной или разделенных дозах, предпочтительно от приблизительно 10 до приблизительно 200 мг в сутки. В настоящее время в Соединенных Штатах Geodon одобрен для лечения шизофрении в форме капсул для перорального введения, содержащих зипразидон в форме моногидрата гидрохлорида. Эти капсулы доступны в виде лекарственных форм, содержащих 20, 40, 60 и 80 мг лекарственного вещества зипразидона. Обычная суточная доза для лечения шизофрении, основанная на массе пациента приблизительно 70 кг, составляет предпочтительно от приблизительно 20 мг 2 раза в сутки до приблизительно 100 мг лекарственного вещества зипразидона 2 раза в сутки, более предпочтительно от приблизительно 20 мг 2 раза в сутки до приблизительно 80 мг 2 раза в сутки. Однако очевидно, что лечащий врач-специалист в данной области может варьировать дозировку и режим введения лекарственного вещества зипразидона по сравнению с указанными выше интервалами и режимами в зависимости от конкретных обстоятельств конкретного пациента. Следующие примеры иллюстрируют настоящее изобретение. Следует понимать, однако, что данное изобретение, как оно полностью описано здесь и изложено в формуле изобретения, не должно быть ограничено подробностями следующих примеров. Примеры Пример 1. Синтез зипразидона. Стадия 1. Ацилирование 6-хлор-1,3-дигидро-2H-индол-2-она по Фриделю-Крафтсу. Объединяли метиленхлорид (310 л) и алюминия хлорид (172,3 кг). Добавляли хлорангидрид хлоруксусной кислоты (66,7 кг), и полученную в результате смесь перемешивали в течение 45 мин. Добавляли 6-хлор-1,3-дигидро-2H-индол-2-он (61,8 кг).Реакционную смесь перемешивали при 28-32 С в течение 19,5 ч и затем охлаждали до 15-20 С. Воду (805 л) охлаждали до 5-10 С. Реакцию гасили медленным добавлением реакционной смеси к холодной воде. После того как погашение было закончено, смесь кипятили с обратным холодильником и метиленхлорид удаляли перегонкой при атмосферном давлении при 43-57 С. Полученную в результате смесь охлаждали до 15-20 С и перемешивали в течение 1 ч. Твердые вещества отделяли фильтрацией и промывали водой (14 л), затем метанолом (114 л). Твердые вещества сушили в подходящей сушилке. 6-Хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он, выход: 91,3 кг (101,4%). Примечание. Выход по массе, превышающий 100%, получился вследствие небольших количеств остаточных солей, которые удаляли на следующей стадии. Полученный в результате 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он использовался на следующей стадии частями; обработка одной из них подробно описана ниже.- 15007866 Стадия 2. Восстановление 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-она смесью трифторуксусная кислота/силан. Трифторуксусную кислоту (278 кг) и (74,2 кг) объединяли и медленно перемешивали при 24-28 С. К перемешиваемой смеси загружали триэтилсилан (77,9 кг). В процессе данного добавления допускали небольшое выделение теплоты из реакционной среды и в ходе реакционного периода температуру поддерживали между 50-62 С. Реакционную смесь перемешивали в течение 8 ч, охлаждали до 38 С и отбирали пробы для определения окончания реакции. Реакционную смесь перемешивали при 50-54 С в течение дополнительных 3 ч. После того как было определено, что реакция завершилась, реакционную смесь охлаждали до 18 С и гасили водой (594 л). Полученную в результате взвесь перемешивали в течение 30 мин при 10-15 С и твердые вещества отделяли фильтрацией. Продукт вымывали из резервуара, и осадок продукта промывали водой (83 л), затем метанолом (76 л). В каждой из двух партий равной величины объединяли тетрагидрофуран (742 л), Darco KB-B (1,9 кг) и влажный остаток продукта и нагревали их до кипения с обратным холодильником. Полученную в результате смесь перемешивали при кипячении с обратным холодильником в течение 30 мин и фильтровали через фильтр Sparkler (предварительно покрытый ускорителем фильтрования (filteraid при 50-60 С для удаления угля. Резервуар и фильтр Sparkler промывали горячим тетрагидрофураном (38 л). После фильтрации две партии объединяли. Раствор концентрировали в вакууме и перемешивали при 4-5 С в течение 1 ч. Твердые вещества отделяли фильтрацией и промывали холодным тетрагидрофураном (38 л). Твердые вещества сушили в вакууме при 45-73 С до достижения усушки на 0,45%, с получением 6-хлор 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, выход: 60,1 кг (85,9%). Полученный в результате 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он объединяли с веществом сопоставимого качества и осуществляли следующую стадию. Стадия 3. Сочетание 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она и 3-(1-пиперазинил)-1,2 бензизотиазола моногидрохлорида. Воду (780 л) и карбонат натрия (126,0 кг) объединяли и смесь перемешивали до растворения. Добавляли 3-(1-пиперазинил)-1,2-бензизотиазола моногидрохлорид (155,0 кг) и 6-хлор-5-(2-хлорэтил)-1,3-дигидро 2H-индол-2-он (150,4 кг), и реакционную смесь нагревали до кипения с обратным холодильником (100 С). Через 24 и 48 ч от реакционной взвеси отбирали пробы для анализа на окончание реакции. Реакцию нашли завершенной после анализа второго образца. Добавляли воду (1251 л) и взвесь охлаждали до температуры между 18-22 С. Твердые вещества отделяли фильтрацией и промывали водой (302 л). Увлажненные водой твердые вещества объединяли с изопропанолом (940 мл) и полученную в результате смесь перемешивали в течение приблизительно 2 ч при температуре окружающей среды. Твердые вещества отделяли фильтрацией,промывали изопропанолом (89 л) и сушили в вакууме при температуре менее 43 С с получением 5-[2-[4(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2H-индол-2-она, выход: 202,8 кг (80,8%). Полученный в результате 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро 2H-индол-2-он делили на две части. С этими партии раздельно осуществляли следующую дополнительную очистку, и получали в результате вещество сопоставимого качества. Обработка для одной из этих партий подробно описана ниже. Стадия 3R. Очистка 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2H-индол-2-она. Объединяли 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2H-индол-2-он(51 кг), ускоритель фильтрации (4 кг) и тетрагидрофуран (2678 л). Смесь нагревали до кипения с обратным холодильником (65 С) в течение 1 ч, фильтровали, поддерживая температуру выше 55 С, и промывали тетрагидрофураном (570 л). Фильтрат, обогащенный продуктом, частично концентрировали в вакууме. Объединяли 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2 Н-индол 2-он (51 кг), ускоритель фильтрации (4 кг) и тетрагидрофуран (2678 л). Смесь нагревали до кипения с обратным холодильником (65 С) в течение 1 ч, фильтровали, поддерживая температуру выше 55 С, и промывали тетрагидрофураном (570 л). Фильтрат, обогащенный продуктом, объединяли с частично сконцентрированной смесью, указанной выше, и концентрировали в вакууме. Полученную в результате смесь охлаждали до 0-5 С. Твердые вещества отделяли фильтрацией, промывали отфильтрованным тетрагидрофураном (113 л) и сушили в вакууме при температуре менее 41 С, с получением зипразидона в форме свободного основания с выходом: 79,3 кг (77,7%). Часть партии объединяли с веществом сопоставимого качества, которое перекристаллизовывали отдельно, и с этой партией осуществляли следующую стадию. Пример 2. Образование соли моногидрата гидрохлорида зипразидона при кристаллизации. Тетрагидрофуран (2715 л), воду (307 л) и 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6 хлор-1,3-дигидро-2H-индол-2-он (100,0 кг) объединяли, нагревали до кипения с обратным холодильником(64 С) и перемешивали в течение 30 мин. Раствор фильтровали и промывали тетрагидрофураном (358 л). Воду (203 л) и концентрированную соляную кислоту (29 л) объединяли и перемешивали при температуре окружающей среды. Полученный в результате водный раствор соляной кислоты загружали к раствору 5-[2-[4-(2,3-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2H-индол-2-она в течение 27 мин. Реакционную смесь охлаждали до температур между 1 и 5 С в течение 2 ч. Смесь перемешива- 16007866 ли при температуре между 1 и 5 С в течение 10 ч. Твердые вещества отделяли фильтрацией, промывали холодным тетрагидрофураном (358 л) и сушили до содержания воды 4,1%. Гидрохлорида моногидрат зипразидона, выход: 108,6 кг (96,0% выход по массе). Твердые вещества перемалывали на мельнице фирмы Bauermeister. Пример 3. Очистка 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она для удаления 5-(2-хлорэтил)1,3-дигидро-2H-индол-2-она. В 100 мл круглодонную колбу, оснащенную магнитной мешалкой и обратным холодильником, загружали 4,0 г (17,4 ммоль) 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она (соединение 3) и 36 мл ацетонитрила и добавляли 4,0 мл воды. Эту смесь мягко нагревали и перемешивали в течение ночи (18 ч при 78 С). Нагревание затем прекращали, взвесь охлаждали до 0-5 С и перемешивали в течение еще 1 ч. Продукт собирали фильтрацией, промывали небольшой порцией ацетонитрила, и продукт сушили под вакуумом при 50 С с получением 3,77 г (94,3% выход) 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2 она. Уровень дехлор-примеси был снижен от 1280 до 230 млн-1. Пример 4. Экспериментальное определение коэффициента очистки по соединению 6 (1,3-дигидро 2H-индол-2-он). Выбрали партию 6-хлор-1,3-дигидро-2 Н-индол-2-она, которая имела очень высокое содержание 1,3-дигидро-2 Н-индол-2-она. Ее выбрали умышленно, чтобы было легче измерить более высокие уровни примеси и определить коэффициент очистки для этой примеси. Дополнительной причиной такой стратегии начать с вещества с большим содержанием примеси с целью определения коэффициента очистки от примеси было стремление избежать очистки вещества до уровня ниже предела аналитической детекции в ходе синтеза, что приводит к нулевому значению в конечном продукте. Поскольку коэффициент очистки представляет собой отношение, не имеет смысла деление на ноль. (Материал с высоким содержанием примеси использовали для данного эксперимента, но не использовали затем в исследованиях с людьми). С партией 6-хлор-1,3-дигидро-2H-индол-2-она, содержащего 4000 млн-1 1,3-дигидро-2H-индол-2-она,осуществляли стандартный процесс синтеза в соответствии с примерами 1 и 2, приведенными выше. После первых двух стадий синтеза измеряли уровень соответствующей дехлор-примеси, используя описанный способ. Было обнаружено, что 1700 млн-1 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она (соединение 8 на схеме 2, выше) присутствовало в 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-оне (соединение 3 на схеме 1 выше). Процесс продолжали до моногидрата гидрохлорида 5-[2-[4-(1,2-бензотиазол-3-ил)-1-пиперазинил]этил]-6-хлор-1,3-дигидро-2H-индол-2-она, в котором было обнаружено присутствие 600 млн-1 5-[2[4-(1,2-бензотиазол-3-ил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она (соединение 9 на схеме 2 выше). Таким образом, коэффициент очистки за весь синтез по дехлор-аналогам составлял от 4000 до 600 млн-1, или приблизительно 6-кратное уменьшение. Незначительные отклонения от опыта к опыту в процессе могут привести к небольшим различиям в выходе и качестве полученных веществ. Следовательно,допустима 20%-ная ошибка в воспроизводимости результатов образования примесей, т.е. если имеется 500 млн-1 в одном опыте, то в других экспериментах ожидают между 400 и 600 млн-1. В случае синтеза,описанного в примерах 1 и 2, с 5 стадиями процесса, дополнительная экспериментальная ошибка может привести к 2-кратному различию в уровне примеси. Таким образом, для установления верхнего предела,с которым лекарственное средство назначают для применения людям, использовали заниженный 3 кратный коэффициент очистки. Следовательно, чтобы гарантировать, что получаемый продукт не содержит более 100 млн-1 5-[2-[4-(1,2-бензотиазол-3-ил)-1-пиперазинил]этил]-1,3-дигидро-2H-индол-2-она(соединение 9), установили предел, составляющий 300 млн-1, 1,3-дигидро-2 Н-индол-2-она (соединение 6) в 6-хлор-1,3-дигидро-2 Н-индол-2-оне (соединение 1). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Композиция, содержащая зипразидон и количество дехлорзипразидона, выбранное из количества,не превышающего приблизительно 1000 миллион-1 (млн-1), не превышающего приблизительно 500 млн-1 и не превышающего приблизительно 100 млн-1. 2. Композиция по п.1, где зипразидон представляет собой зипразидон в форме свободного основания, моногидрат гидрохлорида зипразидона, дигидрат мезилата зипразидона или тригидрат мезилата зипразидона. 3. Фармацевтическая композиция для лечения у млекопитающего расстройства или состояния, выбранного из шизофрении, тревоги, боли при мигрени, синдрома Туретта, глаукомы, ишемической ретинопатии, деменции альцгеймеровского типа, биполярного расстройства, расстройства настроения, агорафобии, социальной фобии, панического расстройства, посттравматического стрессового расстройства, острого стрессового расстройства, тревожного расстройства, вызванного веществами, неуточненных (NOS) тревожных расстройств, дискинезий, поведенческого проявления задержки психического развития, расстройства поведения и аутистического расстройства, содержащая количество композиции по п.1, эффективное для лечения указанного расстройства или состояния, и фармацевтически приемлемый носитель. 4. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, выбранное из количества, составляющего не более чем приблизительноA) 1000 млн-1,Б) не более чем приблизительно 500 млн-1, иB) не более чем приблизительно 100 млн-1, при котором: а) получают один или более чем один образец одной или более чем одной партии 6-хлор-1,3 дигидро-2H-индол-2-она; б) определяют уровень примеси оксиндола в каждом образце со стадии (а); в) выбирают партию 6-хлор-1,3-дигидро-2H-индол-2-она, которая содержит оксиндол на уровне для (А) - не выше приблизительно 0,3%, на основании определения или определений, проведенных на стадии (б),для (Б) - не выше приблизительно 0,15%, на основании определения или определений, проведенных на стадии (б), и для (В) - не выше приблизительно 0,03%, на основании определения или определений, проведенных на стадии (б), и г) используют партию, выбранную на стадии (в), для синтеза указанной композиции зипразидона. 5. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона не более чем приблизительно 1000 млн-1, при котором: а) осуществляют ацилирование композиции, содержащей 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, хлорангидридом хлоруксусной кислоты по методу ацилирования Фриделя-Крафтса,синтезируя композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он; б) обрабатывают композицию, полученную на стадии (а), восстанавливая в содержащейся в ней хлорацетильной группе оксо с образованием композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3 дигидро-2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; в) выделяют образец композиции, полученной на стадии (б); г) определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в образце, выделенном на стадии (в); д) определяют, превышает ли или не превышает количество, определенное на стадии (г), приблизительно 0,28%; и е) если количество, определенное на стадии (г), превышает приблизительно 0,28%, то композицию,полученную на стадии (б), очищают путем перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%, и из очищенной таким образом композиции синтезируют композицию зипразидона; или ж) если количество, определенное на стадии (г), не превышает приблизительно 0,28%, синтезируют композицию зипразидона из композиции, полученной на стадии (б). 6. Способ, в котором применяется высокоэффективная жидкостная хроматография (ВЭЖХ) для определения количества 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, при котором: а) готовят раствор образца из указанной композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3 дигидро-2 Н-индол-2-он, путем растворения части указанной композиции в органическом растворителе с последующим разбавлением органическим растворителем растворенной части таким образом, что получают концентрацию (мас./об.), составляющую, исходя из массы указанной части и объема растворителя,приблизительно 1 мг/мл; б) пропускают раствор образца через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую, по существу, из смеси 0,05 М KН 2 РО 4 с рН=5,5-6,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.) при температуре колонки от приблизительно 30 до 40 С с детекцией в УФ-свете при 254 нм УФ; в) детектируют пик, появляющийся на хроматограмме, полученной на стадии (б), между 8 и 10 мин; г) определяют площадь пика (обозначаемую Ас), детектированного на стадии (в); д) готовят стандарт из композиции, состоящей, по существу, из 5-(2-хлорэтил)-1,3-дигидро-2Hиндол-2-она, путем растворения и разбавления части указанной композиции в органическом растворителе таким образом, чтобы концентрация 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она (мас./об.) приблизительно была равна, исходя из массы части и объема растворителя, выбранному значению содержания,при котором или выше которого требуется детекция 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он; е) пропускают стандарт через колонку ВЭЖХ, содержащую стационарную фазу с привитыми цианогруппами, используя мобильную фазу, состоящую, по существу, из смеси 0,05 М KН 2 РО 4 с рН =5,56,5:ацетонитрил:метанол (75:13-17:8-12 об./об./об.), при температуре колонки от приблизительно 30 до 40 С с детекцией в УФ-свете при 254 нм УФ; ж) определяют площадь пика (обозначаемую Apur1) пика на хроматограмме, полученной на стадии (е); и з) рассчитывают количество 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она в композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-он, для чего: 1) рассчитывают коэффициент отклика для 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она по следующей формуле:WS2 = масса части композиции, используемой на стадии (а); иDF = коэффициент разбавления раствора образца. 7. Способ синтеза композиции зипразидона, которая содержит количество дехлорзипразидона не более приблизительно 1000 млн-1, при котором: а) восстанавливают композицию, содержащую 6-хлор-5-(хлорацетил)-1,3-дигидро-2H-индол-2-он и примесь 5-(хлорацетил)-1,3-дигидро-2H-индол-2-она, путем обработки триэтилсиланом в присутствии сильной кислоты с получением композиции, содержащей 6-хлор-5-(2-хлорэтил)-1,3-дигидро-2H-индол-2 она и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; и б) синтезируют композицию, содержащую зипразидон, из композиции, полученной на стадии (а). 8. Способ по п.7, при котором дополнительно: 1) выделяют до стадии (б) образец композиции, полученной на стадии (а), и в указанном образце определяют количество примеси 5-(2-хлорэтил)-1,3-дигидро-2 Н-индол-2-она; 2) определяют, превышает ли количество, определенное в (1), приблизительно 0,28% или не превышает; и 3) если количество, определенное в (1), превышает приблизительно 0,28%, то композицию, полученную на стадии (а), очищают путем перекристаллизации и/или ресуспендирования до количества примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, не превышающего приблизительно 0,28%; и затем переходят к осуществлению стадии (б), используя композицию, полученную на стадии (а), очищенную таким образом; или 4) если количество в (1) не превышает приблизительно 0,28%, тогда переходят к осуществлению стадии (б). 9. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающего количества, выбранного изB) приблизительно 100 млн-1,при котором: а) очищают композицию, содержащую 6-хлор-1,3-дигидро-2H-индол-2-он и примесь оксиндола, до получения композиции, содержащей количество указанной примеси оксиндола, составляющее для (А) - приблизительно 0,3%,для (Б) - приблизительно 0,15% и для (В) - приблизительно 0,03%; и б) используют композицию, полученную на стадии (а), для синтеза композиции зипразидона. 10. Способ синтеза композиции зипразидона, содержащей количество дехлорзипразидона, не превышающее приблизительноB) 100 млн-1,при котором а) осуществляют перекристаллизацию и/или ресуспендирование композиции, содержащей 6-хлор-5(2-хлорэтил)-1,3-дигидро-2H-индол-2-он и примесь 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она, до получения композиции, содержащей не более чем приблизительно для (А) - 0,3%,для (Б) - 0,15% и для (В) - 0,03%,указанной примеси 5-(2-хлорэтил)-1,3-дигидро-2H-индол-2-она; и б) используют композицию, полученную на стадии (а), для синтеза композиции зипразидона. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: A61P 25/00, A61K 31/496
Метки: зипразидона, синтез, композиции, контролируемый
Код ссылки
<a href="https://eas.patents.su/20-7866-kontroliruemyjj-sintez-ziprazidona-i-ego-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Контролируемый синтез зипразидона и его композиции</a>
Композиции, комплексы включения фармацевтически приемлемой соли зипразидона и циклодекстрина и соли зипразидона

Номер патента: 1731
Опубликовано: 27.08.2001
Авторы: Джонсон Кевин Чарльз, Шанкер Рави Мисор, Ким Йесук
МПК: A61K 47/48, A61P 25/18, C07D 417/14...
Метки: композиции, зипразидона, соли, циклодекстрина, фармацевтически, включения, приемлемой, комплексы
Формула / Реферат:
1. Композиция, содержащая фармацевтически приемлемую соль зипразидона, имеющего формулу и циклодекстрин. 2. Композиция по п.1, отличающаяся тем, что указанный циклодекстрин выбран из g-циклодекстрина, гидроксипропил b-циклодекстрина (ГПБЦЦ) и сульфобутилового эфира b-циклодекстрина (СБЭЦД). 3. Композиция по п.2, отличающаяся тем, что указанный циклодекстрин выбран из ГПБЦЦ и СБЭЦД. 4. Композиция, содержащая сухую смесь соли зипразидона и...
Контролируемый ядерный синтез в конфигурации с обращенным полем и прямое преобразование энергии

Номер патента: 6320
Опубликовано: 29.12.2005
Авторы: Ростокер Норман, Монкхорст Хендрик Дж.
МПК: G21D 7/02, G21B 1/00, H05H 1/12...
Метки: преобразование, синтез, конфигурации, прямое, полем, энергии, обращенным, контролируемый, ядерный
Формула / Реферат:
1. Способ преобразования энергии продуктов ядерного синтеза в электрическую энергию, согласно которому инжектируют ионы вдоль спиральной траектории внутрь, по существу, цилиндрической полости, образованной множеством электродов, которые размещены относительно друг друга с интервалами, формируют в полости электрическое поле, имеющее мультипольную структуру более чем с двумя полюсами, для замедления ионов в результате их взаимодействия с...
Составы на основе зипразидона

Номер патента: 2223
Опубликовано: 28.02.2002
Авторы: Аренсон Даниэл Рей, Хосбергер Энджела Кэрол Гэтлин, Расейди Байджэн, Буш Фрэнк Роберт
МПК: A61K 31/496, A61P 25/18, C07D 263/58...
Метки: основе, составы, зипразидона
Формула / Реферат:
1. Композиция, включающая в себя частицы кристаллического свободного основания 5-[2-[4-(1,2-бензизотиазол-3-ил)-1-пиперазинил]этил]-6-хлоро-1,3-дигидро-2Н-индол-2-она (зипразидона) или кристаллического гидрохлорида зипразидона, имеющие средний размер частиц, равный или меньше чем приблизительно 85 мкм, и фармацевтически приемлемый растворитель или носитель. 2. Композиция, как определено по п.1, отличающаяся тем, что указанная композиция включает...
Композиция зипразидона в виде суспензии

Номер патента: 3907
Опубликовано: 30.10.2003
Авторы: Аренсон Даниэл Рей, Кви Хонг
МПК: A61K 31/496
Метки: композиция, суспензии, виде, зипразидона
Формула / Реферат:
1. Композиция в виде суспензии, включающая свободное основание зипразидона или фармацевтически приемлемую кислотно-аддитивную соль зипразидона, воду, полисорбат, агент вязкости и коллоидный диоксид кремния. 2. Композиция по п.1, в которой указанной кислотно-аддитивной солью зипразидона является гидрохлорид зипразидона. 3. Композиция по п.1, в которой указанным полисорбатом является полисорбат 20, 21, 40, 60, 61, 65, 80, 81, 85 или 120. 4....
Эффективный синтез 1,4-дигидро 2н-3,1-бензоксазин-2-она

Номер патента: 1784
Опубликовано: 27.08.2001
Авторы: Фрей Лайза Ф., Тилльер Ричард Д., Грабовски Эдвард Дж.Дж.
МПК: C07C 271/28, C07D 265/18
Метки: 2н-3,1-бензоксазин-2-она, 1,4-дигидро, эффективный, синтез
Формула / Реферат:
1. Способ получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы включающий следующие стадии: 1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы в органическом растворителе с основанием при температуре примерно от 0 до 25шС, в атмосфере инертного газа для получения промежуточного карбамата формулы где R представляет собой арильную боковую цепь хлорформиата, которую определяют как фенил или нафтил, необязательно...
Предыдущий патент: Дезинтоксикационный инфузионный раствор
Следующий патент: Композиции для лечения флавивирусных и пестивирусных инфекций и способы их применения
Случайный патент: Устойчивые к раздавливанию таблетки оксикодона, предназначенные для предотвращения случайного неправильного применения и недозволенного употребления