Новый способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой









Формула / Реферат
1. Способ синтеза соединения формулы (VII) в рацемической или оптически активной форме

в которой R представляет собой атом водорода или метильную группу,
отличающийся тем, что соединение формулы (VIII)

реагирует с соединением формулы (IX) в рацемической или оптически активной форме, в форме свободного основания или соли

в которой R является таким, как определено выше,
в присутствии соли переходного металла или лантаноида, в растворителе, для получения соединения формулы (X) в рацемической или оптически активной форме

которое преобразуют в соединение формулы (VII) при действии донора водорода.
2. Способ синтеза по п.1, отличающийся тем, что соединение формулы (IX) имеет конфигурацию (S).
3. Способ синтеза по п.2, отличающийся тем, что R представляет собой атом водорода, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой соединение формулы (XI), частный случай соединений формулы (VII)

которое может быть N-метилировано для получения ивабрадина формулы (I)

который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты.
4. Способ синтеза по п.2, отличающийся тем, что R представляет собой метильную группу, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой ивабрадин формулы (I), частный случай соединений формулы (VII)

который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты.
5. Способ синтеза по п.1, отличающийся тем, что соединение формулы (IX) находится в рацемической форме.
6. Способ синтеза по п.5, отличающийся тем, что R представляет собой атом водорода, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой рацемическое соединение формулы (XII), частный случай соединений формулы (VII)

которое может быть N-метилировано для получения рацемического соединения формулы (XIII)

разделение оптических изомеров которого дает ивабрадин формулы (I)

который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты.
7. Способ синтеза по п.5, отличающийся тем, что R представляет собой метильную группу, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой рацемическое соединение формулы (XIII), частный случай соединений формулы (VII)

разделение оптических изомеров которого дает ивабрадин формулы (I)

который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты.
8. Способ синтеза по любому из пп.1-7, отличающийся тем, что соль переходного металла или лантаноида, применяемую для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), выбирают из хлорида меди(I), бромида меди(I), йодида меди(I), трифторметансульфоната иттрия(III), трифторметансульфоната лантана(III), трифторметансульфоната празеодима(III), трифторметансульфоната неодима(III), трифторметансульфоната самария(III), трифторметансульфоната европия(III), трифторметансульфоната гадолиния(III), трифторметансульфоната тербия(III), трифторметансульфоната диспрозия(III), трифторметансульфоната гольмия(III), трифторметансульфоната эрбия(III) и трифторметансульфоната лютеция(III).
9. Способ синтеза по любому из пп.1-8, отличающийся тем, что растворитель, применяемый для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), выбирают из спиртовых растворителей, диметилсульфоксида, N,N-диметилформамида и N-метилпирролидона.
10. Способ синтеза по любому из пп.1-9, отличающийся тем, что донор водорода, применяемый для осуществления преобразования соединения формулы (X) до соединения формулы (VII), выбирают из тетраборгидрида натрия, цианоборгидрида натрия, комплекса боран-морфолин и комплекса боран-диметиламин.
11. Соединение формулы (VIII)

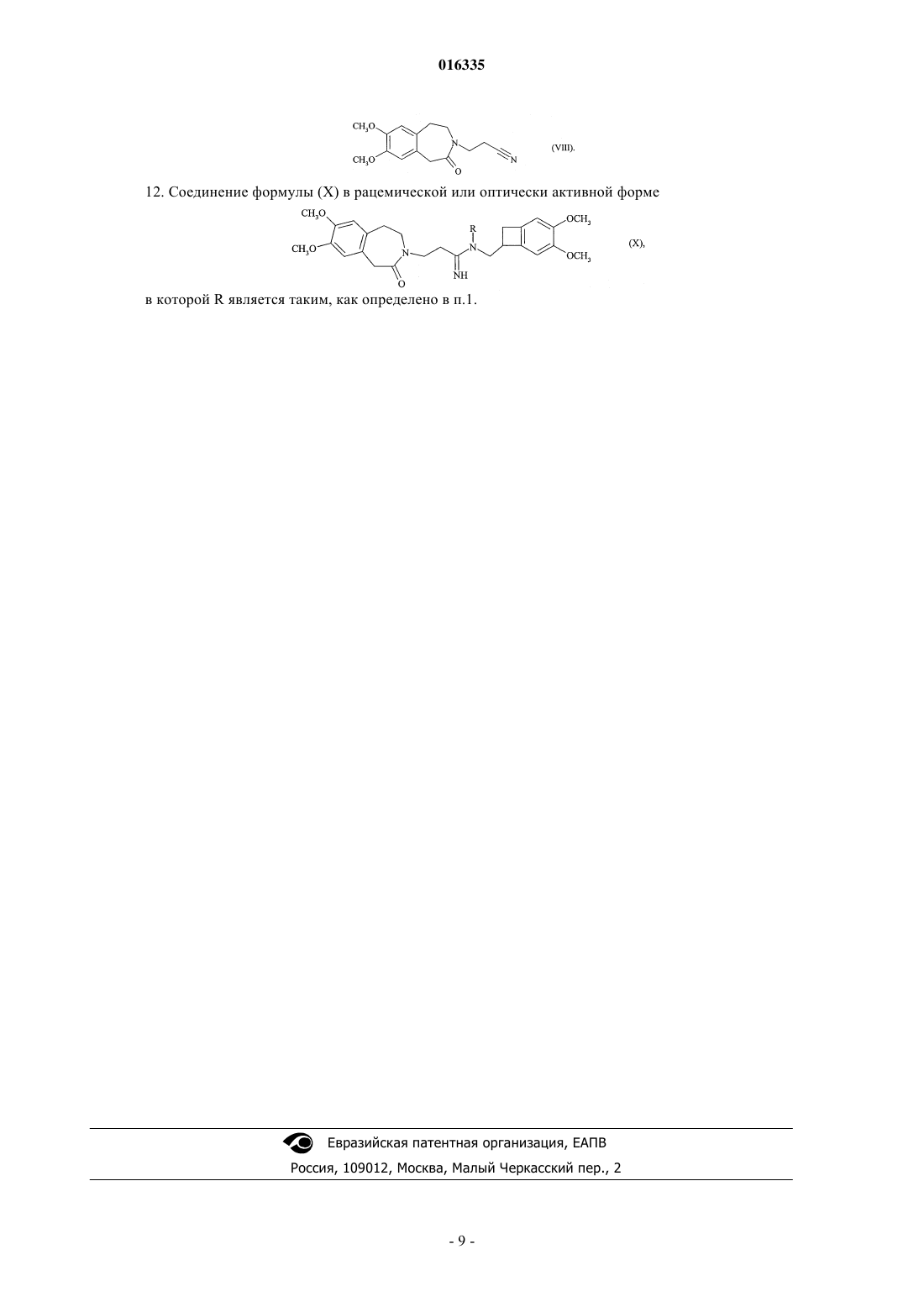
12. Соединение формулы (X) в рацемической или оптически активной форме

в которой R является таким, как определено в п.1.
Текст
НОВЫЙ СПОСОБ СИНТЕЗА ИВАБРАДИНА И ЕГО СОЛЕЙ ПРИСОЕДИНЕНИЯ С ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТОЙ Изобретение относится к способу синтеза ивабрадина формулы (I) и его солей присоединения с фармацевтически приемлемой кислотой(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ (FR) 016335 Настоящее изобретение относится к способу синтеза ивабрадина формулы (I) или 3-3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8 диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-она, его солей присоединения с фармацевтически приемлемой кислотой, и его гидратов. Ивабрадин, его соли присоединения с фармацевтически приемлемой кислотой и более особенно его гидрохлорид имеют очень ценные фармакологические и терапевтические свойства, особенно брадикардические свойства, делающие эти соединения полезными в лечении или предотвращении различных клинических состояний миокардиальной ишемии, таких как бронхиальная астма, инфаркт миокарда и связанных с нею нарушений сердечного ритма, а также в различных патологиях, включающих нарушения сердечного ритма, особенно суправентрикулярные нарушения сердечного ритма, и в сердечной недостаточности. Получение и терапевтическое применение ивабрадина, его солей присоединения с фармацевтически приемлемой кислотой и более особенно его гидрохлорида были описаны в описании европейского патента ЕР 0534859. В настоящем патенте описывается синтез гидрохлорида ивабрадина, начиная с соединения формулы (II) которое разделяют для получения соединения формулы (IV) для получения соединения формулы (VI) каталитическая гидрогенизация которого дает ивабрадин, который потом преобразовывают в его гидрохлорид. Недостаток этого пути синтеза состоит в том, что он приводит к ивабрадину с выходом порядка только 0,6%. Ввиду фармацевтической ценности этого соединения важно, чтобы было возможно получить его эффективным способом синтеза, который приводит к ивабрадину с хорошим выходом.-1 016335 Настоящее изобретение относится к способу синтеза соединения формулы (VII) в рацемической или оптически активной форме в которой R представляет собой атом водорода или метильную группу,отличающемуся тем, что соединение формулы (VIII) реагирует с соединением формулы (IX) в рацемической или оптически активной форме, в форме свободного основания или соли в которой R является таким, как определено выше,в присутствии соли переходного металла или лантаноида, в растворителе, для получения соединения формулы (X) в рацемической или оптически активной форме которое преобразуют в соединение формулы (VII) при действии донора водорода. В предпочтительном варианте осуществления изобретения соединение формулы (IX) находится в оптически активной форме, более особенно в конфигурации (S). В случае, где R представляет собой атом водорода, продуктом реакции соединения формулы (X) с донором водорода является соединение формулы (XI), частный случай соединений формулы (VII) которое может быть N-метилировано для получения ивабрадина формулы (I) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. В случае, где R представляет собой метильную группу, продукт реакции соединения формулы (X) с донором водорода представляет собой ивабрадин формулы (I), частный случай соединений формулы который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной,-2 016335 трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной,малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. В другом предпочтительном варианте осуществления изобретения соединение формулы (IX) находится в рацемической форме. В случае, где R представляет собой атом водорода, продукт реакции соединения формулы (X) с донором водорода представляет собой рацемическое соединение формулы (XII), частный случай соединений формулы (VII) которое может быть N-метилировано для получения рацемического соединения формулы (XIII) разделение оптических изомеров которого дает ивабрадин формулы (I) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной,трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной,малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. В случае, где R представляет собой метильную группу, продукт реакции соединения формулы (X) с донором водорода после этого представляет собой рацемическое соединение формулы (XIII), частный случай соединений формулы (VII) разделение оптических изомеров которого дает ивабрадин формулы (I): которое может необязательно быть преобразовано в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной кислоты, бромисто-водородной кислоты, серной кислоты,фосфорной кислоты, уксусной кислоты, трифторуксусной кислоты, молочной кислоты, пировиноградной кислоты, малоновой кислоты, янтарной кислоты, глутаровой кислоты, фумаровой кислоты, винной кислоты, малеиновой кислоты, лимонной кислоты, аскорбиновой кислоты, щавелевой кислоты, метансульфоновой кислоты, бензолсульфоновой кислоты и камфорной кислоты, и в его гидраты. Среди солей переходных металлов или лантаноидов, которые могут быть применены для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), здесь могут быть упомянуты, не подразумевая какое-либо ограничение, хлорид меди(I), бромид меди(I), йодид меди(I),трифторметансульфонат иттрия(III), трифторметансульфонат лантана(III), трифторметансульфонат празеодима(III), трифторметансульфонат неодима(III), трифторметансульфонат самария(III), трифторметансульфонат европия(III), трифторметансульфонат гадолиния(III), трифторметансульфонат тербия(III),трифторметансульфонат диспрозия(III), трифторметансульфонат гольмия(III), трифторметансульфонат эрбия(III) и трифторметансульфонат лютеция. Предпочтение отдают соли переходного металла, приме-3 016335 няемой для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX),которой является хлорид меди(I). Среди растворителей, которые могут быть применены для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), здесь могут быть упомянуты, не подразумевая какоелибо ограничение: спиртовые растворители, особенно метанол, этанол и изопропанол; диметилсульфоксид (DMSO);N-метилпирролидон (NMP). Предпочтение отдают растворителю, применяемому для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), которым является метанол. Среди доноров водорода, которые могут быть применены для осуществления преобразования соединения формулы (X) в соединение формулы (VII), здесь могут быть упомянуты, не подразумевая какое-либо ограничение, тетраборгидрид натрия, цианоборгидрид натрия, а также комплексы боранморфолин и боран-диметиламин. Среди растворителей, которые могут быть применены для осуществления преобразования соединения формулы (X) в соединение формулы (VII), здесь могут быть упомянуты, не подразумевая какое-либо ограничение: спиртовые растворители, особенно метанол, этанол и изопропанол;N-метилпирролидон (NMP). Соединения формул (VIII) и (X) являются новыми продуктами, которые пригодны в качестве интермедиатов синтеза в химической или фармацевтической промышленности, особенно в синтезе ивабрадина, его солей присоединения с фармацевтически приемлемой кислотой и его гидратов, и как таковые они образуют нераздельную часть настоящего изобретения. Примеры ниже иллюстрируют изобретение. В описании приняты следующие обозначения:DMF: N,N-диметилформамид; ИК: инфракрасный. Точки плавления (т.пл.) были измерены с использованием прибора microKofler (MK). Пример 1. 3-(7,8-Диметокси-2-оксо-1,2,4,5-тетрагидро-3 Н-3-бензазепин-3-ил)пропаннитрил. 2 г (9 ммоль) 7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-она растворяли в 30 мл DMF. К получившемуся раствору добавляли при 25 С 432 мг (10,8 ммоль, 1,2 экв.) гидрида натрия в виде 60% суспензии в масле. Осуществляли перемешивание на протяжении 30 мин при температуре окружающей среды и после этого добавляли раствор 0,9 мл (10,8 ммоль, 1,2 экв.) 3-бромпропионитрила в 10 мл DMF. После осуществляли нагревание при 50 С на протяжении 24 ч и после этого выпаривали растворитель. Остаток вносили в дихлорметан, промывали водой, сушили над MgSO4, фильтровали и выпаривали досуха. Получали 4,1 г остатка, который очищали флэш-хроматографией на 300 г силикагеля(элюент = дихлорметан/этанол: 95/5). Извлекли полученные 630 мг названного продукта в форме масла,и 1 г непрореагировавшего 7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-она (белое твердое вещество, т.пл.=196-198 С). Выход = 26%. ИК (чистый):= 2247, 1648, 1609, 1518, 1246, 1220, 1104 см-1. Пример 2. 3-[3-([(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламино)пропил]7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. Стадия 1. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси-2 оксо-1,2,4,5-тетрагидро-3 Н-3-бензазепин-3-ил)пропанимидамид. В атмосфере азота 630 мг (3,27 ммоль, 1,5 экв.) 1-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5 триен-7-ил]метанамингидрохлорида растворяли в 10 мл метанола. К получившемуся раствору добавляли 0,46 мл (3,27 ммоль, 1,5 экв.) триэтиламина и 260 мг (2,62 ммоль) хлорида меди (I) (чистота: 90%). После этого по каплям добавляли 600 мг (2,18 ммоль) 3-(7,8-диметокси-2-оксо-1,2,4,5-тетрагидро-3 Н-3 бензазепин-3-ил)пропаннитрила, растворенного в 10 мл метанола. Нагревание в колбе с обратным холодильником осуществляли на протяжении 12 ч, после этого охлаждали до температуры окружающей среды и добавляли 5 мл 35% водного раствора гидроксида натрия и 30 мл дихлорметана. Органическую фазу экстрагировали, сушили над MgSO4, фильтровали и после этого выпаривали досуха. Получали 1,08 г коричневого масла, содержащего 47% предполагаемого продукта. Это масло используют без очистки в следующей стадии.-4 016335 Стадия 2. 3-[3-([(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламино)пропил]-7,8 диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. 1 г продукта, полученного на стадии 1 (содержащего 47% амидина), растворяли в 15 мл метанола; получившийся раствор после этого охлаждали до 0 С и добавляли 100 мг (2,61 ммоль, 1,2 экв.) тетраборгидрида натрия. Перемешивание осуществляли в течение ночи при температуре окружающей среды и после этого добавляли 5,3 мл 20% водного раствора гидроксида натрия и 20 мл дихлорметана. Осуществляли энергичное перемешивание на протяжении 15 мин. Органическую фазу после этого экстрагировали, промывали водой, сушили над MgSO4, фильтровали и после этого выпаривали досуха. Получали 1 г масла, которое очищали флэш-хроматографией на 100 г силикагеля (элюент = дихлорметан/этанол/NH4OH:90/10/1) для получения 300 мг предполагаемого продукта в форме масла. Выход = 30% (за 2 стадии). ИК (чистый):= 3302, 1649 см-1. Пример 3. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. Стадия 1. 3-[3-([(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламино)пропил]-7,8 диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он гидрохлорид. 300 мг (0,65 ммоль) амина, полученного в примере 2, стадия 2, растворяли в 10 мл ацетонитрила. К получившемуся раствору добавляли 0,65 мл (1,3 ммоль, 2 экв.) 2 М раствора соляной кислоты в диэтиловом эфире. Осуществляли перемешивание при 25 С в течение 15 мин и после этого выпаривали досуха. Продукт кристаллизовали из 20 мл ацетона. Твердое вещество отфильтровывали и сушили. Было получено 230 мг белых кристаллов. Выход = 71%, т.пл.=204-206 С. Стадия 2. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он. 180 мг (0,36 ммоль) гидрохлорида, полученного на стадии 1, растворяли в смеси 10 мл метанола и 5 мл дихлорметана. К получившемуся раствору добавляли 0,04 мл (0,54 ммоль, 1,5 экв.) формальдегида(37% в воде) и крупинку бромкрезолового зеленого. Добавляли 1N водный раствор соляной кислоты до рН 4 (желто-окрашенный раствор) и после этого осуществляли перемешивание при 25 С в течение 30 мин. После этого добавляли 23 мг (0,36 ммоль) цианоборгидрида натрия, осуществляли перемешивание при 25 С в течение 12 ч, в то же время поддерживая рН 4, и после этого осуществляли выпаривание досуха. Получали 250 мг масла, которое очищали флэш-хроматографией на 100 г силикагеля (элюент = дихлорметан/этанол/NH4OH: 90/10/1), для получения 100 мг названного продукта в форме бесцветного масла, которое кристаллизовали при температуре окружающей среды. Выход = 58%, т.пл.=98-100 С. Пример 4. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он гидрохлорид. Гидрохлорид продукта, полученного в примере 3, стадия 2, получали путем последующей стадии,описанной в патенте ЕР 0534859 (пример 2, стадия Е). Пример 5. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. Стадия 1. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси-2 оксо-1,2,4,5-тетрагидро-3 Н-3-бензазепин-3-ил)-N-метилпропанимидамид. В атмосфере азота 691 мг (2,83 ммоль, 1,5 экв.) 1-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5 триен-7-ил]-N-метилметанамин гидрохлорида растворяли в 10 мл метанола. К получившемуся раствору добавляли 0,4 мл (2,83 ммоль, 1,5 экв.) триэтиламина и 224 мг (2,26 ммоль) хлорида меди (I) (чистота: 90%). После этого по каплям добавляли 520 мг (1,89 ммоль) 3-(7,8-диметокси-2-оксо-1,2,4,5-тетрагидро 3H-3-бензазепин-3-ил)пропаннитрила, растворенного в 10 мл метанола. Нагревание в колбе с обратным холодильником осуществляли на протяжении 24 ч, после этого охлаждали до температуры окружающей среды и добавляли 5 мл 35% водного раствора гидроксида натрия и 30 мл дихлорметана. Органическую фазу экстрагировали, сушили над MgSO4, фильтровали и после этого выпаривали досуха. Получали 1,08 г коричневого масла, содержащего 46% предполагаемого продукта. Это масло используют без очистки в следующей стадии. Стадия 2. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. 1 г продукта, полученного на стадии 1 (содержащего 46% амидина), растворяли в 15 мл метанола и после этого добавляли при температуре окружающей среды 86 мг (2,26 ммоль, 1,2 экв.) тетраборгидрида натрия. Осуществляли перемешивание в течение ночи при температуре окружающей среды и после этого добавляли 5 мл 20% водного раствора гидроксида натрия и 20 мл дихлорметана. Осуществляли энергичное перемешивание на протяжении 15 мин. Органическую фазу после этого экстрагировали, промывали водой, сушили над MgSO4, фильтровали и после этого выпаривали досуха. Получили 1 г масла, которое очищали флэш-хроматографией на 100 г силикагеля (элюент = дихлорметан/этанол/NH4OH: 90/10/1) для-5 016335 получения 210 мг названного продукта в форме масла. Выход = 24% (за 2 стадии). ИК (чистый):= 1633, 831-672 см-1. Пример 6. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он гидрохлорид. Гидрохлорид продукта, полученного в примере 5, стадия 2, получали при помощи последующей стадии, описанной в патенте ЕР 0534859 (пример 2, стадия Е). Пример 7. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он гидрохлорид. Начиная с рацемического (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метанамина и следуя протоколу,описанному последовательностью примеров 2 и 3,получали 3-3-(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил](метил)амино]пропил-7,8-диметокси 1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. 2,1 г этого рацемического соединения после этого разделяли на колонке 60 см 60 мм, упакованной 2,1 кг фазы Chiralpak AD (размер частиц 20 мкм). Применяемым элюентом является смесь этанол/ацетонитрил/диэтиламин (10/90/0,1 по объему) при скорости потока 50 мл/мин. Использовали встроенный ультрафиолетовый детектор при длине волны 280 нм. Получали 0,95 г энантиомера конфигурации (R) в форме белой пенки и после этого 0,95 г энантиомера конфигурации (S) также в форме белой пенки. Гидрохлорид энантиомера конфигурации (S) после получали при помощи последующей стадии,описанной в патенте ЕР 0534859 (пример 2, стадия Е). Пример 8. 3-3-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он гидрохлорид. Начиная с рацемического (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)-N-метилметанамина и следуя протоколу, описанному в примере 5, получали 3-3-(3,4-диметоксибицикло[4.2.0]окта-1,3,5 триен-7-ил)метил](метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он. 2,1 г этого рацемического соединения после этого разделяли на колонке 60 см 60 мм, упакованной 2,1 кг фазы Chiralpak AD (размер частиц 20 мкм). Применяемым элюентом является смесь этанол/ацетонитрил/диэтиламин (10/90/0,1 по объему) при скорости потока 50 мл/мин. Использовали встроенный ультрафиолетовый детектор при длине волны 280 нм. Получали 0,95 г энантиомера конфигурации (R) в форме белой пенки и после этого 0,95 г энантиомера конфигурации (S) также в форме белой пенки. Гидрохлорид энантиомера конфигурации (S) после получали при помощи последующей стадии,описанной в патенте ЕР 0534859 (пример 2, стадия Е). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ синтеза соединения формулы (VII) в рацемической или оптически активной форме в которой R представляет собой атом водорода или метильную группу,отличающийся тем, что соединение формулы (VIII) реагирует с соединением формулы (IX) в рацемической или оптически активной форме, в форме свободного основания или соли в которой R является таким, как определено выше,в присутствии соли переходного металла или лантаноида, в растворителе, для получения соединения формулы (X) в рацемической или оптически активной форме которое преобразуют в соединение формулы (VII) при действии донора водорода. 2. Способ синтеза по п.1, отличающийся тем, что соединение формулы (IX) имеет конфигурацию (S). 3. Способ синтеза по п.2, отличающийся тем, что R представляет собой атом водорода, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой соединение формулы(XI), частный случай соединений формулы (VII) которое может быть N-метилировано для получения ивабрадина формулы (I) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной,трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной,малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. 4. Способ синтеза по п.2, отличающийся тем, что R представляет собой метильную группу, и тем,что продукт реакции соединения формулы (X) с донором водорода представляет собой ивабрадин формулы (I), частный случай соединений формулы (VII) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. 5. Способ синтеза по п.1, отличающийся тем, что соединение формулы (IX) находится в рацемиче-7 016335 ской форме. 6. Способ синтеза по п.5, отличающийся тем, что R представляет собой атом водорода, и тем, что продукт реакции соединения формулы (X) с донором водорода представляет собой рацемическое соединение формулы (XII), частный случай соединений формулы (VII) которое может быть N-метилировано для получения рацемического соединения формулы (XIII) разделение оптических изомеров которого дает ивабрадин формулы (I) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной,трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной,малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. 7. Способ синтеза по п.5, отличающийся тем, что R представляет собой метильную группу, и тем,что продукт реакции соединения формулы (X) с донором водорода представляет собой рацемическое соединение формулы (XIII), частный случай соединений формулы (VII) разделение оптических изомеров которого дает ивабрадин формулы (I) который может необязательно быть преобразован в его соли присоединения с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной,трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной,малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. 8. Способ синтеза по любому из пп.1-7, отличающийся тем, что соль переходного металла или лантаноида, применяемую для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), выбирают из хлорида меди(I), бромида меди(I), йодида меди(I), трифторметансульфоната иттрия(III), трифторметансульфоната лантана(III), трифторметансульфоната празеодима(III), трифторметансульфоната неодима(III), трифторметансульфоната самария(III), трифторметансульфоната европия(III), трифторметансульфоната гадолиния(III), трифторметансульфоната тербия(III), трифторметансульфоната диспрозия(III), трифторметансульфоната гольмия(III), трифторметансульфоната эрбия(III) и трифторметансульфоната лютеция(III). 9. Способ синтеза по любому из пп.1-8, отличающийся тем, что растворитель, применяемый для осуществления реакции между соединением формулы (VIII) и соединением формулы (IX), выбирают из спиртовых растворителей, диметилсульфоксида, N,N-диметилформамида и N-метилпирролидона. 10. Способ синтеза по любому из пп.1-9, отличающийся тем, что донор водорода, применяемый для осуществления преобразования соединения формулы (X) до соединения формулы (VII), выбирают из тетраборгидрида натрия, цианоборгидрида натрия, комплекса боран-морфолин и комплекса борандиметиламин. 11. Соединение формулы (VIII) 12. Соединение формулы (X) в рацемической или оптически активной форме
МПК / Метки
МПК: C07D 223/16
Метки: кислотой, солей, новый, приемлемой, ивабрадина, способ, синтеза, фармацевтически, присоединения
Код ссылки
<a href="https://eas.patents.su/10-16335-novyjj-sposob-sinteza-ivabradina-i-ego-solejj-prisoedineniya-s-farmacevticheski-priemlemojj-kislotojj.html" rel="bookmark" title="База патентов Евразийского Союза">Новый способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой</a>
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8836
Опубликовано: 31.08.2007
Авторы: Дюбюффе Тьерри, Лекув Жан-Пьер
МПК: C07K 5/02, C07K 5/06, C07D 209/42...
Метки: синтеза, солей, фармацевтически, периндоприла, способ, приемлемых, новый
Формула / Реферат:
1. Способ синтеза периндоприла формулы (I) и его фармацевтически приемлемых солей, который характеризуется тем, что соединение формулы (II) подвергают реакции с соединением формулы (III) в которой R1 представляет собой имидазолильную, бензимидазолильную или тетразолильную группу, получая соединение формулы (IV) которое подвергают реакции с соединением формулы (V) в которой R2 представляет собой атом водорода, или бензил, или линейную или...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 12034
Опубликовано: 30.06.2009
Авторы: Дюбюффе Тьерри, Лекув Жан-Пьер
МПК: C07K 5/06
Метки: приемлемых, фармацевтически, новый, синтеза, солей, способ, периндоприла
Формула / Реферат:
1. Способ промышленного синтеза периндоприла формулы (I) и его фармацевтически приемлемых солей, отличающийся тем, что сложный бензиловый эфир формулы (IIa) или (IIb) или аддитивная соль сложного эфира формулы (IIa) или (IIb) с минеральной кислотой или органической кислотой взаимодействует с соединением формулы (III) в присутствии конденсирующего агента, который выбирают из следующих реагентов и пар реагентов: гидрохлорид...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8668
Опубликовано: 29.06.2007
Авторы: Дюбюффе Тьерри, Лекув Жан-Пьер
МПК: C07K 5/06, C07K 5/02, C07D 209/42...
Метки: приемлемых, фармацевтически, новый, синтеза, солей, периндоприла, способ
Формула / Реферат:
1. Способ синтеза соединений формулы (I) и их фармацевтически приемлемых солей, характеризующийся тем, что соединение формулы (II) в которой Вn представляет собой бензильную группу, подвергают реакции с соединением формулы (III), имеющим S-конфигурацию в которой X представляет собой атом галогена и ВОС представляет собой трет-бутоксикарбонильную группу, в присутствии основания, получая после снятия защиты с функциональной аминогруппы...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 8626
Опубликовано: 29.06.2007
Авторы: Лекув Жан-Пьер, Дюбюффе Тьерри
МПК: C07K 5/02, C07K 5/06, C07D 209/42...
Метки: синтеза, новый, периндоприла, способ, солей, приемлемых, фармацевтически
Формула / Реферат:
1. Способ синтеза соединений формулы (I) и их фармацевтически приемлемых солей, характеризующийся тем, что соединение формулы (II) в которой R1 представляет собой бензил или линейную или разветвленную C1-С6-алькильную группу, подвергают реакции с соединением формулы (III), имеющим S-конфигурацию в которой X представляет собой атом галогена и R2 представляет собой защитную группу для функциональной аминогруппы, в присутствии основания, получая...
Новый способ синтеза периндоприла и его фармацевтически приемлемых солей

Номер патента: 9066
Опубликовано: 26.10.2007
Авторы: Дюбюффе Тьерри, Ланглуа Паскаль
МПК: C07K 5/02, C07D 209/42, A61P 9/00...
Метки: фармацевтически, периндоприла, солей, новый, синтеза, приемлемых, способ
Формула / Реферат:
1. Способ синтеза периндоприла формулы (I) а также его фармацевтически приемлемых солей, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (II) подвергают реакции с соединением формулы (III) где R1 - это защитная группа для кислотной группы представляющая собой атом водорода или бензильную группу, a R2 - это защитная группа для группы амина, представляющая собой трет-бутоксикарбонильную группу, с получением соединения...
Предыдущий патент: Горелка для концентрата
Следующий патент: Способ уменьшения осаждения продукта в передающей линии между петлевыми реакторами
Случайный патент: Расширительная матрица для изготовления металлических емкостей и система матриц