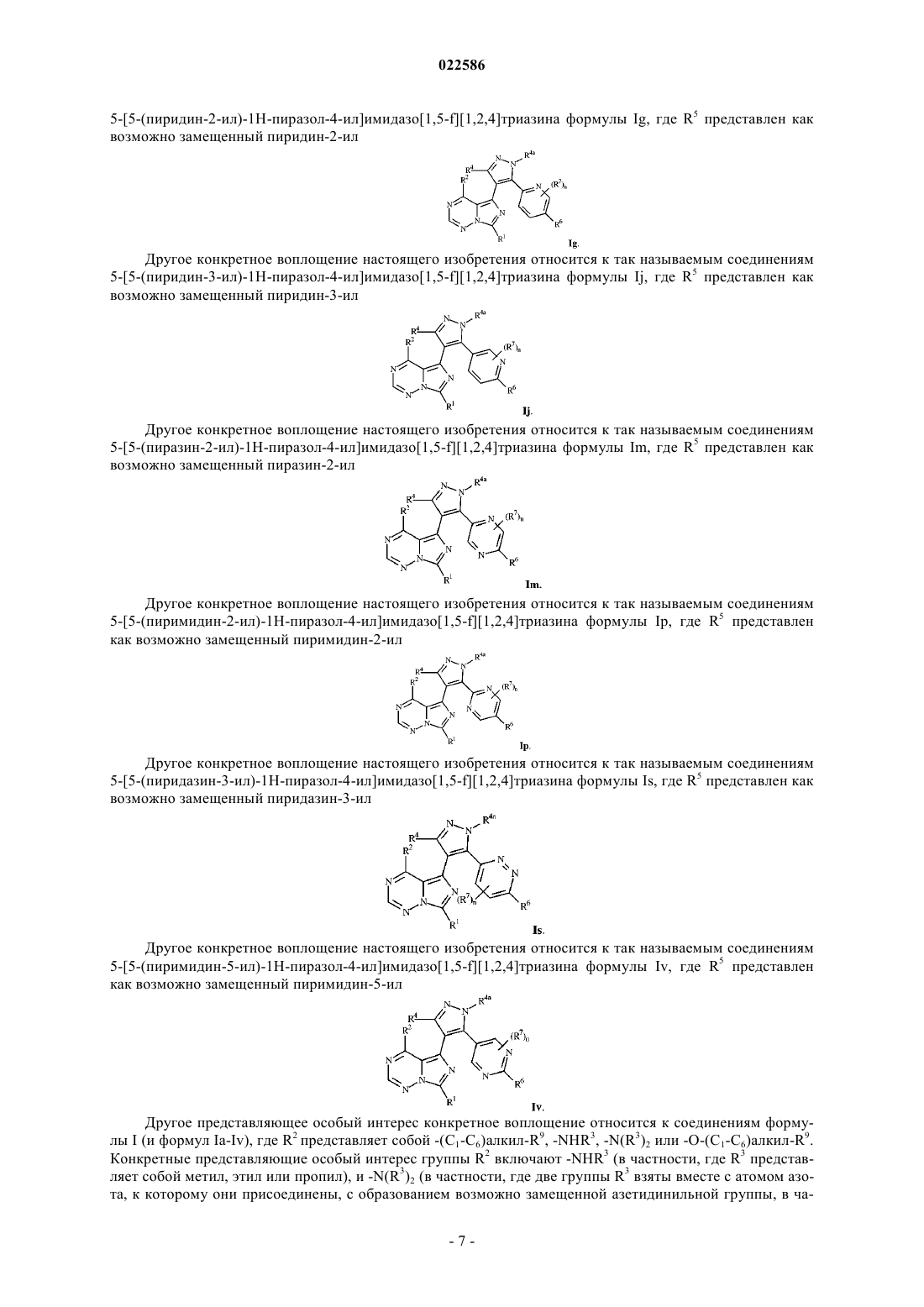
Имидазо[5,1-f][1,2,4]триазины для лечения неврологических расстройств
Номер патента: 22586
Опубликовано: 29.01.2016
Авторы: Верхоэст Патрик Роберт, Хилал Кристофер Джон, Чэппи Томас Аллен, Хамфри Джон Майкл, Янг Эдди




























Формула / Реферат
1. Соединение формулы I

или его фармацевтически приемлемая соль, где
R1 представляет собой водород, (С1-С6)алкил, (С3-С15)циклоалкил или -(С1-С6)алкил-ОН;
R2 представляет собой -(С1-С6)алкил-R9, -NHR3, -N(R3)2, -О-(С1-С6)алкил-R9, (С3-С15)циклоалкил или (3-14-членный)гетероциклил, включающий 1 гетероатом, выбранный из О, S и N; где указанные группировки (С3-С15)циклоалкил или (3-14-членный)гетероциклил могут быть возможно замещены заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из (С1-С6)алкила, (С1-С6)алкокси и галогена;
каждый R3 независимо выбран из группы, состоящей из групп -(С1-С6)алкил-R9 и -(С3-С15)циклоалкил-R9, либо, когда R2 представляет собой -N(R3)2, тогда оба указанных R3 могут быть взяты вместе с атомом азота, к которому они присоединены, с образованием 4-6-членного гетероциклического кольца, возможно замещенного заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из фтора, -ОН, групп -О-(С1-С6)алкил, NH2, -NH-(С1-С6)алкил, -N[(С1-С6)алкил]2, (С1-С6)алкил, -OR8, -O-(C=O)-OR8, -SR8, NH-(C=O)-OR8, -O-(C=O)-N(R8)2, -NH-(C=O)-N(R8)2, -N[(С1-С6)алкил](C=O)-OR8, -N[(С1-С6)алкил](C=O)-N(R8)2 и (С3-С15)циклоалкил;
каждый R4 представляет собой водород;
каждый R4a независимо выбран из группы, состоящей из (С1-С6)алкила и (С3-С15)циклоалкила;
R5 представляет собой

где n равен 0, 1, 2, 3 или 4;
каждый R6 независимо выбран из группы, состоящей из водорода, галогена, (С1-С6)алкила, -CF3, -CHF2,
-CH2F, групп -CF2-(С1-С6)алкил, -CN, -NO2, -(C=O)-R8, -(C=O)-OR8, -OR8, -SR8, -S(O)R8, -S(O)2R8, NH2, -NH-(С1-С6)алкил, -N[(С1-С6)алкил]2 и (С3-С15)циклоалкил;
каждый R7 независимо выбран из группы, состоящей из галогена, (С1-С6)алкила, -CN, -CF3, -CHF2, -CH2F, групп -О-(С1-С6)алкил и (С3-С15)циклоалкил;
каждый R8 там, где он присутствует, независимо выбран из группы, состоящей из водорода, (С1-С6)алкила, (С3-С15)циклоалкила, -CF3 и -CHF2; и
каждый R9 независимо выбран из группы, состоящей из водорода, групп (С1-С6)алкил, (С3-С15)циклоалкил и (С6-С10)арил; где каждая из указанных группировок (С3-С15)циклоалкил и (С6-С10)арил может быть возможно замещена заместителями в количестве от одного до трех, независимо выбранными из (С1-С6)алкила, (С1-С6)алкокси и галогена.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где
R1 представляет собой -(С1-С6)алкил;
R2 представляет собой -NHR3 или -N(R3)2;
каждый R3 независимо выбран из группы, состоящей из групп -(С1-С6)алкил-R9 и -(С3-С15)циклоалкил-R9; либо, когда R2 представляет собой -N(R3)2, тогда оба указанных R3 могут быть взяты вместе с атомом азота, к которому они присоединены, с образованием 4-6-членного гетероциклического кольца, возможно замещенного заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из фтора, -ОН, групп -О-(С1-С6)алкил, NH2, -NH-(С1-С6)алкил, -N[(С1-С6)алкил]2, (С1-С6)алкил, -OR8, -O-(C=O)-OR8, -SR8, -NH-(C=O)-OR8, -O-(C=O)-N(R8)2, -NH-(C=O)-N(R8)2, -N[(С1-С6)алкил](C=O)-OR8, -N[(С1-С6)алкил](C=O)-N(R8)2 и (С3-С15)циклоалкил;
R4 представляет собой водород;
R4a представляет собой (С1-С6)алкил;
R5 представляет собой

где n равен 0, 1, 2, 3 или 4.
3. Соединение по любому из пп.1-2 или его фармацевтически приемлемая соль, где R5 представляет собой

4. Соединение по любому из пп.1-2 или его фармацевтически приемлемая соль, где R5 представляет собой

5. Соединение по любому из пп.1-2 или его фармацевтически приемлемая соль, где R5 представляет собой

6. Соединение по любому из пп.1-2 или его фармацевтически приемлемая соль, где R5 представляет собой

7. Соединение по п.1 или его фармацевтически приемлемая соль, где R5 представляет собой

8. Соединение по п.1 или его фармацевтически приемлемая соль, где R5 представляет собой

9. Соединение по п.1 или его фармацевтически приемлемая соль, где R5 представляет собой

10. Соединение по любому из пп.1-9 или его фармацевтически приемлемая соль, где R2 представляет собой -(С1-С6)алкил-R9, -NHR3, -N(R3)2 или -О-(С1-С6)алкил-R9.
11. Соединение по любому из пп.1-10 или его фармацевтически приемлемая соль, где R2 представляет собой -N(R3)2 или -NHR3.
12. Соединение по п.1 или его фармацевтически приемлемая соль, где R2 представляет собой (С3-С15)циклоалкил или (3-14-членный)гетероциклил, включающий 1 гетероатом, выбранный из О, S и N.
13. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль, где R6 выбран из группы, состоящей из водорода, галогена, -CF3, -CHF2 и -CH2F.
14. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль, где R6 выбран из группы, состоящей из -(C=O)-R8, -(C=O)-OR8, -OR8, -SR8, -S(O)R8, -S(O)2R8, NH2, групп -NH-(С1-С6)алкил и -N[(С1-С6)алкил]2.
15. Соединение по любому из пп.1-12 или его фармацевтически приемлемая соль, где R6 представляет собой (С1-С6)алкил или (С3-С15)циклоалкил.
16. Соединение по п.1, где указанное соединение представляет собой
4-(азетидин-1-ил)-7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]имидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-7-метил-5-{1-метил-5-[4-(трифторметил)фенил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-7-метил-5-{1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-5-[5-(4-хлорфенил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-5-[5-(5-хлорпиридин-2-ил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазин;
5-{5-[4-(дифторметил)фенил]-1-метил-1H-пиразол-4-ил}-N,7-диметилимидазо[5,1-f][1,2,4]триазин-4-амин;
7-метил-N-(d3)метил-5-{1-метил-5-[4-(трифторметил)фенил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин-4-амин;
N,7-диметил-5-{1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин-4-амин;
4-(азетидин-1-ил)-5-{5-[3-фтор-5-(трифторметил)пиридин-2-ил]-1-метил-1Н-пиразол-4-ил}-7-метилимидазо[5,1-f][1,2,4]триазин;
N,7-диметил-5-{1-метил-5-[4-(трифторметокси)фенил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин-4-амин;
4-(азетидин-1-ил)-5-{5-[4-(дифторметил)фенил]-1-метил-1H-пиразол-4-ил}-7-метилимидазо[5,1-f][1,2,4]триазин;
4-(3-фторазетидин-1-ил)-7-метил-5-{1-метил-5-[4-(трифторметил)фенил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-5-[5-(4-бромфенил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазин;
4-(азетидин-1-ил)-5-{5-[4-(дифторметокси)фенил]-1-метил-1H-пиразол-4-ил}-7-метилимидазо[5,1-f][1,2,4]триазин;
4-азетидин-1-ил-7-метил-5-{1-метил-5-[5-(трифторметил)пиразин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин;
4-азетидин-1-ил-5-[5-(5-бромпиридин-2-ил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазин или
N,7-диметил-5-{1-метил-5-[5-(трифторметил)пиразин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин-4-амин;
или его фармацевтически приемлемая соль.
17. Применение соединения формулы I или его фармацевтически приемлемой соли по п.1 для лечения нарушения познавательной способности, ассоциированного с шизофренией, у человека.
18. Фармацевтическая композиция, обладающая активностью ингибитора PDE2, содержащая соединение формулы I или его фармацевтически приемлемую соль по п.1 вместе с фармацевтически приемлемым носителем.
19. 4-(Азетидин-1-ил)-7-метил-5-{1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин или его фармацевтически приемлемая соль.
20. 4-(Азетидин-1-ил)-7-метил-5-{1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-ил}имидазо[5,1-f][1,2,4]триазин.
21. Фармацевтическая композиция, обладающая активностью ингибитора PDE2, содержащая терапевтически эффективное количество соединения по п.19 вместе с фармацевтически приемлемым носителем.
22. Применение соединения формулы I или его фармацевтически приемлемой соли по п.1 для лечения мигрени.
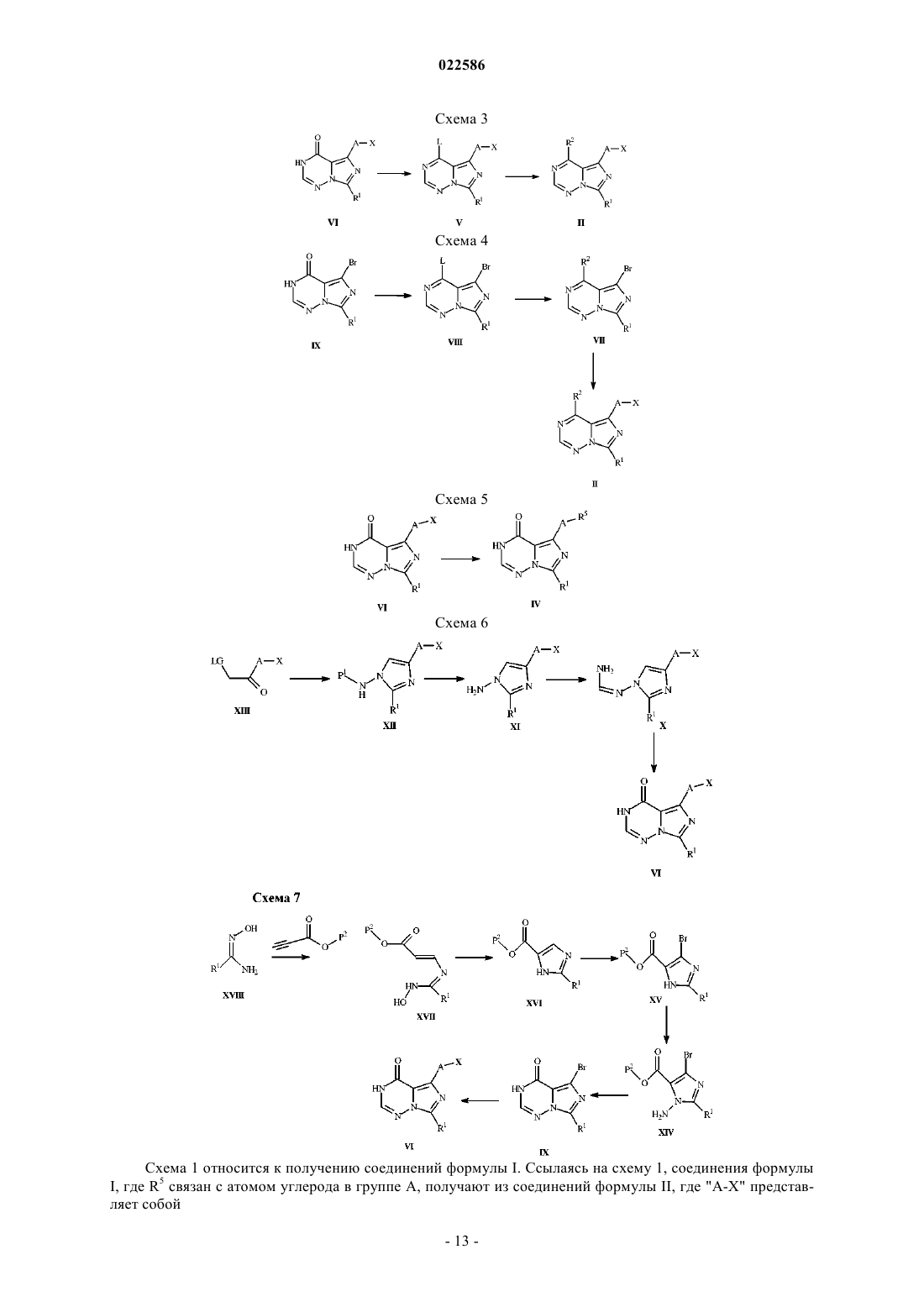
Текст
ИМИДАЗО[5,1-f][1,2,4]ТРИАЗИНЫ ДЛЯ ЛЕЧЕНИЯ НЕВРОЛОГИЧЕСКИХ РАССТРОЙСТВ Настоящее изобретение относится к соединениям формулы (I) Чэппи Томас Аллен, Хамфри Джон Майкл, Верхоест Патрик Роберт, Янг Эдди, Хилал Кристофер Джон (US) Поликарпов А.В. (RU) и их фармацевтически приемлемым солям, к способам их получения, к промежуточным соединениям, используемым для их получения, и к композициям, содержащим эти соединения; и к применению этих соединений в способе лечения заболевания или состояния, выбранного из группы, состоящей из расстройств центральной нервной системы, нарушений познавательной способности, шизофрении, деменции и других расстройств, у млекопитающего. Область изобретения Данное изобретение относится к имидазо[5,1-f][1,2,4]триазинам, которые являются селективными ингибиторами PDE2. Изобретение также относится к промежуточным соединениям для получения этих соединений; к фармацевтическим композициям, содержащим эти соединения; и к применению этих соединений в способах лечения ряда расстройств центральной нервной системы (CNS) или других расстройств. Изобретение также относится к способам лечения нейродегенеративных или психиатрических расстройств, включая психоз, нарушение познавательной способности, шизофрению, депрессию, деменцию и другие расстройства, у млекопитающего. Предпосылки изобретения Фосфодиэстеразы (PDE) представляют собой класс внутриклеточных ферментов, вовлеченных в гидролиз нуклеотидов циклический аденозинмонофосфат (сАМР) и циклический гуанозинмонофосфат(cGMP) в их соответствующие нуклеотидмонофосфаты. Эти циклические нуклеотиды служат в качестве вторичных переносчиков в некоторых клеточных путях, регулируя массив внутриклеточных процессов в пределах нейронов центральной нервной системы, включая активацию сАМР- и cGMP-зависимых протеинкиназ, которые продуцируют последующее фосфорилирование белков, вовлеченных в регулирование синаптической передачи сигналов, синаптической пластичности, нейрональной дифференциации и выживания. До настоящего времени был идентифицирован только один ген для PDE2, PDE2A; однако сообщалось о многочисленных альтернативно сплайсированных изоформах PDE2A, которые включаютPDE2A1, PDE2A2 и PDE2A3. PDE2A была идентифицирована как уникальное семейство на основании первичной аминокислотной последовательности и характерной ферментативной активности. Последовательность человеческой PDE2A3 была выделена в 1997 г. (Rosman et al. Isolation and characterization ofhuman cDNAs encoding cGMP-stimulated 3',5'-cyclic nucleotide phosphodiesterase, Gene, 191 (1): 89-95,1997). Ингибирование PDE2A демонстрирует улучшенную познавательную функцию в многочисленных доклинических моделях познавательной эффективности, что отражает улучшения распознающей памяти,социальных взаимодействий и рабочей памяти, которые являются недостаточными при шизофрении(Boess et al. Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance, Neuropharmacology, 47(7): 1081-92, 2004). Ингибирование PDE2A также уменьшало степень нарушения познавательной способности, которое развивается при старении и болезни Альцгеймера(Brandon et al. Potential CNS Applications for Phosphodiesterase Enzyme Inhibitors, Annual Reports in Medicinal Chemistry 42: 4-5, 2007). Однако это соединение продемонстрировало значительную активность в отношении других изоформ PDE и обладало высокой степенью клиренса и ограниченной способностью проникать в головной мозг и поэтому не рассматривалось как перспективное в клинических условиях. Было продемонстрировано, что ингибиторы PDE2 показали эффективность в доклинических моделях тревоги и депрессии (Masood et al. Anxiolytic effects of phosphodiesterase-2 inhibitors associated withantidepressant therapy increases cyclic GMP signaling in rat hippocampus, Neurosci. Lett., 466(3): 149-53,2009). Белок PDE2A, экспрессируемый в дорсальном роговидном отростке спинного мозга и в дорсальных корешках ганглиев, позволяет PDE2A модулировать уровни циклических нуклеотидов в этих областях во время лечения невропатической и воспалительной боли (Schmidtko et al. cGMP Produced by NO-Sensitivefrom cGMP-Dependent Protein Kinase I, The Journal of Neuroscience, 28(34): 8568-8576, 2008). Было продемонстрировано, что на периферии экспрессия PDE2A в эндотелиальных клетках играет критическую роль в регулировании эндотелиальной барьерной функции. Уровни экспрессии PDE2A в эндотелиальных клетках возрастают в ответ на воспалительные цитокины, такие как TNF-альфа, в условиях сепсиса и острого респираторного дистресс-синдрома и вносят вклад в нарушение эндотелиальной барьерной функции. Было продемонстрировано, что ингибирование PDE2A обращает дефициты проницаемости при сепсисе и увеличивает степени выживания в животных моделях сепсиса и эндотоксикозаhyperpermeability, Blood, 105: 3569-3576, 2005; Kayhan et al. Adenosine deaminase inhibitor erythro-9-[2hydroxyl-3-nonyl]adenine decreases intestinal permeability and protects against experimental sepsis: prospective, randomized laboratory investigation, Critical Care, 12(5): R125, 2008). Были опубликованы сведения о ряде имидазотриазинов как ингибиторах киназ, такие как публикация международной заявки WO 2011005909, озаглавленная "Process for preparation of substituted imidazo[5,1-f][1,2,4]triazine derivatives"; публикация патента США US 20090286768, озаглавленная "Substi-1 022586tuted imidazopyrazines and imidazotriazines as ACK1 inhibitors and their preparation"; публикация международной заявки WO 200911748, озаглавленная "Preparation of mTOR inhibitor salt forms"; публикация международной заявки WO 2009008992, озаглавленная "Preparation of imidazo[1,5-a]pyrazin-8-amine for use incombination therapy of cancers and cancer metastasis"; публикация патента США US 20080139582, озаглавленная "Preparation of substituted pyrazolopyrimidinamines as inhibitors of Bruton's tyrosine kinase"; публикация международной заявки WO 2007106503, озаглавленная Imidazo[1,5-a]pyrazin-8-amine in combinedinhibitors"; публикация международной заявки WO 2007087395, озаглавленная "Preparation of ethynyl- ortreatment of cancer and other diseases"; публикация патента США US 20070112005, озаглавленная "Preparation of substituted imidazopyrazines and related compounds as mTOR inhibitors"; публикация патента СШАUS 20060019957, озаглавленная "Preparation of imidazotriazines as protein kinase inhibitors;" и публикация международной заявки WO 2005097800, озаглавленная "Preparation of 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors". Краткое изложение сущности изобретения Настоящее изобретение относится к новым соединениям формулы или их фармацевтически приемлемым солям, где(С 3-С 15)циклоалкил или (3-14-членный)гетероциклил могут быть возможно замещены заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из (С 1-С 6)алкила, (С 1 С 6)алкокси и галогена; каждый R3 независимо выбран из группы, состоящей из групп -(С 1-С 6)алкил-R9 и -(С 3 С 15)циклоалкил-R9, либо, когда R2 представляет собой -N(R3)2, тогда оба указанных R3 могут быть взяты вместе с атомом азота, к которому они присоединены, с образованием 4-6-членного гетероциклического кольца, возможно замещенного заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из фтора, -ОН, групп -О-(С 1-С 6)алкил, NH2, -NH-(С 1-С 6)алкил, -N[(С 1-С 6)алкил]2,(С 1-С 6)алкил, -OR8, -O-(C=O)-OR8, -SR8, NH-(C=O)-OR8, -O-(C=O)-N(R8)2, -NH-(C=O)-N(R8)2, -N[(С 1 С 6)алкил](C=O)-OR8, -N[(С 1-С 6)алкил](С=О)-N(R8)2 и (С 3-С 15)циклоалкил; каждый R4 представляет собой водород; каждый R4a независимо выбран из группы, состоящей из (С 1-С 6)алкила и (С 3-С 15)циклоалкила;-2 022586 где n равен 0, 1, 2, 3 или 4; каждый R6 независимо выбран из группы, состоящей из водорода, галогена, (С 1-С 6)алкила, -CF3,-CHF2, -CH2F, групп -CF2-(C1-C6)алкил, -CN, -NO2, -(C=O)-R8, -(C=O)-OR8, -OR8, -SR8, -S(O)R8, -S(O)2R8,NH2, -NH-(С 1-С 6)алкил, -N[(С 1-С 6)алкил]2 и (С 3-С 15)циклоалкил; каждый R7 независимо выбран из группы, состоящей из галогена, (С 1-С 6)алкила, -CN, -CF3, -CHF2,-CH2F, групп -О-(С 1-С 6)алкил и (С 3-С 15)циклоалкил; каждый R8 там, где он присутствует, независимо выбран из группы, состоящей из водорода, (С 1 С 6)алкила, (С 3-С 15)циклоалкила, -CF3 и -CHF2; и каждый R9 независимо выбран из группы, состоящей из водорода, групп (С 1-С 6)алкил, (С 3-С 15)циклоалкил и (С 6-С 10)арил; где каждая из указанных группировок (С 3-С 15)циклоалкил и (С 6-С 10)арил может быть возможно замещена заместителями в количестве от одного до трех, независимо выбранными из (С 1 С 6)алкила, (С 1-С 6)алкокси и галогена. Как он используется в данном описании, термин "алкил" определен как включающий насыщенные алифатические углеводороды, включая прямые цепи и разветвленные цепи. Предпочтительно алкильная группа имеет от 1 до 6 атомов углерода. Например, как он используется в данном описании, термин "(С 1 С 6)алкил", также как и алкильные группировки других групп, упоминаемых в данном описании (например, (С 1-С 6)алкокси), относится к линейным или разветвленным радикалам из 1-6 атомов углерода (например, метилу, этилу, н-пропилу, изопропилу, н-бутилу, изобутилу, втор-бутилу, трет-бутилу), возможно замещенным 1-5 подходящими заместителями. Везде, где в данной заявке используется численный диапазон, например, когда в определении "алкила" используется диапазон от 1 до 6, это означает, что алкильная группа может содержать 1 атом углерода, 2 атома углерода, 3 атома углерода и так далее, и включая 6 атомов углерода. Как он используется в данном описании, термин "алкенил" определен как включающий алифатические углеводороды, имеющие по меньшей мере одну двойную связь углерод-углерод, включая прямые цепи и разветвленные цепи, имеющие по меньшей мере одну двойную связь углерод-углерод. Предпочтительно алкенильная группа имеет от 2 до 6 атомов углерода. Более предпочтительно алкенильная группа имеет от 2 до 4 атомов углерода, например, как он используется в данном описании, термин "(С 2 С 6)алкенил" означает ненасыщенные радикалы с прямой или разветвленной цепью из 2-6 атомов углерода, включая, но не ограничиваясь ими, этенил, 1-пропенил, 2-пропенил (аллил), изопропенил, 2-метил-1 пропенил, 1-бутенил, 2-бутенил и тому подобное, возможно замещенные 1-5 подходящими заместителями. Когда соединения формулы I содержат алкенильную группу, эта алкенильная группа может существовать в виде чистой Е (entgegen) формы, чистой Z (zusammen) формы или любой их смеси. Как он используется в данном описании, термин "алкинил" определен как включающий алифатические углеводороды, имеющие по меньшей мере одну тройную связь углерод-углерод, включая прямые цепи и разветвленные цепи, имеющие по меньшей мере одну тройную связь углерод-углерод. Предпочтительно алкинильная группа имеет от 2 до 6 атомов углерода. Например, как он используется в данном описании, термин "(С 2-С 6)алкинил" используется в данном описании как означающий алкинильные радикалы с прямой или разветвленной цепью, как определено выше, имеющие от 2 до 6 атомов углерода и одну тройную связь, возможно замещенные 1-5 подходящими заместителями. Как он используется в данном описании, термин "циклоалкил" определен как включающий насыщенные или ненасыщенные (неароматические) моноциклические или бициклические углеводородные кольца (например, моноциклические, такие как циклопропил, циклобутил, циклопентил, циклогексил,циклогептил, циклооктил или циклононил, или бициклические, включая мостиковые или конденсированные системы, такие как бицикло[2.2.1]гептанил, бицикло[3.2.1]октанил или бицикло[5.2.0]нонанил и так далее), возможно замещенные 1-5 подходящими заместителями. Циклоалкильная группа имеет от 3 до 15 атомов углерода. В одном воплощении циклоалкил может возможно содержать одну, две или более некумулятивных неароматических двойных или тройных связей и от одной до трех оксогрупп. Предпочтительно бициклоалкильная группа имеет от 6 до 15 атомов углерода. Бициклоалкил возможно замещен 1-5 подходящими заместителями. В одном воплощении бициклоалкил может возможно содержать одну,две или более некумулятивных неароматических двойных или тройных связей. Как он используется в данном описании, термин "арил" определен как включающий полностью углеродные моноциклические группы или полициклические группы из конденсированных колец, имеющие системы сопряженных пи-электронов. Арильная группа имеет 6, 8 или 10 атомов углерода в кольце(ах). В более общепринятом смысле арильная группа имеет 6 или 10 атомов углерода в кольце(ах). В наиболее общепринятом смысле арильная группа имеет 6 атомов углерода в кольце. Например, как он используется в данном описании, термин "(С 6-С 10)арил" означает ароматические радикалы, содержащие от 6 до 10 атомов углерода, такие как фенил, нафтил, тетрагидронафтил, инданил и тому подобное. Арильная группа возможно замещена 1-5 подходящими заместителями. Как он используется в данном описании, термин "гетероарил" определен как включающий ароматические гетероциклические моноциклические группы или полициклические группы из конденсированных колец с одним или более гетероатомами, выбранными из О, S и N, по меньшей мере в одном кольце. Гетероарильная группа имеет от 5 до 14 кольцевых атомов, включая от 1 до 13 атомов углерода и от 1 до-3 022586 5 гетероатомов, выбранных из О, S и N. Предпочтительно гетероарильная группа имеет от 5 до 10 кольцевых атомов, включая от одного до четырех гетероатомов. Гетероарильная группа также содержит от одной до трех оксогрупп. Более предпочтительно гетероарильная группа имеет от 5 до 8 кольцевых атомов, включая один, два или три гетероатома. Представляющие особый интерес моноциклические гетероарилы включают гетероарилы с 5 кольцевыми атомами, включая от одного до трех гетероатомов, или гетероарилы с 6 кольцевыми атомами, включая один или два гетероатома азота. Представляющие особый интерес конденсированные бициклические гетероарилы включают два конденсированных 5- и/или 6-членных моноциклических кольца, содержащих от одного до четырех гетероатомов. Подходящие гетероарилы включают пиридил, пиразинил, пиримидинил, пиридазинил, тиенил, фурил, имидазолил, пирролил, оксазолил (например, 1,3-оксазолил, 1,2-оксазолил), тиазолил (например,1,2-тиазолил, 1,3-тиазолил), пиразолил, тетразолил, триазолил (например, 1,2,3-триазолил, 1,2,4-триазолил), оксадиазолил (например, 1,2,3-оксадиазолил), тиадиазолил (например, 1,3,4-тиадиазолил), хинолил, изохинолил, бензотиенил, бензофурил, индолил, пиридон, пиримидон, пиразинон, пиримидинон и тому подобное. Гетероарильная группа возможно замещена 1-5 подходящими заместителями. Как он используется в данном описании, термин "гетероциклил" определен как включающий моноциклическую, мостиковую полициклическую или конденсированную полициклическую, насыщенную или ненасыщенную неароматическую 3-14-членную кольцевую систему из 1-13 атомов углерода и включающую от 1 до 5 гетероатомов, выбранных из О, S и N. Гетероциклическая группа также включает от одной до трех оксогрупп. Примеры таких гетероциклоалкильных колец включают азетидинил, тетрагидрофуранил, имидазолидинил, пирролидинил, пиперидинил, пиперазинил, оксазолидинил, тиазолидинил,пиразолидинил, тиоморфолинил, тетрагидротиазинил, тетрагидротиадиазинил, морфолинил, оксетанил,тетрагидродиазинил, оксазинил, оксатиазинил, индолинил, изоиндолинил, хинуклидинил, хроманил, изохроманил, бензоксазинил, 2-азабицикло[2.2.1]гептанон, 3-азабицикло[3.1.0]гексан, 3-азабицикло[4.1.0]гептан и тому подобное. Дополнительные примеры указанных гетероциклоалкильных колец включают тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, имидазолидин-1-ил, имидазолидин-2-ил, имидазолидин-4 ил, пирролидин-1-ил, пирролидин-2-ил, пирролидин-3-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин 3-ил, пиперидин-4-ил, пиперазин-1-ил, пиперазин-2-ил, 1,3-оксазолидин-3-ил, изотиазолидин, 1,3-тиазолидин-3-ил, 1,2-пиразолидин-2-ил, 1,2-тетрагидротиазин-2-ил, 1,3-тетрагидротиазин-3-ил, 1,2-тетрагидродиазин-2-ил, 1,3-тетрагидродиазин-1-ил, 1,4-оксазин-2-ил, оксазолидинон и тому подобное. Гетероциклоалкильное кольцо возможно замещено 1-5 подходящими заместителями. Предпочтительные гетероциклы включают 5 и 6-членные моноциклические кольца или 9 и 10-членные конденсированные бициклические кольца. Как он используется в данном описании, термин "галоген" или " группа галоген" определен как включающий фтор, хлор, бром или йод. Как было указано выше, соединения формулы I могут существовать в форме фармацевтически приемлемых солей, таких как, например, соли присоединения кислот и соли присоединения оснований соединений формулы I. Выражение "фармацевтически приемлемая(ые) соль(и)", как оно используется в данном описании, если не указано иное, включает соли присоединения кислот или оснований, которые могут присутствовать в соединениях формулы I. Фармацевтически приемлемые соли соединения формулы (I) включают его соли присоединения кислот и оснований. Подходящие соли присоединения кислот образуются из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, эсилат, формиат, фумарат, глуцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изетионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглутамат,сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинафоат. Подходящие соли с основаниями образуются из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина,глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Также могут быть образованы гемисоли кислот и оснований, например соли гемисульфат и соли гемикальция. Обзор подходящих солей приведен в Handbook of Pharmaceutical Salts: Properties, Selection, and Useby Stahl и Wermuth (Wiley-VCH, 2002). Способы получения фармацевтически приемлемых солей соединения формулы I известны специалистам в данной области. Как они используются в данном описании, термины "соединение формулы I" и "соединение формулы I или его фармацевтически приемлемые соли" определены как включающие все формы соединения формулы I, включая его гидраты, сольваты, изомеры, кристаллические и некристаллические формы,изоморфы, полиморфы, метаболиты и пролекарства. Соединения формулы I или их фармацевтически приемлемая соль могут существовать в несольватированных и сольватированных формах. Когда растворитель или вода сильно связаны, комплекс будет-4 022586 обладать хорошо определяемой стехиометрией независимо от влажности. Однако, когда растворитель или вода связаны слабо, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях нормальным будет являться отсутствие стехиометрии. Соединения формулы I могут существовать в виде клатратов или других комплексов. В объем изобретения включены такие комплексы, как клатраты, комплексы включения лекарственное средствохозяин, где в противоположность вышеупомянутым сольватам лекарственное средство и хозяин присутствуют в стехиометрических или нестехиометрических количествах Также включены комплексы формулы I, содержащие два или более органических и/или неорганических компонента, которые могут находиться в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированными, частично ионизированными или неионизированными(обзор таких комплексов см. вHaleblian J.K., J. Pharm. Sci. 1975, 64, 1269-1288). Также в объем изобретения включены метаболиты соединений формулы I, то есть соединения, образующиеся in vivo при введении лекарственного средства. Метаболиты формулы I включают соединения, где R1 представляет собой гидроксиалкил. Соединения формулы I могут иметь асимметричные атомы углерода. Связи углерод-углерод соединений формулы I могут быть изображены с использованием прямой линии, сплошного клина или пунктирного клина. Использование прямой линии для изображения связей с асимметричными атомами углерода предназначено для указания на то, что включены все возможные стереоизомеры (например, отдельные энантиомеры, рацемические смеси и так далее) по данному атому углерода. Использование сплошного или пунктирного клина для изображения связей с асимметричными атомами углерода предназначено для указания на то, что включен только один показанный стереоизомер. Возможно, что соединения формулы I могут содержать более чем один асимметричный атом углерода. В таких соединениях использование прямой линии для изображения связей с асимметричными атомами углерода предназначено для указания на то, что включены все возможные стереоизомеры. Например, если не указано иное, предполагается, что соединения формулы I могут существовать в виде энантиомеров и диастереомеров или в виде рацематов и их смесей. Использование прямой линии для изображения связей с одним или более асимметричных атомов углерода в соединении формулы I и использование сплошного или пунктирного клина для изображения связей с другими асимметричными атомами углерода в одном и том же соединении предназначено для указания на то, что присутствует смесь диастереомеров. Стереоизомеры формулы I включают цис- и транс-изомеры, оптические изомеры, такие как R- и Sэнантиомеры, диастереомеры, геометрические изомеры, вращательные изомеры, конформационные изомеры и таутомеры соединений формулы I, включая соединения, проявляющие более чем один тип изомерии, и их смеси (такие как рацематы и пары диастереомеров). Также включены соли присоединения кислот или соли присоединения оснований, где противоион является оптически активным, например Dлактат или L-лизин, или рацемическим, например DL-тартрат или DL-аргинин. Когда кристаллизуется любой рацемат, возможны кристаллы двух разных типов. Первый тип представляет собой рацемическое соединение (истинный рацемат), упоминавшееся выше, где получается одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где получаются две формы кристалла в эквимолярных количествах, каждая из которых содержит отдельный энантиомер. Соединения формулы I могут проявлять феномен таутомеризма и структурного изомеризма. Например, соединения формулы I могут существовать в нескольких таутомерных формах, включая енольную и иминную форму, и кето- и енаминную форму и геометрические изомеры и их смеси. Все такие таутомерные формы включены в объем соединений формулы I. В растворе таутомеры существуют в виде смесей набора таутомеров. В твердой форме обычно преобладает один таутомер. Несмотря на то что может быть описан один таутомер, настоящее изобретение включает все таутомеры соединения формулы I. Настоящее изобретение также включает соединения, меченные изотопами, которые идентичны соединениям, представленным в формуле I выше, за исключением того факта, что один или более атомов заменен(ы) атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов, которые могут быть включены в соединения формулы I, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как, без ограничения ими, 2 Н, 3 Н, 13 С, 14 С, 15N, 17 О, 18 О, 32 Р, 35S, 18F и 36Cl. Некоторые меченные изотопами соединения формулы I, например соединения, в которые включены радиоактивные изотопы,такие как 3 Н и 14 С, полезны в анализах распределения лекарственных средств и/или субстратной ткани. Особенно предпочтительными являются изотопы тритий, то есть 3 Н, и углерод-14, то есть 14 С, в силу легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, то есть 2 Н, может обеспечить определенные терапевтические преимущества, являющиеся результатом большей метаболической стабильности, например увеличенный период полувыведения in vivo или пониженные дозировки, и поэтому может быть предпочтительным в определенных обстоятельствах. Меченные изотопами соединения формулы I могут в общем быть получены путем осуществления проце-5 022586 дур, раскрытых на схемах и/или в примерах и подготовительных примерах, представленных ниже, посредством замены не меченного изотопом реагента на меченный изотопом реагент. Конкретное воплощение настоящего изобретения относится к соединениям формулы I или их фармацевтически приемлемой соли, гдеR1 представляет собой -(С 1-С 6)алкил (более конкретно метил или этил; еще более конкретно метил);R2 представляет собой -NHR3 или -N(R3)2; каждый R3 независимо выбран из группы, состоящей из групп -(С 1-С 6)алкил-R9 и -(С 3 С 15)циклоалкил-R9 (более конкретно -(С 1-С 6)алкил-R9 и еще более конкретно метил); либо, когда R2 представляет собой -N(R3)2, тогда оба указанных R3 могут быть взяты вместе с атомом азота, к которому они присоединены, с образованием 4-6-членного гетероциклического кольца, возможно замещенного заместителями в количестве от одного до трех, независимо выбранными из группы, состоящей из фтора,-ОН, групп -О-(С 1-С 6)алкил, NH2, -NH-(С 1-С 6)алкил, -N[(С 1-С 6)алкил]2, (С 1-С 6)алкил, -OR8, -O-(C=O)OR8, -SR8, -NH-(C=O)-OR8, -O-(C=O)-N(R8)2, -NH-(C=O)-N(R8)2, -N[(С 1-С 6)алкил](C=O)-OR8, -N[(С 1 С 6)алкил](C=O)-N(R8)2 и (С 3-С 15)циклоалкил (более конкретно одним или двумя заместителями, где указанный(е) заместитель(и) представляет(ют) собой фтор, -ОН, -О-(С 1-С 6)алкил, NH2, -NH-(С 1-С 6)алкил,-N[(C1-С 6)алкил]2, (С 1-С 6)алкил или -NH-(C=O)-OR8; еще более конкретно, где указанный(е) заместитель(и) представляет(ют) собой фтор, метокси, или метилкарбамат);R4a представляет собой (С 1-С 6)алкил (более конкретно метил или этил; еще более конкретно метил); где n равен 0, 1, 2, 3 или 4 (более конкретно, где n равен 0, 1 или 2) (более конкретно, где R7 представляет собой хлор, фтор, метил, метокси или циано; и еще более конкретно, где R6 представляет собой хлор, бром, метил, этил, метокси, -CF3, -CF2CH3, -OCF3, -OCHF2, NO2, -(C=O)-CH3, (С 3-С 15)циклоалкил или изопропил). Конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-(1Hпиразол-4-ил)имидазо[1,5-f][1,2,4]триазина формулы Ia, где формула I содержит группу A1R5 Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-(5-фенил-1H-пиразол-4-ил)имидазо[1,5-f][1,2,4]триазина формулы Id, где R5 представлен как возможно замещенный фенил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям-6 022586 5-[5-(пиридин-2-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Ig, где R5 представлен как возможно замещенный пиридин-2-ил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-[5-(пиридин-3-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Ij, где R5 представлен как возможно замещенный пиридин-3-ил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-[5-(пиразин-2-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Im, где R5 представлен как возможно замещенный пиразин-2-ил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-[5-(пиримидин-2-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Ip, где R5 представлен как возможно замещенный пиримидин-2-ил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-[5-(пиридазин-3-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Is, где R5 представлен как возможно замещенный пиридазин-3-ил Другое конкретное воплощение настоящего изобретения относится к так называемым соединениям 5-[5-(пиримидин-5-ил)-1H-пиразол-4-ил]имидазо[1,5-f][1,2,4]триазина формулы Iv, где R5 представлен как возможно замещенный пиримидин-5-ил Другое представляющее особый интерес конкретное воплощение относится к соединениям формулы I (и формул Ia-Iv), где R2 представляет собой -(С 1-С 6)алкил-R9, -NHR3, -N(R3)2 или -О-(С 1-С 6)алкил-R9. Конкретные представляющие особый интерес группы R2 включают -NHR3 (в частности, где R3 представляет собой метил, этил или пропил), и -N(R3)2 (в частности, где две группы R3 взяты вместе с атомом азота, к которому они присоединены, с образованием возможно замещенной азетидинильной группы, в ча-7 022586 стности, возможно замещенной заместителями, выбранными из фтора, дифтора или метокси). Другие более конкретные R2 включают -(С 1-С 6)алкил-R9 и -О-(С 1-С 6)алкил-R9, в частности, где R9 представляет собой водород. Отдельно изобретение также включает те соединения, где R2 представляет собой -OR8, в частности, где R8 представляет собой метил или этил. Другое представляющее особый интерес воплощение относится к соединениям формулы I (и формул Ia-Iv), где R2 представляет собой (С 3-С 15)циклоалкил или (С 1-С 14)гетероциклил, включающий 1 гетероатом, выбранный из О, S и N; более конкретно, где (С 1-С 14)гетероциклил содержит один атом азота. Другое представляющее особый интерес воплощение относится к соединениям формулы I (и формул Ia-Iv), где R6 представляет собой галоген, -CF3, -CHF2 или -CH2F; более конкретно, где R6 представляет собой галоген. Альтернативные представляющие особый интерес соединения включают фторметильные заместители -CF3, -CHF2 или -CH2F. Другое представляющее особый интерес воплощение относится к соединениям формулы I (и формул Ia-Iv), где R6 представляет собой -(C=O)-R8, -(C=O)-OR8, -OR8, -SR8, -S(O)R8, -S(O)2R8, NH2, -NH-(С 1 С 6)алкил, -N[С 1-С 6)алкил]2; более конкретно, где R6 представляет собой -(C=O)-R8, -(C=O)-OR8 или -OR8. Отдельно настоящее изобретение рассматривает так называемые аминосоединения NH2, -NH-(С 1-С 6)алкил или -N[(С 1-С 6)алкил]2. Отдельно настоящее изобретение рассматривает так называемые тиосоединения -SR8, -S(O)R8 или -S(O)2R8. Другое представляющее особый интерес воплощение относится к соединениям формулы I (и формул Ia-Iv), где R6 представляет собой (С 1-С 6)алкил или (С 3-С 15)циклоалкил; более конкретно, где R6 представляет собой (С 1-С 6)алкил. Отдельно настоящее изобретение рассматривает кольцевые соединения (С 3 С 15)циклоалкил. Каждое из вышеуказанных воплощений предназначено быть интерпретированным как отдельное воплощение, а также как воплощение, взятое в комбинации с каждым из предыдущих вышеуказанных воплощений (например, каждое воплощение R6 взято вместе с R2 и с формулами Ia-Iv). В другом воплощении изобретение также относится к соединениям, раскрытым как примеры 1-86 в разделе "примеры" настоящей заявки, и к их фармацевтически приемлемым солям. В другом воплощении изобретение относится к соединению формулы I, где указанное соединение представляет собой 4-(азетидин-1-ил)-7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]имидазо[5,1-f][1,2,4]триазин; 4-(азетидин-1-ил)-7-метил-5-1-метил-5-[4-(трифторметил)фенил]-1H-пиразол-4-илимидазо[5,1-f]N,7-диметил-5-1-метил-5-[5-(трифторметил)пиразин-2-ил]-1H-пиразол-4-илимидазо[5,1-f][1,2,4] триазин-4-амин; или их фармацевтически приемлемые соли. В другом воплощении изобретение относится к соединениям формулы I, которые будут получены,выбранным из следующей группы: 5-(5-(3-фтор-5-(трифторметил)пиридин-2-ил)-1-метил-1H-пиразол-4-ил)-N,7-диметилимидазо[5,1-f][1,2,4]триазин; 4-(азетидин-1-ил)-5-(4-(5,6-диметилпиридазин-3-ил)-3-метил-1H-пиразол-5-ил)-7-метилимида-9 022586 зо[5,1-f][1,2,4]триазин; 4-(азетидин-1-ил)-5-(4-(2,4-бис-(трифторметил)пиримидин-5-ил)-3-метил-1H-пиразол-5-ил)-7-метилимидазо[5,1-f][1,2,4]триазин; 4-(азетидин-1-ил)-5-(1-(4-фтор-2-метилпиримидин-5-ил)-2-метил-1H-имидазол-5-ил)-7-метилимидазо[5,1-f][1,2,4]триазин; или их фармацевтически приемлемые соли. Настоящее изобретение также относится к композициям, содержащим соединение формулы I или его приемлемую соль (например, фармацевтическим композициям). Соответственно в одном воплощении изобретение относится к фармацевтической композиции, содержащей соединение формулы I, включающей фармацевтически приемлемый носитель и возможно по меньшей мере один дополнительный медицинский или фармацевтический агент. В одном воплощении дополнительный медицинский или фармацевтический агент представляет собой агент против шизофрении, как описано ниже. Фармацевтически приемлемый носитель может содержать любой традиционный фармацевтический носитель или эксципиент. Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители (такие как гидраты и сольваты). Фармацевтические композиции могут, если желательно, содержать дополнительные ингредиенты, такие как корригенты, связующие вещества, эксципиенты и тому подобное. Так, для перорального введения могут быть использованы таблетки, содержащие различные эксципиенты, такие как лимонная кислота, вместе с различными разрыхлителями, такими как крахмал, альгиновая кислота и некоторые комплексные силикаты, и со связующими агентами, такими как сахароза, желатин и аравийская камедь. Кроме того, для целей таблетирования часто являются полезными смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа могут также быть использованы в мягких и твердых заполненных желатиновых капсулах. Неограничивающие примеры материалов, таким образом, включают лактозу или молочный сахар и высокомолекулярные полиэтиленгликоли. Когда для перорального введения желательны водные суспензии или эликсиры, активное соединение в них может быть объединено с различными подсластителями или корригентами, красящими веществами или красителями и, если желательно, эмульгирующими агентами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их комбинации. Фармацевтическая композиция может, например, находиться в форме, подходящей для перорального введения, в виде таблетки, капсулы, пилюли, порошка, композиции замедленного высвобождения,раствора или суспензии, для парентеральной инъекции в виде стерильного раствора, суспензии или эмульсии, для местного введения в виде мази или крема или для ректального введения в виде суппозитория. Типичные формы для парентерального введения включают растворы или суспензии активного соединения в стерильных водных растворах, например водных растворах пропиленгликоля или декстрозы. Такие лекарственные формы могут быть подходящим образом забуферены, если желательно. Фармацевтическая композиция может находиться в формах стандартной дозы, подходящих для однократного введения точных дозировок. Для специалиста в данной области очевидно, что композиция может быть приготовлена в виде препарата в субтерапевтической дозировке, так что представлены множественные дозы. В одном предпочтительном воплощении композиция содержит терапевтически эффективное количество соединения формулы I и включает фармацевтически приемлемый носитель. В настоящем изобретении также раскрыт способ лечения шизофрении или психоза у млекопитающего, предпочтительно человека, включающий введение указанному млекопитающему (предпочтительно человеку) терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В настоящем изобретении также раскрыт способ лечения PDE2-опосредованного расстройства,включающий введение нуждающемуся в этом млекопитающему (предпочтительно человеку) количества соединения формулы I, эффективного для ингибирования PDE2; более предпочтительно введение количества соединения формулы I, эффективного для селективного ингибирования PDE2. В настоящем изобретении также раскрыт способ лечения неврологических расстройств (таких как мигрень; эпилепсия; болезнь Альцгеймера; болезнь Паркинсона; повреждение головного мозга; удар; цереброваскулярные заболевания (включая церебральный артериосклероз, церебральную амилоидную ангиопатию, наследственную церебральную лихорадку и гипоксию-ишемию головного мозга); нарушений познавательной способности (включая амнезию, сенильную деменцию, ВИЧ (вирус иммунодефицита человека), ассоциированную деменцию, деменцию, ассоциированную с болезнью Альцгеймера, деменцию, ассоциированную с болезнью Хантингтона, деменцию телец Леви, васкулярную деменцию, деменцию, связанную с приемом лекарственных препаратов, делирий и умеренное ухудшение познавательной способности); умственную неполноценность (включая синдром Дауна и синдром хрупкой Ххромосомы); расстройств сна (включая гиперсомнию, расстройство сна, связанное с нарушением циркадного ритма, инсомнию, парасомнию и потерю сна) и психиатрические расстройства (такие как тревога (включая острое стрессовое расстройство, генерализованное тревожное расстройство, социальное тре- 10022586 вожное расстройство, паническое расстройство, посттравматическое стрессовое расстройство и обсессивно-компульсивное расстройство); симулятивное расстройство (включая острую галлюциногенную манию); расстройства импульсивного контроля (включая компульсивный азарт и перемежающееся импульсивное расстройство); расстройства настроения (включая биполярное расстройство I типа, биполярное расстройство II типа, манию, смешанное аффективное состояние, большую депрессию, хроническую депрессию, сезонную депрессию, психотическую депрессию и послеродовую депрессию); психомоторные расстройства; психотические расстройства (включая шизофрению, шизоаффективное расстройство,шизофрениформ и бредовое расстройство); лекарственную зависимость (включая наркотическую зависимость, алкоголизм, амфетаминовую зависимость, кокаиновую наркоманию, никотиновую зависимость и синдром отмены лекарств); расстройства приема пищи (включая анорексию, булимию, расстройство в виде склонности к кутежам, гиперфагию и пагофагию); и педиатрические психиатрические расстройства(включая расстройство с дефицитом внимания, расстройство дефицит внимания/гиперактивность, буйство и аутизм) у млекопитающего, предпочтительно человека, включающий введение указанному млекопитающему терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. В настоящем изобретении также раскрыт способ лечения шизофрении. В настоящем изобретении также раскрыт способ лечения нарушения познавательной способности,ассоциированного с шизофренией. Термин "терапевтически эффективное количество", как он используется в данном описании, относится к такому количеству вводимого соединения, которое будет ослаблять в определенной степени один или более симптомов подлежащего лечению расстройства. В отношении лечения шизофрении терапевтически эффективное количество относится к такому количеству, которое оказывает эффект ослабления в определенной степени (или предпочтительно устранения) одного или более симптомов, ассоциированных с шизофренией. Термин "лечить", как он используется в данном описании, если не указано иное, означает реверсирование, ослабление, ингибирование развития или предупреждение расстройства или состояния, в отношении которого применяется такой термин, либо одного или более симптомов такого расстройства или состояния. Термин "лечение", как он используется в данном описании, если не указано иное, относится к акту лечения, как "лечить" определено непосредственно выше. Термин "лечение" также включает адъювантное и неоадъювантное лечение субъекта. Введение соединения формулы I может быть осуществлено любым способом, который делает возможной доставку соединения в место его действия. Эти способы включают пероральные пути, интраназальные пути, ингаляционные пути, интрадуоденальные пути, парентеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или инфузию), местное и ректальное введение. Согласно настоящему изобретению соединения формулы I могут быть предпочтительно введены пероральными путями. Режимы дозирования могут быть скорректированы таким образом, чтобы обеспечить оптимальный желаемый ответ. Например, может быть введен одиночный болюс, несколько разделенных доз могут быть введены с течением времени, либо доза может быть пропорционально уменьшена или увеличена в соответствии с требованиями терапевтической ситуации. Особенно целесообразно готовить парентеральные композиции в форме стандартной дозы для легкости введения и стандартности дозировок. Форма стандартной дозы, как этот термин используется в данном описании, относится к физически дискретным единицам, пригодным в качестве единичных дозировок для субъекта-млекопитающего, подлежащего лечению; каждая единица содержит заданное количество активного соединения, рассчитанное таким образом, чтобы обеспечивать желаемый терапевтический эффект, в сочетании с необходимым фармацевтическим носителем. Требования для форм стандартной дозы по изобретению диктуются и непосредственно зависят от (а) уникальных характеристик химиотерапевтического агента и конкретного терапевтического или профилактического эффекта, который должен быть достигнут, и (б) ограничений, существующих в области приготовления такого активного соединения для лечения чувствительности у индивидуумов. В одном воплощении настоящего изобретения соединения формулы I могут предпочтительно быть использованы для лечения людей. Следует отметить, что величины дозировок могут изменяться в зависимости от типа и тяжести состояния, подлежащего облегчению, и могут включать однократные или множественные дозы. Следует также понимать, что для любого конкретного субъекта специфические схемы введения нужно регулировать с течением времени в соответствии с индивидуальными потребностями и профессиональным суждением специалиста, осуществляющего введение или контролирующего введение композиций, и что диапазоны дозировок, приведенные в данном описании, являются только иллюстративными и не предназначены для ограничения объема или практики заявленной композиции. Например, дозы могут быть скорректированы на основании фармакокинетических или фармакодинамических параметров, которые могут включать клинические эффекты, такие как токсические эффекты и/или лабораторные величины. Таким образом, настоящее изобретение охватывает повышение дозы для пациента, как определено спе- 11022586 циалистом. Определение соответствующих дозировок и схем введения химиотерапевтическогоагента хорошо известно в релевантных областях и, как очевидно, будет находиться в компетенции специалиста при его знакомстве с представленной в данном описании информацией. Количество вводимого соединения формулы I будет зависеть от подлежащего лечению субъекта,тяжести расстройства или состояния, скорости введения, диспозиции соединения и предписания лечащего врача. Однако эффективная дозировка находится в диапазоне от примерно 0,01 до примерно 50 мг на кг массы тела в сутки, предпочтительно от примерно 0,01 до примерно 5 мг/кг/сутки в разовой или разделенных дозах. Для человека массой 70 кг это будет соответствовать количеству от примерно 0,7 мг до примерно 3500 мг/сутки, предпочтительно от примерно 5 мг до примерно 2000 мг/сутки. В некоторых обстоятельствах более чем адекватными могут оказаться уровни дозировок ниже нижнего предела вышеуказанного диапазона, тогда как в других случаях могут быть использованы большие дозы, не вызывая никакого опасного побочного действия, при условии, что такая большая доза сначала разделена на несколько меньших доз для введения на протяжении суток. Как он используется в данном описании, термин "комбинированная терапия" относится к введению соединения формулы I вместе по меньшей мере с одним дополнительным фармацевтическим или медицинским агентом (например, агентом против шизофрении) либо последовательно, либо одновременно. Как уже отмечалось выше, соединения формулы I могут быть использованы в комбинации с одним или более дополнительными агентами против шизофрении, которые описаны ниже. Когда используют комбинированную терапию, один или более дополнительных агентов против шизофрении могут быть введены последовательно или одновременно с соединением по изобретению. В одном воплощении дополнительный агент против шизофрении вводят млекопитающему (например, человеку) перед введением соединения по изобретению. В другом воплощении дополнительный агент против шизофрении вводят млекопитающему после введения соединения по изобретению. В другом воплощении дополнительный агент против шизофрении вводят млекопитающему (например, человеку) одновременно с введением соединения по изобретению. Изобретение также относится к фармацевтической композиции для лечения шизофрении у млекопитающего, включая человека, которая содержит определенное количество соединения формулы I, как оно определено выше (включая гидраты, сольваты и полиморфы указанного соединения или его фармацевтически приемлемые соли), в комбинации с одним или более (предпочтительно от одного до трех) агентами против шизофрении, такими как зипразидон, риспердон, оланзапин, кветиапин, арипипразол,асенапин, блонансерин или лоперидон, где количества активного агента и комбинации, взятой как целое,терапевтически эффективны для лечения шизофрении. Подробное описание изобретения Соединения формулы I могут быть получены в соответствии со следующими реакционными схемами и сопровождающим их обсуждением. Если не указано иное, радикалы с R1 по R8, А и n и структурная формула I на реакционных схемах и в последующем обсуждении являются такими, как определено выше. В общем соединения по данному изобретению могут быть получены способами, которые включают способы, аналогичные способам, известным в области химии, особенно в свете содержащегося там описания. Ряд способов для изготовления соединений по данному изобретению предложены как дополнительные признаки изобретения и проиллюстрированы следующими реакционными схемами. Другие способы могут быть описаны в экспериментальном разделе. Схема 1 Схема 1 относится к получению соединений формулы I. Ссылаясь на схему 1, соединения формулыI, где R5 связан с атомом углерода в группе А, получают из соединений формулы II, где "А-Х" представляет собой и X представляет собой Н, Cl, Br, I или трифлат, посредством катализируемой палладием реакции сочетания с реагентом формулы R5a-R5g В зависимости от типа используемой реакции Z может представлять собой Br, В(ОН)2 или B(OR)2,либо группировку триалкилолова. Например, когда X представляет собой галоген или трифлат и реагентR5 представляет собой бороновую кислоту или эфир бороновой кислоты, может быть использована реакция Сузуки (Suzuki A. J. Organomet. Chem. 1999, 576, 147-168; Miyaura N. and Suzuki A., Chem. Rev. 1995,95, 2457-2483; Littke A.F. et al. J. Am. Chem. Soc. 2000, 122, 4020-4028). Более конкретно, гетероароматический йодид, бромид или трифлат формулы II объединяют с 1-3 эквивалентами арил- или гетероарилбороновой кислоты или эфира бороновой кислоты и подходящим основанием, например 2-5 эквивалентами карбоната натрия, в подходящем органическом растворителе, таком как этанол. Добавляют палладиевый катализатор, такой как 0,01 эквивалента тетракис(трифенилфосфин)палладия(0), и реакционную смесь нагревают до температур в диапазоне от 60 до 100 С в течение промежутка времени от 1 до 24 ч. В некоторых случаях в реакции Сузуки может быть целесообразным использовать от 1 до 2 эквивалентов хлорида меди(I) и от 1 до 2 эквивалентов бромида калия в 1,2-диметоксиэтане в качестве растворителя. Альтернативно, реакция сочетания может быть осуществлена посредством взаимодействия соединения формулы II, где X представляет собой Н, и Z представляет собой Br, с 1-3 эквивалентами реагента (R5)-Br в присутствии от 0,01 до 0,5 эквивалентов димера хлорида аллилпалладия и подходящего основания,такого как от 2 до 4 эквивалентов карбоната калия, в подходящем органическом растворителе, таком как 1,4-диоксан. Взаимодействие может быть осуществлено при температурах в диапазоне от 100 до 160 С в течение промежутка времени от 24 до 72 ч. Когда X представляет собой галоген или трифлат и Z представляет собой триалкилолово, может быть использовано сочетание Стилла (Stille) (Farina V. et at. Organic Reactions 1997, 50, 1-652). Более конкретно, соединение формулы II, где X представляет собой бромид, йодид или трифлат, может быть объединено 1,5-3 эквивалентами R5 станнана в присутствии палладиевого катализатора, такого как 0,05 эквивалентов дихлорбис-(трифенилфосфин)палладия(II), в подходящем органическом растворителе, таком как толуол, и реакционная смесь может быть нагрета до температур в диапазоне от 100 до 130 С в течение промежутка времени от 12 до 36 ч. Когда X представляет собой Br, I или трифлат и Z представляет собой Br или I, может быть использовано сочетание Негиши(Negishi) (Erdik E. Tempahedron 1992, 48, 9577-9648). Более конкретно, соединение формулы II, где X представляет собой бромид, йодид или трифлат, может быть подвергнуто трансметаллированию путем обработки от 1 до 1,1 эквивалентами алкиллитиевого реагента, а затем раствором от 1,2 до 1,4 эквивалентов хлорида цинка в подходящем растворителе, таком как тетрагидрофуран, при температуре в диапазоне от-65 С. После нагревания до температуры между 10 и 30 С реакционная смесь может быть обработана реагентом R-Z и нагрета при 50-70 С с добавлением катализатора, такого как тетракис(трифенилфосфин)палладий(0). Взаимодействие может быть осуществлено в течение промежутка времени в диапазоне от 1 до 24 ч. Ни одно из этих взаимодействий не лимитировано использованием растворителя, основания или катализатора, описанных выше, поскольку могут быть использованы многие другие условия. Альтернативно, ссылаясь на схему 1, полярность реакции сочетания АХ 1 с любым из R5a-R5g может быть обращена. В таком случае группа X в АХ 1 представляет собой группировку бороновой кислоты,бороната или триалкилолова, и группа Z в R5a-R5g представляет собой Cl, Br, I или трифлат. Химизм, ис- 14022586 пользуемый для создания связи углерод-углерод, является таким же, как описано выше. Схема 2 относится к альтернативному получению соединений формулы I. Ссылаясь на схему 2, соединение формулы I, где R2 представляет собой N(R3)2 или HNR3, может быть получено из соединения формулы III, где L представляет собой триазол или хлор, посредством взаимодействия с 1,1-4 эквивалентами первичного или вторичного амина, H2NR3 или HN(R3)2, возможно в присутствии основания, такого как карбонат цезия, в соответствующем органическом растворителе, таком как дихлорметан или N,Nдиметилформамид. Подходящие температуры для вышеуказанного взаимодействия находятся между 0 и 100 С. Подходящее время реакции составляет от 20 мин до 48 ч. Альтернативно, соединение формулы I,где R2 представляет собой арил или гетероарил, может быть получено из соединения формулы III посредством взаимодействия с соответствующим арил- или гетероарилйодидом, бромидом, производным триалкилолова, производным цинка, бороновой кислотой или эфиром бороновой кислоты, как описано для превращения соединения формулы II в соединение формулы I на схеме 1, с получением соединения формулы I. Альтернативно, соединение формулы I, где R2 представляет собой алкил, может быть получено из соединения формулы III, где L представляет собой хлор, посредством взаимодействия с соответствующим цинковым реагентом в реакции Негиши, как описано выше для превращения соединения формулы II в соединение формулы I на схеме 1 (см. также Hendricks R.Т. et al. Bioorg. Med. Chem. Lett. 2009, 19, 410-414.) Альтернативно соединение формулы I, где R2 представляет собой алкил, циклоалкил,или гетероциклил, может быть получено из соединения формулы III, где L представляет собой хлор, посредством взаимодействия с соответствующим реагентом Гриньяра (Grignard) в присутствии ацетилацетоната железа(III) (как описано Ottesen L.K. et al. Organic Lett. 2006, 8, 1771-1773). Ссылаясь на схему 2, соединение формулы I, где R2 представляет собой -C1-С 6 алкил-R9 или -OR8,может быть непосредственно получено из соединения формулы IV посредством алкилирования с соответствующим галогенидом или посредством реакции Мицунобу (Mitsunobu) с необходимым спиртом(см. Bodendiek S.В. et al. Eur. J. Med. Chem. 2009, 44, 1838-1852; Khattab A.F. et al. Synth. Commun. 2006,36, 2751-2761; Smith G. et al. J. Med. Chem. 2008, 51, 8057-8067). Соединения формулы III, где L представляет собой хлор или 1H-1,2,4-триазол-1-ил, могут быть получены из соединения формулы IV посредством обработки оксихлоридом фосфора, возможно в присутствии 1H-1,2,4-триазола. Более конкретно, от 2 до 4 эквивалентов оксихлорида фосфора и от 8 до 11 эквивалентов 1H-1,2,4-триазола в соответствующем органическом растворителе, таком как ацетонитрил или дихлорметан, при температуре между -10 и 5 С обрабатывают от 12 до 15 эквивалентов триэтиламина или N,N-диизопропилэтиламина. После добавления 1 эквивалента имидазотриазинона формулы IV реакционную смесь можно поддерживать при температурах в диапазоне от 25 С до температуры флегмообразования в течение промежутка времени от 2 до 24 ч, с получением соединения формулы III, где L представляет собой 1H-1,2,4-триазол. Если 1H-1,2,4-триазол отсутствует в реакционной смеси, взаимодействие в этом случае может быть осуществлено в толуоле в качестве растворителя, продукт представляет собой соединение формулы III, где L представляет собой хлор. Не является необходимым во всех случаях выделять промежуточное соединение формулы III, которое может быть непосредственно подвергнуто взаимодействию с соответствующим аминным реагентом. Соединения формулы III, где L представляет собой трифлат, могут быть получены из соединения формулы IV стандартными методами (см.Shireman В.Т. et al. Bioorg. Med. Chem. Lett. 2008, 18, 2103-2108). Эти реакции не ограничены использованием описанных выше растворителя или основания, поскольку могут быть использованы многочисленные другие условия. Схема 3 относится к получению соединений формулы II, где R2 представляет собой HNR3 или 3N(R )2. Соединения формулы II могут быть превращены в соединения формулы I согласно методам со схемы 1. Ссылаясь на схему 3, соединение формулы II может быть получено из соединения формулы V методами, аналогичными методам превращения соединения формулы III в соединение формулы I на схеме 2. Соединения формулы V, где L представляет собой хлор или 1H-1,2,4-триазол-1-ил, могут быть получены из соединений формулы VI, где X представляет собой H, Cl, Br или I, методами, аналогичными превращению соединения формулы IV в соединение III на схеме 2. Схема 4 относится к альтернативному получению соединений формулы II, где R2 представляет собой HNR3 или N(R3)2 и X представляет собой Н или Cl. Соединения формулы II могут быть превращены в соединения формулы I в соответствии с методами со схемы 1. Ссылаясь на схему 4, соединения формулы II могут быть получены из бром-соединения формулы VII посредством катализируемого палладием сочетания с соответствующим образом замещенным гетероароматическим соединением формулы ZAXB(OH)2 или В(OR)2, сочетание по связи углерод-углерод может быть осуществлено в условиях, описан- 15022586 ных выше для реакции Сузуки. Для специалиста понятно, что эта химия также будет эффективна, если X представляет собой R5; в этом случае продукт будет представлять собой соединение формулы I. Соединение формулы VII, где R2 представляет собой HNR3 или N(R3)2, могут быть получены из соединения формулы VIII методами, аналогичными превращению соединения формулы III в соединение формулы I на схеме 2. Соединения формулы VIII могут быть получены из соединения формулы IX методами, аналогичными превращению соединений формулы IV в соединения формулы III на схеме 2. Схема 5 относится к получению соединений формулы IV. Соединения формулы IV могут быть превращены в соединения формулы I в соответствии с методами со схемы 2. Ссылаясь на схему 5, соединение формулы IV может быть получено из соединения формулы VI, где X представляет собой H, Cl, Br, I,трифлат, бороновую кислоту, боронат или триалкилолово, таким же образом, как описано для превращения соединения формулы II в соединение формулы I на схеме 1. Схема 6 относится к получению соединений формулы VI, где "А-Х" представляет собой и X представляет собой H, Cl, Br, I, трифлат, бороновую кислоту, боронат или триалкилолово. Соединение формулы VI может быть превращено в соединение формулы I, используя методы, описанные на схеме 3, а затем на схемах 1 или 5, а затем на схеме 2. Ссылаясь на схему 6, соединение формулы VI может быть получено из соединения формулы X посредством воздействия от 1,1 до 3 эквивалентов 1,1'карбонилдиимидазола или 1,1'-карбонилди(1,2,4-триазола) в таком растворителе, как 1,4-диоксан или тетрагидрофуран, при температурах в диапазоне от 40 до 70 С в течение промежутка времени от 1 до 4 ч. Соединение формулы X может быть получено из соединения формулы XI посредством обработки от 2 до 5 эквивалентами формамидинацетата в таком растворителе, как 2-бутанол, при температурах в диапазоне от 60 до 100 С в течение промежутка времени от 1 до 12 ч. Соединение формулы XI может быть получено из соединения формулы XII посредством удаления защитной группы Р 1, где Р 1 может представлять собой -(С=О)арил, -(С=О)алкил, -(С=О)О-(С 1-С 4)алкил или другую подходящую защитную группу, известную специалисту в данной области, посредством щелочного гидролиза амидной или карбаматной группы, или, в случае, когда Р 1 представляет собой -(С=О)О-трет-бутил, посредством взаимодействия с избытком трифторуксусной кислоты в растворителе дихлорметане при температуре в диапазоне от 15 до 35 С. Соединение формулы XII может быть получено из соединения формулы XIII, где X представляет собой H, Cl, Br, I, трифлат, бороновую кислоту, боронат или триалкилолово и где LG представляет собой Cl, Br или I, посредством взаимодействия с 1,1-2 эквивалентами N-защищенного R1 имидогидразидного соединения формулы где Р 1 представляет собой -(С=О)арил, -(С=О)алкил, -(С=О)О-(С 1-С 4)алкил или другую соответствующую защитную группу, известную специалисту в данной области. Это взаимодействие может быть осуществлено в растворителе, таком как 2-метилтетрагидрофуран и/или 1,2-диметоксиэтан, при температурах в диапазоне от 60 до 90 С, в присутствии основания, такого как 2-4 эквивалента N,N-диизопропилэтиламина. Имидогидразидный реагент XIX может быть получен посредством взаимодействия требуемого ацилгидразина с соответствующим иминоэфиром в соответствии с методом Hurtaud D. et al. Synthesis 2001, 2435-2440. Схема 7 относится к получению соединений формулы VI, где X представляет собой Н или Cl. Соединения формулы VI могут быть превращены в соединения формулы I в соответствии с методами, раскрытыми на схеме 3, а затем схемах 1 или 5, а затем схеме 2. Соединение формулы IX может быть использовано для получения соединений формулы I, как представлено на схеме 4, а затем на схеме 1. Ссылаясь на схему 7, соединение формулы VI может быть синтезировано из соединения формулы IX посредством катализируемого палладием взаимодействия с соответствующим образом замещенным гетероароматическим соединением формулы ZAX В этом случае X представляет собой Н или Cl в соединениях ZAX1; Z представляет собой B(OH)2 или B(OR)2. Реакция сочетания углерод-углерод может быть осуществлена аналогично реакции, описанной для превращения соединения VII в соединение формулы II на схеме 4.- 16022586 Соединение формулы IX может быть получено посредством взаимодействия соединения формулыXIV, где Р 2 представляет собой (С 1-С 4)алкил, с 1-30 эквивалентами формамида при температуре в диапазоне от 100 до 180 С в течение промежутка времени от 2 до 20 ч. Соединение формулы XIV может быть получено из соединения формулы XV посредством депротонирования с помощью основания, такого как бис-(триметилсилил)амид лития, и последующего аминирования с помощью такого реагента, как O-(4 нитробензоил)гидроксиламин или О-(дифенилфосфинил)гидроксиламин. Примечание: О-(дифенилфосфинил)гидроксиламин является высокоэнергетическим веществом, которое показало способность к взрывному разложению при окружающих условиях. За его использованием следует осторожно следить. Соединение формулы XV может быть получено посредством бромирования соединения формулы XVI в соответствии с методом Grange Т.L. et al. Tempahedron Lett. 2007, 48, 6301-6303, используя бром в таком растворителе, как N,N-диметилформамид, при температуре в диапазоне от 60 до 90 С, под воздействием основания, такого как бикарбонат калия. Соединения формулы XVI могут быть получены посредством циклизации соединения формулы XVII, осуществляемой, например, посредством нагревания в микроволновом реакторе при температуре от 150 до 190 С в течение промежутка времени от 30 мин до 3 ч, в соответствующем инертном растворителе, таком как 1,4-диоксан. Соединения формулы XVII получают посредством взаимодействия пропиолатного эфира с N'-гидроксиимидамидом формулы XVIII, которое может быть осуществлено при температуре флегмообразования в таком растворителе, как метанол или этанол, в течение периода времени от 2 до 24 ч. Соединения формулы XVIII легко получаются из соответствующих нитрилов (как сообщается Yang X. et al. J. Med. Chem. 2010, 53, 1015-1022). Соединения формулы I, которые имеют хиральные центры, могут существовать в виде стереоизомеров, таких как рацематы, энантиомеры или диастереомеры. Традиционные методики для получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата, используя, например, хиральную жидкостную хроматографию высокого давления (ЖХВД). Альтернативно рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например спиртом, или, в случае, когда соединение содержит кислотную или основную группировку, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученную диастереомерную смесь можно разделить посредством хроматографии и/или фракционной кристаллизации и один или оба диастереоизомера превратить в соответствующий(ие) чистый(ые) энантиомер(ы) способами, хорошо известными специалисту в данной области. Хиральные соединения формулы I (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с помощью хроматографии, обычно ЖХВД,на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана,содержащего от 0 до 50% изопропанола, обычно от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрирование элюата дает обогащенную смесь. Стереоизомерные конгломераты могут быть разделены обычными методами, известными специалисту в данной области (см., например, "Stereochemistry of Organic Compounds" by E. L. Eliel (Wiley, New York, 1994), описание которого включено в данное описание посредством ссылки во всей его полноте). Подходящие стереоселективные методы хорошо известны специалисту в данной области. Когда соединение формулы I содержит алкенильную или алкениленовую группу, возможны геометрические цис/транс (или Z/E) изомеры. Цис/транс изомеры могут быть разделены обычными методами, хорошо известными специалисту в данной области, например хроматографией и фракционной кристаллизацией. Соли по настоящему изобретению могут быть получены в соответствии с методами, известными специалисту в данной области. Соединения формулы I, которые являются основными по своей природе, способны образовывать широкое разнообразие солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, часто на практике бывает желательно сначала выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли и затем просто превратить последнюю обратно в соединение в форме свободного основания путем обработки щелочным реагентом, и затем превратить указанное свободное основание в соль присоединения фармацевтически приемлемой кислоты. Соли присоединения кислот соединения по данному изобретению в форме основания могут быть получены посредством обработки соединения в форме основания, по существу, эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. В результате выпаривания растворителя получают целевую соль в виде твердого вещества. Целевая соль с кислотой также может быть осаждена из раствора свободного основания в органическом растворителе путем добавления к раствору соответствующей минеральной или органической кислоты. Если соединение по изобретению представляет собой основание, целевая фармацевтически приемлемая соль может быть получена с помощью любого подходящего метода, известного из уровня техники,например путем обработки свободного основания неорганической кислотой, такой как соляная кислота,бромисто-водородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное,или органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, мин- 17022586 дальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота,гликолевая кислота, салициловая кислота, изоникотиновая кислота, уксусная кислота, молочная кислота,пантотеновая кислота, бивинная кислота, аскорбиновая кислота, 2,5-дигидроксибензойная кислота, фумаровая кислота, глюконовая кислота, сахарная кислота, муравьиная кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, n-толуолсульфоновая кислота и памоевая (то есть 1,1'-метилен-бис-(2-гидрокси-3-нафтоат кислота, пиранозидиловая кислота, такая как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота, ароматическая кислота, такая как бензойная кислота или циннамовая кислота, сульфоновая кислота, такая как nтолуолсульфоновая кислота или этансульфоновая кислота, и тому подобное. Те соединения формулы I, которые являются кислотными по своей природе, способны образовывать соли оснований с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных металлов или щелочно-земельных металлов и, в частности, соли натрия и калия. Эти соли получают обычными методами. Химическими основаниями, которые используются в качестве реагентов для получения фармацевтически приемлемой соли основания по данному изобретению,являются те, которые образуют нетоксичные соли оснований с кислотными соединениями формулы I. Эти соли могут быть получены любым подходящим методом, например посредством обработки свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочно-земельного металла или тому подобное. Эти соли также могут быть получены посредством обработки соответствующего кислотного соединения водным раствором, содержащим желаемые фармакологически приемлемые катионы, и затем выпаривания полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно они могут быть получены посредством смешивания вместе растворов в низших спиртах кислотного соединения и желаемого алкоксида щелочного металла и затем выпаривания полученного раствора досуха таким же образом, что и ранее. В любом случае предпочтительно использовать стехиометрические количества реагентов для того, чтобы быть уверенными в завершенности реакции и максимальных выходах целевого конечного продукта. Изобретение также включает соединения формулы I, меченные изотопами, где один или более атомов заменены атомом, имеющим такое же атомное число, но атомную массу или массовое число, отличное от атомной массы или массового числа, обычно обнаруживаемого в природе. Меченные изотопами соединения формулы I могут в общем быть получены традиционными методами, известными специалистам в данной области, или способами, аналогичными тем, которые описаны в данной заявке, используя соответствующий меченный изотопом реагент вместо немеченого реагента, обычно используемого. В следующих примерах и подготовительных примерах "DMSO" означает диметилсульфоксид, "н." означает нормальный, "М" означает молярный, "мл" означает миллилитр, "ммоль" означает миллимоли,"мкмоль" означает микромоли, "экв." означает эквивалент, "С" означает градусы Цельсия, "Па" означает Паскали, "УФ" означает ультрафиолет, "МГц" означает мегаГерц. Экспериментальные процедуры Эксперименты в общем проводили в инертной атмосфере (азот или аргон), особенно в случаях, где использовали чувствительные к кислороду или влаге реагенты или промежуточные соединения. Имеющиеся в продаже растворители и реагенты обычно использовали без дополнительной очистки, включая безводные растворители, где целесообразно (обычно Sure-Seal продукты от Aldrich Chemical Company,Milwaukee, Wisconsin). Продукты обычно сушили под вакуумом перед использованием в последующих реакциях или биологическом тестировании. Данные масс-спектрометрии получали посредством оборудования для жидкостной хроматографии-масс-спектрометрии (ЖХ-МС), химической ионизации при атмосферном давлении (ХИАД) или газовой хроматографии-масс-спектрометрии (ГХ-МС). Данные химического сдвига для ядерного магнитного резонанса (ЯМР) выражены в частях на миллион (м.д., ) со ссылкой на остаточные пики из используемых дейтерированных растворителей. Для ссылающихся на синтезы процедур в других примерах или методах реакционные условия (длительность реакции и температура) могут варьировать. В общем за взаимодействиями следовали тонкослойная хроматография или масс-спектрометрия с последующей обработкой, когда целесообразно. Процедуры очистки могут варьировать между экспериментами: в общем, растворители и соотношения растворителей, использованные для элюантов/градиентов, выбирали таким образом, чтобы обеспечить соответствующие величины Rf или времени удерживания. Пример 1(1-Метил-1H-пиразол-5-ил)бороновую кислоту (2,0 г, 16 ммоль), 1-бром-4-метилбензол (1,96 мл,15,9 ммоль), карбонат натрия (5,05 г, 47,6 ммоль) и дихлорбис-(трифенилфосфин)палладий(II) (557 мг,0,794 ммоль) объединяли в смеси воды (20 мл) и 1,2-диметоксиэтана (100 мл) и нагревали при 80 С в течение 18 ч. После охлаждения реакционной смеси ее фильтровали через целит и концентрировали в вакууме. Остаток распределяли между водой и этилацетатом и водный слой экстрагировали дополнительным количеством этилацетата. Объединенные органические слои сушили над сульфатом магния,фильтровали и концентрировали при пониженном давлении. В результате хроматографии на силикагелеN-Йодсукцинимид (95%, 756 мг, 3,19 ммоль) добавляли к раствору 1-метил-5-(4-метилфенил)-1Hпиразола (500 мг, 2,90 ммоль) в ацетонитриле (15 мл) и реакционную смесь оставляли перемешиваться в течение 1 ч при 85 С. В результате удаления растворителя в вакууме получили остаток, который подвергали хроматографии на силикагеле (градиент: от 20 до 50% этилацетата в гептане) с получением продукта в виде коричневого масла. Выход: 630 мг, 2,11 ммоль, 73%. ЖХ-МС m/z 299.2 (М+1). 1H ЯМР (400 МГц, CDCl3)2.44 (br s, 3 Н), 3.83 (s, 3 Н), 7.26-7.33 (m, 4H), 7.57 (s, 1H).C. Получение 1-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил 1 этанона Трибутил(1-этоксивинил)станнан (95%, 1,39 мл, 3,88 ммоль) добавляли к смеси 4-йод-1-метил-5-(4 метилфенил)-1H-пиразола (768 мг, 2,58 ммоль), тетракис(трифенилфосфин)палладия(0) (298 мг, 0,258 ммоль) и хлорида лития (98%, 279 мг, 6,45 ммоль) в N,N-диметилформамиде (20 мл) и эту реакционную смесь перемешивали при 90 С в течение 18 ч. После охлаждения смесь фильтровали через целит и концентрировали в вакууме; в результате хроматографии на силикагеле (градиент: от 10 до 50% этилацетата в гептане) получили продукт в виде бесцветного масла. Выход: 460 мг, 2,15 ммоль, 83%. ЖХ-МС m/z 215.3 (М+1). 1H ЯМР (400 МГц, CDCl3)2.17 (s, 3 Н), 2.45 (br s, 3 Н), 3.69 (s, 3 Н), 7.28 (br АВ квартет,JAВ=8 Гц, АВ=28 Гц, 4 Н), 7.99 (s, 1H).D. Синтез 2-бром-1-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил 1 этанона Бром (97%, 0,104 мл, 1,97 ммоль) добавляли к раствору 1-[1-метил-5-(4-метилфенил)-1H-пиразол-4 ил]этанона (420 мг, 1,96 ммоль) в ледяной уксусной кислоте (10 мл) и эту реакционную смесь интенсивно перемешивали в течение 2 ч при 80 С. После удаления растворителя при пониженном давлении остаток разбавляли этилацетатом, промывали насыщенным водным раствором бикарбоната натрия, промывали насыщенным раствором хлорида натрия и сушили над сульфатом натрия. В результате фильтрования и концентрирования фильтрата в вакууме получили остаток, который очищали путем хроматографии на силикагеле (градиент: от 10 до 30% этилацетата в гептане) с получением продукта в виде бесцветного масла. Выход: 353 мг, 1,20 ммоль, 61%. ЖХ-МС m/z 293.1 (М+1). 1H ЯМР (400 МГц, CDCl3)2.46 (s,3 Н), 3.71 (s, 3 Н), 3.97 (s, 2 Н), 7.31 (br АВ квартет, JAВ=8 Гц, АВ=23 Гц, 4 Н), 8.05 (s, 1H). Стадия 2. Синтез N'-этанимидоилацетогидразида Смесь гидроксида натрия (2,59 г, 64,8 ммоль) в безводном этаноле (300 мл) перемешивали в течение 20 мин при 50 С, чтобы вызвать растворение. Этот раствор охлаждали до 0 С и постепенно добавляли гидрохлорид этилэтанимидоата (8,0 г, 65 ммоль); выпавшие в осадок соли удаляли путем фильтрования и фильтрат обрабатывали ацетогидразидом (4,80 г, 64,8 ммоль) при комнатной температуре. Эту смесь нагревали до 80 С в течение 10 мин, затем оставляли охлаждаться на 18 ч. Осадок собирали фильтрованием и промывали диэтиловым эфиром с получением продукта в виде белого твердого вещества. Выход: 4,4 г, 38 ммоль, 59%. 1 Н ЯМР (400 МГц, DMSO-d6), характеристические пики:1.78 (s, 3 Н), 1.95N'-Этанимидоилацетогидразид (147 мг, 1,28 ммоль) и бикарбонат натрия (99%, 181 мг, 2,13 ммоль) добавляли к раствору 2-бром-1-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]этанона (250 мг, 0,853 ммоль) в ацетонитриле (9 мл) и эту смесь нагревали при 80 С в течение 3 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем разбавляли дихлорметаном и фильтровали через целит. Фильтрат концентрировали в вакууме и остаток очищали хроматографией на силикагеле (элюент: 10% метанола в дихлорметане) с получением продукта в виде желтого масла. Выход: 236 мг, 0,763 ммоль, 89%. ЖХ-МС m/z 310.5 (М+1). 1H ЯМР (400 МГц, CDCl3), предположительно в виде смеси ротамеров:2.10 и 1.77 (2 s, 3 Н), 2.25 и 2.36 (2 s, 3 Н), 2.44 и 2.46 (2 s, 3 Н), 3.69 и 3.72 (2 s, 3 Н), 6.21 (s, 1H),7.22-7.33 (4 Н, m, предполагаемый; частично скрыт за пиком растворителя), 7.96 (br s, 1H). Стадия 4. Синтез 2-метил-4-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-1H-имидазол-1-амина- 19022586 Водную соляную кислоту (1 н., 7,0 мл) добавляли к раствору N-2-метил-4-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-1H-имидазол-1-илацетамида (236 мг, 0,763 ммоль) в метаноле (1,0 мл) и смесь кипятили с обратным холодильником в течение 30 мин. Добавляли дополнительное количество 1 н. водной соляной кислоты (2,0 мл) и нагревание продолжали в течение дополнительных 30 мин. После охлаждения раствор подщелачивали 1 н. водным раствором гидроксида натрия и смесь экстрагировали дважды этилацетатом, содержащим 1% метанола. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением продукта в виде желтого твердого вещества. Выход: 148,5 мг, 0,5555 ммоль, 73%. ЖХ-МС m/z 268.5 (М+1). 1 Н ЯМР (400 МГц, CDCl3)2.39 (s, 3H), 2.45 (br s, 3 Н), 3.70 (s,3 Н), 4.45 (br s, 2H), 6.26 (s, 1H), 7.24-7.32 (m, 4H, предполагаемый; частично скрыт за пиком растворителя), 7.97 (s, 1H). Стадия 5. Синтез N-2-метил-4-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-1H-имидазол-1-ил имидоформамида Гидрохлорид этилимидоформиата (608 мг, 5,55 ммоль) добавляли к раствору 2-метил-4-[1-метил-5(4-метилфенил)-1H-пиразол-4-ил]-1H-имидазол-1-амина (148 мг, 0,554 ммоль) в этаноле (5 мл) и реакционную смесь нагревали при 75 С в течение 66 ч. Добавляли дополнительное количество гидрохлорида этилимидоформиата (300 мг, 2,74 ммоль) и нагревание продолжали в течение 8 ч. Затем проводили финальную загрузку гидрохлорида этилимидоформиата (300 мг, 2,74 ммоль) с последующим поддержанием реакции при 75 С в течение дополнительных 18 ч. Реакционную смесь охлаждали, концентрировали в вакууме и разбавляли этилацетатом. Этот органический слой промывали насыщенным водным раствором бикарбоната натрия, промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. В результате фильтрования и удаления растворителя при пониженном давлении с последующей очисткой путем хроматографии на силикагеле (элюент: 10% метанола в дихлорметане) получили продукт в виде желтого твердого вещества. Выход: 70 мг, 0,24 ммоль, 43%. ЖХ-МС m/z 295.5(М+1). 1H ЯМР (400 МГц, CD3OD), предположительно в виде смеси ротамеров или таутомеров:2.20 и 2.25 (2 s, 3 Н), 2.43 (br s, 3 Н), 3.68 (s, 3 Н), 6.14 и 6.30 (2 s, 1H), 7.26-7.30 (m, 3 Н), 7.35 (br d, J=8 Гц, 2 Н),7.84 и 7.81 (2 s, 1H). Стадия 6. Синтез 7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]имидазо[5,1-f][1,2,4]триазин-4(3H)-она Гидрид натрия (60%-ная дисперсия в минеральном масле, 24 мг, 0,60 ммоль) добавляли к растворуN-2-метил-4-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-1H-имидазол-1-илимидоформамида (70 мг,0,24 ммоль) в 1,4-диоксане (4,0 мл) и смесь нагревали при 75 С в течение 10 мин. Реакционную смесь охлаждали, обрабатывали 1,1'-карбонилдиимидазолом (135 мг, 0,833 ммоль), оставляли перемешиваться при комнатной температуре в течение 30 мин и затем нагревали до 100 С в течение 18 ч. После охлаждения реакционной смеси до комнатной температуры ее гасили водой и разбавляли этилацетатом. Органический слой промывали водой, затем насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. В результате хроматографии на силикагеле(градиент: от 0 до 10% метанола в этилацетате) получили продукт в виде белого твердого вещества. Выход: 55 мг, 0,17 ммоль, 71%. ЖХ-МС m/z 321.5 (М+1). 1H ЯМР (400 МГц, DMSO-d6)2.34 (br s, 3 Н), 2.36(s, 3 Н), 3.71 (s, 3 Н), 7.22 (br d, J=8 Гц, 2 Н), 7.30 (d, J=8,2 Гц, 2 Н), 7.79 (s, 1H), 7.95 (s, 1H). Стадия 7. Синтез 7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-4-(1H-1,2,4-триазол-1-ил) имидазо[5,1-f][1,2,4]триазина 1H-1,2,4-Триазол (162 мг, 2,34 ммоль) пульверизировали, смешивали с дихлорметаном (4,0 мл) и охлаждали до 0 С. Добавляли оксихлорид фосфора (58,2 мкл, 0,624 ммоль), а затем через 1 мин по каплям добавляли триэтиламин (0,349 мл, 2,50 ммоль). Через 10 мин при 0 С ледяную баню удаляли; спустя 5 мин добавляли 7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]имидазо[5,1-f][1,2,4]триазин 4(3H)-он (50 мг, 0,16 ммоль). Реакционную смесь поддерживали при комнатной температуре в течение 4 ч, затем охлаждали до 0 С и гасили водой, затем обрабатывали насыщенным водным раствором бикарбоната натрия. Смесь экстрагировали этилацетатом и объединенные органические слои промывали водой, промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Этот неочищенный материал использовали непосредственно на следующей стадии. ЖХ-МС m/z 372.5 (М+1). 1 Н ЯМР (400 МГц, DMSO-d6)2.27 (br s, 3 Н), 2.70 (s, 3 Н),3.73 (s, 3 Н), 6.86 (br d, J=8,2 Гц, 2H), 7.06 (br d, J=8,0 Гц, 2 Н), 7.37 (s 1H), 8.14 (s, 1H), 8.53 (s, 1H), 8.84 (s,1H). Стадия 8. Синтез 4-(азетидин-1-ил)-7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]имидазо[5,1-f][1,2,4]триазина Азетидин (27,0 мкл, 0,400 ммоль) и карбонат цезия (97%, 202 мг, 0,601 ммоль) добавляли к раствору 7-метил-5-[1-метил-5-(4-метилфенил)-1H-пиразол-4-ил]-4-(1H-1,2,4-триазол-1-ил)имидазо[5,1-f][1,2,4] триазина (материал с предыдущей реакции, менее или равно 0,16 ммоль) в N,N-диметилформамиде (3,0 мл) и реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь разбавляли этилацетатом, промывали водой, промывали насыщенным водным раствором хлорида на- 20022586 трия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. В результате очистки хроматографией на силикагеле (градиент: от 0 до 5% метанола в дихлорметане) с последующей азеотропной перегонкой с гептаном получили продукт в виде белого твердого вещества. Выход: 35 мг, 0,097 ммоль, 61% за 2 стадии. ЖХ-МС m/z 360.2 (М+1). 1H ЯМР (400 МГц, CDCl3)2.20-2.28 4-(Азетидин-1-ил)-7-метил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-илимидазо[5,1-f][1,2,4]триазин Стадия 1. Синтез трет-бутил-2-этанимидоилгидразинкарбоксилата Гидроксид натрия (16,0 г, 400 ммоль) растворяли в абсолютном этаноле (1000 мл) при 60 С. Раствор охлаждали до 0 С и порциями обрабатывали гидрохлоридом этилэтанимидоата (50 г, 400 ммоль); через 10 мин добавляли одной порцией трет-бутилгидразинкарбоксилат (52,9 г, 400 ммоль). Реакционную смесь нагревали до 70 С и перемешивали при 70 С в течение 2,5 ч. Смесь затем охлаждали до 20 С и фильтровали. Фильтрат концентрировали в вакууме и обрабатывали трет-бутилметиловым эфиром(500 мл) и этанолом (20 мл). После внесения затравки смесь оставляли перемешиваться в течение 18 ч,по окончании которых выпавшее в осадок твердое вещество собирали путем фильтрования и промывали охлажденным на льду трет-бутилметиловым эфиром (500 мл). Твердое вещество растворяли в смеси 2 метилтетрагидрофуран:метанол (9:1, 300 мл) и этот раствор концентрировали досуха. Остаток промывали диэтиловым эфиром (3200 мл) и сушили с получением продукта в виде бледно-желтого твердого вещества. Выход: 50,2 г, 290 ммоль, 72%. ЖХ-МС m/z 174.3 (М+1). 1H ЯМР (500 МГц, CD3OD)1.47 (s,9H), 1.88 (s, 3 Н). Стадия 2. Синтез 2-бром-1-(1-метил-1H-пиразол-4-ил)этанонаA. Синтез 1-(1-метил-1H-пиразол-4-ил)этанона 4-Бром-1-метил-1H-пиразол (41,3 мл, 400 ммоль) растворяли в тетрагидрофуране (750 мл) и охлаждали до -78 С. Добавляли по каплям в течение 30 мин раствор N-бутиллития (2,5 М раствор в гексанах,160 мл, 400 ммоль) и полученную смесь перемешивали в течение 1 ч при -78 С. После добавления к этой реакционной смеси по каплям раствора N-метокси-N-метилацетамида (40,9 мл, 400 ммоль) в тетрагидрофуране (100 мл) при -78 С охлаждающую баню оставляли нагреваться до 0 С в течение 4 ч. Реакционную смесь затем гасили насыщенным водным раствором хлорида натрия (50 мл) и летучие вещества удаляли в вакууме. Остаток разбавляли этилацетатом (1000 мл), обрабатывали сульфатом магния и перемешивали в течение 30 мин, затем фильтровали и концентрировали в вакууме. В результате очистки путем хроматографии на силикагеле (материал загружали в минимальном количестве дихлорметана; градиент: от 5 до 100% этилацетата в гептане) получили бледно-желтое масло, которое затвердевало при стоянии. Выход: 28,5 г, 230 ммоль, 57%. 1H ЯМР (500 МГц, CDCl3)2.37 (s, 3 Н), 3.90 (s, 3 Н), 7.83 (s, 1H), 7.84 (s,1H).B. Синтез 2-бром-1-(1-метил-1H-пиразол-4-ил)этанона Раствор 1-(1-метил-1H-пиразол-4-ил)этанона (28,5 г, 230 ммоль) в дихлорметане (400 мл) разбавляли абсолютным этанолом (100 мл) и порциями обрабатывали трибромидом пиридиния (95%, 77,3 г, 230 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, во время которых она затвердевала; смесь разбавляли дихлорметаном (300 мл) и водой (400 мл), обрабатывали сульфитом натрия (5 г) и перемешивали в течение 10 мин. Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток промывали водой (200 мл), собирали фильтрованием,промывали снова водой и сушили с получением продукта в виде не совсем белого твердого вещества. Выход: 41,6 г, 205 ммоль, 89%. 1 Н ЯМР (500 МГц, CDCl3)7.97-7.98 (m, 1H), 7.95 (br s, 1H), 4.17 (s, 2H),3.95-3.96 (m, 3H). Стадия 3. Синтез трет-бутил-[2-метил-4-(1-метил-1H-пиразол-4-ил)-1 Н-имидазол-1-ил]карбамата трет-Бутил-2-этанимидоилгидразинкарбоксилат (17,3 г, 99,9 ммоль), 2-бром-1-(1-метил-1H-пиразол-4-ил)этанон (16,89 г, 83,18 ммоль) и N,N-диизопропилэтиламин (31,9 мл, 183 ммоль) объединяли в охлажденном на льду 2-метилтетрагидрофуране (400 мл) и 1,2-диметоксиэтане (100 мл) и реакционную смесь кипятили с обратным холодильником. Через 2,5 ч реакционную смесь охлаждали и промывали 50%-ным насыщенным водным раствором хлорида натрия (75 мл). Водный слой экстрагировали 2 метилтетрагидрофураном (100 мл) и объединенные органические слои сушили над сульфатом магния,фильтровали и концентрировали в вакууме. Остаток растворяли в теплом этилацетате (60 мл), оставляли охлаждаться до комнатной температуры, затем охлаждали до 5 С в течение 30 мин. Полученное твердое- 21022586 вещество собирали фильтрованием и промывали небольшим количеством холодного этилацетата, затем промывали диэтиловым эфиром с получением продукта в виде бледно-желтого твердого вещества. Выход: 16,0 г, 57,7 ммоль, 69%. ЖХ-МС m/z 278.5 (М+1). 1 Н ЯМР (500 МГц, CDCl3)1.49 (br s, 9H), 2.23 (s,3 Н), 3.84 (s, 3 Н), 6.87 (s, 1H), 7.51 (s, 1H), 7.60 (s, 1H), 8.67 (br s, 1H). Стадия 4. Синтез трифторацетатной соли 2-метил-4-(1-метил-1H-пиразол-4-ил)-1H-имидазол-1-амина Раствор трет-бутил[2-метил-4-(1-метил-1H-пиразол-4-ил)-1H-имидазол-1-ил]карбамата (8,0 г, 29 ммоль) в метиленхлориде (200 мл) и трифторуксусной кислоте (40 мл) перемешивали при комнатной температуре в течение 2,5 ч. После удаления растворителей в вакууме остаток перемешивали в смеси этилацетат/гептан 1:1 в течение 18 ч. Полученное твердое вещество выделяли фильтрованием с получением продукта в виде белого твердого вещества. Выход: 5,3 г, 18 ммоль, 62%. Маточную жидкость концентрировали в вакууме и остаток перемешивали в течение 30 мин в смеси этилацетат/гептан/диэтиловый эфир 1:1:1 (50 мл); в результате фильтрования получили дополнительное количество продукта в виде белого твердого вещества. Объединенный выход: 7,8 г, 26,8 ммоль, 92%. 1 Н ЯМР (400 МГц, DMSO-d6)2.54 (s, 3 Н), 3.89 (s, 3 Н), 6.55 (br s, 2 Н), 7.65 (s, 1H), 7.85 (d, J=0,7 Гц, 1H), 8.11 (br s,1H). Стадия 5. Синтез N-[2-метил-4-(1-метил-1H-пиразол-4-ил)-1H-имидазол-1-ил]имидоформамида Трифторацетатную соль 2-метил-4-(1-метил-1H-пиразол-4-ил)-1H-имидазол-1-амина (103,0 г, 353,7 ммоль) и ацетат формамидина (98%, 131 г, 1,23 моль) объединяли в 2-бутаноле (350 мл). Реакционную смесь нагревали до 100 С в течение 3 ч, по окончании которых ее оставляли охлаждаться до комнатной температуры и разбавляли в смеси 10 н. раствор гидроксида натрия/насыщенный водный раствор хлорида натрия 2:1 (300 мл). После интенсивного перемешивания слои разделяли и водный слой экстрагировали 2-бутанолом (4250 мл). Объединенные органические слои концентрировали в вакууме и полученное твердое вещество суспендировали в ацетонитриле (550 мл), перемешивали в течение 2 ч при комнатной температуре и фильтровали. Собранные твердые вещества промывали безводным ацетонитрилом (3100 мл), затем сушили в вакууме при 40 С в течение 2 ч с получением продукта в виде не совсем белого твердого вещества. Выход: 61,5 г, 301 ммоль, 85%. Маточную жидкость концентрировали досуха, затем растворяли в ацетонитриле (200 мл) и оставляли отстаиваться в течение 18 ч. Полученное твердое вещество выделяли фильтрованием с получением дополнительного количества продукта в виде не совсем белого твердого вещества. Объединенный выход: 64,8 г, 317 ммоль, 90%. 1 Н ЯМР (500 МГц,CD3OD), предположительно в виде смеси ротамеров или таутомеров:2.25 и 2.29 (2 s, 3 Н), 3.88 и 3.88 (2N-[2-Метил-4-(1-метил-1H-пиразол-4-ил)-1H-имидазол-1-ил]имидоформамид (58,3 г, 285 ммоль) объединяли с 1,1'-карбонилдиимидазолом (98%, 59,0 г, 357 ммоль) в тетрагидрофуране (1140 мл) при 63 С и суспензию перемешивали в течение 2,5 ч при 65 С. Смесь охлаждали и концентрировали в вакууме; полученное твердое вещество суспендировали с метанолом (400 мл), нагревали до флегмообразования в течение 20 мин и охлаждали до 7 С. Твердое вещество собирали с получением С 1 в виде бледножелтого твердого вещества. Выход: 45,9 г, 199 ммоль, 70%. ЖХ-МС m/z 231.1 (М+1). 1H ЯМР (500 МГц,DMSO-d6)2.48 (s, 3 Н), 3.88 (s, 3 Н), 7.79 (s, 1H), 8.08 (s, 1H), 8.37 (s, 1H), 11.59 (br s, 1H). Стадия 7. Синтез 4-(азетидин-1-ил)-7-метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина (С 2) Тонкоизмельченный 1H-1,2,4-триазол (278 г, 4,02 моль) смешивали с ацетонитрилом (700 мл), охлаждали до 0 С и обрабатывали по каплям оксихлоридом фосфора (62,4 мл, 669 ммоль), в то же время поддерживая внутреннюю температуру ниже 15 С. Суспензию перемешивали в течение 10 мин, затем медленно обрабатывали по каплям триэтиламином (607 мл, 4,35 моль) при интенсивном перемешивании,в то же время поддерживая внутреннюю температуру ниже 48 С. Реакционную смесь перемешивали в течение 15 мин и охлаждали до 41 С и затем обрабатывали по каплям 7-метил-5-(1-метил-1H-пиразол-4 ил)имидазо[5,1-f][1,2,4]триазин-4(3H)-оном (77,1 г, 335 ммоль). После завершения добавления реакционную смесь нагревали до 73 С в течение 1 ч, затем охлаждали до комнатной температуры, после чего тонкослойная хроматография (элюент: 10% метанола в этилацетате) указала на полное превращение в триазолзамещенное промежуточное соединение. Реакционную суспензию обрабатывали последовательно триэтиламином (279 мл, 2,00 моль) и гидрохлоридом азетидина (94,0 г, 1,00 моль); в течение 10 мин внутренняя температура поднималась от 18 до 38 С. Смесь перемешивали в течение 1 ч, охлаждали до 15-20 С и фильтровали. Осадок на фильтре промывали ацетонитрилом (600 мл) и фильтрат концентрировали в вакууме. Полученную пасту разбавляли водой (650 мл), а затем водным раствором гидроксида натрия (10 н., 450 мл). Эту суспензию экстрагировали дихлорметаном (3350 мл), объединенные органические слои сушили над сульфатом натрия и фильтровали. Этот фильтрат пропускали через набивку силикагеля (230-400 меш, 150 г), элюируя дихлорметаном (1 л), а затем 10%-ным метанолом в этилацетате (1 л). Объединенные элюаты, содержащие продукт, концентрировали в вакууме и остаток промывали трет-бутилметиловым эфиром (350 мл), собирали фильтрованием и промывали диэтиловым эфиром. По- 22022586 лученное твердое вещество растворяли в воде (200 мл) и разбавляли еще раз водным раствором гидроксида натрия (5 н., 250 мл). Смесь экстрагировали дихлорметаном (3250 мл) и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Твердое вещество промывали трет-бутилметиловым эфиром (350 мл) и собирали фильтрованием с получением С 2 в виде бледного рыжевато-коричневого твердого вещества. Выход: 82,15 г, 305 ммоль, 91%. 1 Н ЯМР (500 МГц,CDCl3)2.23-2.30 (m, 2H), 2.65 (s, 3 Н), 3.96 (s, 3H), 3.98-4.07 (m, 4 Н), 7.61 (br s, 1H), 7.61 (br s, 1H), 7.85(s, 1H). Стадия 8. Синтез 4-(азетидин-1-ил)-7-метил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-илимидазо[5,1-f][1,2,4]триазина 4-(Азетидин-1-ил)-7-метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазин (10,0 г, 37,1 ммоль), 2-бром-5-(трифторметил)пиридин (16,8 г, 74,3 ммоль) и измельченный карбонат калия (15,4 г,111 ммоль) объединяли в реакционной колбе, продували азотом и обрабатывали дегазированным 1,4 диоксаном (600 мл). К этой смеси добавляли димер хлорида аллилпалладия(II) (693 мг, 1,86 ммоль) и систему снова продували азотом. Реакционную смесь нагревали до 102 С в течение 36 ч, затем охлаждали и концентрировали в вакууме. Остаток распределяли между этилацетатом (400 мл) и водным раствором соляной кислоты (1 н., 200 мл). Водную фазу нейтрализовали твердым бикарбонатом натрия и экстрагировали этилацетатом (450 мл). Объединенные органические слои промывали 1 н. водной лимонной кислотой, затем насыщенным водным раствором бикарбоната натрия. После обработки активированным углеродом Darco органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток переносили в минимальное количество дихлорметана и концентрировали при пониженном давлении до образования густого масла. Добавляли диэтиловый эфир (100 мл), и при перемешивании этой смеси твердое вещество начинало осаждаться; перемешивание продолжали в течение 1 ч при комнатной температуре и затем белое твердое вещество собирали фильтрованием и промывали диэтиловым эфиром. Дополнительное количество продукта в маточной жидкости выделяли путем концентрирования фильтрата в вакууме и хроматографирования остатка на колонке оксида алюминия (элюент: 70%-ный этилацетат в гептане). Продукт с колонки перекристаллизовывали из теплого 20%-ного этилацетата в гептане с получением дополнительного количества продукта в виде белого твердого вещества. Объединенный выход: 5,3 г, 12,8 ммоль, 35%. Этот материал объединяли с продуктом аналогичной реакции (суммарно 15,5 г, 37,4 ммоль) и дополнительно очищали следующим образом. Материал растворяли в смеси этилацетата (100 мл) и 2-метилтетрагидрофурана (150 мл) при комнатной температуре. Добавляли SiliaBond-тиол (SiliCycle, 1,35 ммоль/г, 15 г) и смесь перемешивали в течение 20 ч, затем фильтровали через целит. Фильтрат обрабатывали активированным углеродом Darco (500 мг) и перемешивали в течение 15 мин, затем фильтровали и концентрировали при пониженном давлении. Полученное масло подвергали азеотропной перегонке в смеси гептана и этилацетата 1:1 с получением не совсем белого твердого вещества, которое смешивали с гептаном (100 мл) и перемешивали при комнатной температуре в течение 6 ч. В результате фильтрования получили продукт в виде белого твердого вещества. Выход стадии очистки: 14,4 г, 34,7 ммоль, 93%. ЖХ-МС m/z 415.0 (М+1). 1 Н ЯМР (400 МГц, CDCl3)2.17-2.26 (m, 2 Н), 2.70 (s, 3 Н), 3.3-3.8 (v br m, 2H), 3.8-4.3 (v br m, 2H), 4.18 (s, 3H), 7.63-7.66 (m, 1H), 7.66N,7-Диметил-5-1-метил-5-[4-(трифторметил)фенил]-1H-пиразол-4-илимидазо[5,1-f][1,2,4]триазин-4-амин Стадия 1. Синтез этил-5-бром-1-метил-1H-пиразол-4-карбоксилата Бромид меди(II) (99%, 20,0 г, 88,6 ммоль) и трет-бутилнитрит (90%, 14,1 мл, 107 ммоль) объединяли в ацетонитриле (65 мл) и нагревали до 65 С. Медленно порциями добавляли этил-5-амино-1-метил 1H-пиразол-4-карбоксилат (10,0 г, 59,1 ммоль) осторожно: газообразование и реакционную смесь поддерживали при 65 С в течение 24 ч. Смесь охлаждали до комнатной температуры, вливали в водную соляную кислоту (3 н., 600 мл), разбавляли этилацетатом (300 мл) и перемешивали в течение 10 мин. Водный слой экстрагировали этилацетатом (150 мл) и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток очищали путем хроматографии на силикагеле (градиент: от 5 до 100% этилацетата в гептане с 5-минутным удерживанием при 32%) с получением продукта в виде бледно-желтого твердого вещества. Выход: 9,10 г, 39,0 ммоль, 66%. ЖХ-МС(s, 1H). Стадия 2. Синтез 5-бром-1-метил-1H-пиразол-4-карбоновой кислоты Суспензию этил-5-бром-1-метил-1H-пиразол-4-карбоксилата (8,00 г, 34,3 ммоль) в тетрагидрофура- 23022586 не (60 мл), воде (20 мл) и этаноле (20 мл) обрабатывали моногидратом гидроксида лития (3,17 г, 75,5 ммоль) и перемешивали в течение 4 ч при комнатной температуре. В результате удаления растворителей при пониженном давлении получили белый твердый остаток, который разбавляли водой (50 мл), промывали диэтиловым эфиром (50 мл) и доводили до рН 2,5 водной 6 н. соляной кислотой. Густую суспензию экстрагировали 2-метилтетрагидрофураном (2125 мл) и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением продукта в виде не совсем белого твердого вещества. Выход: 6,49 г, 31,7 ммоль, 92%. ЖХ-МС m/z 205.2 (М+1). 1H ЯМР (500 МГц,DMSO-d6)3.86 (s, 3 Н), 7.91 (s, 1H), 12.64 (br s, 1H). Стадия 3. Синтез 2-бром-1-(5-бром-1-метил-1H-пиразол-4-ил)этанона Раствор 5-бром-1-метил-1H-пиразол-4-карбоновой кислоты (6,4 г, 31 ммоль) в метаноле (100 мл) помещали в водяную баню, обрабатывали одной порцией метилата натрия (95%, 1,86 г, 32,7 ммоль) и перемешивали в течение 30 мин при комнатной температуре. После удаления летучих веществ в вакууме натриевую соль концентрировали дважды из гептана (100 мл). Затем ее суспендировали в дихлорметане(100 мл) и обрабатывали оксалилхлоридом (3,15 мл, 35,9 ммоль), а затем N,N-диметилформамидом (2 капли). Реакционную смесь перемешивали в течение 20 ч при комнатной температуре, затем концентрировали при пониженном давлении. Твердый остаток суспендировали в ацетонитриле (100 мл), обрабатывали по каплям раствором (триметилсилил)диазометана в диэтиловом эфире (2 М, 39,0 мл, 78,0 ммоль) и перемешивали в течение 3 ч. Смесь охлаждали до 0 С и по каплям добавляли бромистый водород (33%ный в уксусной кислоте, 21,9 мл, 125 ммоль). Через 1 ч при 0 С реакционную смесь концентрировали и твердый остаток смешивали с гептаном (250 мл) и повторно концентрировали. Остаток разбавляли этилацетатом (100 мл) и интенсивно перемешивали насыщенным водным раствором бикарбоната натрия (100 мл). Органический слой сушили над сульфатом магния, фильтровали и концентрировали в вакууме; неочищенный продукт очищали путем хроматографии на силикагеле (градиент: от 12 до 100% этилацетата в гептане) с получением продукта в виде не совсем белого твердого вещества с приблизительно 85%-ной чистотой согласно ЖХ-МС анализу. Выход: 8,10 г, приблизительно 78% (скорректирован с учетом чистоты). ЖХ-МС m/z 282.8 (М+1). 1H ЯМР (500 МГц, CDCl3)3.93 (s, 3 Н), 4.25 (s, 2 Н), 8.01 (s, 1H). Стадия 4. Синтез трет-бутил-[4-(5-бром-1-метил-1H-пиразол-4-ил)-2-метил-1 Н-имидазол-1-ил]карбамата трет-Бутил-2-этанимидоилгидразинкарбоксилат (5,9 г, 34 ммоль), 2-бром-1-(5-бром-1-метил-1H-пиразол-4-ил)этанон (с предыдущей стадии, 8,00 г, приблизительно 24 ммоль) и N,N-диизопропилэтиламин(10,9 мл, 62,6 ммоль) кипятили с обратным холодильником в смеси 2-метилтетрагидрофурана (200 мл) и 1,2-диметоксиэтана (50 мл). Через 2,5 ч реакционную смесь охлаждали и промывали 50%-ным насыщенным водным раствором хлорида натрия (75 мл). Водный слой экстрагировали 2-метилтетрагидрофураном (50 мл) и объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Остаток подвергали хроматографии (градиент: от 0 до 8% метанола в дихлорметане) и очищенный материал (7,5 г) растворяли в диэтиловом эфире (25 мл), обрабатывали гексаном (4 капли) и оставляли кристаллизоваться. Полученное твердое вещество собирали и промывали небольшим количеством холодного диэтилового эфира с получением продукта в виде бледно-розового твердого вещества. Выход: 6,49 г, 18,2 ммоль, 59% за 2 стадии. ЖХ-МС m/z 358.4 (М+1). 1 Н ЯМР (500 МГц, CDCl3)1.49 (br s, 9H), 2.16 (br s, 3H), 3.85 (s, 3 Н), 7.17 (s, 1H), 7.89 (s, 1H), 8.8-9.3 (v br s, 1H). Стадия 5. Синтез трифторацетатной соли 4-(5-бром-1-метил-1H-пиразол-4-ил)-2-метил-1H-имидазол-1-амина трет-Бутил[4-(5-бром-1-метил-1H-пиразол-4-ил)-2-метил-1H-имидазол-1-ил]карбамат (5,00 г, 14,0 ммоль) растворяли в дихлорметане (120 мл), обрабатывали трифторуксусной кислотой (20,9 мл, 281 ммоль) и перемешивали в течение 2,5 ч. После удаления летучих веществ в вакууме масляный остаток разбавляли диэтиловым эфиром (100 мл). Полученную суспензию перемешивали в течение 30 мин при комнатной температуре и затем твердое вещество собирали и промывали диэтиловым эфиром с получением продукта в виде не совсем белого твердого вещества. Выход: 4,98 г, 13,5 ммоль, 96%. ЖХ-МС m/z 256.3 (М+1). 1 Н ЯМР (500 МГц, CD3OD)2.65 (s, 3 Н), 3.95 (s, 3 Н), 7.68 (s, 1H), 7.86 (s, 1H). Стадия 6. Синтез N-[4-(5-бром-1-метил-1H-пиразол-4-ил)-2-метил-1H-имидазол-1-ил]имидоформамида Трифторацетатную соль 4-(5-бром-1-метил-1H-пиразол-4-ил)-2-метил-1 Н-имидазол-1-амина (4,90 г,113,2 ммоль) объединяли с ацетатом формамидина (98%, 4,92 г, 46,3 ммоль) в 2-бутаноле (40 мл) и реакционную смесь нагревали при 100 С в течение 6 ч, затем оставляли охлаждаться до комнатной температуры и перемешивали в течение 18 ч. Не совсем белое твердое вещество собирали фильтрованием и промывали 2-пропанолом, а затем диэтиловым эфиром. Твердое вещество затем растирали с водным гидроксидом аммония (7,5 М, 40 мл); в результате фильтрования получили белое твердое вещество, которое промывали 2-пропанолом, а затем диэтиловым эфиром с получением продукта. Выход: 2,70 г, 9,54 ммоль, 72%. 1 Н ЯМР (500 МГц, CD3OD), предположительно в виде смеси ротамеров или таутомеров:2.26 и 2.31 (2 s, 3 Н), 3.89 и 3.89 (2 s, 3 Н), 7.26 и 7.40 (2 s, 1H), 7.41 и 7.96 (2 br s, 1H), 7.85 и 7.82 (2 s, 1H).- 24022586 Стадия 7. Синтез 5-(5-бром-1-метил-1H-пиразол-4-ил)-7-метилимидазо[5,1-f][1,2,4]триазин-4(3H)она (С 3) 1,1'-Карбонилбис-(1H-1,2,4-триазол) (90%, 2,69 г, 14,8 ммоль) и N-[4-(5-бром-1-метил-1H-пиразол 4-ил)-2-метил-1H-имидазол-1-ил]имидоформамид (2,69 г, 9,50 ммоль) объединяли в 1,4-диоксане (63 мл) и смесь перемешивали в течение 3,5 ч при комнатной температуре, затем нагревали до 50 С в течение 1 ч. Добавляли дополнительное количество 1,1'-карбонилбис-(1H-1,2,4-триазола) (90%, 1,34 г, 7,35 ммоль) и нагревание продолжали в течение 30 мин. После еще одного добавления 1,1'-карбонилбис-(1H-1,2,4 триазола) (90%, 269 мг, 1,48 ммоль) осуществляли нагревание при 50 С в течение дополнительных 75 мин. Реакционную смесь оставляли охлаждаться до комнатной температуры и затем концентрировали до половины ее исходного объема; осадок собирали и промывали этилацетатом с получением белого твердого вещества. Его растворяли в метаноле (50 мл), концентрировали досуха и растирали с водой (25 мл). Собранное твердое вещество промывали 2-пропанолом, а затем диэтиловым эфиром с получением С 3 в виде белого твердого вещества. Выход: 1,95 г, 6,31 ммоль, 66%. ЖХ-МС m/z 309.4 (М+1). 1H ЯМР (500 МГц, DMSO-d6)2.53 (s, 3 Н), 3.87 (s, 3 Н), 7.87 (s, 1H), 8.17 (s, 1H), 11.69 (br s, 1H). Стадия 8. Синтез 5-(5-бром-1-метил-1H-пиразол-4-ил)-7-метил-4-(1H-1,2,4-триазол-1-ил)имидазо[5,1-f][1,2,4]триазина 1H-1,2,4-Триазол (4,49 г, 65,0 ммоль) смешивали с ацетонитрилом (40 мл) и охлаждали до 0 С. Добавляли оксихлорид фосфора (1,78 мл, 19,4 ммоль) с последующим добавлением по каплям триэтиламина (10,9 мл, 78,2 ммоль). Температуру поддерживали при 15-20 С в течение 30 мин после окончания добавления. На этой стадии добавляли 5-(5-бром-1-метил-1H-пиразол-4-ил)-7-метилимидазо[5,1-f][1,2,4] триазин-4(3H)-он (2,0 г, 6,5 ммоль) и реакционную смесь оставляли нагреваться до комнатной температуры, затем нагревали до 70 С в течение 18 ч. Реакционную смесь охлаждали и вливали в раствор фосфата калия с температурой 10 С (97%, 6,56 г, 30,0 ммоль) в воде (30 мл). После перемешивания в течение 5 мин смесь обрабатывали твердым хлоридом натрия (5 г) и перемешивали в течение дополнительных 5 мин. Слои разделяли и водный слой экстрагировали дважды этилацетатом. Объединенные органические слои промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия и фильтровали. В результате удаления растворителей в вакууме получили неочищенный продукт в виде оранжевой пасты (2,1 г; содержал некоторое количество триэтиламина согласно 1 Н ЯМР), который использовали на следующей реакционной стадии без дополнительной очистки. 1H ЯМР (400 МГц, CDCl3), пики продукта:2.86 (s, 3 Н), 3.89 (s, 3 Н), 7.66 (s, 1H), 7.94 (s, 1H), 8.36 (s, 1H), 8.95 (s, 1H). Стадия 9. Синтез 5-(5-бром-1-метил-1H-пиразол-4-ил)-N,7-диметилимидазо[5,1-f][1,2,4]триазин-4 амина (С 4) Метиламин (4,31 мл 2 М раствора в тетрагидрофуране, 8,62 ммоль) добавляли к смеси карбоната цезия (9,78 г, 30,0 ммоль) и 5-(5-бром-1-метил-1H-пиразол-4-ил)-7-метил-4-(1H-1,2,4-триазол-1-ил)имидазо[5,1-f][1,2,4]триазина (с предыдущей реакции, 2,1 г) в N,N-диметилформамиде (12 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Ее гасили смесью воды и насыщенного водного раствора хлорида натрия 1:1, затем экстрагировали этилацетатом (220 мл) и тетрагидрофураном (10 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением С 4. Выход: 1,65 г, 5,12 ммоль, 78% за 2 стадии. ЖХ-МС m/z 322.1[5,1-f][1,2,4]триазин-4-амина 5-(5-Бром-1-метил-1H-пиразол-4-ил)-N,7-диметилимидазо[5,1-f][1,2,4]триазин-4-амин (13,26 г,41,16 ммоль) и [4-(трифторметил)фенил]бороновую кислоту (98%, 9,72 г, 50,2 ммоль) объединяли в этаноле (126 мл) и полученную суспензию обрабатывали раствором фосфата калия (98%, 11,13 г, 51,39 ммоль) в воде (42 мл) и нагревали до 70 С в течение 40 мин, одновременно пропуская через барботер интенсивный поток азота. После добавления тетракис(трифенилфосфин)палладия(0) (482 мг, 0,417 ммоль) реакционную смесь кипятили с обратным холодильником в течение 3,5 ч, затем охлаждали до комнатной температуры и перемешивали в течение дополнительных 16 ч. Смесь фильтровали через набивку ваты и фильтрат концентрировали в вакууме, затем повторно концентрировали с 2-метилтетрагидрофураном (2200 мл). Остаток снова растворяли в 2-метилтетрагидрофуране (150 мл) и экстрагировали водной соляной кислотой (1 М, 70 мл, перемешивали в течение 20 мин). Водный слой (рН примерно 2-3) отбрасывали. [На этой стадии удаляется наибольшая часть дебромированного исходного материала; важно, чтобы значение рН промывки HCl было не ниже 2.] Органический слой затем экстрагировали дважды 1 М водной соляной кислотой: сначала 100 мл (перемешивание в течение 40 мин), затем 75 мл (перемешивание в течение 20 мин). 100 мл Водного слоя подвергали обратной экстракции 2 метилтетрагидрофураном (80 мл, перемешивали в течение 30 мин) для удаления некоторых окрашивающих веществ. Два солянокислотных слоя объединяли и обрабатывали водным раствором гидроксида натрия (5 М, 35,5 мл), рН которого доводили до 6. Полученную смесь экстрагировали 2-метилтетрагидрофураном (130 мл); органический слой пропускали через набивку сульфата натрия (74 г) и кон- 25022586 центрировали в вакууме до объема примерно 150 мл. Его обрабатывали активированным углеродомDarco G-60 (5,03 г) и вращали на роторном испарителе в водяной бане с температурой 50 С в течение 1 ч. Теплый раствор фильтровали через набивку целита, промывая 2-метилтетрагидрофураном, и фильтрат концентрировали при пониженном давлении. Полученную бледно-желтую пену обрабатывали третбутилметиловым эфиром (150 мл) и перемешивали в водяной бане с температурой 50 С в течение 5 мин,затем охлаждали до комнатной температуры при перемешивании в течение 1 ч. Полученную суспензию охлаждали в ледяной бане и выдерживали в течение дополнительных 30 мин. Твердые вещества собирали фильтрованием и промывали охлажденным трет-бутилметиловым эфиром (охлаждали с помощью бани лед-насыщенный водный раствор хлорида натрия; 79 мл), затем суспендировали в гептане (150 мл). Эту смесь концентрировали в вакууме до небольшого объема и повторно концентрировали с гептаном (2150 мл) до конечного объема приблизительно 50 мл. В результате фильтрования получили продукт в виде белого твердого вещества. Выход: 11,91 г, 30,75 ммоль, 75%. ЖХ-МС m/z 388.2 (М+1). 1 Н ЯМР (400 МГц, CDCl3)2.65 (br s, 3 Н), 3.00 (d, J=5,0 Гц, 3 Н), 3.95 (s, 3 Н), 5.49-5.57 (m, 1H), 7.61 (br AB квартет,JAB=8,2 Гц, АВ=41,4 Гц, 4H), 7.73 (s, 1H), 7.91 (br s, 1H). Пример 4 4-(Азетидин-1-ил)-5-[5-(4-хлорфенил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазин Стадия 1. Синтез 4-хлор-7-метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина Оксихлорид фосфора (16,07 г, 104,8 ммоль) добавляли в течение 5 мин к суспензии 7-метил-5-(1 метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазин-4(3H)-она (11,98 г, 52,03 ммоль) в толуоле (180 мл). Добавляли N,N-диизопропилэтиламин (27,04 г, 209,2 ммоль) и реакционную смесь нагревали при 100 С в течение 15 ч. Смесь охлаждали до комнатной температуры, после чего ЖХ-МС анализ показал, что реакция не завершена; добавляли дополнительное количество оксихлорида фосфора (3,98 г, 26,0 ммоль) и реакционную смесь нагревали при 100 С в течение 22 ч. Смесь охлаждали до комнатной температуры,разбавляли дихлорметаном (24 мл) и перемешивали в течение 48 ч при комнатной температуре. Через 50 мин реакционную смесь добавляли к смеси триэтиламина (58 мл), толуола (60 мл) и воды (120 мл), одновременно поддерживая внутреннюю температуру ниже 34 С. Перемешивание продолжали в течение дополнительных 15 мин. Водный слой экстрагировали толуолом (120 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия (200 мл) и сушили путем пропускания через набивку сульфата натрия (71 г). Фильтрат концентрировали в вакууме с получением неочищенного продукта в виде темно-оранжевого твердого вещества (9,77 г); его обрабатывали тетрагидрофураном (60 мл) и нагревали до флегмообразования в течение 15 мин с получением раствора. Его охлаждали до комнатной температуры в течение 30 мин, гранулировали в течение 30 мин, затем охлаждали в ледяной бане и перемешивали в течение 30 мин. Полученное твердое вещество собирали фильтрованием и осадок на фильтре промывали предварительно охлажденным трет-бутилметиловым эфиром (охлажденным с помощью бани лед-насыщенный водный раствор хлорида натрия; 65 мл). Продукт получили в виде яркооранжевого твердого вещества. Выход: 7,59 г, 30,5 ммоль, 59%. ЖХ-МС m/z 249.0 (М+1). 1H ЯМР (400 МГц, CDCl3)2.75 (s, 3 Н), 3.99 (s, 3 Н), 7.90 (s, 1H), 8.00 (s, 1H), 8.12 (s, 1H). Стадия 2. Синтез 4-(азетидин-1-ил)-7-метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина Раствор азетидина (9,21 г, 161 ммоль) в дихлорметане (75 мл) добавляли к раствору 4-хлор-7-метил 5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина (38,14 г, 153,4 ммоль) в дихлорметане (310 мл). После перемешивания в течение 5 мин реакционную смесь обрабатывали водным раствором бикарбоната натрия (0,89 М, 260 мл, 231 ммоль) и интенсивно перемешивали в течение 2 ч. После разделения фаз белое твердое вещество собирали фильтрованием и смешивали с водой и дихлорметаном; оно не- 26022586 полностью растворилось. В результате фильтрования получили вторую смесь вода/дихлорметан, которую объединяли с первым фильтратом. Слои разделяли и водный слой экстрагировали дихлорметаном (3100 мл). Объединенные органические слои промывали насыщенным водным раствором хлорида натрия(250 мл), затем сушили над сульфатом натрия и фильтровали. Фильтрат концентрировали на роторном испарителе при 45 С до тех пор, пока в колбе не начало образовываться твердое вещество. Добавляли при перемешивании трет-бутилметиловый эфир (400 мл) и смесь гранулировали в течение 1 ч. В результате фильтрования получили продукт в виде белого твердого вещества. Выход: 36,07 г, 133,9 ммоль,87%. 1H ЯМР (400 МГц, CDCl3)2.23-2.31 (m, 2H), 2.65 (s, 3 Н), 3.97 (s, 3 Н), 3.98-4.08 (m, 4H), 7.61 (s,1H), 7.62 (s, 1H), 7.85 (s, 1H). Стадия 3. Синтез 4-(азетидин-1-ил)-5-[5-(4-хлорфенил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f][1,2,4]триазина 4-(Азетидин-1-ил)-7-метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазин (10,0 г, 37,1 ммоль), 1-бром-4-хлорбензол (14,2 г, 74,2 ммоль), свежеизмельченный карбонат калия (15,4 г, 111 ммоль) и димер хлорида аллилпалладия(II) (970 мг, 2,60 ммоль) объединяли в реакционной колбе, содержимое которой затем откачивали под вакуумом и продували азотом. Добавляли 1,4-диоксан (180 мл) и реакционную смесь перемешивали при комнатной температуре. Смесь дегазировали под вакуумом и барботировали через нее азот в течение 5 мин. Процедуру вакуумирования/продувки азотом повторяли еще два раза. Реакционную смесь нагревали до 100 С в течение 72 ч, затем охлаждали до комнатной температуры и объединяли с идентичной реакцией, осуществленной на 5,0 г 4-(азетидин-1-ил)-7-метил 5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина. Объединенные реакционные смеси концентрировали в вакууме и затем суспендировали в этилацетате, остаток наносили на набивку силикагеля с верхним слоем целита. Набивку элюировали этилацетатом (1,5 л), а затем смесью этилацетат/метанол 9:1(1 л). Объединенные элюаты концентрировали в вакууме с получением масла (25 г), которое растворяли в этилацетате (500 мл) и экстрагировали водной соляной кислотой (1 М, 300 мл). Водный слой подщелачивали 1 М водным раствором гидроксида натрия и экстрагировали этилацетатом (2250 мл). Два органических слоя объединяли и промывали водной лимонной кислотой (1 М, 200 мл) и слой водной лимонной кислоты экстрагировали этилацетатом (8100 мл). Объединенные органические слои затем промывали смесью насыщенный водный раствор бикарбоната натрия/насыщенный водный раствор хлорида натрия 1:1, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученное твердое вещество перемешивали в горячей смеси гептана (примерно 100 мл) и этилацетата(примерно 15 мл), охлаждали до комнатной температуры и перемешивали в течение 16 ч. Твердое вещество выделяли путем фильтрования с получением белого порошка (10,6 г). Для удаления остаточного палладия (0,3% по анализу QTI Analytical Services) этот материал растворяли в смеси этилацетата (100 мл) и 2-метилтетрагидрофурана (150 мл) при комнатной температуре и обрабатывали SiliCycle-тиолом(SiliCycle, 1,35 ммоль/г, 5 г, 6,75 ммоль активности). Смесь перемешивали в течение 20 ч, затем фильтровали через целит. Фильтрат обрабатывали активированным углеродом Darco (500 мг) и перемешивали в течение 15 мин, а затем фильтровали и концентрировали при пониженном давлении. Полученное масло подвергали азеотропной перегонке со смесью гептана и этилацетата 1:1 с получением белого твердого вещества (9,9 г), которое растирали со смесью гептана (80 мл) и этилацетата (10 мл) при дефлегмации, затем охлаждали до комнатной температуры и перемешивали в течение дополнительных 36 ч. В результате фильтрования получили продукт в виде белого твердого вещества. Выход: 9,48 г, 25,0 ммоль,67%. ЖХ-МС m/z 380.0 (М+1). 1H ЯМР (500 МГц, CDCl3)2.21-2.28 (m, 2H), 2.63 (s, 3 Н), 3.4-4.4 (v br m,4H), 3.90 (s, 3 Н), 7.31-7.36 (m, 4H), 7.64 (s, 1H), 7.80 (s, 1H). Пример 5 4-(Азетидин-1-ил)-5-[5-(5-хлорпиридин-2-ил)-1-метил-1H-пиразол-4-ил]-7-метилимидазо[5,1-f] Синтез указанного в заголовке продукта осуществляли в соответствии с методикой для синтеза 4(азетидин-1-ил)-7-метил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-илимидазо[5,1-f][1,2,4]триазина в примере 2, за исключением того, что 2-бром-5-хлорпиридин использовали вместо 2 бром-5-(трифторметил)пиридина. В данном случае после промывки лимонной кислотой органический слой сушили, фильтровали и концентрировали при пониженном давлении с получением бледно-желтого твердого вещества, которое затем перекристаллизовывали из метанола. Твердое вещество растворяли в 2-метилтетрагидрофуране (300 мл), обрабатывали силикагелем и перемешивали в течение 18 ч. Добавляли активированный углерод Darco (2 г) и эту смесь перемешивали в течение 30 мин, по истечении которых ее фильтровали через набивку целита и концентрировали в вакууме с получением продукта в виде белого твердого вещества. Выход: 17,6 г, 46,2 ммоль, 52%. ЖХ-МС m/z 381.0 (М+1). 1H ЯМР (400 МГц,- 27022586A. Синтез 1-бром-4-(дифторметил)бензола Трифторид (диэтиламино)серы (46 г, 0,29 моль) добавляли порциями в течение 20 мин к раствору 4 бромбензальдегида (37,7 г, 0,204 ммоль) в дихлорметане (170 мл) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Ее затем оставляли охлаждаться до комнатной температуры, перемешивали в течение 18 ч и медленно добавляли в течение 30 мин к перемешиваемому насыщенному водному раствору бикарбоната натрия (377 мл) при 0 С. Бифазную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 15 мин. Водный слой экстрагировали дихлорметаном (280 мл) и объединенные органические слои промывали насыщенным водным раствором хлорида натрия(80 мл), сушили над сульфатом магния и концентрировали в вакууме с получением золотистого масла. Реакцию повторяли еще 10 раз на 60-граммовых партиях 4-бромбензальдегида (суммарное количество исходного материала: 638 г, 3,45 моль) и полученные масла объединяли и очищали путем дистилляции[1,1'-Бис-(дифенилфосфино)ферроцен]дихлорпалладий(II) (25,25 г, 34,5 ммоль) добавляли одной порцией к дегазированной смеси 1-бром-4-(дифторметил)бензола (160 г, 0,77 моль), 4,4,4',4',5,5,5',5'октаметил-2,2'-би-1,3,2-диоксаборолана (392,5 г, 1,55 моль) и ацетата калия (303 г, 3,09 моль) в 1,4 диоксане (2,42 л) и реакционную смесь нагревали до 100 С в течение 18 ч. Смесь затем охлаждали до комнатной температуры и фильтровали через целит, промывая этилацетатом (3 л). Фильтрат концентрировали в вакууме с получением темно-коричневого масла. Реакцию повторяли еще 3 раза на партиях 1 бром-4-(дифторметил)бензола 50 г, 160 и 156 г (суммарное количество исходного материала, 526 г, 2,54 моль) и объединенные неочищенные продукты очищали дважды хроматографией на силикагеле (градиент: от 0 до 3% этилацетата в гептане) с получением желто-белого твердого вещества (803 г). Это вещество перекристаллизовывали из метанола (1,6 л) при -20 С и фильтрат концентрировали до половины его первоначального объема, охлаждали и полученное твердое вещество собирали фильтрованием. Объединенные твердые вещества (426 г) перекристаллизовывали из гептана (500 мл) при -20 С, затем расплавляли и вливали в метанол (200 мл), охлажденный в бане метанол-лед. Смесь фракционировали и фильтровали с получением С 5 в виде твердого вещества. Выход: 250,7 г, 0,987 ммоль, 39%. 1 Н ЯМР (400 МГц,CDCl3)1.37 (s, 12H), 6.65 (t, J=56,4 Гц, 1H), 7.52 (br d, J=8,1 Гц, 2H), 7.92 (br d, J=8,0 Гц, 2 Н). Стадия 2. Синтез 5-5-[4-(дифторметил)фенил]-1-метил-1H-пиразол-4-ил-N,7-диметилимидазо[5-(5-Бром-1-метил-1H-пиразол-4-ил)-N,7-диметилимидазо[5,1-f][1,2,4]триазин-4-амин (13,01 г,40,38 ммоль) и 2-[4-(дифторметил)фенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан (12,75 г, 50,18 ммоль) объединяли в этаноле (126 мл) и полученную суспензию обрабатывали раствором фосфата калия (98%,11,04 г, 50,97 ммоль) в воде (42 мл) и нагревали до 70 С в течение 30 мин, в то же время подавая через барботер интенсивный поток азота. После добавления тетракис(трифенилфосфин)палладия(0) (481 мг,0,416 ммоль) реакционную смесь кипятили с обратным холодильником в течение 4 ч, затем охлаждали до комнатной температуры и перемешивали в течение дополнительных 16 ч. Смесь фильтровали через набивку ваты и фильтрат концентрировали в вакууме, затем повторно концентрировали с 2 метилтетрагидрофураном (2150 мл). Остаток снова растворяли в 2-метилтетрагидрофуране (150 мл) и экстрагировали водной соляной кислотой (1 М, 70 мл, перемешивали в течение 20 мин). Водный слой(рН примерно 2-3) отбрасывали. Органический слой экстрагировали дважды 1 М водной соляной кислотой: сначала 100 мл (перемешивание в течение 1 ч), затем 75 мл (перемешивание в течение 20 мин). Указанный 100-миллилитровый водный слой подвергали обратной экстракции 2-метилтетрагидрофураном(75 мл, перемешивали в течение 20 мин) для удаления светло-желтой окраски; из этого органического слоя осаждалось твердое вещество, которое собирали и промывали трет-бутилметиловым эфиром с получением кристаллов согласно рентгеновскому дифракционному анализу. Анализ рентгеновской дифракции на монокристалле показал, что это вещество представляет собой моногидрат гидрохлоридной соли продукта. Два солянокислотных слоя объединяли и обрабатывали водным раствором гидроксида- 28022586 натрия (5 М, 35,5 мл), рН которого доводили до 6. Полученную смесь экстрагировали 2-метилтетрагидрофураном (150 мл); органический слой пропускали через набивку сульфата натрия (58 г) и концентрировали в вакууме до объема примерно 150 мл. Этот желтый раствор обрабатывали активированным углеродом Darco G-60 (5,03 г) и подвергали вращению на роторном испарителе в водяной бане с температурой 50 С в течение 1,5 ч. Теплый раствор фильтровали через набивку целита и фильтрат концентрировали при пониженном давлении. Полученное светло-желтое твердое вещество обрабатывали трет-бутилметиловым эфиром (250 мл) и подвергали вращению на роторном испарителе в водяной бане с температурой 55 С в течение 1 ч. Примерно 100 мл растворителя было удалено с использованием роторного испарителя, и полученную смесь охлаждали до комнатной температуры при перемешивании в течение 1 ч. Суспензию затем охлаждали в ледяной бане и перемешивали в течение дополнительных 30 мин. Твердые вещества собирали фильтрованием и промывали охлажденным трет-бутилметиловым эфиром(охлажденным на бане лед-насыщенный водный раствор хлорида натрия; 50 мл) с получением продукта в виде порошкообразного белого твердого вещества. Выход: 11,27 г, 30,51 ммоль, 76%. ЖХ-МС m/z 370.2 (М+1). 1H ЯМР (400 МГц, CDCl3)2.65 (br s, 3 Н), 2.98 (d, J=5,1 Гц, 3 Н), 3.94 (s, 3 Н), 5.48-5.55 (m,1H), 6.65 (t, J=56,3 Гц, 1H), 7.52 (br AB квартет, JAB=8,4 Гц, АВ=17,9 Гц, 4 Н), 7.73 (s, 1H), 7.90 (br s, 1H). Пример 7[1,2,4]триазин-4-амина (С 6) Продукт синтезировали способом, аналогичным описанному для получения 4-(азетидин-1-ил)-7 метил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазина в примере 2, за исключением того, что 1-(4-метоксифенил)-N-метилметанамин использовали вместо гидрохлорида азетидина и немного модифицировали обработку: после экстрагирования суспензии дихлорметаном объединенные органические слои промывали 1 н. водным раствором гидроксида натрия, промывали насыщенным водным раствором хлорида натрия и сушили над сульфатом натрия. После фильтрования фильтрат концентрировали в вакууме и пропускали через короткую колонку силикагеля (элюент: 5%-ный метанол в этилацетате). Элюат концентрировали при пониженном давлении и полученное твердое вещество промывали третбутилметиловым эфиром, а затем диэтиловым эфиром с получением С 6. Выход: 36,0 г, 99,1 ммоль, 76%. ЖХ-МС m/z 364.2 (М+1). 1H ЯМР (400 МГц, CDCl3)2.67 (s, 3 Н), 2.84 (s, 3 Н), 3.77 (s, 3 Н), 3.88 (s, 3 Н),4.66 (s, 2 Н), 6.82 (br d, J=8,7 Гц, 2 Н), 7.07 (br d, J=8,6 Гц, 2 Н), 7.58 (s, 1H), 7.62 (s, 1H), 7.89 (s, 1H). Стадия 2. Синтез N-(4-метоксибензил)-N,7-диметил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]1H-пиразол-4-илимидазо[5,1-f][1,2,4]триазин-4-аминаN-(4-Метоксибензил)-N,7-диметил-5-(1-метил-1H-пиразол-4-ил)имидазо[5,1-f][1,2,4]триазин-4 амин (10,0 г, 27,5 ммоль), 2-бром-5-(трифторметил)пиридин (12,4 г, 54,9 ммоль) и порошковый карбонат калия (11,4 г, 82,5 ммоль) объединяли в 1,4-диоксане (90 мл) и кипятили с обратным холодильником в течение 10 мин. Добавляли димер хлорида аллилпалладия (98%, 514 мг, 1,38 ммоль) и реакционную смесь нагревали в течение 22 ч при 160 С в герметично закрытой пробирке, покрытой Q-Tube (Q Labtech). Реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Остаток суспендировали в этилацетате, фильтровали через целит и концентрировали при пониженном давлении. В результате хроматографии на силикагеле (градиент: от 50 до 100% этилацетата в гептане) получили бледно-коричневую пену (7,85 г), которую подвергали кристаллизации из гептана (примерно 100 мл ) и этилацетата (примерно 5 мл) с получением продукта в виде бледно-коричневого порошка. Выход: 7,00 г,13,8 ммоль, 50%. ЖХ-МС m/z 509.1 (М+1). 1 Н ЯМР (500 МГц, CDCl3)2.54 (s, 3 Н), 2.72 (s, 3 Н), 3.75 (s,3 Н), 4.14 (s, 3 Н), 4.34 (br s, 2H), 6.76 (br d, J=8,8 Гц, 2 Н), 6.94 (br d, J=8,5 Гц, 2 Н), 7.39 (d, J=8,3 Гц, 1H),7.72 (s, 1H), 7.76 (dd, J=8,3, 2,2 Гц, 1H), 7.85 (s, 1H), 8.95-8.96 (m, 1H). Стадия 3. Синтез N,7-диметил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4-ил имидазо[5,1-f][1,2,4]триазин-4-аминаN-(4-Метоксибензил)-N,7-диметил-5-1-метил-5-[5-(трифторметил)пиридин-2-ил]-1H-пиразол-4 илимидазо[5,1-f][1,2,4]триазин-4-амин (7,00 г, 13,8 ммоль) растворяли в дихлорметане (46 мл) и обрабатывали трифторуксусной кислотой (40 мл, 520 ммоль) и метоксибензолом (99,7%, 7,0 мл, 64 ммоль). Реакционную смесь нагревали при 40 С в течение 4 ч, затем концентрировали в вакууме. Добавляли водный 1 н. раствор гидроксида натрия и смесь экстрагировали этилацетатом. Органический слой концентрировали при пониженном давлении с получением неочищенного продукта (12 г), который объединяли с неочищенным продуктом от двух дополнительных осуществлений этой реакции (общее количество исходного вещества: 18,09 г, 35,57 ммоль). Объединенный материал растворяли в горячем метаноле, ос- 29
МПК / Метки
МПК: A61K 31/53, A61P 25/18, C07D 487/04
Метки: расстройств, неврологических, лечения, имидазо[5,1-f][1,2,4]триазины
Код ссылки
<a href="https://eas.patents.su/30-22586-imidazo51-f124triaziny-dlya-lecheniya-nevrologicheskih-rasstrojjstv.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазо[5,1-f][1,2,4]триазины для лечения неврологических расстройств</a>
Производные имидазо[1,2-a]пиразина и их применение для профилактики или лечения неврологических, психиатрических и метаболических расстройств и заболеваний

Номер патента: 22012
Опубликовано: 30.10.2015
Авторы: Мартин-Мартин Мария Лус, Макдональд Грегор Джеймс, Пастор-Фернандес Хоакин, Конде-Сейде Сусана, Ванхоф Грета Констанция Петер, Бартоломе-Небреда Хосе Мануэль, Ван Гол Михиль Люк Мария
МПК: A61P 25/00, A61P 3/00, A61K 31/4985...
Метки: заболеваний, метаболических, психиатрических, расстройств, имидазо[1,2-a]пиразина, профилактики, неврологических, производные, лечения, применение
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма,где R1 выбран из группы, состоящей из радикалов формул (а-1), (а-2) и (а-3):где каждый из R6, R7 и R8 независимо выбран из группы, состоящей из фтора, С1-4алкила, С1-4алкилокси и С1-4алкила, замещенного 1, 2 или 3 атомами фтора;R9 представляет собой водород или С1-4алкил;каждый из m1, m2 и m3 независимо выбран из 0, 1, 2, 3 и 4;р2 выбран из 1, 2, 3 и 4;каждый из p1 и р3 независимо выбран из...
Производные индола и их применение для лечения психических и неврологических расстройств

Номер патента: 6104
Опубликовано: 25.08.2005
Авторы: Андерсен Ким, Руланд Томас, Мольтсен Айнер Кнуд, Роттлендер Марио, Миккельсен Иван, Крог-Енсен Кристиан
МПК: A61K 31/497, C07D 401/12
Метки: расстройств, лечения, психических, индола, неврологических, производные, применение
Формула / Реферат:
1. Соединение, представленное общей формулой I где A представляет собой O или S; n равно 2, 3, 4, 5, 6, 7, 8, 9 или 10; m равно 2 или 3; W представляет собой N, C или CH; Q представляет собой N, C или CH; и пунктирная линия представляет возможную связь; R1 представляет собой водород, C1-6-алкил, C2-6-алкенил, C2-6-алкинил, C3-8-циклоалкил-C1-6-алкил, арил-C1-6-алкил или ацил; R2, R3, R4, R5 и R6 независимо представляют собой водород, галоген,...
Аза-гетероциклические соединения, применяемые для лечения неврологических расстройств и выпадения волос

Номер патента: 8723
Опубликовано: 29.06.2007
Авторы: Ву Йонг-Кайан, Норман Марк Х., Гамильтон Грегори С.
МПК: A61K 31/40, A61K 31/41, C07D 207/12...
Метки: применяемые, волос, лечения, выпадения, соединения, расстройств, неврологических, аза-гетероциклические
Формула / Реферат:
1. Фармацевтическая композиция, содержащая: 1) соединение формулы (I) или его фармацевтически приемлемые соли, сложный эфир или сольват, где n представляет собой 1 или 2, X представляет собой либо О, либо S, R1 представляет собой С1-С9алкил с прямой или разветвленной цепью, D представляет собой связь или С1-С10алкилен с прямой или разветвленной цепью, R2 представляет собой тетразол, -СООН, -CONH2, -CN, -SO3H или -ОН; и 2) фармацевтически...
Производные 1,2,4-триазоло[4,3-a]пиридина и их применение для лечения или предупреждения неврологических и психиатрических расстройств

Номер патента: 20672
Опубликовано: 30.12.2014
Авторы: Трабанко-Суарес Андрес Авелино, Тресадерн Гэри Джон, Сид-Нуньес Хосе Мария, Оелрик Даниель, Макдональд Грегор Джеймс, Вега-Рамиро Хуан Антонио
МПК: A61P 25/28, C07D 471/04, A61K 31/437...
Метки: 1,2,4-триазоло[4,3-a]пиридина, применение, предупреждения, психиатрических, лечения, неврологических, расстройств, производные
Формула / Реферат:
1. Соединение формулы (I)или его стереохимически изомерная форма,где n выбран из группы, состоящей из 0 и 1;m выбран из группы, состоящей из 0 и 1;R представляет собой метил;R1 выбран из группы, состоящей из С1-6алкила; (С1-3алкилокси)С1-3алкила; [(С1-3алкилокси)С1-3алкилокси]С1-3алкила;C1-3алкила, замещенного 1-3 галогенозаместителями; незамещенного фенила; (бензилокси)С1-3алкила; незамещенного С3-7циклоалкила; С3-7циклоалкила, замещенного...
7-([1,2,4]триазоло[1,5-α]пиридин-6-ил)-4-(3,4-дихлорфенил)-1,2,3,4-тетрагидроизохинолин, фармацевтическая композиция на его основе и способы лечения неврологических и психических расстройств

Номер патента: 20553
Опубликовано: 30.12.2014
Авторы: Накро Кассоум, Лю Шуан, Молино Брюс Ф.
МПК: A01N 43/60
Метки: 7-([1,2,4]триазоло[1,5-α]пиридин-6-ил)-4-(3,4-дихлорфенил)-1,2,3,4-тетрагидроизохинолин, фармацевтическая, психических, неврологических, основе, лечения, расстройств, способы, композиция
Формула / Реферат:
1. Соединение формулы (I)где атом углерода, обозначенный *, находится в R- или S-конфигурации;или его фармацевтически приемлемая соль либо сольват.2. Соединение по п.1, которое является (+)-стереоизомером.3. Соединение по п.1, которое является (-)-стереоизомером.4. Соединение по п.1, которое находится в S-конфигурации.5. Соединение по п.1, которое находится в R-конфигурации.6. Соединение по п.1, которое является (S)(+)-стереоизомером.7....
Предыдущий патент: Коллоидный продукт, способ его получения и его применение
Следующий патент: Разгрузочно-погрузочный кран, контейнерный терминал и способ разгрузки и погрузки корабля
Случайный патент: Пероральные композиции дезоксипеганина и их применение