Соединения индазола, фармацевтические препараты для ингибирования протеинкиназ и способы их применения
Номер патента: 4460
Опубликовано: 29.04.2004
Авторы: Кания Роберт Стивен, Темпчик-Расселл Анна Мария, Браганза Джон Ф., Томас Кристин, Джонсон Теодор Отто Младший, Луу Хип Те, Тенг Мин, Варни Майкл Дэвид, Хуа Йе, Уоллас Майкл Бреннан, Борчардт Аллен Дж., Криппс Стефан Джеймс, Бендер Стивен Ли, Джонсон Майкл Дэвид, Коллинз Майкл Рэймонд, Палмер Синтия Луиза, Райх Зигфрид Хайнц







Формула / Реферат
1. Соединение формулы I

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является незамещенным C1-C8алкилом, C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 является необязательно замещенным алкилом C1-C8; и
R2 представляет собой 6-12-членный арил или 5-12-членный гетероарил, необязательно замещенный алкилом C1-C6, или группу -Y-X, где Y представляет собой O, S, C=CH2, C=O, C2-C8алкилиден, -NH или -N-(C1-C8алкил), а X является 6-12-членным арилом, 5-12-членным гетероарилом, NH-(C1-C8алкилом), NH-(C3-C8циклоалкилом), NH-(5-12-членным гетероциклоалкилом), NH-(5-12-членным арилом), NH-(5-12-членным гетероарилом), NH-(C1-C8алкоксилом) или NH-(ди-C1-C8алкиламидом) и X необязательно замещен группой OH, галоидом, C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, или группой формулы NH-C(OR11), или C(O)NH-R11, где R11 представляет собой 5-12-членный гетероарил или 6-12-членный арил, необязательно замещенный алкилом C1-C8 или C(O)(C1-C8алкилом), или R11 является C1-C8алкилом, C2-C8алкенилом, C2-C8алкинилом, C1-C8алкоксигруппой, NH-(5-12-членным гетероарилом), или R11 является C1-C8циклоалкилом, необязательно замещенным 6-12-членным арилом;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
2. Соединение формулы I(a)

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8; и
R2 представляет собой 6-12-членный арил или Y-Ar, где Y представляет собой O, S, C=CH2, C=O, CH2, CHCH3, NH или N-(C1-C8алкил) и Ar является 6-12-членным арилом, необязательно замещенным группой OH, галоидом, C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, или группой формулы NH-C(OR11), или C(O)NH-R11, где R11 представляет собой 5-12-членный гетероарил или 6-12-членный арил, необязательно замещенный алкилом C1-C8 или C(O)(C1-C8алкилом), или R11 является C1-C8алкилом, C2-C8алкенилом, C2-C8алкинилом, C1-C8алкоксигруппой, NH-(5-12-членным гетероарилом), или R11 является C3-C8циклоалкилом, необязательно замещенным 6-12-членным арилом;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
3. Соединение формулы II

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является C1-C8алкилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
R4 и R7, каждый независимо, представляет собой атом водорода, группу OH, галоид, C1-C8алкил, C1-C8алкокси, C2-C8алкенил, арилокси, тиоарил, CH2OH, CH2O-(C1-C8алкил), CH2O-арил, CH2-S-(C1-C8алкил) или CH2-S-арил;
R5 и R6, каждый независимо, представляет собой атом водорода, группу OH, галоид, Z-(C1-C8алкил), Z-(C6-C12)арил или Z-CH2CH=CH2, где Z представляет собой O, S, NH или CH2, а каждый алкильный и арильный фрагмент Z-алкила и Z-арила необязательно замещен C1-C8алкилом;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
4. Соединение по п.3, в котором
R1 представляет собой 7-12-членный бициклический гетероарил или группу формулы CH=CH-R3, где R3 является 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
каждый R4 и R7 независимо представляет собой атом водорода или C1-C8алкил и
каждый R5 и R6 независимо представляет собой галоид, Z-(C1-C8алкил) или Z-CH2CH=CH2, где Z представляет собой O или S и алкил необязательно замещен C1-C8алкилом;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
5. Соединение формулы III

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является C1-C8алкилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O, S, C=CH2, C=O, S=O, SO2, CH2, CHCH3, NH или N-(C1-C8алкил);
R8 является C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом, 5-12-членным гетероарилом, C1-C8алкоксилом или C6-C12арилоксилом;
R10 независимо выбирают из атома водорода, галогена и низшего алкила;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
6. Соединение по п.5, в котором
R1 представляет собой 7-12-членный бициклический гетероарил или группу формулы CH=CH-R3, где R3 является 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O, S, C=CH2, C=O, NH или N-(C1-C8алкил);
R8 представляет собой 6-12-членный арил, 5-12-членный гетероарил, C1-C8алкил или C2-C8алкенил и
R10 является атомом водорода или галогена,
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
7. Соединение формулы III(a)

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил, или группу формулы CH=CH-R3, или CH=N-R3, где R3 является C1-C8алкилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O, S, C=CH2, C=O, S=O, SO2, CH2, CHCH3, NH или N-(C1-C8алкил);
R8 является C1-C8алкилом, C2-C8алкенилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членньм арилом, 5-12-членным гетероарилом, C1-C8алкоксилом или C6-C12арилоксилом;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
8. Соединение по п.7, в котором
R1 представляет собой 7-12-членный бициклический гетероарил или группу формулы CH=CH-R3, где R3 является 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O, S, C=CH2, C=O, NH или N-(C1-C8алкил) и
R8 представляет собой 6-12-членный арил, 5-12-членный гетероарил, C1-C8алкил или C2-C8алкенил,
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
9. Соединение по п.7, в котором
R1 представляет собой группу CH=CH-R3, где R3 является 6-12-членной арильной группой, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O или S и
R8 представляет собой 6-12-членный арил или 5-12-членный гетероарил,
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
10. Соединение формулы IV

в которой R1 представляет собой 6-12-членный арил или 5-12-членный гетероарил или группу формулы CH=CH-R3 или CH=N-R3, где R3 является C1-C8алкилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой O, S, C=CH2, C=O, S=O, SO2, CH2, CHCH3, NH или N-(C1-C8алкил);
R9 является C1-C8алкилом, C3-C8циклоалкилом, 5-12-членным гетероциклоалкилом, 6-12-членным арилом, 5-12-членным гетероарилом, C1-C8алкоксилом, C6-C12арилоксилом; C3-C12циклоалкоксилом, NH-(C1-C8алкилом), NH-(6-12-членным арилом), NH-(5-12-членным гетероарилом), N=CH-(C1-C8алкилом), NH(C=O)R11 или NH2, где R11 независимо выбирают из атома водорода, C1-C8алкила, C3-C8циклоалкила, 5-12-членного гетероциклоалкила, 6-12-членного арила и 5-12-членного гетероарила; и
R10 независимо выбирают из атома водорода, галогена и низшего алкила;
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
11. Соединение по п.7, в котором
R1 представляет собой группу CH=CH-R3, где R3 является 6-12-членным арилом или 5-12-членным гетероарилом, и R1 необязательно замещен алкилом C1-C8;
Y представляет собой S или NH и
R9 представляет собой C1-C8алкил, C1-C8алкоксил или NH-(5-12-членный гетероарил);
или его фармацевтически приемлемое пролекарственное производное, или его фармацевтически приемлемая соль.
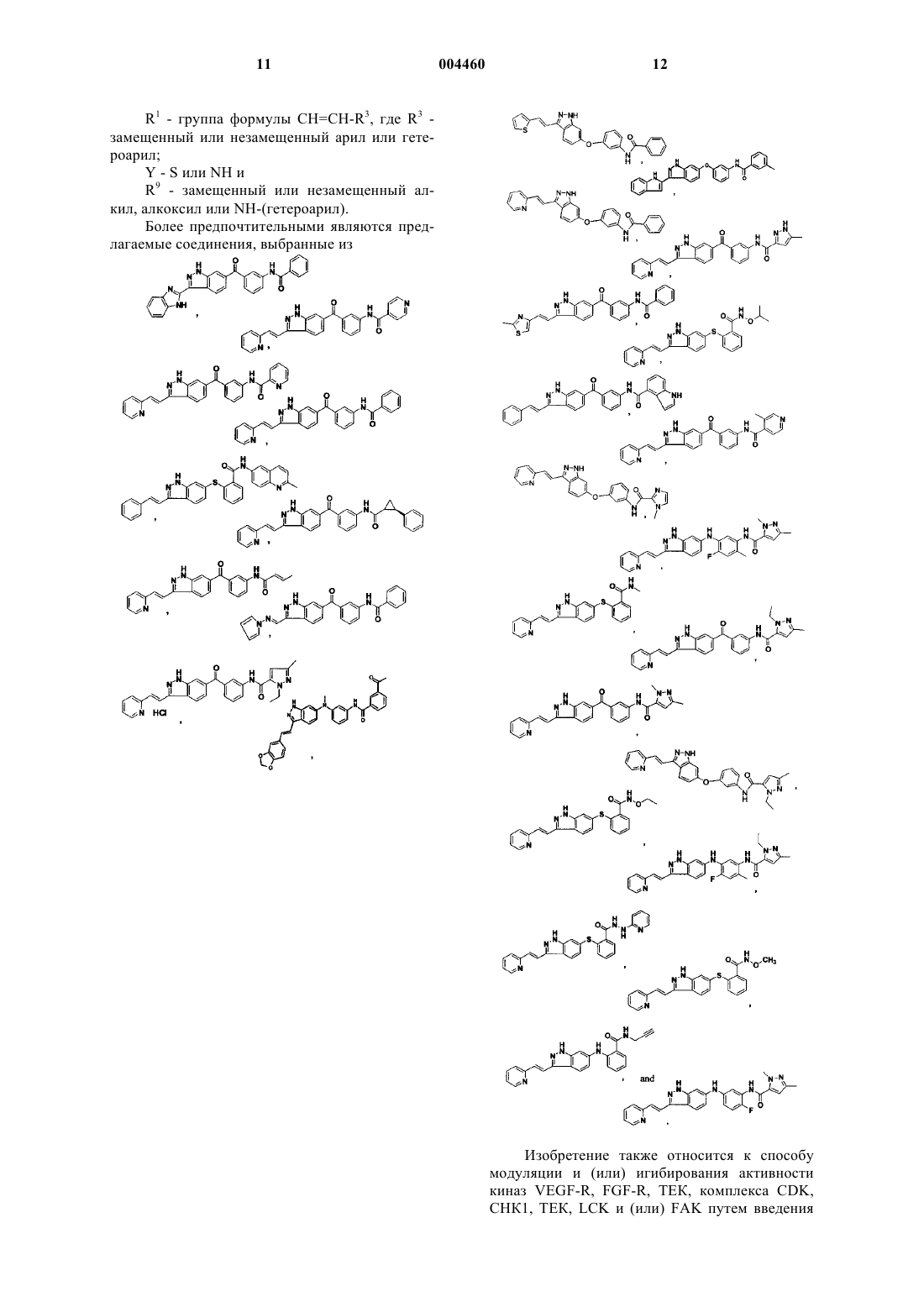
12. Соединение, его фармацевтически приемлемое пролекарственное производное или его фармацевтически приемлемая соль, выбранные из



13. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения по п.1, или его фармацевтически приемлемого пролекарственного производного, или фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или средство доставки.
14. Способ лечения заболеваний млекопитающих, медиируемых активностью протеинкиназы, заключающийся во введении млекопитающему эффективного количества соединения по п.1, или его фармацевтически приемлемого пролекарственного производного, или фармацевтически приемлемой соли.
15. Способ по п.14, при котором заболевание млекопитающего связано с ростом опухоли, пролиферации клеток или ангиогенезом.
16. Способ модуляции активности рецептора протеинкиназы, заключающийся в обеспечении контакта рецептора протеинкиназы с эффективным количеством соединения по п.13, или его фармацевтически приемлемого пролекарственного производного, или фармацевтически приемлемой соли.
17. Способ по п.16, при котором рецептором протеинкиназы является рецептор VEGF.
Текст
1 Область техники, к которой относится изобретение Настоящее изобретение относится к области химии и биологии и, в частности, к соединениям индазола, медиирующим и(или) ингибирующим активность некоторых протеинкиназ, а также к фармацевтическим препаратам, содержащим такие соединения. Изобретение также относится к терапевтическому или профилактическому применению таких соединений и препаратов, а также к способам лечения рака и других заболеваний, связанных с нежелательным ангиогенезом и (или) распространением клеток путем введения эффективных доз указанных соединений. Уровень техники Протеинкиназы представляют собой семейство ферментов, катализирующих фосфорилирование гидроксильной группы специфических остатков тирозина, серина или треонина в белках. Обычно такое фосфорилирование резко нарушает функции белка, при этом протеинкиназы являются ключевыми в регулировании широкого разнообразия клеточных процессов,включая метаболизм, размножение клеток и их выживание. Среди многих различных клеточных функций, для которых, как известно, необходима активность протеинкиназ, некоторые процессы являются привлекательными мишенями для терапевтического вмешательства при определенных заболеваниях. Два примера: ангиогенез и регулирование клеточного цикла, в которых протеинкиназы играют главную роль. Эти процессы существенны для роста твердых опухолей, а также при других заболеваниях. Ангиогенез - это механизм, по которому из существующих сосудов образуются новые капилляры. При необходимости сосудистая система имеет возможность вырабатывать новые сети капилляров с целью поддержания надлежащего функционирования тканей и органов. Однако у взрослых ангиогенез достаточно ограничен и имеет место только при заживлении ран и реваскуляризации эндометрия при менструациях. См. Merenmies et al., Cell GrowthDifferentiation, 8, 3-10 (1997). С другой стороны, нежелательный ангиогенез является признаком нескольких болезней, таких как ретинопатия,псориаз, ревматоидный артрит, возрастная макулярная дегенерация (AMD) и рак (твердые опухоли). Folkman, Nature Med., 1, 27-31 (1995). Протеинкиназы, которые по имеющимся данным участвуют в процессах сосудообразования,включают три члена семейства тирозинкиназ рецепторов роста: VEGF-R2 (рецептор 2 фактора роста сосудистого эндотелия, известный также как KDR (рецептор домена вставки киназы) и как FLK-1); FGF-R (рецептор фактора роста фибробластов); и ТЕК (известный так же какVEGF-R2, который проявляется только на клетках эндотелия, связывает фактор роста со 004460 2 судов VEGF и является посредником в последующей трансдукции сигналов путем активации своей внутриклеточной активности киназ. Таким образом, ожидается, что прямое игибирование активности киназ VEGF-R2 приведет к уменьшению сосудообразования даже в присутствии экзогенного VEGF (см. Strawn et al., Cancer Research, 56, 3540-3545 (1996, как было показано на мутантах VEGF-R2, которые не способствовали трансдукции сигналов. Millaueret al., Cancer Research, 56, 1615-1620 (1996). Далее, представляется, что VEGF-R2 не функционирует у взрослых, кроме как путем обеспечения ангиогенной активности VEGF. Поэтому можно ожидать, что избирательный ингибитор активности киназ VEGF-R2 будет иметь низкую токсичность. Аналогично FGF-R связывает факторы роста сосудов aFGF и bFGF и способствует последующей межклеточной трансдукции сигналов. Недавно было высказано предположение,что такие факторы роста, как bFGF, могут играть важную роль в запуске ангиогенеза в твердых опухолях, достигших определенного размера. Yoshiji et al., Cancer Research, 57, 3924-3928(1997). Однако в отличие от VEGF-R2 FGF-R проявляется в ряде различных типов клеток организма и может быть или не быть важным в других нормальных физиологических процессах взрослого организма. Тем не менее, имеются сообщения о том, что системное введение мелкомолекулярного ингибитора активности киназFGF-R блокирует вызываемый bFGF ангиогенез у мышей без видимой токсичности. Mohammadiet al., EMBOJournal, 17, 5996-5904 (1998). ТЕК (известный также как Tie-2) является рецептором тирозинкиназ, проявляющихся только на клетках эндотелия, которые, как было показано, играют роль в ангиогенезе. Связывание фактора ангиопротеин-1 приводит к автофосфорилированию домена киназы в ТЕК и к процессу трансдукции сигнала, который, как представляется, является посредником взаимодействия клеток эндотелия с опорными клетками периэндотелия, способствуя, таким образом,созреванию вновь образуемых кровеносных сосудов. С другой стороны, представляется, что фактор ангиопротеин-2 является антагонистом действия ангиопротеина-1 на ТЕК и нарушает образование сосудов. Maisonpierre et al., Science,277, 55-60 (1997). В результате описанных выше разработок было предложено лечить ангиогенез с применением соединений, ингибирующих активность киназ VEGF-R2, FGF-R и (или) ТЕК. Например,в публикации ВОИС международной заявкиWO 97/34876 описаны некоторые производные циннолина, являющиеся ингибиторами VEGFR2, которые можно применять для лечения заболеваний, связанных с ненормальным ангиогенезом и (или) повышенной проницаемостью сосудов, таких как рак, диабет, псориаз, ревма 3 тоидные артриты, саркома Капоши, гемоангиома, острые и хронические нефропатии, атерома,артериальный ретиноз, автоиммунные заболевания, острые воспаления и глазные заболевания с разрастанием сосудов сетчатки. Фосфорилаза киназа активирует гликоген фосфорилазу, увеличивая, таким образом, распад гликогенов и выделение глюкозы в печени. Регуляция выработки глюкозы в печени происходит при диабетах типа 2 и является основной причиной голодной гипергликемии, приводящей ко многим вторичным осложнениям для таких пациентов. Поэтому снижение выделения глюкозы из печени приведет к снижению повышенных содержаний глюкозы в плазме. Таким образом, ингибиторы фосфорилаза киназы снижают активность фосфорилазы и гликогенолиза, ослабляя гипергликемию у пациентов. Другой физиологической реакцией наVEGF является сверхпроницаемость сосудов,которая, как считается, играет роль на ранних этапах ангиогенеза. В ишемических тканях,встречающихся, например, в мозгу жертв приступов стенокардии, гипоксия вызывает экспрессию VEGF, приводя к увеличению проницаемости сосудов и, в конечном счете, к отечности окружающих тканей. На крысиной модели приступа стенокардии было показано van Bruggen et al., J. Clinical Invest., 104, 1613-20 (1999),что введение моноклонального антитела вVEGF уменьшает обширность инфаркта. Таким образом, ингибиторы VEGFR, возможно, будут полезны при лечении приступов стенокардии. Помимо своей роли в ангиогенезе, протеинкиназы играют решающую роль в регулировании клеточного цикла. Неконтролируемое размножение клеток является признаком рака. Размножение клеток в ответ на различные стимулы проявляется в виде нарушения регуляции цикла деления клеток - процесса размножения и деления клеток. Обычно опухолевые клетки имеют повреждения генов, которые прямо или косвенно регулируют ход цикла деления клеток. Циклинзависимые киназы (CDK) - это серин-треонин протеинкиназы, играющие определяющую роль в регулировании переходов между различными стадиями клеточного цикла. См.,например, статьи в Science, 274, 1643-1677(1996). Комплексы CDK образуются путем ассоциирования регулирующей субъединицы циклина (например, циклин A, B1, B2, D1, D2,D3 и Е) и субъединицы каталитической киназы(например, CDC2 (CDK1), CDK2, CDK4, CDK5 и CDK6). По своему названию CDK проявляют абсолютную зависимость от субъединицы циклина при фосфорилировании своих целевых субстратов, при этом различные пары киназациклин регулируют развитие на специфических стадиях клеточного цикла. Именно CDK4 в комплексе с D-циклинами играет ведущую роль в запуске цикла деления клетки из положения покоя в положение, при 4 котором клетка становится готовой к делению. Это развитие зависит от разнообразия механизмов регулирования роста, как отрицательных,так и положительных. Считается, что отклонения в этой системе регулирования, в частности такие, которые оказывают влияние на функцииCDK4, переводят клетки в состояние повышенной способности к размножению, типичное для злокачественных образований, в частности, семейных меланом, карцином пищевода, и раковым заболеваниям поджелудочной железы. См.,например, Kamb, Trends in Genetics, 11, 136-140(1995); Kamb et al., Science, 264, 436-440 (1994). В огромном количестве публикаций описаны разнообразные химические соединения,полезные для применения против различных терапевтических мишеней. Например, в международных публикациях ВОИСWO 99/23077 и WO 99/23076 представлены индазолсодержащие соединения, проявляющие ингибирующую активность типа фосфодиэстеразы типа IV, продуцируемой заменой катехолбиоизостеры на индазол. В патенте США 5,760,028 описаны гетероциклические соединения, включающие 3-[1-[3-(имидазолин-2-иламино)пропил]индазол-5-илкарбониламино]-2-(бензилоксикарбониламино)пропионовую кислоту,полезные в качестве антагонистов v3 интегрина и связанных с ним протеин рецепторов, обладающих адгезией к поверхности клетки. В международной публикации ВОИСWO 98/09961 описаны некоторые производные индазола и их применение в качестве ингибиторов фосфодиэстеразы (PDE) типа IV или для продуцирования фактора некроза опухоли (TNF) у млекопитающих. Недавние дополнения к виртуальной библиотеке известных соединений включают такие, которые описывают как противопролиферативные терапевтические средства, ингибирующие CDK. Например, в патенте США 5,621,082 на имя Xiong et al. представлена нуклеиновая кислота, кодирующая ингибитор CDK6, а в европейской патентной публикации 0 666 270 А 2 представлены пептиды и миметики пептидов, действующие в качестве ингибиторов CDK1 и CDK2. В международной публикации ВОИСWO 97/16447 описаны некоторые аналоги хромонов, ингибирующие циклинзависимые киназы, в частности, комплексы CDK/циклин, такие как СDK4/циклинD1, которые можно использовать для ингибирования чрезмерного или ненормального размножения клеток, и, следовательно, для лечения рака. В международной публикации ВОИСCDK. При этом все еще существует необходимость в применении мелкомолекулярных соединений, которые легко синтезировать и которые эффективны при ингибировании одного или нескольких CDK или CDK/циклин комплексов. Так как CDK4 может являться общим активато 5 ром деления большинства клеток, а комплексыCDK4 и циклины типа D циклинов регулируют раннюю фазу G1 клеточного цикла, существует необходимость в эффективных ингибиторахCDK4 и его циклиновых комплексах типа D для лечения одного или нескольких видов опухолей. Кроме того, главенствующая роль циклинаE/CDK2 и циклин B/CDK1 киназ соответственно на стадии G1/S и при переходах G2/M обеспечивают дополнительные мишени для терапевтического воздействия при подавлении нерегулируемого развития клеточного цикла при раковом заболевании. Другая протеинкиназа СНК 1 играет важную роль в качестве контрольной точки развития клеточного цикла. Контрольные точки - это регулирующие системы, координирующие развитие клеточного цикла, влияя на образование,активацию и последующую инактивацию циклинзависимых киназ. Контрольные точки предотвращают развитие клеточного цикла в неподходящее время, поддерживают метаболическое равновесие клеток во время приостановки развития клетки, а также в некоторых случаях могут запускать апоптоз (запрограммированная клеточная смерть), если не удовлетворяются требования контрольной точки. См., например,O'Connor, Cancer Surveys, 29, 151-182 (1997);Science, 246, 629-634 (1989). Один ряд контрольных точек контролирует целостность генома, при этом после обнаружения повреждения ДНК эти "контрольные точки повреждения ДНК" блокируют развитие клеточного цикла на стадиях G1 и G2 и замедляют развитие вплоть до стадии S. O'Connor, Cancer Surveys, 29, 151-182 (1997); Hartwell et al.,Science, 266, 1821-1828 (1994). Это обеспечивает завершение процессов репарации ДНК перед репликацией генома и последующим выделением генетического материала в новые дочерние клетки. Важно, что наиболее часто мутирующий ген при раке у человека - ген подавления опухолей р 53 - продуцирует белок в контрольной точке повреждения ДНК, который блокирует ход клеточного цикла на стадии G1 и (или) запускает апоптоз (запрограммированную клеточную смерть) в результате повреждения ДНК. Hartwell et al., Science, 266, 1821-1828 (1994). Было показано также, что супрессор опухолей р 53 усиливает действие контрольной точки повреждения ДНК на стадии G2 клеточного цикла. См.,например, Bunz et al., Science, 282, 1497-1501(1997). Учитывая определяющее влияние пути супрессора опухолей р 53 при раке у человека, ведутся активные поиски терапевтических воздействий с использованием уязвимостей рака, дефектного по р 53. Одна из возможных уязвимых 6 точек заключается в действии контрольной точки G2 в раковых клетках с дефектом р 53. Поскольку раковые клетки не имеют регулирования со стороны контрольной точки G1, они особенно уязвимы к падению последнего остающегося препятствия, защищающего их от противоракового воздействия средств повреждения ДНК: контрольной точки G2. Контрольная точкаG2 регулируется системой регулирования, которая сохраняется от дрожжей до человека. Важным элементом этой сохранившейся системы является киназа СНК 1, которая осуществляет трансдукцию сигналов от комплекса, чувствительного к повреждению ДНК, для ингибирования активации циклин B/Cdc2 киназы, способствующей развитию митотической энергии. См.,например, Peng et al., Science, 277, 1501-1505(1997). Было показано, что инактивация СНК 1 одновременно прекращает приостановку G2,вызванную повреждением ДНК под влиянием либо противораковых средств, либо эндогенного повреждения ДНК, и приводит к выборочной гибели появившихся в контрольной точке дефектных клеток. См., например, Nurse, Cell, 91,865-867 (1997); Weinert, Science, 277, 1450-1451(1993); и Al-Khodairy et al., Molec. Biol. Cell, 5,147-160 (1994). Избирательное манипулирование регулированием в контрольных очках раковых клеток может иметь более широкое распространение в химиотерапевтических и радиологических методах лечения рака, а также может являться общим признаком "неустойчивости генома" в раке у человека для использования в качестве основы для избирательного разрушения раковых клеток. Ряд факторов определяют СНК 1 в качестве основной мишени при регулировании в контрольной точке повреждения ДНК. Истолкование ингибиторов этих и функционально связанных с ними киназ, таких как Cds1/CHK2 киназа,которая, как было недавно установлено, взаимодействует с СНК 1 при регулировании стадии(1998, могло бы способствовать получению новых ценных терапевтических средств для лечения раковых заболеваний. Связывание рецептора интегрина с ЕСМ вызывает появление межклеточных сигналов,медиируемых FAK (киназа фокальной адгезии),участвующих в подвижности клеток, размножении клеток и их выживании. При раке у человека, чрезмерная экспрессия FAK приводит к росту опухолей и метастатического потенциала,благодаря их роли в путях передачи сигналов,медиируемых интегрином. Тирозин киназы могут быть рецепторного типа (с внеклеточными, трансмембранными и межклеточными доменами) или нерецепторного типа (когда они полностью межклеточные). 7 По меньшей мере, одна из нерецепторных протеин тирозин киназ, а именно, LCK, считается медиатором трансдукции в Т-клетках сигнала от взаимодействия повехностно-клеточного белка (Cd4) со сшитым противо-Cd4 антителом. Более подробное рассмотрение нерецепторных тирозинкиназ приведено в работе Bolen,Oncogene, 8, 2025-2031 (1993), которая включена как часть описания настоящего изобретения в виде ссылки. Кроме описанных выше протеинкиназ многие другие протеинкиназы рассматривали в качестве мишеней для терапии, при этом многочисленные публикации содержат описание ингибиторов активности киназ, как указано ниже:al., Exp. Opin. Invest. Drugs, 7, 553-573 (1998). В публикации ВОИС международной заявки WO 00/18761 описаны некоторые замещенные 3 цианохинолины в качестве ингибиторов протеинкиназ. В то же время, существует необходимость в эффективных ингибиторах протеинкиназ. Кроме того, как понятно специалистам в данной области, желательно, чтобы ингибиторы киназ обладали сродством с киназой или киназами мишеней одновременно с высокой избирательностью в отношении других протеинкиназ. Сущность изобретения Таким образом, цель изобретения - создание сильнодействующих ингибиторов протеинкиназы. Другая цель изобретения - создание эффективных ингибиторов протеинкиназ, обладающих сильным и избирательным средством с одной или несколькими определенными киназами. Указанные и другие цели изобретения, которые станут понятны из приведенного ниже описания, достигаются созданием описанных ниже соединений индазола и их фармацевтически приемлемых пролекарственных производных, фармацевтически активных метаболитов и фармацевтически приемлемых солей (при этом такие соединения, пролекарственные производные, метаболиты и соли вместе называемые средствами), которые модулируют и/или ингибируют активность протеинкиназ. Фармацевтические композиции, содержащие также средства, полезны при лечении заболеваний, медиируемых активностью киназ, таких как рак, а также других заболеваний, связанных с нежелательным ангиогенезом и/или пролифирацией клеток, например, диабетической ретинопатии,неоваскулярной клаукомы, ревматоидных артритов и псориаза. Кроме того, эти средства обладают положительными свойствами при модулировании и/или ингибировании активности 8 киназ, связанной с комплексами VEGF-R, FGFR, CDK, CHK1, LCK, TEK, FAK, и/или киназой фосфорилазы. В общем случае изобретение относится к соединениям формулы IR1 - замещенный или незамещенный арил или гетероарил, или группа формулы CH=CH-R3 или CH=N-R3, гдеR3 - замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил,арил или гетероарил; иR2 - замещенный или незамещенный арил,гетероарил или Y-X, где Y - О, S, C=CH2, C=O,S=O, SO2, алкилиден, NH или N-(C1-C8 алкил) и Х - замещенный или незамещенный Аr, гетероарил, NН-(алкил), NH-(циклоалкил), NH(гетероциклоалкил), NН(арил), NН(гетероарил), NH(алкоксил) или NH-(диaлкилaмид), где Аrарил. Изобретение также относится к фармацевтически приемлемым пролекарствам, фармацевтически приемлемым метаболитам и фармацевтически приемлемым солям соединений формулы I. Описаны также предпочтительные способы приготовления соединений формулы I. Изобретение также относится к соединениям формулы I(а)R1 - замещенный или незамещенный арил или гетероарил или группа формулы СН=СН-R3 или CH=N-R3, где R3 - замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил, арил или гетероарил; иR2 - замещенный или незамещенный арил или Y-Ar, где Y - О, S, С=СН 2, С=O, SO, SO2,СН 2, СНСН 3, NH или N-(C1-C8 алкил), a Ar замещенный или незамещенный арил. Изобретение также относится к фармацевтически приемлемым пролекарствам, фармацевтически активным метаболитам и фармацевтически приемлемым солям соединений формулыI(а). Описаны также предпочтительные способы приготовления соединений формулы I(а). В одном из предпочтительных вариантов осуществления изобретение относится к соединениям формулы IIR1 - замещенный или незамещенный арил или гетероарил или группа формулы СН=СН-R3 или CH=N-R3, где R3 - замещенный или незамещенный алкил, циклоалкил, гетероциклоалкил,арил или гетероарил;- О, S, NH или СН 2, при этом алкильные и арильные составляющие Z-алкила и Z-арила каждая может быть замещенной; и их фармацевтически приемлемые пролекарства, фармацевтически приемлемые метаболиты и фармацевтически приемлемые соли. В предпочтительном варианте формулы II:R1 - замещенный или незамещенный бициклический гетероарил или группа формулы СН=СН-R3, гдеR3 - замещенный или незамещенный арил или гетероарил;R5 и R6 - каждый независимо, галоген, Zалкил, или Z-СН 2 СН=СН 2, где Z - О или S. В другом общем предпочтительном варианте осуществления изобретения предлагаются соединения формулы IIIR1 - замещенный или незамещенный арил или гетероарил или группа формулы СН=СН-R3 или CH=N-R3, где R3 - замещенный или незамещенный алкил, циклоалкил, гетероциклоалкил,арил или гетероарил;R8 - замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил,арил, гетероарил, алкоксил или арилоксил;R10 независимо выбран из водорода, галогена и низшая алкильной группы; и их фармацевтически приемлемые пролекарства, фармацевтически приемлемые метаболиты и фармацевтически приемлемые соли. Более предпочтительно в формуле III: R1 замещенный или незамещенный бициклический гетероарил или группа формулы СН=СН-R3, гдеR3 - замещенный или незамещенный арил или гетероарил; Y - О, S, С=СН 2, С=O, NH или N(C1-C8 алкил); R8 - замещенный или незамещенный арил, гетероарил, алкил и алкенил, и R10 водород или галоген. 10 В другом предпочтительном варианте предлагаются соединения формулы III(а)R1 - замещенный или незамещенный арил или гетероарил или группа формулы СН=СН-R3 или CH=N-R3, гдеR3 - замещенный или незамещенный алкил, циклоалкил, гетероциклоалкил, арил или гетероарил; Y - О, S, C=CH2, C=O, S=O, SO2,СН 2, СНСН 3, NH или N-(C1-C8 алкил);R8 - замещенный или незамещенный алкил, алкенил, циклоалкил, гетероциклоалкил,арил, гетероарил, алкоксил или арилоксил; и их фармацевтически приемлемые пролекарства, фармацевтически приемлемые метаболиты и фармацевтически приемлемые соли. Более предпочтительно в формуле III(а):R1 - замещенный или незамещенный бициклический гетероарил или группа формулы СН=СН-R3, гдеR3 - замещенный или незамещенный арил или гетероарил; Y - О, S, С=СН 2, С=O, NH илиR8 - замещенный или незамещенный арил или гетероарил. В другом предпочтительном варианте предлагаются соединения формулы IVR1 - замещенный или незамещенный арил или гетероарил или группа формулы СН=СН-R3 или CH=N-R3, где R3 - замещенный или незамещенный алкил, циклоалкил, гетероциклоалкил,арил или гетероарил;R9 - замещенный или незамещенный алкил, циклоалкил, гетероциклоалкил, арил,гетероарил, алкоксил, арилоксил, циклоалкоксил, NH-(C1-C8 алкил), NH-(арил), NН(гетероарил), N=CH-(алкил), NH(C=O)R11 илиNH2, где R11 независимо выбран из водорода,замещенного или незамещенного алкила, циклоалкила, гетероциклоалкила, арила и гетероарила; иR10 независимо выбран из водорода, галогена и низшей алкильной группы; и их фармацевтически приемлемые пролекарства, фармацевтически приемлемые метаболиты и фармацевтически приемлемые соли. Более предпочтительно в формуле IV:R1 - группа формулы СН=СН-R3, где R3 замещенный или незамещенный арил или гетероарил;R9 - замещенный или незамещенный алкил, алкоксил или NH-(гетероарил). Более предпочтительными являются предлагаемые соединения, выбранные из Изобретение также относится к способу модуляции и (или) игибирования активности киназ VEGF-R, FGF-R, ТЕК, комплекса CDK,СНК 1, ТЕК, LCK и (или) FAK путем введения 13 соединения формулы I или его фармацевтически приемлемого пролекарства, фармацевтически активных метаболитов или фармацевтически приемлемых солей. Изобретение также предлагает соединения, обладающего избирательной активностью в отношении киназ, т.е. они обладают существенной активностью против одной специфической киназы, обладая меньшей или минимальной активностью против другой киназы. В одном предпочтительном варианте осуществления изобретения предлагаемые соединения - соединения формулы I, обладающие значительно более сильным действием против VEGF рецептора тирозинкиназы, чем против FGF-R1 рецептора тирозинкиназы. Изобретение также направлено на создание способов модуляции активности VEGF рецептора тирозинкиназ без существенной модуляции активности FGF рецептора тирозинкиназ. Предлагаемые соединения также могут успешно использоваться в сочетании с другими известными лечебными средствами. Например,соединения формулы I, II, III или IV, обладающие противоангиогенным действием, могут вводиться совместно с цитотоксическими химиотерапевтическими средствами, такими как таксол, таксотер, винбластин, цисплатин, доксорубицин, адриамицин и подобными с целью обеспечения усиленного противоопухолевого воздействия. Аддитивное или синергетическое усиления терапевтического воздействия можно также обеспечить совместным введением соединений формулы I, II, III или IV, обладающих противоангиогенным действием, с другими противоангиогенными средствами, такими как комбретастатин А-4, эндостатин, приномастат, целекоксиб, рофококсиб, EMD121974, IM862,анти-VEGF моноклональные антитела и антиKDR моноклональные антитела. Изобретение также относится к фармацевтическим препаратам, каждый из которых содержит: эффективную дозу средства, выбранного из соединений формулы I и их фармацевтически приемлемых солей, фармацевтически активных метаболитов, и фармацевтически приемлемых пролекарств, а также фармацевтически приемлемый носитель такое средства. Изобретение также направлено на создание способов лечения рака, а также других заболеваний, связанных с нежелательным ангиогенезом и (или) размножением клеток, при котором вводят эффективное количество указанных агентов пациенту, нуждающемуся в указанном лечении. Предлагаемые соединения формулы I, II,III и IV полезны для медиирования активности протеинкиназ. Более конкретно, эти соединения полезны в качестве противоангиогенных средств, а также в качестве средств для модуляции и (или) игибирования активности протеинкиназ, обеспечивая, таким образом, лечение рака или других заболеваний, связанных с раз 004460 14 множением клеток, медиируемым протеинкиназами. Термин "алкил" в данном случае употребляется в отношении к алкильным группам с прямолинейной или разветвленной цепью,имеющим от одного до двенадцати атомов углерода. Примерами алкильных групп являются метил (Me), этил (Et), n-пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил (t-Bu),пентил, изопентил, трет-пентил, гексил, изогексил и т.п. Термин "низший алкил" означает алкил, имеющий от 1 до 8 атомов углерода (алкилC1-8). Подходящие замещенные алкилы включают фторометил, дифторометил, трифторометил, 2-фтороэтил, 3-фтороэтил, гидроксиметил,2-гидроксиэтил, 3-гидроксипропил и т.п. Термин "алкилиден" относится к двухвалентному радикалу, имеющему от одного до двенадцати атомов углерода. Примеры алкилиденовых групп включают СН 2, СНСН 3, (СН 3)2 и т.п. Термин "алкенил" относится к алкенилгруппам с прямолинейной или разветвленной цепью, имеющим от двух до двенадцати атомов углерода. Примеры алкенилгрупп включают проп-2-енил, бут-2-енил, бут-3-енил, 2-метилпроп-2-енил, гекс-2-енил и т.п. Термин "алкинил" относится к прямоцепным и разветвленным алкинильным группам,имеющим от двух до двенадцати атомов углерода. Термин "циклоалкил" относится к насыщенным или частично ненасыщенным карбоциклическим соединениям, имеющим от трех до двенадцати атомов углерода, включая би- трициклические структуры циклоалкила. Примерами циклоалкила являются циклопропил, циклобутил, циклофенил, циклогексил, циклогептил и т.п."Гетероциклоалкильная" группа означает насыщенный или частично ненасыщенный моноциклический радикал, содержащий атомы углерода, предпочтительно 4 или 5 кольцевых атома углерода и, по меньшей мере, один гетероатом, выбранный из азота, кислорода и серы. Термины "арил" и "гетероарил" относятся к моно- и полициклическими ненасыщенным или ароматическими кольцевым структурам,при этом "арил" означает те из них, которые являются карбоциклами, а "гетероарил" - те из них, которые являются гетероциклами. Примеры ароматических кольцевых структур включают фенил, нафтил, 1,2,3,4-тетрагидронафтил,фурил, тиенил, пирролил, пиридинил, пиразолил, имидазолил, пиразинил, пиридазинил,1,2,3-триазинил,1,2,4-оксадиазолил,1,3,4 оксадиазолил, 1-Н-тетразол-5-ил, индолил, хинолинил, бензофуранил, бензотиофенил (тианафтенил) и т.п. Такие компоненты могут быть замещены слитно-кольцевой структурой или мостиком, например ОСН 2-O. 15 Термин "алкокси" означает радикал -Oалкил. Примерами являются метокси, этокси,пропокси и т.п. Термин "арилокси" представляет -О-арил,где арил определен выше. Термин "циклоалкоксил" представляет -Оциклоалкил, где циклоалкил определен выше. Термин "галоген" представляет хлор, фтор,бром или йод. Термин "гало" представляет хлоро, фторо, бромо или йодо. Обычно различные составные части или функциональные группы переменных в формулах могут в качестве варианта замещаться одним или несколькими подходящими заместителями. Примеры заместителей включают галоген(F, Cl, Вr или I), низший алкил, -ОН, -NO2, -CN,-СО 2 Н, -O-низший алкил, -арил, -арил-низший алкил, -СO2 СН 3, -CONH2, -OCH2CONH2, -NH2,-SO2NH2, галоалкид (например, -СF3, -СН 2 СF3), O-галоалкил (например, -ОСF3, -ОСHF2), и т.п. Термины "содержащий" и "включающий" употребляются в открытом, не ограничивающем смысле. Очевидно, что, хотя соединение формулы I может проявлять таутомерию, начертания формул в данном описании изобретения представляют в явном виде только одну из возможных таутомерных форм. В связи с этим следует отметить, что формула в данном описании изобретения представляют любую таутомерную форму изображаемого соединения, а не ограничены лишь той конкретной таутомерной формой, которая представлена в начертаниях формул. Некоторые предлагаемые соединения могут существовать в виде отдельных стереоизомеров (т.е. практически свободны от других стереоизомеров), рацематов и (или) смесей энантиомеров и (или) диастереомеров. Все подобные отдельные стереоизомеры, рацематы, их смеси входят в объем данного изобретения. Предпочтительно предлагаемые соединения,если они оптически активны, используется в оптически чистом виде. Как должно быть, в общем, понятно специалистам в данной области, оптически чистое соединение, имеющее один хиральный центр(т.е. один несимметричный атом углерода) - это такое соединение, которое состоит в основном из одного из двух возможных энантиомеров (т.е. оно энантиометрически чистое), а оптически чистое соединение, имеющее более одного хирального центра, - это такое соединение, которое одновременно диастереометрически и энатиометрически чистое. Предпочтительно предлагаемые соединения используют в виде, по меньше мере на 90% оптически чистых, т.е. в виде, содержащем не менее 90% одного изомера(80% энантиометрический избыток ("е.е.") или диастереометрический избыток ("d.e.", более предпочтительно не менее 95% (90% е.е. илиd.e.), еще более предпочтительно не менее 16 97,5% (95% е.е. или d.e.), и наиболее предпочтительно не менее 99% (98% е.е. или d.e.). Кроме того, подразумевается, что эти формулы охватывают сольватированные и несольватированные формы указанных структур. Например, формула I включает соединения одинаковой структуры в гидратированном и негидратированном виде. Другие примеры сольватов включают структуры в сочетании с изопропанолом, этанолом, метанолом, ДМСО, этилацетатом, уксусной кислотой или этаноламином. Кроме соединений формулы I, II, III и IV,изобретение охватывает фармацевтически приемлемые пролекарства, фармацевтически активные метаболиты, и фармацевтически приемлемые соли указанных соединений."Фармацевтически приемлемое пролекарство" - это соединение, которое можно превратить в физиологических условиях или путем сольволиза в описанное соединение или в фармацевтически приемлемую соль такого соединения."Фармацевтически активный метаболит" означает фармакологически активный продукт,получаемый путем метаболизма в организме из указанного соединения или его соли. Метаболиты соединения можно определять, пользуясь обычными известными методиками, а их активность определяется тестами, описанными ниже. Пролекарства и активные метаболиты соединения можно определять посредством обычных методик, известных в данной области техники. См., например, Bertolini, Г. et al., J. Med"Фармацевтически приемлемая соль" означает соль, которая сохраняет биологическую эффективность свободных кислот и оснований указанного соединения и которая не является биологически или иным образом нежелательной. Предлагаемое соединение может содержать достаточно кислые, достаточно основные или и те, и другие функциональные группы, и, соответственно, реагировать с рядом неорганических или органических оснований и неорганических или органических кислот с образованием фармацевтически приемлемой соли. Примерами фармацевтически приемлемых солей являются соли, полученные реакцией предлагаемых соединений с минеральной или органической кислотой или с неорганическим основанием, например, соли, включая сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды, бромиды, 17 иодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, форматы, изобутираты, капроаты, гептаноаты, пропиолаты, окасалаты,малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бетен-1,4-диоаты, гексин-1,6 диоаты, бензоаты, хлоробензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты, ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, -гидроксибутираты, гликоляты, тартраты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты, нафталин -2-сульфонаты и манделаты. Если предлагаемое соединение является основанием, необходимую фармацевтически приемлемую соль можно получить любым подходящим известным способом, например, обработкой свободного основания неорганической кислотой, например, соляной кислотой, бромистоводородной кислотой, серной кислотой,азотной кислотой, фосфорной кислотой и т.п. или органической кислотой, например, уксусной кислотой, малеиновой кислотой, янтарной кислотой, миндальной кислотой, фумаровой кислотой, пирувиновой кислотой, гликолевой кислотой, салициловой кислотой, пиранозидиловой кислотой, например, глюкуроновой кислотой, альфа-гидрози кислотой, например, лимонной кислотой или виннокаменной кислотой,аминокислотой, например аспарагиновой кислотой или глютаминовой кислотой, ароматической кислотой, например, бензойной кислотой или коричной кислотой, сульфоновой кислотой,например, р-толуолсульфоновой кислотой или этансульфоновой кислотой и т.п. Если предлагаемое соединение является кислотой, необходимую фармацевтически приемлемую соль можно получить любым подходящим способом, например, обработкой свободной кислоты неорганическим или органическим основанием, например, амином (первичным, вторичным или третичным), гидроксидом щелочного металла или гидроксидом щелочноземельного металла и т.п. Примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глициновая и арганиновая, аммиака, первичного, вторичного и третичного аминов и циклических аминов,таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия,кальция, калия, магния, марганца, железа, меди,цинка, алюминия и лития. В случае твердых препаратов специалистам должно быть понятно, что предлагаемые соединения и соли могут быть в различных кристаллических или полиморфных состояниях,при этом все они входят в объем изобретения и указанных формул. Терапевтически эффективные количества предлагаемых средств можно использовать для лечения заболеваний, медиируемых модуляцией или регуляцией протеинкиназ. Под "эффектив 004460 18 ным количеством" понимается такое количество средства, которое при введении млекопитающему, нуждающемуся в подобном лечении, достаточно для лечения заболевания, медиируемого активностью одной или нескольких протеинкиназ, таких как тирозинкиназ. Таким образом, например, терапевтически эффективное количество соединения формулы I,его соли, активного метаболита или пролекарства представляет собой количество, достаточное для модуляции, регуляции или ингибирования активности одной или нескольких протеинкиназ в такой степени, что заболевание, медиируемое указанной активностью, ослабляется или уменьшается. Количество данного средства, соответствующее такой дозе, меняется в зависимости от таких факторов, как конкретное соединение, вид заболевания и его тяжесть, вес млекопитающего, нуждающегося в лечении, но оно ни в коем случае не может быть обычным способом определено специалистом. "Лечение" означает, по меньшей мере, смягчение характера заболевания млекопитающего, например, человека, который подвергается, по меньшей мере, частично воздействию одной или нескольких протеинкиназ, таких как тирозинкиназы, и включает: предотвращение возникновения заболевания у млекопитающего, в частности, когда установлено,что млекопитающее имеет предрасположенность к такому заболеванию, но заболевание у него еще не диагностировано, модуляцию и(или) игибирование заболевания и (или) смягчение характера заболевания. Предлагаемые средства можно получать,используя схемы реакций и схемы синтеза, описанные ниже, применяя известные приемы и доступные исходные материалы. При применении одного общего процесса синтеза соединения формулы I получают по следующей схеме реакции: 6-Нитроиндазол (соединение V) обрабатывают йодом и основанием, например, NaOH в водно-органической смеси, предпочтительно с диоксаном. Смесь подкисляют, и выделяют продукт фильтраций. К полученному 3-йодо-6 нитроиндазолу в растворе смеси дихлорметана 50% водного КОН при 0 С добавляют реагент защитной группы ("Pg") (где Х = гало), предпочтительно хлорид триметилсилилэтоксиметила (SEM-Cl) и катализатор фазного переноса,например, бромистую соль тетрабутиламмония(ТВАВr). Через 1-4 ч две фазы разбавляют, отделяют органику, высушивают сульфатом натрия, фильтруют и сгущают. Неочищенный продукт очищают силикагелевой хроматографией с получением соединения формулы VI. Обработка соединений формулы VI в соответствующем органическом растворителе с соответствующим R1-металлоорганическим реагентом,предпочтительно R1-бороновой кислотой в присутствии водного основания, например, карбоната натрия и соответствующего катализатора,предпочтительно Рd(РРh3)4 дает после экстрагирования и силикагелевой хроматографии соединения формулы VII. Заместитель R1 можно обменивать в соединениях формулы VII или последующих промежуточных продуктах в ходе выполнения схемы окислительным расщеплением (например, озонолизом) с последующими добавлениями к полученным альдегидным функциональным группам с превращениями Виттига или конденсации (как представлено в примере 42(а-е. Обработка соединений формулы VII восстановителем, предпочтительноSnCl2, обеспечивает после обычной водной обработки и очистки получение соединений фор 004460= NH или N-низший алкил, соединения формулы VIII можно обрабатывать хлоридами, бромидами, йодидами или трифлатами арила или гетероарила в присутствии основания, предпочтительно Сs2 СО 3 и катализатора, предпочтительно Pd-BINAP (где Y = N-низший алкил, с последующим этапом алкилирования) для получения соединений формулы X. Для получения других Y связей добавляют нитрит натрия к соединениям формулы VIII в стандартных условиях охлаждения в кислом водном растворе с последующим добавлением йодида калия и осторожным согреванием. После стандартной обработки и очистки получают йодистые соединения формулы IX. Обработка соединений формулы IX металлоорганическим реагентом, например, бутиллитием способствует обмену лития и галогена. Далее этот промежуточный продукт вводят в реакцию с электрофилом R2, например, карбонилом или трифлатом с возможным посредничеством дополнительных металлов и катализаторов, предпочтительно хлорида цинка иPd(PPh3)4 с получением соединений формулы X. В качестве варианта соединения формулы IX можно обрабатывать элементоорганическими реагентами, такими как органобороновой кислотой в присутствии катализатора, например,Pd(PPh3)4, в атмосфере окиси углерода с получением соединений формулы X. В качестве варианта, в случае производных, в которых Y =NH или S, соединения формулы IX можно обрабатывать соответствующими аминами или тиолами в присутствии основания, предпочтительно Cs2CO3 или К 3 РO4 и катализатора, предпочтительно Pd-BINAP или Pd-(бис-циклогексил) бифенилфосфина с получением соединений формулы X. Можно далее использовать обычные замены функциональных групп, такие как реакции окисления, восстановления, алкилирования, ацилирования, конденсации и удаления защитных групп для приготовления дальнейших производных этого ряда с получением окончательных соединений формулы I. Предлагаемые соединения формулы I можно также получать по общей методике,представленной на следующей схеме: 6-Йодоиндазол(ХI) обрабатывают йодом и основанием, например, NaOH в водноорганической смеси, предпочтительно с диоксаном. Смесь подкисляют, и продукт ХII выделяют фильтрованием. К полученному 3,6 дийодоиндазолу в смеси дихлорметана-50% водного КОН при 0 С добавляют реагент с защитной группой, предпочтительно SEM-Cl и катализатор фазового переноса, например, ТВАВr. Две фазы разбавляют, отделяют органику, высушивают сульфатом натрия, фильтруют и сгущают. Неочищенный продукт очищают силикагелевой хроматографией с получением соединений формулы ХIII. Обработка соединений формулы ХIII в соответствующем органическом растворителе соответствующимR2-металлоорганическим реагентом, например, R2-ZnCl или борным R2-бор реагентом и соответствующим катализатором, предпочтительно Рd(РРh3)4 дает после экстрагирования и силикагелевой хроматографии соединения формулы XIV. Обработка соединений формулы XIV в соответствующем органическом растворителе соответствующимR1- металлоорганическим реагентом (например,борным R1-бор реагентом или R1-ZnCl) в присутствии водного основания, карбоната натрия и подходящего катализатора, предпочтительно Рd(РРh3)4 дает после экстрагирования и силикагелевой хроматографии соединения формулыXV. Можно далее использовать обычные замены функциональных групп, такие как реакции окисления, восстановления, алкилирования,ацилирования, конденсации и удаления защитных групп для получения дальнейших производных этого ряда с получением окончательных соединений формулы I. В качестве варианта соединения формулыI, где R2 - замещенный или незамещенный YAr, где Y - О или S, можно получать по следующей общей схеме Раствор 3-хлороциклогекс-2-енона (XV) в ацетоне при перемешивании, H-R2 и безводный карбонат калия кипятят с обратным холодильником в течение 15-24 ч, остужают и фильтруют. После сгущения и хроматографии фильтрата на силикагеле получают 3-R2-циклогекс-2 енон (XVI). Кетоны XVI можно вводить в реакцию с соответствующим основанием (М-В), предпочтительно бис(триметилсилил)амидом лития и сR1-CO-X (где Х = гало), и после стандартной кислотной обработки и очистки получать соединения формулы XVII. Этот продукт в смеси НОАс/ЕtOН, совмещенный с моногидратом гидразина, нагревают при соответствующей температуре в течение необходимого времени,предпочтительно при 60-80 С в течение 2-4 ч. После охлаждения смесь выливают в насыщенный раствор бикарбоната натрия, экстрагируют органическим растворителем, сгущают и очищают на силикагеле с получением соединений формулы XVIII. Соединения формулы ХVIII можно окислять различными известными способами с получением соединений формулы I. Другие соединения формулы I можно получать способами, аналогичными описанным выше, или пользуясь подробными регламентами, приведенными ниже в примерах. Сродство предлагаемых соединений с рецептором можно улучшать получением многочисленных упакованных копий лиганда, предпочтительно используя "скаффолдинг" (мостки) (scaffolding),образованный составной частью носителя. Показано, что получение таких многовалентных соединений с оптимальным шагом составных частей резко улучшает связывание с рецептором. См., например, Lee et al., Biochem, 23, 4255(1984). Многовалентность и шаг можно регулировать выбором части носителя или единиц линкеров. Такие составные части включают молекулярные носители, содержащие множество функциональных групп, которые можно вводить 23 в реакцию с функциональными группами, связанными с предлагаемыми соединениями. Разумеется, можно использовать различные носители, включая белки, такие как альбумин бычьей сыворотки или HAS, множество пептидов,включая, например, пентапептиды, декапептиды, пентадекапептиды и т.п. Пептиды или белки могут содержать необходимое количество аминокислотных остатков со свободными аминогруппами в боковых цепях, однако, другие функциональные группы, такие как сульфогидрилгруппы или гидроксигруппы можно также использовать для получения устойчивых связей. Соединения, оказывающие сильное регулирующее, модулирующее или ингибирующее действие на активность протеинкиназ, связанных среди прочего с рецепторами FGF, комплексами CDK, ТЕК, СНК 1, LCK, FAK и фосфорилаза киназа, ингибирующие ангиогенез и(или) размножение клеток, целесообразны и составляют одни из предпочтительных вариантов осуществления данного изобретения. Изобретение также направлено на создание способов модуляции или ингибирования активности протеинкиназ, например, в ткани млекопитающего путем введения предлагаемого средства. Активность предлагаемых соединений в качестве модуляторов активности протеинкиназ, а именно активности киназ, может измеряться любыми известными специалистам методами,включая анализы in vivo и (или) in vitro. Примеры подходящих анализов для измерения активности приведены в Parast С. et al., Biochemistry,37, 16788-16801 (1998); Jeffrey et al., Nature, 376,313-320 (July 27, 1995); Публикация ВОИСWO 97/34876 и Публикация ВОИСWO 96/14843. Эти свойства можно оценивать, например, используя одну или несколько методик биологического анализа, описанных в приведенных ниже примерах. Предлагаемые действующие средства могут входить в состав фармацевтических препаратов, как описано ниже. Предлагаемые фармацевтические препараты содержат эффективное количество модулирующего, регулирующего или ингибирующего соединения формулы I, II,III или IV и инертный фармацевтически приемлемый носитель или разбавитель. В одном из вариантов фармацевтических препаратов обеспечены эффективные концентрации предлагаемых средств для реализации терапевтических преимуществ, включающих модуляцию протеинкиназ. Под "эффективными концентрациями" понимаются концентрации, при которых воздействия протеинкиназ, по меньшей мере, регулируются. Эти препараты приготовляются в виде единичных доз, подходящих для данного способа введения, например, парентерально или перорально. Предлагаемое средство вводят в виде обычных доз, приготовленных совмещением терапевтически эффективной дозы средства(например, соединения формулы I) в качестве действующего начала с соответствующими фармацевтическими носителями или разбавителями с использованием стандартных методик. Эти методики могут включать смешивание, гранулирование и прессование или растворение ингредиентов в зависимости от желаемой формы препарата. Применяемый фармацевтический носитель может быть твердым или жидким. Примерами твердых носителей являются лактоза, сахароза,тальк, желатин, агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота и т.п. Примерами жидких носителей являются сироп,арахисовое масло, оливковое масло, вода и т.п. Аналогично, носитель или разбавитель может содержать замедлитель или добавку пролонгированного действия, известные специалистам,такие как моностеарат глицерина или дистеарат глицерина отдельно или с воском, этилцеллюлозой, гидроксипропилметилцеллюлозой, метилметакрилатом и т.п. Можно использовать различные фармацевтические формы. Так, при применении твердого носителя препарат можно таблетировать, помещать в твердые желатиновые капсулы в виде порошка или использовать в виде гранул, либо в виде лепешек или пастилок. Количество твердого носителя может изменяться, но обычно составляет примерно от 25 мг до примерно 1 г. При применении жидкого носителя препарат используется в виде сиропа, эмульсии, мягких желатиновых капсул, стерильного раствора для инъекций или суспензии в ампулах или флаконах, либо в виде неводной жидкой суспензии. Для получения сохранной водорастворимой формы дозы, предлагаемую фармацевтически приемлемую соль растворяют в водном растворе органической или неорганической кислоты, например, в 0,3 М растворе янтарной кислоты или лимонной кислоты. Если нет растворимой формы соли, средство можно растворять в соответствующем сорастворителе или сочетании сорастворителей. Примеры подходящих сорастворителей включают, среди прочего,спирт, пропиленгликоль, полиэтиленгликоль 300, полисорбат 80, глицерин и т.п. в концентрациях, составляющих 0-60% общего объема. В качестве примера, соединение формулы I растворяют в ДМСО и разводят водой. Препарат также может быть в виде раствора соли, являющейся действующим началом, в соответствующем водном носителе, например, в воде или изотоническом физиологическом растворе или растворе декстрозы. Очевидно, что фактическая дозировка средств, применяемых в предлагаемых препаратах, будет меняться в зависимости от применяемого комплекса, конкретной рецептуры препарата, метода введения и конкретного места, организма и заболевания, подлежащего лечению. Учитывая опытный характер средства, специа 25 листы могут подбирать оптимальные дозы для данного набора условий, пользуясь известными тестами по определению дозировки. Для перорального введения примерная суточная доза обычно составляет примерно от 0,001 примерно до 1000 мг/кг веса тела, предпочтительно примерно от 0,001 примерно до 50 мг/кг веса тела,при этом курсы лечения повторяются с соответствующими интервалами. Введение пролекарства обычно дозируется по весовым концентрациям, химически эквивалентным весовым концентрациям полной действующей формы. Предлагаемые препараты могут изготовляться общеизвестными способами, применяемыми для приготовления фармацевтических препаратов, например, с использованием обычных приемов, таких как смешивание, растворение, гранулирование, дражирование, растирание в тонкий порошок, эмульгирование, капсулирование, включение или лиофилизацию. Фармацевтические препараты можно составлять обычным способом с применением одного или нескольких физиологически приемлемых носителей, которые можно выбирать из наполнителей и присадок, обеспечивающих переработку активных соединений в препараты, пригодные для фармацевтического употребления. Правильная рецептура определяется выбранным путем введения. При приготовлении для инъекций предлагаемые средства можно готовить в виде водных растворов предпочтительно в физиологически совместимых буферных растворах, таких как раствор Ханка, раствор Рингера, или физиологический солевой буферный раствор. Для введения через слизистую оболочку в рецептуре используют смачивающие вещества, подобранные для конкретного проницаемого барьера. Такие смачивающие вещества широко известны в технике. Для перорального введения можно составлять рецептуру с соединениями, совмещая активные соединения с фармацевтически приемлемыми носителями, известными в технике. Такие носители позволяют приготовлять предлагаемые соединения в виде таблеток, пилюль,драже, капсул, жидкостей, гелей, сиропов,пульп, суспензий и т.п. для перорального употребления пациентом, проходящим лечение. Фармацевтические препараты для перорального введения можно приготовлять с использованием твердого наполнителя в смеси с активным ингредиентом (средством), при этом можно молоть полученную смесь, а смесь гранул можно перерабатывать при необходимости, добавив соответствующие присадки, для получения таблеток или ядер драже. Подходящие наполнители включают такие наполнители, как сахара, включая лактозу, сахарозу, маннит или сорбит и препараты целлюлозы, например, кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, камедь,метилцеллюлозу,гидроксиметилцеллюлозу, 004460 26 натриевую соль карбоксиметилцеллюлозы или поливинилпирролидон (ПВП). При необходимости можно вводить другие средства, такие как сшитый поливинилпирролидон, агар или альгининовую кислоту либо ее соль, например, альгинат натрия. На ядра драже наносят соответствующие покрытия. С этой целью можно применять концентрированные растворы сахара, которые могут также содержать гуммиарабик, поливинилпирролидон, гель Карбопол, полиэтиленгликоль и (или) двуокись титана, лаковые растворы и соответствующие органические растворители или смеси растворителей. В таблетки или покрытия драже можно добавлять красители или пигменты для маркировки или для обозначения различных сочетаний активных средств. Фармацевтические препараты для перорального введения включают составные капсулы из желатина, а также мягкие капсулы из желатина с пластификатором, например, глицерином или сорбитом. Составные капсулы могут содержать активные ингредиенты в смеси с наполнителями, такими как лактоза, связующими,такими как крахмалы, и (или) смазками, такими как тальк или стеарат магния, а также дополнительно могут содержать стабилизаторы. В мягких капсулах активные средства могут быть растворены или суспендированы в соответствующих жидкостях, таких как жирные масла,жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут вводиться стабилизаторы. Все препараты для перорального введения должны быть в дозировках, пригодных для такого введения. Для трансбуккального введения(через ротовую полость) препараты могут быть в виде таблеток или пастилок, приготовленных обычными способами. Для введения через нос или для введения ингаляцией соединения для предлагаемого применения удобно доставлять в виде аэрозольного распылителя в упаковках под давлением или в виде распылителя с подходящим носителем,например, дихлородифторметаном, трихлорофторметаном, дихлоротетрафторэтаном, углекислым газом или другим подходящим газом. При применении распылителя в упаковке под давлением дозировка может устанавливаться применением клапана, рассчитанного на выдачу определенного количества материала. Капсулы и патроны из желатина для применения в ингаляторе или инсуфляторе и т.п. можно приготовлять в виде порошковой смеси соединения и соответствующей порошковой основы, такой как лактоза или крахмал. Соединения можно приготовлять в виде препаратов для парентерального введения инъекцией, например, введением шарика или непрерывным вливанием. Препараты для инъекций могут быть в виде единичной дозы, например, в ампулах или в упаковках, содержащих ряд доз, с добавлением консерванта. Препараты 27 могут быть в виде суспензий, растворов или эмульсий в маслянистых или водных носителях и могут содержать средства приготовления, такие как взвешивающие, стабилизирующие и(или) диспергирующие добавки. Фармацевтические препараты для парентерального введения включают водные растворы в водорастворимом виде. Кроме того, можно приготовлять суспензии активных средств в виде соответствующих маслянистых суспензий для инъекций. Подходящие липофильные растворители или носители включают жирные масла, такие как кунжутное масло, или синтетические эфиры жирных кислот, такие как этилолеат или триглицериды, либо липосомы. Водные суспензии для инъекций могут содержать вещества для увеличения вязкости, такие как натриевая соль карбоксиметилцеллюлозы, сорбит или декстран. Дополнительно суспензия также может содержать соответствующие стабилизаторы или средства улучшения растворимости соединений с целью приготовления более концентрированных растворов. Для внутриглазного введения соединение формулы I, II, III или IV приготовляют в фармацевтически приемлемом носителе такого вида,чтобы соединение поддерживалось в контакте с глазной поверхностью в течение достаточного времени для проникновения соединения в районы роговицы и внутренние отделы глаза,включая, например, переднюю камеру, заднюю камеру, стекловидное тело, внутриглазную жидкость, жидкую часть стекловидного тела,роговицу, радужно-цилиарную область, хрусталик, сосудистую оболочку с сетчаткой и склеру. Фармацевтически приемлемым офтальмологическим носителем может быть мазь, растительное масло или капсулирующий материал. Предлагаемое соединение можно также вводить путем инъекции непосредственно в стекловидное тело и его жидкую часть. Вместе с тем, активный ингредиент может быть в виде порошка для состава, смешиваемого с соответствующим носителем, например, стерильной не содержащей пирогенов водой непосредственно перед употреблением. Соединения также могут быть в составах для ректального введения, например, в виде свечей или удерживающих клизм, например, содержащих обычные основания для свечей, такие как масло какао или другие глицериды. Кроме описанных выше препаратов, можно также приготовлять соединения в составе вживляемых препаратов. Такие долгосрочные препараты можно вводить имплантацией (например, подкожно, внутримышечно или внутриглазным введением) либо внутримышечной инъекцией. Так, например, соединения можно вводить в состав с соответствующими полимерными или гидрофобными материалами (например, в виде эмульсии в приемлемом масле) или ионообменными смолами либо в виде слаборас 004460 28 творимых производных, например, слаборастворимых солей. Фармацевтический носитель для гидрофобных соединений представляет собой системы сорастворителей в виде бензилового спирта,неполярного ПАВ, смешиваемого с водой органического полимера и водной фазы. Система сорастворителей может быть в виде системы сорастворителей VPD. VPD представляет собой раствор 3 вес./об.% бензинового спирта, 8 вес./об.% неполярного поверхностно-активного полисорбата 80 и 65 вес./об.% полиэтиленгликоля 300 с добавлением абсолютного этанола до полного объема. Система сорастворителей VPD(VPD:5W) содержит VPD, разбавленный в соотношении 1:1 5% водным раствором декстрозы. Эта система сорастворителей хорошо растворяет гидрофобные соединения и сама по себе обладает низкой токсичностью при системном введении. Разумеется, соотношения в системе сорастворителей могут существенно изменяться, не нарушая ее свойства растворимости и токсичности. Далее, можно изменять виды отдельных составных частей сорастворителей: например, можно применять другие малотоксичные неполярные ПАВ вместо полисорбата 80; можно изменять долю полиэтиленгликоля; можно вводить другие биосовместимые полимеры взамен полиэтиленгликоля, например поливинилпирролидон, а другие полисахара и полисахариды могут быть заменены декстрозой. Вместе с тем, можно использовать другие системы доставки гидрофобных фармацевтических соединений. Примерами носителей для доставки или носителей гидрофобных лекарств являются липосомы и эмульсии. Можно также применять некоторые органические растворители, такие как диметилсульфоксид, хотя обычно ценой повышенной токсичности. Кроме того,соединения можно доставлять с помощью системы пролонгированного действия, например полунепроницаемых матриц из твердых гидрофобных полимеров, содержащих терапевтическое средство. Разработаны и известны в технике разнообразные материалы пролонгированного действия. Капсулы пролонгированного действия в зависимости от своей химической природы могут выделять соединения в течение срока от нескольких недель до более 100 дней. В зависимости от химической природы и биологической устойчивости терапевтического реагента можно применять дополнительные меры для стабилизации белка. Фармацевтические препараты могут также содержать соответствующие твердофазные или гелеобразные носители или наполнители. Примерами таких носителей или наполнителей являются карбонат кальция, фосфат кальция, сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоль. Некоторые предлагаемые соединения могут быть в виде солей фармацевтически совмес 29 тимых противоионов. Фармацевтически совместимые соли могут быть образованы со многими кислотами, включая соляную, серную, уксусную, молочную, виннокаменную, яблочную,янтарную и т.п. Соли имеют более высокую растворимость в водных или других протонных растворителях, чем в соответствующие свободноосновные формы. Приготовление предпочтительных предлагаемых соединений подробно описано в следующих ниже примерах, при этом специалисту понятно, что представленные химические реакции можно легко приспособить для приготовления ряда других предлагаемых ингибиторов протеинкиназ. Например, синтез не представленных примерами предлагаемых соединений можно успешно проводить, вводя изменения,очевидные специалистам, например, защищая соответствующим образом взаимовлияющие группы, заменяя одни реагенты на другие известные реагенты, либо производя обычные изменения условия проведения реакций. В то же время другие описанные здесь реакции или известные реакции будут признаны применимыми для приготовления других предлагаемых соединений. Сведения, подтверждающие возможность осуществления изобретения При отсутствии других указаний в приведенных ниже примерах все температуры даны в градусах Цельсия, а все части и проценты - весовые. Реагенты приобретаются у коммерческих поставщиков, таких как Aldrich Chemical Company или Lancaster Synthesis Ltd. и при отсутствии особых указаний они используются без дополнительной очистки. Тетрагидрофуран (ТГФ) и N,N-диметилформамид (ДМФ) приобретают у компании Aldrich в бутылях "Sure seal" и употребляют непосредственно после получения. Если особо не оговорено, все растворители очищают, используя стандартные методы, хорошо известные специалистам в данной области. Приведенные ниже реакции проводят, в общем, при положительном давлении азота или с осушительной трубкой при окружающей температуре (при отсутствии других указаний), в безводных растворителях, при этом используют реакционные колбы с резиновыми разделителями для введения субстратов и реагентов шприцем. Стеклянную посуду сушат в печи и (или) нагреванием. Аналитическую тонкослойную хроматографию (тонкослойная жидкостная хроматография) проводят на пластинах силикагеля на стеклянной подложке 60 F 254 фирмыAnalytech (0,25 мм) с элюированием при соответствующих соотношениях растворителей(об./об.) с указанием вида при необходимости. Реакции анализируют методом тонкослойной хроматографии и оканчивают по расходу исходного материала. Визуализацию пластин производят распылением реагента p-анизальдегид или фосфомо 004460 30 либденовой кислотой (фирма Aldrich Chemical 20 вес.% в этаноле) и активацией нагреванием. При отсутствии других указаний обработку обычно проводят удвоением реакционного объема реакционным растворителем или экстрагирующим растворителем с последующей промывкой указанными водными растворами, используя 25% объема от объема экстрагирования. Растворы продуктов сушат над безводнымNa2SO4 перед фильтрацией и выпариванием растворителей при пониженном давлении в ротационном выпарном аппарате, при этом отдельно указывается, когда растворители удаляют в вакууме. При отсутствии других указаний в примерах флэш-хроматографию (Still et al., J.Org. Chem., 43, 2923 (1978 проводят с применением силикагеля Baker класса "флэш" (47-61 мкм) при соотношении силикагель: неочищенный материал около 20:1 - 50:1 и при окружающем давлении. Спектры 1 Н-ЯМР записывают приборомBruker, работающим на частоте 300 МГц, а спектры 13 С-ЯМР записывают на частоте 75 МГц. Спектры ЯМР получают в виде растворовCDCl3 (данные в ррm [ч/млн]), используя в качестве эталона хлороформ (7,25 ppm и 77,00ppm) или CD3OD (3,4,4,8 ppm и 49,3 ppm), или при необходимости внутри тетраметилсилан(0,00 ppm). При необходимости используют другие растворители для ЯМР. При множественности пиков используют следующие сокращения: s (синглет), d (дуплет), t (триплет), m(мультиплет), br (уширение), dd (дуплет дуплетов), dt (дуплет триплетов). Если даются константы взаимодействия, они приведены в герцах(Гц). Инфракрасные (ИК) спектры записывают на спектрометре Perkin-Elmer FT-IR в виде беспримесных масел, гранул КВr, или растворовCDCl3, при этом они даются в волновых числах(см-1). Массовые спектры получают с помощью жидкостной масс-спектрометрии с вторичной ионизацией (LSIMS) или электрораспылением. Все точки плавления (т.пл.) - без поправок. Пример 1(а). 3-[Е-2-(3,4-Диметоксифенил) винил]-6-(3-метокси-4-гидроксифенил)-1Hиндазол. 3-[Е/Z-2-(3,4-Диметоксифенил)винил]-6[3-метокси-4-(метоксиметокси)фенил]-1Hиндазол (205 мг, 0.461 ммол (теория растворяют в тетрагидрофуране (ТГФ, 10 мл) и обрабатывают водой (10 мл) и трифторуксусной кислотой (TFA, 20 мл). Реакционную смесь перемешивают при 23 С в течение 30 мин. Смесь разбавляют толуолом (100 мл), и летучие материалы удаляют при пониженном давлении (30 мм рт.ст., 35 С) с получением сгущенного объема 5 мл. Снова добавляют толуол (100 мл), и 31 смесь сгущают при пониженном давлении с получением неочищенного материала, все еще содержащего некоторое количество кислоты. Материал делят между этилацетатом и насыщенным раствором бикарбоната натрия, органический материал отделяют, сушат над сульфатом натрия, сливают, и сгущают при пониженном давлении. Остаток - смесь олефиновых изомеров (185 мг, 0,461 ммол (теория отбирают в дихлорметане (50 мл) при 23 С и обрабатывают йодом (80 мг). Смесь перемешивают при 23 С в течение 12 ч. Смесь обрабатывают насыщенным раствором бикарбоната натрия (10 мл) и 5% водным раствором бисульфита натрия(10 мл). Смесь разбавляют этилацетатом (200 мл), и органический материал промывают насыщенным раствором бикарбоната натрия (100 мл), сушат над сульфатом натрия, сливают и сгущают при пониженном давлении с получением неочищенного продукта. Неочищенный продукт очищают на кремнеземе (40 мл, смесь 6:4 - 7:3 этилацетат/гексан), и все фракции,содержащие целевой материал, совмещают,сгущают и осаждают из двухслойной смеси дихлорметан/гексана (1:3) с получением 3-[Е-2(3,4-диметоксифенил)винил]-6-(3-метокси-4 гидроксифенил)-1H-индазола в виде белого твердого вещества (93 мг общий выход): Rf sm 0.42, р 0.35 (смесь 7:3 этилацетат-гексан); ИК Фурье-спектрометрия (тонкопленочн.) 3324,1600, 1514, 1463, 1422, 1264, 1137, 1024, 959,852 см-1; 1 Н ЯМР (CDCl3)10,0 (bs, 1H), 8,08 (d,1H, J=8,4 Гц), 7,59 (s, 1 Н), 7,49 (d, 1H, J=16,6 Гц), 7,45 (dd, 1H, J=1,4, 8,4 Гц), 7,34 (d, 1H,J=16,6 Гц), 7,20-7,12 (m, 4H), 7,03 (d, 1H, J=8,0 Гц), 6,91 (d, 1H, J=8,2 Гц), 5,68 (bs, 1H), 3,99 (s,3H), 3,97 (s, 3H), 3,93 (s, 3H); 13C ЯМР (CDCl3)149,6, 149,5, 146,0, 144,0, 142,6, 140,8, 133,9,131,4, 130,7, 121,7, 121,4, 120,9, 120,4, 120,2,118,6, 115,4, 111,7, 110,8, 109,1, 108,2, 56,4, 56,3,56,2; масс-спектрометрия с высоким разрешением (электрораспыление) [m+H]/z теория 403,1658, найдено 403,1658, [m-H]/z теория 401,найдено 401. Исходный материал получают следующим образом: К 6-аминоиндазолу (40,8 г, 0,3065 мол) в 2 литровой (2 л) круглодонной колбе, содержащей большую магнитную мешалку, добавляют лед(56 г), а затем воду (128 мл), и опускают реакционный сосуд в ледяную баню. К этой суспензии при перемешивании при 0 С добавляют концентрированную водную НСl (128 мл, 1,53 мол). Непосредственно вслед за этим добавляют раствор NaNO2 (23,3 г, 0,338 мол) в воде (96 мл). Через 10 мин перемешивания при 0 С добавляют KI (61 г, 0,368 мол) сначала очень медленно(100 мг за один раз, так как первые малые пор 004460 32 ции KI вызывают бурное выделение газа), а затем быстрее (всего за 5 мин). Удаляют холодную баню, и реакционную смесь согревают до 40 С (выделяется газ). При снижении количества выделяемого газа (30 мин) реакционную смесь согревают до 50 С в течение 30 мин. Далее смесь охлаждают до 23 С и добавляют раствор 3N NaOH (320 мл) для нейтрализации, а затем - 50% насыщенного раствора NaHCO3 (320 мл). Далее суспензию фильтруют через воронку Бюхнера с получением темного красноватокоричневого твердого вещества. Твердое вещество собирают теплым ТГФ (800 мл) и добавляют кремнезем (600 мл безводн.) при перемешивании. К этой суспензии добавляют гексан (1,2 л), и смесь фильтруют в вакууме через слой кремнезема (300 мл) в большом спеченном фильтре. Кремнезем далее промывают 2 л 40% ТГФ в гексане. Фильтраты совмещают и сгущают при пониженном давлении с получением твердого вещества. Далее твердое вещество переводят в порошок этилацетатом (100 мл),фильтруют и высушивают при пониженном давлении с получением 6-йодо-1H-индазола в виде светло-коричневого твердого вещества(2,89 г, 30,1 ммол, 1 экв.). Наблюдается изменение цвета с оранжевого на красный. Добавляют в виде одной порции мезитиленсульфонил хлорид (6,60 г, 30,1 ммол, 1 экв.) и удаляют ледяную баню, после чего дают реакционной смеси согреться до 23 С. Через 40 мин смесь гасят насыщенным хлоридом аммония и делят между водой и этилацетатом. Водную фазу экстрагируют всего 3 раза этилацетатом. Совмещенный органический материал промывают рассолом,сушат над сульфатом натрия и сгущают при пониженном давлении с получением 6-йодо-1(2,4,6-триметилбензолсульфонил)-1 Н-индазола в виде оранжевого твердого вещества (12.8 г,выход 100%, смесь 2:1). 1 Н ЯМР (CDCl3) 8,51 (s,1H), 7,95 (s, 0,66 Н, основной изомер), 7,91 (s,0,33 Н, второстепенный изомер), 7,47 (d, 0,33H,J=8,4 Гц), 7,29 (d, 0,33 Н, J=8,4 Гц), 7,26 (d,0,66H, J=8,9 Гц), 7,18 (d, 0,66 Н, 8,9 Гц), 6,84 (s,2 Н), 2,51 (s, 6H), 2,15 (s, 3 Н). 33 Смесь 6-йодо-1-(2,4,6-триметилбензолсульфонил)-1 Н-индазола (5.78 г, 13,56 ммол,1,00 экв.) и 3-метокси-4-(метоксиметокси) бензолбороновой кислоты (3,45 г, 16,27 ммол, 1,20 экв.) в аргоне растворяют в диоксане (15 мл) и воде (2,0 мл). К этому раствору добавляют триэтиламин (2,83 мл, 20,3 ммол, 1,5 экв.), карбонат калия (2,8 г, 20,3 ммол, 1,5 экв.) и дихлоробис(трифенилфосфин)палладий (476 мг, 0,78 ммол, 0,05 экв.). Реакционную смесь нагревают до 90 С в течение 2 ч и затем охлаждают до 23 С. Смесь делят между этилацетатом (250 мл) и насыщенным раствором бикарбоната натрия(150 мл). Органический материал сушат над сульфатом натрия, сливают и сгущают при пониженном давлении с получением неочищенного 6-(3-метокси-4-метоксиметоксифенил)-1(2,4,6-триметилбензолсульфонил)-1 Н-индазола,который сушат в глубоком вакууме в течение 15 ч и используют без дальнейшей очистки. 3-Метокси-4-(метоксиметокси)бензилбороновую кислоту готовят следующим образом. Готовят в 100-мл колбе раствор 50% КОН в воде(20 г КОН, 7 экв., 20 г льда) в аргоне. К этой быстро перемешиваемой смеси при 0 С (поддерживаемой ледяной баней) добавляют дихлорметан (50 мл), а затем - 4-бромо-2-метоксифенол (10,1 г, 50 ммол, 1,00 экв.), метоксиметилхлорид (МОМСl) (4,00 мл, 42,5 ммол, 1,05 экв.) и бромид тетрабутиламмония (322 мг, 1 ммол, 0,02 экв.). Баню удаляют, и смеси дают медленно согреться до 23 С при энергичном перемешивания в течение 2 ч. Смесь переносят в делительную воронку и разбавляют дихлорметаном (350 мл) и водой (300 мл) для облегчения переноса. Органический материал (теперь нижний слой) отделяют, сушат над сульфатом натрия, сливают и сгущают при пониженном давлении с получением 4-бромо-2-метокси-1(метоксиметокси)бензола в виде желтой жидкости, чистой по 1H ЯМР (11,9 г, 97%): 1H ЯМР(CDCl3)7,0 (s, 3H), 5,13 (s, 2H), 3,84 (s, 3H),3,47 (s, 3H), масс-спектроскопия (El) [m+H]/z теория 235, найдено 235. В 50-мл круглодонной колбе отбирают 4-бромо-2-метокси-1-(метоксиметокси)бензол (4,80 г, 19,4 ммол, 1,00 экв.) в ТГФ (35 мл) и охлаждают до -78 С (20 мин на этот объем). К смеси добавляют n-BuLi (12,75 мл, 1,6 М в гексане, 20,4 ммол, 1,05 экв.), и смесь перемешивают при -78 С в течение 40 мин. Далее ее вводят через канюлю во вторую колбу, содержащую В(ОМе)3 (22 мл, 194 ммол,10 экв.) ТГФ (50 мл) при -78 С. Через 20 мин холодную баню удаляют. После согревания в течение 15 мин (0 С, лед со стороны колбы начинает таять) добавляют к реакционной смеси воду (50 мл) при перемешивании в течение 45 мин. Смесь сгущают при пониженном давлении для удаления большей части ТГФ, а затем делят между этилацетатом (300 мл) и водой (150 мл),которую предварительно подкисляют добавлением небольшого количества 20% лимонной 34 кислоты (10 мл). Органический материал сушат над сульфатом натрия и сгущают при пониженном давлении с получением твердого вещества. После перевода в порошок этилацетатом(10 мл) и гексаном (5 мл) с последующим фильтрованием получают 3-метокси-4-(метоксиметокси)бензолбороновую кислоту в виде белого твердого вещества (3,15 г, 77%): Rf sm 0,59, р 0,18 (смесь этилацетат-гексан 1:1); 1H ЯМР (CDCl3)7,85 (d, 1H, J=8 Гц), 7,72 (s, 1H),7,22 (d, 1H, J=8 Гц), 5,30 (s, 2H), 4,00 (s, 3H),3,55 (s, 3H).(20 мл) и обрабатывают 1N NaOH в МеОН (70 мл с дегазаций барботажем аргона в течение 3-5 мин). Смесь нагревают до 45 С в течение 1 ч и остужают. Смесь нейтрализуют добавлением 1NHCl (50 мл), а затем - насыщенного раствора бикарбоната натрия (200 мл). Продукт в этилацетате (350 мл) сушат над сульфатом натрия и сгущают при пониженном давлении с получением неочищенного 6-(3-метокси-4-метоксиметоксифенил)-1H-индазола. Очистка хроматографией на силикагеле (500 мл кремнезема, 20% этилацетата в бензоле (1,8 л), 30% этилацетата в бензоле (1,8 л дает 6-(3-метокси-4-метоксиметоксифенил)-1H-индазола (1,19 г, 31%): 1 Н ЯМР (CDCl3)7,80 (s, 1H), 7,69 (d, 1H, J = 8,5 Гц), 7,52 (s, 1H), 7,29 (d, 1H, J=8,5 Гц), 7,16 (s,1H), 7,13 (s, 1H), 7,08 (s, 1H), массспектроскопия (электрораспыление) [m+Na]/z теория 337, найдено 337; [m+Cl-]/z теория 349,найдено 349. В 100-мл круглодонной колбе в атмосфере аргона растворяют 6-(3-метокси-4-метоксиметоксифенил)-1H-индазол (1,19 г, 4,18 ммол, 1 экв.) в диоксане (25 мл) и 3N NaOH (14 мл). Эту смесь обрабатывают йодом (1,17 г, 14,60 ммол,1,10 экв.), вводимым примерно 5 порциями (10 мин). Добавляют несколько дополнительных порций (4) йода (по 50 мг каждая) до завершения реакции по показаниям тонкослойной жидкостной хроматографии (смесь 3:7 этилацетат/гексан). Смесь подкисляют 20% лимонной кислотой (25 мл) и добавляют 5% NaHSO3 (20 мл). Смесь делят между этилацетатом (150 мл) и водой (100 мл). Органический материал промывают насыщенным раствором бикарбоната натрия (80 мл) и рассолом (50 мл), сушат над сульфатом натрия и сгущают при пониженном давлении. Очистка кристаллизацией из этилаце 35 тата (3 мл), а затем гексана (7 мл) дает чистый 3-йодо-6-(3-метокси-4-метоксиметоксифенил)1H-индазол в виде твердого вещества (1,33 г, 78 100-мл круглодонной колбе растворяют 3 йодо-6-(3-метокси-4-метоксиметоксифенил)-1Hиндазол (921 мг, 2,245 ммол, 1,00 экв.) в ТГФ(36 мл) и охлаждают до -78 С (8 мин на это количество). Добавляют раствор PhLi (2,5 мл, 1,8 М, 4,49 ммол, 2,00 экв.), и перемешивают смесь в течение 30 мин. Добавляют раствор s-BuLi(3,63 мл, 4,71 ммол, 2,1 экв.), и реакционную смесь перемешивают в течение 1 ч при -78 С. Добавляют чистый ДМФ (1,4 мл, 18 ммол, 8,0 экв.). Далее удаляют холодную баню, а оставляют реакционную смесь на воздухе для медленного согревания до 0 С. По мере таяния льда добавляют насыщенный раствор бикарбоната натрия (20 мл). Продукт экстрагируют в этилацетат (200 мл) из насыщенного раствора бикарбоната натрия (еще 75 мл), сушат над сульфатом натрия, сливают и сгущают при пониженном давлении. Очистка хроматографией на силикагеле (450 мл кремнезема, смесь 4:6 этилацетат/гексан) дает 6-(3-метокси-4-метоксиметоксифенил)-1H-индазол-3-карбальдегид (498 мг, 71%): Rf sm 0.30, р 0,14 (смесь этилацетатгексан 4:6); 1H ЯМР (СDCl3)10,85 (bs, 1H),10,25 (s, 1H), 8,37 (d, 1H, J=8,4 Гц), 7,67 (s, 1H),7,60 (d, 1H, J=8,4 Гц), 6,26 (d, 1H, J=8,7 Гц), 7,19(15 мл) и охлаждают до 0 С. Эту смесь обрабатывают солянокислой солью мезитиленсульфонила (324 мг, 1,48 ммол, 1,05 экв.) и диметиламино пиридином (ДМАП) (181 мг, 1,48 ммол,1,05 экв.). Смесь перемешивают в течение 1 ч при 0 С и гасят добавлением воды. Смесь делят между водой и органическим слоем в виде смеси 1:1 этилацетат/гексан. Органический материал сушат над сульфатом натрия, сливают и сгущают при пониженном давлении с получением неочищенного материала, который очищают хроматографией на силикагеле (50 мл кремнезема, смесь 3:7 этилацетат/гексан) с получением 6-(3-метокси-4-метоксиметоксифенил)-1-(2,4,6 триметилбензолсульфонил)-1H-индазол-3 карбальдегида (374 мг, 54%): Rf sm 0.17, р 0.53 Тонкоизмельченный бромид трифенил(3,4 диметоксибензил)фосфония (1,09 г, 2,22 ммол,4,0 экв.) собирают в виде взвеси в ТГФ (15 мл) и охлаждают до -78 С. К этой смеси добавляют nBuLi (1,04 мл, 1,6 M, 1,66 ммол, 3,0 экв.) с получением красно-оранжевого раствора. Смесь согревают до 23 С в течение 1 ч. Далее этот раствор доливают через канюлю к находящемуся при 0 С раствору 6-(3-метокси-4-метоксиметоксифенил)-1-(2,4,6-триметилбензолсульфонил)-1H-индазол-3-карбальдегида(274 мг, 0,554 ммол, 1,0 экв.) в ТГФ (5 мл). Полученную смесь перемешивают при 0 С в течение 10 мин и гасят насыщенным раствором бикарбоната натрия. Полученную смесь делят между насыщенным раствором бикарбоната натрия и этилацетатом. Органический материал was сгущают при пониженном давлении, и остаток очищают хроматографией на силикагеле (50 мл кремнезема,смесь 3:74:6 этилацетат/гексан) с получением смеси 2,5:1 цис/транс 3-[2-(3,4-диметоксифенил)винил]-6-(3-метокси-4-метоксиметоксифенил)-1-(2,4,6-триметилбензолсульфонил)-1Hиндазола (289 мг, 83%): Rf sm 0,53, р 0,32 (смесь этилацетат-гексан 4:6);1H ЯМР (CDCl3)8,35 (s,0,3H), 8,32 (s, 0,7H), 8,03 (d, 0,3H, J=8,4 Гц),7,60-6,85 (m, H), 6,65 (d, 0,7H, J=8,4 Гц), 6,60 (d,0,7H, J=12,5 Гц), 5,30 (s, 0,6H), 5,29 (s, 1,4H),4,00-3,50 (8 синглеты, 12 Н), 2,72 (s, 1,8H), 2,67 Раствор 1 М КОН (1,0 г, 17,8 ммол) в смеси 1:1 вода/МеОН (всего 18 мл) готовят в атмосфере аргона и дегазируют циклами вакуумирования и продувки аргоном (5 раз). Растворяют в отдельной колбе 3-[2-(3,4-диметоксифенил)винил]-6-(3-метокси-4-метоксиметоксифенил)1-(2,4,6-триметилбензолсульфонил)-1H-индазол(289 мг, 0,461 ммол, 1,0 экв.) в ТГФ (8 мл) в атмосфере аргона. К этому раствору добавляют указанный выше раствор 1M КОН (10 мл, смесь 1:1 вода/МеОН). Реакционную смесь согревают до 30 С и оставляют при перемешивании на 7 ч. Реакционную смесь нейтрализуют добавлением 20% лимонной кислоты (7 мл). Полученную смесь делят между этилацетатом (150 мл) и водой (100 мл). Органический материал отделяют,сушат над сульфатом натрия, сливают, и сгу 37 щают при пониженном давлении с получением цис и транс 3-[2-(3,4-диметоксифенил)винил]-6(3-метокси-4-метоксиметоксифенил)-1Hиндазола (используемого без очистки): Rf sm 0,46, p1 0,17, р 2 0,23 (смесь этилацетат-гексан 1:1); 1H ЯМР изомер (СDCl3)7,55 (s, 1H), 7,37,1 (m, 6H), 7,02 (dd, 1H, J=1,9, 8,3 Гц), 6,85 (d,1H, J=12,5 Гц), 6,78 (d, 1H, J=12,5 Гц), 6,74 (d,1H, J=8,3 Гц), 5,21 (s, 2H), 3,88 (s, 3H), 3,70 (s,3H), 3,43 (s, 3H), 3,42 (s, 3H). массспектроскопия (электрораспыление) [m+H]/z теория 447, найдено 447, [m-H]/z теория 445,найдено 445. Пример 1(b). 3-(Е-Стирил)-6-(3-бензилокси-4-гидроксифенил)-1H-индазол. Пример 1(с) получают аналогично описанию, приведенному в примере 1(а), но вместо 3 метокси-4-(метоксиметокси)бензолбороновой кислоты на этапе (iii) используют 3-аллилокси 4-(метоксиметокси)бензолбороновую кислоту. Масс-спектроскопия (ESI) [M+H]/z теория 429,найдено 429; масс-спектроскопия (ESI) [M-H]/z теория 427, найдено 427. Пример 2(а). 3-(Нафталин-2-ил)-6-(3-метокси-4-гидроксифенил)-1H-индазол. 6-(4-Бензилокси-3-метоксифенил)-3-нафталин-2-ил-1H-индазол (25 мг, 0,055 ммол) растворяют в смеси этилацетата (2 мл), бензола (2 мл) и метанола (2 мл). К этому раствору добавляют палладий на углероде (25 мг, 10 вес.%), и реактор пятикратно вакуумируют и продувают газообразным водородом. Реакционную смесь перемешивают в течение 3 дней (д) при 23 С и фильтруют через пробку из Целита. После сгущения и очистки хроматографией на силикагеле получают 3-(нафталин-2-ил)-6-(3-метокси-4 004460 38 гидроксифенил)-1H-индазол (8 мг, 40%): 1H ЯМР (CDCl3)10,3 (bs, 1H), 8,50 (s, 1H), 8,20 (d,1H, J = 8 Гц), 7,98 (d, 1H, J=8Hz), 7,90 (m, 1H),7,7-6,8 (m, 9H), 3,98 (s, 3H). Macс-спектроскопия (электрораспыление) [m+H]/z теория 367, найдено 367, [m-H]/z теория 365, найдено 365. Исходный материал получают следующим образом: 2-Бромонафталин (117 мг, 0.564 ммол, 6.0 экв.) растворяют в ТГФ (0,75 мл) и охлаждают до -78 С. Смесь обрабатывают n-BuLi (226 мкл,2,5 M, 6,0 экв.) и оставляют для перемешивания при -78 С в течение 30 мин. Смесь затем добавляют через канюлю к свежевысушенному твердому ZnCl2 (139 мг, 0,80 ммол, 8,5 экв.), и полученную смесь согревают до 23 С (желтизна исчезает во время добавления). Через 30 мин при 23 С смесь добавляют через канюлю к смеси 6(4-бензилокси-3-метоксифенил)-3-йодо-1-(2,4,6 триметилбензолсульфонил)-1H-индазола (60 мг,0,094 ммол, 1 экв.) и Рd(РРh3)4 (6 мг, 0,005 ммол,0,05 экв.). Полученный раствор перемешивают в течение 16 ч. Добавляют насыщенный раствор бикарбоната натрия, и делят смесь между насыщенным раствором бикарбоната натрия (15 мл) и этилацетатом (15 мл). Органический материал сушат над сульфатом натрия, сливают и сгущают. Очистка хроматографией на силикагеле (смесь 1:9 -2:8 этилацетат-гексан) дает 6-(4 бензилокси-3-метоксифенил)-3-нафталин-2-ил 1-(2,4,6-триметилбензолсульфонил)-1H-индазол в виде твердого вещества (42 мг, 70%): Пример 2(с) проводят аналогично описанию, приведенному в примере 2(а), но 3,4,5 триметоксифенил бромид используют на этапе Пример 2(d) проводят аналогично описанию, приведенному в примере 2(а), но вместо 2 бромонафтилена на этапе (i) используют 1 фенилсульфонилиндазол. Rf sm 0,20, р 0,15[m+H]/z теория 357,1351, найдено 357,1349. Исходный материал получают следующим образом: 6-(3-Метокси-4-метоксиметоксифенил)1H-индазол-3-карбальдегид (из примера 1(а),этап (vi (20 мг, 0,064 ммол, 1 экв.) растворяют в дегазированной смеси 1:1 МеОН-вода (0,7 мл) и обрабатывают уксусной кислотой (19 мкл, 5 экв.), 1,2-диаминобензолом (8,3 мг, 1,2 экв.) и уксуснокислой медью (II) (18 мг, 1,4 экв.) при 23 С. Смесь перемешивают 30 мин, разбавляют этанолом (3 мл) и водой (2 мл) и обрабатывают барботажным потоком SH2 в течение 3 мин, получая черный осадок. Смесь перемешивают в течение 12 ч. Смесь фильтруют и сгущают. Очистка хроматографией на силикагеле (смесь 6:4 этилацетат-гексан)дает 3-(1H-бензоимидазол-2 ил)-6-(3-метокси-4-метоксиметокси-фенил)-1Hиндазол в виде твердого вещества (14 мг, 54%);N-[3-(2-бензоил-3-стирил-1 Ниндазол-6-илокси)фенил]бензамида (0,09 г, 0,17 ммол) в 2 мл 6N НСl (водной) и 3 мл МеОН нагревают при 65 С примерно в течение 4 ч. Охлажденный раствор осторожно выливают в насыщенный раствор бикарбоната натрия. Осадок фильтруют, собирают и проводят хроматографию на силикагеле с разбавлением смесью гек 41 саны/ЕtOАс (1:1). N-[3-(3-стирил-1 Н-индазол-6 илокси)-фенил]-бензамид получают в виде бежевого твердого вещества (32 мг, 50%): 1 Н ЯМР(ДМСО-d6)13,50 (s, 1H), 10,32 (s, 1H), 8,23 (d,1H, J=8,7 Гц), 7,92 (d, 2H, J=6,8 Гц), 7,72 (d, 2H,J=7,3 Гц), 7,71-7,51 (m, 7H), 7,51 - 7,47 (m, 3H),7,30 (t, 1H, J=7,2 Гц), 7,05 (s, 1H), 7,01 (d, 1H,J=8,7 Гц), 6,86 (dd, 1H, J=8,2, 2,3 Гц). Аналитический расчет для C28H21N3O20,3H2 О: С 76,97; Н 4,98: N 9,62. Найдено: С 76,94; Н 5,13; N 9,40. Исходный материал получают следующим образом:(5,82, 42,1 ммол) в 150 мл ацетона нагревают с обратным холодильником в течение ночи. Охлажденную реакционную смесь фильтруют и сгущают при пониженном давлении. Остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (2:1). При этом получают 3-[3-(бензгидрилиденамино)фенокси]циклогекс-2-енон в виде желтого твердого вещества, (8,82 г, 63%): 1 Н ЯМР (CDCl3)7,78 (d, 2H, J=7,0 Гц),7,50 (d, 1H, J=7,1 Гц), 7,45 (d, 2H, J=7,7 Гц),7,34-7,10 (m, 6H), 6,69 (d, 1H, J=8,0 Гц), 6,61 (d,1H, J=8,0 Гц), 6,38 (s, 1H), 4,89 (s, 1H), 2,55 (t,2H, J=6,2 Гц), 2,34 (t, 2H, J=6,2 Гц), 2,06 (m, 2H). Аналитический расчет для С 25 Н 21NО 20,2 Н 2O: С 80,92; Н 5,81; N 3,78. Найдено: С 81,12; Н 5,81;N 3,72. 3-(Бензгидрилиденамино)фенол готовят следующим образом. При перемешивании раствор бензофенона (15,0 г, 82,8 ммол) и 3 аминофенола (9,03 г, 82,8 ммол) в 25 мл толуола нагревают с обратным холодильником с удалением Н 2 О с помощью ловушки Дина-Старка в течение 3,5 ч. Образующиеся из охлаждаемой реакционной смеси кристаллы собирают вакуумной фильтрацией, промывают гексанами и высушивают на воздухе. При этом получают 3(бензгидрилиденамино)фенол в виде светложелтого твердого вещества (17,3 г, 76%): 1 Н ЯМР (CDCl3)7,64 (d, 2H, J=7,1 Гц), 7,38 (d,1H, J = 7,1 Гц), 7,34 - 7,15 (m, 7H), 7,04 (d, 2H,J=7,2 Гц), 6,88 (t, 1H, J=8,1 Гц), 6,82 (d, 1H, J=8,2 Гц), 6,23 (s, 1H), 6,21 (d, 1H, J=7,8 Гц). Аналитический расчет для C19H15NO: С 83,49; Н 5,53; N 5,12. Найдено: С 83,51; Н 5,65; N 5,03. (ii) Раствор 3-[3-(бензгидрилиден-амино)фенокси]циклогекс-2-енона (4,37 г, 11,89 ммол) в 20 мл ТГФ медленно приливают к растворуLiHMDS (25,0 мл 1,0 M раствора в ТГФ) в 10 мл ТГФ при -78 С. Через пять минут после окончания введения добавляют целиком транс-циннамомил хлорид (1,98 г, 11,89 ммол) и продолжают перемешивание при -78 С в течение 30 мин. Реакцию прекращают насыщенным растворомNH4Cl и экстрагируют EtOAc (2x). Совмещенные органические слои промывают насыщенным раствором NaCl, высушивают (MgSO4) и сгущают при пониженном давлении. Остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (5:1). При этом получают 3-[3-(бензгидрилиденамино)фенол]-6-(3-фенилакрилоил)циклогекс-2 енон в виде желто-оранжевого твердого вещества (3,34 г, 56%): 1 Н ЯМР(СDСl3) : 15,69 (s, 1H),7,80 (d, 2H, J=7,1 Гц), 7,63-7,01 (m, 15H), 6,93 (d,1H, J=15,6 Гц), 6,75 (d, 1H, J=7,6 Гц), 6,66 (d, 1H,J = 8,0 Гц), 6,46 (s, 1H), 4,92 (s, 1H), 2,85 (t, 2H,J=7,2 Гц), 2,62 (t, 2H, J=7,2 Гц). Аналитический расчет для C34H27NO3: С 82,07; Н 5,47; N 2,82. Найдено: С 81,88; Н 5,53; N 2,81. К перемешиваемому раствору 3-[3(бензгидрилиденамино)фенол]-6-(3-фенилакрилоил)циклогекс-2-енона (1,81 г, 3,64 ммол) в 10 мл смеси НОАс/ЕtOН(1:1) добавляют гидрат гидразина (2,0 мл, 41,23 ммол). Раствор нагревают при 75 С в течение 25 мин. После охлаждения реакционную смесь осторожно выливают в насыщенный раствор бикарбоната натрия и экстрагируют ЕtOАс (2 х). Совмещенный органический слой промывают насыщенным раствором NaCl, высушивают (MgSO4) и сгущают при пониженном давлении. Остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (1:1). 3-(3-стирил-4,5-дигидро-1 Н-индазол-6-илокси)фениламин получают в виде желтого твердого вещества (539 мг,45%). 1 Н ЯМР (ДМСО-d6)7,55 (d, 2 Н, J=7,2Hz), 7,38 (t, 2 Н, J=7,2 Гц), 7,27 (t, 1H, J=7,2 Гц),7,05 (m, 3H), 6,38 (d, 1H, J=8,0 Гц), 6,31 (s, 1H),6,23 (d, 1H, J=7,9 Гц), 5,52 (s, 1H), 5,26 (s, 2H),2,92 (t, 2H, J=8,0 Гц), 2,58 (t, 2H, J=8,1 Гц). Аналитический расчет для C21H19N3O0,3H2O: С 75,33; Н 5,90; N 12,55. Найдено: С 75,46; Н 5,96;(54 мкл, 0,31 ммол) в 5 мл СН 2 Сl2, добавляют бензоил хлорид (36 мкл, 0,31 ммол). Через 15 мин реакционную смесь разбавляют СН 2 Сl2 и 43 промывают последовательно 0,5N HCl, насыщенным раствором бикарбоната натрия и рассолом, высушивают (MgSO4) и сгущают при пониженном давлении. К перемешиваемому раствору остатка в 1,4-диоксане добавляют 2,3 дихлоро-5,6-дициано-1,4-бензохинона(35 мг, 0,15 ммол). Через 1 ч реакционную смесь сгущают при пониженном давлении и остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (2:1). При этом получают N-[3-(2-бензоил-3-стирил-1 Ниндазол-6-илокси)фенил]бензамид в виде твердого вещества цвета ржавчины (90 мг, количественно): 1 Н ЯМР (CDCl3)8,13 (s, 1H), 8,02 (d,2H, J=7,0 Гц), 7,94 (d, 1H, J=8,7 Гц), 7,74 (d, 2H,J=6,8 Гц), 7,57-7,19 (m, 17H), 6,84 (d, 1H, J=8,3 Гц). Пример 4(b). N-[3-(3-Стирил-1 Н-индазол 6-илокси)фенил]ацетамид. Пример 4(b) проводят аналогично описанию, приведенному в примере 4(а) выше, но вместо бензоилхлорида на этапе (iv) используют уксусный ангидрид. 1 Н ЯМР(ДМСО-d6)13,08(d, 2H, J=7,3 Гц), 7,52 (s, 2H), 7,44-7,27 (m, 6 Н),7,01 (s, 1H), 6,96 (dd, 1H, J=8,7, 2,1 Гц), 6,78 (d,1H, J=6,9 Гц), 2,01 (s, 3H). Аналитический расчет для C23H19N3O20,25 Н 2 О: С 73,88; Н 5,26; N 11,24. Найдено: С 74,20; Н 5,57; N 10,82. Пример 5(а). [3-(3-Стирил-1 Н-индазол-6 илокси)фенил]амид 5-метилтиазол-2-карбоновой кислоты. Суспензию 3-[1-(5-метилтиазол-2-карбонил)-3-стирил-1H-индазол-6-илоксил]фенил амида 5-метилтиазол-2-карбоновой кислоты (57 мг, 0,10 ммол) и карбонат калия (50 мг, 0,36 ммол) в МеОН перемешивают при 23 С в течение 20 мин. Раствор фильтруют, разбавляют ЕtOАс и промывают рассолом (2 х). Органический слой высушивают (MgSO4) и сгущают при пониженном давлении. При этом получают [3(3-стирил-1 Н-индазол-6-илокси)фенил]амид 5 метилтиазол-2-карбоновой кислоты в виде светло-коричневого твердого вещества с выходом 47%.: 1 Н ЯМР (ДМСО-d6)13,00 (s, 1H), 10,80(t, 2 Н, J=8,6 Гц), 7,53 (s, 2H), 7,41-7,27 (m, 5H),7,04 (s, 1H), 7,00 (d, 1H, J=8,7 Гц), 6,89 (d, 1H,J=8,5 Гц), 2,54 (s, 3H). Аналитический расчет для C26H20N4O2S1,15 Н 2O: С 65,98; Н 4,75; N 11,84; S 6,78. Найдено: С 65,99; Н 4,71; N 11,58;S, 6,76. Исходный материал получают следующим образом: 3-(3-Стирил-4,5-дигидро-1 Н-индазол-6 илокси)фениламин превращают в 3-[1-(5 метилтиазол-2-карбонил)-3-стирил-1H-индазол 6-илоксил]фениламид 5-метилтиазол-2-карбоновой кислоты обработкой 5-метилтиазол-2 карбоновой кислоты и HATU (о-(2-азабензотиазол-1-ил)-N,N,N,'N'-тетраметилурония гексафторофосфат) в ДМФ и обработкой смеси,обработкой DDQ и выделением аналогично примеру 4(а), этап (iv) (50% выход): 1 Н ЯМР(ДМСО-d6)10,85 (s, 1H), 8,45 (d, 1H,J=9,8 Гц), 8,24 (m, 3H), 7,99-7,62 (m, 6 Н), 7,547,34 (m, 5H), 6,96 (d, 1H, J=8,5 Гц), 2,64 (s, 3H),2,54 (s, 3H). Пример 5(b). 3-Метил-N-[3-(3-стирил-1hиндазол-6-илокси)фенил]бензамид.(s, 1H), 6,99 (d, 1H, J=8,5 Гц), 6,87 (d, 1H, J=7,7 Гц), 2,38 (s, 3H). Аналитический расчет для С 29 Н 23N3O20,2 Н 2 О 0,2 гексаны: С 77,78; Н 5,66; Используя N-(3-1-бензоил-3-[2-(4-хлорофенил)винил]-1 Н-индазол-6-илоксилфенил) бензамид, пользуются общим регламентом примера 5(а) для получения целевого соединения в виде кремового твердого вещества с выходом 72%: 1H ЯМР (ДМСО-d6)13,07 (s, 1H), 10,32(s, 1H), 8,24 (d, 1H, J=8,8 Гц), 7,92 (d, 2 Н, J=7,1 Гц), 7,76 (d, 2 Н, J=8,5 Гц), 7,59-7,40 (m, 10 Н),7,05 (s, 1H), 7,00 (d, 1H, J=8,7 Гц), 6,87 (d, 1H,J=7,9 Гц). Аналитический расчет для С 28 Н 20ClN3O20,4 Н 2 О 0,15 гексаны; С 71,41; Н 4,75; N 8,65. Найдено: С 71,62; Н 14,83; N 8,45. Исходный материал получают следующим образом: 45 акрилоил хлорид (приготовленный, как описано ниже), применяют общий регламент примера 4(а), этап (ii). Продукт используют без очистки в опыте с циклизацией гидразином. Пример 4(а) этап (iii), с получением 3-3-[2-(4-хлорофенил) винил]-4,5-дигидро-1 Н-индазол-6-илоксилфениламина в виде желтого твердого вещества с выходом 30%. 1 Н ЯМР (ДМСО-d6)12,45 (s,1H), 7,58 (d, 2H, J=8,5 Гц), 7,43 (d, 2H, J=8,5 Гц),5,52 (s, 1H), 5,26 (s, 2H), 2,92 (t, 2H, J=8,0 Гц),2,58 (t, 2H, J=8,0 Гц). Аналитический расчет дляC21H18CIN3O0,75 Н 2O: С 66,84; Н 5,21; N 11,14. Найдено: С 66,73; Н 4,89; N 11,01. 3-(4-хлорофенил)акрилоил хлорид готовят следующим образом. При перемешивании к суспензии 4-хлоро-транс-коричной кислоты(2,51 г, 13,77 ммол) в бензоле добавляют тионил хлорид (1,1 мл, 15,14 ммол) и каталитическое количество ДМАП. Реакционную смесь нагревают с обратным холодильником в течение 1,5 ч. Летучие материалы удаляют при пониженном давлении. Белый остаток растворяют в Et2O и сгущают снова при пониженном давлении с получением 3-(4-хлорофенил)акрилоил хлорида(2,78 г, количественно) в виде белого твердого вещества: 1 Н ЯМР (CDCl3)7,81 (d, 1H, J=15,6 Гц), 7,54 (d, 2H, J=8,6 Гц), 7,44 (d, 2H, J=8,6 Гц),6,65 (d, 1H, J=15,6 Гц). Пример 6(b) проводят аналогично описанию, приведенному в примере 6(а) выше, но вместо 4-хлор-транс-коричной кислоты используют на этапе (i) 1-SEM-индазол-2-карбоновую кислоту. 1 Н ЯМР (ДМСО-d6)13,19 (s, 1H),11,59 (s, 1H), 10,29 (s, 1H), 8,23 (d, 1H, J=8,7 Гц),7,73-7,38 (m, 9H), 7,12 (s, 1H), 7,03 (d, 2H, J=7,3 Гц), 6,88 (d, 1H, J=7,8 Гц), 2,38 (s, 1H). Массспектрометрия с высоким разрешением [m+H]/z теория: 459,1821, найдено 459,1836. Пример 7. 3-(Стирил-1 Н-индазол-6-илокси)фениламин. Суспензию 3-(3-стирил-4,5-дигидро-1 Ниндазол-6-илокси)фениламина (75 мг, 0,23 ммол) и 90 мг 5% палладия на углероде (Pd/C) нагревают при 155 С. Через 4 ч добавляют еще 5% Pd/C (39 мг). Через 22 ч добавляют еще 5%Pd/C (30 мг). Реакционную смесь фильтруют в горячем состоянии через 26 ч. Катализатор промывают и сгущают фильтрат при пониженном давлении. Остаток подвергают хроматографии на силикагеле с разбавлением смесью гексаны/ЕtOАс(1:1). Соответствующие фракции сгущают и превращают в порошок смесью СН 2 Сl2/гексаны с получением целевого соединения в виде кремового твердого вещества (20 мг, 27%): 1 Н ЯМР(d, 1H, J = 6,5 Гц), 7,06-6,92 (m, 3H), 6,35 (d, 1H,J=8,3 Гц), 6,23 (s, 2H), 5,26 (s, 2H). Аналитический расчет для С 21 Н 17N3O0,15 СН 2 Сl2: С 74,69; Н 5,13; N 12,36. Найдено: С 74,64; Н 5,23; N 12,25. Пример 8(а). 3-(Е-Стирил)-6-фенокси-1Hиндазол.Pd/C (200 мг) в 10 мл тетралина нагревают при 155 С в течение 18 ч. Катализатор удаляют фильтрацией горячего раствора и промывают смесь ТГФ, ЕtOАс и МеОН. Фильтрат сгущают при пониженном давлении и остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (2:1) с получением 3(Е-стирил)-6-фенокси-1H-индазола в виде кремового твердого вещества (110 мг, 55%). 1 Н ЯМР (ДМСО-d6)6,96 (s, 2H), 7,10 (d, 2H, J=7,7 Гц), 7,20 (t, 1H, J=7,1 Гц), 7,30 (t, 1H, J=7,1 Гц),7,44 (m, 6H), 7,71 (d, 2H, J=7,5 Гц), 8,20 (d, 1H,J=9,2 Гц), 12,90 (s, 1H). Аналитический расчет для C21H16N2O0,1H2O: С 80,28; Н 5,20; N 8,92. Найдено: С 80,20; Н 5,21; N 8,93. Исходный материал получают следующим образом:(2,16 г, 23,0 ммол) в 25 мл ацетона добавляют порошковый безводный К 2 СО 3 (3,81 г, 27,6 ммол). После кипячения с обратным холодильником в течение 18 ч смесь охлаждают и фильтруют. Фильтрат сгущают при пониженном давлении и подвергают хроматографии на силикагеле с разбавлением смесью гексаны/ЕtOАс(4:1) с получением 3-феноксициклогекс-2-енона в виде белого твердого вещества: 1 Н ЯМР(301 мг, 1,6 ммол) в 1 мл ТГФ добавляют к перемешиваемому раствору 1,0 М бис (триметилсилил)амида лития в ТГФ (3,2 мл) при -78 С. Через 15 мин добавляют сразу весь циннамомил хлорид (266 мг, 1,6 ммол). Через 15 мин реакционную смесь выливают в 0,5N НСl и экстрагируют ЕtOАс (2 х). Совмещенные органические слои промывают насыщенным раствором NaCl,высушивают (MgSO4), фильтруют и сгущают при пониженном давлении. Хроматография остатка смесью 4:1 гексаны/этилацетат в качестве разбавителя дает 220 мг (43%) 3-фенокси-6-(3 фенилакрилоил)циклогекс-2-енона в виде желтого твердого вещества (220 мг, 43%): 1 Н ЯМР(1:1) добавляют моногидрат гидразина (0,21 мл,4,3 ммол). Реакционную смесь нагревают при 70 С в течение 3 ч, охлаждают, осторожно переливают в насыщенный раствор Na НСО 3 и экстрагируют ЕtOАс (2 х). Совмещенные органические слои промывают насыщенным раствором NaCl, высушивают (MgSO4) и сгущают при пониженном давлении. Остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (2:1) с получением 6 фенокси-3-стирил-4,5-дигидро-1H-индазола (3) в виде кремового твердого вещества (406 мг,36%): 1 Н ЯМР (ДМСО-d6)2.64 (t, 2 Н, J=8,0 Гц),2,95 (t, 2H, J=8,0 Гц), 5,46 (s, 1H), 7,04 (АВ, 2H,J=16,8 Гц), 7,15 (d, 2H, J=8,1 Гц), 7,25 (m, 2H),7,42 (m, 4H), 7,55 (d, 2H, J=7,7 Гц), 12,44 (s, 1H). Аналитический расчет для С 2 Н 18N2O0,2 Н 2 О: С 79,32; Н 5,83, N 8,81. Найдено: С 79,36; Н 5,85; Пример 8(b) проводят аналогично описанию, приведенному в примере 8(а) выше, но вместо фенола на этапе (i) используют 4(метоксиметокси)фенол. 1 Н ЯМР (ДМСО-d6)12,90 (s, 1H), 8,17 (d, 1H, J=8,8 Гц), 7,71 (d, 2H,J=7,6 Гц), 7,50 (s, 3H), 7,41 (t, 2H, J=7,6 Гц), 7,31 48 Аналитический расчет для С 23H20N2O3: С 74,17; Н 5,41, N 7,52. Найдено: С 74,21; Н 5,59; N 7,46. Пример 8(с). 3-(Е-Стирил)-6-фенилсульфанил-1H-индазол. Пример 8(с) проводят аналогично описанию, приведенному в примере 8(а) выше, но на этапе (i) вместо фенола используют тиофенол. 1 Н ЯМР (ДМСО-d6)7,29 (d, 1H, J=8,5 Гц),7,45-7,59 (m, 9 Н), 7,67 (s, 2H), 7,86 (d, 2H, J=7,2 Гц), 8,35 (d, 1H, J=8,5 Гц), 13,30 (s, 1H). Аналитический расчет для C21H16N2S0,25 Н 2O: С 75,76; Н 5,00; N 8,41; S 9,63. Найдено: С 75,79; Н 4,99; N 8,16; S 9,63. Пример 8(d). 6-(3-Бромофенокси)-3-стирил-1 Н-индазол. Пример 8(d) проводят в порядке, аналогичном описанному в примере 8(а) выше, но на этапе (i) вместо фенола используют 3-бромофенол. 1 Н ЯМР (ДМСО-d6)13,08 (s, 1H), 8,23 (d,1H, J=8,8 Гц), 7,72 (d, 2H, J=7,3 Гц), 7,53 (s, 2H),7,43 - 7,35 (m, 4H), 7,30 (t, 2H, J=7,2 Гц), 7,11 (d,1H, J=7,2 Гц), 7,09 (s, 1H), 6,98 (d, 1H, J=8,8 Гц). Аналитический расчет для C21H15BrN2O: С 64,46; Н 3,86; Br 20,42; N 7,16. Найдено: С 64,31; Н 3,99; Br 20,52; N 7,11. Пример 9 (а). 3-(Е-Стирил)-6-[3-гидроксифенокси]-1H-индазол. К перемешиваемому раствору 3-(Е-стирил)-6-[3-(метоксиметокси)фенокси]-1H-индазола (50 мг, 0,13 ммол) в 5 мл CH2Cl2 при -25 С добавляют триметилсилилбромид (75 мкл, 0,57 ммол). Через 1,5 ч добавляют насыщенный раствор NaHCO, и продукт экстрагируют EtOAc(2x). Совмещенные органические слои промывают насыщенным раствором NaCl, высушивают (MgSO4) и сгущают при пониженном давлении. Остаток подвергают хроматографии на силикагеле с элюированием смесью гексаны/ЕtOАс (1:1) с получением, после превращения в порошок с СН 2 Сl2/гексаны 3-(Е-стирил)-6[3-гидроксифенокси]-1H-индазола в виде кремового твердого вещества (22 мг, 50%): 1 Н ЯМР Н ЯМР (СDСl3)3,42 (s, 3H), 5,10 (s, 2H),6,64 (d, 1H, J=8,2 Гц), 6,72 (s, 1H), 6,80 (d, 1H,J=8,3 Гц), 6,98 (s, 1H), 7,00 (d. 1H, J=8,8 Гц),7,19-7,38 (m, 5H), 7,53 (m, 3H), 7,92 (d, 1H, J=8,9 Гц). Аналитический расчет для C23H20N2O3: Пример 9(b) проводят аналогично примеру 9(а), но вместо 3-(Е-стирил)-6-[3-(метоксиметокси)фенокси]-1H-индазола используют 3-(Естирил)-6-[4-(метоксиметокси)фенокси]-1Hиндазол. 1 Н ЯМР (ДMCO-d6)12,95 (s, 1H), 9,58 6-(1-Фенилвинил)-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазол (16,2 мг,0,0358 ммол) растворяют в ТГФ (0,6 мл) и обрабатывают фторидом тетрабутиламмония (TBAF,1M в ТГФ, 0,6 мл). Смесь нагревают до 60 С в аргоне в течение 4 ч. Смесь охлаждают, нейтрализуют избытком насыщенного раствора бикарбоната натрия, и органический материал в этилацетат и сгущают. Эту смесь 3 соединений (с контролем тонкослойной жидкостной хроматографией) обрабатывают смесью ТГФ-вода-TFA(1:1:2, 4 мл) в течение 30 мин. Смесь разбавляют толуолом (20 мл), сгущают, нейтрализуют избытком насыщенного раствора бикарбоната натрия, и органический материал экстрагируют в этилацетат. Органический материал сушат над сульфатом натрия, сливают и сгущают. Очистка хроматографией на силикагеле (2:8 этилацетатгексан) дает 6-(1-Фенилвинил)-3-стирил-1Hиндазола (4,6 мг, 40%): Rf sm 0,62, р 0,24 (смесь этилацетат-гексан 3:7); 1 Н ЯМР (300 МГц,CDCl3)7,99 (d, 1H, J=8,5 Гц), 7,60-7,25 (m,14H), 5,58 (d, 1H, J=1,1 Гц), 5,56 (d, 1H, J = 1,1 Гц); масс-спектрометрия с высоким разрешением (FAB) [m+H]/z теория 323,1548, найдено 323,1545. 50 Исходный материал получают следующим образом: 3,6-Дийодоиндазол (755 мг, 2,04 ммол) добавляют к 50% КОН (2,5 г в 2,5 мл воды) при 0 С и добавляют дихлорметан (4 мл). К этой смеси добавляют бромид тетрабутиламмония(ТВАВr, 6,6 мг, 0,02 ммол, 0,01 экв.) и добавляют по каплям в течение 3 мин 2-(триметилсиланил)этоксиметил хлорид (SEM-Cl, 397 мкл,2,24 ммол, 1,10 экв.). Смесь быстро перемешивают при 0 С в течение 1,5 ч. Добавляют воду(20 мл) и дихлорметан (20 мл), и органический материал отделяют, сушат над сульфатом натрия и сгущают. Хроматографией на силикагеле-42 С в течение 10 мин и добавляют к свежевысушенному хлориду цинка (34 мг, 0,25 ммол, 2,5 экв.). Полученный раствор согревают до 23 С при перемешивании в течение 25 мин. Эту смесь добавляют к смеси неразбавленного 3,6 дийодо-1-[2-(триметилсиланил)этоксиметил]1H-индазола (50 мг, 0,10 ммол, 1 экв.) иPd(PPh3)4 (5 мг, 0,004 ммол, 0,04 экв.). Через 10 мин определяют завершение реакции с контролем по тонкослойной жидкостной хроматографии и прекращают реакцию насыщенным раствором бикарбоната натрия. Органический материал экстрагируют в этилацетат, сушат над сульфатом натрия и сгущают при пониженном давлении. Хроматографией на силикагеле-42 С в течение 7 мин с получением смеси насыщенного красного цвета. Раствор добавляют через канюлю к свежевысушенному хлориду цинка (29 мг, 0,209 ммол, 3,00 экв.), и смесь согревают до 23 С при перемешивании в течение 20 мин. Этот раствор добавляют через канюлю к неразбавленной смеси 3-йодо-6-(1 фенилвинил)-1-[2-(триметилсиланил)этоксиметил]-1 Н-индазола (33,1 мг, 0,0696 ммол, 1,0 экв.) и Рd(РРh3)4 (4 мг, 0,0035 ммол, 0,05 экв.) при 23 С. Этот раствор оставляют при перемешивании на 15 мин, обрабатывают насыщенным раствором бикарбоната натрия и экстрагируют в этилацетат. Органический материал сушат над сульфатом натрия, сливают и сгущают. Очистка хроматографией на силикагеле в двух колонкахTBAF в ТГФ (10,1 мл, 10,1 ммол), а затем - этилендиамин (0,34 мл, 5,04 ммол, 10 экв.). Полученную смесь нагревают до 70 С в течение 5 ч. Реакцию затем прекращают насыщенным раствором NаНСО 3 (10 мл) и экстрагируют смесь 3 х 35 мл ЕtOАс. Собранную вместе фазу ЕtOАс промывают 5 х 20 мл Н 2 О, затем - рассолом (20 52 мл), высушивают Na2SO4, сливают и сгущают при пониженном давлении в пену. Неочищенный материал очищают хроматографией на силикагеле (смесь 9:1 дихлорметан/этилацетат) с получением N-метил-N-(3 стирил-1H-индазол-6-ил)бензол-1,3-диамина в виде пены (120 мг, выход 70%). Rf sm 0,73, Rf p 0,27 (смесь дихлорметан : этилацетат 7:3); 13 С ЯМР (75 МГц, CDCl3)150,3, 148,8, 147,5,147,5, 143,9, 143,4, 137,5, 131,1, 130,3, 129,3,128,9, 128,2, 127,9, 126,7, 121,0, 120,5, 117,0,116,0, 112,6, 109,8, 109,0, 98,3, 40,7; жидкостная хроматография и масс-спектрометрия с вторичной ионизацией (ESI) [M+H]/z теория 341, найдено 341. Аналит. расчет: С 77,62; Н 5,92; N 16,46. Найдено: С 76,16; Н 5,88; N 15,95. Исходный материал получают следующим образом:Pd(PPh3)4 (1,25 г, 1,08 ммол) в атмосфере аргона добавляют толуол (192 мл), МеОН (4 мл) и 2NNaOH (водн.) (32,6 мл, 65,3 ммол). Полученную гетерогенную смесь нагревают до 90 С. Через 8 ч реакционную смесь разбавляют ЕtOАс (150 мл) и водой (50 мл), фазы разделяют, и органическую фазу 2 х 50 мл ЕtOАс. Собранную вместе 53 органическую фазу промывают рассолом (50 мл), затем высушивают Na2SO4, фильтруют и сгущают при пониженном давлении. Неочищенную реакционную смесь очищают хроматографией на силикагеле (смесь 1:9 ЕtOАс: гексан) с получением 6-нитро-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазола в виде желтого твердого вещества (7,65 г, 74%): 13 С ЯМР (75 МГц, CDCl3)148,3, 145,0, 141,3,138,1, 134,2, 130,5, 129,9, 129,8, 129,5, 128,1,127,4, 123,2, 119,8, 117,8, 108,2, 79,7, 68,5, 19,2,0,0; масс-спектроскопия (FAB) [M+Na]/z Теория 418, найдено 418. Аналит. расчет: С 63,77; Н 6,37; N 10,62. Найдено: С 64,04; Н 6,29; N 10,56. 6-Нитро-3-стирил-1-[2-(триметилсиланил) этоксиметил]-1H-индазол (8,1 г, 20,5 ммол) растворяют в ДМФ (75 мл) при 23 С в атмосфере аргона. Добавляют SnCl2 (12,9 г, 67,7 ммол), а затем воду (1,7 мл, 92,2 ммол), и полученную смесь нагревают до 50 С. Через 4 ч добавляют 3N NaOH (45 мл, 135 ммол), а затем - ЕtOАс(100 мл). Полученную эмульсию фильтруют в горячем состоянии через Целит, и слой Целита промывают горячим ЕtOАс (3 х 100 мл). Фильтрат сгущают при пониженном давлении, остаток растворяют в ЕtOАс, промывают рассолом, высушивают Na2SO4, фильтруют и сгущают при пониженном давлении с получением твердого вещества. Неочищенный материал очищают хроматографией на силикагеле (смесь 2:87:3 этилацетат: гексан), с получением 3-стирил-1[2-(триметилсиланил)этоксиметил]-1H-индазол 6-иламина в виде желтого твердого вещества(0,07 г, 0,133 ммол), Pd2(dba)3 (34 мг, 0,0375 ммол) и Cs2CO3 (1,37 г, 4,2 ммол) в атмосфере аргона добавляют толуол (6 мл). Полученную гетерогенную смесь нагревают до 80 С. Через 46 ч реакционную смесь охлаждают до 23 С,разбавляют этилацетатом (ЕtOАс) (20 мл) и фильтруют. Добавляют воду (5 мл), фазы разделяют, и органическую фазу экстрагируют 2 х 50 мл ЕtOАс. Собранный вместе органический материал промывают рассолом, затем высушивают 54 давлении. Неочищенную реакционную смесь очищают хроматографией на силикагеле (разбавление смесью 9:1 гексан : ЕtOАс) с получением (3-нитрофенил)-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазол-6-иламина в виде желтого твердого вещества (7,65 г, 74%): Тонкослойная жидкостная хроматография К (3-нитрофенил)-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазол-6-ил амину (434 мг, 0,89 ммол) в ТГФ (5 мл) после охлаждения до -5 С в атмосфере аргона, добавляют диметилсульфат (0,42 мл, 4,5 ммол), а затем - LiHMDS (1M в ТГФ) (1,8 мл, 1,8 ммол). Через 20 мин реакцию прекращают насыщенным раствором NH4 Сl (водн.) (2 мл), затем экстрагируют 3 х 20 мл EtOAc. Собранный вместе органический материал промывают рассолом(10 мл), высушивают Na2SO4, сливают и сгущают при пониженном давлении. Очистка хроматографией на силикагеле (разбавление смесью гексан : EtOAc 9:1) дает метил-(3-нитрофенил)3-стирил-1-[2-(триметилсиланил)этоксиметил]1H-индазол-6-иламин, в виде масла (367 мг,82%): Тонкослойная жидкостная хроматография(81 мкл, 1,0 ммол), Ас 2O (94 мкл, 1,0 ммол) и ДМАП (катализ.). Реакционная смесь немедленно становится гомогенной. Через 1 ч анализ методом тонкослойной жидкостной хроматографии (СН 2 Сl2: ЕtOAс 4:1) показывает отсутствие исходного материала. Реакцию прекращают насыщенным раствором NaHCO3 (волн.) (2 мл) затем разбавляют ЕtOАс (15 мл), и органическую фазу промывают рассолом (3 мл), сливают и сгущают при пониженном давлении в масло. Масло взвешивают в МеОН (2 мл) и добавляют К 2 СО 3 (83 мг, 0,6 ммол). Полученную смесь перемешивают при 23 С в атмосфере аргона. Через 1 ч реакционную смесь разбавляют ЕtOАс(15 мл), и органическую фазу промывают рассолом (3 мл), сливают и сгущают при пониженном давлении. Неочищенный материал очищают полупрепаративной жидкостной хроматографий с высоким разрешением с получением N-3[метил-(3-стирил-1H-индазол-6-ил)амино] фенилацетамида (8,4 мг, 22%). 1H ЯМР (300 МГц,CDCl3)7,86 (d, 1H, J = 8,68 Гц), 7,58 (d, 1H,J=7,17 Гц), 7,16 - 7,45 (m, 7 Н), 7,15 (d, 1H,J=8,29 Гц), 6,98 (m, 1H), 6,95 (d, 1H, J=1,92 Гц), 004460 Пример 12(b) проводят аналогично описанию, приведенному в примере 12(а) выше, но вместо уксусного ангидрида используют бензоил хлорид. Жидкостная хроматография с массспектрометрией с вторичной ионизацией (массспектрометрия в сочетании с жидкой хроматографией) (ESI) [M+H]/z теория 475, найдено 475. Аналит. расчет С (78,36), Н (5,44), N Пример 12(с) проводят аналогично описанию, приведенному в примере 12(а) выше, но вместо уксусного ангидрида используют карбобензилоксихлорид.N-метил-N-(3-стирил-1Hиндазол-6-ил)бензол-1,3-диамина, приготовленному в примере 11 (26 мг, 0,075 ммол), и 5 метилтиазол-2-карбоновой кислоты (64 мг, 0,45 ммол) в ДМФ (0,375 мл) при 23 С в атмосфере аргона добавляют HATU (171 мг, 0,45 ммол). Через 1 ч анализ методом тонкослойной жидкостной хроматографии (смесь СН 2 Сl2 : ЕtOАс 8:2) показывает отсутствие исходного материала. Реакцию прекращают насыщенным раствором NаНСО 3 (водн.) (2 мл), затем разбавляют ЕtOАс (15 мл), и органическую фазу промывают рассолом (3 мл), сливают и сгущают при пониженном давлении. Масло взвешивают в МеОН(2 мл) и добавляют К 2 СО 3 (62 мг, 0,45 ммол). Полученную смесь перемешивают при 23 С в 57 атмосфере аргона. Через 1 ч анализ методом тонкослойной жидкостной хроматографии(смесь СН 2 Сl2 : ЕtOAс 8:2) показывает отсутствие исходного материала. Реакционную смесь разбавляют ЕtOАс (15 мл), и органическую фазу промывают рассолом (3 мл), сливают и сгущают при пониженном давлении до твердого вещества. Неочищенный материал очищают хроматографией на силикагеле (разбавление смесью СН 2 Сl2 : ЕtOАс 85:15) с получением целевого соединения после очистки полупрепаративной жидкостной хроматографии с высоким разрешением (9,9 мг, 28 %), Rf sm 0,25, Rf p 0,39(ESI) [M+H]/z теория 431, найдено 431. Аналит. расчет: С 78,12; Н 5,15; N 13,01. Найдено: С 77,06; Н 6,91; N 9,88. Исходный материал получают следующим образом: К раствору N-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазол-6-илбензол 1,3-диамина (91 мг, 0,2 ммол) и пиридина (0,081 мл, 1,0 ммол) в СН 2 Сl2 (0,5 мл), охлажденному до -5 С в атмосфере аргона, добавляют бензоилхлорид (0,028 мл, 0,24 ммол). Через 0,5 ч реакцию прекращают насыщенным растворомNaHCO3 (водн.) затем экстрагируют 2 х 5 мл СН 2 Сl2. Собранный вместе органический материал промывают рассолом (5 мл), высушиваютNa2SO4, сливают и сгущают при пониженном давлении с получением масла. Неочищенный материал очищают хроматографией на силикагеле (разбавление смесью гексан : ЕtOАс 3:2) с получением N-(3-3-Стирил-1-[2-(триметилсиланил)этоксиметил]-1H-индазол-6-иламино фенил)бензамида (108 мг, выход 96%). Rf sm 0,35, Rf p 0,44 (этилацетат : гексан 1:1); ИК Фурье-спектрометрия (тонкопленочн,) 3320, 2951,2893, 1657, 1604, 1537, 1493, 1409, 1303, 1248,1074 см-1; жидкостная хроматография с массспектрометрией с вторичной ионизацией (ESI) Метилфенил-3-стирил-1-[2-(триметилсиланил)этоксиметил]-1 Н-индазол-6-иламин превращают в метилфенил-(3-стирил-1H-индазол-6-ил)амин, как описано в примере 11. Массспектроскопия (ESI) [M+H]/z теория 326, найдено 326. Исходный материал получают следующим образом:(1,58 г, 4 ммол) в АсОН (14 мл), воды (3 мл) и концентрированной НСl (1,67 мл), охлажденному до 2 С, добавляют раствор NaNO2 (304 мг,4,4 ммол) в воде (0,5 мл) в течение 5 мин. Полученный раствор насыщенного красного цвета перемешивают при 2 С в течение 0,5 ч, затем добавляют по каплям раствор KI (797 мг, 4,8 ммол) и I2 (610 мг, 2,4 ммол) в воде (1 мл), поддерживая внутреннюю температуру ниже 5 С. Через 2 ч при 2 С реакционную смесь оставляют для перемешивания при 23 С на 17 ч. Реакцию прекращают 3N NaOH (волн.), разбавляют ЕtOАс (50 мл) и Н 2O (15 мл), фазы разделяют, и водную фазу экстрагируют 2 х 15 мл ЕtOАс. Собранную вместе органическую фазу промывают 3 х 20 мл 5% NaHSO3, рассолом (15 мл),высушивают Na2SO4, сливают и сгущают при пониженном давлении. Неочищенную реакционную смесь очищают хроматографией на силикагеле (разбавление смесью 1:1 гексан: ЕtOАс) с получением 6-йодо-3-стирил-1-[2(триметилсиланил)-этоксиметил]-1H-индазола в виде белого твердого вещества (1,3 г, 68%N 13,80. Исходный материал получают следующим образом:Pd(PPh3)4 (0,58 г, 0,5 ммол) при 23 С в атмосфере аргона добавляют 1,4-диоксан (38 мл) и 2NNaOH (водн.) (12,5 мл, 25 ммол). Полученную смесь нагревают до 90 С. Через 2 ч реакционную смесь разбавляют ЕtOАс (100 мл) и водой(70 мл), фазы разделяют, и органическую фазу экстрагируют 2 Х 100 мл ЕtOАс. Собранную вместе органическую фазу промывают рассолом(20 мл), а затем высушивают Na2SO4, фильтруют и сгущают при пониженном давлении. Неочищенную смесь очищают хроматографией на 60 силикагеле (разбавление смесью 9:1 гексан : ЕtOАс) с получением 3-(2-бензо[1,3]диоксол-5 илвинил)-6-нитро-1-[2-(триметилсиланил)этоксиметил]-1H-индазола в виде желтого твердого вещества (4,15 г, выход 94%). ИК Фурьеспектрометрия (тонкопленочн.) 2950, 2898,1523, 1501, 1483, 1446, 1344, 1249, 1080, 1043,927 см-1; 1H ЯМР (300 МГц, CDCl3)8,56 (dd,1H, J=0,68, 1,75 Гц), 8,14 (d, 1H, J=1,78 Гц), 8,13
МПК / Метки
МПК: A61P 35/00, A61K 31/416, C07D 471/04, C07D 231/56
Метки: ингибирования, индазола, применения, препараты, фармацевтические, протеинкиназ, способы, соединения
Код ссылки
<a href="https://eas.patents.su/30-4460-soedineniya-indazola-farmacevticheskie-preparaty-dlya-ingibirovaniya-proteinkinaz-i-sposoby-ih-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения индазола, фармацевтические препараты для ингибирования протеинкиназ и способы их применения</a>
Циклоалкильные соединения, лактамы, лактоны и родственные соединения, содержащие их фармацевтические композиции и способы ингибирования высвобождения и/или синтеза β-амилоидного пептида с помощьюуказанных соединений

Номер патента: 2100
Опубликовано: 24.12.2001
Авторы: Нейц Джеффри, Кви Синтия Л., Ниссен Джеффри С., Портер Варрен Дж., Торсетт Юджин Д., Одия Джеймс Е., Фридман Стефен, Ву Джинг, Дрост Джеймс Дж., Плейсс Майкл А., Макданиел Стейси Л., Танг Джей С., Дрессман Брюс А., Мабри Томас Э., Скотт Уильям Леонард, Стаки Расселл Д., Рил Джон К., Джон Варгес, Латимер Ли Х., Генри Стивен С., Бриттон Томас К.
МПК: A61P 25/28, A61K 31/55, C07D 243/10...
Метки: фармацевтические, циклоалкильные, способы, помощьюуказанных, соединений, лактоны, высвобождения, композиции, beta;-амилоидного, пептида, соединения, лактамы, содержащие, родственные, синтеза, ингибирования
Формула / Реферат:
1. Способ ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, который заключается в том, что в такую клетку вводят соединение или смесь соединений в количестве, эффективном для ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, и указанные соединения имеют формулу I в которой R1 выбирают из группы, включающей C1-С10алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из...
Производные 2-аминопиридинов, способ ингибирования синтазы оксида азота, фармацевтические композиции и способы лечения

Номер патента: 3991
Опубликовано: 25.12.2003
Автор: Лоуи Джон Адамс III
МПК: A61K 31/4439, C07D 213/73, A61P 11/00...
Метки: 2-аминопиридинов, оксида, производные, способы, композиции, способ, ингибирования, синтазы, фармацевтические, азота, лечения
Формула / Реферат:
1. Производные 2-аминопиридинов формулы где n и m в мостиковых кольцах независимо равны 1, 2 или 3 и атом углерода в одном из указанных мостиковых колец может быть заменен гетероатомом, выбранным из O, S и N, при условии, что узловой атом углерода может быть заменен только атомом азота, и R1 и R2 независимо выбраны из C1-C6алкила, который может быть линейным, разветвленным или циклическим или может содержать линейную, циклическую либо...
Антибактериальные соединения карбапенема, содержащие их фармацевтические композиции и способы лечения

Номер патента: 1296
Опубликовано: 25.12.2000
Авторы: Вилкенинг Роберт Р., Близзард Тимоти А., Рэтклифф Рональд В.
МПК: A61K 31/428, C07D 477/14
Метки: способы, соединения, карбапенема, содержащие, антибактериальные, композиции, фармацевтические, лечения
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемые соли, где R1 обозначает Н или метил; СО2М обозначает карбоновую кислоту, карбоксилатный анион, фармацевтически приемлемую сложноэфирную группу или карбоновую кислоту, защищенную защитной группой; Р обозначает водород, гидроксил, F или гидроксил, защищенный гидроксилзащитной группой; каждый R независимо выбирают из -R*; -Q; водорода; галогена; -CN; -NO2; -NRaRb; -ORc; -SRc; -C(O)NRaRb;...
Фармацевтические композиции, содержащие в качестве активного начала жизнеспособные лактобациллы, способы их применения и препарат из микроинкапсулированных лактобацилл

Номер патента: 412
Опубликовано: 24.06.1999
Автор: Форд Лэрри С.
МПК: A61K 35/74
Метки: начала, жизнеспособные, лактобацилл, лактобациллы, способы, активного, композиции, применения, фармацевтические, препарат, качестве, микроинкапсулированных, содержащие
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного начала жизнеспособные лактобациллы и фармацевтически приемлемый носитель для целенаправленной доставки лактобацилл, отличающаяся тем, что единичная доза указанной композиции содержит, по крайней мере, 103 лактобацилл, причем указанные лактобациллы микроинкапсулированы и в результате этого сохраняют жизнеспособность в течение всего срока хранения композиции, а вещество, составляющее...
Усеченные рецепторы фактора некроза опухолей, фармацевтические композиции на их основе и способы применения указанных факторов и композиций

Номер патента: 3900
Опубликовано: 30.10.2003
Авторы: Фишер Эрик Ф., Кифт Гэри Л., Эдвардс Карл К.
МПК: A61P 43/00, A61K 38/17, C07K 14/715...
Метки: указанных, способы, применения, некроза, опухолей, рецепторы, композиции, основе, фактора, фармацевтические, композиций, факторов, усеченные
Формула / Реферат:
1. Усеченный рецептор фактора некроза опухолей (sTNFR), имеющий следующую формулу: R1-[Cys19-Cys103]-R2, где [Cys19-Cys103] представляет собой аминокислотные остатки с 19 по 103 sTNFR-I последовательности SEQ ID NO:2, приведенной на фиг. 1, R1 обозначает метионилированную или неметионилированную аминогруппу Cys19, или аминоконцевой аминокислотный остаток, или последовательность, выбранные из группы, включающей a R2 обозначает карбоксигруппу...
Предыдущий патент: Сложные эфиры l-карнитина или алканоил l-карнитинов, полезные в качестве катионных липидов для внутриклеточной доставки фармакологически активных соединений
Следующий патент: Способ пневматического транспортирования материала
Случайный патент: Композиции и способы для профилактики и лечения ран