Способ получения арипипразола и соответствующие промежуточные продукты и их получение
Номер патента: 12180
Опубликовано: 28.08.2009
Авторы: Атхукуру Венката Субба Рао, Чиннапиллай Раджендиран, Джасти Венкатесварлу, Арава Веера Редди







Формула / Реферат
1. Промежуточный продукт, 6-(4-галогенбутокси)индан-1-он формулы 3

предназначенный для получения арипипразола формулы 1

2. Промежуточный продукт, 6-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]индан-1-он формулы (2)

предназначенный для получения арипипразола формулы 1.
3. Способ получения промежуточного продукта формулы (3), предназначенного для получения арипипразола формулы (1), включающий взаимодействие соединения формулы (11)

с 1,4-дигалогенбутаном формулы (12)

где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90-110шС с образованием промежуточного продукта формулы (3).
4. Способ получения промежуточного продукта формулы (2), предназначенного для получения арипипразола формулы 1, включающий:
(I) взаимодействие соединения формулы (11)

с 1,4-дигалогенбутаном формулы (12)

где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90-110шС с образованием промежуточного продукта формулы (3) и
(II) взаимодействие соединения формулы 3, полученного на стадии (1), с соединением формулы (9)

в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне 80-120шС с получением промежуточного продукта формулы 2.
5. Способ получения арипипразола формулы 1, включающий:
(I) взаимодействие соединения формулы (11) с 1,4-дигалогенбутаном формулы (12), где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90-110шС с образованием промежуточного продукта формулы (3),
(II) взаимодействие соединения формулы (3), полученного на стадии (I), с соединением формулы (9) в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне 80-120шС с образованием промежуточного продукта формулы 2 и
(III) взаимодействие образовавшегося соединения формулы (2) с азидом натрия или триметилсилилазидом в присутствии кислоты при температуре в диапазоне 50-90шС с получением соединения формулы (1) и, в случае необходимости, перекристаллизацию соединения формулы (1).
6. Способ получения по пп.3-5, где на стадии (I) применяют основание, такое как гидрид натрия, метоксид натрия, триэтиламин, карбонат калия, бикарбонат натрия и карбонат натрия, предпочтительно триэтиламин, наиболее предпочтительно карбонат калия.
7. Способ получения по пп.3-6, где применяемый на стадии (I) растворитель выбран из ацетона, хлороформа, метиленхлорида, этилендихлорида, диметилформамида, диметилсульфоксида и ацетонитрила, предпочтительно ацетона и наиболее предпочтительно 1,4-дигалогенбутана.
8. Способ по пп. 3-7, где температура на стадии (I) составляет 90-110шС и наиболее предпочтительно 100-110шС.
9. Способ по пп.3-8, где применяемое на стадии (II) основание представляет собой карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, гидрид натрия, гидрид калия и триэтиламин, наиболее предпочтительно карбонат натрия.
10. Способ по пп.3-9, где применяемый на стадии (II) растворитель выбран из ацетонитрила, ацетона, диметилформамида, диметилсульфоксида, этанола, метанола, н-бутанола и воды, предпочтительно ацетона и воды и наиболее предпочтительно воды.
11. Способ по пп.3-10, где температура на стадии (II) составляет 80-120шС и наиболее предпочтительно составляет 100-120шС.
12. Способ по пп.3-11, где применяемый на стадиях (I) и (II) межфазный катализатор представляет собой хлорид тетрабутиламмония, бромид тетрабутиламмония, хлорид бензилтриэтиламмония, хлорид фенилтриметиламмония.
13. Способ по пп.3-12, где кислота, применяемая на стадии (III), представляет собой серную кислоту, хлорид алюминия, эфират трифторида бора, трифторуксусную кислоту, метансульфоновую кислоту, хлоруксусную кислоту, дихлоруксусную кислоту и трифторметансульфоновую кислоту, предпочтительно трифторуксусную кислоту и метансульфоновую кислоту, наиболее предпочтительно трифторуксусную кислоту.
14. Способ по пп.3-13,где азидный реагент, применяемый на стадии (III), представляет собой триметилсилилазид и азид натрия, предпочтительно азид натрия.
15. Способ по пп.3-14, где температура на стадии (III) составляет 50-90шС и наиболее предпочтительно 60-70шС.
16. Способ по пп.3-15, где количество азида натрия, применяемого на стадии (III), составляет 1-5 моль.экв., предпочтительно 1,5-4 моль.экв. относительно применяемого количества соединения формулы (2).
17. Способ по пп.3-16, где растворитель, применяемый для перекристаллизации на стадии (III), выбран из ацетона, хлороформа, метиленхлорида, этилендихлорида, диметилформамида, диметилсульфоксида и ацетонитрила, предпочтительно ацетона и наиболее предпочтительно метанола.
Текст
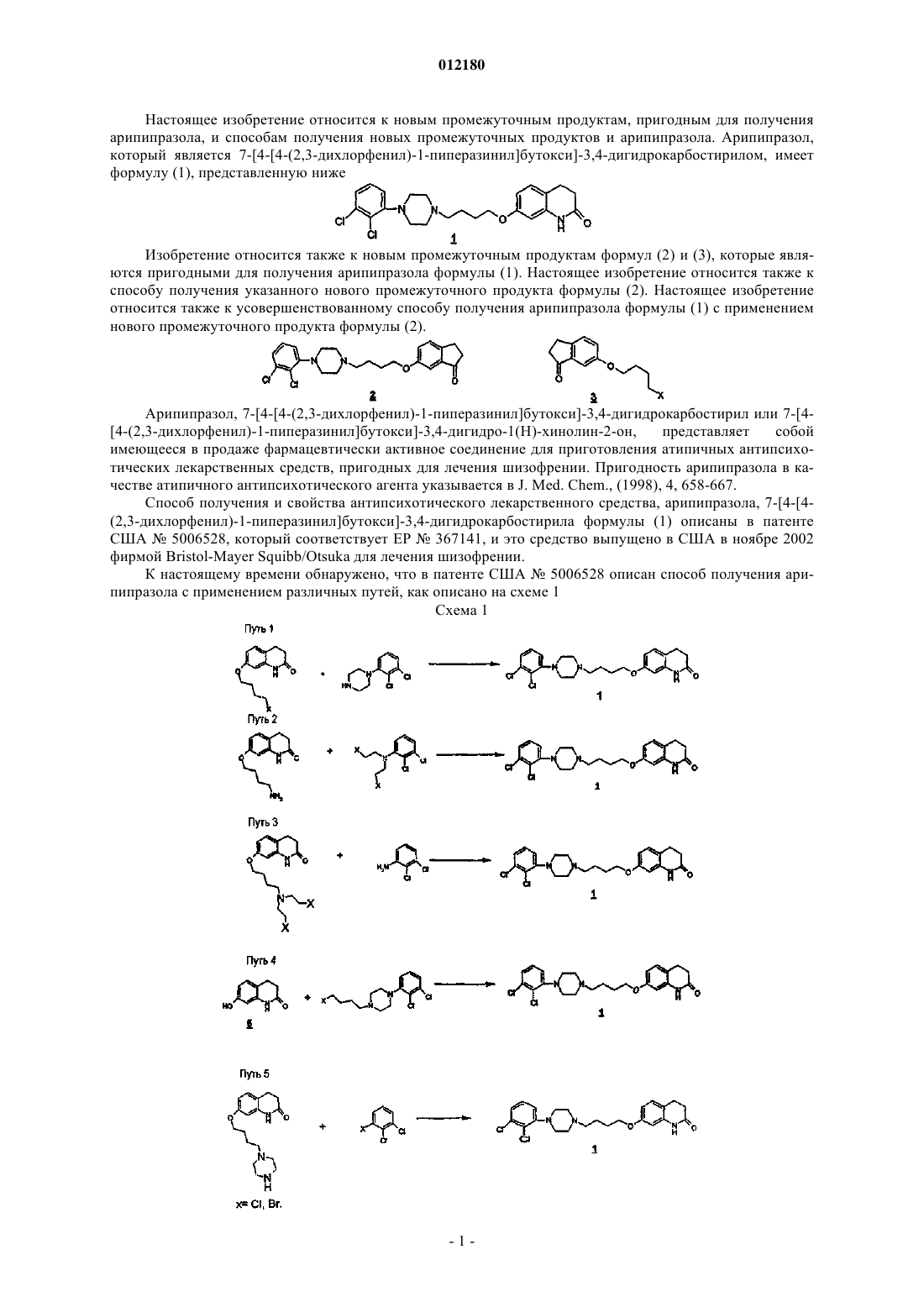
012180 Настоящее изобретение относится к новым промежуточным продуктам, пригодным для получения арипипразола, и способам получения новых промежуточных продуктов и арипипразола. Арипипразол,который является 7-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидрокарбостирилом, имеет формулу (1), представленную ниже Изобретение относится также к новым промежуточным продуктам формул (2) и (3), которые являются пригодными для получения арипипразола формулы (1). Настоящее изобретение относится также к способу получения указанного нового промежуточного продукта формулы (2). Настоящее изобретение относится также к усовершенствованному способу получения арипипразола формулы (1) с применением нового промежуточного продукта формулы (2). Арипипразол, 7-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидрокарбостирил или 7-[4[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидро-1(Н)-хинолин-2-он,представляет собой имеющееся в продаже фармацевтически активное соединение для приготовления атипичных антипсихотических лекарственных средств, пригодных для лечения шизофрении. Пригодность арипипразола в качестве атипичного антипсихотического агента указывается в J. Med. Chem., (1998), 4, 658-667. Способ получения и свойства антипсихотического лекарственного средства, арипипразола, 7-[4-[4(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидрокарбостирила формулы (1) описаны в патенте США 5006528, который соответствует ЕР 367141, и это средство выпущено в США в ноябре 2002 фирмой Bristol-Mayer Squibb/Otsuka для лечения шизофрении. К настоящему времени обнаружено, что в патенте США 5006528 описан способ получения арипипразола с применением различных путей, как описано на схеме 1 Схема 1-1 012180 Далее в патентной заявке WO2003/026659 описан способ получения арипипразола с низкой гигроскопичностью и в патентной заявке WO2004/0663162 описан способ получения арипипразола с высокой чистотой с применением того же пути, как указан в пути 1 (схема 1), и проведением реакции в среде воды. Все указанные выше способы вращаются вокруг того же самого типа промежуточных продуктов,указанных выше. Основной промежуточный продукт 7-гидрокси-2,3-дигидрокарбостирил формулы (5) получают из 3-гидроксианилина формулы (6) и 3-хлорпропионилхлорида формулы (7), как указано на схеме 2. В этом способе помимо целевого продукта формулы (5) образуется также другой, нежелательный изомер формулы (8), который очень трудно удалить, и эта примесь продолжает присутствовать в конечном лекарственном средстве, арипипразоле, и во время процесса его удаления выход целевого продукта становится очень низким. Схема 2 Это привело к необходимости дальнейших исследований с целью разработки нового пути, чтобы исключить образование примеси и максимизировать выход. Далее, для повышения терапевтической эффективности арипипразола, автор изобретения сам прилагал усилия для проведения объективных и предпринимательских исследований с целью разработки нового синтетического пути получения 7-[4-[4(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидрокарбостирила, который мог сделать возможным получение данного соединения с достаточной чистотой и экономически приемлемым выходом. Следовательно, основной задачей настоящего изобретения является создание усовершенствованного способа получения арипипразола, лишенного недостатков известных до настоящего времени способов. Еще одной задачей настоящего изобретения является предложение новых промежуточных продуктов формул (2) и (3), пригодных для получения арипипразола. Еще одной задачей настоящего изобретения является разработка способов получения новых промежуточных продуктов формул (2) и (3), пригодных для получения арипипразола. Этот способ настоящего изобретения иллюстрируется на схеме 3, показанной ниже Схема 3 В соответствии с этим настоящее изобретение предлагает новые промежуточные продукты 6-[4-[4(2,3-дихлорфенил)пиперазин-1-ил]бутокси]индан-1-он формулы (2) и 6-(4-галогенбутокси)индан-1-он формулы (3), которые являются пригодными для получения арипипразола формулы (1). Согласно другому варианту осуществления настоящего изобретения предложены способы получения новых промежуточных продуктов 6-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутокси]индан-1-она формулы (2) и 6-(4-галогенбутокси)индан-1-она формулы (3), которые являются пригодными для получения арипипразола формулы (1). В соответствии с этим настоящее изобретение предлагает способ получения нового промежуточного продукта формулы (3), используемого для получения арипипразола формулы (1), который включает в себя взаимодействие соединения формулы (11) где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в интервале 90-110 С с образованием нового промежуточного продукта формулы (3). Согласно другому варианту осуществления настоящего изобретения, предложен также способ получения другого нового промежуточного продукта формулы (2), используемого для получения арипипразола формулы (1), включающий:(I) взаимодействие соединения формулы (11) с 1,4-дигалогенбутаном формулы (12), где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90110 С с образованием нового промежуточного продукта формулы (3) и(II) взаимодействие нового соединения формулы (3), полученного на стадии (I), с 1-(2,3 дихлорфенил)пиперазином формулы (9) в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне 80-120 С с образованием нового промежуточного продукта формулы (2). Согласно еще одному варианту осуществления настоящего изобретения предложен усовершенствованный способ получения арипипразола формулы (1), включающий:(I) взаимодействие соединения формулы (11) с 1,4-дигалогенбутаном формулы (12), где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90110 С с образованием нового промежуточного продукта формулы (3),(II) взаимодействие нового соединения формулы (3), полученного на стадии (I), с соединением формулы (9) в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне 80-120 С с образованием нового промежуточного продукта формулы 2 и(III) взаимодействие образовавшегося нового соединения формулы (2) с азидом натрия или триметилсилилазидом в присутствии кислот (реакция Шмидта) при температуре между 50 и 90 С с получением соединения формулы (1). На стадии (I) можно применять основание, такое как гидрид натрия, метоксид натрия, триэтиламин,карбонат калия, бикарбонат натрия и карбонат калия, предпочтительно триэтиламин, наиболее предпочтительно карбонат калия. Применяемые растворители можно выбрать из ацетона, хлороформа, метиленхлорида, этилендихлорида, диметилформамида, диметилсульфоксида, ацетонитрила и тому подобное,причем предпочтительным растворителем является ацетон и наиболее предпочтительным является сам 1,4-дигалогенбутан в качестве растворителя. 1,4-Дигалогенбутан можно применять в количестве 1-6 эквивалентов относительно 6-гидроксииндан-1-она. Предпочтительным молярным эквивалентом является 4-6 эквивалентов. Температура реакции может быть предпочтительно между 90 и 110 С и наиболее предпочтительно между 100 и 110 С. Межфазным катализатором, применяемым на стадии (I), является хлорид тетрабутиламмония, бромид тетрабутиламмония, хлорид бензилтриэтиламмония, хлорид фенилтриметиламмония. Основание, применяемое на стадии (II), включает в себя карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, гидрид натрия, гидрид калия и триэтиламин, наиболее предпочтительно карбонат натрия. Растворители, применяемые для реакции на стадии (II), можно выбрать из ацетонитрила, ацетона, диметилформамида, диметилсульфоксида, этанола, метанола, н-бутанола и воды, предпочтительным растворителем является ацетон и вода и наиболее предпочтительной является вода. Температура для взаимодействия на стадии (II) может быть в диапазоне 80-120 С и наиболее предпочтительно в диапазоне 100-120 С. Применяемым межфазным катализатором является хлорид тетрабутиламмония,бромид тетрабутиламмония, хлорид бензилтриэтиламмония, хлорид фенилтриметиламмония. Кислоты, применяемые на стадии (III), включают в себя серную кислоту, хлорид алюминия, эфират трифторида бора, трифторуксусную кислоту, метансульфоновую кислоту, хлоруксусную кислоту, дихлоруксусную кислоту, трихлоруксусную кислоту и трифторметансульфоновую кислоту. Предпочтительными кислотами являются трифторуксусная кислота и метансульфоновая кислота, наиболее предпочтительной кислотой является трифторуксусная кислота. Температура реакции может быть в диапазоне 50-90 С и наиболее предпочтительно в диапазоне 50-70 С. Используемыми в реакции азидами являются азид натрия и триметилсилилазид, наиболее предпочтительно, азид натрия. Количество азида натрия может составлять 1-5 молей, предпочтительно 1,5-4 молярных эквивалентов относительно приме-3 012180 няемого соединения формулы (2). Кристаллизацию можно проводить с применением ацетона, метанола,толуола, этилацетата, метиленхлорида, диметилформамида, диметилсульфоксида и их смеси. Предпочтительным растворителем является ацетон и наиболее предпочтительным растворителем является метанол. Соединение 1-(2,3-дихлорфенил)пиперазин формулы (9) можно получить по методикам, обычно известным в данной области и описанным в патенте Великобритании 850663, и 6-гидроксииндан-1-он формулы (11) можно получить способом, описанным в J. Am. Chem. Soc, (1960), 21, 5202. Настоящее изобретение более конкретно описывается и объясняется в нижеследующих примерах. Должно быть понятно, однако, что настоящее изобретение не ограничивается этими примерами, которые приводятся только для иллюстрации изобретения. Следовательно, могут быть сделаны различные изменения и модификации, не выходящие за пределы объема настоящего изобретения. В этих примерах точки плавления определяли прибором PERKIN ELMER-PYRIS, ИК-спектры регистрировали на инструменте FTIR PERKIN ELMER, дифференциальную сканирующую калориметрию (ДСК) проводили с градиентом температуры 40 С/мин. Масс-спектры регистрировали прибором ЖХ-МС/АР 1 4000. Спектры ядерного магнитного резонанса (ЯМР) регистрировали прибором Брукер 400 Гц в ТМС в качестве внутреннего стандарта. Химический сдвиг указывается в 5 (м.д.). Буквы, обозначенные с, д, дд, т, кв и м, соответственно означают синглет, дублет, дублет дублета, триплет, квартет и мультиплет. Пример 1. Стадия 1. Способ получения нового промежуточного продукта 6-(4-хлорбутокси)индан-1-она формулы (3) Смесь 6-гидроксииндан-1-она формулы (11) (20 г, 0,135 моль), карбоната калия (40 г, 0,289 моль),1,4-дихлорбутана (80 мл, 0,73 моль) и бромида тетрабутиламмония (2 г) перемешивают в течение 1-3 ч при 95 С, затем отгоняют избыток 1,4-дихлорбутана, остаток разбавляют водой (80 мл). Водный слой экстрагируют этилацетатом и экстракт промывают водой, сушат и упаривают при пониженном давлении досуха. Остаток после упаривания очищают кристаллизацией с применением изопропилового простого эфира в качестве растворителя, получая при этом 29,5 г нового 6-(4-хлорбутокси)индан-1-она формулы(д), 138,11 (с), 147,84 (с), 158,49 (с), 206,75 (с). Масс-спектр: m/z (%) = [М+1] 239 (100), 197,1 (15,7), 161,2 (48,8), 149,0 (22,8), 107,1 (27,8). Стадия 2. Способ получения нового 6-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]индан-1-она формулы (2) 6-(4-Хлорбутокси)индан-1-он формулы (3), полученный способом, описанным на стадии (1) (20 г,0,0838 моль), карбонат натрия (40 г, 0,377 моль), бромид тетрабутиламмония (4 г) и гидробромид 1-(2,3 дихлорфенил)пиперазина (28 г, 0,0897 моль) в воде перемешивают в течение 4-6 ч при 95 С. После завершения реакции осажденный продукт отделяют фильтрованием и промывают водой, получая при этом продукт, который при обработке хлороформом и хлористо-водородной кислотой дает 30 г гидрохлорида 6-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]индан-1-она. Продукт при подщелачивании водным аммиаком и экстракции хлороформом с последующей дистилляцией дает 22 г нового 6-[4-[4-(2,3 дихлорфенил)-1-пиперазинил]бутокси]индан-1-она формулы (2). Т.пл.: 95,39 С. ИК (см-1): 1711,81, 1576,39, 1474,07, 1448,34, 1292,80, 1274,96, 1240,72, 994,75, 963,85, 775,15,712,75. МР: : 1,72 (м, 2 Н), 1,84 (м, 2 Н), 2,48 (т, 2 Н), 2,65 (м, 2 Н), 2,71 (т, 2 Н) 3,06 (м, 2 Н), 4,02 (т, 2 Н), 6,95(2) (15 г, 34,63 ммоль), и 90 мл трифторуксусной кислоты и порциями добавляют азид натрия (9 г, 138,46 ммоль) при 65 С. Реакционную смесь выдерживают при 65 С в течение 8-10 ч. После такого периода выдерживания реакционную смесь гасят в 100 г измельченного льда, экстрагируют хлороформом и слой хлороформа подщелачивают водным раствором аммиака, и органический слой отделяют. Органический слой сушат безводным сульфатом натрия и концентрируют при пониженном давлении, получая при этом 19 г остатка. Остаток при кристаллизации с применением смеси метанол/толуол и ацетона дает чистый арипипразол формулы 1. Т. пл.: 136,02 С. ИК (см-1): 1677,95, 1626,97, 1595,06, 1521,51, 1447,24, 1376,84, 1172,27, 960,70, 778,54. ПМР: : 1,69 (м, 2 Н), 1,81 (м, 2 Н), 2,48 (т, 2 Н), 2,62 (м, 6 Н), 2,89 (т, 2 Н), 3,07 (м, 4 Н), 3,95 (т, 2 Н),6,31 (д, 1 Н), 6,53 (дд, 1 Н), 6,96 (кв., 1 Н), 7,02 (д, 1 Н), 7,14 (м, 2 Н), 8,00 (с, 1 Н). 13 С ЯМР: : 23,34 (т), 24,49 (т), 27,20 (т), 31,00 (т), 51,26 (т), 53,20 (т), 58,10 (т), 67,81 (т), 102,25 (д),108,68 (д), 115,55 (с), 118,50 (д), 124,41 (д), 127,34 (д), 127,40 (д), 128,45 (д), 133,91 (с), 138,21 (с), 151,25 Смесь 6-гидроксииндан-1-она (100 г, 0,675 моль), карбоната калия (96 г, 0,695 моль), 1,4 дибромбутана (365 г, 1,69 моль) и бромида тетрабутиламмония в ацетоне вводят в трехгорлую круглодонную колбу, снабженную водяным конденсатором и механической мешалкой. Смесь нагревают до температуры 65 С и выдерживают при 65 С в течение девяти часов и затем из нее отгоняют растворитель, остаток разбавляют водой (400 мл). Водный слой экстрагируют этилацетатом и экстракт промывают водой, сушат и упаривают в роторном испарителе при пониженном давлении, получая при этом остаток, который при кристаллизации из изопропилового простого эфира дает 125 г 6-(4-бромбутокси)индан 1-она формулы 3. Т. пл.: 60,59 С. ИК: (см-1): 1705,61, 1610,51, 1488,19, 1294,21, 1021,54, 836,60, 558,98. ПМР: : 1,94 (м, 2 Н), 2,06 (м, 2 Н), 2,71 (м 2 Н), 3,06 (т, 2 Н), 3,47 (т, 2 Н) 4,01 (т, 2 Н), 7,17 (м, 2 Н), 7,36(д), 138,13 (с), 147,87 (с), 158,50 (с) 206,77 (с). Масс-спектр: m/z (%) = [М+1] 283,4 (55,1), 161,1 (66,1), 149,4 (40,4), 135,4 (100), 106,9 (38,2). Стадия 2. Способ получения 6-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]индан-1-она Смесь 6-(4-бромбутокси)индан-1-она, полученного способом, описанным на указанной выше стадии (1) (100 г, 0,282 моль, чистота 80%) и иодида натрия (42 г, 0,28 моль) в ацетонитриле кипятят с обратным холодильником в течение 30 мин и затем охлаждают до комнатной температуры. К смеси добавляют гидрохлорид 1-(2,3-дихлорфенил)пиперазина (68 г, 0,294 моль) и триэтиламин (77 г, 0,761 моль) и образовавшуюся смесь кипятят с обратным холодильником в течение 4-6 ч. После удаления растворителя полученный таким образом остаток растворяют в хлороформе, раствор промывают водой, обрабатывают хлороформом и хлористоводородной кислотой, получая при этом 140 г гидрохлорида 6-[4-[4-(2,3 дихлорфенил)-1-пиперазинил]бутокси]индан-1-она. Раствор его в хлороформе подщелачивают гидроксидом аммония до рН 9,5 и экстрагируют дважды хлороформом 2500 мл. Объединенный слой хлороформа концентрируют с применением роторного испарителя при пониженном давлении, получая при этом остаток, который при очистке метанолом дает 85 г 6-[4-[4-(2,3-дихлорфенил)-1 пиперазинил]бутокси]индан-1-она формулы 2, который имеет характеристики, идентичные характеристикам продукта, полученного на стадии 2 примера 1. Стадия 3. Способ получения 7-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]-3,4-дигидро-1(Н)хинолин-2-она (арипипразола) Добавляют 6-[4-[4-(2,3-дихлорфенил)-1-пиперазинил]бутокси]индан-1-он, полученный на стадии(2) (15 г, 34,63 ммоль) и 90 мл трифторуксусной кислоты и порциями при 65 С добавляют азид натрия (9-5 012180 г, 138,46 ммоль). Реакционную смесь выдерживают при 65 С в течение 8-10 ч. После выдерживания в течение указанного периода реакционную смесь гасят в 100 г измельченного льда, экстрагируют хлороформом и экстракт в хлороформе подщелачивают водным раствором аммиака и органический слой отделяют. Органический слой сушат безводным сульфатом натрия и концентрируют при пониженном давлении, получая при этом 19 г остатка, который перекристаллизовывают с применением смеси метанол/толуол и ацетона с получением чистого арипипразола формулы 1. Преимущества изобретения 1. Полученный арипипразол имеет высокую чистоту - выше, чем 99%. 2. Предотвращается образование создающей проблемы неудаляемой примеси. 3. Обеспечивается образование новых промежуточных продуктов формул 2 и 3. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Промежуточный продукт, 6-(4-галогенбутокси)индан-1-он формулы 3 предназначенный для получения арипипразола формулы 1 предназначенный для получения арипипразола формулы 1. 3. Способ получения промежуточного продукта формулы (3), предназначенного для получения арипипразола формулы (1), включающий взаимодействие соединения формулы (11) где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90-110 С с образованием промежуточного продукта формулы (3). 4. Способ получения промежуточного продукта формулы (2), предназначенного для получения арипипразола формулы 1, включающий:(I) взаимодействие соединения формулы (11) где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90-110 С с образованием промежуточного продукта формулы (3) и(II) взаимодействие соединения формулы 3, полученного на стадии (1), с соединением формулы (9) в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне-6 012180 80-120 С с получением промежуточного продукта формулы 2. 5. Способ получения арипипразола формулы 1, включающий:(I) взаимодействие соединения формулы (11) с 1,4-дигалогенбутаном формулы (12), где X представляет собой Cl или Br, в присутствии основания и растворителя при температуре в диапазоне 90110 С с образованием промежуточного продукта формулы (3),(II) взаимодействие соединения формулы (3), полученного на стадии (I), с соединением формулы(9) в присутствии основания и межфазного катализатора и растворителя при температуре в диапазоне 80120 С с образованием промежуточного продукта формулы 2 и(III) взаимодействие образовавшегося соединения формулы (2) с азидом натрия или триметилсилилазидом в присутствии кислоты при температуре в диапазоне 50-90 С с получением соединения формулы (1) и, в случае необходимости, перекристаллизацию соединения формулы (1). 6. Способ получения по пп.3-5, где на стадии (I) применяют основание, такое как гидрид натрия,метоксид натрия, триэтиламин, карбонат калия, бикарбонат натрия и карбонат натрия, предпочтительно триэтиламин, наиболее предпочтительно карбонат калия. 7. Способ получения по пп.3-6, где применяемый на стадии (I) растворитель выбран из ацетона,хлороформа, метиленхлорида, этилендихлорида, диметилформамида, диметилсульфоксида и ацетонитрила, предпочтительно ацетона и наиболее предпочтительно 1,4-дигалогенбутана. 8. Способ по пп. 3-7, где температура на стадии (I) составляет 90-110 С и наиболее предпочтительно 100-110 С. 9. Способ по пп.3-8, где применяемое на стадии (II) основание представляет собой карбонат натрия,карбонат калия, бикарбонат натрия, бикарбонат калия, гидрид натрия, гидрид калия и триэтиламин, наиболее предпочтительно карбонат натрия. 10. Способ по пп.3-9, где применяемый на стадии (II) растворитель выбран из ацетонитрила, ацетона, диметилформамида, диметилсульфоксида, этанола, метанола, н-бутанола и воды, предпочтительно ацетона и воды и наиболее предпочтительно воды. 11. Способ по пп.3-10, где температура на стадии (II) составляет 80-120 С и наиболее предпочтительно составляет 100-120 С. 12. Способ по пп.3-11, где применяемый на стадиях (I) и (II) межфазный катализатор представляет собой хлорид тетрабутиламмония, бромид тетрабутиламмония, хлорид бензилтриэтиламмония, хлорид фенилтриметиламмония. 13. Способ по пп.3-12, где кислота, применяемая на стадии (III), представляет собой серную кислоту, хлорид алюминия, эфират трифторида бора, трифторуксусную кислоту, метансульфоновую кислоту,хлоруксусную кислоту, дихлоруксусную кислоту и трифторметансульфоновую кислоту, предпочтительно трифторуксусную кислоту и метансульфоновую кислоту, наиболее предпочтительно трифторуксусную кислоту. 14. Способ по пп.3-13, где азидный реагент, применяемый на стадии (III), представляет собой триметилсилилазид и азид натрия, предпочтительно азид натрия. 15. Способ по пп.3-14, где температура на стадии (III) составляет 50-90 С и наиболее предпочтительно 60-70 С. 16. Способ по пп.3-15, где количество азида натрия, применяемого на стадии (III), составляет 1-5 моль.экв., предпочтительно 1,5-4 моль.экв. относительно применяемого количества соединения формулы(2). 17. Способ по пп.3-16, где растворитель, применяемый для перекристаллизации на стадии (III), выбран из ацетона, хлороформа, метиленхлорида, этилендихлорида, диметилформамида, диметилсульфоксида и ацетонитрила, предпочтительно ацетона и наиболее предпочтительно метанола.
МПК / Метки
МПК: C07D 215/22, C07D 295/088, C07C 49/67
Метки: получение, продукты, получения, арипипразола, промежуточные, соответствующие, способ
Код ссылки
<a href="https://eas.patents.su/8-12180-sposob-polucheniya-aripiprazola-i-sootvetstvuyushhie-promezhutochnye-produkty-i-ih-poluchenie.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения арипипразола и соответствующие промежуточные продукты и их получение</a>
Новые производные пентаэритрита, их получение и применение , а также промежуточные продукты для их синтеза

Номер патента: 1959
Опубликовано: 22.10.2001
Авторы: Хесс Ульрих, Брозиг Хольгер, Виндек Анне-Катрин
МПК: A61P 7/02, C06B 25/00, A61K 31/26...
Метки: получение, пентаэритрита, продукты, применение, новые, производные, синтеза, промежуточные, также
Формула / Реферат:
1. Соединение общей формулы I где R1, R2, R3 представляют собой одинаковые или различные CH2-ONO2, CH2-OR4 или R5, при этом, по крайней мере, один из заместителей R1, R2, R3 идентичен R5, R4 обозначает Н или C1-С6-алканоил, R5 обозначает COR6, R6 обозначает ОН, OR7, NH2, NHR7, NR72, N+R73X-, NR8, NR9R10, NR11R12 или NH-NH2, R7 обозначает неразветвленный или разветвленный C1-С6-алкил, неразветвленный или разветвленный C1-С6-алкенил, арил-,...
Способ получения пиразоло [4,3-d] пиримидин-7-онов и их промежуточные продукты

Номер патента: 3774
Опубликовано: 28.08.2003
Авторы: Данн Питер Джеймс, Леветт Филип Чарльз
МПК: C07D 487/04
Метки: 4,3-d, промежуточные, пиримидин-7-онов, получения, продукты, пиразоло, способ
Формула / Реферат:
1. Способ получения соединения формул (IA) и (IB) включающий реакцию соединения формулы (IIA) и (IIB) соответственно в присутствии -OR, где R, в случае образования (IA), представляет CH2CH3 и R, в случае образования соединения (IB), представляет CH2CH2CH3, где X представляет уходящую группу при условии, что X не является этокси 2. Способ по п.1, где X выбирают из группы, состоящей из арилсульфонилокси, C1-C4алкилсульфонилокси, нитро- или...
Новый продукт, способ и промежуточные продукты получения производных азетидина

Номер патента: 11408
Опубликовано: 27.02.2009
Авторы: Мютти Стефан, Мальпар Жоэль, Крок Вероник, Грондар Люк, Рике-Цапп Йорг, Лавинь Мишель
МПК: C07D 401/12
Метки: способ, продукт, новый, продукты, азетидина, производных, получения, промежуточные
Формула / Реферат:
1. Способ синтеза N-{1-[бис-(4-хлорфенил)метил]азетидин-3-ил}-N-(арил или гетероарил)метилсульфонамида, отличающийся тем, что осуществляют реакцию 1-[бис-(4-хлорфенил)метил]азетидин-3-ола гидробромида с N-(арил или гетероарил)метансульфонамидом в присутствии DIAD, трифенилфосфина в толуоле с образованием N-{1-[бис-(4-хлорфенил)метил]азетидин-3-ил}-N-(арил или гетероарил)метилсульфонамида, который выделяют; причем арил представляет собой...
Способ и промежуточные продукты для получения пиридин-2,3-дикарбоксилатов

Номер патента: 3058
Опубликовано: 26.12.2002
Автор: Ву Вен-Ксуе
МПК: C07C 229/30, C07D 213/79
Метки: пиридин-2,3-дикарбоксилатов, получения, способ, промежуточные, продукты
Формула / Реферат:
1. Способ получения производных пиридин-2,3-дикарбоксилата формулы I где R4 и R6, каждый независимо, представляют собой Н, С1-С6алкил, C1-С6алкенил, фенил или замещенный фенил; R5 представляет собой Н, галоген, С1-С6алкил, необязательно замещенный одной или несколькими С1-С4алкоксигруппами, С1-С6алкенил, фенил или замещенный фенил и R2 и R3, каждый независимо, представляют собой С1-С6алкил, фенил или замещенный фенил, который включает...
Бифункциональные фенилизо(тио)цианаты, способ и промежуточные продукты для их получения

Номер патента: 10872
Опубликовано: 30.12.2008
Авторы: Хампрехт Герхард, Гётц Норберт, Загассер Инго, Зайтц Вернер, Кайл Михаэль, Вольф Бернд, Райнхард Роберт, Пуль Михаэль
МПК: C07C 303/34, C07C 307/06
Метки: продукты, бифункциональные, фенилизо(тио)цианаты, промежуточные, получения, способ
Формула / Реферат:
1. Способ получения фенилизо(тио)цианатов общей формулы I где переменные имеют следующие значения: W означает кислород или серу, Ar означает фенил, который может быть замещен один или несколько раз галогеном; А означает NR1R2, где R1 и R2 означают независимо друг от друга С1-C6-алкил, С2-C6-алкенил или С2-C6-алкинил, либо R1 и R2 образуют вместе насыщенный 6-членный азотный гетероцикл, который может быть замещен С1-С4-алкилом, отличающийся тем,...
Предыдущий патент: Промышленный способ получения высокочистого диарилкарбоната, высокочистый дифенилкарбонат и установка для получения высокочистого диарилкарбоната
Следующий патент: Соединения тиадиазола и их применение
Случайный патент: Тренировочное устройство универсальное