Новые пиперидинкарбоксамидные производные, способ их получения и содержащие их фармацевтические композиции







Формула / Реферат
1. Соединение формулы

где R1 представляет собой атом водорода или метильный радикал;
B представляет собой прямую связь или группу -CH2-;
Z представляет собой фенил, 2,3-дихлорфенил или 2,6-дихлорфенил;
и его соли с неорганическими или органическими кислотами, его сольваты и/или его гидраты.
2. Соединение формулы (I) по п.1 в форме оптически чистых изомеров.
3. Соединение по п.1 или 2, выбранное из
N,N-диметил-1-[2-[4-бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера;
N-метил-1-[2-[4-бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера;
N,N-диметил-1-[2-[4-(2,3-дихлорбензоил)-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, левовращающего изомера;
N,N-диметил-1-[2-[4-(2,6-дихлорфенил)ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера;
N,N-диметил-1-[2-[4-[2-(2,3-дихлорфенил)ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера;
N-метил-1-[2-[4-[2-(2,3-дихлорфенил)ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера;
и их солей с неорганическими или органическими кислотами, их сольватов и/или их гидратов.
4. Соединение по любому из пп.1-3, которое представляет собой N,N-диметил-1-[2-[4-бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамид, правовращающий изомер и его соли с неорганическими или органическими кислотами, его сольваты и/или его гидраты.
5. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы

где B и Z такие, как определено для соединения формулы (I) по п.1, подвергают взаимодействию с соединением формулы

где R1 такой, как определено для соединения формулы (I) по п.1, в присутствии кислоты, в растворителе, а затем образовавшуюся промежуточную иминиевую соль восстанавливают с помощью восстановителя.
6. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы

где B и Z такие, как определено для соединения формулы (I) в п.1, а Y представляет собой метил, фенил, толил или трифторметильную группу, подвергают взаимодействию с соединением формулы
где R1 такой, как определено для соединения формулы (I) в п.1.
7. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы

где R1 такой, как определено для соединения формулы (I) по п.1, подвергают взаимодействию с функциональным производным кислоты формулы
HOOC-B-Z (VI),
где B и Z такие, как определено для соединения формулы (I) по п.1.
8. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пп.1-4 или одну из его фармацевтически приемлемых солей, сольватов и/или гидратов.
9. Фармацевтическая композиция по п.8, содержащая от 0,1 до 1000 мг активного ингредиента в стандартной лекарственной форме, в которой активный ингредиент смешан по меньшей мере с одним фармацевтическим эксципиентом.
10. Применение соединения по любому из пп.1-4 или одной из его фармацевтически приемлемых солей, сольватов и/или гидратов для приготовления лекарственных средств, предназначенных для лечения любой патологии, в которую вовлечены либо нейрокинин A и/или NK2 рецепторы, либо нейрокинин B и/или NK3 рецепторы, либо оба нейрокинин A и нейрокинин B и/или NK2 и NK3 рецепторы.
11. Применение по п.10 для приготовления лекарственных средств, предназначенных для лечения патологий дыхательной, желудочно-кишечной, мочевой, иммунной, сердечно-сосудистой системы и центральной нервной системы, а также боли, мигрени, воспаления, тошноты и рвоты и кожных заболеваний.
12. Применение по п.11 для приготовления лекарственных средств, предназначенных для лечения хронического обструктивного бронхита, астмы, недержания мочи, синдрома раздраженного кишечника, болезни Крона, язвенного колита, депрессии, тревоги, эпилепсии, шизофрении.
13. Лекарственное средство, отличающееся тем, что оно содержит соединение по любому из пп.1-4 или одну из его фармацевтически приемлемых солей, сольватов и/или гидратов.
Текст
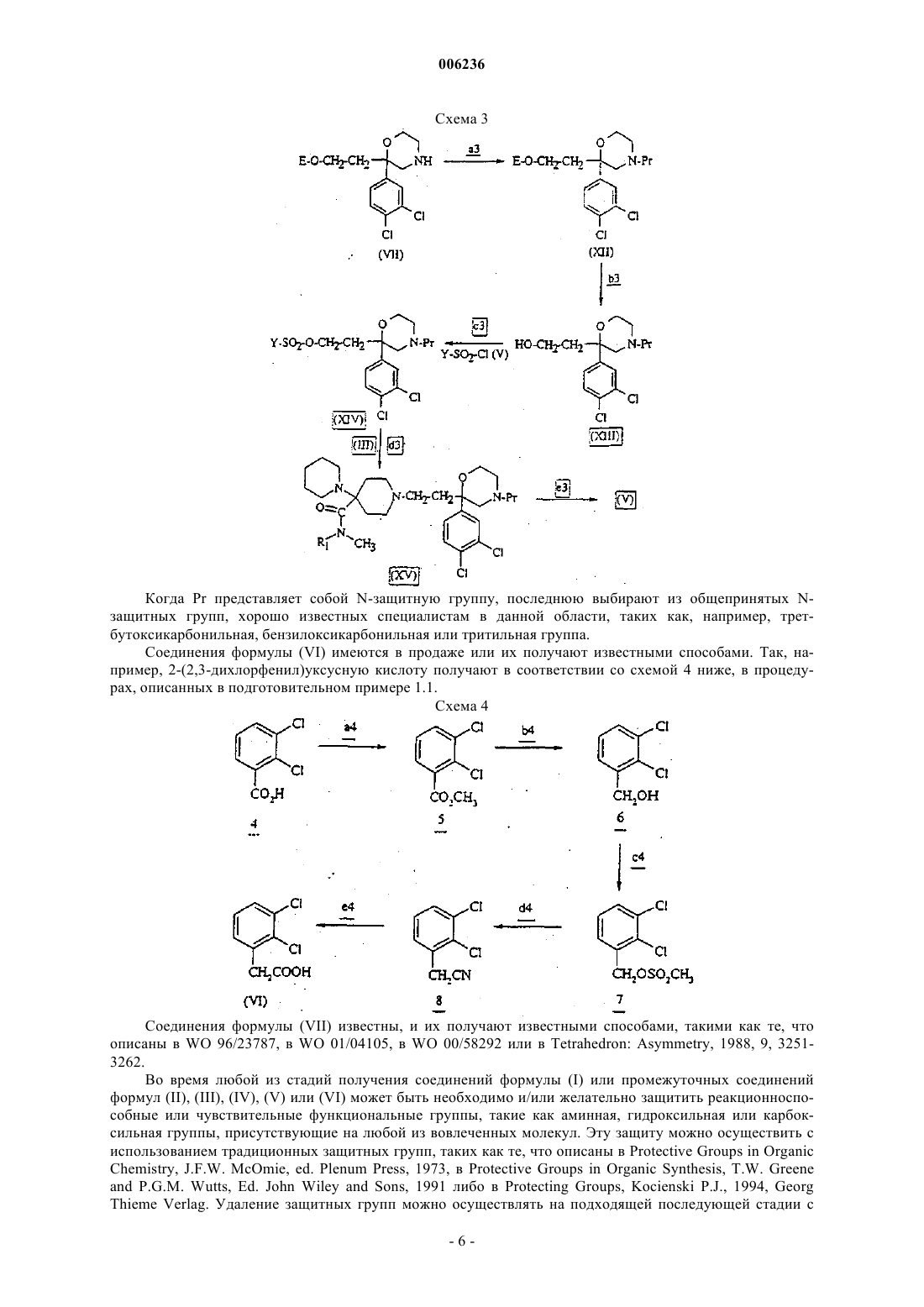
006236 Задачей настоящего изобретения являются новые пиперидинкарбоксамидные производные, способ их получения и фармацевтические композиции, содержащие их в качестве активного ингредиента. Более конкретно, настоящее изобретение относится к новым пиперидинкарбоксамидным производным для терапевтического применения при патологических явлениях, в которые вовлечена тахикининовая система, таких как, например, без ограничения: боль (L. Urban et al., TINS, 1994, 17, 432-438; L.Otsuka and K. Yoshioka, Physiol. Rev. 1993, 73, 229-308). В последние годы проводили многочисленные научные исследования в отношении тахикининов и их рецепторов. Тахикинины распространены как в центральной нервной системе, так и в периферической нервной системе. Рецепторы тахикининов известны и классифицируются на три типа: NK1, NK2,NK3. Вещество Р (SP) представляет собой эндогенный лиганд для NK1 рецепторов, нейрокинин A (NKA) является эндогенным лигандом для NK2 рецепторов, и нейрокинин В (NKB) - лигандом для NK3 рецепторов.NK1, NK2 и NK3 рецепторы выявлены у различных видов. В обзоре С. A. Maggi et al. (J. Autonomic Pharmacol., 1993, 13, 23-93) и обзоре D. Regoli et al. (Pharmacol. Rev., 1994, 46, 551-599) суммированы тахикининовые рецепторы и их антогонисты и описаны фармокологические исследования и применение в терапии человека. В многочисленных патентах или заявках на патент описаны соединения, которые активны в отношении тахикининовых рецепторов. Так, международная заявка WO 96/23787 относится к соединениям формулы где А может представлять собой двухвалентный радикал -О-СН 2-СН 2-;Am, m, Ar1 и Т имеют различные значения. В частности, 1-[2-[4-бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамид (соединение ) описано в примере 65 WO 96/23787. Это соединение обладает высоким сродством к человеческим NK2 рецепторам, но низким сродством к человеческим NK3 рецепторам. Заявка на патент ЕР-А-0776893 относится к соединениям формулы где, в частности, D-E может представлять собой двухвалентный радикал -О-СН 2-СН 2-;L, G, Е, А, В, Ra и Rb имеют различные значения. Патент WO 00/34274 относится к циклогексилпиперидиновым производным, которые являются антагонистами как NK1 рецепторов для вещества Р, так и NK2 рецепторов для нейрокинина А. Теперь обнаружены новые соединения, которые обладают очень высоким сродством как к человеческим NK2 рецепторам для нейрокинина А, так и к человеческим NK3 рецепторам для нейрокинина В, и которые являются антагонистами указанных рецепторов. Более того, соединения по настоящему изобретению обладают хорошей биодоступностью при введении их пероральным путем. Эти соединения могут быть использованы для приготовления лекарственных средств, полезных при лечении любой патологии, в которую вовлечены либо нейрокинин А и/или NK2 рецепторы, либо нейрокинин В и/или NK3 рецепторы, либо оба нейрокинин А и нейрокинин В и/или NK2 и NK3 рецепторы, в частности при лечении патологий дыхательной, желудочно-кишечной, мочевой, иммунной, сердечнососудистой и центральной нервной систем, а также при лечении боли, мигрени, воспаления, тошноты и рвоты и кожных заболеваний. Таким образом, в соответствии с одним аспектом изобретения, в его задачу входят соединения формулыR1 представляет собой атом водорода или метильный радикал; В представляет собой прямую связь или группу -СН 2-;Z представляет собой фенил, 2,3-дихлорфенил или 2,6-дихлорфенил; а также их соли с неорганическими или органическими кислотами, их сольваты и/или их гидраты. Соединения формулы (I) по изобретению включают в себя как оптически чистые изомеры, так и их смеси в любых соотношениях. Таким образом, возможно образовывать соли соединений формулы (I). Эти соли включают в себя соли как с неорганическими, так и с органическими кислотами, которые дают возможность соответствующего разделения или кристаллизации соединений формулы (I), такими как пикриновая кислота, или щавелевая кислота, или оптически активная кислота, например миндальная кислота или камфорсульфоновая кислота, и с теми кислотами, которые образуют фармацевтически приемлемые соли, такие как гидрохлорид, гидробромид, сульфат, кислый сульфат, однозамещенный кислый фосфат, метансульфонат, метилсульфат, оксалат, малеат, фумарат, сукцинат, нафталин-2-сульфонат, глюконат, цитрат, изотионат, бензолсульфонат, п-толуолсульфонат, ацетат. Подразумевается, что выражение атом галогена означает атом хлора, брома, фтора или иода. В соответствии с настоящим изобретением предпочтительны соединения формулы (I) в форме оптически чистых изомеров. Предпочтительны следующие соединения:N-метил-1-[2-[4-[2-(2,3-дихлорфенил)ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамид, правовращающий изомер; а также их соли с неорганическими или органическими кислотами, их сольваты и/или их гидраты. Следующее соединение:N,N-диметил-1-[2-[4-бензоил-2-(3,4-дихлорфенил)-морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамид, правовращающий изомер; а также его соли с неорганическими или органическими кислотами, его сольваты и/или его гидраты особенно предпочтительны. В соответствии с еще одним аспектом настоящее изобретение относится к способу получения соединений формулы (I), их солей, их сольватов и/или их гидратов, отличающемуся тем, что соединение формулы где В и Z такие, как определено для соединения формулы (I), подвергают взаимодействию с соединением формулы где R1 такой, как определено для соединения формулы (I), в присутствии кислоты, в растворителе, а затем образовавшуюся промежуточную иминиевую соль восстанавливают с помощью восстановителя. Возможно, соединение формулы (I) превращают в одну из его солей с неорганическими или органическими кислотами. Реакцию осуществляют в присутствии кислоты, такой как уксусная кислота, в растворителе, таком как метанол или дихлорметан, при температуре между комнатной температурой и температурой дефлегмации растворителя, и в ней in situ образуется промежуточный имин, который химически восстанавливают с использованием, например, цианоборогидрида натрия или триацетоксиборогидрида натрия, или каталитически - с использованием водорода и катализатора, такого как палладий на углероде или никельRaney. В соответствии с одним вариантом способа соединение формулы где В и Z такие, как определено для соединения формулы (I), a Y представляет собой метил, фенил, толил или трифторметильную группу, подвергают взаимодействию с соединением формулы где R1 такой, как определено для соединения формулы (I). Возможно, соединение формулы (I) превращают в одну из его солей с неорганическими или органическими кислотами. Реакцию осуществляют в инертном растворителе, таком как N,N-диметилформамид, ацетонитрил,метиленхлорид, толуол или изопропанол, и в присутствии или в отсутствие основания. Когда используют основание, его выбирают из органических оснований, таких как триэтиламин, N,Nдиизопропилэтиламин или N-метилморфолин, либо из карбонатов или бикарбонатов щелочных металлов, таких как карбонат калия, карбонат натрия или бикарбонат натрия. В отсутствие основания реакцию осуществляют с использованием избытка соединения формулы (III) и в присутствии иодида щелочного металла, такого как иодид калия или иодид натрия. Реакцию осуществляют при температуре между комнатной температурой и 100 С. В соответствии с еще одним вариантом способа соединение формулы где R1 такой, как определено для соединения формулы (I), подвергают взаимодействию с функциональным производным кислоты формулы(VI) где В и Z такие, как определено для соединения формулы (I). Возможно, соединение формулы (I) превращают в одну из его солей с неорганическими или органическими кислотами. В качестве функционального производного кислоты (VI) используют саму кислоту или же одно из функциональных производных, которые взаимодействуют с аминами, например ангидрид, смешанный ангидрид, хлорангидрид или активированный эфир, такой как п-нитрофениловый эфир.-3 006236 Когда используют саму кислоту формулы (VI), процедуру осуществляют в присутствии агента сочетания, используемого в пептидной химии, такого как 1,3-дициклогексилкарбодиимин или бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторфосфат, в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в инертном растворителе, таком как дихлорметан или N,Nдиметилформамид, при температуре между 0 С и комнатной температурой. Когда используют хлорангидрид, реакцию осуществляют в инертном растворителе, таком как дихлорметан или бензол, в присутствии основания, такого как триэтиламин или N-метилморфолин, и при температуре между -60 С и комнатной температурой. Соединения формулы (I), полученные таким образом, могут быть затем выделены из реакционной смеси и очищены общепринятыми способами, например путем кристаллизации или хроматографии. Соединения формулы (I), полученные таким образом, выделяют в форме свободного основания или соли по общепринятым методикам. Когда соединения формулы (I) получают в форме свободного основания, солеобразование осуществляют путем обработки выбранной кислотой в органическом растворителе. Обработкой свободного основания, растворенного, например, в простом эфире, таком как диэтиловый эфир, или в спирте, таком как 2-пропанол, или в ацетоне, или в дихлорметане, или в этилацетате, или в ацетонитриле, раствором выбранной кислоты в одном из вышеупомянутых растворителей получают соответствующую соль, которую выделяют общепринятыми методами. Таким образом получают, например, гидрохлорид, гидробромид, сульфат, трифторацетат, гидросульфат, дигидросульфат, метансульфонат, оксалат, малеат, сукцинат, фумарат, нафталин-2-сульфонат,бензолсульфонат, п-толуолсульфонат, глюконат, цитрат или ацетат. В конце реакции соединения формулы (I) могут быть выделены в форме из их солей, например гидрохлорида или оксалата, в этом случае, если это необходимо, свободное основание может быть получено путем нейтрализации указанной соли неорганическим или органическим основанием, таким как гидроксид натрия или триэтиламин, или карбонатом или бикарбонатом щелочного металла, таким как карбонат или бикарбонат натрия или калия. Соединения формулы (II) получают известными способами, такими как те, что описаны в WO 96/23787. Например, соединение формулы (II) получают в соответствии со схемой 1 ниже, где Е представляет собой атом водорода или О-защитную группу. Схема 1 Когда Е представляет собой защитную группу, последнюю выбирают из традиционных Озащитных групп, хорошо известных специалисту в данной области, таких как, например, тетрагидропиран-2-ил, бензоил или (C1-С 4)алкилкарбонил. На стадии a1 схемы 1 соединение формулы (VII) подвергают взаимодействию с функциональным производным кислоты формулы (VI) способами, описанными ранее, для того чтобы получить соединение формулы (VIII). С соединения формулы (VIII), полученного таким образом, на стадии b1 возможно удаляют защиту способами, известными специалисту в данной области. Например, когда Е представляет собой тетрагидропиран-2-ильную группу, удаление защиты осуществляют путем кислотного гидролиза с использовани-4 006236 ем соляной кислоты в растворителе, таком как диэтиловый эфир, метанол или смесь этих растворителей,либо с использованием пиридиния п-толуолсульфоната в растворителе, таком как метанол, либо, альтернативно, с использованием смолы Amberlyst в растворителе, таком как метанол. Реакцию осуществляют при температуре между комнатной температурой и температурой дефлегмации растворителя. Когда Е представляет собой бензоильную группу или (С 1-С 4)алкилкарбонильную группу, удаление защиты осуществляют путем гидролиза в щелочной среде с использованием, например, гидроксида щелочного металла, такого как гидроксид натрия, гидроксид калия или гидроксид лития, в инертном растворителе,таком как вода, метанол, этанол, диоксан или смесь этих растворителей, при температуре между 0 С и температурой дефлегмации растворителя. На стадии с 1 спирт формулы (IX) окисляют с получением альдегида формулы (II). Реакцию окисления осуществляют с использованием, например, оксалилхлорида, диметилсульфоксида и триэтиламина в растворителе, таком как дихлорметан, и при температуре между -78 С и комнатной температурой. Соединения формулы (III) известны, и их получают известными способами. Например, соединение формулы (III) получают в соответствии со схемой 2 ниже. Схема 2 Стадии а 2 и b2 схемы 2 осуществляют в соответствии с методиками, описанными на стадиях А и В Получения 2.16 в WO 96/23787. На стадии с 2соединение 3 подвергают взаимодействию с метилгалогенидом, предпочтительно метилиодидом, в присутствии сильного основания, такого как гидрид натрия, в растворителе, таком как тетрагидрофуран, и при температуре между комнатной температурой и температурой дефлегмации растворителя, и получают смесь соединения формулы (X), где R1 = Н, и соединения формулы (X), где R1 =CH3, которую разделяют общепринятыми способами, такими как хроматография. С соединения (X) удаляют защиту на стадиях d2 или е 2 известными способами с получением ожидаемых соединений формулы (III). Соединения формулы (IV) получают известными способами, такими как те, что описаны в WO 96/23787. Например, соединение формулы (IX) подвергают взаимодействию с соединением формулы(XI) где Y представляет собой метил, фенил, толил или трифторметильную группу. Реакцию осуществляют в присутствии основания, такого как триэтиламин, пиридин, N,N-диизопропиламин или N-метилморфолин, в растворителе, таком как дихлорметан или толуол, и при температуре между -20 С и температурой дефлегмации растворителя. Соединения формулы (V) получают в соответствии со схемой 3 ниже, где Е представляет собой водород или О-защитную группу, а Рr представляет собой N-защитную группу. Когда Рr представляет собой N-защитную группу, последнюю выбирают из общепринятых Nзащитных групп, хорошо известных специалистам в данной области, таких как, например, третбутоксикарбонильная, бензилоксикарбонильная или тритильная группа. Соединения формулы (VI) имеются в продаже или их получают известными способами. Так, например, 2-(2,3-дихлорфенил)уксусную кислоту получают в соответствии со схемой 4 ниже, в процедурах, описанных в подготовительном примере 1.1. Схема 4 Соединения формулы (VII) известны, и их получают известными способами, такими как те, что описаны в WO 96/23787, в WO 01/04105, в WO 00/58292 или в Tetrahedron: Asymmetry, 1988, 9, 32513262. Во время любой из стадий получения соединений формулы (I) или промежуточных соединений формул (II), (III), (IV), (V) или (VI) может быть необходимо и/или желательно защитить реакционноспособные или чувствительные функциональные группы, такие как аминная, гидроксильная или карбоксильная группы, присутствующие на любой из вовлеченных молекул. Эту защиту можно осуществить с использованием традиционных защитных групп, таких как те, что описаны в Protective Groups in OrganicThieme Verlag. Удаление защитных групп можно осуществлять на подходящей последующей стадии с-6 006236 использованием способов, известных специалисту в данной области, и которые не затрагивают оставшуюся часть вовлеченной молекулы. Разделение рацемических смесей соединений формулы (I) позволяет выделить энантиомеры. Однако предпочтительно осуществлять разделение рацемических смесей от соединения формулы(VII, Е = Н) или, альтернативно, от промежуточного соединения, полезного для получения соединения формулы (VII), способами, описанными в публикациях, указанных выше для получения соединения формулы (VII). Соединения формулы (I) выше также включают такие, в которых один или более чем один атом водорода или углерода заменены их радиоактивными изотопами, например тритием или углеродом-14. Такие меченые соединения полезны в исследовательской работе, работе с метаболизмом или фармакокинетикой либо в биохимических испытаниях в качестве рецепторных лигандов. Соединения по настоящему изобретению были подвергнуты биохимическим тестам. Сродство соединений к тахикининовым рецепторам оценивали in vitro с помощью нескольких биохимических тестов с использованием радиолигандов: 1) связывание [125I]BH-SP (Вещество Р, меченное иодом-125 с помощью реагента Bolton-Hunter) сNK1 рецепторами человеческих лимфобластов (D.G. Payan et al., J. Immunol., 1984,133, 3260-3265); 2) связывание [125l]His-NKA с клонированными человеческими NK2 рецепторами, экспрессированными клетками СНО (клетки яичника китайского хомячка) (Y. Takeda et al., J. Neurochem., 1992, 59, 740745); 3) связывание [125l]His[MePhe7]NKB с клонированными человеческими NK3 рецепторами, экспрессированными клетками СНО (Buell et al., FEBS Letters, 1992, 299, 90-95). Тесты осуществляли в соответствии с X. Emonds-Alt et al. (Eur. J. Pharmacol., 1993, 250, 403-413;Life Sci., 1995, 56, PL 27-32). Соединения по изобретению слабо ингибируют связывание вещества Р с NK1 рецепторами человеческих лимфобластов IM9. Константа ингибирования K1 для рецепторов человеческих лимфобластов больше или равна 810-9 М. Соединения по изобретению сильно ингибируют связывание [125l]His-NKA с клонированными человеческими NK2 рецепторами. Константа ингибирования K1 меньше или равна 510-10 М. Так, соединение примера 1 обладает K1, равной 410-11 М. Соединения по изобретению сильно игибируют связывание [125I]His[MePhe7]NKB с клонированными человеческими NК 3 рецепторами: константа ингибирования K1 меньше или равна 710-10 М. Так, соединение примера 1 обладает K1, равной 410-11 М. Соединениеиз уровня техники ингибирует связывание [125l]His-NKA с клонированными NK2 рецепторами с K1, равной 410-11 М. Оно ингибирует связывание [125I]His[MePhe7]NKB с клонированными человеческими NK3 рецепторами с K1, равной 210-9 М. Соединения по настоящему изобретению были также оценены in vivo на животных моделях. У песчанок поведение вращения индуцировали путем интрастриатального введения специфичного агониста NK2 рецептора [Nle10]NKA (4-10); наблюдали, что одностороннее применение [Nle10]NKA (410) к полосатому телу песчанок приводило к сильным контралатеральным вращениям, которые ингибировали соединениями по изобретению, вводимыми либо интраперитониальным путем, либо пероральным путем. Этот тест осуществляли в соответствии с М. Poncelet et al., Neurosci, Lett., 1993, 149, 40-42. В этом тесте соединения по изобретению являются активными в дозах, варьирующих от 0,1 до 30 мг на кг. Например, соединение примера 1 имеет эффективную дозу 50 (ЭД 50), равную 2,9 мг/кг, при интраперитонеальном пути и ЭД 50, равную 6,5 мг/кг, при пероральном пути. Поведение вращения у песчанок индуцировали путем интрастриатального введения специфичного агониста NK3 рецептора сенктида; наблюдали, что одностороннее применение сенктида к полосатому телу песчанок приводило к сильным контралатеральным вращениям, которые ингибировались соединениями по изобретению, вводимыми либо интраперитониальным путем, либо пероральным путем. Этот тест осуществляли в соответствии с X. Emonds-Alt et al., Life Sci., 1995, 56, PL27-PL32. В данном тесте соединения по изобретению являются активными в дозах, варьирующих от 0,1 до 30 мг на кг. Например,соединение примера 1 имеет ЭД 50, равную 2,8 мг/кг, при интраперитонеальном пути и ЭД 50, равную 4,3 мг/кг, при пероральном пути. У крыс применение агониста NK2 рецепторов к перегородке вызывало увеличение высвобождения ацетилхолина в гиппокампе (тест осуществляли в соответствии с R. Steinberg et al., Eur. J. Neurosci., 1998,10, 2337-2345). Подобным же образом, у морских свинок местное применение агониста NK3 рецепторов к перегородке вызывало увеличение высвобождения ацетилхолина в гиппокампе (тест осуществляли в соответствии N. Marco et al., Neuropeptides, 1998, 32, 481-488). Соединения по изобретению блокировали это увеличение высвобождения ацетилхолина независимо от того, было ли оно вызвано агонистом NK2 рецепторов или агонистом NК 3 рецепторов. Например, соединение примера 1 блокирует это увеличение высвобождения ацетилхолина, вызванное либо агонистом NK2 рецепторов у крыс, либо агонистом NK3 ре-7 006236 цепторов у морских свинок, в дозах 0,1-0,3 мг/кг и 0,3-1 мг/кг соответственно при использовании интраперитонеального пути. У крыс стресс ограничением вызывает увеличение тканевого уровня DOPAC (3,4 дигидроксифенил-уксусной кислоты) в префронтальной коре (тест осуществляли в соответствии с В. A.Morrow et al., Eur. J. Pharmacol., 1993, 238, 255-262). Это увеличение блокируется специфичным антагонистом NK2 рецепторов, таким как саредутант (X. Emonds-Alt et al., Life Sci., 1992, 50, PL101-PL106), и,следовательно, оно опосредовано активацией NK2 рецепторов эндогенным нейрокинином А. Наблюдали,что соединение примера 1, вводимое в концентрации 1 мг/кг интраперитонеальным путем, полностью блокирует это увеличение. У морских свинок обработка галоперидолом, вводимым в дозе 1 мг/кг интраперитонеальным путем,вызывает увеличение числа дофаминэргических нейронов, которые спотанно активны (популяционный ответ) в районе А 10 (VTA, вентральная тегментальная область) головного мозга, измеренное электрофизиологическим способом. Это увеличение опосредовано активацией NK3 рецепторов эндогенным нейрокинином В (С. Gueudet et al., Synapse, 1999, 33. 71-79). Наблюдают, что соединение примера 1, вводимое в концентрации 0,1-1 мг/кг интраперитонеальным путем, блокирует указанное увеличение. Все эти фармакологические результаты показывают, что соединения по изобретению, в частности соединение примера 1, представляют собой смешанные антагонисты NK2 рецепторов и NK3 рецепторов путем блокирования фармакологических эффектов, вызываемых нейрокинином А или нейрокинином В,независимо от того, применяются ли они экзогенно либо провоцируется их эндогенное высвобождение. Более того, эти результаты показывают, что соединения по изобретению хорошо проходят через гематоэнцефалический барьер. Соединения по настоящему изобретению представляют собой, в частности, активные ингредиенты фармацевтических композиций, токсичность которых совместима с их применением в качестве лекарственного средства. Соединения вышеописанной формулы (I) могут быть использованы в суточных дозах от 0,01 до 100 мг на кг массы тела млекопитающего, которого нужно лечить, предпочтительно в суточных дозах от 0,1 до 50 мг/кг. Для людей доза может предпочтительно варьировать от 0,1 до 4000 мг в сутки, более предпочтительно от 0,5 до 1000 мг, в зависимости от возраста субъекта, которого лечат, или типа лечения: профилактического или исцеляющего. Для использования в качестве лекарственных средств соединения формулы (I) обычно вводят в форме единиц дозировки. Указанные единицы дозировки предпочтительно готовят в составе фармацевтических композиций, в которых активный ингредиент смешан с одним или более чем одним фармацевтическим эксципиентом. Таким образом, в соответствии с еще одним из своих аспектов настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение формулы(I) или одну из его фармацевтически приемлемых солей, сольватов и/или гидратов. В фармацевтических композициях по настоящему изобретению для введения с помощью перорального, сублингвального, ингаляционного, подкожного, внутримышечного, внутривенного, чрезкожного,местного или ректального пути активные ингредиенты можно вводить животным и человеческим особям в стандартных формах для введения, в смеси с общепринятыми фармацевтическими носителями. Соответствующие стандартные формы для введения включают формы для введения пероральным путем, такие как таблетки, желатиновые капсулы, порошки, гранулы и пероральные растворы или суспензии,формы для сублингвального и трансбуккального введения, аэрозоли, формы для местного введения, имплантаты, формы для подкожного, внутримышечного, внутривенного, интраназального или внутриглазного введения и формы для ректального введения. Когда твердую композицию готовят в форме таблеток или желатиновых капсул, к активному ингредиенту добавляют в микронизированном или другом виде смесь фармацевтических эксципиентов,которая состоит из разбавителей, таких как, например, лактоза, микрокристаллическая целлюлоза, крахмал, гидрофосфат кальция, связующих веществ, таких как, например, поливинилпирролидон, гидроксипропилметилцеллюлоза, разрыхляющих агентов, таких как поперечно-связанный поливинилпирролидон,поперечно-связанная карбоксиметилцеллюлоза, скользящих веществ, таких как диоксид кремния, тальк,смазывающих веществ, таких как стеарат магния, стеариновая кислота, глицеролтрибегенат, стеарилфумарат натрия. Увлажняющие агенты или поверхностно-активные вещества, такие как лаурил сульфат натрия, полисорбат 80, полоксамер 188, могут быть добавлены в препарат. Таблетки могут быть приготовлены с помощью различных методик: прямого прессования, сухого гранулирования, влажного гранулирования, горячего плавления. Таблетки могут быть без покрытия, либо на них может быть нанесено сахарное покрытие (например, сахарозное), либо они могут быть покрыты различными полимерами или другими соответствующими материалами. Таблетки могут иметь флэш-, замедленное или продолжительное высвобождение благодаря приготовлению полимерных матриц либо использованию специфических полимеров в пленочном покрытии.-8 006236 Желатиновые капсулы могут быть мягкими или твердыми, покрытыми пленочной оболочкой или другим материалом так, чтобы обладать флэш-, продолжительной или замедленной активностью (например, у энтеросолюбильной формы). Они могут содержать не только твердый препарат, приготовленный, как описано выше, для таблеток, но также жидкости или полутвердые вещества. Препарат в форме сиропа или эликсира может содержать активный ингредиент вместе с подсластителем, предпочтительно некалорийным, метилпарабеном и пропилпарабеном в качестве антисептика, а также с усилетелем вкуса и подходящим красящим агентом. Порошки или гранулы, диспергируемые в воде, могут содержать активный ингредиент в смеси с диспергирующими агентами, увлажняющими агентами или суспендирующими агентами, такими как поливинилпирролидон, а также с подсластителями или вкусоароматизаторами. Для ректального введения используют суппозитории, которые готовят со связующими веществами,которые плавятся при температуре прямой кишки, например с масло-какао или полиэтиленгликолями. Для парентерального, интраназального или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные и инъекционные растворы, которые содержат фармакологически совместимые диспергирующие агенты и/или солюбилизирующие агенты, например пропиленгликоль. Так, для приготовления водного раствора, который можно вводить инъекцией внутривенно, возможно применение сорастворителя, такого как, например, спирт, такой как этанол, или гликоль, такой как полиэтиленгликоль или пропиленгликоль, и гидрофильного поверхностно-активного вещества, такого как полисорбат 80 или полоксамер 188. Для приготовления масляного раствора, который можно вводить внутримышечной инъекцией, возможно растворять активный ингредиент в триглицериде или сложном эфире глицерина. Для местного введения можно использовать кремы, мази, гели, глазные примочки и спреи. Для чрезкожного введения возможно применять пластыри в многослойной форме или с резервуаром, в котором активный ингредиент может быть в спиртовом растворе, или спреи. Для введения путем ингаляции используют аэрозоль, который содержит, например, сорбитантриолеат или олеиновую кислоту, а также трихлорфторметан, дихлорфторметан, дихлортетрафторэтан, заменители фреона или любой другой биологически совместимый газ-пропеллент; также возможно применять систему, содержащую активный ингредиент сам по себе или вместе с эксципиентом, в порошкообразной форме. Активный ингредиент может также быть предложен в форме комплекса с циклодекстрином, например -циклодекстрином, 2-гидроксипропилциклодекстрином. Активный ингредиент может быть также приготовлен в форме микрокапсул или микросфер, возможно с одним или более чем одним носителем или добавкой. Из форм с продолжительным высвобождением, которые полезны в случае хронического лечения,могут быть использованы имплантаты. Они могут быть приготовлены в форме масляной суспензии или в форме суспензии микросфер в изотонической среде. В каждой единице дозировки активный ингредиент формулы (I) присутствует в количествах, соответствующих предусмотренным суточным дозам. В общем, каждую единицу дозировки подходящим образом регулируют в соответствии с предусмотренными дозировкой и типом введения, например с помощью таблеток, желатиновых капсул и тому подобного, саше, ампул, сиропов и тому подобного, капель, так что такая единица дозировки содержит от 0,1 до 1000 мг активного ингредиента, предпочтительно от 0,5 до 250 мг, для предстоящего введения от одного до четырех раз в сутки. Хотя указанные дозировки являются примерами средних ситуаций, могут быть отдельные случаи,когда более высокие или более низкие дозировки являются подходящими; такие дозировки также относятся к данному изобретению. В соответствии с обычной практикой, дозировки, подходящие для каждого пациента, определяются врачом в соответствии со способом введения, возрастом, массой и реакцией указанного пациента. В соответствии с одним из своих аспектов, настоящее изобретение относится к применению соединений формулы (I) или одной из их фармацевтически приемлемых солей, сольватов и/или гидратов для приготовления лекарственных средств, предназначенных для лечения любой патологии, в которую вовлечены либо нейрокинин А и/или NK2 рецепторы, либо нейрокинин В и/или NK3 рецепторы, либо оба нейрокинин А и нейрокинин В и/или NK2 и NK3 рецепторы. В соответствии с еще одним из аспектов настоящее изобретение относится к применению соединений формулы (I) или одной из их фармацевтически приемлемых солей, сольватов и/или гидратов для приготовления лекарственных средств, предназначенных для лечения патологий дыхательной, желудочно-кишечной, мочевой, иммунной, сердечно-сосудистой и центральной нервной системы, а также боли,мигрени, воспаления, тошноты, и рвоты и кожных заболеваний. Для примера и не ограничиваясь этим, соединения формулы (I) полезны в качестве анальгетиков, в частности, при лечении травматической боли, такой как послеоперационная боль; невралгии плечевого сплетения; хронической боли, такой как артритическая боль, вызванная-9 006236 остеоартритом, ревматоидным артритом или псориатическим артритом; невропатической боли, такой как постгерпетическая невралгия, тригеминальная невралгия, сегментарная или межреберная невралгия,фибромиалгия, каузалгия, периферическая невропатия, диабетическая невропатия, невропатии, вызванные химиотерапией, невропатии, связанные со СПИДом, затылочная невралгия, невралгия при синдроме коленчатого ганглия или глоссофарингеальная невралгия; фантомной боли людей с ампутированными конечностями; различных форм головной боли, таких как хроническая или острая мигрень, височнонижнечелюстная боль, боль в верхнечелюстной пазухе, лицевая невралгия или зубная боль; боли, испытываемой страдающими раком; боли висцерального происхождения; желудочно-кишечной боли; боли,вызванной сдавлением нерва; боли, вызванной интенсивными занятиями спортом; дисменорреи; менструальной боли; боли, вызванной менингитом или арахноидитом; скелетно-мышечной боли; боли внизу спины, вызванной спинальным стенозом, выпадением диска или ишиалгией; боли, испытываемой страдающими стенокардией; боли, вызванной анкилозирующим спондилоартритом; боли, связанной с подагрой; боли, связанной с ожогами, рубцеванием или зудящим дерматозом; таламической боли; в качестве противовоспалительных агентов, в частности, для лечения воспалений при астме, гриппе, хроническом бронхите (в частности, при обструктивном хроническом бронхите и ХО 3 Л (хроническое обструктивное заболевание легких, кашле, аллергиях, бронхоспазме и ревматоидном артрите; воспалительных заболеваний желудочно-кишечной системы, например болезни Крона, неспецифического язвенного колита, панкреатита, гастрита, воспаления кишечника, расстройств, вызванных нестероидными противовоспалительными средствами, воспалительных и секреторных эффектов, вызванных бактериальными инфекциями, например вызванных Clostridium difficile; воспалительных кожных заболеваний, например герпеса и экземы; воспалительных заболеваний мочевого пузыря, таких как цистит и недержание мочи; офтальмических воспалений, таких как конъюнктивит и витреоретинопатия; дентальных воспалений, таких как гингивит и периодонтит; при лечении аллергических заболеваний, в частности, кожи, таких как крапивница, контактный дерматит, атопический дерматит, и респираторных заболеваний, таких как ринит; при лечении заболеваний центральной нервной системы, в частности, психозов, таких как шизофрения, мания и деменция; расстройств познавательной способности, таких как болезнь Альцгеймера,тревога, деменция, связанная со СПИДом, диабетических невропатий; депрессии; болезни Паркинсона; лекарственной зависимости; злоупотребления веществами; расстройств сознания, нарушений сна, нарушений циркадного ритма, расстройств настроения и эпилепсии; синдрома Дауна; хореи Хантингтона; связанных со стрессом соматических расстройств; нейродегенеративных заболеваний, таких как болезнь Пика или болезнь Крейтцфельдта-Якоба; расстройств, связанных с паникой, фобией или стрессом; при лечении изменений проницаемости гематоэнцефалического барьера во время воспалительных и аутоиммунных процессов центральной нервной системы, например во время инфекций, связанных со СПИДом; в качестве миорелаксанта и антиспазматического средства; при лечении острой или отсроченной и прогнозируемой тошноты и рвоты, например тошноты и рвоты, вызванных лекарствами, такими как агенты, применяемые в химиотерапии в случае рака; лучевой терапией во время облучения грудной клетки или живота при лечении рака или карциноидоза (carcinoidosis); проглатыванием яда; токсинами, появившимися в результате нарушений обмена веществ или инфекционных расстройств, таких как гастрит, или продуцируемых во время бактериальной или вирусной желудочно-кишечной инфекции; во время беременности; во время вестибулярных расстройств, таких как укачивание, головокружение или болезнь Меньера; при послеоперационных заболеваниях; тошноты и рвоты, вызванных диализом или простагландинами; желудочно-кишечными обструкциями; при пониженной желудочно-кишечной двигательной функции; при висцеральной боли, вызванной инфарктом миокарда или перитонитом; при мигрени; при высотной болезни; глотанием опиатных анальгетиков, таких как морфин; при гастроэзофагеальном рефлюксе; при изжоге или чрезмерном потреблении пищи или напитков, при желудочной кислотности, регургитации и изжоге, например эпизодической или ночной изжоге, либо изжоге, вызванной пищей и диспепсией; при лечении заболеваний желудочно-кишечной системы, таких как синдром раздраженной толстой кишки, язвы желудка и двенадцатиперстной кишки, язвы пищевода, диарея, гиперсекреции, лимфомы,гастрит, гастроэзофагеальный рефлюкс, недержание кала и болезнь Гиршспрунга; при лечении кожных заболеваний, таких как псориаз, зуд и ожоги, в частности, солнечная эритема; при лечении заболеваний сердечно-сосудистой системы, таких как гипертензия, сосудистые проявления мигрени, отек, тромбоз, стенокардия, сосудистые спазмы, заболевания кровообращения, вызванные расширением сосудов, болезнь Рейно, фиброз, коллагеновые болезни и атеросклероз, преэклампсия; при лечении мелкоклеточного рака легкого, рака молочной железы, опухолей головного мозга и аденокарцином в мочеполовой сфере; в адъювантном лечении с целью предотвращения метастазов; димиелинизирующих заболеваний, таких как рассеянный склероз или боковой амиотрофический склероз; при лечении заболеваний иммунной системы, связанных с подавлением или стимуляцией функций иммунных клеток, например ревматоидного артрита, псориаза, болезни Крона, диабета, волчанки и реак- 10006236 ций отторжения после трансплантации; при лечении расстройств мочеиспускания, в частности, при поллакиурии, недержании мочи при напряжении, недержании мочи, связанном с непреодолимым позывом, при послеродовом недержании мочи; при лечении гистиоцитарного ретикулеза, например в лимфатических тканях; в качестве анорексигенного агента; при лечении эмфиземы; болезни Рейтера; геморроя; при лечении заболеваний глаз, таких как глаукома, окулярная гипертензия, миоз и избыточная секреция слез; при лечении или предупреждении удара, эпилепсии, черепной травмы, травмы спинного мозга,ишемических повреждений головного мозга, вызванных сосудистым приступом или окклюзией; при лечении нарушений частоты сердечных сокращений и сердечного ритма, в частности, тех, которые вызваны болью или стрессом; при лечении чувствительной кожи и для предупреждения или борьбы с раздражимостью кожи или слизистых оболочек, перхоти, эритемы или зуда; при лечении неврологических кожных нарушений, таких как лишай, пруриго, пруригогинозная токсидермия и сильный зуд нейрогенного происхождения; при лечении язв и всех заболеваний, вызванных Helicobacter pylori или уреазо-положительными грамотрицательными бактериями; при лечении заболеваний, вызванных ангиогенезом, или при которых ангиогенез является симптомом; при лечении глазной и/или пальпебральной (palbebral) боли и/или глазной или пальпебральной дизестезии; в качестве антиперспиранта. Настоящее изобретение также включает в себя способ лечения указанных жалоб в дозах, указанных выше. Фармацевтические композиции по настоящему изобретению могут также содержать другие активные продукты, которые являются полезными для лечения заболеваний или расстройств, указанных выше,например бронходилятаторы, противокашлевые агенты, антигистаминные агенты, противовоспалительные агенты, противорвотные агенты и химиотерапевтические агенты. Следующие подготовительные получения и примеры иллюстрируют изобретение, тем не менее не ограничивая его. Следующие обозначения используют в подготовительных получениях и примерах: ДМФ: диметилформамид ДМСО: диметилсульфоксид ДХМ: дихлорметан ТГФ: тетрагидрофуран Хлористо-водородный эфир: насыщенный раствор соляной кислоты в диэтиловом эфире БОФ: бензотриазол-1-илокситрис(диметиламино)-фосфония гексафторфосфат Т.пл.: температура плавления к.т.: комнатная температура Т. кип.: температура кипения Силикагель Н: 60 Н силикагель, продаваемый Merck (Darmstadt) Спектры протонного ядерного магнитного резонанса (1 Н-ЯМР) записывали при 200 МГц в ДМСО-d6, используя ДМСО-d6 пик в качестве стандарта. Химические сдвигиуказаны в миллионных долях (м.д.). Наблюдаемые сигналы выражали следующим образом:s: синглет, se: широкий синглет, t: триплет, qd: квартет, m: нерастворенный комплекс, mt: мультиплет. Спетры ЯМР подтверждают структуры соединений. Подготовительные получения 1. Подготовительное получение соединений формулы (VI). Подготовительное получение 1.1 2-(2,3-Дихлорфенил)уксусная кислота А) Метиловый эфир 2,3-дихлорбензойной кислоты 6 мл концентрированной серной кислоты добавляют к раствору 25,08 г 2,3-дихлорбензойной кислоты в 125 мл МеОН и затем смесь кипятят с обратным холодильником в течение ночи. Реакционную смесь концентрируют под вакуумом, остаток разводят водой, среду подщелачивают добавлением 10% раствора NaHCO3 и экстрагируют диэтиловым эфиром, органическую фазу промывают дважды водой,- 11006236 сушат над Na2SO4 и растворитель выпаривают под вакуумом. Получают 25,68 г ожидаемого продукта. Б) 2,3-Дихлорбензиловый спирт Суспензию 10,56 г алюмогидрида лития в 125 мл ТГФ охлаждали до 0 С, раствор 25,68 г соединения, полученного на предшествующей стадии, в 100 мл ТГФ добавляли по каплям, температуре давали вернуться до к.т. и смесь оставляли перемешиваться в течение 2 ч при к.т. Реакционную смесь разбавляли добавлением 250 мл ТГФ и гидролизовали добавлением 11 мл воды, 11 мл 4 н. NaOH и 33 мл воды. Ее оставляли стоять в течение ночи при к.т., неорганические соли фильтровали и фильтрат концентрировали под вакуумом. После высушивания под вакуумом при 30 С получали 21,54 г ожидаемого продукта. В) 2,3-Дихлорбензил метансульфонат Раствор 21,54 г соединения, полученного на предшествующей стадии, и 18,6 мл триэтиламина в 150 мл ДХМ охлаждали в ледяной бане, раствор 10,4 мл метансульфонилхлорида в 50 мл ДХМ добавляли по каплям при температуре менее чем 10 С и смесь оставляли перемешиваться, давая температуре вернуться до к.т. Смесь концентрировали под вакуумом, остаток экстрагировали диэтиловым эфиром и среду промывали дважды буферным раствором с рН=2, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Получали 29,25 г ожидаемого продукта. Г) 2,3-Дихлорфенилацетонитрил 10,1 г цианида калия при 97% добавляли к раствору 29,25 г соединения, полученного на предшествующей стадии, в 200 мл ЕtOН и 50 мл воды, и смесь кипятили с обратным холодильником в течение 2 ч. Смесь концентрировали под вакуумом, остаток экстрагировали AcOEt, органическую фазу промывали четыре раза водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток разводили в 200 мл пентана и среде давали кристаллизоваться в течение ночи при перемешивании. Образовавшийся осадок отделяли от жидкости и сушили под вакуумом. Получали 17,17 г ожидаемого продукта. Д) 2-(2,3-Дихлорфенил)уксусная кислота Раствор 24,23 г КОН в 74 мл воды добавляли к раствору 17,17 г соединения, полученного на предшествующей стадии, в 188 мл ЕtOН и затем смесь кипятили в течение ночи с обратным холодильником. Смесь концентрировали под вакуумом, остаток разводили в 100 мл воды, водную фазу промывали три раза диэтиловым эфиром, водную фазу подкисляли до рН = 1 добавлением концентрированного раствораHCl и давали произойти кристаллизации при перемешивании путем охлаждения в ледяной бане. Образовавшийся осадок отделяли от жидкости, промывали водой и сушили под вакуумом при 40 С. Получали 17,17 г ожидаемого продукта. 2. Подготовительное получение соединений формулы (II) Подготовительное получение 2.1 2-[4-Бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]ацетальдегид, единственный изомер(II): В = прямая связь; Z = А) 2-[2-(3,4-Дихлорфенил)морфолин-2-ил]этилбензоат, левовращающий изомер Это соединение получают в соответствии с методикой, описанной в Preparation 1.1 в WO 00/58292. Б) [2(3,4-Дихлорфенил)-2-(2-гидроксиэтил)-морфолин-4-ил](фенил)метанон, единственный изомер Раствор 4 г соединения, полученного на предшествующей стадии, и 1,5 мл триэтиламина в 100 мл ДХМ охлаждали до 0 С, раствор 1,41 г бензоилхлорида в 10 мл ДХМ добавляли по каплям и смесь оставляли перемешиваться в течение 30 мин. Реакционную смесь концентрировали под вакуумом, остаток экстрагировали диэтиловым эфиром, органическую фазу промывали водой, буферным раствором с рН = 2, водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Маслянистый остаток, полученный таким образом, разводили в 70 мл 95% ЕtOН, добавляли 2,5 мл 30% раствора NaOH и смесь оставляли перемешиваться в течение 1 ч при к.т. Смесь концентрировали под вакуумом, остаток экстрагировали AcOEt, органическую фазу промывали три раза водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Получали 4 г ожидаемого продукта. В) 2-[4-Бензоил-2-(3,4-дихлорфенил)морфолин-2-ил]ацетальдегид, единственный изомер Раствор 1,85 г соединения, полученного на предшествующей стадии, и 2,25 мл ДМСО в 25 мл ДХМ охлаждали до -60 С в атмосфере азота, 1,38 мл оксалилхлорида добавляли по каплям и смесь оставляли перемешиваться в течение 2 ч при -60 С. Затем добавляли 4,42 мл триэтиламина и смесь оставляли перемешиваться, позволяя температуре вернуться до к.т. Реакционную смесь разбавляли добавлением ДХМ, органическую фазу промывали водой, 10% раствором Na2 СО 3, дважды водой, насыщенным раствором NaCl, сушили надNa2SO4 и растворитель выпаривали под вакуумом. Получали 1,7 г ожидаемого продукта. Подготовительное получение 2.2 2-[4-(2,3-Дихлорбензоил)-2-(3,4-дихлорфенил)морфолин-2-ил]ацетальдегид, единственный изомер- 12006236 А) (2,3-Дихлорфенил)[2-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолин-4-ил]метатон, единственный изомер 3,3 г БОФ добавляют к раствору 2,5 г соединения, полученного на стадии (А) подготовительного получения 2.1, 1,2 г 2,3-дихлорбензойной кислоты и 0,75 г триэтиламина в 50 мл ДХМ и смесь оставляли перемешиваться в течение 30 мин при к.т. Смесь концентрировали под вакуумом, остаток экстрагировали AcOEt, органическую фазу промывали водой, буферным раствором с рН = 2, водой, сушили надNa2SO4 и растворитель выпаривали под вакуумом. Остаток разводили в 30 мл МеОН, добавляли 3 мл 30% раствора NaOH, и смесь оставляли перемешиваться в течение 30 мин при к.т. Смесь концентрировали под вакуумом, остаток экстрагировали диэтиловым эфиром, органическую фазу промывали водой,сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток хроматографировали на силикагеле Н, элюируя градиентом смеси ДХМ/МеОН от (100/0,1; об./об.) до (100/1; об./об). Получали 1,55 г ожидаемого продукта. Б) 2-[4-(2,3-Дихлорбензоил)-2-(3,4-дихлорфенил)-морфолин-2-ил]ацетальдегид, единственный изомер Раствор 1,5 г соединения, полученного на предшествующей стадии, и 1,5 г ДМСО в 20 мл ДХМ охлаждали до -60 С, 1,25 г оксалилхлорида добавляли по каплям и смесь оставляли перемешиваться в течение 1 ч при -60 С. Затем добавляли 2 г триэтиламина и смесь продолжали перемешивать, давая температуре вернуться до к.т. Реакционную смесь экстрагировали ДХМ, органическую фазу промывали 1 н. раствором HCl, водой, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Получали 1,4 г ожидаемого продукта. Подготовительное получение 2.3 2-[2-(3,4-Дихлорфенил)-4-[2-(2,6-дихлорфенил)ацетил]морфолин-2-ил]ацетальдегид, единственный изомер А) 2-[2-(3,4-Дихлорфенил)-4-[2-(2,6-дихлорфенил)ацетил]морфолин-2-ил]этилбензоат, единственный изомер Раствор 4 г соединения, полученного на стадии (А) подготовительного получения 2.1, в 43 мл ДХМ охлаждали до 0 С, добавляли 2,16 г 2-(2,6-дихлорфенил)уксусной кислоты, затем раствор 3 мл триэтиламина в 50 мл ДХМ и 4,7 г БОФ, после чего смесь оставляли перемешиваться, давая температуре вернуться до к.т. Смесь концентрировали под вакуумом, остаток экстрагировали AcOEt, органическую фазу промывали 2 н. раствором HCl, водой, 10% раствором Na2CO3, водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Получали 6 г ожидаемого продукта. Б) 2-(2,6-Дихлорфенил)-1-[2-(3,4-дихлорфенил)-2-(2 гидроксиэтил)морфолин-4-ил]-1-этанон, единственный изомер Смесь 6 г соединения, полученного на предыдущей стадии, в 100 мл МеОН кипятили с обратным холодильником, добавляли 3,5 мл 30% раствора NaOH и смесь продолжяли кипятить с обратным холодильником в течение 1 ч при перемешивании. Смесь концентрировали под вакуумом, остаток разводили в воде, экстрагировали AcOEt, органическую фазу дважды промывали водой, насыщенным растворомNaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток хроматографировали на силикагеле Н, элюируя ДХМ, а затем градиентом смеси ДХМ/МеОН от (100/1; об./об.) до (100/3; об./об.). Получали 2,42 г ожидаемого продукта. В) 2-[2-(3,4-Дихлорфенил)-4-[2-(2,6-дихлорфенил)ацетил]морфолин-2-ил]ацетальдегид, единственный изомер Смесь 0,6 мл оксалилхлорида в 11 мл ДХМ охлаждали до -60 С, добавляли раствор 1,2 мл ДМСО в 5 мл ДХМ, затем добавляли по каплям раствор 2,42 г соединения, полученного на предыдущей стадии, и 1,6 мл ДМСО в 11 мл ДХМ и смесь оставляли перемешиваться в течение 30 минт при -50 С. Затем добавляли 4,6 мл триэтиламина и смесь оставляли перемешиваться, позволяя температуре вернуться до к.т. Реакционную смесь экстрагировали ДХМ, органическую фазу промывали 2 н. раствором HCl, водой, 10% раствором Na2CO3, водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Получали 2,24 г ожидаемого продукта. Подготовительное получение 2.4 2-[2-(3,4-Дихлорфенил)-4-[2-(2,3-дихлорфенил)ацетил]морфолин-2-ил]ацетальдегид, единственный изомер- 13006236 А) 2-[2-(3,4-Дихлорфенил)-4-[2-(2,3-дихлорфенил)ацетил]морфолин-2-ил]этилбензоат, единственный изомер Это соединение получали в соответствии с методикой, описанной на стадии А подготовительного получения 2.3, из 4,9 г соединения, полученного на стадии А подготовительного получения 2.1, в 52 мл ДХМ, 2,67 г соединения, полученного в подготовительном получении 1.1, раствора 3,62 мл триэтиламина в 36 мл ДХМ и 5,76 г БОФ. Получали 7,11 г ожидаемого продукта. Б) 2-[2,3-Дихлорфенил)-1-[2-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолин-4-ил]-1-этанон, единственный изомер 5 мл 30% раствора NaOH добавляли к раствору 7,11 г соединения, полученного на предыдущей стадии, в 100 мл МеОН и смесь оставляли перемешиваться в течение 1 ч при к.т. Смесь концентрировали под вакуумом, остаток экстрагировали AcOEt, органическую фазу дважды промывали водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток хроматографировали на силикагеле Н, элюируя ДХМ, а затем смесью ДХМ/МеОН (100/1; об./об.). Получали 2,21 г ожидаемого продукта. В) 2-[2-(3,4-Дихлорфенил)-4-[2-(2,3-дихлорфенил)ацетил]морфолин-2-ил]ацетальдегид, единственный изомер Это соединение получали в соответствии с методикой, описанной на стадии В подготовительного получения 2.3, из 0,5 мл оксалилхлорида в 10 мл ДХМ, раствора 1,02 мл ДМСО в 5 мл ДХМ, раствора 2,21 г соединения, полученного на предыдущей стадии, и 1,43 мл ДМСО в 10 мл ДХМ и 4,2 мл триэтиламина. Получали 2,1 г ожидаемого продукта. 3. Подготовительное получение соединений формулы (III) Подготовительное получение 3.1N,N-Диметил-4-(пиперидин-1-ил)пиперидин-4-карбоксамид (III): R1 = -СН 3 А) 1-Бензил-4-циано-4-(пиперидин-1-ил)пиперидин Раствор 5,3 г цианида натрия в 20 мл воды добавляли по каплям и при к.т. к раствору 18,6 г 1 бензилпиперидин-4-она и 12,16 г гидрохлорида пиперидина в 25 мл МеОН и 25 мл воды и смесь оставляли перемешиваться в течение 48 ч при к.т. Образовавшийся осадок отделяли от жидкости, промывали водой и сушили под вакуумом. Получали 27 г ожидаемого продукта. Б) 1 -Бензил-4-(пиперидин-1-ил)пиперидин-4-карбоксамид 28,3 г соединения, полученного на предыдущей стадии, добавляли к 80 мл 95% серной кислоты и смесь нагревали при 100 С в течение 10 мин. После охлаждения до к.т. реакционную смесь выливали на лед, доводили рН до 7 путем добавления 25% раствора NH4OH, экстрагировали ДХМ, органическую фазу промывали водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток разводили в ацетоне,оставляли перемешиваться в течение 2 ч при к.т. и образовавшийся осадок отделяли от жидкости. Получали 20,8 г ожидаемого продукта. В) N,N-Диметил-1-бензил-4-(пиперидин-1-ил)-пиперидин-4-карбоксамид и N-метил-1-бензил-4(пиперидин-1-ил)пиперидин-4-карбоксамид Раствор 9,87 г соединения, полученного на предыдущей стадии, в 120 мл ТГФ добавляли по каплям при к.т. к суспензии 3,6 г гидрида натрия при 60% в масле в 120 мл ТГФ и смесь грели при 60 С в течение 2 ч. После охлаждения до к.т. раствор 8,52 г метилиодида в 60 мл ДМФ добавляли по каплям и смесь оставляли перемешиваться в течение 4 ч при к.т. Реакционную смесь выливали на лед, экстрагировали диэтиловым эфиром, органическую фазу промывали водой, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток хроматографировали на силикагеле Н, элюируя смесью ДXM/MeOH/NH4OH(100/1/0,1; об./об./об.) и выделяли следующие компоненты: наименее полярное соединение: получали 6 г N,N-диметил-1-бензил-4-(пиперидин-1-ил)пиперидин 4-карбоксамида; наиболее полярное соединение: получали 2,6 г N-метил-1-бензил-4-(пиперидин-1-ил)пиперидин-4 карбоксамида. Г) N,N-Диметил-4-(пиперидин-1-ил)пиперидин-4-карбоксамид Смесь 5,9 г наименее полярного соединения, полученного на предыдущей стадии, 3,4 г формиата аммония и 1,5 г 10% палладия на угле в 60 мл МеОН оставляли перемешиваться в течение 3 ч при к.т. Катализатор отфильтровывали через Celite и фильтрат концентрировали под вакуумом. После высушивания под вакуумом при 60 С получали 1,9 г ожидаемого продукта. Подготовительное получение 3.2(III), HCOOH: R1 = Н Смесь 4 г наиболее полярного соединения, полученного на стадии В подготовительного получения 3.1, 2,43 г формиата аммония и 1 г 10% палладия на угле в 50 мл МеОН оставляли перемешиваться в течение 30 мин при к.т. Катализатор отфильтровывали через Celite и фильтрат концентрировали под вакуумом. После высушивания под вакуумом получали 2,6 г ожидаемого продукта. 0,6 г соединения, полученного в подготовительном получении 3.1, добавляли к раствору 0,8 г соединения, полученного в подготовительном получении 2.1, в 15 мл ДХМ, затем добавляли 0,9 г триацетоксиборогидрида натрия и 8 капель уксусной кислоты и смесь оставляли перемешиваться в течение ночи при к.т. Реакционную смесь подщелачивали добавлением 10% раствора Nа 2 СО 3, экстрагировали ДХМ, органическую фазу промывали трижды водой, насыщенным раствором NaCl, сушили над Na2SO4 и растворитель выпаривали под вакуумом. Остаток хроматографировали на силикагеле Н, элюируя градиентом смеси ДХМ/МеОН от (100/0,5; об./об.) до (100/2; об./об.). Полученный продукт разводили в хлористоводородном эфире и растворитель выпаривали под вакуумом. После кристаллизации из смеси пентан/изоэфир получали 0,45 г ожидаемого продукта. 20D = + 14,4 (с = 0,25; МеОН) 1 Н-ЯМР: ДМСО-d6 + TFA (трифторуксусная кислота), 350 К: 5 (м.д.): от 1,3 до 1,8: m, 6H; от 2,0 до 3,3: m: 20H; от 3,.3 до 4,2: m: 8H; от 7,2 до 7,7: m: 8H. Пример 2. Соединение получали в соответствии с процедурой, описанной в примере 1, из 0,58 г соединения,полученного в подготовительном получении 2.1, 15 мл ДХМ, 0,345 г соединения, полученного в подготовительном получении 3.2, 0,65 г триацетоксиборогидрида натрия и 8 капель уксусной кислоты. После кристаллизации из смеси пентан/изоэфир получали 0,6 г ожидаемого продукта. 20D = + 13,6 (с = 0,25; МеОН). Пример 3. Соединение получали в соответствии с процедурой, описанной в примере 1, из 0,75 г соединения,полученного в подготовительном получении 2.2, 20 мл ДХМ, 0,43 г соединения, полученного в подготовительном получении 3.1, 0,7 г триацетоксиборогидрида натрия и 8 капель уксусной кислоты. После кристаллизации из смеси ДХМ/эфир получали 0,8 г ожидаемого продукта. 20D = - 5,4 (с = 0,5; МеОН). Пример 4. Это соединение получали в соответствии с процедурой, описанной в примере 1, из 0,45 г соединения, полученного в подготовительном получении 2.3, 50 мл ДХМ, 0,28 г соединения, полученного в подготовительном получении 3.1, 0,424 г триацетоксиборогидрида натрия и 3 капель уксусной кислоты. После кристаллизации из диэтилового эфира получали 0,419 г ожидаемого продукта. 20D= + 7,6 (с = 0,25; МеОН). Пример 5.N,N-Диметил-1-[2-[4-[2-(2,3-дихлорфенил)-ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4(пиперидин-1-ил)пиперидин-4-карбоксамида дигидрохлорид, правовращающий изомер, дигидрат Это соединение получали в соответствии с процедурой, описанной в примере 1, из 0,5 г соединения,полученного в подготовительном получении 2.4, 7 мл ДХМ, 0,312 г соединения, полученного в подготовительном получении 3.1, 0,47 г триацетоксиборогидрида натрия и 3 капель уксусной кислоты. После кристаллизации из диэтилового эфира получали 0,446 г ожидаемого продукта. 20D= + 8,8 (с = 0,25; МеОН). Это соединение получали в соответствии с процедурой, описанной в примере 1, из 0,6 г соединения,полученного в подготовительном получении 2.4, 60 мл ДХМ, 0,3 г соединения, полученного в подготовительном получении 3.2, 0,56 г триацетоксиборогидрида натрия и 3 капель уксусной кислоты. После кристаллизации из диэтилового эфира получали 0,556 г ожидаемого продукта. 20D = + 8 (с = 0,25; МеОН). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы где R1 представляет собой атом водорода или метильный радикал; В представляет собой прямую связь или группу -СН 2-;Z представляет собой фенил, 2,3-дихлорфенил или 2,6-дихлорфенил; и его соли с неорганическими или органическими кислотами, его сольваты и/или его гидраты. 2. Соединение формулы (I) по п.1 в форме оптически чистых изомеров. 3. Соединение по п.1 или 2, выбранное изN-метил-1-[2-[4-[2-(2,3-дихлорфенил)ацетил]-2-(3,4-дихлорфенил)морфолин-2-ил]этил]-4(пиперидин-1-ил)пиперидин-4-карбоксамида, правовращающего изомера; и их солей с неорганическими или органическими кислотами, их сольватов и/или их гидратов. 4. Соединение по любому из пп.1-3, которое представляет собой N,N-диметил-1-[2-[4-бензоил-2(3,4-дихлорфенил)морфолин-2-ил]этил]-4-(пиперидин-1-ил)пиперидин-4-карбоксамид, правовращающий изомер и его соли с неорганическими или органическими кислотами, его сольваты и/или его гидраты. 5. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы где В и Z такие, как определено для соединения формулы (I) по п.1, подвергают взаимодействию с соединением формулы где R1 такой, как определено для соединения формулы (I) по п.1, в присутствии кислоты, в растворителе,а затем образовавшуюся промежуточную иминиевую соль восстанавливают с помощью восстановителя. 6. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы где В и Z такие, как определено для соединения формулы (I) в п.1, a Y представляет собой метил, фенил,толил или трифторметильную группу, подвергают взаимодействию с соединением формулы где R1 такой, как определено для соединения формулы (I) в п.1. 7. Способ получения соединений формулы (I) по п.1, их солей, их сольватов и/или их гидратов, отличающийся тем, что соединение формулы где R1 такой, как определено для соединения формулы (I) по п.1, подвергают взаимодействию с функциональным производным кислоты формулы(VI),где В и Z такие, как определено для соединения формулы (I) по п.1. 8. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пп.1-4 или одну из его фармацевтически приемлемых солей, сольватов и/или гидратов. 9. Фармацевтическая композиция по п.8, содержащая от 0,1 до 1000 мг активного ингредиента в стандартной лекарственной форме, в которой активный ингредиент смешан по меньшей мере с одним фармацевтическим эксципиентом. 10. Применение соединения по любому из пп.1-4 или одной из его фармацевтически приемлемых солей, сольватов и/или гидратов для приготовления лекарственных средств, предназначенных для лечения любой патологии, в которую вовлечены либо нейрокинин А и/или NK2 рецепторы, либо нейрокинин В и/или NK3 рецепторы, либо оба нейрокинин А и нейрокинин В и/или NK2 и NK3 рецепторы. 11. Применение по п.10 для приготовления лекарственных средств, предназначенных для лечения патологий дыхательной, желудочно-кишечной, мочевой, иммунной, сердечно-сосудистой системы и центральной нервной системы, а также боли, мигрени, воспаления, тошноты и рвоты и кожных заболеваний. 12. Применение по п.11 для приготовления лекарственных средств, предназначенных для лечения хронического обструктивного бронхита, астмы, недержания мочи, синдрома раздраженного кишечника,болезни Крона, язвенного колита, депрессии, тревоги, эпилепсии, шизофрении. 13. Лекарственное средство, отличающееся тем, что оно содержит соединение по любому из пп.1-4 или одну из его фармацевтически приемлемых солей, сольватов и/или гидратов. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: C07D 413/06, A61K 31/5377, A61P 11/00
Метки: содержащие, производные, получения, пиперидинкарбоксамидные, фармацевтические, композиции, способ, новые
Код ссылки
<a href="https://eas.patents.su/18-6236-novye-piperidinkarboksamidnye-proizvodnye-sposob-ih-polucheniya-i-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Новые пиперидинкарбоксамидные производные, способ их получения и содержащие их фармацевтические композиции</a>
Новые бензолсульфонамидные производные, способ их получения и содержащие их фармацевтические композиции

Номер патента: 3426
Опубликовано: 24.04.2003
Авторы: Лавей Жильбер, Симетьер Бернар, Дескомб Жан-Жак, Вербёрен Тони, Симоне Серж
МПК: A61K 31/18, C07C 311/20, A61P 7/02...
Метки: содержащие, получения, способ, бензолсульфонамидные, фармацевтические, производные, композиции, новые
Формула / Реферат:
1. Бензолсульфонамидные производные общей формулы (I) где n обозначает целое число от 1 до 3 включительно, m обозначает целое число от 0 до 6 включительно, Ra обозначает гидрокси-, линейную или разветвленную C1-C6алкокси-, арилокси- или арилалкилоксигруппу, R1 и R2 независимо друг от друга обозначают атом водорода, атом галогена, алкильную группу, линейную или разветвленную C1-C6алкоксигруппу, гидроксигруппу или линейную или разветвленную...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Бушар Эрве, Гризони Серж, Иттэнжер Огюстэн, Ашар Даниель, Майерс Майкл, Букерель Жан, Филош Брюно
МПК: A61P 25/00, A61K 31/397, C07D 205/04...
Метки: получения, содержащие, новые, производные, композиции, способ, 3-аминоазетидина, фармацевтические
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Новые пиперидин-4-сульфонамидные производные, способ их получения и содержащие их фармацевтические композиции

Номер патента: 3096
Опубликовано: 26.12.2002
Авторы: Толлон Катрин, Вийену Николь, Буриньён Мари-Пьер, Дессинге Эме, Пуатевин Кристоф, Пелье Жан-Луи, Вилен Жан-Поль
МПК: A61P 9/10, A61K 31/4525, C07D 405/06...
Метки: пиперидин-4-сульфонамидные, получения, содержащие, новые, производные, способ, фармацевтические, композиции
Формула / Реферат:
1. Соединения формулы (I) где R1 обозначает атом водорода или линейную или разветвленную С1-С6алкильную группу, R2а и R2b могут иметь одинаковые или различные значения и каждый независимо друг от друга обозначает группу, выбранную из ряда, включающего атом водорода, атом галогена, линейную или разветвленную С1-С6алкильную группу, гидроксигруппу, линейную или разветвленную С1-С6алкоксигруппу, линейную или разветвленную С1-С6тригалоалкильную...
Новые производные полигидроксипиразинов, способ их получения и содержащие их фармацевтические композиции

Номер патента: 3550
Опубликовано: 26.06.2003
Авторы: Бушар Эрве, Коммерсон Алан
МПК: A61K 31/495, A61P 3/10, C07D 241/12...
Метки: получения, производные, композиции, фармацевтические, полигидроксипиразинов, способ, содержащие, новые
Формула / Реферат:
1. Соединения общей формулы в которой R1 обозначает стереоизомерные формы цепи -(CHOH)3-CH2-O-COR (II) и либо R2 обозначает атом водорода, а R3 обозначает стереоизомерные формы цепи -CH2-(CHOH)2-CH2-O-COR (III), либо R2 обозначает стереоиэомерные формы цепей -(CHOH)3-CH2-O-COR (II) или -CH2-(CHOH)2-CH2-COR (III), a R3 обозначает атом водорода, и R обозначает радикал -(Алк)i-(Циклоалк), в котором Алк обозначает алкильный...
Новые производные бензола, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 4048
Опубликовано: 25.12.2003
Авторы: Верньер Жан-Клод, Бурри Мартин, Понселе Мартин, Буажегрен Роберт, Поль Раймон, Лер Пьер
МПК: C07D 295/06, A61K 31/55, A61P 25/00...
Метки: бензола, получения, производные, содержащие, композиции, новые, фармацевтические, способ
Формула / Реферат:
1. Соединения формулы где A представляет собой группу, выбранную из следующего: -Cу C-; -CH=CH-; -CH2-CH2-; n равно 1 или 2; X представляет собой атом водорода, хлора или фтора или метильную или метоксигруппу; Y представляет собой атом водорода или атом хлора или фтора; R1 представляет собой циклогексильную группу, монозамещенную, дизамещенную, тризамещенную или тетразамещенную метильной группой; фенильную группу, монозамещенную или...
Предыдущий патент: Средство, отпугивающее насекомых
Следующий патент: Производные аминоалкилбензоилбензофурана, способ их получения и содержащие их композиции
Случайный патент: Способ и устройство для определения характеристик компонентов канала связи под нагрузкой