2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4н-[1,2,4]триазол-3-илсульфанил] уксусная кислота и ее метиловый эфир
Номер патента: 15846
Опубликовано: 30.12.2011
Авторы: Хун Чжи, Гуник Эсмир, Ким Хонг Воо, Жирарде Жан-Люк, Де Ла Роза Марта, Кох Юнг-Хио, Лэнг Стенли











Формула / Реферат
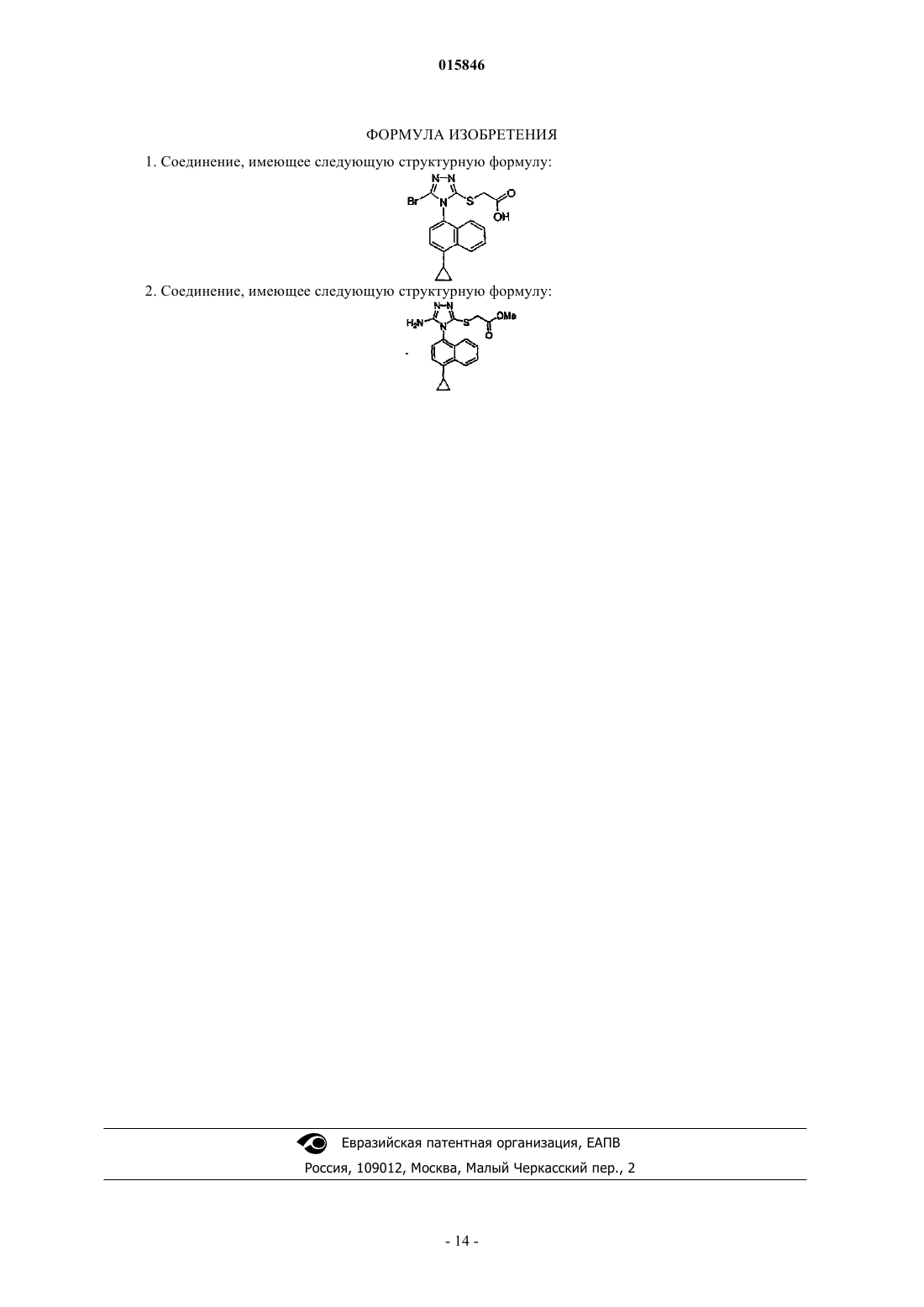
1. Соединение, имеющее следующую структурную формулу:

2. Соединение, имеющее следующую структурную формулу:

Текст
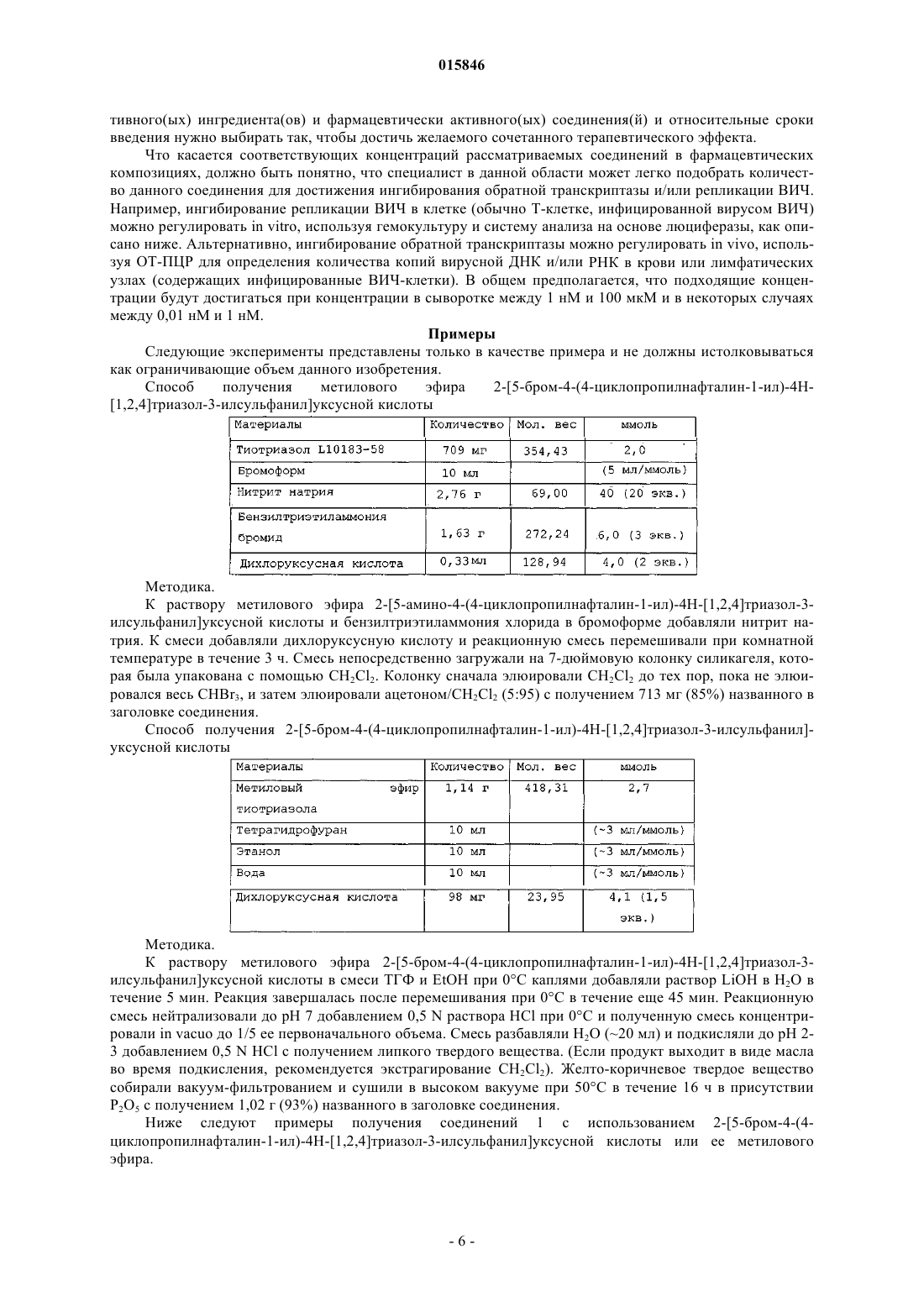
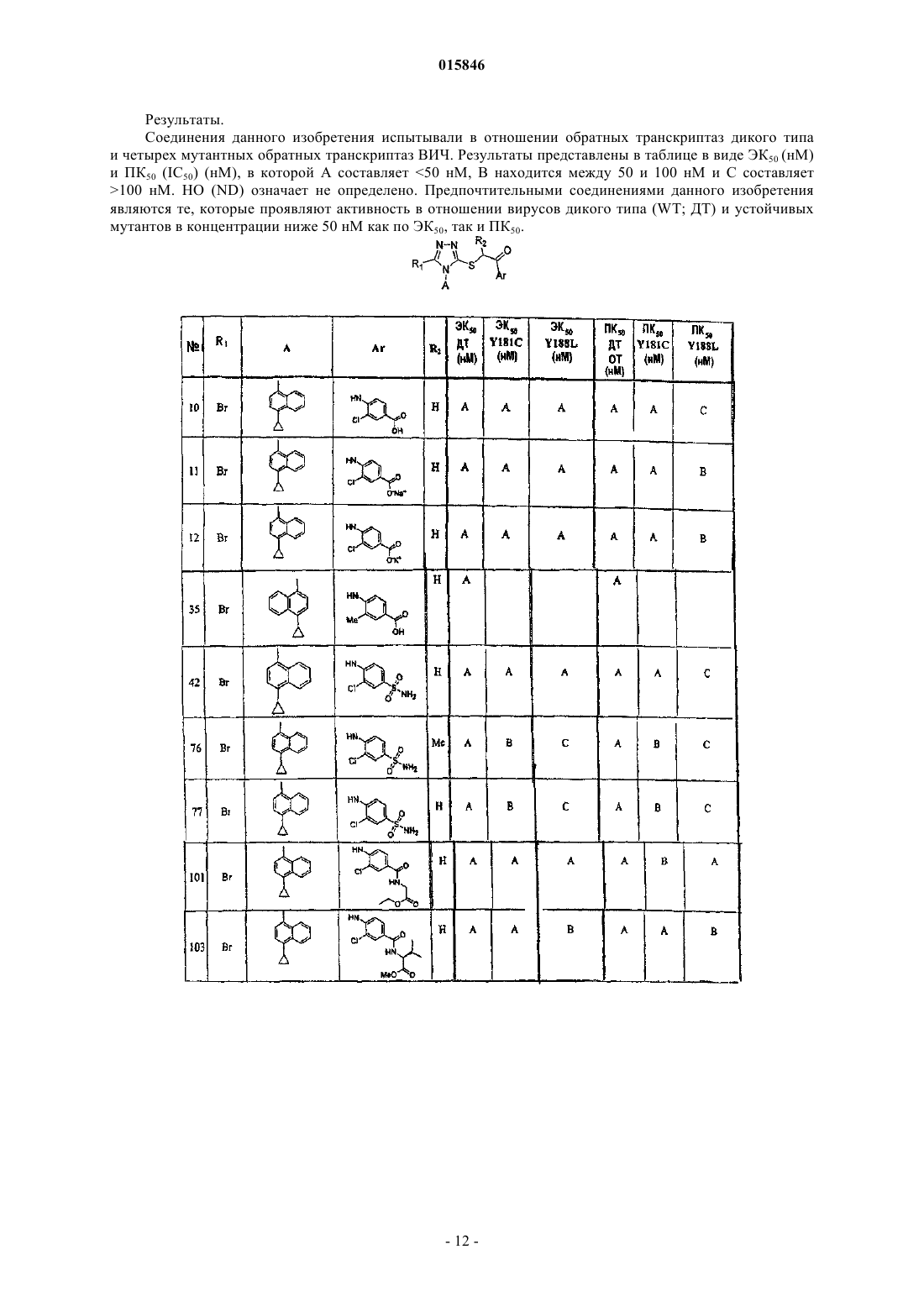
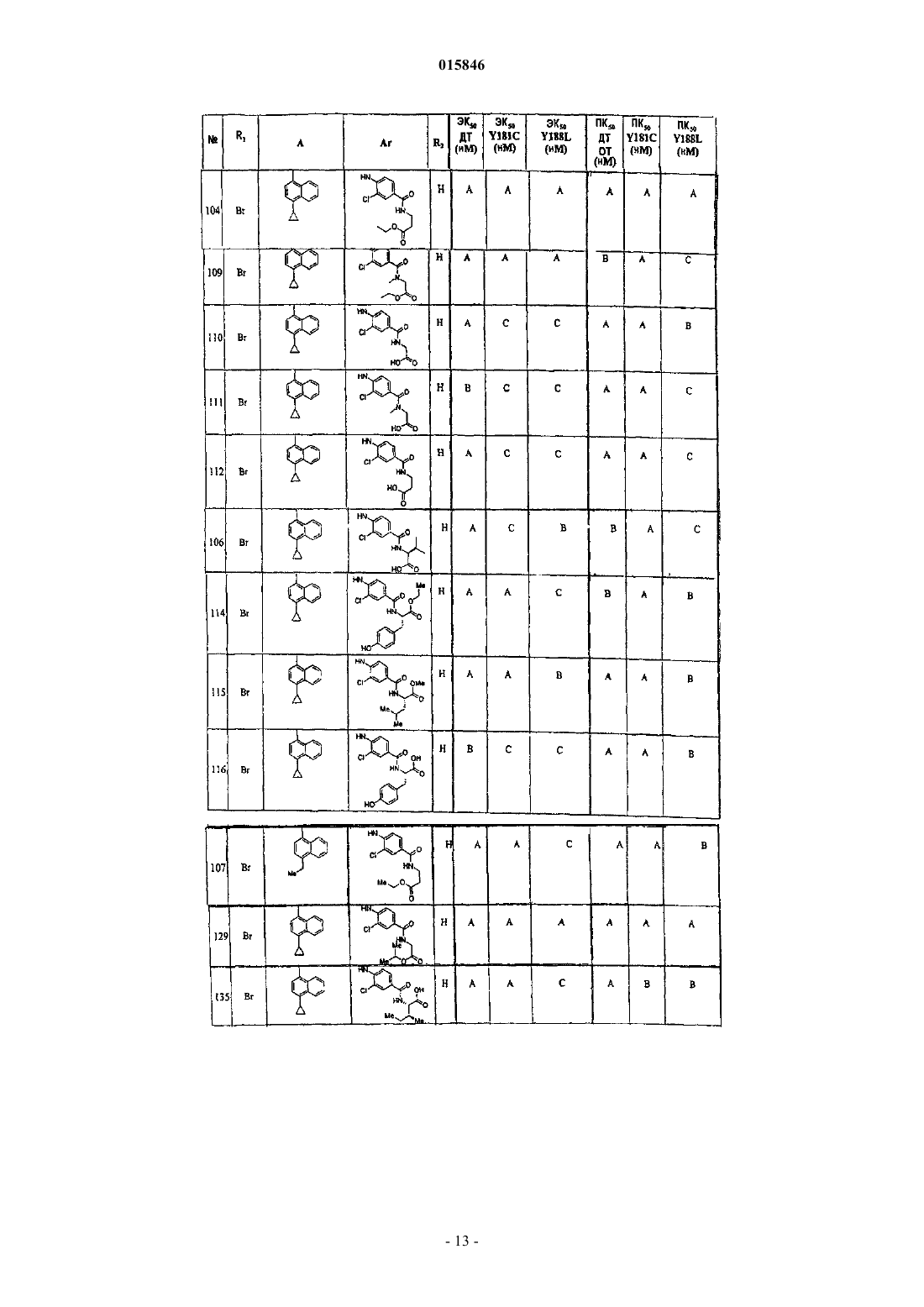
Дата публикации и выдачи патента Номер заявки Представлены 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил] уксусная кислота и ее метиловый эфир, предназначенные для получения S-триазолил-меркаптоацетанилидов, которые ингибируют разные виды обратной транскриптазы ВИЧ и пригодны для лечения вызываемых ВИЧ-инфекций. Жирарде Жан-Люк, Кох Юнг-Хио,Де Ла Роза Марта, Гуник Эсмир, Хун Чжи, Лэнг Стенли, Ким Хонг Воо (US) Медведев В.Н. (RU) 015846 Настоящее изобретение относится к промежуточным соединениям, предназначенным для получения ингибиторов ферментов, которые используются для лечения заболевания, более конкретно, к подавлению in vitro и in vivo обратной транскриптазы ВИЧ в качестве способа лечения инфекции, вызванной ВИЧ. Специалистам известны многочисленные методы лечения в отношении ВИЧ, и среди других фармацевтически активных соединений ингибиторы обратной транскриптазы давали значительный терапевтический эффект у многих инфицированных ВИЧ пациентов. Например, относительно хорошо переносятся антиретровирусные лекарственные средства ламивудин (3 ТС) или зидовудин (AZT). Однако недавно появились многочисленные вирусные штаммы с заметной устойчивостью к этим соединениям. Чтобы преодолеть резистентность, по меньшей мере до некоторой степени, можно применить новые ингибиторы нуклеозидного типа (отдельно или в сочетании с другими ингибиторами нуклеозидного типа),и примеры альтернативных лекарственных средств включают ставудин (d4T), диданозин (ddI),Combivir (известная торговая марка для комбинации ламивудина и зидовудина) и Trizivir (известная торговая марка для комбинации из 3 ТС, AZT и абакавира). К сожалению, развитие устойчивости к одному ингибитору нуклеозидного типа часто сопровождается развитием некоторой степени устойчивости к другому ингибитору нуклеозидного типа, часто делая необходимым переключение на другую группу лекарственных средств. В таких случаях пациент может получать ингибитор протеазы (например, саквинарин, индинавир, нелфинавир и т.д.), обычно в сочетании с другими противоретровирусными средствами. Однако относительно сложный режим введения таких комбинаций часто вызывает неудовольствие у многих пациентов вследствие сложности режима приема и финансовых проблем и соблюдение режима часто меньшее, чем желательно. Совсем недавно лечение ВИЧ фокусировалось на комбинированной терапии, которая включала введение нуклеозидных ингибиторов обратной транскриптазы с ингибиторами протеаз и с ненуклеозидными ингибиторами обратной транскриптазы и тройных комбинаций, включающих нуклеозидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы и ингибиторы протеаз. К сожалению, комбинированная терапия ингибиторами протеаз с нуклеозидными ингибиторами обратной транскриптазы часто плохо переносится и часто приводит к преждевременному окончанию терапии. Поэтому наиболее современные методы лечения включают сочетание нуклеозидных ингибиторов обратной транскриптазы и ненуклеозидных ингибиторов обратной транскриптазы. Ингибиторы ненуклеозидного типа (например, невирамина, делавирдина и эфавиренз) являются структурно неоднородной группой соединений, которые, как полагают, связываются в ненуклеозидный карман из обратных транскриптаз. Они значительно повышают противовирусную эффективность при совместном введении с ингибиторами нуклеозидного типа. Хотя ингибиторы ненуклеозидного типа, повидимому, представляют перспективную группу противовирусных лекарственных средств, все еще остается несколько недостатков. Стоимость известных в настоящее время ингибиторов ненуклеозидного типа относительно высока, и единственная мутация в вирусных обратных транскриптазах может вызвать перекрестную резистентность к широкой группе ненуклеозидных ингибиторов обратной транскриптазы. Поэтому существует острая необходимость получения новых ненуклеозидных ингибиторов обратной транскриптазы, которые обладают сильным противовирусным действием, в частности, против мутантных штаммов ВИЧ, которые проявляют устойчивость к известным в настоящее время ненуклеозидным ингибиторам обратной транскриптазы. Вирус ВИЧ имеет относительно высокую частоту мутации, которая часто приводит к лекарственной резистентности к современным средствам лечения. Были проведены исследования по идентификации спектра мутаций в RT протеинах вирусов, выделенных от пациентов, у которых была безуспешной терапия с применением по меньшей мере одного ННИОТ, и результаты показали, что мутант K103N был наиболее широко распространенным у пациентов, принимающих эфавиренз, тогда как Y181C был преобладающим у пациентов, принимающих невирапин. Другие единичные мутации включали K101E,G190S/A/E и Y188L/C. Некоторые из наиболее часто встречающихся двойных мутаций у пациентов, у которых был неэффективен эфавиренз, включают K103N-P225H, K103N-V108I, K103N-K101Q, K103NL100I, K103N-F227L, V106I-Y188L, K103N-Y188L и K103N-G190A. Существует потребность в получении новых композиций и способов подавления этих и других мутантных обратных транскриптаз. Заявка на данное изобретение относится к работе, раскрытой ранее в находящихся в общей собственности заявках PCT/US02/26186, поданной 23 августа 2002 г., неопубликованной, и PCT/US03/27433,поданной 22 августа 2003 г., которая опубликована как WO 2004/030611 от 15 апреля 2004 г. В патенте США 5939462, Connell et al., описано большое число замещенных гетероциклов, пригодных в качестве антагонистов NPY5, некоторые из которых являются S-триазолилмеркаптоацетанилидами, подобными соединениям с общей структурой 1, приведенной ниже. Simoneau et al. в международной патентной публикации WO 2004/050643 описывают тетразолы и несколько триазолов, имеющих строение, подобное строению соединений данного изобретения, обладающих активностью ингибирования обратной транскриптазы.-1 015846 Краткое описание Было открыто, что обратная транскриптаза (RT, ОТ) ВИЧ может быть ингибирована указанной группой S-триазолилмеркаптоацетанилидов, представляемых общей структурой 1. Неожиданно, что некоторые из этих соединений способны подавлять разные мутантные ОТ, включая K103N, Y181C и где Q выбран из группы, состоящей из СО 2 Н или его соли и CONR'R", где R' и R" независимо выбраны из группы, состоящей из Н,C1-6 алкила, C1-6 алкила, замещенного одним или более из групп OR, CO2R, NHR, NR2 или CF3, где R является Н или C1-6 алкилом, или R' и R" вместе с атомом азота, с которым они связаны, образуют 4-, 5- или 6 членное гетероциклическое кольцо;SO3H или его соли и SO2NR'R", где R' и R" независимо выбраны из группы, состоящей изC1-6 алкила, C1-6 алкила, замещенного одним или более из групп OR, CO2R, NHR, NR2 или CF3, где R является Н или C1-6 алкилом, или R' и R" вместе с атомом азота, с которым они связаны, образуют 4-, 5- или 6 членное гетероциклическое кольцо; Р представляет собой группуR0 выбран из группы, состоящей из Cl, Br, CF3 и метила. Данные соединения ингибируют обратную транскриптазу ВИЧ in vitro и in vivo. Термин "ингибирование обратной транскриптазы" относится к прямому или непрямому снижению образования ДНК по матричной РНК или ДНК с помощью обратной транскриптазы. Прямое ингибирование включает самоуничтожение, конкурентное и неконкурентное ингибирование, аллостерическое ингибирование или связывание ингибитора в ненуклеозидном кармане. Примеры непрямого ингибирования включают истощение нуклеозидов для синтеза ДНК, индукцию или содействие конформационным изменениям и т.д. В соответствии с описанием термин "снижение вирусного воспроизводства" означает, что титр вируса в образце снижается. Снижение может осуществляться разными способами, включая частичное или полное ингибирование репликации вируса, частичное или полное ингибирование процессинга или вирусных протеинов или сборки, ингибирование проникновения в клетки или выхода вируса из инфицированной клетки и/или выведение вируса из системы путем иммунного ответа на вирус. Настоящее изобретение относится к промежуточным соединения 2-[5-амино-4-(4 циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]уксусной кислоты и ее метиловому эфиру следующих структурных формул Синтез соединений. Синтез соединений данного изобретения может быть выполнен в соответствии со следующими методиками, которые, по существу, описаны в WO 2004/030611, WO 2004/050643 и US 5939462. Должно быть признано, однако, что возможны многие альтернативные пути синтеза для соединений данного изобретения. Следующие характерные пути представлены в качестве примера в качестве руководства для специалистов-практиков в области химии органического синтеза. Общая стратегия показана на схеме 1b. Этот подход включает ацилирование анилинов с помощьюS-триазолилмеркаптоуксусных кислот, которые легко приготовить путем алкилирования меркаптотриазолов с помощью -галогенуксусной кислоты или сложного эфира. Подходящие реагенты включают, но не ограничиваются этим, йодуксусную кислоту, метилбромацетат и этилбромпропионат, когда желательно, чтобы R3 являлся метилом. Если используют сложный эфир, его гидролизуют после S-алкилирования для получения свободной карбоновой кислоты. Эти кислота и анилин могут быть связаны с помощью любого из обычных активирующих карбоксил реагентов или смесей реагентов, например карбодиимида в присутствии основания третичного амина, необязательно, с N-гидроксибензотриазолом в качестве катализатора, или тионил- или оксалилхлорида с диметиламинопиридином в качестве катализатора. Эта схема является выгодной, когда анилин является важным относительно триазола. Пример синтеза показан на схеме, ниже, при котором анилин ацилируется с помощью предварительно полученной S-триазолилмеркаптоуксусной кислоты. Схема Фармацевтические композиции. Промежуточные соединения согласно настоящему изобретению предназначены для получения указанного выше соединения 1, обладающего ингибирующей обратную транскриптазу ВИЧ in vitro и in vivo активностью. Когда соединения 1 применяют в составе фармацевтической композиции, предполагается, что соответствующие соединения могут быть смешаны с фармацевтически приемлемыми носителями, наполнителями и другими добавками. Особенно предпочтительно, когда соединения данного изобретения включены в фармацевтическую композицию, которую изготавливают со смешением с одним или более нетоксичными фармацевтически приемлемыми носителями. Фармацевтические композиции могут быть изготовлены по рецептуре для перорального приема в твердой или жидкой форме, для парентеральных инъекций или для ректального введения. Фармацевтические композиции можно применять людям и другим животным перорально, ректально, парентерально, интравагинально, внутрибрюшинно, местно, защечно или в виде жидкости для орального или назального впрыскивания. Термин "парентеральное введение" в соответствии с данным описанием относится к способам введения, которые включают, но не ограничиваются этим, внутривенные,внутримышечные, подкожные и внутрисуставные инъекции и вливание. Фармацевтические композиции для парентеральных инъекций и вливания предпочтительно включают фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки для получения из них стерильных инъекционных растворов или дисперсий непосредственно перед использованием. Примеры подходящих водных и неводных носителей, разбавителей, растворителей и жидких сред включают воду, этанол, полиолы, такие как глицерин,пропиленгликоль, полиэтиленгликоль и т.п.,и их подходящие смеси, растительные масла, такие как оливковое масло, и инъекционные органические сложные эфиры, такие как этилолеат. Надлежащая текучесть может поддерживаться, например, путем использования покрывающих материалов, таких как лецитин,путем сохранения необходимого размера частиц в случае дисперсий или путем использования поверхностно-активных веществ.-3 015846 Композиции могут также содержать такие добавки, как консерванты, улучшающие смачивание вещества, эмульгирующие вещества и диспергирующие вещества. Профилактика действия микроорганизмов может быть обеспечена путем включения разных антибактериальных и противогрибковых средств,например парабена, хлорбутанола, фенолсорбиновой кислоты и т.п. Может быть также желательно включать создающие изотоничность вещества, такие как сахара, хлорид натрия и т.п. Пролонгированное всасывание в инъекционной фармацевтической форме можно придать путем включения веществ, которые замедляют всасывание, таких как моностеарат алюминия и желатин. Чтобы продлить действие соединения, получаемого с использованием промежуточных соединений данного изобретения, может быть желательно замедлить всасывание лекарственного средства после подкожной или внутримышечной инъекции. Это может быть выполнено путем использования жидкой суспензии кристаллического или аморфного вещества с плохой растворимостью в воде. Скорость всасывания соединения тогда зависит от степени его растворения, которая, в свою очередь, может зависеть от размера кристаллов и кристаллической формы. Альтернативно, замедленное всасывание вводимого парентерально соединения данного изобретения может достигаться путем растворения или суспендирования лекарственного средства в масляном носителе. Инъекционные депоформы изготавливают путем формирования унитарных или состоящих из микрочастиц матриц соединения данного изобретения в биоразрушаемых полимерах, включая, но не ограничиваясь этим, полилактид-полигликолид, поли(ортоэфиры) и поли(ангидриды). Скорость высвобождения лекарственного средства можно регулировать путем изменения отношения лекарственного средства к полимеру и природы конкретного применяемого полимера. Инъекционные депопрепараты можно также получать путем заключения данного соединения в липосомы или микроэмульсии, которые совместимы с тканями организма. Твердые дозированные формы для перорального приема включают, но не ограничиваются этим,капсулы, таблетки, пилюли, порошки, драже и гранулы. В таких твердых дозированных формах активное соединение смешано по меньшей мере с одним инертным фармацевтически приемлемым вспомогательным веществом или носителем, таким как цитрат натрия или двухзамещенный фосфат кальция и/или(а) наполнители или разбавители, такие как разные виды крахмала, лактоза, сахароза, глюкоза, маннит и кремниевая кислота; (b) связывающие вещества, такие как карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и аравийская камедь; (с) гигроскопические вещества, такие как глицерин; (d) дезинтеграторы, такие как агар-агар, карбонат кальция, картофельный крахмал или крахмал из тапиоки, альгиновая кислота, некоторые силикаты и карбонат натрия; (е) раствор замедляющих веществ, таких как парафин; (f) ускорители всасывания, такие как соединения четвертичного аммония;(g) улучшающие смачивание вещества, такие как цетиловый спирт и моностеарат глицерина; (h) абсорбенты, такие как каолин и бентонитовая глина и (i) улучшающие скольжение вещества, такие как тальк,стеарат кальция, стеарат магния, твердые полизтиленгликоли, лаурилсульфат натрия и их смеси. Твердые дозированные формы могут также включать буферные вещества. Твердые композиции можно также использовать в качестве заполнителей мягких и твердых желатиновых капсул с использованием таких наполнителей, как лактоза или молочный сахар, а также полиэтиленгликоли с высоким молекулярным весом и т.п. Твердые дозированные формы могут быть изготовлены с покрытиями или оболочками, такими как энтеральные покрытия и другие покрытия, хорошо известные специалистам в области фармацевтической технологии. Они могут, необязательно, содержать замутняющие вещества и могут также состоять из композиции, такой что они высвобождают активный(е) ингредиент(ы) только, или преимущественно, в определенной части кишечного тракта, по желанию замедленным образом. Данные активные соединения могут быть представлены также в микроинкапсулированной форме. Жидкие дозированные формы для перорального приема включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям жидкие дозированные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как вода и другие растворители, солюбилизирующие средства и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид, масла (в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана и их смеси. Пероральные жидкие композиции могут также включать вспомогательные вещества, такие как улучшающие смачивание вещества, эмульгирующие и суспендирующие вещества, подкрашивающие вещества, подсластители,вкусовые добавки и отдушки. Композиции для ректального или вагинального введения предпочтительно представляют суппозитории, которые можно изготовить путем смешивания соединений данного изобретения с подходящими нераздражающими наполнителями или носителями, такими как масло какао, полиэтиленгликоль или другие суппозиторные воски, которые являются твердыми при комнатной температуре, но жидкими при температуре тела и поэтому плавятся в прямой кишке или вагинальной полости и высвобождают активное соединение.-4 015846 Соединения 1 можно вводить также в форме липосом. Как известно специалистам, липосомы обычно получают из фосфолипидов или других липидных веществ. Липосомы обычно образованы моно- или многослойными гидратированными жидкими кристаллами, которые диспергируют в водной среде. Можно использовать любой нетоксичный физиологически приемлемый липид, способный к образованию липосом. Композиции в липосомной форме могут содержать, в дополнение к соединению данного изобретения, стабилизаторы мембраны, консерванты, наполнители и т.п. Предпочтительными липидами являются фосфолипиды и фосфатидилхолины (лецитины) как природные, так и синтетические. Способы формирования липосом известны специалистам. См., например, Prescott, Ed., Methods in Cell Biology,Volume XIV, Academic Press, New York, N.Y. (1976), p. 33 et seq. Соединения 1 можно использовать в форме фармацевтически приемлемых солей, получаемых с неорганическими или органическими кислотами. Под "фармацевтически приемлемыми солями" подразумеваются те соли, которые являются пригодными для использования при контакте с тканями людей и более низших животных без неприемлемой токсичности, раздражения, аллергической реакции и т.п. и соответствуют рациональному соотношению пользы/риска. Фармацевтически приемлемые соли хорошо известны специалистам. Например, S.M. Berge, et al. описывают фармацевтически приемлемые соли подробно в J. Pharmaceutical Sciences, 1977, 66:1 et seq. Данные соли могут быть получены in situ при окончательном выделении и очистке соединений данного изобретения или отдельно путем взаимодействия формы свободного основания с соответствующей кислотой. Типичные соли с присоединением кислоты включают, но не ограничиваются этим, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, цитрат, глюконат, глютамат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид,2-гидроксиэтансульфонат(изотионат),лактат,малеат,метансульфонат,никотинат,2-нафталинсульфонат, оксалат, памоат, пектинат, 3-фенилпропионат, фосфат, пивалат, пропионат, сукцинат, сульфат, тартрат, бикарбонат, п-толуолсульфонат и ундеканоат. Основные содержащие азот группы могут быть также четвертичными с такими реагентами, как галогениды низших алкилов, таких как метил-, этил-, пропил- и бутилхлориды, -бромиды и -йодиды; диалкилсульфаты, подобные диметил-,диэтил- и диамилсульфатам; галогениды с длинной цепью, такие как децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -йодиды; арилалкилгалогениды, подобные бензил- и фенетилбромидам, и другие. Таким образом, получают растворимые в воде или масле или диспергируемые продукты. Аддитивные соли оснований можно получить при конечном выделении и очистке соединений данного изобретения, или позже, путем реакции содержащего группу карбоновой кислоты соединения с подходящим основанием, таким как гидроксид, карбонат или бикарбонат с фармацевтически приемлемым катионом металла или аммония, или органического первичного, вторичного или третичного амина. Фармацевтически приемлемые соли включают, но не ограничиваются этим, соли щелочных и щелочноземельных металлов, таких как литий, натрий, калий, кальций, магний и алюминий и т.п., и нетоксичные соли четвертичного аммония и аминов, включающих аммоний, тетраметиламмоний, тетраэтиламмоний,метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин и т.п. Другие типичные органические амины, пригодные для образования солей с присоединением основания, включают этилендиамин, этаноламин, диэтаноламин, пиперидин, пиперазин, глюкозамин, лейцин и т.п. Фактические дозы активных ингредиентов в фармацевтических композициях могут изменяться так,чтобы получить количество активного(ых) соединения(ий), которое эффективно для достижения желаемой терапевтической реакции у конкретного пациента, для конкретных композиции и способа введения. Выбранный уровень дозы будет зависеть от активности конкретного соединения, пути введения, режима дозировки, тяжести патологического состояния, которое нужно лечить, и состояния здоровья и предыдущей истории заболеваний пациента, которого нужно лечить. Исследования по выбору доз стандартны,и специалист имеет возможность начать с доз данного соединения на уровнях ниже необходимого для достижения желаемого терапевтического эффекта и постепенно повысить дозировку до тех пор, пока не будет достигнут желаемый эффект. Обычно перорально пациенту-млекопитающему назначают уровни дозы от примерно 0,1 до примерно 100, более предпочтительно от примерно 5 до примерно 50 мг активного соединения на 1 кг веса тела в сутки. Если желательно, эффективная суточная доза может быть разделена на несколько доз, например от двух до четырех отдельных доз в сутки. Соединения 1 можно вводить отдельно или в сочетании с другими средствами для лечения ВИЧ. Конкретно, предполагаемые дополнительные соединения включают ингибиторы обратной транскриптазы нуклеозидного типа (например, ламивудин, зидовудин, ставудин, абакавир, тенофовир или диданозин), ингибиторы обратной транскриптазы ненуклеозидного типа (например, невирапин, делавирдин,эфавиренз), ингибиторы протеазы (например, ритонавир, саквинавир, индинавир, нелфинавир), ингибиторы слияния (например, энфувиртид), антагонисты CCR5, иммунотерапевтические средства (например,рибавирин, IL-2) и активные, пассивные и/или терапевтические вакцины. Комбинированная терапия включает введение по меньшей мере одного соединения данного изобретения или его функционального производного и по меньшей мере одного другого фармацевтически активного ингредиента. Активный(е) ингредиент(ы) и фармацевтически активные вещества можно вводить отдельно или вместе, когда их вводят отдельно, это может происходить одновременно или отдельно в любом порядке. Количество ак-5 015846 тивного(ых) ингредиента(ов) и фармацевтически активного(ых) соединения(й) и относительные сроки введения нужно выбирать так, чтобы достичь желаемого сочетанного терапевтического эффекта. Что касается соответствующих концентраций рассматриваемых соединений в фармацевтических композициях, должно быть понятно, что специалист в данной области может легко подобрать количество данного соединения для достижения ингибирования обратной транскриптазы и/или репликации ВИЧ. Например, ингибирование репликации ВИЧ в клетке (обычно Т-клетке, инфицированной вирусом ВИЧ) можно регулировать in vitro, используя гемокультуру и систему анализа на основе люциферазы, как описано ниже. Альтернативно, ингибирование обратной транскриптазы можно регулировать in vivo, используя ОТ-ПЦР для определения количества копий вирусной ДНК и/или РНК в крови или лимфатических узлах (содержащих инфицированные ВИЧ-клетки). В общем предполагается, что подходящие концентрации будут достигаться при концентрации в сыворотке между 1 нМ и 100 мкМ и в некоторых случаях между 0,01 нМ и 1 нМ. Примеры Следующие эксперименты представлены только в качестве примера и не должны истолковываться как ограничивающие объем данного изобретения. Способ получения метилового эфира 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н[1,2,4]триазол-3-илсульфанил]уксусной кислоты Методика. К раствору метилового эфира 2-[5-амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]уксусной кислоты и бензилтриэтиламмония хлорида в бромоформе добавляли нитрит натрия. К смеси добавляли дихлоруксусную кислоту и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь непосредственно загружали на 7-дюймовую колонку силикагеля, которая была упакована с помощью CH2Cl2. Колонку сначала элюировали CH2Cl2 до тех пор, пока не элюировался весь CHBr3, и затем элюировали ацетоном/CH2Cl2 (5:95) с получением 713 мг (85%) названного в заголовке соединения. Способ получения 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]уксусной кислоты Методика. К раствору метилового эфира 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]уксусной кислоты в смеси ТГФ и EtOH при 0 С каплями добавляли раствор LiOH в H2O в течение 5 мин. Реакция завершалась после перемешивания при 0 С в течение еще 45 мин. Реакционную смесь нейтрализовали до рН 7 добавлением 0,5 N раствора HCl при 0 С и полученную смесь концентрировали in vacuo до 1/5 ее первоначального объема. Смесь разбавляли Н 2 О (20 мл) и подкисляли до рН 23 добавлением 0,5 N HCl с получением липкого твердого вещества. (Если продукт выходит в виде масла во время подкисления, рекомендуется экстрагирование CH2Cl2). Желто-коричневое твердое вещество собирали вакуум-фильтрованием и сушили в высоком вакууме при 50 С в течение 16 ч в присутствии Р 2 О 5 с получением 1,02 г (93%) названного в заголовке соединения. Ниже следуют примеры получения соединений 1 с использованием 2-[5-бром-4-(4 циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]уксусной кислоты или ее метилового эфира. 1-Циклопропилнафталин. Циклопропилмагнийбромид (150 мл, 0,5 М в тетрагидрофуране) медленно добавляли к раствору 1-бромнафталина (10 г, 50 ммоль) и [1,3-бис-(дифенилфосфино)пропан]дихлорникеля(II) в тетрагидрофуране (10 мл), перемешиваемому при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и растворитель выпаривали при пониженном давлении. Добавляли EtOAc и раствор хлорида аммония в воде. После экстрагирования органический слой сушили над сульфатом натрия,фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением 1-циклопропилнафталина (6,4 г, 76%). 1-Циклопропил-4-нитронафталин. Нитрит натрия (30 мл) медленно добавляли (в течение 2 ч) к 1-циклопропилнафталину (6,4 г,38 ммоль), перемешиваемому при 0 С. Реакционную смесь перемешивали при 0 С в течение более 30 мин и затем медленно выливали на лед. Добавляли воду с последующим добавлением EtOAc. После экстрагирования органический слой промывали 1% водным раствором NaOH, затем промывали водой,сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением 1-циклопропил-4-нитронафталина (5,2 г, 64%). 1-Амино-4-циклопропил-1-нафталин. Раствор 1-циклопропил-4-нитронафталина (5 г, 23 ммоль) в этаноле (200 мл) перемешивали в атмосфере водорода в присутствии Pd/C (10% нетто, 1,8 г). Реакционную смесь взбалтывали в течение ночи,затем фильтровали через целит. Растворитель выпаривали и остаток очищали хроматографией на силикагеле с получением 1-амино-4-циклопропилнафталина (3,1 г, 73%). 1-Циклопропил-4-изотиоцианонафталин. Тиофосген (1,1 г, 9,7 ммоль) добавляли к раствору 1-амино-4-циклопропилнафталина (1,8 г,9,7 ммоль) и диизопропилэтиламина (2 экв.) в дихлорметане (50 мл), перемешиваемому при 0 С. Реакционную смесь перемешивали в течение 5 мин при этой температуре, затем добавляли 1% раствор HCl в воде и органический слой отделяли, промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и растворитель выпаривали при пониженном давлении. Добавляли гексан и полученный осадок отфильтровывали. Растворитель выпаривали с получением 1-циклопропил-4 изотиоцианатонафталина (1,88 г, 86%). 5-Амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-тиол. Смесь гидрохлорида аминогуанидина (3,18 г, 29 ммоль), 1-циклопропил-4-изотиоцианатонафталина(3,24 г, 14 ммоль) и диизопропилэтиламина (3 экв.) в ДМФ (20 мл) перемешивали при 50 С в течение 15 ч. Растворитель выпаривали, добавляли толуол и растворитель снова выпаривали. Добавляли 2,0 М водный раствор гидроксида натрия (30 мл) и реакционную смесь нагревали при 50 С в течение 60 ч. Реакционную смесь фильтровали и фильтрат нейтрализовывали 2,0 М водным раствором HCl. Снова фильтровали, затем выпаривали растворитель и остаток очищали хроматографией на силикагеле с получением 5-амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-тиола (2,0 г, 49%). 2-[5-Амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид. К раствору 5-амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-тиола (708 мг,2,5 ммоль), K2CO3 (380 мг, 2,5 ммоль) в ДМФ (20 мл) добавляли 2-хлор-N-(2-хлор-4 сульфамоилфенил)ацетамид (710 мг, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. По завершении реакции растворитель выпаривали. Остаток очищали хроматографией на силикагеле с получением 2-[5-амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]-N-(2-хлор-4-сульфамоилфенил)ацетамида (1,26 г, 95%). 2-[5-Бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид. Дихлоруксусную кислоту (180 мкл, 2,2 ммоль) добавляли к суспензии 2-[5-амино-4-(4 циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4-сульфамоилфенил)ацетамида (0,59 г, 1,1 ммоль), нитрита натрия (1,5 г, 22 ммоль) и BTEABr (0,91 г, 3,3 ммоль) в дихлорметане (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, затем экстрагировали дихлорметаном и раствором бикарбоната натрия в воде. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на силикагеле с получением 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]-N-(2-хлор-4-сульфамоилфенил)ацетамида (224 мг, 31%). Методика. К суспензии тиотриазола и карбоната калия в ДМФ добавляли каплями метилхлорацетат при комнатной температуре в течение 5 мин. Реакционную смесь перемешивали при комнатной температуре в течение 24 ч и медленно выливали в перемешиваемый ледяной водный раствор. Желто-коричневый осадок собирали вакуум-фильтрованием и сушили в высоком вакууме при 50 С в течение 16 ч в присутствии Р 2 О 5 с получением 2,24 г (80%) названного в заголовке соединения. Метиловый эфир 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]уксусной кислоты. Методика. К раствору метилового эфира 2-[5-амино-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]уксусной кислоты и бензилтриэтиламмония хлорида в бромоформе добавляли нитрит натрия. К смеси добавляли дихлоруксусную кислоту и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Смесь непосредственно загружали на 7-дюймовую колонку силикагеля, которая была упакована с помощью CH2Cl2. Колонку сначала элюировали CH2Cl2 до тех пор, пока не элюировался весь CHBr3, и затем элюировали ацетоном/CH2Cl2 (5:95) с получением 713 мг (85%) названного в заголовке соединения. Методика. К раствору метилового эфира 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3 илсульфанил]уксусной кислоты в смеси ТГФ и EtOH при 0 С каплями добавляли раствор LiOH в H2O в течение 5 мин. Реакция завершалась после перемешивания при 0 С в течение еще 45 мин. Реакционную смесь нейтрализовали до рН 7 добавлением 0,5 N раствора HCl при 0 С и полученную смесь концентрировали in vacuo до 1/5 ее первоначального объема. Смесь разбавляли H2O (20 мл) и подкисляли до рН 2-3 добавлением 0,5 N HCl с получением липкого твердого вещества. (Если продукт выходит в виде масла во время подкисления, рекомендуется экстрагирование CH2Cl2). Желто-коричневое твердое вещество собирали вакуумфильтрованием и сушили в высоком вакууме при 50 С в течение 16 ч в присутствии Р 2 О 5 с получением 1,02 г (93%) названного в заголовке соединения. 2-[5-Бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид. Методика. К раствору карбоновой кислоты и анилина, представленных выше, в пиридине при 0 С каплями добавляли POCl3 в течение 5 мин. Реакция завершалась после перемешивания при 0 С в течение еще 50 мин. Реакционную смесь гасили добавлением Н 2 О (1 мл), затем концентрировали in vacuo до светлокоричневого масла, которое разбавляли CH2Cl2 (200 мл). Органический слой промывали Н 2 О (150 мл),насыщенным раствором NaHCO3 (150 мл), затем насыщенным раствором соли (150 мл). Органический слой сушили над Na2SO4 и концентрировали досуха. Полученное масло растирали с EtOH с получением светло-желтого твердого вещества. К смеси добавляли Н 2 О, чтобы собрать больше твердого вещества. Светло-желтое твердое вещество собирали вакуумфильтрованием и сушили в высоком вакууме в течение 16 ч с получением 930 мг (72%) продукта. Дополнительно продукт (132 мг, 10%) выделяли экстрагированием фильтрата CH2Cl2 с последующей колоночной хроматографией с помощью ацетона/CH2Cl2 В 50-мл круглодонную колбу загружали 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4-сульфамоилфенил)ацетамид (45 мг, 0,076 ммоль), ЭДХ(29 мг, 0,15 ммоль) и пропионовую кислоту (6,7 мкл, 0,09 ммоль) в смесь 5 мл ТГФ и 5 мл метиленхлорида. К данной смеси добавляли DMAP (18,3 мг, 0,15 ммоль) одной порцией. Реакционную смесь перемешивали при КТ в течение 14 ч. Растворители выпаривали при пониженном давлении, получая плотный маслянистый остаток. Остаток снова растворяли в 20 мл метиленхлорида, затем раствор промывали 20 мл 2,0 М водным раствором HCl. Органический слой сушили над Na2SO4. Растворитель удаляли с помощью роторного испарителя, получая маслянистый остаток. Остаток очищали колоночной хроматогра-9 015846 фией на силикагеле с помощью смеси метанола и метиленхлорида (1:9) и получали 18,5 мг (38%) желаемого продукта в виде белых твердых веществ. 2-[5-Бром-4-(4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 пропионилсульфамоилфенил)лизинамид В 25-мл круглодонную колбу загружали 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4 Н[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4-сульфамоилфенил)ацетамид (50 мг, 0,085 ммоль), ЭДХ(35 мг, 0,18 ммоль), Boc-Lys(Boc)-ОН ДЦГА (47 мг, 0,09 ммоль) в смеси 5 мл ТГФ и 5 мл метиленхлорида. К смеси добавляли DMAP (16 мг, 0,13 ммоль) одной порцией. Реакционную смесь перемешивали при КТ в течение 14 ч. Растворители выпаривали при пониженном давлении, получая плотный маслянистый остаток. Остаток растворяли в 5 мл 4,0 М HCl в диоксане. Реакционную смесь перемешивали при КТ в течение 14 ч. Растворитель выпаривали при пониженном давлении, получая густой маслянистый остаток. Остаток последовательно промывали 10 мл метиленхлорида и 10 мл эфира, получая названное в заголовке соединение в виде светло-желтого твердого вещества (44 мг, 65%). Используя соответствующие исходные вещества, получают следующие соединения 1 по методикам,аналогичным методам, описанным выше: 2-[5-бром-4-(2-хлор-4-(циклопропилметил)фенил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(2-хлор-4-циклобутилфенил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(2-хлор-4-(циклопропилметил)нафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2 хлор-4-сульфамоилфенил)ацетамид,2-[5-бром-4-(2-хлор-4-циклопропилфенил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-трифторметил-4-(2-хлор-4-циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N(2-хлор-4-сульфамоилфенил)ацетамид,2-[5-бром-4-(4-циклопропил-5,6,7,8-тетрагидронафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]N-(2-хлор-4-сульфамоилфенил)ацетамид,2-[5-бром-4-(4-этилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(4-этил-5,6,7,8-тетрагидронафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор 4-сульфамоилфенил)ацетамид,2-[5-бром-4-(5-циклопропилхинолин-8-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(5-циклопропилизохинолин-8-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(5-циклопропилциннолин-8-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(1-метилаценафтен-5-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(2-метилаценафтен-5-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид,2-[5-бром-4-(1,1-диметилаценафтен-5-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-хлор-4 сульфамоилфенил)ацетамид. Ингибирование обратной транскриптазы ВИЧ-1. Соединения 1 подвергают скринингу на ингибирующую активность в отношении вируса иммунодефицита человека типа 1 (ВИЧ-1), используя высокопроизводительное исследование на основе клеток с использованием экспрессии люциферазы светлячков ВИЧ-1 в качестве репортерного гена и псевдотипируют с помощью оболочечного гликопротеина вируса везикулярного стоматита (VSV-G, ВВС-Г). Процедуры эксперимента, по существу, были такими, которые описаны Connor et al. Journal of Virology (1996),70: 5306-5311 (Характеристика функциональных свойств env генов от долгожителей, инфицированных вирусом иммунодефицита человека типа 1), Popik et al. Journal of Virology (2002), 76: 4709-4722 (вирус иммунодефицита человека типа 1 использует совместно локализованные на липидных скоплениях CD4 и хемокиновые рецепторы для продуктивного проникновения в Т-клетки CD4+). В частности, должно быть понятно, что данный вирус содержит две мутации, вызванные в гене ОТ (K103N и Y181C, созданные- 10015846 мутагенезом с помощью ПЦР), которые делают вирус высокорезистентным к современным ненуклеозидным лекарственным препаратам против ВИЧ-1. Материал линий вирусов готовили путем сотрансфицирования плазмидной ДНК, кодирующей ВВС-Г, с вектором pNL4-3Env(-)Luc(+) в клетки 293 Т. Через 64 ч после трансфицирования содержащую вирус среду собирали центрифугированием и хранили замороженной при -80 С. Клетки HeLa инфицировали псевдотипированным вирусом ВВС-Г в присутствии проходящих отбор веществ в 384-луночном формате микротитровальной платы. Через 48 ч после первоначального инфицирования к клеткам добавляли буфер для лизиса и люциферазный реагент для исследования(Promega) и люциферазную активность определяли путем измерения полученной люминесценции с использованием люминометра LJL. Так как люциферазный ген находится в вирусном геноме, уровень его экспрессии непосредственно отражает уровень репликации вируса в присутствии соединения. Чтобы оценить активность соединений 1 в отношении ВИЧ-1 дикого типа, использовали линию клеток HeLa-JC53, которая экспрессирует высокие уровни CD4 и CCR5 (Platt et al., Journal of Virology(1998), 72: 2855-2864: Влияние концентраций на поверхности клеток CCR5 и CD4 на инфицирование макрофаготропных изолятов вируса иммунодефицита человека типа 1). Данную линию клеток модифицировали выделением стабильной линии клеток, которая экспрессирует люциферазу под контролем промотора ВИЧ-1 (длинный концевой повтор, т.е. LTR). Инфицирование ВИЧ-1 данной клеточной линии стимулирует транскрипцию люциферазы из промотора ВИЧ-1 и уровень экспрессии люциферазного гена пропорционален уровню репликации вируса (Harrington et al. Journal of Virology Methods (2000), 88: 111115: Прямое обнаружение инфицирования ВИЧ-1 в крови с использованием исследования клеток центрифугированием с индикатором; Roos et al. Virology (2000), 273: 307-315: Клетки LuSIV: репортерная линия клеток для определения и количественного определения одного цикла репликации ВИЧ и SIV). Методики испытания вирусных инфекций соединений и определения люциферазной активности были такими же, что и для ВВС-Г псевдотипа ВИЧ-1. Для оценки цитотоксичности положительных соединений, выявленных при исследованиях с вирусом ВИЧ-1, использовали два подхода. При первом подходе использовали другую линию модифицированных клеток HeLa-JC53, которая конститутивно экспрессирует люциферазу на высоком уровне без инфицирования вирусом. Уровень экспрессии люциферазы в этих клетках служил в качестве показателя клеточной репликации в присутствии данных соединений 1. Методики испытания соединений 1 и определения люциферазной активности были такими же, что и при испытаниях на инфицирование вирусом. Другое исследование токсичности с использованием клеток HeLa-JC53 и коммерчески доступного для приобретения набора для MTS исследования (Promega), с помощью которого количественно определяют функцию митохондрий в клетках. Используя методы,подобные описанным выше,синтезировали 2-[5-бром-4-(4 циклопропилнафталин-1-ил)-4 Н-[1,2,4]триазол-3-илсульфанил]-N-(2-метил-4-сульфамоилфенил)ацетамид, который был N-4-карбамильным аналогом. Каждое из данных соединений 1 испытывали в отношении набора мутантных обратных транскриптаз ВИЧ, включающего 20 из 22 мутантов, которые обнаружены примерно в 2% или более из образцов от пациентов, которые устойчивы к наиболее широко используемому ненуклеозидному ингибитору ОТ ВИЧ, эфавиренз 4S)-6-хлор-4-(циклопропилэтинил)1,4-дигидро-4-(трифторметил)-2 Н-3,1-бензоксазин-2-он). Для каждого из 20 широко распространенных испытанных мутантов по меньшей мере одно из этих соединений было более чем в 20 раз более активным, чем эфавиренз, или показана ЭК 50 (ЕС 50) менее 1 нМ. В большинстве случаев соединения 1 удовлетворяли обоим критериям. В большинстве случаев все три соединения были более активны, чем эфавиренз. Соединения сравнивали по активности по обратным транскриптазам дикого типа и мутантнымY181C и Y188L. Оба амида значительно превосходили карбоновую кислоту в отношении всех трех ферментов.- 11015846 Результаты. Соединения данного изобретения испытывали в отношении обратных транскриптаз дикого типа и четырех мутантных обратных транскриптаз ВИЧ. Результаты представлены в таблице в виде ЭК 50 (нМ) и ПК 50 (IC50) (нМ), в которой А составляет 50 нМ, В находится между 50 и 100 нМ и С составляет 100 нМ. НО (ND) означает не определено. Предпочтительными соединениями данного изобретения являются те, которые проявляют активность в отношении вирусов дикого типа (WT; ДТ) и устойчивых мутантов в концентрации ниже 50 нМ как по ЭК 50, так и ПК 50. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее следующую структурную формулу: 2. Соединение, имеющее следующую структурную формулу:
МПК / Метки
МПК: C07D 249/14, A61P 31/18, A61K 31/4196, C07D 249/12
Метки: уксусная, кислота, метиловый, эфир, 2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4н-[1,2,4]триазол-3-илсульфанил
Код ссылки
<a href="https://eas.patents.su/15-15846-2-5-brom-4-4-ciklopropilnaftalin-1-il-4n-124triazol-3-ilsulfanil-uksusnaya-kislota-i-ee-metilovyjj-efir.html" rel="bookmark" title="База патентов Евразийского Союза">2-[5-бром-4-(4-циклопропилнафталин-1-ил)-4н-[1,2,4]триазол-3-илсульфанил] уксусная кислота и ее метиловый эфир</a>
Содержащие диариловый эфир соединения мочевины

Номер патента: 15488
Опубликовано: 31.08.2011
Авторы: Ванг Лицзюань Джейн, Кестен Сузанн Росс, Мейерз Марвин Джей, Моррис Марк Энтони, Лазервит Скотт Эдвард, Стифф Кори Майкл, Джонсон Дуглас Скотт, Фей Лоррейн Кэтлин
МПК: A61K 31/444, A61P 29/02, C07D 401/10...
Метки: содержащие, эфир, соединения, мочевины, диариловый
Формула / Реферат:
1. Соединение формулы Iв которой каждый R1 независимо представляет собой водород, -OH, галоген, галогеналкил, -C1-C6-алкил, -O-C1-C6-алкил, -S-C1-C6-алкил, арил, гетероарил, -O-арил, -O-гетероарил, -NH2, -NHC(O)C1-C6-алкил, -(CH2)0-3-C3-C6-циклоалкил, -NHC(O)C3-C6-циклоалкил, -NHC1-C6-алкил, CN, -C(O)NR'R" или -C(O)C1-C6-алкил; причем каждая R1-C1-C6-алкильная группа необязательно замещена -O-C1-C6-алкильной группой или 1-3 -OH...
Сложный эфир-ингибитор spla2

Номер патента: 3277
Опубликовано: 24.04.2003
Авторы: Денни Майкл Лайл, Сойер Джейсон Скотт, Морин Джон Майкл, Солл Дэниел Джон
МПК: A61K 31/404, C07D 209/22, A61P 37/00...
Метки: сложный, эфир-ингибитор, spla2
Формула / Реферат:
1. Соединение N-морфолиноэтиловый эфир ((3-(2-амино-1,2-диоксоэтил)-2-метил-1-(фенилметил)-1H-индол-4-ил)окси)уксусной кислоты.
2. Применение соединения по п.1 в качестве средства для лечения состояний, вызываемых или поддерживаемых избыточным образованием sPLA2.
3. Фармацевтический препарат, содержащий соединение по п.1 в сочетании с носителем или разбавителем.
Ингибитор белка, переносящего эфир холестерила

Номер патента: 9466
Опубликовано: 28.12.2007
Авторы: Сикорски Джеймс А., Гленн Кевин К.
МПК: A61K 45/06, A61P 9/00
Метки: белка, холестерила, ингибитор, переносящего, эфир
Формула / Реферат:
Соединение, имеющее формулу или его диастереомеры, энантиомеры, рацематы, соли и таутомеры. ...
Трипептиды, несущие простой гидроксипролиновый эфир замещённого хинолина, предназначенные для ингибирования протеазы ns3 (гепатит с)

Номер патента: 7742
Опубликовано: 29.12.2006
Авторы: Ллина-Брюне Монсе, Гори Вида Дж.
МПК: C07K 5/08, A61P 31/14
Метки: замещённого, гидроксипролиновый, предназначенные, простой, гепатит, протеазы, несущие, хинолина, ингибирования, трипептиды, эфир
Формула / Реферат:
1. Соединение формулы (I) в котором R1 обозначает гидрокси или NHSO2Ph; R2 обозначает С4-С6циклоалкил; R3 обозначает трет-бутил или С5-С6циклоалкил и R4 обозначает С4-С6циклоалкил; или его фармацевтически приемлемая соль. 2. Соединение формулы (I) по п.1, в котором R1 обозначает NHSO2Ph. 3. Соединение формулы (I) по п.1, в котором R1 обозначает гидрокси. 4. Соединение формулы (I) по одному из пп.1-3, в котором R2 обозначает циклопентил или...
Композиция, включающая ингибитор il-1 и гидрогелевый эфир, и способы ее применения

Номер патента: 1792
Опубликовано: 27.08.2001
Авторы: Бевилаква Майкл П., Коллинс Дэвид С.
МПК: A61P 29/00, A61K 47/36
Метки: применения, композиция, ингибитор, эфир, включающая, способы, гидрогелевый
Формула / Реферат:
1. Фармацевтическая композиция, включающая гидрогелевый эфир, контролирующий высвобождение, и белковый ингибитор интерлейкина-1 (IL-1). 2. Композиция по п.1, отличающаяся тем, что гидрогелевый эфир является гиалуронаном или его солью. 3. Композиция по п.2, отличающаяся тем, что гиалуронан является гиалуроновой кислотой. 4. Композиция по п.2, отличающаяся тем, что гиалуронан является гиланом. 5. Композиция по п.2, отличающаяся тем, что гиалуронан...
Предыдущий патент: Способ и устройство для изготовления наполненной веществом упаковки
Следующий патент: Способ изготовления цепи литьем по газифицируемым моделям
Случайный патент: Фармацевтическая композиция, содержащая производные пиперазина