Азабициклические производные в качестве антагонистов мускаринового рецептора
Номер патента: 9387
Опубликовано: 28.12.2007
Авторы: Салман Мохаммад, Сарма Пакала Кумара Савитхру, Кхугх Анита, Дхармараджан Санкаранарайанан, Кумар Нареш













Формула / Реферат
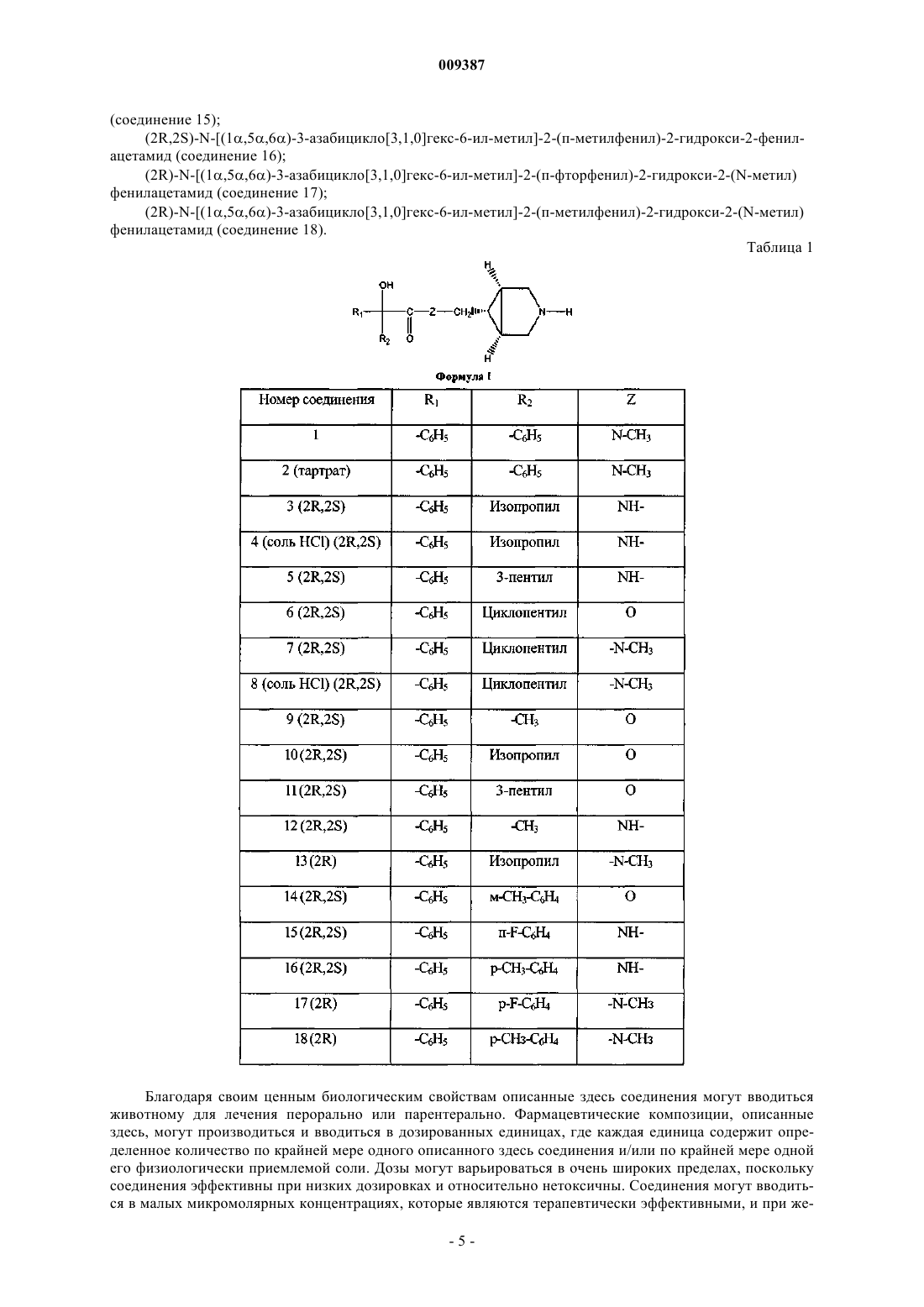
1. 6-Замещенные азабицикло[3,1,0]гексаны, выбранные из следующих соединений:
N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамид (соединение 1);
N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамида тартрат (соединение 2);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамид (соединение 3);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамида гидрохлорид (соединение 4);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(3-пентил)-2-гидрокси-2-фенилацетамид (соединение 5);
(2R,2S)-N-(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-циклопентил-2-гидрокси-2-(N-метил)
фенилацетамид (соединение 7);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-циклопентил-2-гидрокси-2-(N-метил)
фенилацетамида гидрохлорид (соединение 8);
эфир (2R,2S)-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-метил-2-гидрокси-2-фенилуксусной кислоты (соединение 9);
эфир (2R,2S)-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилуксусной кислоты (соединение 10);
эфир (2R,2S)-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(3-пентил)-2-гидрокси-2-фенилуксусной кислоты (соединение 11);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-метил-2-гидрокси-2-фенилацетамид (соединение 12);
(2R,2S)-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-(N-метил)фенилацетамид (соединение 13);
эфир (2R,2S)-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(м-метилфенил)-2-гидрокси-2-фенилуксусной кислоты (соединение 14);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(п-фторфенил)-2-гидрокси-2-фенилацетамид (соединение 15);
(2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(п-метилфенил)-2-гидрокси-2-фенилацетамид (соединение 16);
(2R)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(п-фторфенил)-2-гидрокси-2-(N-метил)
фенилацетамид (соединение 17);
(2R)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(п-метилфенил)-2-гидрокси-2-(N-метил)
фенилацетамид (соединение 18).
2. Способ лечения или профилактики животного или человека, страдающего расстройством или заболеванием дыхательной, мочевыделительной и желудочно-кишечной систем, где заболевание или расстройство опосредованы мускариновыми рецепторами, включающий введение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структуру формулы I

его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров, где
R1 и R2 независимо выбраны из С1-С6алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из С1-С3алкила или галогена;
Z представляет собой кислород или NR3, где R3 представляет собой водород или C1-С3алкил.
3. Способ по п.2, где заболеванием или расстройством является недержание мочи, симптомы нижних мочевыводящих путей (LUTS), бронхиальная астма, хронические обструктивные легочные расстройства (COPD), легочный фиброз, синдром раздраженной кишки, ожирение, диабеты и желудочно-кишечная гиперкинезия.
4. Способ по п.2, где заболеванием или расстройством является недержание мочи, симптомы нижних мочевыводящих путей (LUTS), бронхиальная астма, хронические обструктивные легочные расстройства (COPD), легочный фиброз, синдром раздраженной кишки, ожирение, диабеты и желудочно-кишечная гиперкинезия.
5. Способ получения соединения формулы V

и его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров, где
R1 и R2 независимо выбраны из C1-С6алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из С1-С3алкила или галогена;
R3 представляет собой водород или C1-С3алкил;
где указанный способ включает:
(а) реакцию соединения формулы II с соединением формулы III


с получением защищенного соединения формулы IV, где R1, R2, R3 определены выше, а Р представляет собой защитную группу для аминогруппы

(б) удаление защитной группы в соединении формулы IV в присутствии удаляющего защитную группу агента с получением соединения формулы V, где R1, R2, R3 определены выше.

6. Способ по п.5, где Р представляет собой любую защитную группу для аминогруппы и выбран из группы, состоящей из бензильной и трет-бутилоксикарбонильной групп.
7. Способ по п.5, где реакция соединения формулы II с соединением формулы III с получением соединения формулы IV проводится в присутствии N-метилморфолина, и 1-гидроксибензотриазола, и конденсирующего агента, который выбран из гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC), 1,3-дициклогексилкарбодиимида (DCC) и 1,8-диазабицикло[5,4,0]ундец-7-ена (DBU).
8. Способ по п.5, где реакция соединения формулы II с соединением формулы III проводится в подходящем полярном апротонном растворителе, выбранном из N,N-диметилформамида, диметилсульфоксида, толуола, ксилола и хлороформа.
9. Способ по п.5, где реакция соединения формулы II с соединением формулы III проводится при температуре 0-140шC.
10. Способ по п.5, где удаление защиты в соединении формулы IV проводится удаляющим защиту агентом, выбранным из группы, состоящей из палладия на угле и водорода, формиата аммония палладия на угле, трифторуксусной кислоты (ТФУК) и хлороводородной кислоты.
11. Способ по п.5, где удаление защиты в соединении формулы IV ё получением соединений формулы V проводится в подходящем органическом растворителе, выбранном из группы, состоящей из метанола, этанола, тетрагидрофурана и ацетонитрила.
12. Способ получения соединений формулы VIII

и его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров, где
R1 и R2 независимо выбраны из C1-С6алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из C1-С3алкила или галогена;
где указанный способ включает:
(а) реакцию соединения формулы II с соединением формулы VI (где R' представляет собой защитную группу для гидроксигруппы, выбранную из п-толуолсульфонила и метансульфонила)
с получением защищенного соединения формулы VII, где R1 и R2 определены выше, а Р представляет собой защитную группу для аминогруппы

(б) удаление защитной группы в соединении формулы VII в присутствии удаляющего защитную группу агента с получением соединения формулы VIII, где R1 и R2 определены выше.

13. Способ по п.12, где Р представляет собой любую защитную группу для аминогруппы и выбран из группы, состоящей из бензильной и трет-бутилоксикарбонильной групп.
14. Способ по п.12, где реакция соединения формулы VI с соединением формулы II с получением соединений формулы VII проводится в присутствии конденсирующего агента, который выбран из 1,8-диазабицикло[5,4,0]ундец-7-ена (DBU) или 1,4-диазабицикло[2,2,2]октана (DABCO).
15. Способ по п.12, где реакция соединения формулы VI с соединением формулы II проводится в растворителе, выбранном из бензола, толуола или ксилола.
16. Способ по п.12, где реакция соединения формулы VI с соединением формулы II проводится при температуре 0-140шС.
17. Способ по п.12, где удаление защиты в сoединении формулы VII с получением соединений формулы VIII проводится удаляющим защиту агентом, выбранным из группы, состоящей из палладия на угле и газообразного водорода или формиата аммония и палладия на угле.
18. Способ по п.12, где удаление защиты в соединении формулы VII с получением соединений формулы VIII проводится в подходящем растворителе, выбранном из метанола или этанола.
Текст
009387 Область техники Изобретение относится к антагонистам мускариновых рецепторов, которые полезны, помимо других применений, для лечения различных заболеваний дыхательной, мочевыводящей и желудочно-кишечной систем, опосредованных мускариновыми рецепторами. Конкретно, изобретение относится к производным азабициклических соединений, включая, например, 6-замещенные азабицикло[3,1,0]гексаны, а также к фармацевтическим композициям, содержащим такие соединения, и к способам лечения заболеваний, опосредованных мускариновыми рецепторами. Предшествующий уровень техники Мускариновые рецепторы как члены рецепторов, связанных с G-белком (GPCR), представляют собой семейство из 5 подтипов рецептора (М 1, М 2, М 3, М 4 и М 5) и активируются нейтротрансмиттером ацетилхолином. Эти рецепторы широко распространены в различных органах и тканях и являются критичными для управления центральной и периферической холинэргической нейтротрансмиссией. Было описано распределение подтипов этих рецепторов в мозге и других органах. Например, подтип M1 локализован преимущественно в нервных тканях, таких как кора головного мозга и автономные ганглии, подтипM2 присутствует, главным образом, в сердце, он опосредует холинэергическую брадикардию, а подтипSciences, 22, p. 409 (2001), Eglen и др., описывает биологические потенциалы модулирования лигандами подтипов мускариновых рецепторов в случае различных заболеваний, например болезни Альцгеймера,боли, заболеваний мочевыводящей системы, хронической обструктивной легочной болезни и т.д. Обзор в J. Med. Chem., 43, p. 4333 (2000), Felder и др., описывает терапевтические возможности мускариновых рецепторов в центральной нервной системе и детально разрабатывает структуру и функции мускариновых рецепторов, фармакологию и их терапевтические применения. Фармакологические и медицинские аспекты агонистов и антагонистов ацетилхолина мускаринового типа представлены в обзоре в Molecules, 6, р. 142 (2001).Birdsall и др. в Trends in Pharmacological Sciences, 22, p. 215 (2001) также обобщил последние достижения в определении роли различных подтипов мускариновых рецепторов с использованием мышей при воздействии на различные мускариновые рецепторы. Мускариновые агонисты, такие как мускарин и пилокарпин, и антагонисты, такие как атропин, известны более 100 лет, однако, незначительные успехи в области открытия соединений, селективных к определенному подтипу рецепторов, затрудняют определение специфических функций индивидуальных рецепторов. Хотя классические мускариновые антагонисты, такие как атропин, являются мощными бронхорасширяющими средствами, их клиническое использование ограничено из-за высокой частоты как периферических, так и центральных побочных эффектов, таких как тахикардия, расплывчатое зрение, сухость во рту, запор, слабоумие и т.д. Позднее были разработаны четвертичные производные атропина, такие как ипратропиум бромид, которые лучше переносятся, чем парентерально вводимые предшественники, однако, большинство из них не являются идеальными антихолинэргическими бронхорасширяющими средствами из-за недостаточной селективности к подтипам мускариновых рецепторов. Существующие соединения предлагают ограниченные терапевтические возможности из-за недостатка селективности, приводящей к дозолимитирующим побочным эффектам, таким как жажда, тошнота, мидриазис,и сердечным побочным эффектам, таким как тахикардия, обусловленная воздействием на M2 рецептор.Annual review of Pharmacological Toxicol., 41, p. 691 (2001) описывает фармакологию инфекций мочевыводящих путей. Несмотря на то, что антимускариновые агенты, такие как оксибутинин и толтеродин, неселективно воздействующие на мускариновые рецепторы, на протяжении многих лет использовались для лечения гиперактивности мочевого пузыря, клиническая эффективность этих агентов ограничена из-за побочных эффектов, таких как сухость во рту, помутнение зрения, запор. Считается, что толтеродин, в целом, переносится лучше, чем оксибутинин (Steers и др. в Curr. Opin. Invest. Drugs, 2, 268;Chappie и др. в Urology. 55, 33; Steers и др., Adult and Pediatric Urology, ed. Gillenwatteret и др., рр. 12201325, St. Louis, MO; Mosby. 3rd edition (1996. Существует потребность в развитии новых высокоселективных мускариновых антагонистов, которые могут взаимодействовать с определенными подтипами, таким образом избегаются побочные эффекты. Соединения, имеющие антагонистическую активность в отношении мускариновых рецепторов, были описаны в заявках JP 92921/1994 и 135958/1994; WO 93/16048; US 3176019; GB 940540; ЕР 0325571;WO 97/45414 относятся к 1,4-дизамещенным производным пиперидина; WO 98/05641 описывает фторированные 1,4-дизамещенные производные пиперидина; WO 93/16018 и WO 96/33973 представляют собой другие ссылки ближайшего уровня техники. US 5397800 описывает 1-азабицикло[2,2,1]гептаны. US 5001160 описывает 1-арил-1-гидрокси-1-замещенные-3-(4-замещенные-1-пиперазинил)-2-пропаноны. WO 01/42213 описывает 2-бифенил-4-пиперидинилмочевины. WO 01/42212 описывает карбаматные производные. WO 01/90081 описывает аминоалкиллактам. WO 02/53564 описывает новые хинуклидиновые производные.WO 02/00652 описывает карбаматы, полученные из арилалкиламинов. WO 02/06241 описывает произ-1 009387 водные 1,2,3,5-тетрагидробензо(с)азепин-4-она. Статья в J. Med. Chem., 44, p. 984 (2002) описывает циклогексилметилпиперидинилтрифенилпропиоамидные производные в качестве селективных M3 антагонистов, не взаимодействующие с другими подтипами рецепторов. Сущность изобретения В качестве первого аспекта обеспечиваются азабициклические производные, включая, например, 6 замещенные азабицикло[3,1,0]гексаны, 2,6- и 4,6-дизамещенные производные и 2,4,6-тризамещенные производные в качестве антагонистов мускариновых рецепторов, которые могут быть полезны в качестве безопасных и эффективных терапевтических или профилактических агентов для лечения различных заболеваний дыхательной, мочевыводящей и желудочно-кишечной систем. Также обеспечивается способ получения таких соединений. В другом аспекте обеспечиваются фармацевтические композиции, содержащие такие соединения,вместе с приемлемыми носителями, эксципиентами или разбавителями, которые могут быть полезны для лечения различных заболеваний дыхательной, мочевыводящей и желудочно-кишечной систем. Также обеспечиваются энантиомеры, диастереомеры, фармацевтически приемлемые соли или фармацевтически приемлемые соли этих соединений в комбинации с фармацевтически приемлемым носителем, и необязательно включающие эксципиенты. Другие аспекты изобретения будут установлены в последующем описании и частично будут ясны из описания или могут быть обнаружены при осуществлении изобретения. В соответствии с первым аспектом обеспечиваются соединения, имеющие структуру формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые энантиомеры, диастереомеры, гдеR1 и R2 независимо выбраны из C1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из C1-С 3 алкила или галогена;Z может представлять собой кислород или NR3, где R3 представляет собой водород или C1-C3 алкил. В соответствии со вторым аспектом настоящего изобретения обеспечивается способ лечения или профилактики животного или человека, страдающего расстройством или заболеванием дыхательной,мочевыделительной и желудочно-кишечной систем, где заболевание или расстройство опосредованы мускариновыми рецепторами. Способ включает введение по меньшей мере одного соединения, имеющего структуру формулы I. В соответствии с третьим аспектом настоящего изобретения обеспечивается способ лечения или профилактики животного или человека, страдающего заболеванием или расстройством, связанным с мускариновыми рецепторами, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения, описанного выше, антагониста мускаринового рецептора. В соответствии с четвертым аспектом настоящего изобретения обеспечивается способ лечения или профилактики животного или человека, страдающего заболеванием или расстройством дыхательной системы, таким как бронхиальная астма, хронические обструктивные легочные расстройства (COPD), легочный фиброз и им подобные; мочевыводящей системы, которое вызывает нарушения работы мочевыводящей системы, такие как недержание мочи, симптомы нижних мочевыводящих путей (LUTS) и т.д.; и желудочно-кишечной системы, таким как синдром раздраженной кишки, ожирение, диабеты и желудочно-кишечная гиперкинезия, описанными выше соединениями, где заболевание или нарушение связаны с мускариновыми рецепторами. В соответствии с пятым аспектом настоящего изобретения обеспечивается способ получения описанных выше соединений. Соединения настоящего изобретения обладают значительным потенциалом в плане их активности,которая была определена in vitro связыванием с рецептором и функциональными пробами и в экспериментах in vivo на анестезированных кроликах. Соединения, которые были активны in vitro, были протестированы in vivo. Некоторые из соединений настоящего изобретения являются мощными антагонистами мускариновых рецепторов с высокой аффинностью по отношению к M3 рецепторам. Таким образом,обеспечиваются фармацевтические композиции для возможного лечения заболеваний или нарушений,связанных с мускариновыми рецепторами. Помимо этого, соединения могут вводиться перорально или парентерально. Подробное описание изобретения Описанные здесь соединения могут быть получены методами, представленными следующей последовательностью реакций, показанной на схемах I и II. Соединения формулы V могут быть получены, например, последовательностью реакций, показанной на схеме I. Получение включает реакцию соединения формулы II с соединением формулы III, гдеR1 и R2 независимо выбираются из C1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) выбран из C1-C3 алкила или галогена;R3 представляет собой водород или C1-С 3 алкил и Р представляет собой любую защитную группу для аминогруппы, например бензильную или третбутилоксикарбонильную группы. Реакция между соединением формулы II и соединением формулы III может протекать в присутствии N-метилморфолина, и 1-гидроксибензотриазола, и конденсирующего агента (например, гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC), 1,3-дициклогексилкарбодиимида (DCC) или 1,8-диазабицикло[5,4,0]ундец-7-ена (DBU в растворителе (таком, как N,N-диметилформамид, диметилсульфоксид, толуол, ксилол или хлороформ, при температуре в диапазоне от около 0 до около 140 С) с получением защищенного соединения формулы IV, которое после удаления защитной группы в присутствии удаляющего защитную группу агента (например, палладия на угле и водорода, формиата аммония и палладия на угле, трифторуксусной кислоты (ТФУК) или хлороводородной кислоты) в органическом растворителе (например, метаноле, этаноле, тетрагидрофуране или ацетонитриле, при температуре в диапазоне от около 10 до около 50 С) приводит к получению незащищенного соединения формулы V. Схема II-3 009387 Соединения формулы VIII могут быть получены, например, последовательностью реакций, показанной на схеме II. Получение включает реакцию соединения формулы II с соединением формулы VI,гдеR1 и R2 независимо выбираются из C1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) выбран из С 1-С 3 алкила или галогена;R' представляет собой любую защитную группу для гидроксигруппы, например п-толуолсульфонил или метансульфонил, и Р представляет собой любую защитную группу для аминогруппы, например бензильную или третбутилоксикарбонильную группы. Реакция между соединением формулы II и соединением формулы VI может протекать в присутствии конденсирующего агента (например, 1,8-диазабицикло[5,4,0]ундец-7-ена (DBU) или 1,4-диазабицикло[2,2,2]октана (DABCO в растворителе (таком, как бензол, толуол или ксилол, при температуре в диапазоне от около 0 до около 140 С) с получением защищенного соединения формулы VII, которое после удаления защитной группы в присутствии удаляющего защитную группу агента (например, палладия на угле и водорода, формиата аммония и палладия на угле) в органическом растворителе (например, метаноле или этаноле, при температуре в диапазоне от около 10 до около 50 С) приводит к получению незащищенного соединения формулы VIII. Необходимо понимать, что для приведенной выше схемы, где упомянуты конкретные основания,конденсирующие агенты, защитные группы, удаляющие защиту агенты, растворители, катализаторы,температуры и т.д., могут использоваться другие основания, конденсирующие агенты, защитные группы,удаляющие защиту агенты, растворители, катализаторы, температуры и т.д., известные специалисту в данной области техники. Подобным образом, температура реакции и ее продолжительность могут регулироваться в зависимости от необходимости. Подходящие соли соединений, представленных формулой I, были получены для того, чтобы солюбилизировать соединение в водной среде для биологических испытаний, а также для сопоставления между различными формулировками доз и улучшения биодоступности соединений. Примеры таких солей включают фармацевтически приемлемые соли, такие как соли с неорганической кислотой (например,гидрохлорид, гидробромид, сульфат, нитрат и фосфат), соли с органической кислотой (например, ацетат,тартрат, цитрат, фумарат, малеат, толуолсульфонат и метансульфонат). Когда в качестве заместителя в формуле I присутствует карбоксильная группа, это может быть соль в форме щелочи или соли щелочного металла (например, натрия, калия, кальция, магния и подобных). Эти соли могут быть получены обычными методами, известными из уровня техники, такими как обработка соединения эквивалентным количеством неорганической или органической кислоты или основания в подходящем растворителе. Отдельные соединения представлены ниже: Благодаря своим ценным биологическим свойствам описанные здесь соединения могут вводиться животному для лечения перорально или парентерально. Фармацевтические композиции, описанные здесь, могут производиться и вводиться в дозированных единицах, где каждая единица содержит определенное количество по крайней мере одного описанного здесь соединения и/или по крайней мере одной его физиологически приемлемой соли. Дозы могут варьироваться в очень широких пределах, поскольку соединения эффективны при низких дозировках и относительно нетоксичны. Соединения могут вводиться в малых микромолярных концентрациях, которые являются терапевтически эффективными, и при же-5 009387 лании доза может быть повышена до максимальной дозы, переносимой пациентом. Описанные здесь соединения могут производиться и применяться в виде их энантиомеров, диастереомеров и фармацевтически приемлемых солей. Приведенные далее примеры демонстрируют общие синтетические методики, а также конкретное получение отдельных соединений. Примеры приведены для иллюстрации деталей изобретения и не должны рассматриваться как ограничивающие объем настоящего изобретения. Примеры Различные растворители, такие как ацетон, метанол, пиридин, эфир, тетрагидрофуран, гексаны и дихлорметан, были осушены с использованием различных осушающих реагентов по методикам, известным из уровня техники. ИК-спектры записывали в неупорядоченном виде или в тонких пленках на прибореPerkin Elmer Paragon, спектры Ядерного Магнитного Резонанса (ЯМР) записывали на приборе Varian XL300 MHz с использованием в качестве внутреннего стандарта тетраметилсилана. Пример 1. Получение N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(Nметил)фенилацетамида (соединение 1). Стадия а. Синтез метилового эфира 3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метансульфоновой кислоты. К раствору (3-бензил-3-азабицикло[3,1,0]гекс-6-ил)метанола (полученного, как описано в Synlett,1996; 1097) (5,2 г, 25,6 ммоль) в дихлорметане при 0 С добавляли триэтиламин (10,6 мл, 76,8 ммоль) и метансульфонилхлорид (4 мл, 51,2 ммоль). Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение ночи. К реакционной смеси добавляли насыщенный водный раствор бикарбоната натрия и отделяли органическую фракцию, получая раствор сырого продукта. Раствор промывали водой, насыщенным раствором хлорида натрия, высушивали безводным сульфатом натрия и упаривали, получая сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле,используя в качестве элюента гексан-триэтиламин (99,1), получая целевой продукт в виде бледно-желтой чистой жидкости (2,2 г, 30%). Стадия б. Синтез (3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил)метиламина. К раствору метилового эфира 3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метансульфоновой кислоты(2,4 г, 8,5 ммоль) в метаноле (20 мл) в стальной бомбе добавляли водный 40% раствор метиламина (25 мл). Стальную бомбу закрывали и нагревали до 85-90 С в течение приблизительно 15 ч. Охлаждали до умеренной температуры, затем охлаждали до -78 С и открывали. Смесь переносили в круглодонную колбу,упаривали растворитель, разбавляли водой и хлороводородной кислотой и экстрагировали этилацетатом. Органическую фракцию отделяли и отбрасывали. Водную фракцию подщелачивали добавлением 10% водного раствора гидроксида натрия до рН 12-13. Водную фракцию экстрагировали дихлорметаном и высушивали над безводным сульфатом натрия. Отфильтрованный раствор в дихлорметане упаривали,получая целевое соединение в виде желтой жидкости (1,8 г, 98%). Стадия в. Синтез N-[(1,5,6)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси 2-(N-метил)фенилацетамида. К холодному раствору бензиловой кислоты (1,9 г, 8,33 ммоль, коммерчески доступна) и (3-бензил 3-азабицикло[3,1,0]гекс-6-ил-метил)метиламина (1,8 г, 8,33 ммоль) в диметилформамиде (20 мл) при 0 С добавляли N-метилморфолин (1,8 мл, 16,6 ммоль) и 1-гидроксибензотриазол (1,12 г, 8,33 ммоль), смесь перемешивали в течение приблизительно 45 мин. К смеси добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,6 г, 8,33 ммоль), смесь постепенно нагревали до умеренной температуры и перемешивали в течение ночи. Разбавляли смесь водой и экстрагировали этилацетатом. Органическую фракцию отделяли, промывали водой, насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Органическую фракцию отфильтровывали и упаривали, получая сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле, используя в качестве элюента гексан-этилацетат (от 4:1 до 2:1), получая целевой продукт в виде бесцветного липкого порошка (1,3 г,36%). Стадия г. Синтез N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил) фенилацетамида. К раствору N-[(1,5,6)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(Nметил)фенилацетамида (1,3 г, 3,05 ммоль) в метаноле (20 мл) добавляли катализатор палладий на угле(10 вес.%) и над ним закрепляли 3-ходовой кран для гидрирования, подключенный к наполненному водородом баллону. Откачивали воздух, продували водород. Реакционную смесь перемешивали в течение 5 ч при умеренной температуре. Отфильтровывали катализатор на целите и промывали метанолом. Фильтрат упаривали, получая целевой продукт в виде бесцветной липкой жидкости (0,95 г, 93%). Соединение имело точку плавления 72,4-73,7 С. Данные инфракрасной спектроскопии (DCM): 1627,9 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):8,42-8,29 (м, 10 Н), 4,52 (с, 2 Н), 4,17 (с, 2 Н), 3,94-4,00L(+)винную кислоту (416 мг, 2,77 ммоль) и раствор перемешивали в течение 1 ч при комнатной температуре. Выпадал белый осадок. Его нагревали до 50-55 С в течение 30 мин и упаривали растворитель до половины объема. Добавляли сухой диэтиловый эфир, белый осадок отфильтровывали и промывали большим количеством диэтилового эфира. Получали сухой белый порошок (1,3 г, 96%). Соединение имело точку плавления 101-103 С. Аналогично следующие соединения были получены по методике, описанной в примере 1.(2R)-N-[(1,5,6)-3-Азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-(N-метил)фенилацетамид (соединение 13). Данные инфракрасной спектроскопии (DCM): 1619,7 см-1. Данные 1 Н-ЯМР спектроскопии (D2O):7,25-7,45 (м, 5 Н), 3,45-3,51 (м, 1 Н), 2,80-2,83 (м, 6 Н), 1,94-1,96 (ушс, 3H), 1,24-1,33 (м, 3H), 0,86-0,98 (м,6 Н). Данные масс-спектрометрии: пик m/е 303 (М+Н).(m, 12H). Пример 3. Гидрохлорид (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-циклопентил 2-гидрокси-2-(N-метил)фенилацетамида (соединение 8). К раствору (2R)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-циклопентил-2-гидрокси-2(N-метил)фенилацетамида в дихлорметане (14,0 мл) добавляли раствор хлороводородной кислоты в этаноле (3,5N, 2,1 мл) при 0-5 С и перемешивали в течение приблизительно 30 мин при 20-25 С. Удаляли растворитель при пониженном давлении, остаток переносили в гексан, получая осадок. Осадок отфильтровывали, промывали гексаном, высушивали при пониженном давлении, получая сухой продукт с выходом 90,1%. Данные инфракрасной спектроскопии (DCM): 1617,6 см-1. Данные 1 Н-ЯМР спектроскопии (D2O):7,45-7,52 (м, 5 Н), 3,42-3,50 (м, 4 Н), 3,22-3,29 (м, 2 Н), 2,90 (с, 3H), 1,80 (м, 1 Н), 1,40-1,50 (м, 8 Н), 1,22-1,27(м, 2 Н), 1,10 (м, 1 Н). Данные масс-спектрометрии: пик m/е 329 (М+Н). Пример 4. Получение (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2 гидрокси-2-фенилацетамида (соединение 3). Стадия а. Синтез (3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил)амина. Это соединение было получено по процедуре, описанной в ЕР 0413455. Стадия б. Синтез N-[(1,5,6)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамида. К холодному раствору 2-изопропил-2-гидрокси-2-фенилуксусной кислоты (полученной по методикеJ. Amer. Chem. Soc., 1953; 75: 2654 и J. Org. Chem., 2000; 65: 6283) (1,9 г, 8,33 ммоль) и (3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил)амина, полученного по методике, описанной в ЕР 0413455) (1,8 г, 8,33 ммоль) в диметилформамиде (20 мл) при 0 С добавляли N-метилморфолин (1,8 мл, 16,6 ммоль) и 1-гидроксибензотриазол (1,12 г, 8,33 ммоль), смесь перемешивали в течение приблизительно 45 мин. К смеси добавляли гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (1,6 г, 8,33 ммоль), смесь постепенно нагревали до умеренной температуры и перемешивали в течение ночи. Разбавляли смесь водой и экстрагировали этилацетатом. Органическую фракцию отделяли, промывали водой, насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Органическую фракцию отфильтровывали и упаривали, получая сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле, используя в качестве элюента гексан-этилацетат (от 4:1 до 2:1). Стадия в. Синтез (2R,2S)-]-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамида. К раствору N-[(1,5,6)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2 фенилацетамида (1,3 г, 30,5 ммоль) в сухом метаноле (25,0 мл) в атмосфере азота добавляли 5% палладий на угле (0,2 г), (50 вес.%). Затем при перемешивании добавляли безводный формиат аммония (0,8 г,12,38 ммоль) и реакционную смесь кипятили с обратным холодильником в течение получаса в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, отфильтровывали через слой hyflo. Слой hyflo промывали метанолом (75,0 мл), этилацетатом (25,0 мл) и водой (25,0 мл). Фильтрат концен-7 009387 трировали в вакууме. Остаток разбавляли водой, доводили значение рН полученного раствора до рН 14 добавлением 1N NaOH. Экстрагировали этилацетатом (2x50 мл), этилацетатную фракцию промывали водой и насыщенным раствором хлорида натрия. Высушивали над безводным сульфатом натрия и концентрировали, получая целевое соединение. Данные инфракрасной спектроскопии (DCM): 1654 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,60-7,62 (м, 2 Н), 7,24-7,37 (м, 3H), 6,74 (с, 1H), 3,06-3,16 (м, 2 Н), 2,79-2,94 (м, 5 Н), 1,26-1,31 (м, 2 Н), 1,00(д, J=6 Гц, 3H), 0,72-0,77 (м, 4 Н). Данные масс-спектрометрии: пик m/е 289 (М+1). Пример 5. Получение гидрохлорида (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2 изопропил-2-гидрокси-2-фенилацетамида (соединение 4). К раствору (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2 фенилацетамида (1,4 г, 4,9 ммоль) в дихлорметане (14,0 мл) добавляли раствор хлороводородной кислоты в этаноле (3,5N, 2,1 мл, 7,3 ммоль) при 0-5 С и перемешивали в течение приблизительно 30 мин при 20-25 С. Удаляли растворитель при пониженном давлении, остаток переносили в н-гексан, получая осадок. Осадок отфильтровывали, промывали гексаном, высушивали в вакууме, получая сухой продукт с выходом 95,1% (1,5 г). Соединение имело точку плавления 70 С (начало размягчения). Данные инфракрасной спектроскопии (DCM): 1641,1 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,63-7,65 (м, 2 Н), 7,40-7,47 (м, 3H),3,30-3,37 (м, 4 Н), 3,14-3,16 (м, 2 Н), 2,90-2,93 (м, 1H), 1,74 (с, 2 Н), 1,21-1,23 (м, 1H), 1,00-1,01 (м, 3H),0,81-0,83 (м, 3H). Аналогично, следующие соединения были получены по методике, описанной в примере 4.(соединение 5). Данные инфракрасной спектроскопии (DCM): 1651,7 см-1. Данные 1H-ЯМР спектроскопии (CDCl3):7,61-7,64 (м, 2 Н), 7,27-7,35 (м, 3H), 6,83 (с, 1H), 2,83-3,16 (м, 7 Н), 2,35 (м, 2 Н), 1,90-2,00 (м, 1 Н), 0,781,47 (м, 14 Н). Данные масс-спектрометрии: пик m/е 317 (М+1).(соединение 12). Данные инфракрасной спектроскопии (DCM): 1655,5 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,54-7,56 (м, 2 Н), 7,28-7,37 (м, 3H), 6,76 (ушс, 1 Н), 3,05-3,20 (м, 2 Н), 2,80-2,93 (м, 4 Н), 1,79 (с, 3H), 1,221,32 (м, 2 Н), 0,76-0,80 (м, 1 Н). Данные масс-спектрометрии: пик m/e 261 (M+1).(DBU) (1,6 г, 8,33 ммоль), смесь постепенно нагревали до умеренной температуры и перемешивали в течение ночи. Разбавляли смесь водой и экстрагировали этилацетатом. Органическую фракцию отделяли, промывали водой, насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Органическую фракцию отфильтровывали и упаривали, получая сырой продукт. Сырой продукт очищали колоночной хроматографией на силикагеле. Стадия б. Синтез эфира [(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-циклопентил-2-гидрокси-2-фенилуксусной кислоты. К раствору эфира [(1,5,6)-3-бензил-3-азабицикло[3,1,0]гекс-6-ил-метил]2-циклопентил-2-гидрокси-2-фенилуксусной кислоты в сухом метаноле (25 мл) в атмосфере азота добавляли 5% палладий на угле(0,2 г) (50 вес.%). Затем при перемешивании добавляли безводный формиат аммония (0,8 г, 12,38 ммоль) и реакционную смесь кипятили с обратным холодильником в течение получаса в атмосфере азота. Реакционную смесь охлаждали до комнатной температуры, отфильтровывали через слой hyflo. Слой hyflo промывали метанолом (75,0 мл), этилацетатом (25,0 мл) и водой (25,0 мл). Фильтрат концентрировали в вакууме. Остаток разбавляли водой, доводили значение рН полученного раствора до рН 14 добавлением-8 009387 1N NaOH. Экстрагировали этилацетатом (2x50 мл), этилацетатную фракцию промывали водой и насыщенным раствором хлорида натрия. Высушивали над безводным сульфатом натрия и концентрировали,получая целевое соединение. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,67-7,64 (м, 2 Н), 7,36-7,28 (м, 3H), 4,13-4,05 (м, 2 Н),2,97-2,86 (м, 4 Н), 2,29-1,50 (м, 12 Н). Данные масс-спектрометрии: пик m/е 316,31 (М+1). Аналогично, следующие соединения были получены по методике, описанной в примере 6. Эфир (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-метил-2-гидрокси-2-фенилуксусной кислоты (соединение 9). Данные инфракрасной спектроскопии (DCM): 1729,7 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,55-7,58 (м, 2 Н), 7,29-7,38 (м, 3H), 4,02-4,12 (м, 2 Н), 2,82-2,94 (м, 4 Н), 1,71 (с, 3H), 1,48 (с, 2 Н), 0,930,97 (м, 1 Н). Данные масс-спектрометрии: пик m/e 262 (М+1). Эфир (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилуксусной кислоты (соединение 10). Данные инфракрасной спектроскопии (DCM): 1723,8 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,65-7,67 (м, 2 Н), 7,24-7,37 (м, 3H), 4,05-4,16 (м, 2 Н), 2,81-2,93 (м, 4 Н), 2,61-2,66 (м, 1 Н), 1,29-1,39 (м,3H), 0,94-1,02 (м, 3H), 0,71 (д, J=6 Гц, 2 Н). Данные масс-спектрометрии: пик m/е 290 (М+1). Эфир (2R,2S)-]-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(3-пентил)-2-гидрокси-2-фенилуксусной кислоты (соединение 11). Данные инфракрасной спектроскопии (DCM): 1721,4 см-1. Данные 1 Н-ЯМР спектроскопии (CDCl3):7,64-7,67 (м, 2 Н), 7,29-7,37 (м, 3H), 4,02-4,11 (м, 2 Н), 2,92-3,02 (м, 4 Н), 2,15-2,19 (м, 1 Н), 1,42-1,51 (м, 4 Н),1,09-1,29 (м, 3H), 0,98-1,03 (м, 3H), 0,71-0,76 (м, 3H). Данные масс-спектрометрии: пик m/e 318 (М+1). Эфир (2R,2S)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(м-метилфенил)-2-гидрокси-2 фенилуксусной кислоты (соединение 14). Данные 1 Н-ЯМР спектроскопии (CDCl3):7,43-7,12 (м, 14 Н), 4,18-4,16 (м, 2 Н), 3,03-2,91 (м, 4 Н),2,33-2,28 (м, 3H), 1,30-1,28 (м, 3H). Данные масс-спектрометрии: пик m/e 338,34 (М+1). Биологическая активность Пробы по связыванию с радиолигандом Аффинность тестируемых соединений по отношению к М 2 и М 3 подтипам мускариновых рецепторов определяли изучением связывания с [3H]-N-метилскополамином, используя сердце крысы и подчелюстную железу, соответственно, как описано Moriya и др. (Life Sci., 1999; 64(25): 2351-2358) с незначительными следующими модификациями. Получение мембраны было осуществлено со следующими модификациями: использовали низкоскоростное центрифугирование при 500 g в течение 10 мин при 4 С; использовали 20 ммоль буфера HEPES, 10 ммоль EDTA, рН 7,4; высокоскоростное центрифугирование осуществляли при 40,000 g в течение 20 мин и гомогенат пропускали через фильтровальную марлю до любого вращения. Условия проведения проб были модифицированы следующим образом: объем пробы был 250 л; время инкубации составляло 3 ч; концентрация РЕ была 0,1%; использовали волокнистые фильтры GF/B (Wallac); использовали сцинтиллятор Supermix (Wallac); количество сцинтиллятора составляло 500 л/источник; и использовали счетчик 1450 vicrobeta PLUS (Wallac). Получение мембраны Сублимированные железы и сердце изолировали и непосредственно после умерщвления помещали в ледяной гомогенизирующий буфер (HEPES 20 ммоль, 10 ммоль EDTA, рН 7,4). Ткани гомогенизировали в 10 объемах гомогенизирующего буфер и гомогенат отфильтровывали через два слоя мокрой марли,фильтрат центрифугировали при 500 g в течение 10 мин. Супернатант затем центрифугировали при 40,000 g в течение 20 мин. Полученный таким образом остаток повторно суспендировали в таком же объеме буфера для проб (HEPES 20 ммоль, EDTA 5 ммоль, рН 7,4) и хранили при -70 С до использования. Пробы по связыванию с лигандом Соединения растворяли и разбавляли в ДМСО. Гомогенаты мембран (150-250 г белка) инкубировали в 250 л буфера для проб (HEPES 20 ммоль, рН 7,4) при 24-25 С в течение 3 ч. Неспецифическое связывание определяли в присутствии 1 моль атропина. Инкубацию завершали вакуумным фильтрованием через GF/B волокнистые фильтры (Wallac). Фильтры затем промывали ледяным 50 ммоль Tris HCl буфером (рН 7,4). Фильтровальные матрицы высушивали и подсчитывали связанную радиоактивность,оставшуюся на фильтрах. IC50 и Kd подсчитывали с использованием программы нелинейного построения кривых, используя программное обеспечение G Pad Prism. Значение константы ингибирования Ki посчитывали исходя из изучения конкурентного связывания, используя уравнение ChengPrusoff (Biochem.Pharmacol. 1973; 22: 3099-3108), Ki=IC50/(1+L/Kd), где L представляет собой концентрацию [3H]NMS, использованного в отдельном эксперименте. pKi=-[Log Ki]. Значения Ki для исследованных соединений находились в диапазоне от 0,05 до 136 нмоль для M3 рецептора и в диапазоне от 0,06 до 34,6 нмоль для М 2 рецептора. Функциональные эксперименты с использованием изолированного мочевого пузыря крысы Методика Животных умерщвляли передозировкой уретана, целый мочевой пузырь изолировали, быстро уда-9 009387 ляли и помещали в ледяной Tyrode буфер следующего состава (ммоль/л): NaCl 137; KCl 2,7; CaCl2 1,8;MgCl2 0,1; NaHCO3 11,9; NaH2PO4 0,4; глюкоза 5,55, и длительно продували 95% O2 и 5% CO2. Мочевой пузырь нарезали на продольные полоски (3 мм шириной и 5-6 мм длиной) и помещали в 10-миллилитровые органные бани при 30 С, при этом один конец подсоединяли к основанию держателя тканей, а другой подсоединяли к полиграфу через датчик смещения силы. Каждую ткань подвергали постоянному натяжению в 2 г, позволяли прийти в равновесие в течение 1 ч, в течение которого PSS изменялся каждые 15 мин. В конце периода равновесия стабилизацию ответного сжатия ткани оценивали с помощью 1 моль/л карбахола (Carbachol) последовательно 2-3 раза. Затем получали кривую кумулятивного концентрационного ответа на карбахол (10-9 моль/л до 3 х 10-5 моль/л). После нескольких промываний, когда была достигнута базовая линия, кривую кумулятивного концентрационного ответа получали в присутствии NCE (NCE добавляли за 20 мин до второго CRC). Полученные результаты выражены в % от контрольного Е max. Значения ED50 были рассчитаны путем построения нелинейной регрессионной кривой (Graph Pad Prism). Значения pKB были рассчитаны по формуле pKB=- log [(молярная концентрация антагониста/(отношение доз-1], где отношение доз=ED50 в присутствии антагониста/ED50 в отсутствие антагониста. Эксперименты in vivo на анестезированных кроликах Эффект тестируемых соединений изучался на изменениях кровяного давления, частоты сердечных сокращений и слюноотделения, вызванных карбахолом. Кроликов-самцов весом 1,2-3 кг анестезировали введением уретана (1,5 г/кг) и вводили препарат в виде медленной внутривенной инфузии через крайнюю ушную вену. Катетеризировали трахею, чтобы управлять вентиляцией воздуха. Кровяное давление записывали через бедерную артерию с помощьюStatham P10 EZ датчика давления, присоединенного к полиграфу модели Grass 7D. Частоту сердечных сокращений наблюдали на тахографе, настроенном на волну пульса кровяного давления. Другую бедерную артерию катетеризировали для введения карбахола. Тестируемые соединения и физиологический раствор инфузировали внутривенно через бедерную вену. Мочевой пузырь извлекали посредством срединной лапаротомии, обе уретры идентифицировали,тщательно отделяли и закрепляли. Уретры надрезали проксимально, чтобы обеспечить свободное истекание мочи из почек наружу. Шейку мочевого пузыря аккуратно поддерживали, уретру отсекали от окружающих тканей. Вводили РЕ катетер в мочевой пузырь и закрепляли. Осушали мочевой пузырь, после чего наполняли его 15 мл теплого физиологического раствора (37 С). Другой конец внутрипузырного катетера подсоединяли к полиграфу модели Grass 7D через датчик давления Statham P10 EZ для мониторинга кровяного давления. Подвергаемые воздействию участки поддерживали в тепле и влажности. Период стабилизации параметров после хирургического вмешательства составлял 30-60 мин. Ответ слюноотделения оценивали, помещая предварительно взвешенную абсорбирующую хлопковую марлю в ротовую полость на 2 мин после введения карбахола. Эффект соединения на карбахол (1,5 г/кг, внутриартериально) вызывал изменения кровяного давления, скорости сердечных сокращений и давления в мочевом пузыре. Получали по крайней мере два стабильных ответа. Эти ответы принимались за 100%. Затем изучали эффект увеличения дозы тестируемого соединения или носителя (внутривенно, от 12 до 15 мин до введения карбахола). Изменения в кровяном давлении, слюноотделении и противодействие индуцированной брадикардии выражали в % изменения от значения до обработки. Значения ID50 (доза, требуемая для ингибирования 50% ответа) рассчитывались с использованием нелинейного построения кривых для ответов сигмоидальных доз, используя программное обеспечение Graph Pad Prism, значения были выражены в г/кг. Для тестируемых соединений значения ID50 для кровяного давления находились в диапазоне от около 1,89 до около 4,2 г/кг. Для тестируемых соединений значения ID50 для слюноотделения находились в диапазоне от около 3,7 до около 30,4 г/кг. Поскольку настоящее изобретение было описано в терминах его конкретных воплощений, определенные модификации и эквиваленты ясны специалисту в данной области техники и также включены в объем настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. 6-Замещенные азабицикло[3,1,0]гексаны, выбранные из следующих соединений:(2R)-N-[(1,5,6)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-(п-метилфенил)-2-гидрокси-2-(N-метил) фенилацетамид (соединение 18). 2. Способ лечения или профилактики животного или человека, страдающего расстройством или заболеванием дыхательной, мочевыделительной и желудочно-кишечной систем, где заболевание или расстройство опосредованы мускариновыми рецепторами, включающий введение указанному животному или человеку терапевтически эффективного количества соединения, имеющего структуру формулы I его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров,гдеR1 и R2 независимо выбраны из С 1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из С 1-С 3 алкила или галогена;Z представляет собой кислород или NR3, где R3 представляет собой водород или C1-С 3 алкил. 3. Способ по п.2, где заболеванием или расстройством является недержание мочи, симптомы нижних мочевыводящих путей (LUTS), бронхиальная астма, хронические обструктивные легочные расстройства (COPD), легочный фиброз, синдром раздраженной кишки, ожирение, диабеты и желудочнокишечная гиперкинезия. 4. Способ по п.2, где заболеванием или расстройством является недержание мочи, симптомы нижних мочевыводящих путей (LUTS), бронхиальная астма, хронические обструктивные легочные расстройства (COPD), легочный фиброз, синдром раздраженной кишки, ожирение, диабеты и желудочнокишечная гиперкинезия. 5. Способ получения соединения формулы V и его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров, гдеR1 и R2 независимо выбраны из C1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из С 1-С 3 алкила или галогена;R3 представляет собой водород или C1-С 3 алкил; где указанный способ включает:(а) реакцию соединения формулы II с соединением формулы III с получением защищенного соединения формулы IV, где R1, R2, R3 определены выше, а Р представляет собой защитную группу для аминогруппы(б) удаление защитной группы в соединении формулы IV в присутствии удаляющего защитную группу агента с получением соединения формулы V, где R1, R2, R3 определены выше. 6. Способ по п.5, где Р представляет собой любую защитную группу для аминогруппы и выбран из группы, состоящей из бензильной и трет-бутилоксикарбонильной групп. 7. Способ по п.5, где реакция соединения формулы II с соединением формулы III с получением соединения формулы IV проводится в присутствии N-метилморфолина, и 1-гидроксибензотриазола, и конденсирующего агента, который выбран из гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида(EDC), 1,3-дициклогексилкарбодиимида (DCC) и 1,8-диазабицикло[5,4,0]ундец-7-ена (DBU). 8. Способ по п.5, где реакция соединения формулы II с соединением формулы III проводится в подходящем полярном апротонном растворителе, выбранном из N,N-диметилформамида, диметилсульфоксида, толуола, ксилола и хлороформа. 9. Способ по п.5, где реакция соединения формулы II с соединением формулы III проводится при температуре 0-140C. 10. Способ по п.5, где удаление защиты в соединении формулы IV проводится удаляющим защиту агентом, выбранным из группы, состоящей из палладия на угле и водорода, формиата аммония палладия на угле, трифторуксусной кислоты (ТФУК) и хлороводородной кислоты. 11. Способ по п.5, где удаление защиты в соединении формулы IV с получением соединений формулы V проводится в подходящем органическом растворителе, выбранном из группы, состоящей из метанола, этанола, тетрагидрофурана и ацетонитрила. 12. Способ получения соединений формулы VIII- 12009387 и его фармацевтически приемлемых солей, фармацевтически приемлемых энантиомеров, диастереомеров, гдеR1 и R2 независимо выбраны из C1-С 6 алкила или необязательно замещенного фенила, где необязательный заместитель (заместители) может быть выбран из C1-С 3 алкила или галогена; где указанный способ включает:(а) реакцию соединения формулы II с соединением формулы VI (где R' представляет собой защитную группу для гидроксигруппы, выбранную из п-толуолсульфонила и метансульфонила) с получением защищенного соединения формулы VII, где R1 и R2 определены выше, а Р представляет собой защитную группу для аминогруппы(б) удаление защитной группы в соединении формулы VII в присутствии удаляющего защитную группу агента с получением соединения формулы VIII, где R1 и R2 определены выше. 13. Способ по п.12, где Р представляет собой любую защитную группу для аминогруппы и выбран из группы, состоящей из бензильной и трет-бутилоксикарбонильной групп. 14. Способ по п.12, где реакция соединения формулы VI с соединением формулы II с получением соединений формулы VII проводится в присутствии конденсирующего агента, который выбран из 1,8 диазабицикло[5,4,0]ундец-7-ена (DBU) или 1,4-диазабицикло[2,2,2]октана (DABCO). 15. Способ по п.12, где реакция соединения формулы VI с соединением формулы II проводится в растворителе, выбранном из бензола, толуола или ксилола. 16. Способ по п.12, где реакция соединения формулы VI с соединением формулы II проводится при температуре 0-140 С. 17. Способ по п.12, где удаление защиты в сoединении формулы VII с получением соединений формулы VIII проводится удаляющим защиту агентом, выбранным из группы, состоящей из палладия на угле и газообразного водорода или формиата аммония и палладия на угле. 18. Способ по п.12, где удаление защиты в соединении формулы VII с получением соединений формулы VIII проводится в подходящем растворителе, выбранном из метанола или этанола.
МПК / Метки
МПК: A61P 11/00, C07D 209/52, A61K 31/403, A61P 13/00, A61P 1/00
Метки: мускаринового, азабициклические, качестве, рецептора, антагонистов, производные
Код ссылки
<a href="https://eas.patents.su/14-9387-azabiciklicheskie-proizvodnye-v-kachestve-antagonistov-muskarinovogo-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Азабициклические производные в качестве антагонистов мускаринового рецептора</a>
Замещенные производные азабициклогексана в качестве антагонистов мускаринового рецептора

Номер патента: 9059
Опубликовано: 26.10.2007
Авторы: Силамкоти Арундутт Висванатхам, Гупта Джанг Бахадур, Мехта Анита
МПК: A61P 1/00, A61P 11/00, A61K 31/403...
Метки: антагонистов, азабициклогексана, замещенные, производные, рецептора, качестве, мускаринового
Формула / Реферат:
1. Соединения структурной формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереомеры, N-оксиды, полиморфные формы, где Ar представляет собой арильное или гетероарильное кольцо, имеющее 1-2 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота, арильное или гетероарильное кольца могут быть незамещенными или замещенными от одного до трех заместителями, независимо...
Производные карбамата 1-алкил-1-азониабицикло [2.2.2] октана и их применение в качестве антагонистов мускаринового рецептора

Номер патента: 6505
Опубликовано: 29.12.2005
Авторы: Катена Руис Хуан Лоренсо, Бальса Лопес Долорс, Фернандес Серрат Анна, Фарреронс Гальеми Карлес, Толедо Меса Нативидад, Фернандес Гарсия Андрес, Микель Боно Игнасио-Хосе, Сальседо Рока Каролина, Лагунас Арналь Кармен
МПК: A61P 43/00, C07D 453/06, A61K 31/435...
Метки: производные, качестве, рецептора, 1-алкил-1-азониабицикло, мускаринового, антагонистов, карбамата, применение, октана, 2.2.2
Формула / Реферат:
1. Соединение общей формулы (I) где R1, R2 и R3 означают радикалы, независимо выбранные из группы, состоящей из H, OH, NO2, SH, CN, F, Cl, Br, I, COOH, CONH2, (C1-C4)-алкоксикарбонила, (C1-C4)-алкилсульфанила, (C1-C4)-алкилсульфинила, (C1-C4)-алкилсульфонила, (C1-C4)-алкоксила, необязательно замещенного одним или несколькими атомами F, и (C1-C4)-алкила, необязательно замещенного одним или несколькими атомами F или OH; альтернативно, или R1 и...
Производные 3-азабицикло (3.1.0) гексана в качестве антагонистов опиоидного рецептора

Номер патента: 6708
Опубликовано: 24.02.2006
Авторы: Хек Стивен Дональд, Макхарди Стэнтон Ферст, Лирас Спирос
МПК: C07D 209/52, A61K 31/403, A61K 31/4178...
Метки: производные, качестве, опиоидного, 3-азабицикло, гексана, рецептора, антагонистов, 3.1.0
Формула / Реферат:
1. Соединение, описываемое формулой I, где X представляет собой H, галоген, -OH, -CN, -C1-C4алкил, замещенный атомами галогена в количестве от одного до трех, либо -O(C1-C4алкил), где C1-C4алкил в -O(C1-C4алкиле) необязательно замещен атомами галогена в количестве от одного до трех; Q представляет собой галоген, -OH, -O(C1-C4алкил), -NH2, -N(C1-C4алкил)(C1-C4алкил), -C(=O)NH2, -C(=O)NH(C1-C4алкил), C(=O)N(C1-C4алкил) (C1-C4алкил) или...
Азабициклические, азатрициклические и азаспироциклические производные аминоциклогексана в качестве антагонистов рецепторов nmda, 5ht и нейронных никотиновых рецепторов

Номер патента: 7098
Опубликовано: 30.06.2006
Авторы: Йиргенсонс Айгарс, Парсонс Кристофер Грахам Рафаэль, Калвиньш Ивар, Ванейевс Максимс, Каусс Валерьянс, Даныш Войцех, Хенрих Маркус, Гольд Маркус
МПК: A61P 25/18, A61K 31/40, A61P 25/16...
Метки: азаспироциклические, качестве, рецепторов, антагонистов, азабициклические, нейронных, nmda, аминоциклогексана, никотиновых, производные, азатрициклические
Формула / Реферат:
1. Соединения формулы (1) где R и R1-R5, каждый независимо, выбран из C1-6-алкильных групп, С2-6-алкенильных групп, С2-6-алкинильных групп, С6-12-арил-С1-4-алкильных групп, необязательно замещенных С6-12-арильных групп и в случае R и R2-R5 - из атомов водорода, с условием, что по меньшей мере один из R2 и R3 и по меньшей мере один из R4 и R5 не является водородом, или R и R1 вместе представляют С3-5-алкиленовую или алкениленовую группу, причем...
Производные циклопентена в качестве антагонистов рецептора мотилина

Номер патента: 3252
Опубликовано: 27.02.2003
Авторы: Ксианг Мин, Чен Роберт Х., Биверз Мэри Пэт, Мур Джон Б.Мл.
МПК: C07C 233/41, A61K 31/5375, A61P 1/00...
Метки: антагонистов, качестве, рецептора, производные, циклопентена, мотилина
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет H, C1-5-алкил, замещенный C1-5-алкил (где заместителями являются один или несколько галогенов), амино-C1-5-алкил, C1-5-алкиламино-C1-5-алкил, ди-C1-5-алкиламино-C1-5-алкил, RaRbN-C1-5-алкил (где Ra и Rb независимо выбраны из H и C1-5-алкила или вместе образуют морфолин, пиперазин, пиперидин или N-замещенный пиперидин, где N-заместитель представляет C1-5-алкил или фенил-C1-5-алкил),...
Предыдущий патент: Гидрофильные, реакционные в отношении тиол-группы цианиновые красители, и их коньюгаты с биомолекулами для флюоресцентной диагностики
Следующий патент: Векторы экспрессии и способы их применение
Случайный патент: Автомобильное остекление