Антипикорнавирусные композиции, их получение и применение
Номер патента: 3856
Опубликовано: 30.10.2003
Авторы: Тикхе Джайашри Гириш, Зоу Ру, Драгович Питер Скотт, Веббер Стефен Эван, Мараковиц Джозеф Тимоти, Принс Томас Джей





























Формула / Реферат
1. Соединение формулы I

где Y представляет -N(Ry)-, -C(Ry)2- или -O-, где каждый Ry независимо представляет H или линейный или разветвленный C1-4алкил;
R1 представляет -H, -F, C1-6алкил с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно, замещенный галогеном; -OH, -SH или O-алкильную группу;
R2 и R3, каждый независимо, представляют H;

или

где n является целым числом от 0 до 5, A1 представляет CH или N, A2 и каждый A3 независимо выбраны из C(R41)(R41), N(R41), S, S(O), S(O)2 и O и A4 представляет NH или NR41, где каждый R41 независимо представляет H или C1-4алкил с линейной или разветвленной цепью при условии, что не более двух гетероатомов встречается последовательно в изображенном выше цикле, образованном A1, A2, (A3)n, A4 и C=O, и, по меньшей мере, один из R2 и R3 представляет

R5 и R6, каждый независимо, представляют H, или C1-6арилалкильную группу, арильная часть которой, возможно, замещена галогеном или C1-4алкилом;
R7 и R8, каждый независимо, представляют H, C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом;
R9 представляет пятичленный гетероцикл, имеющий от одного до трех гетероатомов, выбранных из O, N и S; и
Z и Z1, каждый независимо, представляют H или группу CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон;
или его пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват.
2. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.1, где R2 и R3, каждый независимо, представляют H;

где n является целым числом от 0 до 5, каждый R41 независимо представляет H или C1-4алкил с линейной или разветвленной цепью и стереохимия атома углерода, отмеченного звездочкой (*), может быть R или S; при условии, что, по меньшей мере, один из R2 и R3 представляет
 или
или 
3. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.1, где Y представляет -N(Ry)-, где Ry является H или линейным или разветвленным C1-4алкилом.
4. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где Z и Z1, каждый независимо, выбраны из H, -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон.
5. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где R1 представляет H, F или метил.
6. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где, по меньшей мере, один из R2 и R3 представляет

7. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.6, где один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
8. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
9. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где один из R5 и R6 представляет H, а другой представляет незамещенный или замещенный фенилметил.
10. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где R7 и R8, каждый независимо, представляют H, C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом.
11. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где один из R7 и R8 представляет H, а другой представляет 2-пропил, 2-метил-2-пропил, 2-метил-1-пропил, нафтилметил.
12. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где R9 представляет пятичленный гетероцикл, имеющий, по меньшей мере, один гетероатом азота и один гетероатом кислорода.
13. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где R9 выбран из замещенных или незамещенных 1,2-оксазолила, 1,3-оксазолила и 1,2,4-оксадиазолила.
14. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.3, где R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
15. Соединение по п.3 формулы I-A"

где R1, R2, R6, R7, R9, Ry, Z и Z1 такие, как определены в п.3,
или его пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват.
16. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.15, где R2 представляет

17. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.15,
где Ry представляет H или метил;
R1 представляет H, F или метил;
Z и Z1, каждый независимо, выбран из H, -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон;
R2 представляет

R6 представляет незамещенный или замещенный фенилметил;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом; и
R9 представляет 3-изоксазолил шыш 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
18. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.17, где R7 выбирают из 2-пропила, 2-метил-2-пропила, 2-метил-1-пропила и нафтилметила.
19. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.15, где Ry, R1 и Z каждый представляет H, и
R2 представляет CH2CH2C(O)NH2, R6 представляет CH2Ph, R7 представляет CH2CH(CH3)2, Z1 представляет CO2CH2CH3, и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет CH2Ph, R7 представляет CH2CH(CH3)2, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет C(CH3)3, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет C(CH3)3, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет CH(CH3)2, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет C(CH3)3, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z1 представляет CO2CH2CH3 и R9 представляет

или
R2 представляет

R6 представляет

R7 представляет C(CH3)3, Z1 представляет CO2CH2CH3 и R9 представляет

20. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.15, где Ry представляет CH3, R1 и Z каждый представляет H, и
R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет

Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет CH2Ph, R7 представляет CH2CH(CH3)2 и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет

и R9 представляет

R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет CH2CH(CH3)2 и R9 представляет

или
R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет

и R9 представляет

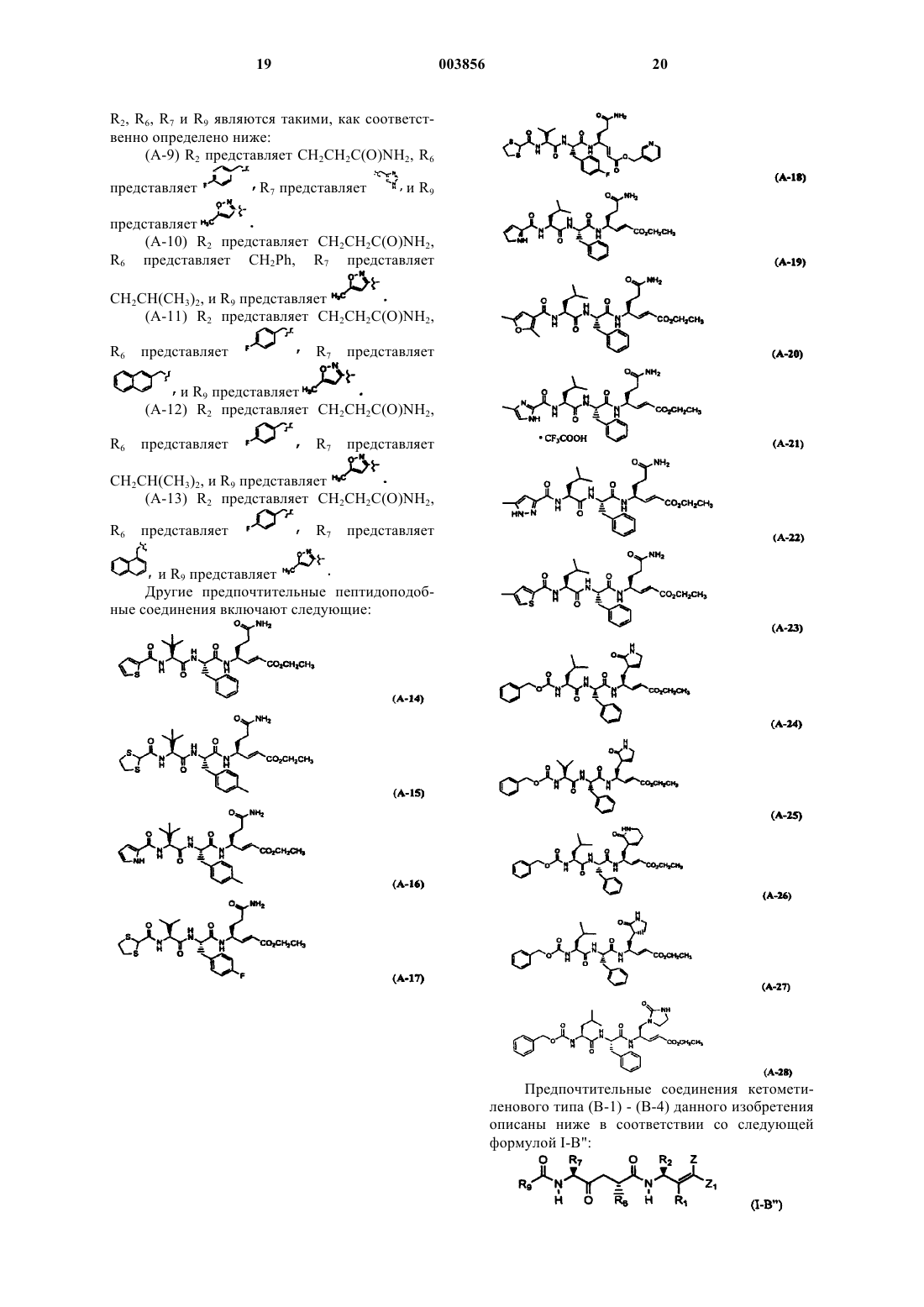
21. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.15, выбранные из группы, состоящей из

22. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.1, где Y представляет -CH2-.
23. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где Z и Z1, каждый независимо, выбран из H, -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон.
24. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где R1 представляет H, F или метил.
25. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где, по меньшей мере, один из R2 и R3 представляет

26. Соединение, пролекарственэря форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.25, где один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
27. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
28. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где один из R5 и R6 представляет H, а другой представляет незамещенный или замещенный фенилметил.
29. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где R7 и R8, каждый независимо, представляют H или C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом.
30. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где один из R7 и R8 представляет H, а другой представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом.
31. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где один из R7 и R8 представляет H, а другой представляет 2-пропил, 2-метил-2-пропил, 2-метил-1-пропил и нафтилметил.
32. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.22, где R9 представляет пятичленный гетероцикл, имеющий, по меньшей мере, один гетероатом азота и один гетероатом кислорода.
33. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.32, где R9 представляет незамещенный или замещенный 1,2-оксазолил, 1,3-оксазолил и 1,2,4-оксадиазолил.
34. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.32, где R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
35. Соединение по п.22 формулы I-B"

где R1, R2, R6, R7, R9, Z и Z1 такие, как определены в п.22,
или его пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват.
36. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.35, где R2 представляет

или

37. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.35,
где R1 представляет H, F или метил;
где Z и Z1, каждый независимо, выбраны из H или группы -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон;
R2 выбирают из

R6 представляет незамещенный или замещенный фенилметил;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом; и
R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
38. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.37, где R7 выбираютиз 2-пропила, 2-метил-2-пропила, 2-метил-1-пропила и нафтилметила.
39. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.35, где R1 представляет H и
R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет CH(CH3)3, Z представляет H, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z представляет H, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z и Z1 вместе представляют

где карбонильная группа находится в цис-положении по отношению к водороду R1, и R9 представляет

или
R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z представляет H, Z1 представляет CO2CH2CH3 и R9 представляет

40. Соединение по п.1 формулы

или его пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват.
41. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.1, где Y представляет -O-.
42. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где Z и Z1, каждый независимо, выбраны из H, -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон.
43. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где R1 представляет H, F или метил.
44. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где, по меньшей мере, один из R2 и R3 представляет

45. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.44, уфх один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
46. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где один из R5 и R6 представляет H, а другой представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом.
47. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где один из R5 и R6 представляет H, а другой представляет незамещенный или замещенный фенилметил.
48. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где R7 и R8, каждый независимо, представляют H, C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом.
49. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где один из R7 и R8 представляет H, а другой представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом.
50. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где один из R7 и R8 представляет H, а другой представляет 2-пропил, 2-метил-2-пропил, 2-метил-1-пропил или нафтилметил.
51. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где R9 представляет пятичленный гетероцикл, имеющий, по меньшей мере, один гетероатом азота и один гетероатом кислорода.
52. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где R9 выбирают из замещенных или незамещенных 1,2-оксазолила, 1,3-оксазолила и 1,2,4-оксадиазолила.
53. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.41, где R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
54. Соединение по п.41 формулы I-C"

где R1, R2, R6, R7, R9, Z и Z1 такие, как определены в п.42,
или его пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват.
55. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.54, где R2 представляет

или

56. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.54,
где R1 представляет H, F или метил;
где Z и Z1, каждый независимо, выбраны из H, -CO2R21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом;
при условии, что Z и Z1 не являются оба H;
или Z и Z1 вместе с атомами, к которым они присоединены, образуют фуранон;
R2 выбирают из

R6 представляет незамещенный или замещенный фенилметил;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом; и
R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов.
57. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.56, где R7 выбирают из 2-пропила, 2-метил-2-пропила, 2-метил-1-пропила и нафтилметила.
58. Соединение, пролекарственная форма, фармацевтически активный метаболит, фармацевтически приемлемая соль или сольват по п.54, где R1 представляет H, Z представляет H и
R2 представляет CH2CH2C(O)NH2, R6 представляет

R7 представляет CH(CH3)2, Z1 представляет CO2CH2CH3 и R9 представляет

R2 представляет

R6 представляет

R7 представляет CH(CH3)2, Z1 представляют CO2CH2CH3 и R9 представляет

59. Соединение, выбранное из группы, состоящей из

или его пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват.
60. Применение соединения, фармацевтически приемлемой соли, пролекарственной формы, фармацевтически активного метаболита или сольвата по п.1 или 59 в качестве антипикорнавирусного агента, антипикорнавирусная активность которого соответствует значению EC50, меньшему или равному 100 мкМ по клеточному культуральному анализу Hl-HeLa.
61. Применение соединения, пролекарственной формы, фармацевтически активного метаболита, фармацевтически приемлемой соли или сольвата по п.1 или 59 в качестве антипикорнавирусного агента, антириновирусная активность которого соответствует значению EC50, меньшему или равному 10 мкМ по клеточному культуральному анализу Hl-HeLa.
62. Соединение, имеющее формулу, выбранную из группы, состоящей из

где R1, R2, Z и Z1 являются такими, как определены в п.1, n равно 1 или 2;
Ry представляет H или линейный или разветвленный C1-4алкил;
R6 представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом;
R9 представляет пятичленный гетероцикл, имеющий от одного до трех гетероатомов, выбранных из O, N и S, или его пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват.
63. Соединение, пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват по п.61,
где R1 представляет H, F или C1-6алкильную группу с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно, замещенной галогеном;
Ry представляет H или метил;
R6 представляет арилалкильную группу, возможно, замещенную галогеном или C1-4алкилом;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом;
R9 представляет пятичленный гетероцикл, имеющий от одного до трех гетероатомов, выбранных из O, N и S, где, по меньшей мере, один из гетероатомов представляет азот, и являющийся незамещенным или замещенным одним или двумя заместителями, выбранными из линейных или разветвленных C1-4алкильных групп и галогенов;
или хую пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват.
64. Соединение, пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват по п.62,
где R6 представляет арилметил;
R7 представляет C1-6алкильную группу, возможно, замещенную нафтилом или имидазолом;
R9 представляет 3-изоксазолил или 5-изоксазолил, незамещенный или замещенный одним или двумя заместителями, выбранными из метила и галогенов, и
где Z представляет H, и Z1 каждый представляет -COOR21, где R21 представляет H, C1-6алкильную группу, возможно, замещенную пиридилом.
65. Соединение, пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват по п.64,
где R1 представляет H или F;
R6 представляет арилметил, и
R7 выбирают из 2-пропила, 2-метил-2-пропила, 2-метил-1-пропила и арилметила.
66. Соединение, пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват по п.65, где R6 представляет фенилметил, где фенильный фрагмент необязательно имеет от одного до трех заместителей, выбранных из галогена, линейного или разветвленного C1-4алкила.
67. Соединение по п.62, выбранное из группы, состоящей из

или его пролекарственная форма, фармацевтически приемлемая соль, фармацевтически активный метаболит или сольват.
68. Фармацевтическая композиция, включающая:
(a) терапевтически эффективное количество, по меньшей мере, одного антипикорнавирусного агента, который представляет соединение, пролекарственную форму, фармацевтически активный метаболит, фармацевтически приемлемую соль или сольват, как определено в п.1 или 59; и
(b) фармацевтически приемлемый носитель, разбавитель, среду для лекарства или эксципиент.
69. Способ лечения болезненного состояния млекопитающего, опосредованного протеазной активностью пикорнавирусов, включающий введение млекопитающему, нуждающемуся в нем, терапевтически эффективного количества, по меньшей мере, одного соединения, пролекарственной формы, фармацевтически активного метаболита, фармацевтически приемлемой соли или сольвата, как определено в п.1 или 59.
70. Способ ингибирования активности 3C протеаз пикорнавирусов, включающий приведение в контакт 3C протеазы пикорнавирусов с эффективным количеством, по меньшей мере, одного соединения, пролекарственной формы, фармацевтически активного метаболита, фармацевтически приемлемой соли или сольвата, как определено в п.1 или 59.
71. Способ по п.70, где 3C протеазой пикорнавирусов является риновирусная протеаза.
72. Соединение формулы

где р является целым числом от 0 до 5;
A11 представляет CH или N, A12 и каждый A13 независимо выбирают из C(R61)(R61), N(R61), S, S(O), S(O)2 и O и A14 представляет NH или NR61, где каждый R61 независимо представляет H, C1-6алкильную группу с линейной или разветвленной, насыщенной или ненасыщенной цепью, замещенной галогеном, или арильную группу, представляющую моно- или бициклический радикал, содержащий 6-10 атомов углерода в кольце, возможно, замещенную галогеном или C1-4алкилом; ацил, при условии, что не более двух гетероатомов встречается последовательно в цикле, образованном A11, A12, (A13)p, A14 и C=O;
каждый R141 независимо представляет H или линейный или разветвленный C1-4алкил;
R52, R53 и R54, каждый независимо, выбирают из H, гидроксила, C1-6алкильной группы с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно замещенной галогеном, или арильной группы, представляющей моно- или бициклический радикал, содержащий 6-10 атомов углерода в кольце, возможно, замещенной галогеном или C1-4алкилом; ацила; или любые два из R52, R53 и R54 вместе образуют H, C1-6алкильной группы с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно, замещенной галогеном, CO2(C1-C6)алкила, C(O)N(C1-C6)алкила и CO2(арила); и
R55 и R56, каждый независимо, представляют H или подходящую защитную группу для азота;
или фармацевтически приемлемая соль упомянутого соединения.
73. Соединение формулы

где p является целым числом от 0 до 5;
R51 представляет H, C1-6алкильную группу с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно, замещенной галогеном, ацил;
R52, R53 и R54, каждый независимо, выбирают из H, гидроксила, C1-6алкильной группы с линейной или разветвленной, насыщенной или ненасыщенной цепью, замещенной галогеном, или арильной группы, представляющей моно- или бициклический радикал, содержащий 6-10 атомов углерода в кольце, возможно, замещенной галогеном или C1-4алкилом, ацила;
или любые два из R52, R53 и R54 вместе образуют =O или =C(R57)(R58), где R57 и R58, каждый независимо, выбирают из H, C1-6алкильной группы с линейной или разветвленной, насыщенной или ненасыщенной цепью, возможно, замещенной галогеном; CO2(C1-C6)алкила, C(O)N(C1-C6)алкила и CO2(арила); и
R55 и R56, каждый независимо, представляют H или подходящую защитную группу для азота;
или фармацевтически приемлемая соль упомянутого соединения.
74. Соединение или фармацевтически приемлемая соль по п.73, где p равно 1 или 2.
75. Соединение или фармацевтически приемлемая соль по п.74, где R52, R53 и R54, каждый независимо, выбирают из H, алкокси, гидрокси и карбонила.
76. Соединение по п.73, выбранное из группы, состоящей из

где PN является подходящей защитной группой для азота, и q равно 1 или 2;
или фармацевтически приемлемая соль упомянутого соединения.
77. Соединение по п.73, выбранное из группы, состоящей из

где BOC представляет т-бутоксикарбонил или фармацевтически приемлемую соль упомянутого соединения.
78. Соединение формулы

где BOC представляет т-бутоксикарбонил.
Текст
1 Перекрестная ссылка на родственную заявку Эта заявка относится к предварительной заявке США 60/083828, поданной 30 апреля 1998 г. от имени Dragovich et al., раскрытие которой включено в данный документ в качестве ссылки. Область и промышленное применение данного изобретения Данное изобретение относится к пептидоподобным и пептидомиметическим соединениям, которые преимущественно ингибируют ферментативную активность 3C протеаз пикорнавирусов, особенно 3C протеаз риновирусов(RVPs), и которые замедляют рост вирусов в клеточной культуре. Данное изобретение также относится к применению таких соединений в фармацевтических композициях и в терапевтическом лечении риновирусных инфекций. Данное изобретение далее относится к способам синтеза таких соединений и соединений, использующихся в таких синтезах. Пикорнавирусы представляют семейство мелких РНК-содержащих вирусов без оболочки,с положительной цепью, которые инфицируют человека и других животных. Эти вирусы включают человеческие риновирусы, человеческие полиовирусы, человеческие вирусы Коксаки,человеческие ЕСНО-вирусы, человеческие и бычьи энтеровирусы, вирусы энцефаломиокардита, вирус менингита, вирусы ног и рта, вирус гепатита А и другие. Человеческие риновирусы являются основной причиной обычной человеческой простуды. В настоящее время на рынке не существует эффективной терапии, которая вылечивает обычную простуду, только лечение,облегчающее симптомы. Лечение пикорнавирусных инфекций может проводиться путем ингибирования протеолитических 3C ферментов. Эти ферменты необходимы для естественного созревания пикорнавирусов. Они отвечают за автокаталитическое расщепление геномного, большого полипротеина на эссенциальные вирусные белки. Членами семейства 3C протеаз являются цистеиновые протеазы, в которых сульфгидрильная группа наиболее часто расщепляет глутаминглициновую амидную связь. Считается, что ингибирование 3C протеаз блокирует протеолитическое расщепление полипротеина, что, в свою очередь, может замедлять созревание и репликацию вирусов путем препятствования продукции вирусной частицы. Следовательно, ингибирование активности этой цистеиновой протеазы селективными небольшими молекулами, способными к специфичному распознаванию, будет представлять важный и полезный подход к обработке и лечению вирусных инфекций этой природы, в особенности, обычной простуды. Недавно были открыты некоторые низкомолекулярные ингибиторы ферментативной активности 3C протеаз пикорнавирусов (т.е. антипикорнавирусные соединения). См., напри 003856 2 мер, патентную заявку США 08/850398, поданную 2 мая 1997 г. Webber et аl.; патентную заявку США 08/991282, поданную 16 декабря 1997 г., Dragovich et al.; и патентную заявку США 08/991739, поданную 16 декабря 1997 г., Webber et al. Эти патентные заявки США,раскрытие которых включено в данный документ в качестве ссылок, описывают некоторые антипикорнавирусные соединения. Однако существует потребность в открытии низкомолекулярных соединений, которые являются особенно эффективными антипикорнавирусными агентами. Таким образом, аспектом данного изобретения является открытие низкомолекулярных соединений, которые являются особенно эффективными антипикорнавирусными агентами. Дальнейшим аспектом данного изобретения является предоставление промежуточных соединений, пригодных для синтеза упомянутых соединений, ингибирующих протеазы, и синтетических методов, применяющихся для таких синтезов. Следующим аспектом данного изобретения является получение высокоэффективных фармацевтических композиций для опосредованного ингибированием 3C протеаз пикорнавирусов лечения заболеваний, таких как обычная простуда. Эти аспекты осуществляются посредством открытия соединений данного изобретения, которые являются ингибиторами 3C протеаз пикорнавирусов, проявляющими особенно сильную антивирусную активность. Неожиданно было обнаружено, что пептидо и пептидомиметические соединения, содержащие пятичленную гетероциклическую группу, обладают высокой ингибирующей активностью по отношению к протеазам риновирусов. Далее было неожиданно обнаружено, что ингибирующая активность по отношению к протеазам риновирусов пептидо и пептидомиметических соединений может быть значительно увеличена путем замещения глутаминподобного фрагмента в некоторых известных соединениях, ингибирующих риновирусы, имеющих боковую цепь,включающую гамма- или дельта-лактам. Ингибиторы настоящего изобретения имеют следующую общую формулу (I) 3 где n является целым числом от 0 до 5,A1 представляет СН или N, A2 и каждый А 3 независимо выбраны из C(R41)(R41), N(R41), S,S(O), S(O)2 и О, и А 4 представляет NH или NR41,где каждый R41 независимо представляет Н или низший алкил, при условии, что не более двух гетероатомов встречается последовательно в изображенном выше цикле, образованном A1,А 2, (А 3)n, А 4 и С=O, и, по меньшей мере, один изR5 и R6, каждый независимо, представляют Н, F, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу или гетероарильную группу;R7 и R8, каждый независимо, представляют Н, алкильную группу, циклоалкильную группу,гетероциклоалкильную группу, арильную группу, гетероарильную группу, -OR17, -SR17,-NR17R18, -NR19NR17R18 или -NR17OR18, где R17,R18 и R19, каждый независимо, представляет Н,алкильную группу, циклоалкильную группу,гетероциклоалкильную группу, арильную группу, гетероарильную группу или ацильную группу, при условии, что, по меньшей мере, один изR7 и R8 независимо представляет алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу, -OR17, -SR17, -NR17R18,-NR19NR17R18 или -NR17OR18;R9 представляет подходящий органический фрагмент иR23 или -C(S)NR21NR22R23, где R21, R22, R23 и R24,каждый независимо, представляет Н, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу, ацильную группу или тиоацильную группу, или где любые два из R21, R22,R23 и R24 вместе с атомом (атомами), к которому они присоединены, образуют гетероциклоалкильную группу, при условии, что Z и Z1 не являются оба Н; или Z1 и R1 вместе с атомами, к которым они присоединены, образуют циклоалкильную или гетероциклоалкильную группу, где Z1 и R1 такие, как определены выше, с исключением фрагментов, которые не могут образовывать циклоалкильную или гетероциклоалкильную группу; 4 или Z и Z1 вместе с атомами, к которым они присоединены, образуют циклоалкильную или гетероциклоалкильную группу, где Z и Z1 такие, как определены выше, с исключением фрагментов, которые не могут образовывать циклоалкильную или гетероциклоалкильную группу. Данное изобретение также относится к пролекарственным препаратам, фармацевтически приемлемым солям, фармацевтически активным метаболитам и фармацевтически приемлемым сольватам соединений формулы (I). В предпочтительных воплощениях соединений формулы (I) R2 и R3, каждый независимо,представляют Н; или где n является целым числом от 0 до 5, каждыйR41 независимо представляет Н или низший алкил и стереохимия атома углерода, отмеченного звездочкой , может быть R или S; при условии, что, по меньшей мере, один из R2 и R3 представляет или Предпочтительно R9 представляет пятичленный гетероцикл, имеющий от одного до трех гетероатомов, выбранных из О, N и S. В других предпочтительных воплощениях варианты формулы (I) являются следующими. Каждый Z и Z1 независимо выбран из Н, F, низшего алкила, -CO2R21 и -C(O)NR21R22, при условии, что Z и Z1 оба не являются Н, где R21 и R22,каждый независимо, представляет Н, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу, ацильную группу или тиоацильную группу, или R21 и R22 вместе с атомом а другой представляет Н. Каждый R5 и R6 независимо выбран из Н, F, алкильной группы, циклоалкильной группы, гетероциклоалкильной группы, арильной группы и гетероарильной группы, более предпочтительно один из R5 и R6 представляет Н, а другой представляет алкил или арил (например, незамещенный или замещенный фенилметил). R7 и R8, каждый незави 5 симо, представляют Н, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу и более предпочтительно один из R7 и R8 представляет Н, а другой представляет алкил(например, 2-пропил, 2-метил-2-пропил или 2 метил-1-пропил) или арилметил (например, незамещенный или замещенный фенилметил или нафтилметил). R9 представляет пятичленный гетероцикл, имеющий от одного до трех гетероатомов, выбранных из О, N и S, более предпочтительно пятичленный гетероцикл, имеющий, по меньшей мере, один гетероатом азота и,по меньшей мере, один гетероатом кислорода(например, незамещенный или замещенный 1,2 оксазолил (т.е. изоксазолил), 1,3-оксазолил (т.е. оксазолил) или оксадиазолил (1,2,3-оксадиазолил, 1,2,4-оксадиазолил или 1,2,5-оксадиазолил). Если R9 представляет оксадиазолил, то предпочтительным является незамещенный и монометилзамещенный 1,2,4-оксадиазолил. В особенно предпочтительных воплощениях R9 представляет 3-изоксазолил или 5-изоксазолил,или незамещенный, или замещенный одной или двумя метильными группами и/или атомами галогена, причем наиболее предпочтительными галогеновыми заместителями являются хлор и фтор. В предпочтительном воплощении соединения пролекарственные препараты, фармацевтически приемлемые соли, фармацевтически активные метаболиты и сольваты обладают антипикорнавирусной активностью со значениемEC50 меньшим или равным 100 мкМ в H1-HeLa клеточном культуральном анализе и более предпочтительно антириновирусной активностью со значением EC50 меньшим или равным 10 мкМ в Hl-HeLa клеточной культуре. В другом аспекте данное изобретение раскрывает промежуточные соединения формулы где р является целым числом от 0 до 5; А 11 представляет СН или N, А 12 и каждыйA13 независимо выбраны из C(R61)(R61), N(R61),S, S(O), S(O)2 и О, и A14 представляет NH илиNR61, где каждый R61 независимо представляет Н, алкил, ацил или арил, при условии, что не более двух гетероатомов встречается последовательно в изображенном выше цикле в формуле II, образованном А 11, A12, (A13)n, A14 и C=O; каждый R141 независимо представляет Н или низший алкил; 6 любые два из R52, R53 и R54 вместе образуют =O или =C(R57)(R58), где каждый R57 и R58 независимо выбран из Н, алкила, СO2(C1-C6)алкила,C(O)N(C1-С 6)алкила и СO2 арила иR55 и R56, каждый независимо, представляет Н или подходящую защитную группу для азота. Данное изобретение также направлено на фармацевтически приемлемые соли соединений формул II и II'. Данное изобретение также относится к фармацевтическим композициям, содержащим терапевтически эффективное количество, по меньшей мере, одного соединения формулы I,или его пролекарственной формы, фармацевтически приемлемой соли, фармацевтически активного метаболита или сольвата. Кроме того,данное изобретение относится к способам ингибирования 3C протеаз пикорнавирусов путем введения терапевтически эффективного количества, по меньшей мере, одного соединения формулы I, или его пролекарственной формы, фармацевтически приемлемой соли, фармацевтически активного метаболита или сольвата. Настоящее изобретение относится к соединениям формулы I где Y, R1, R2, R3, R5, R6, R7, R8, R9, Z и Z1 такие,как определены выше, и к их фармацевтически приемлемым солям, пролекарственным формам,активным метаболитам и сольватам. Предпочтительно, такие соединения, фармацевтически приемлемые соли, пролекарственные формы,активные метаболиты и сольваты обладают антипикорнавирусной активностью, более предпочтительно антириновирусной активностью,соответствующей значению EC50, меньшему или равному 100 мкМ, в H1-HeLa клеточном культуральном анализе, более предпочтительно соответствующей значению EC50, меньшему или равному 10 мкМ, в Hl-HeLa клеточном культуральном анализе. Кроме того, настоящее изобретение относится к предпочтительным соединениям формул где Ry (в формуле I-A) представляет Н или низший алкил, и R1, R2, R3, R5, R6, R7, R8, R9, Z и Z1 такие, как определены выше, и к их фармацев 7 тически приемлемым солям, пролекарственным формам, активным метаболитам и сольватам. Соединения данного изобретения формулI-A, которые упоминаются в данном документе как "пептидоподобные" соединения, I-B, которые упоминаются в данном документе как соединения "кетометиленового типа", и I-C, которые упоминаются в данном документе как "депсипептидные" соединения, различаются в своих основах, что может влиять на специфическое биораспределение или другие физические свойства; тем не менее, каждое из них обладает сильной ингибирующей активностью по отношению к протеазам риновирусов. В предпочтительных воплощениях соединений формул I-A, I-B и I-C, приведенных выше,R1 представляет Н, F или алкильную группу;R2 выбран из одного из следующих фрагментов:R6 представляет алкильную группу, которая имеет в качестве предпочтительного необязательного заместителя арильную группу;R7 представляет алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу или гетероарильную группу;R9 представляет пятичленный гетероцикл,имеющий от одного до трех гетероатомов, выбранных из О, N и S, предпочтительно, где, по меньшей мере, одним из гетероатомов является азот, который является незамещенным или замещенным, где необязательные заместители предпочтительно представляют галоген или низший алкил, и более предпочтительно монохлор или -фтор или метильную группу иZ и Z1, каждый независимо, представляют Н, F, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу, -C(O)R21,-CO2R21, -CN, -C(O)NR21R22, -C(O)NR21OR22,-C(S)R21, -C(S)NR21R22, -NO2, -SOR21, -SO2R21,-SO2NR21R22,-SO(NR21)(OR22),-SONR21,-SО 3R21,-PO(OR21)2,-PO(R21)(R22),-PO(NR21R22)(OR23),-PO(NR21R22)(NR23R24),-C(O)NR21NR22R23 или -С(S)NR21NR22R23, где Z и Z1 не являются оба Н, и где R21, R22, R23 и R24,каждый независимо, представляет Н, алкильную группу, циклоалкильную группу, гетероциклоалкильную группу, арильную группу, гетероарильную группу, ацильную группу или тиоацильную группу или где любые два из R21, R22,R23 и R24 вместе с атомом (атомами), к которому они присоединены, образуют гетероциклоалкильную группу, 003856 8 или Z и Z1 (оба как определены выше) вместе с атомами, к которым они присоединены,образуют гетероциклоалкильную группу. В предпочтительных воплощениях соединения данного изобретения имеют формулы IA', I-B' и I-C'R9 представляет пятичленный гетероцикл,имеющий от одного до трех гетероатомов, выбранных из О, N и S, который является незамещенным или замещенным, где необязательными заместителями предпочтительно являются одна или две низшие алкильные группы и/или галогены. Данное изобретение также относится к пролекарственным формам, фармацевтически приемлемым солям, фармацевтически активным метаболитам и сольватам таких соединений. В предпочтительных воплощениях агентами, ингибирующими RVP, являются соединения любой из стереоспецифических формул I-A", IB" и I-C" 9 где Ry, R1, R2, R6, R7, R9, Z и Z1 такие, как определены выше, и их фармацевтически приемлемые соли, пролекарственные формы, активные метаболиты и сольваты. В предпочтительных воплощениях соединений формул I-A", I-B" или I-C"R2 выбран из одного из следующих фрагментов:R6 представляет арилметил или арилтиометил, где арил предпочтительно является необязательно замещенной фенильной группой;R7 представляет алкильную группу, более предпочтительно он выбран из 2-пропила, 2 метил-2-пропила, 2-метил-1-пропила и арилметил, где арильная группа предпочтительно представляет фенил или нафтил;R9 представляет изоксазолил, оксазолил или оксадиазолил, необязательно замещенный одной или двумя низшими алкильными группами и/или галогенами и-CO2R21, -CN или -C(O)NR21R22, где R21 и R22 такие, как определены выше, или Z и Z1 вместе образуют циклический сложный эфир или амид. Еще более предпочтительно, если агентами, ингибирующими RVP, данного изобретения являются соединения любой из формул I-A'" , IB'" и I-C"' где n, Ry, R1, R20, R6, R7, R9, Z и Z1 такие, как определены выше, и их фармацевтически приемлемые соли, пролекарственные формы, активные метаболиты и сольваты. В предпочтительных воплощениях соединений формул I-A'", I-B'" или I-C"'R6 представляет арилметил или арилтиометил, где арил предпочтительно представляет фенил, незамещенный или замещенный галогеном, низшим алкилом и/или низшим алкокси;R7 представляет алкильную группу и более предпочтительно выбран из 2-пропила, 2-метил 2-пропила, 2-метил-1-пропила, и арилметил, где арильная группа предпочтительно представляет фенил или нафтил;R9 представляет изоксазолил, оксазолил или оксадиазолил, каждый необязательно замещен одной или двумя низшими алкильными группами и/или галогенами и-CO2R21, -CN или -C(O)NR21R22, где R21 и R22 такие, как определены выше, или Z и Z1 вместе образуют циклический сложный эфир или амид. В особенно предпочтительных соединениях данного изобретения общей формулы I (и ее подразделов) R1 представляет Н или F. В другом аспекте данное изобретение направлено на промежуточные соединения формул II и II' где переменные (р, А 11, A12, A13, A14, R51, R52,R53, R54, R55, R56 и R141) такие, как определены выше. Эти соединения используются для синтеза фармацевтически применяемых соединений формулы I. Предпочтительные группы R55 и R56 представляют Н и подходящие защитные группы для азота, например, ВОС (т-бутилоксикарбонил),CBZ (бензилоксикарбонил), FMOC (флюорен-9 метилоксикарбонил), другие алкилоксикарбонилы (например, метилоксикарбонил) и тритил(трифенилметил). Другие подходящие азотзащитные группы могут быть легко подобраны специалистом (см., например, Green and Wutz,Protecting Groups in Chemical Synthesis (2nd ed.),John WileySons, NY (1991. Предпочтительными группами для R52, R53 и R54 являются Н,алкокси, гидрокси и карбонил. Предпочтительные соединения формулы II включают следующие соединения, в которых РN является подходящей защитной группой для азота, и q равно 1 или 2 Другие предпочтительные промежуточные соединения включают следующие соединения, в которых ВОС представляет т-бутилоксикарбонил Из них предпочтительными стереоизомерами являются Особенно предпочтительные промежуточные соединения включают следующие соединения: В соответствии с соглашением, которое применяется используется в данной области,в данном документе в структурных формулах для изображения связи, которая является точкой присоединения фрагмента или заместителя к ядру или основному скелету структуры. Если в химические структуры включены хиральные атомы углерода, и не изображена конкретная ориентация, то подразумевается, что охватываются обе стереоизомерные формы. Подразумевается, что в данном документе термин "алкильная группа" обозначает моновалентный радикал с линейной или разветвленной цепью насыщенных или ненасыщенных атомов углерода и атомов водорода, такой как метил(Me), этил (Et), пропил, изопропил, бутил, изобутил, т-бутил, этенил, пентенил, бутенил, пропенил, этинил, бутинил, пропинил, пентинил,гексинил и т.п., который может быть незамещенным (т.е. содержать только углерод и водород) или замещенным одним или несколькими подходящими заместителями, как определено ниже (например, одним или несколькими галогенами, такими как F, Cl, Вr или I, причем F и Сl являются предпочтительными). Подразумевается, что "низшая алкильная группа" обозначает алкильную группу, имеющую в цепи от 1 до 4 атомов углерода. Подразумевается, что "циклоалкильная группа" обозначает неароматический моновалентный моноциклический, бициклический или трициклический радикал, содержащий 3, 4, 5, 6,7, 8, 9, 10, 11, 12, 13 или 14 циклических атомов углерода, каждый из которых может быть насыщенным или ненасыщенным и который может быть незамещенным или замещенным одним или несколькими подходящими заместителями, как определено ниже, и который может быть сопряжен с одной или несколькими гетероциклоалкильными группами, арильными группами или гетероарильными группами, которые, в свою очередь, могут быть незамещенными или замещенными одним или несколькими заместителями. Иллюстративные примеры циклоалкильных групп включают следующие фрагменты: Подразумевается, что "гетероциклоалкильная группа" обозначает неароматический моновалентный моноциклический, бициклический или трициклический радикал, который является насыщенным или ненасыщенным, содержащий 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15,16, 17 или 18 циклических атомов, включающих 1, 2, 3, 4 или 5 гетероатомов, выбранных из азота, кислорода и серы, где радикал является незамещенным или замещенным одним или несколькими подходящими заместителями, как определено ниже, и который может быть сопряжен с одной или несколькими циклоалкильными группами, арильными группами или гетероарильными группами, которые в свою очередь могут быть незамещенными или замещенными одним или несколькими заместителями. Иллюстративные примеры гетероциклоалкильных групп включают следующие фрагменты: Подразумевается, что "арильная группа" обозначает ароматический моновалентный моноциклический, бициклический или трициклический радикал, содержащий 6, 10, 14 или 18 циклических атомов углерода в кольце, который может быть незамещенным или замещенным одним или несколькими подходящими заместителями, как определено ниже, и который может быть сопряжен с одной или несколькими циклоалкильными группами, гетероциклоалкильными группами или гетероарильными группами, которые, в свою очередь, могут быть незамещенными или замещенными одним или несколькими подходящими заместителями. Иллюстративные примеры арильных групп включают следующие фрагменты: Подразумевается, что "гетероарильная группа" обозначает ароматический моновалентный моноциклический, бициклический или трициклический радикал, содержащий 4, 5, 6, 7, 8,9, 10, 11, 12, 13, 14, 15, 16, 17 или 18 циклических атомов в кольце, включающих 1, 2, 3, 4 или 5 гетероатомов, выбранных из азота, кислорода и серы, который может быть незамещенным или замещенным одним или несколькими подходящими заместителями, как определено ниже, и который может быть сопряжен с одной или несколькими циклоалкильными группами, гетероциклоалкильными группами или арильными группами, которые, в свою очередь, могут быть незамещенными или замещенными одним или несколькими подходящими заместителями. Иллюстративные примеры гетероарильных групп включают следующие фрагменты: 14 Подразумевается, что "гетероцикл" обозначает гетероарильную или гетероциклоалкильную группу (каждая из которых, как определено выше, является необязательно замещенной). Подразумевается, что "ацильная группа" обозначает -C(O)-R радикал, где R является заместителем, как определено ниже. Подразумевается, что "тиоацильная группа" обозначает-C(S)-R радикал, где R является заместителем, как определено ниже. Подразумевается, что "сульфонильная группа" обозначает -SO2-R радикал, где R является заместителем, как определено ниже. Подразумевается, что "гидроксильная группа" обозначает радикал -ОН. Подразумевается, что "аминогруппа" обозначает радикал -NH2. Подразумевается, что "алкиламиногруппа" обозначает радикал -NHRa, где Ra представляет алкильную группу. Подразумевается, что "диалкиламиногруппа" обозначает радикал -NRaRb, где Ra и Rb, каждый независимо, представляют алкильную группу. Подразумевается, что "алкоксильная группа" обозначает радикал -ORa, где Ra представляет алкильную группу. Примеры алкоксильных групп включают метокси, этокси, пропокси и т.п. Подразумевается, что "алкоксикарбонильная группа" обозначает радикал -C(O)ORa, где 15 Подразумевается, что "арилтиогруппа" обозначает радикал -SRc, где Rc представляет арильную группу. Подразумевается, что "гетероарилтиогруппа" обозначает радикал -SRd, где Rd представляет гетероарильную группу. Подразумевается, что термин "подходящий органический фрагмент" обозначает любой органический фрагмент, который, как определяется специалистами в данной области с помощью, например, рутинных анализов, не оказывает неблагоприятного воздействия на ингибиторную активность соединений данного изобретения. Иллюстративные примеры подходящих органических фрагментов включают, не ограничиваясь ими, гидроксильные группы, алкильные группы, оксогруппы, циклоалкильные группы, гетероциклоалкильные группы, арильные группы, гетероарильные группы, ацильные группы, сульфонильные группы, меркаптогруппы, алкилтиогруппы, алкоксигруппы, карбоксильные группы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, карбамоильные группы, арилтиогруппы, гетероарилтиогруппы и т.п. Подразумевается, что термин "заместитель" или "подходящий заместитель" обозначает любой подходящий заместитель, который может быть подобран или выбран, например, с помощью рутинных анализов, специалистами в данной области. Иллюстративные примеры подходящих заместителей включают гидроксильные группы, галогены, оксогруппы, алкильные группы, ацильные группы, сульфонильные группы, меркаптогруппы, алкилтиогруппы, алкоксигруппы, циклоалкильные группы, гетероциклоалкильные группы, арильные группы, гетероарильные группы, карбоксильные группы,аминогруппы, алкиламиногруппы, диалкиламиногруппы, карбамоильные группы, арилоксигруппы, гетероарилоксигруппы, арилтиогруппы,гетероарилтиогруппы и т.п. Подразумевается, что термин "необязательно замещенный" специально определяет,что конкретная группа является незамещенной или замещенной одним или несколькими подходящими заместителями, в случае, если необязательные заместители конкретно определены,этот термин обозначает, что группа является незамещенной или замещенной конкретными заместителями. Как определено выше, различные группы могут быть незамещенными или замещенными (т.е. они являются необязательно замещенными), если здесь не определено иначе(например, путем определения, что конкретная группа является незамещенной). Подразумевается, что "пролекарственная форма" обозначает соединение, которое превращается в физиологических условиях, в результате или сольволиза, или метаболизма, в конкретное соединение, которое является фармацевтически активным. 16 Подразумевается, что "фармацевтически активный метаболит" обозначает фармацевтически активный продукт, который продуцируется в организме в результате метаболизма конкретного соединения. Подразумевается, что "сольват" обозначает фармацевтически приемлемую сольватную форму конкретного соединения, которая сохраняет биологическую активность такого соединения. Примеры сольватов включают соединения данного изобретения в сочетании с водой,изопропанолом, этанолом, метанолом, ДМСО,этилацетатом, уксусной кислотой или этаноламином. Подразумевается, что "фармацевтически приемлемая соль" обозначает соль, которая сохраняет биологическую активность свободной кислоты или основания конкретного соединения, и которая не является биологически или иначе нежелательной. Примеры фармацевтически приемлемых солей включают сульфаты,пиросульфаты, бисульфаты, сульфиты, бисульфиты, фосфаты, моногидрофосфаты, дигидрофосфаты, метафосфаты, пирофосфаты, хлориды,бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутин-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты, динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, сульфонаты,ксилолсульфонаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, гидpoкcибyтиpaты, гликоллаты, тартраты, метансульфонаты, пропансульфонаты, нафталин 1-сульфонаты, нафталин-2-сульфонаты и манделаты. Если соединение данного изобретения является основанием, то целевая соль может быть получена с помощью любого подходящего метода, известного в данной области, включая обработку свободного основания неорганической кислотой, такой как хлористо-водородная кислота; бромисто-водородная кислота; серная кислота; азотная кислота; фосфорная кислота и т.п., или органической кислотой, такой как уксусная кислота; малеиновая кислота; янтарная кислота; миндальная кислота; фумаровая кислота; малоновая кислота; виноградная кислота; щавелевая кислота; гликолевая кислота; салициловая кислота; пиранозидильная кислота, такая как глюкуроновая кислота или галактуроновая кислота; альфа-гидроксикислота, такая как лимонная кислота или винная кислота; аминокислота, такая как аспарагиновая кислота или глутаминовая кислота; ароматическая кислота,такая как бензойная кислота или коричная кислота; сульфоновая кислота, такая как птолуолсульфоновая кислота или этансульфоновая кислота; или т.п. 17 Если соединение данного изобретения является кислотой, то целевая соль может быть получена с помощью любого подходящего метода, известного в данной области, включая обработку свободной кислоты неорганическим или органическим основанием, таким как амин(первичный, вторичный или третичный); гидроксид щелочного или щелочно-земельного металла или т.п. Иллюстративные примеры подходящих солей включают органические соли, полученные с помощью аминокислот, таких как глицин или аргинин; аммиака; первичных, вторичных или третичных аминов; и циклических аминов, таких как пиперидин, морфолин и пиперазин; а также неорганические соли, полученные с помощью натрия, кальция, калия, магния,марганца, железа, меди, цинка, алюминия и лития. В том случае, если соединения, соли или сольваты являются твердыми веществами, специалистам в данной области понятно, что соединения данного изобретения, соли и сольваты могут существовать в разных кристаллических формах, и все они включены в сферу настоящего изобретения и конкретные формулы. Соединения данного изобретения могут существовать в виде индивидуальных стереоизомеров, рацематов и/или смесей энантиомеров и/или диастереомеров. Все такие индивидуальные стереоизомеры, рацематы и их смеси широко включены в сферу настоящего изобретения. Однако предпочтительно, если соединения данного изобретения используются в оптически чистой форме. Специалисты в данной области обычно понимают под оптически чистым соединением такое соединение, которое является энантиомерно чистым. В данной заявке подразумевается, что термин "оптически чистый" обозначает соединение, включающее, по меньшей мере,достаточную (оптическую) активность. Предпочтительно оптически активное количество индивидуального энантиомера для получения требующейся фармакологической чистоты соединения данного изобретения составляет, по меньшей мере, 90% индивидуального изомераI (или любого подраздела формулы) R1 представляет Н или F. В соединениях формулы I предпочтительно R9 является незамещенной или замещенной изоксазолильной группой, где необязательными заместителями являются предпочтительно одна или две метальные группы и/или галогены. Особенно предпочтительные воплощения данного изобретения описаны ниже в соответствии со следующей формулой I-A": Предпочтительные соединения настоящего изобретения включают пептидо (пептидоподобные) соединения (А-1) - (А-8) формулы I-A",приведенной выше, где R1 представляет Н, Z представляет Н, Ry представляет Н, и R2, R6, R7,Z1 и R9 являются такими, как соответственно определено ниже:R7 представляет СН(СН 3)2, Z1 ляет представляет СО 2 СН 2 СН 3, и R9 представляетR7 представляет СН(СН 3)2, Z1 ляет представляет СO2 СН 2 СН 3, и R9 представляет Предпочтительные пептидоподобные соединения формулы I-A далее включают соединения (А-9) - (А-13), приведенные ниже, гдеR2, R6, R7 и R9 являются такими, как соответственно определено ниже: и R9 представляет Другие предпочтительные пептидоподобные соединения включают следующие: Предпочтительные соединения кетометиленового типа (В-1) - (В-4) данного изобретения описаны ниже в соответствии со следующей формулой I-B": или фенилметил (т.е. бензил), где арильная группа необязательно замещена одним, двумя или тремя заместителями, каждый из которых независимо выбран из галогенов, метокси и метила; вместе представляют где карбонильная группа находится в цис-положении по отношению к водороду, соответствующему R1 в формуле I, и R9 представляетR9 представляет Предпочтительные соединения депсипептидного типа (С-1) и (С-2) данного изобретения описаны ниже в соответствии со следующей формулой I-C", где R1 представляет Н:R9 представляет Дополнительные соединения могут быть получены в соответствии со ссылкой на формулу I путем выбора вариантов из следующих заместителей: Настоящее изобретение также направлено на способ ингибирования активности 3C протеаз пикорнавирусов, включающий приведение в контакт протеазы с эффективным количеством соединения формулы I или его фармацевтически приемлемой соли, пролекарственной формы, фармацевтически активного метаболита или сольвата. Например, активность 3C протеаз пикорнавирусов может ингибироваться в ткани млекопитающего путем введения соединения формулы I или его фармацевтически приемлемой соли, пролекарственной формы, фармацевтически активного метаболита или сольвата. Более предпочтительно настоящий способ направлен на ингибирование активности протеазы риновирусов. Подразумевается, что "лечение" или "обработка" обозначает, по меньшей мере, облегчение болезненного состояния у млекопитающего, такого как человек, путем ингибирования активности одной или нескольких 3C протеаз пикорнавирусов, таких как риновирусы человека, полиовирусы человека, вирусы Коксаки человека, вирусы энцефаломиокардита, вирус менингита и вирус гепатита А, и включает: (а) профилактическую обработку млекопитающего,особенно, если обнаружено, что млекопитающее предрасположено к болезненному состоянию, но это состояние еще не диагностировано;(b) ингибирование болезненного состояния; и/или (с) облегчение, полное или частичное,болезненного состояния. Активность соединений данного изобретения, как ингибиторов активности 3C протеаз пикорнавирусов, может быть измерена с помощью любого из подходящих методов, известных специалистам в данной области, включая in vivo и in vitro анализы. Примером подходящего анализа для измерения активности является антивирусный клеточный культуральный Hl-HeLa тест, описанный в данном документе. Введение соединений формулы I и их фармацевтически приемлемых пролекарственных форм, солей, активных метаболитов и сольватов может быть осуществлено в соответствии 23 с любым из принятых способов введения, доступных специалистам в данной области. Иллюстративные примеры подходящих способов введения включают пероральный, назальный, наружный, парентеральный, чрезкожный и ректальный. Интраназальная доставка особенно предпочтительна. Соединение формулы I данного изобретения или его фармацевтически приемлемые соль,пролекарственная форма, активный метаболит или сольват, может быть введено в виде фармацевтической композиции в любой подходящей по мнению специалистов фармацевтической форме. Подходящие фармацевтические формы включают твердые, полутвердые, жидкие или лиофилизированные составы, такие как таблетки, порошки, капсулы, свечи, суспензии, липосомы и аэрозоли. Фармацевтические композиции данного изобретения могут также включать подходящие эксципиенты, разбавители, среды для лекарств и носители, а также другие фармацевтически активные агенты, в зависимости от предполагаемого применения. В предпочтительных воплощениях фармацевтические композиции данного изобретения доставляются интраназально в виде суспензий. Приемлемые методы получения подходящих фармацевтических форм фармацевтических композиций являются известными или могут быть определены обычным способом специалистами в данной области. Например, фармацевтические препараты могут быть получены с помощью традиционных методик фармацевтической химии, включающих такие стадии, как смешивание, гранулирование и компрессование,при необходимости, для таблеточных форм, или смешивание, заполнение и растворение ингредиентов соответственно с получением целевых продуктов для перорального, парентерального,наружного, внутривагинального, интраназального, внутрибронхиального, внутриглазного,внутриушного и/или ректального введения. В фармацевтических композициях могут применяться твердые или жидкие фармацевтически приемлемые носители, разбавители, среды для лекарств или эксципиенты. Иллюстративные твердые носители включают крахмал,лактозу, дигидрат сульфата кальция, каолин,сахарозу, тальк, желатин, пектин, гуммиарабик,стеарат магния и стеариновую кислоту. Иллюстративные примеры носителей включают сироп, кокосовое масло, оливковое масло, солевой раствор и воду. Носитель или разбавитель может включать подходящее для пролонгированного высвобождения вещество, такое как моностеарат глицерина или дистеарат глицерина,одно или с воском. Если используется жидкий носитель, композиция может иметь вид сиропа,эликсира, эмульсии, мягкой желатиновой капсулы, стерильной жидкости для инъекции (например, раствора) или неводной или водной жидкой суспензии. 24 Доза фармацевтической композиции содержит, по меньшей мере, фармацевтически эффективное количество активного соединения(т.е. соединения формулы I или его фармацевтически приемлемой соли, пролекарственной формы, активного метаболита или сольвата) и предпочтительно составлена из одной или нескольких фармацевтических дозировочных единиц. Выбранная доза может быть введена млекопитающему, например, человеку, нуждающемуся в лечении, опосредованном ингибированием активности 3C протеаз пикорнавирусов, с помощью любого известного или подходящего способа введения дозы, включающего наружный, например, в виде мази или крема; пероральный; ректальный, например, в виде свечей; парентеральный путем инъекции; или непрерывно путем внутривагинальной, интраназальной, внутрибронхиальной, внутриушной или внутриглазной инфузии. Подразумевается, что "терапевтически эффективное количество" обозначает количество соединения данного изобретения, которое при введении млекопитающему, нуждающемуся в нем, является достаточным для того, чтобы оказать лечебное действие на болезненные состояния, которые облегчаются в результате ингибирования активности одной или нескольких 3C протеаз пикорнавирусов, таких как риновирусы человека, полиовирусы человека, вирусы Коксаки человека, вирусы энцефаломиокардита,вирус менингита и вирус гепатита А. Количество данного соединения этого изобретения, которое является терапевтически эффективным,варьирует в зависимости от таких факторов, как конкретное соединение, болезненное состояние и степень его тяжести, вид млекопитающего,нуждающегося в нем, это количество может быть установлено специалистом традиционным способом. В качестве иллюстрации, композиция для назальной доставки соединений данного изобретения для лечения риновирусных инфекций может быть получена следующим образом, где все проценты представляют массовые соотношения и суспензию получают в очищенной воде. Соединение формулы I измельчают до получения частиц с уменьшенным размером, так,чтобы D90 10 мкм. Суспензию готовят так, чтобы конечная концентрация содержащегося в ней активного вещества составляла приблизительно от 0,01 до 2%, предпочтительно приблизительно от 0,2 до 2%. Соответствующие консерванты,выбранные из известных в данной области, могут включать, например, хлорид бензалкония/EDTA в соответствующих интервалах конечных концентраций,например,приблизительно 0,02/0,01%. Может быть включен суспендирующий агент, такой как смесь микрокристаллической целлюлозы (конечная концентрация составляет приблизительно 1-1,5%, предпочтительно приблизительно 1,2%) и натрий карбок 25 симетилцеллюлозы (конечная концентрация составляет приблизительно 0,1-0,2%, предпочтительно приблизительно 0,13%). Поверхностно-активное вещество, такое как полисорбат 80,может быть включено с конечной концентрацией, составляющей приблизительно от 0,05 до 0,2%, предпочтительно приблизительно 0,1%. Может быть включен модификатор тонуса с конечной концентрацией, составляющей приблизительно от 4 до 6%, предпочтительно приблизительно 5%. рН конечного раствора доводят соответственно до физиологического интервала, например, 4-6, используя нетоксичные кислоту и/или основание, такие как НСl и/илиNaOH. Пример композиции для назальной доставки соединения данного изобретения примера 17 имеет следующий состав, где проценты представляют массовые соотношения, и суспензию готовят в очищенной воде Активное соединение Консервант Суспендирующий агент Поверхностно-активное вещество Модификатор тонуса Вещество, использующееся для доведения рН подвергают удалению защитных групп и/или дополнительной модификации, получая не содержащий защитных групп, или модифицированный Н. Далее описан альтернативный способ получения промежуточного соединения F. Аминокислоту Е и аминокислоту (или ее соль) I, в которой Р 3 является соответствующей защитной группой для кислорода, подвергают связьобразующему взаимодействию с получением промежуточного соединения J. С молекулы J удаляют защитные группы, получая свободную карбоновую кислоту К, которую затем подвергают амидобразующему взаимодействию с аминоспиртом (или его солью) В с образованием промежуточного соединения F. Соединения данного изобретения формулы(I) могут быть успешно получены с помощью способов настоящего изобретения, включая общие способы, описанные ниже. В каждом из этих общих способов R1, R2, R3, R5, R6, R7, R8,R9, Ry, Z и Z1 такие, как определены выше, и R4 используется (как стенографическое изображение) для обозначения где R7, R8 и R9 такие, как определены выше. В общем способе I, который используют для синтеза пептидоподобных соединений формулы I-A, аминокислота А, в которой P1 является соответствующей защитной группой для азота, подвергается амидобразующей реакции с аминоспиртом (или его солью) В с получением амида С. Затем с соединения С удаляют защитные группы, получая свободный амин (или его соль) D. Амин D и аминокислоту Е, которая может включать или группу R4, или защитную группу для азота (Р 2), подвергают связьобразующему взаимодействию, приводящему к образованию соединения F. Соединение F окисляют до промежуточного соединения G, который затем превращают в ненасыщенный продукт Н. Если в аминокислоте Е или любой группе R (R1-R9), и/или Ry, и/или Z, и/или Z1,использовались защитные группы, продукт Н В общем способе II, который также используют для синтеза пептидоподобных соединений формулы I-A, аминокислоту L, в которойP1 является соответствующей защитной группой для азота, превращают в карбонильное производное М, в котором "Lv" является уходящей группой. Соединение М подвергают взаимодействию, в котором Lv заменяют на R1, получая производное N. Затем производное N превращают в ненасыщенный продукт О. С ненасыщенного соединения О удаляют защитные группы, получая свободный амин (или его соль) Р, или модифицируют по Z или Z1, вначале получая О' и затем после удаления защитных групп Р. Затем промежуточное соединение Р подвергают амидобразующему взаимодействию с карбоновой кислотой K, получая конечный продукт Н. Если в любой группе R (R1-R9),и/или Ry, и/или Z, и/или Z1 использовались защитные группы, продукт Н подвергают удалению защитных групп и/или дополнительной модификации, получая не содержащий защитных групп, или модифицированный Н. Далее описан альтернативный способ получения промежуточного соединения N. Соединение М подвергают взаимодействию, где "Lv" 27 восстанавливают до защищенного аминоспирта В общем способе III, который используют для синтеза пептидоподобных соединений формулы I-A, аминокислоту L, в которой P1 является соответствующей защитной группой для азота, превращают в карбонильное производное М,в котором "Lv" является уходящей группой. С соединения М удаляют защитные группы, получая свободный амин (или его соль) R, который затем подвергают амидобразующему взаимодействию с карбоновой кислотой К, получая промежуточное соединение S. Затем соединениеS непосредственно превращают в карбонильное промежуточное соединение G, или последовательно восстанавливают вначале до спирта F,который окисляется до G. Соединение G подвергают взаимодействию, получая ненасыщенный конечный продукт Н. Если в любой группеR(R1-R9), и/или Ry, и/или Z, и/или Z1, использовались защитные группы, продукт Н подвергают удалению защитных групп и/или дополнительной модификации, получая не содержащий защитных групп, или модифицированный Н. В общем способе IV, который используют для синтеза пептидоподобных соединений формулы I-A, свободный амин (или его соль) Р, получают из промежуточного соединения О, как описано в общем способе II, превращают в амид Т путем взаимодействия с аминокислотой А, в которой P1 является соответствующей защитной группой для азота. Затем с соединения Т удаляют защитные группы, получая свободный амин(или его соль) U, который затем превращают в Н с помощью аминокислоты Е. Если в любой группе R (R1-R9), и/или Ry, и/или Z, и/или Z1,использовались защитные группы, продукт Н подвергают удалению защитных групп и/или дополнительной модификации, получая не содержащий защитных групп, или модифицированный Н. 28 В общем способе V, который используют для синтеза кетометиленовых соединений формулы I-B, оптически активный лактон АА, в котором P4 является соответствующей защитной группой для азота, a R5 и R8 представляют Н(который может быть получен по способу, описанному ниже, или по различным способам,описанным в литературе, включая: (a) Herold etOrg. Chem. 1993, 58, 2286; (f) Pegorier et al., Tetrahedron Lett. 1995, 36, 2753; и (g) Dondoni et al.,J. Org. Chem. 1995, 60, 7927) превращают посредством двухстадийного процесса (основной гидролиз с последующим окислением) в карбоновую кислоту ВВ. Это вещество не выделяют,а подвергают амидобразующему взаимодействию с амином (или его солью) Р, получая конечный продукт СС. Затем проводят удаление защитной группы P4 наряду с любой из дополнительных защитных групп, которые используются в любой из групп R (R1, R2, R3, R6 и/илиR7), и/или Z, и/или Z1, и/или дополнительную модификацию, получая СС, не содержащий защитных групп, или модифицированный. Лактон АА может быть получен в оптически активной форме с помощью следующего общего способа VI (см. Herold et al., J. Org.(не показано). Хлорангидрид подвергают амидобразующему взаимодействию с хиральным амином или хиральным оксазолидоном, получая производное ЕЕ (в котором X1 является хиральным амином или хиральным оксазолидоном). Затем соединение ЕЕ подвергают депротонированию и полученный енолат диастереоселективно алкилируют электрофилом, соответствующим R6, получая соединение FF, в которомR5 представляет Н. Это вещество затем подвергают реакции галолактонизации с получением галогенлактона GG, в котором R5 и R8 представляют Н, и "hal" представляет Вr или I. После чего галогенлактон GG превращают в азид НН,и это вещество затем превращают в лактон АА,в котором Р 4 является соответствующей защитной группой для азота.,-ненасыщенная карбоновая кислота DD может быть получена с помощью общего способа VII (см.: Herold et al., J. Org. Chem. 1989, 54,1178). Альдегид II, который включает R7, взаимодействует с винилмагнийбромидом, давая спирт JJ. Спирт JJ затем превращают в ,ненасыщенную карбоновую кислоту DD посредством трехстадийного процесса следующим образом: (i) обработка диэтилмалонатом в присутствии катализатора Ti(OEt)4 при 160 С в течение 1 ч, (ii) нагревание при 190 С в течение 4 ч и (iii) гидролиз в этанольном растворе КОН при нагревании с обратным холодильником. Карбоновая кислота ВВ может быть также получена с помощью общего способа VIII (см.Hoffman et al., Tetrahedron, 1997, 53, 7119). Аминокислоту KK, которая включает R7 и в которой P4 является соответствующей защитной группой для азота, превращают в -кетоэфирLL. С соединения LL удаляют защитные группы и полученный анион конденсируют с трифлатомMM, который включает R6. Полученный таким образом продукт конденсации обрабатывают трифторуксусной кислотой, получая кетоэфирNN, и это вещество затем гидролизуют, получая карбоновую кислоту ВВ. Если основной гидролиз приводит к эпимеризации, кетоэфир NN может быть подвергнут переэтерификации (аллильный спирт, Ti(Oi-Pr)4) и затем удалению защитных групп в нейтральных условиях(Pd(PPh3)4, морфолин) с получением карбоновой кислоты ВВ. Трифлат MM, в свою очередь, может быть получен из соответствующего спирта путем обработки ангидридом трифторметансульфоновой кислоты и 2,6-лутидином. Лактон АА может быть также получен с помощью общего способа IX (см.: Askin et al., J.Am. Chem. Soc. 1996, 118, 11970). Аминокислоту KK, которая включает R7 и в которой P4 является соответствующей защитной группой для азота, превращают в эпоксид OO (индивидуальный диастереомер) с помощью способа, описанного Luly et al., J. Org. Chem. 1987, 52, 1487. 30 Эпоксид OO подвергают конденсации с анионом, полученным из соединения РР, которое включает R6, и в котором Х 2 является хиральным фрагментом (включая (1S,2R)-1-аминоиндан-2-ол ацетонид), получая продукт конденсации QQ. Затем это вещество подвергают циклизации в кислых условиях, получая лактон АА. Соединение РР может быть получено из соответствующей карбоновой кислоты (не показано) с помощью способа, описанного Askin et al., J. Общий метод X, использующийся при получении депсипептидных соединений формулыI-C, иллюстрирует способ получения промежуточного соединения ТТ. Аминокислота Е и спирт RR, где P5 является соответствующей защитной группой для кислорода, подвергают взаимодействию, приводящему к образованию сложноэфирной связи, получая промежуточное соединение SS. С молекулы SS удаляют защитные группы, получая свободную карбоновую кислоту ТТ, которая может использоваться вместо карбоновой кислоты К в любом из общих способов, описанных выше. Следующие конкретные способы могут быть также использованы для получения различных соединений в соответствии с данным изобретением. Конкретный способ I описывает получение конкретного промежуточного соединения O1,которое может быть использовано как промежуточное соединение О в общих способах, описанных выше. Так, сложный эфир А 1 (полученный как описано Chida et al., J. Chem. Soc.,Chem. Commun. 1992, 1064) подвергают гидролизу, получая кислоту В 1, которую, в свою очередь, превращают в оксазолидинон С 1. Соединение С 1 затем подвергают депротонированию,и полученный енолат диастереоселективно алкилируют, получая аллильное промежуточное соединение D1. Это соединение окисляют посредством озонолиза, и полученный альдегид(не показано) подвергают реакции восстановительного аминирования с получением лактама Е 1. Затем катализируемый кислотой метанолиз Е 1 дает спирт F1. Это промежуточное соединение окисляют с помощью способа Swern (или в других подходящих окисляющих условиях) до альдегида (не показано), который затем подвергают олефинобразующему взаимодействию с получением конкретного промежуточного соединения O1. Конкретный способ II описывает получение конкретного промежуточного соединенияO2, которое может быть использовано как промежуточное соединение О в общих способах,описанных выше. Промежуточное соединениеD1 подвергают последовательно обработке боргидридом/окислению, получая первичный спирт(не показано). Это соединение окисляют с помощью способа Swern (или в других подходящих окисляющих условиях), и полученный альдегид (не показано) подвергают реакции восстановительного аминирования, получая лактамG1. Затем катализируемый кислотой метанолизG1 дает спирт H1. Это промежуточное соединение окисляют с помощью способа Swern (или в других подходящих окисляющих условиях) до альдегида (не показано), который затем подвергают олефинобразующему взаимодействию с получением конкретного промежуточного соединения O2. Приведенные ниже промежуточные соединения P1, P2 и Р 3 могут использоваться в описанных выше общих способах вместо промежуточного соединения О для обеспечения вариантов групп заместителей в положении R2. Синтез промежуточного соединения Р 1 описан ниже. Промежуточное соединение С 1(описано ниже) подвергают депротонированию, и полученный енолат улавливают соответствующим дисульфидом (симметричным или смешанным), получая сульфид p1 (P является подходящей защитной группой для кислорода). Затем защитную группу для кислорода удаляют,получая спирт р 2. Это промежуточное соединение окисляют с помощью способа Swern (или в других подходящих окисляющих условиях), и полученный альдегид (не показано) подвергают реакции восстановительного аминирования,получая лактам р 3. Затем катализируемый кислотой метанолиз р 3 дает спирт р 4. Это промежуточное соединение окисляют с помощью способа Swern (или в других подходящих окисляющих условиях) до альдегида (не показано),который затем подвергают олефинобразующему взаимодействию с получением промежуточного соединения р 5. Это соединение может быть использовано вместо промежуточного соединения О в описанных выше общих синтетических способах; альтернативно, лактамзащитная группа может быть удалена с получением промежуточного соединения Р 1. Для того чтобы синтезировать промежуточное соединение Р 2, промежуточное соединение С 1 подвергают депротонированию, полученный енолят улавливают с помощью соответствующего источника электрофильного кислорода (например, оксазиридина), получая спирт р 6. Это промежуточное соединение алкилируют с помощью алкилгалогенида, несущего подходящие функциональные группы, или трифлата,получая эфир р 7 (Р является подходящей защитной группой для азота). Затем защитную группу для азота удаляют и полученный амин(не показано) подвергают условиям циклизации с получением лактама р 8. После чего катализируемый кислотой метанолиз р 8 дает спирт р 9. Это промежуточное соединение окисляют с помощью способа Swern (или в других подходя 33 щих окисляющих условиях) до альдегида (также не показано), который затем подвергают олефинобразующему взаимодействию с получением промежуточного соединения Р 2. Теперь будет описан способ синтеза конкретного промежуточного соединения Р 3. Промежуточное соединение D1 (описано выше) озонируют и полученный альдегид (не показано) восстанавливают до соответствующего спирта (также не показано). Затем с этого промежуточного соединения удаляют защитные группы, получая соединение р 10 (P1 является подходящей защитной группой для кислорода). Имидную функциональную группу, присутствующую в р 10 подвергают гидролизу с получением карбоновой кислоты р 11, и это промежуточное соединение подвергают конденсации с соответствующим образом защищенным гидроксиламиновым производным, получая амид р 12(Р 2 является подходящей защитной группой для кислорода, которая является стабильной в условиях удаления P1). Затем удаляют защитную группу P1 и полученный спирт (р 13) превращают в соответствующую подходящую группу(галогенид или трифлат, не показано). После чего удаляют защитную группу Р 2 и полученную гидроксамовую кислоту подвергают циклизации с получением промежуточного соединения р 14. Затем катализируемый кислотой метанолиз р 14 дает спирт р 15. Это промежуточное соединение окисляют с помощью способа Swern(или в других подходящих окисляющих условиях) до альдегида (не показано), который затем подвергают олефинобразующему взаимодействию с получением промежуточного соединения Р 3. Конкретный способ III описывает получение промежуточных соединений Q1, Q2 и Q3,которые могут быть использованы в общих спо 003856I1 превращают в молекулу J1, известную из литературы, с помощью модифицированной опубликованной методики (Ikuta et al., J. Med. Chem. 1987, vol. 30, p. 1995). Независимо сложный эфир аминокислоты K1 защищают, получая силиловый эфир L1. Этот эфир восстанавливают с помощью DIBAL (или используя другие подходящие восстанавливающие условия), и полученный альдегид (не показано) подвергают олефинобразующему взаимодействию с промежуточным соединением J1, получая M1. Удаление силильной защитной группы с M1 дает затем спирт N1. Это промежуточное соединение подвергают ряду гидрирующих условий, получая промежуточные соединения Q1, Q2 и Q3. Эти промежуточные соединения могут быть превращены в промежуточные соединения, аналогичные промежуточному соединению O1 (см. конкретный способ I, приведенный выше), с помощью окисления и последующего олефинирования. Специалистам известно, что различные соединения данного изобретения могут быть получены с помощью вышеописанных общих и конкретных способов, а также путем изучения данной области, включая ссылки, расположенные в данном документе, раскрытие которых включено таким образом в качестве ссылки. Кроме того, специалист может получить различные соединения данного изобретения, руководствуясь примером, описанным ниже, или посредством рутинных модификаций синтезов,описанных в данном документе. Примеры Примеры различных предпочтительных соединений формулы I приведены ниже. Структуры соединений следующих примеров подтверждены одним или несколькими из следую 35 щих методов анализа: спектроскопии протонного магнитного резонанса, инфракрасной спектроскопии, элементного микроанализа, массспектрометрии, тонкослойной хроматографии,определении температуры плавления и определении температуры кипения. Если существует какое-либо несоответствие между данной структурной формулой для соединения и его химическим названием, прилагается структурная формула. Спектры протонного магнитного резонанса(1 Н ЯМР) регистрировали с помощью спектрометра Varian UNITY/plus 300 при напряженности поля (рабочей частоте), равной 300 мегагерц(МГц). Значения химических сдвигов приведены в миллионных долях (мд, ) в область понижения поля от сигнала внутреннего стандарта тетраметилсилана. Альтернативно, спектры 1H ЯМР были соотнесены к остаточным сигналам протонного растворителя следующим образом: СНСl3 = 7,26 м.д.; ДМСО = 2,49 м.д.; C6HD5 = 7,15 м.д. Множественность пиков обозначают следующим образом: с = синглет; д = дублет; дд= дублет дублетов; т = триплет; кв = квартет; шир. = широкий резонанс; и м = мультиплет. Константы взаимодействия приведены в герцах. Спектры инфракрасного поглощения (ИК) получали с помощью спектрометра Perkin-Elmer 1600 серии FTIR. Элементные микроанализы проводили с помощью Atlantic Microlab Inc.(Norcross, GA) и получали результаты для установленных элементов в пределах 0,4% от теоретически рассчитанных значений. Колоночную флэш-хроматографию проводили с использованием Silica gel 60 (Merck Art 9385). Аналитическую тонкослойную хроматографию (TLC) проводили с использованием предварительно покрытых пластинок Silica 60 F254 (Merck Art 5719). Температуры плавления (обозначаемые как т.пл.) определяли на приборе Mel-Temp apparatus, они являются нескорректированными. Все реакции проводили в герметично закрытых колбах при слегка положительном давлении азота, если не оговорено иначе. Все коммерческие реагенты использовали в том виде, в котором они были получены от соответственных поставщиков, со следующими исключениями: тетрагидрофуран (ТГФ) перед использованием перегоняли над натрийбензофенонкетилом; дихлорметан (СН 2 Сl2) перед использованием перегоняли над гидридом кальция; безводный хлорид лития получали путем нагревания при 110 С под вакуумом в течение ночи. В данном документе используются следующие аббревиатуры:Et2O относится к диэтиловому эфиру; ДМФ относится к N,N-диметилформамиду; ДМСО относится к диметилсульфоксиду и МТВЕ относится к трет-бутилметиловому эфиру. Другие аббревиатуры включают СН 3 ОН (метанол), EtOH (этанол), EtOAc (этилацетат), DME(S)-Piper-Ala 2S,3'S)-2-амино-3-(2'-оксопиперидин-3'-ил)пропионовая кислота). Кроме того,L обозначает встречающиеся в природе аминокислоты. Для идентификации некоторых промежуточных соединении и конечных продуктов использовали упрощенную систему наименований с использованием аббревиатуры аминокислот. При наименовании соединений, выделенные курсивом аббревиатуры аминокислот представляют модификации по С-концу остатка, а именно:(1) сложные эфиры акриловой кислоты обозначаются как "Е" (транс) пропеноаты; (2) замещенные 3-метилендигидрофуран-2-оны обозначаются как "Е" (транс) 2-(-винил-бутиролактоны); и (3) 5-винилизоксазолы обозначаются как "Е" (транс) пропенизоксазолы. Кроме того, терминология "AA1[COCH2]-АА 2" определяет, что для любой пептидной последовательности две аминокислоты (AA1 и АА 2),обычно связанные амидной связью, заменены на кетометиленовый дипептидный изостерический фрагмент. Терминология "AA1-NCH3-AA2" определяет, что для любой пептидной последовательности амидная связь, которая обычно связывает две аминокислоты (AA1 и AA2), заменена на N-метиламидную связь. Терминология "AA1O-AA2" определяет, что для любой пептидной последовательности амидная связь, которая обычно связывает две аминокислоты (AA1 и АА 2), заменена на эфирную связь. Примеры воплощений в соответствии с данным изобретением описаны ниже."гексан" подразумевается смесь изомеров гексана). Пример 1. Получение соединения сравнения 1: 5-(3'-(Cbz-L-Leu-L-Phe-L-Gln)-Е-пропен) изоксазол.Cbz-L-(Tr-Gln) (0,26 г, 0,50 ммоль, 1 экв.) добавляют к раствору ацетилхлорида (0,25 мл,3,52 ммоль, 7,0 экв.) в СН 3 ОН (5 мл) и перемешивание продолжают при 23 С в течение 1 ч(часа). Растворитель удаляют при пониженном давлении, остаток растворяют в CH2Cl2 (100 мл) и промывают водой (100 мл), насыщеннымNаНСО 3 (100 мл) и насыщенным солевым раствором (100 мл). Органический слой сушат над(20% EtOAc в гексане), получая Cbz-L-(Tr-Gln)ОМе (0,23 г, 84% выход) в виде твердого вещества белого цвета: т. пл.= 139-140 С; ИК (см-1) 1742, 1207; 1H ЯМР (ДМСО-d6)1,16 (т, 1 Н, J = 7,0), 1,77 (м, 1H), 1,97 (м, 1 Н), 3,61 (с, 3 Н), 4,99Cbz-L-(Тr-глутаминол). Хлорид лития (0,24 г, 5,66 ммоль, 2,0 экв.) добавляют к раствору Cbz-L-(Tr-Gln)-OMe (1,50 г, 2,79 ммоль, 1 экв.) в смеси 2:1 ТГФ:ЕtOН (30 мл) и смесь перемешивают при 23 С до тех пор,пока все твердое вещество не растворится (10 мин). Добавляют боргидрид натрия (0,21 г, 5,55 ммоль, 2,0 экв.) и реакционную смесь перемешивают в течение ночи при 23 С. Растворитель удаляют при пониженном давлении, остаток вносят в воду (50 мл) и рН доводят до 2-3 с помощью 10% НСl. Продукт экстрагируют EtOAc(50 мл) и органический слой промывают водой(50 мл) и насыщенным солевым раствором (50 мл), после чего сушат над МgSO4. Органический слой концентрируют и остаток очищают с помощью колоночной флэш-хроматографии(градиентное элюирование, 2050% EtOAc в бензоле), получая Cbz-L-(Тr-глутаминол) (1,02 г, 72% выход) в виде белого стекловидного твердого вещества: т.пл. = 66-70 С; ИК (см-1) 3318, 1699, 1510, 1240; 1H ЯМР (ДМСО-d6)1,40 (м, 1H), 1,72 (м, 1H), 2,26 (м, 2 Н), 3,17-3,50L-(Тr-глутаминол). Суспензию Cbz-L-(Тr-глутаминол) (1,93 г,3,79 ммоль) в СН 3 ОН (25 мл) и Pd/C (10%, 0,19 г) перемешивают в атмосфере водорода (баллон) в течение 4 ч, затем фильтруют через слой целита. Фильтрат концентрируют при понижен 003856 38 ном давлении, получая L-(Тr-глутаминол) в виде белого аморфного твердого вещества (1,38 г,98% выход): т.пл. = 191-193 С; ИК (ст-1) 3255Cbz-L-Leu-L-Phe-L-(Тr-глутаминол). Карбонилдиимидазол (0,17 г, 1,05 ммоль,1,0 экв.) добавляют к раствору Cbz-L-Leu-L-PheOH (0,41 г, 1,0 ммоль, 0,95 экв.) в ТГФ (10 мл) и реакционную смесь перемешивают при 23 С в течение 1 ч. Затем добавляют L-(Тr-глутаминол)(0,39 г, 1,05 ммоль, 1 экв.) и полученный раствор перемешивают в течение ночи. Летучие вещества удаляют при пониженном давлении и остаток очищают с помощью колоночной флэшхроматографии (градиентное элюирование,24% СН 3 ОН в СНСl3), получая Cbz-L-Leu-LPhe-L-(Тr-глутаминол) (0,47 г, 62% выход) в виде белого аморфного твердого вещества: т.пл.Cbz-L-Leu-L-Phe-L-(Тr-Gln)-Н. о-Йодоксибензойную кислоту (0,63 г, 2,25 ммоль, 3,0 экв.) добавляют к раствору Cbz-LLeu-L-Phe-L-(Тr-глутаминол) (0,58 г, 0,75 ммоль, 1 экв.) в ДМСО (7,5 мл) при 23 С. После перемешивания в течение 2 ч ДМСО удаляют при пониженном давлении. Остаток в два раза разбавляют CH2Cl2 и растворитель упаривают для удаления какого-либо остатка ДМСО. Остаток разбавляют EtOAc (30 мл) и фильтруют,фильтрат промывают раствором 5% Na2S2O3/5%NаНСО 3 (30 мл), водой (30 мл) и насыщенным солевым раствором (30 мл) и затем сушат надNa2SO4. Растворитель удаляют при пониженном давлении, получая Cbz-L-Leu-L-Phe-L-(Tr-Gln)H (0,53 г, 92% выход) в виде белого стекловидного твердого вещества, которое сразу используют без дополнительной очистки: 1H ЯМР 39 суспензии бромида изоксазол-5-илметилтрифенилфосфония (0,222 г, 0,525 ммоль, 1,1 экв.) в ТГФ (20 мл) при 0 С, и реакционную смесь перемешивают при 0 С в течение 30 мин. Добавляют раствор Cbz-L-Leu-L-Phe-L-(Tr-Gln)H (0,366 г, 0,477 ммоль, 1 экв.) в ТГФ (10 мл) и реакционную смесь затем перемешивают в течение ночи при 23 С. Растворитель удаляют в вакууме и остаток разбавляют EtOAc (30 мл),промывают водой (30 мл) и затем сушат надMgSO4. Растворитель удаляют при пониженном давлении и остаток очищают с помощью флэшхроматографии на силикагеле (градиентное элюирование, 01% СН 3 ОН в СНСl3), получая 5-3'-(Cbz-L-Leu-L-Phe-L-(Tr-Gln-Е-пропен изоксазол (0,307 г, 70% выход) в виде аморфного твердого вещества: ИК (см-1) 3423, 1678,1568, 1265, 1043, 711; 1H ЯМР (ДМСО-d6)0,77-0,81 (м, 6 Н), 1,21-1,36 (м, 2 Н), 1,40-1,55 (м,1 Н), 1,60-1,80 (м, 2 Н), 2,34-2,45 (м, 2 Н), 2,822,87 (м, 1 Н), 2,91-3,04 (м, 1 Н), 3,95-4,00 (м, 1 Н),4,41-4,50 (м, 1 Н), 4,53-4,60 (м, 1 Н), 4,99 (кв, 2 Н,J = 6,0), 6,19 (д, 1 Н, J = 15,0), 6,36 (дд, 1 Н, J(д, 1 Н, J = 9,0), 7,56-7,63 (м, 5 Н), 7,96 (д, 1 Н, J = 9,0), 8,08 (д, 1 Н, J = 9,0), 8,51 (с, 1 Н), 8,58 (с,1 Н); HRMS (масс-спектрометрия высокого разрешения) рассчитано для C51H53N5O6+Cs 964,3050 (M+Cs), обнаружено 964,3018. Получение продукта: 5-(3'-(Cbz-L-Leu-LPhe-L-Gln)-E-пропен)изоксазол. Трифторуксусную кислоту (1 мл) добавляют к раствору 5-3'-(Cbz-L-Leu-L-Phe-L-(TrGln-Е-пропенизоксазола (0,214 г, 0,257 ммоль) в СН 2 Сl2 (10 мл) и реакционную смесь перемешивают при 23 С в течение ночи. Растворитель удаляют в вакууме и остаток очищают с помощью флэш-хроматографии на силикагеле (градиентное элюирование, 01% СН 3 ОН в СНСl3), получая 5-(3'-(Сbz-L-Leu-L-Phe-LGln)-Е-пропен)изоксазол в виде твердого вещества белого цвета (0,054 г, 36% выход): 1H ЯМРBoc-L-(Tr-Gln)-N(OMe)Me. Изобутилхлорформиат (4,77 мл, 36,8 ммоль, 1,0 экв.) добавляют к раствору Boc-L(Tr-Gln)-ОН (18,7 г, 36,7 ммоль, 1 экв.) и 4 метилморфолина (8,08 мл, 73,5 ммоль, 2,0 экв.) в CH2Cl2 (250 мл) при 0 С. Реакционную смесь перемешивают при 0 С в течение 20 мин (минут), затем добавляют гидрохлорид N,Oдиметилгидроксиламина (3,60 г, 36,7 ммоль, 1,0 экв.). Полученный раствор перемешивают при 0 С в течение 20 мин и при 23 С в течение 2 ч и затем распределяют между водой (150 мл) и СН 2 Сl2 (2150 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют. Остаток очищают с помощью колоночной флэшхроматографии (градиентное элюирование,4020% гексана в EtOAc), получая Boc-L-(TrGln)-N(OMe)Me (16,1 г, 82% выход) в виде белой пены: ИК (см-1) 3411, 3329, 3062, 1701,1659; 1H ЯМР (CDCl3)1,42 (с, 9 Н), 1,63-1,77Boc-L-(Tr-Gln)N(OMe)Me (16,1 г, 30,3 ммоль, 1 экв.) в ТГФ при -78 С и реакционную смесь перемешивают при -78 С в течение 4 ч. Последовательно добавляют метанол (4 мл) и 1,0 М НСl (10 мл) и смесь нагревают до 23 С. Полученную суспензию разбавляют Et2O (150 мл) и промывают 1,0 М НСl (3100 мл), наполовину насыщенным раствором NаНСО 3 (100 мл), и водой (100 мл). Органический слой сушат над МgSO4, фильтруют и концентрируют, получая неочищенныйBoc-L-(Tr-Gln)-Н (13,8 г, 97% выход) в виде белого твердого вещества: т.пл.= 114-116 С; ИК-78 С и полученный раствор перемешивают в течение 20 мин при этой температуре. Неочищенный Boc-L-(Tr-Gln)-Н (10,8 г, 22,9 ммоль, 1 экв.) в ТГФ (50 мл) добавляют через трубочку и реакционную смесь перемешивают 2 ч при-78 С, нагревают до 0 С в течение 10 мин и затем распределяют между 0,5 М HCl (150 мл) и смесью EtOAc и гексана 1:1 (2150 мл). Объединенные органические слои сушат над Na2SO4(т, 3H, J = 7,2), 1,42 (с, 9 Н), 1,70-1,78, (м, 1 Н),1,80-1,96 (м, 1 Н), 2,35 (т, 2 Н, J = 7,0), 4,18 (кв,2 Н, J = 7,2), 4,29 (с, шир, 1H), 4,82-4,84 (м, 1 Н),5,88 (дд, 1H, J = 15,7, 1,6), 6,79 (дд, 1H, J = 15,7,5,3), 6,92 (с, 1H), 7,19-7,34 (м, 15 Н). Аналитически определено (C33H38N2O5) С, Н, N. Получение промежуточного соединения: этил-3-(Boc-L-Phe-L-(Tr-Gln-Е-пропеноат. Раствор НСl в 1,4-диоксане (4,0 М, 15 мл) добавляют к раствору этил-3-(Boc-L-(Tr-GlnЕ-пропеноата (3,26 г, 6,01 ммоль, 1 экв.) в том же растворителе (15 мл) при 23 С. Через 2 ч летучие вещества удаляют при пониженном давлении, получая этил-3-(H2N-L-(Tr-Gln-Епропеонат-HCl. Это вещество растворяют вEDC (1,73 г, 9,02 ммоль, 1,5 экв.). Реакционную смесь перемешивают при 23 С в течение ночи и затем распределяют между водой (100 мл) иCH2Cl2 (2100 мл). Объединенные органические слои сушат над Na2SO4, концентрируют и остаток очищают с помощью колоночной флэшхроматографии (40% EtOAc в гексане), получая этил-3-(Boc-L-Phe-L-(Тr-Gln-Е-пропеноат (3,55 г, 85%) в виде белой пены: ИК (см-1) 3306, 1706,1661; 1H ЯМР (СDСl3)1,29 (т, 3H, J = 7,2), 1,38= 15,8), 6,29 (д, 1H, J = 8,1), 6,64 (дд, 1H, J = 15,8, 5,4), 6,80 (с, шир, 1H), 7,14-7,32 (м,20 Н).Аналитически определено (С 42 Н 47N3O6) С,Н, N. Получение промежуточного соединения: этил-3-(Boc-L-Leu-L-Phe-L-(Tr-Gln-Е-пропеноат. Раствор НСl в 1,4-диоксане (4,0 М, 15 мл) добавляют к раствору этил-3-(Boc-L-Phe-L-[TrGln])-Е-пропеноата (6,40 г, 9,28 ммоль, 1 экв.) в том же растворителе (15 мл) при 23 С. Через 2 ч летучие вещества удаляют при пониженном давлении. Остаток растворяют в CH2Cl2 (100 мл) и последовательно добавляют Boc-L-Leu-OH(2,58 г, 11,1 ммоль, 1,2 экв.), HOBt (1,88 г, 13,9 ммоль, 1,5 экв.), 4-метилморфолин (3,06 мл, 27,8 ммоль, 3 экв.) и EDC (2,67 г, 13,92 ммоль, 1,5 экв.). Реакционную смесь перемешивают при 23 С в течение ночи и затем распределяют между водой (100 мл) и CH2Cl2 (2100 мл). Объединенные органические слои сушат над Na2SO4,концентрируют и остаток очищают с помощью колоночной флэш-хроматографии (2% СН 3 ОН в(дд, 1H, J = 15,6, 5,1), 7,10-7,33 (м, 21 Н). Аналитически определено (C48H58N4O70,33H2 О) С, Н,N. Получение промежуточного соединения: этил-3-5'-метилизоксазол-3'-карбонил)-L-LeuL-Phe-L-(Tr-Gln-Е-пропеноат. Раствор НСl в 1,4-диоксане (4,0 М, 3 мл) добавляют к раствору этил-3-(Boc-L-Leu-L-PheL-(Tr-Gln-Е-пропеноата (0,216 г, 0,27 ммоль, 1 экв.) в том же растворителе (3 мл) при 23 С. Через 2 ч летучие вещества удаляют при пониженном давлении. Остаток растворяют в CH2Cl2(15 мл), охлаждают до 0 С и последовательно добавляют триэтиламин (0,112 мл, 0,81 ммоль,3,0 экв.) и 5-метилизоксазол-3-карбонилхлорид(0,058 г, 0,40 ммоль, 1,5 экв.). Реакционную смесь перемешивают при 0 С в течение 30 мин и затем распределяют между водой (50 мл) иCH2Cl2 (250 мл). Объединенные органические слои сушат над Na2SO4, концентрируют и остаток очищают с помощью колоночной флэшхроматографии (2% СН 3 ОН в CH2Cl2), получая этил-3-5'-метилизоксазол-3'-карбонил)-L-LeuL-Phe-L-(Tr-Gln-Е-пропеноат (0,199 г, 91% выход) в виде белой пены: ИК (см-1) 3286, 1650,1541; 1H ЯМР (СDСl3)0,86 (д, 3H, J = 5,4), 0,89(С 48H53N5O7H2O) С, Н, N. Получение продукта: этил-3-5'-метилизоксазол-3'-карбонил)-L-Leu-L-Phe-L-Gln-Eпропеноат. Триизопропилсилан (0,077 мл, 0,376 ммоль, 1,8 экв.) и трифторуксусную кислоту (3 мл) добавляют последовательно к раствору этил-3-5'-метилизоксазол-3'-карбонил)-L-LeuL-Phe-L-(Tr-Gln-Е-пропеноат (0,185 г, 0,21 ммоль, 1 экв.) в CH2Cl2 (3 мл) при 23 С, получая ярко-желтый раствор. Реакционную смесь перемешивают в течение 30 мин при 23 С, в течение этого времени смесь становится бесцветной. Летучие вещества удаляют при пониженном давлении и полученное белое твердое вещество перетирают с Et2O (10 мл), фильтруют, и сушат на воздухе, получая этил-3-5'-метилизоксазол 3'-карбонил)-L-Leu-L-Phe-L-Glu)-Е-пропеноат(0,87 г, 81% выход) в виде белого твердого ве 43 щества: т.пл. = 223-225 С; ИК (см-1) 3298, 1662,1544, 1457, 1278; 1H ЯМР (ДМСО-d6)0,81 (д,3H, J = 6,0), 0,85 (д, 3H, J = 6,3), 1,23 (т, 3H, J = 6,9), 1,38-1,42 (м, 1 Н), 1,48-1,77 (м, 4 Н), 2,04 (т,2H, J = 7,2), 2,46 (с, 3H), 2,78-2,86 (м, 1 Н), 2,933,00 (м, 1 Н), 4,11 (кв, 2H, J =7,2), 4,36-4,54 (м,3H), 5,63 (д, 1 Н, J = 15,6), 6,56 (с, 1 Н), 6,68 (дд,1 Н,. J = 15,9, 5,4), 6,76 (с, шир, 1 Н), 7,19 (м, 6 Н),8,09 (д, 1 Н, J = 8,1), 8,14 (д, 1 Н, J = 7,8), 8,58 (д,1 Н, J = 7,5). Аналитически определено Получение промежуточного соединения: этил-3-изоксазол-5'-карбонил)-L-Leu-L-Phe-L(Тr-Gln)-Е-пропеноат. Это соединение получают из этил-3-(BocL-Leu-L-Phe-L-(Тr-Gln-Е-пропеноата и изоксазол-5-карбонилхлорида, используя методику,описанную выше (пример 2) для получения этил-3-5'-метилизоксазол-3'-карбонил)-L-LeuL-Phe-L-[Tr-Gln-Е-пропеноата, ИК (см-1) 3282,1643, 1530; 1H ЯМР (CDCl3)0,87 (т, 6 Н, J = 6,6), 1,29 (т, 3H, J = 7,2), 1,49-1,64 (м, 3H), 1,691,80(м, 1 Н), 1,90-1,96 (м, 1 Н), 2,30 (т, 2 Н, J = 7,2), 2,92-2,96 (м, 1 Н), 3,02-3,09 (м, 1 Н), 4,17 (кв,2 Н, J = 7,2), 4,42-4,48 (м, 3H), 5,69 (д, 1 Н, J = 15,3), 6,65 (с, шир, 1 Н), 6,66 (дд, 1 Н, J = 15,9,5,4), 6,76-6,79 (м, 2 Н), 7,00-7,31 (м, 22 Н), 8,24 (с,1 Н). Аналитически определено (C47H51N5O7 0,75 Н 2 О) С, Н, N. Получение этил-3-изоксазол-5'-карбонил)-L-Leu-L-Phe-L-Gln)-Е-пропеноата. Указанное в заголовке соединение получают из этил-3-изоксазол-5'-карбонил)-L-LeuL-Phe-L-(Tr-Gln-Е-пропеноата, используя методику, аналогичную описанной выше (пример 2) для получения этил-3-5'-метилизоксазол-3'карбонил)-L-Leu-L-Phe-L-Gln)-Е-пропеноата,т.пл. = 217-220 С; ИК (см-1) 3302, 1655, 1541; 1H ЯМР (ДМСО-d6)0,81 (д, 3H, J = 6,0), 0,86 (д,3H, J = 6,0), 1,21 (т, 3H, J = 7,2), 1,42-1,75 (м,5 Н), 2,04 (т, 2 Н, J = 7,2), 2,78-2,87 (м, 1 Н), 2,943,01 (м, 1 Н), 4,11 (кв, 2 Н, J = 7,2), 4,37 (м, 1 Н),4,41-4,52 (м, 2 Н), 5,64 (д, 1 Н, J = 15,6), 6,68 (дд,1 Н, J = 15,9, 5,4), 6,76 (с, шир, 1 Н), 7,12-7,19 (м,7 Н), 8,02 (д, 1 Н, J = 8,1), 8,20 (д, 1 Н, J = 8,1),8,74 (д, 1 Н, J = 1,8), 8,94 (д, 1 Н, J = 7,8). Аналитически определено (C28H37N5O7) С, Н, N. Пример 4. Получение соединения А-3: этил-3-5'-метилизоксааол-3'-карбонил)-L(тбутил-Gly)-L-(4-Me-Phe)-L-Gln)-Е-пропеноат.(полученный как описано в примере 2, приведенном выше, 1,37 г, 3,10 ммоль) растворяют в ДМФ (10 мл) при 23 С. Добавляют диизопропилэтиламин (1,08 мл, 6,20 ммоль), затем Boc-L(4-Me-Phe)-ОН (0,87 г, 3,10 ммоль). Реакционную смесь охлаждают до 0 С, добавляют HATU(1,18 г, 3,10 ммоль) и реакционную смесь оставляют нагреваться до комнатной температуры. ДМФ удаляют в вакууме. Остаток растворяют вEtOAc (30 мл) и органическую фазу промывают последовательно 10% раствором НСl (25 мл),насыщенным раствором NаНСО 3 (25 мл), H2O(25 мл) и насыщенным солевым раствором (25 мл). Растворитель сушат (MgSO4), фильтруют и остаток очищают с помощью колоночной флэшхроматографии (градиентное элюирование,00,75% СH3 ОН в СНСl3), получая этил-3(Boc-L-(4-Me-Phe)-L-(Tr-Gln-Е-пропеноат(1,48 г, 68% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1713, 1655, 1491,1175; 1H ЯМР (ДМСО-d6)1,20 (т, 3H, J = 7,0),1,30 (с, 9 Н), 1,62-1,66 (м, 2 Н), 2,23 (с, 3H), 2,32(с, 1 Н). Аналитически определено (С 43 Н 49N3O6) С, Н, N. Получение промежуточного соединения: этил-3-(Boc-L(т-бутил-Gly)-L-(4-Me-Phe)-L(Тr-Gln-Е-пропеноат. Этил-3-(Boc-L-(4-Me-Phe)-L-(Tr-Gln-Епропеноат (1,45 г, 2,06 ммоль) растворяют в 1,4 диоксане (27 мл) и добавляют раствор НСl в 1,4 диоксане (4,0 М, 14 мл). Реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Растворитель удаляют путем упаривания и остаток помещают в EtOAc (50 мл). Органическую фазу промывают насыщенным раствором NaHСО 3 (50 мл) и затем насыщенным солевым раствором (50 мл), сушат (МgSO4) и растворитель удаляют при пониженном давлении, получая 1,23 г не совсем белого аморфного твердого вещества. Это вещество подвергают взаимодействию с Boc-L(т-бутил-Gly)-ОН,используя методику, описанную выше для синтеза этил-3-(Boc-L-(4-Me-Phe)-L-(Tr-Gln-Епропеноата, получая этил-3-(Boc-L(т-бутилGly)-L-(4-Me-Phe)-L-(Tr-Gln)-Е-пропеноат (49% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1655, 1507, 1248, 1171; 1H ЯМР(д, 1 Н, J = 8,1), 8,10 (д, 1 Н, J = 7,7), 8,49 (с, 1 Н). Аналитически определено (C49 Н 60N4O70,4 Н 2 О) С, Н, N. Получение промежуточного соединения: этил-3-5'-метилизоксазол-3'-карбонил)-L(тбутил-Gly)-L-(4-Me-Phe)-L-(Tr-Gln)-E-пропеноат. С этил-3-(Boc-L(т-бутил-Gly)-L-(4-MePhe)-L-(Tr-Gln-Е-пропеноата удаляют защитные группы, используя методику, описанную выше для удаления защитных групп с этил-3(Boc-L-(4-Me-Phe)-L-(Tr-Gln-Е-пропеноата, и полученный амин (0,22 г, 0,30 ммоль) растворяют в CH2Cl2 (3 мл). Добавляют пиридин(0,025 мл, 0,32 ммоль) и реакционную смесь охлаждают до 0 С. Добавляют 5-метилизоксазол-3-карбонилхлорид (0,046 г, 0,32 ммоль). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение одного часа. Растворитель удаляют в вакууме и остаток подвергают колоночной флэш-хроматографии (градиентное элюирование, 01% СН 3 ОН в CH2Cl2), получая этил-35'-метилизоксазол-3'-карбонил)-L(т-бутилGly)-L-(4-Me-Phe)-L-(Tr-Gln-Е-пропеноат (0,19 г, 77% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1651, 1518; 1H ЯМРCH2Cl2 (4 мл) при 23 С. Добавляют трифторуксусную кислоту (0,4 мл) и реакционную смесь перемешивают при комнатной температуре в течение 6 ч. Растворитель удаляют в вакууме и остаток подвергают колоночной флэшхроматографии (градиентное элюирование,02% СН 3 ОН в СН 2 Сl2), получая этил-3-5'метилизоксазол-3'-карбонил)-L(т-бутил-Gly)L-(4-Me-Phe)-L-Gln)-Е-пропеноат (0,085 г, 73% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1661, 1541, 1206; 1H ЯМР(полученный, как описано выше, в примере 2, 1,0 г, 5,0 ммоль), затем Et3N (0,70 мл, 5,0 ммоль). Добавляют карбонилдиимидазол (0,81 г, 5,0 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 20 ч. Растворитель удаляют в вакууме и остаток подвергают колоночной флэш-хроматографии (градиентное элюирование, 01% СН 3 ОН в CH2Cl2), получая этил-3-(Boc-L-(4-F-Phe)-L-(Tr-Gln-Е-пропеноат (1,13 г, 32% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1712, 1666, 1510,1169; 1H ЯМР (ДМСО-d6)1,20 (т, 3H, J = 7,0),1,29 (с, 9 Н), 1,61-1,70 (м, 2 Н), 2,27-2,34 (м, 2 Н),2,74-2,78 (м, 1H), 2,86-2,90 (м, 1H), 4,06-4,13 (м,3H), 4,36-4,40 (м, 1H), 5,58 (д, 1H, J = 15,6), 6,71(дд, 1H, J = 15,6, 5,5), 6,98 (д, 1H, J = 8,1), 7,037,09 (м, 2 Н), 7,14-7,28 (м, 17 Н), 8,06 (д, 1H, J = 8,1), 8,53 (с, 1 Н); LRMS (масс-спектрометрия низкого разрешения) (M+Na) 730. Получение промежуточного соединения: этил-3-(Boc-L(т-бутил-Gly)-L-(4-F-Phe)-L(Tr-Gln-Е-пропеноат. С этил-3-(Boc-L-(4-F-Phe)-L-(Tr-Gln-Епропеноата удаляют защитные группы и подвергают взаимодействию с Boc-L(т-бутилGly)-ОН, используя методики, описанные выше для получения этил-3-(Boc-L(т-бутил-Gly)-L(4-Me-Phe)-L-(Tr-Gln-E-пропеноата, получая этил-3-(Boc-L(т-бутил-Gly)-L-(4-F-Phe)-L(Tr-Gln-Е-пропеноат (54% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1720, 1651, 1506, 1168; 1H ЯМР (ДМСО-d6)0,80 (с, 9 Н), 1,20 (т, 3H, J = 7,0), 1,36 (с, 9 Н),1,53-1,67 (м, 2 Н), 2,23-2,28 (м, 2 Н), 2,79-2,94 (м,2 Н), 3,85 (д, 1 Н, J = 9,9), 4,09 (кв, 2 Н, J = 7,0), 47 4,31-4,35 (м, 1H), 4,53-4,55 (м, 1 Н), 5,46 (д, 1 Н, J= 15,8), 6,36 (д, 1H, J = 9,2), 6,64 (дд, 1 Н, J = 15,8, 5,5), 6,97-7,03 (м, 2 Н), 7,13-7,28 (м, 17 Н),8,08 (д, 1 Н, J = 8,1), 8,14 (д, 1H, J = 8,1), 8,49 (с,1 Н). Аналитически определено (C48H57N4O7F) С,Н, N. Получение промежуточного соединения: этил-3-5'-метилизоксазол-3'-карбонил)-L(тбутил-Gly)-L-(4-F-Phe)-L-(Tr-Gln-E-пропеноат. С этил-3-(Boc-L(т-бутил-Gly)-L-(4-FPhe)-L-(Tr-Gln-Е-пропеноата удаляют защитные группы и подвергают его взаимодействию с 5-метилизоксазол-3-карбонилхлоридом, используя методики, описанные выше для получения этил-3-5'-метилизоксазол-3'-карбонил)-L(тбутил-Gly)-L-(4-Me-Phe)-L-(Tr-Gln-E-пропеноата,получая этил-3-5'-метилизоксазол-3'карбонил)-L(т-бутил-Gly)-L-(4-F-Phe)-L-(TrGln-Е-пропеноат (74% выход) в виде белого аморфного твердого вещества: ИК (см-1) 1659,1535, 1510; 1H ЯМР (ДМСО-d6)0,88 (с, 9 Н),1,20 (т, 3H, J = 7,4), 1,52-1,67 (м, 2 Н), 2,23-2,28(м, 1H), 4,09 (кв, 2 Н, J = 7,0), 4,32-4,36 (м, 1H),4,45 (д, 1H, J = 9,6), 4,50-4,55 (м, 1H), 5,44 (д,1H, J = 15,6), 6,58 (с, 1H), 6,63 (дд, 1 Н, J = 15,6,5,5, 15,6), 6,93-6,99 (м, 2 Н), 7,13-7,28 (м, 17 Н),7,64 (д, 1 Н, J = 9,6), 8,16 (д, 1 Н, J = 8,5), 8,46 (д,1 Н, J = 8,1), 8,51 (с, 1 Н). Аналитически определено (C48H52N5O7F) С, Н, N. Получение этил-3-5'-метилизоксазол-3'карбонил)-L(т-бутил-Gly)-L-(4-F-Phe)-LGln)-Е-пропеноата. С этил-3-[(5'-метилизоксазол-3'-карбонил)L(т-бутил-Gly)-L-(4-F-Phe)-L-(Tr-Gln)]-Епропеноата удаляют защитные группы, используя методику, аналогичную описанной выше для получения этил-3-5'-метилизоксазол-3'карбонил)-L(т-бутил-Gly)-L-(4-Me-Phe)-LGln)-Е-пропеноата,получая этил-3-5'-метилизоксазол-3'-карбонил)-L(т-бутил-Gly)-L(4-F-Phe)-L-Gln)-Е-пропеноат (80% выход) в виде белого аморфного твердого вещества: ИК(10,5 г, 36,5 ммоль, 1 экв.) в СН 3 ОН (150 мл) и полученную мутную реакционную смесь перемешивают при 23 С в течение 3,5 ч. Смесь концентрируют при пониженном давлении до объема 30 мл и затем распределяют между 0,5 М НСl (150 мл) и EtOAc (2150 мл). Объединенные органические слои сушат над МgSO4 и фильтруют под действием силы тяжести. Фильтрат концентрируют при пониженном давлении и остаток сушат под вакуумом, получая трет-бутиловый эфир (4S)-4-(2'-карбоксиэтил)2,2-диметилоксазолидин-3-карбоновой кислоты(10,0 г, 100% выход неочищенного вещества). Это вещество используют без дополнительной очистки: 1H ЯМР (CDCl3, смесь ротамеров)1,49 (с), 1,57 (с), 1,60 (с), 1,84-2,05 (м), 2,39-2,41(м), 3,71-3,74 (м), 3,91-4,05 (м). Получение промежуточного соединения: трет-бутилового эфира (4S,4"S)-4-3'-(4"-бензил-2"-оксооксазолидин-3"-ил)-3'-оксопропил 2,2-диметилоксазолидин-3-карбоновой кислоты. Триэтиламин (8,87 мл, 63,6 ммоль, 3,0 экв.) и пивалоилхлорид (2,61 мл, 21,2 ммоль, 1,0 экв.) добавляют последовательно к раствору третбутилового эфира (4S)-4-(2'-карбоксиэтил)-2,2 диметилоксазолидин-3-карбоновой кислоты(5,80 г, 21,2 ммоль, 1 экв.) в ТГФ (450 мл) при 0 С. Мутную реакционную смесь перемешивают при 0 С в течение 3,5 ч, затем последовательно добавляют хлорид лития (0,988 г, 23,3 ммоль, 1,1 экв.) и (S)-(-)-4-бензил-2-оксазолидинон (3,57 г, 20,1 ммоль, 0,95 экв.). После нагревания до 23 С и перемешивания в течение 19 ч реакционную смесь распределяют между 0,5 М НСl (150 мл) и EtOAc (2150 мл). Объединенные органические слои промывают полунасыщенным Na2CO3 (150 мл), сушат над МgSO4 и фильтруют под действием силы тяжести. Фильтрат концентрируют при пониженном давлении и остаток очищают с помощью колоночной флэш-хроматографии (30% EtOAc в гексане), получая трет-бутиловый эфир (4S,4"S)-43'-(4"-бензил-2"-оксооксазолидин-3"-ил)-3'оксопропил-2,2-диметилоксазолидин-3-карбоновой кислоты (7,17 г, 83%) в виде бесцветного масла: ИК (см-1) 2978, 1783, 1694; 1H ЯМР(триметилсилил)амида (16,6 мл 1,0 М раствора в ТГФ, 16,6 ммоль, 1,0 экв.) в том же растворителе (150 мл) при -78 С. Реакционную смесь перемешивают в течение 20 мин при -78 С и затем добавляют аллилйодид (4,55 мл, 49,8 ммоль, 3,0 экв.). После перемешивания в течение дополнительных 3 ч при -78 С реакционную смесь держат при -45 С в течение 2 ч и затем распределяют между смесью полунасыщенного NH4Cl и 5% Na2S2O3 2:1 (300 мл) и смесью EtOAc и гексана 1:1 (2200 мл). Объединенные органические слои промывают Н 2 О (200 мл), сушат над МgSO4 и фильтруют под действием силы тяжести. Фильтрат концентрируют при пониженном давлении и остаток очищают с помощью колоночной флэш-хроматографии (15% EtOAc в гексане),получая трет-бутиловый эфир(С 26 Н 36 Н 2O6) С, Н, N. Получение промежуточного соединения: трет-бутилового эфира (1S',3S)-3-(1'-(2",4"димeтоксибeнзил)-2'-oкcoпирролидин-3'-ил)-1 гидроксиметилпропилкарбамовой кислоты. Озон барботируют через раствор третбутилового эфира (2'S,4S,4"S)-4-2'-(4"-бензил 2"-оксооксазолидин-3"-карбонил)пент-4'-енил 2,2-диметилоксазолидин-3-карбоновой кислоты-78 С, пока сохраняется синее окрашивание. Реакционную смесь затем очищают аргоном,пока она не станет бесцветной. Добавляют метилсульфид (6,67 мл, 90,8 ммоль, 10 экв.), смесь перемешивают при -78 С в течение 3,5 ч и держат при 0 С в течение еще 1 ч. После распределения реакционной смеси между H2O (200 мл) и смесью EtOAc и гексана 1:1 (2200 мл), объединенные органические слои сушат над МgSO4 и 50 фильтруют под действием силы тяжести. Фильтрат концентрируют при пониженном давлении и остаток немедленно используют без дополнительной очистки. Полученный выше остаток растворяют в смеси ТГФ и EtOH 2:1 (240 мл) при 23 С и последовательно добавляют гидрохлорид 2,4 диметоксибензиламина (7,40 г, 36,3 ммоль, 4,0 экв.), ацетат натрия (2,98 г, 36,2 ммоль, 4,0 экв.) и цианоборгидрид натрия (1,14 г, 18,1 ммоль,2,0 экв.). Полученную суспензию перемешивают в течение 18 ч при 23 С и затем распределяют между 0,5 М НСl (400 мл) и EtOAc (2200 мл). Объединенные органические слои промывают полунасыщенным NаНСО 3 (300 мл), сушат над Na2SO4 и концентрируют при пониженном давлении. Остаток пропускают через короткую колонку с силикагелем (элюируя 50% EtOAc в гексане), получая трет-бутиловый эфир (3'S,4S)4-1'-(2",4"-диметоксибензил)-2'-оксопирролидин-3'-илметил)-2,2-диметилоксазолидин-3'карбоновой кислоты, содержащий примесь (S)(-)-4-бензил-2-оксазолидинона. Это вещество растворяют в СН 3 ОН (100 мл) и добавляют TsOHH2O (0,345 г, 1,81 ммоль,0,20 экв.). Реакционную смесь нагревают до 50 С и держат при этой температуре в течение 2,5 ч. После охлаждения до 23 С реакционную смесь концентрируют при пониженном давлении до объема 20 мл и распределяют между полунасыщенным NаНСО 3 (150 мл) и смесьюCH2Cl2 и СН 3 ОН 9:1 (2150 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют при пониженном давлении. Остаток очищают с помощью колоночной флэшхроматографии (3% СН 3 ОН в CH2Cl2), получая трет-бутиловый эфир(м, 2 Н), 7,09-7,12 (м, 1 Н). Аналитически определено (C21H32N2O6) С, Н, N. Получение промежуточного соединения: этил-3-Вос-L-N-2,4-диметоксибензил)-(S)пиррол-Ala)-Е-пропеноат. ДМСО (0,270 мл, 3,80 ммоль, 3 экв.) добавляют по каплям к -78 С раствору оксалилхлорида (0,166 мл, 1,90 ммоль, 1,5 экв.) в CH2Cl2(14 мл). Реакционную смесь перемешивают 20 мин, затем раствор трет-бутилового эфира(1S,3'S)(1'(2",4"-диметоксибензил)-2'-оксопирролидин-3'-ил)-1-гидроксиметилпропил карбамовой кислоты (0,518 г, 1,27 ммоль, 1 экв.) в СН 2 Сl2 (13 мл) добавляют через трубочку вдоль стенки реакционного сосуда. После перемешивания в течение 20 мин добавляют по кап 51 лям триэтиламин (1,06 мл, 7,60 ммоль, 6 экв.) и реакционную смесь перемешивают в течение 1,5 ч. Добавляют уксусную кислоту (0,479 мл,8,37 ммоль, 6,6 экв.) и реакционную смесь нагревают до 0 С в течение 5 мин, затем разбавляют МТВЕ (200 мл) и промывают водой, насыщенным NаНСО 3 и насыщенным солевым раствором (25 мл каждого). Органическую фазу сушат над Na2SO4 и концентрируют, получая неочищенный альдегид в виде пены (0,516 г,колич. выход), который используют без дополнительной очистки. Натрий бис(триметилсилил)амид (1,23 мл 1,0 М раствора в ТГФ, 1,23 ммоль, 1 экв.) добавляют к раствору триэтилфосфоноацетата (0,244 мл, 1,23 ммоль, 1 экв.) в ТГФ (15 мл) при -78 С и полученный раствор перемешивают в течение 20 мин при этой температуре. Неочищенный альдегид (полученный выше, 0,500 г, 1,23 ммоль, 1 экв.) в ТГФ (13 мл) добавляют через трубочку вдоль стенки реакционного сосуда и реакционную смесь перемешивают в течение 45 мин при -78 С, нагревают до 0 С в течение 7 мин и распределяют между 0,5 М НСl (20 мл) и МТВЕ (250 мл). Объединенные органические слои сушат над МgSO4 и концентрируют. Остаток очищают с помощью колоночной флэшхроматографии (60% EtOAc в гексане), получая этил-3-Вос-L-N-2,4-диметоксибензил)-(S)пиррол-Аlа)-Е-пропеноат (0,356 г, 61%) в виде белой пены: Rf = 0,43 (60% EtOAc в гексане); ИК (см-1) 3307, 1708, 1678; 1H ЯМР (СDСl3)1,28 (т, 3H, J = 7,2), 1,43 (с, 9 Н), 1,52-1,70 (м,2 Н), 1,98-2,09 (м, 1 Н), 2,21-2,34 (м, 1 Н), 2,482,59 (м, 1 Н), 3,16-3,24 (м, 2 Н), 3,80 (с, 6 Н), 4,18(С 25 Н 36N2O70,25H2O) С, Н, N. Получение промежуточного соединения: этил-3-Boc-L-(4-F-Phe)-L-N-2,4-диметоксибензил)-(S)-пиррол-Аlа)-Е-пропеноат. Это вещество получают из этил-3-Boc-LN-2,4-диметоксибензил)-(S)-пиррол-Ala)-Епропеноата и Boc-L-(4-F-Phe)-ОН, используя методику, подобную описанной выше для получения этил-3-(Boc-L-(4-Me-Phe)-L-(Tr-Gln-Епропеноата (пример 4), Rf = 0,34 (60% EtOAc в гексане); ИК (см-1) 3258, 1705, 1666; 1H ЯМР 52 Получение промежуточного соединения: этил-3-Boc-L-Val-L-(4-F-Phe)-L-[(N-2,4-диметоксибензил)-(S)-пиррол-Ala]-Е-пропеноат. Это соединение получают из этил-3-BocL-(4-F-Phe)-L-N-2,4-диметоксибензил)-(S)пиррол-Ala)-Е-пропеноата и Boc-L-Val-OH,используя методику, подобную описанной выше для получения этил-3-(Boc-L-Phe-L-(Tr-Gln-Епропеноата (пример 2), Rf = 0,24 (60% EtOAc в гексане); ИК (см-1) 3284, 1713, 1678 шир, 1643; 1(С 39 Н 53FN4O9) С, Н, N. Получение промежуточного соединения: этил-3-(5'-метилизоксазол-3'-карбонил)-L-ValL-(4-F-Phe)-L-N-2,4-диметоксибензил)-(S)пиррол-Ala)-Е-пропеноат. Это соединение получают из этил-3-ВосL-Val-L-(4-F-Phe)-L-[(N-2,4-диметоксибензил)(S)-пиррол-Ala)-Е-пропеноата и изоксазол-5 карбонилхлорида, используя методику, описанную выше (пример 2), для получения этил-35'-мeтилизoкcaзoл-3'-карбонил)-L-Leu-L-PheL-(Tr-Gln-Е-пропеноата: Rf = 0,36 (5% СН 3 ОН в СН 2 Сl2); ИК (см-1) 3284, 1717, 1650; 1H ЯМР(0,104 г, 0,458 ммоль, 1,3 экв.) нагревают с обратным холодильником 9 ч и затем оставляют охлаждаться до комнатной температуры в течение 8 ч. Реакционную смесь разбавляют CH2Cl2NаНСО 3 и 1N NaOH 2:1 (20 мл). Органическую фазу сушат над MgSO4 и упаривают. Остаток очищают с помощью колоночной флэш 53 хроматографии(градиентное элюирование 23% СН 3 ОН в CH2Cl2), получают этил-3-(5'метилизоксазол-3'-карбонил)-L-Val-L-(4-F-Phe)L-S)-пиррол-Аlа)-Е-пропеноат (0,117 г, 56%) в виде твердого вещества белого цвета: т.пл.= 219-220 С; Rf = 0,23 (5% СН 3 ОН в СН 2 Сl2); ИК Получение промежуточного соединения: этил-3-Boc-L-(4-F)-Phe-L-(Тr-Gln)-Е-пропеноат. Это соединение получают из этил-3-(BocL-(Tr-Gln-E-пропеноата и Boc-L-(4-F)-Phe-OH,используя методику, подобную описанной выше(пример 2), для получения этил-3-(Boc-L-Phe-L(Tr-Gln-Е-пропеноата: ИК (см-1) 3328, 1707,1506, 1168; 1H ЯМР (CDCl3)1,29 (т, 3H, J = 7,2), 1,37 (с, 9 Н), 1,66-1,78 (м, 1H), 1,88-1,98 (м,1 Н), 2,32 (т, 2 Н, J = 6,6), 2,85-2,92 (м, 1H), 2,973,04 (м, 1H), 4,18 (кв, 2H, J = 7,2), 4,52 (м, 1H),4,96 (м, 1H), 5,60 (д, 1H, J = 15,6), 6,56 (д, 1H, J = 8,1), 6,66 (дд, 1H, J = 15,6, 5,1), 6,80 (с, шир, 1H),6,92-6,98 (м, 2H), 7,08-7,12 (м, 2H), 7,17-7,23 (м,6 Н), 7,24-7,33 (м, 10 Н). Аналитически определено (С 42 Н 46FN3O6) С, Н, N. Получение промежуточного соединения: этил-3-Вос-L-Vаl-L-(4-F)-Phe-L-(Тr-Gln)-Епропеноат. Это соединение получают из этил-3-BocL-(4-F)-Phe-L-(Tr-Gln)-Е-пропеноата и Boc-LVal-OH по способу, описанному выше (пример 2), для получения этил-3-Вос-L-Leu-L-Phe-L(Тr-Gln)-Е-пропеноата: ИК (см-1) 3319, 1657,1511, 1172; 1H ЯМР (СDСl3)0,78 (д, 3H, J = 6,9), 0,87 (д, 3H, J = 6,9), 1,29 (т, 3H, J = 7,2),1,37 (с, 9 Н), 1,69-1,79 (м, 1H), 1,93-2,06 (м, 2H),2,33 (т, 2H, J = 7,2), 2,97-3,04 (м, 1H), 3,73-3,77 54 2 Н), 7,18-7,31 (м, 16 Н), 8,02 (с, 1H). Аналитически определено (C47H55FN4O7) С, Н, N. Получение промежуточного соединения: этил-3-(5'-метилизоксазол-3'-карбонил)-L-ValL-(4-F)-Phe-L-(Tr-Gln)-Е-пропеноат. Это соединение получают из этил-3-ВосL-Vаl-L-(4-F)-Рhе-L-(Tr-Gln)-Е-пропеноата и 5 метилизоксазол-3-карбонилхлорида по способу,подобному описанному выше (пример 2), для получения этил-3-[5'-метилизоксазол-3'-карбонил)-L-Leu-L-Phe-L-(Tr-G-Zn)-Е-пропеноата: ИК (см-1) 3319, 1657, 1511, 1172; 1H ЯМРDCC (0,765 г, 3,71 ммоль, 1,05 экв.), бензиловый спирт (0,347 мл, 3,35 ммоль, 0,95 экв.) и DMAP (0,022 г, 0,18 ммоль, 0,05 экв.) последовательно добавляют к раствору Boc-L-(4-FPhe)-OH (1,0 г, 3,53 ммоль, 1 экв.) в CH2Cl2 (15 мл). После перемешивания в течение 18 ч осадок удаляют путем фильтрации и фильтрат разбавляют МТВЕ (75 мл), промывают 10% KHSO4 и насыщенным солевым раствором (10 мл каждого), сушат над Na2SO4 и упаривают. Очистка 55 остатка с помощью колоночной флэшхроматографии (12% EtOAc в гексане) дает BocL-(4-F-Phe)-OBn (0,992 г, 79%) в виде твердого вещества белого цвета. Спектральные данные 1Boc-L-(4-F-Phe)-OBn (2,0 г, 5,36 ммоль, 1 экв.) перемешивают 1 ч в смеси CH2Cl2 (20 мл) и TFA (10 мл), затем добавляют еще TFA (10 мл) и реакционный раствор перемешивают дополнительно еще час. Летучие вещества упаривают и остаток растворяют в DMF (30 мл), добавляют Boc-L(т-бутил-Gly)-ОН (1,24 г, 5,36 ммоль, 1 экв.) и раствор охлаждают до 0 С. Последовательно добавляютN,Nдиизопропилэтиламин (2,80 мл, 16,1 ммоль, 3 экв.) и HATU (2,04 г, 5,37 ммоль, 1 экв.). После перемешивания в течение 20 мин реакционную смесь оставляют нагреваться до комнатной температуры в течение 1 ч, затем разбавляют МТВЕ(500 мл) и промывают 5% KHSO4 (100 мл), насыщенным NаНСО 3 (50 мл) и насыщенным солевым раствором (50 мл). Органическую фазу сушат и упаривают. Остаток очищают с помощью колоночной флэш-хроматографии (20%(м, 5 Н). Аналитически определено C27H35FN2O5) С, Н, N. Получение промежуточного соединения: этил-3-Boc-L(т-бутил-Gly)-L-(4-F-Phe)-LN-2,4-диметоксибензил)-(S)-пиррол-Ala)-Епропеноат. Раствор НСl в 1,4-диоксан (4,0 М, 8 мл) добавляют к раствору этил-3-Boc-L-(N-2,4 диметоксибензил)-(S)-пиррол-Ala)-Е-пропеноата (0,309 г, 0,648 ммоль, 1 экв.) в 1,4-диоксан (8 мл). После перемешивания в течение 1,5 ч летучие вещества упаривают, получая неочищенную соль амина в виде пены. Палладий на угле (10%, 200 мг) добавляют к раствору Boc-L(т-бутил-Gly)-L-(4-F-Phe)OBn (2,04 г, 4,19 ммоль) в EtOAc (200 мл). Атмосферу заменяют на водород через баллон. После перемешивания в течение 3 ч атмосферу заменяют на аргон и реакционную смесь фильтруют через фильтровальную бумагу Whatman 3 и 5. Фильтрат упаривают, получая белую пену. Эту пену объединяют с неочищенной солью амина (полученной выше) в DMF (5 мл) и охлаждают до 0 С. Последовательно добавляютN,N-диизопропилэтиламин (0,339 мл, 1,95 ммоль, 3 экв.) и HATU (0,247 г, 0,650 ммоль, 1 экв.). После перемешивания в течение 20 мин реакционную смесь оставляют нагреваться до комнатной температуры в течение 1 ч, затем разбавляют МТВЕ (100 мл) и промывают 5%KHSO4, насыщенным NаНСО 3 и насыщенным солевым раствором (15 мл каждого). Органическую фазу сушат и упаривают. Остаток очищают с помощью колоночной флэшхроматографии (60% EtOAc в гексане), получая этил-3-Вос-Е(т-бутил-Gly)-L-(4-F-Phe)-LN-2,4-диметоксибензил)-(S)-пиррол-Ala)-Епропеноат (0,364 г, 74%) в виде белой пены: Rf= 5,9). Аналитически определено (C40H55FN4O9) С, Н, N. Получение промежуточного соединения: этил-3-(5'-метилизоксазол-3'-карбонил)-L(тбутил-Gly)-L-(4-F-Phe)-L-N-2,4-диметоксибензил)-(S)-пиррол-Ala)-Е-пропеноат. Это соединение получают из этил-3-ВосL(т-бутил-Gly)-L-(4-F-Phe)-L-N-2,4 диметоксибензил)-(S)-пиррол-Ala]-Е-пропеноата и изоксазол-5-карбонилхлорида, используя методику, описанную выше (пример 2),для получения этил-3-(5'-метилизоксазол-3'карбонил)-L-Leu-L-Phe-L-(Tr-Gln)-Е-пропеноата; Rf = 0,60 (10% СН 3 ОН в СНСl3); ИК (см-1) 3295, 1713, 1666, 1643; 1H ЯМР (CDCl3)1,07(дд, 1 Н, J = 15,6, 5,3), 6,79-6,88 (м, 2 Н), 6,92 (д,1 Н, J = 9,0), 7,09-7,22 (м, 3H), 7,37 (д, 1 Н, J = 9,0), 8,27 (д, 1 Н, J = 6,2). Аналитически определено (C40H50FN5O90,25H2O) С, Н, N. Получение продукта: этил-3-(5'-метилизоксазол-3'-карбонил)-L(т-бутил-Gly)-L-(4-FPhe)-L-S)-пиррол-Ala)-Е-пропеноат. Это соединение получают из этил-3-(5'метилизоксазол-3'-карбонил)-L(т-бутил-Gly)L-(4-F-Phe)-L-N-2,4-диметоксибензил)-(S)пиррол-Ala)-Е-пропеноата, используя методику, описанную выше (пример 6), для получения этил-3-(5'-метилизоксазол-3'-карбонил)-L-ValL-(4-F-Phe)-L-S)-пиррол-Ala)-Е-пропеноата: Получение промежуточного соединения: трет-бутиловый эфир (2'S,4S,4"S)-4-2'-(4"бензил-2"-оксооксазолидин-3"-карбонил)-5'гидроксипентил)-2,2-диметилоксазолидин-3 карбоновой кислоты. Раствор борантетрагидрофуранового комплекса (0,96 мл 1,0 М раствора в ТГФ, 0,96 ммоль, 1 экв.) добавляют к 0 С раствору третбутилового эфира (2'S,4S,4"S)-4-2'-(4"-бензил 2"-оксооксазолидин-3"-карбонил)пент-4'-енил 2,2-диметилоксазолидин-3-карбоновой кислоты(полученного, как описано в примере 6, 0,455 г,0,963 ммоль, 1 экв.) в ТГФ (3 мл). После перемешивания в течение 30 мин, добавляют воду (3 мл) и тетрагидрат пербората натрия (0,148 г,0,962 ммоль, 1 экв.) и баню со льдом удаляют. После дополнительного часа реакционную смесь разбавляют МТВЕ (125 мл), промывают водой (15 мл) и насыщенным солевым раствором (215 мл), сушат над Nа 2SO4 и концентрируют. Остаток очищают с помощью колоночной флэш-хроматографии (50% EtOAc в гексане),получая трет-бутиловый эфир (2'S,4S,4"S)-4-2'(4"-бeнзил-2"-oкcooкcaзoлидин-3"-кapбoнил)-5'гидpoкcипeнтил-2,2-диметилоксазолидин-3 карбоновой кислоты (0,339 г, 72%) в виде бесцветного стекловидного вещества: Rf = 0,41(С 26 Н 38N2O70,5 Н 2O) С, Н, N. Получение промежуточного соединения: трет-бутиловый эфир (1S',3'S)-2-(1'-(2",4"димeтокcибeнзил)-2'-oкcoпипepидин-3'-ил)-1 гидроксиметилэтилкарбамовой кислоты. Трет-бутиловый эфир (2'S,4S,4"S)-4-2'(4"-бензил-2"-оксооксазолидин-3"-карбонил)-5'гидpoкcипeнтил-2,2-димeтилоксазолидин-3 карбоновой кислоты (3,88 г, 7,92 ммоль, 1 экв.) растворяют в Et3N (3,97 мл, 28,51 ммоль, 3,6 экв.). Смесь охлаждают до -12 С и добавляют 58 раствор комплекса триоксид серы-пиридин (5,04 г, 31,67 ммоль, 4 экв.) в ДМСО (150 мл) с такой скоростью, чтобы поддерживать температуру в интервале 8-17 С. Раствор перемешивают при 23 С в течение 3 ч. Реакционную смесь охлаждают в бане с водой со льдом и гасят путем добавления H2O (150 мл). Полученный раствор экстрагируют EtOAc (2 х 150 мл). Объединенные органические слои промывают 5% лимонной кислотой (100 мл), насыщенным солевым раствором (100 мл), сушат над Na2SO4 и фильтруют. Растворитель удаляют при пониженном давлении и остаток сушат под вакуумом, получая белую пену (3,53 г). К раствору этого вещества (3,53 г, 7,22 ммоль, 1 экв.) в смеси ТГФ и EtOH 2:1 (120 мл) добавляют гидрохлорид 2,4-диметоксибензиламина (5,88 г, 28,89 ммоль, 4 экв.), NaOAc (2,37 г, 28,89 ммоль, 4 экв.) и NaBH3CN (0,908 г, 14,45 ммоль, 2 экв.). Реакционную смесь перемешивают в течение ночи (20 ч) и затем разбавляют МТВЕ (200 мл). Органический слой промывают 10% KHSO4 (100 мл), насыщенным NаНСО 3(100 мл), насыщенным солевым раствором (100 мл), сушат над Na2SO4 и концентрируют, получая бледно-желтую пену. К раствору этой пены (3,34 г, 7,22 ммоль, 1 экв.) в СН 3 ОН (50 мл) добавляют птолуолсульфоновую кислоту (0,275 г, 1,44 ммоль, 0,2 экв.). Реакционную смесь перемешивают при 50 С в течение 2,5 ч и затем разбавляют СН 2 Сl2 (100 мл). Органический слой промывают насыщенным NаНСО 3 (100 мл), сушат над Na2SO4 и концентрируют. Остаток очищают с помощью колоночной флэш-хроматографии(3% СН 3 ОН в CH2Cl2), получая трет-бутиловый эфир (1S',3'S)-2-(1'-(2",4"-диметоксибензил)-2'оксопиперидин-3'-ил)-1-гидроксиметилэтил карбамовой кислоты в виде белой пены (1,33 г,44% с трех стадий): 1H ЯМР (СDСl3):1,44 (с,9 Н), 1,71-1,85 (м, 2 Н), 1,92-1,98 (м, 2 Н), 2,402,48 (м, 1 Н), 2,71-2,78 (м, 1 Н), 3,19-3,32 (м, 2 Н),3,45-3,69 (м, 4 Н), 4,11-4,20 (м, 2 Н), 4,68 (м, 1 Н),5,47 (м, 1 Н), 6,44 (с, 1 Н), 7,20-7,33 (м, 2 Н). Получение продукта: этил-3-(5'метилизоксазол-3'-карбонил)-L-Val-L-(4-F-Phe)L-S)-пипер-Ala)-Е-пропеноат. трет-Бутиловый эфир (1S',3'S)-2-(1'-(2",4"диметоксибензил)-2'-оксопиперидин-3'-ил)-1 гидроксиметилэтилкарбамовой кислоты превращают в продукт этил-3-(5'-метилизоксазол 3'-карбонил)-L-Val-L-(4-F-Phe)-L-S)-пиперAla)-Е-пропеноат по способу, аналогичному использующемуся для превращения третбутилового эфира Получение промежуточного соединения: транс-6-метил-гепт-4-еноевая кислота. Раствор изобутиральдегида (9,59 г, 133 ммоль, 1 экв.) в ТГФ (50 мл) добавляют по каплям через дополнительную воронку к раствору винилмагний бромида (133 мл 1,0 М раствора в ТГФ, 133 ммоль, 1,0 экв.) в ТГФ (300 мл) при 0 С. После завершения добавления реакционную смесь перемешивают в течение 30 мин при 0 С и затем добавляют этилмалонилхлорид(17,0 мл, 133 ммоль, 1,0 экв.). После перемешивания в течение 1 ч при 0 С реакционную смесь распределяют между насыщенным NH4Cl (150 мл) и смесью EtOAc и гексана 1:1 (2200 мл). Объединенные органические слои сушат надNa2SO4 и концентрируют. Остаток очищают путем фильтрации через силикагель (элюируя 5% EtOAc в гексане), получая промежуточный малоновый эфир (11,5 г, 40% выход). Это вещество не характеризуют, но объединяют (неразбавленное) с Ti(OEt)4 (1,13 мл, 5,39 ммоль, 0,10 экв.) и нагревают до 190 С в течение 4 ч, затем охлаждают до 60 С, последовательно добавляютEtOH (50 мл) и 6,0 М КОН (50 мл) и коричневую реакционную смесь нагревают с обратным холодильником 4 ч. После охлаждения до 23 С,реакционную смесь фильтруют через стекловату и фильтрат распределяют между водой (150 мл) и Et2O (2150 мл). Водный слой затем подкисляют до рН 2 (определяют по индикаторной бумаге) концентрированной НСl и экстрагируют смесью EtOAc и гексана 1:1 (2150 мл). Объединенные органические слои сушат над Na2SO4,концентрируют и остаток перегоняют при пониженном давлении, получая транс-6-метилгепт-4-еновую кислоту (3,58 г, 47%) в виде бесцветной жидкости: т. кип. 107-112 С (давление 1 Торр); ИК (см-1) 2960, 1711; 1H ЯМР (CDCl3)0,96 (д, 6 Н, J = 6,5), 2,18-2,45 (м, 5 Н), 5,31-5,50N,N-диметилформамид (0,03 мл, 0,39 ммоль,0,016 экв.) в бензоле (60 мл) при 23 С. Реакционную смесь перемешивают при 23 С в течение 2 ч и затем концентрируют при пониженном давлении. Полученное масло растворяют в ТГФ(20 мл) и добавляют через трубочку к раствору(1R,2R)-(-)-псевдоэфедрина (3,87 г, 23,4 ммоль,1 экв.) и триэтиламина (3,92 мл, 28,1 ммоль, 1,2 экв.) в ТГФ (150 мл) при 0 С. Реакционную смесь перемешивают при 0 С в течение 30 мин,затем распределяют между полунасыщеннымNH4Cl (150 мл) и EtOAc (2150 мл). Объединенные органические слои сушат над Na2SO4,концентрируют и остаток очищают с помощью колоночной флэш-хроматографии (градиентное элюирование 4050% EtOAc в гексане), получая транс-6-метил-гепт-4-еноевой кислоты (2Rгидрокси-1R-метил-2-фенилэтил)метиламид(м), 2,82 (с), 2,92 (с), 3,99-4,04 (м), 4,32-4,42 (м),4,44-4,49 (м), 4,55-4,62 (м), 5,32-5,49 (м), 7,247,42 (м). Аналитически определено (C18H27NO2) С, Н, N. Получение промежуточного соединения: транс-6-метил-2S-(4-фторбензил)-гепт-4-еноевой кислоты (2R-гидрокси-1R-метил-2-фенилэтил)метиламид. н-Бутиллитий (32,5 мл 1,6 М раствор в гексане, 52,0 ммоль, 3,1 экв.) добавляют к суспензии безводного хлорида лития (7,18 г, 169 ммоль, 10 экв.) и диизопропиламина (7,80 мл,55,7 ммоль, 3,3 экв.) в ТГФ (250 мл) при -78 С. Реакционную смесь перемешивают в течение 30 мин при -78 С, держат при 0 С в течение 5 мин и затем снова охлаждают до -78 С. Через трубочку добавляют транс-6-метил-гепт-4-еноевой кислоты (2R-гидрокси-1R-метил-2-фенилэтил) метиламид (4,91 г, 17,0 ммоль, 1 экв.) в ТГФ (50 мл) и полученный раствор перемешивают при-78 С в течение 1,75 ч, держат при 0 С в течение 20 мин, перемешивают при 23 С в течение 5 мин и затем снова охлаждают до 0 С. Добавляют раствор 4-фторбензилбромида (6,34 мл, 50,9 ммоль, 3 экв.) в ТГФ (15 мл) и реакционную смесь перемешивают при 0 С в течение 30 мин,затем ее распределяют между полунасыщенным(200 мл, 2150 мл). Объединенные органические слои сушат над Na2SO4 и концентрируют. Остаток очищают с помощью колоночной флэш-хроматографии (градиентное элюирование 2040% EtOAc в гексане), получая транс-6
МПК / Метки
МПК: A61P 31/16, A61K 38/05, C07K 5/027, C07D 413/12
Метки: композиции, применение, антипикорнавирусные, получение
Код ссылки
<a href="https://eas.patents.su/30-3856-antipikornavirusnye-kompozicii-ih-poluchenie-i-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Антипикорнавирусные композиции, их получение и применение</a>
Бициклические антагонисты тахикининов, их получение и применение в фармацевтической композиции

Номер патента: 697
Опубликовано: 28.02.2000
Авторы: Аркамоне Федерико, Маджи Карло Альберто, Джаннотти Данило, Куартара Лаура
МПК: C07K 7/22, A61K 38/12
Метки: антагонисты, применение, тахикининов, получение, композиции, фармацевтической, бициклические
Формула / Реферат:
1. Бициклические соединения общей формулы где X1, Х2, Х3, Х4, Х5 и Х6, одинаковые или различные, представляют собой группу -NR'CO- или -CONR'-, где R' является Н или C1-3-алкилом; Y представляет собой группу, выбранную из -NRCO-, -CONR- или -SS-, где R является Н или C1-3-алкилом; по меньшей мере, одна из групп R1, R2, R3 и R4, одинаковых или различных, является гидрофильной, а остальные группы являются гидрофобными; m и n, одинаковые...
Производные карбоксамидотиазолов, их получение, содержащие их фармацевтические композиции

Номер патента: 3093
Опубликовано: 26.12.2002
Авторы: Буажегрэн Робер, Олльеро Доминик, Биньон Эрик, Броден Роже, Молимар Жан-Шарль
МПК: A61K 31/427, A61P 1/06, A61K 31/405...
Метки: фармацевтические, производные, карбоксамидотиазолов, получение, композиции, содержащие
Формула / Реферат:
1. Соединение формулы в которой R6a означает хлор или метил; R10, R11, R13 означают, каждый независимо друг от друга, водород, метил, этил, гидроксил, ацетилоксигруппу, метоксигруппу, этоксигруппу, метилтиогруппу, трифторметил, аминогруппу или галоген; при условии, что один или два из заместителей R10, R11, R13 отличаются от водорода; или его соли или сольваты. 2. Соединение по п.1 формулы в которой один или два из заместителей R10a, R11a,...
Производные бензофурана, их получение и применение

Номер патента: 3781
Опубликовано: 28.08.2003
Авторы: Дансер Роберт, Педерсен Хенрик, Роттлендер Марио, Руланд Томас, Бегесе Клаус Петер, Андерсен Ким
МПК: A61K 31/343, C07D 307/87
Метки: производные, применение, получение, бензофурана
Формула / Реферат:
1. Изобензофуран общей формулы I где R1 представляет собой водород, галоген, трифторметил, трифторметилсульфонилокси, C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-8-циклоалкил, C1-6-алкокси, гидрокси, формил, ацил, амино, C1-6-алкиламино, C2-12-диалкиламино, ациламино, C1-6-алкоксикарбониламино, аминокарбониламино, C1-6-алкиламинокарбониламино, C2-12-диалкиламинокарбониламино, нитро, циано, COOH или COO-C1-6-алкил; R2 и R3, каждый независимо,...
Новые таксоиды, их получение и фармацевтические композиции, их содержащие

Номер патента: 1516
Опубликовано: 23.04.2001
Авторы: Бушар Эрве, Бурза Жан-Доминик, Коммерсон Ален
МПК: A61K 31/335, C07D 305/14, A61P 35/00...
Метки: новые, композиции, таксоиды, фармацевтические, получение, содержащие
Формула / Реферат:
1. Новые таксоиды общей формулы (I) в которой Z означает радикал общей формулы (II) в которой R1 означает радикал бензоил или алкоксикарбонил; R3 означает фенил; R4 означает радикал гидрокси или радикал алкокси, содержащий от 1 до 4 атомов углерода с прямой или разветвленной цепью, алканоилокси, алканоильная часть которого содержит от 2 до 4 атомов углерода с прямой или разветвленной цепью; R5 означает радикал алкокси, содержащий от 1 до...
Новые таксоиды,их получение и содержащие их фармацевтические композиции

Номер патента: 1533
Опубликовано: 23.04.2001
Авторы: Бушар Эрве, Коммерсон Алан
МПК: A61K 31/335, A61P 35/00, C07D 305/14...
Метки: получение, фармацевтические, новые, композиции, содержащие, таксоиды,их
Формула / Реферат:
1. Новые таксоиды общей формулы в которой Z обозначает радикал общей формулы в которой R1 обозначает бензоильный радикал или радикал R2-О-СО-, в котором R2 представляет собой алкильный радикал с 1-8 атомами углерода и R3 обозначает фенил или a - или b -нафтил; и либо R4 обозначает атом водорода, R6 и R7 вместе образуют кетонную функцию и R и R5 вместе образуют связь, либо R4 обозначает атом водорода или радикал алкокси с 1-6 атомами...
Предыдущий патент: Измерение магнитных полей с использованием струны, закрепленной на обоих концах
Следующий патент: Пептидная фармацевтическая композиция и способ повышения эластичности ткани
Случайный патент: Переворачиваемый инструмент для сельскохозяйственных чизель-культиваторов и тому подобного