Производные полимиксина
Номер патента: 24792
Опубликовано: 31.10.2016
Авторы: Вадман Сьёрд Николаас, Дюперчи Эстер, Браун Памела, Доусон Майкл Джон, Саади Мона





















Формула / Реферат
1. Соединение формулы (I)

где X представляет собой -C(O)-, -NHC(O)-, -OC(O)-, -CH2- или -SO2-;
R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток фенилаланина, лейцина или валина;
R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток лейцина, изолейцина, фенилаланина, треонина, валина или норвалина;
R3 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток треонина или лейцина;
R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток α,γ-диаминомасляной кислоты или серина;
R5 представляет собой группу (а)-(g), где:
(a) представляет собой С1-12алкил(С3-8циклоалкил), где алкил или циклоалкил замещен (i) одной, двумя или тремя гидроксильными группами, или (ii) одной -NR6R7 группой, или (iii) одной -NR6R7 группой и одной или двумя гидроксильными группами;
(b) представляет собой С2-12алкил, где алкил замещен (i) одной, двумя или тремя гидроксильными группами, или (ii) одной -NR6R7 группой на конце алкильной цепи, или (iii) одной -NR6R7 группой и двумя гидроксильными группами;
(c) представляет собой С0-12алкил(С4-6гетероциклил), где С4-6гетероциклил содержит 1 или 2 атома азота и, необязательно, содержит дополнительный кольцевой гетероатом, выбранный из кислорода и серы;
(d) представляет собой С3-8циклоалкил и циклоалкил замещен (i) одной, двумя или тремя гидроксильными группами или (iii) одной -NR6R7 группой и одной или двумя гидроксильными группами;
(e) представляет собой С3-12алкил, где алкил замещен одной -NR6R7 группой и одной гидроксильной группой;
(f) представляет собой С6-12алкил, замещенный одной -NR6R7 группой; и
(g) представляет собой С5циклоалкил, замещенный одной -NR6R7 группой;
R6 представляет собой водород или С1-4алкил;
R7 представляет собой водород или С1-4алкил;
R8 представляет собой метил или водород,
или фармацевтически приемлемая соль указанного соединения.
2. Соединение по п.1, отличающееся тем, что R5 представляет собой С1-12алкил(С3-8циклоалкил) и C1-12алкил выбран из группы, состоящей из метила, этила, н-пропила, изопропила, бутила, н-бутила и трет-бутила, где алкил или циклоалкил замещен (i) одной, двумя или тремя гидроксильными группами, или (ii) одной -NR6R7 группой, или (iii) одной -NR6R7 группой и одной или двумя гидроксильными группами.
3. Соединение по п.1 или 2, отличающееся тем, что R5 представляет собой C1-12алкил(С3-8циклоалкил), где алкил или циклоалкил замещен одной -NR6R7 группой.
4. Соединение по любому из пп.1-3, отличающееся тем, что С3-8циклоалкил представляет собой C5- или С6циклоалкил.
5. Соединение по п.1, отличающееся тем, что R5 представляет собой С2-12алкил, например разветвленный С2-12алкил, где алкил замещен (i) одной, двумя или тремя гидроксильными группами, или (ii) -NR6R7 группой на конце алкильной цепи, или (iii) одной -NR6R7 группой и двумя гидроксильными группами.
6. Соединение по п.5, отличающееся тем, что R5 представляет собой С2-12алкил, замещенный -NR6R7 группой на конце алкильной цепи.
7. Соединение по п.5, отличающееся тем, что R5 представляет собой С2-12алкил, замещенный одной, двумя или тремя гидроксильными группами.
8. Соединение по п.7, отличающееся тем, что R5 представляет собой С2-12алкил, замещенный одной гидроксильной группой.
9. Соединение по п.1, отличающееся тем, что R5 представляет собой С1-12алкил(С4-6гетероциклил).
10. Соединение по п.1, отличающееся тем, что R5 представляет собой С3-8циклоалкил и циклоалкил замещен (i) одной, двумя или тремя гидроксильными группами или (iii) одной -NR6R7 группой и одной гидроксильной группой.
11. Соединение по п.1, отличающееся тем, что R5 представляет собой С3-10алкил, где алкил замещен одной -NR6R7 группой и одной гидроксильной группой.
12. Соединение по п.1, отличающееся тем, что R5 представляет собой С6-12алкил, замещенный одной -NR6R7 группой.
13. Соединение по п.1, отличающееся тем, что R5 представляет собой С5циклоалкил, замещенный одной -NR6R7 группой.
14. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R6 представляет собой водород или метил.
15. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R7 представляет собой водород или метил.
16. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток фенилаланина, например остаток D-фенилаланина.
17. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток лейцина.
18. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R3 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток треонина.
19. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток α,γ-диаминомасляной кислоты.
20. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что R8 представляет собой метил.
21. Соединение формулы (I) по любому предшествующему пункту, отличающееся тем, что X представляет собой -C(O)-.
22. Соединение по п.1, выбранное из группы, состоящей из
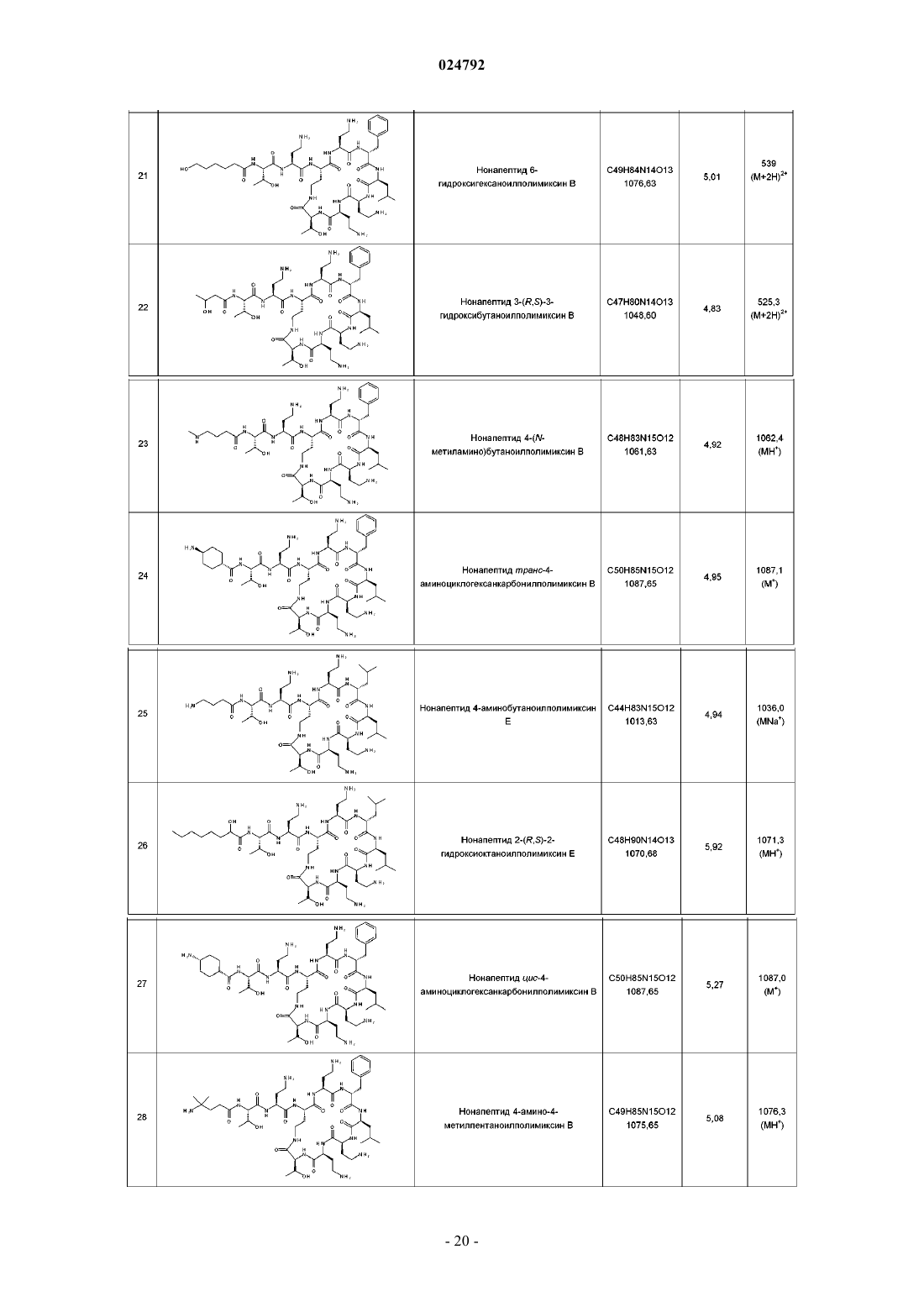
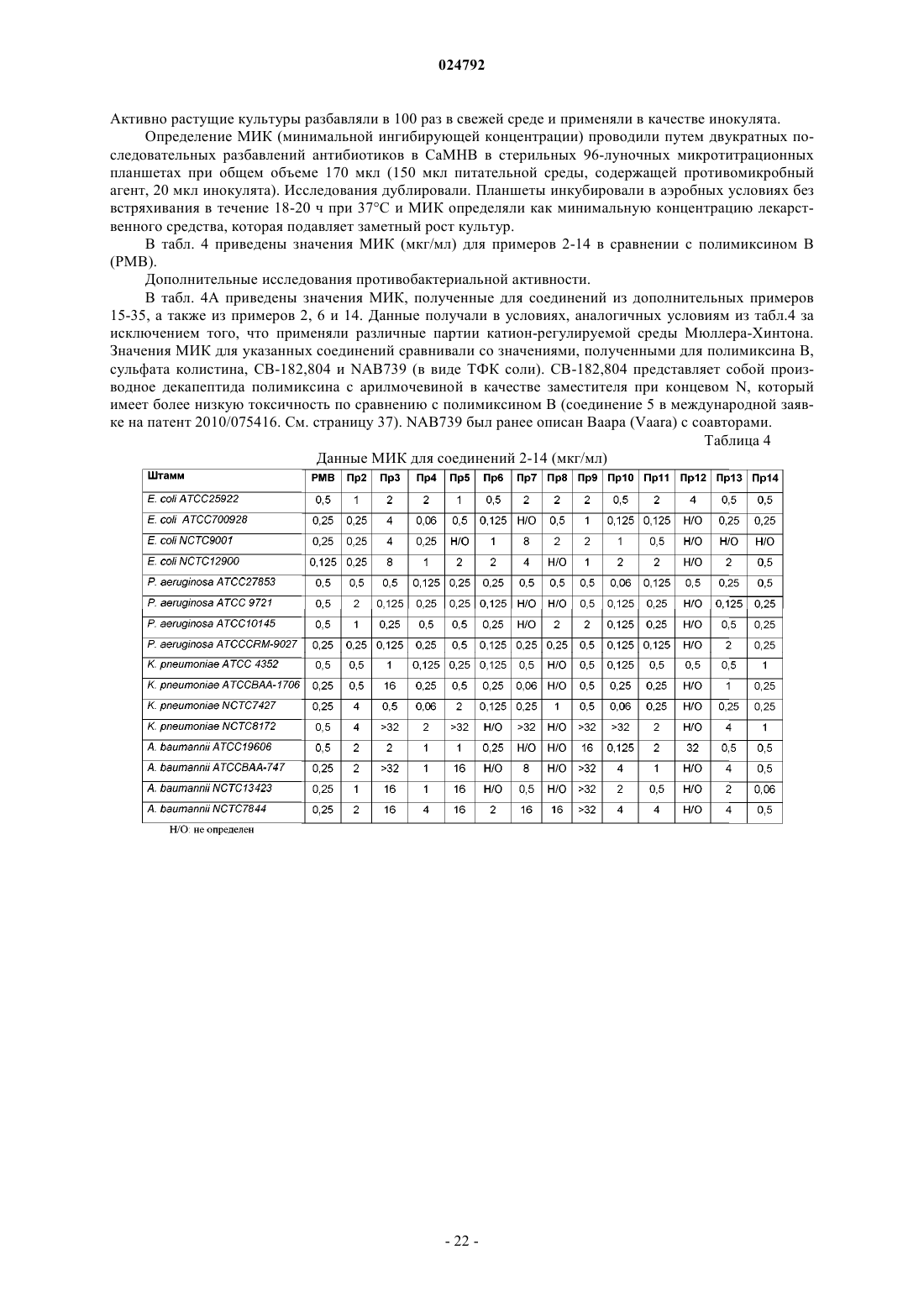
нанопептида 2-гидроксиоктаноилполимиксина B;
нанопептида 3-аминопропаноилполимиксина B;
нанопептида 3-(N,N-диметиламино)пропаноилполимиксина B;
нанопептида 4-аминобутаноилполимиксина B;
нанопептида 6-аминогексаноилполимиксина B;
нанопептида 8-гидроксиоктаноилполимиксина B;
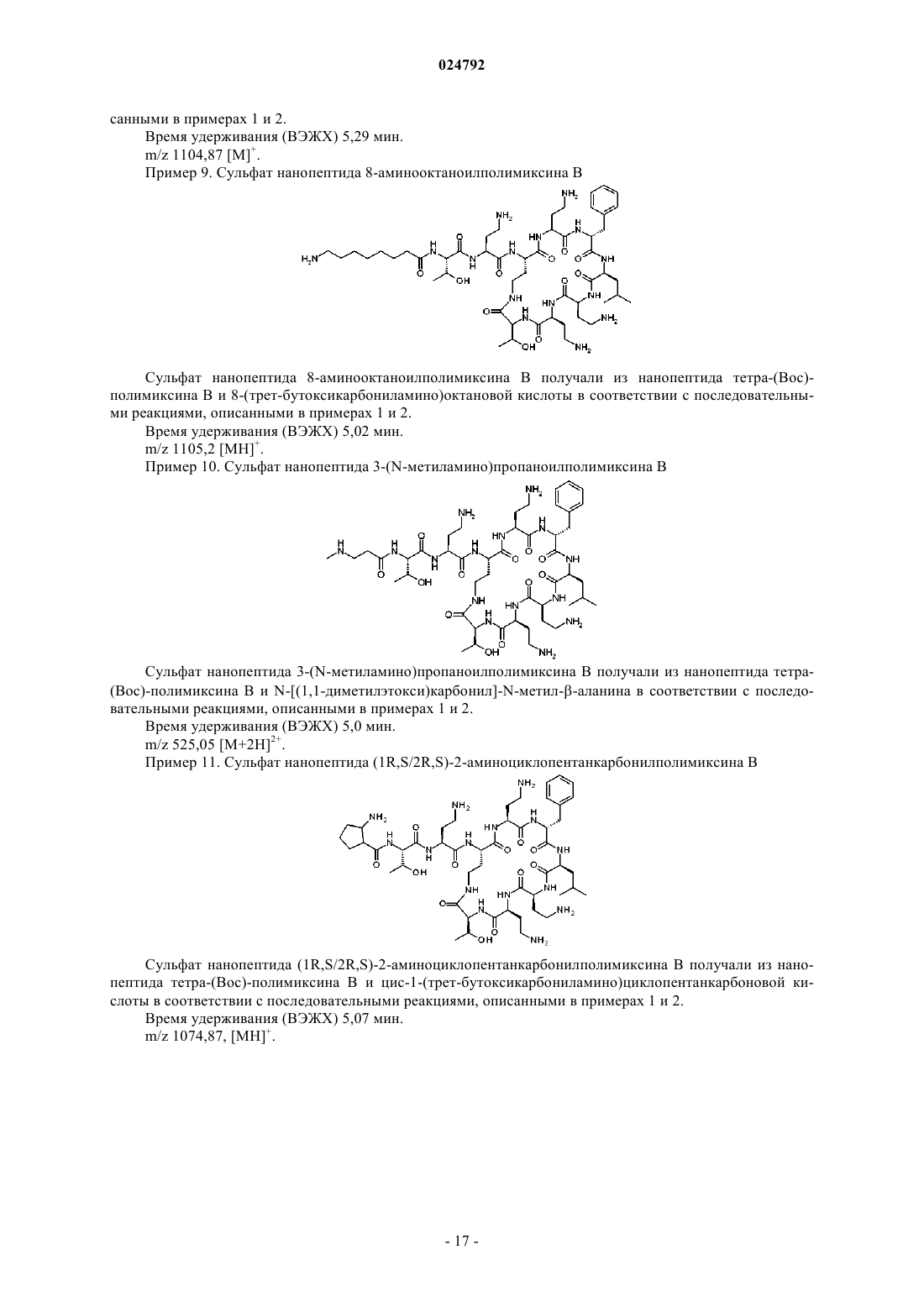
нанопептида 8-аминооктаноилполимиксина B;
нанопептида 3-(N-метиламино)пропаноилполимиксина B;
нанопептида 2-аминоциклопентанкарбонилполимиксина B;
нанопептида 3-аминопропаноилколистина (полимиксина Е);
нанопептида 3-пирролидин-3-карбонилполимиксина B;
нанопептида 3-амино-3-циклогексанпропаноилполимиксина В;
нанопептида 5-аминопентаноилполимиксина B;
нанопептида 3-гидроксиоктаноилполимиксина B;
нанопептида 4-(N,N-диметиламино)бутаноилполимиксина B;
нанопептида 7-аминогептаноилполимиксина B;
нанопептида 4-морфолинилбутаноилполимиксина B;
нанопептида 6-гидроксигексаноилполимиксина B;
нанопептида 3-гидроксибутаноилполимиксина B;
нанопептида 4-(N-метиламино)бутаноилполимиксина B;
нанопептида 4-аминобутаноилполимиксина Е;
нанопептида 2-гидроксиоктаноилполимиксина Е;
нанопептида 4-амино-5-метилгексаноилполимиксина B;
нанопептида 3-(1-пирролидин-2-ил)пропионилполимиксина B;
нанопептида транс-4-гидроксициклогексанкарбонилполимиксина B;
нанопептида 3-гидроксипропаноилполимиксина B;
нанопептида (2-гидрокси-2-циклогексил)этаноилполимиксина B;
нанопептида 2-аминооктаноилполимиксина В или
из фармацевтически приемлемых солей указанных соединений.
23. Фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль по любому из пп.1-22 вместе с фармацевтически приемлемым носителем.
24. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-22 или композиция по п.23 для применения для лечения.
25. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-22 или композиция по п.23 для применения для лечения бактериальной инфекции.
26. Соединение по п.25 для лечения инфекций, вызванных бактериями, устойчивыми ко многим видам противобактериальных препаратов.
27. Соединение по п.25 или 26 для лечения инфекций, вызванных грамотрицательными бактериями.
28. Соединение формулы (I) или его фармацевтически приемлемая соль по любому из пп.1-22 или композиция по п.23 для применения для лечения Acinetobacter инфекции или Klebsiella инфекции.
Текст
KATSUMA NAOKO ET AL.: "Development of Des Предложены соединения и применение указанных соединений для лечения, например для лечения микробных инфекций, в частности, вызванных грамотрицательными бактериями. Соединения представляют собой производные полимиксина, представленные формулой (I) и фармацевтически приемлемые соли указанных соединений, где X представляет собой -NHC(O)-, -C(O)-,-OC(O)-, -CH2- или -SO2-; R5 представляет собой С 0-12 алкил(С 4-6 гетероциклил), С 2-12 алкил или С 0-12 алкил(С 3-8 циклоалкил), где алкил или циклоалкил содержат одну, две или три гидроксильные группы, -NR6R7 группу или одну -NR6R7 группу и одну или две гидроксильные группы; R1-R4 и R6-R8 такие, как определено в описании. Саади Мона, Дюперчи Эстер, Браун Памела, Доусон Майкл Джон (GB), Вадман Сьрд Николаас (DE) Заявка на данное изобретение испрашивает приоритет на основании заявки на патент США 61/561361, поданной 18 ноября 2011 г. (18.11.2011), содержание которой включено в настоящее описание во всей полноте посредством ссылки. Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим указанные соединения, и применению указанных соединений и фармацевтических композиций для лечения, например для лечения микробных инфекций, в частности, вызванных грамотрицательными бактериями. У восприимчивых людей некоторые грамотрицательные бактерии могут вызывать серьезные осложнения, а также развитие инфекций, таких как пневмония, инфекции мочевых путей, раневые инфекции, ушные инфекции, глазные инфекции, внутрибрюшинные инфекции и инфекции ротовой полости, и сепсис. Лечение серьезных бактериальных инфекций в клинической практике может быть осложнено устойчивостью к антибиотикам. В последние годы наблюдается рост количества инфекций, вызванных грамотрицательными бактериями, которые устойчивы ко многим видам противобактериальных препаратов, включая антибиотики широкого спектра действия, такие как аминогликозиды, цефалоспорины и даже карбапенемы. Следовательно, существует потребность в новых противомикробных препаратах,эффективных против грамотрицательных бактерий, в частности, против полирезистентных грамотрицательных бактерий. Полимиксины представляют собой класс антибиотиков, производимых грамположительными бактериями Bacillus polymyxa. Открытые в конце 1940-х гг. полимиксины, в частности полимиксин В и полимиксин Е (колистин), применяли для лечения грамотрицательных бактерий. Тем не менее, указанные антибиотики обладают побочными эффектами, такими как нефротоксичность. Следовательно, их применение в терапии ограничивается лечением в крайнем случае. В международной заявке на патент 2008/017734 предпринята попытка решить указанную проблему токсичности путем применения производных полимиксина, несущих по меньшей мере два, но не больше трех положительных зарядов. Сообщается, что указанные соединения являются эффективными противобактериальными агентами пониженной токсичности. В описании делается предположение, что снижение количества положительных зарядов снижает аффинность соединения к выделенной почечной ткани крыс, что, в свою очередь, может приводить к снижению нефротоксичности. Неожиданно авторы настоящего изобретения установили, что некоторые альтернативные виды полимиксинов, включая производные с четырьмя или более зарядами, обладают подходящей противобактериальной активностью, а также меньшей токсичностью, особенно нефротоксичностью. Краткое описание изобретения Таким образом, в настоящем изобретении предложено соединение формулы (I)R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток фенилаланина, лейцина или валина;R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток лейцина, изолейцина,фенилаланина, треонина, валина или норвалина;R3 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток треонина или лейцина;R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток ,-диаминомасляной кислоты или серина;R5 представляет собой С 0-12 алкил(С 4-6 гетероциклил) или С 2-12 алкил или С 0-12 алкил(С 3-8 циклоалкил),где алкил или циклоалкил содержит:i) одну, две или три гидроксильные группы, илиiii) одну -NR6R7 группу и одну или две гидроксильные группы;R6 представляет собой водород или С 1-4 алкил;R7 представляет собой водород или С 1-4 алкил;R8 представляет собой водород или метил,или фармацевтически приемлемая соль указанного соединения. Соединения формулы (I) характеризуются тем, что пептидная часть их молекул содержит только девять аминокислот, в то время как природные полимиксины содержат 10 аминокислот. Подробное описание изобретения Неожиданно было установлено, что соединения формулы (I) имеют более низкую токсичность, чем исходные полимиксины, при этом сохраняя подходящую для применения противобактериальную активность. Известно, что нанопептид полимиксин, не содержащий ацильной цепи, имеет пониженную токсичность, но при этом обладает недостаточной для применения противобактериальной активностью. Тем не менее, исследование длин цепей простых ацильных производных нанопептида полимиксина В(K. Okimura et. al., Bull. Chem. Soc. Jpn, 2007, 80, 543) свидетельствует о значимости длины цепи для противобактериальной активности, причем оптимальная цепь содержит примерно восемь атомов углерода, и демонстрирует слишком малую активность ацетильных производных против E.coli и Salmonellatyphimurium. Это соответствует результатам для ацильных декапептидов (Р.С de Visser et. al., J. PeptideRes, 2003, 61, 298), в которых аналоги пентаноила и бутаноила демонстрировали заметное снижение активности. Неожиданно было установлено наличие существенной противобактериальной активности совместно со сниженной токсичностью у нанопептидов полимиксинов B, заявленных в настоящем изобретении,включая замещенные нанопептиды с короткими ацильными цепями, в частности цепями, содержащими аминозаместитель. Предположительно токсичность соединений полимиксинов является результатом сурфактантподобного взаимодействия с мембранами эукариотических клеток. Кроме того, нефротоксичность полимиксинов может являться результатом того, что они задерживаются в клетках почек и, таким образом, в большей степени накапливаются, а не выводятся из организма. Не желая быть связанными какой-либо теорией, полагают, что соединения согласно настоящему изобретению содержат группу R5, которая содержит заместитель, который нарушает гидрофобность алкильного компонента. Авторы настоящего изобретения полагают, что данное нарушение изменяет гидрофильно-гидрофобный баланс молекул, что делает их менее подходящими для выстраивания в липидные бислои, образующие мембраны. В свою очередь, данная непригодность для выстраивания в мембраны может приводить к снижению времени нахождения в них и, следовательно, к снижению токсичности. Термин "нанопептид полимиксин", применяемый в настоящем описании, относится к полимиксину В или полимиксину Е с 2-10 аминокислотами. Термин "остаток аминокислоты" (например, остаток лейцина и т.п.), применяемый в настоящем описании, относится к аминокислоте, от которой отняли молекулу воды и которая образует связь с другим фрагментом (например, с другой аминокислотой) через карбонильный конец молекулы, а также образует связь с другим фрагментом (например, с другой аминокислотой) через концевой атом азота. Образуемые связи могут являться, например, амидными связями. Термин "алкил", применяемый в настоящем описании, относится к прямой или разветвленной алкильной цепи, включая, но не ограничиваясь ими, метил, этил, н-пропил, изопропил, бутил, н-бутил и трет-бутил. В одном из вариантов реализации алкил относится к прямой алкильной цепи. Алкил в качестве связующей молекулы (т.е. алкильный заместитель) также относится к фрагментам алкилена, включая изомеры с разветвленными и прямыми цепями. Разветвления могут содержать на концевой алкильный радикал, такой как -CH3. Термин "гетероциклил", применяемый в настоящем описании, относится к насыщенному карбоциклическому кольцу, содержащему по меньшей мере один кольцевой атом азота, например 1 или 2 кольцевых атома азота, к кольцу, содержащему только 1 кольцевой атом азота и возможно дополнительно содержащему гетероатом, выбранный из кислорода и серы. Примеры С 4-6 гетероциклических групп включают азетидин, пирролидинил, пиперидинил, пиперазинил и морфолинил. В одном из вариантов реализации гетероциклическая группа связана с остальной молекулой через атом азота. В термине"С 4-6 гетероциклил" выражение С 4-6 означает общее количество кольцевых атомов, включая атомы углерода и гетероатомы. В одном из вариантов реализации R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток фенилаланина, например D-фенилаланина, или лейцина, например D-лейцина. В одном из вариантов реализации R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток лейцина. В одном из вариантов реализации R3 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток треонина.-2 024792 В одном из вариантов реализации R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток ,-диаминомасляной кислоты (Dab) или серина, например L-Dab или D-Ser. В одном из вариантов реализации X представляет собой -C(=О). В одном из вариантов реализации R5 представляет собой азетидин, пирролидинил или пиперидинил. В одном из вариантов реализации С 2-12 алкильный фрагмент R5 представляет собой С 2 алкил,С 3 алкил, С 4 алкил, С 5 алкил, С 6 алкил, С 7 алкил, С 8 алкил, С 9 алкил, С 10 алкил, С 11 алкил или С 12 алкил. В одном из вариантов реализации С 2-12 алкильный фрагмент R5 представляет собой С 3-10 алкил, например С 4-8 алкил. В одном из вариантов реализации R5 представляет собой С 3-8 циклоалкил, например С 5 циклоалкил или С 6 циклоалкил. В одном из вариантов реализации R5 содержит один заместитель. В одном из вариантов реализации R5 содержит два заместителя. В одном из вариантов реализации R5 содержит три заместителя. В одном из вариантов реализации R5 содержит одну, две или три гидроксильные группы, например одну гидроксильную группу. В одном из вариантов реализации R содержит одну аминогруппу, например С 2-12 алкил, содержащий одну аминогруппу, такой как С 2-4 алкил, содержащий одну аминогруппу. В одном из вариантов реализации R5 содержит одну, две или три гидроксильные группы, например,одну гидроксильную группу. В одном из вариантов реализации R5 содержит одну аминогруппу и одну гидроксильную группу. В одном из вариантов реализации R5 содержит одну аминогруппу и две гидроксильные группы. В одном из вариантов реализации, если R5 содержит одну или более гидроксильные группы, алкильная цепь представляет собой C5-12. В одном из вариантов реализации R5 содержит не более одной аминогруппы. В одном из вариантов реализации, если R5 содержит более одного заместителя, заместители находятся при разных атомах углерода. В одном из вариантов реализации по меньшей мере один R5 заместитель (например, один заместитель) находится в С 2 алкиле, С 3 алкиле, С 4 алкиле, С 5 алкиле, С 6 алкиле, С 7 алкиле, С 8 алкиле, С 9 алкиле,С 10 алкиле, С 11 алкиле или С 12 алкиле. В одном из вариантов реализации по меньшей мере один R5 заместитель (например, один заместитель) находится при концевом атоме углерода прямой алкильной цепи или алкильного разветвления, например прямой алкильной цепи. Если заместитель находится при концевом атоме углерода прямой алкильной цепи (или при концевом атоме углерода разветвления), остальная часть алкильной цепи (или алкильного фрагмента разветвления) образует алкиленовую связь. Таким образом, термин "алкил", применяемый в настоящем описании, представляет собой обобщенный термин, охватывающий случаи, в которых часть или весь алкильный фрагмент представляет собой алкиленовый фрагмент. Термин "концевой атом углерода", применяемый в настоящем описании, относится к атому углерода, который при отсутствии заместителей образовывал бы фрагмент -CH3. В одном из вариантов реализации по меньшей мере один (например, один) заместитель находится не при концевом атоме углерода, т.е. -CH(заместитель)-. В одном из вариантов реализации R6 представляет собой водород. В одном из вариантов реализации R6 представляет собой C1-4 алкила, например, С 1 алкил, С 2 алкил,С 3 алкил или С 4 алкил, например метил. В одном из вариантов реализации R7 представляет собой водород. В одном из вариантов реализации R7 представляет собой C1-4 алкила, например, С 1 алкил, С 2 алкил,С 3 алкил или С 4 алкил, например метил. В одном из вариантов реализации и R6, и R7 представляют собой метил. В одном из вариантов реализации R6 представляет собой Н и R7 представляет собой метил. В одном из вариантов реализации R5 представляет собой -CH(OH)(CH2)5CH3, -CH2NH2,-CH2CH2NH2, -CH2CH2CH2NH2, -(CH2)5NH2, -(CH2)7NH2, -CH2CH2NHCH3, -CH2CH2N(CH3)2 и -(CH2)7 ОН.-3 024792 В одном из вариантов реализации R8 представляет собой метил. В одном из вариантов реализацииR представляет собой водород. В одном из вариантов реализации соединение представляет собой соединение формулы (Ia) 8 или его фармацевтически приемлемую соль. Если R1 (вместе с прилегающими группами) представляет собой фенилаланин, R2 (вместе с прилегающими группами) представляет собой лейцин, R3 (вместе с прилегающими группами) представляет собой треонин, R4 (вместе с прилегающими группами) представляет собой ,-диаминомасляную кислоту и R8 представляет собой метил (и вместе с прилегающими группами представляет собой треонин),соединение формулы (Ia) представляет собой нанопептид полимиксин, содержащий 2-10 аминокислот полимиксина В. Если R1 (вместе с прилегающими группами) представляет собой лейцин, R2 (вместе с прилегающими группами) представляет собой лейцин, R3 (вместе с прилегающими группами) представляет собой треонин, R4 (вместе с прилегающими группами) представляет собой ,-диаминомасляную кислоту и R8 представляет собой метил (и вместе с прилегающими группами представляет собой треонин), соединение формулы (Ia) представляет собой нанопептид полимиксин, содержащий 2-10 аминокислот полимиксина Е. В одном из вариантов реализации соединение формулы (I) имеет три положительных заряда. В одном из вариантов реализации соединение формулы (I) имеет четыре или пять положительных зарядов, например четыре. В одном из вариантов реализации соединение формулы (I) имеет пять положительных зарядов. В одном из вариантов реализации соединение формулы (I) имеет шесть положительных зарядов. В одном из вариантов реализации соединение выбрано из нанопептида 2-гидроксиоктаноилполимиксина B; нанопептида 2-аминоэтаноилполимиксина B; нанопептида 3-аминопропаноилполимиксина B; нанопептида 3-(N,N-иметиламино)пропаноилполимиксина B; нанопептида 4-аминобутаноилполимиксина B; нанопептида 6-аминогексаноилполимиксина B; нанопептида 8-гидроксиоктаноилполимиксина B; нанопептида 8-аминооктаноилполимиксина B; нанопептида 3-(N-метиламино)пропаноилполимиксина B; нанопептида 2-аминоциклопентанкарбонилполимиксина B; нанопептида 3-аминопропаноилколистина (полимиксина Е); нанопептида 3-пирролидин-3-карбонилполимиксина B; нанопептида 3-амино-3-циклогексанпропаноилполимиксина В или из фармацевтически приемлемых солей указанных соединений. Дополнительно или альтернативно, соединение выбрано из нанопептида 5-аминопентаноилполимиксина B; нанопептида гидроксиацетилполимиксина B; нанопептида 3-гидроксиоктаноилполимиксина B; нанопептида 4-(N,N-иметиламино)бутаноилполимиксина B; нанопептида 7-аминогептаноилполимиксина B; нанопептида 4-морфолинилбутаноилполимиксина B; нанопептида 6-гидроксигексаноилполимиксина B; нанопептида 3-гидроксибутаноилполимиксина B; нанопептида 4-(N-метиламино)бутаноилполимиксина B; нанопептида транс-4-аминоциклогексанкарбонилполимиксина B; нанопептида 4-аминобутаноилполимиксина Е; нанопептида 2-гидроксиоктаноилполимиксина Е; нанопептида цис-4-аминоциклогексанкарбонилполимиксина B; нанопептида 4-амино-4-метилпентаноилполимиксина B;B,включая,например,нанопептид 4-(S)-аминопентаноилполимиксин B; нанопептида транс-4-гидроксициклогексанкарбонилполимиксина B; нанопептида 3-гидроксипропаноилполимиксина B; нанопептида (2-гидрокси-2-циклогексил)этаноилполимиксина B; нанопептида 2-аминооктаноилполимиксина В или из фармацевтически приемлемых солей указанных соединений. Примеры солей соединения формулы (I) включают все фармацевтически приемлемые соли, включая, но не ограничиваясь ими, соли присоединения сильных неорганических кислот, такие как соли на основе HCl или HBr, и соли присоединения сильных органических кислот, например соль метансульфоновой кислоты. Другие примеры солей включают сульфаты и ацетаты, такие как трифторацетат или трихлорацетат. В одном из вариантов реализации предложены соединения согласно настоящему описанию в виде сульфата. Также соединение согласно настоящему описанию может быть получено в форме пролекарства. Пролекарства могут содержать противобактериальное соединение, представленное в настоящем описании, в котором одна или более аминогруппы защищены группой, которая может быть удалена in vivo,для высвобождения биологически активного соединения. В одном из вариантов реализации пролекарство представляет собой "аминное пролекарство". Примеры аминных пролекарств включают сульфометил,описанный, например, в Bergen et. al., Antimicrob. Agents and Chemotherapy, 2006, 50, 1953, или HSO3FMOC, описанный, например, в Schechter et. al., J. Med. Chem. 2002, 45(19), 4264, и соли указанных соединений. Другие примеры аминных пролекарств приведены Krise и Oliyai в Biotechnology:Pharmaceutical Aspects, 2007, 5(2), 101-131. В одном из вариантов реализации предложены соединения согласно настоящему описанию в виде пролекарства. Настоящее описание охватывает сольваты соединений формулы (I). Примеры сольватов включают гидраты. Соединения согласно настоящему изобретению включают соединения, в которых указанный атом заменен природным или искусственным изотопом. В одном из вариантов реализации изотоп является стабильным. Таким образом, соединения согласно настоящему изобретению включают, например, соединения, содержащие дейтерий, и т.п. В настоящем изобретении предложены соединения, содержащие 2-10 аминокислот полимиксина B,или их производные, описанные ниже, в которых концевой N аминокислоты, соответствующей остатку 2 в полимиксине B, модифицирован группой R5-Х-. Примеры R5 и X описаны выше. В соединениях согласно настоящему изобретению отсутствует остаток 1 полимиксина В. Вариант соединения представляет собой соединение, в котором одна или более, например от 1 до 5,например, 1, 2, 3 или 4, аминокислоты замещены другой аминокислотой. Аминокислота находится в положении, выбранном из 2, 3, 6, 7 или 10 (по нумерации остатков в полимиксине В). Замещение может обеспечиваться при помощи другой аминокислоты или стереоизомера. В положении 2 вариант может содержать заместитель D-Ser. В положении 3 вариант может содержать заместитель Ser. В положении 6 вариант может содержать заместитель Leu или Val. В положении 7 вариант может содержать заместитель Не, Phe, Thr, Val или Nva (норвалин). В положении 10 вариант может содержать заместитель Leu. Полимиксин Е представляет собой полимиксин В с заместителем Leu в положении 6. Для удобства соединения согласно настоящему изобретению представляют формулой (I), где аминокислоты в положениях 2, 3, 6, 7 ил 10 определяются природой групп R8, R4, R1, R2 и R3, соответственно. Соединения согласно настоящему изобретению, включающие примеры, описанные выше, являются биологически активными. Соединения формулы (I) могут быть получены путем обычного синтеза пептидов с применением способов, известных специалистам в данной области. Подходящие способы включают жидкофазный синтез, описанный в Yamada et. al., J. Peptide Res. 64, 2004, 43-50, или твердофазный синтез, описанный вde Visser et. al., J. Peptide Res, 61, 2003, 298-306 и Vaara et. al., Antimicrob. Agents and Chemotherapy, 52,2008. 3229-3236. Указанные способы включают подходящие способы введения защиты и стадию циклизации. Альтернативно, соединения могут быть получены из легкодоступных полимиксинов, например,путем удаления N-концевой аминокислоты полимиксина (остаток 1). Указанный способ приведен в настоящем описании для получения соединений на основе остатков 2-10 полимиксинов В и Е. В настоящем изобретении также предложен способ получения конкретного соединения формулы (I) путем взаимодействия соединения формулы (II) или его защищенного производного,где R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток фенилаланина,лейцина или валина;R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток лейцина, изолейцина,фенилаланина, треонина, валина или норвалина;R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток треонина или лейцина;R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляют собой остаток ,-диаминомасляной кислоты или серина; с соединением формулы (III) или его защищенным производным,где R5 определен выше для соединений формулы (I);X1 представляет собой группу, которая после присоединения к соединениям формулы (II) превращается или может быть превращена в -NHC(O)-, -C(O)-, -OC(O)-, -CH2- или -SO2;L представляет собой уходящую группу;m равняется 0 или 1,или его фармацевтически приемлемой солью,возможно с последующим снятием защиты с получением соединения формулы (I). В целом, соединения формулы (II) применяют в форме, в которой все свободные аминогруппы,участие которых в целевой реакции нежелательно, защищены подходящей защитной группой, например,трет-бутилоксикарбонильной (Вос), 9-флуоренилметоксикарбонильной (FMOC) или других групп, подходящих для защиты аминогруппы, таких как группы, описанные в "Protective groups in OrganicSynthesis" by Theodora W. Green and Peter G.M. Wuts, Wiley, New York, 1999. Химические реакции, требуемые для снятия защиты с получением соединения формулы (I), могут быть проведены с применением стандартных способов, таких как способы, описанные в "Protectivegroups in Organic Synthesis" by Theodora W. Green and Peter G. M. Wuts, Wiley, New York, 1999. Соединения формулы (I), в которых X представляет собой -NHC(=O)-, могут быть получены с применением соединения формулы (III), которое соответствует изоцианату, такому как где R5 определен выше. Реакция может быть проведена в подходящем растворителе, таком как дихлорметан, возможно в присутствии основания, такого как триэтиламин или N-этилдиизопропиламин (ДИПЭА). Альтернативно, соединения формулы (I), в которых X представляет собой -NHC(=O)-, могут быть получены с применением соединения формулы (IIIb) где R5 определен выше,в присутствии основания, описанного в Gallon et. al., J. Org. Chem., 2005, 70, 6960. Соединения формулы (I), в которых X представляет собой -C(=О)-, -OC(=О)- или -SO2-, могут быть получены с применением соединения формулы (III), где R5 описано выше, X1 представляет собой-C(=О)-, -OC(=О)- или -SO2- и L представляет собой уходящую группу, например, Cl или Br. Реакция может быть проведена в подходящем растворителе, таком как полярный апротонный растворитель, например дихлорметан. Соединения формулы (I), в которых X представляет собой -C(=О)-, могут быть получены с применением соединения формулы (IIIc) где R5 определен выше, например, в присутствии связующего агента, такого как HATU(гексафторфосфат бензотриазол-1-илокси-трис-пирролидинофосфония), в щелочных условиях в полярном растворителе. Соединения формулы (I), в которых X представляет собой -CH2-, могут быть получены с применением соединения формулы (IIId): где R5 определен выше, например, в присутствии восстановителя, такого как триацетоксиборгидрид натрия, цианоборгидрид натрия или цианоборгидрид на полимерной подложке, в растворителе, таком как метанол, дихлорметан, ДМФ, в условиях, описанных в March's Advanced Organic Chemistry, Wiley,2001. В одном из аспектов настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I), например терапевтически эффективное количество указанного соединения, и фармацевтически приемлемое вспомогательное вещество, разбавитель и/или носитель (включая комбинации указанных соединений). Способы введения (доставки) включают, но не ограничиваются ими, один или более из: перорального (например, в виде сухого порошка/сыпучего мелкодисперсного состава, таблетки, капсулы или в виде раствора или суспензии для проглатывания), буккального, подъязычного. Композиции согласно настоящему изобретению включают, композиции в форме, особенно подходящей для парентерального, перорального, буккального, ректального, местного, имплантированного,офтальмологического, назального, ректального или мочеполового применения. В одном из аспектов настоящего изобретения агенты доставляют перорально, поэтому агент находится в форме, подходящей для пероральной доставки. В некоторых случаях может быть возможна доставка соединений согласно настоящему изобретению при помощи местного, парентерального (например, при помощи инъекции) или трансдермального способа, включая мукозный (например, при помощи назального спрея или аэрозоля для ингаляции), назальный, гастроинтестинальный, интраспинальный, внутрибрюшинный, внутримышечный, внутривенный, внутриматочный, внутриглазной, внутрикожный, внутричерепной, внутритрахеальный, внутривагинальный, интрацеребровентрикулярный, внутримозговой, подкожный, офтальмологический (включая,интравитреальный или внутрикамерный). В зависимости от различных систем доставки или различных способов введения к композиции/составу могут предъявляться различные требования. Например, фармацевтическая композиции согласно настоящему изобретению может быть получена в форме для доставки с применением мининасоса или при помощи мукозного способа, например, в виде назального спрея, аэрозоля для ингаляции или раствора для проглатывания, или при помощи парентеральных способов, для которых композицию получают в форме для инъекции, например, внутривенного, внутримышечного или подкожного способа. Альтернативно, состав может быть получен в форме для обоих способов доставки. При необходимости,фармацевтические композиции могут быть введены путем ингаляции, в виде суппозитория или пессария,местно в виде лосьона, раствора, крема, мази или присыпки, при помощи кожного пластыря, перорально в виде таблеток, содержащих вспомогательные вещества, такие как крахмал или лактоза, или в виде капсул или вагинальных суппозиториев, индивидуально или в смеси с вспомогательными веществами, или в виде эликсиров, растворов или суспензий, содержащих ароматизаторы или красители, а также они могут быть введены парентерально, например внутривенно, внутримышечно или подкожно. Для парентерального введения наиболее подходят композиции в виде стерильных водных растворов, которые могут содержать другие вещества, например количества солей или сахаридов, в частности моносахарида, достаточные для получения раствора изотонического крови. Примеры парентерального введения включают один или более из: внутривенное, внутриартериальное, внутрибрюшинное, интратекальное, внутрижелудочковое, внутриуретральное, внутригрудинное, внутричерепное, внутримышечное или подкожное введение и/или введение путем инфузии. В одном из вариантов реализации состав соединений согласно настоящему изобретению получен в виде липосомного состава. Липосомы могут варьироваться в размерах от нескольких до десятков микрометров, однослойные липосомы совместно с различными лигандами для целевой доставки, присоединенными к их поверхности, как правило, имеют размер, относящийся к нижнему диапазону, и за счет этого поверхностного присоединения способны накапливаться в участках патологии для лечения заболевания. Липосомы представляют собой искусственно полученные пузырьки, состоящие из липидного бислоя. В одном из вариантов реализации состав адаптирован для доставки при помощи инфузии или медленной инъекции. В одном из вариантов реализации состав адаптирован для доставки при помощи болюсной инъекции. Для буккального или подъязычного введения композиции могут быть получены в виде таблеток или пастилок, полученных при помощи обычных способов. Соединения согласно настоящему могут быть введены (например, перорально или местно) в виде таблеток, капсул, вагинальных суппозиториев, эликсиров, растворов или суспензий, которые могут содержать ароматизаторы или красители, с немедленным, отсроченным, модифицированным, замедленным, прерывистым или контролируемым высвобождением. Для лечения людей и применения в ветеринарии соединения согласно настоящему также могут быть получены в форме, подходящей для перорального или буккального введения, например в форме растворов, гелей, сиропов, жидкостей для полоскания ротовой полости или суспензий, а также в виде сухого порошка для смешивания с водой или другим подходящим носителем перед применением, возможно с ароматизаторами и красителями. Также могут быть применены твердые композиции, такие как таблетки, капсулы, леденцы, пастилки, драже, порошки, пасты, гранулы, пилюли, или предварительно приготовленные смеси. Твердые и жидкие композиции для перорального применения могут быть получены при помощи способов, хорошо известных в данной области. Такие композиции также могут содержать один или более фармацевтически приемлемые носители и вспомогательные вещества, которые могут находиться в твердой или жидкой форме. Таблетки могут содержать вспомогательные вещества, такие как микрокристаллическая целлюлоза,лактоза, цитрат натрия, карбонат кальция, сульфат кальция, двухосновный фосфат кальция, глицин, маннит, прежелатинированный крахмал, кукурузный крахмал, картофельный крахмал, разрыхлители, такие как крахмалгликолят натрия, кроскармеллоза натрия и некоторые комплексные силикаты, и связующие для гранулирования, такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (ГГГМЦ), гидроксипропилцеллюлоза (ГПЦ), сахароза, желатин и гуммиарабик. Кроме того, могут быть добавлены смазочные агенты, такие как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Твердые композиции подобного вида также могут быть введены в капсулах из желатина или ГПМЦ(гидроксипропилметилцеллюлозы). Подходящие вспомогательные вещества включают микрокристаллическую целлюлозу, лактозу, карбонат кальция, сульфат кальция, двухосновный фосфат кальция, маннит,прежелатинированный крахмал, кукурузный крахмал, картофельный крахмал и высокомолекулярные полиэтиленгликоли. В случае водных суспензий и/или эликсиров агент может быть смешан с различными подсластителями или ароматизаторами, пигментами или красителями, эмульгаторами и/или суспендирующими агентами и разбавителями, такими как вода, этанол, пропиленгликоль и глицерин, и комбинациями указанных соединений. Капсулы могут быть заполнены порошком (только лекарственным средством или в смеси с выбранным(и) наполнителем(ями или жидкостью, каждый из которых содержат одну или более соли согласно настоящему изобретению и возможно носитель. Если капсула заполнена порошком, соли согласно настоящему изобретению и/или носитель могут быть измельчены вплоть до микрочастиц с получением материала с соответствующим размером частиц. Альтернативно, при необходимости, таблетка или капсула может быть помещена в другую капсулу(предпочтительно капсулу из ГПМЦ, такую как Capsugel) с получением составов вида таблетки в капсуле или капсулы в капсуле, которые при введении пациенту приводят контролируемое растворение в желудочно-кишечном тракте, что обеспечивает действие, аналогичное кишечно-растворимому покрытию. Таким образом, в одном из аспектов настоящего изобретения предложен твердый состав соли соединения согласно настоящему изобретению, например состав с кишечно-растворимым покрытием. В другом аспекте настоящего изобретения предложен твердый состав, содержащий защитную капсулу в качестве наружного слоя, например состав вида таблетки в капсуле или капсулы в капсуле. Кишечно-растворимое покрытие может обеспечивать улучшенный профиль стабильности по сравнению с составами без покрытия. В ветеринарии соединения согласно настоящему изобретению также могут быть введены перорально в виде жидких лекарственных форм, таких как раствор, суспензия или дисперсия активного ингредиента с фармацевтически приемлемым носителем или вспомогательным веществом. Для лечения людей и применения в ветеринарии соединения согласно настоящему также могут быть получены в виде суппозиториев или пессариев, например, содержащих обычные суппозиторные основы. В одном из вариантов реализации предложен состав для местного введения, включая состав для ингаляции. Подходящие средства для ингаляции включают ингалируемые порошки, дозированные аэрозоли,содержащие газы-пропелленты, или ингалируемые растворы, не содержащие газы-пропелленты. Ингалируемые порошки согласно настоящему изобретению, содержащие активное соединение, могут состоять исключительно из описанных выше активных соединений или из смеси описанных выше активных соединений с физиологически приемлемым вспомогательным веществом. Указанные ингалируемые порошки могут содержать моносахариды (например, глюкозу или арабинозу), дисахариды (например, лактозу, сахарозу, мальтозу), олиго- и полисахариды (например, декстраны), многоатомные спирты (например, сорбит, маннит, ксилит), соли (например, хлорид натрия, карбонат кальция) или смеси указанных соединений друг с другом. Моно- и дисахариды, в частности, но не исключительно, лактозу или глюкозу предпочтительно применяют в виде гидратов. Для осаждения в легких требуются частицы размером меньше 10 мкм, например 1-9 мкм, предпочтительно 0,1-5 мкм, особенно предпочтительно 1-5 мкм. Газы-пропелленты, которые могут быть применены для получения ингалируемых аэрозолей, известны в данной области. Подходящие газы-пропелленты выбирают из углеводородов, таких как н-пропан, н-бутан или изобутан, и галогенированных углеводородов, таких как хлорированные и/или фторированные производные метана, этана, пропана, бутана, циклопропана или циклобутана. Описанные выше газы-пропелленты могут быть применены индивидуально или в смесях. Наиболее подходящие газы-пропелленты представляют собой галогенированные производные алканов, выбранные из TG11, TG12, TG134 а и TG227. Из перечисленных выше галогенированных углеводородов для применения в составах согласно настоящему изобретению подходят TG134a(1,1,1,2-тетрафторэтан) и TG227 (1,1,1,2,3,3,3-гептафторпропан) и их смеси. Ингалируемые аэрозоли, содержащие газы-пропелленты, также могут содержать другие ингредиенты, такие как сорастворители, стабилизаторы, поверхностно-активные вещества (ПАВ), антиоксиданты,смазочные агенты и рН-регуляторы. Все указанные ингредиенты известны в данной области. Ингалируемые аэрозоли, содержащие газы-пропелленты, согласно настоящему изобретению могут содержать вплоть до 5 мас.% активного соединения. Например, аэрозоли согласно настоящему изобретению могут содержать от 0,002 до 5 мас.%, от 0,01 до 3 мас.%, от 0,015 до 2 мас.%, от 0,1 до 2 мас.%, от 0,5 до 2 мас.% или от 0,5 до 1 мас.% активного соединения. Соли согласно настоящему изобретению также могут быть применены в сочетании с другими терапевтическими агентами. Таким образом, в другом аспекте настоящего изобретения предложена композиция, содержащая соль согласно настоящему изобретению вместе с дополнительным терапевтическим агентом. Например, композиция может представлять собой композицию соли соединения формулы (I) и антибиотика, такого как ванкомицин, фосфомицин, рифамицин, бета-лактам (например, цефалоспорин или карбапенем), аминогликозид, макролид, тетрациклин, липопептид или оксазолидинон, и/или противовоспалительного агента, такого как стероид. Композиции могут быть получены в виде объединенного состава или упакованных вместе отдельных составов для одновременного или последовательного введения. В одном из вариантов реализации предложены соли согласно настоящему изобретению в сочетании с дополнительным терапевтическим агентом. Следует понимать, что необязательно вводить все соединения/соли из композиции при помощи одного способа. Таким образом, если лечение включает применение нескольких активных компонентов,указанные компоненты могут быть введены различными способами. Индивидуальные компоненты указанных композиций могут быть введены раздельно, последовательно или одновременно или в виде объединенных фармацевтических составов при помощи любого подходящего способа. При последовательном введении сперва может быть введена как соль согласно настоящему изобретению, так и дополнительный терапевтический агент. При одновременном введении соединения могут быть введены в одной и той же или в различных фармацевтических композициях. Описанные выше композиции могут быть получены в форме фармацевтического состава, подходящего для применения, таким образом, фармацевтические составы, содержащие композиции, описанные выше, вместе с фармацевтически приемлемым носителем или вспомогательным веществом составляют другой аспект настоящего изобретения. Следует понимать, что при объединении в одном составе два соединения/соли должны являться устойчивыми и совместимыми друг с другом и с другими компонентами состава. При получении в виде отдельных составов они могут быть получены в виде любого подходящего состава, известного для указанных соединений в данной области. Композиции могут содержать 0,01-99% активного соединения. Например, композиция для местного применения, как правило, содержит 0,01-10%, предпочтительно 0,01-1%, активного соединения. В случае применения соли согласно настоящему изобретению в сочетании с дополнительным терапевтическим агентом, активным против того же заболевания, дозы каждого из соединения/соли могут быть аналогичными или отличными от доз, применяемых в случае индивидуального использования соединения/соли. Соответствующие дозы могут быть легко определены специалистами в данной области. Также следует понимать, что количество соли согласно настоящему изобретению, требуемое для лечения, варьируется в зависимости от природы состояния, подлежащего лечению, возраста и состояния пациента и определяется лечащим врачом или ветеринаром. Как правило, врач определяет дозировку, наиболее подходящую для конкретного пациента. Конкретные дозы и частота дозирования для любого конкретного пациента могут изменяться и зависят от различных факторов, включающих активность конкретной применяемой соли, метаболическую устойчивость и продолжительность действия указанной соли, возраст, массу тела, общее состояние здоровья,пол, диету, режим и время введения, скорость выведения, сочетание лекарственных средств, серьезность конкретного состояния и особенности конкретного пациента, проходящего лечение. В случае перорального или парентерального введения людям суточная доза агента может быть представлена одной или несколькими дозами. В случае системного введения суточная доза при лечении взрослого человека находится в диапазоне 2-100 мг/кг массы тела, например 5-60 мг/кг массы тела, и может быть введена при помощи 1-4 ежедневных доз в зависимости, например, от способа введения и состояния пациента. Если композиция представлена в виде лекарственных форм, каждая лекарственная форма предпочтительно содержит от 100 мг до 1 г активного ингредиента. Продолжительность лечения определяется ответной реакцией, а не составляет произвольное количество дней. В одном из вариантов реализации лечение продолжают в течение 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,14, 15, 16, 17, 18, 19, 20, 21 или более дней. Как описано выше, соли согласно настоящему изобретению могут быть применены для лечения или профилактики у людей и/или животных. В настоящем изобретении также предложен способ получения фармацевтической композиции, который включает смешивание соли согласно настоящему изобретению или его фармацевтически приемлемого производного с фармацевтически приемлемым вспомогательным веществом, разбавителем и/или носителем. В другом аспекте настоящего изобретения предложено соединение формулы (I), его фармацевтически приемлемая соль или композиция, содержащее указанное соединение, для применения для лечения, в частности,лечения инфекции, такой как бактериальная инфекция, например инфекция, вызванная грамотрицательными бактериями. В одном из вариантов реализации соединения и композиции согласно настоящему изобретению подходят для применения для лечения пневмонии, инфекций мочевых путей, раневых инфекций, ушных инфекций, глазных инфекций, внутрибрюшинных инфекций, а также чрезмерного бактериального роста и/или сепсиса. В одном из вариантов реализации соединения подходят для применения для лечения инфекций, вызванных бактериями, устойчивыми ко многим видам лекарственных средств. Примеры грамотрицательных бактерий включают, но не ограничиваются ими, Escherichia spp.,Klebsiella spp., Enterobacter spp., Salmonella spp., Shigella spp., Citrobacter spp., Morganella morganii,Yersinia pseudotuberculosis и другие энтеробактерии, Pseudomonas spp., Acinetobacter spp., Moraxella,Helicobacter, Stenotrophomonas, Bdellovibrio, уксуснокислые бактерии, Legionella и альфапротеобактерии, такие как Wolbachia и многие другие. Другие известные группы грамотрицательных бактерий включают цианобактерии, спирохеты, зеленые серные и несерные бактерии. Важные с медицинской точки зрения грамотрицательные бактерии кокки включают три вида, которые вызывают заболевания, передающиеся половым путем (Neisseria gonorrhoeae), менингит (Neisseriameningitidis) и респираторные симптомы (Moraxella catarrhalis). Важные с медицинской точки зрения грамотрицательные палочковидные бактерии включают множество видов. Некоторые из них вызывают респираторные затруднения (Hemophilus influenzae, KlebsiellaAcinetobacter baumannii, которые приводят к бактериемии, вторичному менингиту и вентиляторноассоциированной пневмонии, приобретаемым в отделениях интенсивной терапии больничных учреждений. В одном из вариантов реализации соединения и композиции согласно настоящему изобретению подходят для применения для лечения инфекции, вызванной одной или более из следующих грамотрицательных бактерий: Е.coli, S.enterica, Klebsiella: K.pneumoniae, K.oxytoca; Enterobacter: E.cloacae,E.aerogenes, E.agglomerans, Acinetobacter: A.calcoaceticus, A.baumannii; Pseudomonas aeruginosa,Stenotrophomonas maltophila, Providencia stuartii, Proteus, P.mirabilis, P.vulgaris. В одном из вариантов реализации соединения формулы (I), их фармацевтически приемлемые соли или композиции, содержащие указанные соединения, подходят для лечения Pseudomonas инфекций,включая P.aeruginosa инфекции, например кожные инфекции, инфекции мягких тканей, желудочнокишечные инфекции, инфекции мочевых путей, пневмонию и сепсис. В одном из вариантов реализации соединения формулы (I), их фармацевтически приемлемые соли или композиции, содержащие указанные соединения, подходят для лечения Klebsiella инфекций, вклю- 10024792 чая K. pneumoniae инфекции, например пневмонию, инфекции мочевых путей, менингит и сепсис. В одном из вариантов реализации соединения формулы (I), их фармацевтически приемлемые соли или композиции, содержащие указанные соединения, подходят для лечения Е. coli инфекций, включая Е.coli инфекции, например бактериемию, холецистит, холангит, инфекции мочевых путей, неонатальный менингит и пневмонию. В одном из вариантов реализации соединения формулы (I), их фармацевтически приемлемые соли или композиции, содержащие указанные соединения, подходят для длительного лечения. В одном из аспектов настоящего изобретения предложено соединение формулы (I) или композиция,содержащая указанное соединение, для получения лекарственных средств для лечения одного или более состояний, описанных выше. В одном из аспектов настоящего изобретения предложен способ лечения, включающий стадию введения терапевтически эффективного количества соединения формулы (I), его фармацевтически приемлемой соли или композиции, содержащей указанное соединение, пациенту (человеку или животному),нуждающемуся в данном лечении, например для лечения инфекций, описанных выше. При технической возможности варианты реализации могут быть объединены и, таким образом, настоящее изобретение охватывает все изменения/сочетания вариантов реализации, описанных в настоящем описании. При технической возможности преимущества, указанные для соединений формулы (I), в равной степени могут относиться и к другим соединениям согласно настоящему изобретению. В описании использованы следующие обозначения:Dab - ,-диаминомасляная кислота; ДИПЭА - N,N-диизопропилэтиламин. Примеры Промежуточное соединение 1. Нанопептид полимиксин В. Смесь полимиксина В (20 г), иммобилизованного папаина (185 ELU/г), калий-фосфатного буфера(25 мМ; рН 7, 1,25 л), хлорида калия (30 мМ), ЭДТА (10 мМ) и цистеина (1 мМ) инкубировали при 37C в течение 18 ч при осторожном перемешивании. Протекание реакции отслеживали при помощи ЖХ-МС в условиях, описанных в табл. 1. Иммобилизованный папаин удаляли при помощи фильтрования и фильтрат концентрировали в вакууме с получением твердого остатка, который повторно суспендировали в 10% водном растворе метанола и выдерживали при комнатной температуре в течение ночи. Надосадочную жидкость декантировали и концентрировали в вакууме. Нанопептид полимиксин В очищали от примесей при помощи ТФЭ (твердофазной экстракции) на колонке с силикагелем С 18, элюируя 0-10% водными растворами метанола. В результате выпаривания соответствующих фракций получали продукт в виде белого твердого вещества. Селективное введение Boc-защиты свободных у-аминогрупп Dab остатков нанопептида полимиксина В проводили с применением способа Н. O'Dowd et. al., Tetrahedron Lett., 2007, 48, 2003-2005. Нанопептид полимиксин В (промежуточное соединение 1 7,5 г, 7,78 ммоль) суспендировали в воде (65 мл) при обработке ультразвуком. Добавляли диоксан (65 мл) и триэтиламин (65 мл) и смесь охлаждали во льду в течение 10 мин перед добавлением 1-(Boc-оксиимино)-2-фенилацетонитрила (Вос-ON) (7,67 г; 31,15 ммоль). Протекание реакции отслеживали при помощи ЖХ-МС и реакция завершалась в течение 30 минут, после чего реакцию гасили путем добавления 20% раствора метанольного аммиака (50 мл). Жидкую фазу декантировали и твердый остаток очищали при помощи хроматографии (0-20% растворы метанола в дихлорметане в качестве элюента) на силикагеле с получением нанопептида тетра-(Boc)полимиксина В в виде белого твердого вещества (2,5 г, 24%). ТСХ, Rf 0,2 (10% раствор метанола в дихлорметане).m/z 1362,8 [МН]+. Промежуточное соединение 3. Нанопептид колистин (полимиксин Е). Колистин (полимиксин Е, 5 г) обрабатывали иммобилизованным папаином (185 ELU/г), калийфосфатным буфером (25 мМ; рН 7, 1,25 л), хлоридом калия (30 мМ), ЭДТА (10 мМ) и цистеином (1 мМ) при 37C в течение 32 ч при осторожном перемешивании с получением нанопептида колистина (полимиксина Е). Протекание реакции отслеживали при помощи ЖХ-МС в условиях, описанных для Промежуточного соединения 1 в табл. 1. Иммобилизованный папаин удаляли при помощи фильтрования и фильтрат концентрировали в вакууме с получением твердого остатка, который повторно суспендировали в 10% водном растворе метанола и выдерживали при комнатной температуре в течение ночи. Надосадочную жидкость декантировали и концентрировали в вакууме. Нанопептид колистин (полимиксин Е) очищали от примесей при помощи ТФЭ на колонке с силикагелем С 18 (10 г), элюируя 0-25% водными растворами метанола. В результате выпаривания соответствующих фракций получали продукт в виде Нанопептид колистин (полимиксин Е) (2,5 г, 2,69 ммоль) суспендировали в воде (35 мл) при обработке ультразвуком. Добавляли диоксан (35 мл) и триэтиламин (35 мл) и смесь охлаждали во льду в течение 10 мин перед добавлением 1-(Boc-оксиимино)-2-фенилацетонитрила (Boc-ON) (2,65 г; 10,76 ммоль). Протекание реакции контролировали при помощи ЖХ-МС и реакция завершалась в течение 10 минут, после чего реакцию гасили путем добавления 20% раствора метанольного аммиака (25 мл). Жидкую фазу декантировали, твердый остаток повторно растворяли в воде и последовательно экстрагировали дихлорметаном и изобутанолом. Основываясь на результатах ЖХ-МС, декантированную жидкость и экстракты на основе дихлорметана и изобутанола объединяли, а затем концентрировали в вакууме с получением желтой смолы, которую загружали в колонку для флэш-хроматографии (Si 60 А - 35-70). Содержимое колонки элюировали 0-20% растворами метанола (с 2% аммиака) в дихлорметане. Из фракций, элюированных 7-10% растворами метанола (с 2% аммиака) в дихлорметане, получали нанопептид тетра-(Boc)-колистин (полимиксин Е) в виде белого твердого вещества (1,18 г, 33%). 2-Гидроксиоктановую кислоту (1,16 г, 7,34 ммоль) растворяли в дихлорметане (2 мл). Затем к реакционной смеси добавляли N,N-диизопропилэтиламин (1,19 мл, 7,34 ммоль) и гексафторфосфат 2-(1H-7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) (2,79 г, 7,34 ммоль). После перемешивании при комнатной температуре в течение 30 мин добавляли промежуточное соединение 2 (2,0 г,1,47 ммоль). Через 16 ч при помощи ЖХ-МС устанавливали завершение реакции, реакционную смесь выпаривали досуха и очищали при помощи колоночной хроматографии на силикагеле (0-10% растворы метанола в дихлорметане в качестве элюента). Соответствующие фракции концентрировали с получением нанопептида [2-(R,S)-2-гидроксиоктаноил][тетра-(Boc)]полимиксина В в виде бесцветной маслянистой жидкости (1,28 г, 58%). ТСХ, Rf 0,6 (10% раствор МеОН в дихлорметане).b) Титульное соединение: трифторацетат нанопептида [2-(R,S)-2-гидроксиоктаноил][тетра(Boc)]полимиксина В. Нанопептид [2-(R,S)-2-гидроксиоктаноил][тетра-(Boc)]полимиксин В (1,28 г, 0,85 ммоль) растворяли в дихлорметане (2 мл). Добавляли трифторуксусную кислоту (3,9 мл, 51,02 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч, после чего при помощи ЖХ-МС устанавливали завершение реакции. Реакционную смесь концентрировали в вакууме с получением трифторацетата нанопептида [2-(R,S)-2-гидроксиоктаноил]-полимиксина В в виде бесцветной маслянистой жидкости (1,3 г, 93%). ТСХ, Rf на исходном уровне (10% раствор МеОН в дихлорметане). К соединению из примера 1 (1,3 г) добавляли воду (1 мл) и смесь обрабатывали ультразвуком в течение 5 мин. К полученной суспензии добавляли 1 М раствор NaHCO3 (20 мл) до рН 9. Затем смесь пропускали через 10 г колонку С 18 SPE, последовательно элюируя 0, 40, 50, 60, 70, 80 и 100% водными растворами метанола. Данные ЖХ-МС для каждой фракции показали, что целевой продукт содержался во фракциях, элюированных 60, 70 и 80% водными растворами метанола. Указанные фракции объединяли и выпаривали с получением белого твердого вещества (0,5 г), к которому добавляли 0,1 М раствор H2SO4(30 мл) до рН 7. Добавляли трет-бутанол (10 мл), смесь перемешивали при комнатной температуре в течение 16 ч и лиофилизировали с получением сульфата нанопептида [2-(R,S)-2-гидроксиоктаноил]полимиксина В в виде белого твердого вещества (0,52 г). По результатам проведения ВЭЖХ в соответствии с условиями, приведенными в табл.2, определили время удерживание, равное 5,93 мин. Сульфат нанопептида 2-аминоэтаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 2-(трет-бутоксикарбониламино)уксусной кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,99 мин. Сульфат нанопептида 3-аминопропаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 3-(трет-бутоксикарбониламино)пропионовой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,97 мин. Сульфат нанопептида 3-(N,N-диметиламино)пропаноилполимиксина В получали из нанопептида тетра-(Boc)-полимиксина В и 3-(N,N-диметиламино)пропионовой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 5,01 мин. Сульфат нанопептида 4-аминобутаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 4-(трет-бутоксикарбониламино)бутановой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,97 мин. Сульфат нанопептида 6-аминогексаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 6-(трет-бутоксикарбониламино)гексановой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,97 мин. Сульфат нанопептида 8-гидроксиоктаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 8-гидроксиоктановой кислоты в соответствии с последовательными реакциями, опи- 16024792 Сульфат нанопептида 8-аминооктаноилполимиксина В получали из нанопептида тетра-(Boc)полимиксина В и 8-(трет-бутоксикарбониламино)октановой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 5,02 мин. Сульфат нанопептида 3-(N-метиламино)пропаноилполимиксина В получали из нанопептида тетра(Boc)-полимиксина В и N-[(1,1-диметилэтокси)карбонил]-N-метилаланина в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 5,0 мин. Сульфат нанопептида (1R,S/2R,S)-2-аминоциклопентанкарбонилполимиксина В получали из нанопептида тетра-(Boc)-полимиксина В и цис-1-(трет-бутоксикарбониламино)циклопентанкарбоновой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 5,07 мин. Сульфат нанопептида 3-аминопропаноилколистина (полимиксина Е) получали из нанопептида тетра-(Boc)-колистина (полимиксина Е) (промежуточного соединения 4) и -Bocаланина в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,98 мин. Сульфат нанопептида [3-(R,S)-пирролидин-3-карбонил]полимиксина В получали из нанопептида тетра-(Boc)-полимиксина В и 3-(N-трет-бутоксикарбонил)пирролидинкарбоновой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 4,91 мин. Сульфат нанопептида [3-(R,S)-3-амино-3-циклогексанпропаноил]полимиксина В получали из нанопептида тетра-(Boc)-полимиксина В и 3-(трет-бутоксикарбониламино)-3-циклогексанпропионовой кислоты в соответствии с последовательными реакциями, описанными в примерах 1 и 2. Время удерживания (ВЭЖХ) 5,24 мин.m/z 1116,78, [МН]+. Дополнительные примеры 15-35 Дополнительные соединения из примеров 15-35 получали с применением способов получения, описанных выше в примерах 1 и 2. Таким образом, соединение, содержащее заместитель при концевом N нанопептида полимиксина B, получали из нанопептида тетра-(Boc)-полимиксина В (промежуточное соединение 2) и соответствующей карбоновой кислоты в присутствии связующего агента (например,HATU) и основания (например, ДИПЭА) (как описано в примере 1 а) с последующей обработкой кислотой (например, ТФК) (как описано в примере 1 а) и получением соответствующего продукта (как описано в примере 2). Аналогично, соединение, содержащее заместитель при концевом N нанопептида полимиксина Е, получали из нанопептида тетра-(Boc)-колистина (полимиксина Е) (промежуточное соединение 4) и соответствующей карбоновой кислоты в присутствии связующего агента (например, HATU) и основания (например, ДИПЭА) (как описано в примере 1b) с последующей обработкой кислотой (например,ТФК) (как описано в примере 1b) и превращением в сульфат (как описано в примере 2). Дополнительные соединения из примеров 15-35 приведены в табл. 3. Время удерживания и значения масс, приведенные в таблице, получали с применением условий проведения ЖХ-МС, описанных выше в табл. 2. Соединения выделяли в виде сульфатов приведенных соединений. Таблица 3 Противобактериальная активность. Для оценки эффективности и спектра действия соединений исследование противобактериальной чувствительности проводили против четырех штаммов каждого из четырех грамотрицательных патогенов, Escherichia coli, Pseudomonas aeruginosa, Klebsiellapneumoniae и Acinetobacter baumannii. За день до исследования 3-4 колонии отбирали с планшета со свежим агаром Мюллера-Хинтона(МХА) и переносили в 10 мл катион-регулируемую среду Мюллера-Хинтона (СаМНВ). Культуры инкубировали при 37C и 250 об/мин в течение 18-20 ч, после чего разбавляли в 100 раз свежим СаМНВ. Субкультуры продолжали выращивать до достижения OD600 0,2-0,3, соответствующего 10-10 КОЕ/мл. Активно растущие культуры разбавляли в 100 раз в свежей среде и применяли в качестве инокулята. Определение МИК (минимальной ингибирующей концентрации) проводили путем двукратных последовательных разбавлений антибиотиков в СаМНВ в стерильных 96-луночных микротитрационных планшетах при общем объеме 170 мкл (150 мкл питательной среды, содержащей противомикробный агент, 20 мкл инокулята). Исследования дублировали. Планшеты инкубировали в аэробных условиях без встряхивания в течение 18-20 ч при 37C и МИК определяли как минимальную концентрацию лекарственного средства, которая подавляет заметный рост культур. В табл. 4 приведены значения МИК (мкг/мл) для примеров 2-14 в сравнении с полимиксином В(РМВ). Дополнительные исследования противобактериальной активности. В табл. 4 А приведены значения МИК, полученные для соединений из дополнительных примеров 15-35, а также из примеров 2, 6 и 14. Данные получали в условиях, аналогичных условиям из табл.4 за исключением того, что применяли различные партии катион-регулируемой среды Мюллера-Хинтона. Значения МИК для указанных соединений сравнивали со значениями, полученными для полимиксина B,сульфата колистина, СВ-182,804 и NAB739 (в виде ТФК соли). СВ-182,804 представляет собой производное декапептида полимиксина с арилмочевиной в качестве заместителя при концевом N, который имеет более низкую токсичность по сравнению с полимиксином В (соединение 5 в международной заявке на патент 2010/075416. См. страницу 37). NAB739 был ранее описан Ваара (Vaara) с соавторами. Таблица 4 Данные МИК для соединений 2-14 (мкг/мл) Таблица 4 А Данные МИК для Дополнительных примеров 15-35 и примеров 2, 6 и 14 (мкг/мл) Оценивали противобактериальную активность in vitro соединений из примеров 2 и 6 против 500 грамотрицательных штаммов бактерий по сравнению с колистином. Планшет содержал по 100 клинических штаммов каждого из A.baumannii, E.coli, K.pneumoniae и P.aeruginosa. Планшет отражал эпидемиологическую картину, существующую в Европе и США, и включал ряд соответствующих штаммов с определенными фенотипами, устойчивыми к клинически применяемым в настоящее время противобактериальным агентам. Указанные устойчивые штаммы включали 20 А.baumannii, 22 Е.coli, 25 K.pneumoniae и 20 P.aeruginosa штаммов. Результаты исследования приведены в табл. 4 В. Все соединения исследовали вплоть до максимальной концентрации 64 мкг/мл, за исключением колистина, который оценивали вплоть до максимальной концентрации 16 мкг/мл. Таблица 4 В Значения МИК (мкг/мл) для примеров 2 и 6 и колистина в отношении 400 грамотрицательных клинических штаммов и 100 грамотрицательных штаммов с определенными устойчивыми фенотипами Эффективность in vivo против инфекции бедра, вызванной Е.coli, у мышей. Эффективность 8 соединений согласно настоящему изобретению (примеры 2, 4-8, 10 и 11) in vivo оценивали на модели распространения инфекции бедра, вызванной Е.coli, у мышей. Результаты приведены в табл. 5. Использовали группы из 5 самок беспатогенных CD-1 мышей массой 222 г. У животных вызывали нейтропению путем внутрибрюшинного введения циклофосфамида в дни -4 (150 мг/кг) и -1 (100 мг/кг). В день 0 животным в правое бедро внутримышечно вводили 105 КОЕ/мышь штамма АТСС 25922 Escherichia coli. Через 1 ч у 5 мышей определяли количество КОЕ и остальным мышам (по пять на группу) подкожно вводили лекарственное средство через 1 и 6 ч после инфицирования. В каждом исследовании на каждое из исследуемых соединений использовали по две группы, получающие различные дозы, 1,5 и 5 мг/кг дважды в сутки соответственно. Соединения из примеров 2, 4-8, 10, 11 и полимиксин В получали в физрастворе в концентрации 2 мг/мл и, по мере необходимости, рН раствора доводили до 6-7 путем добавления 0,1 М раствора H2SO4 или 4,2% раствора NaHCO3. Через 24 ч после инфицирования мышей безболезненно умерщвляли. Мышцы правого бедра каждого из животных отбирали, гомогенизировали,последовательно разбавляли и высевали в агаре с сердечно-мозговой вытяжкой + 0,5% уголь (мас./об.) для определения значения КОЕ. Через 24 ч после инфицирования для каждой группы определяли снижение общего количества КОЕ в правом бедре по сравнению с контрольным количеством. Соединения 2 и 6 при дозировании 10 мг/кг/день продемонстрировали эффективность, сравнимую с эффективностью полимиксина B, приводящую к снижению количества бактерий более чем в 3log10 раз. Дополнительные исследования эффективности in vivo против инфекции бедра, вызванной Е.coli, у мышей Эффективность соединения из примера 14 in vivo оценивали на модели распространения инфекции бедра, вызванной Е. coli, у мышей с применением способов, описанных в примерах выше. Результат приведен в табл. 5 А в сравнении с результатом для полимиксинам В. Таблица 5 А Эффективность in vivo против инфекций бедра,вызванных АТСС 25922 Е.coli, у нейтропенических мышей Соединение 14 при дозировании 10 мг/кг/день продемонстрировало эффективность, сравнимую с эффективностью полимиксина B, проводящую к снижению количества бактерий более чем в 3log10 раз. Дополнительные исследования эффективности in vivo против инфекции бедра, вызванной Klebsiellapneumoniae, у мышей. Эффективность трех соединений согласно настоящему изобретению (примеры 2, 6 и 14) in vivo оценивали на модели распространения инфекции бедра, вызванной АТСС 10031 Klebsiella pneumoniae, у мышей при помощи способа, описанного выше, с применением колистина (полимиксина Е) в качестве препарата сравнения. Результаты приведены в табл. 5 В. Соединения 2, 6 и 14 при дозировании 10 мг/кг/день продемонстрировали эффективность, сравнимую с эффективностью колистина, проводящую к снижению количества бактерий примерно в 2log10 раза. Таблица 5 В Эффективность in vivo против инфекций бедра, вызванных АТСС 10031 K.pneumoniae, у нейтропенических мышей Исследования фармакокинетики и почечного клиренса. Фармакокинетику и почечный клиренс 3 соединений (примеры 2, 4 и 6) согласно настоящему изобретению и полимиксина В оценивали на крысах. Растворы лекарственных средств готовили в концентрации 4 мг/мл в физрастворе и рН доводили до 6-7 путем добавления соответствующего объема 0,1 М раствора H2SO4 или 4,2% раствора NaHCO3. Рас- 25024792 творы стерилизовывали посредством фильтрации и хранили при -80C до применения. В день эксперимента растворы лекарственных средств разбавляли до 1 мг/мл стерильным физраствором. Акклиматизацию групп из 3 самцов крыс Спрага-Доули проводили в течение по меньшей мере 4 дней перед исследованием. Крыс анестезировали изофлураном и в яремную вену вводили канюлю. Через день после хирургического вмешательства крысам внутривенно через канюлю струйно вводили раствор в количестве 1 мг/кг с последующим промыванием физраствором. При помощи канюли вручную отбирали кровь перед введением соединения и через 0,08, 0,25, 0,5, 1, 3, 6, 8 и 24 ч после введения. Плазму собирали при помощи центрифугирования сразу после отбора крови. Суточные образцы мочи собирали до введения соединения и в интервалы 0-4 ч, 4-6 ч и 6-24 ч после введения. Образцы плазмы и мочи замораживали при -20C. Определение концентраций лекарственных средств в плазме и моче проводили при помощи жидкостной хроматографии-масс-спектрометрии (ЖХ-МС/МС). Перед анализом образцы плазмы и мочи подготавливали следующим способом. Образцы плазмы размораживали в день исследования и смешивали с 3 объемами ацетонитрила, содержащими 0,1% (об./об.) муравьиной кислоты, и 100 нг/мл внутреннего стандарта. После центрифугирования надосадочные жидкости переносили на 96-луночный планшет и разбавляли 1:1 водой, готовой для проведения ЖХ-МС/МС. Образцы мочи очищали при помощи твердофазной экстракции (ТФЭ) с применением картриджей Oasis HLB (Waters, UK), элюируя 100% раствором метанола. Аликвоту переносили на 96-луночный планшет и разбавляли 1:1 водой перед добавлением раствора внутреннего стандарта, готового для проведения ЖХ-МС/МС. Фармакокинетические параметры определяли при помощи анализа без учета компартментов с применением WinNonLin v5.3. Выведение с мочой определяли как процент выведенного с мочой лекарственного средства в неизмененном виде в первые 24 ч после инъекции. Таблица 6 Фармакокинетика полимиксина В и PMBN производных Интересно, что все соединения демонстрируют более высокие значения выведения с мочой по сравнению с полимиксином В. В ранних исследованиях было показано, что полимиксин Е (колистин) подвергается активной почечной канальцевой реабсорбции (Li et al., Antimicrob. Agents andChemotherapy, 2003, 47(5); Yousef et. al., Antimicrob. Agents and Chemotherapy, 2011, 55(9. He желая быть связанными теорией, полагают, что более высокие значения выведения с мочой могут свидетельствовать о пониженной почечной канальцевой реабсорбции, которая, в свою очередь, приводит к снижению нефротоксичности соединений. Исследование токсичности in vitro в клетках почек. Токсичность соединений в клетках почек оценивали при помощи исследования in vitro с применением клеточной линии HK-2, иммортализованной линии проксимальных клеток почечного канальца,полученных из почек здорового человека. Конечным результатом, определяющим токсичность соединений, являлось снижение содержания резазурина, соответствующее метаболической активности клеток.- 26024792 Клетки выращивали в 150 см 2 колбах в 25 мл KSF (keratinocyte serum-free -бессывороточной среды для культивирования кератиноцитов) с добавками (с 5 нг/мл ЭФР (эпидермального фактора роста) и 50 мкг/мл ВРЕ (bovine pituitary extract - экстракта гипофиза быка. Клетки выдерживали при 70% конфлюентности, проводя не более 25 пассажей. День 1: Среду удаляли и клетки промывали 10 мл DPBS (Dulbecco's phosphate-buffered saline - фосфатно-солевым буфером Дульбекко). Затем в колбу добавляли 6 мл 0,25% раствора трипсина с ЭДТА и клетки возвращали в инкубатор. Через 1-2 мин инкубации в колбу добавляли 14 мл среды для инактивации трипсина. Суспензию клеток переносили в центрифужную пробирку и клетки осаждали центрифугированием при 1000 об/мин в течение 6 мин. Затем клеточную массу повторно суспендировали в свежей среде с добавлением ЭФР и ВРЕ. Подсчитывали количество клеток и клетки разбавляли до 46875 клеток/мл в свежей среде с добавлением ЭФР и ВРЕ. В каждую лунку добавляли 7500 клеток в объеме 160 мкл и инкубировали при 37C в течение 24 ч. День 2: Исследуемые соединения готовили непосредственно в среде. Получали девять различных концентраций от 1000 до 1,95 мкг/мл при двукратном разбавлении в свежей среде. Микротитрационные планшеты вынимали из инкубатора и среду заменяли 100 мкл разбавленных растворов соединений. Каждую концентрацию готовили в трех экземплярах и в каждый планшет добавляли положительный и отрицательный контроль. Затем планшеты инкубировали в течение 24 ч при 37C в увлажненной атмосфере с 5% СО 2. День 3: Реагент, содержащий резазурин, (CellTiter-Blue, Promega) разбавляли в ФСБ (1:4) и добавляли в каждую лунку в количестве 20% (об./об.). Затем планшеты инкубировали при 37C в течение 2 ч и определяли продукт по снижению флуоресценции. Перед исследованием данных при помощи GraphPad Prism вычитали фоновые значения среды. Концентрации соединений откладывали на графике в виде логарифмических значений для построения кривой зависимости от дозы и определения значений IC50 (табл.7). Таблица 7 Данные IC50 для полимиксина В и примеров 2-14 Средние значения вплоть до 6 независимых исследований. Дополнительные исследования токсичности in vitro в клетках почек. Токсичность соединений из дополнительных примеров в клетках почек оценивали при помощи исследования in vitro с применением клеточной линии HK-2, как описано в примере выше. Значения IC50 указанных соединений приведены в табл. 7 А. Для сравнения также оценивали токсичность колистина,СВ 182,804 (соединение 5 из международной заявки на патент 2010/075416) и NAB739 в клетках почек. Средние значения вплоть до 6 независимых исследований. В максимальной концентрации отмечали снижение растворимости. Дополнительные исследования нефротоксичности in vivo. Модель нефротоксичности полимиксинов (на основе Yousef et al., Antimicrob. Agents Chemother.,2011, 55 (9):4044-4049) проверяли на крысах. Соединения из примеров 2, 6 и 14 исследовали в данной модели и сравнивали с колистином (в виде сульфата). После одной недели акклиматизации самцам крыс Спрага-Доули хирургически в яремную вену вводили канюлю и, по мере необходимости, содержали по отдельности в заранее определенных стандартных клетках или метаболических клетках. Колистин и соединения из примеров готовили в солевом растворе. Соединения при помощи канюли вводили в яремную вену два раза в день с интервалом 7 в течение семи дней. Дозу постепенно увеличивали в течение трех дней вплоть до максимальной дозы, которую затем вводили до завершения исследования. Суточные образцы мочи собирали (во льду) до введения дозы и через 4 и 7 дней. Режим дозирования приведен в табл. 8. Таблица 8 Режим дозирования, применяемый в исследовании нефротоксичности in vivo. Дозы указаны в мг/кг лекарственного средства Активность N-ацетилD-глюкозаминидазы (NAG) в моче определяли при помощи спектрофотометрии с применением набора NAG реактивов, произведенного Roche Applied Science. Биомаркеры повреждения почек исследовали при помощи Kidney Injury Panel II из системы для проведения исследования Multi-Spot (Meso Scale Discovery). Примеры 2, 6 и 14 при дозировании 8 мг/кг продемонстрировали значительное снижение почечных биомаркеров NAG, альбумина и цистатина С по сравнению с колистином при таком же режиме дозирования (см. фиг. 1-3). Ответ был аналогичен ответу, вызванному колистином при максимальной концентрации 2 мг/кг. На фиг. 1 приведены концентрации NAG (нг/24 ч) в дни 0, 4 и 7 для соединений 2, 6 и 14 и колистина. На левом графике слева направо приведены колистин (2 мг/кг дважды в день), колистин (8 мг/кг дважды в день), соединение 2 (8 мг/кг дважды в день) и 6 (8 мг/кг дважды в день). На правом графике приведены колистин (2 мг/кг дважды в день), колистин (8 мг/кг дважды в день), соединение 14 (8 мг/кг дважды в день). На фиг. 2 приведены концентрации альбумина (нг/24 ч) в дни 0, 4 и 7 для соединений 2, 6 и 14 и колистина. На левом графике слева направо приведены колистин (2 мг/кг дважды в день), колистин (8 мг/кг дважды в день), соединение 2 (8 мг/кг дважды в день) и 6 (8 мг/кг дважды в день). На правом графике приведены колистин (2 мг/кг дважды в день), колистин (8 мг/кг дважды в день), соединение 14 (8 мг/кг дважды в день). На фиг. 3 приведены концентрации цистатина С (нг/24 ч) в дни 0, 4 и 7 для соединений 2, 6 и 14 и колистина. На левом графике слева направо приведены колистин (2 мг/кг дважды в день), колистин(8 мг/кг дважды в день), соединение 2 (8 мг/кг дважды в день) и 6 (8 мг/кг дважды в день). На правом графике приведены колистин (2 мг/кг дважды в день), колистин (8 мг/кг дважды в день), соединение 14R1 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток фенилаланина, лейцина или валина;R2 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток лейцина, изолейцина,фенилаланина, треонина, валина или норвалина;R3 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток треонина или лейцина;R4 совместно с карбонильной группой и атомом азота, расположенным в альфа-положении относительно атома углерода, к которому он присоединен, представляет собой остаток ,-диаминомасляной кислоты или серина;R5 представляет собой группу (а)-(g), где:(a) представляет собой С 1-12 алкил(С 3-8 циклоалкил), где алкил или циклоалкил замещен (i) одной,двумя или тремя гидроксильными группами, или (ii) одной -NR6R7 группой, или (iii) одной -NR6R7 группой и одной или двумя гидроксильными группами;(b) представляет собой С 2-12 алкил, где алкил замещен (i) одной, двумя или тремя гидроксильными группами, или (ii) одной -NR6R7 группой на конце алкильной цепи, или (iii) одной -NR6R7 группой и двумя гидроксильными группами;(c) представляет собой С 0-12 алкил(С 4-6 гетероциклил), где С 4-6 гетероциклил содержит 1 или 2 атома азота и, необязательно, содержит дополнительный кольцевой гетероатом, выбранный из кислорода и серы;(d) представляет собой С 3-8 циклоалкил и циклоалкил замещен (i) одной, двумя или тремя гидроксильными группами или (iii) одной -NR6R7 группой и одной или двумя гидроксильными группами;(e) представляет собой С 3-12 алкил, где алкил замещен одной -NR6R7 группой и одной гидроксильной
МПК / Метки
МПК: C07K 7/62, A61K 38/12
Метки: полимиксина, производные
Код ссылки
<a href="https://eas.patents.su/30-24792-proizvodnye-polimiksina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные полимиксина</a>
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Филош Брюно, Ашар Даниель, Иттэнжер Огюстэн, Букерель Жан, Бушар Эрве, Гризони Серж, Майерс Майкл
МПК: A61K 31/397, A61P 25/00, C07D 205/04...
Метки: способ, 3-аминоазетидина, получения, содержащие, новые, фармацевтические, композиции, производные
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Харшинг Ласло Габор, Сабо Геза, Мартонне Марко Бернадетт, Леваи Дьердь, Шимиг Дьюла, Линг Иштван, Грефф Зольтан, Сенаши Габор, Баркоци Йожеф, Вег Миклош, Гиглер Габор, Раткаи Зольтан
МПК: A61P 25/00, A61K 31/551, C07D 243/02...
Метки: активного, композиции, ингредиента, производные, 2,3-бензодиазепина, качестве, эти, содержащие, фармацевтические
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Производные 2н-пиридазин-3-она, фармацевтические композиции, содержащие эти производные, и способ получения активного ингредиента

Номер патента: 6246
Опубликовано: 27.10.2005
Авторы: Баркоци Йожеф, Шимиг Дьюла, Рацне Байногель Юдит, Сенаши Габор, Котаи Надь Петер, Эдьед Андраш, Паллаги Каталин, Леваи Дьёрди, Вельман Янош, Гачальи Иштван, Миклошне Ковач Анико, Шмидт Эва
МПК: C07D 413/14, A61K 31/50
Метки: эти, содержащие, получения, производные, фармацевтические, 2н-пиридазин-3-она, способ, ингредиента, композиции, активного
Формула / Реферат:
1. Производное 2H-пиридазин-3-она формулы где R означает атом водорода или C1-4-алкильную группу, X и Y независимо представляют собой атом водорода, атом галогена или группу формулы при условии, что один из X и Y всегда представляет собой группу формулы II, а другой радикал представляет собой атом водорода или атом галогена, где в формуле II n имеет значение 1 или 2, и его фармацевтически приемлемые соли присоединения кислот. 2. Производное...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Бергер Якоб, Кларк Робин Дуглас, Чжао Шухай
МПК: C07D 265/36, A61K 31/536, A61P 25/18...
Метки: качестве, 5-нт-6, модуляторов, фармацевтические, эти, способ, получения, содержащие, композиции, производные, применение, бензоксазина
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Полициклические производные аминокислот, содержащая их фармацевтическая композиция и способ лечения, использующий эти производные

Номер патента: 19879
Опубликовано: 30.07.2014
Авторы: Маринелли Бретт, Лю Цинюнь, У Вэньсюэ, Цзинь Хайхун, Туноори Ашок, Самала Лакшама, Чжан Чэньминь, Девасагаярадж Арокиасами, Ван Ин, Чжан Хаймин, Ши Чжи-Цай
МПК: A61K 31/53, A61K 31/4965, A61K 31/4192...
Метки: использующий, композиция, полициклические, способ, эти, содержащая, лечения, аминокислот, производные, фармацевтическая
Формула / Реферат:
1. Соединение формулыили его фармацевтически приемлемая соль или сольват, гдеА представляет собой необязательно замещенный фенил, нафтил или 5-6-членный гетероцикл, включающий 1-3 гетероатома, выбранных из О, N и S, причем необязательное замещение означает замещение одной или более группами С1-6-алкоксила, амино, циано, галогена, гидроксила или фенила, причем фенильная группа необязательно замещена одной или более группами С1-6-алкоксила, амино,...
Предыдущий патент: Новые макроциклические соединения в качестве ингибиторов фактора xia
Следующий патент: Гетероарильные соединения, применяемые в качестве ингибиторов e1 активирующих ферментов
Случайный патент: Игровой автомат