Производные пиразолов, способ их получения и применение в терапии
Номер патента: 20364
Опубликовано: 30.10.2014
Авторы: Табар Мишель, Демазо Паскаль, Абекасси Пьер-Ив














Формула / Реферат
1. Соединение формулы (I)

в которой X означает хлор или фтор,
в форме основания или кислотно-аддитивной соли с фармацевтически приемлемой кислотой.
2. Соединение формулы (I) по п.1, отличающееся тем, что X означает хлор, в форме основания или кислотно-аддитивной соли с фармацевтически приемлемой кислотой.
3. Способ получения соединения формулы (I) по любому из пп.1, 2, отличающийся тем, что соединение

подвергают взаимодействию с соединением

где X представляет хлор или фтор,
в присутствии диизопропилэтиламина в инертной среде, затем на второй стадии удаляют защитную группу амина паратолуолсульфоновой кислотой.
4. Способ по п.3, отличающийся тем, что инертной средой является апротонная аполярная среда.
5. Способ по п.4, отличающийся тем, что апротонной аполярной средой является тетрагидрофуран.
6. Соединение формулы

где X означает хлор или фтор.
7. Соединение формулы

где X означает хлор или фтор.
8. Противораковое лекарственное средство, отличающееся тем, что оно содержит соединение формулы (I) по любому из пп.1, 2 или аддитивную соль этого соединения с фармацевтически приемлемой кислотой.
9. Противораковая фармацевтическая композиция, отличающаяся тем, что она включает соединение формулы (I) по любому из пп.1, 2 или фармацевтически приемлемую соль этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент.
10. Применение соединения формулы (I) по любому из пп.1, 2 в качестве лекарственного средства для лечения рака.
Текст
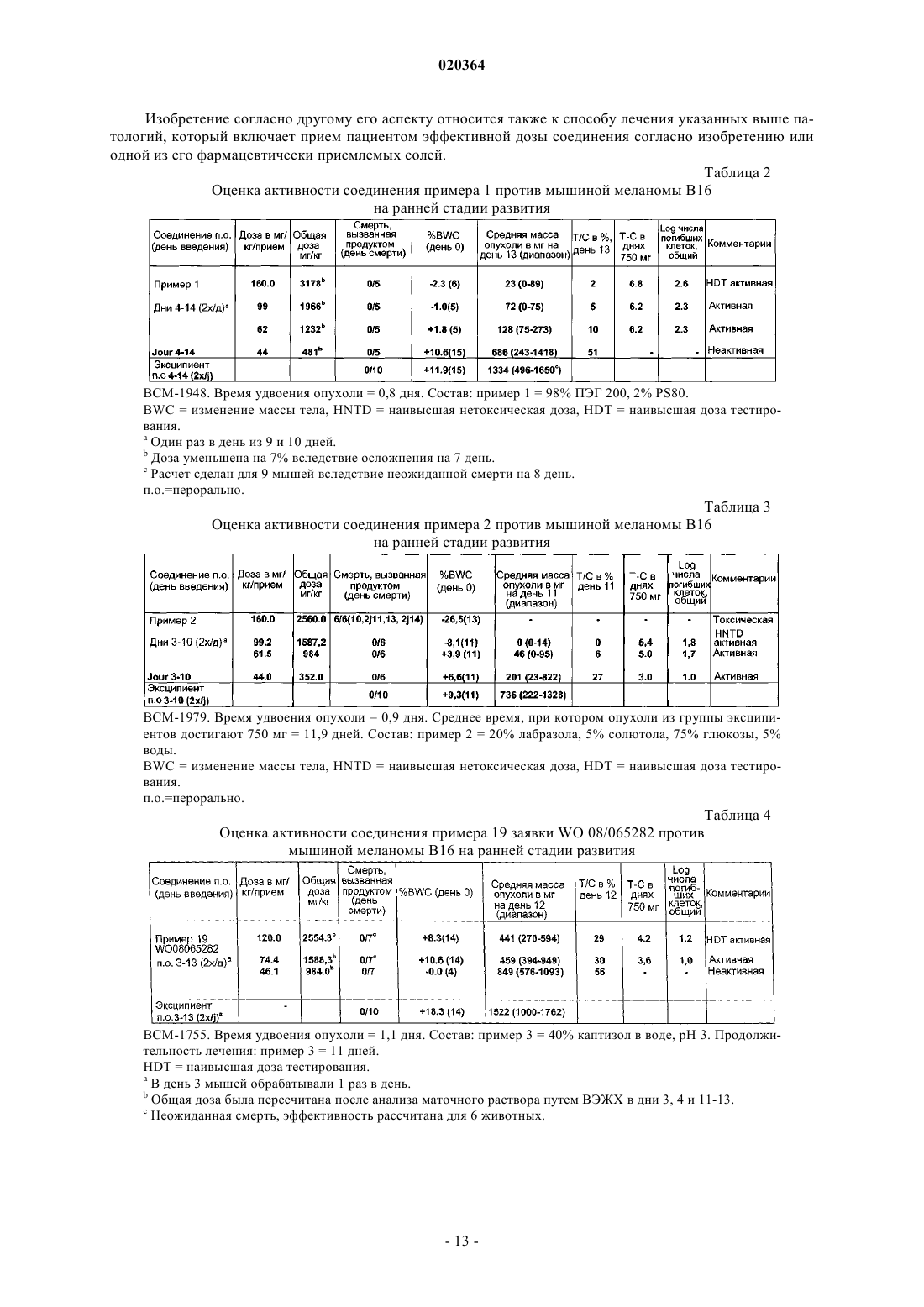
ПРОИЗВОДНЫЕ ПИРАЗОЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ В ТЕРАПИИ Изобретение относится к производным пиразолов общей формулы (I) в которой X означает хлор или фтор, а также к способу получения и применению в терапии. Настоящее изобретение относится к производным пиразолов, к способу их получения и применению их в терапии. Более конкретно и согласно первому аспекту изобретение относится к новым замещенным специфическим пиразолам, проявляющим противораковую активность посредством модуляции активности протеинов, в частности протеинкиназ. Протеинкиназы представляют собой семейство ферментов, которые катализируют фосфорилирование гидроксильных групп специфических остатков протеинов, таких как остатки тирозина, серина или треонина. Такое фосфорилирование может широко изменять функцию протеинов; таким образом, протеинкиназы играют важную роль в регулировании разнообразных клеточных процессов, включающих в себя, в частности, метаболизм, клеточную пролиферацию, дифференцировку, клеточную миграцию или выживаемость клеток. Среди различных клеточных функций, в которые вовлечена активность протеинкиназы, некоторые процессы представляют собой привлекательные цели для лечения раковых заболеваний, а также других болезней. Таким образом, одна из задач изобретения заключается в том, чтобы предложить композиции, обладающие противораковой активностью, проявляемой, в частности, в воздействии на киназы. Среди киназ, в отношении которых исследуется модуляция, можно назвать KDR, Tie2, VEGFR-1 (FLT1),VEGFR-3 (Flt4), PDGFR, FGFR. Предпочтительными являются киназы KDR и/или Tie2. Известны соединения из заявки на патент, опубликованной под номером WO 08/065282, отвечающие следующей общей формуле (I): в которой: 1) A и Ar выбирают независимо из группы, состоящей из арила, гетероарила, замещенного арила,замещенного гетероарила; 2) L выбирают из группы, состоящей из NH-CO-NH и O-CO-NH; 3) R1 выбирают из группы, состоящей из Н, Re, COR6, SO2R6, где R6 выбирают из Н, OR7, NR8R9,алкила, циклоалкила, гетероциклила, замещенного гетероциклила, арила, замещенного арила, гетероарила, замещенного гетероарила, где R7 выбирают из Н, фенила, алкила и R8 и R9 выбирают независимо из группы, состоящей из Н, алкила, циклоалкила, гетероциклила, замещенного гетероциклила, арила, замещенного арила, гетероарила, замещенного гетероарила, или же R8 и R9 соединены между собой с образованием 5-8-членного насыщенного цикла, содержащего 0-3 гетероатомов, выбранных из О, S и N; 4) X выбирают из группы, состоящей из O и NH; 5) R3 выбирают из группы, состоящей из Н, алкила, замещенного алкила, циклоалкила, замещенного циклоалкила; 6) R4a выбирают из группы, состоящей из H и (С 1-С 4)алкила; 7) R4b выбирают из группы, состоящей из H и (С 1-С 4)алкила; 8) R5 выбирают из группы, состоящей из Н, галогена, R10, CN, O(R10), ОС(О)(R10), OC(O)N(R10)(R11),OS(O2)(R10), N(R10)(R11), N=C(R10)(R11), N(R10)С(О)(R11), N(R10)С(О)О(R11), N(R12)C(O)N(R10)(R11),N(R12)C(S)N(R10)(R11), N(R10)S(O2)(R11), C(O)(R10), C(O)O(R10), C(O)N(R10)(R11), C=N(R11)(R10),C=N(OR11)(R10), S(R10), S(O)(R10), S(O2)(R10), S(O2)O(R10), S(O2)N(R10)(R11), где R10, R11, R12 независимо выбирают из группы, состоящей из Н, алкила, алкилена, алкинила, арила, гетероарила, циклоалкила, гетероциклила, замещенного алкила, замещенного алкилена, замещенного алкинила, замещенного арила,замещенного гетероарила, замещенного циклоалкила, замещенного гетероциклила. В этом патенте предпочтительно X означает O, R3 означает метил, R4a и R4b означают Н; L означаетNHCONH; А означает фенил; Ar означает фенил; но в этой заявке ни один из примеров не описывает замещение Ar и его влияние на фармококинетику. Настоящее изобретение относится к двум соединениям, включенным в вышеуказанную заявку и отвечающим формуле (I) в которой X означает Cl или F. Соединения формулы (I) могут существовать в форме оснований или аддитивных солей с кислотами. Такие аддитивные соли составляют часть изобретения. Соединения в своих двух таутомерных формах, указанных ниже, также составляют изобретение: Эти соли могут быть получены с фармацевтически приемлемыми кислотами, но соли других кислот, например, полезных для очистки или выделения соединений формулы (I), также составляют часть изобретения. Среди используемых солей можно назвать, в частности, гидрохлорид. Соединения формулы (I) могут существовать также в форме гидратов или сольватов, а именно в форме ассоциаций или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также составляют часть изобретения. Среди соединений формулы (I) согласно изобретению можно назвать, в частности, следующие соединения: 4-3-[3-(2-хлор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамид и его гидрохлорид; 4-3-3-(2-фтор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамид и его гидрохлорид. В соответствии с изобретением соединения формулы (I) можно получить согласно описанному ниже способу. На представленных ниже схемах исходные соединения и реагенты, если способ их получения не описан, являются коммерчески доступными или описанными в литературе или же их можно получить способами, которые описаны в литературе или известны специалисту. Согласно другому из своих аспектов изобретение относится также к соединениям формул: в которых X означает F или Cl. Эти соединения полезны в качестве промежуточных для получения соединений формулы (I). Следующие примеры описывают получение соединений согласно изобретению. Эти примеры не являются ограничивающими и предназначены только для иллюстрации настоящего изобретения. Способ синтеза согласно примерам. Синтез аминопиразольной части. 4-3-[3-(2-хлор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3 Суспензию 1,64 г (3,48 ммоль) 4-3-[3-(2-хлор-4-трифторметилфенил)уреидо]-5-фторбензиламино 1 Н-пиразол-3-карбоксамида в 50 мл этанола перемешивают при комнатной температуре в атмосфере аргона. Затем добавляют по каплям 35 мл (35 ммоль) раствора соляной кислоты в диэтиловом эфире(1 н.). Реакционная среда становится прозрачным раствором. После 10 ч перемешивания при комнатной температуре растворители выпаривают в роторном испарителе при пониженном давлении. Полученный остаток перемешивают в 50 мл диэтилового эфира в течение 30 мин. После фильтрации и сушки в сушильном шкафу получают 1,8 г гидрохлорида 4-3-[3-(2-хлор-4 трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамида в виде бледно-желтых кристаллов. МС: Время удерживания Tr (мин) = 1,01; [М+Н]+ m/z = 471; [M-H]- m/z = 469. 1 Н ЯМР (400 МГц, ДМСО-d)м.д.: 4,32 (с, 2 Н); 6,89 (д шир., J=9,6 Гц, 1 Н); 7,16 (с шир., 1 Н); 7,187,61 (м, 4 Н); 7,68 (д шир., J=8,9 Гц, 1 Н); 7,86 (с шир., 1 Н); 8,44 (д, J=8,9 Гц, 1 Н); 8,81 (м шир., 1 Н); 10,17 Раствор 5,9 г (9,5 ммоль) 4-3-[3-(2-хлор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Нпиразол-3-(2,4-диметоксибензиламида) и 4,5 г (24 ммоль) паратолуолсульфоновой кислоты в 400 мл толуола нагревают с обратным холодильником в течение 2 ч. После декантации толуольный раствор отделяют от желтой смолы. Смолу разбавляют 150 мл метанола и 500 мл этилацетата. Затем добавляют 500 мл воды. После этого раствор охлаждают, затем подщелачивают 100 мл водного раствора едкого калия (10 н.). После декантации водный слой экстрагируют раствором 400 мл этилацетата и 100 мл метанола. Органические слои объединяют и промывают 100 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют при пониженном давлении с получением твердого вещества бледно-желтого цвета, которое очищают на слое из 200 г диоксида кремния, элюируя раствором дихлор-3 020364 метана/метанола/ацетонитрила 80/10/10 (по объему): получают 1,77 г 4-3-[3-(2-хлор-4 трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамида в виде белого твердого вещества. МС: Время удерживания Tr (мин) = 4,29; [М+Н]+ m/z = 471; [M-H]- m/z = 469. 1(35,2 ммоль) диизопропилэтиламина в 300 мл тетрагидрофурана перемешивают при комнатной температуре в атмосфере аргона. Добавляют 3,85 г (35 ммоль) сульфата магния и 12,70 г (35,2 ммоль) 1-(2-хлор 4-трифторметилфенил)-3-(3-фтор-5-формилфенил)мочевины. После этого реакционную смесь нагревают с обратным холодильником в течение 14 ч. Затем смесь охлаждают до 25C и после этого медленно добавляют 10,08 г (160 ммоль) цианборгидрида натрия. После перемешивания в течение 12 ч при комнатной температуре смесь концентрируют досуха в ротационном испарителе. Полученную смолу перемешивают с 300 мл воды и 500 мл водного раствора едкого натра (1 н.). Эту суспензию перемешивают в течение 30 мин с 1 л дихлорметана. После фильтрации через фриттированное стекло 3 полученный твердый продукт промывают водой 2 раза по 500 мл, затем сушат в сушильном шкафу под вакуумом при 40C. Твердый продукт перекристаллизовывают из 800 мл метанола в горячем состоянии и получают 8,3 г 4-3-[3-(2-хлор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-(2,4-диметоксибензиламида) в виде белого порошка. МС: Время удерживания Tr (мин) = 4,97; [М+Н]+ m/z = 621; [M-H]- m/z = 619. 1 Н ЯМР (400 МГц, ДМСО-d)м.д.: 3,73 (с, 3 Н); 3,81 (с, 3 Н); 4,19 (д, J=6,7 Гц, 2 Н); 4,33 (д, J=6,7 Гц,2 Н); 5,71 (т шир., J=6,7 Гц, 1 Н); 6,46 (дд, J=8,3, 2,3 Гц, 1 Н); 6,55 (д, J=2,3 Гц, 1 Н); 6,80 (д шир., J=9,6 Гц,1 Н); 7,04 (с шир., 1 Н); 7,08 (с, 1 Н); 7,10 (д, J=8,9 Гц, 1 Н); 7,43 (дт, J=11,4, 2,3 Гц, 1 Н); 7,67 (д шир.,J=8,9 Гц, 1 Н); 7,86 (с шир., 1 Н); 7,94 (т шир., J=6,7 Гц, 1 Н); 8,45 (д, J=8,9 Гц, 1 Н); 8,62 (м уш., 1 Н); 9,79 (м уш., 1 Н); 12,60 (м уш., 1 Н). Температура плавления (по Кофлеру) = 168C. 1-(2-Хлор-4-трифторметилфенил)-3-(3-фтор-5-формилфенил)мочевина Раствор 33,15 г (92,68 ммоль) 1-(2-хлор-4-трифторметилфенил)-3-(3-циано-5-фторфенил)мочевины в 600 мл тетрагидрофурана перемешивают при -10C в атмосфере аргона. Затем добавляют равномерно"капля за каплей" 230 мл 20%-ного раствора гидрида диизобутилалюминия в толуоле. Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Затем добавляют при -10C дополнительные 80 мл гидрида диизобутилалюминия. После перемешивания в течение 14 ч при комнатной температуре реакционную среду концентрируют досуха в роторном растворителе с получением густого масла, к которому медленно добавляют при перемешивании 500 г льда и 100 мл 100%-ной уксусной кислоты. Полученную суспензию фильтруют. Полученный твердый продукт обрабатывают этилацетатом 2 раза по 800 мл и органический раствор промывают 600 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния, фильтруют, концентрируют досуха в ротационном растворителе и сушат в сушильном шкафу в вакууме при 40C с получением 29,57 г 1-(2-хлор-4-трифторметилфенил)-3-(3-фтор 5-формилфенил)мочевины в виде порошка бледно-желтого цвета. МС: Время удерживания Tr (мин) = 4,86; [М+Н]+ m/z = 361; [M-H]- m/z = 359. 1 Н ЯМР (400 МГц, ДМСО-d)м.д.: 7,35 (ддд, J=8,4, 2,3, 1,8 Гц, 1 Н); 7,67-7,75 (м, 2 Н); 7,78 (т,J=1,8 Гц, 1 Н); 7,88 (д, J=1,8 Гц, 1 Н); 8,45 (д, J=8,8 Гц, 1 Н); 8,72 (с шир., 1 Н); 9,98 (д, J=1,8 Гц, 1 Н); 10,07 Раствор 15 г (110,2 ммоль) 5-фтор-3-циананилина в 150 мл тетрагидрофурана перемешивают при комнатной температуре в атмосфере аргона. Затем добавляют 17,5 мл (121,22 ммоль) 2-хлор-4 трифторметилфенилизоцианата. Реакционную среду нагревают с обратным холодильником в течение 3 ч, затем концентрируют досуха в ротационном испарителе. Полученный твердый остаток перекристаллизовывают в горячем состоянии из 60 мл этилацетата и получают 33,25 г 1-(2-хлор-4 трифторметилфенил)-3-(3-циано-5-фторфенил)мочевины в виде белого твердого вещества. МС: Время удерживания Tr (мин) = 4,97; [М+Н]+ m/z = 356. Температура плавления (по Кофлеру) = 286C. 5-Фтор-3-циананилин является коммерческим продуктом. Гидрохлорид 4-амино-1 Н-пиразол-3-(2,4-диметоксибензиламида) Суспензию 6,12 г (20 ммоль) 4-нитро-1 Н-пиразол-3-(2,4-диметоксибензиламида) в 340 мл этанола перемешивают при комнатной температуре. Затем добавляют 15,8 г (70 ммоль) дигидрата хлорида олова в течение 5 мин. Реакционную смесь перемешивают 14 ч при комнатной температуре, затем концентрируют досуха в ротационном испарителе. Полученный остаток перемешивают с 330 мл насыщенного водного раствора гидрокарбоната натрия и 300 мл дихлорметана. После декантации органический слой экстрагируют дихлорметаном 2 раза по 150 мл. Органические слои объединяют, промывают 150 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния. После концентрирования в ротационном испарителе получают 4,57 г гидрохлорида 4-амино-1 Н-пиразол-3-(2,4-диметоксибензиламида) в виде твердого вещества фиолетового цвета. МС: IE: [M]+ m/z = 276; пик молекулярного иона m/z = 151. 1 Н ЯМР (400 МГц, ДМСО-d)м.д.: 3,73 (с, 3 Н); 3,81 (с, 3 Н); 4,31 (д, J=6,2 Гц, 2 Н); 4,56 (м уш., 2 Н); 6,46 (дд, J=8,3, 2,9 Гц, 1 Н); 6,55 (д, J=2,9 Гц, 1 Н); 7,06-7,13 (м, 2 Н); 7,84 (т, J=6,2 Гц, 1 Н); 12,50 (м уш.,1 Н). Температура плавления (прибор Buchi): 186C. 4-Нитро-1 Н-пиразол-3-(2,4-диметоксибензиламид)(76,4 ммоль) 1-гидроксибензотриазола в 50 мл диметилформамида перемешивают при комнатной температуре. Добавляют 11,7 г (70,03 ммоль) 2,4-диметоксибензиламина, затем добавляют маленькими порциями 10,2 г 4-нитро-3-пиразолкарбоновой кислоты. После 16 ч перемешивания при комнатной температуре реакционную среду выливают в 500 мл воды. Суспензию фильтруют, затем промывают водой 2 раза по 250 мл. Полученный твердый продукт сушат в сушильном шкафу в вакууме при 40C с получением 18,58 г 4-нитро-1 Н-пиразол-3-(2,4-диметоксибензиламида) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d)м.д.: 3,75 (с, 3 Н); 3,80 (с, 3 Н); 4,35 (д, J=5,9 Гц, 2 Н); 6,50 (дд,J=8,3, 2,4 Гц, 1 Н); 6,56 (д, J=2,4 Гц, 1 Н); 7,21 (д, J=8,3 Гц, 1 Н); 8,71 (с шир., 1 Н); 8,88 (т шир., J=5,9 Гц,1 Н); 14,13 (м уш., 1 Н). МС (ES+/-); Время удерживания Tr (мин) = 3,23; [М+Н]+ m/z = 307; [M-H]- m/z = 305. Температура плавления (по Кофлеру) = 192C. 4-Нитро-3-пиразолкарбоновая кислота является коммерческим продуктом.(0,40 ммоль) 4-3-[3-(2-фтор-4-трифторметилфенил)уреидо]-5 фторбензиламино-1 Н-пиразол-3-карбоксамида в 40 мл этанола перемешивают при комнатной температуре в атмосфере аргона. Затем добавляют по каплям 20 мл (40 ммоль) раствора соляной кислоты в диэтиловом эфире (1 н.). Реакционная среда становится прозрачным раствором. После 12 ч перемешивания при комнатной температуре растворители выпаривают в роторном испарителе при пониженном давлении. Полученный остаток перемешивают в 200 мл диэтилового эфира в течение 30 мин. После фильтрации и сушки в сушильном шкафу получают 1,75 г гидрохлорида 4-3-[3-(2-фтор-4 трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамида в виде бледно-желтых кристаллов. 1 Раствор 22,3 г (36,89 ммоль) 4-3-[3-(2-фтор-4-трифторметилфенил)уреидо]-5-фторбензиламино 1 Н-пиразол-3-(2,4-диметоксибензиламида) и 17,54 г (92,22 ммоль) паратолуолсульфоновой кислоты в 600 мл толуола нагревают с обратным холодильником в течение 14 ч. После декантации толуольный раствор отделяют от желтой смолы. Смолу перемешивают в течение 2 ч в 280 мл воды и 100 мл едкого натра (10 н.). Суспензию фильтруют. Полученный твердый продукт промывают водой 3 раза по 350 мл,затем сушат и получают твердый продукт кремового цвета, который очищают на слое из 350 г диоксида кремния, элюируя раствором дихлорметана/метанола/ацетонитрила 95/2,5/2,5 (по объему): получают 3,85 г 4-3-[3-(2-фтор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-карбоксамида в виде белого твердого вещества. 1 Н ЯМР (400 МГц, ДМСО-d6)м.д.: 4,18 (д, J=5,9 Гц, 2 Н); 5,68-5,77 (м, 1 Н); 6,80 (д, J=8,8 Гц, 1 Н); 7,01-7,08 (с шир., 1 Н); 7,04 (с, 1 Н); 7,06 (с, 1 Н); 7,24 (с шир., 1 Н); 7,40 (дт, J=11,4, 2,0 Гц, 1 Н); 7,54 (д,J=8,3 Гц, 1 Н); 7,69 (д, J=11,5 Гц, 1 Н); 8,41 (т, J=8,4 Гц, 1 Н); 8,92 (с шир., 1 Н); 9,40 (с шир., 1 Н); 12,55 (с шир., 1 Н). МС: Время удерживания Tr (мин) = 4,09; [М+Н]+ m/z = 455; [М-Н]- m/z = 453. Температура плавления (по Кофлеру) = 233C. 4-3-[3-(2-Фтор-4-трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-(2,4 диметоксибензиламид) Раствор 11,40 г (36,45 ммоль) гидрохлорида 4-амино-1 Н-пиразол-3-(2,4-диметоксибензиламида) и 6,65 мл (40,09 ммоль) диизопропилэтиламина в 360 мл тетрагидрофурана перемешивают при комнатной температуре в атмосфере аргона. Добавляют 4,35 г (36,45 ммоль) сульфата магния и 13,80 г (35,2 ммоль) 1-(2-фтор-4-трифторметилфенил)-3-(3-фтор-5-формилфенил)мочевины. Затем реакционную среду нагре-6 020364 вают с обратным холодильником в течение 14 ч. Смесь охлаждают до 25C, затем медленно добавляют 11,46 г (182,25 ммоль) цианоборгидрида натрия. После 72 ч перемешивания при комнатной температуре смесь концентрируют досуха в роторном испарителе. Полученную смолу перемешивают с 400 мл воды и 500 мл водного раствора едкого натра (1 н.). Эту суспензию перемешивают в течение 1 ч, затем фильтруют через фриттированное стекло 3, полученное твердое вещество промывают водой 3 раза по 500 мл, затем сушат в сушильном шкафу в вакууме при 40C с получением 22,45 г 4-3-[3-(2-фтор-4 трифторметилфенил)уреидо]-5-фторбензиламино-1 Н-пиразол-3-(2,4-диметоксибензиламида) в виде твердого розоватого вещества. 1 Раствор 11,90 г (34,87 ммоль) 1-(2-фтор-4-трифторметилфенил)-3-(3-циано-5-фторфенил)мочевины в 150 мл тетрагидрофурана перемешивают при -2C в атмосфере аргона. Затем добавляют равномерно"капля за каплей" 86 мл 20%-ного раствора гидрида диизобутилалюминия в гексане. Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Затем добавляют при -2C дополнительные 60 мл гидрида диизобутилалюминия. После перемешивания в течение 3 ч при комнатной температуре реакционную среду концентрируют досуха в роторном растворителе с получением густого масла, к которому медленно добавляют при перемешивании 500 г льда и 300 мл 100%-ной уксусной кислоты. Полученную суспензию фильтруют. Полученный твердый продукт промывают водой 4 раза по 150 мл, центрифугируют и сушат в сушильном шкафу в вакууме при 40C с получением 13,95 г 1-(2-фтор-4 трифторметилфенил)-3-(3-фтор-5-формилфенил)мочевины в виде твердого вещества бледно-желтого цвета. 1 Н ЯМР (400 МГц, ДМСО-d6)м.д.: 7,33-7,38 (м, 1 Н); 7,57 (д, J=8,8 Гц, 1 Н); 7,69-7,75 (м, 2 Н); 7,79 Раствор 6,15 г (45,18 ммоль) 5-фтор-3-циананилина в 90 мл тетрагидрофурана перемешивают при комнатной температуре в атмосфере аргона. Затем по каплям добавляют 4,3 мл (36,14 ммоль) дифосгена,затем 30 мл (135,54 ммоль) триэтиламина. После нагревания реакционной среды с обратным холодильником в течение 3 ч медленно добавляют раствор 7,10 г (39,64 ммоль) 4-амино-3 фтортрифторметилбензола в 10 мл тетрагидрофурана. Нагревание с обратным холодильником поддерживают более 2 ч. Затем среду перемешивают в 100 мл воды, после этого экстрагируют 100 мл этилацеатата. Органический слой промывают 100 мл насыщенного раствора хлорида натрия, сушат над сульфатом магния и концентрируют досуха в ротационном испарителе. Полученный твердый продукт перекристаллизовывают в горячем состоянии из 90 мл ацетонитрила и получают 10,35 г 1-(2-фтор-4 трифторметилфенил)-3-(3-циано-5-фторфенил)мочевины в виде твердого вещества кремового цвета. 1 Н ЯМР (400 МГц, ДМСО-d6)м.д.: 7,47 (д, J=8,3 Гц, 1 Н); 7,57 (д, J=8,6 Гц, 1 Н); 7,69 (с, 1 Н); 7,707,75 (м, 2 Н); 8,37 (т, J=8,4 Гц, 1 Н); 9,17 (с шир., 1 Н); 9,63 (с шир., 1 Н). МС: Время удерживания Tr (мин) = 1,1; [М+Н]+ m/z = 341. Температура плавления (по Кофлеру) = 253C. Продукты согласно изобретению полезны в качестве агентов, ингибирующих одну или несколько реакций, катализируемых киназой. KDR и/или Tie2 являются киназами, в отношении которых продукты согласно изобретению особенно полезны в качестве ингибиторов. Причины, по которым эти киназы были выбраны, изложены ниже.Factor Receptor 2), экспрессируется главным образом в эндотелиальных клетках. Такой рецептор связывается с проангиогенным фактором роста VEGF и служит, таким образом, медиатором трансдукционного сигнала путем активации своего внутриклеточного домена киназы. Прямое ингибирование активности киназы VEGF-R2 позволяет уменьшить явление ангиогенеза в присутствии экзогенного VEGF (VascularEndothelial Growth Factor: васкулярного эндотелиального фактора роста) (Strawn et al., Cancer Research,1996, vol. 56, p. 3540-3545). Такой процесс был доказан, в частности, с помощью мутантных VEGF-R2(Millauer et al., Cancer Research, 1996, vol. 56, p. 1615-1620). Рецептор VEGF-R2, кажется, не имеет никакой другой функции у взрослого человека, кроме функции, связанной с ангиогенной активностью VEGF. Дополнительно к такой центральной роли в динамическом ангиогенном процессе полученные недавно результаты указывают на то, что экспрессия VEGF способствует выживаемости опухолевых клеток после химио- и радиотерапии, подчеркивая потенциальный синергизм ингибиторов KDR с другими агентами (Lee et al., Cancer Research, 2000, vol. 60, p. 5565-5570).Tie2 (TEK) представляет собой член семейства рецепторов тирозинкиназы, специфичной к эндотелиальным клеткам. Tie2 является первым рецептором с тирозинкиназной активностью, известным одновременно как агонист (ангиопоэтин 1 или Ang1), который стимулирует аутофосфорилирование рецептора и клеточную сигнализацию [S. Davis et al. (1996), Cell, 87, 1161-1169], и антагонист (ангиопоэтин 2 или Ang2) [Р.С. Maisonpierre et al. (1997), Science, 277, 55-60]. Ангиопоэтин 1 может проявлять синергизм с VEGF на последних стадиях неоангиогенеза [Asahara Т. Circ. Res. (1998), 233-240]. Испытания с нокаутом генов и трансгенные манипуляции экспрессии Tie2 или Ang1 приводят к животным, которые проявляют недостаточность васкуляризации [D.J. Dumont et al. (1994), Genes Dev. 8, 1897-1909 et C. Suri(1996), Cell. 87, 1171-1180]. Связь Ang1 с его рецептором ведет к аутофосфорилированию домена киназыTie2, которая является существенной для неоваскуляризации, а также для рекрутирования и взаимодействия сосудов с перицитами и гладкими мышечными клетками; такие явления способствуют созреванию и стабильности новообразованных сосудов [Р.С. Maisonpierre et al. (1997), Science, 277, 55-60]. АвторыLin et al. (1997), J. Clin. Invest. 100, 8; 2072-2078 et Lin P. (1998), PNAS 95, 8829-8834 показали ингибирование роста и опухолевой васкуляризации, а также уменьшение метастазов в легком при инфицировании аденовирусами или инъекциях внеклеточного домена Tie2 (Tek) в моделях ксенотрансплантов опухоли молочной железы и меланомы. По нижеизложенным причинам ингибиторы Tie2 могут применяться в случаях, когда неоваскуляризация или ангиогенез происходит несоответствующим образом, т.е. при раках вообще, а также при конкретных видах рака, таких как саркома Капоши или инфантильная гемоангиома, при ревматиодном артрите, остеоартрите и/или при ассоциированных с ними болях, воспалительных заболеваниях кишечника, таких как геморрагический ректоколит или болезнь Крона, патологиях глаз, таких как мышечная дистрофия, связанная с возрастом, диабетической ретинопатии, хроническом воспалении, псориазе. Ангиогенез представляет собой процесс образования новых капиллярных сосудов от уже существующих сосудов. Опухолевый ангиогенез (формирование новой кровеносной сети), нежелательный при росте опухоли, является также одним из существенных факторов метастатического обсеменения (Oncogene. 2003 May 19:22(20):3172-9; Nat Med. 1995 Jan; 1(1):27-31). Такая неоваскуляризация обусловлена миграцией, а затем пролиферацией и дифференцировкой эндотелиальных клеток под влиянием ангиогенных факторов, секретируемых раковыми клетками и стволовыми клетками (Recent Prog. Horm. Res. 2000, 55:15-35; 35-6). Система ангиопоэтин 1/рецептор Tie2 играет главенствующую роль в созревании сосудов, обеспечивая рекрутирование периэндотелиальных клеток для стабилизации сосудистой структуры (Cell. 1996Dec. 27; 87(7):1161-9. Recent Prog. Horm. Res. 2004; 59:51-71). Таким образом, было доказано, что введение в растворимой рекомбинантной форме внеклеточного домена рецептора Tie2 (exTek) ингибирует ангиогенез в опухоли на моделях мышиных опухолей, а также распространение метастазов (Pengnian Lin,Jake A. Buxton, Ann Acheson, Czeslaw Radziejewski, Peter C. Mainsonpierre, George D. Yancopoulos, KeithUSA. 1998 Jul. 21; 95(15):8829-34; Cecilia Melani, Antonelle Stoppacciaro, Chiara Foroni, Federica Felicetti,Alessandra Car et MarioP. Colombo, Cancer immunol Immunother. 2004 Jul.; 53(7):600-8). В культуре эндотелиальных клеток стимуляция Tie2 активирует PI3-киназный путь и р 42/р 44-киназные пути, задействованный в пролиферации и миграции клеток; путь синтеза PAF (Cell Signal. 2006 Apr. 14; ahead of print),вовлеченный в провоспалительную активность. Стимуляция Tie2 стимулирует Akt-путь и ингибирует апоптоз (Laura M. DeBusk, Dennis E. Hallahan, Pengnian Chorles Lin, Exp. Cell. Res. 2004 Aug. 1; 298(1):167-77), сигнальный трансдукционный путь, известный своей важностью для выживаемости клеток. Добавление Extek (растворимый рецептор Tie2) ингибирует образование псевдотрубочек эндотелиальных клеток на Матригеле (Cecilia Melani, Antonelle Stoppacciaro, Chiara Foroni, Federica Felicetti,-8 020364Alessandra Care et MarioP. Colombo, Cancer immunol Immunother. 2004 Jul; 53(7): 600-8). Эти исследования указывают на то, что система Tie2/Ангиопоэтин необходима на первых стадиях образования сосудистых узелков в тканях взрослого человека и что функция рецептора Tie2 состоит в увеличении продолжительности жизни эндотелиальных клеток в процессе формирования сети кровеносных сосудов. Кроме того, ангиопоэтин-1 стимулирует пролиферацию лимфатических эндотелиальных клеток, а также лимфоангиогенез (развитие новых лимфатических сосудов), преимущественный путь метастазированияMaekawa, Yoshishige Kimura, Masako Ohmura, Takeshi Miyamoto, Shiro Nozawa, Gou Young Koh, Kari Alitalo and Toshio Suda, Blood. 2005 Jun. 15; 105(12):4649-56. Среди ферментов, играющих определенную роль в ангиогенезе, можно назвать также PDGFRangiogenic sprouting and vascular network formation, Nature, 454, 656-660 (20058). Процессы ангиогенеза играют ведущую роль в развитии многочисленных солидных опухолей. Кроме того, было доказано, что вероятность появления метастазов увеличивается очень сильно с усилением васкуляризации первичной опухоли (Br. J. Cancer. 2002 May 20; 86(10):1566-77. Недавно потенциальная роль проангиогенных агентов при лейкемиях и в лимфомах также была подтверждена документально. В общем, было показано, что клеточные клоны при таких патологиях могут быть либо естественным образом разрушены самой иммунной системой, либо превращаются в ангиогенный фенотип, который благоприятствует выживанию клеток и их пролиферации. Такое изменение фенотипа вызвано суперэкспрессией ангиогенных факторов, в частности макрофагами, и/или мобилизацией этих факторов из внеклеточной матрицы (Thomas D.A., Giles F.J., Cortes J., Albitar M., KantarjianH.M., Acta Haematol, (2001), vol. 207, p. 106-190). Существует корреляция между процессом ангиогенеза костного мозга и "экстрамедуллярными поражениями" при CML (хронический миеломоноцитарный лейкоз). Различные исследования показывают,что ингибирование ангиогенеза могло бы служить терапией выбора для лечения данной патологии (LeukRes. 2006 Jan.; 30(1):54-9; Histol Histopathol. 2004 oct.; 19(4):1245-60. Кроме того, имеется сильное предположение, что активация системы Tie2/ангиопоэтин задействована в развитии ангиогенеза костного мозга у пациентов, больных множественной миеломой (Blood. 2003 Jul. 15; 102(2):638-45. Определение активности соединений - Протоколы испытаний. 1. KDR. Ингибирующий эффект соединений определяли в тесте in vitro на фосфорилирование субстрата методом сцинтилляции (96-луночный планшет basic Flash Plate). Цитоплазмический домен (остатки 790-1356) человеческого фермента KDR был клонирован в форме слитого GST в вектор экспрессии бакуловируса pFastBac. Белок был экспрессирован в клетках SF21,очищен и активирован аутофосфорилированием. Субстрат состоял из остатков 658-850 PLC, экспрессированной и очищенной в форме слитого белка GST. Киназную активность KDR измеряли в буфере, содержащем 20 мМ MOPS, 10 мМ MgCl2, 10 мМMnCl2, 1 мМ DTT, рН 7,4. Соединения первоначально разводили в 100% ДМСО, затем готовили раствор 10 Х в смеси ДМСО 30% - буфер 70%. Вносили 10 мкл раствора 10 Х, затем 70 мкл буфера, содержащего 150 нг (1,6 пмоль) фермента KDR, при 4C. Реакцию запускали добавлением 20 мкл раствора, содержащего 2 мкг (41 пмоль) PLC-субстрата, 0,5 мкКи 33P[ATP] и 2 мкМ немеченного АТР. Планшет встряхивали. После 30 мин инкубирования при 37C инкубационный буфер удаляли и лунки промывали три раза 300 мкл PBS. Радиоактивность измеряли в каждой лунке, используя счетчик радиоактивностиTrilux-Wallac. Фоновый шум определяли измерением радиоактивности в четырех разных лунках, содержащих АТР (немеченный и радиоактивно меченый) и субстрат, в отсутствие фермента и соединения. Контрольную активность измеряли в четырех разных лунках, содержащих все реагенты, но в отсутствие соединения. Ингибирование активности KDR с помощью соединения согласно изобретению выражали в процентах ингибирования по отношению к контрольной активности, определяемой в отсутствие соединения. 2. Tie2. Ингибирующий эффект соединений определяли в тесте in vitro на фосфорилирование субстрата методом сцинтилляции (96-луночный планшет basic Flash Plate). Кодирующая последовательность человеческого Tie2, соответствующая аминокислотам внутриклеточного домена 774-1124, была введена в вектор экспрессии бакуловируса pFastBac в форме слитого бел-9 020364 ка GST. GST-Tie2 очищали и активировали аутофосфорилированием. Субстрат состоял из остатков 658850 PLC, экспрессированной и очищенной в форме слитого белка GST. Киназную активность Tie2 измеряли в буфере MOPS 20 мМ, рН 7,4, содержащем 10 мМ MgCl2,10 мМ MnCl2, 1 мМ DTT. Соединения первоначально разводили в 100% ДМСО, затем готовили раствор 10 Х в смеси ДМСО 30% - буфер 70%. Вносили 10 мкл раствора 10 Х, затем 70 мкл буфера, содержащего 100 нг (1,5 пмоль) фермента Tie2, при 4C. Реакцию запускали добавлением 20 мкл раствора, содержащего 2 мкг (41 пмоль) PLC-субстрата, 0,5 мкКи 33 Р[АТР] и 2 мкМ холодного АТР. Планшет встряхивали. После 30 мин инкубирования при 37C инкубационный буфер удаляли и лунки промывали три раза 300 мкл PBS. Радиоактивность измеряли в каждой лунке, используя счетчик радиоактивностиTrilux-Wallac. Фоновый шум определяли измерением радиоактивности в четырех разных лунках, содержащих АТР (холодный и радиоактивно меченый) и субстрат, в отсутствие фермента и соединения. Контрольную активность измеряли в четырех разных лунках, содержащих все реагенты, но в отсутствие соединения. Измеряли величину ингибирования активности Tie2 и выражали в процентах ингибирования от контрольной активности, определяемой в отсутствие соединения. 3. Скрининг соединений путем измерения фосфорилирующей активности киназы FLT1 в присутствии субстрата PLC с помощью радиоактивного мечения Киназную активность FLT1 измеряли таким же способом, что и ингибирование Tie2, используя реакционную смесь, состоящую из 70 мкл киназного буфера, содержащего 13 нМ фермента FLT1 на лунку. Рассчитывали величину ингибирования активности FLT1 и выражали в процентах ингибирования по отношению к контрольной активности, определяемой в отсутствие соединения. Процент ингибирования, соответствующий каждой концентрации соединений, рассчитывали следующим образом:% ингибирования = (среднее значение СРМ обработанных лунок - среднее значение СРМ контрольных лунок)/(СРМ необработанных лунок - среднее значение СРМ контрольных лунок)100. 4. Скрининг соединений путем измерения фосфорилирующей активности киназы PDGFR в присутствии субстрата PLC с помощью радиоактивного мечения. Тест был осуществлен аналогично тесту, проведенному с ферментом FLT1, но вместо 13 нМ FLT1 использовали 16 нМ фермента PDGFR и в 96-луночных планшетах смешивали 18 мкл ферментаGST-PDGFR в концентрации 4 мг/мл (при чистоте 80%), партия VLT802. 5. Скрининг соединений путем измерения фосфорилирующей активности киназы FGFR в присутствии субстрата PLC с помощью радиоактивного мечения. Тест был осуществлен аналогично тесту, проведенному с ферментом FLT1, но вместо 13 нМ FLT1 использовали 27 нМ фермента FGFR и в 96-луночных планшетах смешивали 18 мкл ферментаGST-FGFR в концентрации 1,1 мг/мл (при чистоте 100%), партия JCE3666. Результаты. Соединения согласно примерам изобретения находятся в концентрации, ингибирующей активность киназы на 50%, как правило, между 0,1 нМ и 2 мкМ в отношении киназы KDR и/или Tie2, предпочтительно между 0,1 и 500 нМ, более предпочтительно между 0,1 и 50 нМ. Величины концентраций, приведенные в табл. 1, указаны в качестве иллюстрации. Таблица 1 Соединения согласно изобретению были объектом фармакологических исследований, позволивших определить их печеночный клиренс. Оценка внутреннего клиренса соединений с использованием гепатоцитов человека: протокол эксперимента.NB: Тест in vitro, описанный ниже, осуществленный с клетками печени человека, был использован для предварительного выявления важного фармакокинетического параметра: влияние на печеночный метаболизм данного соединения после его введения в организм человека. Условия культивирования. Инкубирование криоконсервированных человеческих гепатоцитов человека (IVT: IVT-TLN-180608,приобретенных от In Vitro Technologies, inc, Baltimore, Maryland, США), и свежих человеческих гепатоцитов (от Biopredic: HEP200239) проводили в планшетах, содержащих 48 лунок, покрытых коллагеном. Условия эксперимента. Кинетические процессы запускали добавлением в культуральную среду соединений в конечной концентрации 5 мкМ в отсутствие или в присутствии кетоконазола 10 мкМ (ингибитор CYP3A4). Инкубационный объем составлял 100 мкл, продолжительность инкубирования: 0-24 ч (обычные кинетические точки: 0-0,5-1-2-4-6-8-24 ч). Кинетические процессы останавливали добавлением ацетонитрила/воды с использованием кортикостерона в качестве внутреннего стандарта. Клетки открепляли, затем лизировали. Внутриклеточные и внеклеточные среды соединяли и замораживали (-20C) для хранения до анализа ЖХ-МС/МС. Метод анализа ЖХ-МС/МС. Объединенные внутриклеточные и внеклеточные среды оттаивали, подвергали воздействию ультразвуком, перемешивали на вортексе и центрифугировали при 3000g в течение 20 мин. Супернатанты инжектировали и анализировали методом ЖХ-МС/МС. Обработка данных и "классификация". Максимальную первоначальную скорость in vitro рассчитывали для метаболитов, специфичных для цитохром Р 450, и выражали в нмоль/ч/млн клетокV=[концентрация в Т(n) - концентрация в Т(n-1)]/[Т(n)-Т(n-1)]. Определяли внутренний клиренс и выражали в мл/ч/млн клетокAUCO-24 ч: рассчитано с помощью программы WinNonlin, используя некомпартментный анализ с моделированием внутривенной болюсной инъекции. Классификация внутреннего клиренса была представлена следующим образом:Clint0,040 мл/ч 10-6 клеток: промежуточный внутренний клиренс,0,40Clint0,120 мл/ч 10-6 клеток: низкий внутренний клиренс,Clint0,040 мл/ч 10-6 клеток: высокий внутренний клиренс. Демографичская информация о доноре. Препараты на основе криоконсервированных человеческих гепатоцитов от фирмы IVT: IVT, группаTLN: мужчина, европеоидная раса, 25 лет. Препараты на основе свежих человеческих гепатоцитов: от фирмы Biopredic: НЕР 200239: женщина,раса не установлена, 58 лет. На основании этого теста величина внутреннего клиренса соединения, описанного в примере 1, составляет 0,057 мл/ч/млн клеток (классифицируется как промежуточная величина с небольшой межиндивидуальной вариабельностью). Величина внутреннего клиренса соединения, описанного в примере 2, составляет 0,055 мл/ч/млн клеток (классифицируется как промежуточная величина с небольшой межиндивидуальной вариабельностью). Другие исследования, состоящие в определении активности in vivo соединений согласно изобретению, были проведены на опухолях толстой кишки. Действие соединений согласно изобретению на опухоли толстой кишки изучалось на меланоме В 16. В качестве сравнения тестировали продукт примера 19 заявки WO 08/065282. Эффективность продукта in vivo может определяться по различным показателям, так, ее можно определить по проценту торможения роста опухоли % T/C, который представляет собой отношение между средней массой опухолей из группы, подвергшейся лечению (T), и средней массой опухолей из контрольной группы (C) на 12- или 13-й день лечения. Продукт считается активным, если отношение T/C ниже 42%, и считается, что продукт обладает высокой противоопухолевой активностью, если T/C ниже 10%. (Corbett T.H. and coll., Cancer Research, 42, 1707-1715 (1982). Чтобы показать эффективность соединения, можно также определить log10 числа погибших клеток,который находят по следующей формуле:log10 числа погибших клеток= Т-С (дни)/3,32Td,в которой Т-С означает продолжительность роста клеток, представляющую собой средний период времени, выраженный в днях, в течение которого опухоли группы (Т), подвергшейся лечению, и опухоли контрольной группы (С) достигают заранее заданного значения (например, 750 мг) и Td означает период времени, выраженный в днях, необходимый для удвоения объема опухоли у контрольных животныхMethods in Cancer Research, 17, 3-51, New-York, Academic Press Inc. (1979)]. Продукт считают активным,если log10 числа погибших клеток выше 2,8. Эффективность соединений в отношении солидных опухолей может быть определена экспериментально следующим способом. Животным, участвующим в эксперименте, обычно мышам-самкам линии C57BL/6, прививали билатерально подкожным путем от 30 до 60 мг фрагмента опухоли человека В 16 (референсная опухоль) в день 0. Прежде чем животных, являющихся носителями опухоли, подвергнуть различным обработкам и контролю, их рандомизировали. В случае лечения опухолей согласно изобретению начинали лечение на ранней стадии, спустя 3-4 дня после имплантации. Животные, подвергшиеся лечению соединениями,имели массу примерно 20 г. Животных, являющихся носителями опухоли, также подвергали таким же обработкам, но с использованием только одного эксципиента для того, чтобы можно было отделить токсический эффект на опухоль самого эксципиента от эффекта, связанного с химиотерапией. Соединения вводили пероральным путем в дозах, указанных в таблицах, с использованием эксципиентов, указанных в таблицах, при двухразовом введении в день. Эти введения осуществляли в течение 8-11 дней в зависимости от исследования, после имплантации опухоли. Опухоли измеряли два или три раза в неделю до тех пор, пока опухоль не достигла примерно 2 г или пока животное не умерло, если оно выживало до достижения массы опухоли 2 г. Животных умерщвляли и проводили вскрытие. Противоопухолевую активность определяли по различным нормативным параметрам. В следующих таблицах приведены в качестве примеров результаты, полученные с соединениями согласно изобретению, используемыми в оптимальной дозе. Таким образом, изобретение относится к лекарственным средствам, которые содержат соединение формулы (I) или аддитивную соль этого соединения с фармацевтически приемлемой кислотой. Согласно другому аспекту изобретение относится к соединениям, отвечающим формуле (I), для применения их в качестве лекарственного средства при лечении рака. Соединения согласно изобретению могут быть также использованы для получения лекарственных средств, в частности для лекарственных средств, являющихся ингибиторами ангиогенеза. Эти лекарственные средства находят свое применение в терапии, в частности для лечения раковых опухолей, в частности солидных опухолей. Согласно другому аспекту изобретение относится к фармацевтическим композициям, содержащим в качестве активного начала соединение согласно изобретению. Эти фармацевтические композиции содержат эффективную дозу по меньшей мере одного соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент. Упомянутые эксципиенты выбирают, в зависимости от лекарственной формы и желаемого способа введения, из обычных эксципиентов, которые известны специалисту. В фармацевтических композициях согласно изобретению, предназначенных для перорального, сублингвального, подкожного, внутримышечного, внутривенного, топического, локального, интратрахеального, интраназального, трансдермального или ректального введения, активное начало формулы (I), описанной выше, или его соль может быть введено в виде единичной формы введения в смеси с классическими фармацевтическими эксципиентами животным и людям для лечения нарушений или заболеваний,перечисленных выше. Соответствующие единичные формы введения включают формы для перорального ведения, такие как таблетки, мягкие или твердые желатиновые капсулы, порошки, гранулы и растворы или суспензии для приема внутрь, формы для сублингвального, буккального введения, формы для трансдермального,подкожного, внутримышечного или внутривенного введения. В качестве примера единичная форма введения соединения согласно изобретению в форме таблетки может содержать следующие компоненты: Для перорального пути доза активного начала, вводимая ежедневно, может достигать 1200 мг/кг за один или несколько приемов. Возможны особые случаи, когда подходят более высокие или более низкие дозировки, такие дозировки не выходят за рамки изобретения. Согласно обычной практике дозировка, подходящая для каждого пациента, определяется врачом в зависимости от способа введения, веса и восприимчивости указанного пациента. Изобретение согласно другому его аспекту относится также к способу лечения указанных выше патологий, который включает прием пациентом эффективной дозы соединения согласно изобретению или одной из его фармацевтически приемлемых солей. Таблица 2 Оценка активности соединения примера 1 против мышиной меланомы B16 на ранней стадии развития ВСМ-1948. Время удвоения опухоли = 0,8 дня. Состав: пример 1 = 98% ПЭГ 200, 2% PS80.b Доза уменьшена на 7% вследствие осложнения на 7 день. с Расчет сделан для 9 мышей вследствие неожиданной смерти на 8 день. п.о.=перорально. Таблица 3 Оценка активности соединения примера 2 против мышиной меланомы B16 на ранней стадии развития ВСМ-1979. Время удвоения опухоли = 0,9 дня. Среднее время, при котором опухоли из группы эксципиентов достигают 750 мг = 11,9 дней. Состав: пример 2 = 20% лабразола, 5% солютола, 75% глюкозы, 5% воды. Таблица 4 Оценка активности соединения примера 19 заявки WO 08/065282 против мышиной меланомы B16 на ранней стадии развития ВСМ-1755. Время удвоения опухоли = 1,1 дня. Состав: пример 3 = 40% каптизол в воде, рН 3. Продолжительность лечения: пример 3 = 11 дней.b Общая доза была пересчитана после анализа маточного раствора путем ВЭЖХ в дни 3, 4 и 11-13. с Неожиданная смерть, эффективность рассчитана для 6 животных. в которой X означает хлор или фтор,в форме основания или кислотно-аддитивной соли с фармацевтически приемлемой кислотой. 2. Соединение формулы (I) по п.1, отличающееся тем, что X означает хлор, в форме основания или кислотно-аддитивной соли с фармацевтически приемлемой кислотой. 3. Способ получения соединения формулы (I) по любому из пп.1, 2, отличающийся тем, что соединение где X представляет хлор или фтор,в присутствии диизопропилэтиламина в инертной среде, затем на второй стадии удаляют защитную группу амина паратолуолсульфоновой кислотой. 4. Способ по п.3, отличающийся тем, что инертной средой является апротонная аполярная среда. 5. Способ по п.4, отличающийся тем, что апротонной аполярной средой является тетрагидрофуран. 6. Соединение формулы где X означает хлор или фтор. 7. Соединение формулы где X означает хлор или фтор. 8. Противораковое лекарственное средство, отличающееся тем, что оно содержит соединение формулы (I) по любому из пп.1, 2 или аддитивную соль этого соединения с фармацевтически приемлемой кислотой. 9. Противораковая фармацевтическая композиция, отличающаяся тем, что она включает соединение формулы (I) по любому из пп.1, 2 или фармацевтически приемлемую соль этого соединения, а также по меньшей мере один фармацевтически приемлемый эксципиент. 10. Применение соединения формулы (I) по любому из пп.1, 2 в качестве лекарственного средства для лечения рака. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: C07D 231/38, A61K 31/415, A61P 35/00, C07C 275/30
Метки: способ, терапии, пиразолов, производные, получения, применение
Код ссылки
<a href="https://eas.patents.su/15-20364-proizvodnye-pirazolov-sposob-ih-polucheniya-i-primenenie-v-terapii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразолов, способ их получения и применение в терапии</a>
Производные азакарболинов, способ их получения и применение в терапии

Номер патента: 18945
Опубликовано: 29.11.2013
Авторы: Муркрофт Нейл, Ли Жунхуа, Бедель Оливье, Левит Михаил, Гуйон Тьерри, Миньяни Серж, Бабэн Дидье, Арендт Кристофер, Папэн Давид
МПК: A61K 31/437, C07D 471/14, A61P 35/00...
Метки: способ, получения, производные, терапии, азакарболинов, применение
Формула / Реферат:
1. Соединения общей формулы (I)в которой R3, R4 могут независимо один от другого обозначать:1) Н;2) F;3) Cl;4) Br;5) I;6) OR2a;7) NR1aR1b;8) COR2a;9) CO2R2a;10) CO(NR1aR1b);11) С1-C10-алкил, линейный, или разветвленный, или циклический (С3-С7), необязательно моно-, или ди-, или тризамещенный R2a, R2b, R2c;12) С2-С6-алкенил, линейный или разветвленный, необязательно моно-, или ди-, или тризамещенный R2a, R2b, R2c;13) арил, выбранный из фенила,...
Производные 6-гетероарилпиридоиндолона, способ их получения и применение в терапии

Номер патента: 14873
Опубликовано: 28.02.2011
Авторы: Чиапетти Паола, Мюно Иветт, Жегам Самир, Казелла Пьер, Бурри Бернар, Вермют Камилль Жорж, Дерок Жан-Мари
МПК: A61P 35/00, C07D 471/04, A61K 31/4375...
Метки: способ, производные, получения, терапии, применение, 6-гетероарилпиридоиндолона
Формула / Реферат:
1. Соединение формулы (I)в которойR1 обозначает атом водорода или (С1-С4)алкильную группу, CN, CF3 или CHF2;R2 обозначает атом водорода или (С1-С4)алкильную группу;R3 обозначает фенил, не замещенный или замещенный одним или несколькими заместителями, выбираемыми независимо из атома галогена, (С1-С4)алкильной группы, (С1-С4)алкоксильной группы;R4 обозначает гетероциклический радикал, выбираемый изR5 обозначает атом водорода, атом галогена,...
Производные диоксан-2-алкилкарбамата, способ их получения и применение в терапии

Номер патента: 8218
Опубликовано: 27.04.2007
Авторы: Медеско Флоренс, Абуабделла Ахмед, Баз Мишель, Хорнер Кристиан, Ли Адриен Так, Даргазанли Джихад
МПК: C07D 319/06, A61P 25/08, A61K 31/335...
Метки: производные, применение, способ, терапии, получения, диоксан-2-алкилкарбамата
Формула / Реферат:
1. Соединение, соответствующее формуле (I) в которой R1 представляет собой фенильную или нафталинильную группу, возможно замещенную одним(ой) или более атомами галогена или группами гидрокси, циано, нитро, (С1-С3)алкил, (С1-С3)алкокси, трифторметил, трифторметокси, бензилокси, (С3-С6)циклоалкил-O- или (С3-С6)циклоалкил(С1-С3)алкокси; R2 представляет собой либо группу общей формулы CHR3CONHR4, в которой R3 представляет собой атом водорода или...
Производные пиридино-пиридинонов, способ их получения и применение в терапии

Номер патента: 19362
Опубликовано: 31.03.2014
Авторы: Мартен Валери, Белльверг Патрис, Волль-Шаллье Сесиль, Лассалль Жильбер, Маккорт Гари, Сави Пьер
МПК: A61K 31/4375, C07D 471/04, A61P 35/00...
Метки: получения, пиридино-пиридинонов, способ, производные, терапии, применение
Формула / Реферат:
1. Соединение формулы (I)в которой R1 означает (C1-C4)алкильную группу;R2 означает -(СН2)n'-В, где n'=0, 1, 2, 3, 4 и В является (С3-С5)циклоалкильной группой или (С1-С4)алкильной группой, необязательно замещенной одним или несколькими атомами фтора, (С1-С4)алкоксигруппой;U означает карбонильную группу или группу -СН2-;Y, Z, V и W обозначают, независимо один от другого, группу -СН- или атом углерода, замещенный группой R7, или гетероатом, такой...
Производные 2-карбамид-4-фенилтиазола, способ их получения и их применение в терапии
Номер патента: 11076
Опубликовано: 30.12.2008
Авторы: Жегам Самир, Казелла Пьер, Фресс Пьер, Флутар Даниель
МПК: A61K 31/551, A61K 31/427, A61K 31/496...
Метки: применение, 2-карбамид-4-фенилтиазола, производные, способ, получения, терапии
Формула / Реферат:
1. Соединение общей формулы (I) в которой (i) R1 выбран из -О-(C1-C8)алкила; (ii) R2 выбран из группы, состоящей (C1-C8)алкила, перфтор(C1-C4)алкила, (С3-C6)циклоалкила и -О-(C1-C8)алкила; (iii) Y обозначает атом водорода или атом галогена; (iv) R3 обозначает: (a1) группу формулы -(CH2)р-А, в которой р обозначает 0, 1, 2, 3 или 4, и, если р обозначает 2, 3 или 4, то А обозначает группу формулы или -NR4R5, в которой R6 выбран из группы,...
Предыдущий патент: Способ изомеризации парафиновых углеводородов с4-с7
Следующий патент: Клапанное устройство газового прибора
Случайный патент: Геотехническое изделие и способ его изготовления