Противораковое соединение и содержащая его фармацевтическая композиция
Номер патента: 21084
Опубликовано: 30.04.2015
Авторы: Гонтье Сильви, Шев Мишель, Клерк Франсуа, Комбо Сесиль, Лашо Сильветт, Шио Лоран, Карри Жан-Кристоф, Крик Ален











Формула / Реферат
1. Соединение формулы (I) в виде основания или фармацевтически приемлемой соли

2. Соединение по п.1 в левовращающей форме.
3. Соединение по п.2, имеющее удельное вращение [α]D=-38,6±0,7 при концентрации 0,698 мг/мл в метаноле.
4. Применение соединения по пп.1-3 в качестве селективного ингибитора киназ Aurora А и В.
5. Применение соединения по пп.1-3 в качестве противоракового средства.
6. Применение соединения по пп.1-3 для получения лекарственного средства для лечения рака.
7. Фармацевтическая композиция для лечения рака, содержащая соединение по пп.1-3, а также по меньшей мере один фармацевтически приемлемый эксципиент.
8. Лекарственное средство для лечения рака, содержащее соединение по пп.1-3.
9. Способ получения соединения по пп.1-3, включающий
взаимодействие 3 следующих соединений:

,
где PG обозначает защитную группу NH-группы бензимидазола,
с получением соединения

снятие защиты с NH-группы бензимидазола с получением соединения формулы (I).
10. Способ по п.9, дополнительно включающий выделение левовращающего соединения.
11. Способ по п.9 или 10, в котором взаимодействие 3 соединений осуществляют в спирте, в частности 1-бутаноле, при кипячении с обратным холодильником.
12. Способ по одному из пп. 9-11, в котором PG обозначает группу

13. Соединение, которое представляет собой

причем PG обозначает группу

14. Соединение, которое представляет собой

причем PG обозначает группу

15. Применение соединения по п.13 или 14 в качестве промежуточного соединения при получении соединения по пп.1-3.
Текст
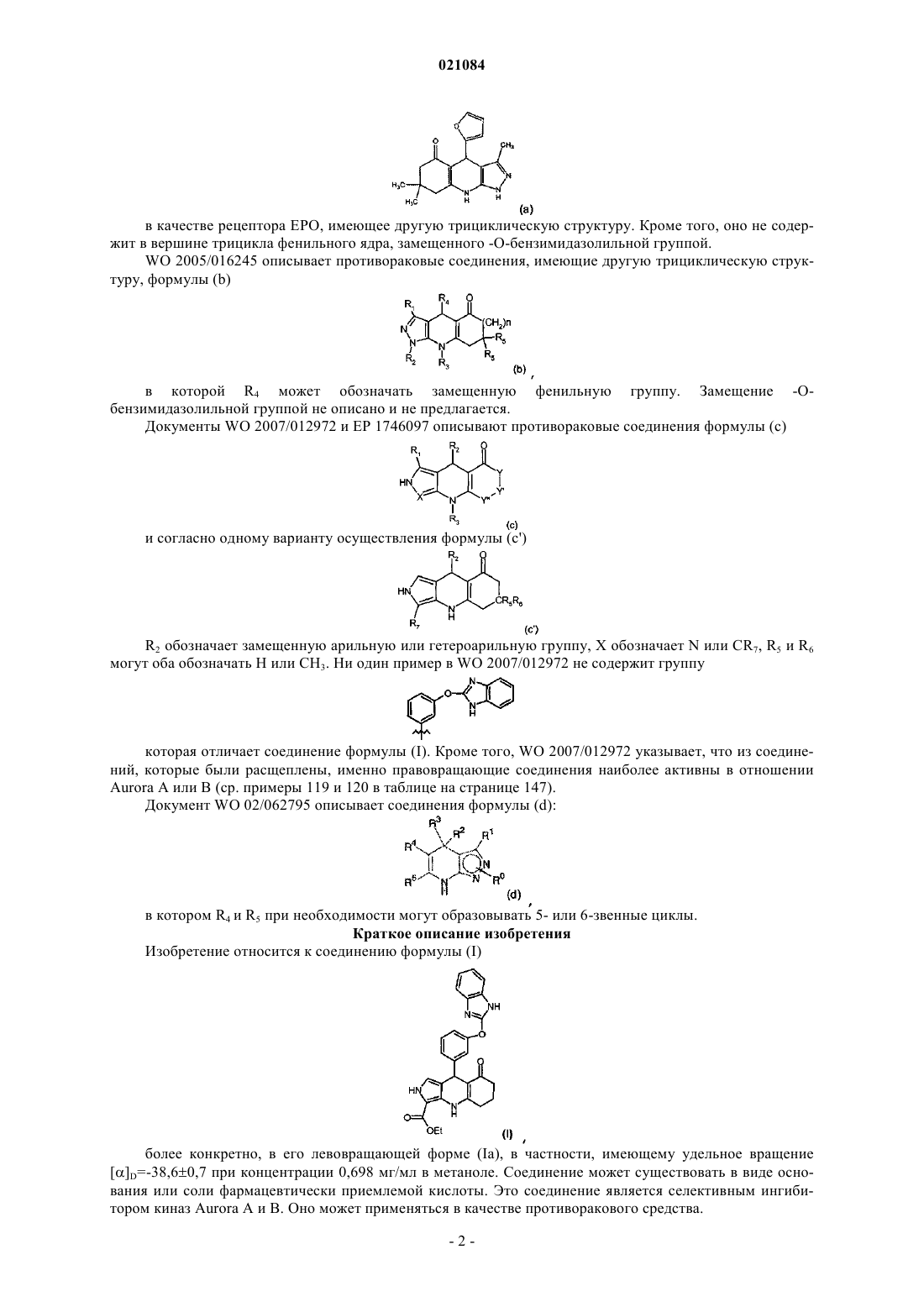
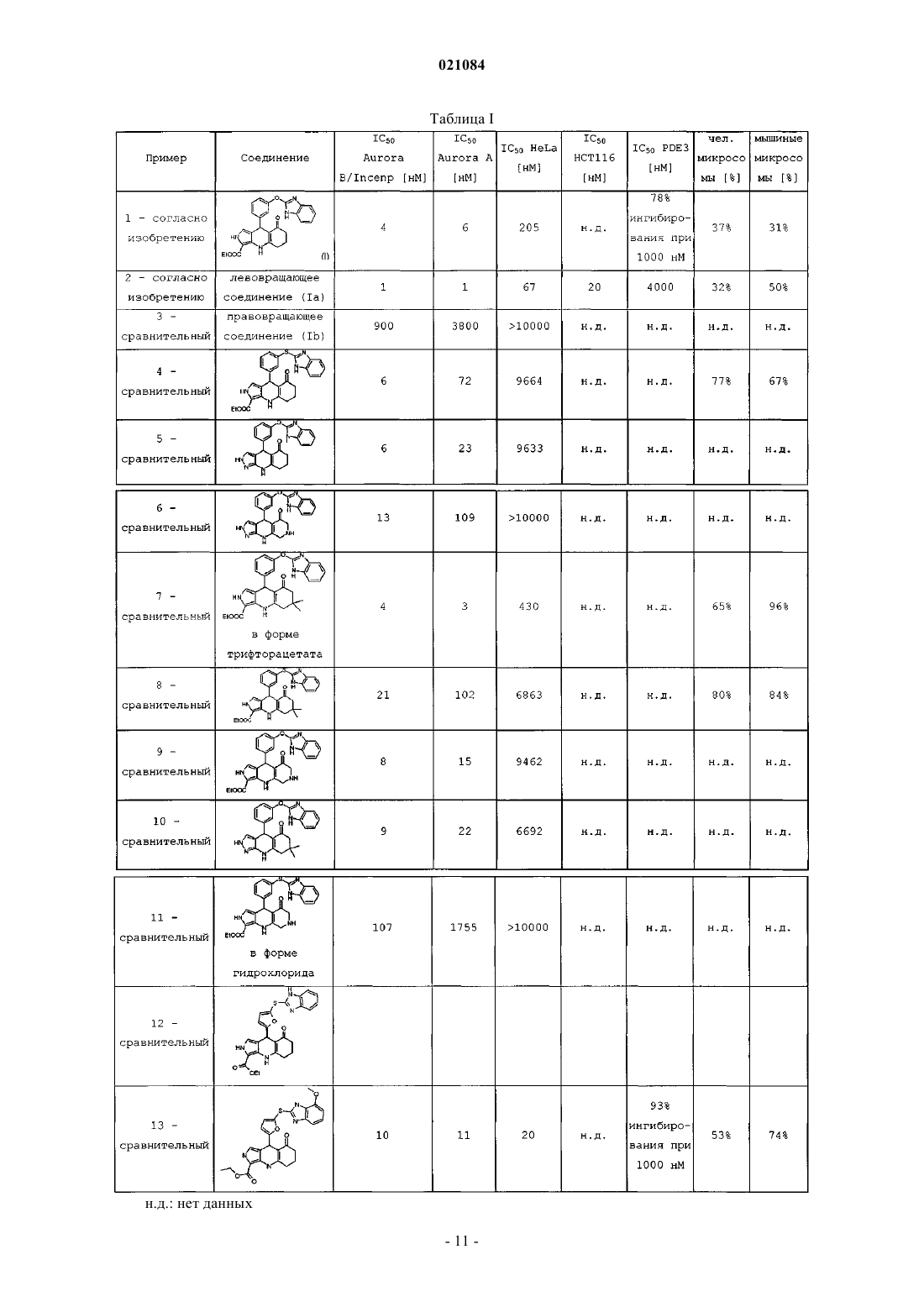
ПРОТИВОРАКОВОЕ СОЕДИНЕНИЕ И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ(Ia), в частности, имеющему удельное вращение []D=-38,60,7 при концентрации 0,698 мг/мл в метаноле. Соединение может существовать в виде основания или соли присоединения кислоты,в частности фармацевтически приемлемой кислоты. Это соединение является селективным ингибитором киназ Aurora А и В. Оно может также применяться в качестве противоракового средства. а также к фармацевтической композиции, содержащей это соединение. Предпочтительно это соединение является левовращающим соединением (Ia). Это соединение находит применение в качестве противоракового средства. Изобретение относится также к способу получения соединения (I) или (Ia), a также к некоторым промежуточным соединениям указанного способа. Техническая проблема Многие стратегии лечения рака направлены на ингибирование киназ типа Aurora, в частности Aurora А и В, которые вовлечены в регуляцию митоза; см. Nature Reviews 2004, 4, 927-936; Cancer Res. 2002,94, 1320; Oncogene 2002, 21, 6175; Mol.Cell.Biol. 2009, 29(4), 1059-1071; Expert Opin. Ther. Patents 2005,15(9), 1169-1182; Clin. Cancer Res. 2008, 14(6), 1639). Определенные соединения - ингибиторы Aurora (например, MLN-8237 от фирмы Millenium, AZD1152 от фирмы Astra-Zeneca или SNS-314 от фирмы Sunesis) проходят в настоящее время клинические испытания. MLN-8237 является селективным в отношении Aurora А, тогда как AZD-1152 селективен в отношение Aurora В. Так как регуляция обеих киназ, Aurora А и В, при раке ослаблена, одновременное ингибирование Aurora А и В дает преимущества по сравнению с селективным ингибированием той или иной из киназ. Кроме того, существуют мультикиназные соединения, такие как соединение АТ-9283 от фирмы Astex, которые ингибируют несколько киназ, в том числе Aurora А и В. Трудно прогнозировать,что этот тип соединений, что ингибирование киназ Aurora можно реально использовать в клинике, так как ингибирование других киназ, кроме Aurora А и В, способно вызывать побочные эффекты. Поэтому одной проблемой, которую намерено решить изобретение, является разработать соединение, которое было бы мощным и селективным ингибитором Aurora А и В. Фермент фосфодиэстераза с циклическим нуклеотидом PDE3 играет важную роль в сигнализации посредством циклических нуклеотидов сАМР и cGMP, которая имеет место в миоцитах гладких мышц сердца и сосудов. Ингибирование PDE3 малыми молекулами оказывает инотропное и сосудорасширяющее действие, которое может оказаться полезным на короткое время при лечении некоторых кардиомиопатий, имеющих недостатки сердечной контракции. Однако было показано, что долговременное воздействие таких молекул повышает смертность у пациентов с нарушениями такого типа. Кроме того, использование ингибиторов PDE3 у пациентов, не имеющих патологии этого типа, таких как больные раком,может вызвать нежелательные воздействия на сердечный ритм. Поэтому в рамках противораковой терапии важно не ингибировать PDE3 (см. Exp.Opin.Invest.drugs 2002, 11, 1529-1536 "Inhibitors of PDE3 asis wrong with positive inotropic drugs Lessons from basic science and clinical trials"). Другой технической проблемой, которую намерено решить изобретение, это то, чтобы соединение - ингибитор Aurora А и В не ингибировало фермент PDE3. Важно также, чтобы противораковое средство имело метаболическую стабильность (см. раздел 10.2.2 в "Chimie pharmaceutique" G.L.Patrick, De Boeck, ed. 2003, ISBN=2-7445-0154-9). Действительно,неудовлетворительная фармакокинетика фармацевтических соединений является одной из главных причин неудач их разработки (Curr. Pharm. 2005, 11, 3545 "Why drugs fail - a study on side effects in new chemical entities"). Кроме того, метаболизм часто является главной детерминантой клиренса, взаимодействия лекарственных средств, межиндивидуальной вариабельности фармакокинетики, а также клинической эффективности и токсичности (Curr.Drug Metab. 2004, 5(5), 443-462 "Human hepatocytes in primary culture:the choice to investigate drug metabolism in man"). Другой технической проблемой, которую намерено решить изобретение, является то, чтобы соединение - ингибитор Aurora А и В имело хорошую химическую и метаболическую стабильность. Уровень техники В журнале Bioorg.Med.Chem.Lett. 2002, 2, 1481-1484 в табл. II описывается соединение 6 А, имеющее другую трициклическую структуру.WO 01/36422 описывает соединения, имеющие другую трициклическую структуру. в качестве рецептора ЕРО, имеющее другую трициклическую структуру. Кроме того, оно не содержит в вершине трицикла фенильного ядра, замещенного -О-бензимидазолильной группой.WO 2005/016245 описывает противораковые соединения, имеющие другую трициклическую структуру, формулы (b) в которой R4 может обозначать замещенную фенильную группу. Замещение -Обензимидазолильной группой не описано и не предлагается. Документы WO 2007/012972 и ЕР 1746097 описывают противораковые соединения формулы (с) и согласно одному варианту осуществления формулы (c')R2 обозначает замещенную арильную или гетероарильную группу, X обозначает N или CR7, R5 и R6 могут оба обозначать H или CH3. Ни один пример в WO 2007/012972 не содержит группу которая отличает соединение формулы (I). Кроме того, WO 2007/012972 указывает, что из соединений, которые были расщеплены, именно правовращающие соединения наиболее активны в отношенииAurora А или В (ср. примеры 119 и 120 в таблице на странице 147). Документ WO 02/062795 описывает соединения формулы (d): в котором R4 и R5 при необходимости могут образовывать 5- или 6-звенные циклы. Краткое описание изобретения Изобретение относится к соединению формулы (I) более конкретно, в его левовращающей форме (Ia), в частности, имеющему удельное вращение[]D=-38,60,7 при концентрации 0,698 мг/мл в метаноле. Соединение может существовать в виде основания или соли фармацевтически приемлемой кислоты. Это соединение является селективным ингибитором киназ Aurora А и В. Оно может применяться в качестве противоракового средства. Изобретение относится также к фармацевтической композиции, содержащей это соединение, а также по меньшей мере один фармацевтически приемлемый эксципиент, и к лекарственному средству, содержащему это соединение. Изобретение относится также к способу получения соединения, включающему: взаимодействие 3 следующих соединений, где PG обозначает защитную группу NH-группы бензимидазола с получением соединения: снятие защиты с NH-группы бензимидазола с получением соединения формулы (I); при необходимости, выделение левовращающего соединения. Взаимодействие 3 соединений осуществляют в спирте, в частности в 1-бутаноле, при кипячении реакционной смеси с обратным холодильником. Следующие промежуточные соединения также являются частью изобретения:PG представляет собой группу Описание изобретения Изобретение относится к соединению формулы (I) Это соединение может существовать в рацемической форме или в виде двух энантиомеров: левовращающего (Ia) и правовращающего (Ib). Левовращающее соединение (Ia) имеет намного большую селективную ингибирующую активность по отношению к киназам Aurora А и В, чем правовращающий энантиомер (Ib). Левовращающее соединение (Ia) обладает также более высокой антипролиферативной активностью, чем правовращающий энантиомер (Ib) (см. табл. I). Все три соединения (I), (Ia) и (Ib) могут существовать в виде основания или соли присоединения кислоты. Предпочтительно, соль получена с фармацевтически приемлемой кислотой (см. P.Stahl,C.Wermuth; Handbook of Pharmaceutical Salts; Wiley Ed. ISBN-13: 978-3906390260, ISBN-10: 3906390268),но и соль кислоты другого типа, полезная, например, для очистки или выделения, также составляет часть изобретения. Соединения (I) и (Ia) могут применяться как противораковые средства или для получения лекарства для лечения рака. Более конкретно, рак является раком, в который вовлечены киназы А и/или В. Соединения (I), (Ia) и (Ib) получают согласно схеме I ниже. Этап 1. Защищают NH-группу 2-галогенобензимидазола (Hal= Br или Cl) с помощью защитной группы PG, с получением A1. PG-X обозначает реагент, позволяющий ввести защитную группу PG. Более конкретно, PG может представлять собой дигидропиран ( в этом случае PG-X обозначает 3,4 дигидро-2 Н-пиран ( Этап 2. А 1 приводят во взаимодействие с 3-формилфенолом в присутствии основания, позволяющего получить соответствующий ион фенолята, чтобы получить В 1. Основание может быть гидридом щелочного металла, как, например, NaH. Реакция проводится в апротонном полярном растворителе, таком как DMF. Этап 3. Совокупность В 1, 3-амино-2-этоксикарбонилпиррола и 1,3-циклогександиона приводят во взаимодействие, например, в спирте (например, 1-бутаноле) при температуре кипения реакционной среды с обратным холодильником, чтобы получить С 1. Этап 4. Снимают защиту с NH-группы С 1, получая соединение (I). Условия снятия защиты зависят от природы PG. Например, в случае, когда PG обозначает дигидропиран, используют сильную кислоту. Этап 5. Разделяют, например, с помощью хиральной хроматографии оба энантиомера (Ia) и (Ib). Для каждого из этапов 1-5 можно взять на вооружение особые условия, описанные в примере 1. Примеры Аналитические методы Метод ЖХ/МС-А. Анализируемые продукты разделяют на колонке ВЭЖХ модель Aquity Beh C18 1,7 мкм 2,150 мм(Waters), термостатированной при 70C, и элюируют при скорости течения 1 мл/мин градиентом ацетонитрила, содержащего 0,1% муравьиной кислоты (растворитель В) в воде, содержащей 0,1% муравьиной кислоты (растворитель А): программа элюирования: изократическое элюирование при 5% растворителя В в течение 0,15 мин, градиент от 5 до 100% растворителя В за 3,15 мин, затем возвращение к начальным условиям за 0,1 мин. Продукты детектируют детектором с диодной матрицей модели PDA Acquity, работающим в видимой и ультрафиолетовой области спектра (Waters, рассматриваемый диапазон длин волн 192-400 нм), датчиком светорассеяния, модель Sedex 85 (Sedere, распылительный газ: азот, температура распыления: 32C, давление распыления 3,8 бар) и масс-спектрометром Acquity SQD (Waters, работающий в положительном и отрицательном режиме, анализируемый диапазон масс от 80 до 800 аем). Метод ЖХ/МС-В. Спектры были получены на приборе Waters UPLC-SQD в режиме положительной и/или отрицательной ионизации электрораспылением (ES+/-), в следующих условиях жидкостной хроматографии: колонка: ACQUITY ВЕН С 18 1,7 мкм, 2,150 мм; Т колонки: 50C; скорость пропускания: 1 мл/мин; растворители: А: H2O (0,1% муравьиной кислоты) В: CH3CN (0,1% муравьиной кислоты); градиент (2 мин): от 5 до 50% В за 0,8 мин; 1,2 мин: 100% В; 1,85 мин 100% В; 1,95 мин 5% В. 1H-ЯМР. Спектры сняты на спектрометре марки Brker, причем продукт растворяют в DMCO-d6. Химические сдвигивыражены в ppm. ИК. Инфракрасный спектр зарегистрирован на спектрометре Nicolet Nexus с таблеткой KBr с разрешением 2 см-1. Измерение удельного вращения. Удельное вращение измеряли при помощи поляриметра 341 Perkin-Elmer. Элементный анализ. Элементный анализ осуществляли на анализаторе ЕА 1108 Thermo. Измерение активности в отношении Aurora А и В. Способность фермента ингибировать киназную активность оценивается по измерению остаточной киназной активности фермента в присутствии разных концентраций испытуемого соединения (обычно от 0,17 до 10000 нМ). Выводится кривая доза-ответ, которая позволяет определить IC50 (концентрация, ингибирующая на 50%). Измерение киназной активности путем радиационного анализа количества радиоактивного фосфата (33 Р), введенного в фрагмент белка NuMA (Nuclear Mitotic Apparatus protein) после 30 мин инкубации при 37C. Испытуемое соединение сначала растворяют до разных концентраций в диметилсульфоксиде (DMCO). Реакция проводится в лунках микротитровального планшета типа FlashPlateNuMA, 1 мкМ АТФ и 0,2 Ci АТФ 33P в буфере 50 мМ Tris-HCl pH 7,5; 10 мМ MaCl2; 50 мМ NaCl, 1 мМ дитиотреитола. Конечная процентное содержание DMCO составляет 3%. После гомогенизации перемешиванием планшет инкубируют в течение 30 мин при 37C. Затем содержимое лунок удаляют и лунки промывают буфером PBS. Затем измеряют радиоактивность с помощью счетчика типа TRILUXI450 Microbeta (WALLAC). В каждом планшете было предусмотрено 8 контрольных лунок: 4 положительных контроля (максимальная киназная активность), для которых измерение проводится в присутствии фермента и субстрата и в отсутствие соединения по изобретению, и 4 отрицательных контроля (фоновый шум), для которых измерение проводится в отсутствие фермента, субстрата и испытуемого соединения. Результаты измерений приведены в табл. I.Aurora A. Используемый рекомбинантный человеческий фермент Aurora A экспрессируется в полной форме с меткой полигистидин в N-терминальном положении и продуцируется в E.coli. Фрагмент (аминокислоты 1701-2115) человеческого белка NuMA с меткой полигистидин в С-терминальной позиции экспрессируется в рекомбинантной форме в E.coli.Aurora B/Incenp. Цельный человеческий фермент Aurora В коэкспрессируется с фрагментом человеческого белка Incenp (aa 821-918) в бакуловирусной системе и экспрессируется в клетках насекомых. Aurora В имеет метку полигистидина в N-терминальном положении, тогда как фрагмент Incenp имеет метку глутатион-Sтрансфераза (GST) в N-терминальном положении. Эти два белка образуют комплекс, называемый AuroraB/Incenp. Фрагмент (аа 1701-2115) человеческого белка NuMA с меткой полигистидин в С-терминальном положении экспрессируется в рекомбинантной форме в E.coli. Этот фрагмент используется в качестве субстрата. Измерение клеточной пролиферации. Клетки (линии опухолевых клеток HeLa (индекс: АТСС CCL-2) и НСТ 116 (индекс: АТСС CCL-247) приводят в контакт с испытуемым соединением на 96 ч, в последние 24 ч добавляют 14 С-тимидин. Пролиферацию клеток оценивают по количеству 14 С-тимидина, поглощенного клетками. Испытуемое соединение переводят в маточный раствор 10 мМ в DMCO, этот маточный раствор используют для получения серии последовательных разбавлений, обычно от 10000 мкМ до 0,3 мкМ, полученные в результате последовательного разбавления растворы, в свою очередь, разбавляют в отношении 1/50 в среде клеточной культуры (раствор 20), которая будет использоваться для разбавления 1/20 в планшетах с клеточной культурой. Конечные концентрации испытуемого соединения будут обычно составлять от 10000 до 0,3 нМ. День 0. Клетки засеивают в 96-луноные планшеты Cytostar в 180 мкл культурной среды. Затем планшеты помещают на 4 ч в инкубатор при 37C и 5% СО 2. Затем испытуемые продукты добавляют в объем 10 мкл на лунку, исходя из раствора 20. Этот раствор содержит 2% DMCO в культурной среде. Таким образом, конечная концентрация DMCO равна 0,1%. Затем планшеты помещают на 72 ч в инкубатор при 37C/5% СО 2. День 3. Через 72 ч в культурную среду добавляют 10 мкл на лунку 14 С-тимидина с уровнем радиоактивности 10 Ci/мл. После этого планшеты помещают на 24 ч в инкубатор при 37C и 5% CO2. День 4. Включение 14 С-тимидина измеряют счетчиком радиоактивности Micro-Beta (Perkin-Elmer) по истечении этих 24 ч "импульса". Полная продолжительность обработки клеток испытуемым продуктом составляет 96 ч. Расчет процентной доли ингибирования, IC50, проводится в Excel по формуле: где X = результат измерения для образца;lanc = результат измерения в лунках без клеток. Расчет IC50 проводится с использованием программы XLfit (IDBS, Великобритания) с помощью формулы 205, с параметром D (число Хилла), фиксированном на значении 1. Результаты приведены в табл. I. Оценка эффекта соединений по изобретению на активность фермента PDE3. Эффект соединений согласно изобретению на активность фермента PDE3 оценивался фирмойCEREP (Le bois l'Evque, 86600 Celle l'Evescault, France; http://www.cerep.fr) согласно своему стандартному протоколу (см. Bender, AT., Beavo, J.A. Pharmacol Rev. 2006, 58, 488-520: фермент PDE3A является рекомбинантным, экспрессируется в клетках Sf9, субстратом является сАМР, остаточный АМРс измеряют методом HTRF (гомогенная флуоресценция с временным разрешением). Эталонным ингибитором в испытании является милринон, IC50 которого составляет 270 нМ. Процентные доли остаточной активности приведены по отношению к контролю без ингибитора). Результаты выражены либо в концентрации,вызывающей 50%-ное ингибирование (IC50), либо в процентной доле ингибирования, измеренной при фиксированной концентрации соединения. Результаты даны в табл. I. Измерение химической стабильности соединений Химическую стабильность соединений определяли в различных средах: соляная кислота 0,05 н. в смеси вода/ацетонитрил 50/50 (об./об.); гидроксид натрия 0,05 н. в смеси вода/ацетонитрил 50/50 (об./об.); буфер фосфат натрия 25 мМ, рН 7,4 в смеси вода/ацетонитрил 50/50 (об./об.); буфер фосфат натрия 25 мМ, рН 7,4 в смеси вода/ацетонитрил 50/50 (об./об.), содержащей 1% (вес./об.) гидрохлорида бензиламина; буфер фосфат натрия 25 мМ, рН 7,4 в смеси вода/ацетонитрил 50/50 (об./об.), содержащей 1%(об./об.) 2-меркаптоэтанола. Растворы соединений разбавляли в исследуемых средах до конечной концентрации 100 мкМ путем разбавления 10 мМ маточного раствора в DMCO. Растворы хранили при 20C в течение 48 ч, и с течением времени методом ВЭЖХ измеряли концентрацию исследуемых соединений(t=0, 1, 6, 12, 24 и 48 ч). ВЭЖХ-анализ осуществляли на приборе Agilent, система 1100, снабженном детектором с диодной матрицей, на колонке Luna С 18, 304,6 мм, 3 мкм (Phenomenex), с элюированием градиентом ацетонитрила (растворитель В) в воде, содержащей 0,5% (об./об.) муравьиной кислоты (растворитель А) при скорости 1,5 мл/мин и температуре 25C. Программа элюирования состоит в градиенте от 10 до 90% растворителя В за 5 мин, с последующим изократическим элюированием одну минуту при 90% растворителя В и возвращением в исходные условия за 1 мин). Концентрация исследуемых продуктов оценивается по высоте и площади характеристического пика исследуемого продукта на хроматограмме на максимальной длине волны для каждого продукта. Площадь и высота, измеряемые каждый раз при отборе проб, приводятся к площади и высоте, полученным для образца в нулевой момент времени. Если наблюдается ухудшение, время полувыведения измеряют, исходя из полученной кривой времяконцентрация. Результаты приведены в табл. II. Оценка метаболизма в присутствии микросомальных препаратов печени человека и мышей. Если микросомальные препараты остаются важными для определения метаболической стабильности фармацевтического соединения, то первичная культура гепатоцитов позволяет более тонкую оценку его внутреннего клиренса и лучшие корреляции vitro-vivo, позволяющие предвидеть печеночный клиренс у человека. Соединения по изобретению (5 мкМ) инкубируют при физиологически приемлемой температуре на микросомальных фракциях печени человека и мышей (1 мг/мл белков), разбавленных фосфатным буфером, в присутствии бычьего сывороточного альбумина (1 мг/мл БСА) и никотинамидадениндинуклеотидфосфата, восстановленная форма (1 мМ NADPH). Для прекращения инкубации добавляют 4 объема ацетонитрила, содержащего кортикостерон в качестве внутреннего стандарта (IS). Образцы центрифугируют и супернатант анализируют комбинацией жидкостной хроматографии с тандемной массспектрометрией (ЖХ/МС-МС). Анализ ЖХ/МС-МС осуществляют на масс-спектрометре API4000QTRAP (Sciex), оборудованном хроматографической системой серии 1100 (Agilent) и автоматическим инжектором Pal CTC. Данные собирают и анализируют с помощью программы Analyst 1.4.1. Образцы разделяют на колонке Polaris C18, 3 мкм, элюируя при скорости протекания 0,7 мл/мин с градиентом ацетонитрила (растворитель В) в воде, содержащей 0,1% муравьиной кислоты (растворитель А). Программа элюирования состоит в градиенте растворителя В от 20 до 90% за 2 мин, изократическом элюировании при 90% растворителя В в течение 0,9 мин и возвращении к начальным условиям за 0,1 мин. Площадь хроматографических пиков соединения и внутреннего эталона интегрируются с помощью алгоритма Analyst-Classic. Метаболическая стабильность продуктов по изобретению оценивается сравнением отношений интегралов (ионные токи соединения/ионные токи IS), измеренных через 0 мин (t0) и 20 мин (t20) инкубации. Затем метаболическая стабильность выражается в процентах исчезновения по следующей формуле: Результаты приведены в табл. III. Оценка клиренса в присутствии человеческих гепатоцитов. Соединения согласно изобретению (0,5 или 5 мкМ) инкубировали в инкубаторе, поддерживающем физиологически приемлемую температуру в течение 24 ч в 48-луночных планшетах, покрытых коллагеном, в присутствии свежих или криоконсервированных человеческих гепатоцитов (200000 клеток на лунку), полученных от специфических доноров. Инкубацию осуществляли с культурной средой (НАМF12 - William E). В разные моменты времени (0, 0,5, 1, 2, 4, 6, 8 и 24 ч) из каждой лунки отбирали 100 мкл и процесс останавливали добавлением 700 мкл смеси ацетонитрил/вода 70/30 (об./об.), содержащей кортикостерон в качестве внутреннего стандарта (IS). Затем клетки разделяли и внутри- и внеклеточную среды смешивали и хранили замороженными при -20C до анализа. После размораживания образцы центрифугировали при 300g в течение 20 мин и супернатанты анализировали комбинацией жидкостной хроматографии и тандемной масс-спектрометрии (ЖХ/МС-МС). Анализ по ЖХ/МС-МС проводится на масс-спектрометре API4000-QTRAP (Sciex), оборудованном хроматографической системой серии 1100(Agilent) и автоматическим инжектором Pal CTC. Данные собирают и анализируют с помощью программы Analyst 1.4.1. Образцы разделяют на колонке Polaris C18, 3 мкм, элюируя при скорости протекания 0,7 мл/мин с градиентом ацетонитрила (растворитель В) в воде, содержащей 0,1% муравьиной кислоты(растворитель А). Программа элюирования состоит в градиенте растворителя В от 20 до 90% за 2 мин,изократическом элюировании при 90% растворителя В в течение 0,9 мин и возвращении к начальным условиям за 0,1 мин. Концентрация продуктов по изобретению определяется интегрированием характеристических ионных токов продуктов, отнесенных к внутреннему эталону (IS). Полученные отношения соединение/IS относят к калибровочным стандартам с известными концентрациями, что позволяет определить концентрацию продуктов по изобретению. Затем определяют внутренний клиренс (выраженный в млч-110-6 клеток), исходя из кинетических кривых (концентрация/время), используя программу WinNonLin (5.0). Результаты представлены в табл. IV. Пример 1. Получение этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси)фенил]-4,5,6,7,8,9-гексагидро 2H-пирроло[3,4-b]хинолин-3-карбоксилата (I) В реактор на 10 л в атмосфере аргона при перемешивании вводят 2,5 л ТГФ, 180 г 2 хлорбензимидазола (1,18 моль) и 325 мл 3,4-дигидро-2H-пирана (6,56 моль, 3 экв.). Реактор нагревают до растворения (температура среды: 40C). Затем вводят 6,3 г пара-толуолсульфоновой кислоты (0,033 моль, 0,028 экв.). Температуру поддерживают на уровне от 49 до 52C в течение 2,5 ч. Охлаждают до 12C и добавляют 7,65 г метилата натрия (0,142 моль, 0,12 экв.), продолжая перемешивание суммарного 15 мин. Затем температуру доводят до 18C, добавляют 5 л н-гептана и профильтровывают через 300 гClarcel FLO-M, фракцию фильтра промывают 5 л н-гептана. Фильтрат концентрируют досуха при пониженном давлении и получают 292,6 г 2-хлор-1-(тетрагидропиран-2-ил)-1 Н-бензимидазола в виде слегка желтоватого масла (выход количественный). 1 В 2 трехгорлые колбы объемом 2 л, оборудованные каждая холодильником, термометром и верхне-7 021084 приводной мешалкой, в атмосфере аргона вводят N,N-диметилформамид (0,4 л на колбу) и 3 гидроксибензальдегид (68,5 г в колбу 1; 64,2 г в колбу 2; 1,08 моль). Затем порциями добавляют гидрид натрия (60%-ная дисперсия в минеральном масле) (колба 1: 26 г; колба 2: 24 г; 1,25 моль, 1,2 экв.), максимальная температура при добавлении составляет 32C. Затем вводят 2-хлор-1-(тетрагидропиран-2-ил)1 Н-бензимидазол (А 1), оценочная концентрация 85%) (колба 1: 151 г в 0,5 л N,N-диметилформамида; колба 2: 142 г в 0,5 л N,N-диметилформамида; 1,05 моль, 0,97 экв.). Затем реакционную смесь доводят до кипения (температура 140C, время повышения температуры 40 мин) и кипячение с обратным холодильником продолжают в течение 1 ч. Затем нагрев прекращают и смесь оставляют охлаждаться в течение 1,5 ч. Содержимое обеих 2 колб соединяют. Все это медленно смешивают на 5 л смеси воды со льдом. Полученную водную фазу экстрагируют затем 4 раза, используя каждый раз по 2,5 л этилацетата (AcOEt). Затем органические фазы объединяют, промывают 3 л воды, 2 л насыщенного раствора NaCl и, наконец,сушат в течение ночи путем добавления MgSO4. Полученную органическую фазу затем фильтруют через спеченное стекло (пористость 4) и концентрируют досуха при пониженном давлении, получая 385 г масла каштанового цвета (ЖХ/МС-А, tr (время удержания) = 1,86 мин, МС в положительном режиме:m/z=323,16). Фракцию весом 158 г неочищенного материала, полученного выше, солюбилизируют при высокой температуре в 1,5 л смеси н-гептан/AcOEt (8/2 по объему), объединяют с 500 г оксида кремния (70-30 меш), и все это перемешивают 45 мин. Полученную суспензию фильтруют через целит, промывают 3 л смеси н-гептан/AcOEt (8/2 по объему). Полученную органическую фазу концентрируют досуха при пониженном давлении. Остаток суспендируют в 200 мл изопропилового эфира путем механического перемешивания и обработки ультразвуком, затем фильтруют через фильтр Шотта (пористость 3). Полученную твердую субстанцию промывают 2 раза, используя каждый раз по 40 мл изопропилового эфира, и сушат при пониженном давлении при 40C в течение 16 ч, получая 68 г твердой субстанции. Аналогичная обработка, примененная к остатку неочищенного продукта, позволяет получить 87,8 г твердой фазы. Полученные твердые продукты объединяют и гомогенизируют, получая 155,8 г 3-[1-(тетрагидропиран-2 ил)-1 Н-бензимидазол-2-илокси]бензальдегида в виде светло-бежевых кристаллов (ЖХ/МС-А, tr=1,87 мин, МС в положительном режиме m/z=323,13). МС(ЖХ/МС-В): tr=1,00 мин; [M+H]+: m/z 323; 1H-ЯМР(д, J=7,3 Гц, 1H); 7,73 (т, J=7,8 Гц, 1H); 7,78-7,83 (м, 1H); 7,87 (д, J=7,8 Гц, 1H); 7,96 (с, 1H); 10,05 (с, 1H). Промывка использованных ранее фаз оксида кремния 2 л смеси н-гептан/AcOEt (1/1 по объему) позволяет получить 67 г материала после концентрирования досуха при пониженном давлении. Этот материал экстрагируют 2 л смеси н-гептан/AcOEt (9/1 по объему), объединяют с 285 г оксида кремния (70-30 меш), перемешивают и обрабатывают ультразвуком в течение 1 ч. Затем суспензию фильтруют через целит, твердую фазу промывают 2 л смеси н-гептан/AcOEt (9/1 по объему). Фильтрат концентрируют досуха при пониженном давлении и остаток растирают в 400 мл смеси н-гептан/этанол (95/5 по объему),фильтруют через спеченное стекло (пористость 3) и сушат при пониженном давлении, получая 35 г 3-[1(тетрагидропиран-2-ил)-1 Н-бензимидазол-2-илокси]бензальдегида в виде светло-бежевых кристаллов. В колбу Эрленмейера объемом 2 л при перемешивании магнитной мешалкой добавляют 50 г гидрохлорида 3-амино-2-этоксикарбонилпиррола и 0,204 л 2 Н гидроксида натрия. Смесь перемешивают в течение 15 мин при температуре окружающей среды (ТА), затем экстрагируют 3 раза, используя каждый раз по 0,3 л дихлорметана. Органические фазы объединяют, сушат над MgSO4 и концентрируют досуха при пониженном давлении. Остаток растирают с н-пентаном, фильтруют и сушат при пониженном давлении до постоянного веса, получая 36,4 г 3-амино-2-этоксикарбонилпиррола в виде твердой субстанции каштанового цвета. В трехгорлую колбу на 2 л, снабженную верхнеприводной мешалкой, термометром и холодильником, вводят 1,2 л 1-бутанола, 145 г 3-[1-(тетрагидропиран-2-ил)-1 Н-бензимидазол-2-илокси]бензальдегида (0,405 моль, В 1), 62,4 г 3-амино-2-этоксикарбонилпиррола (1 экв., 0,405 моль), 46,8 г 97%-ного 1,3 циклогександиона (1 экв., 0,405 моль) и 70,5 мл N,N-диизопропилэтиламина (1 экв.) и доводят до кипения с обратным холодильником (продолжительность повышения температуры 55 мин, кипячение продают 30 мин, температура 114C). Затем среду охлаждают до ТА и концентрируют досуха при пониженном В колбу на 2 л вводят 224 г масла каштанового цвета, содержащего этил-8-оксо-9-3-[1(тетрагидропиран-2-ил)-1 Н-бензимидазол-2-илокси]фенил-4,5,6,7,8,9-гексагидро-2H-пирроло[3,4-b] хинолин-3-карбоксилат (пример 1.3), 0,7 л этанола и 0,243 л 2 Н соляной кислоты. Смесь перемешивают 16 ч при температуре окружающей среды, затем фильтруют через спеченное стекло (пористость 4). Фильтрат концентрируют досуха при пониженном давлении и остаток растирают с 0,5 л изопропилового эфира. Полученную твердую фазу сушат при пониженном давлении до постоянного веса, получая 253 г твердой субстанции каштанового цвета, содержащей этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси) фенил]-4,5,6,7,8,9-гексагидро-2H-пирроло[3,4-b]хинолин-3-карбоксилат. (ЖХ/МС-А, tr=1,46 мин, МС в положительном режиме m/z=469,29). Аналогичная операция, проведенная с 54 г масла каштанового цвета,содержащего этил-8-оксо-9-3-[1-(тетрагидропиран-2-ил)-1 Н-бензимидазол-2-илокси]фенил-4,5,6,7,8,9 гексагидро-2H-пирроло[3,4-b]хинолин-3-карбоксилат (С 1), позволяет получить 60 г коричневой твердой субстанции, содержащей этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси)фенил]-4,5,6,7,8,9-гексагидро-2Hпирроло[3,4-b]хинолин-3-карбоксилат.(ЖХ/МС-А, tr=1,48 мин, МС в положительном режиме m/z=469,29). Аликвотную часть 0,8 г полученного материала можно очистить хроматографией на картридже с 50 г оксида кремния (10-90 мкм) (Biotage SNAP, KP-Sil), элюируя путем изократического элюирования дихлорметаном 20 мин, затем градиентом от 0 до 1% (по объему) изопропанола в дихлорметане за 1 ч и,наконец, изократическим элюированием смесью дихлорметан/изопропанол (99/1 по объему) в течение 20 мин. Фракции, содержащие ожидаемый продукт, объединяют, получая 0,21 г желтой твердой субстанции. Продукты 2 аналогичных хроматографических разделений, проведенных на таком же масштабе,кристаллизуют в ацетонитриле, получая в сумме 0,16 г этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси) фенил]-4,5,6,7,8,9-гексагидро-2H-пирроло[3,4-b]хинолин-3-карбоксилата в виде бежевых кристаллов.(ЖХ/МС-А, tr=1,61 мин, МС в положительном режиме m/z=469,28). 1H-ЯМР (400 МГц, DMCO-d6): 1,29 (т, J=7,0 Гц, 3H); 1,80-1,97 (м, 2H); 2,19-2,27 (м, 2H); 2,55-2,69 (м, 1H); 2,81 (дв.т, J=4,8 и 17,2 Гц,1H); 4,26 (квадруплет, J=7,0 Гц, 2H); 5,11 (с, 1H); 6,73 (д, J=3,3 Гц, 1H); 7,02-7,16 (м, 5H); 7,25 (т, J=7,9 Гц, 1H); 7,31-7,38 (м, 2H); 8,34 (с, 1H); 11,33 (шир.с, 1H); 12,26 (шир.с, 1H). Элементный анализ: С=68,72%; Н=5,10%; N=11,82%; Н 2 О=0,38%. Пример 2. Левовращающий энантиомер (Ia) этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси)фенил]4,5,6,7,8,9-гексагидро-2H-пирроло[3.4-b]хинолин-3-карбоксилата. Левовращающий энантиомер получают очисткой сырого продукта примера D1, на хиральной колонке Welk-01RR, 10 мкМ, 80350 мм (Regis, США), элюируя смесью н-гептан/дихлорметан/этанол/ триэтиламин (50/47,5/2,5/0,1 по объему). Элюирование продуктов контролируют УФ-спектроскопией с длиной волны источника 265 нм. При каждой операции вводят описанный в примере D1 сырой продукт в количестве 10 г. В этих условиях пик, соответствующий левовращающему энантиомеру, элюируется при времени удерживания в интервале от 50 до 80 мин. Фракции очищенного левовращающего энантиомера,соответствующие операциям, необходимым, чтобы очистить 310 г сырого продукта, описанного в примере D1, собирают, гомогенизируют и концентрируют досуха при пониженном давлении, получая 50 г твердой бежевой субстанции. Масс-спектр (ЖХ/МС-В): tr=0,77 мин; [М+Н]+: m/z 469; [М-Н]-: m/z 467. 1H-ЯМР (400 МГц, DMCO-d6): 1,29 (т, J=7,1 Гц, 3H); 1,79-1,97 (м, 2H); 2,19-2,27 (м, 2H); 2,55-2,66 (м,1H); 2,81 (дт, J=4,9 и 17,1 Гц, 1H); 4,26 (кв, J=7,1 Гц, 2H); 5,12 (с, 1H); 6,73 (д, J=3,4 Гц, 1H); 7,02-7,16 (м,5H); 7,25 (т, J=8,3 Гц, 1H); 7,29-7,41 (м, 2H); 8,32 (с, 1H); 11,31 (шир.с, 1H); 12,26 (шир.с, 1H). ИК: основные полосы: 1678, 1578, 1525, 1442, 1188, 1043 и 743 см-1. Удельное вращение: []D=-38,60,7 при С=0,698 мг/мл в метаноле. Элементный анализ: С=68,18%; Н=5,92%; N=11,22%; Н 2 О=1,25%. Пример 3. Правовращающий энантиомер (Ib) этил-8-оксо-9-[3-(1 Н-бензимидазол-2-илокси)фенил 4,5,6,7,8,9-гексагидро-2H-пирроло[3,4-b]хинолин-3-карбоксилата. Правовращающий энантиомер получен очисткой очищенного продукта из примера D1 путем хроматографии на хиральной колонке Welk01SS, 10 мкМ, 60350 мм (Regis, США), элюируя смесью н-гептан/этанол (7/3, затем 6/4 по объему). Элюирование продуктов детектируется УФ-спектроскопией. Фракции правовращающего энантиомера собирают, гомогенизируют и концентрируют досуха при пониженном давлении, получая 1,9 г желтого порошка. МС (ЖХ/МС-В): tr=0,77 мин; [М+Н]+: m/z=469,2; [М-Н]-: m/z=467,2. Удельное вращение:[]D=+53,11,1 при С=3,6 мг/мл в метаноле. Примеры 4-13. Соединения примеров 4-11 были получены согласно указаниям в документе WO 2007/012972 (см. способ в п.26 формулы). Трициклические дигидропиридиновые продукты формулы (II) могут быть получены согласно схеме II: Схема II Смесь 1 эквивалента пиразола (X=N) или пиррол-2-карбоксилата (X=COOEt), 1 эквивалента альдегида R-CHO и 1 эквивалента производного дикетона (Y=CH2, CMe2, N-Boc) кипятят с обратным холодильником в спирте, таком как этанол или 1-бутанол, в течение периода времени от получаса до нескольких часов. Затем смесь охлаждают до температуры окружающей среды. Желаемые продукты выделяют фильтрацией или же растворитель выпаривают досуха. При необходимости неочищенный продукт очищают хроматографией на силикагеле или же высокоэффективной жидкостной хроматографией(ВЭЖХ). Когда Y обозначает N-Boc, защитная группа может быть удалена при помощи обработки раствором трифторуксусной кислоты в дихлорметане (50/50) или же раствором соляной кислоты в диоксане (схема Альдегиды общей формулы (III), использованные при получении соединения 4 (R=H), могут быть получены согласно схеме IV. R обозначает один (n=1) или несколько заместителей (n может варьироваться от 2 до 4) бензимидазольного ядра, выбранных из: Н, F, Cl, Br, ОН, SH, CF3, OCF3, OCH3, SCF3,SCH3, OCHF2, OCH2F, SCH2F, (C1-C6) алкила, O-аллила, фенила, возможно замещенного одним или несколькими атомами галогена. Схема IV Этап 1. Защищают альдегидную группу 3-йодобензальдегида с помощью спиртовой защитной группы, более конкретно, диолом (например, этиленгликоль) в присутствии кислоты, такой как паратолуолсульфоновая кислота, в инертном растворителе, такой как толуол, и при температуре от 20C и до температуры кипения реакционной среды. Этап 2. Полученное промежуточное соединение приводят во взаимодействие с продуктом формулы(IV) в присутствии комплекса палладия, такого как бис(дибензилиденацетон)палладий, производного фосфина, такого как фениловый эфир бис(дифенил)фосфина, и основания, такого как трет-бутилат натрия, в инертном растворителе, таком как толуол, и при температуре в интервале от 20C до температуры кипения реакционной среды. Этап 3. Снимают защиту с альдегидной группы в присутствии водного раствора кислоты, такой как соляная кислота, возможно в растворителе, таком как ацетон, и при температуре в интервале от 20C до температуры кипения реакционной среды. В табл. I приведено сравнение активности ингибирования Aurora А и В, антипролиферативной активности в отношении линий HeLa и НСТ 116, активности ингибирования PDE3, а также метаболизма. Установлено, что соединение (I) или (Ia) являются сильными ингибиторами киназ Aurora А и В, а также обладают очень хорошей активностью по отношению линиями клеток HeLa и НСТ 116. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) в виде основания или фармацевтически приемлемой соли 2. Соединение по п.1 в левовращающей форме. 3. Соединение по п.2, имеющее удельное вращение []D=-38,60,7 при концентрации 0,698 мг/мл в метаноле. 4. Применение соединения по пп.1-3 в качестве селективного ингибитора киназ Aurora А и В. 5. Применение соединения по пп.1-3 в качестве противоракового средства. 6. Применение соединения по пп.1-3 для получения лекарственного средства для лечения рака. 7. Фармацевтическая композиция для лечения рака, содержащая соединение по пп.1-3, а также по меньшей мере один фармацевтически приемлемый эксципиент. 8. Лекарственное средство для лечения рака, содержащее соединение по пп.1-3. 9. Способ получения соединения по пп.1-3, включающий взаимодействие 3 следующих соединений:,где PG обозначает защитную группу NH-группы бензимидазола,с получением соединения снятие защиты с NH-группы бензимидазола с получением соединения формулы (I). 10. Способ по п.9, дополнительно включающий выделение левовращающего соединения. 11. Способ по п.9 или 10, в котором взаимодействие 3 соединений осуществляют в спирте, в частности 1-бутаноле, при кипячении с обратным холодильником. 12. Способ по одному из пп. 9-11, в котором PG обозначает группу 13. Соединение, которое представляет собой причем PG обозначает группу 15. Применение соединения по п.13 или 14 в качестве промежуточного соединения при получении соединения по пп.1-3.
МПК / Метки
МПК: A61K 31/4745, C07D 471/04, C07D 235/26, A61P 35/00
Метки: композиция, фармацевтическая, противораковое, соединение, содержащая
Код ссылки
<a href="https://eas.patents.su/14-21084-protivorakovoe-soedinenie-i-soderzhashhaya-ego-farmacevticheskaya-kompoziciya.html" rel="bookmark" title="База патентов Евразийского Союза">Противораковое соединение и содержащая его фармацевтическая композиция</a>
Активирующее ppar соединение и содержащая его фармацевтическая композиция

Номер патента: 9374
Опубликовано: 28.12.2007
Авторы: Ямазаки Юкийоси, Тома Цитому, Озава Хидефуми, Ода Соити, Абе Казутойо, Окуда Аюму, Араки Такааки, Нисикава Масахиро
МПК: A61K 31/4184, A61K 31/424, A61K 31/423...
Метки: активирующее, фармацевтическая, содержащая, соединение, композиция
Формула / Реферат:
1. Соединение, общей формулы (1) (где каждый из R1 и R2, которые могут быть одинаковыми или отличаются друг от друга, представляет собой атом водорода, метильную группу или этильную группу; каждый из R3a, R3b, R4a и R4b, которые могут быть одинаковыми или отличаются друг от друга, представляет собой атом водорода, атом галогена, нитрогруппу, гидроксильную группу, C1-4алкильную группу, трифторметильную группу, C1-4алкоксильную группу,...
Соединение 3-аминокарбазола, фармацевтическая композиция, его содержащая, и способ их получения

Номер патента: 19068
Опубликовано: 30.12.2013
Авторы: Гарофало Барбара, Драгоне Патриция, Ализи Мария Алессандра, Фурлотти Гвидо, Мауджери Катерина, Мангано Джорджина, Гульельмотти Анджело, Колетта Изабелла, Каццолла Никола
МПК: A61P 29/00, C07D 209/88, A61K 31/403...
Метки: содержащая, соединение, 3-аминокарбазола, получения, фармацевтическая, композиция, способ
Формула / Реферат:
1. Соединение 3-аминокарбазола, отличающееся тем, что имеет общую формулу (I), приведенную нижев которой R1 представляет собой атом галогена, метильную группу или тригалогенметильную группу, нитрогруппу или цианогруппу;R2 представляет собой линейную или разветвленную гидроксиалкильную группу, содержащую от 1 до 8 атомов углерода, или линейную или разветвленную карбонилалкильную группу, содержащую от 1 до 8 атомов углерода,его фармацевтически...
Конденсированное бициклическое соединение – ингибитор mtor, фармацевтическая композиция, его содержащая, и способ лечения рака

Номер патента: 15463
Опубликовано: 31.08.2011
Авторы: Арнольд Ли Д., Пэникер Биджой, Дон Хань-Цин, Кру Эндрю-Филип, Ван Цзин, Биард Джеймз Девид, Сью Кэм В., Хонда Аяко, Малвихилл Марк Джозеф, Вернер Дуглас С., Малвихилл Кристен Мишель, Коут Хизер, Тэвэрес Пола А.Р., Волк Браян, Цзинь Мейчжун, Вэн Цинхуа, Чэнь Синь, Бхарадвадж Апурба
МПК: A61K 31/4985, A61P 35/00, C07D 487/04...
Метки: mtor, фармацевтическая, соединение, композиция, содержащая, бициклическое, способ, лечения, конденсированное, ингибитор, рака
Формула / Реферат:
1. Соединение

или его фармацевтически приемлемая соль.
2. Фармацевтическая композиция, включающая соединение по п.1 вместе с фармацевтически приемлемым носителем.
3. Способ лечения лимфомы или рака яичников, включающий введение эффективного количества соединения или соли по п.1 пациенту, нуждающемуся в этом.
Пестицидная композиция, содержащая синтетическое соединение, полезное в качестве средства для образования клубней у бобовых растений, и инсектицидное соединение

Номер патента: 17507
Опубликовано: 30.01.2013
Авторы: Хунгенберг Хайке, Вор Жан-Пьер, Тилерт Вольфганг
МПК: A01P 7/04, A01N 43/16, A01P 3/00...
Метки: бобовых, образования, пестицидная, композиция, качестве, синтетическое, инсектицидное, клубней, соединение, содержащая, средства, растений, полезное
Формула / Реферат:
1. Пестицидная композиция, содержащая:а) соединение общей формулы (I)где n равно 2 или 3;А представляет собой -С(О)-;В представляет собой фенилен;С представляет собой -О-;D представляет собой линейную углеводородную цепь, содержащую 11 атомов углерода, которая является насыщенной или содержит ненасыщенную связь между 4 и 5 атомами углерода;Е и G независимо друг от друга представляют собой заместитель NHR20;R1-R7 представляют собой Н;R8...
Пестицидная композиция, содержащая синтетическое соединение, полезное в качестве агента образования клубней бобовых растений, и фунгицидное соединение

Номер патента: 18062
Опубликовано: 30.05.2013
Авторы: Вор Жан-Пьер, Сюти-Хайнце Анн
МПК: A01P 3/00, A01N 43/16, A01P 21/00...
Метки: синтетическое, соединение, содержащая, качестве, композиция, клубней, образования, растений, фунгицидное, агента, полезное, пестицидная, бобовых
Формула / Реферат:
1. Пестицидная композиция, содержащая:а) соединение общей формулы (I)где n равно 2 или 3;A представляет собой -С(O)-;B представляет собой фенилен;C представляет собой -O-;D представляет собой линейную углеводородную цепь, содержащую 11 атомов углерода, которая является насыщенной или содержит ненасыщенную связь между 4 и 5 атомами углерода;E и G независимо друг от друга представляют собой заместитель NHR20;R1-R7 представляют собой Н;R8...
Предыдущий патент: Флуопирам для контроля первичных инфекций настоящей мучнистой росы
Следующий патент: Способ регенерации экстракционного раствора в процессах экстракции металлов
Случайный патент: Новые варианты маннаназы