Получение производных соединения 1,3-дифенилпроп-2-ен-1-он






Формула / Реферат
1. Способ получения производных соединения 1,3-дифенилпроп-2-ен-1-он, замещённых группой карбоксиалкилокси или карбоксиалкилтио, включающий следующие стадии:
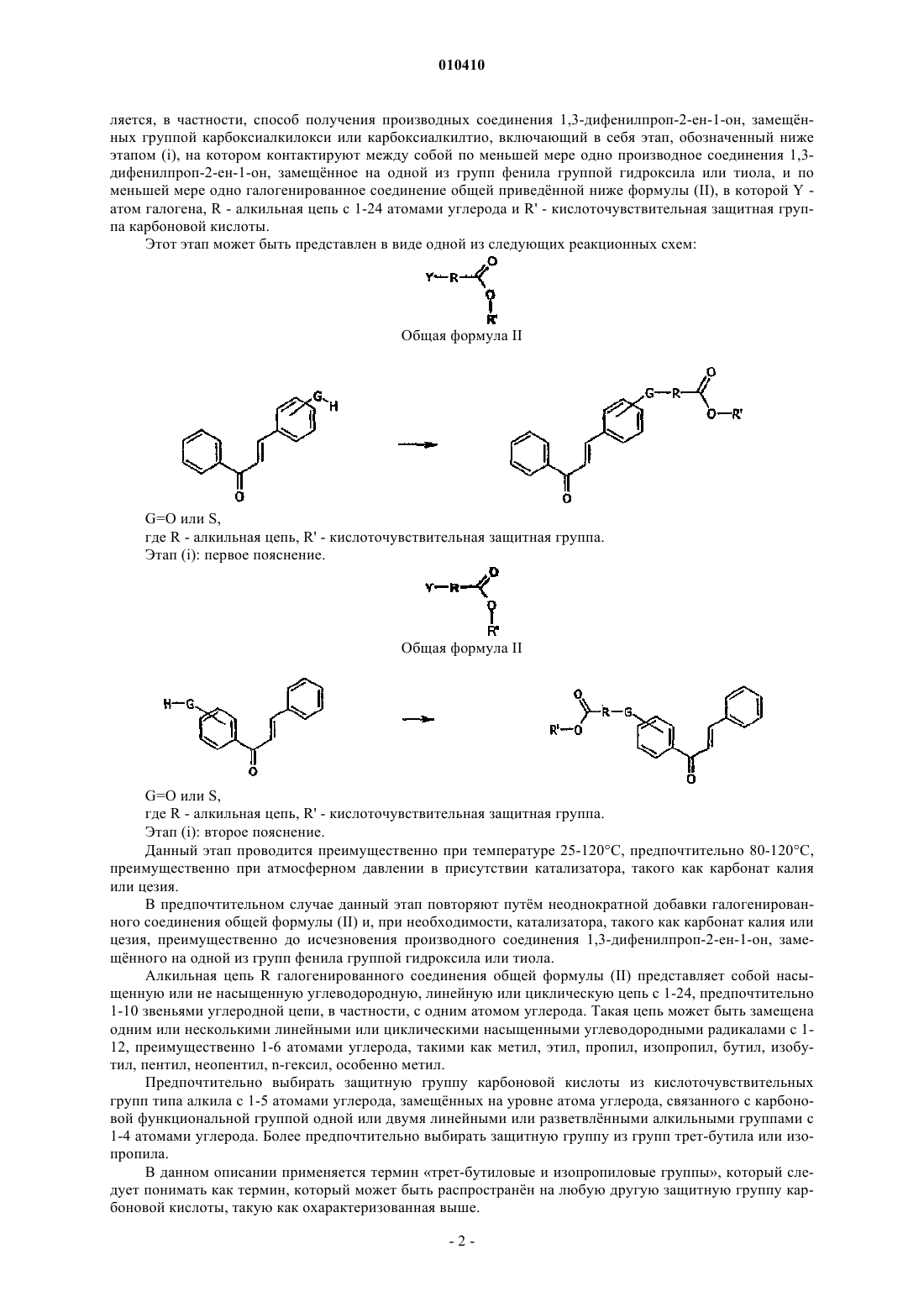
(i) контактирование по меньшей мере одного производного соединения 1,3-дифенилпроп-2-ен-1-он, замещённого на одной из двух групп фенила группой гидроксила или тиола и по меньшей мере одним галогенированным соединением общей формулы (II)

где Y - атом галогена, R - алкильная цепь с 1-24 атомами углерода и R' - кислоточувствительная защитная группа карбоновой кислоты, выбранная из трет-бутиловой и изобутиловой групп;
(ii) кислотный гидролиз (ацидолиз) сложного эфира, полученного на этапе (i) с трифторуксусной кислотой.
2. Способ по п.1, отличающийся тем, что группа R представляет собой алкильную цепь с 1-10 атомами углерода, замещённую при необходимости одним или несколькими линейными или циклическими насыщенными углеводородными радикалами с 1-12 атомами углерода.
3. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится при температуре 25-120шС, предпочтительно 80-120шС.
4. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится в присутствии катализатора.
5. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится с применением карбоната калия или цезия в качестве катализатора.
6. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится при неоднократной добавке галогенированного соединения общей формулы (II) и при необходимости катализатора.
7. Способ по любому из предшествующих пунктов, отличающийся тем, что производное соединения 1,3-дифенилпроп-2-ен-1-он, замещённое группой гидроксила или тиола, используемое на этапе (i), получают реакцией Кляйзена-Шмидта в кислой или щелочной среде, проводимой между соединением типа ацетофенона и производным тио- или гидроксибензальдегида или между производным тио- или гидроксиацетофенона и производным типа бензальдегида.
8. Способ по любому из предшествующих пунктов, отличающийся тем, что количество трифторуксусной кислоты, используемой на этапе (ii), составляет 1-20 эквивалентов, предпочтительно 8-12 эквивалентов.
9. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (ii) проводят при температуре 0-100шС, предпочтительно 18-25шС.
10. Способ по любому из предшествующих пунктов, отличающийся тем, что полученный продукт соответствует общей формуле

где X1 - галоген, или группа -R1, или группа, представленная формулой -G1-R1;
Х2 - атом водорода, или тионитрозо, или алкилокси, или алкилкарбонилокси, или алкилтио, или алкилкарбонилтио;
Х3 - группа -R3 или -G3-R3;
Х4 - галоген, или тионитрозо, или -R4, или -G4-R4;
Х5 - группа -R5 или -G5-R5;
R1, R3, R4, R5, одинаковые или разные, означают атом водорода или алкильную группу, насыщенную или не насыщенную функциональной группой карбоновой кислоты;
G1, G3, G4, G5, одинаковые или разные, означают атом кислорода или серы;
при этом группы X1, Х3, Х4 и Х5 соответствуют формуле -G-R, в которой R означает алкильный радикал, содержащий функциональную группу карбоновой кислоты.
Текст
010410 Область техники, к которой относится изобретение Настоящее изобретение относится к новому способу получения производных соединения 1,3 дифенилпроп-2-ен-1-он, замещнных на одной из двух фенильных групп группой карбоксиалкилокси или группой карбоксиалкилтио. Уровень техники Соединения 1,3-дифенилпропен-1-он получают, как правило, реакцией конденсации альдегида с кетоном в соответствии с реакцией Кляйзена-Шмидта (March J., 1992 "Advanced Organic Chemistry", FourthEdition, 940, Wiley Interscience). Традиционно соединения 1,3-дифенилпроп-2-ен-1-он, замещнные группой карбоксиалкилокси,получают указанным методом из исходных материалов (альдегида и кетона), выбираемых таким образом, чтобы они замещались группой карбоксиалкилокси или соответствующим сложным эфиром. Такая последовательность этапов может быть упрощнно представлена в виде одной из следующих реакционных схем: где R - алкил, R' - водород, алкил. Однако из-за кислотного характера полученного соединения и частого присутствия в реакционной среде вторичных продуктов и не прореагировавших исходных материалов усложняется очистка перекристаллизацией или хроматографией на силикагеле, а также значительно снижается выход. Следовательно, использование такого механизма синтеза для получения соединений, указанных в примерах 1 и 3 настоящего изобретения, не позволяет в целом достигать выхода свыше 10%. Сущность изобретения Авторы изобретения разработали легко осуществляемый способ, позволяющий получать соединения 1,3-дифенил-2-ен-1-он, замещнные группой карбоксиалкилокси или группой карбоксиалкилтио, с высоким выходом. Данный способ отличается от описанного выше способа синтеза тем, что функциональная группа карбоксиалкилокси или карбоксиалкилтио вводится в виде трет-бутилового или изопропилового сложного эфира в результате реакции с производным соединения 1,3-дифенил-2-ен-1-он, замещнным группой гидроксила или тиола. Последнюю получают, как правило, реакцией КляйзенаШмидта. В одном из воплощений способа согласно изобретению в нем применяется кислоточувствительная защитная группа карбоновой кислоты, например, трет-бутилового или изопропилового типа. Авторами изобретения было установлено, что данная группа может вводиться и расщепляться в условиях, отвечающих химической структуре соединений дифенил-1,3-проп-2-ен-1-он. Авторы использовали эти преимущества для разработки нового способа синтеза, являющегося объектом настоящего изобретения. Полученные таким образом соединения 1,3-дифенилпроп-2-ен-1-он, замещнные группой карбоксиалкилокси или группой карбоксиалкилтио, представляют большой интерес для фармацевтической и косметической областей. Действительно, эти соединения обладают одновременно свойствами промоторов PPAR, антиоксидантов и противовоспалительных средств и в связи с этим они обладают большим терапевтическим или профилактическим потенциалом и могут применяться, в частности, при лечении или профилактике патологий сосудов мозга, сердечно-сосудистых заболеваний, синдрома X, рестеноза,диабета, ожирения, гипертонии, воспалительных заболеваний, раковых болезней или неоплазий (доброкачественных или злокачественных опухолей), нейродегенеративных заболеваний, дерматологических заболеваний и нарушений, относящихся к окислительному стрессу, при профилактике или лечении в целом старческих явлений и, в частности, старения кожи (появление морщин и пр.), в частности, в косметике. Целью настоящего изобретения является создание способа получения производных соединения 1,3 дифенилпроп-2-ен-1-он, замещнных на одной из двух групп фенила группой карбоксиалкилокси или карбоксиалкилтио, являющегося простым в применении и позволяющего достигать высокие выходы. Указанная и другие цели достигаются с помощью настоящего изобретения, объектом которого яв-1 010410 ляется, в частности, способ получения производных соединения 1,3-дифенилпроп-2-ен-1-он, замещнных группой карбоксиалкилокси или карбоксиалкилтио, включающий в себя этап, обозначенный ниже этапом (i), на котором контактируют между собой по меньшей мере одно производное соединения 1,3 дифенилпроп-2-ен-1-он, замещнное на одной из групп фенила группой гидроксила или тиола, и по меньшей мере одно галогенированное соединение общей приведнной ниже формулы (II), в которой Y атом галогена, R - алкильная цепь с 1-24 атомами углерода и R' - кислоточувствительная защитная группа карбоновой кислоты. Этот этап может быть представлен в виде одной из следующих реакционных схем:G=O или S,где R - алкильная цепь, R' - кислоточувствительная защитная группа. Этап (i): второе пояснение. Данный этап проводится преимущественно при температуре 25-120 С, предпочтительно 80-120 С,преимущественно при атмосферном давлении в присутствии катализатора, такого как карбонат калия или цезия. В предпочтительном случае данный этап повторяют путм неоднократной добавки галогенированного соединения общей формулы (II) и, при необходимости, катализатора, такого как карбонат калия или цезия, преимущественно до исчезновения производного соединения 1,3-дифенилпроп-2-ен-1-он, замещнного на одной из групп фенила группой гидроксила или тиола. Алкильная цепь R галогенированного соединения общей формулы (II) представляет собой насыщенную или не насыщенную углеводородную, линейную или циклическую цепь с 1-24, предпочтительно 1-10 звеньями углеродной цепи, в частности, с одним атомом углерода. Такая цепь может быть замещена одним или несколькими линейными или циклическими насыщенными углеводородными радикалами с 112, преимущественно 1-6 атомами углерода, такими как метил, этил, пропил, изопропил, бутил, изобутил, пентил, неопентил, n-гексил, особенно метил. Предпочтительно выбирать защитную группу карбоновой кислоты из кислоточувствительных групп типа алкила с 1-5 атомами углерода, замещнных на уровне атома углерода, связанного с карбоновой функциональной группой одной или двумя линейными или разветвлнными алкильными группами с 1-4 атомами углерода. Более предпочтительно выбирать защитную группу из групп трет-бутила или изопропила. В данном описании применяется термин трет-бутиловые и изопропиловые группы, который следует понимать как термин, который может быть распространн на любую другую защитную группу карбоновой кислоты, такую как охарактеризованная выше.-2 010410 Оптимально, чтобы производное соединения 1,3-дифенилпроп-2-ен-1-он, замещнное группой гидроксила или тиола и используемое в рамках настоящего изобретения на описанном выше этапе (i), получали в кислой или щлочной среде реакцией Кляйзена-Шмидта между соединением типа ацетофенона с производным тио- или гидроксибензальдегида или между производным тио- или гидроксиацетофенона с производным типа бензальдегида. Способ согласно изобретению включает в себя проводимый после описанного этапа (i) также этап(ii) для получения производного карбоксиалкилокси или карбоксиалкилтио соединения 1,3 дифенилпроп-2-ен-1-он реакцией кислотного гидролиза (ацидолиза) сложного эфира, полученного на этапе (i). Этот этап (ii) может быть представлен в виде одной из следующих реакционных схем:G=O или S,где R - алкильная цепь, R' - защитная группа. Этап (ii): второе пояснение. Предпочтительно, чтобы этот этап ацидолиза проводился контактированием производного соединения 1,3-дифенилпроп-2-ен-1-он, которое замещено группой алкилоксикарбонилалкилокси или алкилоксикарбонилалкилтио, с трифторуксусной кислотой. Как правило, количество трифторуксусной кислоты составляет от 1 до 20 эквивалентов, в частности от 8 до 12. Целесообразно этот этап проводить при температуре 0-100 С, предпочтительно 18-25 С, преимущественно при атмосферном давлении. Далее более подробно описывается получение соединений 1,3-дифенилпроп-2-ен-1-он, замещнных группой карбоксиалкилокси или группой карбоксиалкитио. Особенность этого способа заключается в комбинации двух этапов проведения синтеза: синтеза трет-бутиловых или изопропиловых сложных эфиров (или любой другой защитной группы) на основе соединений 1,3-дифенилпроп-2-ен-1-он, замещнных на одной из групп фенила группой гидроксила или тиола, с последующей реакцией ацидолиза промежуточных сложных эфиров. Группа трет-бутилоксикарбонилалкила или изопропилоксикарбонилалкила вводится просто с достижением высокого выхода реакцией алкилирования между химическим предшественником (гидрокси 1,3-дифенилпроп-2-ен-1-он или его серосодержащий аналог) и галогенированным производным. Полученный сложный трет-бутиловый или изопропиловый эфирный интермедиат может быть легко очищен,в частности, хроматографией на силикагеле или перекристаллизацией. Сложный трет-бутиловый или изопропиловый эфир расщепляется до соответствующей кислоты под действием трифторуксусной кислоты. Этот метод, адаптированный для расщепления группы третбутила или изопропила, позволяет производить полное преобразование сложного эфира в соответствующую кислоту. Авторы изобретения установили, что данный метод совместим с (позволяет получить) химической структурой соединений 1,3-дифенилпроп-2-ен-1-он. Соответственно он не образует продуктов разложения и позволяет получать кислоты с более высоким выходом, чем традиционно достигаемый. Поскольку три этапа могут осуществляться в рамках настоящего изобретения, то их можно представить следующим образом. Первый этап: синтез химического предшественника. Химический предшественник типа гидрокси-1,3-дифенилпроп-2-ен-1-он или его серосодержащий аналог может быть получен обычной реакцией Кляйзена-Шмидта в кислой или щлочной средеG=O или S Соединения типа ацетофенон, гидроксиацетофенон (или его серосодержащий аналог), бензальдегид и гидроксибензальдегид (или его серосодержащий аналог), используемые в данной реакции, могут быть при необходимости замещены на группах фенила. Такие заместители выбираются, в частности, из атома галогена, алкильного радикала, группы тионитрозо и радикала алкилокси или алкилтио. Условия проведения такой реакции в кислой или щлочной среде известны среднему специалисту в данной области и могут изменяться в широком диапазоне. Контактирование этих двух соединений предпочтительно проводить стехиометрически. Оно проводится преимущественно при комнатной температуре (от около 18 до 25 С) и атмосферном давлении. В щлочной среде реакцию проводят преимущественно в присутствии сильного основания, такого как гидроксид щлочного металла, например гидроксид натрия, или алкоголят щлочного металла, такой как этилат натрия. В кислой среде реакцию предпочтительно проводить в присутствии сильной кислоты, такой как соляная. Синтез в щлочной среде целесообразно осуществлять следующим образом. Одномолярный эквивалент кетона и одномолярный эквивалент альдегида растворяют в водноспиртовом растворе гидроксида натрия в количестве 20 молярных эквивалентов. Смесь перемешивают в течение 6-48 ч, преимущественно 16-20 ч, при температуре 0-100 С, преимущественно 18-25 С. Затем среду подкисляют (для достижения pH, равного, в частности, около 2), в частности, соляной кислотой. Требуемое соединение гидрокси-1,3-дифенилпроп-2-ен-1-он (или его серосодержащий аналог) можно получить осаждением или экстракцией в системе тврдое тело/жидкость после выпаривания реакционной среды. Оно может быть затем очищено хроматографией на силикагеле или перекристаллизацией. Синтез в кислой среде оптимально проводить следующим образом. Одномолярный эквивалент кетона и одномолярный эквивалент альдегида растворяют в растворе этанола, насыщенном газообразной соляной кислотой. Смесь выдерживают при перемешивании при температуре 0-100 С, преимущественно 18-25 С, в течение 1-24 ч, преимущественно 1-6 ч, предпочтительно при атмосферном давлении. Растворитель удаляют, в частности, выпариванием при пониженном давлении, 1,3-дифенилпроп-2-ен-1-он очищают, в частности, хроматографией на силикагеле. Второй этап: получение трет-бутилового или изопропилового сложного эфира.-4 010410 где G - атом кислорода или серы, Y - атом галогена, R - алкильная цепь преимущественно с 1-6 атомами углерода, R' - трет-бутил или изопропил. Реакция может проводиться следующим образом. Гидрокси-1,3-дифенилпроп-2-эн-1-он (или серосодержащий аналог) в количестве 1 молярного эквивалента растворяют в растворителе, предпочтительно ацетонитриле или ацетоне. Добавляют 1-10 эквивалентов, предпочтительно 1-6 эквивалентов, карбоната калия или цезия, затем добавляют производное типа галогенированного трет-бутилового или изопропилового сложного эфира (в количестве 1-20 молярных эквивалентов, предпочтительно 4-8, особо предпочтительно 6). Смесь нагревают при (интенсивном) перемешивании при температуре 25-120 С, предпочтительно 100 С, в течение 1-24 ч, предпочтительно 10-14 ч, особо предпочтительно 10 ч, как правило, при атмосферном давлении. Растворитель удаляют, в частности, выпариванием при пониженном давлении. При необходимости реакционную среду снова приводят в реакцию с 3-6 молярными эквивалентами галогенированного производного и 3-5 молярными эквивалентами карбоната калия или цезия, причм эта операция может повторяться до полного использования сырьевого материала. Соединение 1,3-дифенилпроп-2-ен-1-он, замещнное группой третбутилоксикарбонилалкилокси, трет-бутилоксикарбонилалкилтио, изопропилоксикарбонилалкилокси или изопропилоксикарбонилалкилтио, очищают, в частности, хроматографией на силикагеле. Третий этап: получение кислоты из трет-бутилового или изопропилового сложного эфира.G и R имеют приведнные выше значения.G и R имеют приведнные выше значения.R' означает трет-бутил или изопропил. Соединение 1,3-дифенилпроп-2-ен-1-он, замещнное трет-бутилоксикарбонилалкилокси, третбутилоксикарбонилалкилтио, изопропилоксикарбонилалкилокси или изопропилоксикарбонилалкилтио, в количестве 1 молярного эквивалента растворяют в растворителе, таком как дихлорметан. Добавляют 120, предпочтительно 8-12, особо предпочтительно 10 эквивалентов кислоты, предпочтительно трифторуксусной кислоты. Смесь выдерживают при интенсивном перемешивании при температуре 0-100 С,предпочтительно 18-25 С, в течение 1-24 ч, предпочтительно 8-14 ч, особо предпочтительно 12 ч, преимущественно при атмосферном давлении. Растворитель удаляют, в частности, выпариванием при пониженном давлении. Соединение 1,3-дифенилпроп-2-ен-1-он, замещнное группой карбоксиалкилокси или карбоксиалкилтио, очищают, в частности, хроматографией на силикагеле. Реакцию проводят преимущественно при атмосферном давлении. Данный способ может эффективно применяться для получения соединений, описанных в заявке на патент ФранцииFR 0208571, поданной заявителем 8 июля 2002 г. Эти соединения соответствуют общей формулеR1, R3, R4, R5, одинаковые или разные, означают атом водорода или алкильную группу, замещнную или не замещнную функциональной группой карбоновой кислоты;G1, G3, G4, G5, одинаковые или разные, означают атом кислорода или серы; при этом группы X1, Х 3, Х 4 и Х 5 соответствуют формуле -G-R, в которой R означает алкильный радикал, содержащий функциональную группу карбоновой кислоты. Данный способ синтеза позволяет эффективно получать соединения общей формулы (I), в которой ни одна из групп X1, Х 2, Х 3 и Х 4 не является группой гидрокси или тиола. Предпочтительно получать этим способом соединения общей формулы (I), в которых по меньшей мере одна группа R3, R4, R5 представляет собой алкильную группу, содержащую функциональную группу карбоновой кислоты. Предпочтительно R4 представляет собой алкильный радикал с функциональной группой карбоновой кислоты. Предпочтительно R4 представляет собой алкильный радикал с функциональной группой карбоновой кислоты, а Х 3 и Х 5 являются не замещнными алкилами. Более предпочтительно Х 4 представляет собой группу карбоксидиметилметилокси или карбоксидиметилметилтио. Предпочтительно G4 представляет собой атом кислорода, a R4 - алкильный радикал с функциональной группой карбоновой кислоты. Предпочтительно G4 представляет собой атом кислорода, R4 - алкильный радикал с функциональной группой карбоновой кислоты и Х 3, Х 5 - не замещнные алкилы. Более предпочтительно Х 4 представляет собой группу карбоксидиметилметилокси. Более предпочтительно Х 4 представляет собой группу карбоксидиметилметилтио. Производные общей формулы (I), такие как указанные выше, могут принимать конформацию цис или транс. Предпочтительно Х 3, Х 4 и Х 5 не являются атомами водорода. Согласно настоящему изобретению термин алкил означает насыщенный углеводородный, линейный, разветвлнный или циклический, галогенированный или не галогенированный радикал, содержащий, в частности, 1-24, предпочтительно 1-10 атомов углерода, такой как метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, неопентил, n-гексил. Особо предпочтительны группы с содержанием 1-2 или 2-7 атомов углерода. Наиболее предпочтительны группы метила и этила. Термин тионитрозо означает группу нитрозо, связанную с ароматическим кольцом атомом серы. Термин галоген означает атом хлора, или брома, или йода, или фтора. Термин алкилокси означает алкильную цепь, связанную с кольцом атомом кислорода. Алкильная цепь соответствует приведнному выше определению. Термин алкилтио означает алкильную цепь, связанную с ароматическим кольцом атомом серы(тиоэфирная связь). Алкильная цепь соответствует приведнному выше определению. Ниже приводятся соединения или их промежуточные продукты (сложные эфиры), с соответствующими формулами, получаемые преимущественно способом согласно изобретению 1-[4-хлорфенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-он Другие отличия и преимущества настоящего изобретения приводятся ниже при описании примеров,которые предназначены для пояснения и не являются ограничительными. Примеры 1) Описание общих методов проведения синтеза. Общий метод 1. Синтез соединений гидрокси-1,3-дифенилпроп-2-ен-1-он. Гидроксиацетофенон (или его серосодержащий аналог) в количестве 1 молярного эквивалента и альдегид в количестве 1 молярного эквивалента или гидроксибензальдегид (или его серосодержащий аналог) в количестве 1 молярного эквивалента и кетон в количестве 1 молярного эквивалента растворяют в растворе этанола, насыщенном газообразной соляной кислотой. Смесь перемешивали при комнатной температуре в течение 1-6 ч, затем удаляют растворитель выпариванием при пониженном давлении. Очищают гидрокси 1,3-дифенилпроп-2-ен-1-он хроматографией на силикагеле или перекристаллизацией. Общий метод 2. Алкилирование соединений гидрокси-1,3-дифенилпроп-2-ен-1-он. Гидрокси-1,3-дифенилпропен-1-он (или его серосодержащий аналог) в количестве 1 молярного эк-9 010410 вивалента растворяют в ацетонитриле, затем добавляют галогенированное производное в количестве 3-6 молярных эквивалентов и карбонат калия в количестве 3-5 молярных эквивалентов. Реакционную среду выдерживают в течение около 10 ч при активном перемешивании с использованием дефлегматора. Соли удаляют фильтрованием. При необходимости реакционная среда повторно вводится в реакцию с 3-6 молярными эквивалентами галогенированного производного и 3-5 молярными эквивалентами карбоната калия. Эта операция может повторяться до полного использования исходного материала. Растворитель и избыток реагента удаляют выпариванием при пониженном давлении, целевой продукт очищают хроматографией на силикагеле. Общий метод 3. Ацидолиз трет-бутиловых сложных эфиров соединений 1,3-дифенилпроп-2-ен-1-он с применением трифторуксусной кислоты. трет-Бутиловый сложный эфир соединения 1,3-дифенилпроп-2-ен-1-он в количестве 1 молярного эквивалента растворяют в дихлорметане, добавляют 10 молярных эквивалентов трифторуксусной кислоты, смесь выдерживают в течение 12 ч при перемешивании при комнатной температуре. Полученный продукт очищают хроматографией на силикагеле или перекристаллизацией. 2) Примеры. Пример 1. Синтез 1-[4-хлорфенил]-3-[3,5-диметил-4-карбоксидиметилметилоксифенил]проп-2-ен-1 она Способ позволяет получать это соединение при общем выходе 56%. Синтез химического предшественника. 1-[4-Хлорфенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 1). Это соединение синтезировали с использованием 4-хлорацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 95/5). Выход: 91%. Ядерный магнитный резонанс (ЯМР): 1 Н CDCl3ppm (частей на миллион): 2,30 (s, 6 Н), 7,32 (s, 2 Н),7,34 (d, J=15,25 Гц, 1 Н), 7,47 (d, J=8,86 Гц, 2 Н), 7,75 (d, J=15,26, 1H), 7,97 (d, J=8,86 Гц, 2 Н). Синтез трет-бутилового сложного эфира. 1-[4-Хлорфенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1 он (соединение 2). Это соединение синтезировали с использованием 1-[4-хлорфенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-она (соединение 1) и бромизобутирата трет-бутила в соответствии с описанным выше общим методом 2. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 90/10). Выход: 70%. 3). Это соединение синтезировали с использованием соединения 1-[4-хлорфенил]-3-[3,5-диметил-4 трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-она (соединение 2) в соответствии с описанным выше общим методом 3. Очистка производилась хроматографией на силикагеле (дихлорметан/метанол: 98/2). Выход: 88%. ЯМР 1H DMSOppm: 1,39 (s, 6H), 2,22 (s, 6H), 7,58 (s, 2H), 7,67-7,62 (m, 3 Н), 7,82 (d, J=15,5 Гц,1 Н), 8,17 (d, 2H), 12,96 (s, 1H). Способ позволяет получать это соединение при общем выходе 20%. Синтез химического предшественника. 1-[2-Метоксифенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 4). Это соединение синтезировали на основе 2-метоксиацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 80/20). Выход: 66%. ЯМР 1H CDCl3ppm: 2,27 (s, 6 Н), 3,87 (s, 3 Н), 6,97-7,05 (m, 2 Н), 7,19 (d, 1H, J=15,96 Гц), 7,22 (s,2H), 7,44 (s, 1H), 7,51 (d, 1H, J=15,48 Гц), 7,56 (dd, 1H, J=1,86 Гц, J=7,5 Гц). Синтез трет-бутилового сложного эфира. 1-[2-Метоксифенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен 1-он (соединение 5). Это соединение синтезировали с использованием соединения 1-[2-метоксифенил]-3-[3,5-диметил-4 гидроксифенил]проп-2-ен-1-она (соединение 4) и бромизобутирата трет-бутила в соответствии с описанным выше общим методом 2. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 80/20). Выход: 41%. 1-[2-Метоксифенил]-3-[3,5-диметил-4-карбоксидиметилметилоксифенил]проп-2-ен-1-он (соединение 6). Это соединение синтезировали с использованием соединения 1-[2-метоксифенил]-3-[3,5-диметил-4 трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-она (соединение 5) в соответствии с описанным выше общим методом 3. Очистка производилась хроматографией на силикагеле (дихлорметан/метанол: 98/2). Выход: 70%. ЯМР 1 Н DMSOppm: 1,38 (s, 6 Н), 2,19 (s, 6 Н), 3,93 (s, 3 Н), 7,05 (m, 1 Н), 7,20 (d, J=8,31 Гц, 1 Н), 7,25 Способ позволяет получать это соединение при общем выходе 49%. Синтез химического предшественника. 1-[4-Метилтиофенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 7). Это соединение синтезировали с использованием 4-метилтиоацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 80/20). Выход: 86%. ЯМР 1H DMSOppm: 1,22 (s, 6 Н), 2,54 (s, 3 Н), 7,36 (d, J=8,20 Гц, 2 Н), 7,48 (s, 2 Н), 7,62 (d, J=15,7 Гц, 1H), 7,74 (d, J=15,7 Гц, 1H), 8,10 (d, J=8,20 Гц, 2 Н), 8,92 (s, 1H). Синтез трет-бутилового сложного эфира. 1-[4-Метилтиофенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2 ен-1-он (соединение 8). Это соединение синтезировали с использованием 1-[4-метилтиофенил]-3-[3,5-диметил-4 гидроксифенил]проп-2-ен-1-он (соединение 7) и бромизобутирата трет-бутила. 1-[4-Метилтиофенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он растворяют в ацетонитриле,затем добавляют карбонат калия в количестве 3 молярных эквивалентов и бромизобутират трет-бутила в количестве 3 молярных эквивалентов. Смесь выдерживают при перемешивании в течение 12 ч при 80 С,затем доводят температуру до комнатной. Соли удаляют фильтрованием. После этого добавляют 3 мо- 12010410 лярных эквивалента карбоната калия и 3 молярных эквивалента бромизобутирата трет-бутила. Реакция в смеси продолжится 12 ч, затем температуру доводят до комнатной. Соли удаляют фильтрованием. Добавляют 3 молярных эквивалента карбоната калия и 3 молярных эквивалента бромизобутирата третбутила. Смесь выдерживают при перемешивании в течение 12 ч при 80 С. Соли удаляют фильтрованием,разбавители - выпариванием при пониженном давлении. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 90/10). Выход: 74%. Ацидолиз трет-бутилового сложного эфира. 1-[4-Метилтиофенил]-3-[3,5-диметил-4-карбоксидиметилметилоксифенил]проп-2-ен-1-он (соединение 9). Это соединение синтезировали на основе соединения 1-[4-метилтиофенил]-3-[3,5-диметил-4-третбутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-он (соединение 8) в соответствии с описанным выше общим методом 3. Очистка производилась хроматографией на силикагеле (дихлорметан/метанол: 98/2). Выход: 81%. ЯМР 1 Н DMSOppm: 1,39 (s, 6H), 2,22 (s, 6H), 2,57 (s, 3H), 7,40 (d, J=8,55 Гц, 2H), 7,57 (s, 2H), 7,62 Способ позволяет получать это соединение при общем выходе 24%. Синтез химического предшественника. 1-[4-Гексилоксифенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 10). Данное соединение синтезировали на основе 4-гексилоксиацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Продукт кристаллизовался в конце реакции, из него удаляют влагу. Выход: 63%. ЯМР 1 Н DMSOppm: 0,88 (m, 3 Н), 1,28-1,43 (m, 6 Н), 1,72 (m, 2H), 2,21 (s, 6H), 4,05 (t, J=6,42 Гц,2 Н), 7,40 (d, J=8,43 Гц, 2 Н), 7,48 (s, 2 Н), 7,57 (d, J=15,24 Гц, 1H), 7,72 (d, J=15,24 Гц, 1H), 8,12 (d, J=8,43 Гц, 2H), 8,89 (s, 1H). Синтез сложного трет-бутилового эфира.- 13010410 Это соединение синтезировали на основе 1-[4-гексилоксифенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 10) и трет-бутил бромизобутирата в соответствии с описанным выше общим методом 2. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 95/5). Выход: 75%. Ацидолиз трет-бутилового сложного эфира. 1-[4-Гексилоксифенил]-3-[3,5-диметил-4-карбоксидиметилметилоксифенил]проп-2-ен-1-он (соединение 12). Это соединение синтезировали на основе соединения 1-[4-гексилоксифенил]-3-[3,5-диметил-4-третбутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-она (соединение 11) в соответствии с описанным выше общим методом 3. Очистка производилась перекристаллизацией в среде метанола. Выход: 51%. ЯМР 1 Н DMSOppm: 0,88 (t, J=6,33 Гц, 3 Н), 1,30 (m, 4 Н), 1,39 (s, 6 Н), 1,44 (m, 2 Н), 1,73 (m, 2 Н),2,22 (s, 6 Н), 4,06 (t, J=6,30 Гц, 2 Н), 7,06 (d, J=8,61 Гц, 2 Н), 7,56 (s, 2H), 7,58 (d, J=15,5 Гц, 1H), 7,82 (d,J=15,5 Гц, 1 Н), 8,13 (d, J=6,61 Гц, 2 Н). Способ позволяет получать это соединение при общем выходе 22%. Синтез химического предшественника. 1-[2-Метилокси-4-хлорфенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 13). Это соединение синтезировали на основе 4-хлор-2-метоксиацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 85/15). Выход: 72%. ЯМР 1 Н DMSOppm: 2,21 (s, 6 Н), 3,90 (s, 3 Н), 7,12 (m, 1 Н), 7,23 (d, J=15,5 Гц, 1 Н), 7,29 (d, J=1,80 Гц, 1H), 7,38 (d, J=15,5 Гц, 1H), 7,41 (s, 2 Н), 7,48 (d, J=7,98 Гц, 1H). Синтез трет-бутилового сложного эфира. 1-[2-Метилокси-4-хлорфенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-он (соединение 14). Это соединение синтезировали на основе 1-[2-метилокеи-4-хлорфенил]-3-[3,5-диметил-4 гидроксидиметилметилоксифенил]проп-2-ен-1-она (соединение 13) и трет-бутилбромизобутирата в соот- 14010410 ветствии с описанным выше общим методом 2. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 90/10). Выход: 43%. Ацидолиз трет-бутилового сложного эфира.(соединение 15). Это соединение синтезировали на основе соединения 1-[2-метокси-4-хлорфенил]-3-[3,5-диметил-4 трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-она (соединение 14) в соответствии с описанным выше общим методом 3. Очистка производилась хроматографией на силикагеле (дихлорметан/метанол: 98/2). Выход: 70%. ЯМР 1H DMSOppm: 1,38 (s, 6H), 2,19 (s, 6H), 3,89 (s, 3H), 7,12 (dd, J=7,98, J=1,71 Гц, 1 Н), 7,23 (d,J=15,56 Гц, 1 Н), 7,29 (d, J=1,71 Гц, 1 Н), 7,38 (d, J=15,7 Гц, 1 Н), 7,41 (s, 2H), 7,48 (d, J=7,98 Гц, 1 Н). Способ позволяет получать это соединение при общем выходе 21%. Синтез химического предшественника. 1-[4-Бромфенил]-3-[3,5-диметил-4-гидроксифенил]проп-2-ен-1-он (соединение 16). Это соединение синтезировали на основе 4-бромацетофенона и 3,5-диметил-4 гидроксибензальдегида в соответствии с описанным выше общим методом 1. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 90/10). Выход: 37%. ЯМР 1 Н DMSOppm: 2,30 (s, 6H), 7,32 (s, 2H), 7,56-7,66 (m, 3 Н), 7,75 (d, J=15,27 Гц, 1H), 7,90 (d,J=8,70 Гц, 2 Н), 9,82 (s, 1H). Синтез сложного трет-бутилового эфира. 1-[4-Бромфенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1 он (соединение 17). Это соединение синтезировали на основе 1-[4-бромфенил]-3-[3,5-диметил-4-гидроксифенил]проп-2 ен-1-она (соединение 16) и трет-бутилабромизобутирата в соответствии с описанным выше общим методом 2. Очистка производилась хроматографией на силикагеле (циклогексан/этилацетат: 90/10). Выход: 75%. 18). Это соединение синтезировали на основе 1-[4-бромфенил]-3-[3,5-диметил-4-трет-бутилоксикарбонилдиметилметилоксифенил]проп-2-ен-1-она (соединение 17) в соответствии с описанным выше общим методом 3. Очистка производилась хроматографией на силикагеле (дихлорметан/метанол: 98/2). Выход: 38%. ЯМР 1 Н DMSOppm: 1,39 (s, 6H), 2,22 (s, 6 Н), 7,58 (s, 2H), 7,65 (d, J=15,39 Гц, 1H), 7,84-7,77 (m,3 Н), 8,09 (d, J=8,19 Гц, 2 Н), 13,01 (s, 1H).SM (ES-MS): 417,2 (M-1). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения производных соединения 1,3-дифенилпроп-2-ен-1-он, замещнных группой карбоксиалкилокси или карбоксиалкилтио, включающий следующие стадии:(i) контактирование по меньшей мере одного производного соединения 1,3-дифенилпроп-2-ен-1-он,замещнного на одной из двух групп фенила группой гидроксила или тиола и по меньшей мере одним галогенированным соединением общей формулы (II) где Y - атом галогена, R - алкильная цепь с 1-24 атомами углерода и R' - кислоточувствительная защитная группа карбоновой кислоты, выбранная из трет-бутиловой и изобутиловой групп;(ii) кислотный гидролиз (ацидолиз) сложного эфира, полученного на этапе (i) с трифторуксусной кислотой. 2. Способ по п.1, отличающийся тем, что группа R представляет собой алкильную цепь с 1-10 атомами углерода, замещнную при необходимости одним или несколькими линейными или циклическими насыщенными углеводородными радикалами с 1-12 атомами углерода. 3. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится при температуре 25-120 С, предпочтительно 80-120 С. 4. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится в присутствии катализатора. 5. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится с применением карбоната калия или цезия в качестве катализатора. 6. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (i) проводится при неоднократной добавке галогенированного соединения общей формулы (II) и при необходимости катализатора. 7. Способ по любому из предшествующих пунктов, отличающийся тем, что производное соединения 1,3-дифенилпроп-2-ен-1-он, замещнное группой гидроксила или тиола, используемое на этапе (i),получают реакцией Кляйзена-Шмидта в кислой или щелочной среде, проводимой между соединением типа ацетофенона и производным тио- или гидроксибензальдегида или между производным тио- или гидроксиацетофенона и производным типа бензальдегида. 8. Способ по любому из предшествующих пунктов, отличающийся тем, что количество трифторуксусной кислоты, используемой на этапе (ii), составляет 1-20 эквивалентов, предпочтительно 8-12 эквивалентов. 9. Способ по любому из предшествующих пунктов, отличающийся тем, что этап (ii) проводят при температуре 0-100 С, предпочтительно 18-25 С. 10. Способ по любому из предшествующих пунктов, отличающийся тем, что полученный продукт соответствует общей формулеR1, R3, R4, R5, одинаковые или разные, означают атом водорода или алкильную группу, насыщенную или не насыщенную функциональной группой карбоновой кислоты;G1, G3, G4, G5, одинаковые или разные, означают атом кислорода или серы; при этом группы X1, Х 3, Х 4 и Х 5 соответствуют формуле -G-R, в которой R означает алкильный радикал, содержащий функциональную группу карбоновой кислоты.
МПК / Метки
МПК: C07C 69/712, C07C 323/52, C07C 51/09, C07C 319/14, C07C 67/31, C07C 59/88
Метки: производных, соединения, 1,3-дифенилпроп-2-ен-1-он, получение
Код ссылки
<a href="https://eas.patents.su/18-10410-poluchenie-proizvodnyh-soedineniya-13-difenilprop-2-en-1-on.html" rel="bookmark" title="База патентов Евразийского Союза">Получение производных соединения 1,3-дифенилпроп-2-ен-1-он</a>
Получение арилалкилкарбаматных производных и их применение в терапии

Номер патента: 8801
Опубликовано: 31.08.2007
Авторы: Абуабделла Ахмед, Альмарио Гарсия Антонио, Хурнер Кристан, Раве Антуан
МПК: A61K 31/27, A61K 31/33, C07C 269/00...
Метки: производных, арилалкилкарбаматных, получение, терапии, применение
Формула / Реферат:
1. Соединение формулы (I) в которой n представляет собой целое число в интервале от 1 до 7; А выбран из одной или более чем одной группы X, Y и/или Z; X представляет собой С1-2-алкиленовую группу, возможно замещенную одной или более чем одной С1-12-алкильной, С3-7-циклоалкильной или С3-7-циклоалкил-С1-6-алкиленовой группой; Y представляет собой либо С2-алкениленовую группу, возможно замещенную одной или более чем одной С1-12-алкильной,...
Получение производных бициклогексана

Номер патента: 469
Опубликовано: 26.08.1999
Автор: Роби Роджер Л.
МПК: C07D 487/00, C07C 61/12
Метки: производных, бициклогексана, получение
Формула / Реферат:
1. Способ получения (+)-2-аминобицикло[3.1.0]гексан-2,6-карбоновой кислоты или ее фармацевтически приемлемой соли, отличающийся тем, что включает гидролиз (-)-2-спиро-5'-гидантоинбицикло[3.1.0]гексан-6-карбоновой кислоты или ее соли и необязательное образование фармацевтически приемлемой соли. 2. Способ по п.1, отличающийся тем, что гидролиз проводят в присутствии кислоты или основания в качестве катализатора. 3. Способ по п.2, отличающийся...
Промежуточные соединения для синтеза производных цианопирролидина

Номер патента: 8226
Опубликовано: 27.04.2007
Авторы: Такахаси Масато, Фукусима Хироси, Камео Казуя, Хиратате Акира
МПК: C07D 207/16
Метки: соединения, производных, цианопирролидина, синтеза, промежуточные
Формула / Реферат:
1. Промежуточное соединение, представленное формулой где - R1 представляет атом фтора; R2 - атом водорода или атом фтора; R3 и R4 представляют, каждый, атом водорода; Ra - цианогруппу или аминокарбонильную группу; Re - атом водорода, -C(=X)-Y-Rc или Rb, где Rb представляет трет-бутоксикарбонильную группу, тритильную группу, о-нитробензолсульфенильную группу, бензилоксикарбонильную группу, флуоренилоксикарбонильную группу, аллилоксикарбонильную...
Новые соединения, их применение и получение

Номер патента: 8148
Опубликовано: 27.04.2007
Авторы: Рингберг Эрик, Йенссон Маттиас, Нильссон Бьерн, Шеберг Биргер
МПК: A61K 31/444, A61K 31/497, A61P 25/00...
Метки: получение, применение, новые, соединения
Формула / Реферат:
1. Соединение формулы (I) где nх равно 2-4, предпочтительно 2; каждый из R0 и R1 независимо означает Н или СН3; R2 означает Н, C1-C4-алкил, 2-гидроксиэтил, 2-цианоэтил или тетрагидропиран-2-ил, C1-C4-ацил или C1-C4-алкоксикарбонил; каждый из R3-R5 независимо означает Н, галоген, метил или метокси, при условии, что по меньшей мере один из R3-R5 означает водород; каждый из X, Y и z независимо означает СН или N; A1 означает О, СН или СН2; A2...
Способы и промежуточные соединения для получения замещенных производных хроманола

Номер патента: 2204
Опубликовано: 28.02.2002
Авторы: Рэггон Джеффри В., Раггери Сэлли Г., Пископио Энтони Д., Кейрон Стивен, Келли Сара И., Даггер Роберт В., Хокинс Джоэл М., Кэстэлди Майкл Дж.
МПК: C07C 49/245, C07D 311/22, C07F 5/04...
Метки: соединения, хроманола, замещенных, получения, производных, промежуточные, способы
Формула / Реферат:
1. Способ получения соединения формулы или энантиомера указанного соединения, где в указанном соединении формулы Х группировка R3-замещенной бензойной кислоты присоединена по атомам углерода 6 или 7 хроманового кольца; R1 представляет собой -(CH2)qCHR5R6, где q является числом от 0 до 4; каждый R2 и R3 независимо выбран из группы, которую составляют Н, фторо, хлоро, (С1-С6)алкил, (С1-С6)алкокси, фенилсульфинил, фенилсульфонил и...
Предыдущий патент: Применение декстрансульфата для ингибирования немедленной воспалительной реакции, опосредованной кровью
Следующий патент: 5, 6-диалкил-7-аминотриазолопиримидины, способ их получения и их применение для борьбы с патогенными грибами, а также содержащие их средства
Случайный патент: Способ каталитического частичного окисления углеводородов в синтез-газ