Способ получения левоцетиризина и его промежуточных соединений
Номер патента: 16523
Опубликовано: 30.05.2012
Авторы: Зупет Рок, Павлин Дарья, Коленц Иванка, Тихи Ярослав, Пецавар Аница










Формула / Реферат
1. Способ получения левоцетиризина или его фармацевтически приемлемой соли, включающий следующие стадии:
i) реакции промежуточного соединения формулы (II)

с производным дигликолевой кислоты формулы

или X-CO-CH2-O-CH2-R, где X представляет собой группу ОН или галоген, и R представляет собой СООН; СО-галоген; СООМ, где М представляет собой щелочной или щелочно-земельный металл или N(R1)4; CONH2; CONR1R2, COOR1 или CN, где R1 и R2 независимо выбраны из Н, линейного или разветвленного насыщенного алифатического углеводородного радикала, включающего от 1 до 6 атомов углерода, такого как метил, этил, изопропил, трет-бутил, или линейного или разветвленного алкила, замещенного арилом, такого как бензил или трифенилметил, и замещенной или незамещенной арильной группы, с получением промежуточного соединения формулы (IV)

где R является таким, как определено выше;
ii) восстановления промежуточного соединения формулы (IV) с помощью селективного восстанавливающего агента с получением продукта формулы (V)

где R является таким, как определено выше;
iii) в случае, если R представляет собой не СООН, преобразования промежуточного соединения (V) в левоцетиризин,
iv) возможно, преобразования левоцетиризина в его фармацевтически приемлемую соль.
2. Способ по п.1, где R в промежуточном соединении (IV), полученном на стадии (i), представляет собой СООН и где промежуточное соединение (IV) преобразуют в соединение формулы (IV), где R представляет собой группу, которая может быть преобразована в СООН.
3. Способ по любому из пп.1 и 2, где селективный восстанавливающий агент выбран из группы, состоящей из NaBH4, возможно в присутствии карбоновых кислот, таких как уксусная кислота, трифторуксусная кислота, муравьиная кислота, или в присутствии сульфоновых кислот; NaBH3CN, возможно в присутствии карбоновых кислот, таких как уксусная кислота, пропановая кислота, трифторуксусная кислота; NaBH3OCOR3, где R3 представляет собой метил и трифторметил; боранов, таких как комплексы боран-растворитель, где растворитель выбран из тетрагидрофурана (Н3В-ТГФ), диметилсульфида (H3B-SMe2, BMS), диэтилового эфира (Н3В-диэтиловый эфир); (R4)3O×BF4/NaBH4, где R4 представляет собой метил, этил и пропил.
4. Соединение формулы (IV)

где R является таким, как определено в п.1.
5. Соединение по п.4, где R представляет собой СООН.
6. Применение соединения по п.4 или 5 для получения левоцетиризина.
Текст
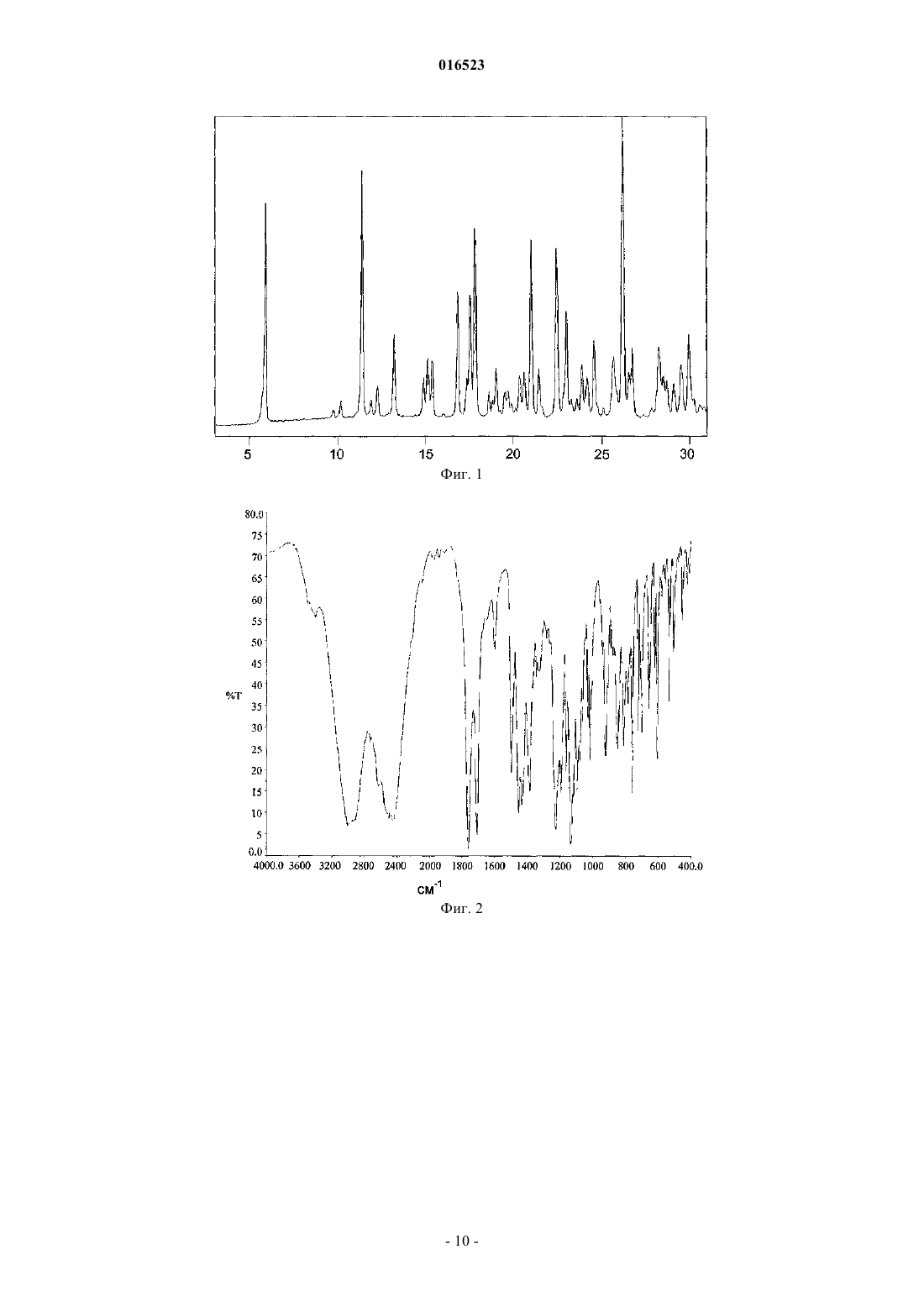
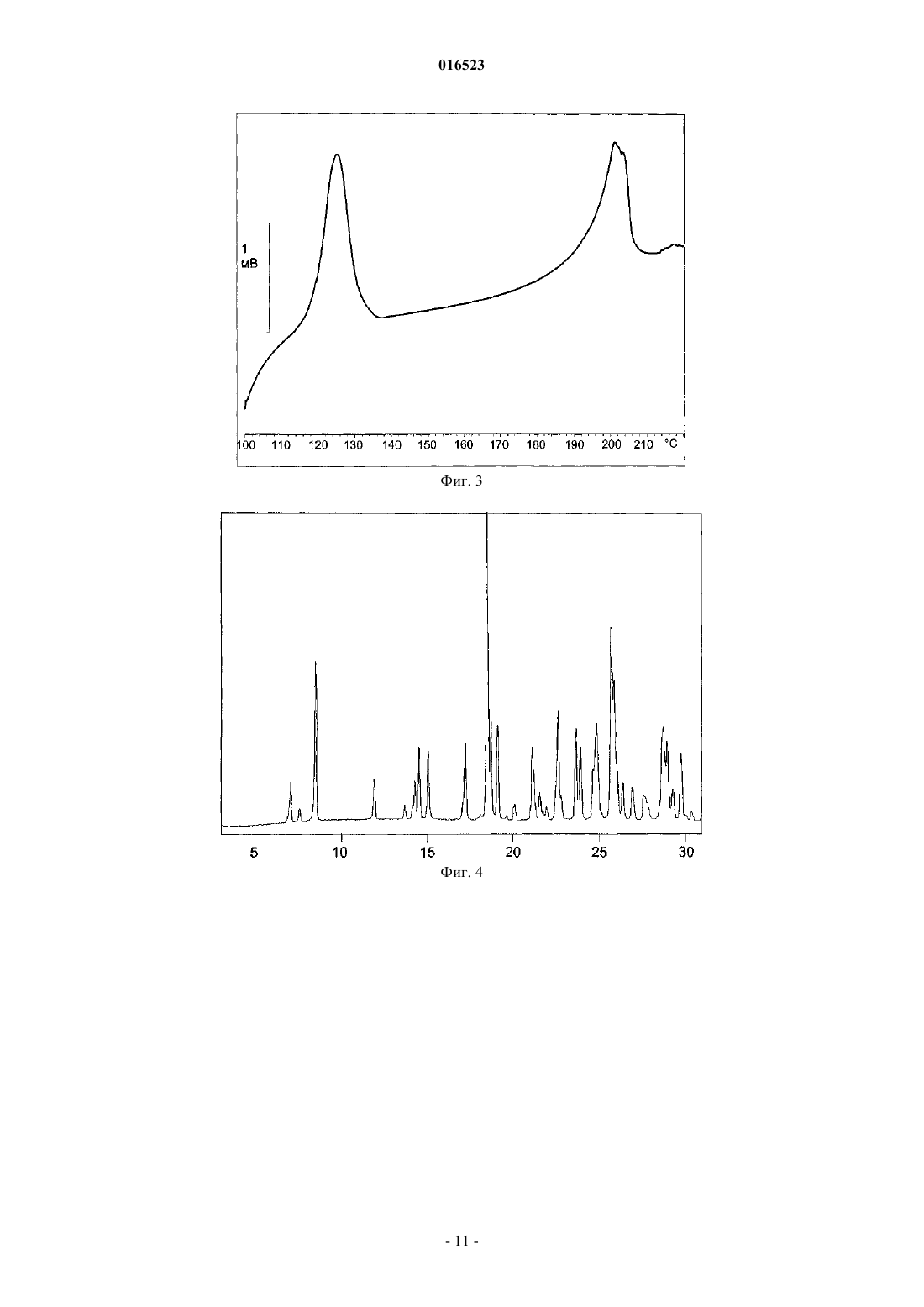
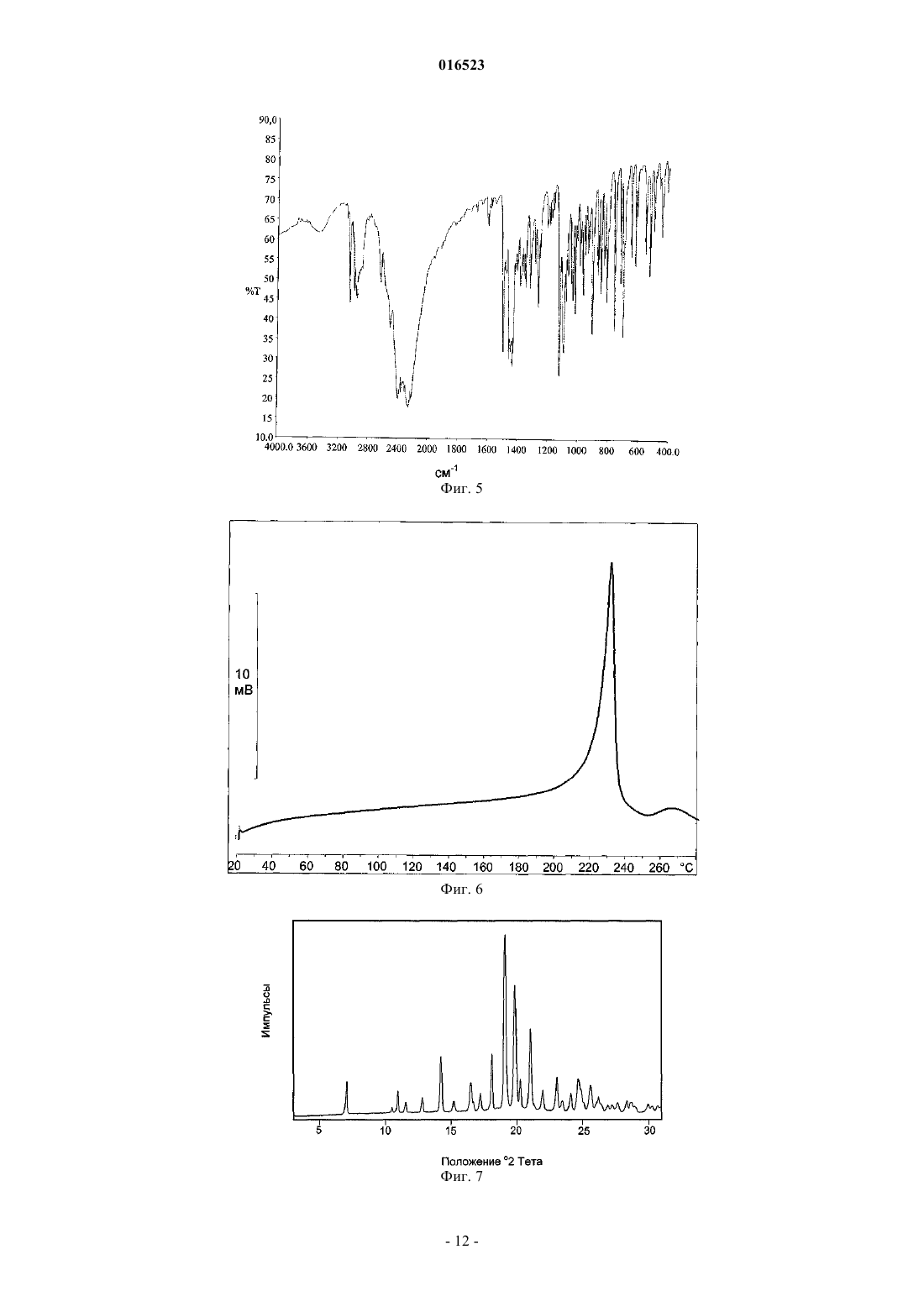
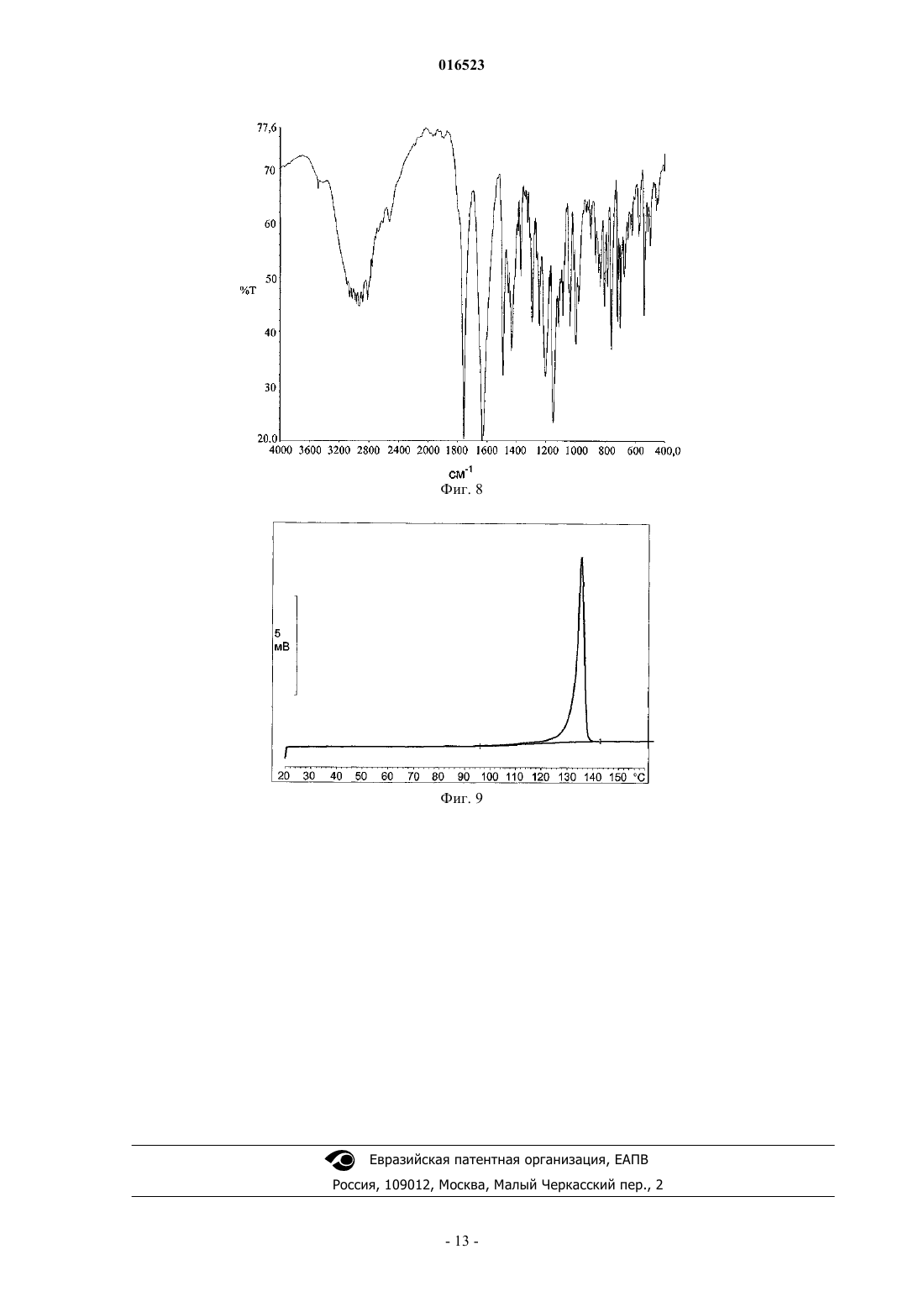
СПОСОБ ПОЛУЧЕНИЯ ЛЕВОЦЕТИРИЗИНА И ЕГО ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ В настоящем изобретении описан новый способ получения левоцетиризина и его фармацевтически приемлемых солей присоединения кислоты с использованием дигликолевой кислоты или ее производных и новые промежуточные соединения, используемые при данном способе. 016523 Область изобретения В настоящем изобретении описан новый способ получения левоцетиризина и его промежуточных соединений, а также его фармацевтически приемлемых солей и эфиров. Предшествующий уровень техники Было показано,что левовращающая[2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]уксусная кислота, также известная под непатентованным названием левоцетиризин, является полезной в качестве терапевтического агента для лечения аллергических заболеваний. Левоцетиризин и его соли, включая его дигидрохлорид, известны и эффективны при лечении аллергий, включая, но не ограничиваясь указанным, хронический и острый аллергический ринит, аллергический конъюнктивит, зуд, крапивницу и тому подобное. Левоцетиризин принадлежит ко второму поколению антагонистов рецептора гистамина Н 1, которые, как считают, имеют значительные преимущества по сравнению с соединениями первого поколения. Исследования показали, что левоцетиризин обеспечивает безопасное и эффективное симптоматическое облегчение сезонных аллергий. Левоцетиризин также применяют для лечения хронической идиопатической крапивницы. В GB 2225321 описан способ получения цетиризина в левовращающей форме, правовращающей форме или в виде их смеси, включающий гидролиз энантиомерно чистого [2-[4-[(4 хлорфенил)фенилметил]-1-пиперазинил]этокси]ацетонитрила. Гидролиз протекает в водной, спиртовой или водно-спиртовой среде с помощью основания или кислоты; кислоту, полученную таким образом,преобразуют в ее дигидрохлорид. Оптически активное исходное вещество 1-[(4 хлорфенил)фенилметил]пиперазин получают путем разделения соответствующего рацемического соединения, предпочтительно путем преобразования в его диастереоизомерную соль с винной кислотой. Выход разделения достаточно низок, а именно всего лишь 12,7%. Полученное оптически активное промежуточное соединение далее подвергают преобразованию с хлорэтоксиацетонитрилом с выходом 69%. В ЕР 0617028 и ЕР 0955295 раскрыт способ получения оптически активного 1-[(4 хлорфенил)фенилметил]пиперазина и его преобразования в цетиризин в левовращающей форме или правовращающей форме, либо в его производное. Этот способ получения показан на приведенной ниже схеме: Недостаток раскрытой реакции состоит в том, что она требует защиты N,N-бис(2 галогеноэтил)амина и последующего удаления защитной группы с полученного промежуточного соединения. Получение цетиризина при большинстве известных синтезов в его левовращающей форме осуществляют из энантиомерно чистого 1-[(4-хлорфенил)фенилметил]пиперазина. Таким образом, оказывается,что весьма желательна разработка новых путей получения его энантиомеров с улучшенной оптической чистотой и хорошими выходами. Полиморфная форма I кристаллической левовращающей соли дигидрохлорида цетиризина и ее аморфная форма раскрыты в WO 2004/050647 и WO 2004/065360. Кристаллическую форму получают путем кристаллизации из растворителя, содержащего кетон, такой как ацетон, метилэтилкетон, диметилкетон, 2-пентанон и их смеси. Аморфную форму получают выпариванием растворителя. Все еще существует потребность в эффективном синтезе левоцетиризина и в новых промежуточных соединениях, используемых при данном способе, пригодных для крупномасштабного производства. Краткое описание графических материалов На фиг. 1 представлена порошковая рентгеновская дифрактограмма сольвата левоцетиризина дигидрохлорида с уксусной кислотой. На фиг. 2 представлен спектр FT-IR (инфракрасный спектр с Фурье-преобразованием Fourier Transform Infra Red) сольвата левоцетиризина дигидрохлорида с уксусной кислотой. На фиг. 3 представлена термограмма ДСК (дифференциальной сканирующей калориметрии) сольвата левоцетиризина дигидрохлорида с уксусной кислотой. На фиг. 4 представлена порошковая рентгеновская дифрактограмма[2-[4-[(4-хлорфенил)фенилметил]-1 пиперазинил]этокси]ацетонитрила дигидрохлорида. На фиг. 7 представлена порошковая рентгеновская дифрактограмма R-2-(2-(4-4 хлорфенил)(фенил)метил)пиперазин-1-ил-2-оксоэтокси)уксусной кислоты. На фиг. 8 представлен спектр FT-IR R-2-(2-(4-4-хлорфенил)(фенил)метил)пиперазин-1-ил-2-1 016523 оксоэтокси)уксусной кислоты. На фиг. 9 представлена термограмма ДСК R-2-(2-(4-4-хлорфенил)(фенил)метил)пиперазин-1-ил-2 оксоэтокси)уксусной кислоты. Описание изобретения В настоящем изобретении предложен новый эффективный синтез левоцетиризина и его фармацевтически приемлемых солей и новые промежуточные соединения, используемые при данном способе. В соответствии с первым аспектом настоящее изобретение относится к способу получения левоцетиризина, включающему приведенные ниже стадии:i) реакции промежуточного соединения формулы (II) с производным дигликолевой кислоты формулы или X-CO-CH2-O-CH2-R, где X представляет собой группу ОН или галоген и R представляет собой СООН или группу, которая может быть преобразована в COOH, предпочтительно R выбран из группы,состоящей из СООН; СО-галоген; СООМ, где М представляет собой щелочной или щелочно-земельный металл или N(R1)4; CONH2; CONR1R2, COOR1, CN, CHO, CH2OH или CH(OR1)2, где R1 и R2 независимо выбраны из Н, линейного или разветвленного насыщенного алифатического углеводородного радикала,включающего от 1 до 6 атомов углерода, такого как метил, этил, изопропил, трет-бутил, или линейного или разветвленного алкила, замещенного арилом, такого как бензил или трифенилметил, и замещенной или незамещенной арильной группы, с получением промежуточного соединения формулы (IV)ii) в случае получения промежуточного соединения формулы (IV), где R представляет собой СООН,его, возможно, могут преобразовывать в соединение формулы (IV), где R представляет собой группу,которая может быть преобразована в COOH;iii) восстановления промежуточного соединения формулы (IV) с помощью селективного восстанавливающего агента с получением продукта формулы (V)iv) в случае, если R представляет собой не СООН, преобразования промежуточного соединения (V) в левоцетиризин; иv) возможно, преобразования левоцетиризина в его фармацевтически приемлемую соль. Производное дигликолевой кислоты может представлять собой ангидрид дигликолевой кислоты формулы или производное формулы X-CO-CH2-O-CH2-R, где X представляет собой ОН или галоген, и R представляет собой COOH или группу, которая может быть преобразована в СООН. Предпочтительно R выбран из группы, состоящей из СООН; СО-галоген; СООМ, где М представляет собой щелочной или щелочно-земельный металл или N(R1)4; CONH2; CONR1R2, COOR1, CN, CHO, CH2OH или CH(OR1)2, гдеR1 и R2 независимо выбраны из Н, линейного или разветвленного насыщенного алифатического углеводородного радикала, включающего от 1 до 6 атомов углерода, такого как метил, этил, изопропил, трет-2 016523 бутил, или линейного или разветвленного алкила, замещенного арилом, такого как бензил или трифенилметил, и замещенной или незамещенной арильной группы. Термин "низший алкил" означает линейные или разветвленные насыщенные алифатические углеводородные радикалы, включающие от 1 до 6 атомов углерода, такие как метил, этил, изопропил, третбутил и т.д.; либо линейный или разветвленный алкил, замещенный арилом, такой как бензил или трифенилметил. Термин "арил" относится к замещенным или незамещенным арильным группам. Для получения соединения формулы (IV) используют избыток или, по существу, эквимолярные количества производного дигликолевой кислоты, предпочтительно используют 1,5 молярный избыток,наиболее предпочтительно 1,1 молярный избыток. Реакцию могут проводить в органическом растворителе, воде или их смеси. В случае использования воды в качестве растворителя процесс могут проводить в присутствии катализатора фазового перехода. Пригодные органические растворители включают полярные или неполярные органические растворители, такие как диметилформамид, диметилсульфоксид,ацетонитрил; галогенированные углеводороды, такие как хлороформ, дихлорметан, тетрахлорид углерода, 1,2-дихлорэтан и тому подобное; эфиры, такие как диэтиловый эфир, диоксан, тетрагидрофуран, 1,3 диметоксиэтан и тому подобное, ароматические углеводороды, такие как бензол, толуол, ксилол. Реакцию могут проводить в отсутствие растворителя. Реакцию предпочтительно проводить в присутствии тетра-н-бутиламмония бромида или тетра-нбутиламмония йодида. Температура реакции может составлять от примерно 0 С до точки кипения реакционной смеси, причем предпочтительным является интервал от примерно 60 С до точки кипения реакционной смеси. Время реакции, достаточное для практически полного завершения реакции, обычно составляет от 1 до 24 ч. При проведении реакции реагенты тщательно смешивают, предпочтительно, добавляя порционно при перемешивании раствор дигликолевой кислоты или производного дигликолевой кислоты в растворителе к раствору амина формулы (II), также в растворителе, и смешивание продолжают с применением или без применения нагревания в течение времени, достаточного для завершения реакции, с образованием желаемого продукта формулы (IV) в реакционной смеси. Продукт могут выделять или очищать с помощью общепринятых методик, таких как экстракция, хроматография или полное или частичное выпаривание растворителя общепринятыми способами и выделение твердого вещества или масла, которое отделяется или остается в виде остатка. Соединение формулы (IV) далее восстанавливают с помощью селективного восстанавливающего агента с получением продукта формулы (V). Селективные восстанавливающие агенты могут выбирать из группы, состоящей из NaBH4, возможно в присутствии карбоновых кислот, таких как уксусная кислота,трифторуксусная кислота, муравьиная кислота, или в присутствии сульфоновых кислот; NaBH3CN, возможно в присутствии карбоновых кислот, таких как уксусная кислота, пропановая кислота, трифторуксусная кислота; NaBH3OCOR3, где R3 представляет собой метил и трифторметил; боранов, таких как комплексы боран-растворитель, где растворитель выбирают из тетрагидрофурана (Н 3 В-ТГФ), диметилсульфида (H3B-SMe2, BMS), диэтилового эфира (Н 3 В-диэтиловый эфир); (R4)3OBF4/NaBH4, где R4 представляет собой метил, этил и пропил. Пригодный растворитель, используемый в фазе восстановления, представляет собой инертный органический растворитель, который может быть выбран из группы, состоящей из эфиров, таких как диоксан, тетрагидрофуран, трет-бутилметиловый эфир, диэтиловый эфир, диизопропиловый эфир, 2 метилтетрагидрофуран; карбоновых кислот, таких как уксусная кислота; галогенированных углеводородов; ароматических углеводородов. Предпочтительными растворителями являются ТГФ и диоксан. Поскольку относительная активность диборана имеет схожие значения как в отношении карбоновых кислот, так и амидов, предпочтительно защищать карбоново-кислотную группу эфирной группой или галогеноангидридной группой. Соответственно, в случае использования боранов в качестве селективного восстанавливающего агента предпочтительно, чтобы R в соединении формулы (IV) являлся иным, чем группа СООН. Как правило, отношение восстанавливающего агента к соединению формулы (IV) составляет от 6:1 до 1:1, предпочтительноот 3:1 до 1:1, более предпочтительно от 1,75:1 до 1:1, наиболее предпочтительно от 1,2:1 до 1:1. Реакцию, как правило, проводят таким образом, что раствор амида формулы (IV) в растворителе добавляют к раствору борана при температуре от 0 С до комнатной температуры. Полученную в результате смесь нагревают от комнатной температуры до точки кипения реакционной смеси и поддерживают при этой температуре от 0,5 часа до нескольких часов до доведения реакции до практически полного завершения. После завершения реакции продукт выделяют и очищают общепринятыми способами, такими как экстракция и кристаллизация. Очистка путем экстракции предпочтительно включает несколько стадий. На первой стадии экстракции предпочтительно используют щелочную экстракцию при рН 9-12. Для этой цели реакционную смесь смешивают с водой, а затем экстрагируют галогенированным или не галогенированным растворителем, не смешивающимся с водой, таким как этилацетат, метиленхлорид или толуол.-3 016523 На второй стадии экстракции рН предпочтительно доводят до рН 4-5, более предпочтительно до 4,2-4,8. Данную стадию могут проводить с использованием такой же системы растворителей, как на первой стадии, то есть вода/галогенированный или не галогенированный растворитель, не смешивающийся с водой,такой как этилацетат, метиленхлорид или толуол. В случае, когда группа R в продукте формулы (V) не является ОН, преобразование промежуточного соединения (V) в левоцетиризин осуществляют способами, известными в данной области техники. Соединение формулы (II), используемое в качестве исходного соединения в соответствии с настоящим изобретением, могут получать в соответствии со способом, раскрытым в настоящем изобретении,или любым другим известным способом, таким как раскрыто в GB 2225321, ЕР 0617028, IN 501/MUM/04. Другой аспект изобретения представляет собой новое соединение формулы (IV) где R является таким, как определено выше. Промежуточное соединение формулы (IV) могут использовать в качестве промежуточного соединения для получения левоцетиризина. Особенно предпочтительной является R-2-(2-(4-4-хлорфенил)(фенил)метил)пиперазин-1-ил-2 оксоэтокси)уксусная кислота, которая характеризуется порошковой рентгеновской дифрактограммой 3,имеющей пики приблизительно при следующих значениях: Исходное соединение (II), (-)-1-[(4-хлорфенил)фенилметил]пиперазин, могут эффективно получать путем взаимодействия R-(-)-4-хлорбензгидриламина и бис(2-галогеноэтил)амина гидрохлорида. Было неожиданно обнаружено, что рацемизация во время этой реакции не происходит. Эта реакция показана на приведенной ниже реакционной схеме 1. Реакционная схема 1 Могут использовать бис(2-галогеноэтил)амина гидрохлорид, где X представляет собой атом хлора,брома или йода. Реакцию, как правило, проводят в присутствии органического основания, которое выступает одновременно в качестве растворителя и акцептора хлорида водорода. Органические основания могут выбирать из группы, состоящей из первичных, вторичных, третичных алкиламинов, таких как Nэтилдиизопропиламин, триэтиламин, диэтиламин, бутилциклогексиламин, диизопропиламин, дибутиламин, или гетероциклических аминов, таких как пирролидин, пиперидин или их алкилзамещенное производное, либо их смеси. Смесь различных оснований могут смешивать в любом соотношении, предпочтительно используют смесь N-этилдиизопропиламина и диэтиламина в объемном отношении от 1:1 до 1:0,01, наиболее предпочтительно 1:0,08. Реакцию, как правило, проводят при температуре от 50 С до температуры дефлегмации реакционной смеси, предпочтительно от 80 С до температуры дефлегмации реакционной смеси. Реакция может длиться от 2 до 24 ч, предпочтительно от 6 до 8 ч. Молярное соотношение обоих реагентов, а именно -(-)-4-хлорбензгидриламина и бис(2 хлорэтил)амина гидрохлорида, может варьировать от 1:1 до 1:2, предпочтительно от 1:1,4 до 1:1,7. Было обнаружено, что при использовании избытка бис(2-хлор)этиламина выходы могут улучшаться. Как правило, после завершения реакции реакционную смесь концентрируют. К остатку добавляют-4 016523 воду и этилацетат. Перед очисткой с использованием колоночной хроматографии значение рН доводят до щелочного с помощью раствора гидроксида натрия. Значение рН реакционной смеси перед очисткой должно составлять более 9, предпочтительно между 10 и 11. Реакцию предпочтительно проводят путем взаимодействия -(-)-4-хлорбензгидриламина с бис(2 хлорэтил)амина гидрохлоридом в N-этилдиизопропиламине в присутствии диэтиламина при температуре дефлегмации в течение нескольких часов, выделение могут проводить путем экстракции в системе растворителей, состоящей из воды и органического растворителя, такого как этилацетат, трихлорметан, дихлорметан, с получением неочищенного (-)-1-[(4-хлорфенил)фенилметил]-4-[(4-метилфенил)сульфонил]пиперазина. Полученный продукт могут дополнительно очищать путем разделения с использованием хроматографии на силикагеле и, наконец, кристаллизовать из гексана с получением продукта с чистотой согласно ВЭЖХ (высокоэффективной жидкостной хроматографии) более чем 98%. Промежуточное соединение формулы (II), полученное способом по настоящему изобретению, можно использовать для преобразования в левоцетиризин и, возможно, его фармацевтически приемлемые соли с помощью любой известной методики. Левоцетиризин, возможно, в форме фармацевтически приемлемой соли, такой как дигидрохлорид,могут также получать способом, характеризующимся реакцией, показанной на реакционной схеме 2. Реакционная схема 2(-)-1-[(4-Хлорфенил)фенилметил]-4-пиперазин формулы (II) подвергают реакции с 2-(2 хлорэтокси)ацетонитрилом в присутствии акцептора кислот, такого как карбонат щелочного металла, и,возможно, в присутствии небольшого количества йодида щелочного металла для ускорения реакции. Реакцию могут проводить в инертном растворителе, например в спирте, таком как этанол, нбутиловый спирт, ароматическом углеводороде, таком как толуол, ксилол, галогенированных алканах,таких как дихлорметан, нитрилах, таких как ацетонитрил. Апротонные растворители особенно предпочтительны. Наиболее предпочтительным растворителем является ацетонитрил. В связи с настоящим изобретением авторы изобретения обнаружили, что выходы данной реакции могут быть существенно улучшены благодаря выделению продукта[2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]ацетонитрила формулы (IIIa) в форме его гидрохлорида. Форму дигидрохлорида получают путем введения газообразного HCl в реакционную смесь или путем добавления газообразного HCl в реакционном растворителе в смесь до достижения значения рН между 3 и 0,5, предпочтительно ниже 1, наиболее предпочтительно 1. Реакция, проведенная в соответствии с настоящим изобретением, протекает с выходами более 90%, предпочтительно 95%. Эта реакция не вызывает рацемизацию, поэтому в случае использования оптически чистого исходного соединения оптическая чистота сохраняется. Обнаружено, что в этом отношении апротонные растворители обладают особым преимуществом. Было обнаружено, что чистота [2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]ацетонитрила формулы (IIIa) может быть улучшена путем суспендирования и перемешивания продукта в спиртах или кетонах при температуре от 0 С до температуры кипения. Спирты, используемые для мацерации, могут представлять собой C1-С 5-спирты, предпочтительно метанол и этанол. Кетоны для суспендирования и перемешивания могут представлять собой ацетон, МЭК (метилэтилкетон) или метилизобутилкетон. Как правило, после полной мацерации получают продукт, имеющий хроматографическую чистоту более 98%, и содержание индивидуальной примеси менее 0,2%. Реакция протекает с лучшими выходами в случае использования молярного избытка 2-(2 хлорэтокси)ацетонитрила. Оптимальное молярное соотношение между промежуточным соединением формулы (II) и 2-(2-хлорэтокси)ацетонитрилом может варьировать от 1:1,1 до 1:4, предпочтительно от 1:1,5 до 1:3, наиболее предпочтительно от 1:1,5 до 1:2. Реакцию проводят при температуре между 60 и 200 С, предпочтительно при температуре между 80 и 120 С, в течение примерно 7-24 ч, как показано на схеме 2. 2-(2-Хлорэтокси)ацетонитрил могут получать любым путем, известным в данной области техники,-5 016523 например, описанным в Suomen Kemistilehti (1944), 17 В, 17-19, Croatica Chemica Acta (1986), 59(19), 30711. Чистота 2-(2-хлорэтокси)ацетонитрила, вступающего в реакцию, согласно ВЭЖХ должна составлять по меньшей мере 95%. Нитрильное промежуточное соединение (IIIa) гидролизуют путем либо основного, либо кислотного гидролиза, либо путем гидролиза, катализируемого ферментом. В случае основного гидролиза нитрильное промежуточное соединение формулы (IIIa) нагревают при температуре от 20 до 110 С, предпочтительно от 60 до 90 С, в присутствии неорганического основания, такого как гидроксид щелочного металла, в растворителе, выбранном из воды или спирта, такого как метанол, этанол, пропанол, 2-пропанол или их смесь. Предпочтительно в качестве гидроксида щелочного металла используют KOH или NaOH, и в качестве растворителя используют смесь воды с метанолом или этанолом. Наиболее предпочтительно используют KOH в смеси метанол/вода. В ходе своей работы авторы изобретения обнаружили, что избыток гидроксида щелочного металла может вызвать рацемизацию. Поэтому наиболее оптимальное массовое отношение гидроксида щелочного металла к промежуточному соединению формулы (IIIa) должно составлять от 1:1 до 4:1, предпочтительно от 1,5:1 до 3:1. Образовавшийся левоцетиризин присутствует в реакционной смеси в форме соли со щелочным металлом, из которой кислоту высвобождают путем подкисления реакционной смеси неорганической кислотой, предпочтительно соляной кислотой. Затем левоцетиризин в виде кислоты экстрагируют органическим растворителем, таким как дихлорметан, толуол, этилацетат, предпочтительно дихлорметаном. Авторы изобретения обнаружили, что значение рН водного раствора перед экстракцией может влиять на выходы экстракции. Следовательно, его нужно поддерживать между 4 и 5, предпочтительно между 4,2 и 4,8. После полной экстракции левоцетиризин могут преобразовывать в дигидрохлорид левоцетиризина путем введения газообразного HCl в экстракт дихлорметана. Для обеспечения лучшего выхода и качества продукта дихлорметан могут выпаривать, а остаток растворять в другом растворителе, таком как спирт, ароматический углеводород, сложный эфир, простой эфир или кетон. Предпочтительными растворителями являются толуол, этилацетат, пропилацетат, изопропилацетат, бутилацетат и ацетон. Наиболее предпочтительным растворителем является ацетон. Для осаждения в этот раствор вводят газообразный HCl или ацетон, насыщенный газообразным HCl. Как правило, описанный способ основного гидролиза дает выходы более 90%, причем чистота полученного продукта согласно ВЭЖХ (площадь) составляет более 98% или даже более 99%. В случае кислотного гидролиза промежуточное соединение формулы (IIIa) нагревают в присутствии неорганической кислоты, такой как соляная кислота, предпочтительно в водной среде, при температуре между 60 С и температурой дефлегмации реакционной смеси. Затем левоцетиризин в форме кислоты экстрагируют из реакционной смеси органическим растворителем, таким как дихлорметан, толуол,этилацетат, предпочтительно дихлорметаном. Затем свободную кислоту левоцетиризин преобразуют в соль дигидрохлорид путем растворения ее в кетоновом растворителе и введения HCl в газообразной форме в реакционную смесь и/или путем введения раствора газообразного HCl в реакционном растворителе, пока рН не достигнет значения между 0,5 и 3, предпочтительно между 0,5 и 1. Кетоновый растворитель могут выбирать из ацетона, метилэтилкетона, диизобутилкетона, диизопропилкетона, предпочтительно ацетона. Полученная соль может существовать в полиморфной форме, раскрытой в WO 2004/050647 или IPCOM 000146553D. Левоцетиризина дигидрохлорид можно дополнительно перекристаллизовать из смеси растворитель/антирастворитель. В качестве растворителя можно использовать карбоновую кислоту или воду, а в качестве антирастворителя можно использовать кетон, сложный эфир или простой эфир. Предпочтительным растворителем является уксусная кислота или вода, а предпочтительным антирастворителем является ацетон, этилацетат, изопропилацетат или бутилацетат. Наиболее предпочтительно в качестве растворителя используют уксусную кислоту, а в качестве антирастворителя используют ацетон. Чистота полученного продукта составляет более 99,5%. Порошковая рентгеновская дифрактограмма полученного таким способом сольвата левоцетиризина дигидрохлорида с уксусной кислотой имеет приведенные ниже пики приблизительно при следующих значениях: Важно контролировать размер частиц левоцетиризина дигидрохлорида в процессе его получения. Средний размер частицы для полученных частиц составляет от 5 до 200 мкм, предпочтительно между 20 и 150 мкм. Если проводить перемешивание, кристаллизация из органических растворителей может также приводить к образованию частиц большего размера, например, имеющих средний диаметр выше 200 мкм, которые необходимо перемалывать или обрабатывать любым другим способом, приводящим к уменьшению размера частиц, перед их применением в фармацевтических препаратах. При перемалывании могут получать частицы, имеющие средний диаметр менее 3 мкм. Для этой цели, как правило, используют струйные мельницы, шаровые мельницы или молотковые мельницы в качестве перемалывающего оборудования. Однако недостаточно контролировать только средний размер частиц, но также нужно контролировать распределение частиц по размеру. Средний размер частиц и распределение частиц по размеру важно для обеспечения того, чтобы технологический процесс был промышленно применимым, то есть не вызывал сегрегации ингредиентов смеси для таблетирования в случае, если эту смесь не таблетируют или не прессуют сразу после получения смеси для таблетирования. Настоящее изобретение проиллюстрировано приведенными ниже примерами без ограничения ими. Примеры Пример 1.R-2-(2-(4-4-хлорфенил)(фенил)метил)пиперазин-1-ил-2-оксоэтокси)уксусная кислота Вариант А. 2 г R-1-4-хлорфенил)(фенил)метил)пиперазина и 0,88 г ангидрида дигликолевой кислоты растворяли в 107 мл ацетонитрила. Реакционную смесь кипятили с обратным холодильником в течение 12 ч. После завершения реакции растворитель выпаривали, и полученный остаток растворяли в 20 мл воды с добавлением 2 мл 1 М NaOH, а затем добавляли 10 мл дихлорметана. Суспензию перемешивали в течение 20 мин и отделяли органическую фазу. К водной фазе добавляли другую порцию 10 мл дихлорметана, рН суспензии доводили до 4,5-5. Водную фазу снова экстрагировали дважды 210 мл дихлорметана. Органические фазы собирали, высушивали над безводным сульфатом натрия, фильтровали и выпаривали. Полученный неочищенный продукт можно использовать на следующей стадии без дальнейшей очистки. Вариант Б. 2 г R-1-4-хлорфенил)(фенил)метил)пиперазина и 0,88 г ангидрида дигликолевой кислоты растворяли в 10 мл диметилсульфоксида. К раствору добавляли 20 мл тетрабутиламмония бромида. Раствор перемешивали в течение еще 10 ч до завершения реакции. Раствор разбавляли 67 мл воды и добавляли 15 мл изопропилацетата. Фазы разделяли, и водный слой повторно экстрагировали изопропилацетатом. Органический слой смешивали с деминерализованной водой (40 мл) и доводили рН суспензии до 10 с помощью 2 М NaOH. Слои разделяли и к водной фазе добавляли 40 мл изопропилацетата. Доводили рН суспензии до 3,5. Слои разделяли, и водную фазу повторно экстрагировали дважды изопропилацетатом. Органическую фазу промывали водой и выпаривали до сухости. Кристаллизовали 2,5 г продукта (ВЭЖХ(площадь) составляет 98,5%). Продукт высушивали в вакууме при 50 С. Пример 2. Левоцетиризина дигидрохлорид Маслянистый продукт из предшествующей стадии растворяли в 20 мл диоксана, к раствору добавляли 1,3 г NaBH4, суспензию охлаждали до 10 С и добавляли при интенсивном перемешивании 2,1 г уксусной кислоты в 7 мл диоксана. Реакционную смесь кипятили с обратным холодильником в течение двух часов, охлаждали до комнатной температуры, и твердые вещества отфильтровывали. Фильтрат выпаривали, к остатку добавляли 20 мл воды и доводили рН до 5. Водную фазу экстрагировали три раза 10 мл дихлорметана. Органические экстракты объединяли, высушивали над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Полученный неочищенный левоцетиризин растворяли в 10 мл ацетона, добавляли 2 мл 36% HCl и перемешивали в течение одного часа. Продукт отфильтровывали и высушивали.-7 016523 Пример 3. 1 г продукта R-2-(2-(4-4-хлорфенил)(фенил)метил)пиперазин-1-ил-2-оксоэтокси)уксусной кислоты растворяли в 10 мл сухого тетрагидрофурана при 0 С. При интенсивном перемешивании добавляли по каплям 0,23 мл оксалилхлорида. По прошествии получаса, когда преобразование кислоты в хлорангидрид было завершено, добавляли 0,8 мл комплекса диметилсульфида борана (10 М, вычислено по BH3) в три порции. После завершения реакции реакционную смесь наливали в воду (250 мл) и дополнительно перемешивали в течение получаса. К суспензии добавляли 100 мл этилацетата, рН доводили до 10, и фазы разделяли. К водной фазе добавляли 100 мл метиленхлорида, рН доводили концентрированной соляной кислотой до 4,2, и фазы разделяли: органический слой промывали водой, высушивали сульфатом натрия и выпаривали до сухого состояния. Масляный остаток растворяли в 30 мл ацетона и вводили газообразный HCl, пока значение рН не стало менее 1. Реакционную смесь нагревали до 60 С и кипятили с обратным холодильником в течение 20 мин. Смесь охлаждали до 30-35 С, а затем фильтровали и осадок промывали 20 мл ацетона. Затем его высушивали при 50-55 С при пониженном давлении (-0,08-0,1 МПа) в течение 8 ч. Получили 720 мг левоцетиризина дигидрохлорида. Пример 4. Перекристаллизация левоцетиризина дигидрохлорида 75 г левоцетиризина дигидрохлорида при 0,17% содержании соединения (II) растворяли в 225 мл уксусной кислоты при 80 С. Раствор охлаждали до 50 С, и при этой температуре постепенно добавляли 562 мл ацетона в течение 1,5 ч. После добавления ацетона суспензию охлаждали до 0 С и перемешивали при этой температуре в течение по меньшей мере 1 ч. Продукт левоцетиризина дигидрохлорид отфильтровывали. Продукт высушивали в вакуумной сушилке при 50 С. Получили 58 г продукта, чистота которого согласно ВЭЖХ (площадь) составляла 99,9%. Содержание соединения (II) находилось ниже предела обнаружения, который составлял 0,005%. Способ анализа: ВЭЖХ; колонка RP18; фосфатный буфер (рН 7), ацетонитрил/метанол; градиентный метод; УФ детектор, 230 нм. Пример 5. Получение левоцетиризина дигидрохлорида сольвата с уксусной кислотой 50 г левоцетиризина дигидрохлорида суспендировали в 100 мл уксусной кислоты и нагревали до растворения вещества. Затем ее медленно охлаждали при перемешивании до 50 С до появления суспензии и после этого при данной температуре к суспензии добавляли 100 мл ацетона. После добавления ацетона суспензию охлаждали до комнатной температуры, осадок отфильтровывали, и продукт промывали ацетоном. Продукт высушивали в вакуумной сушилке при 50 С. Получили 43,1 г продукта, чистота которого согласно ВЭЖХ (площадь) составляла 99,9%. Способ анализа: ВЭЖХ; колонка RP18; фосфатный буфер (рН 7), ацетонитрил/метанол; градиентный метод; УФ детектор, 230 нм. Пример 6. Получение левоцетиризина дигидрохлорида из левоцетиризина дигидрохлорида сольвата с уксусной кислотой 10 г сольвата левоцетиризина дигидрохлорида с уксусной кислотой суспендировали в 60 мл ацетона и перемешивали при комнатной температуре в течение одного часа. После одного часа суспензию отфильтровывали и фильтровальный осадок промывали ацетоном и высушивали в вакуумной сушилке. Масса сухого продукта составляла 8,9 г. Чистота согласно ВЭЖХ (площадь) составляла 99,9%. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения левоцетиризина или его фармацевтически приемлемой соли, включающий следующие стадии:i) реакции промежуточного соединения формулы (II) с производным дигликолевой кислоты формулы или X-CO-CH2-O-CH2-R, где X представляет собой группу ОН или галоген, и R представляет собой СООН; СО-галоген; СООМ, где М представляет собой щелочной или щелочно-земельный металл илиN(R1)4; CONH2; CONR1R2, COOR1 или CN, где R1 и R2 независимо выбраны из Н, линейного или разветвленного насыщенного алифатического углеводородного радикала, включающего от 1 до 6 атомов углерода, такого как метил, этил, изопропил, трет-бутил, или линейного или разветвленного алкила, замещенного арилом, такого как бензил или трифенилметил, и замещенной или незамещенной арильной группы, с получением промежуточного соединения формулы (IV)ii) восстановления промежуточного соединения формулы (IV) с помощью селективного восстанавливающего агента с получением продукта формулы (V)iii) в случае, если R представляет собой не СООН, преобразования промежуточного соединения (V) в левоцетиризин,iv) возможно, преобразования левоцетиризина в его фармацевтически приемлемую соль. 2. Способ по п.1, где R в промежуточном соединении (IV), полученном на стадии (i), представляет собой СООН и где промежуточное соединение (IV) преобразуют в соединение формулы (IV), где R представляет собой группу, которая может быть преобразована в СООН. 3. Способ по любому из пп.1, 2, где селективный восстанавливающий агент выбран из группы, состоящей из NaBH4, возможно в присутствии карбоновых кислот, таких как уксусная кислота, трифторуксусная кислота, муравьиная кислота, или в присутствии сульфоновых кислот; NaBH3CN, возможно в присутствии карбоновых кислот, таких как уксусная кислота, пропановая кислота, трифторуксусная кислота; NaBH3OCOR3, где R3 представляет собой метил и трифторметил; боранов, таких как комплексы боран-растворитель, где растворитель выбран из тетрагидрофурана (Н 3 В-ТГФ), диметилсульфида (H3BSMe2, BMS), диэтилового эфира (Н 3 В-диэтиловый эфир); (R4)3OBF4/NaBH4, где R4 представляет собой метил, этил и пропил. 4. Соединение формулы (IV) где R является таким, как определено в п.1. 5. Соединение по п.4, где R представляет собой СООН. 6. Применение соединения по п.4 или 5 для получения левоцетиризина.
МПК / Метки
МПК: C07D 295/185, C07D 295/088
Метки: промежуточных, получения, соединений, левоцетиризина, способ
Код ссылки
<a href="https://eas.patents.su/14-16523-sposob-polucheniya-levocetirizina-i-ego-promezhutochnyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения левоцетиризина и его промежуточных соединений</a>
Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений

Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Вуд Альберт Шо, Девриз Кейт Майкл, Негри Джоанна Тереза, Леветт Филип Чарльз
МПК: C07D 487/04, C07D 401/12, C07D 401/14...
Метки: соединений, промежуточных, получения, способ, пиридилсульфонильных, пиразоло[4,3-d]пиримидин-7-он-3
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом; R1 (который может быть...
Способ получения промежуточных соединений, применимых для получения противораковых соединений

Номер патента: 5561
Опубликовано: 28.04.2005
Авторы: Сантафьянос Динос Пол, Норрис Тимоти, Лехнер Ричард Шелтон
МПК: C07D 239/94
Метки: применимых, получения, способ, противораковых, соединений, промежуточных
Формула / Реферат:
1. Способ получения соединения формулы 3 где R1 и R2, каждый независимо, выбраны из C1-C10алкила и C1-C10алкокси, причем указанные алкил и алкокси необязательно имеют до 2 заместителей, независимо выбранных из гидрокси и C1-C6алкокси; где соединение формулы 3 получают обработкой соединения формулы 5 где R1 и R2 определены как указано выше, тионилхлоридом в безводном дихлорметане. 2. Способ по п.1, где как R1, так и R2 являются...
Пироглютаматы и их применение для разделения оптических изомеров промежуточных продуктов синтеза декстроцетиризина и левоцетиризина

Номер патента: 14248
Опубликовано: 29.10.2010
Авторы: Паласьо Магали, Ате Селаль
МПК: C07D 207/28, C07D 295/088
Метки: оптических, пироглютаматы, применение, декстроцетиризина, синтеза, изомеров, разделения, продуктов, промежуточных, левоцетиризина
Формула / Реферат:
1. Способ получения (S)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамида или (R)-2-[4-(4-хлорбензгидрил)ииперазин-1-ил]этоксиацетамида, включающий химическое разделение смеси (R)- и (S)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамидов в присутствии (S)-пирролидон-5-карбоновой кислоты или (R)-пирролидон-5-карбоновой кислоты и кристаллизацию.2. Способ по п.1, характеризующийся тем, что химическое разделение проводят в присутствии...
Пироглютаматы и их применение для разделения оптических изомеров промежуточных продуктов синтеза декстроцетиризина и левоцетиризина

Номер патента: 12575
Опубликовано: 30.10.2009
Авторы: Ате Селаль, Паласьо Магали
МПК: C07D 207/28, C07D 295/088
Метки: пироглютаматы, промежуточных, применение, изомеров, продуктов, оптических, левоцетиризина, декстроцетиризина, синтеза, разделения
Формула / Реферат:
1. Соединение, выбранное из группы, включающей соль (S)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамида с (S)-пирролидон-5-карбоновой кислотой, соль (R)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамида с (S)-пирролидон-5-карбоновой кислотой, соль (S)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамида с (R)-пирролидон-5-карбоновой кислотой или соль (R)-2-[4-(4-хлорбензгидрил)пиперазин-1-ил]этоксиацетамида с...
Способ получения циклопропанкарбоновых кислот и их промежуточных соединений

Номер патента: 683
Опубликовано: 28.02.2000
Авторы: Клемменсен Пер Дауселль, Колинн-Андерсен Ханс, Винкельманн Иб
МПК: C07C 61/35, C07D 307/93
Метки: циклопропанкарбоновых, кислот, промежуточных, способ, получения, соединений
Формула / Реферат:
1. Способ получения соединений общей формулы I где R' представляет Н, а два атома водорода циклопропанового кольца находятся в цис-положении по отношению друг к другу, включающий взаимодействие между соединением общей формулы II и соединением СF3-CClХ2, где Х представляет атом галогена, в частности атом хлора или брома, в инертной среде в присутствии Zn и при подходящей температуре от 0 до 150шС, предпочтительно от 20 до 100шС, в...
Предыдущий патент: Способ оценки фактора качества формации
Следующий патент: Система обогрева
Случайный патент: Подгузник одноразового использования