Способ предсказания агрегации белков и конструирования ингибиторов агрегации
Номер патента: 16884
Опубликовано: 30.08.2012
Авторы: Вендрусколо Микеле, Колхофф Кай Й., Сурдо Хесус















Формула / Реферат
1. Способ предсказания потенциальных пептидных последовательностей, ингибирующих агрегацию белков, включающий следующие стадии:
a) идентификация пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени;
b) тестирование, образует ли указанная пептидная последовательность часть β-слоя, причем стадия тестирования включает подстадии идентификации в базе данных белковых структур группы гетерологичных белков, содержащей пептидные последовательности, родственные указанной пептидной последовательности; и идентификации в указанной группе тех белков, в которых указанные родственные пептидные последовательности образуют часть β-слоя;
c) если на стадии b) достигнут положительный результат, извлечение из указанной базы данных белковых структур остатков, которые образуют соседние цепи указанного β-слоя;
d) идентификация остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка,
где подстадия идентификации включает сравнение указанных родственных пептидных последовательностей с остатками, содержащимися в строках "SHEET" в файле PDB для представляющего интерес белка.
2. Способ по п.1, где указанные родственные пептидные последовательности включают указанную пептидную последовательность и фрагменты указанной пептидной последовательности.
3. Способ по п.1 или 2, где указанные родственные пептидные последовательности включают последовательности, содержащие консервативные замены одной или более аминокислот в указанной пептидной последовательности.
4. Способ по любому из пп.1-3, где подстадия идентификации включает идентификацию тех остатков в указанных родственных пептидных последовательностях, которые образуют друг с другом водородные связи; предпочтительно в целях идентификации тех остатков, которые образуют водородные связи друг с другом, вычисляют эвклидово расстояние между каждой парой остатков, которые отделены по меньшей мере тремя остатками, и считают, что водородная связь образуется, если расстояние составляет менее 3,075 ангстрем.
5. Способ по любому из предшествующих пунктов, где остатки, идентифицированные на стадии d), представляют собой те остатки, боковые цепи которых взаимодействуют с указанной пептидной последовательностью посредством водородных связей.
6. Способ по любому из предшествующих пунктов, где на стадии идентификации используют профиль склонности к агрегации.
7. Способ по любому из пп.1-5, где стадию идентификации проводят экспериментально.
8. Способ по любому из предшествующих пунктов, дополнительно включающий стадии тестирования, взаимодействуют ли остатки, идентифицированные на стадии d), с одним или более другими белками; где предпочтительно стадию тестирования:
(i) проводят на множестве белков, гетерологичных указанному белку-мишени, и/или
(ii) проводят с использованием базы данных белковых структур, которые необязательно опосредуют важные клеточные процессы,
и она необязательно включает подстадии идентификации группы белков, содержащихся в указанной базе данных, которые содержат родственные пептидные последовательности, сходные с указанной пептидной последовательностью; и идентификации в указанной группе тех белков, в которых указанные родственные пептидные последовательности взаимодействуют с указанными идентифицированными остатками.
9. Способ по любому из предшествующих пунктов, где часть области агрегации, идентифицированная на стадии а), представляет собой часть спирали, петлю, β-поворот или β-изгиб.
10. Способ по любому из предшествующих пунктов, включающий получение ингибирующего агрегацию белка пептида, содержащего остатки, идентифицированные на стадии d).
11. Способ по любому из предшествующих пунктов, дополнительно включающий стадии:
e) синтеза пептидной библиотеки, причем члены указанной библиотеки содержат остатки, идентифицированные на стадии d); и
f) определения аффинности членов указанной библиотеки в отношении указанного белка-мишени.
12. Способ получения пептида, ингибирующего агрегацию белка, включающий следующие стадии:
a) идентификация пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени;
b) тестирование, образует ли указанная пептидная последовательность часть β-слоя;
c) если на стадии b) достигнут положительный результат, извлечение из указанной базы данных белковых структур остатков, которые образуют соседние цепи указанного β-слоя;
d) идентификация остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка;
e) синтез пептидной библиотеки, причем члены указанной библиотеки содержат остатки, идентифицированные на стадии d); и
f) определение аффинности членов указанной библиотеки к белку-мишени.
13. Способ по п.11 или 12, включающий идентификацию в указанной библиотеке пептида, который проявляет высокую аффинность к белку-мишени относительно контролей, в качестве пептида, ингибирующего агрегацию белка, где предпочтительно способ дополнительно включает:
(i) выделение пептида, идентифицированного в указанной библиотеке, или
(ii) синтез пептида, идентифицированного в указанной библиотеке.
14. Способ по п.10, включающий определение способности указанного пептида к одному или более из следующих:
i) стабилизация белка в отношении агрегации;
ii) снижение скорости потери активности белка при хранении;
iii) снижение опосредуемой агрегацией иммуногенности белка;
iv) повышение выхода белков в системах трансляции in vitro;
v) повышение стабильности в растворе состава для терапевтического применения;
vi) ингибирование одного или более клеточных процессов;
vii) предотвращение олигомеризации или мультимеризации белка.
15. Способ конструирования соединения для (А) стабилизации белка в отношении агрегации или (В) повышения стабильности состава для терапевтического применения в растворе, включающий стадии предсказания пептидов, ингибирующих агрегацию белков по любому из пп.1-9 со стадией тестирования, проводимой на множестве белков, гетерологичных указанному белку-мишени, и применения остатка(ов), идентифицированного на стадии d) этого предсказания, для конструирования соответственно (А) соединения для стабилизации белка в отношении агрегации или (В) соединений с повышенной стабильностью в растворе.

Текст
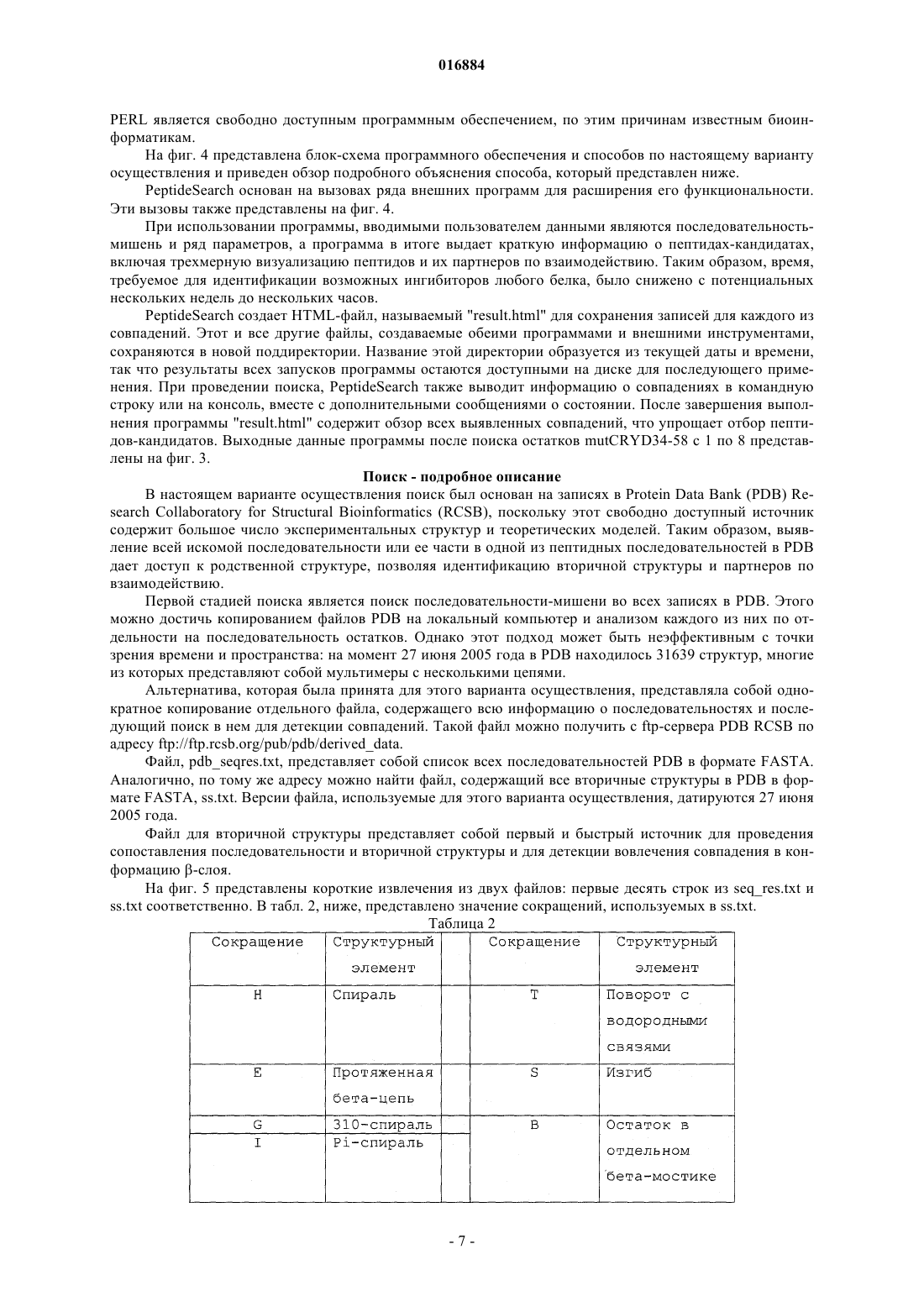
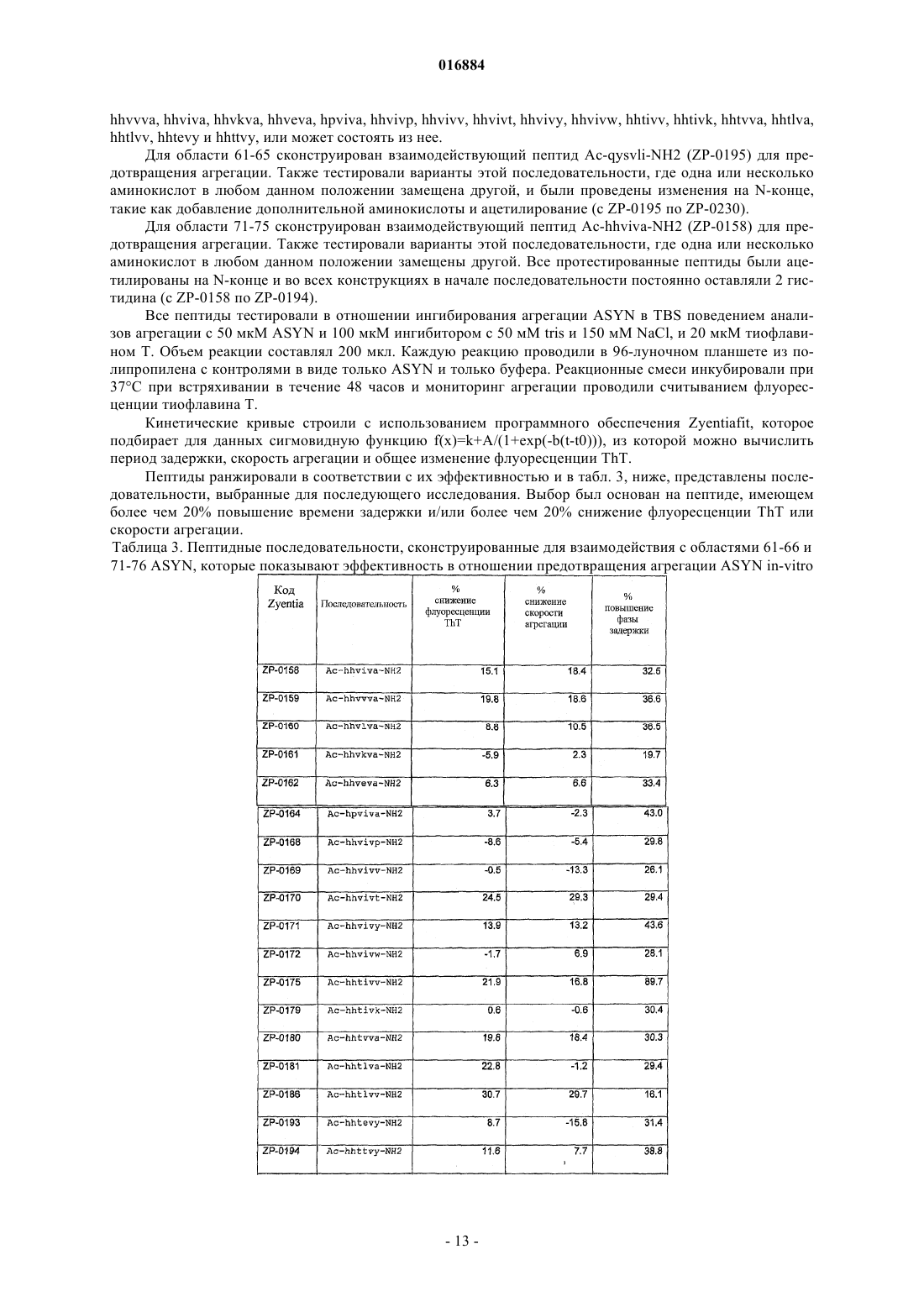
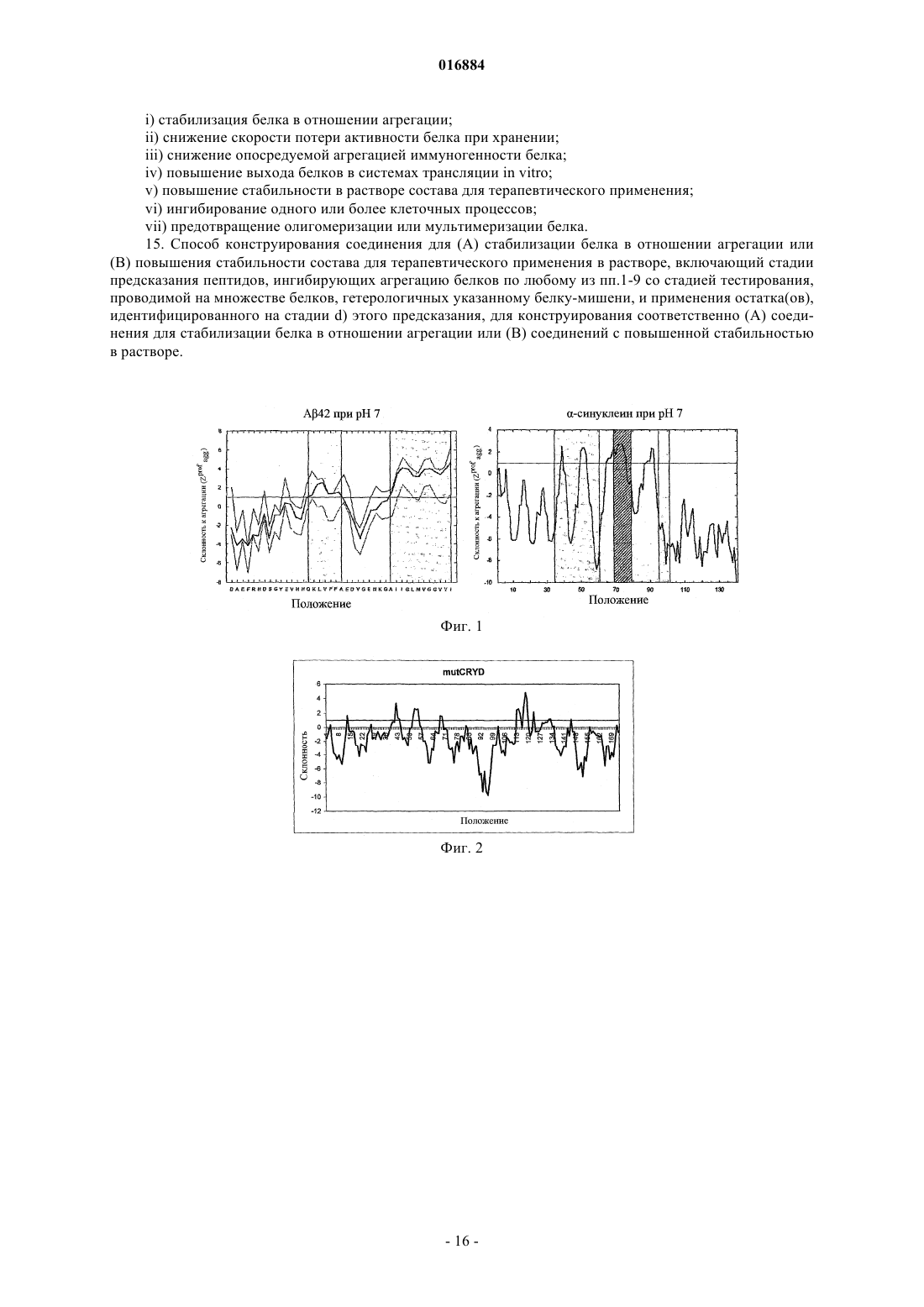
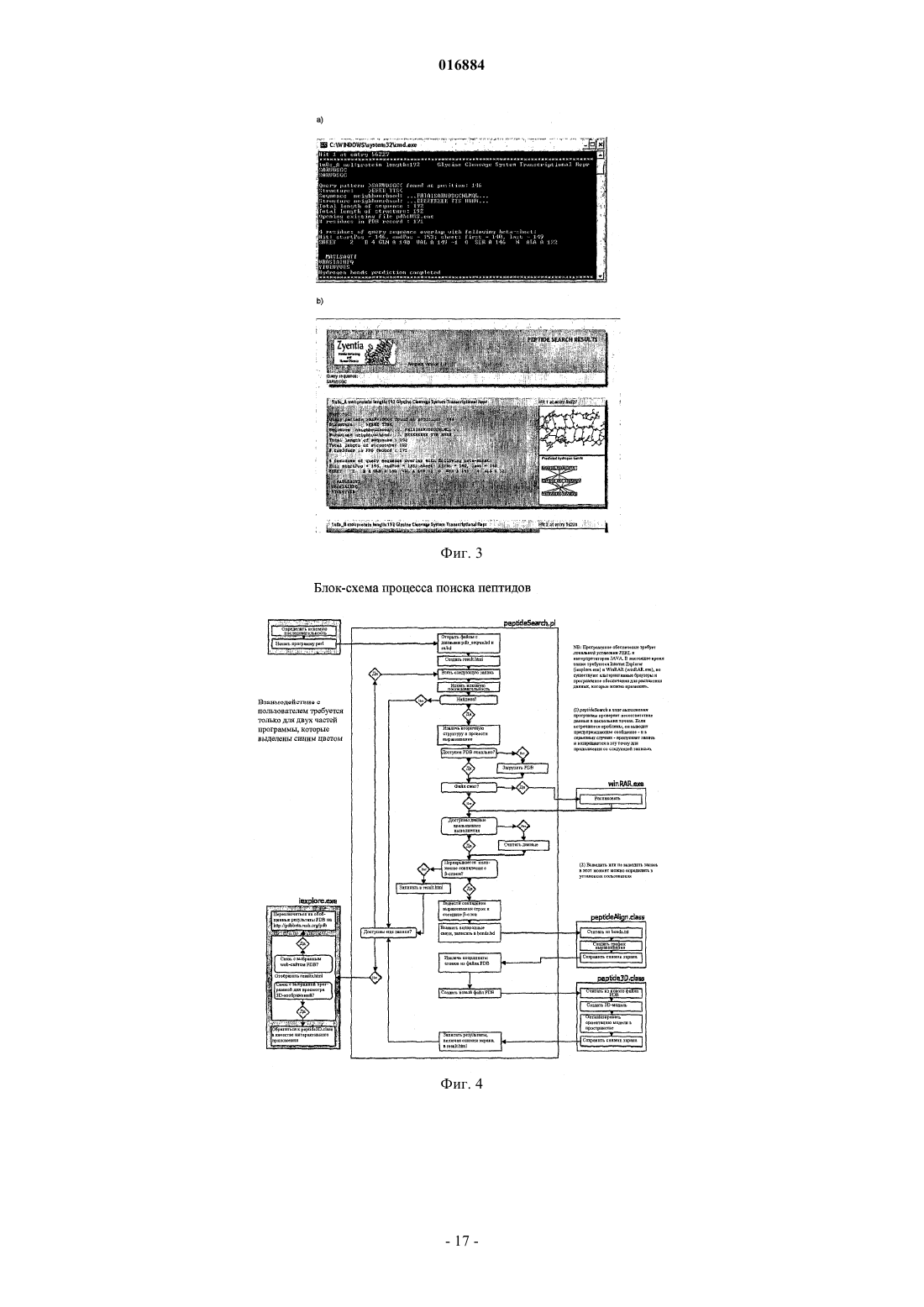
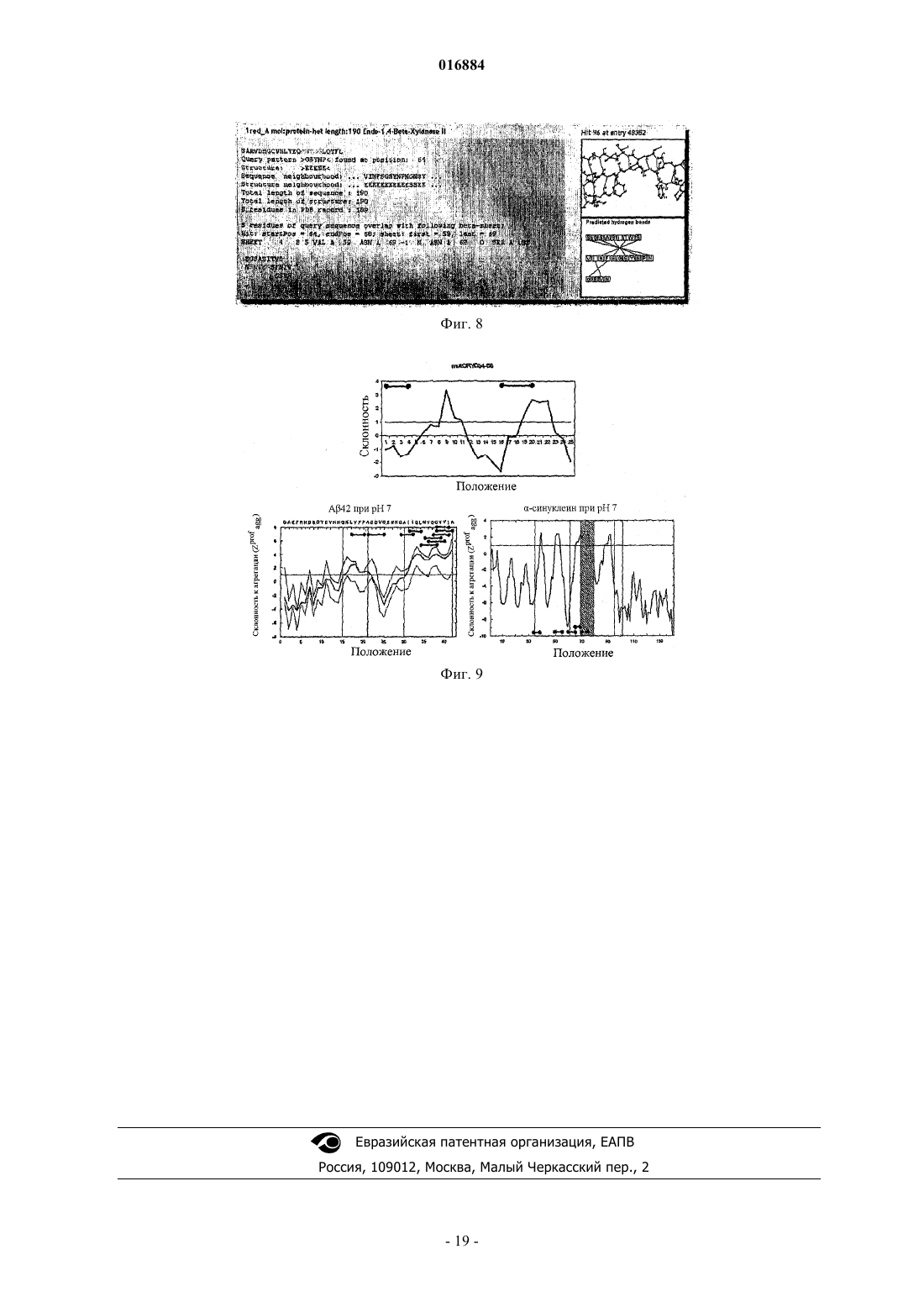
СПОСОБ ПРЕДСКАЗАНИЯ АГРЕГАЦИИ БЕЛКОВ И КОНСТРУИРОВАНИЯ ИНГИБИТОРОВ АГРЕГАЦИИ Изобретение относится к способам предсказания агрегации белков и конструирования ингибиторов агрегации. Один из таких способов предсказания потенциальных пептидных последовательностей, ингибирующих агрегацию белка, включает стадии: а) идентификации пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени; b) тестирования образования указанной пептидной последовательностью слоя; с) если на стадии b) достигнут положительный результат, извлечения соседних цепей слоя; d) идентификации остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка. Также настоящее изобретение относится к способом конструирования соединений с применением остатков, идентифицированных в указанном выше способе; к соединениям, продуцируемым этими способами, и к компьютерным программам для осуществления указанных выше способов. 016884 Изобретение относится к способам предсказания агрегации белков и конструирования ингибиторов агрегации. В частности, в качестве неисключающих примеров, оно относится к способам, которые помогают конструировать соединения для стабилизации белков относительно агрегации, таким образом, потенциально повышая время полужизни белков, снижая иммуногенность белков и повышая выход в системах трансляции in vitro. Предпосылки изобретения Выявлено, что отложения неправильно свернутых белков в клетках или во внутриклеточном пространстве играют роль в ряде тяжелых медицинских нарушений, к которым относятся заболевания, такие как болезнь Альцгеймера, болезнь Паркинсона и диабет 2 типа. Средства, затрачиваемые системами медицинского обслуживания по всему миру для лечения этих медицинских состояний, являются огромными, так же как и влияние на жизни больных и их семей. Вероятно, с увеличением продолжительности жизни количество случаев будет постоянно возрастать. Для решения этой нарастающей проблемы разрабатывают новые способы терапии на основе препятствия возможности образования агрегатов белков на ранних стадиях. Типичный жизненный цикл белка в клетке начинается с синтеза полипептида в рибосоме и продолжается от исходно несвернутого состояния путем сворачивания, который может включать один или несколько промежуточных продуктов, до биологически активного нативного состояния белка. Для большинства белков это нативное состояние соответствует плотно свернутой конформации, хотя существуют некоторые исключения, одним из которых является -синуклеин, который в нативном состоянии является несвернутым (Uversky VN (2002) Natively unfolded proteins: A point where biology waits for physics, Protein Sci. 11:739-756). Жизненный цикл завершается денатурацией и деградацией. Клетка обладает сложными механизмами контроля качества, которые способствуют процессу сворачивания белка. Первым из них является непосредственно рибосома. Во-вторых, белок поддерживают белки теплового шока и шапероны, которые действуют в качестве катализатора или промотора для правильного свертывания белка или для повторного свертывания неправильно свернутых белков (Evans MS,Clarke TF IV, Clark PL (2005) Conformations of Co-Translational Folding Intermediates, Prot, Pept. Let.12(2): 189-195). Если повторного сворачивания не происходит, белки с нарушенным сворачиванием перерабатываются системой убиквитин-протеосома. На первой стадии убиквитин присоединяется к нарушенным структурам. Эта метка маркирует полипептидную цепь для направления на деградацию, а эту задачу выполняет протеосома. Более подробное описание процессов сворачивания и нарушенного сворачивания можно найти в Dobson CM (2003) Protein folding and misfolding, Nature 426: 884-890 и Vendruscolo M,Zurdo J, MacPhee CE, Dobson CM (2003) Protein folding and misfolding: a paradigm of self-assembly andregulation in complex biological systems, Phil. Trans. R. Soc. Lond. A 361: 1205-1222). Однако контроль качества в клетке по многим причинам может нарушаться, что ведет к накоплению белков с нарушенным сворачиванием. Затем белки могут агрегировать, образуя плотные структуры, называемые амилоидными фибриллами с центральной областью, состоящей из сплошных скоплений -слоев (Dobson CM (2005)Prying into prions, Nature 435: 747-749). В живой ткани отложение белков (часто в форме амилоидных агрегатов) часто ассоциировано с различными заболеваниями, многие из которых связаны со старением. Например, эти заболевания включают нейродегенеративные заболевания, такие как болезнь Паркинсона, болезнь Альцгеймера и губкообразные энцефалопатии, а также системные (такие как амилоидозы легкой цепи иммуноглобулина или транстиретина) и периферические тканевые нарушения (такие как диабет 2 типа). У человека известно более 30 различных нарушений, ассоциированных с отложением белка. В частности, в развитом мире, где продолжает непрерывно расти продолжительность жизни, серьезную проблему для общества представляет постоянно увеличивающееся количество людей, страдающих этими заболеваниям. Согласно оценкам, в 2000 году только в США приблизительно 4,5 миллионов человек страдали болезнью Альцгеймера, а к 2050 году количество случаев может возрасти до 16 миллионов (Hebert LE,Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer Disease in the U.S. Population: PrevalenceEstimates С using the 2000 Census, Arch. Neurol. 60: 1119-1122). Риск людей заболеть этим нейродегенеративным заболеванием оценивается как 1 из 10 для людей старше 60 лет и практически 1 из 2 для людей старше 85 лет (Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, Hebert LE, Hennekens CH, Taylor JO (1989) Prevalence of Alzheimer Disease in a Community Population of Older Persons.Higher, than Previously Reported, Jama 262: 2551-2556). Влияние на систему здравоохранения является очень значительным, и некоторые авторы предсказывают, что нейродегенеративные заболевания могут стать основной причиной смерти (Lozano AM, Kalia SK (2005) New Movements in Parkinsons's, Sci. Am.,291(1): 58-65). Кроме того, склонность биомолекул к образованию агрегатов в растворе всегда была одной из основных проблем при создании лекарственных средств. Терапевтические молекулы должны быть как растворимыми, так и реакционноспособными и не должны образовывать агрегаты при введении в относи-1 016884 тельно высоких концентрациях или при хранении в течение длительных периодов времени. Во многих случаях, выявление условий, при которых такие полипептиды являются достаточно стабильными, требуют много времени и является дорогостоящим, а иногда даже невозможным при доступных в настоящее время способах. Таким образом, выявление способов препятствования процессу сворачивания в целях предотвращения образования агрегатов может повысить эффективность разработки лекарственных средств. Сущность изобретения Таким образом, для предпочтительного содействия решению указанных выше задач желательно иметь возможность создавать соединения, которые взаимодействуют с патологическим белком или с терапевтической молекулой в растворе, так чтобы соединение конкурентно связывало и блокировало наиболее важные участки, запускающие процесс агрегации. Одно из приближений к достижению этой цели может включать конструирование образованных из пептидов молекул, которые будут препятствовать процессу агрегации. Таким образом, в широком смысле, один из аспектов настоящего изобретения относится к способу конструирования пептидов, ингибирующих агрегацию белков, включающему идентификацию пептидных последовательностей, боковые цепи которых будут взаимодействовать со склонной к агрегации областью в белке-мишени. Первый аспект настоящего изобретения относится к способу предсказания потенциальных пептидных последовательностей, ингибирующих агрегацию белков, включающему стадии:a) идентификации пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени;b) тестирования, образует ли указанная пептидная последовательность части -слоя;c) если на стадии b) достигнут положительный результат, извлечение соседних цепей того слоя;d) идентификации остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка. Предпочтительно стадию тестирования проводят на множестве белков, гетерологичных белкумишени. Таким образом, даже когда область агрегации в белке-мишени не образует часть -слоя, можно идентифицировать соответствующие пептидные последовательности из сходных или идентичных последовательностей, встречающихся в других белках, которые образуют часть -слоя. Это тестирование можно проводить с использованием базы данных белковых структур и предпочтительно тестирования образования указанной пептидной последовательностью части -слоя в любой из известных белковых структур, присутствующих в этой базе данных белков. В вариантах осуществления этого изобретения используют данные из Protein Data Bank (PDB) из Research Collaboratory for StructuralBioinformatics (RCSB), который содержит большое количество экспериментальных структур и теоретических моделей (Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, BournePE (2000) The Protien Data Bank, Nuc. Acids Res. 28: 235-242). Ha момент 27 июня 2005 года в PDB было 31639 структур. Однако можно использовать другие структурные базы данных или банки данных. Первой стадией предсказания пептида, ингибирующего агрегацию, является идентификация одной или нескольких областей агрегации в белке-мишени и пептидных последовательностей, образующих по меньшей мере часть этой области. Предпочтительным способом идентификации этой области является использование профиля агрегации амилоида. Этот теоретический способ предсказания горячих точек агрегации в полипептидных цепях описан в DuBay KF, Pawar АР, Chiti F, Zurdo J, Dobson CM, Vendruscolo M (2004) Prediction of theAbsolute Aggregation Rates of Amyloidogenic Polipeptide Chains, J. Mol. Biol. 341: 1317-1326 и в Pawar АР,DuBay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (2005) Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases, J. Mol. Biol. 350: 379-392. В способе предоставлен алгоритм, который, на основе присущих аминокислотам свойств, можно использовать для вычисления профиля склонности к амилоидной агрегации для любого белка. После получения профиля амилоидной агрегации для белка-мишени можно идентифицировать области агрегации с учетом частей белка, для которых склонность к агрегации в профиле превышает заданную величину (например, 1). Альтернативно или дополнительно, области агрегации можно идентифицировать посредством экспериментальных измерений, например, посредством систематической мутации каждого из остатков в пептиде или белке, или их фрагменте и синтеза фрагментов таких пептидов или белков, или фрагментов и анализа их склонности к агрегации в анализах in vitro. Предпочтительно стадия тестирования включает подстадии: идентификации содержащейся в базе данных белковых структур группы белков, содержащей пептидные последовательности, родственные пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени; и идентификации в этой группе тех белков, в которых указанные родственные пептидные последова-2 016884 тельности образуют часть -слоя. Для повышения возможного количества совпадений (т.е. идентифицированных белков, в которых родственные пептидные последовательности образуют часть -слоя) родственные пептидные последовательности предпочтительно включают представляющую интерес пептидную последовательность и фрагменты указанной пептидной последовательности. Альтернативно или дополнительно, для повышения возможного количества совпадений родственные пептидные последовательности могут включать последовательности, которые включают консервативные замены одной или нескольких аминокислот в представляющей интерес пептидной последовательности. В рамках этой стадии консервативная замена представляет собой замену, которая сохраняет связанные с агрегацией свойства подлежащей замене аминокислоты. В частности, консервативные заместители могут быть выбраны из аминокислот со склонностью к агрегации при рН 7,0, отличающимися друг от друга в пределах 0,2. Альтернативно или дополнительно консервативные заместители могут быть выбраны из остатков со сходными свойствами (например, основные, кислые, гидрофобные, полярные, ароматические). Более предпочтительно родственные пептидные последовательности включают одну или несколько из указанных выше пептидных последовательностей как в прямом, так и обратном порядке. В одном из вариантов осуществления настоящего аспекта подстадия идентификации группы белков, содержащихся в базе данных, включает сравнение родственных пептидных последовательностей с остатками, содержащимися в строках "SHEET" в файле PDB для рассматриваемого белка. Альтернативно или дополнительно подстадия идентификации группы белков, содержащихся в базе данных, включает идентификацию тех остатков в родственных пептидных последовательностях, которые образуют друг с другом водородные связи. Одним из способов, которым можно идентифицировать остатки, образующие друг с другом водородные связи, является вычисление эвклидова расстояния между каждой парой остатков, которые отделены по меньшей мере тремя остатками, причем полагают, что водородные связи образуются, если это расстояние составляет менее 3,2 Ангстрем, а более предпочтительно, если это расстояние составляет менее 3,075 Ангстрем. В одном из вариантов осуществления эти эвклидовы расстояния вычисляют с использованием записей "АТОМ" из файла PDB для рассматриваемого белка. Предпочтительно способ включает дополнительную стадию отображения идентифицированных пар остатков, которые образуют водородные связи друг с другом, и образованных водородных связей. Для проверки достоверности результатов при любом из указанных выше способов идентификации групп белков результаты обоих способов можно использовать для перекрестной проверки остатков,идентифицированных другим способом. Такую перекрестную проверку, альтернативно или дополнительно, можно проводить другими способами идентификации групп белков. Способ предпочтительно включает дополнительную стадию отображения остатков, идентифицированных на стадии d), а также он может включать стадию отображения информации о белке, в котором идентифицированы эти остатки. Это отображение также может принимать форму 3-мерного расположения идентифицированных остатков в -слое. Предпочтительно способ включает дополнительную стадию модификации остова соседних цепей и боковых цепей потенциальной пептидной последовательности, ингибирующей агрегацию белка, не принимающих участие во взаимодействии с указанной пептидной последовательностью, образующей часть области агрегации в белке-мишени, для максимизации взаимодействия с указанной пептидной последовательностью и повышения потенциальных свойств ингибирования агрегации потенциальной ингибирующей агрегацию последовательности. После идентификации одной или нескольких пептидных последовательностей или "матриц" можно сконструировать и синтезировать пептидную библиотеку, которая вносит вариабельность в области матрицы, не вовлеченные непосредственно во взаимодействие с рассматриваемой областью агрегации. Затем эту библиотеку можно подвергать скринингу с использованием запатентованных биохимических анализов агрегации и цитотоксичности для исследования изменений скоростей агрегации и токсичности белков в присутствии различных соединений. Предпочтительно библиотеку создают посредством добавления модификаций к аминокислотной последовательности(ям)-кандидату, которые повышают свойства, такие как стабильность и растворимость. Как описано в настоящем документе, способ, например, может включать: е) синтез пептидной библиотеки, причем члены указанной пептидной библиотеки содержат остатки, идентифицированные на стадии d), и f) определение аффинности связывания членов указанной библиотеки с белком-мишенью. В библиотеке можно идентифицировать один или несколько пептидов, которые связываются с белком-мишенью с высокой аффинностью относительно контролей. Такие пептиды могут представлять собой пептиды-кандидаты, ингибирующие агрегацию белка. Пептид, для которого выявлено, что он с высокой аффинностью связывается с мишенью, можно выделять, очищать и/или синтезировать.-3 016884 Пептидные последовательности, предсказанные или идентифицированные, как описано выше, можно подвергать скринингу в отношении препятствия клеточным процессам (т.е. токсикологию). Например, способ может включать: тестирование наличия взаимодействия остатков, идентифицированных на стадии d) , указанной выше, с одним или несколькими не являющимися мишенью белками, находящимися в Protein Data Bank (или любой другой базе данных белков), предпочтительно не являющимися мишенью белками, которые могут опосредовать важные клеточные процессы, такие как метаболические каскады, структурными белками гомеостаза ионов, белками, вовлеченными в ответ на стресс, регуляцию экспрессии генов, репарацию ДНК и т.д. Стадию тестирования можно проводить на множестве белков, гетерологичных указанному белкумишени, предпочтительно с использованием базы данных белковых структур. Например, тестирование можно проводить посредством идентификации в базе данных групп белков, которые содержат родственные пептидные последовательности, сходные с тестируемой пептидной последовательностью; и идентификации в группе тех белков, в которых родственные пептидные последовательности взаимодействуют с остатками, идентифицированными на стадии d), представленной выше. Можно идентифицировать пептидные последовательности-кандидаты, ингибирующие агрегацию белка, которые не взаимодействуют с белками, опосредующими важные клеточные процессы. Эффективность идентифицированной пептидной последовательности, как описано выше, можно определять в модели заболевания, связанного с нарушением сворачивания белка. Модели заболеваний, связанных с нарушением сворачивания белка, хорошо известны в данной области. Пригодные модели включают клетки, которые сверхэкспрессируют склонный к агрегации белок, и модели с трансгенными животными, такие как модели на мышах или дрозофилах, которые сверхэкспрессируют склонный к агрегации белок и которые могут подвергаться другим воздействиям, таким как окислительный стресс, и т.д., или могут не подвергаться им. Например, см.: Junn E, Mouradian MM, Human alpha-synuclein over-expression increases intracellularParkinson's disease, Drug Discov Today. 2006 Feb; 11 (3-4): 119-26. Склонные к агрегации белки включают следующие белки, предшественники или их фрагменты: синуклеин (либо дикого типа, либо любая из мутантных форм, ассоциированных с болезнью Паркинсона), хантингтин (а также другие белки с протяженными полиглутаминовыми или полиаланиновыми повторами), амилоидный бета-пептид (А 42), прионный белок, островковый амилоидный полипептид(hIAPP) или амилин, супероксиддисмутаза, Tau, альфа-1-антитрипсин и другие серпины, лизоцим, витронектин, кристаллинов, альфа-цепь фибриногена, аполипопротеин AI, цистатин С, гельзолин, лактоферрин, кератоэпителин, кальцитонин, предсердный натрийуретический фактор, пролактин, кератин,медин (или полноразмерный лактадгерин), легкие цепи иммуноглобулинов, транстиретин (TTR), сывороточный амилоидный апобелок A (SAA), бета-2-микроглобулин, тяжелые цепи иммуноглобулинов или любой другой белок, ассоциированный с каким-либо нарушением сворачивания белка. Можно определять способность пептидной последовательности, идентифицированной, как описано выше, к одному или более из следующих:ii Снижение скорости потери активности белка при хранении;iii. Снижение опосредуемой агрегацией иммуногенности белка;iv. Повышение выхода белков в системах трансляции in vitro;v. Повышение стабильности в растворе состава для терапевтического применения;vi. Ингибирование одного или более клеточных процессов;vii. Предотвращение олигомеризации или мультимеризации белка. Дополнительные аспекты настоящего изобретения относятся к способам конструирования ингибиторов агрегации для лечения заболеваний, связанных с нарушением сворачивания белка, с использованием остатка(ов), идентифицированного на стадии d) указанного выше первого аспекта; конструирования соединений для стабилизации белков (например, биофармацевтических соединений, антител, ферментов и т.д.) в отношении агрегации в составах, носителях и других растворах с использованием идентифицированного остатка(ов); конструирования соединений для повышения срока хранения таких белков с использованием идентифицированного остатка(ов); конструирования соединений для снижения иммуногенности белков вследствие агрегации с использованием идентифицированного остатка(ов) и конструирования соединений для повышения выхода белков в системах трансляции in vitro с использованием идентифицированного остатка(ов). Другой аспект настоящего изобретения относится к компьютерной программе, которая, при выпол-4 016884 нении на компьютере, осуществляет способ по любому из указанных выше аспектов. Другой аспект настоящего изобретения относится к носителю компьютерных данных, содержащему компьютерную программу в соответствии с предыдущим аспектом. Дополнительный аспект настоящего изобретения относится к компьютеру, настроенному на выполнение способа по любому из указанных выше аспектов способа. Предпочтительно этот компьютер представляет собой компьютер общего назначения, который настроен для доступа к базам данных, содержащим информацию об известных белках, для применения в целях получения предсказания. Такие базы данных могут храниться локально, например, на жестком диске или в памяти, но предпочтительно они хранятся дистанционно и доступны через каналы связи, такие как сеть или интернет. Краткое описание иллюстраций Варианты осуществления этого изобретения далее описаны на основании прилагаемых иллюстраций, где на фиг. 1 представлены профили амилоидной агрегации А 42 и -синуклеина при рН 7; на фиг. 2 представлен профиль амилоидной агрегации мутантной формы -кристаллина D при рН 7; на фиг. 3 представлены выходные данные программы PeptideSearch для остатков с 1 по 8mutCRYD34-58; на фиг. 4 представлена блок-схема процесса поиска пептидов; на фиг. 5 представлены извлечения из файлов pdbseqres.txt и ss.txt, полученных из PDB; на фиг. 6 представлено предсказание водородных связей в трех соседних цепях в -слое; на фиг. 7 представлен пример выходных данных программы для предсказания водородных связей между остатками; на фиг. 8 представлены обобщенные результаты для совпадения при поиске пептидов; и на фиг. 9 представлены профили склонности к агрегации для mutCRYD34-58, А 42 и -синуклеина с указанием положений, по которым были выявлены совпадения коротких пептидов в PDB. Подробное описание предпочтительных вариантов осуществления Далее описаны варианты осуществления настоящего изобретения. Способы настоящего варианта осуществления будут продемонстрированы на мутантной форме -кристаллина D (mutCRYD). Для целей иллюстрации также рассмотрены следующие два белка: -синуклеин, который вовлечен в болезнь Паркинсона, и А 42, который ассоциирован с болезнью Альцгеймера.mutCRYD распространен в клетках хрусталика глаза человека. При нарушении сворачивания он может формировать агрегаты, которые проявляются в качестве катаракт, ведущих к неясному зрению или слепоте (Heon Е, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL (1999) The(2004) Dying to see, Sci. Am., 291(4): 52-59). -Кристаллин D и мутантная форма отличаются тремя остатками, R58H, R36S и R14C, которые, как было выявлено, усиливают агрегацию (Pande A, Pande J, Asherie-Синуклеин представляет собой белок из 140 остатков, который встречается в качестве основного компонента в тельцах Леви (плотные отложения, встречающиеся в головном мозге пациентов с болезнью Паркинсона, которые могут вызывать нейродегенерацию - Spillantini MG, Schmidt МЛ, Lee VMY, Trojanowski JQ, Jakes R, Goedert M (1997) -Synuclein in Lewy bodies, Nature. 388: 839-840).A42 представляет собой небольшой гидрофобный пептид из 42 остатков, который может играть непосредственную роль в прерывании синаптической функции при болезни Альцгеймера, наиболее распространенной форме патологии нервов (Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298:789-790). Склонность к образованию плотных подобных -слою структур, т.е. к агрегации, варьирует между белками, в зависимости от аминокислотного состава и последовательности. Целью способа настоящего варианта осуществления была идентификация пептидных последовательностей-кандидатов, которые могут позволить снизить скорость образования и количество агрегатов посредством блокирования склонных к агрегации областей белков-мишеней. Идентификация областей агрегации Так называемые области, чувствительные к агрегации, в частях двух белков: -синуклеина и А 42 были предсказаны Pawar АР, DuBay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM (2005) Prediction of"agregation-prone" and "agregation-susceptible" regions in proteins associated with neurodegenerative diseases,J. Mol. Biol. 350: 379-392. В более общем смысле, в указанной выше статье предложен алгоритм, который позволяет вычислять профиль склонности к агрегации для белка и который, как было показано, приводит к результатам,которые тесно согласуются с широким диапазоном экспериментальных измерений. Для любой данной пептидной последовательности он позволяет вычислить склонность каждого отдельного остатка или усредненную склонность для окна из нескольких аминокислот. В табл. 1 приведена склонность для каждой аминокислоты отдельно при рН 7,0.-5 016884 Таблица 1. Склонность к агрегации для каждой из 20 встречающихся в природе аминокислот отдельно при рН 7,0 На фиг. 1 представлены профили склонности к агрегации для -синуклеина и А 42, вычисленные в соответствии с этим алгоритмом (и представленные в указанной выше статье). На фигуре слева представлен профиль склонности к агрегации А 42 при рН 7. Профиль агрегации для неизменной последовательности А 42 дикого типа нанесен на график в виде средней линии, вместе с максимальными и минимальными значениями склонности, полученными после тестирования в отношении какой-либо возможной мутации в каждом аминокислотном положении на протяжении последовательности. Линия Zprofagg = 1 изображена для облегчения идентификации обеспечивающих агрегацию областей. Те же области, полученные экспериментальными способами, представлены в виде затемненных областей. Аналогично, на фигуре справа представлен профиль амилоидной агрегации -синуклеина при рН 7,также включающий линию Zprofagg = 1 для облегчения идентификации областей, способствующих агрегации. На этой фигуре светло-серым оттенком представлена большая область белка, которая, как полагают,является структурированной в фибриллы; более темным серым оттенком показана более высоко амилоидогенная область NAC; и штриховыми линиями показана область 69-79, которая, как было выявлено,является особенно амилоидогенным сегментом в этой области NAC. Считают, что части белка, для которых показатель пересекает порог (например, Zprofagg = 1 на фиг. 1), имеют наиболее высокую склонность к агрегации и полагают, что они являются центральными участками агрегата и образования фибрилл in vivo. Предсказанные центральные участки агрегации из фиг. 1 используют в этом варианте осуществления в качестве последовательностей-мишеней для детекции любых партнеров по взаимодействию, которые могут действовать в качестве ингибиторов. Способы по настоящему варианту осуществления будут продемонстрированы на mutCRYD. На фиг. 2 показан профиль склонности к агрегации mutCRYD, усредненный для семи остатков. Как в случае профилей склонности к агрегации, представленных на фиг. 1, для облегчения отличия между областями, которые следует рассматривать как чувствительные к агрегации, и другими частями белка, на уровне значения склонности к агрегации 1,0 добавлена прямая. Для дальнейшего поиска были отобраны остатки с 34 по 58. Эта область содержит два пика, для которых предсказаны горячие точки агрегации, и содержит две мутации. Для полного анализа рассматривают все области со склонностью к агрегации, превышающей 1,но выбранный короткий фрагмент является достаточным для демонстрации используемого способа. Искомая последовательность из 24 остатков представляет собой SARVDSGCWMLYEQPNYSGLQYFL, и она обозначена как mutCRYD34-58. Поиск - обзор Хотя поиск последовательности в одном файле является стандартным способом и его можно проводить с помощью большинства редактирующих текст программ или средств командной строки, в файле для коротких последовательностей вполне могут существовать сотни или тысячи совпадений. Рассмотрение каждого из них вручную, поиск информации о вторичной структуре, а затем проверка web-сайтаPDB для получения большей информации и применение 3D-средства для визуализации структуры и поиска взаимодействий между остатками в целях извлечения пептидов, которые тесно связаны водородными связями, может отнять недели или месяцы для каждого белка. Таким образом, для управления процессом было разработано проприетарное программное обеспечение. Программное обеспечение было разработано в виде большого числа небольших инструментов,которые затем поэтапно были включены в одну комплексную программу, названную PeptideSearch. Для написания PeptideSearch был выбран язык программирования PERL, поскольку все используемые данные были доступны в качестве обычных текстовых файлов, a PERL предоставляет мощные процедуры для анализа этих файлов, и извлечения и переработки содержащейся в них информации. ТакжеPERL является свободно доступным программным обеспечением, по этим причинам известным биоинформатикам. На фиг. 4 представлена блок-схема программного обеспечения и способов по настоящему варианту осуществления и приведен обзор подробного объяснения способа, который представлен ниже.PeptideSearch основан на вызовах ряда внешних программ для расширения его функциональности. Эти вызовы также представлены на фиг. 4. При использовании программы, вводимыми пользователем данными являются последовательностьмишень и ряд параметров, а программа в итоге выдает краткую информацию о пептидах-кандидатах,включая трехмерную визуализацию пептидов и их партнеров по взаимодействию. Таким образом, время,требуемое для идентификации возможных ингибиторов любого белка, было снижено с потенциальных нескольких недель до нескольких часов.PeptideSearch создает HTML-файл, называемый "result.html" для сохранения записей для каждого из совпадений. Этот и все другие файлы, создаваемые обеими программами и внешними инструментами,сохраняются в новой поддиректории. Название этой директории образуется из текущей даты и времени,так что результаты всех запусков программы остаются доступными на диске для последующего применения. При проведении поиска, PeptideSearch также выводит информацию о совпадениях в командную строку или на консоль, вместе с дополнительными сообщениями о состоянии. После завершения выполнения программы "result.html" содержит обзор всех выявленных совпадений, что упрощает отбор пептидов-кандидатов. Выходные данные программы после поиска остатков mutCRYD34-58 с 1 по 8 представлены на фиг. 3. Поиск - подробное описание В настоящем варианте осуществления поиск был основан на записях в Protein Data Bank (PDB) Research Collaboratory for Structural Bioinformatics (RCSB), поскольку этот свободно доступный источник содержит большое число экспериментальных структур и теоретических моделей. Таким образом, выявление всей искомой последовательности или ее части в одной из пептидных последовательностей в PDB дает доступ к родственной структуре, позволяя идентификацию вторичной структуры и партнеров по взаимодействию. Первой стадией поиска является поиск последовательности-мишени во всех записях в PDB. Этого можно достичь копированием файлов PDB на локальный компьютер и анализом каждого из них по отдельности на последовательность остатков. Однако этот подход может быть неэффективным с точки зрения времени и пространства: на момент 27 июня 2005 года в PDB находилось 31639 структур, многие из которых представляют собой мультимеры с несколькими цепями. Альтернатива, которая была принята для этого варианта осуществления, представляла собой однократное копирование отдельного файла, содержащего всю информацию о последовательностях и последующий поиск в нем для детекции совпадений. Такой файл можно получить с ftp-сервера PDB RCSB по адресу ftp://ftp.rcsb.org/pub/pdb/deriveddata. Файл, pdbseqres.txt, представляет собой список всех последовательностей PDB в формате FASTA. Аналогично, по тому же адресу можно найти файл, содержащий все вторичные структуры в PDB в формате FASTA, ss.txt. Версии файла, используемые для этого варианта осуществления, датируются 27 июня 2005 года. Файл для вторичной структуры представляет собой первый и быстрый источник для проведения сопоставления последовательности и вторичной структуры и для детекции вовлечения совпадения в конформацию -слоя. На фиг. 5 представлены короткие извлечения из двух файлов: первые десять строк из seqres.txt и-7 016884 Очистка файлов с последовательностями и структурами Два указанных выше файла были получены автоматически с помощью PDB с использованием данных из всех записей PDB. Информация о вторичной структуре в файлах PDB определяется рядом различных программ. Однако два файла имеют неравную длину. На извлечениях, представленных на фиг. 5, показана одна из причин этого: строки заголовка FASTA в файле с последовательностями являются более длинными, включая более подробную информацию. Учитывая источник данных, можно ожидать, что для каждой последовательности должна существовать одна запись о структуре и наоборот. Также, сначала, кажется, что количество строк в файлах может быть идентичным, что очень удобно для сопоставления содержания двух файлов. Однако ни одно из этих предположений не является истинным: количества строк не всегда совпадают. Скрипт PERL, просматривающий файл с извлечением всех строк заголовков FASTA, возвратил 74560 записей для pdbseqres.txt и 69903 записей для ss.txt. Существует ряд других проблем, которые необходимо было решить перед тем, как использовать файлы в качестве входных данных. Например, в этих двух файлах не было согласовано использование ID. В файле с последовательностями используютсяID, такие как 1tsv, а различные цепи в нем представлены как 1thl, lthl1, 1thl2 или как 1pr2 А, 1pr2 В,в то время как в ss.txt вместо этого используются 1TSV:, 1THL:, 1THL1, 1PR2:A, 1PR2:B. Также в файле с последовательностями существует много ID, которые не имеют соответствующей записи в файле со вторичными структурами, и наоборот: такие записи, как 1J3W, 10G8 и IVGS, содержатся в ss.txt, но не в pdb-seqres.txt. Они были заменены на ID 2CVZ, 2BUH и 2CV4 из Protein Data Bank, который учитывается только в pdbseqres.txt. Для удаления всех уникальных записей из обоих файлов понадобился ряд скриптов PERL. Полученные после этого файлы содержали 68383 совпадающих записи. Тем не менее, остается потенциальная проблема, состоящая в том, что некоторые из последовательностей не совпадают по длине. Программу PeptideSearch настроили для указания таких несоответствий во время выполнения. Сопоставление искомой последовательности с последовательностями Представляющие интерес экспериментальные структуры представляют собой структуры, в которых встречается аминокислотная последовательность, состоящая из искомой строки. Это означает, что необходим поиск точных совпадений строки в прямом направлении, для mutCRYD34-58:SARVDSGCWMLYEQPNYSGLQYFL а также для инвертированной последовательности, здесь:LFYQLGSYNPQEYLMWCGSDVRAS. Очевидно, что нахождение в PDB точного совпадения последовательностей для длинных пептидов значительно менее вероятно, чем для коротких. Действительно, при поиске совпадений для полного белка, очень вероятно, что будет найдено только одно: сам белок. Даже при сосредоточении на чувствительной области белка может быть желательным поиск совпадений с помощью участков из аминокислот, которые просто являются слишком длинными для того, чтобы дать точные совпадения в PDB. Поскольку структура мутантной формы -кристаллина D не включена в PDB, то не существует результата для полной последовательности из 24 остатков. Для повышения выхода совпадений применяли два дополнительных способа. Во-первых, белковую последовательность-мишень разделяли на две подстроки и партнеры по взаимодействию подвергали поиску по этим подстрокам. Это позволяет идентифицировать пептиды, которые связываются с частью последовательности. Затем их можно тестировать по отдельности, например, в сочетании с другими пептидами, или конструировать в более длинный пептид, соединяя несколько таких пептидов вместе. В программе PeptideSearch, пользователь может установить минимальную длину n и максимальную длину m каждой искомой последовательности. После испытаний было выявлено, что длина искомой последовательности в диапазоне от 5 до 8 остатков приводит к достаточному количеству совпадений, выявляя партнеров, которые все еще были достаточно длинными для того, чтобы все еще быть пригодными. Однако также можно использовать подстроки другой длины. Программа настроена для автоматического проведения полного поиска по всем непрерывным подстрокам исходной последовательности с длиной в данном диапазоне. Таким образом, общее количество искомых последовательностей q, которые ищут для полной искомой последовательности длины l, составляет: Во-вторых, программа позволяет использовать регулярные выражения. Это позволяет вносить вариабельность в поисковую последовательность. Таким образом, PeptideSearch позволяет определять консервативные замены, которые учитываются при поиске по файлу с аминокислотными последовательностями.-8 016884 Например, в соответствии с табл. 1, Gln (Q) и Asn (N) обладают сходной склонностью к агрегации,и, таким образом, одним вариантом увеличения количества пригодных совпадений может быть учет их обеих при поиске. Тогда, регулярное выражение в строке поиска, например, QANT может представлять собой [QN]A[.QN]T. Аналогично можно использовать другие замены, исходя из склонности остатков к агрегации. Два способа можно комбинировать, так что каждая возможная подстрока вплоть до минимальной длины превращается в регулярное выражение, которое учитывает консервативные замены, описанные выше. Выявление элементов вторичной структуры Данные как от pdbseqres.txt, так и от ss.txt считываются один раз при запуске программы и сохраняются в больших массивах. Затем записи из обоих массивов обрабатываются совместно. Поскольку, как отмечалось выше, длина аминокислотной последовательности и структура не всегда совпадают, номера строк нельзя использовать в качестве указателя. Вместо этого, для того, чтобы убедиться, что поиск учитывает записи, которые соответствуют друг другу, и что информация в обоих файлах остается синхронизированной, в качестве ориентира служат строки заголовков FASTA. Если записи разделяются на несколько строк, их объединяют, удаляя разрывы строк между ними. Это создает две строковых переменных для каждой записи: одну для полной аминокислотной последовательности и одну для соответствующей вторичной структуры. Если на этом этапе две строки обладают неравной длиной, то данную информацию более не рассматривают как достоверную. В этом случае программа отображает предупреждение с извещением пользователя о несовместимости и предлагает исследование совпадения вручную.PERL предоставляет простую для использования процедуру поиска строки последовательности на встречаемость искомой последовательности, либо поиском абсолютных совпадений, либо сопоставлением регулярных выражений. Если существуют какие-либо совпадения, то PeptideSearch извлекает их из строки последовательности и выравнивает их с соответствующим участком строки вторичной структуры. Это дает возможность первой визуальной проверки того, сколько остатков в подобранных последовательностях находятся в структуре протяженного -слоя. Поскольку окружение структуры также может представлять интерес, программа настроена также для вывода ряда остатков и информации о структуре как слева, так и справа от совпадения. Размер этого окна может быть установлен в параметрах программы. В идеальном случае, эту информацию можно просто использовать для отбора всех тех записей, для которых файл вторичной структуры указывает на наличие -слоев, и, затем, далее исследовать только их. Однако, для используемых файлов, это может привести к множеству ложноположительных, а также ложноотрицательных результатов. Основная причина этого состоит в том, что предсказание в ss.txt часто является неточным. Это было выявлено при сравнении выравнивания с существующей структурой в файле PDB. Таким образом, получается, что лучше рассматривать каждое совпадение в последовательности независимо от информации в ss.txt. Это повышает количество совпадений, которые необходимо обрабатывать, далее требуя доступа к PDB, но также это приводит к тому, что ни один важный пептид-кандидат не упускается. Получение файлов PDB После детекции искомой последовательности в одной из записей, требуется получить больше информации и необходимо преобразовать существующий файл PDB. PDB ID извлекается из строки заголовка FASTA. В PeptideSearch определяется локальная директория PDB. Если файл PDB, который относится к этому ID, доступен в локальной директории, в сжатом или несжатом состоянии, тогда используется эта локальная копия. В ином случае PeptideSearch использует одну из его операций для автоматического доступа к зеркалу ftp-сервера RCSB Protein Data Bank в CambridgeCrystallographic Data Centre (CCBC), ftp://pdb.ccdc.cam.ac.uk/rcsb/data/structures/divided/pdb/, и загружает сжатую копию файла PDB. Затем этот .Z-архив извлекается посредством системного вызова программыWinRAR. Обработка файлов PDB Перед открытием файла PDB программа проверяет, использовался ли тот же PDB ID для предсказания совпадения. Это может происходить в случае, когда существует несколько совпадений в одной аминокислотной последовательности или когда структура в файле представляет собой мультимер, так что существуют аналогичные совпадения на каждой цепи. Если ID является таким же, тогда данные все еще содержатся в массивах данных программы и их можно повторно использовать, экономя значительное количество времени. В ином случае, открывается локальная копия файла PDB, и данные считываются в память. Существуют две части каждого файла, которые представляют интерес. Первая часть представляет собой строки, которые можно использовать для идентификации перекрывания искомой последовательности с -слоями. Они начинаются с метки "SHEET", которая показывает положения в -слоях.-9 016884 Вторая относится к записям "АТОМ", которые дают координаты каждой части молекулы. Эта информация позволяет предсказывать водородные связи, поскольку знание того, какие остатки в действительности взаимодействуют, обеспечивает важную информацию и дает исследователю больше свободы при конструировании ингибиторных пептидов. Например, остатки, боковые цепи которых не вовлечены во взаимодействие, можно заменить другими, которые дают пептид с лучшими биохимическими свойствами. Для каждой цепи в -слое существует одна строка "SHEET". Она содержит индекс первого и последнего остатка в цепи. Если эта цепь не является первой в слое, тогда строка также содержит два остатка, где эта и предыдущая цепи совпадают, т.е. индексы одной пары остатков, для которых выявлена связь водородной связью. Полную информацию о записях "SHEET" и "АТОМ" можно найти наhttp://www.rcsb.org/pdb/docs/format/pdbguide2.2/guide2.2frame. html. Извлечение информации из этих строк и начальная и конечная точки совпадения в аминокислотной последовательности обеспечивают тест на перекрывание: если стартовый индекс слоя не превышает индекс конца совпадения и индекс конца слоя не является меньшим, чем начало совпадения, тогда существует перекрывание, в случае которого вычисляется количество перекрывающихся остатков. В идеальном случае, количество перекрывающихся остатков не должно быть слишком маленьким,поскольку оно определяет длину потенциального ингибиторного пептида. Ингибитор, который состоит только из небольшого количества остатков, может оказаться не очень эффективным, и будет трудно сконструировать стабильный пептид с благоприятными свойствами. Программа позволяет пользователю определять порог и исключает все совпадения с перекрыванием, более коротким, чем этот порог из вывода в "result.html". Затем -цепь, которая перекрывается с искомой последовательностью, и соседние цепи (либо одна,либо две, в зависимости от положения первой цепи в слое) отображаются в выравнивании, в котором учитывается ориентация (параллельная или антипараллельная) и совмещение цепи. Например, если пептидная последовательность представляет собой "ADDYYTATGHWYAT" и цепи проходят для поиска остатков с 1 по 4, с 6 по 10 и с 12 по 14, соответственно, в антипараллельной структуре -слоя с совмещением в 3 и 7, и 9 и 13, тогда выравнивание дает в результате:YAT Затем в "result.html" остатки искомой последовательности в этом выравнивании будут показаны красным цветом. Если поиск осуществлялся, например, на наличие "ATGHW", который был выявлен в индексах с 7 по 11 в пептидной последовательности, представленной выше, тогда остатки "HGTA" при выравнивании будут выделены цветом. Выравнивание обеспечивает быстрый скрининг совпадений на совпадения с большим перекрыванием между искомой последовательностью и -слоем. Оно показывает, сколько и какие из остатков формируют -слой, и, следовательно, сколько и какие из остатков могли бы блокироваться, если бы их партнеры по взаимодействию были включены в короткие ингибиторные пептиды. Несмотря на то что указанный выше способ поиска обеспечивает один путь идентификации пептидов-кандидатов, он не является полностью надежным при доступных текущих данных. В частности, индексы остатков, находящиеся в строках "SHEET", относятся к индексам в строках "АТОМ", но не в записях "SEQRES" в том же файле. Protein Data Bank гарантирует, что записи "АТОМ" протестированы на отсутствие ошибок. Однако данные в "pdbseqres.txt" были получены из записей "SEQRES", и их индексы часто не совпадают. Например, запись "SEQRES" может начинаться с МЕТ, в то время как первая запись "АТОМ" представляет собой SER, или последовательности могут быть одинаковыми, но запись"АТОМ" начинается с индекса остатка 21, а не 1. Это означает, что, хотя указанный выше способ, демонстрирующий выравнивание, может воспроизводить относительные положения цепи -слоя корректно, идентификатор остатков может быть взят из неправильной части аминокислотной последовательности. Также это означает, что идентификация перекрывания с -слоем может быть некорректной в первом положении: искомая последовательность в действительности может быть расположена в другом в структуре.peptideAlign.class Для того чтобы убедиться, что такое событие можно выявлять, при исследовании обобщенных результатов для этого совпадения позднее, был разработан следующий способ выравнивания с использованием записей "АТОМ". Это обеспечило альтернативный способ выявления пептидов-кандидатов или перекрестной проверки результатов, полученных при поиске "SHEET", описанном выше. Этот способ выравнивания также включает детекцию и отображение водородных связей между остатками, делая выравнивание значительно более информативным. Для поиска в данных "АТОМ" был изобретен следующий алгоритм: для всех пар атомов, которые отделены по меньшей мере тремя остатками и которые находятся в пределах представляющей интерес части -слоя, вычисляется эвклидово расстояние между ними (с использованием стандартной формулы где d представляет собой эвклидово расстояние, и х, у и z представляют собой разности соответствующих координат). Если вычисленное расстояние составляет менее 3,075 Ангстрем, то индексы в этой паре остатков перемещают в массив для хранения водородных связей. Предел длины был выбран из литературы и он является достаточно большим для детекции большинства водородных связей между атомами остова и боковыми цепями, и в то же время он является достаточно маленьким для поддержания малого количества ложноположительных результатов. После завершения предсказания, информация о цепях и связях записывается в файл, называемый"bonds.txt". Этот файл используется в качестве файла для хранения промежуточных данных для объединения информации с другим приложением: "peptideAlign.class", основанным Java инструментом, который был создан для изображения графического выравнивания -слоя. Хотя PERL обладает мощными процедурами для обработки больших количеств текста, Java оказывается легче для использования графических функций. Таким образом, свойства обоих языков программирования были объединены посредством использования системного вызова "PeptideSearch.pl" для внешней связи с PeptideAlign. Последний создает снимок экрана выравнивания, который затем программа PERL включает в обзор текущего совпадения в "result.html". Применение графических изображений облегчает указание на водородные связи между остатками,которое можно осуществлять простым проведением линий. PeptideAlign не учитывает непосредственно антипараллельную форму -слоя, но графические выходные данные указывают на то, следует ли инвертировать ориентацию цепи, поскольку в этом случае водородные связи пересекаются. Например, синими линиями, указывающими на водородные связи на фиг. 6, показано, что ориентацию средней цепи необходимо инвертировать. Для правильной ориентации цепей и сдвига их относительно друг друга для минимизации общей длины связей используют дополнительный алгоритм.PeptideAlign считывает информацию о пептидах и связях из "bonds.txt" вместе с названием для ожидаемого выходного файла с изображением. Названия файлов с изображениями имеют формуhitX.png, где X указывает на текущий номер совпадения. Программа переводит информацию в файле в графическое представление и возвращает управление PeptideSearch. Пример выходных данных представлен на фиг. 7: файл содержит путь и название файла, под которым предполагается сохранить снимок экрана, количества атомов и связей в файле с последующими ID остатков для всех атомов и информацией о том, какие из них соединены связями; графическое представление справа показывает, какие остатки связаны друг с другом. Минимальный файл PDB Выравнивание, проводимое поиском "АТОМ", является корректным, благодаря отбору по PDB. Если оно соответствует выравниванию, проводимому PeptideSearch, тогда запись "SEQRES" также согласуется. В ином случае, пользователю необходимо будет проверить графический обзор визуально в отношении перекрывания. Если оно отсутствует, требуется проверка файла PDB вручную, чтобы видеть существует ли отличающийся -слой, который содержит часть искомой последовательности или всю ее. Следует отметить, что, в настоящее время, невозможно создать аминокислотную последовательность заново из записи "АТОМ" и провести ее поиск в отношении наличия искомой строки. Это является следствием того, что в записи часто отсутствуют остатки. Например, в pdblp9w.ent после остатка 108 следует остаток 121. Брешь можно объяснить рядом причин, например недостаточными экспериментальными данными, низким разрешением ЯМР или рентгенографии, или высокоподвижными боковыми цепями, которые приводят к размытым сигналам, что делает невозможным точное определение положений атомов. Другое объяснение этого просто состоит в том, что в научной работе, в которой получены данные, иногда интерес представляет только конкретная часть белка, например активный участок, и, таким образом, не предоставляется какого-либо предсказания об оставшейся структуре. Однако, когда доступны полные данные, такой способ поиска может быть подходящей отдельной альтернативой поиску по"SHEET", рассмотренному выше. После нахождения совпадения в -слое можно видеть, какие из остатков взаимодействуют, и построить остов для конструирования пептидов-кандидатов. Например, остатки, которые не взаимодействуют с остатками в искомой последовательности, можно заменять другими аминокислотами, обеспечивая больший диапазон пептидов, подлежащих конструированию и тестированию. Например, в коротком выравнивании, представленном на фиг. 6, SARV взаимодействует с VTY. Водородные связи предсказаны между А и V, и V и Y. Однако Т по-видимому не взаимодействует с искомой последовательностью вообще, так что пептид VXY можно использовать в качестве остова для создания пептидов-кандидатов, причем X является заменяемым различными аминокислотами. В реальных условиях такой остов, наиболее вероятно, является слишком коротким, чтобы быть эффективным,таким образом, предпочтительным является поиск пептидов с длинами из 5 остатков и более. Для каждого успешного совпадения PeptideSearch создает новый файл .pdb, содержащий координаты всех атомов, входящих в -слой. Для этого он извлекает эту часть из полной структуры, которая со- 11016884 держит -цепи с фрагментом искомой последовательности и ее партнерами по взаимодействию. Файл можно открыть с помощью любой из обычных программ для просмотра 3D-изображений, таких какRasmol или vmd, и он позволяет эффективно проверить предсказанные водородные связи и пространственную ориентацию боковых цепей.Peptide3D.class Для предварительного просмотра структуры в новом файле .pdb в записи "results.html" включаются снимки экрана. Для осуществления этого необходима программа для просмотра 3D-изображений, которая может обеспечить прорисовку молекул, помещение их в информативной ориентации в пространстве,и получение снимка экрана, все это без необходимости взаимодействия с пользователем.Results.html После завершения PeptideSearch анализа всех возможных подстрок и мутаций во всех записях вpdbseqres.txt он открывает окно браузера посредством системного вызова и предоставляет все совпадения, которые были выявлены, в виде обзора. Этот обзор предоставляет ссылки на страницу отчета PDB для ID PDB, связанным с каждым совпадением, и ссылку на Peptide3D, которая применяется в качестве апплета для просмотра структуры в формате 800600 пикселей. Результаты Выполнение программы с mutCRYD34-58 в качестве входных данных, с установкой минимальной и максимальной длины подстрок на пять, с обеспечением отсутствия мутаций и с исключением всех совпадений, в которых перекрывание с -слоем является меньшим четырех, дало 153 точных совпадения вpdb seqres.txt, 47 из которых были в -слоях. Только восемь из этих результатов были длиннее трех остатков и последние были включены в "result.txt". Фактически, этот набор в действительности дает только два отличающихся совпадения, поскольку первые два и последние шесть совпадений являются практически идентичными, так как их источником являются различные цепи мультимера. Первое из этих совпадений относится к последовательности SARVD (остатки mutCRYD34-58 с 1 по 5), которая была выявлена в предполагаемом транскрипционном репрессоре системы отщепления глицина (PDB ID 1U8S). Для этого совпадения результат представлен на фиг. 3. Перекрывание из четырех остатков представлено на фиг. 6. Второе совпадение было выявлено для последовательности PNYSG (остатки mutCRYD34-58 с 15 по 20) в Endo-1, 4-Beta-Xylanase II. Обобщенные результаты для этого совпадения из файла HTML представлены на фиг. 8. На фиг. 9 цветом выделены два совпадения в профиле склонности к агрегации mutCRYD34-58. Расположения точных совпадений рядом с горячими точками агрегации двух белков -синуклеина и А 42,или вблизи них, также представлены в соответствующих профилях на фиг. 9. Для -синуклеина длина подстрок была установлена в диапазоне от 6 до 7, а для А 42 она была установлена на 5. Для mutCRYD первое совпадение может быть не очень эффективным в качестве кандидата для ингибитора агрегации, поскольку он не находится в области, чувствительной к агрегации. Однако второе совпадение имеет потенциал в отношении блокирования в пептиде инициации образования агрегатов в этой области. Для других двух белков, каждый из партнеров по взаимодействию, соответствующий показанным совпадениям, обладает потенциалом к действию в качестве ингибитора агрегации. Количество совпадений для -синуклеина значительно возрастает при установке длины подстроки на 5. Результаты анализа -синуклеина являются объектом дополнительной патентной заявки настоящего заявителя. С использованием описанного выше программного обеспечения сконструирована серия пептидов для взаимодействия с участками 61-66 (EQVTN) и 71-76 (VTGVT) -синуклеина. В этом анализе идентифицирован пептид, состоящий из последовательности D-аминокислот, которая взаимодействует с областью -синуклеина между остатками 61-66 (EQVTN). Один из подходящих пептидов содержит или последовательность D-аминокислот QYSVLI (ZP-0195, описанную в таблице 3,ниже), или состоит из нее. Другие подходящие пептиды могут содержать последовательность Dаминокислот QYSVLI с одной, двумя или тремя аминокислотными заменами, или состоять из нее. Например, пептид может состоять из последовательности D-аминокислот, выбранной из группы, состоящей из: qykvli, qysvpi, qyspli, qypvli, rysvli, qysvli, qytvli, pysvli или qysvlv. Пептид может содержать один, два или три дополнительных N-концевых остатка. Например, пептид может содержать последовательность, выбранную из группы, состоящей из:ekysvli и drysvli, или состоять из нее. Также в анализе идентифицирован пептид, состоящий из последовательности D-аминокислот, которая взаимодействует с областью -синуклеина между остатками 71-76 (VTGVT). Например, пептид может состоять из последовательности D-аминокислот hhviva (ZP-0158) или он может содержать последовательность D-аминокислот hhviva с одной, двумя или тремя аминокислотными заменами, или состоять из нее. Предпочтительно N-концевые остатки гистидина не замещены. Например, пептид может содержать последовательность, выбранную из группы, состоящей изhhvvva, hhviva, hhvkva, hhveva, hpviva, hhvivp, hhvivv, hhvivt, hhvivy, hhvivw, hhtivv, hhtivk, hhtvva, hhtlva,hhtlvv, hhtevy и hhttvy, или может состоять из нее. Для области 61-65 сконструирован взаимодействующий пептид Ac-qysvli-NH2 (ZP-0195) для предотвращения агрегации. Также тестировали варианты этой последовательности, где одна или несколько аминокислот в любом данном положении замещена другой, и были проведены изменения на N-конце,такие как добавление дополнительной аминокислоты и ацетилирование (с ZP-0195 по ZP-0230). Для области 71-75 сконструирован взаимодействующий пептид Ac-hhviva-NH2 (ZP-0158) для предотвращения агрегации. Также тестировали варианты этой последовательности, где одна или несколько аминокислот в любом данном положении замещены другой. Все протестированные пептиды были ацетилированы на N-конце и во всех конструкциях в начале последовательности постоянно оставляли 2 гистидина (с ZP-0158 по ZP-0194). Все пептиды тестировали в отношении ингибирования агрегации ASYN в TBS поведением анализов агрегации с 50 мкМ ASYN и 100 мкМ ингибитором с 50 мМ tris и 150 мМ NaCl, и 20 мкМ тиофлавином Т. Объем реакции составлял 200 мкл. Каждую реакцию проводили в 96-луночном планшете из полипропилена с контролями в виде только ASYN и только буфера. Реакционные смеси инкубировали при 37 С при встряхивании в течение 48 часов и мониторинг агрегации проводили считыванием флуоресценции тиофлавина Т. Кинетические кривые строили с использованием программного обеспечения Zyentiafit, которое подбирает для данных сигмовидную функцию f(x)=k+A/(1+exp(-b(t-t0, из которой можно вычислить период задержки, скорость агрегации и общее изменение флуоресценции ThT. Пептиды ранжировали в соответствии с их эффективностью и в табл. 3, ниже, представлены последовательности, выбранные для последующего исследования. Выбор был основан на пептиде, имеющем более чем 20% повышение времени задержки и/или более чем 20% снижение флуоресценции ThT или скорости агрегации. Таблица 3. Пептидные последовательности, сконструированные для взаимодействия с областями 61-66 и 71-76 ASYN, которые показывают эффективность в отношении предотвращения агрегации ASYN in-vitro Конструирование соединений на основе результатов Совпадения, идентифицированные способами или программными средствами, описанными выше,можно использовать в качестве матриц для конструирования ингибиторов или стабилизаторов агрегации. Для тестирования библиотек соединений на их аффинность к последовательности-мишени на основе этих матриц можно использовать молекулярную динамику или другие пригодные компьютерные способы. В компьютерных способах в целях максимизации взаимодействия между ингибитором и агрегирующим полипептидом-мишенью для множества ингибиторов на основе матрицы можно использовать конкретные процедуры минимизации энергии и силовых полей (см. Das В, Meirovitch H, Navon IM, Performance of hybrid methods for large-scale unconstrained optimization as applied to models of proteins, J Comput. Chem. 2003;24:1222-31 и de Bakker PI, DePristo MA, Burke DF, Blundell TL, Ab initio construction ofpolypeptide fragments: Accuracy of loop decoy discrimination by an all-atom statistical potential and the AMBER force field with the Generalized Born solvation model, Proteins, 2003; 51 (1): 21-40). Отобранные библиотеки соединений можно тестировать в анализах агрегации in vitro в целях идентификации основных соединений, которые могут ингибировать агрегацию полипептидов-мишеней. Когда необходимо, чтобы стабилизатор/ингибитор агрегации был устойчивым к другим протеазам,можно использовать ретро-энантио-производные (с реверсией последовательности C-N и Dаминокислот). Идентифицированный стабилизатор/ингибитор агрегации можно подвергать слиянию с другими белками/пептидами. Белок/пептид, слитый со стабилизатором/ингибитором агрегации, может действовать в качестве носителей для направления доставки в конкретные области организма, конкретные органы, конкретные типы клеток и т.д.; облегчать внутриклеточную доставку; облегчать проникновение через гематоэнцефалический барьер; повышать время полужизни в плазме и/или взаимодействовать с другим белком или рецептором. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ предсказания потенциальных пептидных последовательностей, ингибирующих агрегацию белков, включающий следующие стадии:a) идентификация пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени;b) тестирование, образует ли указанная пептидная последовательность часть -слоя, причем стадия тестирования включает подстадии идентификации в базе данных белковых структур группы гетерологичных белков, содержащей пептидные последовательности, родственные указанной пептидной последовательности; и идентификации в указанной группе тех белков, в которых указанные родственные пептидные последовательности образуют часть -слоя;c) если на стадии b) достигнут положительный результат, извлечение из указанной базы данных белковых структур остатков, которые образуют соседние цепи указанного -слоя;d) идентификация остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех- 14016884 остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка,где подстадия идентификации включает сравнение указанных родственных пептидных последовательностей с остатками, содержащимися в строках "SHEET" в файле PDB для представляющего интерес белка. 2. Способ по п.1, где указанные родственные пептидные последовательности включают указанную пептидную последовательность и фрагменты указанной пептидной последовательности. 3. Способ по п.1 или 2, где указанные родственные пептидные последовательности включают последовательности, содержащие консервативные замены одной или более аминокислот в указанной пептидной последовательности. 4. Способ по любому из пп.1-3, где подстадия идентификации включает идентификацию тех остатков в указанных родственных пептидных последовательностях, которые образуют друг с другом водородные связи; предпочтительно в целях идентификации тех остатков, которые образуют водородные связи друг с другом, вычисляют эвклидово расстояние между каждой парой остатков, которые отделены по меньшей мере тремя остатками, и считают, что водородная связь образуется, если расстояние составляет менее 3,075 ангстрем. 5. Способ по любому из предшествующих пунктов, где остатки, идентифицированные на стадии d),представляют собой те остатки, боковые цепи которых взаимодействуют с указанной пептидной последовательностью посредством водородных связей. 6. Способ по любому из предшествующих пунктов, где на стадии идентификации используют профиль склонности к агрегации. 7. Способ по любому из пп.1-5, где стадию идентификации проводят экспериментально. 8. Способ по любому из предшествующих пунктов, дополнительно включающий стадии тестирования, взаимодействуют ли остатки, идентифицированные на стадии d), с одним или более другими белками; где предпочтительно стадию тестирования:(i) проводят на множестве белков, гетерологичных указанному белку-мишени, и/или(ii) проводят с использованием базы данных белковых структур, которые необязательно опосредуют важные клеточные процессы,и она необязательно включает подстадии идентификации группы белков, содержащихся в указанной базе данных, которые содержат родственные пептидные последовательности, сходные с указанной пептидной последовательностью; и идентификации в указанной группе тех белков, в которых указанные родственные пептидные последовательности взаимодействуют с указанными идентифицированными остатками. 9. Способ по любому из предшествующих пунктов, где часть области агрегации, идентифицированная на стадии а), представляет собой часть спирали, петлю, -поворот или -изгиб. 10. Способ по любому из предшествующих пунктов, включающий получение ингибирующего агрегацию белка пептида, содержащего остатки, идентифицированные на стадии d). 11. Способ по любому из предшествующих пунктов, дополнительно включающий стадии:e) синтеза пептидной библиотеки, причем члены указанной библиотеки содержат остатки, идентифицированные на стадии d); иf) определения аффинности членов указанной библиотеки в отношении указанного белка-мишени. 12. Способ получения пептида, ингибирующего агрегацию белка, включающий следующие стадии:a) идентификация пептидной последовательности, образующей по меньшей мере часть области агрегации в белке-мишени;b) тестирование, образует ли указанная пептидная последовательность часть -слоя;c) если на стадии b) достигнут положительный результат, извлечение из указанной базы данных белковых структур остатков, которые образуют соседние цепи указанного -слоя;d) идентификация остатков в цепях, расположенных рядом с указанной пептидной последовательностью, боковые цепи которых взаимодействуют с указанной пептидной последовательностью, т.е. тех остатков, которые образуют потенциальную пептидную последовательность, ингибирующую агрегацию белка;e) синтез пептидной библиотеки, причем члены указанной библиотеки содержат остатки, идентифицированные на стадии d); иf) определение аффинности членов указанной библиотеки к белку-мишени. 13. Способ по п.11 или 12, включающий идентификацию в указанной библиотеке пептида, который проявляет высокую аффинность к белку-мишени относительно контролей, в качестве пептида, ингибирующего агрегацию белка, где предпочтительно способ дополнительно включает:(ii) синтез пептида, идентифицированного в указанной библиотеке. 14. Способ по п.10, включающий определение способности указанного пептида к одному или более из следующих:ii) снижение скорости потери активности белка при хранении;iii) снижение опосредуемой агрегацией иммуногенности белка;iv) повышение выхода белков в системах трансляции in vitro;v) повышение стабильности в растворе состава для терапевтического применения;vi) ингибирование одного или более клеточных процессов;vii) предотвращение олигомеризации или мультимеризации белка. 15. Способ конструирования соединения для (А) стабилизации белка в отношении агрегации или(В) повышения стабильности состава для терапевтического применения в растворе, включающий стадии предсказания пептидов, ингибирующих агрегацию белков по любому из пп.1-9 со стадией тестирования,проводимой на множестве белков, гетерологичных указанному белку-мишени, и применения остатка(ов),идентифицированного на стадии d) этого предсказания, для конструирования соответственно (А) соединения для стабилизации белка в отношении агрегации или (В) соединений с повышенной стабильностью в растворе.
МПК / Метки
МПК: G06F 19/16
Метки: конструирования, способ, ингибиторов, предсказания, белков, агрегации
Код ссылки
<a href="https://eas.patents.su/20-16884-sposob-predskazaniya-agregacii-belkov-i-konstruirovaniya-ingibitorov-agregacii.html" rel="bookmark" title="База патентов Евразийского Союза">Способ предсказания агрегации белков и конструирования ингибиторов агрегации</a>
Способ ингибирования агрегации амилоидных белков и визуализации амилоидных отложений с использованием производных изоиндолина

Номер патента: 4405
Опубликовано: 29.04.2004
Авторы: Лай Йингджи, Оджелли-Зэфрэн Коринн Элизабет, Уокер Лари Кразуэлл, Сэккэб Аннетт Тереза
МПК: C07D 209/44, A61P 43/00, A61K 31/4035...
Метки: отложений, амилоидных, агрегации, производных, ингибирования, визуализации, белков, использованием, способ, изоиндолина
Формула / Реферат:
1. Соединения, имеющие формулу I или их фармацевтически приемлемые соли, где X представляет собой фенил или замещенный фенил; Y представляет собой фенил, замещенный фенил, пиридил или замещенный пиридил; где замещенный фенил и замещенный пиридил могут иметь от 1 до 4 заместителей, каждый из которых независимо выбран из -OC1-C12алкила, галогена, -C1-C6алкила, фенила, -CO2H, -CO2R1, -NO2, -CF3, -CN, -NR1R2, -(CH2)nCO2H, -(CH2)nCO2R1, -SO2NR1R2,...
Ингибиторы агрегации амилоидных белков, их применение (варианты), композиция на их основе и способ визуализации амилоидных отложений

Номер патента: 4632
Опубликовано: 24.06.2004
Авторы: Барвиан Марк Роберт, Лай Йингджи, Оджелли-Зэфрэн Коринн Элизабет, Кимура Такенори, Глейз Шелли Энн, Сэккэб Аннетт Тереза, Жуанг Ниан, Уокер Лари Кразуэлл, Ясунага Томоюки, Кейли Джон Стивен, Суто Марк Джеймс, Хачийя Шуничиро, Бигг Кристофер Франклин
МПК: A61K 31/195, C07D 233/54, A61P 25/28...
Метки: визуализации, способ, белков, ингибиторы, применение, варианты, отложений, композиция, амилоидных, агрегации, основе
Формула / Реферат:
1. Соединение формулы I где Ra представляет собой водород, C1-C6алкил или алкил; n равен числу от 1 до 5 включительно; R1, R2, R3, R4, R5, R6 и R7 независимо представляют собой водород, галоген, -OH, -NH2, NRbRc, -CO2H, -CO2C1-C6алкил, -NO2, -OC1-C12алкил, C1-C8алкил, -CF3, -CN, -OCH2фенил, -OCH2-замещенный фенил, -(CH2)m-фенил, -O-фенил, -O-замещенный фенил, -CH=CH-фенил, R8 представляет собой COOH, тетразолил, -SO2Rd или -CONHSO2Rd; Rb и...
Способы основанного на структуре конструирования лекарственных средств для индентификации ингибиторов d – ala -d – ala – лигазы в качестве антибактериальных лекарств

Номер патента: 7612
Опубликовано: 29.12.2006
Авторы: Ала Пол Дж., Перола Эмануэле, Коннелли Патрик Р., Навиа Мануэл А., Мэджи Эндрю С., Гриффит Джеймс П., Али Джанид А., Мо Скотт Т., Фэрман Карлос Х.
МПК: C12Q 1/37
Метки: лекарственных, лекарств, структуре, способы, конструирования, индентификации, лигазы, основанного, ингибиторов, качестве, средств, антибактериальных
Формула / Реферат:
1. Способ оценки потенциала химического объекта к взаимодействию с молекулой или молекулярным комплексом, включающим связывающий карман, определенный структурными координатами аминокислот Lys144, Glu180, Lys181, Leu183, Glu187, Asp257 и Glu270 D-Ala-D-Ala-лигазы E. coli, в соответствии с фиг. 8; или с гомологом указанной молекулы или молекулярного комплекса, где указанный гомолог содержит связывающий карман, который имеет среднее квадратичное...
Индолизины в качестве ингибиторов киназных белков

Номер патента: 7983
Опубликовано: 27.02.2007
Авторы: Амендола Шелли, Олдоус Дэвид Джон, Саунесс Джон Эдвард, Немесек Консепсьон, Уолш Роджер Джон Айтчисон, Вено Коринн, Венслер Сильви, Рэтклифф Эндрю Джеймс, Турайратнам Сукантини, Мэйджид Тахир Надим
МПК: C07D 209/00, A61K 31/495, C07D 221/00...
Метки: киназных, белков, качестве, индолизины, ингибиторов
Формула / Реферат:
1. Соединения общей формулы (I) необязательно дополнительно замещенные в насыщенном цикле одним или несколькими алкильными заместителями, где R1 означает водород, R4, -C(=Y)-NHR4, -SO2NHR4, -C(=Z1)-R4, -SO2-R4 или -C(=Z1)-OR4; R2 означает водород, цианогруппу, галоген или -CуC-R5; R3 означает водород, ацил, алкоксикарбонил, алкил, ароил, арил, арилоксикарбонил, карбоксильную группу, циклоалкенил, циклоалкил, гетероароил, гетероарил,...
Димерные ингибиторы ингибиторов белков апоптоза (iap)

Номер патента: 12810
Опубликовано: 30.12.2009
Авторы: Кондон Стефен М., Риппин Сюзан Р., Лапорте Мэттью Г., Денг Йиджун
МПК: A61K 31/40, C07D 209/20, A61P 35/00...
Метки: димерные, белков, ингибиторов, апоптоза, iap, ингибиторы
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль в котором R5a и R5b независимо представляют собой Н, алкил, циклоалкил или необязательно замещенный алкил, где заместители алкила выбраны из группы, состоящей из гидрокси, алкокси, арилокси и амино; либо R5a и R5b вместе образуют алкилен, гидроксизамещенный алкилен; алкенилен или -CH2SSCH2-; R7a и R7b независимо представляют собой Н или низший алкил; R8a и R8b независимо...
Предыдущий патент: 4-(4-трифторметил-3-тиобензоил)пиразолы и их применение в качестве гербицидов
Случайный патент: Прозрачное оконное стекло с нагреваемым покрытием