Соединения 1,5-дифенилпирролидин-2-она как лиганды св-1
















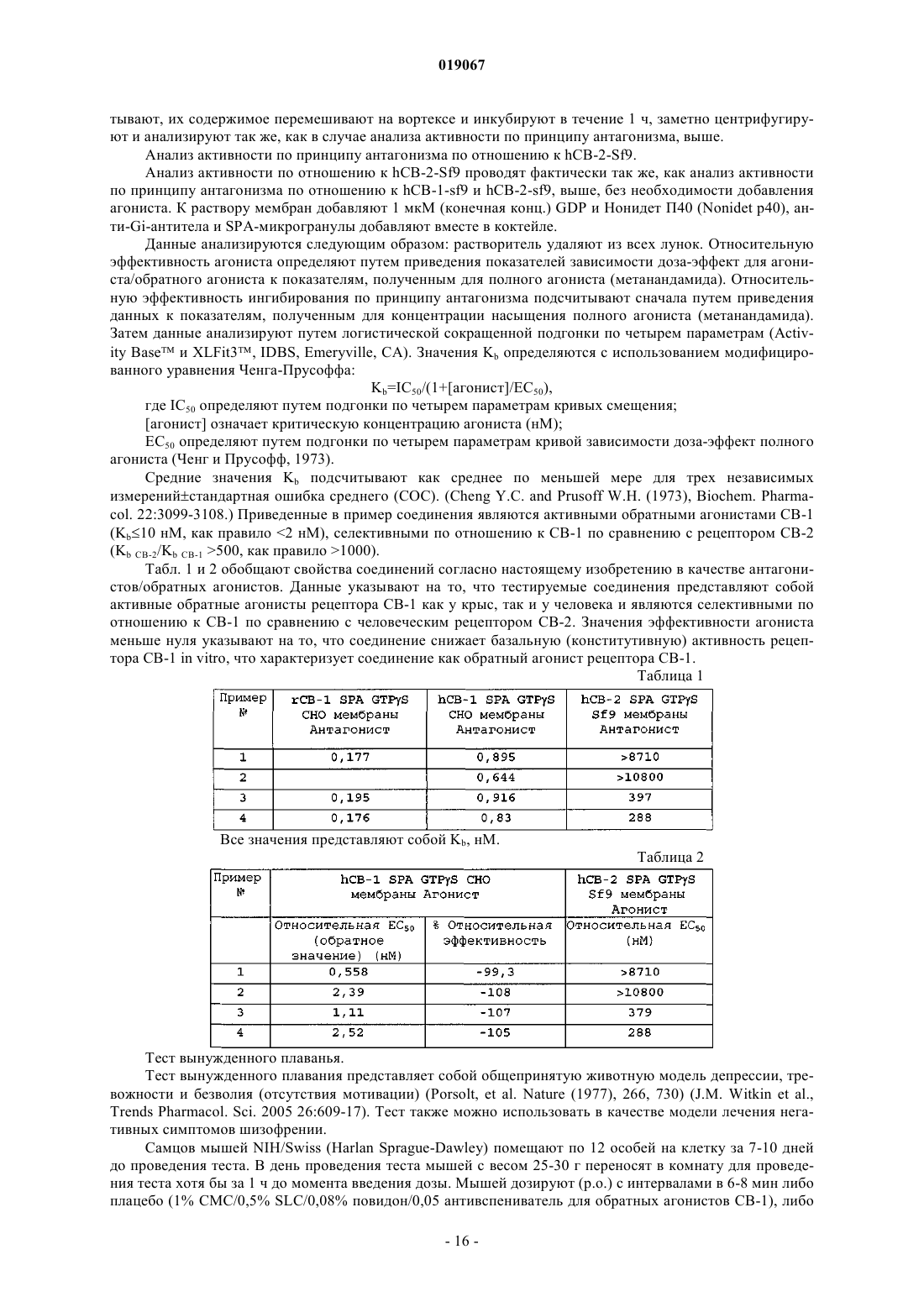



Формула / Реферат
1. Соединение формулы

где R1 выбирают из группы, включающей трифторметил и трифторметокси;
R2 выбирают из группы, включающей водород, галоген и трифторметокси;
R3 представляет собой водород;
R4 выбирают из группы, включающей трифторметил и циклопропил;
при условии, что если R1 представляет собой трифторметил, то R2 представляет собой трифторметокси;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, которое представляет собой (3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-он, или его фармацевтически приемлемая соль.
3. Соединение по п.1, которое представляет собой (3R,5R)-3-[1-(2-циклопропилпиримидин-4-ил)-1-метилэтиламин]-5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-он, или его фармацевтически приемлемая соль.
4. Соединение по п.1, которое представляет собой (3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-фторфенил)-1-(4-трифторметоксифенил)пирролидин-2-он, или его фармацевтически приемлемая соль.
5. Соединение по п.1, которое представляет собой (3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-фенил-1-(4-трифторметоксифенил)пирролидин-2-он, или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 или 5 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент.
7. Применение соединения по любому из пп.1-4 или 5, его фармацевтически приемлемой соли для лечения нарушения, выбранного из расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, ожирения, шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическим средством, или для лечения, направленного на прекращение курения.
8. Применение соединения по любому из пп.1-4 или 5 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения нарушения, выбранного из расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, ожирения, шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическим средством, или для лечения, направленного на прекращение курения.
Текст
Обратные агонисты рецептора СВ-1 - соединения формулы Шаус Джон Менерт (US) Медведев В.Н. (RU) и фармацевтические составы для лечения ожирения или когнитивных нарушений, связанных с шизофренией.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Каннабиоидный рецептор СВ-1 (СВ-1) в основном присутствует в центральной и периферической нервных системах и в меньшем количестве в некоторых периферических органах. Каннабиоидный рецептор СВ-2 (СВ-2) обнаруживается в основном в иммунной системе. Были описаны фармакологические и терапевтические возможности лигандов каннабиоидных рецепторов (Pacher, et al. Pharmacol. Rev.(2006), 58, 389. Была показана эффективность антагонистов/обратных агонистов рецептора СВ-1 в снижении потребления пищи и уменьшении массы тела на животных моделях ожирения. В результате различных исследований было показано, что антагонисты/обратные агонисты СВ-1 также усиливают действие антипсихотических веществ и могут быть эффективными при лечении как негативных, так и когнитивных симптомов шизофрении. Кроме того, действие в отношении уменьшения массы тела, обусловленное действием антагонистов/обратных агонистов СВ-1, было продемонстрировано на животных моделях увеличения массы тела, вызванного лечением антипсихотическими средствами, и поэтому антагонисты/обратные агонисты СВ-1 могут также быть эффективными в контроле связанного с лечением увеличения массы тела и метаболического синдрома, проявляющихся на фоне лечения антипсихотическими средствами. Более того, на животных моделях приема алкоголя было показано, что антагонисты/обратные агонисты рецептора СВ-1 снижают потребление алкоголя и, таким образом, могут быть полезны в лечении алкоголизма и/или злоупотребления психоактивными веществами. Соединения, действующие на рецептор СВ-2, могут оказывать действие на иммунную систему. Поэтому при разработке терапевтических агентов, действующих на рецептор СВ-1, требуется их высокая селективность по отношению к рецептору СВ-1 по сравнению с рецептором СВ-2 во избежание нежелательных эффектов. Ряд антагонистов/обратных агонистов рецептора СВ-1 центрального действия был изучен при лечении ожирения и/или других нарушений. Так, например, в международной публикацииWO 2007/020502 рассматриваются некоторые замещенные соединения пирролидин-2-она в качестве антагонистов СВ-1. Пероральное введение, как правило, является предпочтительным способом введения агентов для лечения ожирения и/или шизофрении. Чтобы соединение имело хорошую биодоступность при пероральном введении, оно, как правило, должно быть хорошо растворимо в воде и иметь достаточную метаболическую стабильность, чтобы минимизировать его пресистемный метаболизм в печени. Эндогенные лиганды каннабиоидных рецепторов и комплементарный сайт, с которым они связываются на рецепторе СВ-1, являются высоко липофильными. Следовательно, известные лиганды рецептора СВ-1 также проявляют свойство липофильности, что приводит к их низкой растворимости. Кроме того, многие лиганды рецептора СВ-1 метаболически нестабильны. Указанные свойства растворимости и/или метаболические характеристики многих соединений-лигандов СВ-1 приводят к их ограниченной абсорбции и биодоступности при пероральном введении. Окислительный метаболизм некоторых соединений может приводить к формированию реактивных электрофильных промежуточных веществ метаболизма. Такие промежуточные вещества способны реагировать с другими нуклеофилами в теле пациента, такими как белки, глутатион, ДНК, РНК и т.д., что может приводить к нежелательным токсическим эффектам. На фармакокинетические свойства терапевтического агента может влиять совместное введение других агентов. Эти так называемые межлекарственные взаимодействия могут приводить либо к повышению, либо к снижению содержания терапевтического агента в плазме крови, что приводит к проблемам переносимости агента или его эффективности. Соединения, которые выводятся метаболическим путем с использованием насыщаемых механизмов (например, CYP3A4, CYP2D6, CYP2C9 и CYP1A2), в особенности подвержены таким межлекарственным взаимодействиям. В свою очередь, соединения, которые ингибируют такие насыщаемые механизмы, способны вызывать такие межлекарственные взаимодействия. Некоторые известные антагонисты/обратные агонисты СВ-1 имеют указанные недостатки. По-прежнему остается необходимость в антагонистах или обратных агонистах рецептора СВ-1,имеющих высокую селективность к СВ-1 по сравнению с СВ-2, проявляющих высокую активность в условиях in vivo (малые значения нМ для Kb), имеющих приемлемую биодоступность, которые не формируют реактивные метаболические промежуточные продукты и которые имеют низкий потенциал для межлекарственных взаимодействий. Соединения согласно настоящему изобретению обладают некоторыми или всеми такими преимуществами. Настоящее изобретение предлагает соединения формулы (I)R4 выбирают из группы, включающей трифторметил и циклопропил; при условии, что если R1 представляет собой трифторметил, то R2 представляет собой трифторметокси; и фармацевтически приемлемые соли указанных соединений. Специалисту в данной области очевидно, что соединения согласно настоящему изобретению могут существовать в формах, имеющих разные точки присоединения отдельных атомов водорода, и являются,таким образом, таутомерными. Отдельные таутомеры, также как и их смеси, рассматриваются в рамках соединений формулы (I), как если бы они были специально проиллюстрированы. Каждая из таутомерных форм может существовать, превращаться в другую форму и подвергаться таутомеризации при определенных условиях. Другой аспект настоящего изобретения обеспечивает фармацевтическую композицию, которая содержит соединение согласно формуле (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество. Один из вариантов реализации этого аспекта настоящего изобретения обеспечивает фармацевтическую композицию, которая содержит соединение согласно формуле (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или вспомогательное вещество, причем оптическая чистота отдельного стереоизомера соединения формулы (I) или его соли больше чем 90%, предпочтительно больше чем 95%, например больше чем 99%. Другой аспект настоящего изобретения обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль для применения в терапии. Один из вариантов реализации этого аспекта настоящего изобретения обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль для применения при лечении расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, увеличением массы тела, ожирения,шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и/или увеличения массы тела,связанного с лечением антипсихотическими средствами, или для лечения, направленного на прекращение курения. В другом своем аспекте настоящее изобретение относится к применению соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства для лечения расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, увеличения массы тела,ожирения, шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики,связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическими средствами, или для лечения, направленного на прекращение курения. Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, адаптированную для лечения расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, ожирения,шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическим средством, или для лечения, направленного на прекращение курения, содержащую соединение формулы (I) или его фармацевтически приемлемую соль совместно с фармацевтически приемлемым носителем, разбавителем или вспомогательным веществом. Соединения формулы (I) представляют собой селективные обратные агонисты или антагонисты рецептора СВ-1. Данные соединения в особенности селективны по отношению к рецептору СВ-1 по сравнению с рецептором СВ-2. Соединения, которые являются обратными агонистами (или антагонистами) рецептора СВ-1, можно использовать для лечения и/или предупреждения патологических состояний,связанных с рецептором СВ-1. Такие патологические состояния включают, например, расстройства пищевого поведения, связанные с чрезмерным потреблением пищи, ожирение, шизофрению, в особенности негативную симптоматику, связанную с шизофренией, например когнитивные нарушения, связанные с шизофренией, злоупотребление алкоголем, алкогольную зависимость, лечение, направленное на прекращение курения, и увеличение массы тела, связанное с лечением антипсихотическими веществами. См. DSM-IV-TR., Diagnostic и Statistical Manual of Mental Disorders. Revised, 4th Ed., Text Revision (2000). Специалисту в данной области очевидно, что существуют альтернативные номенклатуры, нозологические системы и системы классификации патологических физиологических состояний и что эти системы меняются вместе с научным прогрессом в области медицины. Соединения формулы (I) могут также быть полезны для уменьшения массы тела у субъекта независимо от того, можно ли поставить указанному субъекту клинический диагноз ожирение. Необходимость применения соединения в профилактических целях определяют с помощью общеизвестных факторов риска. Эффективное количество определенного соединения определяет, в конечном счете, врач, назначающий лечение, но оно зависит от таких факторов, как конкретное нарушение, которое собираются лечить, тяжесть указанного нарушения и наличие других заболеваний или патологиче-2 019067 ских состояний, выбранный способ введения, прием других лекарственных препаратов и проведение другого лечения, также необходимого пациенту, и других факторов, существенных для заключения врача. Величина терапевтической или профилактической дозы соединения формулы (I) будет, конечно,варьировать в зависимости от веса пациента и его возраста, природы и тяжести патологического состояния, которое собираются лечить, определенного выбранного соединения и желаемого способа введения. Дозу можно вводить в виде однократной суточной дозы или же полную суточную дозу можно вводить в виде нескольких отдельных доз, например два, три или четыре раза в день. Кроме того, в зависимости от свойств определенного выбранного для введения соединения и/или характеристик лекарственной формы (например, модифицированной кинетики высвобождения) дозу можно вводить реже, например один раз в неделю, два раза в неделю, один раз в месяц и т.д. Единичную дозу можно соответственно вводить чаще или реже. При введении трансдермальным путем или в случае продолжительного введения внутривенного раствора режим дозирования будет, конечно, непрерывным в отличие от прерывистого режима введения. При использовании выше по тексту и далее в описании изобретения нижеперечисленные термины,если иное не указано, имеют следующие значения. При использовании в данном патенте термин "(С 1-С 3)алкил" относится к метилу, этилу, пропилу и изопропилу. Термин "галоген" относится к фтору, хлору или брому. Термин "обратный агонист(агонисты)" относится к таким соединениям, которые обладают отрицательной внутренней активностью и способны обращать конститутивную активность рецептора. Обратные агонисты подавляют или обращают активность агонистов. Термин "ожирение" относится к наличию большого количества телесного жира. Человек считается страдающим ожирением, если он или она имеют индекс массы тела (ИМТ) 30 кг/м 2 или больше. В общем, считается, что человек с ИМТ=27-30 имеет избыточный вес. Условно считается, что люди с нормальным весом имеют ИМТ от 19,9 до 26,9. Ожирение может быть обусловлено любой причиной, генетической или связанной с факторами окружающей среды. Примеры нарушений, которые могут способствовать ожирению или быть его причиной, включают избыточное питание, пониженную физическую активность и патологические состояния, связанные со сниженной метаболической активностью. Изобретение не ограничивается конкретным определением ожирения в соответствии с ИМТ стандартом, все такие определения следует рассматривать как эквиваленты. Термин "фармацевтический" или "фармацевтически приемлемый" при использовании в данном патенте в качестве определения означает фактически не токсичный и фактически безвредный для реципиента. Под термином "фармацевтическая композиция" также понимают, что носитель, растворитель,вспомогательные вещества и/или соль должны быть совместимы с активным компонентом композиции(например, соединением формулы (I. Также необходимо понимать, что фармацевтическая композиция согласно настоящему изобретению содержит одно или более соединений формулы (I) и, при желании,может также содержать один или более других активных компонентов для конкретной фармацевтической композиции. Термин "предупреждение/предотвращение" (ожирения или увеличения массы тела) относится к предотвращению состояния ожирения при условии, что лечение проводят до возникновения указанного состояния. Кроме того, если лечению подвергают уже страдающего ожирением субъекта, то такое лечение направлено на предупреждение или предотвращение дальнейшего прогрессирования увеличения массы тела. В качестве примера можно привести предотвращение дальнейшего увеличение массы тела у человека, получающего лечение антипсихотическими средствами. Аббревиатуры при использовании в данном изобретении имеют следующие значения:"BSA" означает бычий сывороточный альбумин."Tr" означает время удерживания. В то время как все соединения согласно настоящему изобретению можно применять в качестве обратных агонистов (или антагонистов) СВ-1, определенные классы являются предпочтительными, как,например, соединения, имеющие любые из следующих перечисленных наборов заместителей. 1) R1 представляет собой -OCF3 или -CF3.-3 019067 2) R2 представляет собой водород, фтор, хлор, трифторметокси. 3) R2 представляет собой трифторметокси. 4) R2 представляет собой трифторметокси и R3 представляет собой водород. 5) R3 представляет собой водород. 6) R4 представляет собой -CF3. 7) R1 представляет собой -OCF3 и R2 представляет собой водород, фтор, хлор, трифторметокси. 8) R1 представляет собой -OCF3; R2 представляет собой трифторметокси и R3 представляет собой водород. 9) R1 представляет собой -OCF3; R2 представляет собой водород, -OCF3; R3 представляет собой водород и R4 представляет собой -CF3. Конкретные предпочтительные соединения согласно настоящему изобретению описаны в данном изобретении в разделе "Примеры" и включают свободные основания и фармацевтически приемлемые соли указанных соединений. Общие схемы Соединения согласно настоящему изобретению могут быть получены в соответствии с нижеследующими схемами синтеза при использовании способов, общеизвестных и признанных в данной области. Подходящие условия реакций для этапов, представленных на этих схемах, известны в данной области, и специалист в данной области сможет подобрать подходящие замены растворителям и сопутствующим реагентам. Подобным образом, специалистам в данной области ясно, что промежуточные соединения синтеза при необходимости или при желании можно изолировать и/или очистить в соответствии с различными известными способами и что часто возможно использовать различные промежуточные вещества непосредственно в последующих этапах синтеза с небольшой очисткой или без нее. Кроме того,специалистам в данной области техники очевидно, что в некоторых обстоятельствах порядок введения веществ не является критичным. Определенный порядок стадий, необходимых для получения соединений формулы (I), зависит от конкретного синтезируемого соединения, исходного соединения и относительной лабильности уходящих групп, что также очевидно для специалиста в области химии. Все заместители, если иное не указано, такие же, как определено ранее, и все реагенты известны и используются в данной области. Схема I Согласно схеме I соединение формулы (II) можно получить в соответствии со способом, описанным Андрейчиковым и коллегами (Андрейчиков и др., Журнал органической химии 22(10), c. 2208-13 (1986,согласно которому смесь амина формулы (1) и альдегида формулы (2) обрабатывают сложным эфиром пировиноградной кислоты (3), где Q1 представляет собой C1-3 алкильную группу, в подходящем растворителе, таком как ледяная НОАс. Подходящие сложные эфиры пировиноградной кислоты включают этил пируват. Реакцию можно проводить в диапазоне температур от комнатной температуры до температуры кипения растворителя. В некоторых случаях продукт (II) может выпадать в осадок в ходе реакции или при добавлении растворителя, в котором продукт плохо растворим. Такие растворители включают изопропиловый спирт, воду и их смеси. Если формируется осадок, соединение формулы (II) можно выделить путем фильтрации и вакуумной сушки или фильтрации и хроматографии. Альтернативно, соединение можно изолировать путем концентрирования реакционной смеси и хроматографии осадка или путем водной экстракции, концентрирования и хроматографии органического экстракта. Схема II Согласно схеме II соединение формулы (III) можно получить при добавлении к соединению формулы (II) воды, возможно в присутствии кислоты. Эту реакцию можно также проводить в присутствии дополнительных растворителей, таких как тетрагидрофуран, уксусная кислота или этанол или их смесей. Часто удобно проводить реакцию в присутствии хотя бы одного эквивалента 2,5-4 019067 диметокситетрагидрофурана. Как только соединение формулы (III) сформировалось, его можно изолировать путем добавления воды и охлаждения с образованием осадка и дальнейшего отделения осадка путем фильтрации или добавления смеси растворителей, таких как толуол и изопропил ацетат, и промывки водой и насыщенным раствором водного бикарбоната натрия. Органический слой можно высушить с помощью десиканта, такого как сульфат натрия, и сконцентрировать с получением продукта в виде неочищенной смеси. Органический слой также можно использовать непосредственно в следующей реакции без дополнительной концентрации или очистки. Схема III Согласно схеме III соединение формулы (IV) можно получить путем обработки раствора соединения формулы (III) соединением формулы (4) в подходящем растворителе, таком как толуол, метанол,изопропил ацетат, или смеси растворителей. Эту реакцию можно также проводить в присутствии катализатора, такого как НОАс. Соединение формулы (IV) можно при желании изолировать в соответствии со способами, известными в данной области, такими как осаждение или хроматография на силикагеле. Схема IV Согласно схеме IV соединение формулы (Ia) можно получить при обработке соединения формулы(IV) в подходящих восстановительных условиях. Подходящие восстановительные условия включают обработку NaCNBH3 в присутствии НОАс в таком растворителе, как тетрагидрофуран. Соединение формулы (Ia) можно изолировать таким способом, как водная экстракция или осаждение продукта. Дальнейшую очистку можно осуществлять с использованием таких способов, как хроматография на силикагеле. В процессе синтеза соединения формулы (Ia) промежуточные соединения как формулы (III), так и формулы (IV) можно непосредственно использовать в последующих реакциях без очистки необработанных промежуточных веществ. Отдельные энантиомеры соединений формулы (Ia) в целом являются предпочтительными по сравнению с соответствующими рацемическими смесями. Индивидуальные энантиомеры можно получить путем разделения соединений формулы (Ia) с использованием способов, таких как продуктивная хроматография на неподвижной хиральной фазе. Энантиомеры можно также получить способом разделения,который включает образование соли рацемической смеси с оптически активной кислотой и очистку желаемой диастереоизомерной соли. Желательную диастереомерную соль можно очистить путем кристаллизации. Альтернативно, любое из промежуточных соединений формулы (II), (III) или (IV) можно расщепить с получением по существу отдельного энантиомера, который можно затем преобразовать согласно способам, описанным выше, с получением соединения формулы (Ia) в энантиомерно чистой форме,например в виде соединения формулы (I). Промежуточные соединения формулы (II), (III) или (IV) можно получить путем расщепления соответствующего рацемического соединения с использованием способов,таких как продуктивная хроматография на хиральной неподвижной фазе. Альтернативный и часто предпочтительный способ получения очищенных энантиомеров соединений формулы (III) приведен на схеме V. Рацемическое соединение формулы (III) реагирует с соединением формулы (5), в котором Q2 представляет собой водород или галоген, с образованием диастереомерной смеси соединений формул (Va) и (Vb). Предпочтительные соединения формулы (5) включают R-альфаметилбензиламин или S-альфа-метилбензиламин. Эту реакцию можно осуществить в соответствии со схемой III, как описано выше, путем смешивания соединения формулы (III) и соединения (5) в инертном растворителе. Эту реакцию можно также провести в присутствии катализатора, такого как НОАс. Диастереомеры формул (Va) и (Vb) затем разделяют с использованием способов, таких как хроматография на силикагеле или кристаллизация из инертных растворителей, таких как изопропанол или метанол/KOH. Желаемый диастереомер (обозначенный (Va на схеме V затем гидролизуют с образованием чистого энантиомера формулы (IIIa). Подходящие условия для гидролиза включают обработку раствора желаемого диастереомера в НОАс с водной соляной кислотой или трифторуксусной кислотой и водой и,возможно, 2,5-диметокситетрагидрофураном. В некоторых примерах неочищенное соединение (IIIa) может содержать значительное количество димера формулы (VI). В соответствии со схемой V рацемическое соединение формулы (III) может представлять собой неочищенный продукт, образованный в результате процесса, представленного на схеме II. Кроме того, препарат очищенного энантиомера формулы (IIIa) (возможно включающий соединение формулы (VI можно использовать в процессе, приведенном на схеме III, непосредственно после реакции гидролиза без дополнительной очистки. На схеме V (R)-энантиомер соединения (5) выбран для иллюстрации процесса. Специалисту в данной области очевидно, что в этой реакции также можно использовать (S)-энантиомер соединения (5). Можно выбрать для использования определенный энантиомер -(R)- или (S)-энантиомер - в зависимости от того, из которого из них получится более легко изолируемый желаемый диастереомер. Схема VI Согласно схеме VI соединение формулы (IVb) можно также получить путем обработки соединения формулы (VI) соединением (4) в таких же условиях, какие описаны для реакции соединения (III) с соединением (4). (Схема (III Схема VII Согласно схеме VII соединение (4) получают путем обработки соединения (9), в котором R10 представляет собой водород, -СН 3, -СН 2 СН 3 или -С(О)СН 3, ацетонитрилом в присутствии кислоты с получением соединения формулы (10). Подходящие кислоты включают серную кислоту или подходящие ки-6 019067 слоты Льюиса, такие как эфират трехфтористого бора. После получения вышеуказанной смеси реакцию нагревают, охлаждают примерно до комнатной температуры и гасят водным гидроксидом натрия. Соединение (10) изолируют с помощью стандартных способов, таких как экстракция подходящим растворителем, таким как этилацетат, и концентрация органического слоя. Соединение (10) нагревают в растворе водной соляной кислоты до примерно 100 С. Соединение экстрагируют диэтиловым эфиром. Водный слой защелачивают с помощью водного гидроксида натрия и экстрагируют диэтиловым эфиром. Органические слои объединяют, сушат над сульфатом натрия, фильтруют и концентрируют с образованием соединения (4). Препараты и примеры Условия для проведения высокоэффективной жидкостной хроматографии (ВЭЖХ). Способы, ссылки на которые имеются в разделах препаратов и примеров. Перемешивают 3-(трифторметокси)бензальдегид (25,2 г, 132 ммоль) и этил пируват (15,3 г, 132 ммоль) в ледяной уксусной кислоте (125 мл) при температуре окружающей среды в течение 10 мин. Добавляют по капле 4-(трифторметил)анилин (46,7 г, 290 ммоль) в течение 15 мин при непрерывном перемешивании, нагревают раствор до 30 С и перемешивают в течение примерно 22 ч. Охлаждают раствор до 26 С, добавляют изопропанол (125 мл) и воду (125 мл). Перемешивают раствор при комнатной температуре в течение 15 мин, фильтруют осадок и промывают смесью изопропиловый спирт/вода (1:1)(559 мл, 5,50 моль) и этил пирувата (605 мл, 5,50 моль) в ледяной уксусной кислоте (5,0 л). Наблюдают экзотермический эффект до 43 С. Перемешивают при температуре окружающей среды в течение 18 ч. Фильтруют осадок и промывают отфильтрованный осадок ледяной уксусной кислотой (500 мл). Сушат в вакууме в течение 3 ч для получения-5-фенил-1-(4-трифторметоксифенил)-3-(4 трифторметоксифениламин)-1,5-дигидропиррол-2-она (1999 г, 74%) в виде кристаллического твердого вещества белого цвета. 1 Н ЯМР (400 МГц, ДМСО-d6):8,43 (с, 1 Н), 7,74 (д, J=12 Гц, 2H), 7,19-7,38 (м, 11 Н), 6,42 (с, 1 Н),6,08 (с, 1 Н). Смешивают 3-фторбензальдегид (10,0 мл, 94,3 ммоль), 4-(трифторметокси)анилин (31,9 мл, 235,7 ммоль) и этил пируват (10,4 мл, 94,3 ммоль) в ледяной уксусной кислоте (150 мл). Перемешивают при температуре окружающей среды в течение 18 ч. Фильтруют осадок и промывают гексанами для получения Смешивают этанол (120 мл), ледяную уксусную кислоту (15 мл), воду (3,0 мл, 164,7 ммоль), трифторуксусную кислоту (6,2 мл, 82,4 ммоль), -5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)-3(4-трифторметилфениламин)-1,5-дигидропиррол-2-он (30,0 г, 54,9 ммоль) и 2,5-диметокситетрагидрофуран (10,7 мл, 82,4 ммоль). Нагревают раствор до 50 С и перемешивают реакционную смесь в течение примерно 18 ч. Прекращают нагревать раствор, добавляют воду (35 мл) и охлаждают реакционную смесь до -19 С. Фильтруют суспензию и промывают твердое вещество смесью вода/метанол 1:4(20 мл). Переносят фильтрат в сортировочную воронку и промывают 6% солевым раствором (280 мл), к органической фазе добавляют 6% солевой раствор (100 мл), метанол (40 мл), диэтиловый эфир (100 мл) и насыщенный раствор бикарбоната натрия (43 мл). Разделяют слои, добавляют метанол (60 мл) к органической фазе и концентрируют раствор примерно до 1 объема, содержащего -5-(3 трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2,3-дион. Добавляют метанол (60 мл) и(R)-4-хлор-альфа-метилбензиламин (7,8 мл, 55,0 ммоль) и перемешивают при комнатной температуре в течение 24 ч. Завершение реакции контролируют с помощью ВЭЖХ (способ 4), затем охлаждают раствор до -7 С и продолжают перемешивание при этой температуре в течение 72 ч. Добавляют предварительно размешанный раствор гидроксида калия (0,69 г, 10,5 ммоль) в метаноле (11 мл) к реакционной смеси, нагревают раствор до 10 С и перемешивают в течение дополнительных 4 ч. Охлаждают раствор до -7 С, фильтруют суспензию и промывают полученный в результате продукт метанолом (5 мл 3). Сушат твердое вещество в вакууме для получения 1-(4-трифторметилфенил)-3-[1(R)-(4-хлорфенил)этиламин]-5(R)-(3-трифторметоксифенил)-1,5-дигидропиррол-2-она (12,3 г, 47,7%) в виде твердого вещества белого цвета. Добавляют уксусную кислоту (464 мл, 8,09 моль), 2,5-диметокситетрагидрофуран (393 мл, 3,03 моль), воду (2,27 л) и трифторуксусную кислоту (153 мл, 2,02 моль) последовательно к раствору -5 фенил-1-(4-трифторметоксифенил)-3-(4-трифторметоксифениламин)-1,5-дигидропиррол-2-она (1000 г,2,02 моль) в THF (8,43 л). Перемешивают реакционную смесь при температуре окружающей среды в течение 18 ч. Добавляют толуол (4,0 л) и изопропил ацетат (2,0 л). Промывают смесь водой (8,0 л) и насыщенным раствором гидрокарбоната натрия (6,0 л). Удаляют водный слой. К органическому слою добавляют (R)-(+)метил бензиламин (390 мл, 3,03 моль). Перемешивают раствор при температуре окружающей среды в течение 3 ч. Концентрируют реакционную смесь для получения смеси 1-(4 трифторметоксифенил)-3-R)-1-фенилэтиламин)-(5)-(S)-фенил-1,5-дигидропиррол-2-она и 1-(4 трифторметоксифенил)-3-R)-1-фенилэтиламин)-(R)-5-фенил-1,5-дигидропиррол-2-она в виде черного масла. Растворяют смесь (888 г, 2,02 моль) в изопропаноле (2,0 л) и охлаждают до -7 С. Фильтруют осадок и промывают холодным изопропанолом. Сушат в вакууме в течение 12 ч для получения 1-(4 трифторметоксифенил)-3-R)-1-фенилэтиламин)-(R)-5-фенил-1,5-дигидропиррол-2-она (130 г, 29%) в виде твердого вещества почти белого цвета. 1 Н ЯМР (400 МГц, ДМСО-d6):7,64 (д, J=8 Гц, 2 Н), 7,34 (д, J=8 Гц, 2 Н), 7,24 (м, 4 Н), 7,11 (м, 4 Н),6,98 (д, J=4 Гц, 2 Н), 5,78 (м, 2 Н), 5,13 (с, 1 Н), 4,30 (м, 1 Н), 1,44 (д, J=4 Гц, 3 Н).(23,4 г, 45,7 ммоль) в THF (45 мл). Перемешивают реакционную смесь при 30 С в течение 18 ч. Наблюдают образование значительного количества(R)-5-(3-фторфенил)-1-(4 трифторметоксифенил)пирролидин-2,3-диона (LC-MS ESI m/z: 352 (М-Н)-, способ 1). Выливают реакционную смесь в раствор толуола (200 мл) и изопропил ацетата (50 мл) в воде (100 мл) и перемешивают в течение 5 мин. Разделяют слои и промывают органический слой водой (50 мл) и 5% раствором гидрокарбоната натрия (50 мл). К органическому слою добавляют (R)-(+)метил бензиламин (7,7 мл, 59,9 ммоль). Перемешивают раствор при температуре окружающей среды в течение 18 ч. Добавляют (R)-(+)-метил бензиламин (3,0 мл, 24,8 ммоль) и нагревают до 30 С в течение 3 ч. Промывают реакционную смесь водой и солевым раствором. Концентрируют реакционную смесь для получения смеси 1-(4 трифторметоксифенил)-3-R)-1-фенилэтиламин)-5(S)-(3-фторфенил)-1,5-дигидропиррол-2-она и 1-(4 трифторметоксифенил)-3-R)-1-фенилэтиламин)-5(R)-(3-фторфенил)-1,5-дигидропиррол-2-она. Растворяют смесь в изопропаноле (70 мл) и перемешивают при температуре окружающей среды в течение 72 ч. Фильтруют осадок и промывают изопропанолом для получения 1-(4-трифторметоксифенил)-3-R)-1 фенилэтиламин)-5(R)-(3-фторфенил)-1,5-дигидропиррол-2-она (6,30 г, 30%) в виде твердого вещества белого цвета. Охлаждают раствор THF (200 мл) и бромидом метилмагния (3,0 М в диэтиловом эфире, 81 мл, 243 ммоль) до 0 С. Добавляют раствор метилового эфира трифторметилпиримидин-4-карбоновой кислоты(16,7 г, 81 ммоль) в THF (100 мл) в течение 5 мин. Перемешивают в течение 30 мин при температуре 0 С,затем медленно наливают в насыщенный раствор водного хлорида аммония. Экстрагируют водный слой диэтиловым эфиром, объединяют органический слои, сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении для получения 2-(2-трифторметилпиримидин-4-ил)пропан-2-ола(16,7 г, 100%) в виде прозрачного бесцветного масла. МС (m/z): 207,2 (М+Н)+. Препарат 8. N-[1-Метил-1-(2-трифторметилпиримидин-4-ил)этил]ацетамид Нагревают раствор ацетонитрила (300 мл) и 2-(2-трифторметилпиримидин-4-ил)пропан-2-ола (16,5 г, 80 ммоль) до 90 С. В отдельном реакционном сосуде охлаждают ацетонитрил (70 мл) до 0 С и добавляют серную кислоту (19,2 мл, 360 ммоль) с такой скоростью, чтобы температура не превышала 10 С. Добавляют охлажденный раствор серной кислоты к нагретому раствору 2-(2-трифторметилпиримидин-4 ил)пропан-2-ола и ацетонитрила и перемешивают при 90 С в течение 60 мин. Охлаждают до комнатной температуры, концентрируют до примерно 1/3 исходного объема и добавляют водный 5 н. NaOH (150 мл). Растирают с этилацетатом и солевым раствором, затем экстрагируют водный слой этилацетатом (3 Х). Концентрируют при пониженном давлении объединенные органические слои для получения N-[1-метил 1-(2-трифторметилпиримидин-4-ил)этил]ацетамида (15,4 г, 78%) в виде твердого вещества желтого цвета. МС (m/z): 248,0 (М+Н)+. Нагревают раствор N-[1-метил-1-(2-трифторметилпиримидин-4-ил)этил]ацетамида (15,4 г, 62 ммоль) и водного 5 н. HCl (150 мл) до 100 С в течение 20 ч, затем охлаждают до комнатной температуры. Экстрагируют реакционную смесь диэтиловым эфиром (2 Х), затем защелачивают водный слой с помощью водного 5 н. NaOH. Экстрагируют водный слой диэтиловым эфиром (3 Х), сушат объединенные органические слои над сульфатом натрия, фильтруют и концентрируют при пониженном давлении для получения 1-метил-1-(2-трифторметилпиримидин-4-ил)этиламина (7,2 г, 56%) в виде коричневого масла. 1 Ссылка: Dujardin, G; Rossignol, S.; Brown, E. Synthesis, 1998, 763-770. Добавляют этил хлороксоацетат (67 мл, 600 ммоль) к раствору ацетата палладия (1,35 г, 6 ммоль), этил винилэфира (315 мл, 3,3 моль) и триэтиламина (125 мл, 900 ммоль) при температуре окружающей среды. Нагревают до 55 С в течение 24 ч, охлаждают до комнатной температуры, затем растирают реакционную смесь с диэтиловым эфиром и водой. Органический слой концентрируют при пониженном давлении для получения этилового эфира Ссылка: Riley, T.A.; Hennen, W.J.; Dalley, N.K.; Wilson, В.Е.; J. Heterocyclic Chem., 1987, 24, 955-964. Нагревают смесь циклопропилкарбамидин гидрохлорида (2,05 г, 17 ммоль), этилового эфира (Е)-4 этокси-2-оксо-бут-3-еновой кислоты (4,39 г, 25,5 ммоль), этанола (12 мл) и этоксида натрия (1,16 г, 17 ммоль) в микроволновой электропечи при 140 С в течение 20 мин. Концентрируют реакционную смесь при пониженном давлении и растирают осадок с этилацетатом и солевым раствором. Отделяют органический слой и концентрируют при пониженном давлении. Очищают осадок с помощью хроматографии на силикагеле (10-30% этилацетат/гексан) для получения этилового эфира 2-циклопропилпиримидин-4 карбоновой кислоты (1,5 г, 46%) в виде светло-желтого масла. МС (m/z): 193,0 (М+Н)+. Препарат 12. 2-(2-Циклопропилпиримидин-4-ил)пропан-2-ол Охлаждают раствор THF (150 мл) и бромида метилмагния (3,0 M) в диэтиловом эфире (52 мл, 243 ммоль) до 0 С. Добавляют раствор этилового эфира 2-циклопропилпиримидин-4-карбоновой кислоты(9,9 г, 52 ммоль) в THF (50 мл) в течение 5 мин. Перемешивают в течение 45 мин при температуре 0 С,затем медленно выливают в насыщенный водный раствор хлорида аммония. Экстрагируют водный слой диэтиловым эфиром, сушат объединенные органические слои над сульфатом натрия, фильтруют и концентрируют при пониженном давлении для получения 2-(2-циклопропилпиримидин-4-ил)пропан-2-ола(9,15 г, 100%) в виде твердого вещества оранжевого цвета. МС (m/z): 179,0 (М+Н)+. Препарат 13. N-[1-(2-Циклопропилпиримидин-4-ил)-1-метилэтил]ацетамид Нагревают раствор ацетонитрила (150 мл) и 2-(2-циклопропилпиримидин-4-ил)пропан-2-ола (8,85 г,50 ммоль) до 90 С. В отдельном реакционном сосуде охлаждают ацетонитрил (50 мл) до 0 С и добавляют серную кислоту (11,9 мл, 223 ммоль) с такой скоростью, чтобы температура не превышала 10 С. Добавляют охлажденный раствор серной кислоты к нагретому раствору 2-(2-циклопропилпиримидин-4 ил)пропан-2-ола и ацетонитрила и перемешивают при 95 С в течение 4 дней. Охлаждают до комнатной температуры и добавляют водный 5 н. NaOH (100 мл). Экстрагируют водный слой этилацетатом (3 Х). Концентрируют при пониженном давлении объединенные органические слои для получения N-[1-(2 циклопропилпиримидин-4-ил)-1-метилэтил]ацетамида (4,5 г, 41%) в виде желтого масла. МС (m/z): 220,0 (М+Н)+. Препарат 14. 1-(2-Циклопропилпиримидин-4-ил)-1-метилэтиламин Нагревают раствор N-[1-(2-циклопропилпиримидин-4-ил)-1-метилэтил]ацетамида (4,5 г, 20,5 ммоль) и водной 5 н. HCl (125 мл) до 100 С в течение 18 ч. Охлаждают смесь до комнатной температуры. Экстрагируют реакционную смесь диэтиловым эфиром (2 Х), затем защелачивают водный слой с помощью водного 5 н. NaOH. Экстрагируют водный слой диэтиловым эфиром (3 Х), сушат объединенные органические слои над сульфатом натрия, фильтруют и концентрируют при пониженном давлении для получения 1-(2-циклопропилпиримидин-4-ил)-1-метилэтиламина (1,3 г, 36%) в виде темно-желтого масла. 1 Н ЯМР (400 МГц, CDCl3):8,45 (д, J=5,6 Гц, 1 Н), 7,12 (д, J=5,6 Гц, 1 Н), 2,30 (м, 1 Н), 1,76 ушир.с,2 Н), 1,41 (с, 6 Н), 1,09 (м, 2 Н), 1,02 (м, 2 Н). Препарат 15. Добавляют трифторуксусную кислоту (7,56 мл, 100 ммоль) к раствору 1-(4-трифторметилфенил)-3[(R)-1-(4-хлор-фенил)этиламин]-5(R)-(3-трифторметоксифенил)-1,5-дигидропиррол-2-она(10,8 г,20,0 ммоль) в уксусной кислоте (100 мл) и воде (5 мл). Перемешивают при температуре окружающей среды в течение 60 мин. Наблюдают образование значительного количества (R)-5-(3 трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2,3-диона (LC-MS ESI m/z: 426 (M+Na)+,Tr=2,76 мин, способ 2). Разбавляют реакционную смесь толуолом (200 мл) и водой (150 мл). Промывают органический слой водой и насыщенным раствором водного бикарбоната натрия. Фильтруют органический слой через сульфат натрия. Добавляют уксусную кислоту (9,17 мл, 160 ммоль) и 1-метил-1-(2 трифторметилпиримидин-4-ил)этиламин (4,51 г, 22 ммоль) к этому раствору толуола, содержащему (R)5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2,3-дион. Нагревают до 55 С в течение 18 ч. Концентрируют реакционную смесь при пониженном давлении для получения темнофиолетового масла. Очищают осадок с помощью хроматографии на силикагеле (25% этилацетат/гексан) для получения (R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-1-(4-трифторметилфенил)5-(3-трифторметоксифенил)-1,5-дигидропиррол-2-она (7,70 г, 65%) в виде фиолетовой пены. Указанное в заголовке соединение получают, по сути, согласно способу получения препарата 15 с использованием 1-(2-циклопропилпиримидин-4-ил)-1-метилэтиламина. Выход 38%, LC-MS ESI m/z: 563 (М+Н)+, Tr=3,46 мин, способ 2. Пример 1. ной кислоте (60 мл) и THF (15 мл) до 0 С и добавляют цианоборгидрид натрия (1,60 г, 25,4 ммоль). Снимают с охлаждающей бани через 5 мин и перемешивают в течение 60 мин при температуре окружающей среды. Разбавляют реакционную смесь этилацетатом и медленно выливают в насыщенный раствор водного бикарбоната натрия. Отделяют органический слой и концентрируют при пониженном давлении. Осадок очищают с помощью хроматографии на силикагеле (15-40% этилацетат/гексан) для получения(3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-трифторметоксифенил)-1-(4 трифторметилфенил)пирролидин-2-она (5,20 г, 69%) в виде фиолетовой пены. Чтобы избавиться от фиолетовой окраски, растворяют (3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3 трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-он в метаноле (150 мл) и добавляют активированный уголь. Перемешивают при температуре окружающей среды в течение 20 мин, фильтруют и концентрируют при пониженном давлении с получением белой пены (4,6 г, 88%). 1 Указанное в заголовке соединение получают, по сути, согласно способу получения примера 1, проводя реакцию при температуре -10 С и используя 1 экв. цианоборгидрида натрия. Выход 42%. 1 Н ЯМР (400 МГц, CDCl3):8,46 (д, J=4,8 Гц, 1 Н), 7,44 (д, J=8,4 Гц, 2 Н), 7,36 (д, J=8,8 Гц, 2 Н), 7,287,24 (м, 2 Н), 7,08 (д, J=7,5 Гц, 1 Н), 7,03 (д, J=7,9 Гц, 1 Н), 6,98 (с, 1 Н), 5,00 (дд, J=6,2, 10,1 Гц, 1 Н), 3,36(0,23 мл, 1,8 ммоль), уксусной кислоты (0,34 мл, 6,0 ммоль) и трифторуксусной кислоты (0,23 мл, 3,0 ммоль) при температуре 35 С в течение 18 ч. Наблюдают образование значительного количества (R)-5(3-фторфенил)-1-(4-трифторметоксифенил)пирролидин-2,3-диона (LC-MS ESI m/z: 354 (М+Н)+, Tr=2,65 мин, способ 2). Наливают реакционную смесь в насыщенный раствор водного бикарбоната натрия и разбавляют 10 мл смеси толуол/изопропил ацетат (1:1). Отделяют органический слой и фильтруют через сульфат натрия. Добавляют уксусную кислоту (0,69 мл, 12,0 ммоль) и 1-метил-1-(2 трифторметилпиримидин-4-ил)этиламин (339 мг, 1,65 ммоль) к этому раствору, содержащему (R)-5-(3 фторфенил)-1-(4-трифторметоксифенил)пирролидин-2,3-дион. Нагревают до 55 С в течение 24 ч. Концентрируют реакционную смесь с получением темного фиолетового масла. Наблюдают образование значительного количества 3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-1-(4-трифторметоксифенил)-5(R)-(3-фторфенил)-1,5-дигидропиррол-2-она (LC-MS ESI m/z: 541 (М+Н)+, Tr=3,32 мин, способ 2). Растворяют неочищенный 3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-1-(4 трифторметоксифенил)-5(R)-(3-фторфенил)-1,5-дигидропиррол-2-он в уксусной кислоте (6 мл) и THF(1,5 мл), охлаждают до 0 С и добавляют цианоборгидрид натрия (189 мг, 3,0 ммоль). Снимают с охлаждающей бани через 5 мин и перемешивают в течение 30 мин при температуре окружающей среды. Разбавляют реакционную смесь этилацетатом и медленно наливают в насыщенный раствор водного бикарбоната натрия. Концентрируют органический слой при пониженном давлении и очищают осадок с помощью хроматографии на силикагеле (10-35% этилацетат/гексан) для получения (3R,5R)-3-[1-метил-1(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-фторфенил)-1-(4-трифторметоксифенил)пирролидин-2- 13019067 Указанное в заголовке соединение получают, по сути, согласно способу получения соединения примера 3. Выход 47%. 1(10 мл). Добавляют моногидрат п-толуолсульфоновой кислоты (1,49 г, 7,71 ммоль) и нагревают до 45 С до тех пор, пока раствор не станет гомогенным. Охлаждают до температуры окружающей среды и добавляют зародыш кристаллизации. Оставляют при температуре окружающей среды на 64 ч. Фильтруют осадок и промывают гептаном. Сушат в вакууме в течение 4 ч для получения тозилата (3R,5R)-3-[1 метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-трифторметоксифенил)-1-(4 трифторметилфенил)пирролидин-2-она (5,06 г, 86%) в виде белого порошка.(1 мл). Добавляют моногидрат п-толуолсульфоновой кислоты (20,0 мг, 104 ммоль) с получением гомогенного раствора. Добавляют тозилат (3R,5R)-3-[1-метил-1-(6-трифторметилпиридин-3-ил)этиламин]-5(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-она (1 мг) для инициации кристаллизации. Оставляют раствор на воздухе и позволяют выпариваться до высыхания в течение 18 ч для получения тозилата(3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3 трифторметоксифенил)-1-(4-трифторметилфенил) пирролидин-2-она (77 мг, 97%) в виде твердого вещества почти белого цвета. Пример 6. Адипат(3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4-ил)этиламин]-5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-он (105 мг, 0,177 ммоль) в метаноле (0,8 мл) и этилацетате (2 мл). Добавляют адипиновую кислоту (30 мг, 0,20 ммоль) и выпаривают при комнатной температуре в течение ночи для получения адипата (3R,5R)-3-[1-метил-1-(2-трифторметилпиримидин-4 ил)этиламин]-5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-она в количественном вещественном балансе (129 мг, 100%) в виде засахаренного твердого вещества.ES/MS m/z: 593,0 [М+Н]+. Как было отмечено, соединения согласно настоящему изобретению представляют собой селективные и высокоактивные обратные агонисты или антагонисты рецептора СВ-1 и, в силу указанных фармакологических свойств, можно применять при лечении различных нарушений. Следующие способы анализа можно использовать для определения активности предложенных соединений по отношению к рецептору СВ-1, их селективности к рецептору СВ-1 и их активности на животных моделях различных нарушений, которые, как предполагается, можно лечить по принципу обратного агонизма или антагонизма по отношению к рецептору СВ-1. Следует отметить, что чистый антагонист, по определению, подавляет лиганд-опосредованную активацию рецептора (т.е. блокирует зависимую от агониста стимуляцию рецептора). Некоторые рецепторы, включая рецептор СВ-1, могут передавать сигнал даже в отсутствие агонистов (эндогенных/экзогенных), это свойство называют базальной активностью рецептора, или конститутивной активностью. В случае таких рецепторов обратные агонисты не только подавляют зависимую от агонистов стимуляцию рецептора, но и снижают/подавляют базальную активность рецептора. Поскольку рецепторы СВ-1 имеют базальную сигнальную активность, обратные агонисты являются предпочтительными по сравнению с чистыми антагонистами для применения в качестве терапевтических агентов при лечении нарушений, СВ-1-опосредованных. Соединения согласно настоящему изобретению являются селективными обратными агонистами или антагонистами рецептора СВ-1. Функциональный анализ активности по отношению к CB-1 и СВ-2 in vitro. Соединения согласно настоящему изобретению можно тестировать на предмет функциональной активности по отношению к рецепторам СВ-1 и СВ-2 по принципу как агонизма, так и антагонизма, с использованием сцинтилляционного анализа близости (SPA), основанного на анализе связывания GTP-35S. Все компоненты для проведения анализа готовят в аналитическом буфере (20 мМ HEPES, 100 мМNaCl, 5 мМ MgCl2, pH 7,4) при комнатной температуре. Полулогарифмические разведения тестируемого соединения готовятся в аналитическом буфере, содержащем BSA (0,125% конечная конц.) для анализа активности по принципу агонизма. Для анализа активности по принципу антагонизма разведения тестируемых соединений готовят аналогичным образом, но также включают 80% эффективной дозы полного агониста (метанандамида). Интенсивность связывания GTP-35S при анализе активности по принципу антагонизма можно измерять в 96-луночном планшете с использованием модифицированного способа"захвата" антител, описанного ранее (DeLapp, N.W, et al. (1999), J. Pharmacol. Exp. Ther 289:946-955.) Активность по принципу агонизма по отношению к рецептору СВ-2 можно измерить с помощью аналогичного способа при использовании hCB-2-Sf9 мембран. Активность по принципу агонизма по отношению к рецептору СВ-1 может быть измерена с помощью способа "захвата" мембран при использовании hCB-1CHO мембран. Все инкубации проводятся при комнатной температуре. Анализ активности по принципу антагонизма. Анализ активности по принципу антагонизма по отношению к hCB-1-CHO и rCB-1-CHO.hCB-1-CHO или rCB-1-CHO мембраны (Applied Cell Sciences, Rockville, MD) и GDP (1 мкМ конечная конц.) добавляют к ледяному аналитическому буферу и гомогенизируют. Ряд концентраций соединения, GTP35S (500 нМ конечная конц.) и мембраны помещают в лунки аналитического планшета и инкубируют в течение 30 мин при комнатной температуре. Затем добавляют смесь, содержащую детергент Нонидет П 4 (Nonidet p40) (0,2% конечная конц.), поликлональные антитела IgG Gi-3 кролика (производитель Covance, Princeton, NJ) и 1,25 мг SPA-микрогранул с антителами против Ig кролика (GEHealthcare, Piscataway, NJ). Планшеты запечатывают, их содержимое перемешают на вортексе и инкубируют в течение дополнительных 2 ч. Планшеты затем центрифугируют при 700 г в течение 10 мин и измеряют радиоактивность. Анализы активности по принципу антагонизма по отношению к hCB-1-Sf9 и hCB-2-Sf9.hCB-1-Sf9 и hCB-2-Sf9 мембраны (Perkin Elmer, Boston, MA) обрабатывают фактически так же, как описано выше, с 1 мкМ (конечная конц.) GDP для hCB-1-Sf9 и 0,05 мкМ (конечная конц.) GDP для hCB2-Sf9. Анализ проводят по существу таким же образом, как описано выше для СНО мембран. Разбавленные мембраны предварительно инкубируют с тестируемым соединением в течение 15 мин с последующим добавлением GTP35S и дальнейшей инкубацией в течение 35 мин. Последовательно добавляют Нонидет П 40 (Nonidet p40) и aHTM-Gi-антитела с инкубациями в течение 15 мин после каждого добавления. Затем добавляют SPA-микрогранулы, планшеты запечатывают и их содержимое перемешивают на вортексе и затем инкубируют при комнатной температуре в течение 1 ч. Анализ активности по принципу агонизма. Анализ активности по принципу агонизма по отношению к hCB-1-CHO.Louis, МО) смешивают и готовят на льду, как для анализа активности по принципу антагонизма, выше. Разбавленные тестируемые соединения, GDP35S (500 нМ конечная конц.) и мембраны смешивают на аналитическом планшете и инкубируют в течение 30 мин. Затем добавляют 1 мг на лунку SPAмикрогранул с агглютинином из проростков пшеницы (GE Healthcare, Piscataway, NJ), планшеты запеча- 15019067 тывают, их содержимое перемешивают на вортексе и инкубируют в течение 1 ч, заметно центрифугируют и анализируют так же, как в случае анализа активности по принципу антагонизма, выше. Анализ активности по принципу антагонизма по отношению к hCB-2-Sf9. Анализ активности по отношению к hCB-2-Sf9 проводят фактически так же, как анализ активности по принципу антагонизма по отношению к hCB-1-sf9 и hCB-2-sf9, выше, без необходимости добавления агониста. К раствору мембран добавляют 1 мкМ (конечная конц.) GDP и Нонидет П 40 (Nonidet p40), анти-Gi-антитела и SPA-микрогранулы добавляют вместе в коктейле. Данные анализируются следующим образом: растворитель удаляют из всех лунок. Относительную эффективность агониста определяют путем приведения показателей зависимости доза-эффект для агониста/обратного агониста к показателям, полученным для полного агониста (метанандамида). Относительную эффективность ингибирования по принципу антагонизма подсчитывают сначала путем приведения данных к показателям, полученным для концентрации насыщения полного агониста (метанандамида). Затем данные анализируют путем логистической сокращенной подгонки по четырем параметрам (Activity Base и XLFit3, IDBS, Emeryville, CA). Значения Kb определяются с использованием модифицированного уравнения Ченга-Прусоффа:Kb=IC50/(1+[агонист]/EC50),где IC50 определяют путем подгонки по четырем параметрам кривых смещения;[агонист] означает критическую концентрацию агониста (нМ);EC50 определяют путем подгонки по четырем параметрам кривой зависимости доза-эффект полного агониста (Ченг и Прусофф, 1973). Средние значения Kb подсчитывают как среднее по меньшей мере для трех независимых измеренийстандартная ошибка среднего (СОС). (Cheng Y.C. and Prusoff W.H. (1973), Biochem. Pharmacol. 22:3099-3108.) Приведенные в пример соединения являются активными обратными агонистами СВ-1(Kb10 нМ, как правило 2 нМ), селективными по отношению к СВ-1 по сравнению с рецептором СВ-2(Kb СВ-2/Kb СВ-1 500, как правило 1000). Табл. 1 и 2 обобщают свойства соединений согласно настоящему изобретению в качестве антагонистов/обратных агонистов. Данные указывают на то, что тестируемые соединения представляют собой активные обратные агонисты рецептора СВ-1 как у крыс, так и у человека и являются селективными по отношению к СВ-1 по сравнению с человеческим рецептором СВ-2. Значения эффективности агониста меньше нуля указывают на то, что соединение снижает базальную (конститутивную) активность рецептора СВ-1 in vitro, что характеризует соединение как обратный агонист рецептора СВ-1. Таблица 1 Все значения представляют собой Kb, нМ. Таблица 2 Тест вынужденного плаванья. Тест вынужденного плавания представляет собой общепринятую животную модель депрессии, тревожности и безволия (отсутствия мотивации) (Porsolt, et al. Nature (1977), 266, 730) (J.M. Witkin et al.,Trends Pharmacol. Sci. 2005 26:609-17). Тест также можно использовать в качестве модели лечения негативных симптомов шизофрении. Самцов мышей NIH/Swiss (Harlan Sprague-Dawley) помещают по 12 особей на клетку за 7-10 дней до проведения теста. В день проведения теста мышей с весом 25-30 г переносят в комнату для проведения теста хотя бы за 1 ч до момента введения дозы. Мышей дозируют (р.о.) с интервалами в 6-8 мин либо плацебо (1% СМС/0,5% SLC/0,08% повидон/0,05 антивспениватель для обратных агонистов СВ-1), либо тестируемым соединением, и помещают в чистые клетки (по 4 мыши на клетку). Для проведения теста каждую мышь помещают в отдельный чистый пластиковый цилиндр (около 10 см в диаметре 25 см высотой), заполненный на 6 см водой с температурой 22-25 С, на 6 мин. Записывается длительность периода неподвижности в течение последних 4 мин. Мышь считается неподвижной, когда она не совершает плавательных движений или же делает только те движения, которые необходимы для удержания головы над водой. Время неподвижности (в секундах) анализируют методом ANOVA с использованием теста Данетта. Минимальная эффективная доза (MED) считается самой низкой дозой тестируемого соединения, при которой наблюдают статистически значимое уменьшение времени неподвижности по сравнению с контролем плацебо. Биодоступность. Способы оценки биодоступности хорошо известны в данной области. В частности, они описаны вMedicinal Research Reviews, Vol. 21,5, 382-396 (2001). Биодоступность соединений по существу можно оценить следующим образом. Используются группы по три или четыре крысы Sprague-Dawley (самцы) весом 250-450 г или собаки Beagle (женского или мужского пола весом приблизительно 10 кг). I.v. дозу в соответствии с исследованием не обязательно вводить животным натощак. Собакам дозу вводят i.v. по канюлированной головной вене, а кровь отбирают из яремной вены. Животные сначала получают дозу 0,25 мг/кг i.v. и затем отбирают образцы крови (0,2 мл) при использовании EDTA в качестве антикоагулянта в 0,0830, 0,25,0,50, 1, 2, 4, 8, 12, 24, 48, 72, 96 и 120 ч. Затем хотя бы через два дня и через 18-24 ч голодания животных дозируют по 1,0 мг/кг путем перорального принудительного кормления. В ходе исследования общий объем собранной крови (мл) не должен превышать 1% от общего веса тела в граммах. При необходимости получения больших объемов крови объем крови образца замещают гепаринизированной цельной кровью от животного-донора. В случае запланированного перекрестного исследования крысы получают объем гепаринизированной цельной крови каждый день проведения исследования после взятия последнего образца, приблизительно равный отобранному в ходе исследования объему. Концентрацию соединения в плазме крови измеряют с помощью способов LC/MS/MS. Данные затем анализируются с помощью стандартного фармакокинетического анализа без компартментализации. Биодоступность при пероральном введении рассчитывают как: Проанализировали соединение примера 5 и было показано, что оно имеют следующую биодоступность у крыс при введении перорально: натощак, особи мужского пола крыс SD;IV доза: 0,25 мг/кг, композиция: 20% МЕОР/80% дистиллированной воды в объемном отношении; РО доза: 1 мг/кг, композиция: 1% NaCMC/0,5% SLS/0,05% антивспенивателя в дистиллированной воде; биодоступность при пероральном введении в среднем составляет 5119% (среднееСО, n=3 крысы) и рассчитана на основании AUC0-24 ч. Метод "отпечатков пальцев" для сравнения с CYP человека. Метод "отпечатков пальцев" является общепринятым способом и используется в качестве показателя потенциального риска межлекарственных взаимодействий в области фармацевтики. Соединения согласно настоящему изобретению можно проанализировать в соответствии с общеизвестными способами по существу следующим образом. Соединения инкубируются при 37 С в конечной концентрации 4 мкМ с объединенными разнородными микросомами человеческой печени и 1 мМ NADPH (никотин аденин динуклеотид фосфатом) (конечная конц.) в течение 0 и 30 мин как в отсутствие ингибиторов CYP, так и с каждым ингибитором CYP по отдельности в течение 30 мин. Каждый ингибитор является специфическим для определенного цитохрома Р 450. Специфические ингибиторы для CYP 2C9, 2D6 и 3 А были представлены сульфафеназолом,хинидином и кетоконазолом, соответственно. Кетоконазол (CYP3A) приготовили в концентрации 25 мМ в ДМСО и затем разбавили в буфере до конечной концентрации 10 мкМ. Хинидин (CYP2D6) приготовили в концентрации 5 мМ в 50/50 смеси ацетонитрил/вода и затем разбавили в буфере до конечной концентрации 10 мкМ. Сульфафеназол (CYP2C9) приготовили в концентрации 100 мМ в ДМСО и затем разбавили в буфере до конечной концентрации 5 мкМ. Образцы анализировали с помощью LC-MS с электрораспылением положительных или отрицательных ионов с использованием хроматографа Acquity фирмы "Waters" для ультра-высокоэффективной ЖХ, совместно с массспектрометром MicroMass Q-Tof-2. Данные анализировали с помощью MetaboLynx версии 4.1. В присутствии ингибитора уменьшение площади пика метаболита менее чем на 30% (по сравнению с контрольной инкубацией без ингибиторов) соответствует низкому риску возникновения межлекарственных взаимодействий, уменьшение площади пика на 30-70% соответствует среднему риску возникновения межлекарственных взаимодействий и уменьшение более чем на 70% соответствует высокому риску возникновения межлекарственных взаимодействий. Соединения, полученные в примерах, анализировали, по существу, как описано выше, и были определены следующие показатели их схожести с CYP. Риск возникновения межлекарственных взаимодействий Эффективность на моделях питания in vivo. Способность соединения согласно настоящему изобретению вызывать снижение массы тела можно анализировать на моделях питания крыс, по существу как описано далее. У самцов крыс Long-Evans вызывают алиментарное ожирение (АО) путем поддержания их с момента окончания грудного вскармливания на рационе, включающем примерно 40% жиров, примерно 39% углеводов и примерно 21% белка, без ограничения в течение хотя бы 12 недель. Группам крыс вводят тестируемое соединение или плацебо(р.о., один раз в день) в течение двух недель. Активность соединения определяют как дозу, необходимую для получения различий в 17 г по сравнению с группой плацебо после лечения в течение двух недель(Т 17 активность). Она отражает минимальное биологически достоверное снижение веса тела на 3-4% по сравнению с весом животных, получавших плацебо в течение двух недель. Модель увеличения массы тела в связи с лечением антипсихотическими препаратами. Способность соединения согласно настоящему изобретению способствовать сохранению/уменьшению массы тела на фоне лечения антипсихотическими средствами можно исследовать на модели питания крыс, по существу, как описано далее. Линию взрослых женских особей крыс SpragueDawley поддерживают на нормальном корме для грызунов Purina LabDiet 5001 (12,3% жира) и воде, без ограничения. Одна группа крыс (n7) получает плацебо (1% молочная кислота) с 1 по 14 день, в то время как остальные крысы получают оланзапин (2 мг/кг, р.о.). На протяжении двухнедельного лечения отслеживают прием пищи, измеряют вес тела и изменения жировой массы. Через 14 дней приема лекарства животных, получающих лечение оланзапином, разделяют на группы (n8), одну группу лечат тестируемым соединением в концентрации 0,3 мг/кг совместно с оланзапином, вторую группу лечат тестируемым соединением в концентрации 1 мг/кг совместно с оланзапином и последнюю группу лечат плацебо совместно с оланзапином в течение с 15-го по 28-й день. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулыR4 выбирают из группы, включающей трифторметил и циклопропил; при условии, что если R1 представляет собой трифторметил, то R2 представляет собой трифторметокси; или его фармацевтически приемлемая соль. 2. Соединение по п.1,которое представляет собой(3R,5R)-3-[1-метил-1-(2 трифторметилпиримидин-4-ил)этиламин]-5-(3-трифторметоксифенил)-1-(4 трифторметилфенил)пирролидин-2-он, или его фармацевтически приемлемая соль. 3. Соединение по п.1, которое представляет собой (3R,5R)-3-[1-(2-циклопропилпиримидин-4-ил)-1 метилэтиламин]-5-(3-трифторметоксифенил)-1-(4-трифторметилфенил)пирролидин-2-он, или его фармацевтически приемлемая соль. 4. Соединение по п.1,которое представляет собой(3R,5R)-3-[1-метил-1-(2 трифторметилпиримидин-4-ил)этиламин]-5-(3-фторфенил)-1-(4-трифторметоксифенил)пирролидин-2-он,или его фармацевтически приемлемая соль. 5. Соединение по п.1,которое представляет собой трифторметилпиримидин-4-ил)этиламин]-5-фенил-1-(4-трифторметоксифенил)пирролидин-2-он, или его фармацевтически приемлемая соль. 6. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 или 5 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент. 7. Применение соединения по любому из пп.1-4 или 5, его фармацевтически приемлемой соли для лечения нарушения, выбранного из расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, ожирения, шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическим средством, или для лечения, направленного на прекращение курения. 8. Применение соединения по любому из пп.1-4 или 5 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения нарушения, выбранного из расстройства пищевого поведения, связанного с чрезмерным потреблением пищи, ожирения, шизофрении, когнитивных нарушений, связанных с шизофренией, негативной симптоматики, связанной с шизофренией, злоупотребления алкоголем, алкогольной зависимости и увеличения массы тела, связанного с лечением антипсихотическим средством, или для лечения, направленного на прекращение курения.
МПК / Метки
МПК: A61K 31/506, A61P 25/18, C07D 403/12
Метки: лиганды, св-1, 1,5-дифенилпирролидин-2-она, соединения
Код ссылки
<a href="https://eas.patents.su/20-19067-soedineniya-15-difenilpirrolidin-2-ona-kak-ligandy-sv-1.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 1,5-дифенилпирролидин-2-она как лиганды св-1</a>
Лиганды ванилоидных рецепторов

Номер патента: 10380
Опубликовано: 29.08.2008
Авторы: Никси Томас, Петтус Липинг Х., Норман Марк Х., Жу Джиаванг, Ванг Хаи-Линг, Дохерти Элизабет М., Кэйтон Джоди, Фэлси Джеймс Ричард, Рзаса Роберт Майкл, Стек Маркиан, Чакрабарти Партха П., Огнянов Вассил И., Фотш Кристофер Х., Чен Нинг, Халм Кристофер, Домингэз Селиа
МПК: C07D 239/34, C07D 401/12, C07D 239/38...
Метки: рецепторов, ванилоидных, лиганды
Формула / Реферат:
1. Соединение общей формулы где J обозначает О или S; Rd обозначает Н; R1 обозначает: (i) фенил, необязательно замещенный одним или более заместителями, выбранными из группы, включающей -С(СН3)3, СН3, CF3, F, Cl, Br, CN, ОН, -OCH3, -C(O)H, -CH2OH, -OCF3, NO2, NH2, -О(СН2)2морфолин, -ОСН2ОСН3, пиперидинил, -OBz, -SMe, пирролидинил, пиридинил, -OSO2CF3, -NHC(O)CH3, фенил, фторфенил, -СН=СН2, -NHC(O)OC(CH3)3, -C(O)OMe, -NHSO2Me, -NHSO2Ph, ...
Циклопентадиенильно-флуоренильные лиганды с углеродными мостиками

Номер патента: 14645
Опубликовано: 30.12.2010
Авторы: Гладиш Джон, Кириллов Евгений, Разави Аббас
МПК: C08F 4/64, C07F 17/00, C08F 10/00...
Метки: циклопентадиенильно-флуоренильные, лиганды, мостиками, углеродными
Формула / Реферат:
1. Способ получения каталитического компонента общей формулыRa2C(3,6-Rb2-Flu)(2-Rc-4-Rd-C5H2)MQ2,где Ra2C является мостиком с одним углеродным атомом и либо оба Ra представляют собой водород, либо один Ra является водородом, а другой Ra является незамещенным или замещенным фенилом, либо оба Ra являются замещенными фенильными группами;Rb, Rc и Rd независимо выбирают из Н или алкила, имеющего от 1 до 12 атомов углерода, или арильных групп,...
Азабициклические лиганды рецепторов 5нт1

Номер патента: 3400
Опубликовано: 24.04.2003
Автор: Брайт Джин Майкл
МПК: C07D 471/04, A61P 25/00, A61K 31/495...
Метки: рецепторов, 5нт1, азабициклические, лиганды
Формула / Реферат:
1. Соединение формулы где R3, R4 и Z независимо выбраны из водорода, галогена (например хлора, фтора, брома или иода), (C1-C4)алкила, возможно замещенного атомами фтора в количестве от одного до трех, (C1-C4)алкокси, возможно замещенного атомами фтора в количестве от одного до трех, и (C1-C4)алкокси(C1-C4)алкила, где каждая алкильная группировка может быть замещена атомами фтора в количестве от одного до трех; W представляет собой...
Лиганды рецепторов 5-нт и их применение

Номер патента: 7183
Опубликовано: 25.08.2006
Авторы: Уэлч Уиллард Маккауэн, Чьянг Юан-Чинг Фиби, Новомайсл Уильям Альберт
МПК: A61K 31/506, A61P 25/28, C07D 239/46...
Метки: рецепторов, применение, лиганды, 5-нт
Формула / Реферат:
1. Соединение формулы (IA) где X и Y представляют собой CR, a Z представляет собой N или X представляет собой N, a Y и Z представляют собой CR, где R для каждого случая представляет собой водород; W представляет собой окси; по меньшей мере один из R1a, R1b, R1d и R1e независимо выбран из группы, состоящей из галогена, нитро, амино, циано, -C(O)NH2 и (C1-C4)алкила, либо R1a и R1b, взятые вместе, образуют пяти- или шестичленное ароматическое или...
Лиганды crth2 рецептора и их применение

Номер патента: 14729
Опубликовано: 28.02.2011
Авторы: Ульвен Тронд, Гримструп Мари, Рист Эйстейн, Ресевё Жан-Мари, Хёгберг Томас, Костенис Эви, Фримурер Томас
МПК: A61K 31/42, A61K 31/415, A61K 31/4166...
Метки: лиганды, применение, рецептора, crth2
Формула / Реферат:
1. Применение соединения формулы (I) или его соли, гидрата или сольватагде А обозначает карбоксильную группу -СООН или тетразолильную группу формулыA1 обозначает водород или метил;кольцо Ar1обозначает фенильное кольцо, при этом возможные заместители выбраны из фтора, хлора, брома, йодо, циано, нитро, трифторметила, трифторметилтио, (C1-C3-алкил)-SO2, NH2SO2, (C1-C3-алкил)-NHSO2-, (C1-C3-алкил)2NSO2-, (C1-C6)алкила, (C1-C6)алкокси, циклоалкила,...
Предыдущий патент: Устройство для обнаружения космических объектов
Следующий патент: Соединение 3-аминокарбазола, фармацевтическая композиция, его содержащая, и способ их получения
Случайный патент: Фосфолипазы, кодирующие их нуклеиновые кислоты и способы их производства и применения