Гетероциклические азотсодержащие соединения, их применение и содержащая их фармацевтическая композиция
Номер патента: 21799
Опубликовано: 30.09.2015
Авторы: Ледуссаль Бенуа, Пьер Камилль, Роулендс Дэвид, Рено Эмили, Гурдель Мари-Эдит, Кебси Адель, Лампила Максим














Формула / Реферат
1. Соединение, выбранное из группы, состоящей из
транс-8-(аминометил)-2-карбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-2-диметилкарбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-2-метилкарбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-1-(2-аминоэтил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-2-(2-аминоэтил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-2-(2-пиридинил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7Н-пиразоло[3,4-е][1,3]диазепин-6(5Н)-она,
транс-8-(аминометил)-5,6-дигидро-6-оксо-5-(сульфоокси)-4,7-метано-4Н-пиразоло[3,4-е][1,3]диазепин-2(8Н)уксусной кислоты,
транс-8-(аминометил)-5,6-дигидро-6-оксо-5-(сульфоокси)-4,7-метано-4Н-пиразоло[3,4-е][1,3]диазепин-2(8Н)ацетамида,
или его фармацевтически приемлемая соль.
2. Применение любого из соединений по п.1 в изготовлении лекарственного средства, используемого в качестве антибактериального средства.
3. Фармацевтическая композиция, содержащая лекарственное средство, как оно определено в п.2, в качестве действующего начала.
Текст
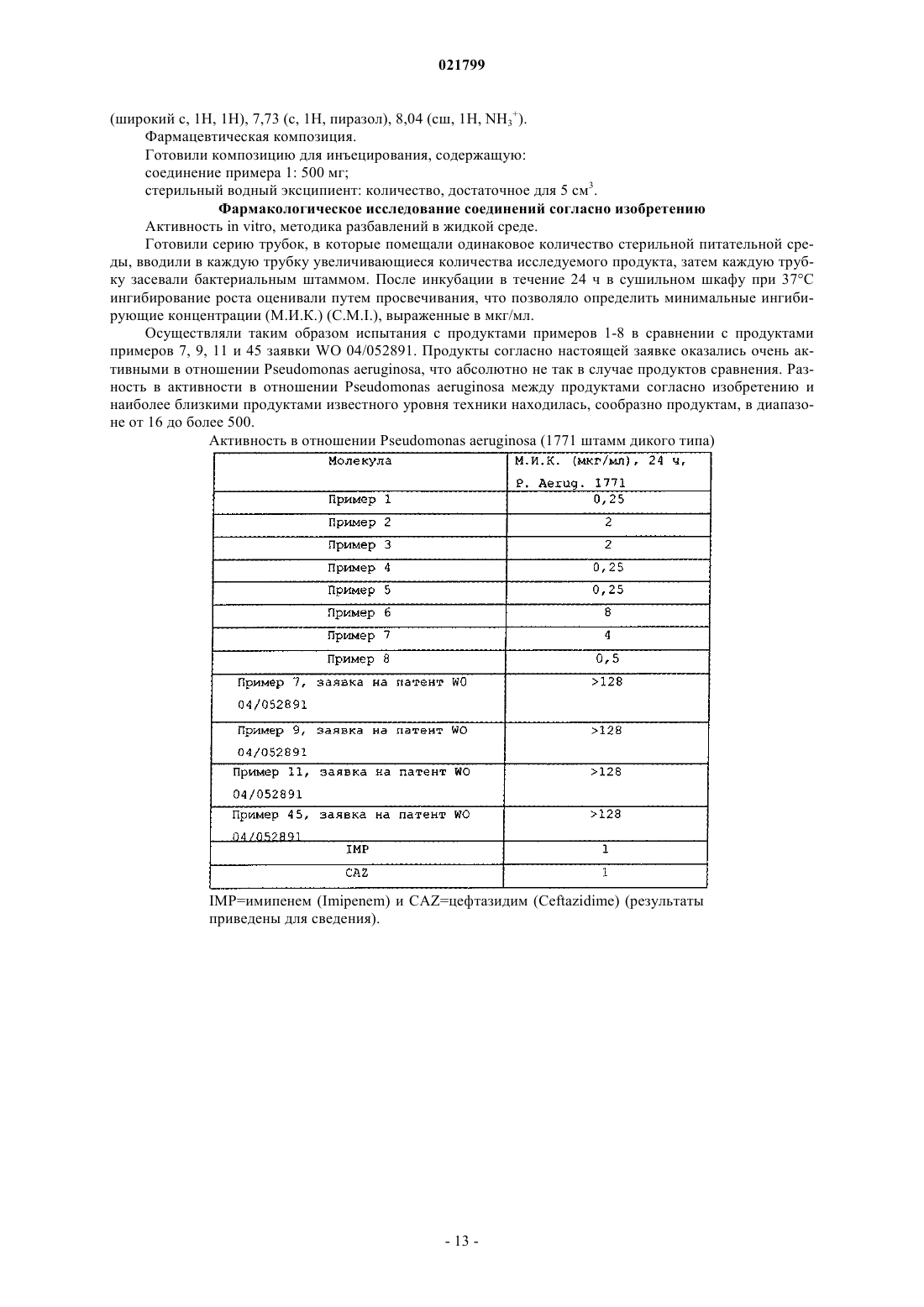
ГЕТЕРОЦИКЛИЧЕСКИЕ АЗОТСОДЕРЖАЩИЕ СОЕДИНЕНИЯ, ИХ ПРИМЕНЕНИЕ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ Изобретение касается гетероциклических азотсодержащих соединений, указанных в описании изобретения, в свободной форме и в форме цвиттерионов и солей с фармацевтически приемлемыми основаниями и неорганическими или органическими кислотами, их получения и их применения в качестве антибактериальных лекарственных средств.(71)(73) Заявитель и патентовладелец: АСТРА ЗЕНЕКА ХОЛДИНГ ФРАНС САС (FR) Изобретение касается гетероциклических азотсодержащих соединений, их получения и их применения в качестве антибактериальных лекарственных средств. В заявке WO 04/052891 описаны, в частности, соединения, отвечающие следующей формуле:R выбран из группы, образованной алкильным радикалом, содержащим от 1 до 6 атомов углерода,возможно, замещенным одним или несколькими атомами галогена или пиридильным радикалом, радикалом -СН 2-алкенил, содержащим в общей сложности от 3 до 9 атомов углерода, полиалкоксиалкильной группой, содержащей от 1 до 4 атомов кислорода и от 3 до 10 атомов углерода, арильным радикалом,содержащим от 6 до 10 атомов углерода, или аралкильным радикалом, содержащим от 7 до 11 атомов углерода, при этом цикл арильного или аралкильного радикала, возможно, замещен радикалом ОН, NH2,NO2, алкилом, содержащим от 1 до 6 атомов углерода, алкоксигруппой, содержащей от 1 до 6 атомов углерода, или одним или несколькими атомами галогена;R6 и R7 индивидуально выбраны из группы, образованной атомом водорода, алкильным радикалом,содержащим от 1 до 6 атомов углерода, алкоксигруппой, содержащей от 1 до 6 атомов углерода, арильным радикалом, содержащим от 6 до 10 атомов углерода, аралкильным радикалом, содержащим от 7 до 11 атомов углерода, и алкильным радикалом, содержащим от 1 до 6 атомов углерода, замещенным пиридильным радикалом;R3 и R4 вместе образуют фенил или 5- или 6-членный гетероцикл ароматической природы, содержащий от 1 до 4 гетероатомов, выбранных среди азота, кислорода и серы, замещенный одной или несколькими группами R', при этом R' выбран из группы, образованной радикалами -(O)a-(CH2)b-(O)aCONR6R7, -(O)a-(CH2)b-OSO3H, -(O)а-(CH2)b-SO3H, -(O)a-SO2R, -(O)a-SO2-CHal3, -(O)a-(CH2)b-NR6R7, -(O)a(CH2)b-NH-COOR, -(CH2)b-COOH, -(CH2)b-COOR, -OR", OH, -(СН 2)b-фенил и -(CH2)b-гетероцикл, 5- или 6-членный, ароматической природы, содержащий от 1 до 4 гетероатомов, выбранных среди азота, кислорода и серы, при этом фенил и гетероцикл, возможно, замещены одним или несколькими галогенами,алкилом, содержащим от 1 до 6 атомов углерода, алкоксигруппой, содержащей от 1 до 6 атомов углерода, или CF3, при этом R, R6 и R7 являются такими, как определенные перед этим, R" выбран из группы,образованной алкильными радикалами, содержащими от 1 до 6 атомов углерода, замещенными одной или несколькими гидроксильными группами, защищенными гидроксильными группами, оксогруппой,галогеном или цианогруппой, причем а равно 0 или 1, b является целым числом в интервале от 0 до 6,разумеется, что, когда R' представляет собой ОН, R1 обозначает радикал CONR6R7, в котором R6 или R7 представляет собой алкоксигруппу, содержащую от 1 до 6 атомов углерода;R2 выбран из группы, образованной атомом водорода, атомом галогена и радикалами R, S(O)mR,OR, NHCOR, NHCOOR и NHSO2R, при этом R является таким, как определенный перед этим, m равно 0,1 или 2;X обозначает двухвалентную группу -C(O)-В-, связанную с атомом азота посредством атома углерода, В обозначает двухвалентную группу -О-(СН 2)n"-, связанную с карбонилом посредством атома кислорода, группу -NR8-(CH2)n"- или -NR8-O-, связанную с карбонилом посредством атома азота, n" равно 0 или 1, и R8 выбран из группы, образованной атомом водорода, радикалами ОН, R, OR, Y, OY, Y1, OY1,Y2, OY2, Y3, O-CH2-CH2-S(O)m-R, SiRaRbRc и OSiRaRbRc, при этом Ra, Rb и Rc обозначают индивидуально алкильный радикал, линейный или разветвленный, содержащий от 1 до 6 атомов углерода, или арильный радикал, содержащий от 6 до 10 атомов углерода, при этом R и m являются такими, как определенные перед этим;n равно 1 или 2,а также соли этих соединений с основаниями или неорганическими или органическими кислотами. Асимметрические атомы углерода, содержащиеся в соединениях формулы (I), могут независимо одни от других иметь конфигурацию R, S или RS, и соединения формулы (I) существуют, следовательно,в форме чистых энантиомеров, или чистых диастереоизомеров, или в форме смеси энантиомеров, в частности, рацемических смесей, или смесей диастереоизомеров. Кроме того, так как заместители Rl, R2 или R4, взятые по отдельности, с одной стороны, и X, с другой стороны, могут быть в положении цис и/или транс по отношению к циклу, с которым они связаны,соединения формулы (I) находятся в форме цис-изомеров, или транс-изомеров, или смесей. Кроме того, в заявке WO 02/100860 описаны родственные соединения. Заявитель обнаружил, что среди соединений, описанных в заявке 04/052891, некоторые особенные соединения, ни одно из которых не описано в экспериментальной части этой заявки, обладают совершенно неожиданными антибактериальными свойствами. Исключительное свойство соединений согласно изобретению заключается в том, что они демонстрируют отличную активность по отношению к Pseudomonas aeruginosa, бактериальному штамму, часто встречающемуся в нокозомиальных инфекциях, а также у пациентов, страдающих муковисцидозом. Эта интересная и неожиданная активность отсутствует у самых близких соединений, полученных в заявке WO 02/100860. Она проиллюстрирована дальше, в экспериментальной части. Кроме того, соединения согласно изобретению показывают себя активными на образцах животных инфекций, включая штаммы, по обыкновению устойчивые к обычно используемым антибиотикам. Соединения согласно изобретению способны противодействовать основным механизмам устойчивости бактерий, которыми являются -лактамазы, выводящие насосы и мутации поринов. Согласно настоящему изобретению предложены следующие соединения: и их фармацевтически приемлемые соли. Согласно настоящему изобретению также предложено применение любого из соединений по настоящему изобретению в изготовлении лекарственного средства, используемого в качестве антибактериального средства. Согласно настоящему изобретению также предложена фармацевтическая композиция, содержащая определенное выше лекарственное средство в качестве действующего начала. Соединения по настоящему изобретению могут существовать в свободной форме и в форме цвиттерионов и солей с фармацевтически приемлемыми основаниями и неорганическими или органическими кислотами. Среди солей кислот соединений по настоящему изобретению можно назвать, между прочим, соли,образованные с неорганическими кислотами, такими как соляная, бромисто-водородная, йодистоводородная, серная или фосфорная кислоты, или с органическими кислотами, такими как муравьиная,уксусная, трифторуксусная, пропионовая, бензойная, малеиновая, фумаровая, янтарная, винная, лимонная, щавелевая, глиоксиловая, аспарагиновая кислоты, алкансульфоновыми кислотами, такими как метансульфоновая и этансульфоновая кислоты, арилсульфоновыми кислотами, такими как бензолсульфо-2 021799 кислота и паратолуолсульфокислота. Среди солей оснований соединений по настоящему изобретению можно назвать, между прочим,соли, образованные с неорганическими основаниями, такими как, например, гидроксид натрия, калия,лития, кальция, магния или аммония, или с органическими основаниями, такими как, например, метиламин, пропиламин, триметиламин, диэтиламин, триэтиламин, N,N-диметилэтаноламин, трис(гидроксиметил)аминометан, этаноламин, пиридин, пиколин, дициклогексиламин, морфолин, бензиламин, прокаин, лизин, аргинин, гистидин, N-метилглюкамин, или соли фосфония, такие как алкилфосфониевые, арилфосфониевые, алкиларилфосфониевые, алкениларилфосфониевые, или четвертичные аммониевые соли, такие как соль тетра-н-бутиламмония. Асимметрические атомы углерода, присутствующие в соединениях по настоящему изобретению,могут независимо одни от других иметь конфигурацию R, S или RS, и, следовательно, соединения по настоящему изобретению находятся в форме чистых энантиомеров, или чистых диастереоизомеров, или в форме смеси энантиомеров, в частности, рацематов, или смесей диастереоизомеров. Среди соединений по настоящему изобретению объектом изобретения, совсем конкретно, являются соединения, описанные далее в экспериментальной части, названия которых следуют ниже: в свободной форме, форме цвиттерионов и солей с фармацевтически приемлемыми основаниями и неорганическими или органическими кислотами, в форме их возможных изомеров или диастереоизомеров либо смесей. Как указано выше, соединения по настоящему изобретению обладают отличной антибиотической активностью по отношению к Pseudomonas aeruginosa, а также к образцам инфекций животных, вызванных штаммами, устойчивыми к действию обычно используемых антибактериальных средств. Эта замечательная и неожиданная антибиотическая активность не была обнаружена для соединений, описанных в заявке WO 04/052891, и, в частности, для структурно близких соединений. Это иллюстрируется дальше. Эти свойства делают указанные соединения, в свободной форме и в форме цвиттерионов, или в форме солей фармацевтически приемлемых кислот и оснований, пригодными к применению в качестве лекарственных средств для лечения тяжелых инфекций, вызванных Pseudomonas, в частности нозокомиальных инфекций и, вообще, основных инфекций у субъектов группы риска. В частности, такими инфекциями являются инфекции дыхательных путей, например острая пневмония или хронические инфекции нижних дыхательных путей, инфекции крови, например сепсисы, острые или хронические инфекции мочевых путей, инфекции слуховой системы, например злокачественный наружный отит или хронический гнойный отит, инфекции кожи или мягких тканей, например дерматиты, инфицированные раны,фолликулит, пиодермит, не поддающиеся лечению формы угрей, инфекции глаз, например язва роговицы, инфекции нервной системы, в частности менингиты и абсцесс мозга, сердечные инфекции, такие как эндокардит, инфекции костей и суставов, такие как стеносуставной пиоартроз, вертебральный остеомиэлит, лобковый симфизит, инфекции желудочно-кишечного тракта, такие как некрозирующий энтероколит и периректальные инфекции. Таким образом, объектом настоящего изобретения являются в качестве лекарственных средств, в частности антибиотических лекарственных средств, соединения по настоящему изобретению, такие как определенные выше, в свободной форме и в форме цвиттерионов и солей с фармацевтически приемлемыми основаниями и неорганическими или органическими кислотами. Среди соединений по настоящему изобретению, совсем конкретно, объектом изобретения, в качестве лекарственного средства, являются соединения, названия которых следуют ниже: в свободной форме, форме цвиттерионов и солей с фармацевтически приемлемыми основаниями и неорганическими или органическими кислотами, в форме их возможных изомеров или диастереоизомеров либо смесей. Объектом изобретения являются также фармацевтические композиции, содержащие в качестве действующего начала по меньшей мере одно из соединений согласно изобретению, таких как определенные выше. Эти композиции могут быть введены оральным, ректальным или парентеральным путем, в частности, внутримышечно, или местно, путем топического нанесения на кожу и слизистые оболочки. Композиции согласно изобретению могут быть твердыми или жидкими и могут находиться в фармацевтических формах, обычно используемых в медицине человека, как, например, простые или дражированные таблетки, желатиновые капсулы, гранулы, суппозитории, препараты, пригодные для инъецирования, мази, кремы, гели; их получают согласно обычным способам. Действующее начало или действующие начала могут быть введены в них с эксципиентами, обычно используемыми в этих фармацевтических композициях, такими как тальк, гуммиарабик, лактоза, крахмал, стеарат магния, масло какао, водные или неводные растворители, жиры животного или растительного происхождения, парафиновые производные, гликоли, различные смачивающие, диспергирующие или эмульгирующие средства, консерванты. Эти композиции могут находиться, в частности, в форме лиофилизата, предназначенного для растворения в подходящем растворителе перед самым употреблением, например стерильной апирогенной воде. Вводимая доза варьирует сообразно излечиваемому заболеванию, конкретному пациенту, пути введения и рассматриваемому продукту. Она может находиться, например, в интервале от 0,250 до 10 г в день для человека при пероральном приеме, с продуктом, описанным в примере 1, 4 или 5, или находиться в интервале от 0,25 до 10 г в день при внутримышечном или внутривенном введении. Продукты соединений по настоящему изобретению могут быть также использованы в качестве дезинфицирующих средств для хирургических инструментов. Следующие примеры иллюстрируют изобретение. Пример 1. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-карбамоил-4,8-дигидро-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Стадия А. Транс-8-(гидроксиметил)-4,8-дигидро-6-оксо-5-(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-он. Сложный эфир, транс-4,5,6,8-тетрагидро-6-оксо-5-(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-8-метилкарбоксилат, описанный в заявке WO 2004/052891 (пример 1, стадия К) (5 г, 15,2 ммоль), растворяли в безводной смеси метанол/тетрагидрофуран 1/1 (100 мл) в атмосфере азота. Затем по порциям добавляли NaBH4 (2,3 г, 60,9 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь обрабатывали 10%-ным водным раствором NaH2PO4 (100 мл). После выпаривания досуха реакционную смесь извлекали в воду. Образовавшийся осадок перемешивали в течение ночи во льду, затем фильтровали и сушили при пониженном давлении в присутствии P2O5 с получением желаемого соединения (3,30 г, 11,0 ммоль, 72%) в форме белого порошка. МС (ЭС(+: m/z [М+Н]+=301. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)=3,18-3,50 (АВХ, 2 Н, N-CH2-CH-N), 3,65-3,76 (АВХ, 2 Н, NCH-CH2-ОН), 4,34 (т, 1 Н, N-CH-CH2-OH), 4,46 (д, 1 Н, N-CH2-CH-N), 4,88 (с, 2 Н, CH2-Ph), 7,29-7,43 (м,5 Н, Ph), 7,66 (с, 1 Н, Н пиразол), 12,72 (широкий, 1 Н, ОН). Стадия В. 1,1-Диметил-транс-4,5,6,8-тетрагидро-6-оксо-5-(фенилметокси)-4,7-метано-7 Нпиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Спирт, полученный на стадии А примера 1 (1,73 г, 5,76 ммоль), растворяли в безводном пиридине(35 мл) в атмосфере азота при 0C, затем по каплям добавляли метансульфонилхлорид (1,78 мл, 23 ммоль). После 2 ч 30 мин перемешивания при комнатной температуре реакционную смесь обрабатывали насыщенным водным раствором хлорида аммония (100 мл), затем экстрагировали этилацетатом. Объединенные органические фазы промывали затем насыщенным водным раствором хлорида аммония, сушили, затем концентрировали при пониженном давлении с получением желаемого димезилзамещенного производного в форме желтого масла. Димезилзамещенный промежуточный продукт растворяли в безводном диметилформамиде (45 мл) в атмосфере азота в присутствии азида натрия (1,12 г, 17,3 ммоль). Реакционную смесь грели при 70C в течение 24 ч. Для того чтобы конверсия была полной, возможно, добавляли 1 экв. азида. Когда реакция завершалась полностью, смесь обрабатывали 10%-ным водным раствором NaH2PO4 (100 мл), затем экстрагировали дихлорметаном. Объединенные органические фазы сушили, затем концентрировали при пониженном давлении с получением желаемого азида в форме желтого масла. Промежуточный продукт в атмосфере азота вводили в реакцию в абсолютном этаноле (17,5 мл), затем последовательно добавляли ди-трет-бутилкарбонат (1,38 г, 6,34 ммоль), триэтилсилан (1,38 мл, 8,64 ммоль) и 10% гидроксида палладия на угле (52 мг). После ночи при комнатной температуре реакционную смесь фильтровали, затем концентрировали с получением неочищенного желтого масла. Этот сырой продукт очищали хроматографией на колонке с диоксидом кремния (градиентное элюированиеCH2Cl2/MeOH от 100/0 до 95/5 по 1%), что приводило к желаемому соединению (1,36 г, 3,40 ммоль, 34%) в форме белого твердого вещества. МС (ЭС(+: m/z [М+Н]+=401. 1H-ЯМР (400 МГц, МеОН-d4):(м.д.)=1,51 (с, 9 Н, С(CH3)3), 3,21-3,59 (м, 4 Н, N-CH2-CH-N et N-CHCH2-NHBoc), 4,36 (м, 1 Н, N-CH-CH2-OH), 4,46 (м, 1 Н, N-CH2-CH-N), 4,99 (АВ, 2 Н, CH2-Ph), 7,41-7,52 (м,5 Н, Ph), 7,63 (с, 1 Н, Н пиразол). Стадия С. 1,1-Диметилэтил-транс-2-карбамоил-4,5,6,8-тетрагидро-6-оксо-5-(фенилметокси)-4,7 метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. В атмосфере азота амин, полученный на стадии В примера 1 (100 мг, 0,250 ммоль), растворяли в дихлорметане (13 мл). При 0C добавляли триэтиламин (70 мкл, 0,500 ммоль), после чего по каплям быстро добавляли дифосген (45 мкл, 0,376 ммоль). После 2 ч 30 мин перемешивания при 0C быстро добавляли гидроксид аммония (20%-ный водный, 0,4 мл) и среду тщательно перемешивали при комнатной температуре в течение 1 ч. Среду переливали в ампулу для декантации, ополаскивали дихлорметаном (5 мл), затем промывали 10%-ным водным раствором фосфата натрия (10 мл). Водную фазу экстрагировали дихлорметаном (10 мл). Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили, затем концентрировали при пониженном давлении с получением после хроматографии на колонке с диоксидом кремния (элюент CH2Cl2/этилацетат 70/30) желаемого производного (94 мг, 0,212 ммоль, 85%) в форме бежевого твердого вещества. МС (ЭС(+: m/z [M+H]+=443. 1(с, 1 Н, Н пиразол). Стадия D. Пиридиниевая соль 1,1-диметилэтил-транс-2-карбамоил-4,5,6,8-тетрагидро-6-оксо-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамата. В атмосфере азота производное, полученное на стадии С (94 мг, 0,212 ммоль), растворяли в диметилформамиде (0,3 мл) и дихлорметане (0,9 мл), затем добавляли катализатор с 10% палладия на угле с концентрацией 50% в воде (68 мг, 0,032 ммоль). После продувки вакуум/азот реакционную смесь помещали в атмосферу водорода до исчезновения исходного продукта, регистрируемого ВЭЖХ. Тогда реакционную смесь концентрировали в вакууме, затем выпаривали совместно безводным дихлорметаном,наконец, сушили при пониженном давлении в присутствии Р 2 О 5 в течение 2 ч с получением желаемого дибензилзамещенного промежуточного продукта. Дибензилзамещенное производное извлекали в безводный пиридин (0,6 мл) в присутствии комплекса пиридин/триоксид серы (68 мг, 0,425 ммоль). Затем реакционную смесь перемешивали при комнатной температуре вплоть до полной конверсии, определяемой методом ВЭЖХ, затем выпаривали досуха после обработки добавлением воды. Реакционноспособ-5 021799 ный сырой продукт хроматографировали на колонке с диоксидом кремния (градиентное элюированиеCH2Cl2/MeOH от 100/0 до 80/20 по 5%) с получением желаемого продукта (50 мг, 0,093 ммоль, 43%) в форме белого твердого вещества. МС (ЭС(-: m/z [М-]=431. 1 Н-ЯМР (400 МГц, MeOH-d4):(м.д.)=1,52 (с, 9 Н, С(CH3)3), 3,41-3,53, 3,62-3,75 (м, 4 Н, N-CH2-CHN et CH-CH2-NHBoc), 4,64 (м, 1 Н, СН-CH2-NHBoc), 4,98 (д, 1 Н, N-CH2-CH-N), 8,00 (м, 2H, Py), 8,28 (с,1 Н, Н пиразол), 8,74 (м, 1 Н, Py), 8,95 (м, 2 Н, Py). Стадия Е. Натриевая соль 1,1-диметилэтил-транс-2-карбамоил-4,5,6,8-тетрагидро-6-оксо-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамата. Суспензию 6 г смолы DOWEX 50WX8 в растворе 2 н. гидроксида натрия (30 мл) перемешивали в течение 1 ч, затем выливали в хроматографическую колонку. Колонку кондиционировали деминерализованной водой до нейтрального pH, затем смесью вода/ТГФ 90/10. Производное, полученное на стадии D примера 1 (49 мг, 0,091 ммоль), растворяли в минимальном количестве метанола, наносили на колонку,затем элюировали смесью вода/ТГФ 90/10. Фракции, содержащие субстрат, объединяли, замораживали и лиофилизировали с получением желаемой натриевой соли (44 мг, 0,091 ммоль, 100%) в форме бежевого твердого вещества. МС (ЭС(-: m/z [М-Н]-=431. 1 Н-ЯМР (400 МГц, MeOH-d4):(м.д.)=1,52 (с, 9 Н, С(CH3)3), 3,41-3,53, 3,62-3,75 (м, 4 Н, N-CH2-CHN et CH-CH2-NHBoc), 4,64 (м, 1 Н, CH-CH2-NHBoc), 4,98 (д, 1 Н, N-CH2-CH-N), 8,29 (с, 1 Н, Н пиразол). Стадия F. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-карбамоил-4,8-дигидро-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Раствор трифторуксусной кислоты (2,4 мл) в дихлорметане (2,4 мл) в атмосфере азота по каплям добавляли к охлажденному до 0C раствору натриевой соли, полученной на стадии Е (42 мг, 0,092 ммоль), в дихлорметане (1,2 мл). Реакционную смесь поддерживали при перемешивании в течение 1 ч при комнатной температуре. Смесь выпаривали досуха и извлекали в воду с получением бежевого осадка. Осадок фильтровали, затем промывали этанолом с получением желаемого производного (12 мг, 0,026 ммоль, 28%) в форме бежевого твердого вещества. 1 Н-ЯМР (400 МГц, MeOH-d6):(м.д.)=3,18 (м, 1 Н, N-CH2-CH-N), 3,40-3,47 (м, 3H, N-CH2-CH-N et(широкий, 1 Н, CONH2), 8,09 (широкий, 3H, NH3+), 8,26 (с, 1 Н, Н пиразол). Пример 2. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-диметилкарбамоил-4,8 длгидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Стадия А. 1,1-Диметилэтил-транс-4,5,6,8-дигидро-2-диметилкарбамоил-6-оксо-5-(фенилметокси)4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Действуя, как указано на стадии С примера 1, использование производного, полученного на стадии В примера 1 (200 мг, 0,501 ммоль), дихлорметана (26 мл, триэтиламина (140 мкл, 1,00 ммоль), дифосгена(91 мкл, 0,751 ммоль) и диметиламина (водный, 40 мас.%, 0,634 мл, 5,01 ммоль) приводило после хроматографирования на колонке с диоксидом кремния (элюент CH2Cl2/MeOH 99/1) к желаемому производному (170 мг, 0,361 ммоль, 72%) в форме бежевого твердого вещества. МС (ЭС(+: m/z [M+H]+=471. 1H-ЯМР (400 МГц, CDCl3):(м.д.)=1,20 (с, 9 Н, С(CH3)3), 2,80 (дд, 1 Н, N-CH2-CH-N), 2,93 (с, 6 Н,N(CH3)2), 3,09 (м, 2 Н, CH-CH2-NHBoc, N-CH2-CH-N), 3,51 (м, 1 Н, CH-CH2-NHBoc), 3,74 (д, 1 Н, N-CH2CH-N), 4,33 (м, 1H, CH-CH2-NHBoc), 4,69 (AB, 2H, CH2-Ph), 4,90 (широкий, 1H, NH), 7,12-7,18 (м, 5 Н,Ph), 7,72 (с, 1 Н, H пиразол). Стадия В. Пиридиниевая соль 1,1-диметилэтил-транс-4,5,6,8-тетрагидро-2-диметилкарбамоил-6 оксо-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамата. Действуя, как указано на стадии D примера 1, использование производного, полученного на стадии А (176 мг, 0,374 ммоль), диметилформамида (0,5 мл), дихлорметана (1,6 мл) и катализатора с 10% палладия на угле с концентрацией 50% в воде (119 мг, 0,032 ммоль) приводило к желаемому дибензилзамещенному промежуточному продукту. Дибензилзамещенный промежуточный продукт, пиридин (1,1 мл) и комплекс пиридин/триоксид серы (119 мг, 0,748 ммоль) приводили, после хроматографирования на колонке с диоксидом кремния(градиентное элюирование CH2Cl2/MeOH от 100/0 до 80/20 по 5%), к желаемому производному (167 мг,0,309 ммоль, 83%) в форме бежевого твердого вещества. МС (ЭС(-: m/z [M-H]-=459. 1 Н-ЯМР (400 МГц, MeOH-d4):(м.д.)=1,52 (с, 9 Н, С(СН 3)3), 3, 23 (с, 6 Н, N(CH3)2, 3,41-3,53, 3,563,65 (м, 4 Н, N-CH2-CH-N et CH-CH2-NHBoc), 4,64 (м, 1 Н, CH-CH2-NHBoc), 4,98 (д, 1 Н, N-CH2-CH-N),8,07 (м, 2 Н, Ру), 8,20 (с, 1 Н, Н пиразол), 8,60 (м, 1 Н, Ру), 8,88 (м, 2 Н, Py). Стадия С. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-диметилкарбамоил-4,5,6,8 тетрагидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Действуя, как указано на стадии Е примера 1, использование производного, полученного на стадии В (167 мг, 0,309 ммоль), смолы DOWEX 50WX8 (20 г) и 2 н. гидроксида натрия (100 мл) приводило к ожидаемой натриевой соли (139 мг, 0,288 ммоль, 93%). Действуя, как указано на стадии F примера 1, натриевая соль (139 мг, 0,288 ммоль), дихлорметан (4 мл) и тетрафторуксусная кислота (7,9 мл) в дихлорметане (7,9 мл) приводили к неочищенному производному, которое извлекали в воду (приблизительно 2 мл), затем замораживали и лиофилизировали с получением желаемого производного (143 мг, 0,288 ммоль, 100%) в форме бежевого твердого вещества. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)-3,07 (с, 6 Н, N (CH3)2), 3,23-3,27, 3,37-3,42 (м, 4 Н, N-CH2-CHN et CH-CH2-NH3+), 4,68 (м, 1 Н, СН-СН 2-NH3+), 4,85 (д, 1 Н, N-CH2-CH-N), 8,11 (широкий, 3H, NH3+), 8,19(с, 1 Н, Н пиразол). Пример 3. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-метилкарбамоил-4,8 дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Стадия А. 1,1-Диметилэтил-транс-4,5,6,8-тетрагидро-2-метилкарбамоил-6-оксо-5-(фенилметокси)4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Действуя, как указано на стадии С примера 1, реакцию, вовлекающую производное, полученное на стадии В примера 1 (200 мг, 0,501 ммоль), дихлорметан (26 мл), триэтиламин (140 мкл, 1,00 ммоль), дифосген (91 мкл, 0,751 ммоль) и раствор метиламина (40 мас.% водный, 0,437 мл, 5,01 ммоль), повторяли 2 раза. Неочищенные продукты объединяли и получали, после хроматографирования на колонке с диоксидом кремния (CH2Cl2/AcOEt от 100/0 до 80/20), желаемое производное (170 мг, 0,372 ммоль, 60%). МС (ЭС(+: m/z [М+Н]+=457. 1 Н-ЯМР (400 МГц, CDCl3):(м.д.)=1,49 (с, 9 Н, С(СН 3)3), 3,02 (д, 3H, NH-CH3), 3,10 (АВ, 1 Н, N-CH2CH-N), 3,34-3,38 (м, 2 Н, N-CH2-CH-N et CH-CH2-NHBoc), 3,8 (широкий, 1 Н, CH-CH2-NHBoc), 4,00 (д,1 Н, N-CH2-CH-N), 4,56-4,60 (м, 1H, CH-CH2-NHBoc), 4,88-5,06 (AB, 2H, N-O-CH2-Ph), 5,10 (широкий, 1 Н,NH), 6,95 (широкий, 1 Н, NH), 7,42-7,75 (м, 5 Н, Ph), 8,07 (с, 1 Н, H пиразол). Стадия В. Пиридиниевая соль 1,1-диметилэтил-транс-4,5,6,8-тетрагидро-2-метилкарбамоил-6 оксо-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамата. Действуя, как указано на стадии D примера 1, использование производного, полученного на стадии А (160 мг, 0,350 ммоль), диметилформамида (0,51 мл), дихлорметана (1,52 мл), катализатора с 10% палладия на угле с концентрацией 50% в воде (112 мг, 0,052 ммоль) и гидрирование в течение 2 ч 15 мин приводило к желаемому дибензилзамещенному промежуточному продукту. Использование дибензилзамещенного промежуточного продукта, пиридина (1,0 мл) и комплекса пиридин/триоксид серы (111 мг, 0,699 ммоль) приводило после хроматографирования на колонке с диоксидом кремния (элюирование (CH2Cl2/MeOH от 100/0 до 80/20) к желаемому производному (120 мг,0,224 ммоль, 64%) в форме бежевого твердого вещества. МС (ЭС(+: m/z [М+Н]+=447 и (ЭС(-: m/z [М-Н]-=445. 1H-ЯМР (400 МГц, CDCl3):(м.д.)=1,48 (с, 9 Н, С(СН 3)3), 3,01 (д, 3H, NH-CH3), 3,25 (широкий, 1 Н,N-CH2-CH-N), 3,40 (широкий, 1 Н, CH-CH2-NHBoc), 3,7 (широкий, 1 Н, N-CH2-CH-N), 3,85 (широкий, 1 Н,СН-CH2-NHBoc) 4,60 (широкий, 1 Н, N-CH2-CH-N), 5,03 (с, 1 Н, СН-СН 2-NHBoc), 5,40 (широкий, 1 Н, NH),7,10 (широкий, 1 Н, NH), 7,87-7,91 (м, 2 Н, пиридин), 8,20 (с, 1 Н, Н пиразол), 8,36 (т, 1 Н, пиридин), 8,94 (д,2 Н, пиридин). Стадия С. Натриевая соль 1,1-диметилэтил-транс-4,5,6,8-тетрагидро-2-метилкарбамоил-6-оксо-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамата. Действуя, как указано на стадии Е примера 1, использование производного, полученного на стадии В (120 мг, 0,228 ммоль), осажденного в минимальном количестве воды, смолы DOWEX 50WX8 (20 г) и 2 н. гидроксида натрия (70 мл) приводило к желаемой натриевой соли (100 мг, 0,213 ммоль, 93%) в форме белого лиофилизата. МС (ЭС(-:m/z [М-Н]-=445. 1et CH-CH2-NHBoc), 4,60 (м, 1 Н, N-CH2-CH-N), 5,10 (с, 1 Н, CH-CH2-NHBoc), 8,23 (с, 1 Н, H пиразол). Стадия D. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-метилкарбамоил-4,8 дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Действуя, как указано на стадии F примера 1, использование натриевой соли, полученной на стадии С (94 мг, 0,2 ммоль), дихлорметана (3 мл) и трифторуксусной кислоты (2 мл) приводило к неочищенному производному, которое извлекали в воду (10 мл), затем замораживали и лиофилизировали. Получали желаемое производное (95 мг, 0,196 ммоль, 98%) в форме коричневого твердого вещества. МС (ЭС(-: m/z [М-Н]-=345 и ЭС(+: m/z [M+H]+=447. 1 Стадия А. Транс-1-(2-трет-бутоксикарбониламиноэтил)-4,5,6,8-тетрагидро-6-оксо-5(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-метилкарбоксилат. Транс-2-(2-трет-бутоксикарбониламиноэтил)-4,5,6,8-тетрагидро-6-оксо-5-(фенилметокси)-4,7 метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-метилкарбоксилат. Сложный эфир, транс-4,5,6,8-тетрагидро-6-оксо-5-(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-8-метилкарбоксилат, описанный в заявке WO 2004/052891 (пример 1, стадия К) (1,13 г,3,44 ммоль) растворяли в безводном диметилформамиде (4,0 мл) в присутствии карбоната калия (712 мг,5,16 ммоль) и 2-(Вос-амино)этилбромида (770 мг, 3,44 ммоль). Реакционную смесь грели при 55C. Дополнительные количества K2CO3 (2712 мг, 25,16 ммоль) и бромида (2770 мг, 23,44 ммоль) по истечении дополнительных 4 и 14 ч. Реакцию проводили еще 8 ч при 55C. Суспензию охлаждали, фильтровали и промывали этилацетатом. Органическую фазу промывали 10%-ным раствором винной кислоты,затем сушили и концентрировали при пониженном давлении. Сырой продукт очищали хроматографией на диоксиде кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 90/10), что приводило к производному, замещенному у N1 (380 мг, 0,81 ммоль, 23%), а также к изомеру, замещенному у N2 (475 мг,1,01 ммоль, 29%). Производное, замещенное у N1: МС (ЭС(+: m/z [М+Н]+=472. 1H-ЯМР (400 МГц, CDCl3):(м.д.)=1,45 (с, 9 Н, С(СН 3)3), 3,4 8-3,53 (м, 4 Н, N-CH2-CH-N, СН 2-СН 2NHBoc), 3,85 (с, 3H, СН 3), 3,97 (д, 1 Н, N-CH2-CH-N), 4,18 (м, 2 Н, СН 2 СН 2-NHboc), 4,95 (АВ, 2 Н, CH2-Ph),5,29 (с, 1 Н, СН-СО 2 Ме), 7,2 5 (с, 1 Н, Н пиразол), 7,38-7,4 3 (массив, 5 Н, Ph). Стадия В. Транс-1-(2-трет-бутоксикарбониламиноэтил)-8-(гидроксиметил)-4,5,6,8-тетрагидро-5(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-он. Действуя, как указано на стадии А примера 1, использование сложного эфира, замещенного у N1,полученного на стадии А (47 5 мг, 1,0 ммоль), NaBH4 (76+76 мг, 2,0+2,0 ммоль), тетрагидрофурана (12,5 мл) и метанола (12,5 мл) при 0C приводило после хроматографирования на колонке с диоксидом кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 90/10) к желаемому производному (321 мг, 0,72 ммоль, 72%). 1 Н-ЯМР (400 МГц, CDCl3):(м.д.)=1,42 (с, 9 Н, С(СН 3)3), 3,26-3,32 (м, 3H, N-CH2-CH-N, СН 2-СН 2NHBoc), 3,50 (м, 2 Н, N-CH2-CH-N, СН 2-CH2-NHboc), 3,95 (д, 1 Н, N-CH2-CH-N), 4,06 (м, 3H, CH2-CH2NHBoc, CH-СН 2-ОН), 4,62 (м, 1 Н, СН-CH2-ОН), 4,95 (АВ, 2 Н, CH2-Ph), 5,28 (широкий, 1 Н, NH), 7,36-7,44(м, 6 Н, Ph+H пиразол). Стадия С. 1,1-Диметил-транс-1-(2-трет-бутоксикарбониламиноэтил)-4,5,6,8-тетрагидро-6-оксо-5(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Действуя, как указано на стадии В примера 1, использование спирта, полученного на стадии В (320 мг, 0,72 ммоль), в дихлорметане (20 мл), метансульфонилхлорида (83+55 мкл, 1,08+0,72 ммоль) и триэтиламина (151+100 мкл, 1,08+0,72 ммоль) приводило, после очистки хроматографией на колонке с диоксидом кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 90/10), к желаемому мезилзамещенному производному (229 мг, 0,44 ммоль, 61%). 1 Н-ЯМР (400 МГц, CDCl3):(м.д.)=1,46 (с, 9 Н, С(СН 3)3), 3,17 (с, 3H, SO2Me), 3,23 (д, 1 Н, N-CH2CH-N), 3,37 (дд, 1 Н, N-CH2-CH-N), 3,54 (м, 2 Н, CH2-CH2-NHBoc), 3,97 (д, 1 Н, N-CH2-CH-N), 4,07 (м, 2 Н,СН 2-СН 2-NHBoc), 4,62 (м, 2 Н, CH2-OMs), 4,87 (м, 1 Н, CH-CH2-OMs), 4,95 (АВ, 2 Н, CH2-Ph), 5,06 (широкий, 1 Н, NH), 7,38-7,45 (м, 6 Н, Ph, H пиразол). Мезилзамещенное производное (300 мг, 0,575 ммоль) в диметилформамиде (4 мл) и NaN3 (75+75 мг, 1,15+1,15 ммоль) приводили к желаемому азиду. 1H-ЯМР (400 МГц, CDCl3):(м.д.)=1,43 (с, 9 Н,С (СН 3)3), 3.24 (д, 1 Н, N-CH2-CH-N), 3,31 (дд, 1 Н, NCH2-CH-N), 3,49 (м, 2 Н, СН 2-СН 2-NHBoc), 3,75 (м, 2 Н, CH2-N3), 3,94 (д, 1 Н, N-CH2-CH-N), 3,99 (м, 2 Н,CH2-CH2-NHBoc), 4,68 (дд, 1 Н, CH-CH2-N3), 4,91 (АВ, 2 Н, CH2-Ph), 5,17 (широкий, 1 Н, NH), 7,33-7,41 (м,6 Н, Ph, H пиразол). Триметилфосфин (раствор 1 M в тетрагидрофуране, 748 мкл, 0,75 ммоль) добавляли при 0C к раствору полученного выше азида (320 мг, 0,575 ммоль) в тетрагидрофуране (2,5 мл) и толуоле (2,5 мл). Этот раствор перемешивали 2 ч при комнатной температуре, затем охлаждали до 0C и добавляли к нему раствор Boc-ON (212 мг, 0,86 ммоль) в тетрагидрофуране (2 мл). Раствор перемешивали 1 ч при комнатной температуре, затем гидролизовали добавлением насыщенного раствора NaHCO3, затем экстрагировали этилацетатом. Объединенные органические фазы сушили, затем концентрировали. Остаток очищали хроматографией на колонке с диоксидом кремния (градиентное элюирование циклогексан/этилацетат от 60/40 до 30/70) с получением желаемого производного (220 мг, 0,41 ммоль, 70%). МС (ЭС(+: m/z [M+H]+=543.(АВ, 2 Н, CH2-Ph), 5,29 (широкий, 2 Н, 1H), 7,35-7,43 (м, 6 Н, Ph, H пиразол). Стадия D. Натриевая и трифторацетатная соль транс-8-(аминометил)-1-(2-аминоэтил)-4,8-дигидро 5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Действуя, как указано на стадии D примера 1, использование соединения, полученного на стадии С(210 мг, 0,387 ммоль), в диметилформамиде (1 мл) и дихлорметана (3 мл), Pd/C (50% Н 2 О, 75+40 мг) приводило к желаемому дибензилзамещенному производному. Использование дибензилзамещенного промежуточного продукта, комплекса пиридин/триоксид серы (123 мг, 0,775 ммоль) и пиридина (2 мл) приводило, после очистки хроматографией на колонке с диоксидом кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 80/20), к желаемой пиридиниевой соли (230 мг, 0,387 ммоль, 100%). Действуя, как указано на стадии С примера 2, использование полученной выше пиридиниевой соли(230 мг, 0,387 ммоль), раствора 2 н. гидроксида натрия (50 мл) и смолы DOWEX 50WX8 (18 г) приводило к желаемой натриевой соли (121 мг, 0,22 ммоль, 56%) в форме белого порошка. МС (ЭС(-: m/z [M-H]-=531. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)=1,37 (с, 9 Н, С(СН 3)3), 1,41 (с, 9 Н, С(CH3)3), 3,20-3,33 (м, 5 Н,N-CH2-CH-N, CH-CH2-NHBoc, СН 2-СН 2-NHBoc), 3,43 (м, 1 Н, СН-СН 2-NHBoc), 3,99 (м, 2 Н, СН 2-СН 2NHBoc), 4,44 (дд, 1 Н, CH-CH2-NHBoc), 4,65 (д, 1 Н, N-CH2-CH-N), 6,92 (широкий, 1 Н, NH), 7,11 (широкий, 1 Н, NH), 7,43 (с, 1 Н, Н пиразол). Использование натриевой соли (55 мг, 0,099 ммоль) в дихлорметане (1,5 мл) и смеси трифторуксусной кислоты (3 мл) и дихлорметана (3 мл) приводило к желаемой натриевой и трифторацетатной соли(широкий, 3H, NH3+), 8,19 (широкий, 3H, NH3+). Пример 5. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-(2-аминоэтил)-4,8-дигидро 5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Стадия А. Транс-2-(2-трет-бутоксикарбониламиноэтил)-8-(гидроксиметил)-4,8-дигидро-5(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-он. Действуя, как указано на стадии А примера 1, использование сложного эфира, замещенного у N2,полученного на стадии А примера 4 (623 мг, 1,32 ммоль), NaBH4 (300 мг, 7,92 ммоль), тетрагидрофурана(13 мл) и метанола (13 мл) при 0C приводило, после хроматографии на колонке с диоксидом кремния(градиентное элюирование CH2Cl2/MeOH от 98/2 до 90/10), к желаемому производному (250 мг, 0,58 ммоль, 43%). МС (ЭС(+: m/z [M+H]+=444. 1 Н-ЯМР (400 МГц, CDCl3):(м.д.)=1,40 (с, 9 Н, С(СН 3)3), 3,24 (д, 1 Н, N-CH2-CH-N), 3,31 (дд, 1 Н, NCH2-CH-N), 3,35 (м, 1 Н, СН 2-СН 2-NHBoc), 3,48 (м, 1 Н, CH2-CH2-NHBoc), 3,89-4,11 (м, 5 Н, CH2-CH2NHBoc, N-CH2-CH-N, СН-СН 2-ОН), 4,61 (дд, 1 Н, N-CH-CH2-N), 4,92 (АВ, 2 Н, СН 2-Ph), 5,18 (широкий,1 Н, NH), 7,21 (с, 1 Н, Н пиразол), 7,33-7,42 (м, 5 Н, Ph). Стадия В. 1,1-Диметил-транс-2-(2-трет-бутоксикарбониламиноэтил)-4,5,6,8-тетрагидро-6-оксо-5(фенилметокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Действуя, как указано на стадии С примера 4, использование спирта, полученного на стадии А (450 мг, 1,05 ммоль), в дихлорметане (30 мл), метансульфонилхлорида (131 мкл, 1,68 ммоль) и триэтиламина(237 мкл, 1,68 ммоль) приводило к желаемому мезилзамещенному производному (532 мг, 1,02 ммоль,97%). МС (ЭС(+: m/z [M+H]+=522. 1 Н-НМР (400 МГц, CDCl3):(м.д.)=1,45 (с, 9 Н, С (СН 3)3), 3,15 (с, 3H, SO2CH3), 3,20 (д, 1 Н, N-CH2CH-N), 3,40 (дд, 1 Н, N-CH2-CH-N), 3,50 (м, 2 Н, CH2-CH2-NHboc), 3,98 (д, 1 Н, N-CH2-CH-N), 4,13 (м, 2 Н,СН:-СН 2-NHBoc), 4.61 (м, 2 Н, CH2-OMs), 4,88 (м, 1 Н, CH-CH2-OMs), 4,95 (АВ, 2 Н, CH2-Ph), 7,24 (с, 1 Н, Н пиразол), 7,37-7,45 (м, 5 Н, Ph). Использование мезилзамещенного промежуточного продукта (532 мг, 1,05 ммоль) в диметилформамиде (7,5 мл) и NaN3 (615 мг, 9,45 ммоль) приводило к желаемому азиду (566 мг, 1,05 ммоль). 1H-ЯМР (400 МГц, CDCl3):(м.д.)=1,41 (с, 9 Н, С (СН 3)3), 3,20 (д, 1 Н, N-CH2-CH-N), 3,35 (дд, 1 Н, NCH2-CH-N), 3,44 (м, 2 Н, СН 2-СН 2-NHBoc), 3,65 (м, 2 Н, CH2-N3), 3,95 (д, 1 Н, N-CH2-CH-N), 4,09 (м, 2 Н,CH2-CH2-NHBoc), 4.71 (дд, 1 Н, CH-CH2-N3), 4,92 (АВ, 2 Н, CH2-Ph), 4,98 (широкий, 1 Н, NH), 7,21 (с, 1 Н,Н пиразол), 7,33-7,41 (м, 5 Н, Ph). Использование указанного выше азида (565 мг, 1,05 ммоль), триметилфосфина (раствор 1 M в тетрагидрофуране, 1,36 мл, 1,36 ммоль), Boc-ON (388 мг, 1,58 ммоль), тетрагидрофурана (5,5 мл) и толуола-9 021799 МС (ЭС(+: m/z [M+H]+=543. Н-ЯМР (400 МГц, CDCl3):(м.д.)=1,45 (с, 9 Н, С(СН 3)3), 1,46 (с, 9 Н, С(CH3)3), 3,10 (д, 1 Н, N-CH2CH-N), 3,29 (дд, 1 Н, N-CH2-CH-N), 3,37 (м, 1 Н, CH-CH2-NHBoc), 3,49 (м, 2 Н, CH2-CH2-NHBoc), 3,69 (м,1 Н, CH-CH2-NHBoc), 3,94 (д, 1 Н, N-CH2-CH-N), 4,10 (м, 2 Н, CH2-CH2-NHBoc), 4,58 (дд, 1 Н, CH-CH2NHBoc), 4,91 (широкий, 1 Н, NH), 4,92 (АВ, 2 Н, CH2-Ph), 5,13 (широкий, 1 Н, NH), 7,20 (с, 1 Н, Н пиразол),7,37-7,44 (м, 5 Н, Ph). Стадия С. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-(2-аминоэтил-4,8-дигидро-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Действуя, как указано на стадии D примера 1, использование соединения, полученного на стадии В(85 мг, 0,157 ммоль), в смеси диметилформамид/дихлорметан (1/3, 2 мл) и Pd/C (50% Н 2 О, 30 мг) приводило к желаемому дибензилзамещенному производному. Использование полученного дибензилзамещенного промежуточного продукта, комплекса пиридин/триоксид серы (50 мг, 0,314 ммоль) и пиридина (0,75 мл) приводило, после очистки хроматографией на колонке с диоксидом кремния (градиентное элюирование CH2Cl2/MeOH от 98/2 до 80/20), к желаемой пиридиниевой соли (85 мг, 0,139 ммоль, 86%). Действуя, как указано на стадии С примера 2, использование пиридиниевой соли (85 мг, 0,139 ммоль), раствора 2 н. гидроксида натрия 42 (мл) и смолы DOWEX 50WX8 (8,5 г) приводило к желаемой натриевой соли (75 мг, 0,135 ммоль, 86%) в форме кремового порошка. МС (ЭС(-: m/z [М-]=531. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)=1,37 (с, 9 Н, С(СН 3)3), 1,40 (с, 9 Н, С(СН 3)3), 3,17-3,32 (м, 5 Н,N-CH2-CH-N, CH-CH2-NHBoc, СН 2-СН 2-NHBoc), 3,60 (м, 1 Н, CH-CH2-NHBoc), 4,04 (м, 2 Н, CH-CH2NHBoc), 4,31 (дд, 1 Н, CH-CH2-NHBoc), 4,65 (с, 1 Н, N-CH2-CH-N), 6,94 (широкий, 2 Н, NH), 7,65 (с, 1 Н, Н пиразол). Использование натриевой соли (75 мг, 0,135 ммоль) в дихлорметане (2 мл) и смеси трифторуксуснои кислоты (4 мл) и дихлорметана (4 мл) приводило к натриевой и трифторацетатнои соли (35 мг, 0,059 ммоль, 44%) в форме кремового порошка. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)=3,20-3,41 (м, 6 Н, N-CH2-CH-N, СН-СН 2-NNH3+, СН 2-СН 2+NH3 ), 4,30 (м, 2 Н, СН 2-СН 2-NH3+), 4,63 (дд, 1 Н, СН-CH2-NH3+), 4,77 (д, 1 Н, N-CH2-CH-N), 7,85 (с, 1 Н, Н пиразол), 8,04 (широкий, 3H, NH3+), 8,17 (широкий, 3H, NH3+). Пример 6. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-(2-пиридинил)-4,8-дигидро 5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Стадия А. 1,1-Диметилэтил-транс-4,5,6,8-тетрагидро-2-(2-пиридинил)-6-оксо-5-(фенилметокси)4,7-метано-7H-пиразоло[3,4-е][1,3]диазепин-8-ил]метил]карбамат. Производное, полученное на стадии В примера 1 (0,500 г, 1,248 ммоль), 2-бромпиридин (217 мг,1,373 ммоль), L-пролин (32 мг, 0,275 ммоль), йодид меди (24 мг, 0,125 ммоль) и карбонат калия (345 мг,2,497 ммоль) суспендировали в безводном диметилсульфоксиде (1,875 мл). Реакцию затем осуществляли в атмосфере азота в запаянной трубке при 100C в течение 48 ч. Реакционную смесь затем обрабатывали водой, затем экстрагировали дихлорметаном. Органическую фазу затем сушили, потом концентрировали. Полученный таким образом сырой продукт очищали тогда хроматографией на диоксиде кремния(элюент CH2Cl2/MeOH 98/2 затем 95/5) с получением желаемого продукта (91 мг, 0,189 ммоль, 15%). МС (ЭС(+: m/z [M+H]+=477. 1(м, 6 Н, Ph, пиридин), 7,90 (м, 2 Н, пиридин), 8,42 (д, 1 Н, пиридин), 8,51 (с, 1 Н, пиразол). Стадия В. Натриевая соль 1,1-диметилэтил-транс-4,5,6,8-тетрагидро-2-(2-пиридинил)-6-оксо-5(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3] диазепин-8-ил]метил]карбамата. Действуя, как указано на стадии D примера 1, использование производного, полученного на стадии А (90 мг, 0,189 ммоль), смеси диметилформамид/дихлорметан 1/3 (2,0 мл) и катализатора с 10% палладия на угле с концентрацией 50% в воде (36 мг) приводило после 3 дней в атмосфере водорода к желаемому дибензилзамещенному промежуточному продукту. Использование дибензилзамещенного промежуточного продукта, пиридина (0,73 мл) и комплекса пиридин/триоксид серы (60 мг, 0,378 ммоль) приводило, после хроматографии на колонке с диоксидом кремния (элюент CH2Cl2/MeOH 90/10) к желаемому производному (63 мг). Неочищенный продукт затем извлекали в пиридин (0,73 мл), в атмосфере азота, в присутствии комплекса SO3/пиридин (60 мг, 0,378 ммоль). Реакционную смесь затем перемешивали при комнатной температуре вплоть до полной конверсии по ВЭЖХ (72 ч). После обработки добавлением Н 2 О, смесь фильтровали, затем выпаривали досуха. Полученный таким образом сырой продукт очищали хроматографией на колонке с диоксидом кремния (элюент CH2Cl2/МеОН 90/10). Полученный таким образом продукт был чистым (63 мг). Суспензию 8,5 г смолы DOWEX 50WX8 в растворе 2 н. гидроксида натрия (43 мл) перемешивали в течение 1 ч, затем выливали в хроматографическую колонку. Колонку кондиционировали деминерализо 1 ванной водой до нейтрального pH. Полученное производное (63 мг) растворяли в минимальном количестве метанола и воды, нанесенном на колонку, затем элюировали H2O. Фракции, содержащие субстрат,объединяли, замораживали и лиофилизировали с получением желаемой натриевой соли (55 мг, 0,112 ммоль, 60%) в форме желтого порошка. МС (ЭС(-: m/z [M-H]-=465. 1 Н-ЯМР (400 МГц, MeOD-d4):(м.д.)=1,53 (s, 9 Н, tBu), 1,54 (м, 4 Н, N-CH2-CH-N, N-CH-CH2NHBoc), 4,58 (дд, 2 Н, N-CH-CH2-HBoc), 5,02 (д, 1 Н, N-CH2-CH-N), 7,34 (м, 1 Н, пиридин), 7,97 (м, 2 Н,пиридин), 8,47 (д, 1 Н, пиридин), 8,65 (с, 1 Н, Н пиразол). Стадия С. Натриевая и трифторацетатная соль транс-8-(аминометил)-2-(2-пиридинил)-4,8-дигидро 5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4-е][1,3]диазепин-6(5 Н)-она. Действуя, как указано на стадии F примера 1, использование натриевой соли, полученной на стадии В (55 мг, 0,112 ммоль), безводного дихлорметана (1,92 мл) и смеси трифторуксусная кислота/дихлорметан 1/1 (7,68 мл) приводило к неочищенному производному, которое извлекали в воду, затем промывали простым диэтиловым эфиром. Нерастворимое вещество фильтровали и сушили при пониженном давлении с получением желаемого продукта (20 мг, 0,04 ммоль, 35%) в форме бежевого порошка. МС (ЭС(+: m/z [М+Н]+=367. 1(дд, 2 Н, N-CH-CH2-NH3+), 4,92 (м, 1 Н, N-CH2-CH-N), 7,35 (м, 1 Н, пиридин), 7,83 (д, 1 Н, пиидин), 7,95 (м,1 Н, пиридин), 8,49 (м, 1 Н, пиридин), 8,61 (с, 1 Н, Н пиразол). Пример 7. Натриевая и трифторацетатная соль транс-8-(аминометил)-5,6-дигидро-6-оксо-5(сульфоокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)уксусной кислоты. Стадия А. 1,1-Диметилэтил-транс-5,6-дигидро-8-(трет-бутоксикарбониламинометил)-6-оксо-5(фенилметокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)ацетат. Производное, полученное на стадии В примера 1 (0,200 г, 0,5 ммоль), растворяли в безводном диметилформамиде (0,5 мл), затем добавляли трет-бутилбромацетат (234 мг, 1,2 ммоль) и карбонат калия(138 мг, 1 ммоль). Реакцию проводили затем в атмосфере азота в запаянной трубке при 75C. За реакцией следили при помощи ВЭЖХ. Когда конверсия была полной, реакционную смесь обрабатывали Н 2 О, затем экстрагировали дихлорметаном. Объединенные органические фазы затем сушили над сульфатом натрия, фильтровали, потом концентрировали. Полученный таким образом сырой продукт затем очищали хроматографией на диоксиде кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 95/5) с получением желаемого продукта (186 мг, 0,36 ммоль, 72%) в форме смеси 2 изомеров N1/N2 в соотношении приблизительно 1/2. Изомер N2: 1(с, 2 Н, CH2Bn), 6,95 (м, 1 Н, NHBOC), 7,36-7,43 (м, 5 Н, Ph), 7,68 (с, 1 Н, пиразол). Стадия В. Натриевая соль 1,1-диметилэтил-транс-5,6-дигидро-8-(третбутоксикарбониламинометил)-6-оксо-5-(сульфоокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин 2(8 Н)уксусной кислоты. Соединение, полученное на стадии А (186 мг, 0,362 ммоль) растворяли в смеси дихлорметан/диметилформамид 3/1 (4,12 мл). После продувки вакуум/азот добавляли катализатор с 10% палладия на угле с концентрацией 50% в воде (74 мг). После новой продувки вакуум/азот реакционную смесь помещали в атмосферу водорода и перемешивали при комнатной температуре. За ходом реакции следили методом ВЭЖХ. После исчезновения исходного продукта (3 ч 30 мин) смесь концентрировали, выпаривали совместно с безводным дихлорметаном, наконец, выдерживали при пониженном давлении в присутствии Р 2 О 5 в течение 1 ч. Затем сырой продукт извлекали в пиридин (1,39 мл), в атмосфере азота, в присутствии комплекса SO3/пиридин (115 мг, 0,724 ммоль). Затем реакционную смесь перемешивали при комнатной температуре до полной конверсии, определяемой методом ВЭЖХ (24 ч). После обработки добавлением Н 2 О смесь фильтровали, затем выпаривали досуха. Полученный таким образом сырой продукт очищали хроматографией на диоксиде кремния (градиентное элюирование CH2Cl2/MeOH от 95/5 до 80/20). Таким образом, получали желаемый продукт (117 мг). Суспензию 20 г смолы DOWEX 50WX8 в растворе 2 н. гидроксида натрия (100 мл) перемешивали в течение 1 ч, затем выливали в хроматографическую колонку. Колонку кондиционировали деминерализованной водой до нейтрального pH. Полученное производное (117 мг, 0,233 ммоль) растворяли в минимальном количестве воды, нанесенном на колонку, затем элюировали H2O. Фракции, содержащие субстрат, объединяли, замораживали и лиофилизировали с получением желаемой натриевой соли (66 мг,0,126 ммоль, 35%) в форме белого порошка. Изомер N2: МС (ЭС(-: m/z [M-H]-=502. 1 Н-ЯМР (400 МГц, ДМСО-d6):(м.д.)=1,42 (с, 9 Н, С (СН 3)3), 3, 20-3,35 (м, 4 Н, N-CH2-CH-N, N-CHCH2-NHBoc), 4,32 (дд, 2 Н, N-CH-CH2-NHBoc), 4,81 (м, 1 Н, N-CH2-CH-N), 4,85 (с, 2 Н, СН 2 СО 2 С(СН 3)3), 6,- 11021799 99 (м, 1 Н, NHBOC), 7,67 (с, 1 Н, пиразол). Стадия С. Натриевая и трифторацетатная соль транс-8-(аминометил)-5,6-дигидро-6-оксо-5(сульфоокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)уксусной кислоты. Действуя, как указано на стадии F примера 1, использование натриевой соли, полученной на стадии В (66 мг, 0,126 ммоль), безводного дихлорметана (2,3 мл) и смеси трифторуксусная кислота/дихлорметан 1/1 (9,2 мл) приводило к сырому производному, которое извлекали в воду, потом промывали простым эфиром и гексаном. Водную фазу потом замораживали и лиофилизировали с получением желаемого продукта (54 мг, 0,111 ммоль, 88%) в форме желтого твердого вещества. Продукт представлял собой смесь изомеров N1/N2 в соотношении 28/72. Изомер N2: МС (ЭС(-: m/z [М-Н]-=346. 1H-HMP (400 МГц, MeOD-d4):(м.д.)=3,36-3,56 (m, 4H, N-CH2-CH-N, N-CH-CH2-NH3+), 4,78 (дд,1 Н, N-CH-CH2-NH3+), 4,92 (дд, 1 Н, N-CH2-CH-N), 4,99 (с, 2 Н, СН 2 СО 2 Н), 7,80 (с, 1 Н, пиразол). Пример 8. Натриевая и трифторацетатная соль транс-8-(аминометил)-5,6-дигидро-6-оксо-5(сульфоокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)ацетамида. Стадия А. Транс-5,6-дигидро-8-(трет-бутоксикарбониламинометил)-6-оксо-5-(фенилметокси)-4 Н 4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)ацетамид. Производное, полученное на стадии В примера 1 (1 г, 2,5 ммоль), растворяли в безводном диметилформамиде (2,5 мл). Добавляли 2-бромацетамид (829 мг, 6 ммоль) и карбонат калия (692 мг, 5 ммоль). Реакционную смесь перемешивали в атмосфере азота в запаянной трубке при 75C. После ночи добавляли 2-бромацетамид (1 экв.) и K2CO3 (1 экв.), и реакцию проводили в течение 4 дней (приблизительно 60% превращения). Реакционную смесь обрабатывали Н 2 О, затем экстрагировали дихлорметаном. Объединенные органические фазы затем сушили над сульфатом натрия, фильтровали, потом концентрировали. Полученный таким образом сырой продукт очищали хроматографией на диоксиде кремния (градиентное элюирование CH2Cl2/MeOH от 100/0 до 95/5) с получением желаемого продукта (188 мг, 0,41 ммоль, 16%) в форме смеси изомеров N1/N2 в соотношении приблизительно 1/2. Изомер N2: МС (ЭС(+:m/z [M+H]+=457. 1H-ЯМР (400 МГц, ДМСО-d6):(м.д.)=1,39 (с, 9 Н, С(СН 3)3), 3,12-3,33 (м, 4 Н, N-CH2-CH-N, N-CHCH2-NHBoc), 4,31 (м, 1 Н, N-CH-CH2-NHBoc), 4,40 (м, 1 Н, N-CH2-CH-N), 4,66 (с, 2 Н, CH2CONH2), 4,89 (с,2 Н, CH2Bn), 6,99 (м, 1 Н, NHBOC), 7,58-7,62 (м, 5 Н, Ph), 7,66 (с, 1 Н, пиразол). Стадия В. Натриевая соль транс-5,6-дигидро-8-(трет-бутоксикарбониламинометил)-6-оксо-5(сульфоокси)-4 Н-4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)ацетамида. Действуя как на стадии В примера 7, соединение, полученное на стадии А (188 мг, 0,411 ммоль),гидрировали, затем сульфатировали в присутствии комплекса SO3/пиридин (131 мг, 0,823 ммоль) в пиридине (1,58 мл), в атмосфере азота, при комнатной температуре в течение 4 дней. Полученный сырой продукт очищали хроматографией на диоксиде кремнияCH2Cl2/MeOH/NH4OH 80/20/1) с получением желаемого продукта (23 мг, 0,044 ммоль, 11%) в форме смеси изомеров N1/N2 в соотношении приблизительно 1/2. Изомер N2: МС (ЭС(+: m/z [M+H]+=447. 1H-ЯМР (400 МГц, ДМСО-d6):(м.д.)=1,41 (с, 9 Н, С (CH3)3), 3,24-3,32 (м, 4 Н, N-CH2-CH-N, N-CHCH2-NHBoc), 4,36 (м, 1 Н, N-CH-CH2-NHBoc), 4,67 (м, 1 Н, N-CH2-CH-N), 4,69 (с, 2 Н, CH2CONH2), 7,02 (м,1 Н, NHBOC), 7,40 (с, 2 Н, NH2), 7,65 (с, 1 Н, пиразол). Ионный обмен осуществляли на смоле DOWEX 50WX8 (4 г), как указано на стадии В примера 7, с получением после лиофилизации желаемой натриевой соли (17 мг, 0,126 ммоль, 35%) в форме бежевого порошка. МС (ЭС(-: m/z [M-H]+=445. Стадия С. Натриевая и трифторацетатная соль транс-8-(аминометил)-5,6-дигидро-6-оксо-5-(сульфоокси)-4 Н 4,7-метанопиразоло[3,4-е][1,3]диазепин-2(8 Н)ацетамида. Соединение, полученное на стадии В (17 мг, 0,036 ммоль) суспендировали в безводном дихлорметане (0,07 мл) в атмосфере азота. Затем по каплям добавляли трифторуксусную кислоту (0,027 мл), после чего реакцию проводили при комнатной температуре в течение трех часов. После выпаривания досуха продукт извлекали затем в воду, замораживали, потом лиофилизировали с получением желаемого продукта (17 мг, 0,035 ммоль, 98%) в форме бежевого твердого вещества, представлявшего собой смесь изомеров N1/N2 в соотношении приблизительно 1/2. Изомер N2: МС (ЭС(-: m/z [M-H]-=345. 1(широкий с, 1 Н, 1H), 7,73 (с, 1 Н, пиразол), 8,04 (сш, 1 Н, NH3+). Фармацевтическая композиция. Готовили композицию для инъецирования, содержащую: соединение примера 1: 500 мг; стерильный водный эксципиент: количество, достаточное для 5 см 3. Фармакологическое исследование соединений согласно изобретению Активность in vitro, методика разбавлений в жидкой среде. Готовили серию трубок, в которые помещали одинаковое количество стерильной питательной среды, вводили в каждую трубку увеличивающиеся количества исследуемого продукта, затем каждую трубку засевали бактериальным штаммом. После инкубации в течение 24 ч в сушильном шкафу при 37C ингибирование роста оценивали путем просвечивания, что позволяло определить минимальные ингибирующие концентрации (М.И.К.) (C.M.I.), выраженные в мкг/мл. Осуществляли таким образом испытания с продуктами примеров 1-8 в сравнении с продуктами примеров 7, 9, 11 и 45 заявки WO 04/052891. Продукты согласно настоящей заявке оказались очень активными в отношении Pseudomonas aeruginosa, что абсолютно не так в случае продуктов сравнения. Разность в активности в отношении Pseudomonas aeruginosa между продуктами согласно изобретению и наиболее близкими продуктами известного уровня техники находилась, сообразно продуктам, в диапазоне от 16 до более 500. Активность в отношении Pseudomonas aeruginosa (1771 штамм дикого типа) ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, выбранное из группы, состоящей из транс-8-(аминометил)-2-карбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-2-диметилкарбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Нпиразоло[3,4-е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-2-метилкарбамоил-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-1-(2-аминоэтил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-2-(2-аминоэтил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-2-(2-пиридинил)-4,8-дигидро-5-(сульфоокси)-4,7-метано-7 Н-пиразоло[3,4 е][1,3]диазепин-6(5 Н)-она,транс-8-(аминометил)-5,6-дигидро-6-оксо-5-(сульфоокси)-4,7-метано-4 Н-пиразоло[3,4 е][1,3]диазепин-2(8 Н)уксусной кислоты,транс-8-(аминометил)-5,6-дигидро-6-оксо-5-(сульфоокси)-4,7-метано-4 Н-пиразоло[3,4 е][1,3]диазепин-2(8 Н)ацетамида,или его фармацевтически приемлемая соль. 2. Применение любого из соединений по п.1 в изготовлении лекарственного средства, используемого в качестве антибактериального средства. 3. Фармацевтическая композиция, содержащая лекарственное средство, как оно определено в п.2, в качестве действующего начала.
МПК / Метки
МПК: A61K 31/437, C07D 471/18, A61P 31/04
Метки: содержащая, гетероциклические, соединения, азотсодержащие, композиция, фармацевтическая, применение
Код ссылки
<a href="https://eas.patents.su/15-21799-geterociklicheskie-azotsoderzhashhie-soedineniya-ih-primenenie-i-soderzhashhaya-ih-farmacevticheskaya-kompoziciya.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероциклические азотсодержащие соединения, их применение и содержащая их фармацевтическая композиция</a>
Азотсодержащие гетероциклические производные, содержащая их фармацевтическая композиция, их применение и способ лечения

Номер патента: 7537
Опубликовано: 27.10.2006
Авторы: Андерсен Ким, Мольтсен Айнер Кнуд, Смит Гаррик Пол, Банг-Андерсен Бенни, Руланд Томас, Пюшль Аск
МПК: A61K 31/44, A61K 31/4965, A61P 25/00...
Метки: лечения, композиция, содержащая, способ, производные, фармацевтическая, применение, гетероциклические, азотсодержащие
Формула / Реферат:
1. Соединение, представленное общей формулой I где Y означает N, С или СН; X означает S; m означает 1 или 2; р означает 0, 1, 2, 3, 4, 5, 6, 7 или 8; q означает 0, 1, 2, 3 или 4; s означает 1 или 2; пунктирная линия означает возможную связь; каждый R1 независимо выбран из группы, представленной C1-6-алкилом, или два R1, присоединенные к одному и тому же атому углерода, могут образовывать 3-6-членный спироприсоединенный циклоалкил; каждый R2...
Гетероциклические соединения, содержащая их фармацевтическая композиция и их применение для лечения pask-опосредованного заболевания

Номер патента: 21275
Опубликовано: 29.05.2015
Авторы: Макколл Джон М., Маккёрн Джон, Ромеро Доннал Л., Клэр Майкл
МПК: A61K 31/497, A61P 3/00, A61K 31/498...
Метки: композиция, соединения, лечения, фармацевтическая, содержащая, pask-опосредованного, заболевания, применение, гетероциклические
Формула / Реферат:
1. Соединение структурной формулы (IV)или его соль или сложный эфир,где Rz выбран из OH, NR8R9, NR8OR9;R1 выбран из арила и гетероарила, представляющего собой 3-7-членное ненасыщенное гетеромоноциклическое кольцо или бициклическую систему конденсированных колец, в которой по меньшей мере одно из конденсированных колец является ароматическим, которое(ая) содержит один или два гетероатома, независимо выбранных из O, S и N; любой из которых может...
Азотсодержащие гетероароматические соединения в качестве ингибиторов фактора ха, фармацевтическая композиция и способ лечения

Номер патента: 3056
Опубликовано: 26.12.2002
Авторы: Кван Мими Лайфен, Хан Кви, Каччиола Джозеф, Пруитт Джеймс Рассел, Февиг Джон Мэттью, Пинто Дональд Джозеф Филлип, Росси Карен Анита, Орват Майкл Джеймс
МПК: A61P 7/02, C07D 207/34, A61K 31/41...
Метки: фармацевтическая, фактора, азотсодержащие, ингибиторов, композиция, ха, качестве, лечения, соединения, гетероароматические, способ
Формула / Реферат:
1. Соединение формулы I или его стереоизомер или фармацевтически приемлемая соль, где кольцо М содержит, кроме J, 0-3 атома N, при условии, что если М содержит 2 атома N, тогда R1b отсутствует, и если М содержит 3 атома N, тогда R1a и R1b отсутствуют; J представляет собой N или NH; D выбран из CN, C(=NR8)NR7R9, NHC(=NR8)NR7R9, NR8CH(=NR7), C(O)NR7R8 и (CR8R9)tNR7R8, при условии, что D присоединен к Е в мета- или пара-положении относительно G, Е...
Применение пептидного соединения для добавочной терапии к антагонисту дофамина и фармацевтическая композиция, его содержащая

Номер патента: 15566
Опубликовано: 31.10.2011
Автор: Штёр Томас
МПК: A61P 25/18, A61K 31/165
Метки: соединения, композиция, антагонисту, фармацевтическая, содержащая, добавочной, пептидного, дофамина, терапии, применение
Формула / Реферат:
1. Применение соединения, имеющего формулу (IIb)в которойAr является фенилом, который незамещен или замещен по меньшей мере одной галогенной группой;R3 является CH2-Q, где Q является алкокси, содержащим 1-6 атомов углерода, и R1 является алкилом, содержащим 1-6 атомов углерода,или его фармацевтически приемлемой соли для получения фармацевтической композиции для предупреждения, облегчения или/и лечения болезни, которую лечат антагонистом...
Комбинация гетероциклического соединения и антиоксиданта, ее применение и содержащая ее фармацевтическая композиция для лечения или предотвращения ожирения

Номер патента: 12966
Опубликовано: 26.02.2010
Авторы: Бертело Паскаль, Кастейя Луи, Кэньяр Даньель-Анри, Пенико Люк, Дакке Катрин
МПК: A61K 31/15, A61K 31/428
Метки: комбинация, композиция, лечения, фармацевтическая, антиоксиданта, ожирения, гетероциклического, предотвращения, соединения, применение, содержащая
Формула / Реферат:
1. Комбинация, включающая 2-этокси-3-{4-[2-(6-[(гидроксиимино)(фенил)метил]-2-оксо-1,3-бензотиазол-3(2Н)-ил)этокси]фенил}пропионовую кислоту, ее энантиомеры и диастереоизомеры, а также ее соль присоединения с фармацевтически приемлемой кислотой или основанием и коэнзим Q10.2. Фармацевтическая композиция, содержащая 2-этокси-3-{4-[2-(6-[(гидроксиимино)(фенил)метил]-2-оксо-1,3-бензотиазол-3(2Н)-ил)этокси]фенил}пропионовую кислоту, ее энантиомеры и...
Предыдущий патент: Бензоксазины, бензотиазины и родственные соединения, обладающие ингибирующей nos активностью
Следующий патент: Система и способ для определения отношения (vp/vs) скоростей продольной и поперечной волн в области, удаленной от буровой скважины
Случайный патент: Тетрациклические соединения, способ их получения и содержащие их фармацевтические композиции