Производные пиразола
Номер патента: 24648
Опубликовано: 31.10.2016
Авторы: Пфлигер Филипп, Галлей Гуидо, Гелламалла Седрик, Норкросс Роджер







Формула / Реферат
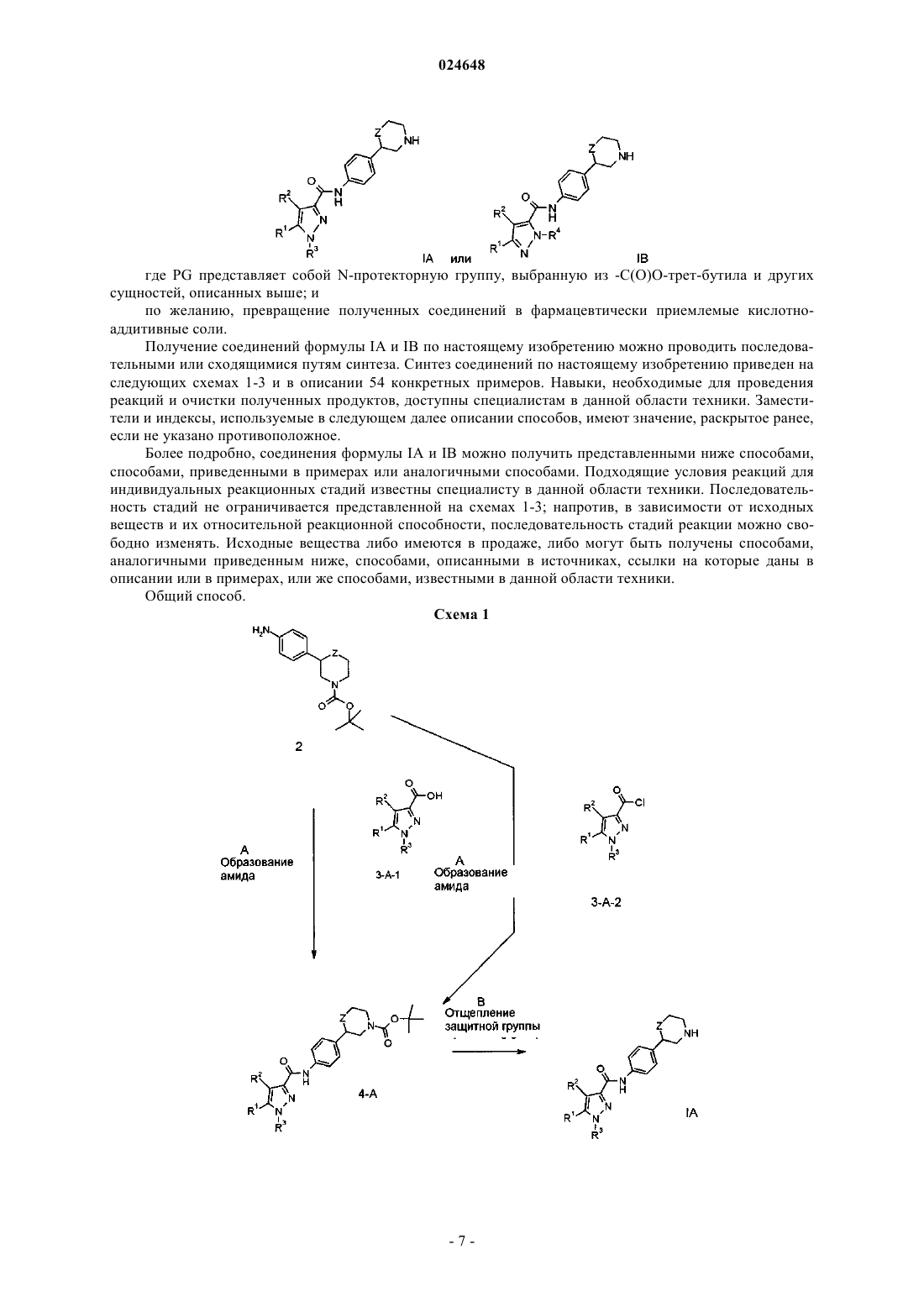
1. Соединение формулы

где R1 представляет собой водород или фенил, возможно содержащий в качестве заместителя галоген, CN или C1-4алкоксигруппу или C1-4алкоксигруппу, замещенную галогеном;
R2 представляет собой водород или C1-4алкил;
R3 представляет собой водород или C1-4алкил, или представляет собой фенил, возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или C1-4алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или C1-4алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя C1-4алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу или C1-4алкил, замещенный галогеном;
Z представляет собой связь, -CH2- или -O-,
или его фармацевтически приемлемые кислотно-аддитивные соли.
2. Соединение формулы

где R1 представляет собой водород или фенил, возможно содержащий в качестве заместителя галоген, CN или C1-4-алкоксигруппу или C1-4алкоксигруппу, замещенную галогеном;
R2 представляет собой водород или C1-4алкил;
R4 представляет собой водород, C1-4алкил или фенил;
Z представляет собой связь, -CH2- или -O-,
или его фармацевтически приемлемые кислотно-аддитивные соли
3. Соединение формулы IA-1 по п.1

где R представляет собой водород, галоген, CN или C1-4алкоксигруппу или C1-4алкоксигруппу, замещенную галогеном;
R2 представляет собой водород или С1-4алкил;
R3 представляет собой водород или С1-4алкил;
Z представляет собой связь, -CH2- или -O-;
n равно 1 или 2, если n=2, каждый R может быть определен независимо от другого,
или его фармацевтически приемлемые кислотно-аддитивные соли.
4. Соединение формулы IA-1 по п.1, причем такими соединениями являются следующие:
(S)-N-(4-(морфолин-2-ил)фенил)-5-фенил-1H-пиразол-3-карбоксамид,
(S)-1-метил-N-(4-(морфолин-2-ил)фенил)-5-фенил-1H-пиразол-3-карбоксамид,
(S)-5-(3-цианофенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-5-(3-цианофенил)-4-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-5-(5-циано-2-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-5-(3-циано-4-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид или
(S)-5-(3-(дифторметокси)фенил)-1-этил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид.
5. Соединение формулы IB-1 по п.2

где R представляет собой водород, галоген, CN или С1-4алкоксигруппу или С1-4алкоксигруппу, замещенную галогеном;
R2 представляет собой водород или C1-4алкил;
R4 представляет собой водород или С1-4алкил;
Z представляет собой связь, -CH2- или -O-;
n равно 1 или 2, если n=2, то каждый R может быть определен независимо от другого,
или их фармацевтически приемлемые кислотно-аддитивные соли.
6. Соединение формулы IB-1 по любому из пп.2 или 5, причем такими соединениями являются следующие:
(S)-3-(3-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(4-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-метоксифенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-1-метил-N-(4-(морфолин-2-ил)фенил)-3-фенил-1H-пиразол-5-карбоксамид,
(S)-4-метил-N-(4-(морфолин-2-ил)фенил)-3-фенил-1H-пиразол-5-карбоксамид,
(S)-3-(4-метоксифенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(2-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(2-метоксифенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(2-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3,4-диметоксифенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-3-(4-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-3-(2-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(4-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-3-(3-хлорфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-3-(3-метоксифенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-хлорфенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-1-метил-N-(4-(морфолин-2-ил)фенил)-3-фенил-1H-пиразол-5-карбоксамид,
(S)-3-(4-цианофенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(4-фторфенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-метоксифенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-цианофенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-цианофенил)-1-этил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(4-цианофенил)-1-этил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-цианофенил)-N-(4-(пиперидин-3-ил)фенил)-1H-пиразол-5-карбоксамид,
(R)-3-(3-цианофенил)-N-(4-(пиперидин-3-ил)фенил)-1H-пиразол-5-карбоксамид,
(rac) 3-(3-цианофенил)-N-(4-(пирролидин-3-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-(дифторметокси)фенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-(дифторметокси)фенил)-1-этил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид,
(S)-3-(3-циано-2-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид или
(S)-3-(3-(дифторметокси)фенил)-1-метил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-5-карбоксамид.
7. Соединение формулы IA-2 по п.1

где R1 представляет собой водород;
R2 представляет собой водород или С1-4алкил;
R3 представляет собой фенил, возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или C1-4алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или C1-4алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя C1-4алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу или C1-4-алкил, замещенный галогеном;
Z представляет собой связь, -CH2- или -O-,
или их фармацевтически приемлемые кислотно-аддитивные соли.
8. Соединение формулы IA-2 по любому из пп.1 или 7, причем такими соединениями являются следующие:
(S)-1-(4-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(R)-1-(4-фторфенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-1-(5-хлорпиридин-2-ил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-N-(4-(морфолин-2-ил)фенил)-1-(5-(трифторметил)пиридин-2-ил)-1H-пиразол-3-карбоксамид,
(S)-1-(4-цианофенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(R)-N-(4-(морфолин-2-ил)фенил)-1-(5-(трифторметил)пиридин-2-ил)-1H-пиразол-3-карбоксамид,
(S)-1-(4-(дифторметокси)фенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(R)-1-(4-(дифторметокси)фенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-N-(4-(морфолин-2-ил)фенил)-1-(5-(трифторметил)пиримидин-2-ил)-1H-пиразол-3-карбоксамид,
(S)-1-(6-хлорпиразин-2-ил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-1-(3-хлорпиразин-2-ил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-1-(5-хлорпиразин-2-ил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид,
(S)-N-(4-(морфолин-2-ил)фенил)-1-(6-(трифторметил)пиримидин-4-ил)-1H-пиразол-3-карбоксамид,
(S)-N-(4-(морфолин-2-ил)фенил)-1-(6-(трифторметил)пиразин-2-ил)-1H-пиразол-3-карбоксамид,
(S)-1-(5-цианопиразин-2-ил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид или
(S)-N-(4-(морфолин-2-ил)фенил)-1-(2-(трифторметил)пиримидин-4-ил)-1H-пиразол-3-карбоксамид.
9. Соединение формулы IB-2 по п.2

где R1 представляет собой водород;
R2 представляет собой водород или С1-4алкил;
R4 представляет собой водород, С1-4алкил или фенил;
Z представляет собой связь, -CH2- или -O-,
или их фармацевтически приемлемые кислотно-аддитивные соли.
10. Соединение по любому из пп.2 или 9, причем таким соединением является (S)-N-(4-(морфолин-2-ил)фенил)-1-фенил-1H-пиразол-5-карбоксамид.
11. Фармацевтическая композиция для связывания с TAAR1, включающая соединение по любому из пп.1-10 и фармацевтически приемлемый носитель и/или вспомогательное вещество.
12. Применение фармацевтической композиции, включающей соединение по любому из пп.1-10 и фармацевтически приемлемый носитель и/или вспомогательное вещество для лечения психических расстройств, неврологических заболеваний, нейродегенеративных расстройств, метаболических расстройств и сердечно-сосудистых заболеваний.
13. Применение соединения по любому из пп.1-10 в качестве терапевтически активного вещества, связывающего TAAR1.
14. Применение соединения по любому из пп.1-10 в качестве терапевтически активного вещества в лечении психических расстройств, неврологических заболеваний, нейродегенеративных расстройств, метаболических расстройств и сердечно-сосудистых заболеваний.
15. Применение соединения по любому из пп.1-10 для изготовления лекарственных средств для терапевтического и/или профилактического лечения психических расстройств, неврологических заболеваний, нейродегенеративных расстройств, метаболических расстройств и сердечно-сосудистых заболеваний.
16. Применение по любому из пп.12, 14 или 15, где заболевание выбрано из депрессии, тревожного невроза, биполярного расстройства, синдрома дефицита внимания и гиперактивности, расстройств, связанных со стрессом, шизофрении, болезни Паркинсона, болезни Альцгеймера, эпилепсии, мигрени, гипертонии, наркотической зависимости и токсикомании, расстройств приема пищи, диабета, диабетических осложнений, тучности, дислипидемии, расстройств потребления и усвоения энергии, расстройств и нарушений функции гомеостаза температуры тела, расстройств сна и суточного биоритма.
Текст
где R1 представляет собой водород или фенил, возможно содержащий в качестве заместителя галоген, CN илиC1-4 алкоксигруппу или C1-4 алкоксигруппу, замещенную галогеном; R2 представляет собой водород или C1-4 алкил;R3 представляет собой водород или C1-4 алкил или представляет собой фенил, возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или C1-4 алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или C1-4 алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя C1-4 алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу илиC1-4 алкил, замещенный галогеном; Z представляет собой связь, -CH2- или -O-, либо соединениям формулы IB где R1 представляет собой водород или фенил, возможно содержащий в качестве заместителя галоген, CN илиC1-4 алкоксигруппу или C1-4 алкоксигруппу, замещенную галогеном; R2 представляет собой водород или C1-4 алкил;R4 представляет собой водород, C1-4 алкил или фенил; Z представляет собой связь, -CH2- или -O-, или к их фармацевтически приемлемым кислотно-аддитивным солям. Было показано, что соединения формулы IA и IB обладают высоким сродством по отношению к рецепторам следовых аминов TAAR1. Эти соединения можно применять для лечения психических расстройств, неврологических заболеваний, нейродегенеративных расстройств, метаболических расстройств и сердечно-сосудистых заболеваний. Настоящее изобретение относится к соединениям формулы где R1 представляет собой водород или фенил, возможно содержащий в качестве заместителя галоген, CN или низшую алкоксигруппу или же низшую алкоксигруппу, замещенную галогеном;R2 представляет собой водород или низший алкил;R3 представляет собой водород или низший алкил или же представляет собой фенил, возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или низшая алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или низший алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя низший алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу или низший алкил, замещенный галогеном;R4 представляет собой водород, низший алкил или фенил;Z представляет собой связь, -CH2- или -O-,или к их фармацевтически приемлемым кислотно-аддитивным солям. Настоящее изобретение включает все рацемические смеси, все их соответствующие энантиомеры и/или оптические изомеры. Кроме того, все таутомерные формы соединений формулы IA и IB также охвачены настоящим изобретением. К настоящему времени было обнаружено, что соединения формулы IA и IB обладают сильным сродством к рецепторам следовых аминов (TAARs), в частности TAAR1. Предложенные соединения можно применять для лечения депрессии, тревожного невроза, биполярного расстройства, синдрома дефицита внимания с гиперактивностью (СДВГ), расстройств, связанных со стрессом, психических расстройств, таких как шизофрения, неврологических заболеваний, таких как болезнь Паркинсона, нейродегенеративных расстройств, таких как болезнь Альцгеймера, эпилепсии,мигрени, гипертонии, наркотической зависимости и токсикомании, а также метаболических расстройств,таких как расстройства приема пищи, диабета, диабетических осложнений, тучности, дислипидемии,расстройств потребления и усвоения энергии, расстройств и нарушения функции гомеостаза температуры тела, расстройств сна и суточного биоритма и сердечно-сосудистых заболеваний. Некоторые физиологические эффекты (т.е. сердечно-сосудистые эффекты, гипотензия, индукция седативного эффекта), о которых сообщалось в отношении соединений, способных связываться с адренергическими рецепторами (WO 02/076950, WO 97/12874 или EP 0717037), могут быть расценены как нежелательные побочные эффекты для лекарственных средств, предназначенных для лечения заболеваний центральной нервной системы, описанных выше. Таким образом, желательно получить лекарственные средства, селективные к TAAR1 рецепторам, но не к адренергическим рецепторам. Объекты по настоящему изобретению демонстрируют селективность к TAAR1 рецепторам над адренергическими рецепторами, в частности хорошую селективность над человеческими и крысиными адренергическими рецепторами альфа 1 и альфа 2. Классические биогенные амины (серотонин, норадреналин, адреналин, дофамин, гистамин) играют важную роль как нейротрансмиттеры в центральной и периферической нервной системе [1]. Их синтез и хранение, а также их деградация и обратный захват после высвобождения жестко регулируются. Известно, что дисбаланс уровня биогенных аминов является причиной измененной функции мозга при многих патологических состояниях [2-5]. Другой класс эндогенных аминосоединений, так называемые следовые амины (TAs), в значительной мере пересекается с классическими биогенными аминами в отношении структуры, метаболизма и внутриклеточной локализации. TAs включают паратирамин,-фенилэтиламин, триптамин и октопамин, и они присутствуют в нервной системе млекопитающих, в целом, при более низких концентрациях, по сравнению с классическими биогенными аминами [6]. Их дисрегуляцию связывают с различными психическими заболеваниями, такими как шизофрения и депрессия [7], а также с другими состояниями, такими как синдром дефицита внимания с гиперактивностью, головная боль, болезнь Паркинсона, наркотическая зависимость и токсикомания, а также расстройства приема пищи [8, 9]. На протяжении долгого времени существование TA-специфичных рецепторов лишь гипотетически предполагалось на основании анатомически дискретных высокоаффинных TA-связывающих сайтов в ЦНС человека и других млекопитающих [10, 11]. Таким образом, считалось, что фармакологические эффекты TAs опосредованы хорошо известным механизмом классических биогенных аминов как за счет запуска их высвобождения или ингибирования их обратного захвата, так и за счет перекрестной реакции с их рецепторными системами [9, 12, 13]. Эта точка зрения существенно изменилась в связи с недавним открытием нескольких членов нового семейства GPCRs, рецепторов следовых аминов (TAARs) [7, 14]. У человека присутствует 9 генов TAAR (включая 3 псевдогена), у мышей - 16 генов (включая 1 псевдоген). Гены TAAR не содержат интронов (с одним исключением: TAAR2 содержит 1 интрон) и расположены друг за другом в одном и том же сегменте хромосомы. Такое филогенетическое родство рецепторных генов, в согласии с тщательным сравнением сходства GPCR-фармакофоров и фармакологическими данными, указывает на то, что эти рецепторы образуют три различных подсемейства [7, 14]. TAAR1 представляет собой первый подкласс, состоящий из четырех генов (TAAR1-4), высококонсервативных у человека и грызунов. TAs активируют TAAR1 через Gas. Было показано, что дисрегуляция TAs вносит свой вклад в этиологию многих заболеваний, таких как депрессия, психоз, синдром дефицита внимания с гиперактивностью, наркотическая зависимость и токсикомания, болезнь Паркинсона, головная боль, расстройства приема пищи, метаболические расстройства, и поэтому лиганды TAAR1 обладают большим потенциалом для лечения подобных заболеваний. Таким образом, имеется повышенный интерес к расширению знаний о рецепторах следовых аминов. Используемые ссылки: Объектами настоящего изобретения являются новые соединения формулы IA и IB, а также их фармацевтически приемлемые соли, их применение для изготовления лекарственных средств для лечения заболеваний, связанных с биологической функцией рецепторов следовых аминов, их получение, а также лекарственные средства на основе соединения по настоящему изобретению для контроля или профилактики заболеваний, таких как депрессия, тревожный невроз, биполярное расстройство, синдром дефицита внимания с гиперактивностью, расстройства, обусловленные стрессом, психические расстройства, такие как шизофрения, неврологические расстройства, такие как болезнь Паркинсона, нейродегенеративные расстройства, такие как болезнь Альцгеймера, эпилепсия, мигрень, наркотическая зависимость и токсикомания, а также метаболические расстройства, такие как расстройства приема пищи, диабет, диабетические осложнения, тучность, дислипидемия, расстройства потребления и усвоения энергии, расстройства и нарушение функции гомеостаза температуры тела, расстройства сна и суточного биоритма, а также сердечно-сосудистые расстройства. Предпочтительными показаниями к применению соединений по настоящему изобретению являются депрессия, психоз, болезнь Паркинсона, тревожность и синдром дефицита внимания с гиперактивностью (СДВГ), а также диабет. В данном тексте термин "низший алкил" обозначает насыщенную группу с линейной или разветвленной цепью, содержащую от 1 до 7 атомов углерода, такую как метил, этил, пропил, изопропил,н-бутил, изобутил, 2-бутил, трет-бутил и т.п. Предпочтительными алкильными группами являются группы, содержащие 1-4 атома углерода. В данном тексте термин "низшая алкоксигруппа" обозначает группу, в которой алкильный остаток таков, как определено выше, и он присоединен через атом кислорода. Термин "галоген" обозначает хлор, йод, фтор и бром. В данном тексте термин "низшая алкоксигруппа, замещенная галогеном" обозначает алкоксигруппу, раскрытую выше, в которой по меньшей мере один атом водорода замещен галогеном. Термин "фармацевтические приемлемые кислотно-аддитивные соли" охватывает соли неорганических и органических кислот, таких как хлороводородная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота, п-толуолсульфоновая кислота и т.п. Одним из осуществлений настоящего изобретения являются соединения формулы IA-1 где R представляет собой водород, галоген, CN или низшую алкоксигруппу, или же низшую алкоксигруппу, замещенную галогеном;R2 представляет собой водород или низший алкил;R3 представляет собой водород или низший алкил;n равно 1 или 2; если n=2, каждый R может быть определен независимо от другого,или их фармацевтически приемлемые кислотно-аддитивные соли,например следующие соединения: Еще одним осуществлением настоящего изобретения являются соединения формулы IB-1 где R представляет собой водород, галоген, CN или низшую алкоксигруппу, или же низшую алкоксигруппу, замещенную галогеном;R2 представляет собой водород или низший алкил;R4 представляет собой водород или низший алкил;n равно 1 или 2, если n=2, каждый R может быть определен независимо от другого; или их фармацевтически приемлемые кислотно-аддитивным солям,например следующие соединения: Одним из осуществлений настоящего изобретения являются соединения формулы IA-2R2 представляет собой водород или низший алкил;R3 представляет собой фенил, возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или низшая алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или низший алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя низший алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу или низший алкил, замещенный галогеном;Z представляет собой связь, -CH2- или -O-,или их фармацевтически приемлемые кислотно-аддитивные соли,например следующие соединения: Еще одним осуществлением настоящего изобретения являются соединения формулы IB-2R2 представляет собой водород или низший алкил;R4 представляет собой водород, низший алкил или фенил;Z представляет собой связь, -CH2- или -O-,или их фармацевтически приемлемые кислотно-аддитивные соли,например следующее соединение: Предложенные соединения формулы IA и IB и их фармацевтически приемлемые соли можно получить способами, известными в данной области техники, например способами, описанными ниже, включающими: а) отщепление N-протекторной группы от соединений формулы где PG представляет собой N-протекторную группу, выбранную из -C(O)O-трет-бутила и других сущностей, описанных выше; и по желанию, превращение полученных соединений в фармацевтически приемлемые кислотноаддитивные соли. Получение соединений формулы IA и IB по настоящему изобретению можно проводить последовательными или сходящимися путям синтеза. Синтез соединений по настоящему изобретению приведен на следующих схемах 1-3 и в описании 54 конкретных примеров. Навыки, необходимые для проведения реакций и очистки полученных продуктов, доступны специалистам в данной области техники. Заместители и индексы, используемые в следующем далее описании способов, имеют значение, раскрытое ранее,если не указано противоположное. Более подробно, соединения формулы IA и IB можно получить представленными ниже способами,способами, приведенными в примерах или аналогичными способами. Подходящие условия реакций для индивидуальных реакционных стадий известны специалисту в данной области техники. Последовательность стадий не ограничивается представленной на схемах 1-3; напротив, в зависимости от исходных веществ и их относительной реакционной способности, последовательность стадий реакции можно свободно изменять. Исходные вещества либо имеются в продаже, либо могут быть получены способами,аналогичными приведенным ниже, способами, описанными в источниках, ссылки на которые даны в описании или в примерах, или же способами, известными в данной области техники. Общий способ. Схема 1 заместители раскрыты выше. Стадия А. Получение амида можно осуществить реакцией конденсации между амином 2 и соединениями хорида кислоты 3-А-2 в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан, или эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или ТВМЕ, в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин. Примеры подходящих аминов 2 включают N-защищенные производные морфолина, такие как 2-а[CAS 1002726-96-6], производные пиперидина, такие как 2-b [CAS 875798-79-1], производные пирролидина, такие как 2-c [CAS 908334-28-1]. Предпочтительными условиями являются триэтиламин в THF при комнатной температуре в течение 18 ч. Как вариант, получение амида можно осуществить реакцией конденсации между амином 2 и карбоновыми кислотами 3-А-1 в присутствии сшивающего реагента, такого как DCC, EDC, TBTU, HBTU илиN-метилморфолин, в галогенированных растворителях, таких как DMF, дихлорметан или 1,2-дихлорэтан,или эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или ТВМЕ. Предпочтительными условиями являются HBTU с N-метилморфолином в DMF при 60C в течение 18 ч. Стадия В. Удаление N-протекторной ВОС группы можно осуществить с помощью минеральных кислот, таких как HCl, H2SO4 или Н 3 РО 4, или органических кислот, таких как CF3COOH, CHCl2COOH,НОАс или п-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, THF, диоксан,MeOH, EtOH или Н 2 О, при 0-80C. Предпочтительными условиями являются HCl в диоксане при 60C в течение 1-20 ч. Схема 2 Условия процесса такие же, как описано на схеме 1. Схема 3R2 представляет собой водород и низший алкил;R3 представляет собой фенил возможно содержащий один или более заместителей, выбранных из следующих: галоген, цианогруппа или низшая алкоксигруппа, замещенная галогеном, или представляет собой пиридинил, возможно содержащий в качестве заместителя галоген или низший алкил, замещенный галогеном, или представляет собой пиримидинил, возможно содержащий в качестве заместителя низший алкил, замещенный галогеном, или представляет собой пиразинил, возможно содержащий в качестве заместителя галоген, цианогруппу или низший алкил, замещенный галогеном; иZ представляет собой связь, -CH2- или -O-; Стадия А. Получение амида 6 можно осуществить реакцией конденсации между амином 2 и 1H-пиразол-3-карбоновой кислотой 5 с помощью селективного сшивающего агента, такого как 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиний хлорид, в растворителе, таком как метанол,этанол или изопропанол, при температуре от 0 до 50C в течение 1-24 ч. Предпочтительными условиями являются использование 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4 метилморфолиния хлорида в метаноле в течение 1 ч при 0C с последующим перемешиванием 18 ч при комнатной температуре. Примеры подходящих аминов 2 включают N-защищенные производные морфолина, такие как 2-а[CAS 1002726-96-6], производные пиперидина, такие как 2-b [CAS 875798-79-1], производные пирролидина, такие как 2-c [CAS 908334-28-1]. Стадия В. Образование связи C-N можно осуществить обработкой арилгалида 7 или гетероарилгалида 7 пиразолом 6 в присутствии палладиевого или медного катализатора, лиганда и основания, в растворителях, таких как диоксан, DME, THF, толуол, DMF и DMSO, при повышенной температуре, например, с помощью катализируемой палладием реакции Бухвальда-Хартвига. Предпочтительными условиями являются каталитический комплекс трис-(дибензилиденацетон)дипалладия с хлороформом, катализатор 9,9-диметил-4,5-бис-(дифенилфосфино)ксантен (ксантфос) и карбонат цезия в диоксане, в запаянной трубке, при нагревании при 100C в течение ночи согласно модификации способа по van Leeuwen et al. (Tetrahedron. Lett. 1999, 40, 3789-3790). В случае, когда арилгалид 7 или гетероарилгалид 7 активируют, подготавливая к нуклеофильному замещению за счет присутствия электрон-акцепторных заместителей, предпочтительно за счет присутствия трифторметильной группы, конденсацию с пиразолом 6 можно осуществить проведением реакции с этими соединениями в присутствии основания, такого как диизопропилэтиламин, триэтиламин, карбонат калия или гидрид натрия, в растворителе, таком как изопропанол, диоксан, диметилсульфоксид, диметилацетамид или диметилформамид, при температуре между 50 и 140C в течение 1-24 ч. Предпочтительными условиями являются нагревание смеси соединений 6 и 7 с карбонатом калия в диметилацетамиде при 120C в течение 20 ч. Стадия С. Образование амида можно осуществить реакцией конденсации между амином 2 соединением хорида кислоты 8 а в галогенированных растворителях, таких как дихлорметан или 1,2-дихлорэтан,или в эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или ТВМЕ, в присутствии органического основания, такого как триэтиламин или N,N-диизопропилэтиламин. Предпочтительными условиями являются триэтиламин в THF при комнатной температуре в течение 18 ч. Как вариант, образование амида можно осуществить реакцией конденсации между амином 2 и карбоновыми кислотами 8b в присутствии сшивающего агента, такого как DCC, EDC, TBTU, HBTU илиN-метилморфолин, в галогенированных растворителях, таких как DMF, дихлорметан или 1,2-дихлорэтан,или эфирных растворителях, таких как диэтиловый эфир, диоксан, THF, DME или ТВМЕ. Предпочтительными условиями являются HBTU с N-метилморфолином в DMF при 60C в течение 18 ч. Стадия D. Удаление N-протекторной ВОС группы можно осуществить с помощью минеральных кислот, таких как HCl, H2SO4 или Н 3 РО 4, или органических кислот, таких как CF3COOH, CHCl2COOH,НОАс или п-толуолсульфоновая кислота, в растворителях, таких как CH2Cl2, CHCl3, THF, диоксан,MeOH, EtOH или Н 2 О, при 0-80C. Предпочтительными условиями являются HCl в диоксане при 60C в течение 1-20 ч. Тот же общий способ, описанный на схеме 3 можно применять для получения соединений формулыIB-2. Выделение и очистка соединений. Выделение и очистку соединений и промежуточных продуктов, описанных в данном тексте, можно осуществить, по желанию, любым удобным способом выделения или очистки, таким как, например,фильтрация, экстракция, кристаллизация, колоночная хроматография, тонкослойная хроматография, толстослойная хроматография, препаративная жидкостная хроматография низкого или высокого давления или комбинация этих способов. Конкретные примеры подходящих способов разделения и выделения можно привести посредством ссылки на способы получения и примеры, данные ниже. При этом, разумеется, также можно применять другие эквивалентные способы разделения или выделения. Рацемические смеси хиральных соединений формулы I можно разделять с помощью хиральной ВЭЖХ. Соли соединений формулы IA и IB. Соединения формулы IA и IB являются основными, и их можно перевести в соответствующие кислотно-аддитивные соли. Такое превращение можно осуществить обработкой с помощью, по меньшей мере, стехиометрического количества подходящей кислоты, такой как хлороводородная кислота, бромоводородная с кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органических кислот,таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малоновая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота,метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота и т.п. Как правило, свободное основание растворяют в инертном органическом растворителе, таком как диэтиловый эфир, этилацетат, хлороформ, этанол или метанол и т.п., и добавляют кислоту в таком же растворителе. Температуру поддерживают в интервале между 0 и 50C. Получаемая соль осаждается самопроизвольно или ее можно извлечь из раствора с помощью менее полярного растворителя. Кислотно-аддитивные соли основных соединений формулы IA и IB можно переводить в соответствующие свободные основания обработкой, по меньшей мере, стехиометрическим эквивалентом подходящего основания, такого как гидроксид натрия или калия, карбонат калия, бикарбонат натрия, аммиак и т.п.a) (S)-трет-Бутил 2-(4-(5-фенил-1H-пиразол-3-карбоксамидо)фенил)морфолин-4-карбоксилат. В круглодонной колбе на 25 мл (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилат (100 мг,359 мкмоль, экв.: 1.00), 3-фенил-1H-пиразол-5-карбоновую кислоту (87.9 мг, 467 мкмоль, экв.: 1.3)(CAS-1134-49-2), N-метилморфолин (109 мг, 118 мкл, 1.08 ммоль, экв.: 3) и HBTU (204 мг, 539 мкмоль,экв.: 1.5) объединяли с DMF (3.75 мл). Реакционную смесь перемешивали при 60C в течение 16.5 ч. Эту смесь вливали в воду (10 мл) и экстрагировали дважды с помощью EtOAc. Органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали в вакууме. Неочищенное вещество очищали хроматографией на колонке. (6 г силикагеля (63-200 А), элюент: гептан/EtOAc 2:1) с получением указанного в заголовке соединения в виде белого твердого вещества (120 мг, 74.5%).(S)-трет-бутил-2-(4-(5-фенил-1H-пиразол-3-карбоксамидо)фенил)морфолин-4 карбоксилата (120 мг, 268 мкмоль, экв.: 1.00) в диоксане (0.5 мл) добавляли 4 М HCl в диоксане (1.00 мл,4.01 ммоль, экв.: 15). Реакционную смесь перемешивали при 60C в течение 2 ч. К этой смеси затем добавляли 10 мл диоксана, и полученную суспензию отфильтровывали, промывали эфиром и высушивали в высоком вакууме с получением целевого соединения в виде белого твердого вещества (82.3 мг, 79.9%).(S)-2-(4-Бромфенил)морфолин (36.3 г, 150 ммоль) и N,N-диизопропилэтиламин (23.3 г, 31.4 мл,180 ммоль) в THF (360 мл) обрабатывали с помощью ди-трет-бутилдикарбоната (39.3 г, 180 ммоль). Реакционную смесь перемешивали в течение 17 ч при КТ, концентрировали в вакууме, разбавляли этилацетатом, промывали 1 М лимонной кислотой (2100 мл), высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенное вещество кристаллизовали из гексана с получением 47.1 г(18.5 г, 192 ммоль). Полученную темно-коричневую смесь перемешивали при 90C в течение 18 ч. Желто-коричневую реакционную смесь разбавляли толуолом (700 мл), охлаждали до КТ и экстрагировали дважды водой. Органический слой отделяли, высушивали над сульфатом магния и концентрировали в вакууме. Неочищенный продукт разбавляли с помощью 300 мл гексана, перемешивали в течение 1 ч и отфильтровывали с получением оранжевого твердого вещества (68 г), которое очищали хроматографией на колонке (1.3 кг силикагеля, 20% этилацетат/гептан). Объединенные и концентрированные фракции суспендировали в гексане, перемешивали в течение 17 ч, отфильтровывали и высушивали в высоком вакууме, с получением 54.1 г (89%) желтого твердого вещества.MS (ISP): 443.3 ([М+Н]+). Стадия d). (S)-трет-Бутил-2-(4-аминофенил)морфолин-4-карбоксилат. Суспензию (S)-трет-бутил-2-(4-(дифенилметиленамино)фенил)морфолин-4-карбоксилата (54.1 г,122 ммоль), формиата аммония (116 г, 1.83 моль) и Pd/C 5% (6.5 г, 3.06 ммоль) в метаноле (930 мл) перемешивали при 60C в течение 2 ч. Реакционную смесь фильтровали и концентрировали. Остаток растворяли в этилацетате и воде. Органическую фазу экстрагировали дважды с помощью 0.5 М HCl. Объединенные водные фазы подщелачивали с помощью 2 М NaOH и экстрагировали дважды с помощью DCM. Органические фазы высушивали над сульфатом магния, фильтровали и высушивали в вакууме, с получением 31.95 г грязно-белого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-хлорфенил)-1H-пиразол-3-карбоновой кислоты (CAS-595610-50-7) вместо 5-фенил-1H-пиразол-3 карбоновой кислоты. Белое твердое вещество. MS (ISP): 383.12 ([M+Н]+). Пример 3. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(4-фторфенил)-1H-пиразол-3-карбоновой кислоты (CAS-870704-22-6) вместо 5-фенил-1H-пиразол-3 карбоновой кислоты. Белое твердое вещество. MS (ISP): 367.15 ([М+Н]+). Пример 4. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-метоксифенил)-1H-пиразол-3-карбоновой кислоты (CAS-834868-54-1) вместо 5-фенил-1H-пиразол 3-карбоновой кислоты. Белое твердое вещество. MS (ISP): 379.17 ([М+Н]+). Пример 5. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 1-метил-3-фенил-1H-пиразол-5-карбоновой кислоты (CAS-10250-64-3) вместо 5-фенил-1H-пиразол-3 карбоновой кислоты. Белое твердое вещество. MS (ISP): 363.18 ([М+Н]+). Пример 6. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 4-метил-5-фенил-2 Н-пиразол-3-карбоновой кислоты (CAS-879770-33-9) вместо 5-фенил-1H-пиразол-3 карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 363.5 ([M+H]+). Пример 7. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(4-метоксифенил)-2 Н-пиразол-3-карбоновой кислоты (CAS- 27069-16-5) вместо 5-фенил-1H-пиразол-3 карбоновой кислоты. Белое твердое вещество. MS (ISP): 379.4 ([M+H]+). Пример 8. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(2-фторфенил)-2 Н-пиразол-3-карбоновой кислоты (CAS-859155-87-6) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Белое твердое вещество. MS (ISP): 367.1 ([M+H]+). Пример 9. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(2-метоксифенил)-2 Н-пиразол-3-карбоновой кислоты (CAS- 834868-54-1) вместо 3-фенил-1H-пиразол 5-карбоновой кислоты. Белое твердое вещество. MS (ISP): 379.4 ([M+H]+). Пример 10. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(2-хлорфенил)-1H-пиразол-5-карбоновой кислоты (CAS-890621-13-3) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Белое твердое вещество. MS (ISP): 383.2 ([M+H]+). Пример 11. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3,4-диметоксифенил)-2 Н-пиразол-3-карбоновой кислоты (CAS-909857-88-1) вместо 3-фенил-1Hпиразол-5-карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 409.3 ([M+H]+). Пример 12. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью(R)-2-(4-Бромфенил)морфолин (6 г, 24.8 ммоль) и N,N-диизопропилэтиламин (3.84 г, 5.19 мл,29.7 ммоль) в THF (60 мл) обрабатывали ди-трет-бутилдикарбонатом (6.49 г, 29.7 ммоль). Реакционную смесь перемешивали в течение 17 ч при КТ, концентрировали в вакууме, разбавляли этилацетатом, промывали 1 М лимонной кислотой, высушивали над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенное вещество кристаллизовали из смеси гептан/этилацетат с получением 8.48 г (87%)(R)-трет-бутил-2-(4-бромфенил)морфолин-4-карбоксилата в виде белого твердого вещества.(2.12 г, 22.1 ммоль). Полученную темно-коричневую смесь перемешивали при 90C в течение 18 ч. Желто-коричневую реакционную смесь разбавляли толуолом (100 мл), охлаждали до КТ и экстрагировали дважды водой. Органический слой отделяли, высушивали над сульфатом магния и концентрировали в вакууме. Неочищенный продукт разбавляли с помощью 50 мл гексана, перемешивали в течение 1 ч и отфильтровывали, с получением желтого твердого вещества (7.4 г) которое очищали хроматографией на колонке (50 г силикагеля, этилацетат/гептан от 5 до 15%). Объединенные и концентрировали фракции суспендировали в гексане, перемешивали в течение 17 ч, отфильтровывали и высушивали в высоком вакууме, с получением 6.15 г (86%) желтого твердого вещества.MS (ISP): 443.4 ([M+H]+). Стадия d). (R)-трет-Бутил-2-(4-аминофенил)морфолин-4-карбоксилат. Суспензию (R)-трет-бутил-2-(4-(дифенилметиленамино)фенил)морфолин-4-карбоксилата (6 г,13.6 ммоль), формиата аммония (12.8 г, 203 ммоль) и Pd/C 5% (721 мг, 0.339 ммоль) в метаноле (103 мл) перемешивали при 60C в течение 2 ч. Реакционную смесь фильтровали и концентрировали. Полученный остаток растворяли в этилацетате и воде. Органическую фазу экстрагировали дважды с помощью 0.5 М HCl. Объединенные водные фазы подщелачивали с помощью 2 М NaOH и экстрагировали дважды с помощью DCM. Органические фазы высушивали над сульфатом магния, фильтровали и высушивали в вакууме, с получением 3.04 г грязно-белого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(4-хлорфенил)-1H-пиразол-5-карбоновой кислоты (CAS-54006-63-2) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 383.1 ([M+H]+). Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-хлорфенил)-2-метил-2 Н-пиразол-3-карбоновой кислоты Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 1-метил-5-фенил-1H-пиразол-3-карбоновой кислоты (CAS-10199-53-8) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 363.2 ([M+H]+). Пример 19. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-цианофенил)-1H-пиразол-3-карбоновой кислоты (CAS-1242427-10-6) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Светло-коричневое твердое вещество. MS (ISP): 374.0 ([M+H]+). Пример 21. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Светло-коричневое твердое вещество. MS (ISP): 388.0 ([M+H]+). Получение 3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислоты. Стадия а). (Z)-Этил-4-(4-цианофенил)-2-гидрокси-4-оксобут-2-еноат. В сухой колбе в атмосфере аргона добавляли порциями натрий (317 мг, 13.8 ммоль, экв.: 1.00) к этанолу (9.0 мл). (Температура повышалась до 60C). Реакционную смесь охлаждали при 0C. После этого добавляли по каплям диэтилоксалат (2.01 г, 1.87 мл, 13.8 ммоль, экв.: 100), а затем 4-ацетилбензонитрил (2 г, 13.8 ммоль, экв.: 1.00) в этаноле (3.00 мл). Получали белое твердое вещество. Реакционную смесь перемешивали на магнитной мешалке в течение ночи и отслеживали ход реакции с помощью ТСХ. Затем реакционную смесь концентрировали в вакууме. Полученный остаток охлаждали при 0C и в колбу добавляли воду. К этому раствору добавляли 1 М HCl (рН 3), затем полученный раствор экстрагировали два раза с помощью EtOAc. Органический слой промывали три раза с помощью 20 мл солевого раствора. Полученный органический слой высушивали над MgSO4, фильтровали и концентрировали в вакууме с получением грязно-белого твердого вещества. Это грязно-белое твердое вещество смешивали с эфиром при 0C. Суспензию фильтровали с получением белого твердого вещества(2.082 г, 61.6%). Стадия b). Этил-3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоксилат. В атмосфере аргона растворяли (Z)-этил-4-(4-цианофенил)-2-гидрокси-4-оксобут-2-еноат (500 мг,2.04 ммоль, экв.: 1.00) в этаноле (10 мл) при КТ. Добавляли по каплям метилгидразин (95.9 мг, 110 мкл,2.04 ммоль, экв.: 1.00) (раствор становился желтым). Этот раствор перемешивали в течение ночи при КТ с последующим нагреванием 6 ч при 50C, охлаждали до КТ и концентрировали в вакууме. Остаток непосредственно очищали хроматографией на колонке (20 г) смесью гептан/EtOAc: 9/1 с получением желаемого пиразола (173 мг, 33.2%) в виде белого твердого вещества.MS (ISP): 256.3 ([M+H]+). Стадия с). 3-(4-Цианофенил)-1-метил-1H-пиразол-5-карбоновая кислота. К раствору этил-3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоксилата (70 мг, 274 мкмоль, экв.: 1.00) в THF (5 мл) и MeOH (1.00 мл) добавляли 1 М LiOH (548 мкл, 548 мкмоль, экв.: 2). Эту смесь перемешивали в течение примерно 8 ч при КТ, затем обрабатывали водой и 1 н. HCl (рН 3). Эту смесь экстрагировали два раза этилацетатом. Полученные органические слои объединяли, промывали солевым раствором и высушивали над MgSO4, фильтровали и концентрировали с получением желаемого соединения(55 мг, 88.3%) в виде белого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(4-Фторфенил)-2-метил-1H-пиразол-3-карбоновой кислоты гидро Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-метоксифенил)-2-метил-1H-пиразол-3-карбоновой кислоты Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(3-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Светло-коричневое твердое вещество. MS (ISP): 388.0 ([M+H]+). Получение 3-(3-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислоты. По аналогии с 3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислотой, как описано в примере 21. Пример 25. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(3-цианофенил)-1-этил-1H-пиразол-5-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 402.1 ([M+H]+). Получение 3-(3-цианофенил)-1-этил-1H-пиразол-5-карбоновой кислоты. По аналогии с 3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислотой, как описано в примере 21. Пример 26. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 3-(4-цианофенил)-1-этил-1H-пиразол-5-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Грязно-белое твердое вещество. MS (ISP): 402.1 ([M+H]+). Получение 3-(4-цианофенил)-1-этил-1H-пиразол-5-карбоновой кислоты. По аналогии с 3-(4-цианофенил)-1-метил-1H-пиразол-5-карбоновой кислотой, как описано в примере 21. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-цианофенил)-4-метил-1H-пиразол-3-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Коричневое твердое вещество. MS (ISP): 388.1 ([M+H]+). Получение 5-(3-цианофенил)-4-метил-1H-пиразол-3-карбоновой кислоты. Стадия а). (Z)-4-(3-Цианофенил)-1-этокси-3-метил-1,4-диоксобут-2-ен-2-олат лития. При перемешивании на магнитной мешалке к 1 М раствору LiHMDS в THF (7.94 мл, 7.94 ммоль,экв.: 1.00) в Et2O (50 мл) при -78C добавляли по каплям раствор 3-пропионилбензонитрила (1.264 г,7.94 ммоль, экв.: 1.00) в Et2O (10.0 мл) в атмосфере аргона. Перемешивали смесь при той же температуре еще в течение 45 мин, после чего добавляли по каплям диэтилоксалат (1.22 г, 1.13 мл, 8.34 ммоль, экв.: 1.05). Реакционную смесь нагревали до КТ и перемешивали в течение 3 суток. Образовавшийся преципитат собирали фильтрацией, промывали диэтиловым эфиром и высушивали в вакууме с получением желаемой литиевой соли в виде желтого твердого вещества (929 мг, 44.1%). Стадия b). Этил-5-(3-цианофенил)-4-метил-1H-пиразол-3-карбоксилат. К раствору (Z)-4-(3-цианофенил)-1-этокси-3-метил-1,4-диоксобут-2-ен-2-олата лития (400 мг,1.51 ммоль, экв.: 1.00) в этаноле (10 мл) добавляли гидразин гидрохлорид (113 мг, 1.65 ммоль, экв.: 1.093) при КТ с получением оранжевого раствора. Полученную смесь перемешивали в течение ночи при этой же температуре. Через 1 сутки растворитель удаляли при пониженном давлении и к смеси добавляли солевой раствор. Этот раствор экстрагировали два раза с помощью AcOEt и объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали с получением желаемого соединения в виде желтой смолы (114 мг, 26.6%).MS (ISP): 256.0 ([M+H]+). Стадия с). 3-(3-Цианофенил)-1-метил-1H-пиразол-5-карбоновая кислота. К раствору этил-5-(3-цианофенил)-4-метил-1H-пиразол-3-карбоксилата (100 мг, 392 мкмоль, экв.: 1.00) в THF (5 мл) в MeOH (1.00 мл) добавляли 1 М LiOH (2.35 мл, 2.35 ммоль, экв.: 6). Эту смесь перемешивали в течение ночи. К полученному остатку добавляли воду и 1 н. HCl (рН 1), эту водную фазу экстрагировали два раза этилацетатом, полученные органические слои объединяли и промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали в вакууме с получением желаемого соединения (52 мг, 52.6%) в виде желтого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(5-циано-2-фторфенил)-1H-пиразол-3-карбоновой кислоты вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Коричневое твердое вещество. MS (ISP): 392.0 ([M+H]+). Получение 5-(5-циано-2-фторфенил)-1H-пиразол-3-карбоновой кислоты. По аналогии с 5-(3-цианофенил)-4-метил-1H-пиразол-3-карбоновой кислотой, как описано в примере 21. Пример 29. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 1-фенил-1H-пиразол-5-карбоновой кислоты (CAS-1133-77-3) вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Белое твердое вещество. MS (ISP): 349.1 ([M+H]+). Пример 31. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 47 с помощью 2-бром-5-хлорпиридина вместо 2-хлор-5-(трифторметил)пиримидина на стадии b). Грязно-белое твердое вещество. MS (ISP): 384.2 ([M+H]+). Пример 33. Указанное в заголовке соединение получали по аналогии с примером 47 с помощью 2-бром-5-(трифторметил)пиридина вместо 2-хлор-5-(трифторметил)пиримидина на стадии b). Белое твердое вещество. MS (ISP): 418.2 ([M+H]+). Пример 34. Указанное в заголовке соединение получали по аналогии с примером 47 с помощью 2-бром-бензонитрила вместо 2-хлор-5-(трифторметил)пиримидина на стадии b). Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-цианофенил)-1H-пиразол-3-карбоновой кислоты (CAS-1242427-10-6) вместо 3-фенил-1H-пиразол-5 карбоновой кислоты. Белое твердое вещество. MS (ISP): 372.0 ([M+H]+). Пример 37. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью Указанное в заголовке соединение получали по аналогии с примером 1 с помощью трет-бутилового эфира 3-(4-аминофенил)-пирролидин-1-карбоновой кислоты (CAS-908334-28-1) вместо (S)-трет-бутил-2(4-аминофенил)морфолин-4-карбоксилата и 5-(3-цианофенил)-1H-пиразол-3-карбоновой кислоты (CAS1242427-10-6) вместо 3-фенил-1H-пиразол-5-карбоновой кислоты. Белое твердое вещество. MS (ISP): 357.8 ([M+H]+). Указанное в заголовке соединение получали по аналогии с примером 1 с помощью 5-(3-циано-4 фторфенил)-1H-пиразол-3-карбоновой кислоты (полученной, как указано ниже: a-d) вместо 3-фенил-1Hпиразол-5-карбоновой кислоты. Белое твердое вещество. MS (ISP): 392.1 ([M+H]+). Получение 5-(3-циано-4-фторфенил)-1H-пиразол-3-карбоновой кислоты. а) (Z)-4-(3-Бром-4-фторфенил)-1-этокси-1,4-диоксобут-2-ен-2-олат лития. При перемешивании на магнитной мешалке к 1 М раствору LiHMDS в THF (9.22 мл, 9.22 ммоль,экв.: 1) добавляли Et2O (31.2 мл) при -78C с получением желтого раствора. К этой смеси добавляли по каплям в атмосфере аргона раствор 1-(3-бром-4-фторфенил)этанона (2 г, 9.22 ммоль, экв.: 1.00) в Et2O(15.6 мл). Эту смесь затем перемешивали при той же температуре еще в течение 45 мин, после чего добавляли по каплям диэтилоксалат (1.41 г, 1.31 мл, 9.68 ммоль, экв.: 1.05). Реакционную смесь нагревали до КТ и перемешивали еще в течение 2 суток. Образовавшийся преципитат собирали фильтрацией, промывали диэтиловым эфиром и высушивали в вакууме с получением желаемой литиевой соли в виде светло-желтого твердого вещества (2.677 г, 89.9%).b) Этил-5-(3-бром-4-Фторфенил)-1H-пиразол-3-карбоксилат. К раствору (Z)-4-(3-бром-4-фторфенил)-1-этокси-1,4-диоксобут-2-ен-2-олата лития (600 мг,1.86 ммоль, экв.: 1.00) в этаноле (25 мл) добавляли гидразин моногидрат (139 мг, 2.03 ммоль, экв.: 1.093) при КТ с получением белой суспензии, через 1 ч эта суспензия превращалась в раствор. Полученную смесь перемешивали в течение ночи. Через 1 сутки реакция была завершена. После перемешивания растворитель удаляли при пониженном давлении и к смеси добавляли солевой раствор, этот раствор экстрагировали два раза с помощью AcOEt и объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением этил-5-(3-бром-4-фторфенил)-1H-пиразол-3-карбоксилата в виде белого твердого вещества (460 мг, 79.1%).c) Этил-5-(3-циано-4-фторфенил)-1H-пиразол-3-карбоксилат. Смесь этил-5-(3-бром-4-фторфенил)-1H-пиразол-3-карбоксилата (300 мг, 958 мкмоль, экв.: 1.00),цианида цинка (65.2 мг, 556 мкмоль, экв.: 0.58) и Pd(PPh3)4 (111 мг, 95.8 мкмоль, экв.: 0.1) нагревали при 160C в DMF (2 мл) (держали над микрофильтром) в течение 30 мин в микроволновом шкафу. Эту смесь делили между EtOAc (40 мл) и 2 н. NH4OH (40 мл). Органическую фазу экстрагировали с помощью 2 н.NH4OH, промывали солевым раствором, высушивали над MgSO4 и концентрировали в вакууме. Неочищенную смесь очищали хроматографией на колонке (10 г), элюент:гептан/EtOAc: 95/5, с получением желаемого нитрильного соединения в виде белого кристаллического твердого вещества (180 мг, 72.5%).d) 5-(3-Циано-4-фторфенил)-1H-пиразол-3-карбоновая кислота. К раствору этил-5-(3-циано-4-фторфенил)-1H-пиразол-3-карбоксилата (180 мг, 694 мкмоль, экв.: 1.00) в THF (5.00 мл) и MeOH (1 мл) добавляли 1 М LiOH (4.17 мл, 4.17 ммоль, экв.: 6). Эту смесь перемешивали в течение ночи. После добавления LiOH раствор становился оранжевым. К полученному остатку добавляли воду и 1 н. HCl (рН 1), эту водную фазу экстрагировали два раза этилацетатом; полученные органические слои объединяли и промывали солевым раствором. Затем высушивали над MgSO4,фильтровали и концентрировали с получением желаемого соединения (45 мг, 22.4%) в виде белого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 1 с помощью(220 мг, 3.21 ммоль, экв.: 1.093) при КТ с получением оранжевой суспензии. Полученную смесь перемешивали в течение ночи при этой же температуре. Через 1 сутки реакция была завершена. После перемешивания растворитель удаляли при пониженном давлении и к этой смеси добавляли солевой раствор,этот раствор экстрагировали два раза с помощью AcOEt и объединенные органические слои высушивали над MgSO4, фильтровали и концентрировали с получением желаемого соединения в виде светлокоричневого твердого вещества (630 мг, 79.9%).b) 5-(3-Дифторметоксифенил)-2H-пиразол-3-карбоновая кислота. К раствору метилового эфира 5-(3-дифторметоксифенил)-2H-пиразол-3-карбоновой кислоты(620 мг, 2.31 ммоль, экв.: 1.00) в THF (10 мл) и MeOH (2.00 мл) добавляли 1 М LiOH (13.9 мл,13.9 ммоль, экв.: 6) с получением коричневого раствора. Эту смесь перемешивали в течение ночи. После добавления LiOH этот раствор становился. К полученному остатку добавляли воду и 1 н. HCl (рН 1), эту водную фазу экстрагировали два раза этилацетатом, полученные органические слои объединяли и промывали солевым раствором. Затем высушивали над MgSO4, фильтровали и концентрировали с получением желаемого соединения (510 мг, 86.8%) в виде светло-желтого твердого вещества. а) (S)-трет-Бутил-2-(4-(3-(3-(дифторметокси)фенил)-1H-пиразол-5-карбоксамидо)фенил)морфолин 4-карбоксилат. В круглодонной колбе на 25 мл, 3-(3-(дифторметокси)фенил)-1H-пиразол-5-карбоновую кислоту(515 мг, 2.03 ммоль, экв.: 1.1) (получение описано в примере 40), (S)-трет-бутил-2-(4 аминофенил)морфолин-4-карбоксилат (513 мг, 1.84 ммоль, экв.: 1.00) (получение описано в примере 1),N-Метилморфолин (559 мг, 608 мкл, 5.53 ммоль, экв.: 3) и HBTU (1.05 г, 2.76 ммоль, экв.: 1.5) объединяли с DMF (2 мл) с получением желтого раствора. Реакционную смесь перемешивали в течение ночи при 60C. Эту смесь вливали в воду (10 мл) и экстрагировали дважды с помощью EtOAc. Органические слои промывали с помощью NaHCO3, солевого раствора, высушивали над MgSO4, фильтровали и концентрировали в вакууме с получением коричневой неочищенной смеси. Смесь разбавляли гептаном, перемешивали в течение 15 мин и эту суспензию фильтровали. Полученное твердое вещество промывали несколько раз гептаном с получением желаемого соединения в виде коричневого твердого вещества (550 мг,58.0%).(S)-трет-бутил-2-(4-(3-(3-(дифторметокси)фенил)-1-этил-1H-пиразол-5 карбоксамидо)фенил)морфолин-4-карбоксилат. К смеси (S)-трет-бутил-2-(4-(3-(3-(дифторметокси)фенил)-1H-пиразол-5-карбоксамидо)фенил)морфолин-4-карбоксилата (70 мг, 136 мкмоль, экв.: 1.00) и карбоната калия (41.4 мг, 299 мкмоль, экв.: 2.2) в DMF (2 мл) добавляли йодэтан (25.5 мг, 13.2 мкл, 163 мкмоль, экв.: 1.2) и перемешивали в течение ночи при КТ. К полученной смеси добавляли воду, органическую фазу экстрагировали водой и солевым раствором, затем органический слой высушивали над MgSO4, отфильтровывали и концентрировали в вакууме с получением неочищенного соединения в виде смеси изомеров, которую разделяли хроматографией на колонке (картридж на 10 г) с получением (S)-трет-бутил-2-(4-(5-(3-(дифторметокси)фенил)-1 этил-1H-пиразол-3-карбоксамидо)фенил)морфолин-4-карбоксилата (17 мг, 23.0%) и (S)-трет-бутил-2-(4(3-(3-(дифторметокси)фенил)-1-этил-1H-пиразол-5-карбоксамидо)фенил)морфолин-4-карбоксилатаc) (S)-5-(3-Дифторметокси)фенил)-1-этил-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3-карбоксамид гидрохлорид. К раствору (S)-трет-бутил-2-(4-(5-(3-(дифторметокси)фенил)-1-этил-1H-пиразол-3-карбоксамидо)фенил)морфолин-4-карбоксилата (17 мг) в диоксане (40.8 мкл) добавляли 4 М HCl в диоксане (117 мкл,470 мкмоль, экв.: 15). Реакционную смесь перемешивали при 60C в течение ночи. К этой смеси добавляли 2 мл диэтилового эфира и перемешивали в течение 15 мин при комнатной температуре. Эту смесь фильтровали и концентрировали в высоком вакууме с получением желаемого гидрохлорида в виде белого твердого вещества (9 мг, 60.0%).(40.8 мкл) добавляли 4 М HCl в диоксане (117 мкл, 470 мкмоль, экв.: 15). Реакционную смесь перемешивали при 60C в течение ночи. К этой смеси добавляли 2 мл диэтилового эфира и перемешивали в течение 15 мин при комнатной температуре. Эту смесь фильтровали и концентрировали в высоком вакууме с получением желаемого гидрохлорида в виде белого твердого вещества (15 мг, 63%). Указанное в заголовке соединение получали по аналогии с примером 39 с помощью 1-(3-бром-2 фторфенил)этанона вместо 1-(3-бром-4-фторфенил)этанона. Белое твердое вещество. Указанное в заголовке соединение получали по аналогии с примером 42 с помощью метилиодида вместо йодэтана. Белое твердое вещество. MS (ISP): 429.1 ([M+H]+). Пример 45. а) Этил-2-хлор-2-(2-(4-(дифторметокси)фенил)гидразон)ацетат. 4-(Дифторметокси)анилин (796 мг, 5 ммоль) растворяли в тетрафторборной кислоте (2.38 г, 1.7 мл,13.0 ммоль) и воде (2 мл). После охлаждения до 0C медленно добавляли раствор нитрита натрия(345 мг, 5.0 ммоль) в воде (0.75 мл). Эту смесь перемешивали в течение 30 мин, собирали густой преципитат фильтрацией и промывали диэтиловым эфиром (около 3 мл). Полученное светло-розовое твердое вещество растворяли в 1.5 мл ацетона и добавляли 5 мл диэтилового эфира. После перемешивания в течение 15 мин с охлаждением полученное белое твердое вещество фильтровали, промывали диэтиловым эфиром и высушивали в высоком вакууме в течение 15 мин с получением 4-дифторметоксифенилдиазония тетрафторбората. Эту соль диазония (851 мг, 3.3 ммоль) добавляли к раствору этил-2-хлор-3-оксобутирата (494 мг,420 мкл, 3 ммоль) в пиридине (0.8 мл) и воде (0.8 мл). Полученную очень густую суспензию перемешивали при -5C в течение 30 мин. Твердое вещество фильтровали, промывали ледяной водой и высушивали в вакууме с получением оранжевого твердого вещества (0.67 г, 76%).(4 мл) и добавляли 2,5-норборнадиен (906 мг, 1 мл, 9.83 ммоль) и триэтиламин (587 мг, 808 мкл,5.8 ммоль). Реакционную смесь перемешивали при 70C в течение 30 мин и оставляли перемешиваться при комнатной температуре в течение ночи. Полученное твердое вещество отфильтровывали и промывали толуолом. Органическую фракцию выпаривали, полученный остаток растворяли в ксилоле (12 мл) и нагревали при температуре флегмы в течение 2 ч. Растворитель выпаривали и полученный остаток очищали хроматографией на колонке (50 г силикагеля, дихлорметан) с получением 387 мг (69%) светложелтого твердого вещества.c) 1-(4-Дифторметокси)фенил)-1H-пиразол-3-карбоновой кислоты. К раствору этил-1-(4-(дифторметокси)фенил)-1H-пиразол-3-карбоксилата (350 мг, 1.24 ммоль) в смеси THF (3.1 мл), метанола (1.6 мл) и воды (1.6 мл) добавляли гидроксид лития гидрат (89 мг,3.72 ммоль). Этот раствор нагревали до 80C в течение 2 ч. Большую часть органического растворителя удаляли при пониженном давлении. Добавляли раствор бикарбоната натрия и этилацетат, органический слой отделяли. Водный слой подкисляли добавлением 25% водной хлороводородной кислоты и эту смесь экстрагировали 2 раза этилацетатом. Органические слои объединяли, высушивали (MgSO4) и выпаривали. Полученный продукт высушивали в вакууме и непосредственно использовали на следующей стадии.N-метилморфолин (119 мг, 130 мкл, 1.18 ммоль) объединяли с DMF (2 мл) с получением светло-желтого раствора. Реакционную смесь перемешивали при 50C в течение 17 ч. Реакционную смесь вливали в 25 мл воды и дважды экстрагировали этилацетатом. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (20 г силикагеля, этилацетат в гептане от 30 до 50%) с получением грязно-белого твердого вещества(S)-трет-Бутил-2-(4-(1-(4-(дифторметокси)фенил)-1H-пиразол-3-карбоксамидо)фенил)морфолин-4 карбоксилат (130 мг, 0.25 ммоль) растворяли в диоксане (0.6 мл) и добавляли раствор HCl в диоксане (4 М, 0.12 мл, 3.8 ммоль). Реакционную смесь перемешивали в течение ночи при 60C. После охлаждения добавляли эфир, полученное твердое вещество отфильтровывали, промывали эфиром и высушивали в вакууме с получением (S)-1-(4-(дифторметокси)фенил)-N-(4-(морфолин-2-ил)фенил)-1H-пиразол-3 карбоксамида гидрохлорида (90 мг, 79%) в виде грязно-белого твердого вещества. гидро Указанное в заголовке соединение получали по аналогии с примером 45 с помощью(S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилат (1.39 г, 5 ммоль). Этот раствор охлаждали до 0C и к реакционной смеси добавляли по каплям 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4 метилморфолиний хлорид (1.8 г, 6.5 ммоль), растворенный в 5 мл метанола, в течение 1 ч. Реакционную смесь перемешивали при 0C в течение 2 ч, затем в течение ночи при комнатной температуре. Растворитель выпаривали, полученный остаток растворяли в дихлорметане и адсорбировали на силикагеле. Полученное вещество очищали флэш-хроматографией (силикагель, 20 г, EtOAc в гептане от 30 до 50%) с получением белого твердого вещества (1.61 г; 86%), которое использовали на следующей стадии.(33 мг,0.089 ммоль) и 2-хлор-5-(трифторметил)пиримидин (16.2 мг, 0.089 ммоль) растворяли в DMSO (0.7 мл) и добавляли карбонат калия (24.5 мг, 0.177 ммоль). Реакционную смесь помещали на шейкер Bchi на время 20 ч при 120C. После охлаждения смеси добавляли воду с последующей двукратной экстракцией этилацетатом. Объединенные органические слои высушивали над MgSO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (силикагеля, 5 г, EtOAc в гептане от 25 до 50%) с получением грязно-белого твердого вещества (11 мг, 24%).(S)-трет-Бутил-2-(4-(1-(5-(трифторметил)пиримидин-2-ил)-1H-пиразол-3 карбоксамидо)фенил)морфолин-4-карбоксилат (11 мг, 21.2 мкмоль) растворяли в диоксане (80 мкл), добавляли раствор HCl в диоксане (79.6 мкл, 318 мкмоль) и эту реакционную смесь перемешивали при 60C в течение 2 ч. После охлаждения добавляли диэтиловый эфир, полученное твердое вещество фильтровали и промывали диэтиловым эфиром с получением (S)-N-(4-(морфолин-2-ил)фенил)-1-(4(трифторметокси)фенил)-1H-пиразол-4-карбоксамида гидрохлорида (7 мг, 70%) в виде грязно-белого твердого вещества. Указанное в заголовке соединение получали по аналогии с примером 47 с помощью 2,6 дихлорпиразина вместо 2-хлор-5-(трифторметил)пиримидина на стадии b). Желтое твердое вещество.(90 мг,0.24 ммоль) и 2,3-дихлорпиразин (43 мг, 0.29 ммоль) растворяли в диметилацетамиде (2 мл) и добавляли карбонат калия (67 мг, 0.48 ммоль). Реакционную смесь помещали на шейкер Bchi на время 16 ч при 80C. Для завершения реакции вносили дополнительное количество 2,3-дихлорпиразина (10 мг) и продолжали нагревать еще в течение 2 ч при 120C. После охлаждения смеси добавляли воду с последующей двукратной экстракцией этилацетатом. Объединенные органические слои высушивали над MgSO4 и выпаривали. Неочищенное вещество очищали флэш-хроматографией (силикагель, 10 г, EtOAc в гептане от 25 до 50%) с получением грязно-белой смолы (48 мг, 42%).(340 мкл, 1.36 ммоль) и реакционную смесь перемешивали при 60C в течение 90 мин. Этот растворитель выпаривали и полученный остаток перекристаллизовывали из смеси этилацетата и этанола с получением светло-желтого твердого вещества (27 мг, 70%). Указанное в заголовке соединение получали по аналогии с примером 49 с помощью 2,5-дихлорпиразина вместо 2,3-дихлорпиразина на стадии а). Грязно-белое твердое вещество. MS (ISP): 385.2 ([35ClM+H]+), 387.2 ([37ClM+H]+). ([M+H]+). Пример 51. Указанное в заголовке соединение получали по аналогии с примером 49 с помощью 4-хлор-6-(трифторметил)пиримидина вместо 2,3-дихлорпиразина на стадии а). Белое твердое вещество. MS (ISP): 419.2 ([M+H]+). Пример 52. Указанное в заголовке соединение получали по аналогии с примером 49 с помощью 2-йод-6-(трифторметил)пиразина вместо 2,3-дихлорпиразина на стадии а). Светло-желтое твердое вещество. MS (ISP): 419.2 ([M+H]+). Указанное в заголовке соединение получали по аналогии с примером 49 с помощью 5-бромпиразин-2-карбонитрила вместо 2,3-дихлорпиразина на стадии а). Светло-желтое твердое вещество. MS (ISP): 376.3 ([M+H]+). Пример 54. Указанное в заголовке соединение получали по аналогии с примером 49 с помощью 4-хлор-2-(трифторметил)пиримидина вместо 2,3-дихлорпиразин на стадии а). Светло-зеленое твердого вещества. MS (ISP): 419.2 ([M+H]+). Соединения формулы I и их фармацевтически приемлемые аддитивные соли обладают ценными фармакологическими свойствами. В частности, было обнаружено, что соединения по настоящему изобретению обладают высоким сродством к рецепторам следовых аминов (TAARs), в частности TAAR1. Эти соединения исследовали в соответствии с тестом, приведенным ниже. Материалы и методы. Конструирование плазмид экспрессии TAAR и получение стабильно трансфицированных клеточных линий. Для конструирования плазмид экспрессии кодирующие последовательности TAAR1 человека, крысы и мыши были амплифицированы из геномной ДНК, в целом, как описано в литературе (Lindemann etal. [14]). Использовали систему Expand High Fidelity PCR System (Roche Diagnostics) с 1.5 мМ Mg2+ и очищенные продукты ПЦР клонировали в вектор клонирования pCR2.1-TOPO (Invitrogen) согласно инструкции производителя. Продукты ПЦР субклонировали в вектор pIRESneo2 (BD Clontech, Palo Alto,Калифорния), верифицировали векторы экспрессии по последовательности, после чего вводили их в клеточные линии. Клетки HEK-293 (АТССCRL-1573) культивировали в целом так, как описано в литературе(Lindemann et al. (2005. Для получения стабильно трансфицированных клеточных линий HEK-293 клетки трансфицировали с помощью плазмид экспрессии pIRESneo2, содержащих TAAR-кодирующие последовательности (описанные выше) с помощью Lipofectamin 2000 (Invitrogen) согласно инструкции производителя, и через 24 ч после трансфекции в культуральную среду добавляли 1 мг/мл G418 (Sigma,Buchs, Швейцария). По истечении примерно 10-дневного периода культивирования клоны выделяли,наращивали и тестировали на восприимчивость к следовым аминам (все соединения были заказаны в компании Sigma) с помощью иммуноферментной системы (ИФА) cAMP Biotrak Enzyme immunoassay(Amersham), следуя процедуре ИФА без ацетилирования, рекомендованной производителем. Моноклональные линии клеток, которые демонстрировали стабильную EC50 на протяжении периода культивирования, составляющего 15 пассажей, были использованы во всех ниже описанных исследованиях. Радиолигандный иммуноанализ на TAAR1 крысы. Получение мембран и связывание радиолиганда. Клетки HEK-293, стабильно экспрессирующие крысиный TAAR1, выдерживали при 37C и 5% СО 2 в высокоглюкозной среде DMEM, содержащей фетальную сыворотку коровы (10%, термоинактивированную в течение 30 мин при 56C), пенициллин/стрептомицин (1%) и 375 мкг/мл генетицин (Gibco). Клетки высвобождали из культуральных флаконов с помощью трипсина/EDTA, собирали, промывали дважды ледяным PBS (без Са 2+ и Mg2+), осаждали при 1000 об/мин в течение 5 мин при 4C, замораживали и хранили при -80C. Замороженные гранулы суспендировали в 20 мл HEPES-NaOH (20 мМ, рН 7.4),содержащем 10 мМ EDTA, и гомогенизировали с помощью Polytron (PT 6000, Kinematica) при 14000 об/мин в течение 20 с. Гомогенат центрифугировали при 48000g в течение 30 мин при 4C. Затем супернатант удаляли и отбрасывали, осадок ресуспендировали в 20 мл HEPES-NaOH (20 мМ, рН 7.4),содержащем 0.1 мМ EDTA, с помощью Polytron (20 с при 14000 об/мин). Эту процедуру повторяли и конечный осадок ресуспендировали в HEPES-NaOH, содержащем 0.1 мМ EDTA, и гомогенизировали с помощью Polytron. Как правило, аликвоты мембран по 2 мл хранили -80C. Для каждой новой партии мембран определяли константу диссоциации (Kd) по кривой насыщения. Использовали радиолиганд 3WO 2008/098857) в концентрации, равной вычисленной величине Kd, которая составляла обычно около 2.3 нМ, что давало конечное связывание около 0.2% радиолиганда и специфическое связывание, составляющее примерно 85% от общего связывания. Неспецифическое связывание определяли как количество 3[Н]-(S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламина, связавшегося в присутствии 10 мкМ немеченного лиганда. Все соединения тестировали в широком диапазоне концентраций (от 10 пМ до 10 мкМ) в дупликатах. Тестовые соединения (20 мкл/на лунку) переносили на 96-луночный планшет с глубокими лунками (TreffLab) и добавляли 180 мкл HEPES-NaOH (20 мМ, рН 7.4), содержащего MgCl2[H]-(S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламина в концентрации 3.3Kd в нМ и 500 мкл мембран (ресуспендированных в концентрации 50 мкг белка на 1 мл). Проводили инкубацию в 96-луночных планшетах с глубокими лунками в течение 1 ч при 4C. Инкубацию прерывали путем быстрой фильтрации через планшет Unifilter-96 (Packard Instrument Company) и стеклянные фильтры GF/C(Perkin Elmer), предварительно замоченные в течение 1 ч в полиэтиленимине (0.3%), промывали 3 раза по 1 мл холодным связывающим буфером. После добавления 45 мкл Microscint 40 (PerkinElmer) планшетUnifilter-96 закрывали пленкой и через 1 ч считывали радиоактивность с помощью сцинтилляционного счетчика для микропланшетов TopCount (Packard Instrument Company). Радиолигандный иммуноанализ на мышиных TAAR1. Получение мембран и связывание радиолиганда. Клетки HEK-293, стабильно экспрессирующие мышиный TAAR1, выдерживали при 37C и 5% CO2 в высокоглюкозной среде DMEM, содержащей фетальную сыворотку коровы (10%, термоинактивированной в течение 30 мин при 56C), пенициллин/стрептомицин (1%) и 375 мкг/мл генетицин (Gibco). Клетки высвобождали из культуральных флаконов с помощью трипсина/EDTA, собирали, промывали дважды ледяным PBS (без Ca2+ и Mg2+), осаждали при 1000 об/мин в течение 5 мин при 4C, замораживали и хранили при -80C. Замороженные гранулы суспендировали в 20 мл HEPES-NaOH (20 мМ, рН 7.4),содержащем 10 мМ EDTA, и гомогенизировали с помощью Polytron (PT 6000, Kinematica) при 14000 об/мин в течение 20 с. Гомогенат центрифугировали при 48000g в течение 30 мин при 4C. Затем супернатант удаляли и отбрасывали, осадок ресуспендировали в 20 мл HEPES-NaOH (20 мМ, рН 7.4),содержащем 0.1 мМ EDTA, с помощью Polytron (20 с при 14000 об/мин). Эту процедуру повторяли и конечный осадок ресуспендировали в HEPES-NaOH, содержащем 0.1 мМ EDTA, и гомогенизировали с помощью Polytron. Как правило, аликвоты мембран по 2 мл хранили -80C. Для каждой новой партии мембран определяли константу диссоциации (Kd) по кривой насыщения. Использовали радиолиганд 3WO 2008/098857) в концентрации, равной вычисленной величине Kd, которая составляла обычно около 0.7 нМ, что давало конечное связывание около 70% от общего связывания. Неспецифическое связывание определяли как количество 3[Н]-(S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламина, связавшегося в присутствии 10 мкМ немечненого лиганда. Все соединения тестировали в широком диапазоне концентраций (от 10 пМ до 10 мкМ) в дупликатах. Тестовые соединения (20 мкл/на лунку) переносили на 96-луночный планшет с глубокими лунками (TreffLab) и добавляли 180 мкл HEPES-NaOH (20 мМ, рН 7.4), содержащего MgCl2 (10 мМ) и CaCl2 (2 мМ) (связывающий буфер), 300 мкл радиолиганда 3[H]-(S)-4-[(этилфениламино)метил]-4,5-дигидрооксазол-2-иламина в концентрации 3.3Kd в нМ и 500 мкл мембран (ресуспендированных в концентрации 60 мкг белка на 1 мл). Проводили инкубацию в 96-луночных планшетах с глубокими лунками в течение 1 ч при 4C. Инкубацию прерывали быстрой фильтрацией через планшет Unifilter-96 (Packard Instrument Company) и стеклянные фильтры GF/C(Perkin Elmer), предварительно замоченные в течение 1 ч в полиэтиленимине (0.3%), промывали 3 раза по 1 мл холодным связывающим буфером. После добавления 45 мкл Microscint 40 (PerkinElmer) планшетUnifilter-96 закрывали пленкой и через 1 ч считывали радиоактивность с помощью сцинтилляционного счетчика для микропланшетов TopCount (Packard Instrument Company). Соединения характеризуются величинами Ki (мкМ) в мышиных или крысиных TAAR1 в диапазоне 0.01 мкМ, приведенными в нижеследующей таблице. Соединения формулы IA и IB и фармацевтически приемлемые соли соединений формулы IA и IB можно применять в качестве лекарственных средств, например в форме фармацевтических препаратов. Такие фармацевтические препараты можно вводить орально, например в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Кроме того, введение можно проводить также ректально, например в форме суппозиториев, или парентерально, например в форме растворов для инъекций. Соединения формулы IA и IB можно процессировать совместно с фармацевтически инертными неорганическими или органическими носителями для получения фармацевтических препаратов. В качестве таких носителей для таблеток, таблеток, покрытых оболочкой, драже и твердых желатиновых капсул,например, можно использовать лактозу, кукурузный крахмал или его производные, тальк, стеариновые кислоты или ее соли и т.п. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые вещества и жидкие полиолы и т.п. При этом в зависимости от природы действующего вещества в случае мягких желатиновых капсул носители, как правило, не требуются. Подходящими носителями для изготовления растворов и сиропов являются, например,вода, полиолы, глицерин, растительное масло и т.п.
МПК / Метки
МПК: A61K 31/4245, A61P 25/00, C07D 413/14, C07D 413/12
Метки: производные, пиразола
Код ссылки
<a href="https://eas.patents.su/30-24648-proizvodnye-pirazola.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиразола</a>
Производные пиразола, минеральное удобрение в твердом виде, обработанное неорганической или органической кислотой и ингибитором нитрификации на основе производного пиразола, способ получения обработанного минерального удобрения и способ внесения удобрения

Номер патента: 2098
Опубликовано: 24.12.2001
Авторы: Барт Томас, Риттингер Штефан, Гольд Рандалл Ивэн, Эрхард Клаус, Дрессель Юрген, Лайбольд Эдгар, Рибер Норберт, Хорхлер Фон Локквенг Клаус
МПК: C05G 3/08, C07D 231/16
Метки: неорганической, внесения, виде, получения, органической, производного, основе, минеральное, нитрификации, производные, кислотой, удобрение, ингибитором, обработанного, минерального, пиразола, удобрения, способ, твердом, обработанное
Формула / Реферат:
1. Производные пиразола общей формулы где а) R1 и R2 означают метил и R3 - водород, или б) R1 означает хлор, R2 - метил и R3 - водород, или в) R1 - хлор, метил, R2 - метил, R3 - гидроксиметил, причем в случаях а) и б) производное пиразола имеется в виде аддитивной соли с фосфорной кислотой. 2. Минеральное удобрение в твердом виде, обработанное, по меньшей мере, одной неорганической или органической кислотой и, по меньшей мере, одним...
Производные 1н-тиено [2,3-c] пиразола, предназначенные для использования в качестве ингибиторов киназы

Номер патента: 11032
Опубликовано: 30.12.2008
Авторы: Вианелло Паола, Тезеи Даниа, Фанчелли Даниеле, Бинди Симона, Виольо Серджо, Варази Марио
МПК: A61P 17/06, A61P 19/02, A61K 31/4162...
Метки: киназы, ингибиторов, качестве, использования, 1н-тиено, пиразола, 2,3-c, производные, предназначенные
Формула / Реферат:
1. Способ лечения клеточных пролиферативных расстройств, вызванных и/или связанных с измененной активностью протеинкиназы, включающий введение нуждающемуся в нем млекопитающему эффективного количества производного 1Н-тиено[2,3-c]пиразола формулы (I) где R представляет собой фенил или 5- или 6-членную гетероарильную группу, содержащую 1 или 2 гетероатома, выбранных из N, S или О, где R необязательно замещен в любом из его свободных положений 1-6...
Производные пиразола для лечения инфекции вируса иммунодефицита человека (вич)

Номер патента: 7184
Опубликовано: 25.08.2006
Авторы: Стаппл Пол Энтони, Селби Мэттью Данкан, Маубрей Чарлз Эрик, Прайс Дейвис Энтони, Джоунз Лин Хауард
МПК: A61K 31/415, A61K 31/4155, A61K 31/4162...
Метки: производные, пиразола, лечения, вич, иммунодефицита, инфекции, человека, вируса
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемые соль, сольват или производное, где либо R1 представляет собой Н, С1-С6алкил, С3-С7циклоалкил, фенил, бензил, галогено, -CN, -OR7, -CO2R10, -CONR5R10, R8 или R9, причем указанные С1-С6алкил, С3-С7циклоалкил, фенил и бензил возможно замещены галогено, -CN, -OR10, S(O)xR10, -CO2R10, -CONR5R10, -OCONR5R10, -NR5CO2R10, -NR10R11, -NR5COR10, -SO2NR5R10,-NR5CONR5R10, -NR5SO2R10 или R10; и R2...
Производные пиразола, фармацевтические композиции и их применение

Номер патента: 12431
Опубликовано: 30.10.2009
Авторы: Сюн Юйшэн, Го Цзянь, Парми Эмма Р., Броканьер Линда, Лян Жуй
МПК: A61K 31/4155, A61K 31/415, C07D 231/12...
Метки: применение, композиции, фармацевтические, производные, пиразола
Формула / Реферат:
1. Соединение, представленное формулой I или его фармацевтически приемлемая соль, где каждый R1 означает Н или выбран из группы, состоящей из: (a) галогена или ОН, (b) C1-6алкила или OC1-6алкила, необязательно замещенного 1-5 галогеногруппами вплоть до образования пергалогеналкильной группы; каждый R2 выбран из R1, определенного выше, или две R2 группы, взятые вместе, могут образовывать конденсированную 5-6-членную циклическую структуру,...
Производные пиразола

Номер патента: 179
Опубликовано: 24.12.1998
Авторы: Сибата Мицуру, Сакамото Масаси, Ямамото Хироси, Камано Хидеки
МПК: A01N 43/56, C07D 409/10
Метки: пиразола, производные
Формула / Реферат:
1. Производное пиразола формулы (I): где R1 представляет C1-C4 алкильную группу, C2-С4 алкенильную группу или C2-C4 галогеналкенильную группу; R2 представляет атом водорода, C1-C4 алкильную группу, C1-C4 галогеналкильную группу или С2-С4 алкоксиалкильную группу; Х представляет C1-C4 алкильную группу, C1-C4 галогеналкильную группу, C2-C4 алкоксиалкильную группу, атом галогена, C1-C4 алкоксигруппу или C1-C4 галогеналкоксигруппу; р равно...
Предыдущий патент: Ингибиторы hsp90
Следующий патент: Оценка уровней текучей среды в системе погружного винтового насоса
Случайный патент: Система и способ для определения местоположения аномалии в пласте, окружающем скважину