Способ получения (1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4as,7as)-октагидро-6h-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты






Формула / Реферат
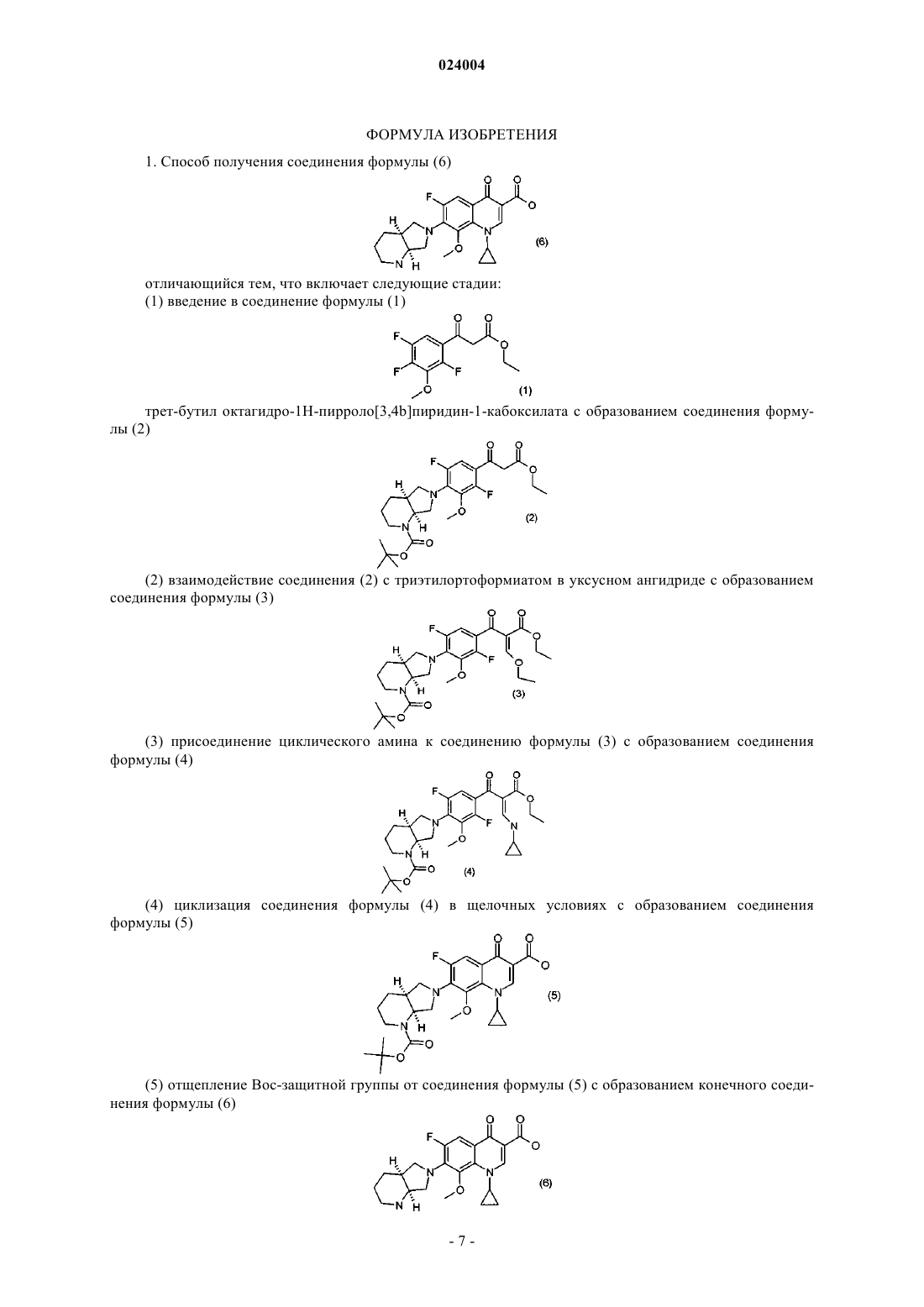
1. Способ получения соединения формулы (6)

отличающийся тем, что включает следующие стадии:
(1) введение в соединение формулы (1)

трет-бутил октагидро-1Н-пирроло[3,4b]пиридин-1-кабоксилата с образованием соединения формулы (2)

(2) взаимодействие соединения (2) с триэтилортоформиатом в уксусном ангидриде с образованием соединения формулы (3)

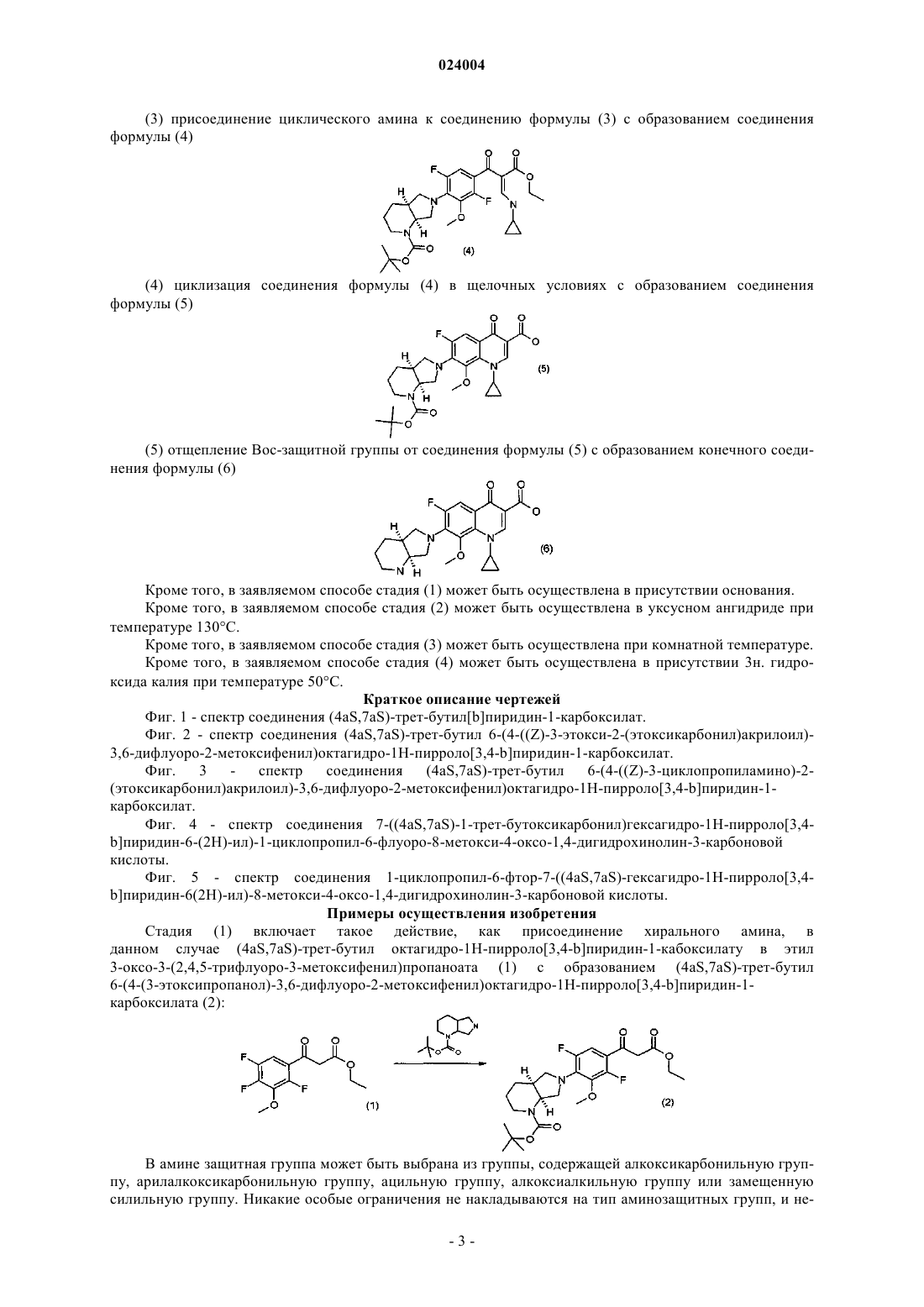
(3) присоединение циклического амина к соединению формулы (3) с образованием соединения формулы (4)

(4) циклизацию соединения формулы (4) в щелочных условиях с образованием соединения формулы (5)

(5) отщепление Boc-защитной группы от соединения формулы (5) с образованием конечного соединения формулы (6)

2. Способ по п.1, отличающийся тем, что стадию (1) осуществляют в присутствии основания.
3. Способ по п.1, отличающийся тем, что стадию (2) осуществляют в уксусном ангидриде при температуре 130°С.
4. Способ по п.1, отличающийся тем, что стадию (3) осуществляют при комнатной температуре.
5. Способ по п.1, отличающийся тем, что стадию (4) осуществляют в присутствии 3н. гидроксида калия при температуре 50°С.
Текст
Данное изобретение относится к способам получения такого химического соединения,как(1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4 аS,7 аS)-октагидро-6 Нпирроло[3,4b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты, который заключается в присоединении гетероциклического амина, который содержит защитную группу, к этил-3-оксо-3-(2,4,5-трифлуоро-3-метоксифенил)пропаноату, следующим взаимодействием с триэтилортоформиатом, присоединении циклического амина, последующей циклизации и получении целевого продукта. Заявляемый способ получения является технологически простым в сравнении с аналогом и не требует специальных сложных технических операций, что, в свою очередь, упрощает способ получения данного химического соединения и удешевляет стоимость готового продукта, промышленное производство по заявленному способу имеет низкую степень опасности. Область техники Настоящее изобретение относится к способам получения химического соединения (1-циклопропил 6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)-октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3 хинолинкарбоновой кислоты. Предшествующий уровень техники Производные хинолинкарбоновой кислоты широко используют как синтетические антибактериальные лекарства в медицине. Соединения из группы фторхинолонов имеют бактерицидное действие и проявляют активность по широкому спектру грамположительных и грамотрицательных микроорганизмов,анаэробных, кислотоустойчивых и атипичных бактерий: Mycoplasma spp., Chlamydia spp., Legionella spp. Противомикробное средство на основе (1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты эффективно по отношению к большинству штаммов микроорганизмов, резистентных к бета-лактамным антибиотикам и макролидам. Способы получения соединения 1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты описаны в заявкеDE 42004144 А 1 (опубл. 15.07.1993). Соединение получают двумя способами. По первому способу (1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)-октагидро-6 Нпирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты получают из солей производных хинолон- и нафтиридонкарбоновой кислоты (кислотно-аддитивные соли, щелочные соли, щелочноземельные соли, серебряные и гуанидина соли соответствующих кислот), которые содержат циклические амины, путем взаимодействия с соединениями, содержащими галоген, при наличии средства, связывающего кислоту. По второму способу эти же производные хинолинкарбоновой кислоты подвергают взаимодействию с акцептором Михаэля, например диалкиловым эфиром ацетилендикарбоновой кислоты, аллилового эфира пропиоловой кислоты. В этих способах рацемические промежуточные соединения взаимодействуют с энантиомерно чистым вспомогательным реагентом, полученные диастереомеры разделяют с помощью хроматографии, снова отщепляют вспомогательную хиральную группу из полученного диастереомера. Рацемические бициклические амины имеют возможность превращаться при взаимодействии с энантиомерными кислотами или сульфокислотами в смесь диастереомерных солей, которые разделяют фракционной кристаллизацией на диастереомерно чистые соли. Молярное соотношение амина и энантиомерно чистой кислоты может изменяться в широком диапазоне. Обработкой этих солей гидроксидами щелочных и щелочно-земельных металлов выделяют энантиомерно чистые амины. Аналогически проводят расщепление рацематов основных промежуточных соединений, которые образуются при получении рацемических бициклических аминов с энантиомерно чистыми кислотами. Рацемические амины и промежуточные соединения могут быть разделены хроматографически на хиральных носителях, могут быть переведены химическим связыванием с хиральным ацильным остатком в смесь диастереомеров,которые разделяют дистилляцией, кристаллизацией или хроматографией на диастереомерно чистые ацильные производные, из которых выделяют омылением энантиомерно чистые амины. Недостатком указанных способов является то, что в процессе осуществления способов образуется как рацемическая смесь промежуточных соединений, так и рацемическая смесь конечных соединений, которые необходимо разделять хроматографически, что, в свою очередь, требует больших затрат растворителей и времени. Известен способ получения производных 3-хинолинкарбоновой кислоты, в которых соединения общей формулы подвергают взаимодействию с диэтиловым эфиром малоновой кислоты в среде растворителя в присутствии метилат магния с образованием соответствующих сложных эфиров, в которых соответствующие заместители X1, Х 2, Х 3, Hal, представляющие различные заместители, в том числе хлор, фтор, метоксигруппу, могут быть одинаковыми или разными, подвергаются частичному омылению и декарбоксилированию в водной среде в присутствии каталитических количеств серной кислоты или п-толуолсульфокислоты. Полученные соединения подвергают взаимодействию с триэтиловым эфиром ортомуравьиной кислоты в присутствии уксусного ангидрида. Образующиеся промежуточные соединения дальше подвергают взаимодействию с циклопропиламином, затем следует стадия циклизации, после которой присоединяют необходимый амин (заявка ЕР 0167763 А 1, опубл. 15.01.1986). Недостатком этого способа является неудовлетворительный выход продукта. Кроме того, при щелочном омылении могут образовываться побочные продукты, которые способны образовывать полимеры, что является нежелательным. Также омыление в кислых условиях приводит к выделению фтористого водорода, который вызывает коррозию производственной установки и загрязнений продукта комплексными фторидами металлов. В патенте на изобретение UA 41323 С 2 описан способ получения производных 3 хинолонкарбоновой кислоты, который включает стадии взаимодействия галогенангидрида кислоты с эфиром карбоновой кислоты в растворителе в присутствии щелочного агента, взаимодействия с гетероциклическим амином, щелочного омыления и выделения продукта в свободной форме или в виде соли. Галогенид кислоты формулы где X1, Х 2, Hal представляют собой хлор или фтор; А является СН, CF, CCI,подвергают омылению с аминоакриловой кислотой, затем с циклопропиламином следующим омылением, что приводит к циклизации, которая происходит в присутствии карбоната калия. Каждую стадию проводят без предварительного выделения и очистки промежуточных продуктов. Как гетероциклический амин используют соединение Выделение конечного продукта осуществляют таким образом, что после щелочного омыления реакционную смесь нейтрализуют кислотой и отделяют образовавшийся продукт. Недостатком этого способа является то, что при взаимодействии с эфиром акриловой кислоты образуются побочные продукты,которые способны присоединяться к ненасыщенной связи акриловой кислоты по образованию продуктов полимеризации, которые являются ядовитыми и взрывоопасными. Кроме того, проведение следующих стадий способа без выделения и очистки промежуточных продуктов приводит к образованию смеси побочных продуктов, которые могут участвовать в последующих стадиях способа, что, в свою очередь,приводит к образованию небольшого количества конечного продукта. Конечный продукт содержит значительное количество примесей и поэтому возникает необходимость значительных затрат на его отделение от примесей. Поэтому осуществление способа требует значительных затрат на реагенты, растворители и является технологически сложным. Описание изобретения Задачей изобретения является усовершенствование способа получения соединения (1-циклопропил 6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)-октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3 хинолинкарбоновой кислоты путем изменения действий и реагентов в способе получения этого соединения. Задача решается способом получения (1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4aS,7aS)октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты, который включает следующие стадии: трет-бутил октагидро-1H-пирроло[3,4b]пиридин-1-кабоксилата с образованием соединения формулы (2)(2) взаимодействие соединения (2) с триэтилортоформиатом в уксусном ангидриде с образованием соединения формулы (3)(3) присоединение циклического амина к соединению формулы (3) с образованием соединения формулы (4)(4) циклизация соединения формулы (4) в щелочных условиях с образованием соединения формулы (5)(5) отщепление Boc-защитной группы от соединения формулы (5) с образованием конечного соединения формулы (6) Кроме того, в заявляемом способе стадия (1) может быть осуществлена в присутствии основания. Кроме того, в заявляемом способе стадия (2) может быть осуществлена в уксусном ангидриде при температуре 130 С. Кроме того, в заявляемом способе стадия (3) может быть осуществлена при комнатной температуре. Кроме того, в заявляемом способе стадия (4) может быть осуществлена в присутствии 3 н. гидроксида калия при температуре 50 С. Краткое описание чертежей Фиг. 1 - спектр соединения (4aS,7aS)-трет-бутил[b]пиридин-1-карбоксилат. Фиг. 2 - спектр соединения (4aS,7aS)-трет-бутил 6-(4-Z)-3-этокси-2-(этоксикарбонил)акрилоил)3,6-дифлуоро-2-метоксифенил)октагидро-1 Н-пирроло[3,4-b]пиридин-1-карбоксилат. Фиг. 3 спектр соединения(4aS,7aS)-трет-бутил 6-(4-Z)-3-циклопропиламино)-2(этоксикарбонил)акрилоил)-3,6-дифлуоро-2-метоксифенил)октагидро-1H-пирроло[3,4-b]пиридин-1 карбоксилат. Фиг. 4 - спектр соединения 7-4aS,7aS)-1-трет-бутоксикарбонил)гексагидро-1 Н-пирроло[3,4b]пиридин-6-(2 Н)-ил)-1-циклопропил-6-флуоро-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты. Фиг. 5 - спектр соединения 1-циклопропил-6-фтор-7-4aS,7aS)-гексагидро-1 Н-пирроло[3,4b]пиридин-6(2 Н)-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты. Примеры осуществления изобретения Стадия (1) включает такое действие, как присоединение хирального амина, в данном случае (4aS,7aS)-трет-бутил октагидро-1 Н-пирроло[3,4-b]пиридин-1-кабоксилату в этил 3-оксо-3-(2,4,5-трифлуоро-3-метоксифенил)пропаноата (1) с образованием (4aS,7aS)-трет-бутил 6-(4-(3-этоксипропанол)-3,6-дифлуоро-2-метоксифенил)октагидро-1 Н-пирроло[3,4-b]пиридин-1 карбоксилата (2): В амине защитная группа может быть выбрана из группы, содержащей алкоксикарбонильную группу, арилалкоксикарбонильную группу, ацильную группу, алкоксиалкильную группу или замещенную силильную группу. Никакие особые ограничения не накладываются на тип аминозащитных групп, и не-3 024004 которые группы могут быть применены в реакции до тех пор, пока не начнут ингибировать взаимодействие между соединением (1) и аминосоединением. Целесообразно применять трет-бутоксикарбонильную группу и 2,2,2-трихлороэтоксикарбонильную, преимущество имеет трет-бутоксикарбонильная группа. Для реакции используют один или более эквивалент амина. Реакцию осуществляют в присутствии основания, поскольку HF образуется во время проведения этой стадии и может ингибировать реакцию с соединением (1), образуя соль с амином. Стадия (2) включает такое действие, как взаимодействие соединения (2) с триэтилортоформиатом в уксусном ангидриде, что приводит к образованию соединения формулы (3): Алкилортоформиат и уксусный ангидрид используют в эквивалентном количестве. Алкилортоформиат может содержать от одной до шести алкильных групп. Предпочитают такие соединения, как этилортоформиат и метилортоформиат. Алкилортоформиат используют как реагент и растворитель одновременно. Реакцию осуществляют в интервале температур от комнатной до температуры кипения растворителя от 1 до 6 ч. Стадия (3) включает взаимодействие соединения формулы (3) с циклическим амином с образованием соединения формулы (4): Реакцию осуществляют в присутствии органического основания, которое может быть выбрано из группы органических, например триметиламин, триэтиламин, 4-(диметиламино)пиридин, и неорганических, таких как аммоний, карбонат калия, карбонат натрия, гидроксид натрия, гидроксид калия. Предпочтение отдают третичным аминам, в частности триэтиламину. Аминовое соединение может быть кислой солью. Кислую соль могут образовывать неорганические кислоты, такие как соляная кислота, серная кислота, азотная кислота, бромисто-водородная, фтористо-водородная, йодисто-водородная кислоты,а также органические кислоты, такие как толуолсульфоновая, бензолсульфоновая, метансульфоновая(сульфонкислоты могут содержать атом галогенов или алкильную группу в качестве заместителя), трифторуксусная кислота, малеиновая и фумаровая кислоты. Отсутствуют также ограничения в выборе растворителя, и много растворителей используются до тех пор, пока они не начнут ингибировать реакцию. Могут быть использованы толуол, N,N-диметилацетамид, N,N-диметилформамид, диметилсульфоксид,N-метилпирролидон. Предпочтительно использовать толуол. Реакцию проводят при комнатной температуре от 30 мин до 6 ч, в зависимости от того, образовался ли конечный продукт и израсходовались ли исходные соединения. Стадия (4) включает циклизацию соединения формулы (4) с образованием соединения формулы (5): Стадию (4) проводят в присутствии основания, межфазный катализатор может быть комбинированный, хотя соединение (4) не всегда требуется выделять и очищать. В реакции применяют как органические основания, такие как триметиламин, триэтиламин,4-(диметиламино)пиридин, так и неорганические основания, такие как аммоний, карбонат калия, карбонат натрия, гидроксид натрия, гидроксид калия. Предпочтительно использовать гидроксид калия. Основание преимущественно используют в количестве, необходимом для захвата фтористого водорода, который генерируется при замыкании кольца, и для гидролиза сложного эфира. Основание можно непосредственно добавлять в реакционную смесь или можно добавлять в реакционную смесь в виде водного раствора. Основание или раствор не являются необходимой формой примеси с реакционным растворителем. В качестве растворителей используют толуол, N,N-диметилацетамид, N,N-диметилформамид, диметилсульфоксид, N-метилпирролидон. Предпочтительно использовать толуол. В качестве катализатора используют тетрабутиламмоний бромид. Реакцию можно проводить в температурном режиме от комнатной температуры до температуры кипения реакционной смеси. Время проведения реакции зависит от времени преобразования исходных соединений в конечные и может составлять от 1 до 24 ч. При осуществлении заявленного способа соединение (5) может быть выделено и очищено обычными методами. В одном из методов рН реакционной смеси регулируют добавлением соответствующей кислоты и смесь перемешивают при охлаждении со льдом. Таким образом, выпадающие кристаллы фильтруют. В другом методе рН реакционной смеси регулируют добавлением соответствующей кислоты и добавлением соответствующего растворителя в реакционную смесь для извлечения данного соединения. Полученный экстракт концентрируют и соединение (5) перекристаллизовывают из соответствующего растворителя в вышеупомянутых процессах в свободной форме или в форме соли. Примеры солей включают соли неорганических кислот, таких как соляная кислота, серная кислота, азотная кислота,бромисто-водородная, фтористо-водородная, йодисто-водородная кислоты, а также органические кислоты, такие как толуолсульфоновая, бензолсульфоновая, метансульфоновая, трифторуксусная, трихлоруксусная, уксусная, муравьиная и фумаровая кислоты, и соли щелочных и щелочно-земельных металлов,такие как натрий, калий, кальций или литий. Даже когда соединение является смесью свободной формы и соли, соединение может быть выделено в форме сольвата. Сольват может быть образован с водой, этанолом, пропанолом, ацетонитрилом, ацетоном или может быть образован поглощением воды. Стадию (5) проводят с отщеплением трет-бутилкарбоксилата от соединения формулы (5) с образованием соединения формулы (6): Стадию (5) осуществляют в растворителе при нагревании с соляной кислотой. Ниже приведены примеры, которые показывают один из возможных вариантов осуществления заявляемого способа получения соединения моксифлоксацин-(1-циклопропил-6-фтор-1,4-дигидро-8 метокси-7-[(4aS,7aS)-октагидро-6 Н-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты. Пример 1. Получение (4aS,7aS)-трет-бутил[b]пиридин-1-карбоксилата.(4aS,7aS)-трет-Бутил октагидро-1 Н-пирроло[3,4-b]пиридин-1-карбоксилат (4,10 г, 18,1 ммоль) добавляют к раствору, содержащему (5 г, 18,1 ммоль) этил 3-оксо-3-(2,4,5-трифлуоро-3 метоксифенил)пропаноата, ацетонитрила (50 мл), триэтиламина (5,1 мл, 2 экв.), и смесь перемешивают при 25 С три дня и при 50 С 4:00. После этого реакционную смесь охлаждают, растворитель выпаривают при пониженном давлении. К остатку добавляют толуол (50 мл), насыщают солевым раствором (30 мл). Органический остаток сушат над сульфатом магния, растворитель удаляют при пониженном давлении. Получают соединение в виде желтого-зеленого цвета 7,295 г (84%). Соединение было идентифицировано с помощью физико-химических методов. Спектр ЯМР-спектроскопии соединения приведен на фиг. 1. Пример 2. Получение (4aS,7aS)-трет-бутил 6-(4-Z)-3-этокси-2-(этоксикарбонил)акрилоил)-3,6-дифлуоро-2 метоксифенил)октагидро-1H-пирроло[3,4-b]пиридин-1-карбоксилата.(4aS,7aS)-трет-Бутил[b]пиридин-1-карбоксилат (1,033 г, 2,14 ммоль) растворяют в уксусном ангидриде (1,21 мл, 6 экв.) и триэтилортоформиате (2,13 мл, 6 экв.), реакционную смесь перемешивают 14 ч при температуре 130 С. После этого реакционную смесь охлаждают, растворитель удаляют при низком давлении. К остатку добавляют толуол (10 мл), реакционную смесь кипятят дважды. Уксусную кислоту,которая содержится в жидкости, нейтрализуют бикарбонатом натрия и образовавшийся неорганический продукт отфильтровывают. Растворитель, оставшийся при фильтрации, изымают при пониженном давлении, образуя осадок желто-оранжевого цвета 933 мг (81%). Соединение было идентифицировано с помощью физико-химических методов. Спектр ЯМР-спектроскопии соединения приведен на фиг. 2. Пример 3. Получение (4aS,7aS)-трет-бутил 6-(4-Z)-3-циклопропиламино)-2-(этоксикарбонил)акрилоил)-3,6 дифлуоро-2-метоксифенил)октагидро-1 Н-пирроло[3,4-b]пиридин-1-карбоксилата. Циклопропиламин (390 мг, 6,83 ммоль) добавляют к раствору, содержащему (4aS,7aS)-трет-бутил 6-(4-Z)-3-этокси-2-(этоксикарбонил)акрилоил)-3,6-дифлуоро-2-метоксифенил)октагидро-1 Нпирроло[3,4-b]пиридин-1-карбоксилат (787 мг, 1,45 ммоль), толуол (15,2 мл), триэтиламин (0,22 мл,-5 024004 1,1 экв.), и реакционную смесь перемешивают 10 мин при температуре 25 С. Органический остаток промывают водой (210 мл) и солевым раствором (10 мл) и сушат над сульфатом магния. Растворитель удаляют при пониженном давлении. Получают (4aS,7aS)-трет-бутил[b]пиридин-1-карбоксилат 789 мг (99%) желто-оранжевого цвета. Пример 4. Получение 7-4aS,7aS)-1-трет-бутоксикарбонил)гексагидро-1 Н-пирроло[3,4-b]-пиридин-6-(2H-ил)1-циклопропил-6-флуоро-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты. Тетрабутиламмоний бромид (ТВАВ, 8 мг) добавляют к раствору, содержащему (4aS,7aS)-третбутил 6-(4-(Z)3-циклопропиламино)-2-(этоксикарбонил)акрилоил)-3,6-дифлуоро-2-метоксифенил)октагидро-1 Н-пирроло[3,4-b]пиридин-1-карбоксилат (736 мг, 1, 34 ммоль), толуол (14,8 мл) и 3 н. гидрохлорида калия (2,23 мл, 5 экв.), и реакционную смесь перемешивают 4:00 при температуре 50 С. 3 н. гидрохлорид калия (2,23 мл, 5 экв.) еще добавляют к реакционной смеси и перемешивают 2:00. Затем реакционную смесь выливают на лед, немного подкисляют 3 н. раствором соляной кислоты до образования суспензии, в которую добавляют воду (15 мл) и солевой раствор (5 мл) к разделению. Водный остаток экстрагируют толуолом (220 мл), таким образом, восстанавливают органические компоненты и все органические слои объединяют. Объединенные органические остатки сушат над сульфатом натрия, растворитель удаляют на роторе при пониженном давлении. Остаток кристаллизуют, растворитель удаляют при пониженном давлении. Затем остаток растворяют в толуоле (1,5 мл) и гексане (15 мл). Смесь перемешивают 3:00 при 25 С. Полученные осадки отфильтровывают и сушат. В результате получают 558 мг (83%) соединения с желто-оранжевыми кристаллами. Соединение было идентифицировано с помощью физико-химических методов. Спектр ЯМР-спектроскопии соединения приведен на фиг. 4. Пример 5. Получение 1-циклопропил-6-флуоро-7-4aS,7aS)-гексагидро-1 Н-пирроло[3,4-b]пиридин-6(2H)-ил)8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоновой кислоты. Суспендируют в этаноле кислоту (727 мг, 1,45 ммоль) при температуре 22-30 С, обрабатывают 21,70 мг соляной кислоты, (37 мас.%) и кипятят с обратным холодильником в течение 2 ч. После завершения преобразования большую часть спирта отгоняют. Соль, которая образовалась, осаждают, нагревают раствор до 40 С и добавляют дихлорметан. Осаждение кислоты проводят, растворяя хлороводородную соль в смеси растворителей EtOH/вода, 1:1. При температуре 0-7 С добавляют порциями 30% раствор гидроксида натрия до тех пор, пока величина рН не достигнет 12,5. Через 4-48 ч вещество, выпавшее в осадок, отфильтровывают, промывают водой и сушат в вакууме. Способ дает порошок от белого до желтоватого цвета 518 мг (89%) с температурой плавления 324-325 С. Соединение было идентифицировано с помощью физико-химических методов. Спектр ЯМР-спектроскопии соединения приведен на фиг. 5. Как видно из приведенных примеров, при осуществлении заявленного способа получения 1-циклопропил-6-флуоро-7-4aS,7aS)-гексагидро-1 Н-пирроло[3,4-b]пиридин-6(2 Н)-ил)-8-метокси-4 оксо-1,4-дигидрохинолин-3-карбоновой кислоты используются более доступные действующие реагенты,при проведении стадий в заявленном способе в результате присоединения хирального амина, содержащего защитную группу, на первой стадии, не образуются рацемические смеси, которые необходимо разделять, очищать на хроматографической колонке с силикагелем. Таким образом, в заявленном способе образуются энантиомерно чистые промежуточные соединения, которые легко кристаллизуют, очищают и используют на следующих стадиях. Благодаря этому повышаются выходы как промежуточных соединений, так и конечного продукта. В заявленном способе отпадает необходимость дополнительных затрат на растворители, силикагель, реагенты, время, что является экономически выгодным. Сами технологические операции при действиях, как правило, проходят в обычных условиях (без дополнительного нагрева или значительного снижения температуры) и без необходимости точного соблюдения значений рН, способ осуществляется без использования дорогостоящих растворителей. Указанное выше приводит к тому, что заявленный способ получения является технологически простым по сравнению с аналогом и не требует специальных сложных технических операций, в свою очередь, упрощает способ получения данного химического соединения, удешевляет стоимость готового продукта, промышленное производство по заявленному способу имеет низкую степень экологической опасности. Приведенные примеры осуществления заявленного способа получения 1-циклопропил-6-флуоро-74aS,7aS)-гексагидро-1 Н-пирроло[3,4-b]пиридин-6(2 Н)-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3 карбоновой кислоты лишь иллюстрируют изобретение, но не ограничивают его. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы (6) отличающийся тем, что включает следующие стадии: трет-бутил октагидро-1 Н-пирроло[3,4b]пиридин-1-кабоксилата с образованием соединения формулы (2)(2) взаимодействие соединения (2) с триэтилортоформиатом в уксусном ангидриде с образованием соединения формулы (3)(3) присоединение циклического амина к соединению формулы (3) с образованием соединения формулы (4)(4) циклизация соединения формулы (4) в щелочных условиях с образованием соединения формулы (5)(5) отщепление Boc-защитной группы от соединения формулы (5) с образованием конечного соединения формулы (6) 2. Способ по п.1, отличающийся тем, что стадию (1) осуществляют в присутствии основания. 3. Способ по п.1, отличающийся тем, что стадию (2) осуществляют в уксусном ангидриде при температуре 130 С. 4. Способ по п.1, отличающийся тем, что стадию (3) осуществляют при комнатной температуре. 5. Способ по п.1, отличающийся тем, что стадию (4) осуществляют в присутствии 3 н. гидроксида калия при температуре 50 С.
МПК / Метки
МПК: C07D 471/04
Метки: получения, способ, 1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4as,7as)-октагидро-6h-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой, кислоты
Код ссылки
<a href="https://eas.patents.su/10-24004-sposob-polucheniya-1-ciklopropil-6-ftor-14-digidro-8-metoksi-7-4as7as-oktagidro-6h-pirrolo34-bpiridin-6-il-4-okso-3-hinolinkarbonovojj-kisloty.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения (1-циклопропил-6-фтор-1,4-дигидро-8-метокси-7-[(4as,7as)-октагидро-6h-пирроло[3,4-b]пиридин-6-ил]-4-оксо-3-хинолинкарбоновой кислоты</a>
Новая кристаллическая форма n-[4- [2- ( 2-амино-4,7-дигидро-4-оксо-3h-пирроло[ 2,3-d]пиримидин-5-ил) этил] бензоил] -l-глутаминовой кислоты и способ ее получения

Номер патента: 4684
Опубликовано: 24.06.2004
Авторы: Ройтцель-Эденс Сюзн Мари, Снорек Шэрон Ван Ден Берг, Челиус Эрик Кристофер
МПК: C07D 487/04, A61P 35/00, A61K 31/519...
Метки: бензоил, кислоты, форма, способ, новая, 2,3-d]пиримидин-5-ил, l-глутаминовой, получения, 2-амино-4,7-дигидро-4-оксо-3h-пирроло, этил, кристаллическая, n-[4
Формула / Реферат:
1. Гидратная кристаллическая форма динатриевой соли N-[4-[2-(2-амино-4,7-дигидро-4-оксо-3H-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил]-L-глутаминовой кислоты ("гептагидратная кристаллическая форма"), характеризующаяся спектром дифракции рентгеновских лучей, который включает максимум, соответствующий межплоскостному расстоянию d: 7,78+ 0,04 Е, полученным измерением при 22+2шC и 20-80% относительной влажности с использованием медного источника...
Применение комбинации из 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина и химиотерапевтического агента для получения лекарственного средства для лечения рака желудка

Номер патента: 23697
Опубликовано: 29.07.2016
Авторы: Абуруб Актхам, Васудеван Венкатрагхаван, Энглер Томас Альберт, Чедид Марсио
МПК: A61K 31/4985, A61K 31/4745, A61K 31/513...
Метки: лечения, желудка, лекарственного, получения, применение, агента, рака, средства, 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина, химиотерапевтического, комбинации
Формула / Реферат:
Применение комбинации из 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1Н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина или фармацевтически приемлемой соли или сольвата указанного соединения и химиотерапевтического агента, выбранного из группы, состоящей из 5-фторурацила и цисплатина, для получения лекарственного средства для лечения рака желудка.
Применение комбинации 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина с химиотерапевтическим агентом для лечения злокачественных опухолей

Номер патента: 18447
Опубликовано: 30.08.2013
Авторы: Абуруб Актхам, Чедид Марсио, Энглер Томас Альберт, Васудеван Венкатрагхаван
МПК: A61K 31/4745, A61K 31/24, A61K 31/4985...
Метки: химиотерапевтическим, комбинации, 7-(2,5-дигидро-4-имидазо[1,2-а]пиридин-3-ил-2,5-диоксо-1н-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина, применение, злокачественных, опухолей, лечения, агентом
Формула / Реферат:
1. Применение комбинации 7-(2,5-дигидро-4-имидазо[1,2-a]пиридин-3-ил-2,5-диоксо-1H-пиррол-3-ил)-9-фтор-1,2,3,4-тетрагидро-2-(1-пиперидинилкарбонил)пирроло[3,2,1-jk][1,4]бензодиазепина или фармацевтически приемлемой соли или сольвата указанного соединения с химиотерапевтическим агентом, выбранным из группы, состоящей из СРТ-11, пеметрекседа, гемцитабина, этопозида, доксорубицина, цисплатина и карбоплатина, для получения лекарственного средства...
Полиморфная форма iii n-[2-(диэтиламино)этил]-5-[(5-фтор-1,2-дигидро-2-оксо-3н-индол-3-илиден)метил]-2,4-диметил-1н-пиррол-3-карбоксамида и способ ее получения

Номер патента: 20067
Опубликовано: 29.08.2014
Авторы: Мохамад Несрин, Латц Рюдигер, Бёзе Роланд, Штригель Ханс-Гюнтер
МПК: C07D 403/06
Метки: форма, получения, полиморфная, способ, n-[2-(диэтиламино)этил]-5-[(5-фтор-1,2-дигидро-2-оксо-3н-индол-3-илиден)метил]-2,4-диметил-1н-пиррол-3-карбоксамида
Формула / Реферат:
1. Полиморфная форма III Сунитиниба, характеризующаяся пиками на XRPD диаграмме при 6,3±0,2; 22,2±0,2 и 26,4±0,2 градусах 2-тета.2. Полиморфная форма III Сунитиниба по п.1, характеризующаяся следующими характеристиками:кристаллографическая система: моноклинная;пространственная группа: Р 21/n;параметры элементарной ячейки:а = 4,97560(10) Å;b = 28,1365(6) Å;с= 14,5880(3) Å;β = 93,5130(10)°;объем ячейки V: 2038,42(7)...
Производные октагидро-6, 10-диоксо-6н-пиридазино [1,2-а] [1,2] диазепин-1-карбоновой кислоты, способ их получения и их применение для получения терапевтически активных соединений

Номер патента: 3280
Опубликовано: 24.04.2003
Авторы: Крок Вероник, Колладан Колетт, Руссель Патрик, Ларкин Джон Патрик
МПК: C07D 487/04
Метки: получения, способ, активных, октагидро-6, 1,2-а, кислоты, терапевтически, производные, применение, 10-диоксо-6н-пиридазино, 1,2, соединений, диазепин-1-карбоновой
Формула / Реферат:
1. Соединения общей формулы (I) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS, в формуле которых R представляет собой атом водорода, радикал алкил, содержащий от 1 до 4 атомов углерода, a R1 и R2 означают H или защитную группу аминофункции. 2. Соединения по п.1, в которых аминогруппа защищена в форме фталимида, формулы (IA1) имеющие конфигурацию SR или находящиеся в виде смеси SR+SS. 3. Соединения формулы (I) согласно любому из...
Предыдущий патент: Способ получения эстетрола
Следующий патент: Частицы фосфата, полифосфата и метафосфата алюминия, их использование в качестве пигментов в красках и способ их получения
Случайный патент: Способ и устройство для определения удельного сопротивления формации, через которую проходит обсаженная скважина