Соединения 4-замещенного-3-бензилоксибицикло[3.1.0]гексана в качестве антагонистов mglur 2/3
Номер патента: 22196
Опубликовано: 30.11.2015
Авторы: Орнштейн Пол Лесли, Дрессман Брюс Энтони, Тромикзак Эрик Джордж, Ветман Татьяна Натали, Митч Чарльз Ховард, Чаппелл Марк Дональд, Фивуш Адам Майкл






Формула / Реферат
1. Соединение формулы

где каждый из R1 и R2 независимо представляет собой водород, C1-С3 алкоксикарбонилоксиметил, C1-С3 алкилкарбонилоксиметил или С3-6 циклоалкилкарбонилоксиметил;
R3, независимо в каждом случае, представляет собой метил, фтор или хлор;
R4 представляет собой гидроксил, метилкарбониламино, гидроксиметилкарбониламино, метоксикарбониламино, имидазол-2-илсульфанил, тиазол-2-илсульфанил, 1,2,4-триазол-3-илсульфанил, 1-метил-1,2,4-триазол-3-илсульфанил или 1-метил-1,2,4-триазол-5-илсульфанил; и
n представляет собой 1 или 2;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что n представляет собой 2, обе группы R3 представляют собой хлор и указанные хлоргруппы находятся в 3 и 4 положениях фенила, или его фармацевтически приемлемая соль.
3. Соединение по любому из пп.1 или 2, отличающееся тем, что каждый из R1 и R2 представляет собой водород, или его фармацевтически приемлемая соль.
4. Соединение по любому из пп.1 или 2, отличающееся тем, что оба R1 и R2 отличны от водорода, или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.1 или 2, отличающееся тем, что R1 и R2 одинаковы и отличны от водорода, или его фармацевтически приемлемая соль.
6. Соединение по п.5, отличающееся тем, что каждый из R1 и R2 представляет собой изопропилоксикарбонилоксиметил.
7. Соединение по п.1, представляющее собой (1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновую кислоту, или его фармацевтически приемлемая соль.
8. Соединение по п.1, представляющее собой бис-(изопропоксикарбонилоксиметил) (1R,2S,3S,4R,5S,6R)-4-амино-3-[(3,4-дихлорфенил)метокси]-2-гидроксибицикло[3.1.0]гексан-4,6-дикарбоксилат, или его фармацевтически приемлемая соль.
9. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения депрессивных расстройств у млекопитающего.
10. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли для лечения чрезмерной сонливости у млекопитающего.
11. Применение по любому из пп.9 или 10, где млекопитающее представляет собой человека.
12. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемой соли в совместной, раздельной или последовательной комбинации с ингибитором обратного захвата серотонина для лечения депрессивных расстройств у млекопитающего.
13. Применение по п.12, отличающееся тем, что указанный ингибитор обратного захвата серотонина представляет собой флуоксетин.
14. Применение по п.12 или 13, где млекопитающее представляет собой человека.
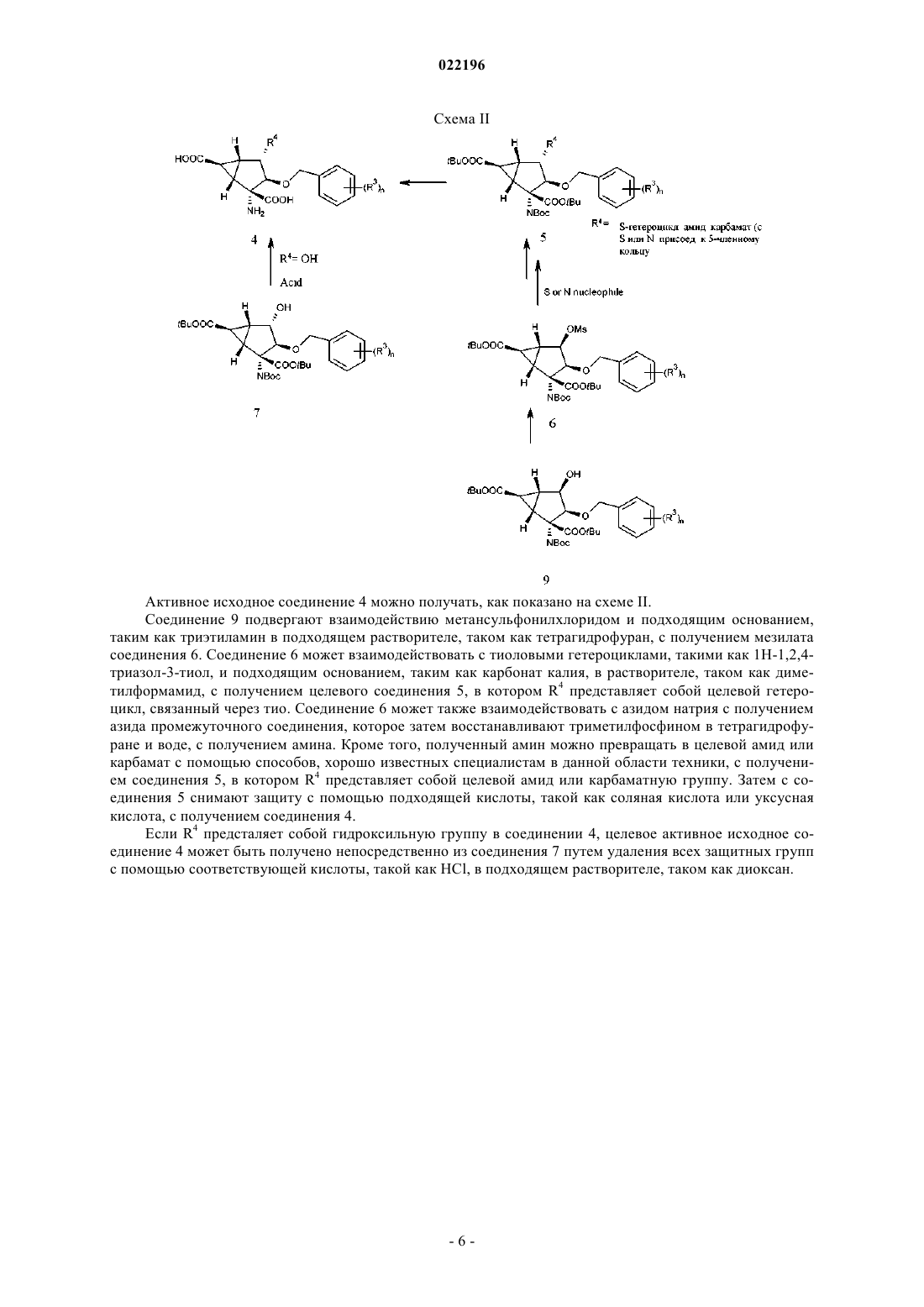
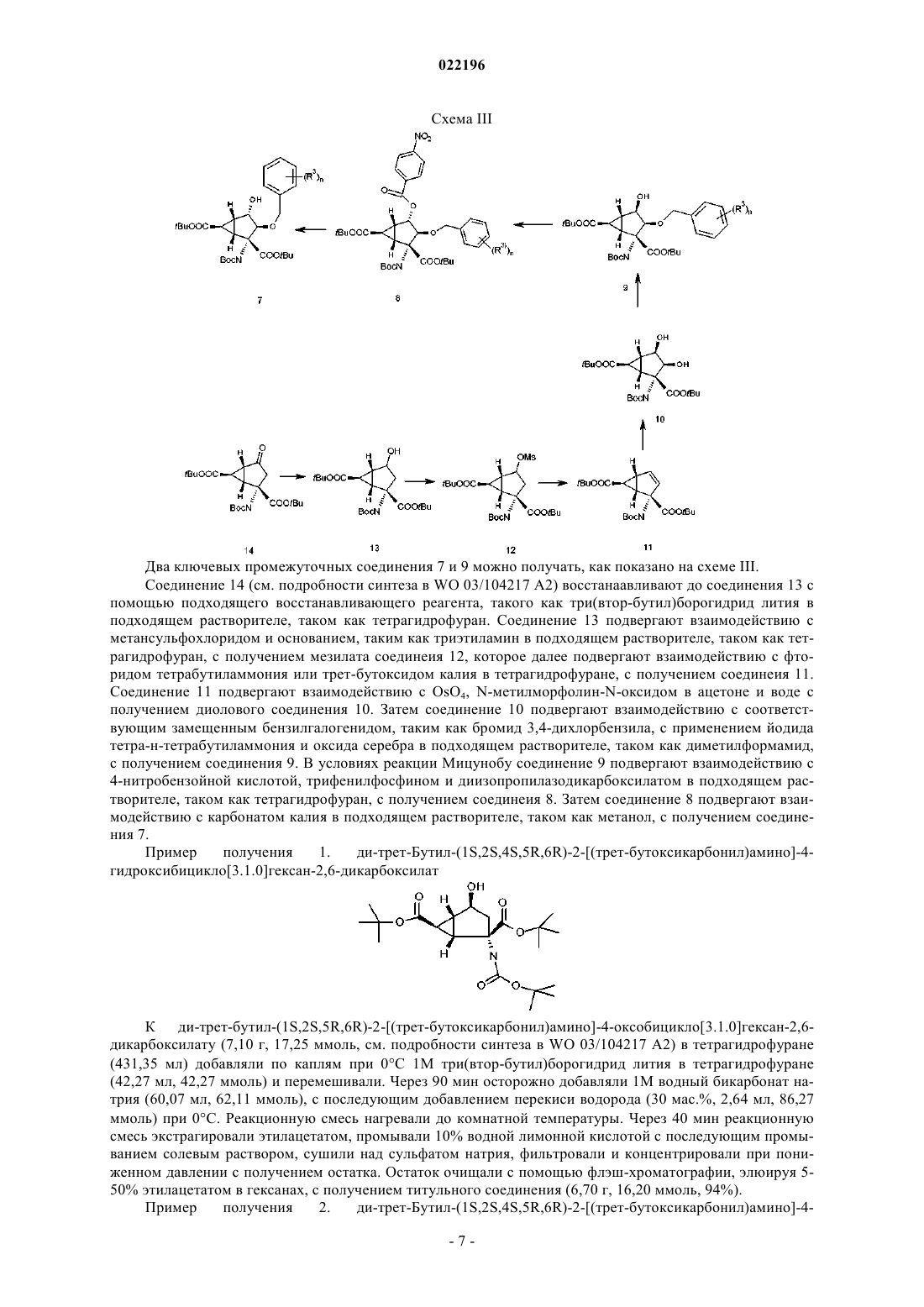
Текст
(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Глутамат представляет собой основной возбуждающий нейромедиатор в головном мозге и вовлечен в большое количество физиологических процессов, опосредованных по меньшей мере 11 различными рецепторами, каждый со своей фармакологией. Подтипы 2 и 3 метаботропного глутаматного рецептора(также известные как mGlu2 и mGlu3) часто объединяют как mGlu рецепторы II группы на основании гомологии их последовательностей, сходного связывания вторичного мессенджера и сходных фармакологических характеристик. Антагонисты mGlu2/3 рецепторов обладали значительным фармакологическим действием на животных моделях в отношении депрессивных расстройств и расстройств чрезмерной сонливости. В связи с этим полагают, что антагонисты mGlu2/3 могут подходить для лечения депрессивных расстройств, таких как большое депрессивное расстройство (БДР), монополярная депрессия, дистимия и/или циклотимия, и/или подходить для лечения расстройств чрезмерной сонливости, таких как чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). В патенте США 5916920 описаны некоторые соединения 3-монозамещенногобицикло[3.1.0]гексана в качестве модуляторов метаботропного рецептора глутамата, подходящие для лечения различных расстройств, в том числе в качестве антидепрессивных агентов. В патенте США 7157594 описаны различные соединения 3-монозамещенного-бицикло[3.1.0]гексана в качестве антагонистов mGlu рецептора II группы для применения для лечения различных расстройств, включающих депрессивные симптомы. В заявке на патент США 2007/0021394 А 1 описаны различные соединения 3 монозамещенного-бицикло[3.1.0]гексана в качестве антагонистов mGlu рецептора II группы и их пролекарства для применения для лечения различных расстройств, включая депрессию. В настоящем изобретении предложено семейство соединений 4-замещенного-3 фенилсульфанилметилбицикло[3.1.0]гексана, обладающих высокой антагонистической активностью в отношении mGlu2 и mGlu3 рецепторов. Соединения согласно настоящему изобретению также являются селективными по отношению к указанным mGlu2 и mGlu3 рецепторам, в частности, по сравнению с другими mGlu рецепторами. Также посредством изучения некоторых соединений на животных моделях было показано, что соединения согласно настоящему изобретению могут подходить для лечения депрессивных расстройств (которые могут включать большое депрессивное расстройство (БДР), монополярную депрессию, дистимию и/или циклотимию) и расстройств чрезмерной сонливости (которые могут включать чрезмерную дневную сонливость (EDS), гиперсомнию, связанную с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими,расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическую гиперсомнию и/или чрезмерную сонливость, связанную с невосстанавливающим сном (NRS). Антидепрессивный и стимулирующие эффекты указанного механизма также позволяют предположить воздействие на симптомы депрессивных расстройств, таких как усталость, которые иначе трудно поддаются лечению существующими антидепрессантами. В настоящем изобретении предложены соединения формулы I где каждый из R1 и R2 независимо представляет собой водород, C1-С 3 алкоксикарбонилоксиметил, C1-С 3 алкилкарбонилоксиметил или С 3-6 циклоалкилкарбонилоксиметил;n представляет собой 1 или 2; или их фармацевтически приемлемые соли. Особенностью настоящего изобретения является то, что соединения формулы I, где оба R1 и R2 представляют собой водород (дикислотные соединения), являются терапевтически активными соединениями in vivo, тогда как соединения, в которых R1, или R2, или оба отличны от водорода, представляют-1 022196 собой пролекарства их терапевтически активных дикислотных аналогов. Соединения, в которых R1 илиR2 или оба отличны от водорода гидролизуются in vivo с получением терапевтически активного дикислотного аналога. При пероральном введении указанные пролекарственные соединения, в частности дисложноэфирные пролекарства, обеспечивают повышенную биодоступность указанного дикислотного метаболита по сравнению с пероральным введением дикислотных соединений (оба R1 и R2 представляют собой водород), но указанные дикислотные соединения обеспечивают лучшую активность при внутривенном, внутримышечном или подкожном введении. В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или наполнителем. Кроме того, в указанном аспекте изобретения предложена фармацевтическая композиция, подходящая для лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми наполнителями, носителями или разбавителями. Согласно другому варианту реализации указанного аспекта изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, наполнителем или разбавителями, и возможно другие терапевтические ингредиенты. Согласно еще одному варианту реализации указанного аспекта изобретения указанная фармацевтическая композиция дополнительно содержит второй терапевтический агент, представляющий собой лекарственное средство, подходящее для лечения депрессивных расстройств, например ингибитор обратного захвата серотонина, например флуоксетин и/или циталопрам. Согласно еще одному варианту реализации указанного аспекта изобретения предложена фармацевтическая композиция, подходящая для лечения расстройств чрезмерной сонливости, например чрезмерной дневной сонливости (EDS), гиперсомнии, связанной с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна,вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатической гиперсомнии и/или чрезмерной сонливости, связанной с невосстанавливающим сном (NRS), содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми наполнителями, носителями или разбавителями. В настоящем изобретении также предложен способ лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии, у млекопитающего, включающий введение млекопитающему, которое нуждается в подобном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно другому варианту реализации указанного аспекта изобретения указанный способ дополнительно включает введение второго терапевтического агента в совместной, раздельной или последовательной комбинации,который представляет собой лекарственное средство, подходящее для лечения депрессивных расстройств, например ингибитор обратного захвата серотонина, например флуоксетин и/или циталопрам. Согласно другим вариантам реализации изобретения предложены способы лечения расстройств чрезмерной сонливости, включающие введение млекопитающему, которое нуждается в подобном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно другим вариантам реализации указанного аспекта изобретения указанная чрезмерная сонливость возникает вследствие любого одного или более следующих расстройств: чрезмерная дневная сонливость(EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Согласно одному конкретному варианту реализации указанных способов лечения указанное млекопитающее представляет собой человека. В настоящем изобретении также предложено соединение формулы I или его фармацевтически приемлемая соль для применения в терапии. В данном аспекте изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для применения для лечения депрессивных расстройств. Согласно дополнительным вариантам реализации указанное депрессивное расстройство представляет собой любое из большого депрессивного расстройства (БДР), монополярной депрессии, дистимии и/или циклотимии. Согласно другим вариантам реализации аспекта настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемая соль для применения в совместной, раздельной или последовательной комбинации с ингибитором обратного захвата серотонина, например флуок-2 022196 сетином и/или циталопрамом, для лечения депрессивных расстройств. Кроме того, указанный аспект изобретения включает соединение формулы I или его фармацевтически приемлемую соль для применения для лечения расстройств чрезмерной сонливости. Согласно конкретным вариантам реализации аспекта изобретения чрезмерная сонливость возникает вследствие любого одного или более следующих расстройств: чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна(включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Согласно одному конкретному варианту реализации указанного аспекта изобретения указанные применения осуществляют у млекопитающих, в частности человека. Согласно другому аспекту настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения депрессивных расстройств, например большого депрессивного расстройства, монополярной депрессии, дистимии и/или циклотимии. Согласно другому варианту реализации указанного аспекта изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли и второго терапевтического агента, подходящего для лечения депрессивных расстройств, например ингибитора обратного захвата серотонина, например флуоксетина и/или циталопрама, для получения лекарственного средства для лечения депрессивных расстройств. Согласно другому варианту реализации изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения расстройств чрезмерной сонливости. Согласно конкретным вариантам реализации указанного аспекта изобретения указанное лекарственное средство подходит для лечения любого одного или более следующих расстройств: чрезмерная дневная сонливость (EDS), гиперсомния, связанная с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна(включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна, вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сон-бодрствование), идиопатическая гиперсомния и/или чрезмерная сонливость, связанная с невосстанавливающим сном (NRS). Соединения согласно настоящему изобретению содержат основные и кислотные фрагменты и соответственно взаимодействуют с множеством органических и неорганических кислот и оснований с получением фармацевтически приемлемых солей. Фармацевтически приемлемые соли каждого из соединений согласно настоящему изобретению включены в объем настоящей заявки. В настоящей заявке термин"фармацевтически приемлемая соль" относится к любой соли соединения согласно изобретению, которая, по существу, нетоксична для живых организмов. Такие соли включают перечисленные в Journal ofPharmaceutical Science, 66, 2-19 (1977), известные специалистам в данной области. Предпочтительные классы соединений согласно настоящему изобретению представляют собой соединения, в которых: 1) оба R1 и R2 представляют собой водород; 2) R1 или R2 или оба отличны от водорода; 3) оба R1 и R2 отличны от водорода; 4) R1 и R2 одинаковы и отличны от водорода; 5) каждый из R1 и R2 представляет собой изопропоксикарбонилоксиметил; 6) n представляет собой 2; 7) n представляет собой 2, обе группы R3 представляют собой хлор, и группы хлора находятся в 3 и 4 положениях фенила; 8) R4 представляет собой гидроксил. Следует понимать, что следующие предпочтительные соединения представляют собой комбинацию указанных выше предпочтительных выборок для заданных заместителей с предпочтительными выборками для других заместителей. Примеры таких комбинаций включают, но не ограничиваются ими, следующие предпочтительные классы соединений: 9) предпочтительные соединения согласно любому из абзацев 1-5 (предпочтительные выборки дляR1 и R2), в которых n представляет собой 2, обе группы R3 представляют собой хлор и указанные группы хлора находятся в 3 и 4 положениях фенила (абзац 7); 10) предпочтительные соединения согласно любому из абзацев 1-5 (предпочтительные выборки дляR1 и R2), в которых R4 представляет собой гидроксил (абзац 8); 11) предпочтительные соединения согласно любому из абзацев 1-5 предпочтительные выборки для R1 и R2), в которых n представляет собой 2, обе группы R3 представляют собой хлор и указанные группы хлора находятся в 3 и 4 положениях фенила (абзац 7), и в которых R4 представляет собой гидроксил (абзац 8). Конкретные предпочтительные соединения согласно изобретению представляют собой соединения,описанные в примерах, включая их свободные основания и фармацевтически приемлемые соли. Некоторыми предпочтительными соединениями являются(1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновая кислота или ее фармацевтически приемлемая соль и бис(изопропоксикарбонилоксиметил)(1R,2S,3S,4R,5S,6R)-4-амино-3-[(3,4-дихлорфенил)метокси]-2 гидроксибицикло[3.1.0]гексан-4,6-дикарбоксилат или или его фармацевтически приемлемая соль (т.е. соединения примеров 1, 10 и 15 и их различные фармацевтически приемлемые соли). В настоящей заявке сокращения определяются следующим образом:"БСА" означает бычий сывороточный альбумин."DMEM" означает минимальную среду Игла, модифицированную по способу Дульбекко."HBSS" означает сбалансированный солевой раствор Хенкса."HPLC" означает жидкостную хроматографию высокого давления."IC50" означает концентрацию, при которой достигается 50% от максимального ингибирования."в.в." означает внутривенный или внутривенно."LC/MS" означает жидкостную хроматографию с последующей масс-спектроскопией."mFST" означает тест принудительного плавания на мышах; животная модель для антидепрессивной активности."ЯМР" означает ядерный магнитный резонанс."tBu" означает фрагмент третичного-бутила. Общая химия Соединения согласно настоящему изобретению можно получать согласно следующим схемам синтеза посредством способов, хорошо известных и применяемых в данной области техники. Подходящие условия реакций для стадий указанных схем хорошо известны в данной области техники и подходящие заменители для растворителей и сореагентов известны в рамках данной области техники. Кроме того,специалистам в данной области техники будет понятно, что синтетические промежуточные вещества можно выделять и/или очищать с помощью различных хорошо известных способов при необходимости или желании и настолько часто, чтобы было возможно применять различные промежуточные вещества непосредственно на последующих стадиях синтеза с небольшой очисткой или без очистки. Кроме того,специалисту в данной области техники будет понятно, что в некоторых случаях порядок включения фрагментов не важен. Конкретный порядок стадий, требуемый для получения соединений формулы I,зависит от конкретного синтезируемого соединения, исходного соединения и относительной реакционной способности замещенных фрагментов, как хорошо известно квалифицированному химику. Все заместители, если не указано иное, такие как определено выше, и все реагенты хорошо известны и применяемы в данной области техники. Соединения 1, представляющие собой пролекарство, можно получать как показано на схеме I, гдеR1, R2, R3, R4 и n такие, как определено ранее, оба R1 и R2 не являются водородом в соединении 1. Соединения 1 можно получать с применением химии, приведенной на схеме I. Соединение 4 подвергают взаимодействию с реагентом, защищающим аминогруппу, таким как дитрет-бутилдикарбонат, в условиях, хорошо известных специалистам в данной области техники, с получением соединения 3. Если указанные группы R1 и R2 в соединении 2 одинаковы, соединение 3 подвергают взаимодействию с достаточным количеством подходящего хлор- или йодметилалкилкарбоната и подходящими реагентами, такими как йодид натрия и карбонат цезия, в подходящем растворителе, таком как диметилформамид, с получением целевого дисложноэфирного соединения 2, в котором R1 и R2 одинаковы. Если R1 и R2 в соединении 2 различны, путем регулирования количества первого хлорметилалкилкарбоната до примерно 1 экв. карбоновую кислоту на 5-членном кольце можно сначала превращать вR2 моносложный эфир. Указанное соединение R2 моносложного эфира можно дополнительно подвергать взаимодействию с 1 экв. другого хлорметилалкилкарбоната. Группу свободной карбоновой кислоты на 3 членном кольце можно затем превращать в R1 сложный эфир с получением целевого дисложного эфира с различными R1 и R2. Для получения R2 моносложного эфира на 5-членном кольце соединения 2 соединение 3 подвергают взаимодействию с примерно 1 экв. подходящего хлорметилалкилкарбоната и подходящими реагентами, такими как йодид натрия и карбонат цезия, в подходящем растворителе, таком как диметилформамид, с получением целевого R2 моносложного эфира соединения 2, в котором R1 представляет собой водород. Для получения R1 моносложного эфира на 3-членном кольце группу карбоновой кислоты на 5-членном кольце следует сначала защитить, поскольку она более реакционноспособна. Более конкретно, группа карбоновой кислоты на 5-членном кольце в соединении 3 может взаимодействовать с -хлор-4-метокситолуолом, йодидом натрия и бикарбонатом натрия в подходящем растворителе,таком как диметилформамид, с получением 4-метоксилбензил моносложного эфира. Затем указанную группу карбоновой кислоты на 3-членном кольце защищенного соединения 4-метоксилбензил моносложного эфира подвергают взаимодействию с подходящим хлорметилалкилкарбонатом с получением целевого R1 сложного эфира на указанном 3-членном кольце. Дисложный эфир обрабатывают подходящей кислотой, такой как трифторуксусная кислота, со снятием защиты с 4-метоксилбензиловой и N-третбутоксикарбониловой групп, с получением целевого R1 моносложного эфира соединения 1. Затем с соединения 2, включая R2 моносложный эфир и дисложный эфир с одинаковыми или различными R1 и R2,снимают защиту с помощью подходящей кислоты, такой как соляная кислота в диоксане, с получением целевого соединения 1 или фармацевтически приемлемой соли. Активное исходное соединение 4 можно получать, как показано на схеме II. Соединение 9 подвергают взаимодействию метансульфонилхлоридом и подходящим основанием,таким как триэтиламин в подходящем растворителе, таком как тетрагидрофуран, с получением мезилата соединения 6. Соединение 6 может взаимодействовать с тиоловыми гетероциклами, такими как 1H-1,2,4 триазол-3-тиол, и подходящим основанием, таким как карбонат калия, в растворителе, таком как диметилформамид, с получением целевого соединения 5, в котором R4 представляет собой целевой гетероцикл, связанный через тио. Соединение 6 может также взаимодействовать с азидом натрия с получением азида промежуточного соединения, которое затем восстанавливают триметилфосфином в тетрагидрофуране и воде, с получением амина. Кроме того, полученный амин можно превращать в целевой амид или карбамат с помощью способов, хорошо известных специалистам в данной области техники, с получением соединения 5, в котором R4 представляет собой целевой амид или карбаматную группу. Затем с соединения 5 снимают защиту с помощью подходящей кислоты, такой как соляная кислота или уксусная кислота, с получением соединения 4. Если R4 предсталяет собой гидроксильную группу в соединении 4, целевое активное исходное соединение 4 может быть получено непосредственно из соединения 7 путем удаления всех защитных групп с помощью соответствующей кислоты, такой как HCl, в подходящем растворителе, таком как диоксан. Два ключевых промежуточных соединения 7 и 9 можно получать, как показано на схеме III. Соединение 14 (см. подробности синтеза в WO 03/104217 A2) восстанаавливают до соединения 13 с помощью подходящего восстанавливающего реагента, такого как три(втор-бутил)борогидрид лития в подходящем растворителе, таком как тетрагидрофуран. Соединение 13 подвергают взаимодействию с метансульфохлоридом и основанием, таким как триэтиламин в подходящем растворителе, таком как тетрагидрофуран, с получением мезилата соединеия 12, которое далее подвергают взаимодействию с фторидом тетрабутиламмония или трет-бутоксидом калия в тетрагидрофуране, с получением соединеия 11. Соединение 11 подвергают взаимодействию с OsO4, N-метилморфолин-N-оксидом в ацетоне и воде с получением диолового соединения 10. Затем соединение 10 подвергают взаимодействию с соответствующим замещенным бензилгалогенидом, таким как бромид 3,4-дихлорбензила, с применением йодида тетра-н-тетрабутиламмония и оксида серебра в подходящем растворителе, таком как диметилформамид,с получением соединения 9. В условиях реакции Мицунобу соединение 9 подвергают взаимодействию с 4-нитробензойной кислотой, трифенилфосфином и диизопропилазодикарбоксилатом в подходящем растворителе, таком как тетрагидрофуран, с получением соединеия 8. Затем соединение 8 подвергают взаимодействию с карбонатом калия в подходящем растворителе, таком как метанол, с получением соединения 7. Пример получения 1. ди-трет-Бутил-(1S,2S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат(431,35 мл) добавляли по каплям при 0 С 1 М три(втор-бутил)борогидрид лития в тетрагидрофуране(42,27 мл, 42,27 ммоль) и перемешивали. Через 90 мин осторожно добавляли 1 М водный бикарбонат натрия (60,07 мл, 62,11 ммоль), с последующим добавлением перекиси водорода (30 мас.%, 2,64 мл, 86,27 ммоль) при 0 С. Реакционную смесь нагревали до комнатной температуры. Через 40 мин реакционную смесь экстрагировали этилацетатом, промывали 10% водной лимонной кислотой с последующим промыванием солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя 550% этилацетатом в гексанах, с получением титульного соединения (6,70 г, 16,20 ммоль, 94%). Пример получения 2. ди-трет-Бутил-(1S,2S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-4-7 022196 Метансульфохлорид (224,61 мкл, 2,90 ммоль) добавляли к ди-трет-бутил-(1S,2S,4S,5R,6R)-2-[(третбутоксикарбонил)амино]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилату (400 мг, 967 мкмоль) и триэтиламину (472 мкл, 3,39 ммоль) в тетрагидрофуране (4,84 мл) при 0 С и перемешивали. Охлаждающую баню удаляли и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Разбавляли водой и насыщенным водным бикарбонатом натрия, затем экстрагировали этилацетатом. Объединенные органические вещества промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (600 мг, 1,22 ммоль, 100%). Пример получения 3. ди-трет-Бутил-(1S,2S,5R,6S)-2-[(трет-бутоксикарбонил)амино]бицикло[3.1.0] гекс-3-ен-2,6-дикарбоксилат К ди-трет-бутил-(1S,2S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-4-[(метилсульфонил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилату (600,00 мг, 1,22 ммоль) в тетрагидрофуране (2,03 мл) добавляли 1 М фторида тетрабутиламмония в тетрагидрофуране (3,84 мл, 3,84 ммоль) и нагревали с обратным холодильником в течение 3 ч. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали этилацетатом, промывали солевым раствором, сушили над сульфатом натрия,фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя 5-75% этилацетатом в гексанах, с получением титульного соединения (0,23 г, 0,58 ммоль, 48%). Пример получения 4. ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3,4 дигидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат(2,43 г, 17,97 ммоль), ди-трет-бутил-(1S,2S,5R,6S)-2-[(трет-бутоксикарбонил)амино]бицикло[3.1.0]гекс-3 ен-2,6-дикарбоксилату (3,23 г, 8,17 ммоль), ацетону (80,66 г, 102,09 мл, 1,39 моль) и воде (40,83 мл, 40,83 г, 2,27 моль). Перемешивали при комнатной температуре. Через 5 ч к реакционной смеси добавляли насыщенный водный раствор тиосульфата натрия (10 мл). Концентрировали до половинного объема при пониженном давлении. Разбавляли концентрат водой, экстрагировали этилацетатом (350 мл). Объединенные органические слои промывали солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя 5-50% этилацетатом в гексанах, с получением титульного соединения(987,35 мг, 4,26 ммоль) в диметилформамиде (17 мл) при комнатной температуре. Перемешивали в течение ночи. Разбавляли диэтиловым эфиром и гексанами (1:1), фильтровали черех целит и затем промывали эфиром и гексанами (1:1). Фильтрат промывали водой, солевым раствором, сушили над сульфатом магния и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя смесью 0-30% этилацетат:гексаны, с получением титульного соединения (1,7 г, 2,84 ммоль, 78%): MS (m/z): 610/612 (M+Na). Пример получения 6. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-[(4-нитрофенил)карбонил]оксибицикло[3.1.0]гексан-2,6-дикарбоксилат Трифенилфосфин (12,66 г, 48,26 ммоль) и 4-нитробензойную кислоту (8,19 г, 48,26 ммоль) добавляли к ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилату (14,20 г, 24,13 ммоль) и тетрагидрофурану (241,28 мл,2,97 моль) при комнатной температуре. Смесь охлаждали на ледяной бане. Добавляли диизопропилазодикарбоксилат (9,57 мл, 9,76 г, 48,26 ммоль) в тетрагидрофуране (20 мл) и реакционную смесь постепенно нагревали до комнатной температуры в течение ночи. Разбавляли диэтиловым эфиром и затем промывали водным карбонатом натрия. Снова экстрагировали водный слой диэтиловым эфиром. Объединяли все эфирные экстракты, промывали водой, солевым раствором, сушили над сульфатом магния и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэшхроматографии, элюируя 10-100% этилацетатом в гексанах, с получением титульного соединения (9 г,12,2 ммоль, 51%): MS (m/z): 759 (M+1). Пример получения 7. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору ди-трет-бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-[(4-нитрофенил)карбонил]оксибицикло[3.1.0]гексан-2,6-дикарбоксилата (9,00 г,12,20 ммоль) и метанола (122,01 мл, 3,01 моль) добавляли карбонат калия (5,11 г, 36,60 ммоль) и затем перемешивали при комнатной температуре. Через 1 ч реакционную смесь разбавляли этилацетатом, промывали водой (75 мл), солевым раствором (75 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя смесью 10-60% этилацетат:гексаны, с получением титульного соединения (5,3 г, 9,01 ммоль, 74%). К ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилату (2,00 г, 3,40 ммоль), триэтиламину (1,66 мл, 1,20 г,11,89 ммоль) и тетрагидрофурану (33,98 мл, 30,11 г, 417,61 ммоль) добавляли при 0 С метансульфохлорид (0,841 мл, 1,25 г, 10,87 ммоль) и перемешивали. Реакционную смесь постепенно нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь разбавляли насыщенным водным бикарбонатом натрия, затем экстрагировали этилацетатом (3100 мл). Объединенные органические экстракты промывали солевым раствором, сушили над сульфатом магния, фильтровали и затем концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэшхроматографии, применяя элюирование 5-60% этилацетатом в гексанах, с получением титульного соединения (1,40 г, 2,10 ммоль, 62%). Пример получения 9. ди-трет-Бутил-(1R,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-(1 Н-1,2,4-триазол-3-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат К 1H-1,2,4-триазол-3-тиолу (424,79 мг, 4,20 ммоль) и диметилформамиду (10,50 мл, 9,93 г, 135,80 ммоль) при комнатной температуре добавляли карбонат калия (1,45 г, 10,50 ммоль) и перемешивали. Через 5 мин к реакционной смеси добавляли ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(третбутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-[(метилсульфонил)окси]бицикло[3.1.0]гексан-2,6 дикарбоксилат (1,40 г, 2,10 ммоль) и перемешивали при 85 С. Через 2 дня реакционную смесь охлаждали до комнатной температуры, разбавляли насыщенным водным бикарбонатом натрия, затем экстрагировали этилацетатом (3 Х). Объединенные органические вещества промывали водой, солевым раствором,фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя 5-75% этилацетатом в гексанах, с получением титульного соединения (978,0 мг, 1,46 ммоль, 69%). MS (m/z): 671/669 (M-1). Пример получения 10. ди-трет-Бутил-(1R,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-(1,3-тиазол-2-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат(0,127 г, 0,893 ммоль, предварительно обработанный посредством фильтрации через щелочной оксид алюминия) и оставляли перемешиваться при -78 С в течение 15 мин. Смесь оставляли нагреваться до комнатной температуры и затем перемешивали в течение 30 мин при комнатной температуре. Выливали неочищенную реакционную смесь в воду и доводили рН до 6 с помощью 1N водной HCl и экстрагировали в этилацетат. Органический слой сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Очищали с помощью обращенно-фазовой флэш-хроматографии (100 г С 18 колонка, элюируя смесью 45-95% ацетонитрил/вода (с 0,1% трифторуксусной кислотой в обоих), соединение элюировали при 89% ацетонитрила). Желаемые фракции объединяли и нейтрализовали водным бикарбонатом. Экстрагировали этилацетатом, органический слой сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением титульного соединения в виде белой пены (0,16 г, 0,23 ммоль, 26%). MS (m/z) 473/475/477 (M+l). Пример получения 12. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-азидо-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилат Азид натрия (8,29 г, 127,45 ммоль) добавляли к ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(третбутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-[(метилсульфонил)окси]бицикло[3.1.0]гексан-2,6 дикарбоксилату (16,99 г, 25,49 ммоль) и 15-краун-5 (508,57 мкл, 561,46 мг, 2,55 ммоль) в диметилформамиде (254,90 мл, 240,96 г, 3,30 моль) при 85 С и перемешивали. Через 7 дней реакционную смесь охлаждали до комнатной температуры, затем разбавляли водой и экстрагировали этилацетатом (3 Х). Объединенные органические вещества промывали водой, солевым раствором, сушили над сульфатом магния,фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали с помощью флэш-хроматографии, элюируя 2-60% этилацетатом в гексанах, с получением титульного соединения (11,2 г, 18,3 ммоль, 72%). MS (m/z) 635/637 (M+l). Пример получения 13. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-амино-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилат К ди-трет-бутил-(1S,2R,3R,4S,5R,6S)-4-азидо-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилату (8,00 г, 13,04 ммоль) в тетрагидрофуране (43,46 мл) и воде (130,39 мл) при комнатной температуре добавляли 1 М раствор триметилфосфина в тетрагид- 11022196 рофуране (19,56 мл, 19,56 ммоль) и перемешивали в течение ночи. К реакционной смеси добавляли насыщенный водный бикарбонат натрия, затем экстрагировали этилацетатом (3 Х). Объединенные органические вещества промывали солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (7,42 г, 12,63 ммоль, 97%). К перемешиваемому охлажденному раствору ди-трет-бутил-(1S,2R,3R,4S,5R,6S)-4-амино-2-[(третбутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилата (2,30 г,3,91 ммоль) и триэтиламина (709,32 мкл, 514,97 мг, 5,09 ммоль) в дихлорметане (19,57 мл) при 0 С добавляли ацетилхлорид (334,31 мкл, 368,75 мг, 4,70 ммоль) в дихлорметане (5 мл), затем перемешивали. Через 90 мин разбавляли водой и экстрагировали дихлорметаном (325 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением титульного соединения (2,40 г, 3,81 ммоль, 97%). MS (m/z): 653 (M+l). Пример получения 15. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-[(метоксикарбонил)амино]бицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]-4-[(метилсульфонил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилата (0,80 г, 0,84 ммоль) в диметилформамиде (1,68 мл) добавляли 2-метил-1H-1,2,4-триазол-3-тион (0,126 г, 1,09 ммоль) и карбонат калия (0,35 г, 2,52 ммоль). После нагревания в течение 2 дней при 80 С раствор выливали в солевой раствор и экстрагировали этилацетатом три раза, объединенные органические фазы сушили сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Очищали с помощью флэш-хроматографии, элюируя 10% этилацетатом/CH2Cl2, с получением титульного соединения в виде масла (0,576 г, 0,84 ммоль, 100%). MS (m/z): 685/687/689 (M+l). Пример получения 17. ди-трет-Бутил-(1R,2R,3S,4S,5R,6R)-2-(трет-бутоксикарбониламино)-3-[(3,4 дихлорфенил)метокси]-4-(1 Н-имидазол-2-илсульфанил)бицикло[3.1.0]гексан-2,6-дикарбоксилат Указанное соединение получали, по существу, как описано в примере получения 20 путем применения ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]4-[(метилсульфонил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилата. Очищали с помощью флэшхроматографии, элюируя 30-50% этилацетат:гексаны, с получением титульного соединения с 38% выходом. MS (m/z): 671/673/675 (M+1). Пример получения 18. Диэтил-(1S,2R,3R,4S,5R,6S)-4-[(2-ацетоксиацетил)амино]-2-(третбутоксикарбониламино)-3-[(3,4-дихлорфенил)метокси]бицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору диэтил-(1S,2R,3R,4S,5R,6S)-4-амино-2-(трет-бутоксикарбониламино)-3-[(3,4 дихлорфенил)метокси]бицикло[3.1.0]гексан-2,6-дикарбоксилата (0,47 г, 0,89 ммоль) и диизопропилэтиламина (0,32 мл, 1,83 ммоль) в дихлорметане (9 мл) при комнатной температуре добавляли ацетоксиацетилхлорид (0,116 мл, 1,08 ммоль). Через 2,5 ч раствор промывали насыщенным NaHCO3 (2), солевым раствором (1), сушили над MgSO4, фильтровали и конц. при пониженном давлении. Очищали с помощью флэш-хроматографии, элюируя градиентом от 0 до 100% этилацетата в гексанах в течение 30 мин, с получением титульного соединения в виде белого твердого вещества (0,43 г, 0,68 ммоль, 76%). MS (m/z): 531 (М+Н-ВОС). Пример получения 19. (1S,2R,3R,4S,5R,6S)-2-(трет-бутоксикарбониламино)-3-[(3,4-дихлорфенил) метокси]-4-[(2-гидроксиацетил)амино]бицикло[3.1.0]гексан-2,6-дикарбоновая кислота К раствору диэтил-(1S,2R,3R,4S,5R,6S)-4-[(2-ацетоксиацетил)амино]-2-(трет-бутоксикарбониламино)-3 -[(3,4-дихлорфенил)метокси]бицикло[3.1.0]гексан-2,6-дикарбоксилата (0,42 г, 0,67 ммоль) в тетрагидрофуране (7,0 мл) при комнатной температуре добавляли 1 М водный гидроксид лития (4,0 мл, 4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь подкисляли до приблизительно рН 1 с помощью 5N водной HCl. Экстрагировали этилацетатом (325 мл). Объединенные органические слои сушили, фильтровали и конц. при пониженном давлении. HPLC анализ неочищенной реакционной смеси показал присутствие исходного вещества. Добавляли 1 М водный гидроксид лития (4,0 мл, 4,0 ммоль) к раствору неочищенной реакционной смеси в тетрагидрофуране (7,0 мл) и нагревали при 50 С. Через 17 ч реакционную смесь подкисляли до приблизительно рН 1 с помощью 5N водной HCl. Экстрагировали этилацетатом (325 мл). Объединенные экстракты сушили, фильтровали и конц. при пониженном давлении. Очищали с помощью препаративной обращенно-фазовой высокоэффективной жидкостной хроматографии, с применением градиента от 95% вода (с 0,03% HCl)/5% ацетонитрил до 5% вода (с 0,03%HCl)/95% ацетонитрил в течение 40 мин, с получением (1S,2R,3R,4S,5R,6S)-2-(третбутоксикарбониламино)-3-[(3,4-дихлорфенил)метокси]-4-[(2-гидроксиацетил)амино]бицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,181 г, 0,34 ммоль, 51%) в виде белого твердого вещества. MS (m/z): 531(1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновую кислоту (2 г, 5,32 ммоль) растворяли в диоксане (10,63 мл), затем обрабатывали насыщенным водным бикарбонатом натрия (10,63 мл, 6,90 ммоль) и ди-трет-бутилдикарбонатом (5,80 г, 26,58 ммоль). Двухфазную смесь перемешивали при комнатной температуре в течение 18 дней, затем концентрировали для удаления растворителя. Полученный остаток растворяли в ацетонитриле (с применением 1N HCl) и экстрагировали дважды для удаления ди-трет-бутилдикарбоната. Слой ацетонитрила концентрировали. Полученный остаток растворяли в этилацетате, затем дважды промывали 1N HCl, однократно промывали солевым раствором, сушили над сульфатом натрия и концентрировали досуха, с получением титульного соединения (2,1 г, 4,41 ммоль, 83%). MS (m/z): 474 (M-1). Пример получения 21. (1S,2R,3R,4S,5R,6S)-4-(ацетиламино)-2-[(трет-бутоксикарбонил)амино]-3[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоновая кислота(1S,2R,3R,4S,5R,6S)-4-(ацетиламино)-2-амино-3-[(3,4-дихлорбензил)окси]бицикло[3.1.0]гексан-2,6 дикарбоновой кислоты, с получением титульного соединения с 74% увыходом. MS (m/z): 515/517 (M-1). Пример получения 22. бис([(1-Метилэтокси)карбонил]оксиметил)(1S,2R,3S,4S,5R,6R)-2-[(третбутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат карбонат цезия (2,84 г, 8,72 ммоль). Смесь перемешивали в течение ночи при комнатной температуре в атмосфере азота. Реакционную смесь разбавляли этилацетатом и водой. Слои разделяли. Водный слой дважды экстрагировали этилацетатом. Объединенные органические слои экстрагировали солевым раствором, сушили над сульфатом магния, фильтровали и концентрировали. Очищали остаток с помощью флэш-хроматографии, элюируя от 0 до 75% этилацетатом в гексане, с получением титульного соединения в виде пены (940 мг, 1,33 ммоль, 33%). MS (m/z): 732 (M+Na). Соединения согласно примерам получения 23-26 можно получить, по существу, как описано в примере получения 22. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (5,30 г, 9,01 ммоль) добавляли к воде (30,02 мл) и уксусной кислоте (30,02 мл). Реакционную смесь нагревали с обратным холодильником до завершения расходования исходного вещества. Охлаждали и концентрировали на роторном испарителе. Концентрат растирали с диэтиловым эфиром, осадок собирали, затем сушили в вакуумной печи с получением титульного соединения (3,30 г, 8,77 ммоль, 97%). MS (m/z): 376/378 (M+l). Соединения согласно примерам 2-3 можно получить, по существу, как описано в примере 1. ди-трет-Бутил-(1S,2R,3R,4S,5R,6S)-4-(ацетиламино)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4 дихлорбензил)окси]бицикло[3.1.0]гексан-2,6-дикарбоксилат (2,40 г, 3,81 ммоль) добавляли к уксусной кислоте (7,99 г, 7,62 мл, 133,05 ммоль) и воде (3,81 мл, 3,81 г, 211,60 ммоль) и нагревали в микроволновом реакторе до 140 С. Через 15 мин охлаждали до комнатной температуры, разбавляли водой и фильтровали для сбора твердых веществ. Твердые вещества последовательно промывали водой (250 мл) и диэтиловым эфиром (250 мл) и затем сушили в вакуумной печи с получением титульного соединения Уксусную кислоту (2,0 мл) и воду (2,0 мл) добавляли к ди-трет-бутил-(1R,2R,3S,4S,5R,6R)-2-(третбутоксикарбониламино)-3-[(3,4-дихлорфенил)метокси]-4-[(2-метил-1,2,4-триазол-3-ил)сульфанил]бицикло[3.1.0]гексан-2,6-дикарбоксилату (0,305 г, 0,445 ммоль) в сосуде для микроволнового реактора и герметизировали и смесь нагревали до 140 С в течение 25 мин. Смесь охлаждали и концентрировали досуха и затем добавляли 5N соляную кислоту (2 мл), ацетонитрил (2 мл) и переносили в виалу. Лиофилизировали в течение ночи с получением титульного соединения в виде белого твердого вещества (0,227 г, 0,445 ммоль; 95%). MS (m/z): 473/475/477 (M+1). Соединения согласно примерам 7-8 можно получить, по существу, как описано в примере 6. К смеси (1S,2R,3R,4S,5R,6S)-2-(трет-бутоксикарбониламино)-3-[(3,4-дихлорфенил)метокси]-4-[(2 гидроксиацетил)амино]бицикло[3.1.0]гексан-2,6-дикарбоновая кислота (0,032 г, 0,06 ммоль) в воде (0,3 мл) добавляли 4N хлороводород (3,0 мл, 12,0 ммоль) в 1,4-диоксане. Твердое вещество промывали ацетонитрилом и диэтиловым эфиром на пористой фильтровальной воронке. Последовательно промывали твердое вещество ацетонитрилом и диэтиловым эфиром и затем дополнительно сушили в вакуумной печи в течение ночи при 50 С с получением титульного соединения в виде белого твердого вещества (0,018 г, 0,04 ммоль, 65%). MS (m/z): 433 (М+Н). Пример 10. Гидрохлорид бис([(1-метилэтокси)карбонил]оксиметил)(1S,2R,3S,4S,5R,6R)-2-амино- 17022196 Добавляли бис([(1-метилэтокси)карбонил]оксиметил)(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат в 4,0 М растворе хлороводорода в диоксане (14,47 мл, 57,87 ммоль) и перемешивали в течение 45 мин. Добавляли этилацетат и концентрировали реакционную смесь до образования пены. Пену растирали с этилацетатом с получением тврдого вещества. Концентрировали и сушили в вакууме при 40 С в течение ночи с получением титульного соединения в виде беловатого твердого вещества (1,77 г, 2,75 ммоль, 95%). MS[3.1.0]гексан-2,6-дикарбоксилата (1000 г, 2,43 моль) в сухом тетрагидрофуране (10 л) по каплям при 0 С в атмосфере азота добавляли 1 М три-(втор-бутил)борогидрида лития в тетрагидрофуране (2,67 л, 2,67 моль). Через 2 ч при 0 С добавляли 2 М Na2CO3 в воде (850 г, 8,02 моль) при 0 С в течение 2 ч с последующим добавлением 35% водной перекиси водорода (740 мл, 8,99 моль) в воде (3,3 л). Через 40 мин смесь нагревали до комнатной температуры, после чего органическую фазу отделяли. Органическую фазу разбавляли метил-трет-бутиловым эфиром (3,5 л) и слои разделяли снова. Водную фазу экстрагировали метил-трет-бутиловым эфиром (3,5 л). Все объединенные органические фазы последовательно промывали 1 М водным Na2SO3 (2,67 л), водой (2,67 л) и солевым раствором (2,67 л), сушили над сульфатом натрия, фильтровали и концентрировали до объема 3 л. Осажденное твердое вещество фильтровали и промывали гептаном (1,3 л) с получением титульного соединения (864,76 г). Маточные растворы концентрировали досуха и остаток растирали гептаном (500 мл) и фильтровали с получением дополнительного титульного соединения (130,64 г, всего 995,4 г, выход 99%). MS (m/z): 436 (М+23). Стадия 2. ди-трет-Бутил-(1S,2S,5R,6S)-2-[(трет-бутоксикарбонил)амино]бицикло[3.1.0]гекс-3-ен 2,6-дикарбоксилат[3.1.0]гексан-2,6-дикарбоксилата (995,4 г, 2,41 моль) в сухом тетрагидрофуране (8,4 л) добавляли триэтиламин (735 мл, 5,30 моль) по каплям при 0 С в атмосфере N2 в течение 40 мин. Метансульфохлорид (373 мл, 4,82 моль) добавляли при 0 С в течение 50 мин. Реакционную смесь нагревали до 23 С. Через 3 ч реакционную смесь фильтровали для удаления гидрохлоридной соли триэтиламина. Фильтрационный осадок промывали сухим тетрагидрофураном (22 л). Фильтрат (13 л) разделяли на две равные части. К одной половине фильтрата добавляли по каплям 1 М трет-бутоксид калия в тетрагидрофуране (3,6 л, 3,6 моль), к раствору, полученному на предыдущей стадии (неочищенный V=6,5 л, 1,2 моль) при 15 С в течение 2 ч 15 мин, затем оставляли нагреваться до 23 С. Через 2 ч 30 мин реакционную смесь гасили путем выливания в 10% водную уксусную кислоту (950 мл). Слои разделяли и водный слой экстрагировали этилацетатом (1,7 л). Органические слои объединяли и концентрировали в вакууме с получением твердого вещества, которое растворяли в ацетоне (1,45 л) и добавляли к воде (7,3 л). Полученную суспензию перемешивали в течение ночи при 23 С. Отфильтровывали осадок, промывали водой (21 л) и сушили в вакуумной печи при 40 С с получением титульного соединения (450,67 г, выход 95%). MS (m/z): 418(600,00 г, 1,52 моль) в ацетоне (12,00 л) и воде (1,80 л). Смесь перемешивали при 23 С в течение ночи. Анализ тонкослойной хроматографии (гексан/этилацетат 3:2; пятно: РМА) реакционной смеси показал завершение расходования ди-трет-бутил-(1S,2S,5R,6S)-2-[(трет-бутоксикарбонил)амино]бицикло[3.1.0]гекс-3-ен-2,6-дикарбоксилата. Реакционную смесь гасили насыщенным водным Na2S2O3 (1 л) и экстрагировали метил-трет-бутиловым эфиром (15 л). Органическую фазу промывали водным растворомMgSO4, фильтровали и концентрировали в вакууме. Остаток суспендировали в гептане (1,5 л), фильтровали, промывали гептаном (2500 мл) и получали твердое вещество. Твердое вещество сушили в вакууме с получением титульного соединения (450 г, 69% выход). Стадия 4. ди-трет-Бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3,4-дигидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата (450 г, 1,05 моль) в дихлорметане (4,50 л) при комнатной температуре последовательно добавляли 3,4-дихлорбензилбромид (144,73 мл, 238,80 г, 995,32 ммоль), хлорид тетра-н-бутиламмония (58,24 г, 209,54 ммоль) и 50% мас./мас. водный раствор гидроксида натрия (829,80 мл, 15,72 моль). Через 7 ч реакционную смесь разбавляли водой (1 л) и разделяли фазы. Органическую фазу экстрагировали солевым раствором (500 мл), сушили над безводным MgSO4, фильтровали и концентрировали. Остаток очищали с помощью хроматографии (2,5 кг SiO2; элюент: гексан/этилацетат от 12:1 до 0:1) с получением титульного соединения (539 г, 72% выход). Стадия 5. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-[(4-нитрофенил)карбонил]оксибицикло[3.1.0]гексан-2,6-дикарбоксилат К раствору ди-трет-бутил-(1S,2R,3S,4R,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата (539,00 г, 751,00 ммоль) в тетрагидрофуране (6,76 л) добавляли трифенилфосфин (393,96 г, 1,50 моль) и 4-нитробензойную кислоту (251,02 г,1,50 моль). Смесь охлаждали до 10 С, после чего по каплям добавляли диизопропилазодикарбоксилат(40% об/об в толуоле, 744,41 мл, 759,30 г, 1,50 моль) в течение 15 мин и оставляли нагреваться до 23 С. Через 18 ч смесь разбавляли метил-трет-бутиловым эфиром (2 л), последовательно промывали вод. нас.NaHCO3 (22 л) и солевым раствором (1 л) и концентрировали в вакууме. Остаток суспендировали в гептане (5 л) при 40 С в течение 30 мин, затем охлаждали до 23 С. Полученное твердое вещество фильтровали и промывали последовательно гептаном (2500 мл), гептан/метил-трет-бутиловый эфир (31 л). Объединенные фильтраты концентрировали в вакууме и остаток очищали с помощью хроматографии (2,5 кг SiO2; элюент: гексан/этилацетат от 95:5 до 75:25), с последующей перекристаллизацией в гептане (10 л/кг), с получением титульного соединения в виде белого твердого вещества (273 г, 49% выход). Стадия 6. ди-трет-Бутил-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат[3.1.0]гексан-2,6-дикарбоксилата (273,00 г, 370,11 ммоль) в метаноле (3,28 л) при комнатной температуре. Через 16 ч реакционную смесь концентрировали в вакууме. Остаток растворяли в метил-третбутиловом эфире (2 л) и затем промывали водой (0,8 л), солевым раствором (0,8 л), сушили над MgSO4 и концентрировали в вакууме, с получением титульного соединения в виде белого твердого вещества (215 г, 98% выход). Стадия 7. Гидрохлорид (1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Смесь воды (430 мл) и 37% HCl в воде (299.9 мл, 3.65 моль) добавляли к суспензии ди-трет-бутил(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилата (215 г, 365,3 ммоль) в 1,4-диоксане (73,1 мл). Полученную суспензию перемешивали при 100 С в течение 4,5 ч, охлаждали до 25 С и концентрировали в вакууме с получением белого твердого вещества. Последовательно растирали с метил-трет-бутиловым эфиром (2 л) и гексанами (2 л) и затем фильтровали для сбора твердых веществ. Сушили в вакууме в течение 16 ч. Твердое вещество снова промывали метил-трет-бутиловым эфиром (21 л) и гексаном (1 л), затем сушили в вакууме при 50 С в течение ночи с получением титульного соединения в виде белого твердого вещества (140,4 г, 93% выход). MS (m/z): 376/378 (M+1). Стадия 8. (1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоновая кислота(1S,2R,3S,4S,5R,6R)-2-амино-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (140 г, 339 ммоль) в 1,4-диоксане (203,56 мл) и воды (0,6 мл/моль-чист.-LR, 203,56 мл),при комнатной температуре. Через 4,5 ч к реакционной смеси добавляли дополнительные 2-(трет- 21022196 бутоксикарбонилоксиимино)-2-фенилацетонитрил (25,07 г, 101,78 ммоль) и триэтиламин (23,64 мл, 17,17 г, 169,63 ммоль) и перемешивали при 50 С в течение ночи. Охлаждали до 23 С, разбавляли водой (500 мл) и промывали метил-трет-бутиловым эфиром (6500 мл). Водную фазу разбавляли 10% вод. HCl (300 мл; конеч. рН 2) и экстрагировали этилацетатом (2500 мл). Этилацетатные экстракты сушили надMgSO4 и концентрировали в вакууме с получением титульного соединения (148,3 г, 92% выход). Стадия 9. бис-([(l-Метилэтокси)карбонил]оксиметил)-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат(148,30 г, 311,35 ммоль) в диметилформамиде (2,49 л) при 23 С последовательно добавляли карбонат калия (172,12 г, 1,25 моль), хлорметил изопропил карбонат (118,03 мл; 135,73 г, 871,79 ммоль) и иодид натрия (9,33 г, 62,27 ммоль). Перемешивали в течение 18 ч. Разбавляли метил-трет-бутиловым эфиром (2 л) и водой (2 л). Разделяли слои и последовательно промывали органическую фазу водой (200 мл) и солевым раствором (200 мл). Сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью хроматографии (2,5 кг SiO2; элюент: гексан/этилацетат от 3:1 до 1:1), с получением титульного соединения (167 г, 76% выход). MS (m/z): 608/610 (М-100 (-CO2tBu. Стадия 10. бис-([(1-Метилэтокси)карбонил]оксиметил)-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат бис-([(1-Метилэтокси)карбонил]оксиметил)-(1S,2R,3S,4S,5R,6R)-2-[(трет-бутоксикарбонил)амино]-3-[(3,4-дихлорбензил)окси]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоксилат (165,50 г, 233,58 ммоль) обрабатывали 4 М HCl в 1,4-диоксане (1,16 л, 4,63 моль) при 23 С и перемешивали в течение 2 ч. Летучие вещества удаляли в вакууме. Полученный остаток суспендировали в метил-трет-бутиловом эфире (1200 мл) в течение ночи. Полученное твердое вещество фильтровали, промывали метил-третбутиловым эфиром (2100 мл) и сушили в вакууме при 1 мбар/45 С в течение 6 часов с получением титульного соединения в виде белого твердого вещества (134 г, 89% выход). MS (m/z): 608/610 (M+1). Литературные данные (Witkin, Jeffrey M., и Eiler, William J.A. (2006), Antagonism of MetabotropicDevelopment Research, т. 67, с. 757-769; и Yasuhara, Akito и Chaki, Shigeyuki, (2010) Metabotropic Glutamate Receptors: Potential Drug Targets for Psychiatric Disorders, The Open Medicinal Chemistry Journal, т. 4,с. 20-36) и данные, полученные в неклинических исследованиях на животных, подтверждают роль антагонистов mGlu2/3 в лечении депрессивных расстройств и расстройств чрезмерной сонливости. В частности, с помощью грызунов, контролируемых ЭЭГ, было обнаружено, что антагонисты рецепторов mGlu 2/3 эффективны на моделях депрессивных расстройств у грызунов и стимулируют бодрствование без проявления непропорциональной или клинически значимой гиперактивности или чрезмерной компенсационной гиперсомнии. Повышенная концентрация внимания свидетельствует о повышенной внимательности, улучшенной когнитивной функции и вероятно уменьшенной усталости. Поскольку описанные ранее расстройства представляют собой распространнные сопутствующие клинические состояния, антагонисты mGlu2/3 рецептора могут быть особенно эффективны у конкретных групп пациентов, таких как пациенты с большим депрессивным расстройством, монополярной депрессией, дистимией и/или циклотимией или любым расстройством чрезмерной сонливости. Расстройства чрезмерной сонливости могут включать, но не ограничиваются ими, чрезмерную дневную сонливость (EDS), гиперсомнию, связанную с синдромом обструктивного апноэ во сне или нарколепсией, расстройства циркадного ритма сна (включая, но не ограничиваясь ими, расстройство сна, вызванное сменным режимом работы, расстройство сна,- 22022196 вызванное сменой часового пояса (расстройство джетлаг), расстройство сна, вызванное задержкой фазы сна, расстройство сна, вызванное ранним наступлением фазы сна и синдром не-24-часового цикла сонбодрствование), идиопатическую гиперсомнию и/или чрезмерную сонливость, связанную с невосстанавливающим сном (NRS). Для демонстрации характеристик настоящих соединений, типичные соединения могут быть протестированы в следующих in vitro и in vivo анализах. Анализы цАМФ антагонистов mGlu2 и mGlu3 рецепторов. Антагонистическую активность анализировали на рекомбинантных клетках AV12, стабильно экспрессирующих человеческие mGlu2 или mGlu3 рецепторы и крысиный глутаматный транспортер ЕААТ 1(транспортер возбуждающих аминокислот 1 типа). Клеточную линию выдерживали путем культивирования в DMEM с высоким уровнем глюкозы и гидрохлорида пиридоксина, с добавлением 5% диализированной фетальной бычьей сыворотки (FBS), 1 мМ пирувата натрия, 1 мМ HEPES и 1 мМ L-глутамина; генетицин и гигромицин В применяли в качестве антибиотиков. Сливные культуры выращивали при 37 С в атмосфере, содержащей 6,5% СО 2, и пассажировали дважды в неделю. Клетки собирали с помощью 0,25% трипсина, суспендировали в замораживающей среде (FBS с 10% ДМСО) в концентрации 107 клеткок/мл и аликвоты хранили в жидком азоте. За 24 ч до анализа клетки высеивали с плотностью 8,000-10,000 клеток на лунку в 96-луночные черные планшеты для культуры тканей с половинным объемом лунок (Costar 3875) в 50 мкл DMEM с высоким уровнем глюкозы и гидрохлорид пиридоксина, с добавлением 5% диализированой FBS, 1 мМ пирувата натрия, 1 мМ HEPES, 100 мкг/мл ампициллина и 250 мкМ (mGlu2) или 125 мкМ (mGlu3) L-глутамина. Обращение ингибирования выработки цАМФ, стимулированной форсколином, с помощью исследуемых соединений измеряли с применением технологии гомогенных исследований флуоресценции с разрешением по времени (HTRF; Cisbio cat62 АМ 4 РЕВ). Среду удаляли и клетки инкубировали со 100 мкл цАМФ стимулирующего буфера (STIM) в течение 30 мин при 37 С. (STIM буфер содержал 500 млHBSS (сбалансированный солевой раствор Хенкса), 1000 мл DPBS (фосфатно-солевой буфер Дульбекко),0,034% БСА, 1,67 мМ HEPES и 500 мкМ IBMX (изобутилметилксантин) (Sigma I5879. Соединения исследовали с помощью 10-точечной кривой концентрация-эффект с применением 3 последовательных разведений, с последующим 40-кратным разбавлением в STIM буфере. DCG IV (Tocris 0975) служит в качестве контрольного агониста. Конечная реакционная смесь содержала 1 мкМ (для mGlu2) или 3 мкМ(для mGlu3) форсколина (Sigma F6886), DCG IV при его ЕС 90 и до 25 мкМ исследуемого соединения. Клетки инкубировали при 37 С в течение 20 мин. Для измерения уровня цАМФ конъюгат цАМФ-d2 и конъюгат анти-цАМФ-криптата в лизисном буфере инкубировали с обработанными клетками при комнатной температуре в течение 1 ч (mGlu2) или 1,5 ч (mGlu3). Сигнал HTRF детектировали с применением планшет-ридера EnVision (Perkin-Elmer), с вычислением отношения флуоресценции при 665-620 нМ. Необработанные данные преобразовывали в количество цАМФ (пикомоль/лунка) с применением стандартной кривой цАМФ, полученной для каждого эксперимента. Относительные значения IC50 вычисляли сверху вниз кривой концентрация-эффект с применением программного обеспечения для четырехпараметрического логистического построения кривой (ActivityBase v5.3.1.22).FLIPR и цАМФ анализы на селективность к mGlu рецептору. Относительную антагонистическую активность соединений согласно изобретению в отношении других человеческих mGlu рецепторов можно оценить с помощью либо цАМФ анализа, либо флуорометрического аназиза кальциевого ответа (см., например, Fell et al., JPET (в печати. Вкратце, в указанных исследованиях применяли индивидуальные AV12 клеточные линии, содержащие крысиный глутаматный транспортер ЕААТ 1 и стабильно экспрессирующие человеческие mGlu1, 2, 3, 4, 5 и 8 рецепторы. Рецепторы mGlu1 и 5 являются Gq-сопряженными, поэтому они естественно передают сигнал на фосфолипазу С, приводя к выработке ответа в виде кальциевого потока, который можно применять для измерения активации рецептора с использованием флюориметрического планшетного ридера (FLIPR, Molecular Devices). Клеточные линии, экспрессирующие mGlu2, 3, 4 и 8 рецепторы, разработаны для экспрессирования субъединицы G15, чтобы указанные Gi-сопряженные рецепторы вырабатывали ответ в виде потока кальция, аналогично клеточным линиям, экспрессирующим рецепторы mGlu1 и 5. mGlu6 рецептор тестировали в цАМФ формате с использованием способов, аналогичных способам, разработанным для mGlu2 и mGlu3, приведенным выше. Указанные клеточные линии выдерживали так же, как описано выше, за исключением того, что количества L-глутамина и подборки агентов (генетицин, гигромицин В, зеоцин и бластицидин) могут зависеть от клеточной линии. Сливные культуры пассажировали дважды в неделю. Уровни внутриклеточного кальция контролировали с помощью FLIPR до и после добавления тестируемых соединений и красителей Fluo-3 AM (Invitrogen) или Calcium 4 (Molecular Devices) в зависимости от клеточной линии. Клетки высеивали в планшет за 24 ч до анализа в различных концентрациях глутамина и при различной плотности клеток на лунку в зависимости от клеточной линии. Среду удаляли и клетки инкубировали с 8 мкМ красителя (50 мкл на лунку) в течение 90 или 120 мин (в зависимости от клеточной линии) при 25 С. FLIPR анализ с двумя добавлениями, генерирующий 11-точечную кри- 23022196 вую концентрация-эффект для агониста глутамата, проводили перед каждым экспериментом для подтверждения соответствующей восприимчивости клеток. Результаты анализировали с применениемGraphPad Prism v4.03 для расчета концентраций глутамата, необходимых для получения ЕС 90 (анализ антагониста) и ЕС 10 (анализ потенциирующего фактора) ответов. Соединения исследовали на каждом mGlu рецепторе в FLIPR анализе с двумя добавлениями с применением 10-точечного профиля концентрация-эффект, начиная с конечной концентрации 25 мкМ для анализа агониста и 12,5 мкМ для анализов потенциирующего фактора и антагониста. В первом добавлении выявляли какую-либо агонистическую активность и во втором добавлении, состоящем из 100 мкл выбранных концентраций (в зависимости от клеточной линии) глутамата в буфере для анализа, получали ЕС 10 или ЕС 90 глутаматный ответ. Агонистические эффекты агониста количественно определяли как процентное отношение стимуляции, вызванной только соединением к максимальному глутаматному ответу. Антагонистические эффекты количественно определяли путем расчета процентного отношения ингибирования ЕС 90 глутаматного ответа, вызванного соединением. Потенциирующие эффекты количественно определяли как процентное отношение увеличения при наличии ЕС 10 ответа в глутамате и ECmax ответа. Все данные рассчитывали в виде относительных значений IC50 или EC50 с применением программного обеспечения для четырехпараметрического логистического построения кривой (ActivityBasev5.3.1.22). Антагонистическую активность в mGlu6 клетках измеряли с использованием цАМФ согласно способу, аналогичному описанному выше для mGlu2 и mGlu3 активности, за исключением того, что контрольным агонистом был L-AP4 (Tocris). Для измерения mGlu6 агонистической активности рассчитывают степень, в которой соединение ингибирует выработку цАМФ, стимулированную форсколином. Относительные значения IC50 и EC50 вычисляли сверху вниз кривой концентрация-эффект с применением программного обеспечения для четырехпараметрического логистического построения кривой (ActivityBasev5.3.1.22). Соединения, приведенные в примерах, в которых оба R1 и R2 представляют собой водород, тестировали, по существу, как описано выше, и обнаружили, что они обладают высокой антагонистической активностью в отношении mGlu2 и mGlu3 рецепторов. Также обнаружили, что соединения, приведенные в примерах, в которых оба R1 и R2 представляют собой водород, являются селективными антагонистамиmGlu2 и mGlu3 рецепторов в отличие от других подтипов mGlu рецепторов и обладают значенями IC50 в отношении mGlu2 и mGlu3 рецепторов менее 70 нМ и 50 нМ соответственно, в то время как значенияIC50 в отношении других тестируемых mGlu рецепторов были значительно выше (см. табл. 1).ND = не определено Кроме того, некоторые соединения по настоящему изобретению показали отсутствие существенной активности в отношении других физиологически значимых рецепторов, таких как, но не ограничиваясь ими, hERG канал, серотониновые рецепторы (особенно 5-НТ 2 А и 5-HT2B), мускариновые рецепторы (особенно М 2) и iGluR рецепторы (особенно iGluR5). Соединение согласно примеру 1 тестировали с использованием известных способов анализа и обнаружили, что оно не обладает заметной активностью в отношении указанных рецепторов. Следовательно, полагают, что физиологически значимые дозы соединений согласно изобретению обеспечивают существенное ингибирование mGlu2 и mGlu3 рецепторов in vivo, при этом, по существу,не взаимодействуя с другими mGlu рецепторами или другими физиологически значимыми рецепторами,и, таким образом, обеспечивают требуемую фармакологию, предотвращая возникновение нежелательных побочных эффектов, связанных с нецелевой активностью. Тест вынужденного плавания у мышей (mFST).mFST является доказанным in vivo анализом на антидепрессивную активность (Li et al., J.Pharmacol. Exp. Ther. 319(l):254-9, 2006). Мыши, обработанные известными клинически эффективными антидепрессантами (селективными ингибиторами обратного захвата серотонина и/или трициклическими антидепрессантами), демонстрировали поведение, выраженное в уменьшении времени неподвижности после помещения в сосуд с водой, поведение, связанное с отчаяньем. mFST применяли для определения потенциальной антидепрессивной активности новых mGlu2/3 антагонистов, в частности так, как описано в ранее опубликованных способах (см., например, Li et al., J. Pharmacol. Exp. Ther. 319(l):254-9, 2006). Вкратце, применяли мышей NIH-Swiss мужского пола (Harlan Sprague-Dawley, Indianapolis, IN), весящих 25-30 г. Животных, размещенных в одной группе, изымали из вивария в исследовательскую зону в отдельные клетки и позволяли адаптироваться к новой среде в течение по меньшей мере 1 ч перед тестированием. Соединения, в которых оба R1 и R2 представляют собой водород, растворяли в воде с минимальным количеством NaOH, добавленным для растворения, и вводили и.п. Соединения, в которых R1 и/илиR2 отличны от водорода, получали в день использования в 2,0-2,5% N-метилпирролидиноне и затем суспендировали в 1% НЕС, 0,25% Tween 80 и 0,05% Dow antifoam и вводили перорально. Мышей помещали в цилиндр (диаметр 10 см; высота 25 см), наполненный 6 см воды (22-25 С), на 6 мин. Подсчитывали продолжительность неподвижности в течение последних 4 мин из 6 мин периода исследования. Мышь записывали как неподвижную, если отсутствовали плавательные движения или совершались только те движения, которые необходимы для поддержания головы над водой. Типичные соединения тестировали, по существу, как описано выше, и обнаружили, что они значительно уменьшают время неподвижности у мышей дикого типа (см. табл. 2). Следовательно, полагают,что соединения согласно настоящему изобретению обладают антидепрессивной активностью in vivo. Таблица 2 Согласно другим исследованиям изучали мышей с делециями рецепторов (mGlu2 нокаутные мыши); указанных мышей разводили путем применения скрещивания "гетерозиготагетерозигота" и применяли в качестве однопомтников для -/- и +/+ мышиных контрольных групп (Taconic Farms). Обнаружили, что соединение согласно примеру 12 (10 мг/кг и.п., за 30 мин) значительно уменьшает время неподвижности у mGlu2+/+ мышей, но не у mGlu2-/- мышей. Также обнаружили, что соединение согласно примеру 10/15 (30 мг/кг п.о., за 120 мин) уменьшает время неподвижности у mGlu2+/+ мышей, но не уmGlu2-/- мышей. Данные результаты также демонстрируют, что mGlu2 рецепторы участвуют в антидепрессивном действии соединений согласно изобретению. Соединения согласно изобретению могут также быть протестированы с другими соединениями,подходящими для лечения депрессивных расстройств, например СИОЗС, на их способность усиливать антидепрессивное действие по сравнению с каждым соединением в отдельности. Соединение согласно примеру 10 (17 мг/кг п.о.) исследовали в тесте вынужденного плавания у мышей в отдельности и в комбинации с флуоксетином (10 мг/кг и.п), и обнаружили значительное увеличение антидепрессивного действия относительно действия любого соединения по отдельности, как показано в табл. 3, приведенной ниже. Кроме того, исследование уровня активного дикислотного фрагмента соединения согласно примеру 10 (т.е. того же соединения, что и свободное основание согласно примеру 1) в мозгу и плазме, показало отсутствие увеличения степени воздействия, что подтверждает обнаружение того, что повышенная антидепрессивная активность обеспечивается не только благодаря увеличению воздействия соединения в центральной нервной системе. Контроль бодрствования и поведения у крыс. Типичные соединения согласно настоящему изобретению тестировали на крысах на способность увеличивать количество времени в состоянии бодрствования без нежелательных побочных эффектов,таких как БДГ-фаза, двигательная недостаточность в состоянии бодрствования (непропорциональная гипер- или гиполокомоция) и/или рикошетная гиперсомния. Тестируемых животных постоянно контролировали с помощью электроэнцефалограмм (ЭЭГ), электромиограмм (ЭМГ) и измерения общего времени бодрствования, рикошетной гиперсомнии и интенсивности двигательной активности в течение бодрствования. Способы проведения подобных исследований хорошо известны в данной области техники (см., например, способы, описанные в Edgar D.M., Seidel W.F. Modafinil induces wakefulness withoutusing electrophysiological recordings in rats. J. Neurosci Methods. 2009; 184(1): 10-8). Исследования проводили следующим образом. Подготовка животного. Взрослых крыс Wistar мужского пола (приблизительно 270-300 г на момент операции) хирургически подготавливали для постоянной регистрации ЭЭГ, ЭМГ, температуры тела и проводили следующие действия. Крыс хирургически снабжали черепным имплантатом, состоящим из четырех ввинчивающийся электродов из нержавеющей стали, для регистрации ЭЭГ (два фронтальных[3,9 мм перед теменем и 2,0 мм медиолатерально] и два затылочных [6,4 мм за теменем, 5,5 мм медиолатерально]) и двух проводов из нержавеющей стали, покрытых тефлоном, для регистрации ЭМГ (расположенных под затылочной трапециевидной мышцей). Перед операцией вся проводка была припаяна к миниатюрному разъему (Microtech, Boothwyn, РА). Устройство имплантата фиксировали к черепу с помощью комбинации ввинчивающийся электродов из нержавеющей стали для ЭЭГ, цианакрилата, нанесенного между разъемом имплантата и черепом, и стоматологического акрила. Температуру тела и двигательную активность контролировали посредством миниатюрного передатчика (Minimitter PDT4000 г,Philips Respironics, Bend или) хирургически помещенного брюшную полость. На восстановление отводили по меньшей мере 3 недели. Условия при регистрации. Каждую крысу размещали отдельно внутри микроизолирующей клетки,модифицированной вставленной поликарбонатной крышкой-фильтром, позволяющей увеличить вертикальный габарит. Гибкий кабель, минимально затрудняющий движения, подсоединяли одним концом к коммутатору, размещенному вверху клетки и другим концом к черепному имплантату животного. Каждая клетка размещена в отдельном вентилируемом отсеке камеры для регистрации сна-бодрствования из нержавеющей стали. Еда и вода были доступны ad libitum и комнатную температуру поддерживали примерно 231 С. В ходе исследования поддерживали 24-часовой цикл свет-темнота (LD 12:12) с помощью люминесцентного света. Относительная влажность составляла приблизительно 50%. Животных не тревожили в течение по меньшей мере 30 ч до и после каждой обработки. Дизайн клинического исследования и дозировки. Соединения, в которых оба R1 и R2 представляют собой водород, растворяли в воде с минимальным количеством NaOH, добавленного для растворения, и вводили и.п. в объеме 1,0 мл на 1 кг массы тела. Соединения, в которых R1 и/или R2 отличны от водорода, вводили п.о. в объеме 2 мл на 1 кг массы тела в одном из двух возможных носителях: i) 2,5% Nметил-2-пирролидинона в гидроксиэтилцеллюлозе или ii) 10% камедь с 0,05% Dow Corning Antifoam в воде. Носитель или одну из доз соединения вводили псевдослучайно, чтобы ни одна из крыс не получила одинаковую обработку дважды, и ни одна из крыс не получила более двух из 8 обработок в каком-либо исследовании. Каждую крысу забирали из ее клетки на примерно минуту для взвешивания и обработки. По меньшей мере 6-дневный "промывочный" период предшествовал и следовал после каждой обработки. Сбор данных. Распознавание сна и бодрствования может быть автоматизировано (например, Van(Х 10,000, полоса пропускания 1-30 Гц), ЭМГ усиливали и интегрировали (полоса пропускания 110-100 Гц, RMS интегрирование) и одновременно контролировали неспецифическую двигательную активность(LMA). Состояния возбуждения разделяли на 10-секундные периоды не-REM сна, REM сна, бодрствования или тета-доминирующего бодрствования. Двигательную активность (LMA) регистрировали в виде числа отсчтов в минуту и определяли с помощью коммерчески доступных телеметрических примников(ER4000, Minimitter, Bend, OR). Статистический анализ. Возраст и массу тела суммировали по среднему, минимуму и максимуму относительно экспериментальных групп. Все животные, обладающие по меньшей мере одним выходным данным, включали в обобщающие результаты (например, мы включали подходящие данные из экспериментальных групп животных, для которых были применимы телеметрические данные, но не применимы ЭЭГ данные). Период наблюдений после обработки разделяли на 2 постдозировочных интервала (первые 7 ч и первые 19 ч), при этом время дозирования определяли как начальное время = 0. Выходные данные объединяли по каждому периоду путем вычисления либо среднечасовых, либо суммарных значений за каждый период. Каждое выходное значение за каждый период анализировали посредством анализа ковариации с использованием экспериментальных групп и даты обработки в качестве факторов и соответствующий интервал перед обработкой, ранее 24 ч, в качестве ковариаты. Скорректированное среднее и изменение относительно среднего носителя, а также соответствующие среднеквадратические ошибки суммировали для каждой экспериментальной группы. Скорректированные Р-значения множественного сравнения Даннета показаны для каждого выходного значения за каждый период. Не все выходные значения анализировали за все периоды, как показано в табл. 1, что, таким образом, влияет на коэффициент ошибок первого рода с поправкой на эффект множественных сравнений. По сущетсву, для множественного тестирования не применяли дополнительных корректировок. Определение эффективности. Пороговую эффективную дозу оценивали как наименьшую дозу, при которой общее время бодрствования превышало на 50 мин контроль носителем за первые 7 ч после обработки. Более точное определение можно выполнить путем проведения последовательного изучения доз с меньшим шагом в районе эффективной дозы. Определение нежелательных эффектов. Оценивали два потенциальных нежелательных эффекта: рикошетную гиперсомнию и повышенную моторную активность (Edgar D.M., Seidel W.F., 1997).(i) Рикошетную гиперсомнию можно измерить как уменьшение степени бодрствования в течение 819 ч после обработки эффективной дозой. Биологически значимое понижение определяют как более чем 50%-ное общее снижение в течение первых 7 ч. Таким образом, если бодрость увеличилась на 100 мин в течение первых 7 ч, тогда уменьшение общей бодрости на 50 мин или более относительно контроля носителем в течение 8-19 ч после обработки будет рассматриваться как биологически значимое. Средние групповые изменения, показанные в табл. 2, подтверждают отсутствие рикошетной гиперсомнии.(ii) Повышенную моторную активность определяли как среднее увеличение относительно контроля носителем, превышающее 5 LMA отсчтов в минуту ЭЭГ-определенного бодрствования при эффетивной пороговой дозе, для которой эффект является дозозависимым. Все средние групповые увеличенния в табл. 2 были до 5 отсчтов в минуту бодрствования и не зависят от дозы. Соединения, приведенные в примерах, тестировали, по существу, как описано, и обнаружили, что они улучшают бодрствование без получения значительной рикошетной гиперсомнии или повышенной моторной активности. Соединения, приведенные в примерах, в которых оба R1 и R2 представляют собой водород (введенные и.п.), тестировали, по существу, как описано, и обнаружили, что они являются эффективными при дозировках 10 мг/кг или менее. Соединение примера 10 тестировали, по существу, как описано, и обнаружили, что оно обладает профилем общего времени бодрствования и интенсивностью двигательной активности, приведенными в табл. 4. Таблица 4 Общее время бодрствования в течение первых 7 ч Общее время бодрствования в течение 8-19 ч Интенсивность двигательной активности (note 1) Статистика выходных значений: средние значения определяют разницу относительно контроля носителем. СО = средняя квадратическая ошибка среднего; Р = Р-значение, скорректированное для множественного сравнения для переменной эффективности. Нескорректированные Р-значения показаны для измерений "нежелательного действия" (общее время бодрствования в течение 8-19 ч и интенсивность двигательной активности). Общее время бодрствования дано в минутах. Прим. 1. Интенсивность двигательной активности (LMA) = отсчт LMA в минуту ЭЭГ-определенного бодрствования, усредненного за 7 первых часов после обработки. Хотя соединения, используемые в способах согласно настоящему изобретению, можно вводить непосредственно без какого-либо состава, соединения, как правило, вводят в форме фармацевтических композиций, содержащих по меньшей мере одно соединение формулы I или его фармацевтически приемлемую соль в качестве активного ингредиента и по меньшей мере один фармацевтически приемлемый носитель, разбавитель и/или наполнитель. Указанные композиции можно вводить различными способами, в том числе пероральным, подкожным, внутривенным и внутримышечным способами. Указанные фармацевтические композиции и способы их получения хорошо известны в данной области техники. См,например, Remington: The Science и Practice of Pharmacy (University of the Sciences in Philadelphia, ed., 21sted., Lippincott WilliamsWilkins Co., 2005). Соединения формулы I, где R1 и/или R2 отличны от водорода, предпочтительны для перорального введения с целью повышения биодоступности, тогда как соединения формулы I, где оба R1 и R2 представляют собой водород, предпочтительны для в.в. или и.п. введения. Композиции предпочтительно получают в единичной лекарственной форме, дозировка которой содержит от примерно 1 до примерно 600 мг, чаще от примерно 30 до примерно 300 мг, например между примерно 50 и примерно 250 мг активного ингредиента. Термин "единичная лекарственная форма" относится к физически дискретным единицам, походящим в качестве единичных доз для человеческих субъектов и других млекопитающих, причем каждая единица содержит заданное количество активного вещества, вычисленного для получения желаемого терапевтического эффекта, в комбинации по меньшей мере с одним подходящим фармацевтически приемлемым носителем, разбавителем и/или наполнителем. Соединения формулы I, в целом, эффективны в широком диапазоне дозировок. Например, суточные дозы,как правило, подпадают в диапазон от примерно 0,01 до примерно 10 мг/кг, чаще от примерно 0,3 до 5,0 мг/кг и, например, между 0,5 и 3,0 мг/кг от массы тела. В некоторых случаях уровни дозы ниже границы указанного диапазона могут быть более чем адекватными, в то время как в других случаях можно применять большие дозы без получения каких-либо неблагоприятных побочных эффектов, и, следовательно,приведенный выше диапазон доз не следует понимать как ограничивающий каким-либо образом объем изобретения. Следует понимать, что количество конкретного вводимого соединения должно быть определено врачом с учетом значимых обстоятельств, включая состояние, подлежащее лечению, выбранный способ введения, конкретное вводимое соединение или соединения, возраст, вес и ответ отдельного пациента, и тяжести симптомов пациента. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы
МПК / Метки
МПК: A61P 25/24, A61K 31/21, C07C 229/50
Метки: качестве, 4-замещенного-3-бензилоксибицикло[3.1.0]гексана, соединения, mglur, антагонистов
Код ссылки
<a href="https://eas.patents.su/30-22196-soedineniya-4-zameshhennogo-3-benziloksibiciklo310geksana-v-kachestve-antagonistov-mglur-2-3.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 4-замещенного-3-бензилоксибицикло[3.1.0]гексана в качестве антагонистов mglur 2/3</a>
Соединения 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана в качестве антагонистов mglur 2/3

Номер патента: 21724
Опубликовано: 31.08.2015
Авторы: Ли Ренуа, Смит Стефон Корнелл, Ветман Татьяна Натали, Митч Чарльз Говард
МПК: C07C 323/58, A61K 31/10
Метки: 4-замещенного-3-фенилсульфанилметилбицикло[3.1.0]гексана, mglur, качестве, антагонистов, соединения
Формула / Реферат:
1. Соединение формулыгде каждый из R1 и R2 независимо представляет собой водород, C1-C3 алкоксикарбонилоксиметил, C1-C5 алкилкарбонилоксиметил или C3-6 циклоалкилкарбонилоксиметил;R3 независимо в каждом случае представляет собой метил, фтор или хлор;R4 представляет собой гидроксил, амино, метилкарбониламино или 1,2,4-триазолилтио иn представляет собой 1 или 2;или его фармацевтически приемлемая соль.2. Соединение по п.1, отличающееся тем, что...
Производные 3-азабицикло (3.1.0) гексана в качестве антагонистов опиоидного рецептора

Номер патента: 6708
Опубликовано: 24.02.2006
Авторы: Макхарди Стэнтон Ферст, Лирас Спирос, Хек Стивен Дональд
МПК: A61K 31/4178, A61K 31/403, C07D 209/52...
Метки: 3.1.0, производные, рецептора, гексана, антагонистов, 3-азабицикло, опиоидного, качестве
Формула / Реферат:
1. Соединение, описываемое формулой I, где X представляет собой H, галоген, -OH, -CN, -C1-C4алкил, замещенный атомами галогена в количестве от одного до трех, либо -O(C1-C4алкил), где C1-C4алкил в -O(C1-C4алкиле) необязательно замещен атомами галогена в количестве от одного до трех; Q представляет собой галоген, -OH, -O(C1-C4алкил), -NH2, -N(C1-C4алкил)(C1-C4алкил), -C(=O)NH2, -C(=O)NH(C1-C4алкил), C(=O)N(C1-C4алкил) (C1-C4алкил) или...
Бензамиды 4-(аминометил) пиперидина, замещённого гидроксикарбонилфенилом, в качестве антагонистов 5нт4-рецепторов

Номер патента: 9464
Опубликовано: 28.12.2007
Авторы: Гейсен Хенрикус Якобус Мария, Босман Жан-Поль Рене Мари Андре, Мевеллек Лоранс Анн
МПК: C07D 405/12, A61P 1/00
Метки: антагонистов, качестве, 4-(аминометил, замещённого, гидроксикарбонилфенилом, пиперидина, 5нт4-рецепторов, бензамиды
Формула / Реферат:
1. Бензамиды 4-(аминометил)пиперидина, замещенного гидроксикарбонилфенилом формулы (I) их стереоизомеры или фармацевтически приемлемые кислотно-аддитивные и основно-аддитивные соли, в которых -Rl-R2- является двухвалентным радикалом формулы R3 представляет водород, галоген, C1-6алкил или C1-6алкилоксигруппу; R4 представляет водород, галоген, C1-6алкил; R5 представляет водород или C1-6алкил, и радикал -OR5, расположенный в 3- или 4-положении...
Спиропиперидиновые соединения в качестве антагонистов рецептора orl-1

Номер патента: 20391
Опубликовано: 30.10.2014
Авторы: Хименес-Агуадо Альма Мария, Лафуэнте Бланко Селия, Бенито Колладо Ана Белен, Диас Буэсо Нурия, Толедо Эскрибано Мигель Анхель, Мартинес-Грау Мария Анхелес, Педрегал-Терсеро Консепсьон
МПК: C07D 495/20, A61K 31/438, A61P 25/00...
Метки: рецептора, спиропиперидиновые, orl-1, антагонистов, качестве, соединения
Формула / Реферат:
1. Соединение формулыгдеR1 представляет собой фтор или хлор;каждый из R2a и R2b представляет собой водород или фтор;R3 представляет собой водород, метил, гидроксиметил или (С1-С3)алкоксиметил;R4 выбран из группы, состоящей из фтора, хлора, циано, цианометила, (C1-С3)алкила, циклопропила, гидроксиметила, метокси, метоксиметила, аминокарбонилоксиметила, метиламинокарбонилоксиметила, диметиламинокарбонилоксиметила, метилкарбонила, аминокарбонила,...
Спиропиперидиновые соединения в качестве антагонистов рецептора orl-1

Номер патента: 20848
Опубликовано: 27.02.2015
Авторы: Толедо Эскрибано Мигель Анхель, Диас Буэсо Нурия, Мартинес-Грау Мария Анхелес, Бенито Колладо Ана Белен, Хименес-Агуадо Альма Мария, Педрегал-Терсеро Консепсьон, Лафуэнте Бланко Селия
МПК: A61K 31/444, A61P 25/00, C07D 495/20...
Метки: рецептора, orl-1, соединения, антагонистов, качестве, спиропиперидиновые
Формула / Реферат:
1. Соединение формулыгде R1 представляет собой фтор или хлор;каждый из R2a и R2b представляет собой водород или фтор;R3 представляет собой водород, метил, гидроксиметил или (C1-С3) алкоксиметил;R4 выбран из группы, состоящей из фтора, хлора, циано, цианометила, (С1-С3) алкила, циклопропила, гидроксиметила, метокси, циклопропилметокси, аминокарбонилметокси, (C1-С3) алкоксиметила, циклопропилоксиметила, циклопропилметоксиметила,...