Ингибиторы вируса гепатита с
Номер патента: 24171
Опубликовано: 31.08.2016
Авторы: Сринивасу Потхуканури, Лопез Омар Д., Бендер Джон А., Рампулла Ричард А., Гупта Самаямунтула Венката Сатиа Арун Кумар, Белема Маконен, Чен Ки, Минвелл Николас А.





















Формула / Реферат
1. Соединение формулы (II)

или его фармацевтически приемлемая соль,
где каждый D представляет собой NH;
L представляет собой связь или фенил;
каждый из Z1 и Z2 независимо выбран из СН и N;
каждый из Z3 и Z4 независимо выбран из С и N, при условии, что не более двух из Z1, Z2, Z3 и Z4 представляют собой N;
A представляет собой четырех-шестичленное кольцо, необязательно содержащее один, два или три гетероатома, независимо выбранных из атома азота и серы;
R1 и R2 независимо выбраны из атома водорода и CH3, где указанный метил может необязательно образовывать конденсированное трехчленное кольцо с другим кольцевым атомом углерода;
R3 представляет собой -C(O)R5;
R4 представляет собой -C(O)R6;
R5 и R6 независимо представляют собой C1-4алкил, замещенный NHCOOCH3 и/или тетрагидропиранилом, необязательно замещенным двумя группами CH3;
при этом соединение не является [1-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-3-метилбутирил)пирролидин-2-ил]-3H-имидазол-4-ил}фенил)нафталин-1-ил]-1H-имидазол-2-ил}пирролидин-1-карбонил)-2-метилпропил]карбаминовой кислоты метиловым эфиром.
2. Соединение формулы (III)

или его фармацевтически приемлем соль,
где каждый D представляет собой NH;
Q представляет собой фенил;
X представляет собой СН2;
Y представляет собой СН2;
каждый из Z1 и Z2 независимо выбран из CH и N;
каждый из Z3 и Z4 независимо выбран из С и N, при условии, что не более двух из Z1, Z2, Z3 и Z4 представляют собой N;
A представляет собой четырех-шестичленное кольцо, необязательно содержащее один, два или три гетероатома, независимо выбранных из атома азота и серы;
R1 и R2 независимо выбраны из атома водорода и CH3, где указанный метил может необязательно образовывать конденсированное трехчленное кольцо с другим кольцевым атомом углерода;
R3 представляет собой -C(O)R5;
R4 представляет собой -C(O)R6;
R5 и R6 независимо представляют собой C1-4алкил, замещенный NHCOOCH3 и/или тетрагидропиранилом, необязательно замещенным двумя группами CH3;
R7 и R8 независимо представляют собой атомы водорода;
при этом соединение не является [1-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-3-метилбутирил)пирролидин-2-ил]-3H-имидазол-4-ил}фенил)нафталин-1-ил]-1H-имидазол-2-ил}пирролидин-1-карбонил)-2-метилпропил]карбаминовой кислоты метиловым эфиром.
3. Соединение, выбранное из




или его фармацевтически приемлемая соль.
4. Композиция для лечения инфекции вирусом гепатита С (ВГС) у пациента, содержащая соединение по п.1 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
5. Композиция по п.4, дополнительно содержащая один или два дополнительных соединения, обладающих противовирусной активностью в отношении ВГС.
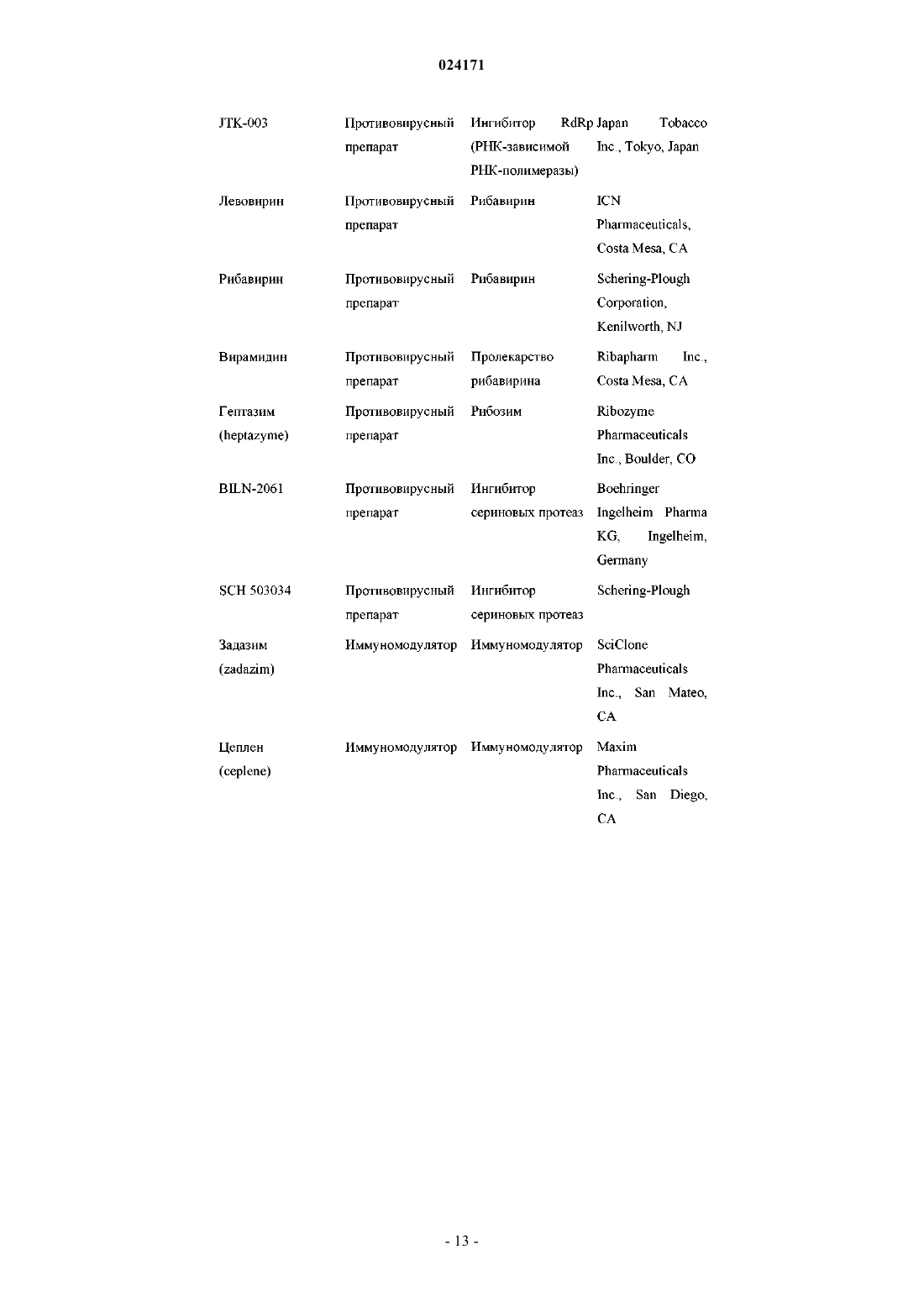
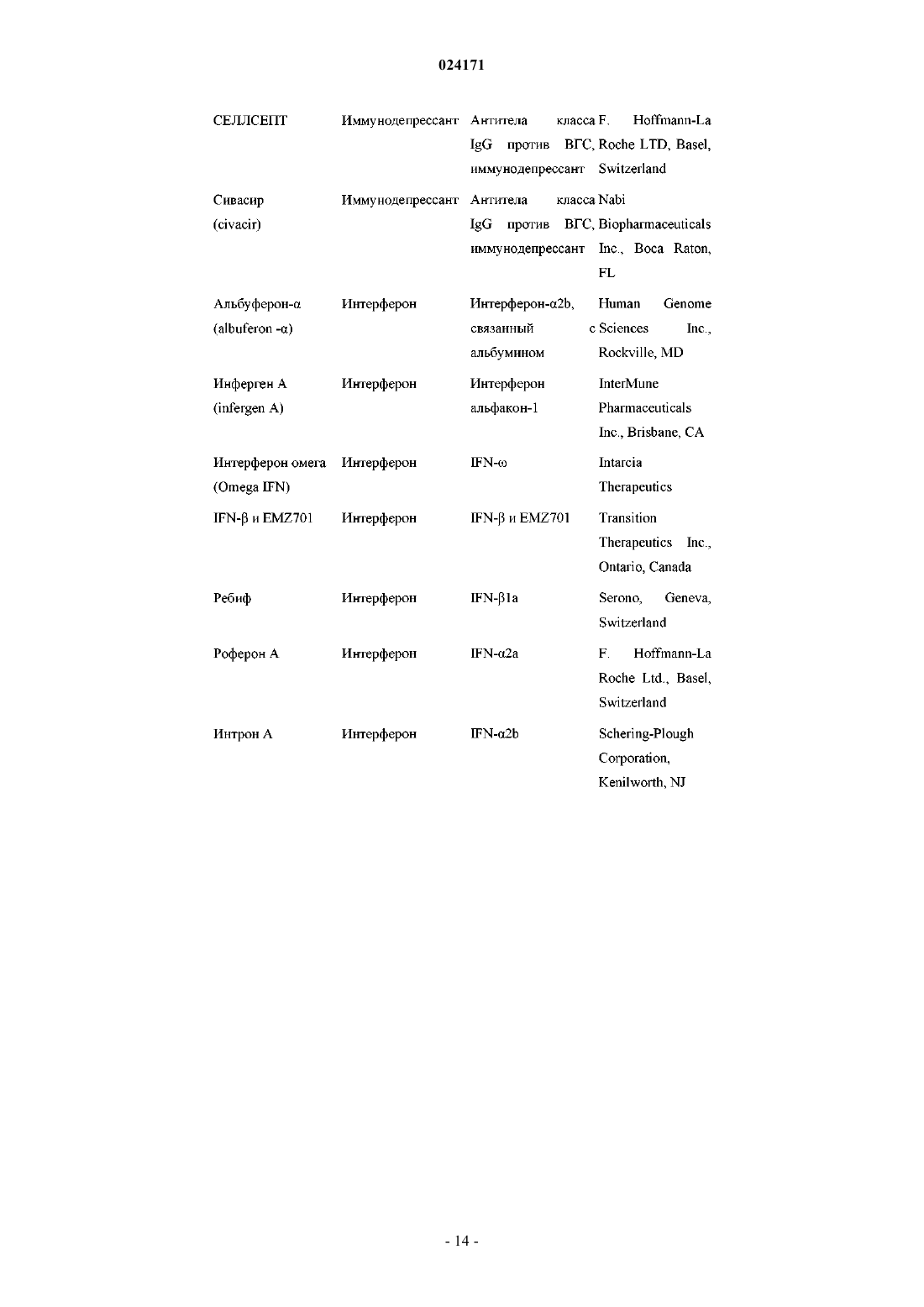
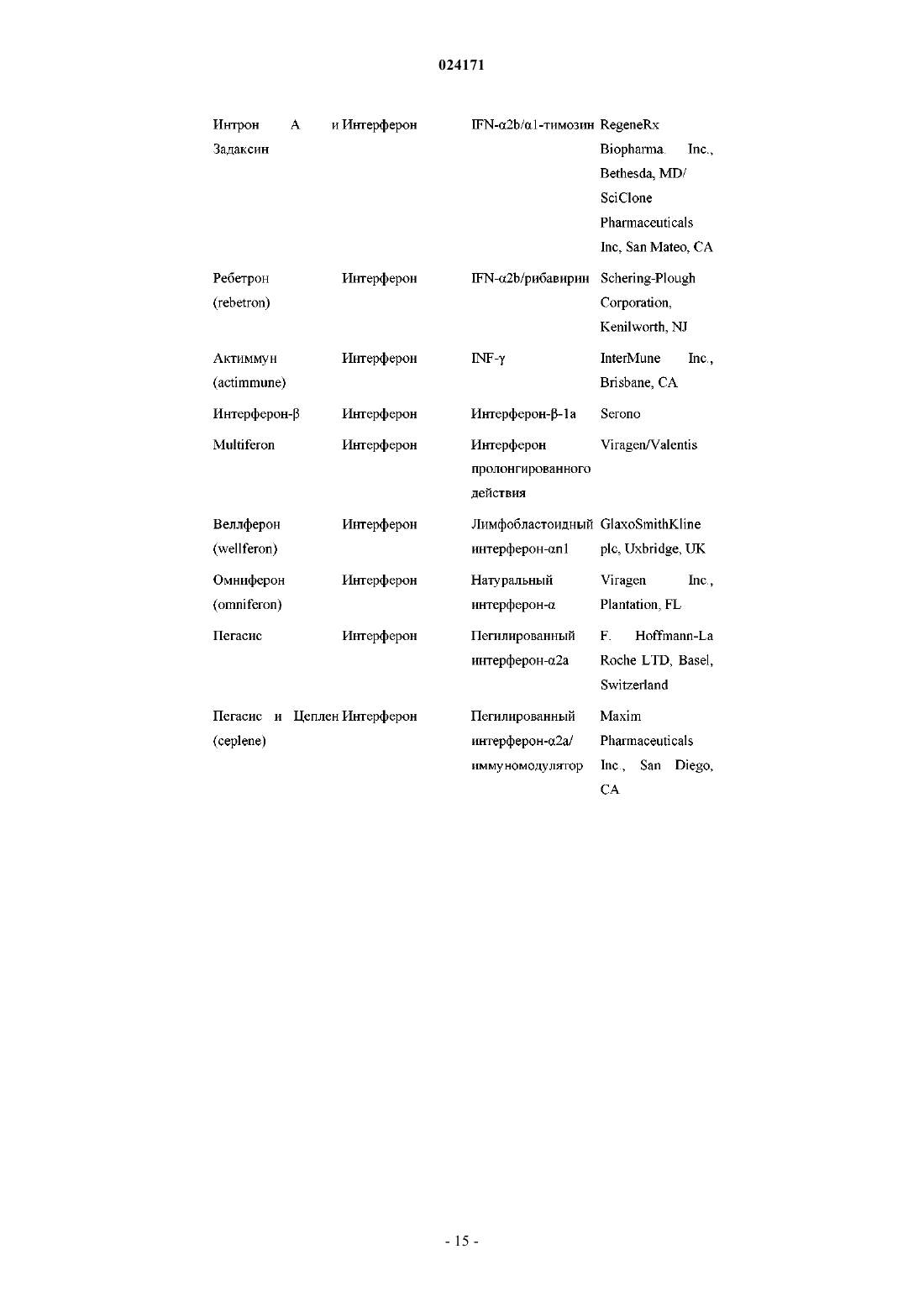
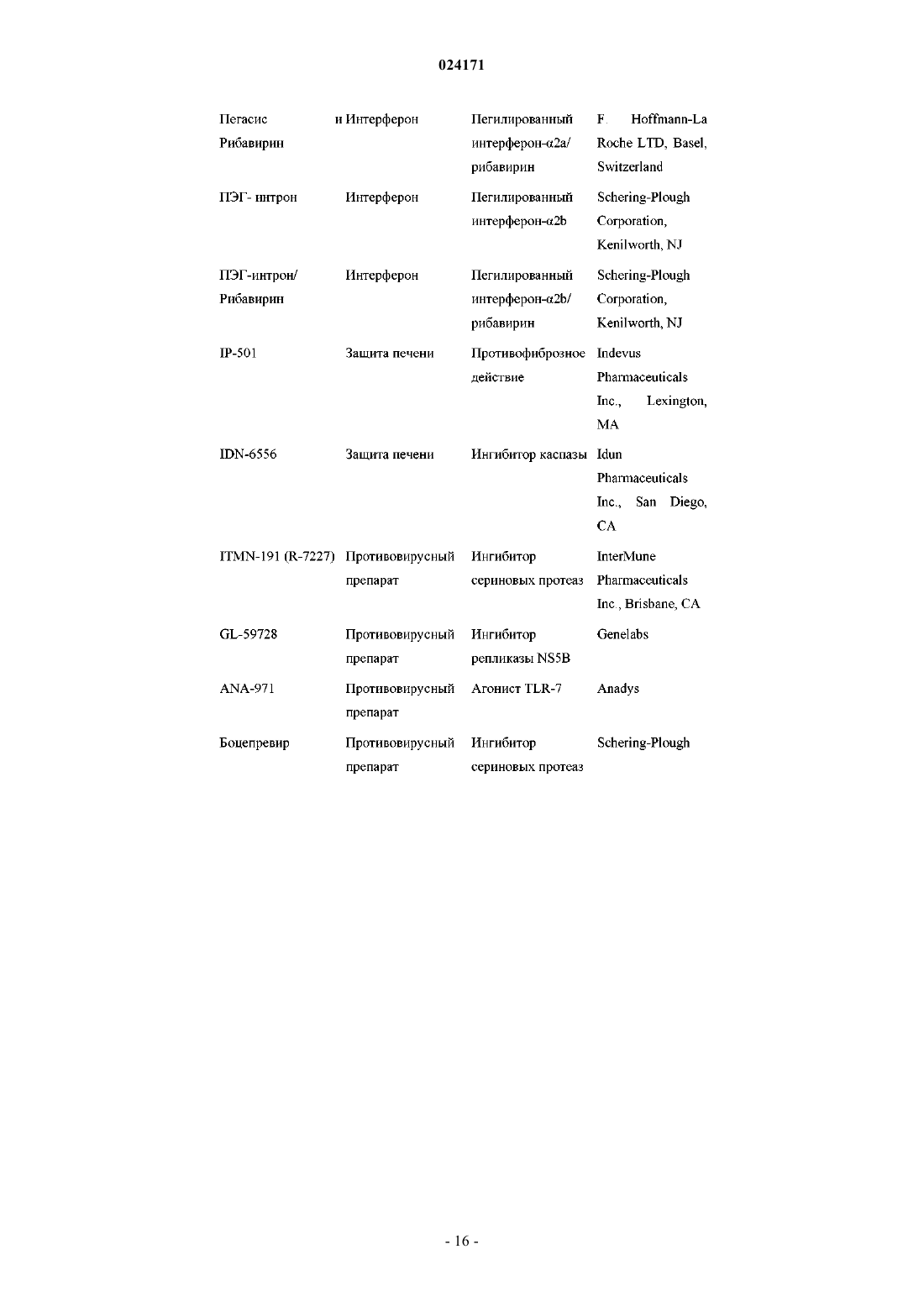
6. Композиция по п.5, в которой по меньшей мере одно из дополнительных соединений представляет собой интерферон или рибавирин.
7. Композиция по п.6, в которой интерферон выбран из интерферона альфа 2B, пегилированного интерферона альфа, пегилированного интерферона ламбда, консенсусного интерферона, интерферона альфа 2A и лимфобластоидного интерферона тау.
8. Композиция по п.5, в которой по меньшей мере одно из дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы ВГС, сериновой протеазы ВГС, полимеразы ВГС, хеликазы ВГС, белка NS4B ВГС, проникновения ВГС в клетку, сборки вирионов ВГС, выхода вирионов ВГС из клетки, белка NS5A ВГС и IMPDH, для лечения ВГС-инфекции.
9. Способ лечения ВГС-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли.
10. Способ по п.9, дополнительно включающий введение одного или двух дополнительных соединений, обладающих противовирусной активностью в отношении ВГС, до введения соединения по п.1 или его фармацевтически приемлемой соли, после указанного введения или одновременно с соединением по п.1 или его фармацевтически приемлемой солью.
11. Способ по п.10, отличающийся тем, что по меньшей мере одно из дополнительных соединений представляет собой интерферон или рибавирин.
12. Способ по п.9, отличающийся тем, что интерферон выбран из интерферона альфа 2B, пегилированного интерферона альфа, пегилированного интерферона ламбда, консенсусного интерферона, интерферона альфа 2A и лимфобластоидного интерферон тау.
13. Способ по п.9, отличающийся тем, что по меньшей мере одно из дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы ВГС, сериновой протеазы ВГС, полимеразы ВГС, хеликазы ВГС, белка NS4B ВГС, проникновения ВГС в клетку, сборки вирионов ВГС, выхода вирионов ВГС из клетки, белка NS5A ВГС и IMPDH, для лечения ВГС-инфекции.
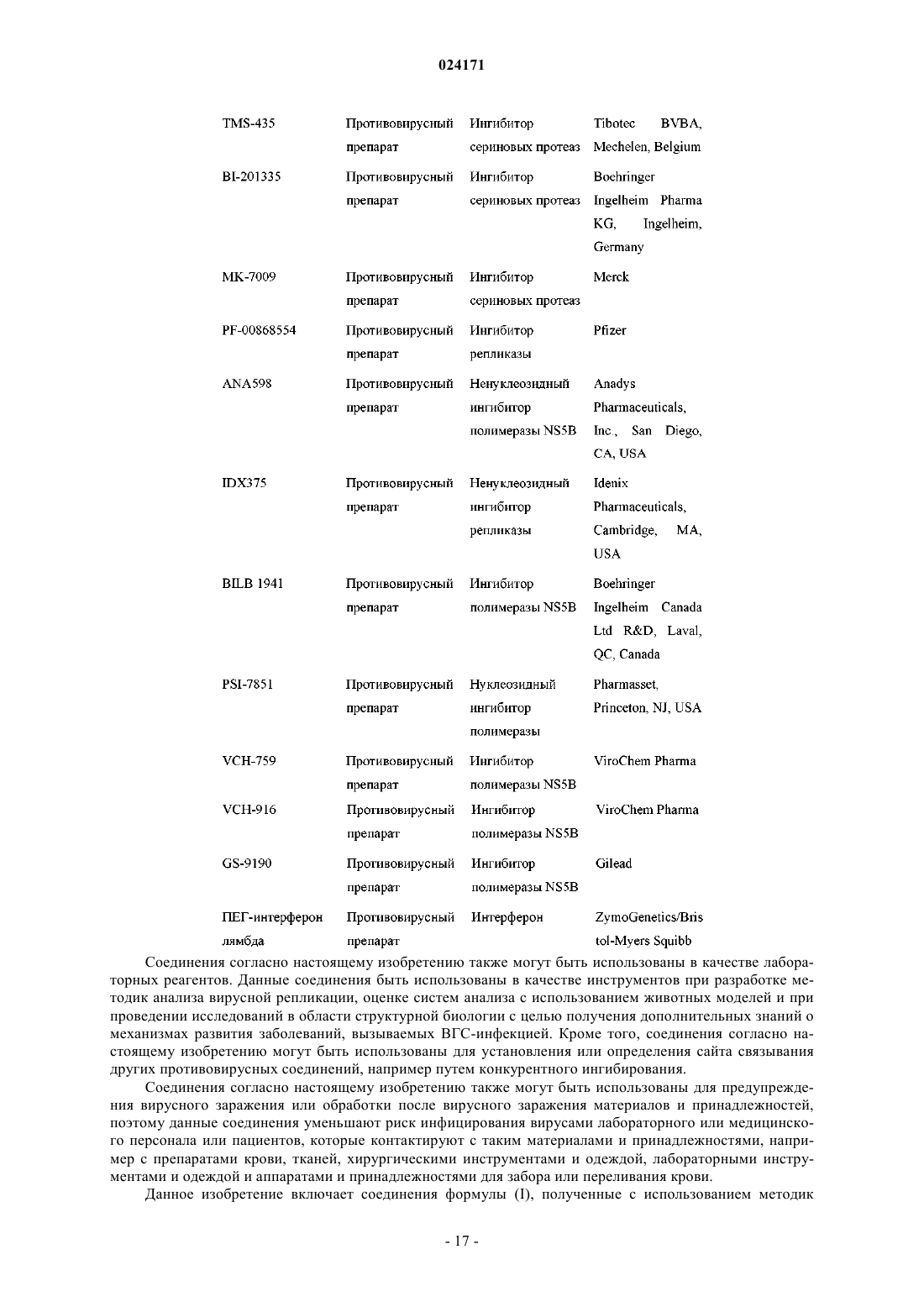
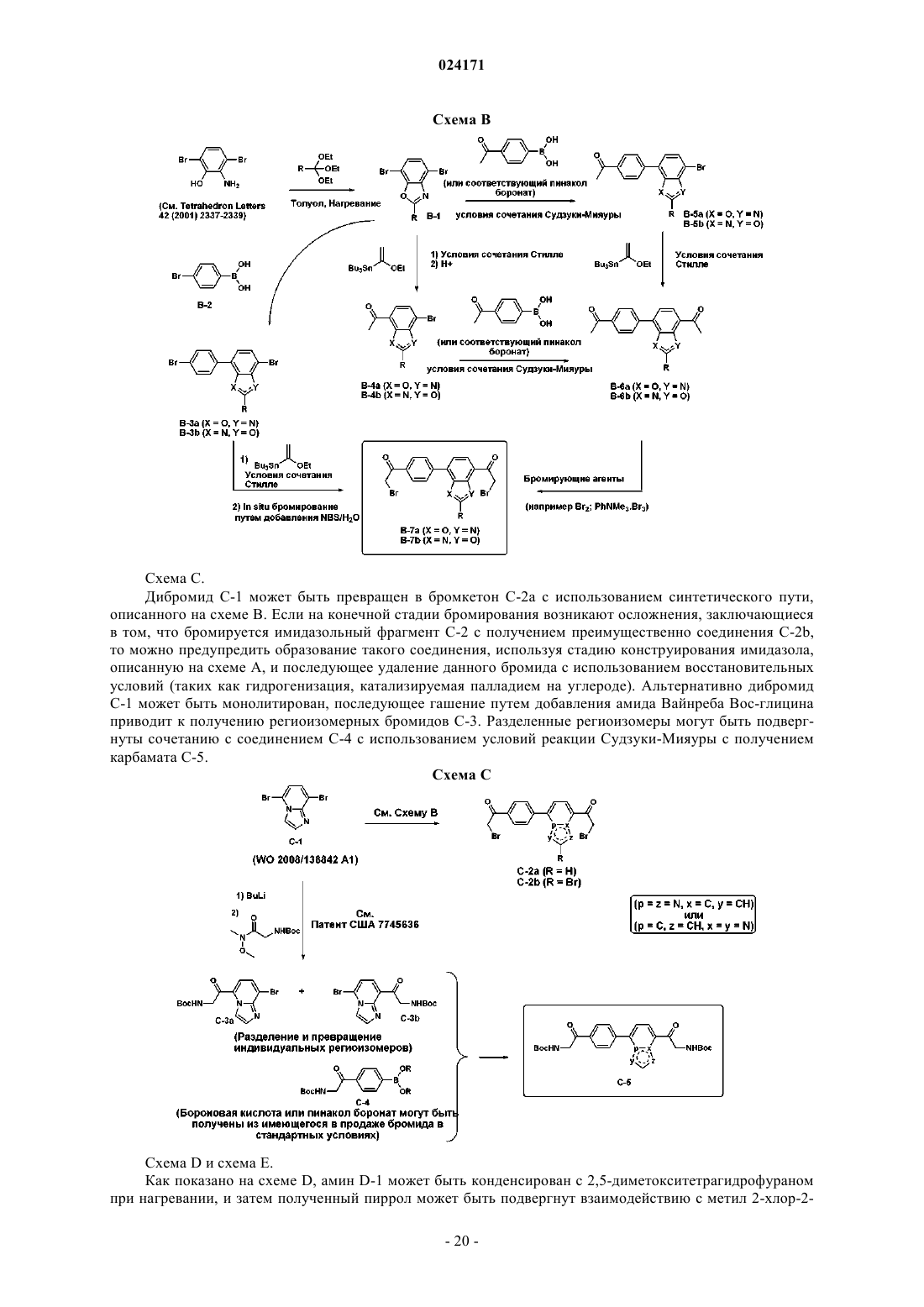
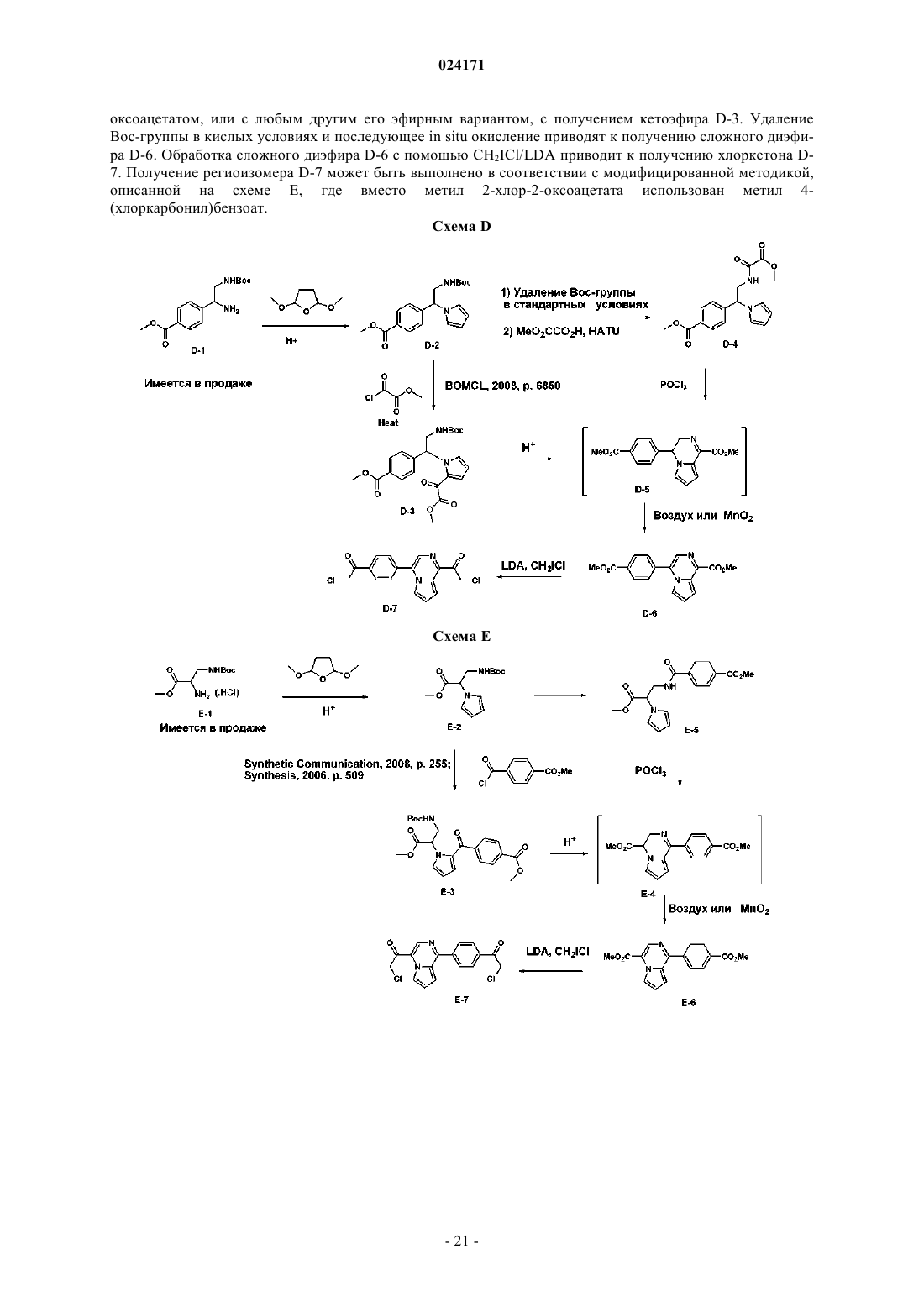
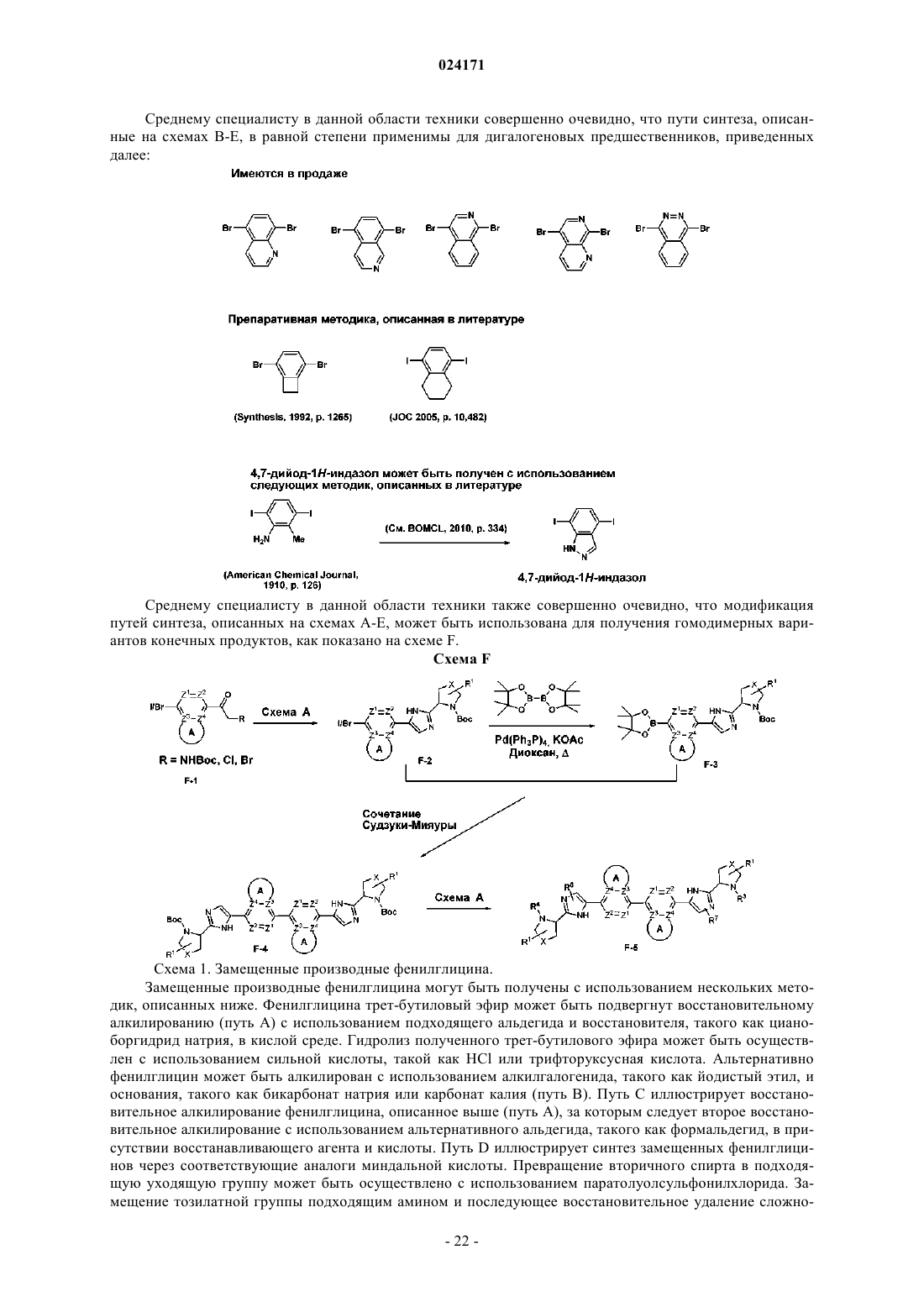
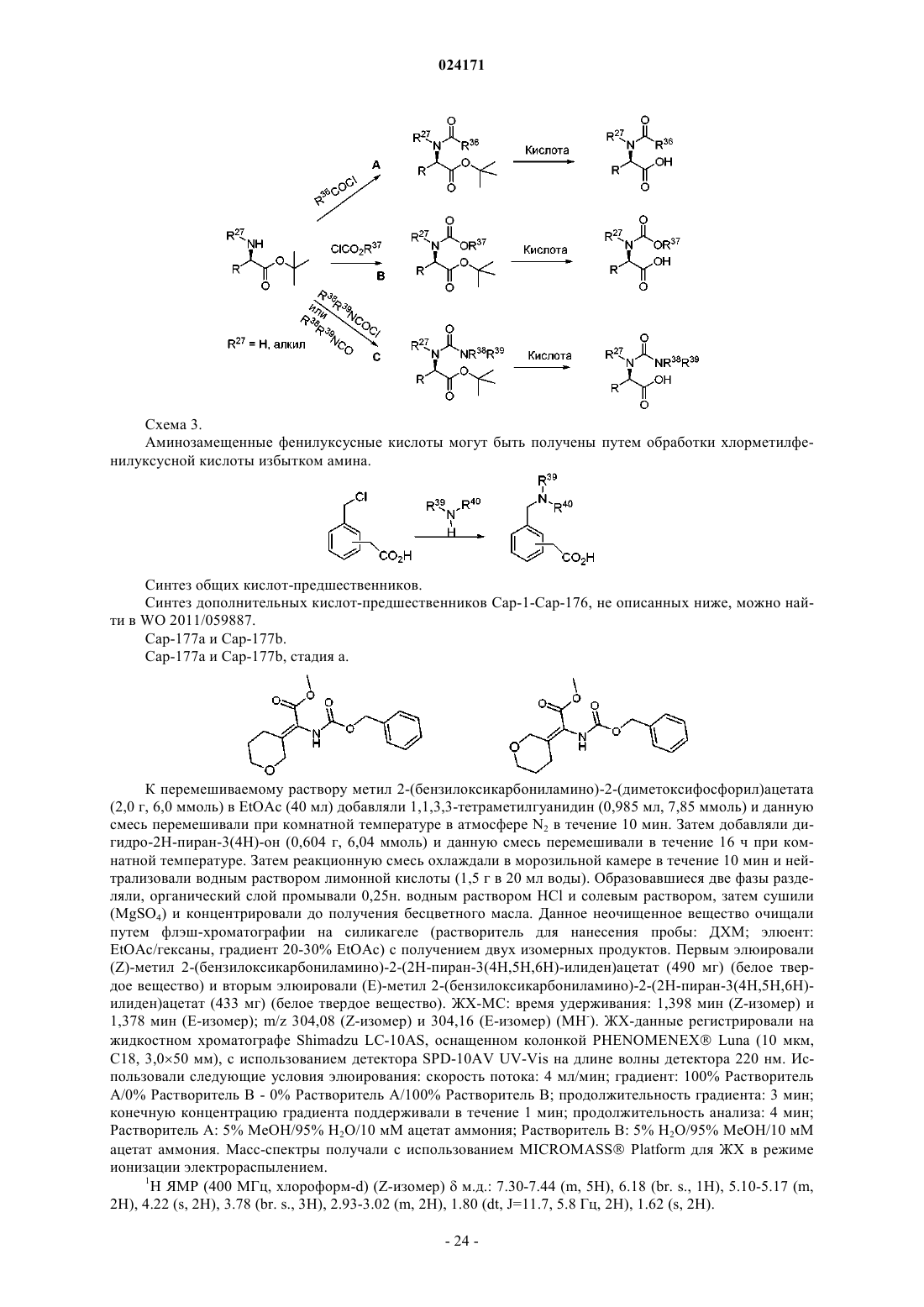
Текст
ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С Настоящее изобретение относится к соединениям, композициям и способам лечения инфекции,обусловленной вирусом гепатита C (ВГС). Также описаны фармацевтические композиции,содержащие указанные соединения, и способы применения данных соединений для лечения ВГСинфекции.(US), Сринивасу Потхуканури, Гупта Самаямунтула Венката Сатиа Арун Кумар (IN) Лыу Т.Н. (RU) Перекрестная ссылка на родственные заявки Заявка на данный патент испрашивает приоритет согласно предварительной заявке на патент США 61/373070, поданной 12 августа 2010 г. Настоящее изобретение в целом относится к противовирусным соединениям и, в частности, к соединениям, которые могут ингибировать функцию белка NS5A, кодируемого вирусом гепатита С (ВГС),к композициям, содержащим указанные соединения, и к способам ингибирования функции белка NS5A. ВГС является самым распространенным патогеном человека, которым инфицировано приблизительно 170 млн человек во всем мире, что примерно в пять раз превышает количество инфицированных вирусом иммунодефицита человека типа 1. У существенной части ВГС-инфицированных индивидуумов в дальнейшем развивается тяжелое прогрессирующее заболевание печени, включая цирроз печени и печеночно-клеточный рак. В настоящее время стандартное лечение ВГС, которое включает использование комбинации пегилированного интерферона и рибавирина, имеет неоптимальную частоту достижения стойкого вирусологического ответа и вызывает многочисленные побочные эффекты. Очевидно, что назрела необходимость в разработке эффективной терапии, предназначенной для решения данной медицинской проблемы. ВГС относится к вирусам, содержащим одноцепочечную (+)РНК. На основе сравнения предсказанной аминокислотной последовательности и обширного сходства в нетранслируемой 5'-области РНК ВГС относят к отдельному роду семейства флавивирусов (Flaviviridae). Вирусные частицы всех членов семейства флавивирусов представляют собой покрытые оболочкой вирионы, геном которых состоит из одноцепочечной (+)РНК, которая кодирует все известные специфичные вирусные белки и транслируется как одна непрерывная открытая рамка считывания. Значительная гетерогенность нуклеотидной и кодируемой аминокислотной последовательности,найденная в различных областях генома ВГС, обусловлена высокой частотой ошибок, возникающих при репликации, выполняемой кодируемой вирусом РНК-зависимой РНК-полимеразой, которая лишена корректирующей активности. Охарактеризовано по меньшей мере шесть основных генотипов и описано более 50 подтипов, имеющих широкое распространение. Клиническое значение генетической гетерогенности ВГС проявляется в тенденции возникновения мутаций при использовании монотерапии, поэтому желательно использовать дополнительные схемы лечения. Возможность модулирующего воздействия генотипов на патогенез и терапию остается невыясненной. Геном ВГС, представляющий собой одноцепочечную РНК, содержит приблизительно 9500 нуклеотидов и имеет единственную открытую рамку считывания (ORF), кодирующую один большой полипротеин, состоящий из приблизительно 3000 аминокислот. В инфицированных клетках данный полипротеин расщепляется в нескольких сайтах клеточными и вирусными протеазами, что приводит к образованию структурных и неструктурных (NS) белков. У ВГС образование зрелых неструктурных белков (NS2, NS3,NS4A, NS4B, NS5A и NS5B) зависит от двух вирусных протеаз. Первая протеаза предположительно является металлопротеазой и расцепляет связь NS2-NS3; вторая протеаза (N-концевая область NS3, в данном описании также именуемая NS3-протеазой) является сериновой протеазой и расщепляет полипротеин во всех сайтах, следующих за NS3, как в цис-положении, а именно по сайту NS3-NS4A, так и в трансположении, по остальным сайтам NS4A-NS4B, NS4B-NS5A, NS5A-NS5B. Белок NS4A, по-видимому,является многофункциональным белком и играет роль кофактора NS3-протеазы и участвует в мембранной локализации NS3 и других компонентов репликации вируса. Образование NS3-NS4A комплекса необходимо для формирования специфической протеазной активности и приводит к увеличению эффективности протеолитического расщепления. Белок NS3 также обладает активностью нуклеозидтрифосфатазы и РНК-хеликазы. NS5B (в данном описании также именуемый ВГС-полимеразой) представляет собой РНК-зависимую РНК-полимеразу и вместе с другими ВГС-белками, включая NS5A, в составе репликазного комплекса участвует в репликации ВГС. В качестве соединений, пригодных для лечения ВГС-инфицированных пациентов, желательно использовать соединения, которые селективно ингибируют репликацию ВГС. В частности, желательно использовать соединения, которые являются эффективными ингибиторами функции белка NS5A. Белок Согласно первому аспекту настоящего изобретения предложено соединение формулы (II) или его фармацевтически приемлемая соль,где каждый D независимо выбран из NH;L представляет собой связь или фенил; каждый из Z1 и Z2 независимо выбран из CH и N; каждый из Z3 и Z4 независимо выбран из C и N,при условии, что не более двух из Z1, Z2, Z3 и Z4 представляют собой N; А представляет собой четырех-шестичленное кольцо, необязательно содержащее один, два или три гетероатома, независимо выбранных из атома азота и серы;R1 и R2 независимо выбраны из атома водорода и CH3, где указанный метил может необязательно образовывать конденсированное трехчленное кольцо с другим кольцевым атомом углерода;R5 и R6 независимо представляют собой C1-4 алкил, замещенный NHCOOCH3 и/или тетрагидропиранилом, необязательно замещенным двумя группами CH3; при этом соединение не является[1-(2-5-[4-(4-2-[1-(2-метоксикарбониламино-3 метилбутирил)пирролидин-2-ил]-3H-имидазол-4-илфенил)нафталин-1-ил]-1H-имидазол-2 илпирролидин-1-карбонил)-2-метилпропил]карбаминовой кислоты метиловым эфиром. Согласно второму аспекту настоящего изобретения предложено соединение формулы (III) или его фармацевтически приемлемая соль,где каждый D представляет собой NH;Y представляет собой CH2; каждый из Z1 и Z2 независимо выбран из CH и N; каждый из Z3 и Z4 независимо выбран из C и N,при условии, что не более двух из Z1, Z2, Z3 и Z4 представляют собой N; А представляет собой четырех-шестичленное кольцо, необязательно содержащее один, два или три гетероатома, независимо выбранных из атома азота и серы;R1 и R2 независимо выбраны из атома водорода и CH3, где указанный метил может необязательно образовывать конденсированное трехчленное кольцо с другим кольцевым атомом углерода;R5 и R6 независимо представляют собой C1-4 алкил, замещенный NHCOOCH3 и/или тетрагидропиранилом, необязательно замещенным двумя группами CH3;R7 и R8 независимо представляют собой атомы водорода; при этом соединение не является[1-(2-5-[4-(4-2-[1-(2-метоксикарбониламино-3 метилбутирил)пирролидин-2-ил]-3H-имидазол-4-илфенил)нафталин-1-ил]-1H-имидазол-2 илпирролидин-1-карбонил)-2-метилпропил]карбаминовой кислоты метиловым эфиром. Согласно третьему аспекту настоящего изобретения предложена композиция, содержащая соединение формулы (II) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Согласно первому варианту осуществления третьего аспекта изобретения данная композиция дополнительно содержит еще одно или два соединения, обладающие противовирусной активностью в отношении ВГС. Согласно второму варианту осуществления третьего аспекта изобретения по меньшей мере одно из указанных дополнительных соединений представляют собой интерферон или рибавирин. Согласно третьему варианту осуществления указанный интерферон выбран из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. Согласно четвертому варианту осуществления третьего аспекта настоящего изобретения предложена композиция, содержащая соединение формулы (II) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и одно или два дополнительных соединения, обладающих противовирусной активностью в отношении ВГС, где по меньшей мере одно из указанных дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое усиливает развитие ответа хелперных T-клеток типа 1, интерферирующей РНК, антисмысловой РНК, имиквимода(imiqimod), рибавирина, ингибитора инозин-5'-монофосфат-дегидрогеназы, амантадина и римантадина. Согласно пятому варианту осуществления третьего аспекта настоящего изобретения предложена композиция, содержащая соединение формулы (II) или его фармацевтически приемлемую соль, фармацевтически приемлемый носитель и одно или два дополнительных соединения, обладающих противовирусной активностью в отношении ВГС, где по меньшей мере одно из указанных дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы ВГС, сериновой протеазы ВГС, полимеразы ВГС, хеликазы ВГС, белка NS4B ВГС, проникновения ВГС в клетку, сборки вирионов ВГС, выхода вирионов ВГС из клетки, белка NS5A ВГС и IMPDH (инозинмонофосфатдегидрогеназы), для лечения ВГС-инфекции. Согласно четвертому аспекту настоящего изобретения предложен способ лечения ВГС-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли. Согласно первому варианту осуществления четвертого аспекта изобретения данный способ дополнительно включает введение еще одного или двух соединений, обладающих противовирусной активностью в отношении ВГС, до введения соединения формулы (II) или его фармацевтически приемлемой соли, после указанного введения или одновременно с соединением формулы (II) или его фармацевтически приемлемой солью. Согласно второму варианту осуществления четвертого аспекта изобретения по меньшей мере одно из указанных дополнительных соединений представляет собой интерферон или рибавирин. Согласно третьему варианту осуществления четвертого аспекта изобретения указанный интерферон выбран из интерферона альфа 2 В, пегилированного интерферона альфа, консенсусного интерферона, интерферона альфа 2 А и лимфобластоидного интерферона тау. Согласно четвертому варианту осуществления четвертого аспекта настоящего изобретения предложен способ лечения ВГС-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли, и одного или двух дополнительных соединений, обладающих противовирусной активностью в отношении ВГС, до введения соединения формулы (II), или его фармацевтически приемлемой соли, после указанного введения или одновременно с соединением формулы (II), или его фармацевтически приемлемой солью, где по меньшей мере одно из указанных дополнительных соединений выбрано из интерлейкина 2, интерлейкина 6, интерлейкина 12, соединения, которое усиливает развитие ответа хелперных T-клеток типа 1, интерферирующей РНК, антисмысловой РНК, имиквимода (imiqimod), рибавирина, ингибитора инозин-5'монофосфат-дегидрогеназы, амантадина и римантадина. Согласно пятому варианту осуществления четвертого аспекта настоящего изобретения предложен способ лечения ВГС-инфекции у пациента, включающий введение пациенту терапевтически эффективного количества соединения формулы (II) или его фармацевтически приемлемой соли и одного или двух дополнительных соединений, обладающих противовирусной активностью в отношении ВГС, до введения соединения формулы (II) или его фармацевтически приемлемой соли, после указанного введения или одновременно с соединением формулы (II) или его фармацевтически приемлемой солью, где по меньшей мере одно из указанных дополнительных соединений является эффективным ингибитором функции мишени, выбранной из металлопротеазы ВГС, сериновой протеазы ВГС, полимеразы ВГС, хеликазы ВГС,белка NS4B ВГС, проникновения ВГС в клетку, сборки вирионов ВГС, выхода вирионов ВГС из клетки,белка NS5 А ВГС и IMPDH, для лечения ВГС-инфекции. Другие варианты осуществления настоящего изобретения могут включать подходящие комбинации двух или более вариантов осуществления и/или аспектов изобретения, раскрытых в данном описании. Другие варианты осуществления и аспекты изобретения будут очевидны из приведенного ниже описания. Соединения согласно настоящему изобретению также существуют в виде таутомеров; поэтому настоящее изобретение также включает все таутомерные формы. Описание настоящего изобретения следует истолковывать в соответствии с законами и принципами образования химических связей. Необходимо понимать, что соединения согласно настоящему изобретению представляют собой соединения, которые являются достаточно стабильными для их использования в качестве фармацевтиче-3 024171 ского агента. Подразумевается, что определение любого заместителя или переменной (например, Rc и Rd) в конкретном положении в молекуле не зависит от его определений в любых других положениях в данной молекуле. Все патенты, заявки на патенты и литературные публикации, цитированные в описании изобретения, включены в данное описание посредством ссылки во всей своей полноте. В случае несоответствия,настоящее описание, включая определения, имеет преимущественную силу. В настоящем описании приведенные ниже термины имеют значения, указанные в определениях этих терминов. В контексте данного описания формы единственного числа существительных включают соответствующие формы множественного числа, если из контекста ясно не следует иное. Если не указано иное, все арильные, циклоалкильные и гетероциклические группы согласно настоящему изобретению могут иметь заместители, описанные в каждом из соответствующих определений. Например, арильная часть арилалкильной группы может иметь заместители, описанные в определении термина "арил". Термин "алкокси" в контексте данного описания относится к алкильной группе, соединенной с основной молекулярной группировкой через атом кислорода. Термин "алкил" в контексте данного описания относится к группе, происходящей от насыщенного углеводорода с нормальной или разветвленной цепью, содержащего от одного до шести атомов углерода. В соединениях согласно настоящему изобретению, у которых X и/или Y представляет собой CH2 и R1 и/или R2 представляет собой алкил, соответственно, указанный алкил может необязательно формировать конденсированное трех-шестичленное кольцо с соседним атомом углерода с образованием одной из приведенных ниже структур: где z имеет значение 1, 2, 3 или 4; или, альтернативно, алкильная группа может формировать четырех- или пятичленное мостиковое кольцо с образованием одной из приведенных ниже структур: или, альтернативно, алкильная группа может формировать спироциклическое трех-шестичленное кольцо с атомом углерода, к которому она присоединена, с образованием одной из приведенных ниже структур: где z имеет значение 1, 2, 3 или 4. Термин "С 2 алкинил" в контексте данного описания относится к Термин "арил" в контексте данного описания относится к фенильной группе или бициклической конденсированной кольцевой системе, у которой одно из колец или оба кольца представляют собой фенильную группу. Бициклические конденсированные кольцевые системы состоят из фенильной группы,конденсированной с четырех-шестичленным ароматическим или неароматическим карбоциклическим кольцом. Арильные группы согласно настоящему изобретению могут быть соединены с основной молекулярной группировкой через любой атом углерода группы, который способен иметь заместители. Типичные примеры арильных групп включают инданил, инденил, нафтил, фенил и тетрагидронафтил, но не ограничены ими. Арильные группы согласно настоящему изобретению необязательно содержат один,два, три, четыре или пять заместителей, независимо выбранных из алкокси, алкоксиалкила, алкоксикарбонила, алкила, алкилкарбонила, второй арильной группы, арилалкокси, арилалкила, арилкарбонила,циано, атома галогена, галогеналкокси, галогеналкила, гетероциклила, гетероциклилалкила, гетероциклилкарбонила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкила, оксо и -P(O)OR2, где каждый R независимо выбран из атома водорода и алкила, и где алкильная часть арилалкила и гетероциклилалкила не содержит заместители, и где вторая арильная группа, арильная часть арилалкила, арильная часть арилкарбонила, гетероциклил и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила необязательно содержат один, два или три заместителя, независимо выбранные из алкокси, алкила, циано, атома галогена, галогеналкокси, галогеналкил и нитро. Термин "арилалкил" в контексте данного описания относится к алкильной группе, содержащей в качестве заместителей одну, две или три арильные группы. Кроме того, алкильная часть арилалкила необязательно содержит в качестве заместителей одну или две дополнительные группы, независимо выбранные из алкокси, алкилкарбонилокси, атома галогена, галогеналкокси, галогеналкила, гетероциклила,гидрокси и -NRcRd, где гетероциклил дополнительно необязательно содержит один или два заместителя,независимо выбранные из алкокси, алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, атома галогена, галогеналкокси, галогеналкил, гидрокси, -NRxRy и оксо. Термин "циклоалкил" в контексте данного описания относится к насыщенной моноциклической или бициклической углеводородной кольцевой системе, содержащей от трех до четырнадцати атомов углерода и не содержащей гетероатомы. Типичные примеры циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил, бицикло[3.1.1]гептил и адамантил, но не ограничены ими. Циклоалкильные группы согласно настоящему изобретению необязательно имеют один, два, три,четыре или пять заместителей, независимо выбранных из алкокси, алкила, арила, циано, атома галогена,галогеналкокси, галогеналкила, гетероциклила, гидрокси, гидроксиалкила, нитро и -NRxRy, где арил и гетероциклил дополнительно необязательно содержат один, два или три заместителя, независимо выбранные из алкокси, алкила, циано, атома галогена, галогеналкокси, галогеналкила, гидрокси, нитро и оксо. Термин "гетероциклил" в контексте данного описания относится к а четырех-, пяти-, шести- или семичленному кольцу, содержащему один, два, три или четыре гетероатома, независимо выбранные из атома азота, кислорода и серы. Четырехчленное кольцо не содержит двойных связей, пятичленное кольцо содержит 0-2 двойные связи, и шести- и семичленное кольцо содержит 0-3 двойные связи. Термин"гетероциклил" также включает бициклические группы, у которых гетероциклическое кольцо конденсировано с другой моноциклической гетероциклической группой или с трех-семичленным ароматическим или неароматическим карбоциклическим кольцом, бициклические группы, у которых гетероциклическое кольцо содержит в качестве заместителя трех-семичленное спироциклическое кольцо, а также мостиковые бициклические группы, такие как 3-оксабицикло[3.2.1]октил, 7-азабицикло[2.2.1]гепт-7-ил, 2 азабицикло[2.2.2]окт-2-ил и 2-азабицикло[2.2.2]окт-3-ил. Гетероциклические группы согласно настоящему изобретению могут быть соединены с основной молекулярной группировкой через любой атом углерода или атом азота группы. Примеры гетероциклических групп включают бензотиенил, фурил,имидазолил, индолинил, индолил, изохинолинил, изотиазолил, изоксазолил, морфолинил, оксазолил,пиперазинил, пиперидинил, пиразолил, пиридинил, пирролидинил, пирролопиридинил, пирролил, хинолинил, тетрагидропиранил, тиазолил, тиенил и тиоморфолинил, но не ограничены ими. Гетероциклические группы согласно настоящему изобретению необязательно имеют один, два, три, четыре или пять заместителей, независимо выбранных из алкенила, алкокси, алкоксиалкила, алкоксикарбонила, алкила,алкилкарбонила, арила, арилалкила, арилкарбонила, циано, атома галогена, галогеналкокси, галогеналкила, второй гетероциклической группы, гетероциклилалкила, гетероциклилкарбонила, гидрокси, гидроксиалкила, нитро, -NRxRy, (NRxRy)алкила и оксо, где алкильная часть арилалкила и гетероциклилалкила не содержит заместители, и где арил, арильная часть арилалкила, арильная часть арилкарбонила, вторая гетероциклическая группа и гетероциклильная часть гетероциклилалкила и гетероциклилкарбонила необязательно содержат один, два или три заместителя, независимо выбранные из алкокси, алкила, циано,атома галогена, галогеналкокси, галогеналкила и нитро. Термин "гетероциклилалкил" в контексте данного описания относится к алкильной группе, содержащей в качестве заместителей одну, две или три гетероциклильные группы. Кроме того, алкильная часть гетероциклилалкила необязательно содержит в качестве заместителей одну или две дополнительные группы, независимо выбранные из алкокси, алкилкарбонилокси, арила, атома галогена, галогеналкокси, галогеналкила, гидрокси и -NRcRd, где дополнительно арил необязательно содержит один или два заместителя, независимо выбранные из алкокси, алкила, незамещенного арила, незамещенного арилалкокси, незамещенного арилалкоксикарбонила, атома галогена, галогеналкокси, галогеналкила, гидрокси и -NRxRy. Термин "-NRcRd" в контексте данного описания относится к двум группам, Rc и Rd, которые соединены с основной молекулярной группировкой через атом азота. Rc и Rd независимо выбраны из атома водорода, алкенилоксикарбонила, алкоксиалкилкарбонила, алкоксикарбонила, алкила, алкилкарбонила,алкилсульфонила, арила, арилалкоксикарбонила, арилалкила, арилалкилкарбонила, арилкарбонила, арилоксикарбонила, арилсульфонила, циклоалкила, циклоалкилоксикарбонила, циклоалкилсульфонила,формила, галогеналкоксикарбонила, гетероциклила, гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила, гетероциклилоксикарбонила, гидроксиалкилкарбонила, (NReRf)алкила, (NReRf)алкилкарбонила, (NReRf)карбонила, (NReRf)сульфонила, -C(NCN)OR' и-C(NCN)NRxRy, где R' выбран из алкила и незамещенного фенила, и где дополнительно алкильная часть арилалкила, арилалкилкарбонила, гетероциклилалкила и гетероциклилалкилкарбонила необязательно содержит в качестве заместителя одну группу -NReRf; и где дополнительно арил, арильная часть арилалкоксикарбонила, арилалкила, арилалкилкарбонила, арилкарбонила, арилоксикарбонила и арилсульфонила, гетероциклил и гетероциклильная часть гетероциклилалкоксикарбонила, гетероциклилалкила, гетероциклилалкилкарбонила, гетероциклилкарбонила и гетероциклилоксикарбонила необязательно содержат один, два или три заместителя, независимо выбранные из алкокси, алкила, циано, атома галогена,галогеналкокси, галогеналкил и нитро. Термин "(NRcRd)алкенил" в контексте данного описания относится к где Rc и Rd являются такими, как определено в данном описании, и каждый Rq независимо представляет собой атом водорода или C1-3 алкил. Термин "(NRcRd)алкил" в контексте данного описания относится к алкильной группе, содержащей в качестве заместителей одну или две группы -NRcRd. Кроме того, алкильная часть (NRcRd)алкила необязательно содержит в качестве заместителей одну или две дополнительные группы, выбранные из алкокси,алкоксиалкилкарбонила, алкоксикарбонила, алкилсульфанила, С 2 алкинила, арилалкоксикарбонила, карбокси, циано, циклоалкила, атома галогена, гетероциклила, гетероциклилкарбонила, гидрокси и(NReRf)карбонила, где дополнительно гетероциклил необязательно содержит один, два, три, четыре или пять заместителей, независимо выбранных из алкокси, алкила, циано, атома галогена, галогеналкокси,галогеналкила и нитро. Термин "-NReRf" в контексте данного описания относится к двум группам, Re и Rf, которые соединены с основной молекулярной группировкой через атом азота. Re и Rf независимо выбраны из атома водорода, алкила, незамещенного арила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного (циклолалкил)алкила, незамещенного гетероциклила, незамещенного гетероциклилалкила,(NRxRy)алкила и (NRxRy)карбонила. Термин "-NRxRy" в контексте данного описания относится к двум группам, Rx и Ry, которые соединены с основной молекулярной группировкой через атом азота. Rx и Ry независимо выбраны из атома водорода, алкоксикарбонила, алкила, алкилкарбонила, незамещенного арила, незамещенного арилалкоксикарбонила, незамещенного арилалкила, незамещенного циклоалкила, незамещенного гетероциклила и(NRx'Ry')карбонила, где Rx' и Ry' независимо выбраны из атома водорода и алкила. Соединения согласно настоящему изобретению имеют асимметрические центры. Данные центры обозначены символами "R" или "S", в зависимости от конфигурации заместителей у хирального атома углерода. Необходимо понимать, что данное изобретение включает все стереохимические изомерные формы, или их смеси, которые обладают способностью ингибировать NS5A. Индивидуальные стереоизомеры соединений могут быть получены путем синтеза из имеющихся в продаже исходных веществ,которые содержат хиральные центры, или путем расщепления полученной смеси энантиомерных продуктов, например путем ее превращения в смесь диастереоизомеров и последующего разделения с использованием перекристаллизации или хроматографических методик или путем прямого разделения энантиомеров на хиральных хроматографических колонках. Исходные соединения с известной стереохимической конфигурацией либо имеются в продаже, либо могут быть получены с использованием методик разделения, известных в данной области техники. Некоторые соединения согласно настоящему изобретению могут существовать в различных стабильных конформационных формах, которые могут быть разделены. Торсионная асимметрия, обусловленная ограниченным вращением вокруг асимметрической простой связи, например, вследствие стерического препятствия или напряжения кольца, может быть использована для разделения разных конформеров. Настоящее изобретение включает каждый конформационный изомер указанных соединений и их смеси. Термин "соединения согласно настоящему изобретению" и эквивалентные выражения включают соединения формулы (I) и их фармацевтически приемлемые энантиомеры, диастереоизомеры и соли. Аналогично промежуточные соединения также включают их соли, если это не находится в противоречии с контекстом. Подразумевается, что настоящее изобретение включает все изотопы атомов, которые присутствуют в соединениях согласно настоящему изобретению. Изотопы представляют собой атомы, имеющие одно и то же атомное число, но разные массовые числа. В качестве неограничивающего примера часто встречающихся изотопов можно привести изотопы водорода, включающие дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Соединения согласно изобретению, меченные изотопами, обычно могут быть получены с помощью стандартных методик, известных специалистам в данной области техники, или с помощью методик, аналогичных методикам, приведенным в данном описании, с использованием подходящих реагентов, меченных изотопами, вместо немеченых реагентов, используемых в других случаях. Такие соединения могут иметь целый ряд возможных применений, например, могут быть использованы в качестве стандартов и реагентов при определении биологической активности. Стабильные изотопы обладают потенциальной способностью благоприятно модифицировать биологические, фармакологические или фармакокинетические свойства содержащих их соединений. Соединения согласно настоящему изобретению могут существовать в виде фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" в контексте данного описания означает соли или цвиттер-ионные формы соединений согласно настоящему изобретению, которые являются растворимыми или диспергируемыми в воде или масле, являются, по результатам медицинской оценки,пригодными для использования в контакте с тканями пациентов, не обладают чрезмерной токсичностью,не вызывают чрезмерного раздражения, аллергической реакции или других проблем или осложнений,соизмеримых с разумным соотношением пользы и риска, и являются эффективными, если используются по назначению. Данные соли могут быть получены в результате взаимодействия подходящего атома азота с подходящей кислотой на заключительных стадиях выделения и очистки соединения или отдельно,после синтеза соединения. Типичные соли присоединения кислот включают ацетат, адипат, альгинат,цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, дигидробромид, дигидрохлорид, дигидроиодид, глицерофосфат, полусульфат, гептаноат, гексаноат,формиат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат,мезитиленсульфонат, метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат,пальмоат, пектинат, персульфат, 3-фенилпроприонат, пикрат, пивалат, пропионат, сукцинат, тартрат,трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат, паратолуолсульфонат и ундеканоат. Примеры кислот, которые могут быть использованы для получения фармацевтически приемлемых солей присоединения, включают неорганические кислоты, такие как соляная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Соли присоединения оснований могут быть получены на заключительных стадиях выделения и очистки соединений в результате взаимодействия карбоксильной группы с подходящим основанием,таким как гидроксид, карбонат или бикарбонат катиона металла, или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы фармацевтически приемлемых солей включают катионы лития, натрия, калия, кальция, магния и алюминия, а также нетоксичные катионы четвертичных аминов, такие как аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин, N,N-диметиланилин, Nметилпиперидин,N-метилморфолин,дициклогексиламин,прокаин,дибензиламин,N,Nдибензилфенетиламин и N,N-дибензилэтилендиамин. Другие типичные органические амины, которые могут быть использованы для получения солей присоединения оснований, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин. Если в терапевтических целях терапевтически эффективное количество соединения формулы (I), а также его фармацевтически приемлемых солей, может быть введено в виде неочищенного химического соединения, активный ингредиент может быть представлен в виде фармацевтической композиции. Соответственно, в изобретении дополнительно предложены фармацевтические композиции, которые включают терапевтически эффективное количество соединений формулы (I), или их фармацевтически приемлемых солей, и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. Термин "терапевтически эффективное количество" в контексте данного описания относится к общему количеству каждого активного компонента, которое является достаточным для достижения значимого положительного результата при введении пациенту, например для достижения уменьшения вирусной нагрузки. Когда данный термин используется в отношении индивидуального активного ингредиента, который вводят в режиме монотерапии, термин относится только к одному этому ингредиенту. Когда данный термин используется в отношении комбинации, он относится к суммарному количеству всех активных ингредиентов, которое обеспечивает терапевтический эффект, независимо от того, вводят активные ингредиенты в комбинации, последовательно или одновременно. Соединения формулы (I) и их фармацевтически приемлемые соли являются такими, как описано выше. Носитель(и), разбавитель(и) или эксципиент(ы) должен (должны) быть приемлемым(и) в смысле его (их) совместимости с другими ингредиентами препарата и не наносить вреда реципиенту. В соответствии с другим аспектом настоящего изобретения также предложен способ получения фармацевтического препарата, включающий смешивание соединения формулы (I), или его фармацевтически приемлемой соли, с одним или более чем одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. Термин "фармацевтически приемлемый" в контексте данного описания относится к соединениям, веществам, композициям и/или лекарственным формам, которые по результатам медицинской оценки подходят для использования в контакте с тканями пациентов, т.е. не проявляют чрезмерной токсичности, не вызывают раздражения,аллергической реакции или других расстройств или осложнений, соизмеримых с приемлемым соотношением польза/риск, и являются эффективными, когда их используют по назначению. Фармацевтические препараты могут быть представлены в стандартных лекарственных формах, со-7 024171 держащих заданное количество активного ингредиента на стандартную дозу. Уровень дозы, который обычно используют для предупреждения и лечения заболевания, вызываемого ВГС-инфекцией, в режиме монотерапии, составляет от приблизительно 0,01 до приблизительно 250 миллиграммов соединений согласно настоящему изобретению на килограмм ("мг/кг") массы тела в сутки, предпочтительно от приблизительно 0,05 до приблизительно 100 мг/кг массы тела в сутки. Обычно фармацевтические композиции согласно настоящему изобретению вводят от приблизительно 1 до приблизительно 5 раз в сутки или альтернативно в виде непрерывной инфузии. Такое введение может быть использовано при лечении хронического или острого заболевания. Количество активного ингредиента, которое может быть использовано в комбинации с носителями с целью получения лекарственной формы для однократного введения, зависит от состояния, которое лечат, тяжести состояния, продолжительности введения, пути введения, скорости экскреции используемого соединения, длительности лечения и возраста, пола, массы и состояния пациента. Предпочтительные препараты в стандартной лекарственной форме содержат суточную дозу или субдозу (уровень дозы указан выше) или подходящую часть суточной дозы активного ингредиента. Лечение можно начинать с назначения маленьких доз, которые существенно меньше оптимальной дозы соединения. Затем дозу постепенно увеличивают, пока не достигается оптимальный в данных обстоятельствах эффект. В общем случае наиболее предпочтительно вводить соединение в концентрации, которая обычно обеспечивает эффективный результат в лечении вирусного заболевания, не вызывая никаких вредных или опасных побочных эффектов. Когда композиции согласно настоящему изобретению содержат комбинацию соединения согласно настоящему изобретению и одного или более чем одного дополнительного терапевтического или профилактического агента, дозы соединения и дополнительного агента обычно составляют от приблизительно 10 до 150% и более предпочтительно от приблизительно 10 до 80% от дозы, которую обычно вводят при использовании режима монотерапии. Фармацевтические препараты могут быть адаптированы для введения любым подходящим путем,например пероральным (включая трансбуккальный или сублингвальный), ректальным, назальным, местным (включая трансбуккальный, сублингвальный или трансдермальный), вагинальным или парентеральным (включая подкожные, внутрикожные, внутримышечные, внутрисуставные, интрасиновиальные, интрастернальные, интратекальные, внутривенные или интрадермальные инъекции или инфузии, а также инъекции или инфузии внутрь пораженных тканей) путем. Такие препараты могут быть получены с помощью методик, известных в области фармации, например путем объединения активного ингредиента с носителем (носителями) или эксципиентом (эксципиентами). Пероральное введение или введение путем инъекции является предпочтительным. Фармацевтические препараты, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, например капсул или таблеток, порошков или гранул, растворов или суспензий в водных или неводных жидкостях, пен или кремов для приема внутрь или жидких эмульсий типа масло в воде или эмульсий типа вода в масле. Например, в препарате, адаптированном для перорального введения, в форме таблетки или капсулы активный лекарственный компонент может быть комбинирован с подходящим для перорального введения нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки изготавливают путем измельчения соединения до получения подходящего тонкого помола и смешивания с измельченным подобным образом фармацевтическим носителем, таким как пригодный для приема внутрь углевод, например крахмал или маннит. Препарат также может содержать ароматизаторы, консерванты, диспергирующие и окрашивающие агенты. Капсулы изготавливают из описанной выше порошковой смеси, которой заполняют желатиновую оболочку. Перед заполнением капсульной оболочки в порошковую смесь могут быть добавлены скользящие и смазывающие вещества, такие как коллоидный кремнезем, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль. Когда капсулы предназначены для приема внутрь, для улучшения доступности лекарства также могут быть добавлены разрыхлители или солюбилизирующие агенты, такие как агар-агар, карбонат кальция или карбонат натрия. Кроме того, если это желательно или необходимо, в смесь также могут быть включены подходящие связующие вещества, смазывающие вещества, разрыхлители и окрашивающие агенты. Подходящие связующие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза,кукурузные подсластители, природные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль и т.п. Смазывающие вещества, используемые в данных лекарственных формах, включают олеат натрия, хлорид натрия и т.п. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бетонит (betonite), ксантановую камедь и т.п. Таблетки изготавливают, например, путем получения порошковой смеси, гранулирования или комкования, добавления смазывающего вещества и разрыхлителя и прессования в форме таблетки. Порошковую смесь получают путем смешивания соединения, измельченного подходящим образом, с разбавителем или основой, описанными выше, и, необязательно, со связующим веществом, таким как карбоксиметилцеллюлоза, алигинат, желирующее вещество или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем ресорбции, таким как четвертичная соль, и/или поглощающим агентом, таким как бетонит, каолин или гидрофосфат кальция. Порошковая смесь может быть гранулирована путем увлажнения связующим веществом, таким как сироп, крахмальная паста, раствор гуммиарабика или растворы целлюлозных или полимерных веществ, и последующего продавливания через сито. В качестве альтернативы гранулированию, порошковая смесь может быть пропущена через таблеточную машину, которая дробит разнородные куски смеси, превращая их в гранулы. Для предотвращения прилипания гранул к формообразующему штампу при производстве таблеток, гранулы могут быть покрыты смазывающим веществом путем добавления стеариновой кислоты, соли стеариновой кислоты,талька или минерального масла. После добавления смазывающего вещества из смеси прессуют таблетки. Соединения согласно настоящему изобретению также могут быть комбинированы с подвижным инертным носителем, затем из полученной смеси сразу прессуют таблетки, минуя стадии гранулирования или комкования. На таблетки могут быть нанесены прозрачные или матовые защитные покрытия, состоящие из изолирующего покрытия шеллаком, покрытия сахаром или полимерным веществом и полирующего покрытия воском. Чтобы отличать разные стандартные дозы, в эти покрытия могут быть добавлены красящие вещества. Жидкие препараты, подходящие для перорального введения, такие как растворы, сиропы и эликсиры, могут быть изготовлены в стандартной лекарственной форме, чтобы принимаемое количество жидкости содержало заданное количество соединения. Сиропы могут быть получены путем растворения соединения в подходящих ароматизированных водных растворах, а эликсиры получают с использованием нетоксичного носителя. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбита, консерванты, вкусовые добавки, такие как мятное масло или натуральные подсластители, или сахарин или другие искусственные подсластители, и тому подобное. При необходимости препараты в стандартных лекарственных формах, предназначенные для перорального введения, могут быть получены путем микрокапсулирования. Данные препараты также могут быть изготовлены в форме препаратов пролонгированного или замедленного высвобождения, например,путем покрытия или внедрения частиц вещества в полимеры, воск и тому подобное. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть введены в форме липосомальных систем доставки, таких как малые моноламеллярные везикулу, большие моноламеллярные везикулы и многослойные везикулы. Липосомы могут быть получены из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть доставлены с помощью моноклональных антител в качестве индивидуальных носителей, к которым присоединены молекулы соединения. Соединения также могут быть присоединены к растворимым полимерам в качестве носителей для направленной доставки лекарства. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенол, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, содержащий в качестве заместителей пальмитоильные остатки. Кроме того, соединения могут быть присоединены к биодеградируемым полимерам, которые можно использовать для достижения контролируемого высвобождения лекарства, например, к полимолочной кислоте,поли-эпсилон-капролактону, полигидроксимасляной кислоте, полиортоэфирам, полиацеталям, полидигидропиранам, полицианоакрилатам и поперечно-сшитым или амфипатичным блок-сополимерам гидрогелей. Фармацевтические препараты, адаптированные для трансдермального введения, могут быть изготовлены в форме отдельных пластин пластыря, которые остаются в непосредственном контакте с эпидермисом реципиента в течение продолжительного периода времени. В этом случае активный ингредиент может быть доставлен из пластыря, например, путем ионтофореза, общее описание которого можно найти в Pharmaceutical Research 1986, 3(6), 318. Фармацевтические препараты, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, паст, гелей, спреев, аэрозолей или масел. Фармацевтические препараты, адаптированные для ректального введения, могут быть изготовлены в виде суппозиториев или в виде клизм. Фармацевтические препараты, адаптированные для назального введения, где в качестве носителя используется твердое вещество, включают крупный порошок, имеющий размер частиц, например, в диапазоне 20-500 мкм, который можно втягивать носом, как нюхательный табак, т.е. путем быстрой ингаляция через носовой ход из контейнера с порошком, который держат близко к носу. Подходящие препараты в форме назального спрея или назальных капель, где в качестве носителя используется жидкость,включают водные или масленые растворы активного ингредиента. Фармацевтические препараты, адаптированные для введения путем ингаляции, включают мелкие частицы пыли или тумана, которые могут быть сформированы с помощью различных типов аэрозольный дозирующих устройств, небулайзеров или инсуффляторов. Фармацевтические препараты, адаптированные для вагинального введения, могут быть изготовлены в форме пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические препараты, адаптированные для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферные агенты, бактериостатические вещества и соли, которые делают препарат изотоничным крови реципиента, для которого он предназначен, а также водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Данные препараты могут находиться в упаковках для одноразового приема или в упаковках для многократного приема, например в запаянных ампулах и пробирках, а также могут храниться в лиофилизированном состоянии (после сублимационной сушки); в лиофилизированные препараты непосредственно перед использованием требуется добавить только стерильный жидкий носитель, например воду для инъекций. Экстемпоральные инъекционные растворы и суспензии могут быть приготовлены из стерильных порошков, гранул и таблеток. Следует понимать, что кроме конкретных ингредиентов, описанных выше, препараты могут содержать другие агенты, используемые в данной области техники, имеющие отношение к данному препарату,например, препараты, подходящие для перорального введения, могут содержать корригенты. Термин "пациент" включает человека и других млекопитающих. Термин "лечение" включает (i) предупреждение заболевания, расстройства или патологического состояния у пациента, который может быть предрасположен к развитию данного заболевания, расстройства и/или патологического состояния, но диагноз еще не поставлен; (ii) ингибирование заболевания, расстройства или патологического состояния, т.е. остановку его развития; и (iii) облегчение заболевания,расстройства или патологического состояния, т.е. регрессию заболевания, расстройства и/или патологического состояния. Соединения согласно настоящему изобретению также могут быть введены вместе с циклоспорином, например циклоспорином А. В клинических исследованиях показано, что циклоспорин А обладает активностью в отношении ВГС (Hepatology 2003, 38, 1282; Biochem. Biophys. Res. Commun. 2004, 313,42; J. Gastroenterol. 2003, 38, 567). В табл. 1 приведены некоторые иллюстративные примеры соединений, которые могут быть введены вместе с соединениями согласно настоящему изобретению. Соединения согласно настоящему изобретению могут быть введены вместе с другими соединениями, обладающими противовирусной активностью в отношении ВГС, в режиме комбинированной терапии, одновременно или раздельно, или в составе композиции, содержащей комбинацию данных соединений. Таблица 1 Соединения согласно настоящему изобретению также могут быть использованы в качестве лабораторных реагентов. Данные соединения быть использованы в качестве инструментов при разработке методик анализа вирусной репликации, оценке систем анализа с использованием животных моделей и при проведении исследований в области структурной биологии с целью получения дополнительных знаний о механизмах развития заболеваний, вызываемых ВГС-инфекцией. Кроме того, соединения согласно настоящему изобретению могут быть использованы для установления или определения сайта связывания других противовирусных соединений, например путем конкурентного ингибирования. Соединения согласно настоящему изобретению также могут быть использованы для предупреждения вирусного заражения или обработки после вирусного заражения материалов и принадлежностей,поэтому данные соединения уменьшают риск инфицирования вирусами лабораторного или медицинского персонала или пациентов, которые контактируют с таким материалами и принадлежностями, например с препаратами крови, тканей, хирургическими инструментами и одеждой, лабораторными инструментами и одеждой и аппаратами и принадлежностями для забора или переливания крови. Данное изобретение включает соединения формулы (I), полученные с использованием методик синтеза, или в результате метаболических процессов, включая метаболические процессы в организме человека или животного (in vivo), или в результате процессов, протекающих in vitro. Аббревиатуры, использованные в настоящем изобретении, включая, в частности, аббревиатуры, использованные в иллюстративных схемах и примерах, приведенных ниже, хорошо известны специалистам в данной области техники. Некоторые из использованных аббревиатур перечислены ниже.Boc или ВОС = трет-бутилоксикарбонил; HATU = О-(7-азабензотриазол-1-ил)-N,N,N',N'тетраметилурония гексафторфосфат; DIEA, или DiPEA, или DIPEA = диизопропилэтиламин; NCS = Nхлорсукцинимид; NBS = N-бромсукцинимид; ДМФА = N,N-диметилформамид; ACN или MeCN = ацетонитрил; OAc = ацетат; EtOAc = этилацетат; Et = этил; Bu = бутил; Ph = фенил; Me = метил; LDA = диизопропиламид лития; Bn = бензил; Ts = тозил; RT или rt = комнатная температура или время удерживания(в зависимости от контекста); ч = час или часы; мин = минута или минуты; ДХМ = дихлорметан; MeOH = метанол; d = сутки; ТГФ = тетрагидрофуран; Et2O = диэтиловый эфир; Bz = бензоил; AIBN = азобисизобутиронитрил; LiHMDS = гексаметилдисилазид лития; Hex = гексаны; EDC = 1-этил-3-(3 диметиламинопропил)карбодиимид; DMAP = 4-N,N-диметиламинопиридин; EtOH = этанол; ДЭАД = азодикарбоновой кислоты диэтиловый эфир; ДМСО = диметилсульфоксид; (S,S) Me-BPE-Rh = (-)-1,2 бис-2S,5S)-2,5-диметилфосфолано)этан(1,5-циклооктадиен)родия (I) тетрафторборат; TBS = третбутилдиметилсилил; мета-СРВА = метахлорпероксибензойная кислота; TBAF = тетрабутиламмония фторид; DBU = 1,8-диазабицикло[5.4.0]ундец-7-ен; ON, или о/n, или on = в течение ночи; АсОН = уксусная кислота; dppf = дифенилфосфиноферроцен; ТФУ = трифторуксусная кислота; pTsA или PTSA = паратолуолсульфоновая кислота; TMSCl = хлортриметилсилан; DDQ = 2,3-дихлор-5,6-дициано-1,4 бензохинон; Tf = трифторметилсульфонил; TMSCN = триметилсилилцианид; n-BuLi = н-бутиллитий иSEM = 2-триметилсилилэтоксиметокси. Соединения и способы согласно настоящему изобретению будут лучше понятны из приведенных ниже схем синтеза, на которых проиллюстрированы методики, которые могут быть использованы для получения соединений согласно настоящему изобретению. Исходные вещества имеются в продаже или могут быть получены с использованием описанных в литературе общепринятых методик, которые известны средним специалистам в данной области техники. Среднему специалисту в данной области техники совершенно очевидно, что определенные выше соединения могут быть синтезированы путем замены подходящих реагентов и агентов в описанных ниже методиках синтеза. Специалисту в данной области техники совершенно очевидно, что для успешного осуществления синтеза стадии избирательного введения и снятия защитных групп (а также порядок самих этих стадий) могут быть выполнены в различном порядке, который зависит от природы переменных. Переменные являются такими, как определено выше, если не указано иное. Схема A иллюстрирует, каким образом ключевые предшественники A-1 и A-3 могут быть превращены в желаемый продукт A-7, и схемы В-Е демонстрируют, каким образом данные ключевые предшественники получают в контексте различных групп гетероциклических соединений. Схема A. Стандартное катализируемое кислотой снятие защитных групп у карбамата A-1 и последующее взаимодействие с защищенной аминокислотой, такой как Вос-пролин, приводят к получению кетоамидаA-2, который может быть циклизирован в имидазол A-5 путем нагревания в присутствии ацетата аммония. Альтернативно имидазол A-5 может быть получен из кетоэфира A-4, который в свою очередь получают из галогенкетона A-3, с использованием аналогичной реакции, а именно путем нагревания с ацетатом аммония. Стандартное катализируемое кислотой снятие защитной Вос-группы и последующая конденсация с кислотой в стандартных для пептидного синтеза условия, например с использованием HATU/DIEA, приводят к получению соединения A-7. Для получения имидазол-функционализированных производных соединения A-7 существуют многочисленные подходы. Например, соединение A-7 может быть прямо галогенизировано такими реагентами, как NCS или NBS, альтернативно галогенирование имидазольной группировки может быть выполнено до стадии снятия защитной Вос-группы. Следует отметить, что в том случае, когда галогеновая группировка представляет собой иодид или бромид, последующая функционализация может быть выполнена с использованием условий сочетания, включающих присутствие металлического катализатора, например с использованием сочетания Судзуки-Мияуры,которые являются общепринятыми в химической литературе. Схема B. Для получения региоизомеров B-6 а и B-6b из бензоксазола B-1 существуют многочисленные подходы. Дибромид B-1 может быть превращен в дикетон B-5 с использованием комбинации условий сочетания Стилле и сочетания Судзуки-Мияуры, где продукт реакции сочетания Стилле обрабатывают кислотой, такой как HCl, для получения кетоновой группировки. Разделение региоизомеров может быть выполнено на промежуточной стадии (бромид B-4 или B-5) или на стадии получения дикетона B-6. Каждый дикетон B-6 может быть превращен в дибромид B-7 с использованием таких реагентов, как бром. Согласно альтернативному подходу бензоксазол B-1 может быть подвергнут сочетанию с бороновой кислотой B-2 с использованием условий реакции Судзуки-Мияуры, полученная региоизомерная смесь может быть разделена, и индивидуальные региоизомеры могут быть превращены в бромкетон B-6 путем комбинации сочетания Стилле и in situ бромирования такими реагентами, как NBS, в присутствии воды. Схема C. Дибромид C-1 может быть превращен в бромкетон C-2 а с использованием синтетического пути,описанного на схеме B. Если на конечной стадии бромирования возникают осложнения, заключающиеся в том, что бромируется имидазольный фрагмент C-2 с получением преимущественно соединения C-2b,то можно предупредить образование такого соединения, используя стадию конструирования имидазола,описанную на схеме A, и последующее удаление данного бромида с использованием восстановительных условий (таких как гидрогенизация, катализируемая палладием на углероде). Альтернативно дибромидC-1 может быть монолитирован, последующее гашение путем добавления амида Вайнреба Вос-глицина приводит к получению региоизомерных бромидов C-3. Разделенные региоизомеры могут быть подвергнуты сочетанию с соединением C-4 с использованием условий реакции Судзуки-Мияуры с получением карбамата C-5. Схема C Схема D и схема E. Как показано на схеме D, амин D-1 может быть конденсирован с 2,5-диметокситетрагидрофураном при нагревании, и затем полученный пиррол может быть подвергнут взаимодействию с метил 2-хлор-2- 20024171 оксоацетатом, или с любым другим его эфирным вариантом, с получением кетоэфира D-3. Удаление Вос-группы в кислых условиях и последующее in situ окисление приводят к получению сложного диэфира D-6. Обработка сложного диэфира D-6 с помощью CH2ICl/LDA приводит к получению хлоркетона D7. Получение региоизомера D-7 может быть выполнено в соответствии с модифицированной методикой,описанной на схеме E, где вместо метил 2-хлор-2-оксоацетата использован метил 4(хлоркарбонил)бензоат. Схема D Среднему специалисту в данной области техники совершенно очевидно, что пути синтеза, описанные на схемах B-E, в равной степени применимы для дигалогеновых предшественников, приведенных далее: Среднему специалисту в данной области техники также совершенно очевидно, что модификация путей синтеза, описанных на схемах A-E, может быть использована для получения гомодимерных вариантов конечных продуктов, как показано на схеме F. Схема F Схема 1. Замещенные производные фенилглицина. Замещенные производные фенилглицина могут быть получены с использованием нескольких методик, описанных ниже. Фенилглицина трет-бутиловый эфир может быть подвергнут восстановительному алкилированию (путь A) с использованием подходящего альдегида и восстановителя, такого как цианоборгидрид натрия, в кислой среде. Гидролиз полученного трет-бутилового эфира может быть осуществлен с использованием сильной кислоты, такой как HCl или трифторуксусная кислота. Альтернативно фенилглицин может быть алкилирован с использованием алкилгалогенида, такого как йодистый этил, и основания, такого как бикарбонат натрия или карбонат калия (путь B). Путь C иллюстрирует восстановительное алкилирование фенилглицина, описанное выше (путь A), за которым следует второе восстановительное алкилирование с использованием альтернативного альдегида, такого как формальдегид, в присутствии восстанавливающего агента и кислоты. Путь D иллюстрирует синтез замещенных фенилглицинов через соответствующие аналоги миндальной кислоты. Превращение вторичного спирта в подходящую уходящую группу может быть осуществлено с использованием паратолуолсульфонилхлорида. Замещение тозилатной группы подходящим амином и последующее восстановительное удаление сложно- 22024171 эфирной бензильной группы приводит к получению замещенных производных фенилглицина. Путь Е иллюстрирует расщепление рацемических замещенных производных фенилглицина путем этерификации с использованием энантиомерно чистого хирального вспомогательного реагента, такого как (без ограничения) (+)-1-фенилэтанол, (-)-1-фенилэтанол, оксазолидинон Эванса или энантиомерно чистый пантолактон. Разделение полученных диастереоизомеров выполняют путем хроматографии (силикагель, ВЭЖХ,кристаллизация и т.д.) и затем удаляют хиральный вспомогательный реагент с получением энантиомерно чистых производных фенилглицина. Путь H иллюстрирует последовательность стадий синтеза, которая пересекается с путем E, где вышеупомянутый хиральный вспомогательный реагент вводят до введения амина. Альтернативно эфир арилуксусной кислоты может быть бромирован с использованием в качестве источника иона бромония, например, брома, N-бромсукцинимида или CBr4. Полученный бензиловый бромид может быть подвергнут замещению с использованием различных моно- или дизамещенных аминов в присутствии основного третичного амина, такого как триэтиламин или основание Хюнига. Гидролиз полученного сложного метилового эфира путем обработки гидроксидом лития при низкой температуре или 6 н. HCl при повышенной температуре приводит к получению замещенных производных фенилглицина. Путь G иллюстрирует еще одну методику. Аналоги глицина могут быть модифицированы с использованием различных арилгалогенидов в присутствии источника палладия(0), такого как палладий бис(трибутилфосфин), и основания, такого как фосфат калия. Полученный сложный эфир затем может быть гидролизован путем обработки основанием или кислотой. Необходимо понимать, что существуют другие известные методики получения производных фенилглицина и данные методики могут быть модифицированы в данном описании с целью получения желаемых соединений. Также необходимо понимать, что конечные производные фенилглицина могут быть очищены путем препаративной ВЭЖХ до энантиомерной чистоты более 98% ее (энантиомерный избыток). Схема 2. Ацилированные производные аминокислот. Согласно другому варианту осуществления настоящего изобретения ацилированные фенилглициновые производные могут быть получены следующим образом. Фенилглициновые производные, у которых карбоксильная группа защищена легко удаляемой сложноэфирной группировкой, могут быть ацилированы с использованием хлорангидрида кислоты в присутствии основания, такого как триэтиламин, с получением соответствующих амидов (путь A). Путь B иллюстрирует ацилирование исходных фенилглициновых производных с использованием подходящего хлороформата, а путь C - с использованием подходящего изоцианата или карбамоилхлорида. Каждое из трех промежуточных соединений, показанных на схемах путей A-C, может быть деблокировано с использованием методик, известных специалистам в данной области техники (а именно путем обработки полученного трет-бутилового эфира сильной кислотой, такой как HCl или трифторуксусная кислота). Схема 3. Аминозамещенные фенилуксусные кислоты могут быть получены путем обработки хлорметилфенилуксусной кислоты избытком амина. К перемешиваемому раствору метил 2-(бензилоксикарбониламино)-2-(диметоксифосфорил)ацетата(2,0 г, 6,0 ммоль) в EtOAc (40 мл) добавляли 1,1,3,3-тетраметилгуанидин (0,985 мл, 7,85 ммоль) и данную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 10 мин. Затем добавляли дигидро-2H-пиран-3(4H)-он (0,604 г, 6,04 ммоль) и данную смесь перемешивали в течение 16 ч при комнатной температуре. Затем реакционную смесь охлаждали в морозильной камере в течение 10 мин и нейтрализовали водным раствором лимонной кислоты (1,5 г в 20 мл воды). Образовавшиеся две фазы разделяли, органический слой промывали 0,25 н. водным раствором HCl и солевым раствором, затем сушили(MgSO4) и концентрировали до получения бесцветного масла. Данное неочищенное вещество очищали путем флэш-хроматографии на силикагеле (растворитель для нанесения пробы: ДХМ; элюент:EtOAc/гексаны, градиент 20-30% EtOAc) с получением двух изомерных продуктов. Первым элюировали(Z)-метил 2-(бензилоксикарбониламино)-2-(2H-пиран-3(4H,5H,6H)-илиден)ацетат (490 мг) (белое твердое вещество) и вторым элюировали (E)-метил 2-(бензилоксикарбониламино)-2-(2H-пиран-3(4H,5H,6H)илиден)ацетат (433 мг) (белое твердое вещество). ЖХ-МС: время удерживания: 1,398 мин (Z-изомер) и 1,378 мин (E-изомер); m/z 304,08 (Z-изомер) и 304,16 (E-изомер) (МН-). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм,C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS Platform для ЖХ в режиме ионизации электрораспылением. 1(Примечание: абсолютную конфигурацию региоизомеров определяли по сдвигам и константам спин-спинового взаимодействия 1 Н-ЯМР-спектров). К перемешиваемому раствору (Z)-метил 2-(бензилоксикарбониламино)-2-(2H-пиран-3(4H,5H,6H)илиден)ацетата (310 мг, 1,015 ммоль) в MeOH (10 мл) добавляли (-)-1,2-бис-2S,5S)-2,5 диметилфосфолано)этан(циклооктадиен)родия(I) тетрафторборат (28,2 мг, 0,051 ммоль) и данную смесь продували с использованием вакуумного насоса азотом и затем водородом, затем реакционную смесь перемешивали в атмосфере водорода (60 psi (413700 Па в течение 2 суток при комнатной температуре. Затем реакционную смесь концентрировали и остаток очищали путем флэш-хроматографии на силикагеле (растворитель для нанесения пробы: ДХМ; элюент: 20% EtOAc в гексанах) с получением (S)-метил 2(бензилоксикарбониламино)-2-S)-тетрагидро-2H-пиран-3-ил)ацетата (204 мг) в виде прозрачного бесцветного масла. ЖХ-МС: время удерживания: 1,437 мин; m/z 307,89 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм,C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS Platform для ЖХ в режиме ионизации электрораспылением. 1E)-метил 2-(бензилоксикарбониламино)-2-(2H-пиран-3(4H,5H,6H)илиден)ацетат) (360 мг, 1,18 ммоль) восстанавливали аналогичным образом с получением (S)-метил 2(бензилоксикарбониламино)-2-R)-тетрагидро-2H-пиран-3-ил)ацетата (214 мг) в виде прозрачного бесцветного масла. ЖХ-МС: время удерживания: 1,437 мин; m/z 308,03 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм,C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS Platform для ЖХ в режиме ионизации электрораспылением. 1 К раствору (S)-метил 2-(бензилоксикарбониламино)-2-S)-тетрагидро-2H-пиран-3-ил)ацетата (200 мг, 0,651 ммоль) и дикарбоновой кислоты диметилового эфира [4525-33-1] (0,104 мл, 0,976 ммоль) вMeOH (10 мл) добавляли 10% Pd/C (69,3 мг, 0,065 ммоль). Данную реакционную смесь продували с использованием вакуумного насоса азотом и затем водородом, затем реакционную смесь перемешивали в атмосфере водорода (55 psi (379225 Па в течение 5 ч при комнатной температуре. Затем реакционную смесь фильтровали через подушку CELITE/силикагель и фильтрат концентрировали до получения бесцветного масла. Данное неочищенное масло очищали путем флэш-хроматографии на силикагеле (растворитель для нанесения пробы: ДХМ; элюент: 30% EtOAc в гексанах) с получением продукта (S)-метил 2-(метоксикарбониламино)-2-S)-тетрагидро-2H-пиран-3-ил)ацетата (132 мг) в виде бесцветного масла. ЖХ-МС: время удерживания: 0,92 мин; m/z 231,97 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм, C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS Platform для ЖХ в режиме ионизации электрораспылением. 1(m, 2H), 3.77 (s, 3H), 3.70 (s, 3H), 3.29-3.38 (m, 1H), 3.23 (dd, J=11.2, 10.4 Гц, 1 Н), 2.03-2.14 (m, 1H), 1.561.75 (m, 3H), 1.32-1.43 (m, 1H). Аналогичным образом из другого диастереоизомера S)-метил 2-(бензилоксикарбониламино)-2R)-тетрагидро-2H-пиран-3-ил)ацетата) получали(S)-метил 2-(метоксикарбониламино)-2-R)тетрагидро-2H-пиран-3-ил)ацетат в виде прозрачного бесцветного масла. ЖХ-МС: время удерживания: 0,99 мин; m/z 231,90 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм, C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS Platform для ЖХ в режиме ионизации электрораспылением. 1Cap-177 а и Cap-177b. К перемешиваемому раствору (S)-метил 2-(метоксикарбониламино)-2-S)-тетрагидро-2H-пиран-3 ил)ацетата (126 мг, 0,545 ммоль) в ТГФ (4 мл) при комнатной температуре добавляли 1 М раствор LiOH(1,090 мл, 1,090 ммоль) в воде. Данную реакционную смесь перемешивали в течение 3 ч при комнатной температуре, нейтрализовали путем добавления 1 М HCl (1,1 мл) и экстрагировали EtOAc (этилацетатом)(310 мл). Органические экстракты сушили, фильтровали и концентрировали с получением (S)-2(метоксикарбониламино)-2-S)-тетрагидро-2H-пиран-3-ил)уксусной кислоты (Cap-177 а) (125 мг) в виде прозрачного бесцветного масла. ЖХ-МС: время удерживания: 0,44 мин; m/z 218,00 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм, C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% H2O/10 мМ ацетат аммония; Растворитель В: 5% H2O/95% MeOH/10 мМ ацетат аммония. Масс-спектры получали с использованием MICROMASS(m, 1H), 3.91 (d, J=11.0 Гц, 1H), 3.71 (s, 3H), 3.33-3.41 (m, 1H), 3.24-3.32 (m, 1H), 2.10-2.24 (m, 1H), 1.741.83 (m, 1H), 1.63-1.71 (m, 2H), 1.35-1.49 (m, 1H). Аналогичным образом из другого диастереоизомера S)-метил 2-(метоксикарбониламино)-2-R)тетрагидро-2H-пиран-3-ил)ацетата) получали(S)-2-(метоксикарбониламино)-2-R)-тетрагидро-2Hпиран-3-ил)уксусную кислоту (Cap-177b) в виде прозрачного бесцветного масла. ЖХ-МС: время удерживания: 0,41 мин; m/z 217,93 (MH+). ЖХ-данные регистрировали на жидкостном хроматографе Shimadzu LC-10AS, оснащенном колонкой PHENOMENEX Luna (10 мкм, C18, 3,050 мм), с использованием детектора SPD-10AV UV-Vis на длине волны детектора 220 нм. Использовали следующие условия элюирования: скорость потока: 4 мл/мин; градиент: 100% Растворитель А/0% Растворитель В - 0% Растворитель А/100% Растворитель В; продолжительность градиента: 3 мин; конечную концентрацию градиента поддерживали в течение 1 мин; продолжительность анализа: 4 мин; Растворитель А: 5% MeOH/95% В реактор для гидрогенизации, содержащий раствор (2S,3S,4S)-2-метил-3,4-дигидро-2H-пиран-3,4 диил диацетата (5 г, 23,34 ммоль) в MeOH (20 мл) добавляли Pd/C (150 мг, 0,141 ммоль). Полученную смесь гидрогенизировали в течение 1 ч при давлении 40 psi (275800 Па) в шейкере Парра. Затем смесь фильтровали и фильтрат концентрировали с получением Cap-178, стадия a (5,0 г) в виде прозрачного масла, которое со временем затвердевало. 1 К раствору Cap-178, стадия a (5,0 г, 23 ммоль) в MeOH (50 мл) добавляли несколько капель метилата натрия. После перемешивания в течение 30 мин при комнатной температуре добавляли метилат натрия (0,1 мл, 23,12 ммоль) и раствор перемешивали в течение ночи при комнатной температуре. Затем под вакуумом удаляли растворитель. Остаток разбавляли бензолом и концентрировали с получением соответствующего диола в виде желтого твердого вещества. Полученное твердое вещество растворяли в пиридине (50 мл) и к данному раствору при -35C добавляли по каплям бензоилхлорид (2,95 мл, 25,4 ммоль). Полученную смесь перемешивали в течение 1 ч при -35C и затем в течение ночи при комнатной температуре. Затем смесь разбавляли Et2O (диэтиловым эфиром) и промывали водой. Водный слой экстрагировали EtOAc (этилацетатом) (2). Объединенные органические слои сушили с использованиемMgSO4 и концентрировали. Данный неочищенный продукт очищали путем флэш-хроматографии (силикагель, 5-15% EtOAc/Нех) с получением Cap-178, стадия b (4,5 г) в виде прозрачного масла, которое медленно кристаллизовалось при продолжительном хранении. ЖХ-МС: вычислено для [M+Na]+ C13H16NaO4: 259,09; найдено: 259,0. 1 К смеси NaH (1,143 г, 28,6 ммоль) (60% в минеральном масле) и CS2 (6 мл) добавляли по каплям в течение 15 мин Cap-178, стадия b (4,5 г, 19 ммоль) в CS2 (40 мл). Полученную смесь перемешивали в течение 30 мин при комнатной температуре. Смесь становилась светло-оранжевой, и образовывалось небольшое количество твердого вещества. Затем добавляли по каплям в течение 20 мин MeI (14,29 мл,229 ммоль). Затем данную смесь перемешивали в течение ночи при комнатной температуре. Затем реакционную смесь осторожно гасили путем добавления насыщенного раствора NH4Cl. Данную смесь экстрагировали EtOAc (этилацетатом) (3). Объединенные органические слои сушили с использованиемMgSO4 и концентрировали. Данный неочищенный продукт очищали путем флэш-хроматографии (силикагель, 6% EtOAc/Нех) с получением Cap-178, стадия c (3,13 г) в виде прозрачного масла. ЖХ-МС: вычислено для [M+Na]+ C15H18NaO4S2: 349,05; найдено: 349,11. 1 К смеси Cap-178, стадия c (3,13 г, 9,59 ммоль) и AIBN (азобисизобутиронитрила) (120 мг, 0,731 ммоль) в бензоле (40 мл) при 80C добавляли гидрид три-н-бутилолова (10,24 мл, 38,4 ммоль). Полученную смесь перемешивали при температуре дефлегмации в течение 20 мин, затем охлаждали до комнатной температуры. Смесь разбавляли диэтиловым эфиром, добавляли 100 мл водного раствора KF (10 г) и данную смесь интенсивно перемешивали в течение 30 мин. Затем два образовавшихся слоя разделяли и водную фазу экстрагировали EtOAc (этилацетатом) (2). Органический слой сушили с использованиемMgSO4 и концентрировали. Данный неочищенный продукт очищали путем флэш-хроматографии (силикагель, дезактивировали с использованием 3% Et3N в гексанах и промывали с использованием 3% Et3N в гексанах с целью удаления производного, содержащего группу трибутилолова, затем элюировали смесью 15% EtOAc/Нех) с получением Cap-178, стадия d (1,9 г) в виде прозрачного масла. 1 К смеси Cap-178, стадия d (1,9 г, 8,63 ммоль) и MeOH (10 мл) добавляли метилат натрия (2 М раствор в метаноле, 2 мл, 4,00 ммоль). Полученную смесь перемешивали в течение 5 ч при комнатной температуре. Растворитель удаляли под вакуумом. Смесь нейтрализовали насыщенным раствором NH4Cl и экстрагировали EtOAc (этилацетатом) (3). Органические слои сушили с использованием MgSO4 и концентрировали с получением Cap-178, стадия e (0,8 г) в виде прозрачного масла. Данный продукт использовали на следующей стадии без дополнительной очистки. 1 К раствору Cap-178, стадия e (0,8 г, 6,89 ммоль) и пиридина (2,23 мл, 27,5 ммоль) в CH2Cl2 (100 мл) добавляли тозил-Cl (2,63 г, 13,77 ммоль). Полученную смесь перемешивали в течение 3 суток при комнатной температуре. Затем к реакционной смеси добавляли воду (10 мл) и данную смесь перемешивали в течение 1 ч при комнатной температуре. Два образовавшихся слоя разделяли и органическую фазу промывали водой и 1 н. водным раствором HCl. Органическую фазу сушили с использованием MgSO4 и концентрировали с получением Cap-178, стадия f (1,75 г) в виде светло-желтого твердого вещества. Данный продукт использовали на следующей стадии без дополнительной очистки. Вычислено для [М+Н]+ В пробирку для микроволновой печи вносили этил 2-(дифенилметиленамино)ацетат (1,6 г, 5,92 ммоль) и Cap-178, стадия f (1,6 г, 5,92 ммоль). Добавляли 10 мл толуола. Пробирку герметично закрывали и добавляли по каплям в атмосфере N2 LiHMDS (1 н. раствор в толуоле, 7,1 мл, 7,10 ммоль). Полученный темно-коричневый раствор нагревали в течение 6 ч при 100C с использованием микроволнового излучения. Затем к смеси добавляли воду и данную смесь экстрагировали EtOAc (этилацетатом) (3). Объединенные органические слои промывали солевым раствором, сушили с использованием MgSO4 и концентрировали с получением диастереоизомерной смеси Сар-3, стадия g (3,1 г) в виде оранжевого масла. Данную неочищенную смесь использовали на следующей стадии без разделения. ЖХ-МС: вычислено для [М+Н]+ C23H28NO3: 366,21; найдено: 366,3. К раствору диастереоизомерной смеси этил Cap-178, стадия g в ТГФ (20 мл) добавляли HCl (2 н. водный раствор, 30 мл, 60,0 ммоль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Затем смесь экстрагировали EtOAc (этилацетатом) и водный слой концентрировали с получением HCl-соли Cap-178, стадия h (1,9 г) в виде оранжевого масла. Данную соль использовали на следующей стадии без дополнительной очистки. ЖХ-МС: вычислено для [М+Н]+ C10H20NO3: 202,14; найдено: 202,1. Раствор HCl-соли Cap-178, стадия h (1,9 г), DIPEA (4,19 мл, 24,0 ммоль) и хлормуравьиной кислоты метилового эфира (1,24 мл, 16,0 ммоль) в CH2Cl2 (20 мл) перемешивали в течение 1 ч при комнатной температуре. Затем смесь разбавляли CH2Cl2 (дихлорметаном) и промывали водой. Органический слой сушили с использованием Na2SO4 и концентрировали. Данный неочищенный продукт очищали путем флэш-хроматографии (силикагель, 0-20% EtOAc/Hex) с получением Cap-178, стадия i (1,1 г) в виде желтого масла. Вычислено для [M+Na]+ C12H21NNaO5: 282,13; найдено: 282,14. 1 К смеси Cap-178, стадия i (1,1 г, 4,2 ммоль) в ТГФ (5 мл) и воды (2 мл) добавляли LiOH (2 н. водный раствор, 6,36 мл, 12,7 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре. Затем смесь нейтрализовали путем добавления 1 н. водного раствора HCl и экстрагировали EtOAc(этилацетатом) (3). Объединенные органические слои сушили с использованием MgSO4 и концентрировали с получением Cap-178, стадия j (0,8 г) в виде прозрачного масла. ЖХ-МС: вычислено для [М+Н]+EDC (219 мг, 1,14 ммоль) в CH2Cl2 (10 мл) добавляли DMAP (13,95 мг, 0,114 ммоль). Полученный раствор перемешивали в течение ночи при комнатной температуре и растворитель удаляли под вакуумом. Остаток переносили в EtOAc, промывали водой, сушили с использованием MgSO4 и концентрировали. Данный неочищенный продукт очищали путем хроматографии (силикагель, 0-15% EtOAc/гексаны) с получением Cap-178, стадия k в виде смеси двух диастереоизомеров. Данную смесь разделяли путем хи- 29
МПК / Метки
МПК: C07D 405/14, A61K 31/4178, C07D 413/14, C07D 403/14, A61K 31/433, C07D 487/04
Метки: ингибиторы, вируса, гепатита
Код ссылки
<a href="https://eas.patents.su/30-24171-ingibitory-virusa-gepatita-s.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы вируса гепатита с</a>
Ингибиторы вируса гепатита с

Номер патента: 21194
Опубликовано: 30.04.2015
Авторы: Белема Маконен, Пател Бхарат П., Тимонко Стивен, Кеннет Дж., Пак Шон К., Натали Джр.
МПК: C07D 209/54
Метки: ингибиторы, гепатита, вируса
Формула / Реферат:
1. Способ получения соединения формулы (III)или его фармацевтически приемлемой соли,где R4 и R5 независимо выбраны из атома водорода, (С1-С6)алкила, (C6-C10)арила, (С6-С10)арил(С1-С6)алкила, гетероциклила и гетероциклил(С1-С2)алкила, где гетероциклил представляет собой 5-6-членный гетероциклил, включающий 1-2 гетероатома, выбранных из N, S и О,включающий обработку соединения формулы (IV)ацетатом аммония в присутствии основания.2. Способ по п.1,...
Ингибиторы вируса гепатита с (hcv)

Номер патента: 15415
Опубликовано: 31.08.2011
Авторы: Венкатарамани Чандрэсикар, Линк Джон О., Граупе Михаель
МПК: A61K 47/00, A61K 31/74, A61K 31/00...
Метки: вируса, ингибиторы, гепатита, hcv
Формула / Реферат:
1. Соединение формулы (I)где Е представляет собой -COCONHR6, где R6представляет собой водород, алкил, циклоалкил, аралкил или гетероаралкил, где ароматическое кольцо необязательно замещено одним или двумя галоидами;R1 представляет собой алкил, циклоалкилалкил, где алифатические или алициклические группы в R1необязательно замещены одним или двумя радикалами Rb, которые независимо выбирают из гидрокси, алкокси, арилокси, гетероарилокси, алкилтио,...
Ингибиторы вируса гепатита с

Номер патента: 18793
Опубликовано: 30.10.2013
Авторы: Белема Маконен, Кэдоу Джон Ф.
МПК: A61K 31/439, A61K 31/4178, A61P 31/14...
Метки: ингибиторы, вируса, гепатита
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, где L выбран из связи,R1 и R2 представляют собойили R1 представляет собойи R2 выбран изгде обозначает точку присоединения к исходной молекуле;R3 и R4 независимо выбраны из водорода и гало;каждый R5 независимо выбран из водорода;каждый R6 независимо выбран из алкила;R6a представляет собой алкил, где алкил необязательно может образовывать конденсированное 3-членное кольцо со смежным...
Ингибиторы вируса гепатита с

Номер патента: 20815
Опубликовано: 30.01.2015
Авторы: Чэнь Ци, Истер Джон А., Ян Чжун, Бендер Джон А., Хевавасам Пиясена, Су Бао-Нин, Сюй Ниннин, Смит Майкл Дж., Минвелл Николас А., Ван Гань, Белема Маконен, Ромине Джефри Ли, Нгуен Ван Н., Лопез Омар Д., Сейнт Лорен Денис Р.
МПК: C07D 405/14, C07D 403/14
Метки: гепатита, ингибиторы, вируса
Формула / Реферат:
1. Соединение формулы (I)или его фармацевтически приемлемая соль, гдеn равно 0,1 или 2;X выбирают из водорода, циано, галогена циклопропила, циклопропилметила, 1-метилциклопропила, аллила, 1-метилпиразол-5-ила, 1-метилимидазол-5-ила и изоксазол-4-ила;R1 представляет собой водород;R2 выбирают из водорода, циклопропила и галогена; илиR1 и R2 вместе образуют -CH=CH- или -CH=CCl-;при условии, что по меньшей мере один из X и R2 не обозначает...
Ингибиторы вируса гепатита с

Номер патента: 22127
Опубликовано: 30.11.2015
Автор: Ромайн Джеффри Ли
МПК: C07D 403/14, A61K 31/4184, A61P 31/12...
Метки: вируса, ингибиторы, гепатита
Формула / Реферат:
1. Соединение, выбранное из:метил (2-((2S,5S)-2-(7-((2-((2S,5S)-1-((2S)-2-(4,4-дифторциклогексил)-2-((метоксикарбонил)амино)ацетил)-5-метил-2-пирролидинил)-1Н-бензимидазол-5-ил)этинил)-1H-нафто[1,2-d]имидазол-2-ил)-5-метил-1-пирролидинил)-2-оксо-1-(тетрагидро-2Н-пиран-4-ил)этил)карбамата;метил...
Предыдущий патент: Гетероарильные производные в качестве модуляторов nachr альфа7
Следующий патент: Способ изготовления основы с маркировкой и основа с маркировкой, изготовленная указанным способом
Случайный патент: Способ стереоселективного синтеза бициклических гетероциклов