Промежуточные соединения для получения ациклических нуклеозидов














Формула / Реферат
1. Соединение формулы

в котором R6 и R7 являются С1-7алкилом или бензилом или R6 и R7 вместе являются -СН2-СН2- или -СН2СН2СН2-, -СН2СН2СН2СН2- и R9 представляет собой водород или спирт-защитную группу.
2. Соединение по п.1, в котором R9 является водородом.
3. Соединение по п.1, в котором спирт-защитной группой является бензильная группа.
4. Соединение по п.1 или 2, в котором R6 и R7 каждый является этилом.
Текст
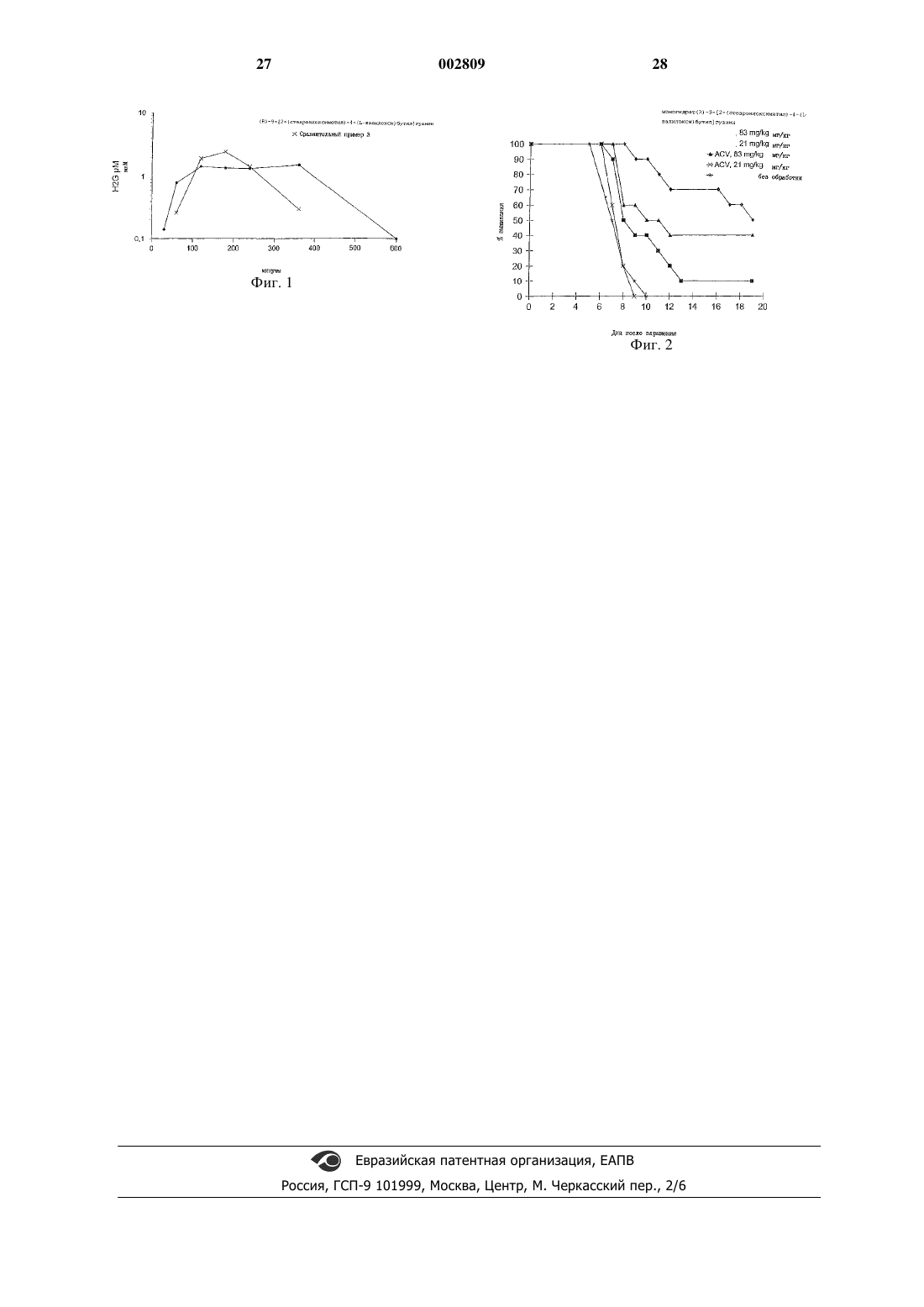
1 Настоящее изобретение относится к новым промежуточным соединениям для получения производных ациклических нуклеозидов, которые могут быть использованы в качестве антивирусных препаратов, применяемых в борьбе с герпесом и ретровирусными инфекциями. Практическое использование ациклических нуклеозидов ограничено их относительно умеренными фармакокинетическими свойствами. В общем плане, для решения такой проблемы был применен ряд подходов с использованием пролекарств, целью которых являлось улучшение биоприменимости ациклических нуклеозидов. Один из таких подходов заключается в получении сложноэфирных производных, особенно алифатических сложных эфиров, одной или более гидроксигрупп ациклической боковой цепи. В европейском патенте ЕР 165 289 описывается перспективный антигерпесный агент 9[4-гидрокси-(2-гидроксиметил)бутил]гуанин,известный под названием H2G. В европейском патенте ЕР 186 640 описывается 6-деокси H2G. В европейском патенте ЕР 343 133 указывается,что такие соединения, особенно R-(-)энантиомер, также обладают активностью против таких ретровирусных инфекций, как HIV. Различные производные H2G, такие, как фосфонаты, алифатические сложные эфиры (например, диацетат и дипропионат), а также простые эфиры по гидроксигруппам ациклической боковой цепи раскрыты в ЕР 343 133. В указанном патенте также описываются способы получения таких производных, заключающиеся в конденсации ациклической боковой цепи по N9-положению типичного 6-галогенированного пуринового фрагмента, или, в альтернативном случае, в замыкании имидазольного цикла пиримидина или фуразано-[3,4-d]-пиримидинового фрагмента, или замыкании пиримидинового кольца имидазольного фрагмента, в том случае, когда ациклическая боковая цепь уже присутствует в предшествующем пиримидиновом или имидазольном фрагментах, соответственно. В соответствии с наиболее широкими аспектами каждого из таких способов ациклическая боковая цепь является предварительно замещенной,однако в специальных примерах показано одностадийное диацилирование H2G с помощью уксусного или пропионового ангидрида и ДМФ.(1989) сообщают об исследовании ряда низших алифатических сложных эфиров ациклического нуклеозида, 9-[4-гидрокси-(3-гидроксиметил)бутил]гуанина, известного также под названием пенцикловир, и его 6-деокси аналога. Фамцикловир,торговый антивирусный агент, представляет собой диацетильное производное 6-деоксипенцикловира.(1987) описывает низшие алифатические сложные эфиры 9-[(1,3-дигидрокси-2-пропокси) 002809 2 метил]гуанина, известного также под названием ганцикловир. Отмечается, что предпочтительным сложным эфиром является дипропионат.(1989) диацетатные и дипропионатные производные H2G, и также моноацетатаные и диацетатные производные 6-деокси H2G. Сообщается, что диацетатные и дипропионатные производные H2G обеспечивают лишь незначительное улучшение биоприменимости, по сравнению с H2G. В международной патентной заявке WO 94/24134, опубликованной 27 октября 1994 г,раскрываются алифатические сложноэфирные пролекарства 6-деокси-N-7 аналога ганцикловира, включающие дипивалоил, дивалероил, моновалероил, моноолеоил и моностеароильные сложные эфиры. В обеих международных заявках на патенты WO 93/07163, опубликованной 15 апреля 1993 г., и WO 94/22887, опубликованной 13 октября 1994 г., описываются моносложноэфирные производные нуклеозидных аналогов - производных мононенасыщенных С 18 или С 20 жирных кислот. В патенте США 5,216,142, от 1 июня 1993 г. также описываются моносложноэфирные производные жирных кислот нуклеозидных аналогов. Второй подход, используемый для получения пролекарств ациклических нуклеозидов,включает получение аминокислотных сложных эфиров по одной или более гидроксигруппам ациклической боковой цепи. В европейском патенте ЕР 99 493 описываются аминокислотные сложные эфиры ацикловира, а в заявке на европейский патент ЕР 308 065, опубликованной 22 марта 1989 г, раскрываются валиновый и изолейциновый эфиры ацикловира. В заявке на европейский патент ЕР 375 329, опубликованной 27 июня 1990 г, раскрываются аминокислотные сложноэфирные производные ганцикловира, включающие дивалин,диизолейцин, диглицин и диаланин сложноэфирные производные. В международной заявке на патент WO 95/09855, опубликованной 13 апреля 1995 г., описываются сложноэфирные производные пенцикловира, включающие моновалин и дивалин сложноэфирные производные. В ДЕ 19526163, опубликованной 1 февраля 1996 г., а также в патенте США 5,543,414 от 6 августа 1996 г., описываются ахиральные аминокислотные сложные эфиры ганцикловира. В заявке на европейский патент ЕР 694 547, опубликованной 31 января 1996 г., раскрывается моно-L-валиновый сложный эфир ганцикловира и его получение из дивалилганцикловира. В заявке на европейский патент ЕР 654 473, опубликованной 24 мая 1995 г., описываются различные бис-аминокислотные сложно 3 эфирные производные 9-[1',2'-бисгидроксиметил)-циклопропан-1'-ил]метилгуанина. В международной патентной заявке WO 95/22330, опубликованной 24 августа 1995 г.,описываются алифатические сложные эфиры,аминокислотные сложные эфиры, а также сложные ацетат/валинатные сложные эфиры ациклического нуклеозида, 9-[3,3-дигидроксиметил-4 гидрокси-бут-1-ил]гуанина. В этой работе указывается, что биоприменимость понижается в том случае, когда один из валиновых эфиров тривалинового производного заменяют на ацетатную сложноэфирную группу. Установлено, что дисложноэфирные производные H2G со специальной комбинацией аминокислотного сложного эфира и сложного эфира жирной кислоты, позволяют значительно улучшить оральную биодоступность в сравнении с родственным соединением (H2G). Задачей настоящего изобретения является создание новых промежуточных соединений общей формулы в которой R6 и R7 являются низшим С 1-7 алкилом или бензилом или R6 и R7 вместе являются-СН 2-СН 2- или -CH2CH2CH2-, -CH2CH2CH2CH2-,и R9 является водородом или группой, защищающей ОН, предназначенных для получения новых ациклических нуклеозидов, обладающих антивирусными свойствами, которые имеют следующую формулу IR3 представляет собой ОН или Н; а также их фармацевтически приемлемые соли. Благоприятное влияние при оральном применении смешанных сложных эфиров жир 002809 4 ных кислот и аминокислот, указанных выше,является особенно неожиданным в сравнении с соответствующим применением сложных эфиров жирных кислот. На основании результатов анализа мочевой регенерации (табл. 1 А) или плазмолекарственного анализа (табл. 1 В) H2G крыс, можно сделать вывод, что ни моно-, или дисложные эфиры жирных кислот H2G не обеспечивают какого-либо улучшения оральной биодоступности в сравнении с родственными соединениямиH2G. Действительно,дистеаратное производное обеспечивает значительно более низкую биодоступность, чем родственное соединение, что может быть следствием нежелательного влияния стеаратного сложного эфира на улучшение оральной биоприменимости H2G. Имеются сообщения о том, что превращение одного или обоих гидроксилов в некоторых других ациклических нуклеозидных аналогах в соответствующий валиновый или дивалиновый сложный эфир улучшают биодоступность. Превращение H2G в соответствующие моно- или дивалил сложноэфирные производные обеспечивает аналогичное улучшение биодоступности по сравнению с родственным соединением. Учитывая тот факт, что жирнокислотные производные H2G оказывают отрицательное воздействие на улучшение биодоступности, неожиданным оказалось то, что смешанные сложноэфирные производные H2G на основе аминокислоты/жирной кислоты обеспечивают улучшенную или сравнимую оральную биодоступность в сравнении с такой характеристикой валинового дисложноэфирного производного H2G, о чем свидетельствуют данные мочевинно-регенерационного или плазмолекарственного анализов, соответственно. Группа R1 Водород Водород Стеароил Валил Валил Валил Группа R2 Водород Стеароил Стеароил Водород Валил Стеароил См. приведенный ниже подробный биологический пример 1. Группа R1 Водород Водород Стеароил Валил Валил Валил Группа R2 Водород Стеароил Стеароил Водород Валил Стеароил Подробности см. в приведенном ниже биологическом примере 2. Указанные ациклические нуклеозиды, получаемые из промежуточных соединений настоящего изобретения, являются сильными антивирусными агентами, особенно в отношении 5 таких герпесных инфекций, которые вызываются вирусом Varicella zoster, вирусом герпеса обыкновенного типа 1 и 2, вирусом Epstein-Barr,Herpes типа 6 (HHV-6) и типа 8 (HHV-8). Такие соединения особенно применимы против инфецирования вирусом Varicella zoster, такого как опоясывающий лишай у пожилых людей, включая постгерпесную невралгию, или куриный сифилис у молодых особей, когда длительность и тяжесть заболевания могут быть снижены за несколько дней.Epstein-Barr вирусные инфекции, поддающиеся лечению соединениями согласно изобретению, включают инфекционную мононуклеоз/железистую лихорадку, которая ранее не поддавалась лечению и которая может вызывать в течение многих месяцев неспособность к обучению у подростков. Соединения формулы I также обладают активностью против некоторых ретровирусных инфекций, особенно таких, как SIV, HIV-1 иHIV-2, а также против инфекций, для которых указан трансактивирующий вирус. Преимущественно группа R3 представляет собой гидроксигруппу или ее таутомерную форму =O так, что основная часть соединений изобретения представляет собой встречающийся в природе гуанин, например, в том случае, когда боковая цепь расщепляется in vivo. С другой стороны, R3 может представлять собой водород,и в этом случае имеют дело, как правило, с более растворимым 6-деоксипроизводным, которое может окисляться in vivo (например, с помощью ксантин оксидазы) в гуаниновую форму. Соединения формулы I могут присутствовать в рацемической форме, т.е. представлять собой смесь 2R и 2S изомеров. Однако, предпочтительно, чтобы соединения формулы I содержали, по крайней мере, 70%, предпочтительно, по крайней мере, 90% R-формы, например,более 95% такой формы. Наиболее предпочтительно, когда соединение формулы I представляет собой энантиомерно чистую R-форму. Предпочтительно, аминокислота группыR1/R2 содержит четное число атомов углерода, и главным образом, представляет собой деканоил(C16), стеароил (C18), или эйкозанил (С 20). Другие подходящие R1/R2 группы включают бутирил, гексаноил, октаноил или бехеноил (С 22). Другие подходящие R1/R2 группы включают производные миристолеиновой, миристэлаидиновой, пальмитолевой, пальмэлаидиновой, н-6 октадеценовой, олеиновой, элаидиновой, гандоиновой, эруковой или брассидиновой кислот. Мононенасыщенные сложные эфиры жирных кислот, как правило, имеют двойную связь в транс-конфигурации, предпочтительно в -6, 9 или -11 положении, в зависимости от их 6 длины. Предпочтительно, R1/R2 группа является производной жирной кислоты, содержащей С 9 С 17 насыщенный или n:9 мононенасыщенный алкил. Насыщенная или ненасыщенная жирная кислота, или R1/R2 могут быть необязательно замещены одинаковыми или различными заместителями, в количестве до пяти, выбранными из группы, состоящей из таких заместителей, как гидрокси, C1-C6 алкил, C1-C6 алкокси, C1-С 6 алкокси C1-C6 алкил, C1-C6 алканоил, амино, гало,циано, азидо, оксо, меркапто и нитро, и др. Наиболее предпочтительными соединениями формулы I являются те, в которых R1 представляет собой -С(O)СНСН(СН 3)2NH2 или С(O)СН(СН(СН 3)СН 2 СН 3)NH2 и R2 представляет собой -C(O)C9-C17 насыщенный алкил. Используемый в тексте термин "низший алкил" относится к алкильным радикалам нормального или разветвленного строения, содержащим 1-7 углеродных атомов, включающим,но не ограничивающимся ими, метил, этил, нпропил, изопропил, н-бутил, изобутил, втор.бутил, трет.-бутил, н-пентил, 1-метилбутил, 2,2 диметилбутил, 2-метилпентил, 2,2-диметилпропил, н-гексил и т.п. Используемый в тексте термин "Nзащитная группа" или "N-защищенная" относится к тем группам, которые предназначены для защиты концевого N-аминокислоты или пептида, или для защиты аминогруппы от нежелательных реакций в ходе операций синтеза. Традиционно используемые N-защитные группы описаны в книге Greene "ProtectiveGroups in Organic Synthesis" (John WileySons,New York, 1981) и на эту работу ссылаются в настоящем описании. N-защитные группы включают такие ацильные группы, как формил,ацетил,пропионил,пивалоил,трет.бутилацетил,2-хлорацетил,2-бромацетил,трифторацетил, трихлорацетил, фталил, онитрофеноксиацетил, -хлорбутирил, бензоил,4-хлорбензоил, 4-бромбензоил, 4-нитробензоил и т.п.; такие сульфонильные группы, как бензолсульфонил, п-толуолсульфонил, и т.п.; такие карбамат-образующие группы, как бензилоксикарбонил, п-хлорбензилоксикарбонил, п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, 2-нитробензилоксикарбонил, п-бромбензилоксикарбонил,3,4-диметоксибензилоксикарбонил, 4-метоксибензилоксикарбонил, 2 нитро-4,5-диметоксибензилоксикарбонил, 3,4,5 триметоксибензил-оксикарбонил, 1-(п-бифенилил)-1-метилэтоксикарбонил, ,-диметил-3,5 диметоксибензилоксикарбонил, бензгидрилоксикарбонил, трет.-бутоксикарбонил, диизопропилметоксикарбонил, изопропилоксикарбонил,этоксикарбонил, метоксикарбонил, аллилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, феноксикарбонил, 4-нитрофеноксикарбонил, флуоренил-9-метоксикарбонил, циклопентилоксикар 7 бонил, адамантилоксикарбонил, циклогексилоксикарбонил, фенилтиокарбонил, и т.п.; такие алкильные группы, как бензил, трифенилметил,бензилоксиметил и т.п.; такие силильные группы, как триметилсилил и т.п. Предпочтительными N-защитными группами являются формил, ацетил, бензоил, пивалоил, трет.бутилацетил, фенилсульфонил, бензил, трет.бутоксикарбонил (ВОС) и бензилоксикарбонил(Cbz). Используемый в тексте термин "активированное сложноэфирное производное" относится к таким галоидоангидридам, как хлорангидриды, а активированные сложные эфиры включают, но не ограничиваются ими, ангидриды производные муравьиновой и уксусной кислоты, ангидриды - производные таких алкоксикарбонилгалогенидов, такие как изобутилоксикарбонилхлорид и т.п., N-гидроксисукцинимид производные сложные эфиры,Nгидроксифталимид - производные сложные эфиры, N-гидроксибензотриазол производные сложные эфиры, N-гидрокси-5-норборнен-2,3 дикарбоксамидпроизводные сложные эфиры,2,4,5-трихлорфенилпроизводные сложные эфиры и т.п. Предпочтительные соединения формулы I включают(R)-2-амино-9-[2-13-докозеноил)оксиметил)-4-(L-изолейцилокси)бутил]пурин, а также их фармацевтически приемлемые соли. Другие предпочтительные соединения включают(R)-2-амино-9-[2-13-докозеноил)-оксиметил)-4-(L-валилокси)-бутил]пурин; а также их фармацевтически приемлемые соли. Другие предпочтительные соединения формулы I включают(R)-9-[4-(додеканоилокси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-(тетрадеканоилокси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-(гексадеканоилокси)-2-[L-валилоксиметил)бутил]гуанин,(R)-9-[4-(октадеканоилокси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-(эйкозаноилокси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-(докозаноилокси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-9-тетрадеценоил)окси)-2-(L-валилоксиметил)бутил]-гуанин,(R)-9-[4-9-гексадеценоил)окси)-2-(L-валилоксиметил)бутил]-гуанин,(R)-9-[4-6-октадеценоил)окси)-2-(L-валилоксиметил)бутил]-гуанин,(R)-9-[4-9-октадеценоил)окси)-2-(L-валилоксиметил)бутил]-гуанин,(R)-9-[4-11-эйкозеноил)окси)-2-(L-валилоксиметил)бутил]гуанин,(R)-9-[4-13-докозеноил)-окси)-2-(L-валилоксиметил)бутил]-гуанин,(R)-2-амино-9-[4-(бутирилокси)-2-(L-валилоксиметил)бутил]пурин,(R)-2-амино-9-[4-(4-ацетилбутирилокси)-2(L-валилоксиметил)-бутил]пурин,(R)-2-амино-9-[4-(гексаноилокси)-2-(L-валилокси)бутил]пурин,(R)-2-амино-9-[4-(октаноилокси)-2-(L-валилоксиметил)бутил]пурин,(R)-2-амино-9-[4-(деканоилокси)-2-(L-валилоксиметил)бутил]пурин,(R)-2-амино-9-[4-(додеканоилокси)-2-(Lвалилоксиметил)бутил]-пурин,(R)-2-амино-9-[4-(тетрадеканоилокси)-2(L-валилоксиметил)бутил]-пурин,(R)-2-амино-9-[4-(гексадеканоилокси)-2(L-валилоксиметил)бутил]-пурин,(R)-2-амино-9-[4-(октадеканоилокси)-2-(Lвалилоксиметил)бутил]-пурин,(R)-2-амино-9-[4-(эйкозаноилокси)-2-(Lвалилоксиметил]бутил-пурин,(R)-2-амино-9-[4-(докозаноилокси)-2-(Lвалилоксиметил)бутил]-пурин,(R)-2-амино-9-[4-9-тетрадеценоил)окси)2-(L-валилоксиметил)-бутил]пурин,(R)-2-амино-9-[4-9-гексадеценоил)окси)2-(L-валилоксиметил)-бутил]пурин,(R)-2-амино-9-[4-6-октадеценоил)окси)2-(L-валилоксиметил)-бутил]пурин,(R)-2-амино-9-[4-9-октадеценоил)окси)2-(L-валилоксиметил)-бутил]пурин,(R)-2-амино-9-[4-11-эйкозеноил)окси)-2(L-валилокси)бутил]-пурин,(R)-2-амино-9-[4-13-докозеонил)оксиметил)-2-(L-валилокси)-бутил]пурин, или фармацевтически приемлемые соли указанных соединений. Соединения формулы I могут образовывать соли. Подходящие фармацевтически при 11 емлемые соли соединений формулы I включают соли органических кислот, главным образом карбоновых кислот, включающих, но не ограничивающихся ими, ацетат, трифторацетат, лактат, глюконат, цитрат, тартрат, малеат, малат,пантотенат, изоэтионат, адипат, альгинат, аспартат, бензоат, бутират, диглюконат, циклопентанат, глюкогептанат, глицерофосфат, оксалат, гептаноат, гексаноат, фумарат, никотинат,пальмоат, пектинат, 3-фенилпропионат, пикрат,пивалат, пропионат, тартрат, лактобионат, пиволат, камфорат, ундеканоат и сукцинат, такие соли органических сульфокислот, как метансульфонат, этансульфонат, 2-гидроксиэтансульфонат, камфорсульфонат, 2-нафталинсульфонат, бензолсульфонат, п-хлорбензолсульфонат, и п-толуолсульфонат; а также соли неорганических кислот, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, бисульфат,полусульфат, тиоцианат, персульфат, а также соли фосфорных и сульфокислот. Удобнее всего использовать соли хлористо-водородных кислот. Соединения формулы I могут быть выделены в виде гидратов, в кристаллической форме,предпочтительно в виде гомогенных кристаллов, и в этом случае дополнительный аспект настоящего изобретения предусматривает соединения формулы I в практически чистой кристаллической форме, содержащей 70%, предпочтительно 90% гомогенного кристаллического материала, например 95% гомогенного кристаллического материала. Соединения формулы I особенно подходят для орального применения, однако, они могут применяться ректально, вагинально, нозально,местно, трансдермально или парентерально,например внутримышечно, внутривенно или эпидурально. Соединения изобретения могут применяться, как таковые, например, в виде капсул, или, как правило, совместно с фармацевтически приемлемым носителем или разбавителем. Оральные рецептуры удобнее всего получать в виде таких форм разовой дозировки, как капсулы или таблетки, с использованием таких традиционных носителей или связующих веществ, как стеарат магния, мел, крахмал, лактоза, воск, камедь или желатин. Для создания рецептур пролонгированного действия могут использоваться липосомы, или такие синтетические или природные полимеры, как НРМС илиPVP. С другой стороны, такие рецептуры могут быть изготовлены в виде назальных или глазных капель, сиропа, геля или крема, содержащих раствор, суспензию, эмульсию, препарат типа масло в воде или вода в масле, в таких традиционных наполнителях, как вода, физиологический раствор, этанол, растительное масло или глицерин, необязательно в присутствии отдуш 002809 12 ки и/или предохранительного агента, и/или эмульгатора. Соединения формулы I могут применяться с дневной дозировкой в интервале 0,1-200 мг/кг/день,преимущественно 0,5-100 мг/кг/день, более предпочтительно 10-50 мг/кг/день, например 10-25 мг/кг/день. Типичная дозировка для нормального взрослого может составлять 50-500 мг, например,300 мг, которую применяют один или два раза в день в случае герпеса и 2-10 раз - в случае HIV инфецирований. В соответствии с обычными правилами антивирусной терапии, соединения могут применяться в комбинации с такими другими антивирусными агентами, как ацикловир, валцикловир,пенцикловир, фамцикловир, ганцикловир и их пролекарства, цидофовир, фоскарнет и аналогичные препараты, предписанные для лечения герпеса, а также AZT, ddI, ddC, d4T, 3TC, фоскарнет, ритонавир, индинавир, саквинавир, делавиридин. Vertex VX 478, Agouron AG1343 и другими препаратами, предписанными для лечения ретровирусных инфекций. Схема С иллюстрирует получение промежуточного соединения согласно изобретению(на схеме соединение 11) и его последующее превращение в биологически-активное соединение формулы I. Схема С В соответствии со схемой С, малонат 1 (R4 и R5 представляют собой низший алкил, бензил или т.п.) алкилируют по реакции с примерно 0,5-2,0 молярными эквивалентами ацеталя 2 (R6 и R7 представляют собой низший алкил, бензил и т.п., или R6 и R7 совместно представляют собой -СН 2CH2- или -СН 2 СН 2 Сн 2-, -или 13 СН 2 СН 2 СН 2 СН 2-, a X1 представляет собой уходящую группу (например, Сl, Вr или I, либо такой сульфонат, как метансульфонат, трифлат, птолуолсульфонат, бензолсульфонат и т.п.) в присутствии 0,5-2,0 молярных эквивалентов основания (например, трет.-бутилата калия, или этилата натрия, либо NaH или КН и т.п.) в среде инертного растворителя (например такого, как ДМФ, ТГФ, диоксан, диоксолан, N-метилпирролидон и т.п.) при температуре в интервале-40-190 С с получением алкилированного малоната 3. В результате восстановления 3, при соотношении 0,5-4,0 молярных эквивалентов сложного эфира к спиртовому восстанавливающему агенту (например, такому, как LiBH4, Ca(BH4)2 или NaBH4, LiAlH4 и т.п.) в среде инертного растворителя (например, ТГФ, метил трет.бутилового эфира или трет.-ВuОН, или т.п.) при температуре в интервале от -20 до примерно 100 С, получали диол 4. В результате энзимной этерификации 4 по реакции с 1,0-20,0 молярными эквивалентами в винилового эфира 5 (R8 представляет собой С 3-С 21 насыщенный или мононенасыщенный, необязательно замещенный алкил) в присутствии липазы (например,липазы PS-30, липазы PPL, липазы CCL и т.п.) или фосфолипазы (например фосфолипазы D и т.п.), получали желаемый стереоизомер сложного эфира 6. Такую реакцию можно проводить в отсутствии растворителя или в присутствии инертного растворителя (например, метил трет.бутилового эфира, толуола или гексана, и т.п.). Реакцию проводили при температуре в интервале от -20 до примерно 80 С. Спиртовый заместитель соединения 6 превращают в удаляемую группу (например такую,как галоген или сульфонат) по реакции с галогенирующим агентом (например таким, какNCS/P(Ph)3/NaI в среде ацетона или аналогичного растворителя) в среде инертного растворителя (например такого, как хлористый метилен,толуол, этилацетат и т.п.) или по реакции с 0,82,0 молярными эквивалентами сульфонил галогенида (например такого, как бензолсульфонилхлорид, толуолсульфонилхлорид или метан сульфонилхлорид и т.п.) в присутствии 1,0-4,0 молярных эквивалентов основания (например такого, как триэтиламин, карбонат калия, пиридин, диметиламинопиридин или этилдиизопропиламин и т.п.) в среде инертного растворителя(например такого, как метиленхлорид, толуол,этилацетат, пиридин, метил трет.-бутиловый эфир и т.п.) при температуре в интервале от 25 С до, примерно, 100 С с получением сложного эфира 7 (Х 2 представляет собой галоген или сульфонатную уходящую группу). По реакции соединения 7 с 0,9-2,0 молярными эквивалентами 2-амино-4-хлорпурина 8 в присутствии 1,0-6,0 молярных эквивалентов основания (например такого, как карбонат ка 002809Mitsunobu сочетания (например, с использованием системы Р(Ph)3/диэтилазидокарбоксилат) спирта 6 с 2-амино-4-хлорпурином 8 получали соединение 9. В результате реакции 9 с 2,0-20 молярными эквивалентами спирта R9 ОН (R9 представляет собой такую спиртозащитную группу, как бензил и т.п.) в присутствии 1,0-6,0 молярных эквивалентов основания (например такого, как трет.-бутилат калия, карбонат калия, NaH, KH,диизопропиламид лития и т.п.) в среде инертного растворителя (например такого, как ТГФ,ДМФ и т.п.), при температуре от -25 С до, примерно, 150 С получали спирт 10. В результате удаления спиртозащитной группы R9 соединения 10 (например, в результате каталитического гидрирования в среде такого инертного растворителя, как этанол, бензиловый спирт, метанол, ТГФ и т.п. в присутствии такого гидрирующего катализатора, как Pd/C или Pd(OH)2 и т.п.), получали замещенный гуанин 11. В результате этерификации соединения 11 по реакции с а) 0,8-2,0 молярными эквивалентами R10 СООН и сшивающим агентом (например ДСС/ДМАР) и т.п. в среде инертного растворителя (например, ТГФ, ДМФ и т.п.) или b) с 0,82,0 молярными эквивалентами активированного производного R10COOH (например такого, как хлорангидрид, или N-гидроксисукцинимидный сложный эфир, или R10C(О)ОС(О)R10 и т.п.) в присутствии 0-3,0 молярных эквивалентов основания (например такого, как пиридин, триэтиламин, этилдиизопропиламин, ДВУ, карбонат калия и т.п.), в среде инертного растворителя (например такого, как метиленхлорид, ТГФ,пиридин, ацетонитрил, ДМФ и т.п.) при температуре от -25 С до, примерно, 100 С, получали сложный эфир 12. Ацетальный заместитель соединения 12 удаляли и полученный в результате альдегид восстанавливали путем реакции соединения 12 с 0,1-10,0 молярными эквивалентами кислоты(например такой, как трифликовая, НСl, уксусная, серная кислоты и т.п.) в среде инертного растворителя (например такого, как ТГФ/Н 2 О,этилацетат/Н 2 О,этаметиленхлорид/Н 2O,нол/Н 2 О, метанол/Н 2 О и т.п.) при температуре в интервале от -25 С до, примерно, 100 С. К сырой смеси добавляли 0,1-10,0 молярных эквивалентов основания (например такого, как бикарбонат натрия, карбонат калия, триэтиламин,пиридин, КОН и т.п.), дополнительное количество инертного растворителя (например такого,как ТГФ, хлористый метилен, этилацетат, метил трет.-бутиловый эфир, изопропанол и т.п.), а также от 0,3 до 5,0 молярных эквивалентов агента восстанавливающего альдегид (например такого, как борогидрид натрия, RaNi/Н 2 и т.п.),при температуре в интервале от -25 до 100 С, с получением спирта 13. В результате реакции 13 с 0,8-3,0 молярными эквивалентами N-защищенной аминокислоты PiNHCH(R11)COOH или ее активированного производного (P1 представляет собой Nзащитную группу, а Рц представляет собой изопропил или изобутил) в среде инертного растворителя (например такого, как ТГФ, диоксан,диоксолан, ДМФ, хлористый метилен и т.п.) при температуре 25-100 С, получали спирт 14. В результате снятия N-защитной группы соединения 14 получали соединение настоящего изобретения формулы I, в которой R3 представляет собой -ОН. В альтернативном случае, соединение 13 может взаимодействовать с симметричным ангидридом, производным от P1NHCH(R11)COOH (например, P1NHCH(R11)С(О)O-С(О)СН(R11)NHP1) с получением соединения 1, в котором R3 представляет собой ОН. Пример 1. Получение (R)-9-[4-диэтокси-2(гидроксиметил)бутил]гуанидина 4,4-диэтокси-2 Трет.-бутилат калия (141,8 г, 1,11 экв.) растворяли в сухом ДМФ (1 л). В течение 5 мин добавляли диэтилмалонат (266 мл, 1,54 экв.). В течение 5 мин добавляли диэтилацеталь бромацетальдегида (172 мл, 1,14 моля). Смесь нагревали до 120 С (внутренняя температура) и перемешивали при 120 С в течение 5 ч. Полученной смеси давали охлаждаться до комнатной температуры, переливали в воду (5 л) и экстрагировали метил-трет.-бутиловым эфиром(МТВЕ, 3 х 600 мл). Органический раствор сушили над MgSO4, фильтровали, концентрировали и перегоняли (0,5 мм, 95-140 С) с получением желаемого диэфира (244 г, 78%) в виде бесцветного масла. Спектр 1H ЯМР (CDCl3)1,19 (т, 6 Н), 1,28LiBH4 (торговый раствор, 2 М в ТГФ, 22,5 мл) и продукт со стадии а) примера 1 (5 г в 15 мл ТГФ, 18,1 ммоля) объединяли друг с другом, 16 нагревали до 60 С и перемешивали при 60 С в течение 4 ч. Реакционной смеси давали охлаждаться до комнатной температуры и реакционный сосуд помещали в баню с холодной водой. Затем, триэтаноламин (5,97 мл, 1 экв.) добавляли с такой скоростью, чтобы температура реакционной смеси поддерживалась в интервале 2025 С. Рассол (17,5 мл) добавляли с такой скоростью, которая позволяла регулировать газовыделение и полученную смесь перемешивали в течение 45 мин при комнатной температуре. Происходило расслаивание системы, и органический слой промывали рассолом (2 х 15 мл). Объединенные промывки рассолом экстрагировали МТВЕ (метил-трет.-бутиловым эфиром, 3 х 20 мл). Объединенные органические экстракты выпаривали и остаток растворяли в МТВЕ (50 мл) и промывали рассолом (25 мл). Слой рассола подвергали обратной экстракции МТВЕ (3 х 25 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали с получением желаемого диола (3,36 г,15,5 ммоля, 97 %) в виде бесцветного масла. Спектр 1H ЯМР (СDСl3)1,22 (т, 6 Н), 1,73(м, 2 Н), 3,69 (м, 2 Н), 3,72 (м, 4 Н), 4,62 (т, 1 Н). с) Получение (2R)-2-ацетоксиметил-4,4 диэтоксибутанола В 1-горлую круглодонную колбу объемом 10 мл загружали продукт со стадии b) примера 1(3,84 г, 20 ммолей), после чего добавляли винилацетат (2,6 г, 30 ммолей) и наконец липазуPS 30 (69 мг, полученную от Amano, Lombard,Illinois). Смесь перемешивали при температуре окружающего воздуха в течение 16 ч. За ходом реакции внимательно следили методом ТСХ(2/1 гексан -EtOAc; окрашивание Ce2 (SO4)3 и обугливание на горячей пластинке; r.f. диола 0,1, моноацетата -0,3, бис-ацетата -0,75). Реакционную смесь разбавляли СН 2 Сl2 и фильтровали через 5-микронный фильтр. Фильтр промывали дополнительным количеством CH2Cl2. Затем фильтрат концентрировали в вакууме с получением желаемого продукта. В 1-горлую круглодонную колбу емкостью 100 мл, снабженную магнитной мешалкой и мембраной, в атмосфере азота загружали сырой продукт со стадии с) примера 1 (4,62 г, 19 ммолей), сухой CH2Cl2 (20 мл) и Et3N (5,62 мл, 40 ммолей). К полученному раствору добавляли хлористый тозил (4,76 г, 25 ммолей). Полученную в результате смесь перемешивали при температуре окружающего воздуха в течение 4 ч. Загружали Н 2O (0,27 г, 15 ммолей) и смесь интенсивно перемешивали в течение 4 ч. Реакционную смесь разбавляли 80 мл 17 органическому слою добавляли 75 мл 5% водного раствора КН 2 РO4. После перемешивания и разделения слоев, водный слой отбрасывали. Органический слой промывали 50 мл насыщенного раствора NаНСО 3, сушили над Na2SO4,фильтровали и концентрировали в вакууме до постоянного веса, в результате чего получали 7,40 г желаемого продукта. Спектр 1H ЯМР (CDCl3)1,17 (т, 6 Н), 1,62 В 1-горлую круглодонную колбу емкостью 50 мл загружали продукт со стадии d) примера 1(3,88 г, 10 ммолей), безводный ДМФ (20 мл), 2 амино-4-хлорпурин (2,125 г 12,5 ммоля) и К 2 СО 3 (4,83 г). Полученную в результате суспензию перемешивали при 40 С в атмосфере N2 в течение 20 ч. Смесь концентрировали для удаления большей части ДМФ на роторном испарителе. Остаток разбавляли EtOAc (50 мл) и Н 2O (50 мл). Реакционную смесь переносили в делительную воронку, встряхивали и отделяли водный слой. Водный слой экстрагировалиEtOAc (25 мл). Органические слои объединяли и промывали 5% КН 2 РO4 (75 мл). Органический слой отделяли и промывали Н 2O (75 мл), рассолом (75 мл), сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 3,95 г сырого продукта. Этот сырой продукт суспендировали с 40 мл метил-трет.-бутилового эфира. Полученную смесь перемешивали в течение ночи при 4 С и фильтровали. Фильтрат концентрировали с получением 3,35 г продукта в виде масла (содержащего 2,6 г целевого продукта в соответствии с данными HPLC анализа). 300 МГц 1H ЯМР (СDСl3)1,19 (м, 6 Н),1,69 (2 Н), 1,79 (с, 1 Н), 2,03 (с, 3 Н), 2,52 (м, 1 Н),3,48 (м, 2 Н), 3,62 (м, 2 Н), 4,04 (м, 2 Н), 4,16 (м,2 Н), 4,61 (т, 1 Н), 5,12 (широкий с, 2 Н), 7,81 (с,1 Н). В 1-горлую круглодонную колбу емкостью 500 мл загружали бензиловый спирт (136 мл),смесь охлаждали до 0 С после чего порциями добавляли КО-трет.-Вu (36 г, 321 ммоль). Смеси давали нагреваться до 40 С и в течение 20 мин смесь перемешивали. К полученной смеси при 0 С добавляли сырой продукт со стадии е) примера 1 (24,7 г, 64,2 ммоля), растворенный в 25 18 мл безводного ТГФ и бензиловый спирт (30 мл). Температуре давали медленно повышаться до 8 С за два часа. Реакционную смесь переливали в 500 мл льда и экстрагировали 500 мл МТВЕ. Органический слой промывали 250 мл рассола,сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 193 г раствора целевого продукта в бензиловом спирте. В соответствии с данными HPLC анализа, было установлено, что раствор содержал 25,96 г целевого продукта. 300 МГц 1H ЯМР (CDCl3)1,22 (м, 6 Н),1,55 (2 Н), 2,18 (м, 1 Н), 3,15 (м, 1 Н), 3,40 (м, 1 Н); 3,51 (м, 2 Н), 3,70 (м, 2 Н), 4,25 (м, 2 Н), 4,63 (т,1 Н), 4,90 (широкий с, 2 Н), 5,25 (м, 1 Н), 5,58 (с,2 Н), 7,35 (м, 3 Н), 7,51 (м, 2 Н), 7,72 (с, 1 Н).g) Получение соединения, указанного в заголовке. В 1-горлую круглодонную колбу емкостью 100 мл загружали сырой продукт со стадии f) примера 1 (9,65 г раствора в бензиловом спирте,содержащего 1,30 г, 3,13 ммоля продукта стадииf) примера 14 (растворенного в абсолютированном EtOH (20 мл). К раствору добавляли 0,45 г 10% Pd/C, суспендированного в 5 мл абсолютированного EtOH. Реакционную колбу откачивали и из баллона с водородом трижды заполняли Н 2. В реакционной колбе создавали давление H2,равное 1 атм. и реакционную смесь перемешивали в течение ночи. Реакционную смесь фильтровали через слой диатомовой земли для удаления Pd/C. Летучие продукты удаляли в вакууме. Остаток смешивали с 25 мл изопропилацетата и затем концентрировали в вакууме. Остаток разбавляли EtOAc (10 мл), вводили затравку целевого продукта, нагревали до дефлегмации и затем добавляли CH3CN (2 мл) и МТВЕ (35 мл). Затем полученную смесь перемешивали в течение 30 мин. Осадок отфильтровывали и сушили до постоянного веса, с получением 600 мг целевого продукта. 300 МГц 1H ЯМР(d2-DМSO)1,16 (м, 6 Н),1,45 (м, 1 Н), 1,61 (м, 1 Н), 2,16 (м, 1 Н), 3,45 (м,2 Н), 3,40 (м, 1 Н), 3,62 (м, 2 Н), 4,02 (м, 2 Н), 4,53(с, 1 Н), MS = (М+Н)+ = 416 (Cl). Аналогичным способом, исходя из соответствующих 4,4-дибензокси-2-этоксикарбонилбутирата и 4-алкилендиокси-2-этоксикарбонилбутирата, получают заявляемые промежуточные соединения, в которых R6 и R7 является бензилом или вместе образуют группы -CH2-CH2-,-CH2CH2CH2-, -CH2-CH2-CH2-CH2-. Пример 2. Получение (R)-9-[4-гидрокси-2(стеароилоксиметил)бутил]гуанина. а) Получение 19 В 1-горлую круглодонную колбу емкостью 25 мл загружали (R)-9-[4-диэтокси-2-(гидроксиметил)бутил]гуадина, полученного по примеру 1 (0,650 г, 2,0 ммоля), пиридин (4 мл) и CH2Cl2(2 мл), ДМАР (10 мг). Полученную смесь охлаждали до -5 С и в течение 5 мин добавляли стеароилхлорид (790 мг, 2,6 ммоля), растворенный в СН 2 Сl2 (0,5 мл). Полученную в результате смесь перемешивали в течение 16 ч при -5 С. Добавляли абсолютированный этанол (0,138 г,3,0 ммоля) и смесь перемешивали еще в течение 1 ч. Затем реакционную смесь концентрировали в вакууме. К остатку добавляли толуол (30 мл) и затем смесь концентрировали в вакууме. К полученному остатку снова добавляли толуол (30 мл) и смесь концентрировали в вакууме. К полученному остатку добавляли 1% КН 2 РO4 (25 мл) и эту смесь экстрагировали СН 2 Сl2 (60 мл). Органический слой отделяли и сушили надNа 2SO4, фильтровали и концентрировали в вакууме до постоянного веса 1,65 г. Сырой продукт подвергали хроматографической очистке на 40 г SiO2, проводя элюирование смесьюCH2Cl2 - EtOH в соотношении 95/5, в результате чего получали 367 мг целевого продукта. 300 МГц 1H ЯМР (СDСl3)0,89 (т, 3 Н),1,26 (м, 30 Н), 1,65 (м, 3 Н), 2,32 (м, 1 Н), 3,45 (м,1 Н), 3,60 (м, 2 Н), 4,08 (м, 2 Н), 4,60 (м, 1 Н), 6,0 В 1-горлую круглодонную колбу емкостью 25 мл загружали продукт со стадии а) примера 2(0,234 г, 0,394 ммоля), растворенный в ТГФ (1,7 мл). В этот раствор добавляли трифликовую кислоту (0,108 г) в 180 мг H2O. Полученную смесь перемешивали в течение ночи при комнатной температуре. К реакционной смеси добавляли насыщенный раствор NаНСО 3 (10 мл),ТГФ (5 мл), СН 2 Сl2 (2 мл) и NaBH4 (0,10 г). Смесь перемешивали в течение 30 мин. К реакционной смеси добавляли 5% раствор КН 2 РO4(30 мл). Полученную смесь экстрагировали 2 х 15 мл CH2Cl2. Органические слои объединяли и сушили над Na2SO4, фильтровали и концентрировали в вакууме. Пример 3. Получение (R)-9-[4-(N-Cbz-Lвалилокси)-2-(стеароилоксиметил)бутил]-гуанина. Раствор N-Cbz-L-валина (169 г, 0,67 моля) в сухом ТГФ (750 мл) готовили в 2-х литровой колбе, снабженной механической мешалкой,термометром и капельной воронкой. В течение 5 мин добавляли раствор дициклогексилкарбодиимида (69,3 г, 0,34 моля) в ТГФ (250 мл) и полученную в результате суспензию перемешивали при 205 С в течение 2 ч. Суспензию 20 фильтровали и осадок на фильтре промывали ТГФ (300 мл). Фильтрат и промывные жидкости загружали в 3-литровую колбу, снабженную мешалкой и термометром. Продукт примера 2(116 г, 0,22 моля) добавляли в виде твердого вещества, споласкивая реактор ТГФ (250 мл). Добавляли 4-(N,N-диметиламино)пиридин (2,73 г, 0,022 моля) и белый шламм перемешивали при 205 С. За 15 мин твердые вещества полностью растворялись, и реакция завершалась за 1 ч(о чем судили по данным HPLC: колонка 4,6 х 250 мм Zorbax RxC8; 85:15 ацетонитрил - 0,2% вод. НСlO4, добавление со скоростью 1 мл/мин; УФ детекция при 254 нм; исходный материал элюирует через 4,1 мин, а продукт реакции через 5,9 мин). Реакцию гасили добавлением воды (5 мл) и полученный раствор концентрировали в вакууме, в результате чего оставалось светло-желтое полутвердое вещество. Это вещество переносили в метанол (1,5 л) и нагревали до начала дефлегмации в течение 30 мин. Полученный раствор охлаждали до 25 С и осадок удаляли фильтрацией. Фильтрат концентрировали в вакууме, в результате чего оставалось вязкое, бледно-желтое масло. Добавляли ацетонитрил (1 л) и полученную в результате белую суспензию перемешивали при 205 С в течение 90 мин. Сырой твердый продукт выделяли фильтрацией, промывали ацетонитрилом (2 х 100 мл) и сушили на воздухе в течение ночи с получением желаемого продукта в виде воскообразного липкого твердого вещества (122 г). Это вещество очищали перекристаллизацией из этилацетата (500 мл) и сушили в вакууме при 30 С с получением целевого продукта в виде белого, воскообразного твердого вещества (104 г). Пример 4. Получение(R)-9-[4-(Lвалилокси)-2-(стеароилоксиметил)бутил]гуанина. Раствор, полученный в примере 3 (77 г) в теплом (40 С) этаноле (2,3 л) загружали в реактор гидрирования совместно с 5% Pd-C (15,4 г). Полученную смесь перемешивали при 40 С при давлении водорода 40 фунтов/дюйм 2 (2,8 кг/см 2) в течение 4 ч, откачивали и гидрировали еще в течение 4-10 ч. Катализатор удаляли фильтрацией и фильтрат концентрировали в вакууме с целью удаления белого твердого вещества. Это вещество перемешивали с этанолом (385 мл) при 45 С в течение 1 ч, затем охлаждали до 0 С и фильтровали. Осадок на фильтре сушили воздухом,затем в вакууме при 35 С с получением целевого соединения в виде белого порошка (46 г). Пример 5. (R)-9-[2-(L-валилоксиметил)-4(стеароилокси)бутил]гуанин. а) (R)-9-[2-(N-Boc-L-валилоксиметил)-4(стеароилокси)бутил]гуанин. Смесь N-Boc-L-валина (528 мг; 2,1 ммоля) и N,N'-дициклогексилкарбодиимида (250 мг; 1,21 мг) в дихлорметане (20 мл) перемешивали в течение ночи при комнатной температуре, дициклогексилмочевину отфильтровывали и экст 21 рагировали небольшим объемом дихлорметана,и фильтрат выпаривали в вакууме до небольшого объема. Добавляли (R)-9-[2-гидроксиметил-4(стеароилокси)бутил]гуанин (340 мг; 0,654 ммоля), 4-диметиламинопиридин (25 мг; 0,205 ммоля) и сухой N,N-диметилформамид (15 мл) и полученную смесь перемешивали в течение 4 ч при 50 С в атмосфере азота. Растворитель выпаривали в вакууме до небольшого объема. В результате колоночной хроматографии на силикагеле, и затем на оксиде алюминия (элюент этилацетат:метанол:вода в соотношении 15:2:1) получали 185 мг (39%) чистого целевого соединения в виде белого твердого вещества. Спектр 1H ЯМР (CDCl3) : 0,85-1,0 (м, 9 Н) 18-СН 3, СН(СН 3)2; 1,25 (с, 28 Н) 4-17-СН 2; 1,44(2,0 г) добавляли к (R)-9-[2-(N-Boc-Lвалилоксиметил)-4-(стеароилокси)бутил]гуанину (180 мг; 0,25 ммоля) и полученный раствор выдерживали при комнатной температуре в течение 1 ч, выпаривали до небольшого объема и дважды лиофилизировали с диоксаном до получения белого аморфного порошка. Выход целевого соединения, полученного в трифторацетатной соли, был количественным. Спектр 1H ЯМР (DMSO-d6) : 0,87 (т, 3 Н) 18-СН 3, 0,98 (дд, 6 Н) СН(СН 3)2; 1,25 (с, 28 Н) 417-СН 2; 1,50 (квинтет, 2 Н) 3-СН 2; 1,68 (кв, 2 Н) 3'-СН 2; 2,19 (м, 1 Н) 2'-СН; 2,26 (т, 2 Н) 2-СН 2; 2,40 (м, 1 Н) СН(СН 3)2; 3,9-4,25 (м, 7 Н) С 1'-СН 2,С 2-СН 2, С 4-СН 2, СН; 6,5 (широкий с, 2 Н) гyaNH2; 7,79 (с, 1 Н) гуаН 8; 8,37 (широкий с,3 Н) NН 3+; 10,73 (широкий с, 1 Н) гyaNH. Спектр 13 Н ЯМР (DMSO-d6) :14,2 (С 18); 17,9/18,3 (2 Val СН 3); 22,3 (С 17); 24,6 (C3); 27,7(100 мг) и IN HCl (520 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч при 1 атм. H2. Реакционную смесь фильтровали и растворитель выпаривали из фильтрата с получением целевого продукта в виде кристаллического твердого вещества (300 мг). Рецептурный пример А. Препарат в виде таблетки Следующие ингредиенты просеивали через сито с размером отверстий 0,15 мм и перемешивали в сухом состоянии: 10 г (R)-9-[2-(стеароилоксиметил)-4-(Lвалилокси)бутил]гуанина 40 г лактозы 49 г кристаллической целлюлозы 1 г стеарата магния Таблетировочную машину использовали для прессования смеси в таблетки, содержащие 250 мг активного ингредиента. Рецептурный пример В. Покрытая энтеросолюбильной оболочкой таблетка Таблетки рецептурного примера А покрывали распылением в машине для нанесения покрытий раствором, содержащим 120 г этилцеллюлозы 30 г пропиленгликоля 10 г моноолеата сорбита в 1000 мл дистиллированной воды. Рецептурный пример С. Препарат с контролируемым высвобождением 50 г (R)-9-[2-(стеароилоксиметил)-4-(Lвалилокси)бутил]гуанина,12 г гидроксипропилметилцеллюлозы(Methocell K15),4,5 г лактозы перемешивали в сухом состоянии и гранулировали с водной пастой повидона. Добавляли стеарат магния (0,5 г) и смесь прессовали в таблетировочной машине в таблетки диаметром 13 мм, содержащие 500 мг активного агента. Рецептурный пример D. Мягкие капсулы 250 г R)-9-[2-(стеароилоксиметил)-4-(Lвалилокси)бутил]гуанина 100 г лецитина 100 г арахисового масла Активный ингредиент диспергировали в лецитине и арахисовом масле и заполняли полученной смесью мягкие желатиновые капсулы. Биологический пример 1. Тестирование на биодоступность на крысах Биодоступность соединения формулы I,полученного из промежуточного соединения настоящего изобретения сравнивали с родственным соединением H2G и производными H2G на крысиной модели. Соединения изобретения и родственные соединения применяли перорально(с помощью катетера вставленного в желудок) на группах из трех индивидуально взвешенных животных, получавших по 0,1 ммоль/кг растворенного пролекарства в водном растворе, на основе арахисового масла или пропиленгликолевом носителе, в зависимости от растворимости активного ингредиента испытуемого соединения. Животных лишали питания в период от 5 ч до применения до, примерно, 17 ч после применения и держали в метаболических клетках. Мочу собирали в течение 24 ч после введения препарата и замораживали до анализа. H2G анализировали в моче с использованием HPLC/УФ анализа по методу Sthle, berg, AntimicrobAgents Chemother, 362, 339-342 (1992), модифицированного следующим образом: образцы после оттаивания разбавляли в соотношении 1:100 дистиллированной Н 2O и фильтровали через фильтр Амикон при центрифугировании со скоростью 3000 об./мин. в течение 10 мин. Дублированные 30 мкл образцы подвергали хроматографическому разделению на HPLC колонке; Zorbax SB-C18; 75-4,6 мм; 3,5 микронов; подвижная фаза 0,05 М NH4PO4, 3-4% метанола, рН 3,3-3,5; 0,5 мл/мин.; 254 нм, время удерживания H2G при концентрации МеОН 4 % и рН 3,33 - 12,5 мин. Биодоступность рассчитывали, как измеренное H2G выделение из каждого животного, усредненное, по крайней мере, от трех животных и выражали как процент усредненного 24 часового выделения H2G из мочи от группы из 4 индивидуально взвешенных крыс,соответственно, получавших внутривенные инъекции по 0,1 ммоль/кг H2G в буферном растворе Рингера, проводя анализ, как описано выше. Соединение в сравнительных примерах получали аналогично описанным в настоящей заявке. Полученные результаты приведены в следующей табл. 2. СоединениеR2 Сравнительный Водород пример 1 Сравнительный Валил пример 2 Сравнительный Валил пример 3 Пример по Валил изобретению Сравнительный Стеароил пример 4 Пример по Валил изобретению Пример по Валил изобретению Пример по Валил изобретению Сравнительный Валил пример 5 Пример по Валил изобретению Пример по Валил изобретению 24 Пример по изобретению Пример по изобретению Пример по изобретению Пример по изобретению Пример по изобретению Пример по изобретению Пример по изобретению Сравнительный пример 6 Сравнительный пример 7 Сравнительный пример 8 Сравнительный пример 9 Сравнительный пример 10 Сравнение биодоступности соединений формулы I со сравнительными примерами показывает, что специфическая комбинация жирных кислот по R1/R2 с аминокислотами по R1/R2 обеспечивает значительно более высокие значения биодоступности, чем соответствующие диаминокислотный сложный эфир или сложный эфир двуосновной жирной кислоты. Так, например, в рамках такой модели, соединение согласно изобретению -(R)-9-[2-(стероилоксиметил)-4-(L-валилокси)бутил]гуанин, демонстрирует на 55% лучшую биодоступность, чем соответствующий дивалиновый сложный эфир сравнительного примера 3, или соответственно, -(R)9-[2-(бутирилоксиметил)-4-(L-валилокси)бутил]гуанин, демонстрирует на 25% лучшую биодоступность, чем соответствующий дивалиновый эфир. Также совершенно очевидно, например, из сравнительных примеров 5, 6 и 7, что только специальные жирные кислоты настоящего изобретения в комбинации с конкретными аминокислотами обеспечивают такое неожиданное увеличение фармакокинетических параметров. Биологический пример 2. Концентрации плазмы у крыс Анализ концентрации плазмы проводили на самцах Sprague Dawley крыс. Животных лишали пищи в течение ночи перед введением препарата, но обеспечивали свободный доступ к воде. Каждое из испытуемых соединений готовили в виде раствора/суспензии в пропиленгликоле с концентрацией, соответствующей 10 мг Н 2G/мл и встряхивали при комнатной температуре в течение восьми часов. Группы крыс (по крайней мере, по 4 крысы в каждой группе) получали оральную дозу в 10 мг/кг (1 мл/кг) каждого из соединений; каждую дозировку применяли через желудочный зонд. Через определенные промежутки времени после приема дозы после дозирования) из хвостовой вены каждого животного получали гепаринизированные образцы крови (0,4 мл/образец). Эти образцы крови немедленно охлаждали на бане со льдом. В течение двух часов сбора, плазму отделяли от эритроцитов центрифугированием и замораживали до последующего анализа. Интересующие компоненты отделяли от белков плазмы осаждением в ацетонитриле. После лиофилизации и реконструкции, концентрации плазмы определяли обратимо-фазной HPLC с флуоресцентной детекцией. Оральное поглощение H2G и других испытуемых соединений определяли путем сравнения площади H2G под кривой для оральной дозировки с соответствующим значением,полученным при введении внутривенной дозыH2G в 10 мг/кг, которую применяли на отдельной группе крыс. Полученные результаты представлены в приведенной выше табл. 1 В. Биологический пример 3. Биоприменимость на обезьянах Соединения согласно изобретению и сравнительному примеру 3 (см. биологический пример 1, приведенный выше) применяли р. о. через желудочный зонд на обезьянах cynomolgus. Применяемые растворы содержали Активный ингредиент согласно 150 мг растворенные в 6,0 изобретению -(R)-9-[2-(стероил- пропиленгликоля, что соотоксиметил)-4-(L-валилокси)буветствует 25 мг/кг или тил]гуанин 0,0295 ммоль/кг Сравнительный пример 3 164 мг растворяли в 7,0 мл воды, что соответствует 23,4 мг/кг или 0,0295 ммоль/кг Образцы крови отбирали через 30 мин, 1,2, 3, 4, 6, 10 и 24 ч. Плазму отделяли центрифугированием со скоростью 2500 об./мин. и образцы инактивировали при 54 С в течение 20 мин перед тем, как их замораживали до последующих анализов. Уровни содержания H2G в плазме регистрировали с помощью HPLC/УФ анализа, как описано выше в примере 30. На фиг. 1 изображено выделение H2G плазмы, как функция времени. Хотя невозможно сделать статистически значимые выводы из единичных опытов на животных, можно предположить, что животные, получившие соединение настоящего изобретения, испытывают, в некоторой степени, более быстрое и более сильное воздействие H2G, чем животные, на которых применяли альтернативное пролекарство 26 Биологический пример 4. Антивирусная активность Мыши зараженные вирусом-1 простого герпеса (HSV-1) служили моделью для определения эффективности антивирусных агентов invivo. На мышах внутрибрюшинно инокулированных HSV-1, при 1000-кратном значенииLD50, применяли рецептуру, содержащую,выпускаемый в настоящее время антигерпесный агент ацикловир (21 и 83 мг/кг в 2% пропиленгликоля в носителе, представляющем собой стерильную воду трижды в день, перорально), или соединение примера 29 (21 и 83 мг/кг в 2% пропиленгликоля, в носителе, представляющем собой стерильную воду, трижды в день, перорально), в течение 5 последовательных дней, начиная применение через 5 дней после инокулирования. Ежедневно оценивали смертность животных. Полученные результаты изображены на фиг. 2, где представлена зависимость степени выживания от времени. В надписи к рисунку соединение изобретения обозначено, как Ex.29, а ацикловир обозначен, как ACV. Процентное количество мышей, переживших заражение HSV-1, оказалось значительно большим в случае применения на них данной дозировки соединения изобретения, по сравнению с применением эквивалентной дозы ацикловира. Представленный выше материал приведен только для иллюстрации, и настоящее изобретение не ограничивается содержанием этого материала. Подразумевается, что различные варианты и изменения, которые очевидны для специалиста в данной области, охватываются объемом притязаний изобретения, как это отражено в прилагаемой формуле изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы в котором R6 и R7 являются С 1-7 алкилом или бензилом или R6 и R7 вместе являются -СН 2 СН 2- или -СН 2 СН 2 СН 2-, -СН 2 СН 2 СН 2 СН 2- и R9 представляет собой водород или спиртзащитную группу. 2. Соединение по п.1, в котором R9 является водородом. 3. Соединение по п.1, в котором спиртзащитной группой является бензильная группа. 4. Соединение по п.1 или 2, в котором R6 и
МПК / Метки
МПК: C07D 473/18, A61K 31/522, A61P 31/22
Метки: получения, соединения, нуклеозидов, промежуточные, ациклических
Код ссылки
<a href="https://eas.patents.su/15-2809-promezhutochnye-soedineniya-dlya-polucheniya-aciklicheskih-nukleozidov.html" rel="bookmark" title="База патентов Евразийского Союза">Промежуточные соединения для получения ациклических нуклеозидов</a>
Производные ациклических нуклеозидов.

Номер патента: 1404
Опубликовано: 26.02.2001
Авторы: Энгельхардт Пер, Йоханссон Нильс Гуннар, Линдборг Бьерн, Жоу Ксиао-Ксионг, Хегберг Марита
МПК: C07D 473/18, A61P 31/12, A61K 31/52...
Метки: нуклеозидов, производные, ациклических
Формула / Реферат:
1. Соединение формулы I в которой a) R1 представляет собой -С(О)СН(СН(СН3)2NH2 или -С(O)СН(СН(СН3)СH2СН3)NН2, a R2 представляет собой -С(O)С3-С21 насыщенный или мононенасыщенный, необязательно замещенный гидроксигруппой, С1-6алкилом, С1-6алкокси-, С1-6алкоксиС1-6алкилом, С1-6алканоилом, амино-, галоид-, циано-, азидо-, меркапто- и нитрогруппой алкил; b) R1 представляет собой -С(O)С3-С21 насыщенный или мононенасыщенный, необязательно...
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61P 5/30, C07D 471/00, A61K 31/35...
Метки: соединения, замещенные, лечения, способы, четырехциклические, конденсированные, методы, гетероатомами, получения, промежуточные, композиции, арилом
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Кроуелл Томас А., Брайант Генри У., Джонс Чарльз Д., Палковиц Алан Д.
МПК: C07C 47/546, A61K 31/33, A61P 19/10...
Метки: промежуточные, снижения, получения, соединения, соединений, нафтильных, применение, холестерина, нафтильные, способ
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Икрами Хуссейн М., Шен Вей Чанг
МПК: C07H 19/048, A61K 31/44, C07D 213/70...
Метки: промежуточные, получения, конюгатов, тканях, жирной, млекопитающего, производного, сохранения, абсорбции, повышения, способ, сульфгидрильную, группу, конъюгаты, крови, пролонгированного, содержащего, способы, соединения, кислоты
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
Способы и промежуточные соединения для получения замещенных производных хроманола

Номер патента: 2204
Опубликовано: 28.02.2002
Авторы: Хокинс Джоэл М., Кейрон Стивен, Пископио Энтони Д., Кэстэлди Майкл Дж., Рэггон Джеффри В., Раггери Сэлли Г., Даггер Роберт В., Келли Сара И.
МПК: C07D 311/22, C07F 5/04, C07C 49/245...
Метки: соединения, замещенных, промежуточные, способы, получения, хроманола, производных
Формула / Реферат:
1. Способ получения соединения формулы или энантиомера указанного соединения, где в указанном соединении формулы Х группировка R3-замещенной бензойной кислоты присоединена по атомам углерода 6 или 7 хроманового кольца; R1 представляет собой -(CH2)qCHR5R6, где q является числом от 0 до 4; каждый R2 и R3 независимо выбран из группы, которую составляют Н, фторо, хлоро, (С1-С6)алкил, (С1-С6)алкокси, фенилсульфинил, фенилсульфонил и...
Предыдущий патент: Бетаины, используемые в качестве адъювантов для проверки чувствительности и антимикробной терапии
Следующий патент: Способ изготовления цепочки и цепочка, изготовленная таким способом
Случайный патент: Быстрорастворимый везикулярный продукт