Антагонисты гонадотропин-высвобождающего фактора.
Номер патента: 828
Опубликовано: 24.04.2000
Авторы: Чу Лин, Виврэтт Мэттью Дж., Гаулет Марк, Эштон Уоллес Т., Фишер Майкл Х.





























Формула / Реферат
1. Соединение формулы

где А представляет C1-C6 алкил, замещенный C1-C6 алкил, С3-С7 циклоалкил, замещенный С3-С7 циклоалкил, С3-C6 алкенил, замещенный С3-С6 алкенил, С3-С6 алкинил, замещенный С3-С6 алкинил, C1-C6 алкокси или C0-C5алкил-S(O)n-С0-С5 алкил, C0-C5 алкил-О-С0-С5алкил, C0-C5 алкил-NR18-С0-С5 алкил, где R18 и C0-C5 алкил, взятые вместе, могут образовывать кольцо

R0 представляет водород, C1-С6 алкил, замещенный C1-C6 алкил, где заместители определены ниже; арил, замещенный арил, аралкил или замещенный аралкил, где заместители являются такими, как они определены ниже для R3, R4 и R5;
R1 представляет

где Y представляет В, С или связь;
B представляет О, S(O)n, C(O), NR18 или C(R11R12)p;
С представляет B(CH2)p-;
R2 представляет водород, C1-C6 алкил, замещенный C1-C6 алкил, аралкил, замещенный аралкил, арил, замещенный арил, алкил-OR11, C1-C6(NR11R12), C1-С6(CONR11R12) или С(NR11R12)NH;
R2 и А, взятые вместе, образуют кольцо из 5-7 атомов;
R3, R4 и R5 независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, С2-С6 алкенил, замещенный С2-С6 алкенил, CN, нитро, C1-С3 перфторалкил, C1-С3 перфторалкокси, арил, замещенный арил, аралкил, замещенный аралкил, R11O(СН2)p-, R11С(O)O(СН2)p-, R11OC(O)(CH2)p-, -(CH2)pS(O)nR17-, (CH2)pC(O)NR11R12 или галоген, где R17 представляет водород, C1-C6алкил, С1-С3 перфторалкил, арил или замещенный арил;
R3 и R4, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов углерода или гетероциклическое кольцо, содержащее 1-3 гетероатомов, выбранных из N, О и S;
R6 представляет водород, C1-C6 алкил, замещенный C1-C6 алкил, арил, замещенный арил, C1-С3 перфторалкил, CN, NO2, галоген, R11O(CH2)p-, NR21C(O)R20, NR21C(O)NR20R21 или SOnR20;
R7 представляет водород, C1-C6 алкил или замещенный C1-C6 алкил, при условии, что, если Х является водородом или галогеном, то R7 отсутствует;
R8 представляет C(O)OR20, С(O)NR20R21, NR20R21, C(O)R20, NR21C(O)R20, NR21C(O)NR20R21, NR20S(O)2R21, NR21S(O)2NR20R21, OC(O)R20, OC(O)NR20R21, OR20, SOnR20, S(O)nNR20R21, гетероциклическое кольцо или бициклическое гетероциклическое кольцо, имеющее 1-4 гетероатомов, выбранных из N, O и S, которые могут быть необязательно замещены R3, R4 и R5, C1-C6 алкил или замещенный C1-C6 алкил; или
R7 и R8, взятые вместе, образуют гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, O и S, которые могут быть необязательно замещены R3, R4 и R5;
R9 и R9a независимо представляют водород, С1=С6алкил, замещенный C1-C6алкил, арил, замещенный арил, аралкил или замещенный аралкил, если m=0; или
R9 и R9a, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или  если mь0; или
если mь0; или
R9 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов, если mь0; или
R10 и R10a независимо представляют водород, C1-C6 алкил, замещенный C1-С6 алкил, арил, замещенный арил, аралкил или замещенный аралкил; или
R10 и R10a, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или 
R9 и R10, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или гетероциклическое кольцо, содержащее один или несколько гетероатомов, если m=0;или
R9 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов, если mь0; или.
R10 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов; или
R10 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов; или
R11 и R12 независимо представляют водород C1-C6 алкил, замещенный C1-C6 алкил, арил, замещенный арил, аралкил, замещенный аралкил, карбоциклическое кольцо с 3-7 атомов углерода или замещенное карбоциклическое кольцо, содержащее 3-7 атомов;
R11 и R12, взятые вместе, могут образовывать необязательно замещенное кольцо с 3-7 атомами;
R13 представляет водород, ОН, NR7R8, NR11SO2(C1-C6 алкил), NR11SO2 (замещенный C1-C6 алкил), NR11SO2 (арил), NR11SO2 (замещенный арил), NR11SO2 (C1-С3 перфторалкил); SO2NR11 (C1-C6алкил), SO2NR11 (замещенный C1-C6 алкил), SO2NR11 (арил), SO2NR11 (замещенный арил), SO2NR11 (С1-С3-перфторалкил), SO2NR11(С(O)C1-C6 алкил); SO2NR11(С(O)-замещенный C1-C6 алкил); SO2NR11С(O) арил);
SO2NR11(С(O)-замещенный арил); S(O)n(C1-C6алкил); S(O)n (замещенный C1-C6алкил), S(O)n(арил), S(O)n(замещенный арил), C1-С3 перфторалкил, C1-С3перфторалкокси, C1-C6алкокси, замещенный C1-С6 алкокси, СООН, галоген, NO2 или CN;
R14 и R15 независимо представляют водород, C1-C6 алкил, замещенный C1-С6 алкил, С2-С6 алкенил, замещенный С2-С6 алкенил, CN, нитро, C1-С3 перфторалкил, C1-С3 перфторалкокси, арил, замещенный арил, аралкил, замещенный аралкил, R11O(CH2)p-, R11C(O)O(СН2)p-, R11OC(O)(CH2)p-, -(CH2)pS(O)nR17, -(СН2)pС(O)NR11R12 или галоген, где R17 представляет водород, C1-C6 алкил, C1-С3 перфторалкил, арил или замещенный арил;
R16 представляет водород, C1-C6алкил, замещенный C1-C6алкил или N(R11R12);
R18 представляет водород, C1-C6алкил, замещенный C1-C6алкил, C(O)OR11, C(O)NR11R12, C(O)R11, S(O)nR11;
R20 и R21 независимо представляют водород, C1-С6 алкил, замещенный C1-C6 алкил, арил, замещенный арил, аралкил, замещенный аралкил, карбоциклическое кольцо, содержащее 3-7 атомов, замещенное карбоциклическое кольцо, содержащее 3-7 атомов, гетероциклическое кольцо или бициклическое гетероциклическое кольцо с 1-4 гетероатомами, выбранными из N, O и S, которые могут быть необязательно замещены R3, R4 и R5; C1-C6алкил, замещенный гетероциклическим кольцом или бициклическим гетероциклическим кольцом с 1-4 гетероатомами, выбранными из N, 0 и S, которые могут быть необязательно замещены R3, R4 и R5;
R20 и R21, взятые вместе, образуют необязательно замещенное кольцо из 3-7 атомов;
Х представляет N, O, S(O)n, C(O), (CR11R12)p, простую связь с R8, С2-С6 алкенил, замещенный С2-С6 алкенил, С2-С6 алкинил или замещенный С2-С6 алкинил, причем, если Х представляет O,S(O)n, С(O) или CR11R12, то может присутствовать только R8;
m=0-3;
n=0-2;
р=0-4;
где алкильные, циклоалкильные, алкенильные и алкинильные заместители выбирают из группы, включающей C1-C6алкил, С3-С7циклоалкил, арил, замещенный арил, аралкил, замещенный аралкил, гидрокси, оксо, циано, C1-С6алкокси, фтор, C(O)OR11, арил C1-С3алкокси, замещенный арил C1-С3 алкокси, а арильные заместители являются такими, как они были определены для R3, R4 и R5;
или их фармацевтически приемлемые аддитивные соли и/или гидраты, или, когда необходимо, их геометрические или оптические изомеры, или их рацемическая смесь.
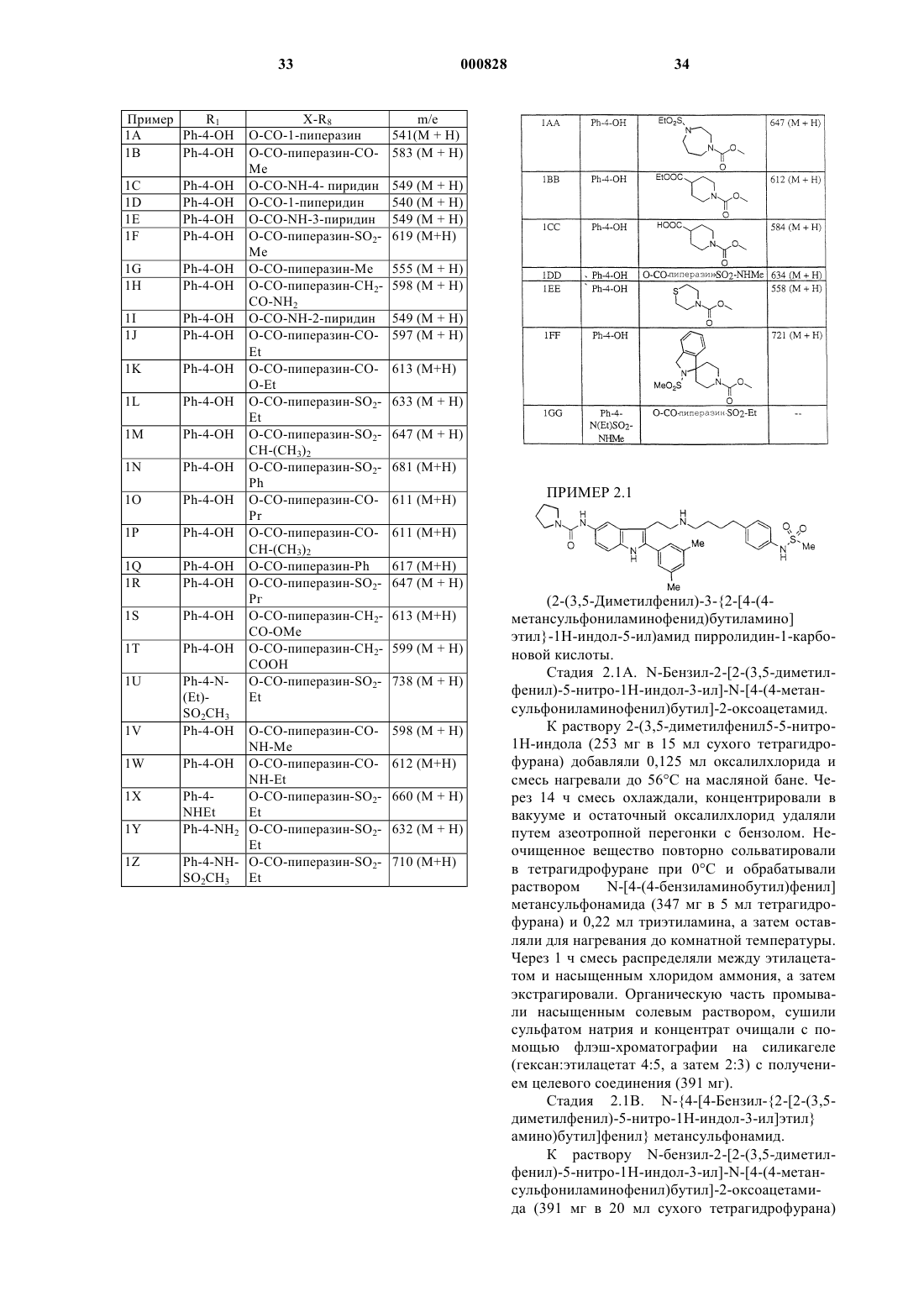
2. Соединение по п.1 формулы

где R1 и X-R8 являются такими, как определены в нижеследующей таблице


или их фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
3. Соединение по п.1 формулы

где X-R7R8 является таким, как он определен в нижеследующей таблице


или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
4. Соединение по п.1 формулы

где X-R7R8 является таким, как он определен в нижеследующей таблице


или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
5. Соединение по п.1 формулы

где X-R8 является таким, как он определен в нижеследующей таблице


или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
6. Соединение по п.1 формулы

или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
7. Соединение по п.1 формулы

где X-R8 является таким, как он определен в нижеследующей таблице

8. Соединение по п.1 формулы

где X-R7R8 является таким, как он определен в нижеследующей таблице

или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
9. Соединение по п.1 формулы

где X-R7R8 является таким, как он определен в нижеследующей таблице


или его фармацевтически приемлемая аддитивная соль и/или гидрат, или, если это приемлемо, его геометрический или оптический изомер или их рацемическая смесь.
10. Фармацевтическая композиция, которая содержит эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
11. Способ ингибирования гонадотропин-высвобождающего гормона у индивидуума, нуждающегося в этом, предусматривающий введение эффективного количества соединения по п.1 индивидууму, страдающему расстройством, ассоциированным с гонодотропин-высвобождающим гормоном.
12. Способ по п.11, где расстройством, ассоциированным с гонадотропин-высвобождающим гормоном, является состояние, ассоциированное с половым гормоном.
13. Способ по п.11, где расстройством, ассоциированным с гонадотропин-высвобождающим гормоном, является гормональнозависимый рак, доброкачественная гипертрофия предстательной железы или миома матки.
14. Способ по п.13, где гормональнозависимым раком является рак предстательной железы, рак матки, рак молочной железы и гонадотропные аденомы гипофиза.
15. Способ по п.12, где состояние, ассоциированное с половыми гормонами, выбирают из группы, включающей эндометриоз, поликистоз яичника, фиброз матки и преждевременное половое развитшх.
16. Способ предупреждения беременности у индивидуума, нуждающегося в этом, предусматривающий введение эффективного количества соединения по п.1.
17. Способ лечения системной красной волчанки у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения по п.1.
18. Способ лечения синдрома раздражения кишечника у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения по п.1.
19. Способ лечения предменструального синдрома у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения по п.1.
20. Способ лечения гирсутизма у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения по п.1.
21. Способ лечения недостаточного роста или дефицита гормона роста у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения, стимулирующего эндогенное продуцирование гормона роста, и эффективное количество соединения по п.1.
22. Способ лечения расстройства сна, такого как приступы апноэ во сне, у индивидуума, нуждающегося в таком лечении, предусматривающий введение эффективного количества соединения по п.1.
23. Фармацевтическая композиция, которая содержит инертный носитель и эффективное количество соединения, стимулирующего эндогенное продуцирование или высвобождение гормона роста, в комбинации с соединением по п.1.
24. Фармацевтическая композиция, изготовленная путем смешивания соединения по п.1 и фармацевтически приемлемого носителя.
25. Способ получения фармацевтической композиции, предусматривающий смешивание соединения по п.1 и фармацевтически приемлемого носителя.
Текст
(GnRH), называемый также фактором высвобождения лютеинизирующего гормона (LHRH),представляет собой декапептид, который играет ключевую роль в репродукции человека. Этот гормон высвобождается из гипоталамуса и воздействует на гипофиз, стимулируя биосинтез и секрецию лютеинизирующего гормона (LH) и фоллирулостимулирующего гормона (FSH). LH,высвобождаемый из гипофиза, является ответственным, в первую очередь, за регуляцию продуцирования половых стероидных гормонов у обоих полов, а FSH регулирует сперматогенез у мужчин и развитие фоллокулов у женщин. Было показано, что GnRH-агонисты и антагонисты являются эффективными при лечении некоторых состояний, которые требуют ингибирования высвобождения LH/FSH. В частности, было показано, что терапия на основе GnRH является эффективной в лечении эндометриоза, фибромы матки, поликистоза яичников, преждевременного полового созревания и некоторых опухолей,ассоциируемых с половыми стероидными гормонами, главным образом, рака предстательной железы, рака молочной железы и рака яичника.GnRH-агонисты и -антагонисты были также использованы в различных методах оплодотворения и были исследованы как возможные контрацептивы как для мужчин, так и для женщин. Было также обнаружено, что они могут быть использованы для лечения гонадотропных аденом гипофиза, расстройств сна, таких как приступы апноэ (остановки дыхания) во время сна,синдрома раздражения кишечника, предменструального синдрома, доброкачественной гиперплазии предстательной железы, гирсутизма как следствие терапии путем введения гормона роста при дефиците гормона роста у детей и мышиных моделей волчанки. Соединения настоящего изобретения могут быть также использованы в комбинации с бифосфонатными агентами (бифосфоновыми кислотами) или другими агентами, такими как, стимуляторы секреции гормона роста, например, МК-0677, для лечения и предупреждения нарушений кальциевого обмена, фосфатного обмена и костного метаболизма, а в частности, для предупреждения разрежения кости в процессе терапии с использованием GnRH-антагониста, а также в комбинации с эстрогенами, прогестеронами, антиэстрогенами, антипрогестинами и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотропных симптомов, таких как приступообразное ощущение жара ("приливы") во время терапии с использованием GnRHантагониста. Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как финастерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4, 7-диметил-4-аза-5 000828 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-5-андростан и 3-оксо-4-аза 4,7-диметил-16-(фенокси)-5-андростан, описанным в WO 93/23420 и WO 95/11254; двойными ингибиторами 5-редуктазы 1 и 5 редуктазы 2, таких как 3-оксо-4-аза-17-(2,5 трифторметилфенилкарбамоил)-5-андростан,описанными в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона; и альфа-1-блокаторами, такими как празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза и прекращения роста с наступлением половой зрелости. Существующие в настоящее время GnRHантагонисты представляют собой GnRHподобные декапептиды, которые из-за их очень низкой пероральной активности обычно вводят внутривенно или подкожно. Они имеют аминокислотные замещения, обычно, в положениях 1,2, 3, 6 и 10. Непептидные GnRH-антагонисты имеют,возможно, преимущества в отношении пероральной активности. Непептидные GnRHантагонисты были описаны в Европейской заявке 0219292, в работе De, В. et al., J. Med. Chem.,32, 2036-2038 (1989), в WO 95/28405, WO 95/29900 и ЕР 0679642 (все Takeda ChemicalIndusrties, Ltd.). В нижеследующих патентах и патентных заявках описаны замещенные индолы, известные специалистам. В патенте США 5030640 описаны альфа-гетероциклические этаноламиноалкилиндолы, которые являются сильнодействующими -агонистами. В патенте США 4544663 описаны индоламиновые производные,которые, как предполагается, могут быть использованы в качестве противозаточных средств, применяемых мужчинами. В WО 90/05721 описаны альфа-амино-индол-3 уксусные кислоты, которые могут быть использованы в качестве средств против диабета, ожирения и атеросклероза. В патенте Франции 2181559 раскрываются индоловые производные, обладающие седативной, нейролептической, аналгезирующей, гипотензивной, антисеротониновой и адренолитической активностью. В патенте Бельгии 879381 описаны 3 аминоалкил-1 Н-индол-5-тиоамидные и карбоксамидные производные, используемые в качестве сердечнососудистых средств для лечения гипертензии, болезни Рейно и мигрени. 3 Краткое описание изобретения Настоящее изобретение относится к соединениям, которые являются непептидными антагонистами GnRH и которые могут быть использованы для лечения ряда состояний, ассоциированных с половыми гормонами, как у женщин, так и мужчин; к способам их получения; а также к способам и фармацевтическим композициям, содержащим указанные соединения и используемым для введения млекопитающим. Поскольку соединения настоящего изобретения являются антагонистами гормона GnRH,то они могут быть использованы для лечения ряда ассоциированных с половыми гормонами состояний, как у женщин, так и мужчин. Такими состояниями являются эндометриоз, фиброма матки, поликистоз яичников, гирсутизм, преждевременное половое созревание и некоторые гормонально-зависимые опухоли, главным образом рак предстательной железы, рак молочной железы и рак яичника, гонадотропные аденомы гипофиза, расстройство сна, такое как приступы апноэ (остановки дыхания) во время сна, синдром раздражения кишечника, предменструальный синдром, доброкачественная гиперплазия предстательной железы. Указанные соединения могут быть также использованы для лечения состояний, связанных с дефицитом гормона роста и при низком росте, а также для лечения системной красной волчанки. Кроме того, соединения настоящего изобретения могут быть использованы для in vitro-оплодотворения и в качестве противозачаточных средств. Эти соединения могут быть также использованы в комбинации с андрогенами, эстрогенами, прогестеронами, антиэстрогенами и антипрогестогенами для лечения эндометриоза и фибромы, и для контрацепции. Они могут быть также использованы в комбинации с тестостероном и другими андрогенами или антипрогестогенами в качестве контрацептивов для мужчин. Эти соединения могут быть также использованы в комбинации с ингибитором ангиотензинконвертирующего фермента, таким как аналаприл или каптоприл; антагонистом рецептора ангиотензина II, таким как лосартан; или с ингибитором ренина для лечения фибромы матки. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с бифосфонатами (бифосфоновыми кислотами) и другими агентами, для лечения и предупреждения нарушений кальциевого обмена, фосфатного обмена и костного метаболизма, а в частности, для предупреждения разрежения кости в процессе терапии с использованием GnRHантагониста и в комбинации с эстрогенами, прогестеронами и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотропных симптомов, таких как приступообразное ощущение жара ("приливы") во время терапии с использованием GnRH-антагониста. 4 Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как финастерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4,7-диметил-4-аза-5 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-5-андростан и 3-оксо-4-аза 4,7-диметил-16-(фенокси)-5-андростан, описанным в WO 93/23420 и WO 95/11254; двойными ингибиторами 5-редуктазы и 1 и 5 редуктазы 2, таких как 3-оксо-4-аза-17-(2,5 трифтор-метилфенилкарбамоил)-5-андростан,описанными в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона, и альфа-1-блокаторами, такими как празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза и прекращения роста с наступлением половой зрелости. Подробное описание изобретения Настоящее изобретение относится к соединениям общей формулы где А представляет C1-C6 алкил, замещенный C1C6 алкил, С 3-С 7-циклоалкил, замещенный С 3 С 7 циклоалкил, С 3-С 6 алкенил, замещенный С 3 С 6 алкенил, С 3-С 6 алкинил, замещенный С 3-С 6 алкинил, C1-С 6 алкокси или С 0-С 5 алкил-S(O)n-C0C5 алкил, С 0-С 5 алкил-О-С 0-С 5 алкил, C0-C5 алкилNR18-C0-C5 алкил, где R18 и C0-C5 алкил взятые вместе, могут образовывать кольцо или простую связь.R0 представляет водород, C1-С 6 алкил, замещенный C1-С 6 алкил, где заместители определены ниже; арил, замещенный арил, аралкил или замещенный аралкил, где заместители являются такими, как они определены ниже дляR2 и А, взятые вместе, образуют кольцо из 5-7 атомов;R3, R4 и R5 независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, С 2 С 6 алкенил, замещенный C2-C6 алкенил, CN, нитро, C1-С 3 перфторалкил, C1-С 3 перфторалкокси,арил, замещенный арил, аралкил, замещенный аралкил,R11 О(СН 2)p-,R11 С(O)O(СН 2)p-,R11OC(O)(CH2)p-,-(CH2)pS(O)nR17,-(СН 2)pС(О)NR11R12 или галоген, где R17 представляет водород, C1-C6 алкил, C1-С 3 перфторалкил, арил или замещенный арил;R3 и R4, взятые вместе, образуют карбоциклическое кольцо из 3-7 атомов углерода или гетероциклическое кольцо, содержащее 1-3 гетероатомов, выбранных из N, O и S;R7 представляет водород, C1-C6 алкил или замещенный C1-C6 алкил, при условии, что, если Х является водородом или галогеном, то R7 отсутствует;R8 представляет C(O)OR20, C(O)NR20R21,NR20R21,C(O)R20,NR21C(O)R20,NR21C(O)NR20R21,NR20S(O)2R21,NR21S(O)2NR20R21, OC(O)R20, ОС(O)NR20R21,OR20, SOnR20, S(O)nNR20R21, гетероциклическое кольцо или бициклическое гетероциклическое кольцо, имеющее 1-4 гетероатомов, выбранных из N, O и S, которые могут быть необязательно замещены R3, R4 и R5; C1-C6 алкил или замещенный C1-C6 алкил; или R7 и R8, взятые вместе, образуют гетероциклическое кольцо, содержащее один или несколько гетероатомов, выбранных из N, O и S,которые могут быть необязательно замещеныR9 и R9a независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, арил,замещенный арил, аралкил или замещенный аралкил, если m0; илиR9 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода и один или несколько гетероатомов,если m0; илиR9 и R10, взятые вместе, образуют карбоциклическое кольцо, содержащее 3-7 атомов углерода, или гетероциклическое кольцо, содержащее один или несколько гетероатомов,если m0; илиR9 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов,если m0; илиR10 и R2, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов;R10 и А, взятые вместе, образуют гетероциклическое кольцо, содержащее 3-7 атомов углерода, и один или несколько гетероатомов; илиR11 и R12 независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, арил,замещенный арил, аралкил, замещенный аралкил, карбоциклическое кольцо с 3-7 атомов углерода или замещенное карбоциклическое кольцо, содержащее 3-7 атомов;R11 и R12, взятые вместе, могут образовывать необязательно замещенное кольцо с 3-7 атомами;R14 и R15 независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, С 2C6 алкенил, замещенный С 2-С 6 алкенил, CN, нитро, C1-С 3 перфторалкил, C1-С 3 перфторалкокси,арил, замещенный арил, аралкил, замещенный аралкил,R11 О(СН 2)p-,R11 С(O)O(СН 2)p-,R11 ОС(О)(СН 2)p-,-(CH2)pS(O)nR17,-(СН 2)pС(O)NR11R12 или галоген,где R17 представляет водород, C1-C6 алкил, C1-С 3 перфторалкил, арил или замещенный арил;R20 и R21 независимо представляют водород, C1-C6 алкил, замещенный C1-C6 алкил, арил,замещенный арил, аралкил, замещенный арал 7 кил, карбоциклическое кольцо, содержащее 3-7 атомов, замещенное карбоциклическое кольцо,содержащее 3-7 атомов, гетероциклическое кольцо или бициклическое гетероциклическое кольцо с 1-4 гетероатомами, выбранными из N,O и S, которые могут быть необязательно замещены R3, R4 и R5; C1-C6 алкил, замещенный гетероциклическим кольцом или бициклическим гетероциклическим кольцом с 1-4 гетероатомами, выбранными из N, O и S, которые могут быть необязательно замещены R3, R4 и R5;R20 и R21, взятые вместе, образуют необязательно замещенное кольцо из 3-7 атомов; Х представляет N, O, S(O)n, C(O),(CR11R12)p, простую связь с R8, С 2-С 6 алкенил,замещенный С 2-С 6 алкенил, C2-C6 алкинил или замещенный С 2-С 6 алкинил, причем, если Х представляет O, S(O)n, С(O) или CR11R12, то может присутствовать только R8;n=0-2; р=0-4; где алкильные, циклоалкильные, алкенильные и алкинильные заместители выбирают из группы, включающей C1-C6 алкил, С 3 С 7 циклоалкил, арил, замещенный арил, аралкил,замещенный аралкил, гидрокси, оксо, циано, C1C6 алкокси, фтор, C(O)OR11, арил C1-C3 алкокси,замещенный арил C1-C3 алкокси, а арильные заместители являются такими, как они были определены для R3, R4 и R5; или их фармацевтически приемлемые аддитивные соли и/или гидраты, либо геометрические или оптические изомеры, если они являются подходящими, или их рацемическая смесь. Если это не оговорено особо, то в описании изобретения и в формуле изобретения будут использованы следующие определения: если какая-либо группа (например, арил,гетероцикл, R1 и т.п.) встречается более одного раза в любом радикале или в формуле I, то его определение в каждом случае является независимым от определения в любом другом случае. Кроме того, комбинации заместителей и/или радикалов являются допустимыми только, если эти комбинации приводят к получению стабильных соединений. Используемый термин "алкил" означает разветвленные или прямые насыщенные алифатические углеводородные группы, имеющие конкретно определенное число атомов углерода,например метил (Me), этил (Et), пропил, бутил,пентил, гексил, гептил, октил, нонанил, децил,ундецил, додецил и их изомеры, такие как изопропил (i-Pr), изобутил(i-Bu), втор-бутил (s-Bu),трет-бутил (t-Bu), изопентан, изогексан и т.п. Термин "арил" включает фенил и нафтил. Предпочтительным арилом является фенил. Термин "галоген" или "галогеновая группа" включает фтор, хлор, бром и йод. Термин "гетероцикл" или "гетероциклическое кольцо" относится ко всем неароматиче 000828 8 ским, гетероциклическим кольцам, содержащим 3-7 атомов и 1-3 гетероатома, выбранных из N,O и S, таким как оксиран, оксетан, тетрагидрофуран, тетрагидропиран, пирролидин, пиперидин, тетрагидропиридин, тетрагидропиримидин,тетрагидротиофен, тетрагидротиопиран, морфолин, гидантоин, валеролактам, пирролидинон и т.п. Используемый в настоящем описании термин "композиция" означает продукт, содержащий конкретные ингредиенты в конкретных количествах, а также любой продукт, который получают непосредственно или опосредованно из комбинации конкретных ингредиентов в конкретных количествах. Кроме того, каждому специалисту хорошо известно, что многие вышеуказанные гетероциклические группы могут существовать в более чем одной таутомерной форме. При этом имеется в виду, что все указанные таутомеры входят в объем настоящего изобретения. Настоящее изобретение также включает оптические изомерные формы, а именно смеси энантиомеров, например рацематы, или диастереомеров, а также отдельные энантиомеры или диастереомеры настоящего изобретения. Эти отдельные энантиомеры обычно обозначаются в соответствии с их оптическим вращением, а именно, символами (+) и (-), и (L) и (D), (1) и (d) или их комбинацией. Эти изомеры могут быть также обозначены в соответствии с их абсолютной пространственной конфигурацией символами (S) и (R), которые означают левовращающий и правовращающий изомер соответственно. Отдельные оптические изомеры могут быть получены с использованием стандартных методов разделения, например, путем обработки соответствующей оптически активной кислотой,разделения диастереомеров, а затем выделения нужного изомера. Кроме того, отдельные оптические изомеры могут быть получены методами асимметрического синтеза. Кроме того, данная химическая формула или название охватывают фармацевтически приемлемые аддитивные соли этих соединений и их сольваты, такие как гидраты. Хотя соединения настоящего изобретения сами по себе являются эффективными, однако,для большей стабильности, для облегчения их кристаллизации, повышения растворимости и придания других желаемых свойств, они могут быть изготовлены и введены в виде их фармацевтически приемлемых аддитивных солей. Соединения настоящего изобретения могут быть введены в форме фармацевтически приемлемых солей. Используемый в настоящем описании термин "фармацевтически приемлемая соль" включает все приемлемые соли. Примерами таких солей являются соли, которые могут быть образованы хлористо-водородной, азотной, серной, фосфорной, муравьиной, уксусной,трифторуксусной, пропионовой, малеиновой, 9 янтарной, малоновой, метансульфоновой кислотой и т.п., и которые могут быть использованы в качестве лекарственной формы для модификации свойств растворимости или гидролизных свойств, либо они могут быть использованы в препаратах пролонгированного действия или пролекарственных препаратах. В зависимости от конкретных функциональных групп, присутствующих в соединении настоящего изобретения, фармацевтически приемлемые соли соединений настоящего изобретения включают соли,образованные катионами, такими как натрий,калий, алюминий, кальций, литий, магний,цинк; а также основаниями, такими как аммиак,этилендиамин, N-метилглутамин, лизин, аргинин,орнитин,холин,N,N'-дибензилэтилендиамин, хлорпрокаин, диэтаноламин, прокаин, N-бензилфенэтиламин, диэтиламин, пиперазин, трис(гидроксиметил)аминометан и гидроксидтетраметиламмония. Эти соли могут быть получены стандартными способами, например,посредством реакции свободного основания с подходящим органическим или неорганическим основанием, либо альтернативно, посредством реакции свободного основания с подходящей органической или неорганической кислотой. Кроме того, в случае присутствия кислотной (-СООН) или спиртовой группы, могут быть использованы фармацевтически приемлемые сложные эфиры, например метил, этил, бутил,ацетат, малеат, пивалоилоксиметил и т.п., которые, как известно, модифицируют свойства растворимости или гидролизные свойства для изготовления препаратов с пролонгированным высвобождением лекарственного средства или пролекарственных препаратов. Соединения настоящего изобретения могут иметь хиральные центры, отличающиеся от тех центров, стереохимия которых описана формулой I, а поэтому они могут существовать в виде рацематов, рацемических смесей и в виде отдельных энантиомеров или диастереомеров, где все указанные изомерные формы, а также их смеси входят в объем настоящего изобретения. Кроме того, некоторые из кристаллических форм соединений настоящего изобретения могут существовать как полиморфы, и, как таковые, они входят в объем настоящего изобретения. Более того, некоторые соединения настоящего изобретения могут образовывать сольваты с водой или с обычно используемыми органическими растворителями. Эти сольваты также входят в объем настоящего изобретения. Соединения настоящего изобретения были получены в соответствии с нижеследующими реакционными схемами. Все заместители являются такими, как они были определены выше,если это не оговорено особо. Схема А Реакционная схема А. Как показано в схеме А, после обработки триптамина (1) N-карбоксифталимидом в инертном органическом растворителе, таком как тетрагидрофуран, при температуре 20-65 С, а предпочтительно 65 С, в течение 12-48 ч получают соответствующееNфталимидотриптамин (2) может быть затем модифицирован путем обработки бромирующим агентом, таким как пербромид гидробромида пиридиния, гидротрибромид пирролидона или т.п., в инертном органическом растворителе,таком как тетрагидрофуран, метиленхлорид,хлороформ или их смеси, при 0-25 С в течение периода времени от 30 мин до 4 ч с получением 2-бромтриптамина (3). Бромид (3) может быть подвергнут реакции с арилбороновой кислотойScr. 1986, 26, 311-314), с палладиевым (O) катализатором, слабой кислотой, такой как водный карбонат натрия или т.п., и с хлоридным соединением, таким как хлорид лития, в инертном растворителе, таком как толуол, бензол, этанол,пропанол или их смеси, при температуре 25100 С, предпочтительно 80 С, в течение 1-6 ч с получением 2-арилтриптаминового производного (4). И наконец, фталимидогруппа может быть удалена путем обработки соединения (4) вод 11 ным гидразином в инертном растворителе, таком как метанол или этанол при температуре 025 С в течение 4-24 ч с получением триптамина Реакционная схема В. Как показано в реакционной схеме В, 2 арилтриптамин может быть конденсирован с карбоновой кислотой типа (6) с использованием сочетающего реагента гидрохлорида 1-(3 диметиламинопропил)-3-этилкарбодиимидаNметилморфолин (МММ), триэтиламина или т.п.,в инертном органическом растворителе, таком как метиленхлорид, хлороформ, диметилформамид, или их смеси, примерно или при комнатной температуре в течение 3-24 ч с получением соответствующего амидного производного (7). Альтернативно, 2-арилтриптамин (5) может быть обработан активным сложным эфиром или хлорангидридом типа (8) в инертном органическом растворителе, таком как метиленхлорид, хлороформ, тетрагидрофуран, диэтиловый эфир или т.п., и третичным аминовым основанием, таким как триэтиламин, диизопропилэтиламин, пиридин или т.п. при температуре 025 С в течение периода времени от 30 мин до 4 ч с получением соединения (7). Схема С 12 Реакционная схема С Как показано в реакционной схеме С, карбониламид соединения (7) может быть восстановлен путем обработки бораном, алюмогидридом лития или эквивалентными исходными гидридными соединениями в инертном органическом растворителе, таком как тетрагидрофуран,диэтиловый эфир, 1,4-диоксан или т.п., при 25100 С, предпочтительно при 65 С, в течение 1-8 ч с получением соответствующего аминного соединения (9). Схема D Реакционная схема D. Как показано в реакционной схеме D, 2 арилтриптамин (5) может быть модифицирован путем обработки альдегидом или кетоном типа(10) в присутствии слабой кислоты, такой как трифторуксусная кислота (TFA), уксусная кислота или т.п., в присутствии или в отсутствии осушителя, такого как молекулярные сита 3 или сульфат магния, и гидридного соединения,такого как боргидрид натрия или цианоборгидрид натрия, в инертном органическом растворителе, таком как метанол, этанол, изопропанол,тетрагидрофуран, дихлорметан, хлороформ или их смеси, при температуре 0-25 С в течение 112 ч с получением соответствующего вторичного или третичного аминового производного (11). Схема Е Реакционная схема Е. Как показано в реакционной схеме Е, в результате обработки арилгидразина или гидрохлорида арилгидразина (12) арилциклопропилкетоном типа (13) в полярном органическом растворителе, таком как метанол, этанол, нпропанол, изопропанол, н-бутанол, трет 13 бутанол, предпочтительно н-бутанол, при температуре 70-120 С в течение 8-24 ч получают 2 арилтриптамин (5). Альтернативно, при обработке арилгидразина или гидрохлорида арилгидразина (12) арилбутиловым кетоном типа(14), содержащим уходящую группу (хлорид,бромид,иодид,O-метансульфонат,Oтрифторметансульфонат или т.п.) в 4 положении, в полярном растворителе, таком как метанол, этанол, н-пропанол, изопропанол, нбутанол, трет-бутанол или их смеси, при комнатной температуре в течение периода времени от 30 мин до 2 ч с последующим нагреванием до температуры 65-100 С в течение 4-24 ч, получают 2-арилтриптамин (5). Схема F Реакционная схема F. Как показано в реакционной схеме F, иодоанилины типа (15) могут быть подвергнуты реакции с арилацетиленами, соответствующим палладиевым (O) катализатором, таким как тетракис(трифенилфосфин)палладий; галогенидом меди (I), таким как бромид меди, в инертном органическом растворителе, таком как триэтиламин, при температуре 50-88 С в течение периода времени от 30 мин до 5 ч, с получением диарилацетилена (16). Ацетилен (16) может быть затем модифицирован путем обработки палладиевым (II) катализатором, таким как хлорид палладия (II) или ацетат палладия (II), в инертном органическом растворителе, таком как ацетонитрил, при температуре 50-82 С в течение периода времени от 30 мин до 6 ч с получением 2-арилиндола (17). Схема G 14 Реакционная схема G. Как показано в реакционной схеме G, после обработки 2-арилиндола (17) оксалилхлоридом, чистым или в инертном органическом растворителе, таком как метиленхлорид, хлороформ, дихлорэтан, тетрагидрофуран или т.п.,при температуре 25-65 С в течение 3-24 ч получают продукт присоединения ацилхлорида (18). Неочищенный продукт (18) может быть подвергнут реакции с амином типа (19) в инертном органическом растворителе, таком как диэтилэфир, тетрагидрофуран, метиленхлорид, хлороформ или т.п., в присутствии аминового основания, такого как триэтиламин, диизопропилэтиламин или пиридин, при температуре 0-25 С в течение периода времени от 30 мин до 4 ч с получением амидного производного (20). Амид(20) может быть затем модифицирован путем обработки восстановителем, таким как боран или алюмогидрид лития, в инертном органическом растворителе, таком как тетрагидрофуран,при повышенной температуре, предпочтительно при нагревании с обратным холодильником в течение 1-5 ч, с получением соединения (21). Схема Н(22b) могут быть восстановлены с получением вторичных аминовых аналогов (7) путем обработки водородом (1 атм) и соответствующим катализатором, таким как палладий-на-угле,гидроксид палладия-на-угле или т.п., в инертном органическом растворителе, таком как тетрагидрофуран, этилацетат, метанол, этанол или их смеси, к которому добавляют слабую кислоту, такую как 30% водная уксусная кислота, в течение периода времени от 10 мин до 3 ч или до тех пор, пока не будет удалена арильная группа, в результате чего получают вторичный амин. Реакционная схема I. Как показано в реакционной схеме I, в результате обработки нитроиндола типа (24) водородом (1 атм) и соответствующим катализатором, таким как никелевый катализатор Ренея(Raney Ni), в инертном органическом растворителе, таком как этанол, метанол или т.п., при комнатной температуре в течение 2-12 ч, получают соответствующее аминоиндольное производное (25). Схема J Реакционная схема J. Как показано в реакционной схеме J, амино- или гидроксииндол (25) может быть модифицирован путем ацилирования в различных условиях. Например, в результате обработки соединения (25) хлорангидридом, ангидридом кислоты или активным сложным эфиром и аминным основанием, таким как триэтиламин,диизопропилэтиламин, пиридин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, тетрагидрофуран или их смеси, при температуре от 0 С до комнатной температуры в течение 1-12 ч получают соответствующие амидное или сложноэфирное производные (26). Альтернативно, (25) может быть подвергнуто взаимодействию с карбоновой кислотой в присутствии одного или нескольких обычно используемых дегидратирующих агентов. Так, например, обработка аминоиндола (25) соответствующей карбоновой ки 000828(NММ), триэтиламин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, диметилформамид или их смеси, при комнатной температуре или примерно при комнатной температуре, в течение 3-24 ч приводит к получению соответствующего амидного или сложноэфирного производного Реакционная схема K. Как показано в реакционной схеме K, производное мочевины или карбаматное производное (25) может быть получено путем обработки карбамоилхлоридом типа (27 а) или, альтернативно, изоцианатным реагентом типа (27b) и аминным основанием, таким как пиридин, триэтиламин,диизопропилэтиламин,Nметилморфолин или т.п., в инертном органическом растворителе, таком как метиленхлорид,хлороформ, диметилформамид, тетрагидрофуран или их смеси, при температуре 0-65 С в течение 1-72 ч с получением соединения (28). Соединение (25) может быть также модифицировано путем обработки би(электрофильным) реагентом, таким как фосген, трифосген, 1,1'карбонилдиимидазол,N,N'-дисукцинимидилкарбокат или т.п., с добавлением или без добавления аминного основания, такого как пиридин,триэтиламин, диизопропилэтиламин, N-метилморфолин, в инертном органическом растворителе,таких как метиленхлорид, хлороформ или т.п., при температуре от -20 С до 0 С в течение периода времени от 20 мин до 2 ч. По истечении этого времени, реакционную смесь обрабатывают соответствующим моно- или дизамещенным амином при -20-25 С в течение 1-5 ч с получением мочевинного или карбаматного аналога (28).(25) может быть модифицирован путем обработки соответствующим сульфонилхлоридом типа (29) или сульфамилхлоридом типа (30) в присутствии аминного основания, такого как пиридин, триэтиламин, диизопропилэтиламин,N-метилморфолин, в инертном органическом растворителе, таком как метиленхлорид, хлороформ, дихлорэтан или их смеси, при температуре -20-25 С в течение периода времени от 20 мин до 2 ч с получением соответствующего Nсульфонамидного (31) или N-сульфамиламидного (32) производного соответственно. Схема М Реакционная схема М. Как показано в реакционной схеме М, 2 арилтриптамин (33) может быть модифицирован путем обработки эпоксидом, таким как соединение (34), в инертном органическом растворителе, таком как метанол, этанол, изопропанол,бутанол, трет-бутанол или их смеси, при температуре 65-110 С в течение 8-20 ч с получением соответствующего аминоспиртового производного (35). Реакционная схема N. Как показано в реакционной схеме N,амидные производные, содержащие кислоту индольного производного, такого как (36), могут быть получены путем обработки соответствующим амином (R11R12NH) и подходящим сочетающим агентом, таким как гексафторфосфат бензотриазол-1-ил-окси-трис(пирролидино)фосфония (Ру-ВОР), гексафторфосфат бензотриазол-1-ил-окси-трис(диметиламино) фосфония(HOBt) и третичного аминного основания, такого как N-метил-морфолин (NММ), триэтиламин или т.п., в инертном органическом растворителе, таком как метиленхлорид, хлороформ, тетрагидрофуран, диметилформамид или их смеси,при комнатной температуре или примерно при комнатной температуре в течение 3-24 ч с получением соответствующего амидного производного (37). Соединения настоящего изобретения могут быть использованы для лечения различных ассоциированных с половыми гормонами состояний у мужчин и женщин. Эффективность этих соединений может быть подтверждена их способностью действовать как антагонисты нейропептидного гормона GnRH, что было продемонстрировано активностью этих соединений в нижеследующих in vitro-анализах. Анализ на связывание с рецепторомGnRH-гипофиза крысы Неочищенные плазматические мембраны,полученные из тканей гипофиза крысы, инкубировали в буфере Трис-НСl (50 мМ, рН 7,5), содержащем альбумин бычьей сыворотки (0,1%),[I-125]D-t-Bu-Ser6-Рго 9-этиламид-GnRН и нужную концентрацию испытуемого соединения. Аналитические смеси инкубировали при 4 С в течение 90-120 мин с последующей быстрой фильтрацией и неоднократным промыванием через стекловолоконный фильтр. Радиоактивность связанных с мембранами радиоактивных лигандов определяли на гамма-счетчике. Исходя из этих данных, вычисляли IC50 радиоактивного лиганда, связывающегося с рецепторами GnRH в присутствии испытуемого соединения. 19 Анализ на ингибирование высвобождения LH Активные соединения, выявленные в анализе на связывание с рецептором GnRH, были затем оценены с помощью in vitro-анализа на высвобождение LH для подтверждения их антагонистической активностиGnRH-индуцированного высвобождения LH). 1. Получение образцов Анализируемые соединения растворяли и разводили в ДМСО. Конечная концентрация ДМСО в среде для инкубирования составляла 0,5%. 2. Анализ Самцы крыс Wistar (150-200 г) были получены из лаборатории Charles River Laboratories(Wilmington, MA). Крыс выдерживали при постоянной температуре (25 С) в условиях смены дня и ночи: 12 ч при свете и 12 ч в темноте. Крысам давали еду и воду ad libitum (по желанию). Затем животных умерщвляли путем обезглавливания, гипофизы удаляли в асептических условиях и помещали в сбалансированный солевой раствор Хэнкса (HBSS) в 50 миллилитровую полипропиленовую центрифужную пробирку. Пробирку для сбора образцов центрифугировали при 250 х g в течение 5 мин и HBSS удаляли путем отсасывания. Гипофизы переносили в одноразовую чашку Петри и измельчали скальпелем. Затем измельченную ткань переносили в 50-миллилитровую одноразовую центрифужную пробирку путем суспендирования фрагментом ткани в трех последовательно добавляемых 10 мл аликвотах HBSS,содержащих 0,2% коллагеназы и 0,2% гиалуронидазы. Диспергирование клеток осуществляли в водяной бане при 37 С, слегка размешивая, в течение 30 мин. По окончании инкубирования,клетки 20-30 раз отсасывали пипеткой, а негидролизованные фрагменты гипофиза оставляли на 3-5 мин для осаждения. Суспендированные клетки удаляли путем отсасывания, а затем подвергали центрифугированию при 1200 х g в течение 5 мин. После этого клетки ресуспендировали в культуральной среде. Негидролизованные фрагменты гипофиза обрабатывали 30 миллилитровыми аликвотами вышеуказанных гидролизирующих ферментов всего за 3 стадии гидролиза с использованием смеси коллагеназы/гиалуронидазы. Полученные клеточные суспензии объединяли, оценивали и разводили до концентрации 3 х 105 кл./мл и 1,0 мл этой суспензии помещали в каждую лунку 24-луночного планшета (Costar, Cambridge, MA). Клетки поддерживали в атмосфере воздуха - 5% CO2 при влажности 95% и при 37 С в течение 3-4 дней. Культуральная среда состояла из DMEM (модифицированной по способу Дульбекко среды Игла), содержащей 0,37% NаНСО 3, 10% лошадиной сыворотки, 2,5% фетальной телячьей сыворотки, 1% заменимых (не основных) аминокислот, 1% глутамина и 0,1% гентамицина. В 20 день эксперимента клетки три раза промывали за полтора часа до начала эксперимента и еще два раза непосредственно перед началом эксперимента средой DMEM, содержащей 0,37%(100 Х), 1% пенициллина/стрептомицина (10000 ед. пенициллина и 10000 микрограммов стрептомицина на 1 мл) и 25 мМ HEPES, рН 7,4. Высвобождение LH инициировали путем добавления в каждую лунку (в дубликатах) 1 мл свежей среды, содержащей испытуемые соединения в присутствии 2 нМ GnRH. Инкубирование проводили в течение 3 ч при 37 С. После инкубирования среду удаляли и центрифугировали при 2000 х g в течение 15 мин для удаления всего клеточного материала. Надосадочную жидкость удаляли и оценивали на содержание LH методом РИА с "двойным" антителом с использованием материалов,предоставленныхDr.A.F.Parlow (Harbor-UCLA Medical Center,Torrance, CA). Соединения формулы I могут быть использованы в ряде участков организма, подвергаемых неблагоприятному действию GnRH. Они могут быть использованы при состояниях, ассоциированных с половыми гормонами; гормонально-зависимом раке; доброкачественной гипертрофии предстательной железы или миоме матки. Введение соединений настоящего изобретения может дать хороший эффект при раке,ассоциированном с половыми гормонами,включая рак предстательной железы, рак матки,рак молочной железы и гонадотропные аденомы гипофиза. Соединения настоящего изобретения могут быть успешно введены при других состояниях, ассоциированных с половыми гормонами, включая эндометриоз, поликистоз яичников, фиброму матки и преждевременное половое развитие. Эти соединения могут быть также использованы в комбинации с ингибитором ангиотензинконвертирующего фермента, таким как эналаприл или каптоприл; антагонистом рецептора ангиотензина II, таким как лосартан; или с ингибитором ренина для лечения фибромы матки. Соединения настоящего изобретения могут быть также использованы в качестве контрацептивов, применяемых как мужчинами, так и женщинами для предохранения от беременности; для in vitro-оплодотворения; при предменструальном синдроме, а также для лечения системной красной волчанки, гирсутизма, синдрома раздражения кишечника и расстройств сна,таких как приступы апноэ во сне. Кроме того, соединения настоящего изобретения могут быть использованы как вспомогательное средство при гормональной терапии детей с дефицитом гормона роста. Эти соединения могут быть введены вместе с гормоном роста или с соединением, стимулирующим эндо 21 генное продуцирование или высвобождение гормона роста. Был разработан ряд соединений,которые стимулируют высвобождение эндогенного гормона роста. Известны пептиды, которые стимулируют высвобождение эндогенного гормона роста, например, фактор высвобождения гормона роста; пептиды, высвобождающие гормон роста, GHRP-6 и GHRP-1 (описанные в патенте США 4411890, в публикации патента РСТWO 89/07110 и в публикации патента РСТWO 89/07111) и GHRP-2 (описанный в публикации патента РСТWO 93/04081), а также гексарелин (J.Endocrinol Invesr., 15(Suppl.4) 45 (1992). Другие соединения, которые стимулируют высвобождение эндогенного гормона роста, описаны, например, в следующих работах: патент США 3239345; патент США 4036979; патент США 4411890; патент США 5206235; патент США 5283241; патент США 5284841; патент США 5310737; патент США 5317017; патент США 5374721; патент США 5430144; патент США.5434261; патент США 5438136; публ. патента ЕРО 0144230; публ. патента ЕРО 0513974; публ. патента РСТWO 94/07486; публ. патента РСТWO 94/08583; публ. патента РСТWO 94/11012; публ. патента РСТWO 94/13696; публ. патента РСТWO 94/19367; публ. патента РСТWO 95/03289; публ. патента РСТWO 95/03290; публ. патента РСТWO 95/09633; публ. патента РСТWO 95/11029; публ. патента РСТWO 95/12598; публ. патента РСТWO 95/13069; публ. патента РСТWO 95/14666; публ. патента РСТWO 95/16675; публ. патента РСТWO 95/16692; публ. патента РСТWO 95/17422; публ. патента РСТWO 95/17423;Natl. Acad. Sci. USA, 92, 7001-7005 (июль 1995). Предпочтительными характерными стимуляторами гормона роста, используемыми в комбинации с соединениями настоящего изобретения, являются следующие соединения: 1)N-[1(R)-[(1,2-Дигидро-1-метансульфонилспиро[3 Н-индол-3,4'-пиперидин]-1'-ил) карбонил]-2-(1 Н-индол-3-ил)этил]-2-амино-2 метилпропанамид; 2) N-[1(R)-[(1,2-Дигидро-1-метанкарбонилспиро[3 Н-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(1H-индол-3-ил)этил]-2-амино-2-метилпропанамид; 3) N-[1(R)-[(1,2-Дигидро-1-бензол-сульфонилспиро[3 Н-индол-3,4'-пиперидин]-1'-ил)карбонил]-2-(1 Н-индол-3-ил)этил]-2-амино-2 метилпропанамид; 4) N-[1(R)-[(1,2-Дигидроспиро[2 Н-1-бензопиран-2,4'-пиперидин]-1'-ил) карбонил]-2-(1 Ниндол-3-ил)этил]-2-амино-2-метилпропа-намид; 5) N-[1(R)-[(2-Ацетил-1,2,3,4-тетрагидроспиро[изохинолин-4,4'-пиперидин]-1'-ил)кар 000828[3 Н-бензотиофен-3,4'-пиперидин]-1'-ил)карбонил]-2-(фенилметилокси)этил]-2-амино-2-метилпропанамид; и их фармацевтически приемлемые соли. Соединения настоящего изобретения могут быть использованы в комбинации с бисфосфонатами (бисфосфоновыми кислотами) и другими агентами для лечения и предупреждения нарушений кальциевого обмена, фосфатного обмена и костного метаболизма, а в частности, для предупреждения разрежения кости в процессе терапии с использованием GnRH-антагониста; и в комбинации с эстрогенами, прогестеронами и/или андрогенами для предупреждения или лечения разрежения кости или гипогонадотроп 23 ных симптомов, таких как приступообразное ощущение жара ("приливы") во время терапии с использованием GnRH-антагониста. Известно, что бисфосфонаты (бисфосфоновые кислоты) ингибируют разрежение кости,а поэтому они могут быть использованы для лечения костного литиаза, как описано в патенте США 4621077 (Rosini et al.). В литературе описаны различные бисфосфоновые кислоты, которые могут быть использованы для лечения и предупреждения заболеваний, связанных с разрежением кости. Характерные примеры таких соединений могут быть найдены в следующих работах: патент США 3251907, патент США 3422137, патент США 3584125, патент США 3940436, патент США 3944599, патент США 3962432, патент США 4054598, патент США 4267108,патент США 4327039, патент США 4407761, патент США 4578376, патент США 4621077, патент США 4624947, патент США 4746654, патент США 4761406, патент США 4922007, патент США 4942157,патент США 5227506, патент США 5270365, публ. патента ЕРО 0252504, и J. Org.Chem., 36, 3843 (1971). Получение бисфосфоновых кислот и галогенбисфосфоновых кислот хорошо известно специалистам. Характерные примеры такого получения могут быть найдены в вышеупомянутых работах, в которых описаны соединения,используемые для лечения нарушений кальциевого или фосфатного обмена, а в частности, используемые в качестве ингибиторов разрежения кости. Предпочтительные бисфосфонаты выбирают из группы следующих соединений: алендроновая кислота, энтидроновая кислота, клодроновая кислота, памидроновая кислота, тилудроновая кислота, ризедроновая кислота, 6-амино 1-гидрокси-гексилиден-бисфосфоновая кислота,и 1-гидрокси-3(метилпентиламино)пропилиденбисфосфоновая кислота; или их любые фармацевтически приемлемые соли. Особенно предпочтительным бисфосфонатом является алендроновая кислота (алендронат) или ее фармацевтически приемлемая соль. Особенно предпочтительным бисфосфонатом является алендронат натрия, включая тригидрат алендроната натрия. Алендронат натрия получил одобрение Регуляторной комиссии на его продажу в США под торговым знаком FOSAMAX. Кроме того, соединения настоящего изобретения могут быть введены одновременно с ингибитором 5-редуктазы 2, таким как финастерид или эпристерид; ингибитором 5 редуктазы 1, таким как 4,7-диметил-4-аза-5 холестан-3-он, 3-оксо-4-аза-4,7-диметил-16(4-хлорфенокси)-5-андростан и 3-оксо-4-аза 4,7-диметил-16-(фенокси)-5-андростан, описанным в WO 93/23420 и WO 95/11254; двой 000828 ных ингибиторов 5-редуктазы 1 и 5 редуктазы 2, таких как 3-оксо-4-аза-17-(2,5 трифторметилфенилкарбамоил)-5-андростан,описанных в WO 95/07927; антиандрогенами,такими как флутамид, казодекс и ацетат ципротерона, и альфа-1-блокаторами, такими как празозин, теразозин, доксазозин, тамсулозин и альфузозин. Кроме того, соединения настоящего изобретения могут быть использованы в комбинации с гормоном роста; фактором высвобождения гормона роста или стимулятором гормона роста для замедления преждевременного полового созревания у детей с дефицитом гормона роста, что позволяет им набирать рост до образования эпифиза и прекращения роста с наступлением половой зрелости. В случае комбинированного лечения с использованием более, чем одного активного агента, где активные агенты применяются в виде отдельных лекарственных препаратов, указанные активные агенты могут быть введены как отдельно, так и в сочетании друг с другом. Кроме того, введение одного агента может быть осуществлено до, одновременно или после введения другого агента. Фармацевтические композиции, содержащие активный ингредиент, могут быть изготовлены в форме, подходящей для перорального введения, например, в виде таблеток, таблетоклепешек, пастилок, водных или масляных суспензий, диспергируемых порошков или гранул,эмульсий, жестких и мягких капсул, сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть получены любым стандартным методом, обычно используемым специалистами для изготовления фармацевтических композиций, и эти композиции могут содержать один или несколько агентов, выбранных из группы, включающей подслащивающие агенты, ароматизирующие агенты, красители, и консерванты, для получения фармацевтических препаратов, имеющих эстетически привлекательный вид и приятные вкусовые качества. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, которые являются подходящими для изготовления таблеток. Такими наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например, крахмал, желатин или аравийская камедь; и замасливающие агенты, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми, либо для замедления дезинтеграции и абсорбции в желудочно-кишечном тракте, они могут быть покрыты стандартными методами, в 25 результате чего могут быть получены препараты пролонгированного действия. Для этого, например, может быть использован инерционный материал, такой как глицерилмоностеарат или глицерилдистеарат. Эти таблетки могут быть покрыты методами, описанными в патентах США 4256108; 4166452 и 4265874, с получением осмотических терапевтических таблеток для регулируемого высвобождения. Препараты для перорального введения могут быть также изготовлены в виде жестких желатиновых капсул, содержащих активный ингредиент, смешанный с инертным твердым разбавителем, например карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, содержащих активный ингредиент, смешанный с водной или масляной средой, например с арахисовым маслом, парафиновым маслом или оливковым маслом. Водные суспензии содержат активный ингредиент в смеси с наполнителями, подходящими для изготовления водных суспензий. Такими наполнителями являются суспендирующие агенты, например натрийсодержащая карбоксиметилцеллюлоза,метилцеллюлоза,гидроксипропилметилцеллюлоза, альгинат натрия,поливинил-пирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие агенты, которыми могут быть природный фосфатид, например лецитин, или продукты конденсации алкилендоксида с жирными кислотами, например полиоксиэтиленстеарат,или продукты конденсации этиленоксида с длинноцепными алифатическими спиртами,например гептадекаэтиленоксицетанол, или продукты конденсации этиленоксида с неполными сложными эфирами, образованными жирными кислотами и гекситом, такие как моноолеат полиоксиэтиленсорбита; или продукты конденсации этиленоксида с неполными сложными эфирами, образованными жирными кислотами и ангидридами гексита, например полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов,например этил или н-пропил,пгидроксибензоат; один или несколько красителей; один или несколько ароматизирующих агентов; и один или несколько подслащивающих агентов, таких как сахароза, сахарин или аспартам. Масляные суспензии могут быть изготовлены путем суспендирования активного ингредиента в растительном масле, например в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле; либо в минеральном масле, таком как парафиновое масло. Масляные суспензии могут содержать загущающий агент, например пчелиный воск, твердый парафин или цетиловый спирт. Для получения перорального препарата с приятными вкусовыми качествами могут быть добавлены подслащивающие агенты, такие как указано выше, а так 000828 26 же ароматизирующие агенты. Для лучшего сохранения, в эти композиции может быть добавлен антиоксидант, такой как аскорбиновая кислота. Диспергируемые порошки и гранулы, подходящие для приготовления водных суспензий путем добавления воды, содержат активный ингредиент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Примеры подходящих диспергирующих или смачивающих агентов и суспендирующих агентов были уже приведены выше. В качестве добавок могут также присутствовать подслащивающие агенты, ароматизирующие агенты и красители. Фармацевтические композиции настоящего изобретения могут быть также изготовлены в виде эмульсий "масло в воде". Масляной фазой может быть растительное масло, например оливковое масло или арахисовое масло, или минеральное масло, например парафиновое масло,или их смеси. Подходящими эмульгирующими агентами могут быть природные фосфатиды,например соевые бобы, лецитин, и сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита,например сорбитанмоноолеат; а также продукты конденсации указанных неполных сложных эфиров с этиленоксидом, например полиоксиэтиленсорбитанмоноолеат. Эти эмульсии могут также содержать подслащивающие и ароматизирующие агенты. Сиропы и эликсиры могут быть изготовлены с использованием подслащивающих агентов,например глицерина, пропиленгликоля, сорбита или сахарозы. Такие препараты могут также содержать средство, смягчающее раздражение,консервант, отдушку и краситель. Фармацевтические композиции могут быть изготовлены в форме стерильных водных или масляных суспензий для инъекций. Эти суспензии могут быть получены стандартными методами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, которые были упомянуты выше. Стерильный препарат для инъекций может быть также получен в виде стерильного инъецируемого раствора или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3 бутандиоле. Из приемлемых наполнителей и растворителей могут быть использованы вода,раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя и суспендирующей среды обычно используют стерильные жирные масла. В этих целях может быть использовано любое мягкое жирное масло, включая синтетические моно- или диглицериды. Помимо этого, для получения препаратов для инъекций используются также жирные кислоты, такие как олеиновая кислота. 27 Соединения формулы I могут быть также изготовлены в форме суппозиториев для ректального введения лекарственного средства. Эти композиции могут быть получены путем смешивания лекарственного средства с подходящим нераздражающим наполнителем, который является твердым при обычных температурах, и жидким при температуре прямой кишки, а поэтому при попадании в прямую кишку он расплавляется с высвобождением лекарственного средства. Такими веществами являются какаомасло и полиэтиленгликоли. Для местного применения используют кремы, мази, гели, растворы или суспензии и т.п., содержащие соединения формулы I. (В этих целях препаратами для наружного применения могут быть растворы для полоскания рта и горла). Соединения настоящего изобретения могут быть введены через нос путем использования носителей, подходящих для местного применения, либо они могут быть введены чрескожно с использованием пластырей для наложения на кожу, хорошо известных каждому специалисту. При введении препарата, изготовленного в форме системы для чрескожной доставки лекарственного средства, введение дозы в процессе лечения, разумеется, будет постоянным, а не периодическим. Соединения настоящего изобретения могут быть также введены в виде суппозитория, изготовленного на соответствующей основе, такой как масло-какао, глицеринированный желатин, гидрогенизированные растительные масла, смеси полиэтиленгликолей различной молекулярной массы со сложными эфирами жирных кислот и полиэтиленгликоля. Схему введения с использованием соединений настоящего изобретения выбирают в зависимости от ряда факторов, включая тип, вид,возраст, вес, пол и состояние здоровья пациента; тяжесть заболевания, подвергаемого лечению; способ введения; функцию почек и печени пациента; и конкретно используемое соединение. Любой врач или ветеринар может легко определить и прописать эффективное количество лекарственного средства, необходимое для предупреждения, подавления или прекращения прогрессирования заболевания или обратного развития заболевания. Для оптимальной оценки пределов нужной концентрации лекарственного средства, в целях определения схемы лечения,дающего желаемый эффект без токсического действия, необходимо учитывать кинетику доступности лекарственного средства для данного участка организма пациента. Для этого необходимо принимать в расчет распределение, равновесие и выведение лекарственного средства. Предпочтительные дозы соединения структурной формулы I, используемые в способе настоящего изобретения, составляют в пределах от 0,01 до 1000 мг в день для взрослого индивидуума. Наиболее предпочтительно, если эти 28 дозы составляют от 0,1 до 500 мг/день. Композиции для перорального введения обычно изготавливают в форме таблеток, содержащих от 0,01 до 1000 мг активного ингредиента, а в частности 0,01; 0,05; 0,1; 0,5; 1,0; 2,5; 5,0; 10,0; 15,0; 25,0; 50,0; 100 и 500 мг активного ингредиента для симптоматической оценки дозы для пациента, подвергаемого лечению. Эффективное количество лекарственного средства обычно достигается при уровнях доз, составляющих от около 0,0002 мг/кг до около 50 мг/кг веса тела пациента в день. А более конкретно, этот интервал составляет от около 0,001 мг/кг до 1 мг/кг веса тела в день. Преимущественно, активный агент настоящего изобретения может быть введен в виде одноразовой суточной дозы, либо эта суточная доза может быть введена в виде дробных доз два, три или четыре раза в день. Количество активного ингредиента, которое может быть объединено с материалами носителя для получения унифицированной лекарственной формы, варьируется в зависимости от индивидуума, подвергаемого лечению и от способа введения лекарственного средства. Следует отметить, однако, что конкретный уровень дозы для каждого конкретного пациента будет зависеть от ряда факторов, включая возраст, вес тела, общее состояние здоровья,пол, режим питания, время введения, способ введения, скорость выведения лекарственного средства из организма, комбинации лекарственных средств и тяжесть конкретного заболевания, подвергаемого терапевтическому лечению. Нижеследующие примеры иллюстрируют получение некоторых соединений настоящего изобретения, однако, они не должны рассматриваться как некое ограничение изобретения. ПРИМЕР 1. 2-(3,5-Диметилфенил)-3-[2-[4-(4-гидроксифенил)бутиламино]этил]-1 Н-индол-5-иловый эфир морфолин-4-карбоновой кислоты. Стадия 1 А. 2-[2-(5-бензилокси-1 Н-индол 3-ил)этил]изоиндол-1,3-дион. К размешанной суспензии гидрохлорида 5 бензилокситриптамина (1,0 г в 10 мл безводного тетрагидрофурана) добавляли триэтиламин (0,50 мл), а затем N-карбэтоксифталимид (750 мг) и смесь нагревали с обратным холодильником на масляной бане. Через 48 ч реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрат концентрировали в вакууме. Полученное твердое вещество суспендировали в смеси гексан/метиленхлорид (2,5:1,50 мл) и(1,3 г). Стадия 1 В. 2-[2-(5-Бензилокси-2-бром-1 Ниндол-3-ил)этил] изоиндол-1,3-дион. К раствору 2-[2-(5-бензилокси-1 Н-индол 3-ил)этил]изоиндол-1,3-диона (800 мг в смеси 25 мл сухого тетрагидрофурана и 25 мл сухого хлороформа) при 0 С добавляли пербромид гидробромида пиридиния (666 мг) и смесь перемешивали при 0 С. Через 23 мин реакцию гасили путем добавления насыщенного бикарбоната натрия и экстрагировали этилацетатом. Органическую часть промывали насыщенным бикарбонатом натрия (3 х) и 0,3 М бисульфата натрия (3 х), а затем сушили сульфатом магния. Концентрат очищали с помощью флэшхроматографии на силикагеле (гексан: этилацетат 7:2), а затем повторно очищали с помощью флэш-хроматографии на силикагеле (метиленхлорид) и получали целевое соединение (632 мг). Стадия 1 С. 2-[2-[5-Бензилокси-2-(3,5 диметилфенил)-1 Н-индол-3-ил]этил]изоиндол 1,3-дион. К раствору 2-[2-(5-бензилокси-2-бром-1 Ниндол-3-ил)этил] изоиндол-1,3-диона (500 мг в смеси 6 мл этанола и 16 мл толуола) добавляли 3,5-диметилфенилбороновую кислоту (205 мг), а затем 2,7 мл 1 М карбоната натрия. К перемешанному раствору добавляли хлорид лития (156 мг), а затем тетракис(трифенилфосфин) палладий (78 мг) и смесь нагревали с обратным холодильником на масляной бане. Через 2 ч смесь охлаждали до комнатной температуры и концентрировали в вакууме. После очистки с помощью флэш-хроматографии на силикагеле(гексан:метиленхлорид:этилацетат 15:8:1, а затем 12:8:1) получали целевое соединение (479 мг). Стадия 1D. 2-[2-[2-(3,5-Диметилфенил)-5 гидрокси-1 Н-индол-3-ил]этил]изоиндол-1,3 дион. К перемешанному раствору 2-[2-[5 бензиолокси-2-(3,5-диметилфенил)-1H-индол-3 ил]этил]изоиндол-1,3-диона (510 мг в 20 мл сухого этилацетата) добавляли 197 мг 10% палладия-на-угле в качестве катализатора. В реакционную колбу подавали баллонный водород, а затем откачивали и снова подавали (3 раза), после чего содержимое перемешивали при комнатной температуре. Через 37 ч реакционную смесь продували азотом, фильтровали через диатомовую землю и концентрировали в вакууме с получением целевого соединения (418 мг). Стадия 1 Е. 3-(2-Аминоэтил)-2-(3,5 диметилфенил)-1 Н-индол-5-ол. К раствору 2-[2-[2-(3,5-диметилфенил)-5 гидрокси-1 Н-индол-3-ил]этил]изоиндол-1,3 диона (418 мг в смеси 7 мл этанола и 7 мл тетрагидрофурана) добавляли 2,5 мл 95% водного раствора гидразина и реакционную смесь перемешивали при комнатной температуре. Через 12(метиленхлорид:метанол:гидроксид аммония,89:11:1) с получением целевого соединения (228 мг). Стадия 1F. 4-(4-Бензилоксифенил)-N-[2-[2(3,5-диметилфенил)-5-гидрокси-1 Н-индол-3 ил]этил]бутирамид. К перемешанному раствору 4-бензилоксифенилмасляной кислоты (159 мг в смеси 2 мл метиленхлорида и 0,5 мл N,N-диметилформамида) добавляли 1-гидроксибензотриазол(110 мг) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (113 мг) и реагенты перемешивали в течение 30 мин. В это время добавляли раствор 3-(2-аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ола (144 мг в 4 мл N,N-диметилформамида) и реакционную смесь перемешивали при комнатной температуре. Через 6 ч смесь концентрировали в вакууме и очищали с помощью флэш-хроматографии на силикагеле (гексан:этилацетат 4:5), в результате чего получали целевое соединение (241 мг). Стадия 1G. 3-[2-[4-(4-Бензилоксифенил) бутиламино]этил]-2-(3,5-диметилфенил)-1 Ниндол-5-ол. К перемешанному раствору 4-(4 бензилоксифенил)-N-[2-[2-(3,5-диметилфенил)5-гидрокси-1 Н-индол-3-ил]этил]бутирамида(241 мг в 10 мл сухого тетрагидрофурана) добавляли 4 мл 1 М раствора борана в тетрагидрофуране и смесь медленно нагревали с обратным холодильником на масляной бане. Через 2 ч смесь охлаждали до комнатной температуры и избыточное количество борана гасили путем осторожного добавления метанола. Смесь концентрировали до половины объема, обрабатывали N,N-диметилэтаноламином (1,4 мл) и нагревали с обратным холодильником на масляной бане. Через 3 ч смесь охлаждали до комнатной температуры и концентрировали в вакууме. После очистки с помощью флэш-хроматографии на силикагеле (метиленхлорид:метанол 92:8) получали целевое соединение (234 мг). Стадия 1 Н. Бензиловый эфир [4-(4 бензилоксифенил)бутил]-[2-[2-(3,5-диметилфенил)-5-гидрокси-lH-индол-3-ил]этил]карбаминовой кислоты. К раствору 3-[2-[4-(4-бензилоксифенил) бутиламино]этил]-2-(3,5-диметилфенил)-1 Ниндол-5-ола (234 мг в 5 мл сухого метиленхлорида) при -78 С добавляли бензилхлорформиат (0,082 мл) и диизопропилэтиламин (0,104 мл) и смесь перемешивали при комнатной температуре. Через 1 ч реакцию гасили путем добавления насыщенного бикарбоната натрия и экстрагировали этилацетатом. Органическую часть промывали насыщенным хлоридом аммония, сушили сульфатом магния и концентрировали в вакууме. После очистки с помощью флэш-хроматографии на силикагеле (гек 31 сан:этилацетат 3:1, а затем 2:1) получали целевое соединение (155 мг). Стадия 1I. 3-(2-[Бензилоксикарбонил)-[4(4-бензилоксифенил)бутил]амино]этил)-2-(3,5 диметилфенил)-1 Н-индол-5-иловый эфир морфолин-4-карбоновой кислоты. К перемешанному раствору бензилового эфира 2-(3,5-диметилфенил)-5-гидрокси-N-(4 бензилоксифенилбутил)-N-(бензилоксикарбонил) триптамин-[4-(4-бензилоксифенил)бутил][2-[2-(3,5-диметилфенил)-5-гидрокси-1 Н-индол 3-ил]этил]карбаминовой кислоты (20 мг в 2 мл сухого метиленхлорида) при 0 С добавляли трифосген (4,8 мг) и пиридин (0,040 мл 10% раствора в метиленхлориде) и реагенты оставляли для размешивания на 20 мин. Эту смесь через канюлю добавляли к раствору морфолина(0,10 мл в 1 мл сухого метиленхлорида) при 0 С, а затем нагревали до комнатной температуры. Через 45 мин реакцию гасили путем добавления 0,3 М бисульфата натрия и экстрагировали этилацетатом. Органическую часть промывали 0,3 М бисульфатом натрия (3 х) и насыщенным солевым раствором, а затем сушили сульфатом магния и концентрировали в вакууме. После очистки посредством флэш-хроматографии на силикагеле (гексан:этилацетат 3:2) получали целевое соединение (20 мг). Стадия 1J. 2-(3,5-Диметилфенил)-3-[2-[4(4-гидрокси-фенил)бутиламино]этил]-1 Н-индол 5-иловый эфир морфолин-4-карбоновой кислоты. К перемешанному раствору 3-(2[бензилоксикарбонил-[4-(4-бензилоксифенил) бутил]амино]этил)-2-(3,5-диметилфенил)-1Hиндол-5-илового эфира морфолин-4-карбоновой кислоты (20 мг в смеси 2 мл тетрагидрофурана и 0,5 мл метанола) добавляли 16,8 мг 10% палладия-на-угле, а затем уксусную кислоту (0,010 мл 30% раствора в воде). В реакционную колбу накачивали баллонный водород, а затем его откачивали и накачивали снова (3 раза), после чего перемешивали при комнатной температуре. Через 1,5 ч реакционную смесь продували азотом, фильтровали через диатомовую землю и концентрировали в вакууме. После очистки с помощью флэш-хроматографии (метиленхлорид:метанол:гидроксид аммония 90:7:1) получали целевое соединение (12 мг). М/е=542 (М+Н). Получение синтетических промежуточных соединений 4-(4-Бензилоксифенил)масляная кислота. Стадия А. Бензиловый эфир 4-(4-бензилоксифенил)масляной кислоты. К перемешанному раствору 4 гидроксифенилмасляной кислоты (810 мг в 8 млN,N-диметилформамида) при 0 С добавляли гидрид натрия [290 мг 80%-ной дисперсии в минеральном масле) и смесь оставляли для нагревания до комнатной температуры. Через 20 мин добавляли бензилбромид (1,2 мл) и смесь перемешивали при комнатной температуре. Че 000828 32 рез 13 ч реакцию гасили путем добавления насыщенного хлорида аммония и экстрагировали этилацетатом. Органическую часть промывали водой (4 х), сушили сульфатом натрия и концентрировали в вакууме. После очистки с помощью флэш-хроматографии на силикагеле (гексан:этилацет 13:1) получали целевое соединение(1,45 г). Стадия В. 4-(4-Бензилоксифенил)масляная кислота. К перемешанному раствору бензилового эфира 4-(4-бензилоксифенил)масляной кислоты(277 мг в смеси 3 мл метанола и 1 мл метиленхлорида) при 0 С добавляли 1,5 мл 5 М гидроксида натрия и смесь нагревали до комнатной температуры. Через 2 ч смесь подкисляли до рН 2 путем добавления водного раствора хлористоводородной кислоты, а водную часть экстрагировали этилацетатом (5 х) и полученные органические экстракты концентрировали в вакууме. После очистки с помощью флэшхроматографии на силикагеле (метиленхлорид:метанол 94:6, а затем 96:4 + 0,25% TFA) получали целевое соединение (196 мг). 3,5-Диметилфенилбороновая кислота. К раствору 5-бром-м-ксилола (1,5 г в 15 мл безводного тетрагидрофурана) при -78 С добавляли 6,4 мл 1,4 М раствора бутиллития в гексане и смесь перемешивали в течение 20 мин. В это время добавляли триизопропилборат (2,8 мл) и смесь оставляли для нагревания до комнатной температуры. Через 1,5 ч реакционную смесь концентрировали в вакууме до 1/3 объема, а затем охлаждали до 0 С и обрабатывали 2 н хлористо-водородной кислотой (9 мл) с последующим нагреванием до комнатной температуры. Через 4 ч смесь подщелачивали путем добавления 2,5 М гидроксида натрия и распределяли между этиловым эфиром (75 мл) и 1,25 М гидроксидом натрия. Органический слой экстрагировали 1,25 М гидроксидом натрия (2 х), после чего водную часть охлаждали до 0 С и подкисляли до рН 3 путем добавления по каплям концентрированной хлористо-водородной кислоты. Белую суспензию растворяли в метиленхлориде, а органическую часть сушили сульфатом магния и концентрировали в вакууме с получением целевого соединения (960 мг). Нижеследующие соединения получали способом, аналогичным способу, описанному выше.(2-(3,5-Диметилфенил)-3-2-[4-(4 метансульфониламинофенид)бутиламино] этил-1 Н-индол-5-ил)амид пирролидин-1-карбоновой кислоты. Стадия 2.1 А. N-Бензил-2-[2-(3,5-диметилфенил)-5-нитро-1 Н-индол-3-ил]-N-[4-(4-метансульфониламинофенил)бутил]-2-оксоацетамид. К раствору 2-(3,5-диметилфенил 5-5-нитро 1 Н-индола (253 мг в 15 мл сухого тетрагидрофурана) добавляли 0,125 мл оксалилхлорида и смесь нагревали до 56 С на масляной бане. Через 14 ч смесь охлаждали, концентрировали в вакууме и остаточный оксалилхлорид удаляли путем азеотропной перегонки с бензолом. Неочищенное вещество повторно сольватировали в тетрагидрофуране при 0 С и обрабатывали растворомN-[4-(4-бензиламинобутил)фенил] метансульфонамида (347 мг в 5 мл тетрагидрофурана) и 0,22 мл триэтиламина, а затем оставляли для нагревания до комнатной температуры. Через 1 ч смесь распределяли между этилацетатом и насыщенным хлоридом аммония, а затем экстрагировали. Органическую часть промывали насыщенным солевым раствором, сушили сульфатом натрия и концентрат очищали с помощью флэш-хроматографии на силикагеле(гексан:этилацетат 4:5, а затем 2:3) с получением целевого соединения (391 мг). Стадия 2.1 В. N-4-[4-Бензил-2-[2-(3,5 диметилфенил)-5-нитро-1 Н-индол-3-ил]этил амино)бутил]фенил метансульфонамид. К раствору N-бензил-2-[2-(3,5-диметилфенил)-5-нитро-1 Н-индол-3-ил]-N-[4-(4-метансульфониламинофенил)бутил]-2-оксоацетамида (391 мг в 20 мл сухого тетрагидрофурана) 35 добавляли 5,5 мл 1 М раствора борана в тетрагидрофуране и смесь медленно нагревали с обратным холодильником на масляной бане. Через 2 ч смесь охлаждали до комнатной температуры и избыток борана гасили путем осторожного добавления метанола. Полученную смесь концентрировали до половины объема, обрабатывали N,N-диметилэтаноламином (1,8 мл) и нагревали с обратным холодильником на масляной бане. Через 3 ч смесь охлаждали до комнатной температуры и концентрировали в вакууме. После очистки посредством флэш-хроматографии на силикагеле (гексан:этилацетат, 3:2) получали целевое соединение (317 мг). Стадия 2.1 С. N-4-[4-(2-[5-Амино-2-(3,5 диметилфенил)-1H-индол-3-ил]этилбензиламино)бутил]фенилметансульфонамид. К перемешанному раствору N-4-[4(бензил-[2-[2-(3,5-диметилфенил)-5-нитро-1 Ниндол-3-ил]этиламино)бутил]фенилметансульфонамида (491 мг в 30 мл абсолютного этанола) добавляли приблизительно 30 мг никелевого катализатора Ренея. В реакционную колбу накачивали баллонный водород, а затем откачивали и накачивали снова (3 раза), после чего содержимое перемешивали при комнатной температуре. Через 2,5 ч реакционную смесь продували азотом, фильтровали через диатомовую землю и концентрировали в вакууме. После очистки с помощью флэш-хроматографии на силикагеле (гексан:этилацетат 2:3, а затем, гексан:этилацетат 1:3) получали целевое соединение (344 мг). Стадия 2.1D. [3-(2-Бензил-[4-(4-метансульфониламинофенил)бутил]аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ил]амид пирролидин-1-карбоновой кислоты. К перемешанному раствору N-4-[4-(2-[5 амино-2-(3,5-диметилфенил)-1H-индол-3-ил] этилбензиламино)бутил]фенилметансульфонамида (15 мг в 1,5 мл безводного метиленхлорида) добавляли 0,01 мл пирролидинкарбамилхлорида и 0,013 мл диизопропилэтиламина и эту смесь перемешивали при комнатной температуре. Через 24 ч добавляли еще 0,02 мл пирролидинкарбамилхлорида и 0,26 мл диизопропилэтиламина. Через 6 дней (всего) реакционную смесь концентрировали в вакууме и очищали с помощью флэш-хроматографии на силикагеле (метилен-хлорид:метанол 95:5) с получением целевого соединения (16 мг). Стадия 2.1 Е. (2-(3,5-Диметилфенил)-3-2[4-(4-метансульфонониламинофенил)бутиламино]этил-1 Н-индол-5-ил)амид пирролидин-1-карбоновой кислоты. К перемешанному раствору [3-(2-бензил[4-(4-метансульфониламинофенил)бутил] аминоэтил)-2-(3,5-диметилфенил)-1H-индол-5 ил] амида пирролидин-1-карбоновой кислоты(0,015 мл 30%-ного раствора в воде). В реакционную колбу подавали баллонный водород, а затем его откачивали и подавали снова (3 раза),после чего содержимое перемешивали при комнатной температуре. Через 25 мин реакционную смесь продували азотом, фильтровали через диатомовую землю и концентрировали в вакууме. После очистки с помощью флэшхроматографии (метиленхлорид:метанол: гидроксид аммония 90:8:1) получали целевое соединение (14 мг). М/е=602 (М+Н). ПРИМЕР 2.2.N-[4-4-2-[2-(3,5-Диметилфенил)-5-(3 этил-2-оксоимидазолидин-1-ил)-lH-индол-3-ил) этил]этиламинобутил)фенил] метансульфонамид. Стадия 2.2 А. 2-(3,5-Диметилфенил)-5 амино-1 Н-индол. К перемешанному раствору 2-(3,5 диметилфенил)-5-нитро-1 Н-индола (2 г в смеси 55 мл тетрагидрофурана и 20 мл метанола) добавляли 347 мг катализатора 10%-ного гидроксида палладия-на-угле, а затем уксусную кислоту (1 мл 30% раствора в воде). В реакционную колбу подавали баллонный водород, а затем откачивали и подавали снова (3 раза), после чего перемешивали при комнатной температуре. Через 25 ч в колбу загружали еще 320 мг катализатора и 1 мл 30%-ной уксусной кислоты и повторно продували водородом. По истечении полного времени реакции (3 дня), смесь продували азотом, фильтровали через диатомовую землю и концентрировали в вакууме. После очистки с помощью флэш-хроматографии (гексан:этилацетат 1:1) было получено целевое соединение (1,16 г). Стадия 2.2 В. Бензиловый эфир [2-(3,5 диметилфенил)-1 Н-индол-5-ил]карбаминовой кислоты. К раствору 2-(3,5-диметилфенил)-5-амино 1 Н-индола (1,16 г в 60 мл безводного метиленхлорида) при -60 С добавляли 0,86 мл бензилхлорформиата, а затем 1 мл диизопропилэтиламина и эту смесь медленно нагревали до 0 С. Через 45 мин реакцию гасили путем добавления насыщенного водного раствора хлорида аммония и смесь экстрагировали метиленхлоридом. Органическую часть промывали насыщенным солевым раствором, сушили сульфатом натрия и концентрировали в вакууме. После очистки с помощью флэш-хроматографии (гексан: метиленхлорид:этилацетат,8:8:1, а затем, 7:7:1) получали целевое соединение (1,68 г). Стадия 2.2 С. Бензиловый эфир [3-бензил[4-(4-нитрофенил)бутил]аминооксалил-2-(3,5 37 диметилфенил)-1 Н-индол-5-ил]карбаминовой кислоты. Целевое соединение (1,81 г) получали, в основном, как описано в стадии А примера 2.1,исходя из бензилового эфира(1,13 г) и бензил-[4-(4 нитрофенил)бутил]амина. Стадия 2.2D. Бензиловый эфир [3-(2 бензил-[4-(4-нитрофенил)бутил]аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ил]карбаминовой кислоты. Целевое соединение (1,38 г) получали, в основном, как описано в стадии В примера 2.1,исходя из бензилового эфира [3-бензил-[4-(4 нитрофенил)бутил]аминооксалил-2-(3,5-диметилфенил)-1 Н-индол-5-ил]карбаминовой кислоты (1,81 г). Стадия 2.2 Е. 3-(2-Бензил-[4-(4-нитрофенил)бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-ил-амин. К раствору бензилового эфира [3-(2 бензил-[4-(4-нитрофенил)бутил]аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ил] карбаминовой кислоты (500 мг в 25 мл безводного метиленхлорида) добавляли 0,50 мл анизола, а затем 0,035 мл бензилового спирта и смесь перемешивали при комнатной температуре. Затем по каплям добавляли 2,94 мл 1 М раствора хлорида алюминия в нитробензоле. Через 11,5 ч реакционную смесь охлаждали до 0 С, гасили путем добавления насыщенного водного раствора бикарбоната натрия и распределяли между этилацетатом и водным раствором тартрата калиянатрия. Органическую часть промывали дополнительным количеством тартратного раствора и сушили сульфатом натрия. Концентрат очищали с помощью флэш-хроматографии на силикагеле(метиленхлорид:метанол, 97:3), в результате чего получали целевое соединение (354 мг). Стадия 2.2F.(32,2 мг в 6 мл сухого метиленхлорида) добавляли 48 мг 1-гидрокси-бензотриазола, а затем 50 мг гидрохлорида 1-(3-диметиламинопропил)-3 этилкарбодиимида и эту смесь перемешивали при комнатной температуре. Через 50 мин добавляли раствор 3-(2-бензил-[4-(4-нитрофенил) бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Ниндол-5-ил-амина (120 мг в 2 мл метиленхлорида) и перемешивание продолжали. Через 4 ч(полные) смесь концентрировали в вакууме и очищали с помощью флэш-хроматографии на силикагеле (метиленхлорид:метанол 95:5) с получением целевого соединения (131 мг). Стадия 2.2G. N-[3-(2-бензил-[4-(4-нитрофенил)бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-N'-этил-этан-1,2 диамин.(20 мг в 3 мл сухого метиленхлорида) при температуре 0 С добавляли 9,6 мг N,N'дисукцинимидилкарбоната, а затем триэтиламин (0,110 мл 10% раствора в метиленхлориде) и эту смесь перемешивали при низкой температуре. Через 30 мин реакцию гасили путем добавления насыщенного водного раствора хлорида аммония и экстрагировали этилацетатом. Органическую часть промывали насыщенным бикарбонатом натрия, сушили сульфатом натрия и концентрировали в вакууме с получением неочищенного целевого соединения (21 мг). Стадия 2.21. 1-[3-(2-[4-(4-Аминофенил) бутил]бензиламиноэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-З-этилимидазолидин-2-он. Целевое соединение (47,5 мг) получали, в основном, как описано в стадии С примера 2.1,исходя из 1-[3-(2-бензил-[4-(4-нитрофенил) бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Ниндол-5-ил]-3-этилимидазолидин-2-она (85 мг). Стадия 2.2I. N-4-[4-(Бензил-2-[2-(3,5 диметилфенил)-5-(3-этил-2-оксоимидазолидин 1-ил)-1 Н-индол-3-ил]этиламино)бутил]фенил амино) метансульфонамид. К раствору 1-[3-(2-[4-(4-аминофенил) бутил]бензиламиноэтил)-2-(3,5-диметилфенил)1 Н-индол-5-ил]-3-этилимидазолидин-2-она (47,5 мг в 1,5 мл сухого метиленхлорида) при 0 С добавляли метансульфонилхлорид (0,066 мл 10% раствора в метиленхлориде), а затем триэтиламин (0,125 мл 10% раствора в метиленхлориде) и эту смесь перемешивали при низкой температуре. В течение 2 ч добавляли дополнительные порции метансульфонилхлорида и триэтиламин и спустя 3 ч реакцию гасили путем добавления насыщенного бикарбоната натрия. Полученную смесь распределяли между этилацетатом и водой, а органическую часть последовательно промывали насыщенным хлоридом аммония и насыщенным бикарбонатом натрия, а затем сушили сульфатом натрия. Концентрат очищали с помощью флэш-хроматографии на силикагеле (метиленхлорид:метанол:гидроксид аммония 95:5:1) и получали целевое соединениеN-4-[4-(бензил-2-[2-(3,5 диметилфенил)-5-(3-этил-2-оксоимидазолидин 1-ил)-1H-индол-3-ил]этиламино)бутил] фенил метансульфонамида (50 мг). М/е=602 (М+Н). Нижеследующие соединения были получены способом, аналогичным описанному выше.N-[4-(4-2-[2-(3,5 диметилфенил)-5-нитро-1 Н-индол-3-ил]этиламинобутил)фенил]метансульфонамида (полученного, в основном, как описано в стадии В примера 2.1; 800 мг в 15 мл тетрагидрофурана и 4 мл воды) при 0 С добавляли раствор 580 мг ди-трет-бутилдикарбоната, а затем 317 мг карбоната калия и полученную суспензию интенсивно перемешивали при 0 С. Через 25 мин реакцию гасили путем добавления избыточного количества насыщенного водного раствора хлорида аммония и смесь экстрагировали этилацетатом. Органическую часть сушили сульфатом натрия и концентрировали в вакууме. Остаток очищали с помощью флэш-хроматографии на силикагеле (гексан:этилацетат,1:1), в результате чего получали целевое соединение (900 мг). Стадия 3.2 В. Трет-бутиловый эфир 2-[2(3,5-диметилфенил)-5-нитро-1 Н-индол-3-ил] этил-[4-(4-амино(диметансульфонил)фенил) бутил]карбаминовой кислоты. К раствору трет-бутилового эфира 2-[2(3,5-диметилфенил)-5-нитро-1 Н-индол-3-ил] этил-[4-(4-метансульфониламинофенил)бутил] карбаминовой кислоты (300 мг в 7 мл метиленхлорида) при 0 С добавляли 0,135 мл триэтиламина и 0,066 мл метансульфонилхлорида и смесь перемешивали при низкой температуре. Через 30 мин реакцию гасили путем добавления 41 насыщенного водного раствора бикарбоната натрия и распределяли между водой и этилацетатом. Органическую часть последовательно промывали насыщенным хлоридом аммония,насыщенным бикарбонатом натрия и насыщенным солевым раствором, а затем сушили сульфатом натрия и концентрировали в вакууме с получением неочищенного целевого соединения(330 мг). Стадия 3.2 С. Трет-бутиловый эфир 2-[5 амино-2-(3,5-диметилфенил)-1H-индол-3-ил] этил-[4-(4-амино(диметансульфонил)фенил) бутил]карбаминовой кислоты. Целевое соединение (286 мг) получали, в основном, как описано в стадии Е примера 2.1,исходя из трет-бутилового эфира 2-[2-(3,5 диметилфенил)-5-нитро-1 Н-индол-3-ил] этил[4-(4-амино(диметансульфонил)фенил)бутил] карбаминовой кислоты (330 мг) и с использованием в качестве катализатора 10%-ного палладия-на-угле. Стадия 3.2D. Трет-бутиловый эфир 2-[2(3,5-диметилфенил)-5-изоцианат-1 Н-индол-3 ил]этил-[4-(4-амино-(диметансульфонил) фенил)бутил]карбаминовой кислоты. К раствору трет-бутилового эфира 2-[5 амино-2-(3,5-диметилфенил)-1H-индол-3-ил] этил-[4-(4-амино(диметансульфонил)фенил) бутил] карбаминовой кислоты (120 мг в 8 мл сухого метиленхлорида) при 0 С добавляли 0,040 мл пиридина, а затем 18,2 мг трифосгена и смесь перемешивали при низкой температуре. Через 20 мин добавляли 0,025 мл триэтиламина,а еще через 10 мин смесь наносили на колонку с силикагелем для очистки с помощью флэшхроматографии (гексан:этилацетат 2:3) и получали целевое соединение (106 мг). Стадия 3.2 Е. Трет-бутиловый эфир 2-[2(3,5-диметилфенил)-5-(5-фенилоксазол-2 иламино)-1H-индол-3-ил]этил-[4-(4-амино(диметансульфонил)фенил)бутил]карбаминовой кислоты. К раствору трет-бутилового эфира 2-[2(3,5-диметилфенил)-5-изоцианат-1 Н-индол-3 ил]этил-[4-(4-амино(диметансульфонил)фенил) бутил]карбаминовой кислоты (30 мг в 0,50 мл сухого метиленхлорида) добавляли 9 мг 2 азидо-1-фенил-этанола, а затем 14 мг трифенилфосфина и смесь перемешивали при комнатной температуре. Через 4 ч смесь наносили на колонку с силикагелем для очистки с помощью флэш-хроматографии (гексан:этилацетат 3:2, затем 1:1, затем 2:3, а затем 1:3), в результате чего получали целевое соединение (13 мг). Стадия 3.2F. Трет-бутиловый эфир 2-[2(3,5-диметилфенил)-5-(5-фенилоксазол-2 иламино)-1 Н-индол-3-ил]этил-4-(4-метансульфониламинофенил)бутил]карбаминовой кислоты. К раствору трет-бутилового эфира 2-[2(3,5-диметилфенил)-5-(5-фенилоксазол-2 илами-но)-1H-индол-3-ил]этил]-[4-(4-амино(диметансульфонил)фенил)бутил]карбаминовой кислоты (13 мг в 0,50 мл тетрагидрофурана) добавляли 0,125 мл 1,25 н раствора гидроксида натрия и эту смесь перемешивали при комнатной температуре. Через 30 мин реакционную смесь распределяли между этилацетатом и водой, а органическую часть последовательно промывали насыщенным хлоридом аммония,насыщенным бикарбонатом натрия и насыщенным солевым раствором. Объединенные органические экстракты сушили сульфатом натрия,концентрировали в вакууме и остаток очищали с помощью флэш-хроматографии на силикагеле(метиленхло-рид:метанол 96:4) с получением целевого соединения (10 мг). Стадия 3.2G. N-[4-(4-2-[2-(3,5-Диметилфенил)-5-(5-фенилоксазол-2-иламино)-1Hиндол-3-ил]этиламинобутил)фенил]метансульфонамид. К раствору трет-бутилового эфира 2-[2(3,5-диметилфенил)-5-(5-фенилоксазол-2-иламино)-1 Н-индол-3-ил]этил-[4-(4-метансульфониламинофенил)бутил] карбаминовой кислоты(10 мл в 0,80 мл метиленхлорида) при 0 С добавляли 0,050 мл анизола, а затем 0,20 мл трифторуксусной кислоты и эту смесь перемешивали при 0 С. Через 3 ч смесь концентрировали в вакууме и остаток кислоты удаляли путем азеотропной перегонки с толуолом. Концентрат очищали с помощью флэш-хроматографии на силикагеле (метиленхлорид:метанол:гидроксид аммония 90:7:1), в результате чего получали целевое соединение (8 мг). М/е=648 (М+Н). Нижеследующие соединения получали способом, аналогичным описанному выше.N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(морфолин-4-карбонил)-1 Н-индол-3-ил] этиламино] бутил]фенил]метансульфонамид. Стадия 4 А. Этиловый эфир 3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 карбоновой кислоты. Смесь 7,60 г (50 ммоль) 4-гидразинбензойной кислоты, 10,55 г (50 ммоль) 3 хлорпропил-3,5-диметилфенилкетона и 200 мл абсолютного этанола перемешивали в атмосфере азота и нагревали с обратным холодильником. Через 12 ч смесь охлаждали и фильтровали. Твердый осадок на фильтре промывали дополнительными небольшими объемами этанола. Фильтрат обрабатывали 4 мл концентрированной серной кислоты и перемешивали при нагревании с обратным холодильником в атмосфере азота в течение 4 дней. Охлажденную смесь перемешивали в ледяной бане, добавляя по каплям раствор этоксида натрия (21 мас.% в этаноле) в атмосфере азота до тех пор, пока смесь не становилась щелочной, на что указывала индикаторная бумага для определения рН. Эту смесь фильтровали и концентрировали в вакууме при 30 С. Остаток распределяли между диэтиловым эфиром и водой при добавлении небольшого количества насыщенного водного раствора хлорида натрия для облегчения разделения слоев. Водную фазу промывали еще 100 мл эфира. Объединенные органические экстракты сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаточную смолу подвергали флэш-хроматографии на силикагелеNH4OH 97:3:0,3, а затем 95:5:0,5). После концентрирования фракций продукта получали 4,03 г чистого продукта в виде твердой пены (продукт является фактически гомогенным, на что указывала ТСХ в СН 2 Сl2-МеОН-концентрированный NH2OH 95:5:0,5). После концентрирования смешанных фракций получали еще 0,93 г продукта, который снова хроматографировали и получали еще 0,77 г чистого соединения для полного выхода 4,80 г (29%). 1 Н-ЯМР 400 МГц (CDCl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl):m/е=337 (М+Н). Стадия 4 В. Этиловый эфир 2-(3,5-диметилфенил)-3-2-[4-(4-метансульфониламинофенил) бутиламино]этил-1 Н-индол-5-карбоновой кислоты. В сухую колбу вводили 672 мг (2,0 ммоль) этилового эфира 3-(2-аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-карбоновой кислоты, 530 мг (2,2 ммоль) 4-[4-(метансульфонамидо)фенил] 44 бутиральдегида, 1,20 г (10 ммоль) и магнитный стержень для перемешивания. Колбу продували азотом, охлаждали до температуры от 10 до-5 С в бане со смесью лед/метанол и перемешивали при постепенном добавлении 4 мл сухогоCDCl3 с помощью шприца. Полученную смесь перемешивали в атмосфере азота в течение 15 мин. Затем мембрану удаляли и быстро добавляли 100 мг (2,6 ммоль) боргидрида натрия. Мембрану сразу же устанавливали и эту систему снова продували азотом. Смесь перемешивали в атмосфере азота при температуре около-5 С, постепенно добавляя шприцем 4 мл сухого метанола. После выдерживания в течение 20 мин при этой температуре реакцию гасили путем постепенного добавления с помощью шприца 1 мл ацетона для разложения избытка боргидрида натрия. Еще через несколько минут смесь удаляли из охлаждающей бани и распределяли между 25 мл этилацетата и 25 мл воды. Органический слой промывали 10 мл насыщенного водного раствора хлорида натрия, а затем сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (элюируяCH2Cl2-MeOH 97:3, а затем 95:5). Собранные фракции продукта концентрировали в вакууме и получали 663 мг (59%) пены; продукт являлся фактически гомогенным, на что указывала ТСХ(CDCl3 + небольшое количество ДМСО-d6) соответствовал предполагаемой структуре. Массспектр (ЭСИ): m/е=562 (М+Н). Стадия 4 С. Этиловый эфир 3-(2-бензилоксикарбонил-[4-(4-метансульфониламинофенил)бутил]аминоэтил)-2-(3,5-диметилфенил)1 Н-индол-5-карбоновой кислоты. Раствор 646 мг (1,15 ммоль) этилового эфира 2-(3,5-диметилфенил)-3-2-[4-(4-метансульфониламинофенил)бутиламино]этил-1 Ниндол-5-карбоновой кислоты в 5 мл сухого метиленхлорида и 5 мл безводного тетрагидрофурана перемешивали в атмосфере азота и охлаждали до -78 С в бане из сухого льда и ацетона при добавлении 200 млN,N-диизопропилэтиламина, а затем с помощью шприца постепенно добавляли 173 мл (207 мг; 1,15 ммоль, исходя из чистоты 95%) бензилхлорформиата. Через 30 мин раствор удаляли из охлаждающей бани и оставляли для нагревания до комнатной температуры. Затем его распределяли между 25 мл этилацетата и 25 мл 5% водного раствора бисульфата калия. Органический слой промывали еще 25 мл 5% бисульфата калия, а затем 10 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили(сульфатом магния), фильтровали и концентрировали в вакууме. Остаток подвергали флэшхроматографии на силикагеле (элюент:СН 2 Сl2 МеОН 98:2) и получали 611 мг (76%) пены; продукт являлся гомогенным, на что указывала ТСХ в CH2Cl2-MeOH 95:5. 1H-ЯМР 500 МГц 45 являлся комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/е=696 (М+Н). Стадия 4D. 3-(2-Бензилоксикарбонил-[4(4-метансульфониламинофенил) бутил]амино 2-(3,5-диметилфенил)-1 Н-индол-5-карбоновая кислота. Раствор 600 мг (0,862 ммоль) этилового эфира 3-(2-бензилоксикарбонил-[4-(4-метансульфониламинофенил)бутил]аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-карбоновой кислоты в 18 мл (9 ммоль) 0,50 н гидроксида калия в метаноле перемешивали при температуре около 60 С по мере постепенного добавления 2,0 мл воды. Перемешивание продолжали при 60-65 С в атмосфере азота в течение 10 ч. Охлажденную смесь, которая содержала белый осадок, концентрировали в вакууме до небольшого объема. Остаточную суспензию распределяли между 25 мл этилацетата и 25 мл 0,5 н хлористоводородной кислоты. После отделения водного слоя, начиналось осаждение в этилацетатной фазе. Осадок снова растворяли путем разбавления 25 мл тетрагидрофурана. Водную фазу подвергали обратному экстрагированию 10 мл этилацетате + 10 мл тетрагидрофурана. Объединенные органические фазы сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаток твердого вещества растирали с диэтиловым эфиром, собирали на фильтре и промывали еще небольшим количеством эфира,в результате чего (после ушки) получали 573 мг(100%) порошка, т.пл. 211,5-213 С; продукт являлся фактически гомогенным, на что указывала ТСХ (СН 2 Сl2-МеОН, 92,5:7,5). 1 Н-ЯМР 500 МГц(6,5 мг, 0,075 ммоль) морфолина и 0,0400 мл сухого метиленхлорида перемешивали в атмосфере азота при комнатной температуре в колбе с притертой пробкой. Через 5,5 ч раствор распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органическую фазу сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью препаративной ТСХ на пластинах с силикагелем GF 21000 микрон (проявленных в CH2Cl2 93:7) и получали аморфное белое твердое вещество с количественным выходом; продукт являлся гомогенным, на что указывала ТСХ в СН 2 Сl2 000828 46 МеОН, 95:5. 1H-ЯМР (500 МГц, CDCl3) из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ):m/е=737,2 (М+Н). Стадия 4F. N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(морфолин-4-карбонил)-1H-индол-3-ил] этиламино]бутил] фенил]метансульфонамид. Смесь 38 мг (0,05 ммоль) бензилового эфира [2-[2-(3,5-диметилфенил)-5-(морфолин-4 карбонил)-1H-индол-3-ил]этил]-[4-[4-(метансульфониламино)фенил]бутил]карбаминовой кислоты, 10 мг 20%-ного гидроксида палладияна-угле и 10 мл 2-метокси-этанола встряхивали в атмосфере водорода (приблизительно 45 фунт/кв.дюйм=3,164 кг/см 2) в автоклаве в течение нескольких часов. Катализатор удаляли путем фильтрации через Целит и фильтрат концентрировали в вакууме. Остаток очищали с помощью препаративной ТСХ на пластинах с силикагелем GF 21000 микрон (проявленных вCH2Cl2 93:7) и получали 17 мг (56%) твердого вещества, т.пл. 138 С с разлож. (с предварительным размягчением); гомогенность продукта подтверждали с помощью ТСХ в CH2Cl2-MeOH,90:10. 1H-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ):m/e=603,3 (М+Н). Получение синтетических промежуточных соединений Стадия А. 4-Хлор-N-метокси-N-метилбутирамид. К раствору 4-хлорбутирилхлорида (10,0 г в 200 мл сухого метиленхлорида) добавляли 10,4 г гидрохлорида N,O-диметилгидроксиламина. Смесь перемешивали в атмосфере азота и поддерживали при температуре ниже 25 С путем охлаждения в ледяной бане при добавлении по каплям триэтиламина (29,1 мл), если это необходимо, примерно в течение 20 мин с образованием осадка. После выдерживания в течение 1,5 ч при комнатной температуре смесь концентрировали в вакууме. Остаток распределяли между 100 мл диэтилового эфира и 100 мл насыщенного водного раствора бикарбоната натрия. Органический слой промывали еще 100 мл насыщенного бикарбоната натрия, а водные фракции подвергали обратной экстракции эфиром. Объединенные органические фазы сушили сульфатом натрия, фильтровали и концентрировали в вакууме с получением 10,5 г (90%) масла, которое являлось достаточно чистым, на что указывал 1 Н-ЯМР (CDCl3). Масс-спектр (РВ-NН 3 Сl):m/e=166 (М+Н). Стадия В. 3-Хлорпропил 3,5-диметилфенилкетон. Раствор 10,2 мл (13,9 г, 72 ммоль) 5-бромм-ксилола в 200 мл безводного тетрагидрофурана перемешивали в атмосфере азота при -78 С при добавлении по каплям 35,8 мл (84 ммоль) 2,5 М н-бутиллития в тетрагидрофуране. После выдерживания в течение 15 мин при -78 С по каплям в течение 25-30 мин добавляли раствор 47 10,0 г (60 ммоль) 4-хлор-N-метокси-Nметилбутирамида в 30 мл безводного тетрагидрофурана. Полученный раствор поддерживали при -78 С в течение 45 мин, а затем быстро нагревали до комнатной температуры. Реакцию гасили путем добавления 40 мл 2 н хлористоводородной кислоты, а затем распределяли между этилацетатом и водой. Органическую фазу промывали насыщенным водным раствором бикарбоната натрия, а затем насыщенным водным раствором хлорида натрия. Органический раствор сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток подвергали флэш-хроматографии и получали 8,91 г(70%) масла , которое имело достаточную чистоту, на что указывал 1 Н-ЯМР (CDCl3). Стадия АА. 4-(4-Нитрофенил)масляной кислоты N-метокси-N-метиламид. Перемешанный раствор 6,29 г (30 ммоль) 4-(4-нитрофенил) масляной кислоты в 90 мл сухого метиленхлорида (выдержанный в атмосфере азота и охлажденный в водяной бане) обрабатывали 4,17 мл (3,03 г, 30 ммоль) триэтиламина, а затем 13,26 г (30 ммоль) реагента ВОР. Через несколько минут добавляли 3,22 г (33 ммоль) гидрохлоридаN,O-диметилгидроксиламина, а затем еще 4,59 мл (3,33 г, 33 ммоль) триэтиламина. Через 2,25 ч раствор разбавляли 200 мл диэтилового эфира и последовательно промывали 3 х 100 мл 2 н хлористоводородной кислоты, 1 х 100 мл и 2 х 50 мл насыщенного водного раствора бикарбоната натрия и 1 х 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали флэшхроматографии на силикагеле (элюируя сначала гексан -EtOAc, 2:1, а затем гексан -EtOAc, 3:2) и получали 6,27 г (83%) кристаллов, т.пл. 39,541,5 С; продукт являлся гомогенным, на что указывала ТСХ гексан-EtOAc 1:1. 1H-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Сl): m/e=253(М+Н). Стадия ВВ. N-метокси-N-метиламид 4-(4 аминофенил) масляной кислоты. Смесь 6,05 г (24 ммоль) N-метокси-Nметиламида 4-(4-нитрофенил)масляной кислоты, 50 мг 10% палладия-на-угле и 200 мл этанола встряхивали в атмосфере водорода (первоначальное давление водорода = 53 фунт/кв.дюйм(3,726 кг/см 2 в течение 1,5 ч, причем за этот период времени поглощение водорода прекращалось и ТСХ указывала на завершение реакции. Смесь фильтровали через Целит в атмосфере азота и фильтрат концентрировали в вакууме с получением 5,29 г масла, которое являлось гомогенным, на что указывала ТСХ 48 Стадия СС. N-метокси-N-метиламид 4-[4(метансульфонамидо)фенил]масляной кислоты. Раствор 5,33 г (24 ммоль) N-метокси-Nметиламида 4-(4-аминофенил)масляной кислоты в 48 мл сухого пиридина перемешивали в атмосфере азота при охлаждении в ледяной бане при добавлении по каплям в течение примерно 15 мин 1,86 мл (2,75 г, 24 ммоль) метансульфонилхлорида. После завершения добавления раствор оставляли для нагревания до комнатной температуры. Через 1,5 ч раствор концентрировали в вакууме при комнатной температуре. Остаток разбавляли 10 мл метиленхлорида и распределяли между смесью 100 мл этилацетат + 100 мл тетрагидрофуран и 100 мл 2 н хлористоводородной кислоты. Органический слой промывали еще 4 х 100 мл 2 н хлористо-водородной кислоты, а затем 50 мл насыщенного водного раствора бикарбоната натрия и, наконец, 20 мл насыщенного водного раствора хлорида натрия. Органическую фазу разбавляли некоторым количеством тетрагидрофурана, сушили сульфатом магния и обрабатывали углем. Смесь фильтровали через Целит и осадок на фильтре промывали дополнительным количеством ТГФ. Фильтрат концентрировали в вакууме и получали 4,39 г (61%) кристаллов, т.пл. 115-117 С; продукт являлся гомогенным, на что указывала ТСХ в CH2Cl2-MeOH, 95:5. 1H-ЯМР (400 МГц,ДМСО-d6) соответствовал предполагаемой структуре. Масс-спектр (РВ-NН 3/Cl): m/e=301(М+Н). Стадия DD. 4-[4-(метансульфонамидо) фенил]бутиральдегид. Смесь 4,20 г (14 ммоль) N-метокси-Nметиламида 4-[4-(метансульфонамидо)фенил] масляной кислоты и 100 мл безводного тетрагидрофурана перемешивали в атмосфере азота при охлаждении в ледяной бане по мере постепенного добавления с помощью шприца 17,5 мл(17,5 ммоль) 1 М алюмогидрида лития в тетрагидрофуране. Через 0,75 ч с помощью шприца осторожно добавляли 70 мл 5%-ного (водного) раствора бисульфата калия. Затем эту смесь удаляли из ледяной бани, разбавляли 150 мл воды и встряхивали со 150 мл этилацетата. Водную фазу молочного цвета экстрагировали еще 50 мл этилацетата. Объединенные органические фракции последовательно промывали 2 х 100 мл 1 н хлористо-водородной кислоты, а затем 50 мл насыщенного водного раствора бикарбоната натрия и, наконец, 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (элюент:гексан-ЕtОАс, 3:2) и получали 2,47 г (73%) масляного продукта, который являлся гомогенным, на что указывала ТСХ (гексан-EtOAc 1:1). После хранения в холодильнике продукт становился твердым (т.пл. 41-44 С). 1H-ЯМР 400 МГц(CDCl3) соответствовал предполагаемой струк 49 туре. Масс-спектр (РВ-NН 3/Cl): m/e=259(M+NH4). Нижеследующие соединения были получены способом, аналогичным описанному выше.N-[4-[4-[2-[5-(1,1-Диметил-2-оксо-2 пирролидин-1-илэтил)-2-(3,5-диметилфенил)1H-индол-3-ил]этиламино]бутил]фенил]метансульфонамид. Стадия 6.1 А. Этиловый эфир 2-[3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 ил]-2-метилпропионовой кислоты. В соответствии со способом, описанным в стадии А примера 4, этил 2-(4-гидразинофенил)2-метилпропионат подвергали реакции с 3 хлорпропил 3,5-диметилфенилкетоном, в результате чего получали целевое соединение в виде твердой пены с 16%-ным выходом; продукт фактически является гомогенным, на что указывала ТСХ в СН 2 Сl2-МеОН-концентрированный NH4OH. 1 Н-ЯМР 500 МГц (СDСl3) соответствовал предполагаемой структуре. Масс-спектр (PB-NH3/Cl): m/e=379 (M+H)+. Стадия 6.1 В. Этиловый эфир 2-(2-(3,5 диметилфенил)-3-2-4-(4-метансульфониламинофенил)бутиламино]этил-1 Н-индол-5-ил)-2 метилпропионовой кислоты. Реакцию восстановительного аминирования этилового эфира 2-[3-(2-аминоэтил)-2-(3, 5 диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты с 4-[4-(метансульфонамидо) фенил]бутиральдегидом проводили способом,описанным в стадии В примера 4, и получали целевое соединение в виде твердой пены с 43%ным выходом; продукт фактически являлся гомогенным, на что указывала ТСХ в CH2Cl2MeOH, 92,5:7,5. 1H-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Массспектр (РВ-NН 3/Сl): m/e=604 (M+H). Стадия 6.1 С. Этиловый эфир 2-[3-(2 бензилоксикарбонил-[4-(4-метансульфониламинофенил)бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты. Реакцию этилового эфира 2-(2-(3,5 диметилфенил)-3-2-[4-(4-метансульфониламинофенил)бутиламино]этил-1 Н-индол-5-ил)2-метилпропионовой кислоты с бензилхлорформиатом проводили способом, описанным в стадии С примера 4, в результате чего получали целевое соединение с выходом 72% в виде твердой пены; продукт являлся гомогенным, на что указывала ТСХ в СН 2 Сl2-МеОН 95:5. 1H-ЯМР 500 МГц являлся комплексным из-за присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/e=738(М+Н). Стадия 6.1D. 2-[3-(2-Бензилоксикарбонил-[4-(4-метансульфониламинофенил)бутил] аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 ил]-2- метилпропионовая кислота. Омыление этилового эфира 2-[3-(2-бензилоксикарбонил-[4-(4-метансульфониламинофенил)бутил]аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты проводили способом, описанным в стадии 1D примера 4, за исключением того, что 51 время реакции увеличивали до 30 ч, в результате чего получали количественный выход целевого соединения в виде порошка, т.пл. 102 С(постепенно; с неполным разложением); продукт являлся гомогенным, на что указывала ТСХ в СН 2 Сl2-МеОН, 92,5:7,5. 1 Н-ЯМР 500 МГц(ДМСО-d6) являлся комплексным вследствие присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ):m/e=710 (М+Н). Стадия 6.1 Е. Бензиловый эфир [2-[5-(1,1 диметил-2-оксо-2-пирролидин-1-илэтил )-2-(3,5 диметилфенил)-1H-индол-3-ил]этил]-[4-(4 метансульфониламинофенил)бутил]карбаминовой кислоты. Смесь 60,4 мг (0,085 ммоль) [3-[2[бензилоксикарбонил-[4-[4-(метансульфониламино)фенил]бутил]амино]этил]-2-(3,5-диметилфенил)-1 Н-индол-5-ил]-2-метилпропионовой кислоты, 46,8 мг (0,09 ммоль) реагента РуВОР,0,042 мл (36,3 мг, 0,51 ммоль) пирролидина и 0,400 мл сухого метиленхлорида перемешивали в атмосфере азота при комнатной температуре в колбе с притертой пробкой. Через 21 ч раствор распределяли между этилацетатом и 0,5 н хлористо-водородной кислотой. Органический слой промывали насыщенным водным раствором бикарбоната натрия, а затем сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью препаративной ТСХ на 4 пластинах с силикагелем GF 1000 микрон (проявленных вCH2Cl2-MeOH, 92,5:7,5; полоса продукта экстрагирована тем же растворителем) и получали 55,0 мг (85%) продукта в виде почти бесцветной твердой пены или стекла; продукт являлся достаточно чистым, как подтверждала ТСХ вCH2Cl2-MeOH, 95:5. 1 Н-ЯМР 500 МГц, (CDCl3) являлся комплексным вследствие присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/e=763,7(M+H)+. Стадия 6.1F. N-[4-[4-[2-[5-(1,1-Диметил-2 оксо-2-пирролидин-1-илэтил)-2-(3,5-диметилфенил)-1H-индол-3-ил]этиламино]бутил] фенил]метансульфонамид. Смесь, содержащую 52,6 мг (0,069 ммоль) бензилового эфира [2-[5-[1,1-диметил-2-оксо-2 пирролидин-1-илэтил)-2-(3,5-диметилфенил)1 Н-индол-3-ил]этил]-[4-(4-метансульфониламинофенил)бутил] карбаминовой кислоты (из стадии 1), 25 мг 20% гидроксида палладия-наугле, 2,5 мл этанола, 2,5 мл этилацетата и 0,010 мл ледяной уксусной кислоты, встряхивали в атмосфере водорода(примерно 45 фунт/кв.дюйм=3,164 кг/см 2) в автоклаве в течение 1,8 ч. Катализатор удаляли путем фильтрации через Целит и фильтрат концентрировали в вакууме. Остаток очищали с помощью препаративной ТСХ на 4 клиновидных пластинах с силикагелем GF Analtech (проявленных в СН 2 Сl2 МеОН-концентрированный 52 7,5:0,75; полоса продукта, экстрагированная тем же растворителем) и получали 38,4 мг (88%) твердой пены кремового цвета; продукт являлся гомогенным, на что указывала ТСХ в CH2Cl2MeOH-конц. NH4OH 92,5:7,5:0,75. 1H-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/e=629,6(М+Н)+. Получение синтетических промежуточных соединений Стадия А. Гидрохлорид этил 2-(4 гидразинофенил)ацетата и гидрохлорид 2-(4 гидразинофенил)уксусной кислоты. Это соединение (смесь этилового сложного эфира и карбоновой кислоты) получали из 13,4 г (75 ммоль) этил 2-(4-аминофенил)ацетата путем реакции диазотирования и восстановления диазониевой соли хлоридом олова (2), как описано в методе L.J.Street, et al., J. Med. Chem.,36, 1529 (1993). Продукт был получен в двух сборах. Первый сбор состоял из 6,40 г порошка,т.пл.200 С. С помощью 1 Н-ЯМР (400 МГц,ДМСО-d6) было обнаружено, что продукт состоял из смеси карбоновой кислоты и этилового сложного эфира приблизительно в молярном отношении 4:3. Масс-спектр (РВ-NН 3/Сl):195(катион арилгидразония сложного этилового эфира). Второй сбор состоял из 4,60 г порошка,т.пл. 180 С. 1H-ЯМР 400 МГц, (ДМСО-d6) указывал на то, что продукт состоял из смеси карбоновой кислоты и этилового сложного эфира приблизительно в молярном отношении 7:1. После уточнения состава смеси в двух сборах,вычисленный полный выход составил 69%. Поскольку этерификация карбоновой кислоты происходит в следующей стадии, то сложный эфир и кислота взаимодействуют с образованием того же самого продукта. Стадия АА. Этил (+/-)-2-(4-нитрофенил) пропионат. К раствору 9,76 г (50 ммоль) (+/-)-2-(4 нитрофенил)пропионовой кислоты в 150 мл абсолютного этанола добавляли 3,0 мл концентрированной серной кислоты. Полученный раствор перемешивали при нагревании с обратным холодильником в атмосфере азота. Через 6 ч раствор охлаждали и интенсивно перемешивали при постепенном добавлении 250 мл насыщенного водного раствора бикарбоната натрия (осторожно: образуется пена). Смесь распределяли между 750 мл этилацетата и 500 мл воды. Органический слой промывали 100 мл насыщенного водного раствора бикарбоната натрия, а затем 100 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 10,86 г (97%) масляного продукта, который является гомогенным (ТСХ:гексан-этилацетат, 9:1). 1H-ЯМР (400 МГц, CDCl3) соответствовал предполагаемой структуре. 53 Стадия ВВ. Этил 2-метил-2-(4-нитрофенил)пропионат. Суспензию 924 мг (23 ммоль) гидрида натрия (60% в масле) в 21 мл сухого N,N-диметилформамида перемешивали в атмосфере азота в ледяной бане при постепенном добавлении в течение около 10 мин раствора 4,68 г (21 ммоль) этил (+/-)-2-(4-нитрофенил)пропионата в 20,5 мл сухого N,N-диметилформамида. Во время добавления появлялась интенсивная фиолетовая окраска. Затем смесь оставляли для нагревания до комнатной температуры. Приблизительно через один час смесь снова охлаждали в ледяной бане при добавлении по каплям с помощью шприца раствора 1,44 мл (3,28 г, 23 ммоль) иодметана в 5 мл сухого N,N-диметилформамида в течение примерно 10 мин, поддерживая при этом внутреннюю температуру при 10-15 С. Эту смесь нагревали до комнатной температуры, после чего цвет этой смеси становился коричневым. Через 1 ч добавляли еще 187 мл (426 мг, 3 ммоль) иодметана. На следующий день смесь представляла собой суспензию из некоторого количества сероватого твердого вещества в золотистой жидкости. Эту суспензию интенсивно перемешивали и гасили путем постепенного добавления 10 мл 5%-ного водного раствора бисульфата калия. Смесь распределяли между 400 мл диэтилового эфира и 400 мл воды. Органический слой промывали еще 3 х 400 мл воды, а затем 50 мл насыщенного водного раствора хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (элюируя гексаном-этилацетатом, 19:1) и получали 4,31 г(87%) масляного продукта, который являлся гомогенным, на что указывала ТСХ в гексанэтилацетат 9:1. 1 Н-ЯМР (400 МГц, CDCl3) предполагаемой структуре. Стадия СС. Этил 2-(4-аминофенил)-2 метилпропионат. Смесь 4,27 г (18 ммоль) этил 2-метил-2-(4 нитрофенил)пропионата, 200 мг 10%-ного палладия-на-угле и 120 мг абсолютного этанола встряхивали в потоке водорода (первоначальное давление водорода 47 фунт/кв.дюйм=3,305 кг/см 2). Катализатор удаляли путем фильтрации через Целит в атмосфере азота и осадок на фильтре промывали дополнительным количеством этанола. Фильтрат концентрировали в вакууме при температуре вплоть до 50 С и получали 3,74 г (100%) масляного продукта, который являлся гомогенным, на что указывала ТСХ в гексан -EtOAc 4:1. 1H-ЯMP (400 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр ЭСИ): m/e=208 (М+Н). Стадия DD. Этил 2-(4-гидразинфенил)-2 метилпропионат. Раствор 3,725 г (18 ммоль) этил 2-(4 аминофенил)-2-метилпропионата в 18 мл концентрированной хлористо-водородной кислоты 54 перемешивали при температуре от -10 до -5 С в бане из льда и ацетона при добавлении по каплям раствора 1,29 г (18,7 ммоль) нитрита натрия в 7,5 мл воды приблизительно в течение 15 мин. Перемешивание продолжали при этой температуре в течение еще 30 мин. Затем небольшое количество нерастворившегося твердого вещества удаляли путем фильтрации в охлажденной колбе-приемнике. После этого фильтрат в течение 10-15 мин по каплям добавляли к раствору 20,3 г (90 ммоль) дигидрата хлорида олова (2) в 14,5 мл концентрированной хлористоводородной кислоты, перемешанному в атмосфере азота в бане из льда и ацетона. Добавление осуществляли так, чтобы внутренняя температура составляла около -5 С. Во время добавления образовывалось смолистое вещество. После завершения добавления перемешивание продолжали в течение 1 ч при температуре от-10 до -5 С. Водную фазу декантировали, а остаток смолистого вещества растворяли в 250 мл этилацетата. Этилацетатный раствор осторожно обрабатывали 250 мл насыщенного водного раствора бикарбоната натрия и встряхивали в делительной воронке. Этилацетатный слой промывали 50 мл насыщенного водного раствора хлорида натрия. Полученную смесь фильтровали,после чего фазы разделяли. Этилацетатную фазу сушили сульфатом магния, фильтровали и концентрировали в вакууме при комнатной температуре с получением 2,59 г (65%) масла. 1 НЯМР (500 МГц, СDСl3) соответствовал предполагаемой структуре и указывал на присутствие лишь небольшого количества примесей. ПРИМЕР 6.2.N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(1 этил-3-метил-2-оксопирролидин-3-ил)-1Hиндол-3-ил]этиламино]бутил]фенил]метансульфонамид. Стадия 6.2 А. -N-Этил-N-(2-гидроксиэтил)-2-(4-нитрофенил)пропионамид. Раствор 1,95 г (10 ммоль) -2-(4-нитрофенил)пропионовой кислоты и 5,46 г (10,5 ммоль) реагента РуВОР в 40 мл сухого метиленхлорида перемешивали в атмосфере азота при охлаждении в бане из воды со льдом. К этому раствору добавляли 4,87 мл (4,46 г, 50 ммоль) 2-(этиламино)этанола. Через 10 мин охлаждающую баню удаляли и раствор оставляли для охлаждения до комнатной температуры. Через 4 дня раствор распределяли между диэтиловым эфиром и 0,5 н хлористо-водородной кислотой. Органический слой промывали насыщенным водным раствором бикарбоната натрия, а затем насыщенным водным раствором хлорида натрия. Органическую фазу сушили 55 сульфатом магния и концентрировали в вакууме. Остаток подвергали флэш-хроматографии на силикагеле (элюируя сначала CH2Cl2-MeOH 99:1, а затем СН 2 Сl2-МеОН 98:2) и получали 1,87 г (70%) светло-желтого вязкого масла, достаточно чистого, что подтверждалось ТСХ вCH2Cl2-MeOH-AcOH 95:5:0,5. 1 Н-ЯМР (500 МГц, CDCl3) являлся комплексным вследствие присутствия ротамеров, но соответствовал предполагаемой структуре. Масс-спектр (РВNH3/Сl): m/e=267,2 (М+Н). Стадия 6.2 В. -1-Этил-3-метил-3-(4 нитрофенил)пирролидин-2-он. Раствор 1,73 г (6,5 ммоль) -N-этил-N-(2 гидроксиэтил)-2-(4-нитрофенил)пропионамида в 17 мл безводного тетрагидрофурана перемешивали в атмосфере азота и охлаждали в ледяной бане при добавлении 1,36 мл (985 мг, 9,75 ммоль) триэтиламина с последующим добавлением по каплям 0,554 мл (819 мг, 7,15 ммоль) метансульфонилхлорида. После завершения добавления смесь удаляли из охлаждающей бани и оставляли для перемешивания при комнатной температуре в колбе с притертой пробкой. Через 44 ч смесь разбавляли 125 мл диэтилового эфира и встряхивали в течение нескольких минут. Эту смесь фильтровали в потоке азота и промывали еще некоторым количеством диэтилового эфира. Фильтрат концентрировали в вакууме и получали 1,85 г неочищенного метансульфоната в виде красноватого масла. В осушенной колбе 312 мг (7,8 ммоль) гидрида натрия (60% в масле) перемешивали в атмосфере азота при добавлении с помощью шприца 10 мл безводного тетрагидрофурана. Полученную суспензию перемешивали в ледяной бане в атмосфере азота при добавлении по каплям раствора неочищенного метансульфоната в 10 мл безводного тетрагидрофурана в течение приблизительно 45 мин. Через 20 ч темно-коричневую смесь гасили путем добавления по каплям небольшого количества ледяной уксусной кислоты до тех пор, пока цвет смеси не менялся на светло-оранжевый. Через несколько минут смесь разбавляли примерно 50 мл эфира и перемешивали еще некоторое время. Нерастворившиеся соли удаляли путем фильтрации (осадок на фильтре промывали еще некоторым количеством эфира) и фильтрат концентрировали в вакууме. Остаточное масло подвергали флэшхроматографии на силикагеле (элюируя гексанEtOAc 2:1) и получали 786 мг (49%) золотистооранжевого масляного продукта, который был достаточно чистым, на что указывала ТСХ гексан -EtOAc 1:1. 1H-ЯМР 500 МГц (CDCl3) соответствовал предполагаемой структуре. Массспектр (РВ-NН 3/Сl): m/e=291,1 (М+Н)+. Стадия 6.2 С. -3-(4-Аминофенил)-1-этил 3-метилпирролидин-2-он. Смесь 770 мг (3,1 ммоль) -1-этил-3 метил-3-(4-нитрофенил)пирролидин-2-она, 50 мг 10% палладия-на-угле и 50 мл абсолютного 56 этанола встряхивали в потоке водорода (первоначальное давление водорода 46 фунт/кв.дюйм=3,234 кг/см 2) в автоклаве в течение 2,5 ч. Катализатор удаляли путем фильтрации через целит в атмосфере азота и осадок на фильтре промывали дополнительным количеством этанола. Фильтрат концентрировали в вакууме и получали 684 мг (количественный выход) ярко окрашенного в светло-розовый цвет масляного продукта; этот продукт в основном являлся гомогенным, что подтверждала ТСХ гексан -EtOAc 1:1. 1H-ЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Масс-спектр (PB-NH3/Cl): m/e=219,1 (М+Н). Стадия 6.2D. -1-Этил-3-(4-гидразинофенил-3-метилпирролидон-2-он. Раствор 676 мг (3,1 ммоль) -3-(4 аминофенил)-1-этил-3-метилпирролидин-2-она(из стадии 3) в 3,1 мл концентрированной хлористо-водородной кислоты перемешивали при температуре около -5 С в бане из льда и ацетона при добавлении по каплям раствора 222 мг (3,2 ммоль) нитрита натрия в 1,25 мл воды в течение около 15 мин. Перемешивание продолжали в течение 45 мин при температуре от -10 до -5 С. Смесь выдерживали на холоде и постепенно добавляли к раствору 3,50 г (15,5 ммоль) дигидрата хлорида олова (2) в 2,5 мл концентрированной хлористо-водородной кислоты, перемешанному в атмосфере азота при 0 С в бане из льда и ацетона. После завершения добавления,перемешивание продолжали в охлаждающей бане в течение около 30 мин, а затем смесь распределяли между 25 мл этилацетата и 4 мл воды. Водный слой промывали еще 10 мл этилацетата. Объединенные этилацетатные фракции осторожно обрабатывали 25 мл насыщенного водного раствора карбоната натрия, в результате чего образовывался тяжелый осадок и выделялось небольшое количество газа. После разбавления дополнительным количеством этилацетата и воды смесь фильтровали. Фильтрат переносили в делительную воронку и этилацетатный слой промывали насыщенным водным раствором хлорида натрия. Органическую фазу сушили сульфатом магния, фильтровали, концентрировали в вакууме при комнатной температуре и получали 396 мг (55%) вязкого желтого масла; в основном одно пятно (ТСХ, СН 2 Сl2 95:5). 1HЯМР (500 МГц, CDCl3) соответствовал предполагаемой структуре. Стадия 6.2 Е. -3-[3-(2-Аминоэтил)-2-(3,5 диметилфенил)-1 Н-индол-5-ил]-1-этил-3-метилпирролидин-2-он. Раствор 395 мг (1,7 ммоль) -1-этил-3-(4 гидразинофенил)-3-метилпирролидин-2-она и 359 мг (1,7 ммоль) 3-хлорпропил 3,5 диметилфенилкетона (примера 4) в 7 мл абсолютного этанола перемешивали при нагревании с обратным холодильником в атмосфере азота в течение 60 ч. Затем раствор охлаждали и концентрировали в вакууме. Остаток очищали с(элюент:CH2Cl2-MeOH 95:5, а затем СН 2 Сl2 МеОН-конц.NН 4OН 95:5:0,5) и получали 71,4 мг продукта в виде примеси, который затем очищали на 4 клиновидных препаративных ТСХпластинах с силикагелем Analtech (проявленных в CH2Cl2-MeOH-конц.NH4OH 95:5:0,5; полосу продукта экстрагировали тем же растворителем), и получали 38,4 мг (5,8%) светлооранжевой твердой смолы; продукт являлся фактически гомогенным, на что указывала ТСХ в СН 2 Сl2-МеОН-конц. NH4OH 90:10:1. 1H-ЯМРm/e=390,1 (М+Н). Стадия 6.2F. -N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(1-этил-3-метил-2-оксопирролидин-3-ил)-1 Н-индол-3-ил]этиламино]бутил] фенил]метансульфонамид. Смесь 37,0 мг (0,095 ммоль) -3-[3-(2 аминоэтил)-2-(3,5-диметилфенил)-1 Н-индол-5 ил]-1-этил-3-метилпирролидин-2-она (из стадии 5), 25,3 мг (0,105 ммоль) 4-[4-(метансульфонамидо)фенил]бутиральдегида (пример 4) и 57,0 мг (0/475 ммоль) безводного сульфата магния продували азотом и охлаждали в бане из льда-метанола примерно при температуре от -10 до -5 С по мере постепенного добавления с помощью шприца 0,300 мл сухого СDСl3. Смесь перемешивали в атмосфере азота при этой температуре в течение 15-20 мин. Мембрану удаляли на период добавления 4,5 мг (0,12 ммоль) боргидрида натрия и раствор снова продували азотом. Смесь перемешивали при температуре от 10 до -5 С при постепенном добавлении 0,200 мл сухого метанола и перемешивание продолжали при этой температуре. Через 25 мин смесь распределяли между 5 мл этилацетата и 5 мл воды. Этилацетатный слой промывали насыщенным солевым раствором, а затем сушили сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали с помощью препаративной ТСХ на клиновидных пластинах с силикагелем GF Analtech (проявленных в СН 2 Сl2-МеОН-конц.NН 4OН 92,5:7,5:0,75). Полосу продукта выделяли (путем экстракции тем же растворителем) и получали 32,3 мг (55%) твердой светло-желтой пены, которая являлась гомогенной, на что указывала ТСХ в CH2Cl2 МеОН-конц.NН 4OН 92,5:7,5:0,75. 1H-ЯМР 500 МГц (CDCl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/e=615,4N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(1 этил-4,4-диметил-5-оксо-4,5-дигидро-1 Нпиразол-3-ил)-1H-индол-3-ил]этиламино]бутил] фенил] метансульфонамид. Стадия 6.3 А. 2-Этил-4,4-диметил-5-(4 нитрофенил)-2,4-дигидропиразол-3-он. Смесь 1,00 г (4 ммоль) метилового эфира 2,2-диметил-3-(4-нитрофенил)-3-оксопропионовой кислоты (Yang C.Y.; Wnek G.E. Polymer,1992, 33, 4191-4196), 3,00 г (20 ммоль) оксалата этилгидразина, 8 мл 2-метоксиэтанола и 4 мл ледяной уксусной кислоты перемешивали в атмосфере азота при мягком нагревании с обратным холодильником в течение 24 ч. Охлажденный раствор концентрировали в вакууме. Остаток распределяли между этилацетатом и водой. Этилацетатную фазу промывали дополнительным количеством воды, а затем насыщенного водного раствора хлорида натрия. Органический раствор сушили сульфатом магния, фильтровали и концентрировали в вакууме с получением 703 мг (67%) светло-желтых кристаллов,т.пл. 121-122 С; продукт являлся гомогенным(М+Н). Стадия 6.3 В. 5-(4-Аминофенил)-2-этил-4,4 диметил-2,4-дигидропиразол-3-он. Реакцию гидрирования 2-этил-4,4 диметил-5-(4-нитрофенил)-2,4-дигидропиразол 3-она проводили, как описано в стадии С примера 6.2, и получали количественный выход продукта в виде светлого коричневато-желтого твердого вещества, т.пл. 118-120,5; продукт являлся гомогенным, на что указывала ТСХ гексан -EtOAc 1:1 и CH2Cl2-MeOH 98:2. 1H-ЯМР 500 МГц (CDCl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ): m/e=232,1(М+Н). Стадия 6.3 С. 2-Этил-5-(4-гидразинофенил)-4,4-диметил-2,4-дигидропиразол-3-он. Это соединение получали из 5-(4 аминофенил)-2-этил-4,4-диметил-2,4-дигидропиразол-3-она способом, описанным в стадии D примера 6.2, за исключением того, что всю реакционную смесь после восстановления хлоридом олова (2) перемешивали в ледяной бане и осторожно обрабатывали избыточным количеством насыщенного раствора карбоната натрия с получением осадка. Этот осадок переносили в делительную воронку и встряхивали с этилацетатметиленхлоридом (2:1). Смесь фильтровали,а затем фазы разделяли. Водную фазу экстрагировали еще несколькими порциями этилацетата. Объединенные органические фракции концентрировали в вакууме и получали продукт с выходом 80% в виде аморфного светлого оранжево-желтого твердого вещества, т.пл. 131,5135 С; с разлож.; неточно определено с помощью ТСХ в CH2Cl2-MeOH 95:5. 1H=ЯМР 500 59 МГц (СDСl3) соответствовал предполагаемой структуре. Стадия 6.3D. 5-[3-(2-Аминоэтил)-2-(3,5 диметилфенил)-1 Н-индол-5-ил]-2-этил-4,4 диметил-2,4-дигидропиразол-3-он. Это соединение получали из 2-этил-5-(4 гидразинофенил)-4,4-диметил-2,4-дигидропиразол-3-она и 3-хлорпропил 3,5-диметилфенилкетона (пример 4), как описано в стадии Е примера 6.2, за исключением того, что время реакции составляло 15 ч. Неочищенный продукт подвергали флэш-хроматографии на силикагеле(92,5:7,5:0,75). 1H-ЯМР 500 МГц (CDCl3) соответствовал предполагаемой структуре. Массспектр (PB-NH3/Cl): m/e=403,2 (М+Н)+. Стадия 6.3 Е. N-[4-[4-[2-[2-(3,5-Диметилфенил)-5-(1-этил-4,4-диметил-5-оксо-4,5-дигидро-1 Н-пиразол-3-ил)-1H-индол-3-ил]этиламино] бутил]фенил]метансульфонамид. Реакцию восстановительного аминирования 4-[4-(метансульфонамидо)фенил] бутиральдегида (пример 4) с 5-[3-(2-аминоэтил)-2(3,5-диметилфенил)-1 Н-индол-5-ил]-2-этил-4,4 диметил-2,4-дигидропиразол-3-оном проводили как описано в способе стадии F примера 6.2, за исключением того, что для проявления препаративных ТСХ-пластин использовали СН 2 Сl2 МеОН, 90:10, а для экстракции продукта использовали СН 2 Сl2-МеОН-конц.NH4OH,90:10:1. Таким образом был получен продукт с выходом 55% в виде очень бледного золотистожелтого стекла; гомогенность подтверждена с помощью ТСХ в CH2Cl2-MeOH, 92,5:7,5. 1HЯМР (500 МГц, СDСl3) соответствовал предполагаемой структуре. Масс-спектр (ЭСИ):m/e=628,5 (M+H)+. Нижеследующие соединения получали способом, описанным в примере 6.1.J.Am.Chem.Soc, 1949, 71, 2272) в 50 мл этанола перемешивали при нагревании с обратным холодильником в атмосфере азота в течение 20 ч. Затем раствор охлаждали и концентрировали в вакууме. Остаток обрабатывали 50 мл 2 н хлористо-водородной кислоты, помешивали в течение нескольких минут, а затем экстрагировали смесью 50 мл диэтилового эфира и 10 мл тетрагидрофурана. Отфильтрованную водную фазу перемешивали при 60 С в атмосфере азота в течение 4 ч. Охлажденный раствор концентрировали в вакууме и остаток распределяли между 100 мл эфира и 50 мл насыщенного раствора бикарбоната натрия (осторожно: образуется пена). Смесь фильтровали, после чего фазы разделяли. Органическую фазу концентрировали и очищали с помощью колоночной хроматографии на силикагеле (элюирование в градиенте 0,5-2% МеОН в СН 2 Сl2), в результате чего получали 1,62 г (25%) темно-коричневатого масла; продукт являлся достаточно чистым, на что указывала ТСХ в CH2Cl2-MeOH 95:5. 1HЯМР (300 МГц, СDСl3) соответствовал предполагаемой структуре. Масс-спектр (FAB):m/e=260 М+Н). Стадия 7.1 В. 4-(1-Бутил-1 Н-имидазол-2 илметил )фениламин. Раствор 259 мг (1 ммоль) 1-бутил-2-(4 нитробензил)-1 Н-имидазола в 3 мл тетрагидрофурана перемешивали в ледяной бане при добавлении по каплям раствора 1,36 г (6 ммоль) дигидрата хлорида олова (2) в 1,8 мл концентрированной хлористо-водородной кислоты в течение 3-4 мин. После удаления ледяной бани,смесь перемешивали в атмосфере азота при комнатной температуре в течение 3,5 ч. Затем ее добавляли к смеси 9 мл 50% гидроксида натрия и приблизительно 35 г льда. После помешивания в течение 2-3 мин продукт экстрагировали 50 мл диэтилового эфира. Органическую фазу сушили (сульфатом натрия), фильтровали и концентрировали в вакууме. Остаток растирали с небольшим количеством эфира и получали кристаллическое твердое вещество, которое со
МПК / Метки
МПК: C07D 413/02, A61K 31/535
Метки: гонадотропин-высвобождающего, антагонисты, фактора
Код ссылки
<a href="https://eas.patents.su/30-828-antagonisty-gonadotropin-vysvobozhdayushhego-faktora.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонисты гонадотропин-высвобождающего фактора.</a>
Производные индола как антагонисты рецептора 5-нт

Номер патента: 304
Опубликовано: 29.04.1999
Авторы: Дэвис Дэвид Томас, Гэстер Лэрэми Мэри, Малхоллэнд Кит Раймонд, Форбес Ян Томсон, Вайман Пол Эдриан, Джоунз Грэхэм Элджин, Дакворт Дэвид Малькольм
МПК: A61K 31/44, C07D 401/12
Метки: 5-нт, производные, рецептора, антагонисты, индола
Формула / Реферат:
1. Соединение формулы (I) или его соль где Р1 и Р2 независимо представляет собой фенил, ароматические или частично насыщенные моноциклические или бициклические гетероциклические кольца, содержащие до трех гетероатомов, выбранных из азота, кислорода или серы; А представляет собой связь, цепь из 1-5 атомов, необязательно замещенных С1-6алкилом, или А представляет собой необязательно замещенный фенил или необязательно замещенное 5-7-членное...
Пиразолпиримидины как антагонисты крф рецепторов.

Номер патента: 803
Опубликовано: 24.04.2000
Авторы: Уилкоуксен Кейт М., Вебб Томас Р., Чен Чен, Маккарти Джеймс Р., Моран Тиренс Дж., Хуанг Чарльз
МПК: A61K 31/495, C07D 487/04
Метки: рецепторов, пиразолпиримидины, антагонисты, крф
Формула / Реферат:
1. Соединение формулы I включая его стереоизомерные формы и фармацевтически приемлемые кислотно-аддитивные соли, где R1 представляет собой NR4R5 или OR5; R2 представляет C1-6-алкил, C1-6-алкилокси или C1-6-алкилтио; R3 представляет водород, C1-6-алкил, C1-6-алкилсульфонил, C1-6-алкилсульфокси или C1-6-алкилтио; R4 представляет водород, C1-6-алкил, моно- или ди(С3-6-циклоалкил) метил, С3-6-циклоалкил, С3-6-алкенил, гидрокси-С1-6-алкил,...
Cпособ получения фактора ix из биологических источников и фактор ix, полученный этим способом

Номер патента: 222
Опубликовано: 24.12.1998
Авторы: Йосич Дьюро, Хоффер Лутц, Морфельд Франк
Метки: источников, полученный, фактор, получения, способом, этим, cпособ, биологических, фактора
Формула / Реферат:
1. Способ получения фактора IX из биологических источников с помощью хроматографии, отличающийся тем, что перед разделением с помощью аффинной хроматографии указанный источник обрабатывается сульфатом аммония в концентрации от 1,5-2,3 моль/л. 2. Способ по п.1, отличающийся тем, что указанным биологическим источником является плазма крови или плазма, истощенная по фактору VIII и фибриногену путем отделения криопреципитата (криообедненная...
Трициклические бензазепиновые антагонисты вазопрессина.

Номер патента: 607
Опубликовано: 29.12.1999
Авторы: Делос Сантос Эфрен Гильермо, Ду Ксумей, Рейч Марвин Фред, Венкатесан Аранапакан Мудумбай, Олбрайт Джей Дональд
МПК: C07D 495/14, A61K 31/55
Метки: антагонисты, бензазепиновые, вазопрессина, трициклические
Формула / Реферат:
1. Трициклические бензазепины общей формулы I где Y представляет связь или фрагмент, выбранный из -СН2-, -СНОН, -СНО-алкил (C1-C6), -СН-S-алкил(С1-C6), -CHNH2, -CHN-алкил(C1-C6), -С[N-алкил(C1-C6)]2, -СНОСО-алкил(С1-C6), -CHNH (CH2)mNH2, -CHNH(CH2)m-NH-алкил(C1-C6), -CHNH (CH2)m-N[алкил(C1-С6)]2; -СНNН(СН2)m-S-алкил(С1-C6), -CHNH(CH2)m-O-алкил(C1-C6), S, О, -NH, -N-алкил (C1-C6), -NCO-алкил (C1-C6); m представляет целое число от 2...
Анализ крови на соотношение активности фактора v для оценки вероятности заболевания тромбоэмболией

Номер патента: 673
Опубликовано: 28.02.2000
Авторы: Ку Дехью Вэйн, Аркел Йэйл С.
МПК: G01N 33/86, C12Q 1/56
Метки: анализ, тромбоэмболией, крови, вероятности, соотношение, оценки, активности, фактора, заболевания
Формула / Реферат:
1. Способ идентификации субъектов с риском тромботических нарушений вследствие дефекта фактора V, включающий a) инкубацию анализируемой плазмы с первым реагентом, содержащим фосфолипидную композицию и контактный активатор; b) добавление второго реагента, содержащего активированный белок С, к первой аликвоте анализируемой плазмы и инкубации первой аликвоты и второй аликвоты; и c) измерение активности фактора V плазмы в первой аликвоте и во...
Предыдущий патент: Отопительный водогрейный котел “люкс-1″
Следующий патент: Антагонисты гонадотропин-высвобождающего фактора.
Случайный патент: Антитела против pcrv