S-нитрозотиолы в качестве агентов для лечения нарушений кровообращения
Номер патента: 3577
Опубликовано: 26.06.2003
Авторы: Триас Адроер Нурия, Пубиль Кой Франсиско, Кабеса Льоренте Лидия, Репольес Молинер Хосе, Салас Перес-Расилья Эдуардо, Мичелена Льягуно Педро, Мурат Морено Хесус, Карбо Банус Марсель.Ли, Серда Риудаветс Хуан Антонио, Феррер Сисо Алисия, Негрье Рофес Кристина


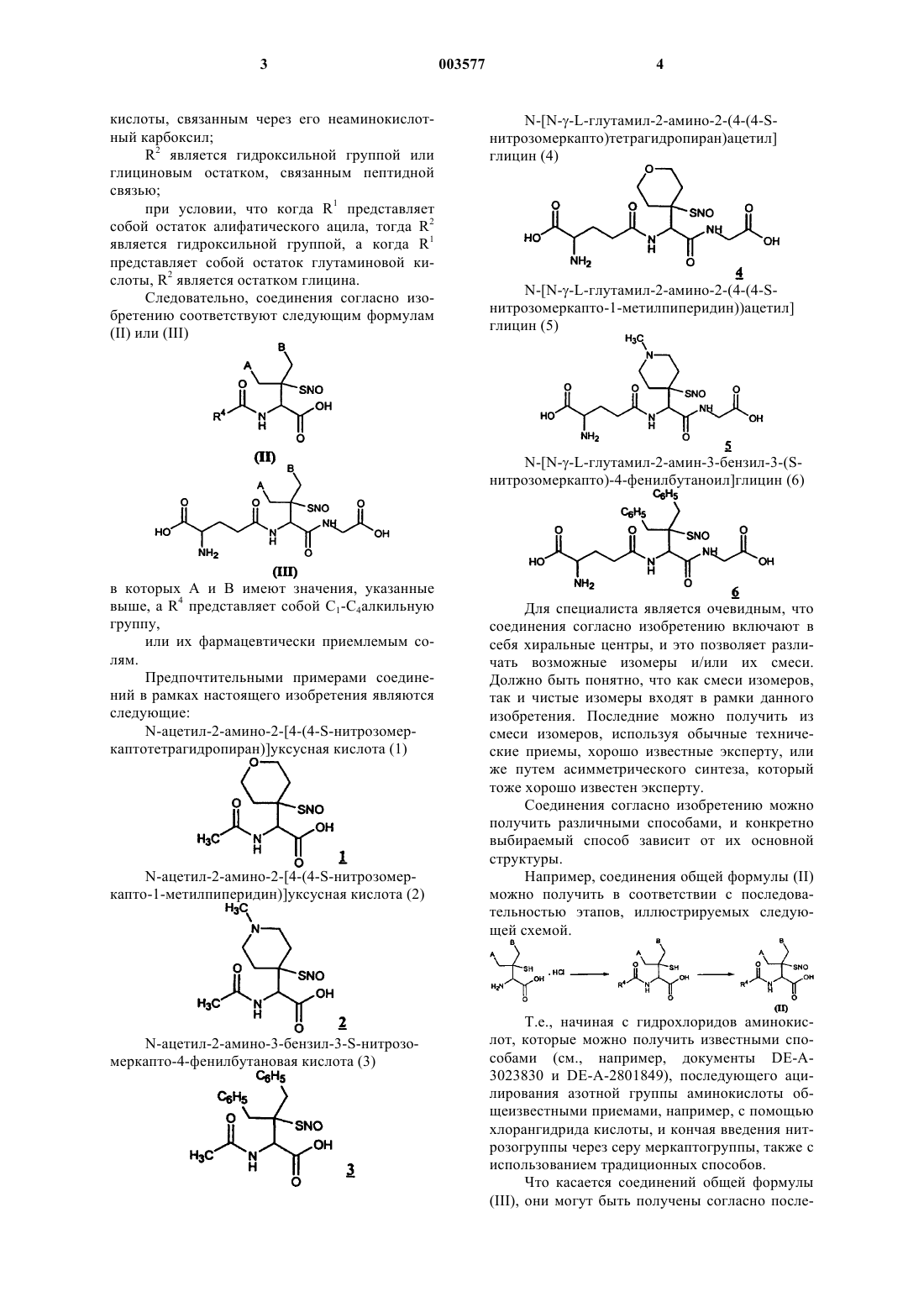





Формула / Реферат
1. S-Нитрозотиоловые производные пеницилламина или глутатиона и их фармацевтически приемлемые соли, которые соответствуют следующей общей формуле (I)

в которой
A и B являются фенильными группами или же вместе образуют остаток -CH2-Q-CH2-, образующий шестичленное кольцо, в котором Q представляет собой атом кислорода или серы, или группу N-R3, в которой R3 является водородом или алкильной группой C1-C4;
R1 является ацильным остатком, который может быть C1-C5 алифатической ацильной группой или остатком глутаминовой кислоты, связанным через его неаминокислотный карбоксил;
R2 является гидроксильной группой или остатком глицина, связанным через его пептидную связь;
при условии, что когда R1 является алифатическим ацильным остатком, тогда R2 является гидроксильной группой, а когда R1 является остатком глутаминовой кислоты, тогда R2 является остатком глицина.
2. S-Нитрозотиолы по п.1, которые соответствуют следующей общей формуле (II)

в которой A и B имеют значения, указанные выше, и R4 представляет собой C1-C4алкильную группу.
3. Соединение по любому из пп.1 или 2, представляющее собой N-ацетил-2-амино-2-[4-(4-S-нитрозомеркаптотетрагидропиран)]уксусную кислоту (1)

4. Соединение по любому из пп.1 или 2, представляющее собой N-ацетил-2-амино-2-[4-(4-S-нитрозомеркапто-1-метилпиперидин)]уксусную кислоту (2)

5. Соединение по любому из пп.1 или 2, представляющее собой N-ацетил-2-амино-3-бензил-3-S-нитрозомеркапто-4-фенилбутановую кислоту (3)

6. S-Нитрозотиолы по п.1, которые соответствуют следующей общей формуле (III)

в которой A и B принимают значения, указанные выше.
7. Соединение по любому из пп.1 или 6, представляющее собой N-[N-y-L-глутамил-2-амино-2-(4-(4-S-нитрозомеркапто)тетрагидропиран)ацетил]глицин (4)

8. Соединение по любому из пп.1 или 6, представляющее собой N-[N-y-L-глутамил-2-амино-2-(4-(4-S-нитрозомеркапто-1-метилпиперидин))ацетил]глицин (5)

9. Соединение по любому из пп.1 или 6, представляющее собой N-[N-y-L-глутамил-2-амино-3-бензил-3-(S-нитрозомеркапто)-4-фенилбутаноил]глицин (6)

10. Применение S-нитрозотиолов по любому из пп.1-9, в качестве активного агента для лечения дисфункций кровообращения.
11. Применение по п.10, в котором указанный агент предназначен для лечения дисфункций сердечно-сосудистой системы.
Текст
1 Область создания изобретения Настоящее изобретение связано с новымиS-нитрозотиоловыми производными пеницилламина или глутатиона, которые обладают сосудорасширяющими свойствами и которые ингибируют агрегацию тромбоцитов и, следовательно, могут быть полезны в изготовлении лекарственных средств для лечения дисфункций системы кровообращения, в частности на уровне сердечно-сосудистой системы. Предпосылки создания изобретения Хорошо известно, что соединения, способные высвобождать окись азота (NO) в организме, часто проявляют тот или иной тип активности в отношении сосудистой системы, например сосудорасширяющую активность или ингибирование агрегации тромбоцитов, что делает их потенциально полезными для лечения различных заболеваний, связанных с дисфункциями системы кровообращения. Кроме того, описано также, что специфические производные, которые содержат Sнитрозотиоловую группу, имеют с медицинской точки зрения преимущественные свойства, благодаря которым они обладают способностью высвобождения окиси азота в организме.Radomski et al., Br.J.Pharmacol., (1992) 107,745-749, описывают способность соединения 3 нитрозоглутатиона (GSNO) ингибировать активность тромбоцитов.(1992), сообщают, что S-нитрозоцистеин способен ингибировать активность тромбоцитов благодаря его антитромботическому действию. У Smith et al., Met. Find. Exp. Cline. Pharmacy., (1994), 16, 5, описано, что GSNO оказывает сильное расслабляющее действие на артериолы. В заявке WO 95/12394 описано применение S-нитрозоаддуктов пептидов, среди прочихS-нитрозо-N-ацетилпеницилламинов (SNAP), в качестве защитных агентов против воспаления сосудов травматической природы. В заявке WO 95/07691 описано применение различных S-нитрозотиолов, в частностиGSNO, в лечении и профилактике действия тромбоцитов и образования тромбозов на поверхности поврежденного сосуда. В заявке WO 93/09806 описаны Sнитрозированные белки или аминокислотные остатки, способные высвобождать NO, которые оказывают расслабляющее действие на мускулатуру и ингибирующее действие на агрегацию тромбоцитов. В патенте ЕР-В 1-412699 описаны Sнитрозотиолы, которые соответствуют следующей общей формуле: и их применение в качестве терапевтических агентов против сердечно-сосудистых заболеваний, в частности, в качестве антигипертензивных агентов (повышенное кровяное давление) и в качестве агентов для лечения грудной жабы. Количество возможных соединений в рамках этой формулы огромно, и данное описание охватывает только несколько десятков таких продуктов. Далее, в данном патенте описаны только данные, относящиеся к общему сосудорасширяющему действию этих соединений, и ничего не сказано об их активности в отношении тромбоцитов.S-Нитрозотиолы, описанные в указанных выше публикациях, сами по себе не решают множества сложных проблем в рамках лечения сосудистых заболеваний, в частности, на уровне сердечно-сосудистой системы. Следовательно,необходимы новые соединения, которые окажутся более сильнодействующими и эффективными. Краткое описание изобретения Объект данного изобретения связан с новыми S-нитрозотиоловыми производными пеницилламина или глутатиона, которые оказывают как мощное сосудорасширяющее действие, так и сильное ингибирующее действие на агрегацию тромбоцитов. Далее, объект данного изобретения связан с применением новых соединений для производства лекарственных средств для лечения расстройств, связанных с дисфункциями системы кровообращения, в частности, на сердечнососудистом уровне. Подробное описание изобретения Соединения согласно изобретению представляют собой S-нитрозотиоловые производные пеницилламина или глутатиона и их фармацевтически приемлемые соли, которые соответствуют следующей общей формуле (I) в которой А и В представляют собой фенильную группу или вместе образуют остаток -CH2-QCH2-, образующий шестичленное кольцо, в котором Q представляет собой атом кислорода,серы или группу N-R3, в которой R3 является водородом или алкильной группой C1-C4;R1 представляет собой ацильный остаток,который может быть алифатической C1-C5 ацильной группой или остатком глутаминовой 3 кислоты, связанным через его неаминокислотный карбоксил;R2 является гидроксильной группой или глициновым остатком, связанным пептидной связью; при условии, что когда R1 представляет собой остаток алифатического ацила, тогда R2 является гидроксильной группой, а когда R1 представляет собой остаток глутаминовой кислоты, R2 является остатком глицина. Следовательно, соединения согласно изобретению соответствуют следующим формулам в которых А и В имеют значения, указанные выше, a R4 представляет собой C1-C4 алкильную группу,или их фармацевтически приемлемым солям. Предпочтительными примерами соединений в рамках настоящего изобретения являются следующие: Для специалиста является очевидным, что соединения согласно изобретению включают в себя хиральные центры, и это позволяет различать возможные изомеры и/или их смеси. Должно быть понятно, что как смеси изомеров,так и чистые изомеры входят в рамки данного изобретения. Последние можно получить из смеси изомеров, используя обычные технические приемы, хорошо известные эксперту, или же путем асимметрического синтеза, который тоже хорошо известен эксперту. Соединения согласно изобретению можно получить различными способами, и конкретно выбираемый способ зависит от их основной структуры. Например, соединения общей формулы (II) можно получить в соответствии с последовательностью этапов, иллюстрируемых следующей схемой. Т.е., начиная с гидрохлоридов аминокислот, которые можно получить известными способами (см., например, документы DE-A3023830 и DE-A-2801849), последующего ацилирования азотной группы аминокислоты общеизвестными приемами, например, с помощью хлорангидрида кислоты, и кончая введения нитрозогруппы через серу меркаптогруппы, также с использованием традиционных способов. Что касается соединений общей формулы(III), они могут быть получены согласно после 5 довательности этапов, проиллюстрированных на следующей схеме. Т.е., начиная с тех же гидрохлоридов аминокислот, что и в приводимом выше случае,затем проводят защиту меркаптогруппы путем образования тиоэфира параметилбензила. Затем следует защита азота аминокислоты с помощью общеизвестной Вос-защиты, и, наконец, с помощью известных приемов твердофазных методик техники (см., например, Barany, G. et al., ThePhase Peptide Synthesis, Freeman, San Francisco,CA (1969); Bodanszky, M. et al., Peptide Synthesis, 2nd ed., Wiley, NY (1976 получают трипептид с остатками глутаминовой кислоты и глицина (С-концевая аминокислота). Образование трипептида включает в себя следующие стадии:a) закрепление С-концевой аминокислоты глицина (Gly) в виде производного третбутоксикарбонила (Воc) на параметилбензилгидриламин/полистирольной смоле;c) введение промежуточной защищенной модифицированной аминокислоты;d) оценка реакции связывания посредством нингидрина (Kaiser E. et al., Anal. Biochem., 34,595 (1970;e) повторение указанной выше стадии для остатка -глутаминовой кислоты (-Glu), которая включена в качестве Воc-производной или бензилзащищенной (Boc-Glu-OBzl);f) ацидолиз с помощью безводного HF для снятия защиты с трипептида и высвобождения его из смолы;g) очистка и анализ свойств конечных продуктов. Сразу после образования трипептида переходили к конечному этапу нитрозирования меркаптогруппы, высвободившейся из защищающей ее группы. Приведенные тесты показали, что Sнитрозированные соединения согласно изобретению обладают сосудорасширяющей активностью, которая, по меньшей мере, сравнима с таковой GSNO, а в некоторых случаях значительно выше ее. Кроме того, они оказывают ингибирующее действие на агрегацию тромбоцитов, которое при сравнении близко или выше 6 действия GSNO, а в некоторых случаях значительно превосходящее его. Это означает, что соединения согласно изобретению можно очень эффективно использовать для изготовления лекарственного средства, обладающего сосудорасширяющим и антитромботическим действием, для лечения дисфункций системы кровообращения, в особенности на сердечно-сосудистом и коронарном уровнях. Следовательно, соединения общей формулы (I), также как и их фармацевтически приемлемые соли, могут быть использованы с применением общеизвестных фармацевтических технологий для получения лекарственных средств,которые можно вводить различными способами. В качестве примера, их можно вводить перорально в качестве лекарственных средств в виде таблеток, капсул, сиропов и суспензий. Парентерально, например, в виде растворов или эмульсий и т.д. Их можно применять также местно в виде кремов, мазей, бальзамов и т.д. или же трансдермально с применением пластырей или повязок. Их можно применять непосредственно к прямой кишке в виде суппозиториев. Указанные лекарственные средства могут включать в себя физиологически приемлемые носители, эксципиенты, активаторы, хелатирующие агенты, стабилизаторы и т.д. В случае инъекций они могут быть включены в качестве физиологически приемлемых буферов, солюбилизирующих агентов или изотонических растворов. Суточная доза может варьировать в зависимости от возраста, веса тела пациентов, способа введения и т.д., обычная суточная доза для взрослого человека может составлять от 0,1 до 500 мг, и такая доза может вводиться либо в виде однократной дозы, либо может быть разделена на несколько доз, вводимых в течение суток. В рабочих примерах (приведенных ниже) детально описаны способы получения различных соединений, соответствующих общей формуле (I). В отношении этих примеров следует сказать, что в компетенции специалиста в данной области решать, следовать ли ему в точности приводимым в примерах методикам при получении заявленных соединений или же получать их путем соответствующих модификаций представленных здесь экспериментальных примеров. Соответственно, представленные здесь экспериментальные примеры не следует интерпретировать как ограничивающие объем данного изобретения, а только в качестве дополнительной, более детальной иллюстрации, которая будет способствовать более глубокому пониманию данного изобретения специалистами. Примеры В экспериментальной части примеров использованы следующие аббревиатуры: этилацетат уксусная кислота трет-бутоксикарбонил бензил дициклогексилкарбодиимид дихлорметанN-этилдиизопропиламин химическая ионизация с прямым вводом диметилформамид диметилсульфоксид-d6 натриевая соль этилендиаминтетрауксусной кислоты этанол диэтиловый эфир бомбардировка быстрыми атомами циклический гуанозин-3',5'-монофосфат жидкостная хроматография высокого давления ацетонитрил метанол масс-спектрометрия Гидрохлориды исходных аминокислот синтезированы в соответствии с описанием патентных заявок Германии DE-A-3023830 и DEА-2801849. Спектры ядерного магнитного резонанса получены на установке Varian Gemini-200. В спектрах 1H-ЯМР указаны рабочая частота и используемый при получении спектров растворитель. Положение сигналов указано в(м.д.) с использованием в качестве контроля сигнала протонов растворителя. Контрольные величины составляют 7,24 м.д. в случае хлороформа и 2,49 м.д. в случае диметилсульфоксида дейтерия. В скобках указано число протонов,соответствующее каждому сигналу, измеряемому путем электронной интеграции, а тип сигнала показан следующими обозначениями: s(широкий сигнал), sc (сигнальный комплекс),d.e. D2O (исчезновение в процессе получения спектра после добавления нескольких капель дейтериевой воды). В 13 С-ЯМР указаны рабочая частота и растворитель, используемый при получении каждого из спектров. Положение сигналов указано в(м.д.), с использованием в качестве контроля сигнала протонов растворителя. Контрольные величины составляют 77,00 м.д. в случае дейтерированного хлороформа и 39,50 м.д. в случае дейтерированного диметилсульфоксида. 8 Далее, эксперименты ядерного магнитного резонанса проводили с помощью прилагаемого протонного теста (APT). Анализы методом HPLC с целью определения чистоты образцов проводили в следующих условиях: А - Общий градиент Колонка: RP-C18, Симметрия 1503,9 мм,5, 100 А Мобильная фаза: Н 2O+Н 3 РO4 0,1%/СН 3 СN+5% Н 2O+0,1% Н 3 РO4 Температура: комнатная температура Скорость потока: 1 мл/мин Вводимый объем: 10 мкл В - Изократический Колонка: RP-C18, Симметрия 1503,9 мм,5, 100 А Мобильная фаза: 50% H2O+H3PO4 0,1% до 50% СН 3 СN + 5% Н 2O +0,1% Н 3 РO4 Температура: комнатная температура Скорость потока: 1 мл/мин Вводимый объем: 10 мкл При получении трипептидов для очистки исходного материала были использованы методика препаративной хроматографии и HPLC, а для подтверждения идентификации продукта масс-спектрометрия и аминокислотный анализ. Степень чистоты трипептидов определяли методом HPLC при следующих условиях: колонка типа С 18 Nucleosyl, 2504 мм, 120 А, 10 мкм; скорость потока: 1 мл/мин; элюенты: А = Н 2O(+0,045% TFA) и В = СН 3 СN (+0,036% TFA). Ультрафиолетовые (UV) спектры получали на спектрофотометре Shimadzu UV-160A. Для каждого из соединений указаны длина волныв нм и молекулярная абсорбцияв см-1 моль-1 л. Для жидкостной хроматографии высокого давления (HPLC) использовали ультрафиолетовый фотодиодный детектор Hewlett-Packard 1040 А. Пример 1. Получение N-ацетил-2-амино-2[4-(4-S-нитрозомеркаптотетрагидрофуран)] уксусной кислоты (1). Стадия 1). В 250 мл стеклянную колбу,снабженную магнитной мешалкой, вводили 4,0 г (17,6 ммоль) гидрохлорида 2-амино-2-[4-(4 меркаптотетрагидропиран)]уксусной кислоты и 60 мл 0,5 н. раствора гидроокиси калия. Затем добавляли 3,07 г (21,8 ммоль) безводного карбоната калия и 60 мл MeCN. Смесь охлаждали на ледяной бане и по каплям добавляли 1,42 мл(19,7 ммоль) уксусной кислоты, растворенной в 25 мл МеСN. В конце добавляли 0,5 н. гидроокись калия, доводя рН до 9-10. Смесь перемешивали при комнатной температуре в течение 2 ч. После этого добавляли 2 н. соляную кислоту до тех пор, пока рН смеси не достигнет 6,0, и 9 при пониженном давлении удаляли растворитель. Полученный твердый остаток сушили под вакуумом над пятиокисью фосфора, затем его суспендировали в 150 мл абсолютного EtOH и перемешивали при комнатной температуре. Нерастворимый остаток фильтровали и промывали абсолютным EtOH. Растворитель удаляли при пониженном давлении. Получали 3,98 г сырого промежуточного продукта в виде N-ацетил-2 амино-2-[4-(4-меркаптотетрагидропиран)]уксусной кислоты, который подвергали различным препаративным обращенно-фазовым хроматографическим очисткам с получением 80 мг продукта с 90% степенью чистоты (градиент A(СН 3). Стадия 2). 80 мг (0,34 ммоль) N-ацетил-2 амино-2-(4-(4-меркаптотетрагидропирануксусной кислоты растворяли в 4000 мкл МеОН и затем добавляли 500 мкл 1 н. раствора НСl и 136 мкл 5 М раствора NaNO2. Полученный в результате раствор в течение 1 мин обрабатывали в ультразвуковой бане и потом добавляли 50 мкл 10 н. NaOH и 14 мкл Н 2O. Из полученного раствора отбирали часть А в виде аликвоты в 470 мкл и к остатку в избытке добавляли воду, замораживали и лиофилизировали, получая 190 мг гигроскопического твердого вещества В, слегка увлажненного.HPLC (градиент А):220 нм: степень чистоты фракции А 68%339 нм: степень чистоты фракции А 95% ЯМР фракция В: 1 Стадия 1). В 250 мл стеклянную колбу,снабженную магнитной мешалкой, вводили 3,69 г (13 ммоль) дихлоргидрата 2-амино-2-[4-(4 меркапто-1-метилпиперидин)]уксусной кислоты и 30 мл 0,5 н. раствора гидроокиси калия. После этого добавляли 2,2 г (16 ммоль) безводного карбоната калия и 30 мл MeCN. Смесь охлажда 003577 10 ли на ледяной бане и по каплям добавляли 1,0 мл (14 ммоль) уксусной кислоты, растворенной в 10 мл MeCN. В конце добавляли 0,5 н. гидроокись калия, доводя рН до 9-10. Смесь перемешивали при комнатной температуре в течение 2 ч. После этого добавляли 2 н. соляную кислоту до тех пор, пока рН смеси не достигнет 6,0, и при пониженном давлении удаляли растворитель. Полученный твердый остаток сушили под вакуумом над пятиокисью фосфора, затем высушенное твердое вещество суспендировали в 100 мл абсолютного EtOH и перемешивали при комнатной температуре. Нерастворимый осадок фильтровали и промывали абсолютным EtOH и растворитель удаляли при низком давлении. Получали 2,8 г сырого промежуточного продукта в виде N-ацетил-2-амино-2-[4-(4-меркаптометилпиперидин)]уксусной кислоты, который подвергали различным препаративным обращенно-фазовым хроматографическим очисткам с получением 0,5 г продукта с 99% степенью чистоты (градиент A HPLC, =210 нм). 1(СН 2), 23,42 (СН 3). Стадия 2). 50 мг (0,20 ммоль) N-ацетил-2 амино-2-(4-(4-меркапто-1-метилпиперидин уксусной кислоты растворяли в 800 мкл 1 н. раствора НСl и затем добавляли 80 мкл 5 М раствора NaNO2. Полученный в результате раствор в течение 1 мин обрабатывали в ультразвуковой бане, и потом добавляли 80 мкл 10 н. NaOH и 40 мкл Н 2O. Отделяли часть полученного раствора в виде 100 мкл аликвоты А, а остаток замораживали и лиофилизировали, получая 190 мг твердого вещества В.HPLC (градиент А): степень чистоты фракции А: 90%;230 нм: степень чистоты фракции В 93%; степень чистоты фракции А 98%;334 нм: степень чистоты фракции В 98%; ЯМР фракции В: 1 Пример 4. Получение N-[NL-глутамил 2-амино-2-(4-(4-S-нитрозомеркапто)тетрагидропиран)ацетил]глицин (4). Стадия 1). В 100 мл стеклянную колбу,снабженную магнитной мешалкой, вводили 2,0 г (5,9 ммоль) 2-амино-3-бензил-3-меркапто-4 фенилбутановой кислоты и 20 мл 0,5 н. раствора гидроокиси калия. После этого добавляли 1,0 г(7,2 ммоль) безводного карбоната калия и 20 мл МеСN. Смесь охлаждали на ледяной бане и по каплям добавляли 0,5 мл (6,5 ммоль) уксусной кислоты, растворенной в 8 мл MeCN. В конце добавляли 0,5 н. гидроокись калия до тех пор,пока рН смеси достигнет значений 12-13. Смесь перемешивали при комнатной температуре в течение 2 ч. После этого добавляли 2 н. соляную кислоту до тех пор, пока рН смеси не достигнет 6,0, и при пониженном давлении удаляли растворитель. Полученный неочищенный остаток суспендировали в 100 мл абсолютного EtOH и перемешивали при комнатной температуре. Нерастворимый остаток фильтровали и промывали абсолютным EtOH. Растворитель удаляли при низком давлении. Остаток перекристаллизовывали в воде. Нерастворимую часть фильтровали и отбрасывали. Фильтрат замораживали и лиофилизировали. Получали 1,64 г промежуточного продукта в виде N-ацетил-2-амино-3-бензил-3 меркапто-4-фенилбутановой кислоты. Выход: 81%. 1(2CHar), 126,44 (CHar), 126,26 (CHar), 60,16 (СН),55,62 (C), 43,70 (СН 2), 42,94 (СН 2), 23,14 (СН 3). Стадия 2). 0,62 г (1,8 ммоль) N-ацетил-2 амино-3-бензил-3-меркапто-4-фенилбутановой кислоты растворяли в 25 мл МеОН, затем добавляли 25 мл 1 н. раствора НСl и раствор из 250 мг NaNO2 в 25 мл Н 2O. Полученный в результате раствор в течение 35 мин перемешивали при комнатной температуре, фильтровали и промывали водой. После высушивания при пониженном давлении в присутствии Р 2O5 получали 0,33 г твердого вещества. Стадия 1). В 250 мл стеклянной колбе,снабженной магнитной мешалкой, перемешивали 5,8 г (25,5 ммоль) гидрохлорида 2-амино-2(4-(4-меркаптотетрагидропирануксусной кислоты, 4,7 г (25,5 ммоль) -бром-параксилола и 100 мл смеси H2O/EtOH 3:1. Добавляли 11 мл триэтиламина и смесь перемешивали в течение 18 ч при комнатной температуре в атмосфере аргона. Затем добавляли еще 50 мл H2O/EtOH 3:1 и перемешивали еще в течение 2 ч. Полученную суспензию отфильтровывали и промывали 100 мл воды и 50 мл EtOH. Получали твердое вещество, которое сушили при пониженном давлении. На полученное твердое вещество добавляли 300 мл концентрированной соляной кислоты, и растворитель удаляли при пониженном давлении. Последнюю обработку соляной кислотой повторяли второй раз, и в результате получали твердое вещество, которое высушивали при пониженном давлении. Получали 1,59 г искомого промежуточного продукта. 1(СН 3). Стадия 2). 1004 мг гидрохлорида 2-амино 2-(4-(4-(параметилбензилмеркапто)тетрагидропирануксусной кислоты (3,029 ммоль) суспендировали в 20 мл tBuOH/H2O 2:1 и добавляли 5% NaOH до тех пор, пока рН не установится равным 9. После этого добавляли 3 эквивалента Вос 2O, и смесь оставляли на 30 мин для прохождения реакции, после чего рН еще раз доводили до 9,0. Реакцию проводили еще 30 мин, после чего вновь производили коррекцию значения рН. Путем сравнения с исходной аминокислотой с помощью тонкослойной хроматографии(TLC) делали заключение о завершении реакции. Продукт выделяли путем двух последовательных экстракций 50 мл гексана и водную фазу обрабатывали 1 н. НСl для достижения рН 2. После этого проводили три стадии экстракции: 60 мл AcOEt каждая. Раствор AcOEt суши 13 ли безводным MgSO4 и,после удаления растворителя путем его упаривания продукт вновь растворяли в DCM и вновь упаривали. После повторения этой стадии еще дважды получали 1191 мг (3,015 ммоль) N-Boc-защищенного продукта. Полученный продукт был охарактеризован следующим образом:a) TLC, в которой в качестве подвижной фазы использовали СНСl3/МеОН/АсОН 85:10:5.Rf = 0,48, сигнал исходного материала не обнаруживался.MeCN в течение 30 мин + изократично при 100% MeCN 5 мин. Полученная в результате степень чистоты соответствовала 96% (220 нм).(2 Н, д, J=8 Гц), 5,54 (1 Н, д, J=11,2 Гц), 4,44 (1 Н,д, J=8,3 Гц), 3,69 (1 Н, д, J= 11,2 Гц), 3,57 (1 Н, д,J=11,2 Гц), 3,75-3,98 (4 Н, шир.с), 2,32 (3 Н, с),1,26-2,19 (4 Н, шир.с), 1,46 (9 Н, с). Стадия 3). Трипептид получен методом твердофазного синтеза, начиная с 1595 мг ВосGlу-ОСН 2-Раm-смолы (Neosystem, ref. NoRP00801) при замещении 0,63 ммоль/г. Рабочий диапазон составляет 1,005 ммоль. Общая методика добавления остаточного азота: 1) Набухание смолы в результате 5 промываний DCM (по 0,5 мин); 2) Удаление Вос-группы с помощью 40%TFA в DCM, 11 мин + 120 мин; 3) Промывка DCM, 50,5 мин; 4) Тестирование нингидрином; 5) Промывка DMF, 31 мин; 6) Связывание в течение 1,5 ч; 7) Промывка DMF, 31 мин; 8) Промывка DCM, 50,5 мин; 9) Тестирование нингидрином. В случае, когда тест на нингидрин на последней стадии оказывался положительным,проводили повторное связывание, повторяя стадии 5-8. Связывание производили с использованием 6 эквивалентов (экв.) DIEA, 3 экв. Восаминокислоты и 3 экв. активного агента, которым в данном случае являлся РуАОр (гексафторфосфат 7-азабензотриазол-1-ил-окситрис(пирролидино)фосфония). Для связывания -Glu использовали Boc-Glu-(-OBzl). Завершая последовательность стадий связывания, пептидил-смолу подвергали ацидолизуHF/паракрезолом (9:1) в течение 1 ч при 0 С. После упаривания образовавшийся осадок экстрагировали безводным EtOEt, а затем выделяли пептид путем экстракции 10% АсОН. Сразу после упаривания неочищенный осадок лиофилизировали и анализировали с помощьюb) масс-спектрометрии с электрораспылением: Мвычисл.= 377, (М+Н+)эксперим.= 378,2. Количество пептида определяли путем аминокислотного анализа: m = 329 мг (0,873 ммоль). Во всех количественных оценках пептидов для гидролиза пробы при пептидном анализе пользовались добавлением извне патронаIlе (изолейцина). Очистку трипептида (в два приема) производили методом препаративной HPLC. При этом использовали следующую систему:- Градиент: Изократический режим: 0% В в течение 40 мин + градиент от 0 до 10% В в течение 40 мин. Поток: 25 мл/мин. Всего получено 227 мг (0,602 ммоль) промежуточного трипептида, который характеризовался а) HPLC в градиенте от 5 до 45% MeCN в течение 20 мин. Полученная в результате степень чистоты соответствовала 95% (220 нм). При возрастающем разведении нанесенного образца минорный пик при 8,4 мин пропорционально уменьшался, вплоть до полного исчезновения. Соответственно, было сделано заключение, что он соответствует агрегации трипептида(а не окислению) и, следовательно, он был признан в качестве значимого продукта.b) Масс-спектрометрией с электрораспылением: Мвычисл.= 377, (М+Н+)эксперим.= 378,2. Стадия 4). К раствору, содержащему 14,2 мг (0,38 ммоль) трипептида, полученного на стадии 3, в 800 мкл 1 н. НСl добавляли 76 мкл 0,5 М раствора NaNO2. После перемешивания в ультразвуковой бане при комнатной температуре в течение 1 мин добавляли 80 мкл 10 н. раствора NaOH. Полученный раствор замораживали и лиофилизировали, получая в результате твердое вещество, имеющее следующие свойства: 1 Стадия 1). В 100 мл стеклянной колбе,снабженной магнитной мешалкой, смешивали 6,4 г (23 ммоль) дигидрохлорида 2-амино-2-(4(4-меркапто-1-метилпиперидинуксусной кислоты, 54,3 г (23 ммоль) -бром-параксилола и 40 мл смеси H2O/EtOH 1:1. Добавляли 13 мл триэтиламина, создавали атмосферу аргона и смесь перемешивали в течение 42 ч при комнатной температуре. Из полученного в результате раствора при пониженном давлении удаляли растворитель и к полученному неочищенному остатку добавляли 20 мл абсолютного EtOH. Образовавшееся твердое вещество фильтровали и промывали абсолютным EtOH и EtOEt, затем сушили при пониженном давлении. Получали 2,11 г промежуточного продукта в виде гидрохлорида 2-амино-2-(4-(4-(параметилбензилмеркапто)-1-метилпиперидинуксусной кислоты. Выход: 29,6%. Спектр ЯМР регистрировали в D2O, а в качестве эталона было взято 4,75 м.д. для HDO. 1(CH2), 22,77 (СН 3). Стадия 2). Начиная с 1016 мг (2,958 ммоль) промежуточного продукта, полученного на предыдущей стадии, и продолжая действовать по схеме, описанной в стадии 2 примера 4,получали 1016 мг (2,490 ммоль) N-Bocзащищенного промежуточного соединения, характеризуемого следующими показателями:Rf = 0,32, сигнал исходного материала не обнаруживался. 16 Стадия 3). Методом, описанным в стадии 3 примера 4, получено трипептидное промежуточное соединение, начиная с 794 мг смолы,указанной выше. Рабочий диапазон составляет 0,5 ммоль. Из-за трудностей солюбилизации Воc-производной, вместо DIEA, в указанной выше реакции был выбран в качестве основанияN-метилморфолин (3 экв.). После введения Glu образовывался трипептид, и связь пептидсмола подвергали ацидолизу тем же способом,что и описанный выше. В результате получали неочищенное вещество в 10% АсОН, которое затем лиофилизировали и количественно оценивали. Получали 137 мг (0,351 ммоль) промежуточного трипептида, характеризуемого следующими показателями:a) HPLC, изократический градиент 0% в течение 10 мин + 5-65% MeCN в течение 30 мин. Показано, что вводимый продукт в кислом растворе элюируется с фронтом, что делает необходимым лиофилизацию аликвоты для удаления кислоты, а затем повторное растворение лиофилизата в 1 мМ НСl. Таким образом пептид можно было хроматографировать, и он элюировался за 6,8 мин в используемом градиенте.b) Масс-спектрометрия с электрораспылением: Мвычисл.= 390 (М+Н+)эксперим. = 391,2. Очистку полученного неочищенного вещества производили методом HPLC, в препаративном диапазоне, причем параметры процесса соответствовали уже описанным в примере 4. Применяли также изократический режим 0% В в течение 40 мин, затем градиент от 0 до 10% В в течение 40 мин. Таким образом получали 107 мг (0,274 ммоль) продукта, имеющего следующие характеристики:a) градиент HPLC от 0 до 0% в течение 10 мин плюс от 0 до 30% в течение 20 мин. Степень чистоты - 94% (220 нм). Подтвердилось,что дополнительный пик на 8,0 минуте в этом случае также соответствует агрегации, и он может быть отнесен к целевому продукту.b) Масс-спектрометрия с электрораспылением: Мвычисл. = 390 (М+Н+)эксперим.= 391,1. Стадия 4). Начиная с 10,0 мг (0,026 ммоль) промежуточного трипептида, полученного на предыдущей стадии, проводили нитрозирование, как описано в стадии 4 примера 4, используя 52 мкл 0,5 М NaNO2. Полученный продукт имеет следующие характеристики: 13 С-ЯМР (50 МГц, DMSO-d6): 171,57 (CON), 170,99 (CO-N), 170,76 (CO-N), 168,20 (СОO), 60,23 (С), 59,53 (СН), 51,57 (СН), 49,02 Стадия 1). В 100 мл стеклянной колбе,снабженной магнитной мешалкой, смешивали 1,25 г (3,7 ммоль) гидрохлорида 2-амино-3 бензил-3-меркапто-4-фенилбутановой кислоты,0,68 г (3,7 ммоль) -бром-параксилола и 40 мл смеси H2O/EtOH 1:1. Добавляли 1,6 мл триэтиламина и растворяли основную часть суспендированного твердого вещества. В атмосфере аргона смесь перемешивали в течение 24 ч при комнатной температуре. Этанол и часть триэтиламина удаляли при пониженном давлении. Полученную водную суспензию фильтровали и получали твердое вещество белого цвета. Затем его очищали, используя обращеннофазовую колоночную хроматографию с СН 3 СNН 2O. К полученному твердому веществу добавляли 100 мл МеОН/НСl 1:1 и растворитель удаляли при пониженном давлении. Получали 0,5 г промежуточного продукта, гидрохлорида 2 амино-3-бензил-3-(4-метилбензилмеркапто)-4 фенилбутановой кислоты. 1(СН-N), 53,64 (C), 42,22 (CH2), 41,65 (CH2),31,90 (СН 2-S), 20,48 (СН 3). Стадия 2). Начиная с двух фракций, полученных на предыдущей стадии (180 мг со степенью чистоты 83% и 530 мг со степенью чистоты 94%), и в соответствии с описанным в стадии 2 примера 4, получали 733 мг (1,45 ммоль) промежуточного N-Boc-защищенного продукта,имеющего следующие характеристики:c) EM-MALDI-TOF (масс-спектр): Подложка из дигидроксибензойной кислоты,Мвычисл.= 505, (М+Н+)эксперим.= 528,585 (M+Na) 18 Стадия 3). Методом, описанным в стадии 3 примера 4, получали промежуточный трипептид, начиная с 0,43 ммоль указанной выше смолы. После ацидолитической стадии выделяли неочищенное вещество, содержащее 14,4 мгa) Аналитическая HPLC с градиентом от 5 до 65% в течение 30 мин, примерная степень чистоты 95%.b) Масс-спектроскопия с электрораспылением Мрасч. = 487 (М+Н+), Мэксперим.= 488,3. Стадия 4). Начиная с 5,7 мг (0,012 ммоль) промежуточного трипептида, полученного на предыдущей стадии, с использованием 24 мкл 0,5 М NaNO2 проводили нитрозирование в соответствии с описанным в стадии 4 примера 4. Полученный продукт имел следующие характеристики:UV:345 нм (Н 2O) 368 см-1 моль-1 л. Пример 7. Тесты по расширению сосудовin vitro. Использованный в данных исследованиях метод по существу является методом, описанным в следующих публикациях:Salas, E. et al., Eur.J.Pharmacol., 1994; 258: 47-55. Соединения тестировали при 5 различных концентрациях, варьирующих от 0,001 до 10 мМ, при этом при исследовании эффекта каждой из концентраций использовали от 6 до 9 артериальных колец. Полученные результаты сравнивали с результатом действия S-нитрозоглутатиона (GSNO), который использовали в качестве контрольного продукта. Результаты приведены в табл. 1 ниже и представлены в виде CE50 (эффективная концентрация 50), которая представляет собой концентрацию каждого из тестируемых соединений,которая вызывает 50% расширение артериального кольца, предварительно сократившегося под действием 1 мкМ норэпинефрина. Таблица 1 Тест на расширение сосудов Соединение Как следует из таблицы, все тестируемые соединения обладают сосудорасширяющей способностью, сходной с активностью контрольного продукта (GSNO) или превосходящей ее, а соединение 2 обладает сосудорасширяющей активностью, значительно более высокой, нежели у контрольного продукта. Пример 8. Тесты in vitro на ингибирование агрегации тромбоцитов. Использованный в данных исследованиях метод, по существу, воспроизводит метод, описанный в следующих источниках:Salas E. et al., Br.J.Pharmacol., 1994; 112: 1071-1076. Соединения тестировали при четырех различных концентрациях, используя тромбоциты от 5 до 23 различных доноров. Полученные результаты сравниваются с таковыми, полученными при действии S-нитрозоглутатиона(GSNO), который использовали в качестве контрольного продукта. Результаты показаны в табл. 2 и выражены в виде CI50 (ингибирующая концентрация 50),представляющей собой концентрацию каждого из тестируемых соединений, при которой происходит 50% ингибирование агрегации, происходящей под действием субмаксимальной концентрации коллагена (1 мкг/мл). Таблица 2 Тесты на ингибирование агрегации тромбоцитов Соединение Из табл. 2 видно, что все тестируемые соединения обладают потенциальной ингибирующей агрегацию тромбоцитов активностью,которая сравнима или же превосходит активность контрольного продукта (GSNO), а соединения 1 и 5 обладают ингибирующей агрегацию тромбоцитов активностью, которая значительно превосходит таковую контрольного продукта. Пример 9. Тесты in vitro по увеличению внутритромбоцитарных уровней цГМФ. Соединения, полученные в примере 5 (5),тестировали in vitro с целью проверки их способности вызывать увеличение уровней цГМФ в тромбоцитах в отмытых препаратах тромбоцитов человека. 20 Использованный в данном тесте метод в основном совпадает с методом, описанным в публикациях, приведенных в примере 8. Соединения тестировали в четырех различных концентрациях, используя тромбоциты 5 разных доноров. Полученные результаты сравнивали с таковыми, полученными при действии GSNO (контрольный продукт), и с исходными значениями. Результаты приведены в таблице 3 и выражены в пмоль/109 тромбоцитов. Таблица 3 Тест на увеличение внутритромбоцитарных уровней цГМФ мкМ 3 1 0,3 0,1 0,03 0 Соединение 5, наподобие GSNO, способствует повышению уровней внутритромбоцитарного цГМФ. Следует отметить, что при значениях, близких к IC50, соединение 5 индуцирует более низкие уровни цГМФ, чем те, которые индуцируются под действием GSNO, используемым в концентрациях, близких к его значению IC50. Пример 10. Тест in vitro на блокирование ингибирования агрегации тромбоцитов. Использованный в данном тесте метод в основном совпадает с методом, описанным в публикациях, приведенных в примере 8, которые можно дополнить следующей ссылкой: Моrо МА. et al., Pr.Nat.Acad.Sc.USA, 1996; 93:1480-1485. Соединение ODQ тестировали in vitro на его способность блокировать ингибирование агрегации тромбоцитов, вызываемой полученными продуктами, на отмытых препаратах тромбоцитов человека. Соединение, полученное в примере 5 (5),тестировали при трех различных концентрациях, в присутствии или в отсутствие ODQ (1 мкМ), используя тромбоциты 5 различных доноров. Полученные результаты сравнимы с таковыми, получаемыми при действии GSMO(контрольный продукт), и с величинами, получаемыми в отсутствие ODQ. Результаты представлены в табл. 4 и 5 и выражены в виде процента ингибирования максимальной агрегации. Таблица 4 Контрольный продукт (GSNO) мкМ 1 0,3 0,1 Соединение ODQ (1 мкМ) способно блокировать ингибирующий эффект GSNO и соединения 5 при использовании их в дозе IC50. При более высоких, чем IC50, дозах эффект ингибирования агрегации блокировался сильнее под действием соединения 5, нежели под действием контрольного продукта. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. S-Нитрозотиоловые производные пеницилламина или глутатиона и их фармацевтически приемлемые соли, которые соответствуют следующей общей формуле (I) в которой А и В являются фенильными группами или же вместе образуют остаток -CH2-Q-CH2-, образующий шестичленное кольцо, в котором Q представляет собой атом кислорода или серы,или группу N-R3, в которой R3 является водородом или алкильной группой C1-C4;R1 является ацильным остатком, который может быть C1-C5 алифатической ацильной группой или остатком глутаминовой кислоты,связанным через его неаминокислотный карбоксил;R2 является гидроксильной группой или остатком глицина, связанным через его пептидную связь; при условии, что когда R1 является алифатическим ацильным остатком, тогда R2 является гидроксильной группой, а когда R1 является остатком глутаминовой кислоты, тогда R2 является остатком глицина. 2. S-Нитрозотиолы по п.1, которые соответствуют следующей общей формуле (II) 22 3. Соединение по любому из пп.1 или 2,представляющее собой N-ацетил-2-амино-2-[4(4-S-нитрозомеркаптотетрагидропиран)]уксусную кислоту (1) 4. Соединение по любому из пп.1 или 2,представляющее собой N-ацетил-2-амино-2-[4(4-S-нитрозомеркапто-1-метилпиперидин)]уксусную кислоту (2) 5. Соединение по любому из пп.1 или 2,представляющее собой N-ацетил-2-амино-3 бензил-3-S-нитрозомеркапто-4-фенилбутановую кислоту (3) 6. S-Нитрозотиолы по п.1, которые соответствуют следующей общей формуле (III) в которой А и В принимают значения, указанные выше. 7. Соединение по любому из пп.1 или 6,представляющее собой N-[N-у-L-глутамил-2 амино-2-(4-(4-S-нитрозомеркапто)тетрагидропиран)ацетил]глицин (4) 8. Соединение по любому из пп.1 или 6,представляющее собой N-[N-у-L-глутамил-2 амино-2-(4-(4-S-нитрозомеркапто-1-метилпиперидинацетил]глицин (5) 9. Соединение по любому из пп.1 или 6,представляющее собой N-[N-у-L-глутамил-2 амино-3-бензил-3-(S-нитрозомеркапто)-4 фенилбутаноил]глицин (6) 24 10. Применение S-нитрозотиолов по любому из пп.1-9, в качестве активного агента для лечения дисфункций кровообращения. 11. Применение по п.10, в котором указанный агент предназначен для лечения дисфункций сердечно-сосудистой системы.
МПК / Метки
МПК: A61P 9/10, A61K 31/198, C07K 5/093, C07D 309/08, C07C 323/07
Метки: s-нитрозотиолы, кровообращения, нарушений, агентов, качестве, лечения
Код ссылки
<a href="https://eas.patents.su/13-3577-s-nitrozotioly-v-kachestve-agentov-dlya-lecheniya-narushenijj-krovoobrashheniya.html" rel="bookmark" title="База патентов Евразийского Союза">S-нитрозотиолы в качестве агентов для лечения нарушений кровообращения</a>
Производные циклической аминокислоты, фармацевтическая композиция на их основе и способ лечения эпилепсии, приступов слабости, гипокинезии, черепных нарушений, нейродегенеративных или невропатологических нарушений,депрессий, состояний тревоги и паники и боли

Номер патента: 2765
Опубликовано: 29.08.2002
Авторы: Ретклифф Джильс С., Хартенштайн Йоганнес, Нин Клер О., Моррелл Эндру И., Орвелл Дейвид Кристофер, Брайэнс Джастин С.
МПК: A61K 31/164, C07C 229/28, A61P 25/28...
Метки: лечения, эпилепсии, состояний, паники, основе, нарушений, тревоги, циклической, способ, нарушений,депрессий, производные, слабости, гипокинезии, аминокислоты, черепных, фармацевтическая, боли, нейродегенеративных, приступов, невропатологических, композиция
Формула / Реферат:
1. Производные циклической аминокислоты общей формулы I в которой 1 - 4 из радикалов R1-R10 означают метил, а остальные - водород, или один из указанных радикалов R1-R10 означает изопропил или трет.-бутил, а остальные - водород, и их фармацевтически приемлемые соли. 2. Производные циклической аминокислоты формулы I по п.1, в которой R5 означает изопропил или трет.-бутил. 3. Производные циклической аминокислоты формулы I по п.1, в которой R3...
Производные индола в качестве эстрогенных агентов

Номер патента: 815
Опубликовано: 24.04.2000
Авторы: Миллер Крис П., Коллини Майкл Д., Тран Бэч Д.
МПК: A61K 31/405, C07D 209/08
Метки: качестве, агентов, эстрогенных, производные, индола
Формула / Реферат:
1. Соединение, имеющее структуру где R1 выбран из Н, ОН, -ОС(=O)(С1-С4алкил), -О(С1-С4алкил) или галогена; R2, R3, R4, R5 и R6 независимо выбраны из Н, ОН, -ОС(=O)(C1-С4алкил), -О(С1-C4алкил), галогена или C1-С6алкила, при условии, что в случае, когда R1 представляет собой Н, R2 не является группой ОН; Х выбран из Н, C1-С6алкила, нитро или галогена; Z выбран из где n равно 1, 2 или 3; Y выбран из: а) группы где R7 и R8...
Замещенные 4-(6-фтор-(1н)-индол-3-ил)-1,2,3,6-тетрагидропиридин для лечения нарушений центральной нервной системы

Номер патента: 1469
Опубликовано: 23.04.2001
Автор: Фэйрхерст Джон
МПК: C07D 513/06, A61P 25/24, A61K 31/475...
Метки: 4-(6-фтор-(1н)-индол-3-ил)-1,2,3,6-тетрагидропиридин, нарушений, нервной, замещенные, центральной, лечения, системы
Формула / Реферат:
1. Соединение формулы где R1 представляет водород, C1-4-алкил, C1-4-алкокси или галоген и R2 представляет водород, C1-4-алкил или C1-4-алкокси, или его соль. 2. Соединение по п.1, где как R1, так и R2 представляют водород или либо R1, либо R2 представляет водород. 3. Соединение по пп.1 или 2, где R1 представляет водород, метил, метокси или фтор и R2 представляет водород или метил. 4. 2,2-Диоксид...
Производные индазоламида в качестве серотонинергических агентов

Номер патента: 2352
Опубликовано: 25.04.2002
Авторы: Бруфани Марио, Каццолла Никола, Пинца Марио, Джаннанджели Марилена, Алиси Алессандра
МПК: C07D 401/12, A61K 31/416
Метки: серотонинергических, качестве, индазоламида, агентов, производные
Формула / Реферат:
1. Индазоламидные соединения общей формулы в которой R6 выбран из группы, включающей фенил C1-3 алкил, С3-7-циклоалкил, 5- или 6-членное гетероциклическое кольцо, в котором от 1 до 2 членов являются одинаковыми или различными гетероатомами, выбранными из группы, состоящей из N и О, диметиламино C1-3 алкил, метокси C1-3 алкил, аминосульфонилметил, арил, замещенный гидроксигруппой; их соли присоединения к фармацевтически приемлемым органическим и...
Нитробензамиды, полезные в качестве антиаритмических агентов

Номер патента: 2439
Опубликовано: 25.04.2002
Авторы: Слэйтер Грэхам Ральф, Вестлэйк Пол Джеффри
МПК: A61K 31/166, C07C 233/78, A61P 9/06...
Метки: антиаритмических, полезные, качестве, агентов, нитробензамиды
Формула / Реферат:
1. Гидратированный гидрохлорид N-[3-[[2-(3,4-диметоксифенил)этил]амино]пропил]-4-нитробензамида характеризующийся тем, что он (i) включает воду в интервале от 1,7 до 2,4 молярных эквивалентов; и/или (ii) имеет точку плавления в интервале 150-154шС; и/или (iii) демонстрирует инфракрасный спектр, содержащий пики при 3510, 3342, 3076, 1665, 1598, 1343, 1330, 1216 и 801 см1; и/или (iv) демонстрирует спектр твердофазного ядерного магнитного...
Предыдущий патент: Производные резорцина
Следующий патент: Способ полимеризации в газовой фазе
Случайный патент: Новое применение пептидных соединений для лечения боли при болезненной диабетической нейропатии