Аналоги изоксазол-3(2н)-она в качестве терапевтических агентов
Номер патента: 20825
Опубликовано: 27.02.2015
Авторы: Бострём Йонас, Ченг Лейфенг, Петтерсен Даниель, Карле Михель, Шелль Петер, Фекс Томас






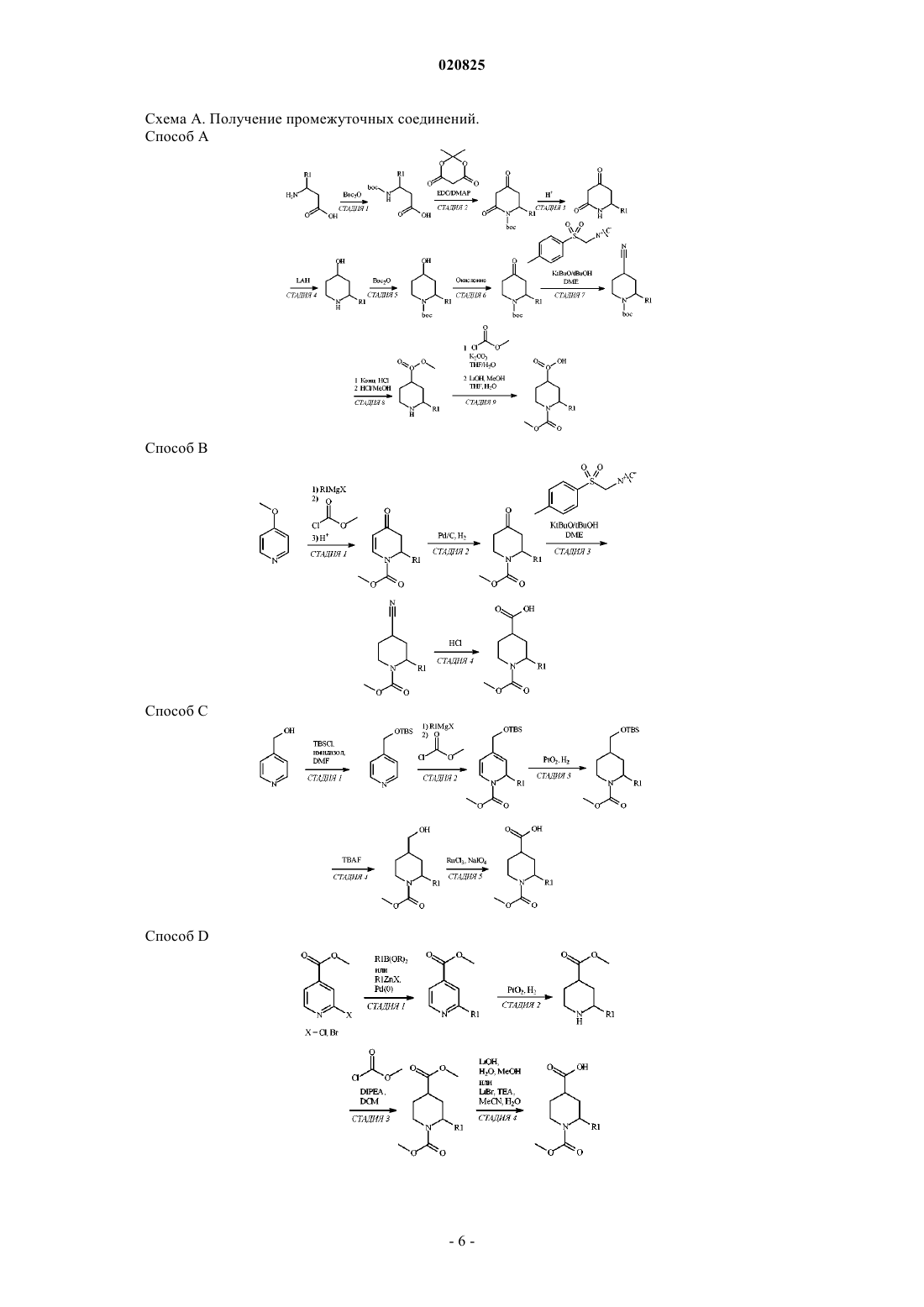
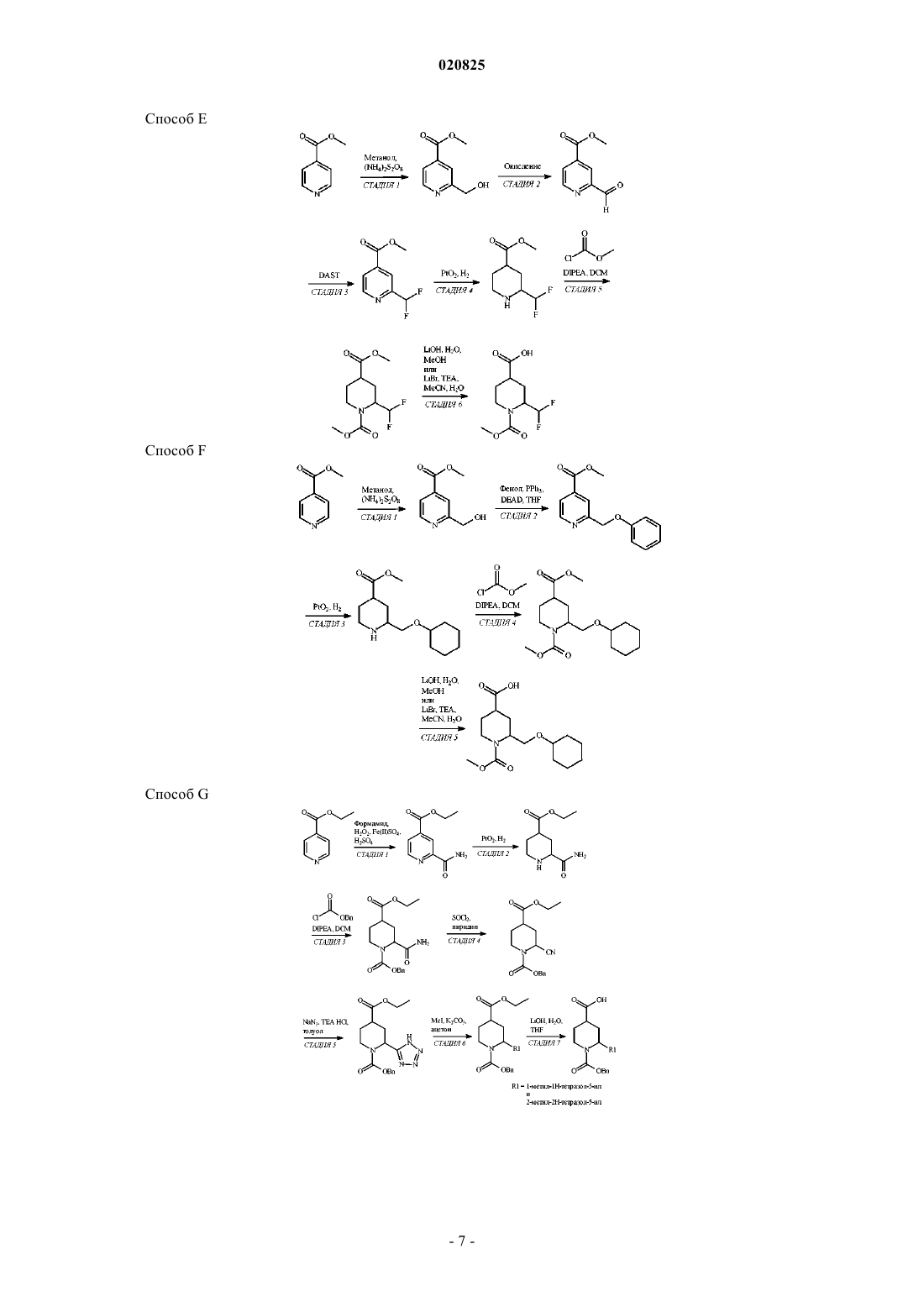
Формула / Реферат
1. Соединение формулы I

или его фармацевтически приемлемая соль,
где R1 представляет собой метил, 2-метил-2-фенилпропил, изобутил, трет-бутил, неопентил, 3,3-диметилбутил, циклогексил, циклогексилметил, где циклогексил, возможно, замещен 1 или 2 атомами фтора, циклогексилоксиметил, фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил, где указанные фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил, возможно, замещены 1, 2 или 3 заместителями, независимо выбранными из фтора, хлора, метила, трифторметила, трифторметокси, трет-бутила или метилсульфонила, и где атомы водорода в фениле, возможно, заменены дейтерием.
2. Соединение по п.1, выбранное из следующих соединений:
5-((2S,4S)-2-бензилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-бензилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-бензилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-бензилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-изобутилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-изобутилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-изобутилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-фенетилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-фенетилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-фенетилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(4-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-неопентилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-неопентилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-метилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-метилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-метилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(2-метил-2-фенилпропил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-метил-2-фенилпропил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(циклогексилметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(циклогексилметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(циклогексилметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,4-дифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,4-дифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-хлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(4-хлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-хлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-трет-бутилфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-трет-бутилфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-(метилсульфонил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((транс-2-(4-(метилсульфонил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(6-(трифторметил)пиридин-3-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(6-(трифторметил)пиридин-3-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(5-трет-бутилтиофен-2-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(5-трет-бутилтиофен-2-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,4-дифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-хлор-2-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-хлор-2-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-хлор-4-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2-хлор-4-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-хлор-3-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-хлор-3-фторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,4-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(2,4-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(2,4-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(2,4-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,5-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,5-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,5-дихлорфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,4,5-трифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,4,5-трифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,4,5-трифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,4,5-трифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(2,4,5-трифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-хлор-3,5-дифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-хлор-3,5-дифторфенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-метил-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-метил-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3-метил-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,5-дифтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,5-дифтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-метил-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-фтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2-фтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-фтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-фтор-4-(трифторметил)фенил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-фенилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-фенилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-циклогексилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-циклогексилпиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-метил-2Н-тетразол-5-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2-метил-2Н-тетразол-5-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(1-метил-1H-тетразол-5-ил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(циклогексилоксиметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(циклогексилоксиметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(циклогексилоксиметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(дифторметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(дифторметил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-((4,4-дифторциклогексил)метил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-((4,4-дифторциклогексил)метил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-((4,4-дифторциклогексил)метил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(4-фторфенетил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(4-фторфенетил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-фторфенетил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(3,3-диметилбутил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,3-диметилбутил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(4-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(3-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(2-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(2-фторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(2-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-(трифторметокси)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-(трифторметокси)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-хлорбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-хлорбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(4-(метилсульфонил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(4-(метилсульфонил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(4-(метилсульфонил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(3,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(3,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4S)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4R)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-(транс-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2R,4S)-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2Н)-он;
5-((2S,4R)-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2Н)-он и
5-(транс-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2Н)-он.
3. Применение соединения по любому из пп.1, 2 для лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной.
4. Применение по п.3, где указанное заболевание или состояние выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени.
5. Применение соединения по любому из пп.1, 2 в изготовлении лекарственного средства для лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной.
6. Применение по п.5, где указанное заболевание выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени.
Текст
или их фармацевтически приемлемой соли, где R1 представляет собой метил, 2 метил-2-фенилпропил, изобутил, трет-бутил, неопентил, 3,3-диметилбутил, циклогексил,циклогексилметил, где циклогексил, возможно, замещен 1 или 2 атомами фтора,циклогексилоксиметил, фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил,где указанные фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил, возможно,замещены 1, 2 или 3 заместителями, независимо выбранными из фтора, хлора, метила,трифторметила, трифторметокси, трет-бутила или метилсульфонила, и где атомы водорода в фениле, возможно, заменены дейтерием. Изобретение также относится к применению указанных соединений для лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной, предпочтительно выбранных из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени, а также к применению этих соединений для изготовления лекарственного средства для лечения или профилактики указанных заболеваний или состояний.(71)(73) Заявитель и патентовладелец: ЭМЕРИТИ ФАРМА АБ (SE) Область изобретения Настоящее изобретение относится к соединениям формулы I, к фармацевтической композиции, содержащей их, и к их применению в лечении заболеваний или состояний, связанных с фибринолизом,например наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени, и для лечения наследственного ангионевротического отека. Предшествующий уровень техники Кровотечение представляет собой распространенную клиническую проблему при травме, операции,расстройствах свертываемости крови, инсульте, меноррагии и заболеваниях печени. Лечение кровотечения включает агенты в первичном и вторичном гемостазе, а также ингибирование фибринолиза. Фибринолитическая система включает неактивный зимоген, плазминоген (PLG), который может быть активирован до активного фермента плазмина (PLN). PLN расщепляет нерастворимый фибрин на растворимые фибриновые фрагменты. Результатом этой активности является растворение фибринового сгустка. Активация плазминогена до плазмина происходит на поверхности сгустка после связывания с фибрином. Медиаторами активации являются урокиназный активатор плазминогена (u-РА) или активатор плазминогена тканевого типа (t-PA). Активация фибринолитического процесса может быть использована для лечения тромботического состояния. Наоборот, ингибирование фибринолиза может быть использовано и используется для лечения состояний кровотечения. Существует несколько возможных мишеней для ингибирования фибринолиза. Примерами являются активация ингибитора 1 активатора плазминогена (PAI-1), ингибирование активности u-РА и/или t-PA, ингибирование активности PLN и активация антиплазмина. Специфическое ингибирование протеолитических сайтов в tPA, uPA и PLN является трудным. Однако контроль кровотечения посредством ингибирования связывания плазмин(огена) фибрина аналогами лизина, как было доказано на людях, является безопасным и эффективным механизмом действия. Ингибирование фибринолиза с помощью лизинового миметика представляет собой подтвержденную концепцию уменьшения кровопотери без повышенного риска тромботических осложнений, например, после операции, при меноррагии, гемофилии и болезни Виллебранда. Потенциальные применения соединений состоят в блокировании плазмин-индуцированного протеолиза как универсального патологического механизма распространения рака и сердечно-сосудистых,воспалительных и многих других заболеваний. Антифибринолитические средства также были успешно использованы для лечения наследственного ангионевротического отека. При этом заболевании кожа или слизистая оболочка вокруг рта, горла и языка быстро набухают. Набухание может возникать и в других местах, таких как конечности или гениталии. По не очень понятным причинам антифибринолитические средства могут быть использованы для профилактики или срочного лечения наследственного ангионевротического отека. Транексамовая кислота, соединение, предложенное на рынке в настоящее время для лечения меноррагии, требует очень высоких и многократных доз и имеет желудочно-кишечные побочные эффекты. Ее применение было описано в "Tranexamis acid. A review of its use in surgery and other indications"; Dunn,C.J.; Goa K.L.; Drugs 1999, June 57 (6): 1005-1032. Задача изобретения Задача изобретения состоит в том, чтобы предложить фармацевтическое соединение для предупреждения или лечения чрезмерного кровотечения, лечебной или профилактической терапии, имеющей одно или более улучшений, таких как повышенная эффективность, селективность, проницаемость, длительность, меньшее количество побочных эффектов и улучшенная биодоступность. Описание изобретения В настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль,где R1 представляет собой метил, 2-метил-2-фенилпропил, изобутил, трет-бутил, неопентил, 3,3 диметилбутил, циклогексил, циклогексилметил, где циклогексил, возможно, замещен 1 или 2 атомами фтора, циклогексилоксиметил, фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил, где указанные фенил, бензил, фенетил, тиофенил, тетразолил или пиридинил, возможно, замещены 1, 2 или 3 заместителями, независимо выбранными из фтора, хлора, метила, трифторметила, трифторметокси, третбутила или метилсульфонила, и где атомы водорода в фениле, возможно, заменены дейтерием. Согласно другому аспекту изобретения предложено соединение, выбранное из следующих соеди-1 020825 5-2R,4S)-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2Н)-он; 5-(транс-2-(2,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-(транс-2-(2,6-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4S)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4R)-2-(3,5-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-(транс-2-(2,4-дифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2H)-он; 5-(транс-2-(3-фтор-5-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-(транс-2-(3-фтор-4-(трифторметил)бензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-(транс-2-(3,4,5-трифторбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-(транс-2-(3,5-ди-трет-бутилбензил)пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2R,4S)-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2 Н)-он; 5-2S,4R)-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2 Н)-он и 5-(транс-2-бензил-2,3,4,5,6-d5-пиперидин-4-ил)изоксазол-3(2 Н)-он. Согласно изобретению также предложено применение соединения, как оно охарактеризовано выше,для лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной. Предпочтительно указанное заболевание или состояние выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени. Также согласно изобретению предложено применение соединения, как оно определено выше, в изготовлении лекарственного средства для лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной. Предпочтительно указанное заболевание выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени. Соединения формулы I могут существовать в стереоизомерных и/или таутомерных формах. Следует понимать, что все энантиомеры, диастереомеры, рацематы, таутомеры и их смеси включены в область изобретения. Различные изомеры могут иметь различную биологическую активность. Возможно, что различные соединения общей формулы I могут демонстрировать самую высокую биологическую активность в другой конфигурации. Например, для одного соединения (2R,4S)-конфигурация может иметь самую высокую биологическую активность, а для другого соединения (2S,4S) может иметь высокую активность. Согласно первому аспекту изобретения предложено соединение формулы II со следующей конфигурацией: Согласно первому аспекту изобретения предложено соединение формулы III со следующей конфигурацией: Согласно первому аспекту изобретения предложено соединение формулы IV со следующей конфигурацией: Согласно первому аспекту изобретения предложено соединение формулы V со следующей конфигурацией: В изобретении также раскрыта фармацевтическая композиция, содержащая по меньшей мере одно соединение и фармацевтически приемлемый носитель или разбавитель. В изобретении также раскрыт способ лечения или профилактики заболевания или состояния, такого как тяжелое кровотечение, наследственные или приобретенные расстройства свертываемости крови, инсульт, меноррагия и заболевания печени, в котором модуляция фибринолиза является благоприятной, и для лечения наследственного ангионевротического отека, включающий введение теплокровному животному, нуждающемуся в таком лечении, терапевтически эффективного количества по меньшей мере одного соединения формулы I. Другие области применения соединения формулы I заключаются в ранозаживлении, уменьшении кровопотери в связи с операцией, включающем стоматологическую операцию, и для лечения пациентов, которые принимают антикоагулянты, например, при подготовке к операции. Соединение также может быть использовано для уменьшения гемотрансфузии, необходимой в различных ситуациях. Соединение также может быть использовано в профилактическом лечении болезни Виллебранда, гемофилии или в случае пациентов, принимающих антикоагулянты. Также в изобретении раскрыт способ, где указанное заболевание или состояние выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени. Изобретение касается лечения или профилактики заболевания или состояния, при котором модуляция фибринолиза является благоприятной, предпочтительно где указанное заболевание или состояние выбрано из наследственных расстройств свертываемости крови, инсульта, меноррагии и заболеваний печени. Соединения формул I-V могут быть получены следующим путем. Примечание. Стереохимические центры, отмеченные звездочками, следует считать определенными для относительных конфигураций обозначенных атомов углерода, а не их абсолютными конфигурациями, если специально не указано иное. Специалисту в данной области будет понятно, что вышеописанные способы не являются единственными способами получения соединений по изобретению и что специалист в данной области может менять порядок последовательностей, условий реакций и другие параметры соответствующим образом. Примеры параметров, которые могут варьироваться, представляют собой использование других растворителей, кислот и оснований и температуры, а также для защиты аминной, гидроксильной или других потенциально реакционноспособных групп. Подходящие защитные группы и детали способов добавления и удаления таких групп, в общем, хорошо известны в данной области; см., например, "Protectivegroups in Organic Synthesis", 3rd edition (1999) by Greene and Wuts. Разделение энантиомеров может быть осуществлено на любой стадии с использованием, например,разделения на колонке, перекристаллизации с использованием хиральной кислоты или основания. Разделение также может быть осуществлено посредством ферментативно селективной реакции. Альтернативно, энантиомеры могут быть стереоспецифически синтезированы. Для применений, способов, лекарственных средств и композиций, упомянутых в данной заявке, используемое количество соединения формул I-V, или его фармацевтически приемлемых солей, или их смесей, и вводимая дозировка могут различаться в зависимости от используемого соединения формул IV, или фармацевтически приемлемых солей, или их смесей; и/или от желаемого способа введения, и/или лечения. Однако, в общем, удовлетворительные результаты получают в том случае, когда соединение в соответствии с формулами I-V, или фармацевтически приемлемые соли, их или смеси вводят в суточной дозировке от примерно 0,1 мг до примерно 400 мг/кг массы тела животного. Такие дозы можно вводить раздельными дозами от 1 до 4 раз в сутки или в форме с замедленным высвобождением. Для человека общая суточная доза может варьироваться, например, от примерно 5 до примерно 7000 мг и более конкретно от примерно 10 до примерно 1500 мг. Стандартные лекарственные формы, подходящие для перорального введения, обычно содержат, например, от примерно 2 до примерно 1400 мг по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей, смешанных по меньшей мере с одним твердым и/или жидким фармацевтическим носителем,смазывающим агентом и/или разбавителем. Однако конкретный дозовый уровень и частота дозировки для любого конкретного субъекта могут варьироваться и обычно зависят от множества факторов, включая, но не ограничиваясь этим, например,биодоступность конкретного(ых) соединения(ий) формул I-V, или фармацевтически приемлемых солей,или их смесей, во вводимой форме; метаболическую стабильность и продолжительность действия конкретного(ых) соединения(ий) формул I-V, или фармацевтически приемлемых солей, или их смесей; вид,возраст, массу тела, общее состояние здоровья, пол и диету субъекта; путь и время введения; скорость экскреции; комбинацию лекарственных средств и тяжесть конкретного состояния. Соединение(я) в соответствии с формулами I-V, или фармацевтически приемлемые соли, или их смеси можно вводить любым путем, подходящим для состояния, подлежащего лечению, и количества соединения(ий) формул I-V, или фармацевтически приемлемых солей, или их смесей, предназначенных для доставки. Соединение(я) в соответствии с формулами I-V, или фармацевтически приемлемые соли, или их смеси можно вводить в форме традиционной фармацевтической композиции любым путем, включая, но не ограничиваясь этим, например, пероральный, внутримышечный, подкожный, местный, интраназаль-8 020825 ный, эпидуральный, интраперитонеальный, интраторакальный, внутривенный, интратекальный, интрацереброветрикулярный, через внутриматочное устройство и путем инъецирования в суставы, путем нанесения в устройство, такое как пластырь или тампон для местного лечения, такого как носовое кровотечение. В одном из воплощений путь введения является пероральным, внутривенным, через внутриматочное устройство или внутримышечным. Соединение формул I-V, или фармацевтически приемлемые соли, или их смеси можно использовать сами по себе или в форме подходящих медицинских препаратов для энтерального или парентерального введения. Приемлемые твердые фармацевтическая композиции включают в себя, но не ограничиваются этим,например, порошки, таблетки, диспергируемые гранулы, капсулы, сашеты и суппозитории. В твердой фармацевтической композиции фармацевтически приемлемые носители включают в себя, но не ограничиваются этим, например, по меньшей мере одно твердое вещество, по меньшей мере одну жидкость и их смеси. Твердый носитель также может представлять собой разбавитель, ароматизатор, солюбилизатор, смазывающий агент, суспендирующий агент, связующий агент, инкапсулирующее вещество и/или агент для разрыхления таблетки. Подходящие носители включают в себя, но не ограничиваются этим, например, карбонат магния; стеарат магния; тальк; лактозу; сахар; пектин; декстрин; крахмал; трагакант; метилцеллюлозу; натрий-карбоксиметилцеллюлозу; низкоплавкий воск; какао-масло и их смеси. Порошок может быть получен, например, путем смешивания тонкоизмельченного твердого вещества по меньшей мере с одним тонкоизмельченным соединением формул I-V, или фармацевтически приемлемыми солями, или их смесями. Таблетка может быть получена, например, путем смешивания по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей в подходящих пропорциях с фармацевтически приемлемым носителем, имеющим необходимые связывающие свойства,и спрессованы в таблетку желаемой формы и размера. Суппозиторий может быть получен, например, путем смешивания по меньшей мере одного соединения формул I-V, или фармацевтически приемлемых солей, или их смеси по меньшей мере с одним подходящим нераздражающим эксципиентом, который является жидким при ректальной температуре, но твердым при температуре ниже ректальной температуры, где нераздражающий эксципиент сначала расплавляют и соединение формулы I диспергируют в данной заявке. Расплавленную гомогенную смесь затем вливают в формы подходящего размера и оставляют охлаждаться и отверждаться. Примерные нераздражающие эксципиенты включают в себя, но не ограничиваются этим, например, какао-масло; обработанный глицерином желатин; гидрирогенизированные растительные масла; смеси полиэтиленгликолей различных молекулярных масс и жирно-кислотные сложные эфиры полиэтиленгликоля. Приемлемые жидкие фармацевтические композиции включают в себя, но не ограничиваются этим,например, растворы, суспензии и эмульсии. Примерные жидкие фармацевтические композиции, подходящие для парентерального введения,включают в себя, но не ограничиваются этим, например, стерильную воду или водные пропиленгликолевые растворы по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей; и водные полиэтиленгликолевые растворы по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей. Водные растворы для перорального введения могут быть получены путем растворения по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и/или загустителей при желании. Водные суспензии для перорального введения могут быть получены путем диспергирования по меньшей мере одного тонкоизмельченного соединения формулы I-V, или фармацевтически приемлемых солей, или их смесей в воде вместе с вязким веществом, таким как, например, природная синтетическая смола, камедь, метилцеллюлоза и натрий-карбоксиметилцеллюлоза. В одном из воплощений фармацевтическая композиция содержит от примерно 0,05 до примерно 99 мас.% по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей. Все проценты по массе основаны на общей композиции. В другом воплощении фармацевтическая композиция содержит от примерно 0,10 до примерно 50 мас.% по меньшей мере одного соединения в соответствии с формулами I-V, или фармацевтически приемлемых солей, или их смесей. Все проценты по массе основаны на общей композиции. Также в данной заявке раскрыт способ получения фармацевтической композиции, включающий смешивание или компаундирование ингредиентов вместе и формирование смешанных ингредиентов в таблетки или суппозитории; инкапсулирование ингредиентов в капсулах или растворение ингредиентов с образованием инъецируемых растворов. Соединение согласно настоящему изобретению может быть использовано в комбинации с другими агентами для лечения состояний, описанных выше. Примерами таких агентов являются нестероидные противовоспалительные агенты, такие как ингибиторы NSAIDS, и гормональное лечение, такое как прогестероновое и/или эстрогеновое, антитромбоцитарные или антикоагулянтные агенты для предупреждения побочных эффектов, факторы свертывания, такие как факторы VII и VIII, фактор Виллебранда. Способ анализа. Способ анализа, используемый для оценки биологической активности соединений согласно изобретению, представлял собой анализ фибринолитической активности в буфере. Измерение ингибирования плазминогена осуществляли в 200 мкл реакционной смеси, содержащей 15 мМ HEPES, рН 7,4, 100 мМ NaCl, 0,008% Tween-80, 13 мкг/мл человеческого Glu-плазминогена(Chromogenix, Италия), 1,7 мг/мл человеческого фибриногена (Aniara, США), 0,02 нМ человеческого одноцепочечного активатора плазминогена тканевого типа (Biopool, Швеция), 1% DMSO и 0,05 NTH ед./мл человеческого тромбина (Sigma, США). Тестируемое вещество добавляли в реакционную смесь в 10% DMSO. Реакцию начинали путем добавления тромбина. Образование и распад фибрина отслеживали в спектрофотометре при 405 нм в течение 15 ч при 37 С. Биологическая активность. Измеряли согласно анализу фибринолитической активности в буфере, описанному выше. Пример 1 продемонстрировал значение IC50 2,85 мкМ. Пример 2 продемонстрировал значение IC50 169 мкМ. Пример 3 продемонстрировал значение IC50 1,16 мкМ. Пример 4 продемонстрировал значениеIC50 0,81 мкМ. Пример 100 продемонстрировал значение IC50 1,93 мкМ. Пример 101 продемонстрировал значение IC50 4,7 мкМ. Пример 102 продемонстрировал значение IC50 2,18 мкМ. Пример 103 продемонстрировал значение IC50 94 мкМ. Пример 104 продемонстрировал значение IC50 1,5 мкМ. Пример 105 продемонстрировал значение IC50 7,2 мкМ. Пример 106 продемонстрировал значение IC50 88,8 мкМ. Пример 107 продемонстрировал значение IC50 1,7 мкМ. Пример 108 продемонстрировал значение IC50 70,5 мкМ. Пример 109 продемонстрировал значение IC50 5 мкМ. Пример 110 продемонстрировал значение IC50 1,2 мкМ. Пример 111 продемонстрировал значение IC50 31,3 мкМ. Пример 112 продемонстрировал значение IC50 5,5 мкМ. Пример 113 продемонстрировал значение IC50 1,1 мкМ. Пример 114 продемонстрировал значение IC50 6,5 мкМ. Пример 115 продемонстрировал значение IC50 0,8 мкМ. Пример 116 продемонстрировал значение IC50 5,6 мкМ. Пример 117 продемонстрировал значение IC50 0,8 мкМ. Пример 118 продемонстрировал значение IC50 225 мкМ. Пример 119 продемонстрировал значение IC50 10,7 мкМ. Пример 120 продемонстрировал значение IC50 0,89 мкМ. Пример 121 продемонстрировал значение IC50 120 мкМ. Пример 122 продемонстрировал значение IC50 4,4 мкМ. Пример 123 продемонстрировал значение IC50 147 мкМ. Пример 124 продемонстрировал значение IC50 1,2 мкМ. Пример 125 продемонстрировал значение IC50 92,9 мкМ. Пример 126 продемонстрировал значение IC50 3,6 мкМ. Пример 127 продемонстрировал значение IC50 1,4 мкМ. Пример 128 продемонстрировал значение IC50 31,4 мкМ. Пример 129 продемонстрировал значение IC50 10,2 мкМ. Пример 130 продемонстрировал значение IC50 1,1 мкМ. Пример 131 продемонстрировал значение IC50 128 мкМ. Пример 132 продемонстрировал значение IC50 2,2 мкМ. Пример 133 продемонстрировал значение IC50 120 мкМ. Пример 134 продемонстрировал значение IC50 0,85 мкМ. Пример 135 продемонстрировал значение IC50 69,1 мкМ. Пример 136 продемонстрировал значение IC50 4,94 мкМ. Пример 137 продемонстрировал значение IC50 1 мкМ. Пример 138 продемонстрировал значение IC50 123 мкМ. Пример 139 продемонстрировал значение IC50 2,9 мкМ. Пример 140 продемонстрировал значение IC50 0,6 мкМ. Пример 141 продемонстрировал значение IC50 69,5 мкМ. Пример 142 продемонстрировал значение IC50 1,8 мкМ. Пример 143 продемонстрировал значение IC50 0,9 мкМ. Пример 144 продемонстрировал значение IC50 88,2 мкМ. Пример 145 продемонстрировал значение IC50 4,3 мкМ. Пример 146 продемонстрировал значение IC50 6,6 мкМ. Пример 147 продемонстрировал значение IC50 44,6 мкМ. Пример 148 продемонстрировал значение IC50 2,7 мкМ. Пример 149 продемонстрировал значение IC50 0,9 мкМ. Пример 150 продемонстрировал значение IC50 87 мкМ. Пример 151 продемонстрировал значение IC50 7,7 мкМ. Рентгеновская дифракция на порошке. Картину рентгеновской дифракции на порошке (обозначается в данной заявке как XRPD или XRD) определяли путем размещения образца на нулевом фоновом держателе, единственном кристалле кремния, и растягивания образца в тонкий слой. Используя тета-2 тета дифрактометр Bruker D8 Advance с детектором VNTEC-1, образец формовали (для улучшения статистика отсчетов) и облучали рентгеновскими лучами, генерированными медной трубкой, работающей при 30 кВ и 50 мА. Использовали автоматически изменяющиеся щели расходимости. Анализ рентгеновской дифракции осуществляли согласно стандартным способам, которые можно найти, например, в Kitaigorodsky, A.I. (1973), Molecular Crystals and Molecules, Academic Press, New York;(1974), X-ray Diffraction Procedures, John WileySons, New York. Данные рентгеновской дифракции на порошке не корректировали с использованием внутреннего стандарта. Измерения осуществляли с изменяющимися щелями. Следует понимать, что значения 2-тета картины рентгеновской дифракции на порошке могут слегка различаться от одной машины к другой, а также зависят от изменений в получении образца и варьирования между партиями, и поэтому приведенные значения не следует считать абсолютными. Также следует понимать, что относительные интенсивности пиков могут варьировать в зависимости от эффектов ориентации, так что интенсивности, показанные во включенной в данной заявке кривой XRD, являются иллюстративными и их не следует использовать для абсолютного сравнения. Соединения могут кристаллизоваться в виде ансольватов или сольватов (включая гидраты и смешанные сольваты). Молекулы органического растворителя могут быть более или менее сильно связанными в структуре. Трансформации могут происходить между ансольватами и сольватами или между стехиометрическими и нестехиометрическими сольватами. Трансформации могут быть либо обратимыми,либо необратимыми. Относительная влажность или присутствие других растворителей является тем, что может влиять на картину рентгеновской дифракции на порошке в большей или меньшей степени в зависимости от количества органического растворителя, присутствующего в структуре. Картины рентгеновской дифракции на порошке типичного образца из примеров 10, 14, 18, 55, 65,104, 115 и 143 показаны на фиг. 1-13. Примеры Перечень аббревиатур, используемых в примерах: АсОН - уксусная кислота,CV - объем колонки,DME - 1,2-диметоксиэтан,DEE - диэтиловый эфир,DMF - N,N-диметилформамид,DIPEA - N,N-диизопропилэтиламин,DCM - дихлорметан,dppf - 1,1'-бис-(дифенилфосфино)ферроцен,DMAP - 4-диметиламинопиридин,EDC - 1-этил-3-(3-диметиламинопропил)карбодиимид,EtOH - этанол,FA - муравьиная кислота,ч - час(ы),мин - минута(ы),МТВЕ - метил-трет-бутиловый эфирIPA - изопропиловый спирт,LAH - алюмогидрид лития,TEA - триэтиламин,THF - тетрагидрофуран,TBDMSCl - трет-бутилдиметилхлорсилан,TBAF - тетрабутиламмония фторид,МеОН - метанол,EtOAc - этилацетат,Ph - фенил,нас. - насыщенный. Получение ссылочных соединений Ссылочное соединение 1. 2-Бензил-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. трет-Бутил-2-бензил-4-цианопиперидин-1-карбоксилат. К раствору 2-бензил-4-оксопиперидин-1-карбоновой кислоты трет-бутилового сложного эфира (15 г, 51,8 ммоль) (от SYNTECH) в DME (250 мл) в атмосфере азота одновременно добавляли толуол-4 сульфонилметилизоцианид (15,2 г, 77,7 ммоль, в 250 мл DME) и трет-бутилат калия (156 мл, 1 M в третбутаноле) в течение 1 ч так, чтобы температура поддерживалась ниже -10 С. Раствор перемешивали при-10 С в течение 2 ч и затем оставляли до достижения им комнатной температуры в течение 16 ч. В реакционную смесь добавляли Н 2 О (400 мл), раствор перемешивали в течение 20 мин и затем экстрагировалиDEE (3) и EtOAc (3). Объединенные органические фазы сушили над Na2SO4 и упаривали. Остаток очищали посредством колоночной хроматографии с использованием смеси EtOAc/гептан (10-60% градиент EtOAc) с получением трет-бутил-2-бензил-4-цианопиперидин-1-карбоксилата (11,85 г, 76%). 1 Н ЯМРHCl (43 мл). После 20 мин перемешивания раствор переносили в реакционные флаконы для микроволновой обработки и нагревали до 140 С в течение 30 мин в одномодовом микроволновом реакторе. Растворители выпаривали. Остаток растворяли в HCl (1,25 М в МеОН, 50 мл) и суспензию кипятили с обратным холодильником в течение 1 ч. Удаление растворителей приводило к получению масла, которое переносили в нас. NaHCO3 (приблизительно 80 мл) и затем нейтрализовали твердым NaHCO3. Водную фазу экстрагировали DCM (3), который сушили с помощью фазоразделителя и упаривали с получением метил-2-бензилпиперидин-4-карбоксилата (7,36 г, 80%) в виде масла. 1H ЯМР (600 МГц, CDCl3)1.18-2.40(М+Н)+. Стадия 3. 2-Бензил-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. К раствору метил-2-бензилпиперидин-4-карбоксилата (7,36 г, 31,55 ммоль) и DIPEA (11,02 мл,63,09 ммоль) в DCM (200 мл) добавляли метилхлорформиат (3,18 мл, 41,01 ммоль) в 100 мл DCM в течение 1 ч. Реакционную смесь перемешивали в течение 40 мин, затем промывали нас. NaHCO3, сушили с помощью фазоразделителя и упаривали. Остаток растворяли в THF (80 мл) с последующим добавлениемLiOH (1,0 г, 42,0 ммоль), МеОН (60 мл) и воды (60 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 72 ч. После выпаривания растворителей остаток переносили в воду. рН корректировали до менее 2 путем добавления HCl (10%). Водную фазу затем экстрагировали DCM (5). Объединенные органические слои сушили с помощью фазоразделителя и упаривали с получением 2-бензил-1-(метоксикарбонил)пиперидин-4-карбоновой кислоты в виде масла. MS m/z 276 (МН)-. Ссылочное соединение 2. 2-Изобутил-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. 3-(трет-Бутоксикарбониламино)-5-метилгексановая кислота. К раствору DLгомолейцина (45 г, 0,31 моль) в 1 н. NaOH (1 л) при 0 С добавляли по каплям раствор (Вос)2 О (87,8 г, 0,403 моль) в 1,4-диоксане (500 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, охлаждали до 0 С и нейтрализовали с помощью 1 н. HCl (приблизительно 1000 мл). Твердое вещество отфильтровывали и сушили под вакуумом с получением 3-(третбутоксикарбониламино)-5-метилгексановой кислоты (48,0 г, 63%) в виде твердого вещества. Стадия 2. трет-Бутил-2-изобутил-4,6-диоксопиперидин-1-карбоксилат. К перемешиваемому раствору 3-(трет-бутоксикарбониламино)-5-метилгексановой кислоты (44,0 г,0,179 моль) в DCM (800 мл) при 0 С добавляли EDC гидрохлорид (51,56 г, 0,269 моль), DMAP (32,8 г,0,269 моль) и кислоту Мелдрума (25,8 г, 0,179 моль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, промывали 1 н. KHSO4 (500 мл), сушили над Na2SO4 и концентрировали. Остаток растворяли в сухом этилацетате (1,5 л) и кипятили с обратным холодильником в течение ночи. Реакционную смесь промывали 1 н. KHSO4 (500 мл), рассолом (500 мл), сушили над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии с использованием петролейного эфира и EtOAc (65:35) в качестве элюента с получением 3-(трет-бутоксикарбониламино)-5-метилгексановой кислоты в виде твердого вещества (35 г, 72%). Стадия 3. 6-Изобутилпиперидин-2,4-дион. К раствору трет-бутил-2-изобутил-4,6-диоксопиперидин-1-карбоксилата (30 г, 0,114 моль) в сухом 1,4 диоксане (300 мл) добавляли HCl (3 М в 1,4 диоксане, 100 мл) и перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали и очищали путем кристаллизации с использованием диэтилового эфира с получением 6-изобутилпиперидин-2,4-диона (14 г, 74%) в виде белого твердого вещества. Стадия 4. 2-Изобутилпиперидин-4-ол. К перемешиваемой на льду охлажденной суспензии LAH (16,1 г, 0,414 моль) в THF (100 мл) добавляли по каплям раствор 6-изобутилпиперидин-2,4-диона (14,0 г, 0,083 моль) в THF (100 мл) в атмосфереN2. Реакционную смесь нагревали до комнатной температуры и перемешивали в атмосфере N2 в течение 48 ч. Реакционную смесь охлаждали до 0 С и гасили водой (16,1 мл), 1 н. NaOH (16,1 мл) и водой (16,1 мл). Реакционную смесь фильтровали и твердое вещество промывали горячим THF (100 мл). Фильтрат концентрировали с получением 2-изобутилпиперидин-4-ола в виде твердого вещества (9 г, неочищенное). Стадия 5. трет-Бутил-4-гидрокси-2-изобутилпиперидин-1-карбоксилат. 2-Изобутилпиперидин-4-ол (9,0 г, 0,057 моль) переносили в 50:50 раствор THF и нас. NaHCO3 (100 мл) и смесь охлаждали до 0 С. Добавляли по каплям раствор (Вос)2 О (13,7 г, 0,063 моль) в THF (50 мл). Полученный раствор перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и экстрагировали с помощью EtOAc (100 мл). Органическую фазу промывали водой(100 мл), рассолом (100 мл), сушили над Na2SO4 и концентрировали. Концентрированный продукт очищали посредством колоночной хроматографии с использованием 20% EtOAc в петролейном эфире с получением смеси двух соединений. Смесь затем очищали посредством препаративной HPLC с получением трет-бутил-4-гидрокси-2-изобутилпиперидин-1-карбоксилата в виде бледно-желтой жидкости (4,5 г,39,5%). Стадия 6. 2-Изобутил-4-оксопиперидин-1-карбоновой кислоты трет-бутиловый сложный эфир. К перемешиваемому раствору трет-бутил-4-гидрокси-2-изобутилпиперидин-1-карбоксилата (4,6 г,0,018 моль) в DCM (50 мл) при 0 С добавляли порциями 1,1,1-триацетокси-1,1-дигидро-1,2 бензйодоксол-3(1 Н)-он (9,1 г, 0,02146 моль), полученный раствор нагревали до комнатной температуры и перемешивали в атмосфере N2 в течение ночи. Реакционную смесь гасили нас. раствором NaHCO3 (50 мл) и фильтровали через набивку Celite. Фильтрат экстрагировали несколько раз с помощью DCM (50 мл), объединенные органические фазы промывали водой (50 мл), рассолом (50 мл), сушили над Na2SO4 и концентрировали. Концентрированный продукт очищали посредством колоночной хроматографии с использованием 10% EtOAc в петролейном эфире с получением продукта с чистотой 90% согласно HPLC. Загрязненный продукт затем очищали посредством препаративной HPLC с получением 2-изобутил-4 оксопиперидин-1-карбоновой кислоты трет-бутилового сложного эфира в виде жидкости (2,1 г, 46%). Стадия 7. трет-Бутил-4-циано-2-изобутилпиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 1, стадия 1, из 2-изобутил-4 оксопиперидин-1-карбоновой кислоты трет-бутилового сложного эфира (4,55 г, 17,8 ммоль), толуол-4- 13020825 сульфонилметилизоцианида (5,2 г, 26,7 ммоль) и трет-бутилата калия (53,4 мл, 1 M в трет-бутаноле), которые давали неочищенный трет-бутил-4-циано-2-изобутилпиперидин-1-карбоксилат. MS m/z 267(М+Н). Стадия 8. 2-Изобутилпиперидин-4-карбоновой кислоты метиловый эфир. Соединение получали, как описано для ссылочного соединения 1, стадия 2, из неочищенного третбутил-4-циано-2-изобутилпиперидин-1-карбоксилата (6,43 г, примерно 24,2 ммоль), что давало в результате неочищенный 2-изобутилпиперидин-4-карбоновой кислоты метиловый эфир. MS m/z 200 (М+Н)+. Стадия 9. 2-Изобутил-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Соединение получали, как описано для ссылочного соединения 1, стадия 3, из неочищенного 2 изобутилпиперидин-4-карбоновой кислоты метилового эфира (2,6 г, 13,0 ммоль), DIPEA (4,55 мл, 26,1 ммоль) и метилхлорформиата (1,31 мл, 17,0 ммоль) и затем LiOH (1,3 экв.), которые давали неочищенную 2-изобутил-1-(метоксикарбонил)пиперидин-4-карбоновую кислоту. MS m/z 242 (М-Н)-. Ссылочное соединение 3. 1-(Метоксикарбонил)-2-фенетилпиперидин-4-карбоновая кислота. Стадия 1. Метил-4-оксо-2-фенетил-3,4-дигидропиридин-1(2 Н)-карбоксилат. 4-Метоксипиридин (9,30 мл, 91,64 ммоль) растворяли в THF (150 мл) в атмосфере азота и охлаждали до -15 С. Добавляли по каплям фенетилхлорид магния (93 мл, 93,47 ммоль, 1 M в THF) и образовывалась суспензия. После перемешивания при -20 С в течение 30 мин добавляли метилхлорформиат (9,23 мл, 119,13 ммоль) в течение 1 мин. Перемешивание продолжали при -10 С в течение 1 ч и затем добавляли HCl (10%). Смесь перемешивали в течение 20 мин и затем концентрировали. Водную фазу экстрагировали эфиром (2) и органическую фазу сушили (MgSO4) и упаривали с получением метил-4-оксо-2 фенетил-3,4-дигидропиридин-1(2 Н)-карбоксилата (21,7 г, 82%) в виде масла. 1H ЯМР (600 МГц, cdcl3)1.98 (m, 2H), 2.54 (m, 2H), 2.69 (m, 1H), 2.81 (m, 1H), 3.83 (s, 3H), 4.62 (m, 1H), 5.33 (m, 1H), 7.12-7.38 (m,5H), 7.72 (m, 1H); MS m/z 260 (M+H)+. Стадия 2. Метил-4-оксо-2-фенетилпиперидин-1-карбоксилат. Метил-4-оксо-2-фенетил-3,4-дигидропиридин-1(2 Н)-карбоксилат (21,7 г, примерно 75 ммоль) гидрировали над Pd/C (5%) в EtOAc при 5 бар (0,5 МПа) в течение 20 ч. Смесь фильтровали через слой диоксида кремния и затем упаривали с получением продукта в виде масла (19,8 г). 1 Н ЯМР (600 МГц, cdcl3)1.69 (m, 1H), 1.80 (m, 1H), 2.26 (m, 2H), 2.41 (m, 1H), 2.47-2.70 (m, 3H), 3.16 (m, 1H), 3.69 (m, 3H), 4.154,48 (br m, 2 Н), 7.14-7.30 (m, 5H). MS 262 m/z (M+H)+. Стадия 3. Метил-4-циано-2-фенетилпиперидин-1-карбоксилат. К раствору метил-4-оксо-2-фенетилпиперидин-1-карбоксилата (19,8 г, 75,7 ммоль) в DME (250 мл) в атмосфере азота одновременно добавляли толуол-4-сульфонилметилизоцианид (16,6 г, 85,0 ммоль, в 250 мл DME) и трет-бутилат калия (197 мл, 1 M в трет-бутанол) в течение 1 ч для того, чтобы температура поддерживалась ниже -10 С. Смесь затем перемешивали при -10 С в течение 2 ч и оставляли до достижения ею температуры окружающей среды в течение 16 ч. К оранжевой реакционной смеси добавляли Н 2 О (400 мл), это перемешивали в течение 20 мин и затем экстрагировали эфиром (3) и EtOAc (3). Органические фазы объединяли, сушили над Na2SO4 и упаривали с получением 24,8 г остатка. В результате флэш-хроматографии с использованием смеси EtOAc/гептан (30-80% градиент EtOAc) получали продукт(13,2 г, 64%) в виде смеси диастереомеров (основной цис-изомер). MS m/z 273 (М+Н)+. транс-Изомер: 1H ЯМР (600 МГц, cdcl3)1.71 (m, 2H), 1.84-2.09 (m, 4H), 2.47-2.68 (m, 2H), 2.71-2.92 (m, 2H), 3.70 (s, 3H),4.12 (br m, 1H), 4.45 (br m, 1H), 7.01-7.40 (m, 5H). цис-Изомер: 1H ЯМР (600 МГц, cdcl3)1.76 (m, 1H),1.88 (m, 1H), 1.92-2.07 (m, 2H), 2.07-2.15 (m, 1H), 2.24-2.38 (m, 1H), 2.55-2.72 (m, 2H), 2.96 (m, 1H), 3.163.29 (m, 1H), 3.69 (s, 3H), 4.10 (m, 1H), 4.35 (m, 1H), 7.15-7.35 (m, 5H). Стадия 4. 1-(Метоксикарбонил)-2-фенетилпиперидин-4-карбоновая кислота. К метил-4-циано-2-фенетилпиперидин-1-карбоксилату (13,2 г, 48,5 ммоль) в реакционном флаконе для микроволновой обработки добавляли 6 М HCl. Смесь нагревали до 100 С в течение 30 мин в одномодовом микроволновом реакторе. Водную фазу экстрагировали с помощью EtOAc и полученную органическую фазу промывали один раз 10% HCl, сушили над MgSO4 и упаривали с получением неочищенного продукта 3,47 г. MS m/z 290 (М-Н)-. Ссылочное соединение 4. 2-(4-трет-Бутилбензил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. 4-трет-Бутилдиметилсилилокси)метил)пиридин. К раствору пиридин-4-илметанола (25,7 г, 0,24 моль) и имидазола (19,8 г, 0,29 моль) в сухом DMF(300 мл) и сухом DCM (33 мл) в атмосфере азота добавляли TBDMSCl (42,6 г, 0,29 моль). Раствор перемешивали 18 ч, в течение которых образовывался осадок. Реакционную смесь концентрировали путем удаления летучих веществ (примерно 100 мл) с последующим добавлением воды (500 мл). Полученную смесь экстрагировали смесью 1:1 гептан:EtOAc (200 мл 3). Объединенные органические фазы промывали рассолом (2), сушили (MgSO4), фильтровали и упаривали с получением 4-третбутилдиметилсилилокси)метил)пиридина (51,70 г, 98%) в виде масла. 1H ЯМР (600 МГц, cdcl3)0.01 (s,6H), 0.82 (s, 9H), 4.63 (s, 2H), 7.14 (m, 2H), 8.43 (m, 2H).THF) в течение 10 мин. Добавляли по каплям метилхлорформиат (2,443 мл, 31,02 ммоль) в течение 10 мин. Реакционную смесь оставляли до достижения ею 0 С в течение 2 ч. Органические растворители выпаривали, реакционную смесь разбавляли этилацетатом, затем промывали 1 н. HCl и рассолом. Органический слой сушили над MgSO4 и упаривали с получением 9 г неочищенного остатка, который очищали 2 равными порциями посредством автоматизированной колоночной хроматографии (Biotage, KP-SIL 340 г), элюент: градиент гептан/EtOAc 0-50%, что давало в результате (6,5 г, 64%) продукт в виде прозрачного масла. 1 Н ЯМР (400 МГц, cdcl3) (комплексный)0.07-0.21 (m, 6H), 0.9-1.30 (m, 18H), 2.0-7.4 (m,15H). MS m/z 431 (М+Н)+. Стадия 3. Метил-2-(4-трет-бутилбензил)-4-трет-бутилдиметилсилилокси)метил)пиперидин-1 карбоксилат. К раствору метил-2-(4-трет-бутилбензил)-4-трет-бутилдиметилсилилокси)метил)пиридин-1(2 Н)карбоксилата (6,5 г, 15,13 ммоль) в этилацетате (50 мл) добавляли оксид платины(IV) (0,07 г, 0,31 ммоль). Суспензию гидрировали в атмосфере Н 2 при 6 бар (0,6 МПа) в течение 20 ч. Смесь фильтровали через целит и растворители выпаривали с получением продукта (6,27 г, 96%) в виде масла. 1H ЯМР (400 МГц, cdcl3)0.07-0.18 (m, 6H), 0.77-1.01 (m, 9H), 1.28-1.37 (m, 9 Н), 1.36-1.87 (m, 4H), 2.58-2.74 (m, 1H),2.88-3.06 (m, 2H), 3.40-3.55 (m, 3H), 3.60-3.72 (m, 3H), 3.78-4.10 (m, 2 Н), 7.14-7.32 (m, 4 Н). MS m/z 434(18,79 мл, 18,79 ммоль, 1 M в THF) и реакционную смесь перемешивали при комнатной температуре в течение 90 мин. Растворители выпаривали, остаток растворяли в этилацетате и промывали нас. NaHCO3(1), затем рассолом (2). Органический слой сушили над MgSO4 и упаривали. Остаток очищали посредством автоматизированной колоночной хроматографии (Biotage, (KP-SIL 340 г) элюент EtOAc/гептан,градиент 20-90% EtOAc) с получением продукта (3,21 г, 70%) в виде масла. 1 Н ЯМР (400 МГц, cdcl3)1.30 (s, 9 Н), 1.42-1.50 (m, 2H), 1.65-1.91 (m, 2H), 2.68 (m, 1H), 2.89-3.07 (m, 2H), 3.39-3.62 (m, 3H), 3.67 (m,3H), 3.87 (m 1H), 4.07 (m, 1H), 7.11 (m, 2H), 7.30 (m, 2H). MS m/z 320 (М+Н)+. Стадия 5. 2-(4-трет-Бутилбензил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. К раствору метил-2-(4-трет-бутилбензил)-4-(гидроксиметил)пиперидин-1-карбоксилата (2,9 г, 9,08 ммоль) в тетрахлориде углерода (18 мл) добавляли перйодат натрия (5,83 г, 27,24 ммоль) и воду (27 мл). К этой смеси добавляли ацетонитрил (18 мл), затем хлорид рутения(III) (0,041 г, 0,20 ммоль). Полученную двухфазную смесь энергично перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли водой и DCM, водный слой экстрагировали DCM (3). Объединенные органические слои сушили над MgSO4 и упаривали с получением продукта в виде масла (3,0 г, 98%). MS m/z 332(М-Н)-. Ссылочное соединение 5. 1-(Метоксикарбонил)-2-неопентилпиперидин-4-карбоновая кислота. Стадия 1. Метил-4-трет-бутилдиметилсилилокси)метил)-2-неопентилпиридин-1(2H)-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 2, начиная с 4-(третбутилдиметилсилилокси)метил)пиридина, полученного, как описано для ссылочного соединения 4, стадия 1, неопентилхлорида магния (40,5 мл, 40,5 ммоль, 1 M в THF) и метилхлорформиата (3,77 мл, 47,9 ммоль). Остаток очищали посредством автоматизированной колоночной флэш-хроматографии на колонке Biotage KP-SIL 340 г. Градиент от 15:1 до 10:1 гептан:EtOAc использовали в качестве подвижной фазы с получением указанного в заголовке соединения (9,3 г, 71%): 1 Н ЯМР (400 МГц, cdcl3)0.00 (s,3H), 0.09 (s, 3H), 0.87 (s, 18H), 1.09-1.81 (m, 2H), 1.94-2.44 (m, 1H), 3.70 (s, 3H), 4.22-4.90 (m, 2H), 5.125.61 (m, 1H), 5.69-5.97 (m, 1H), 6.25-6.79 (m, 1H). MS m/z 354 (M+H)+. Стадия 2. Метил-4-трет-бутилдиметилсилилокси)метил)-2-неопентилпиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 3, начиная с метил-4 трет-бутилдиметилсилилокси)метил)-2-неопентилпиридин-1(2 Н)-карбоксилата, что давало в результате указанное в заголовке соединение. MS m/z 358 (М+Н)+. Стадия 3. Метил-4-(гидроксиметил)-2-неопентилпиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 4, начиная с метил-4 трет-бутилдиметилсилилокси)метил)-2-неопентилпиперидин-1-карбоксилата и TBAF (34,2 мл, 34,2 ммоль, 1 M в THF). Остаток очищали посредством автоматизированной колоночной флэшхроматографии на колонке Biotage KP-SIL 340 г. Градиент от 30 до 100% EtOAc в гептане использовали в качестве элюента, что давало в результате указанное в заголовке соединение. Стадия 5. 1-(Метоксикарбонил)-2-неопентилпиперидин-4-карбоновая кислота. Соединение получали, как описано для ссылочного соединения 4, стадия 5, начиная с метил-4- 15020825(гидроксиметил)-2-неопентилпиперидин-1-карбоксилата, перйодата натрия (15,0 г, 70,3 ммоль) и хлорида рутения(III) (0,11 г, 0,52 ммоль), что давало в результате указанное в заголовке соединение. MS m/z 256 (М-Н)-. Ссылочное соединение 6. 1-(Метоксикарбонил)-2-метилпиперидин-4-карбоновая кислота. Стадия 1. Метил-4-трет-бутилдиметилсилилокси)метил)-2-метилпиридин-1(2 Н)-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 2, начиная с 4-(третбутилдиметилсилилокси)метил)пиридина, полученного, как описано для ссылочного соединения 4, стадия 1, метилхлорида магния (3 М в THF) (13,1 мл, 39,4 ммоль, 1 M в THF) и метилхлорформиата (3,61 мл, 46,7 ммоль). Остаток очищали посредством колоночной хроматографии (Biotage гептан:EtOAc с использованием градиента 0-15% EtOAc; колонка snap 360), что давало в результате указанное в заголовке соединение в виде слегка желтого масла (2,48 г, 23%): 1 Н ЯМР (400 МГц, cdcl3)0.03 (d, 6H), 0.66-1.04MS m/z 298 (M+H)+. Стадия 2. Метил-4-трет-бутилдиметилсилилокси)метил)-2-метилпиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 3, начиная с метил-4 трет-бутилдиметилсилилокси)метил)-2-метилпиридин-1(2 Н)-карбоксилата, что давало в результате указанное в заголовке соединение. MS m/z 302 (М+Н)+. Стадия 3. Метил-4-(гидроксиметил)-2-метилпиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 4, начиная с метил-4 трет-бутилдиметилсилилокси)метил)-2-метилпиперидин-1-карбоксилата и TBAF (10,65 мл, 10,65 ммоль, 1 M в THF). Остаток очищали посредством колоночной хроматографии (Biotage 40-90% градиентEtOAc в гептане, колонка 340 snap), что приводило к получению указанного в заголовке соединения. 1 Н ЯМР (400 МГц, cdcl3)0.95-1.42 (m, 3H), 1.68-1.95 (m, 3H), 3.08 (m, 1H), 3.39-3.53 (m, 2H), 3.61 (m, 1H),3.68 (s, 3H), 3.71-3.87 (m, 1H), 3.87-4.08 (m, 1H), 4.30-4.62 (m, 1 Н). Стадия 5. 1-(Метоксикарбонил)-2-метилпиперидин-4-карбоновая кислота. Соединение получали, как описано для ссылочного соединения 4, стадия 5, начиная с метил-4(гидроксиметил)-2-метилпиперидин-1-карбоксилата, перйодата натрия (3,53 г, 16,5 ммоль) и хлорида рутения(III) (0,025 г, 0,12 ммоль), что давало в результате указанное в заголовке соединение. MS m/z 200(М-Н)-. Ссылочное соединение 7. 1-(Метоксикарбонил)-2-(2-метил-2-фенилпропил)пиперидин-4 карбоновая кислота. Стадия 1. Метил-4-трет-бутилдиметилсилилокси)метил)-2-(2-метил-2-фенилпропил)пиридин 1(2 Н)-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 2, начиная с 4-(третбутилдиметилсилилокси)метил)пиридина, полученного, как описано для ссылочного соединения 4, стадия 1, (2-метил-2-фенилпропил)хлорида магния (0,5 М в THF, 93 мл, 46,6 ммоль) и метилхлорформиата(3,61 мл, 46,7 ммоль) с получением продукта (17,5 г). MS m/z 416 (М+Н)+. Стадия 2. Метил-4-трет-бутилдиметилсилилокси)метил)-2-(2-метил-2-фенилпропил)пиперидин-1 карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 3, начиная с неочищенного метил-4-трет-бутилдиметилсилилокси)метил)-2-(2-метил-2-фенилпропил)пиридин-1(2 Н)-карбоксилата (17,5 г), что давало в результате указанное в заголовке соединение (16,5 г). MS m/z 420 (М+Н)+. Стадия 3. Метил-4-(гидроксиметил)-2-(2-метил-2-фенилпропил)пиперидин-1-карбоксилат. Соединение получали, как описано для ссылочного соединения 4, стадия 4, начиная с неочищенного метил-4-трет-бутилдиметилсилилокси)метил)-2-(2-метил-2-фенилпропил)пиперидин-1-карбоксилата (16,5 г) и TBAF (46,5 мл, 46,6 ммоль, 1 M в THF). Остаток очищали посредством колоночной хроматографии (Biotage 40-65% градиент EtOAc в гептане, колонка 340 snap, 2 раза), что давало в результате указанное в заголовке соединение. MS m/z 306 (М+Н)+. Стадия 4. 1-(Метоксикарбонил)-2-(2-метил-2-фенилпропил)пиперидин-4-карбоновая кислота. Соединение получали, как описано для ссылочного соединения 4, стадия 5, начиная с метил-4(гидроксиметил)-2-(2-метил-2-фенилпропил)пиперидин-1-карбоксилата (8,0 г, 26,2 ммоль), перйодата натрия (16,8 г, 78,6 ммоль) и хлорида рутения(III) (0,12 г, 0,58 ммоль), что давало в результате указанное в заголовке соединение (7,24 г, 87%). MS m/z 318 (М-Н)-. Ссылочное соединение 8. 2-(Циклогексилметил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-бензилизоникотинат. Метил-2-хлоризоникотинат (4,29 г, 25 ммоль) и Pd(PPh3)4 (1,156 г, 1,00 ммоль) растворяли в THF(60 мл) с получением желтого раствора. Затем добавляли бромид бензилцинка(II) (0,5 М в THF) (75 мл,37,50 ммоль). Полученную коричневую смесь нагревали до 60 С в масляной бане в течение 18 ч. Реакционную смесь гасили путем добавления метанола, затем разбавляли этилацетатом и промывали нас. NH4Cl и водой. Объединенные органические слои сушили над MgSO4 и упаривали. Остаток очищали с помо- 16020825(s, 2H), 7.19-7.34 (m, 5 Н), 7.66 (dd, 1H), 7.69 (d, 1H), 8.70 (d, 1H). Стадия 2. Метил-2-(циклогексилметил)пиперидин-4-карбоксилат. Метил-2-бензилизоникотинат (1,29 г, 5,67 ммоль) и PtO2 (0,13 г, 0,57 ммоль) добавляли к уксусной кислоте (50 мл). Реакционную смесь гидрировали в гидрогенизаторе Bchi при 8 бар (0,8 МПа) при комнатной температуре в течение 3 суток. Добавляли метанол (100 мл) и катализатор отфильтровывали. Растворитель выпаривали. Неочищенный продукт распределяли между Na2CO3 (водн.) и этилацетатом. Органическую фазу выделяли, сушили над Na2SO4, фильтровали через Celite и растворитель выпаривали. 1 Н ЯМР (600 МГц, cdcl3)0.78-0.91 (m, 2H), 1.04-1.27 (m, 5H), 1.27-1.38 (m, 1H), 1.48 (ddd, 2H), 1.63 (dd,5H), 1.80-1.93 (m, 2H), 2.36 (tt, 1H), 2.51-2.57 (m, 1H), 2.61 (td, 1H), 3.11 (ddd, 1H), 3.64 (s, 3H). Стадия 3. Диметил-2-(циклогексилметил)пиперидин-1,4-дикарбоксилат. Метил-2-(циклогексилметил)пиперидин-4-карбоксилат (1,79 г, 7,48 ммоль), метилхлорформиат(1,06 г, 11,2 ммоль) и DIPEA (1,93 г, 15,0 ммоль) добавляли к дихлорметану (60 мл) при комнатной температуре и перемешивали в течение 2 ч. Реакционную смесь разбавляли диэтиловым эфиром и промывали водой. Органическую фазу сушили над MgSO4, фильтровали через Celite и растворитель выпаривали. Неочищенный продукт 2,2 г. 1 Н ЯМР (400 МГц, cdcl3)0.71-0.97 (m, 2H), 1.02-1.28 (m, 5H), 1.38 (dt,1H), 1.69 (ddd, 6H), 1.98 (ddd, 3H), 2.52-2.61 (m, 1H), 3.01-3.16 (m, 1H), 3.67 (s, 3H), 3.69 (s, 3H), 3.81-3.94(25 мл) и воде (25 мл). Добавляли LiOH (0,266 г, 11,1 ммоль), полученную смесь перемешивали при комнатной температуре в течение ночи и кипятили с обратным холодильником в течение 30 мин. Реакционную смесь распределяли между 1 М KHSO4 и диэтиловым эфиром. Органическую фазу сушили (Na2SO4),фильтровали через Celite и растворитель выпаривали. Неочищенный продукт снова растворяли в THF(25 мл) и воде (25 мл). В реакционную колбу добавляли LiOH (0,23 г). Реакционную смесь перемешивали в течение ночи. Реакционную смесь обрабатывали, как указано выше, с получением продукта. 1 Н ЯМР(600 МГц, cdcl3)0.85 (ddd, 2H), 1.19 (dddd, 5H), 1.38-1.48 (m, 1H), 1.61 (d, 4H), 1.72 (tt, 2H), 1.97 (qd,3H), 2.56-2.65 (m, 1H), 3.02-3.14 (m, 1H), 3.67 (d, 3H), 3.88 (d, 1H), 4.22 (s, 1H). Ссылочное соединение 9. 2-(3,4-Дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(3,4-дифторфенил)изоникотинат. Смесь метил-2-хлоризоникотината (5 г, 29,14 ммоль), 3,4-дифторфенилбороновой кислоты (5,06 г,32,05 ммоль), карбоната калия (2,416 г, 17,48 ммоль) и PdCl2(dppf) (1,066 г, 1,46 ммоль) перемешивали в метаноле (30 мл) и нагревали в одномодовом микроволновом реакторе при 100 С в течение 30 мин. Растворитель выпаривали в вакууме. Добавляли DCM (400 мл) и 8% NaHCO3 (водн.) (400 мл), встряхивали и фазы разделяли. Водную фазу экстрагировали DCM (400 мл). Объединенные органические фазы сушили с помощью фазоразделителя и упаривали в вакууме. Остаток очищали посредством автоматизированной флэш-хроматографии на трех колонках Biotage KP-SIL 100 г. В качестве подвижной фазы использовали градиент от 5% EtOAc в гептане за 2 CV, затем от 5 до 20% EtOAc в гептане за 9 CV. Метил-2-(3,4 дифторфенил)изоникотинат (5,82 г, 80%) выделяли в виде белого твердого вещества. 1H ЯМР (400 МГц,cdcl3)4.00 (s, 3H), 7.23-7.33 (m, 1H), 7.76-7.84 (m, 2H), 7.90-8.00 (m, 1H), 8.21-8.27 (m, 1H), 8.82 (dd, 1H). Стадия 2. Метил-2-(3,4-дифторфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(3,4-дифторфенил)изоникотинат (5,821 г, 23,36 ммоль) растворяли в хлористом водороде(1,25 М в МеОН) (37,4 мл, 46,72 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали в вакууме. Оставшуюся HCl соль перерастворяли в МеОН (20 мл), добавляли оксид платины(IV) (0,159 г, 0,70 ммоль), реакционную смесь гидрировали в гидрогенизаторе Bchi при 5 бар (0,5 МПа) и комнатной температуре в течение 6 ч. Катализатор отфильтровывали, промывали МеОН и фильтрат упаривали в вакууме с получением метил-2-(3,4-дифторфенил)пиперидин-4-карбоксилата гидрохлорида (6,10 г, 90%) в виде бледного твердого вещества. MS m/z 256 (М+Н)+. Стадия 3. Диметил-2-(3,4-дифторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(3,4-дифторфенил)пиперидин-4-карбоксилата гидрохлорид (6,1 г, 20,91 ммоль) и DIPEA(9,13 мл, 52,28 ммоль) растворяли в DCM (30 мл). Добавляли метилхлорформиат (1,944 мл, 25,09 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч. Добавляли DCM (170 мл). Органическую фазу промывали 0,1 М HCl (2200 мл), нас. NaHCO3(200 мл), сушили с помощью фазоразделителя и упаривали в вакууме, получая диметил-2-(3,4-дифторфенил)пиперидин-1,4 дикарбоксилат (6,58 г, 100%) в виде темного масла. MS m/z 314 (M+H)+. Стадия 4. 2-(3,4-Дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(3,4-дифторфенил)пиперидин-1,4-дикарбоксилат (6,578 г, 21,00 ммоль) растворяли вTHF (40 мл) и воде (0,800 мл). Добавляли бромид лития (14,59 г, 167,97 ммоль) и TEA (11,71 мл, 83,98 ммоль) и реакционную смесь перемешивали при 80 С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и некоторое количество органического растворителя выпаривали в вакууме. Добавляли EtOAc (150 мл) и воду (150 мл), встряхивали и фазы разделяли. Органическую фазу экстрагировали водой (150 мл). Объединенные водные фазы подкисляли 3 М HCl до рН 1 и экстрагировали с помощью EtOAc (2300 мл). Объединенные органические фазы сушили с помощью Na2SO4, фильтровали и упаривали в вакууме с получением 2-(3,4-дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновой кислоты (5,66 г, 90%) в виде светло-коричневого масла. MS m/z 298 (М+Н)+. Ссылочное соединение 10. 2-(4-Фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(4-фторфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (4,5 г, 26,23 ммоль), 4-фторфенилбороновую кислоту (4,51 г, 32,26 ммоль), карбонат калия (2,247 г, 16,26 ммоль) и PdCl2(dppf) (0,380 г, 0,52 ммоль) смешивали в метаноле(30 мл) в двух 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Твердые вещества удаляли фильтрацией и фильтрат упаривали с получением темно-красной суспензии. К остатку добавляли HCl (1,25 М в метаноле, 100 мл). Смесь перемешивали при 45 С в течение 4 ч, затем концентрировали. Добавляли нас. NaHCO3 (100 мл) при охлаждении на льду и экстрагировали с помощью DCM (3100 мл). Объединенные органические фазы промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Неочищенное вещество растворяли в МТВЕ (180 мл) и некоторое количество твердых веществ отфильтровывали. Охлаждали ледяной водой, во время перемешивания добавляли по каплям хлористый водород (4 M в диоксане, 10 мл, 40 ммоль) и образовывалась суспензия. Суспензию перемешивали при 0 С в течение 10 мин. Твердое вещество собирали фильтрацией и промывали МТВЕ с получением метил-2-(4-фторфенил)изоникотината гидрохлорида (6,4 г, 91%) в виде бежевого твердого вещества. 1 Н ЯМР (400 МГц, cd3od) d 4.07 (s, 3H), 7.37-7.51 (m, 2 Н), 8.02-8.13 (m, 2H), 8.37(dd, 1H), 8.71 (d, 1H), 8.97 (dd, 1H). MS m/z 232 (M+H)+. Стадия 2. Метил-2-(4-фторфенил)пиперидин-4-карбоксилат. Метил-2-(4-фторфенил)изоникотината гидрохлорид (6,4 г, 23,91 ммоль) растворяли в метаноле (100 мл) и добавляли оксид платины(IV) (0,271 г, 1,20 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 21 ч. Катализатор отфильтровывали,промывали МеОН и фильтрат упаривали. Добавляли DCM и нас. NaHCO3 и фазы разделяли. Органический слой промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого масла. Остаток очищали посредством автоматизированной флэш-хроматографии на колонке Biotage KP-SIL 100 г. Градиент от 0 до 5% МеОН в DCM (содержащем 1% Et3N) за 10 CV использовали в качестве подвижной фазы и затем элюировали в изократическом режиме 5%-ным МеОН в DCM (содержащим 1% Et3N). Выделяли метил-2-(4-фторфенил)пиперидин-4-карбоксилат (4,2 г, 74%). MS m/z 238(М+Н)+. Стадия 3. Диметил-2-(4-фторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-фторфенил)пиперидин-4-карбоксилат (7,4 г, 31,19 ммоль) растворяли в DCM (100 мл) и добавляли DIPEA (6,52 мл, 37,43 ммоль), затем метилхлорформиат (2,95 мл, 37,43 ммоль) при 0 С. Раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь промывали 0,1 М HCl и нас. NaHCO3. Органическую фазу пропускали через фазоразделитель и упаривали с получением диметил-2-(4-фторфенил)пиперидин-1,4-дикарбоксилата (8,07 г, 97%) в виде коричневого масла. MS m/z 296(М+Н)+. Стадия 4. 2-(4-Фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(4-фторфенил)пиперидин-1,4-дикарбоксилат (8,97 г, 30,38 ммоль) растворяли в ацетонитриле (60 мл) и воде (1,200 мл), затем добавляли бромид лития (21,10 г, 243,00 ммоль). Добавляли триэтиламин (16,84 мл, 121,50 ммоль) и полученную коричневую суспензию кипятили с обратным холодильником в течение 1 ч. Добавляли воду (120 мл) и МТВЕ (120 мл). Органическую фазу экстрагировали водой (225 мл). Объединенный водный слой подкисляли до рН 1 с помощью 6 М HCl и экстрагировали МТВЕ (2150 мл). Объединенный органический слой промывали водой (25 мл), сушили над безводным(30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением оранжевого твердого вещества. Твердое вещество растворяли в МТВЕ, затем добавляли HCl (4 M в диоксане) с получением неочищенного метил-2-(4-хлорфенил)изоникотината гидрохлорида (4,3 г). MS m/z 248 (М+Н)+. Стадия 2. Метил-2-(4-хлорфенил)пиперидин-4-карбоксилат. Метил-2-(4-хлорфенил)изоникотината гидрохлорид (4,46 г, 15,69 ммоль) растворяли в МеОН (40 мл) и добавляли оксид платины(IV) (0,356 г, 1,57 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 1 ч. Катализатор отфильтровывали и промывали МеОН и элюат упаривали с получением коричневого твердого вещества. Твердое вещество растворяли в DCM и промывали 1 М K2CO3 и рассолом. Органический слой сушили с помощью фазоразделителя и упаривали. Остаток растворяли в DCM и добавляли на катионообменные колонки SCX-2 (колонки 410 г). Каждую колонку промывали DCM, МеОН и затем элюировали NaH3/МеОН (1 CV для каждого). NaH3/МеОН слой упаривали, получая метил-2-(4-хлорфенил)пиперидин-4-карбоксилат (3,13 г,79%). MS m/z 254 (М+Н)+. Стадия 3. Диметил-2-(4-хлорфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-хлорфенил)пиперидин-4-карбоксилат (3,13 г, 12,34 ммоль) растворяли в DCM (50 мл),затем добавляли DIPEA (5,39 мл, 30,84 ммоль). К раствору добавляли по каплям метилхлорформиат(1,360 мл, 17,27 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Смесь промывали 0,1 М HCl (2100 мл) и нас. NaHCO3 (100 мл), затем сушили с помощью фазоразделителя и упаривали, получая диметил-2-(4-хлорфенил)пиперидин-1,4-дикарбоксилат (3,97 г, колич.) в виде оранжевого масла. MS m/z 312 (М+Н)+. Стадия 4. 2-(4-Хлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(4-хлорфенил)пиперидин-1,4-дикарбоксилат (3,97 г, 12,73 ммоль) растворяли в ацетонитриле (55 мл) и воде (1,1 мл), затем добавляли бромид лития (8,85 г, 101,87 ммоль). Добавляли Et3N(7,10 мл, 50,94 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 2 ч. Добавляли воду (100 мл) и МТВЕ (150 мл). Фазы разделяли и органический слой экстрагировали водой (2). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и экстрагировали МТВЕ (2). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали с получением 2-(4 хлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновой кислоты (3,49 г, 92%) в виде желтого полутвердого вещества. MS m/z 298 (М+Н)+. Ссылочное соединение 12. 1-(Метоксикарбонил)-2-(3-(трифторметил)фенил)пиперидин-4 карбоновая кислота. Стадия 1. Метил-2-(3-(трифторметил)фенил)изоникотинат. Метил-2-бромизоникотинат (2,71 г, 12,57 ммоль), 3-(трифторметил)фенилбороновую кислоту (3,58 г, 18,85 ммоль), карбонат калия (2,61 г, 18,85 ммоль) и PdCl2(dppf) (0,162 г, 0,25 ммоль) смешивали в метаноле (30 мл) в двух 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли DCM и воду и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Остаток растворяли в DCM и добавляли на катионообменную колонку SCX-2. Колонку промывали DCM, МеОН и затем элюировали смесью NaH3/МеОН (1 CV для каждого). NaH3/МеОН слой упаривали, получая метил-2-(3-(трифторметил)фенил)изоникотинат (2,6 г, 73%). MS m/z 282 (М+Н)+. Стадия 2. Метил-2-(3-(трифторметил)фенил)пиперидин-4-карбоксилат. Метил-2-(3-(трифторметил)фенил)изоникотинат (2,6 г, 9,25 ммоль) растворяли в уксусной кислоте(40 мл) и добавляли оксид платины(IV) (0,210 г, 0,92 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 3 ч. Катализатор отфильтровывали, промывали МеОН и элюат упаривали. Добавляли DCM и 1 М K2CO3 и фазы разделяли. Органический слой промывали рассолом, пропускали через фазоразделитель и упаривали с получением метил-2-(3(трифторметил)фенил)пиперидин-4-карбоксилата (1,4 г, 52%) в виде слегка коричневого масла. MS m/z 288 (М+Н)+. Стадия 3. Диметил-2-(3-(трифторметил)фенил)пиперидин-1,4-дикарбоксилат. Метил-2-(3-(трифторметил)фенил)пиперидин-4-карбоксилат (1,4 г, 4,87 ммоль) растворяли в DCM(50 мл), затем добавляли DIPEA (2,128 мл, 12,18 ммоль). К раствору добавляли по каплям метилхлорформиат (0,537 мл, 6,82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч, промывали 0,1 М HCl (100 мл) и нас. NaHCO3 (100 мл), сушили с помощью фазоразделителя и упаривали, получая диметил-2-(3-(трифторметил)фенил)пиперидин-1,4-дикарбоксилат (1,7 г, колич.) в виде оранжевого масла. MS m/z 346 (М+Н)+. Стадия 4. 1-(Метоксикарбонил)-2-(3-(трифторметил)фенил)пиперидин-4-карбоновая кислота. Диметил-2-(3-(трифторметил)фенил)пиперидин-1,4-дикарбоксилат (1,7 г, 4,92 ммоль) растворяли в ацетонитриле (22 мл) и воде (0,440 мл), затем добавляли бромид лития (3,42 г, 39,38 ммоль). ДобавлялиEt3N (2,74 мл, 19,69 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 2 ч. Добавляли воду (50 мл) и МТВЕ (150 мл). Фазы разделяли и органический слой экстрагировали водой (2 раза). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и экстрагировали МТВЕ (2 раза). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали,- 19020825 получая 1-(метоксикарбонил)-2-(3-(трифторметил)фенил)пиперидин-4-карбоновую кислоту (1,48 г, 91%) в виде слегка желтого полутвердого вещества. MS m/z 332 (М+Н)+. Ссылочное соединение 13. 1-(Метоксикарбонил)-2-(4-(трифторметил)фенил)пиперидин-4 карбоновая кислота. Стадия 1. Метил-2-(4-(трифторметил)фенил)изоникотинат. Метил-2-хлоризоникотинат (5 г, 29,14 ммоль), 4-(трифторметил)фенилбороновую кислоту (8,30 г,43,71 ммоль), карбонат калия (6,04 г, 43,71 ммоль) и PdCl2(dppf) (0,633 г, 0,87 ммоль) смешивали в метаноле (30 мл) в двух 20-мл флаконах для микроволновой обработки. Две капли воды добавляли в один из флаконов, флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Твердые вещества удаляли фильтрацией и фильтрат упаривали с получением темнокрасной суспензии. Добавляли DCM и воду и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Неочищенное вещество растворяли в МТВЕ (180 мл) и твердые вещества отфильтровывали. Во время перемешивания добавляли по каплям хлористый водород(4 M в диоксане) (7,29 мл, 29,14 ммоль) и образовывалась суспензия. Суспензию перемешивали при комнатной температуре в течение 2,5 ч. Твердое вещество собирали фильтрацией и промывали МТВЕ. К твердому веществу добавляли МТВЕ и нас. NaHCO3, органическую фазу сушили (Na2SO4), фильтровали и упаривали с получением метил-2-(4-(трифторметил)фенил)изоникотината (5,75 г, 70%) в виде бежевого твердого вещества. 1 Н ЯМР (400 МГц, cdcl3)4.00 (s, 3H), 7.75 (d, 2H), 7.84 (dd, 1H), 8.18 (d, 2H), 8.33 (s,1H), 8.87 (dd, 1H). MS m/z 282 (M+H)+. Стадия 2. Метил-2-(4-(трифторметил)фенил)пиперидин-4-карбоксилат. Метил-2-(4-(трифторметил)фенил)изоникотинат (4,985 г, 17,73 ммоль) растворяли в уксусной кислоте (45 мл) и добавляли оксид платины(IV) (0,232 г, 1,02 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 5 ч. Добавляли дополнительно оксид платины(IV) (0,116 г, 0,51 ммоль) и гидрирование продолжали при 5 бар (0,5 МПа) в течение 1 ч. Катализатор отфильтровывали и промывали МеОН и элюат упаривали. Добавляли DCM и 10%K2CO3 и фазы разделяли. Органический слой промывали рассолом, пропускали через фазоразделитель и упаривали с получением метил-2-(4-(трифторметил)фенил)пиперидин-4-карбоксилата (5,04 г, 99%) в виде коричневого масла. MS m/z 288 (М+Н)+. Стадия 3. Диметил-2-(4-(трифторметил)фенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-(трифторметил)фенил)пиперидин-4-карбоксилат (4,973 г, 17,31 ммоль) растворяли вDCM (100 мл) и добавляли DIPEA (3,62 мл, 20,77 ммоль), затем метилхлорформиат (1,636 мл, 20,77 ммоль). Раствор перемешивали при комнатной температуре в течение 1 ч 45 мин. Реакционную смесь промывали 0,1 М HCl и нас. NaHCO3. Органическую фазу пропускали через фазоразделитель и упаривали с получением неочищенного диметил-2-(4-(трифторметил)фенил)пиперидин-1,4-дикарбоксилата (6,05 г, 101%) в виде коричневого масла. MS m/z 346 (М+Н)+. Стадия 4. 1-(Метоксикарбонил)-2-(4-(трифторметил)фенил)пиперидин-4-карбоновая кислота. Диметил-2-(4-(трифторметил)фенил)пиперидин-1,4-дикарбоксилат (5,996 г, 17,36 ммоль) растворяли в ацетонитриле (60 мл) и воде (1,200 мл), затем добавляли бромид лития (12,06 г, 138,91 ммоль). Добавляли триэтиламин (9,63 мл, 69,46 ммоль) и полученную коричневую суспензию кипятили с обратным холодильником в течение 1 ч. Добавляли воду и МТВЕ. Органическую фазу экстрагировали водой (2). Объединенный водный слой подкисляли до рН 1 с помощью 3,8 М водн. HCl и затем экстрагировали МТВЕ (2). Объединенный органический слой промывали водой и упаривали. Остатки воды подвергали азеотропному удалению с помощью MeCN. 1-(Метоксикарбонил)-2-(4-(трифторметил)фенил)пиперидин 4-карбоновую кислоту (5,71 г, 99%) выделяли в виде коричневого масла. MS m/z 332 (М+Н)+. Ссылочное соединение 14. 2-(3-трет-Бутилфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. 2-(3-трет-Бутилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан. 1-Бром-3-трет-бутилбензол (4,48 г, 21 ммоль) растворяли в DMSO (150 мл) и добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (5,87 г, 23,10 ммоль), ацетат калия (6,18 г, 63,00 ммоль) и Pd(PPh3)4 (1,213 г, 1,05 ммоль). Полученную коричневую суспензию нагревали до 80 С в атмосфере азота. После 9 ч добавляли воду и диэтиловый эфир и фазы разделяли. Органическую фазу упаривали и воду подвергали азеотропному удалению с помощью MeCN. Остаток очищали с помощью Biotage за два раза (от 0 до 20% EtOAc в гептане, 5 CV; колонка Biotage KP-SIL 340 г) с получением желтого масла. Очищали снова с помощью Biotage (от 0 до 20% EtOAc в гептане, 8 CV; колонка Biotage KP-SIL 340 г). 2-(3-трет-Бутилфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (3,39 г, 62%) выделяли в виде желтого масла. 1 Н ЯМР (600 МГц, cdcl3)1.32 (s, 9H), 1.33 (s, 12H), 7.29 (t, 1H), 7.47-7.50 (m, 1H), 7.61 (d,1H), 7.81 (s, 1H). Стадия 2. Метил-2-(3-трет-бутилфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (1,7 г, 9,91 ммоль), 2-(3-трет-бутилфенил)-4,4,5,5-тетраметил-1,3,2 диоксаборолан (3,393 г, 13,04 ммоль), карбонат калия (2,054 г, 14,86 ммоль) и PdCl2(dppf) (0,215 г, 0,30 ммоль) смешивали в метаноле (14 мл) в 20-мл флаконе для микроволновой обработки. Флакон закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Реакционную смесь суспендировали в метаноле, твердые вещества удаляли фильтрацией и фильтрат упаривали с получением темно-красной суспензии. Добавляли DCM и воду и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали до коричневого твердого вещества. Твердое вещество растворяли в МТВЕ (100 мл) и твердые вещества отфильтровывали. Во время перемешивания добавляли по каплям хлористый водород (4 M в диоксане, 3,50 мл, 13,99 ммоль) и образовывалась суспензия. Суспензию перемешивали при комнатной температуре в течение 3 суток. Твердое вещество собирали фильтрацией и промывали МТВЕ с получением метил-2-(3-трет-бутилфенил)изоникотината гидрохлорида (2,14 г, 70%) в виде слегка коричневого твердого вещества. 1 Н ЯМР (600 МГц, cd3od)1.41 (s, 9H), 4.07 (s, 3H), 7.62 (t, 1H), 7.777.80 (m, 2H), 8.00-8.02 (m, 1H), 8.37-8.40 (m, 1H), 8.69-8.71 (m, 1H), 8.94-8.97 (m, 1H). Стадия 3. Метил-2-(3-трет-бутилфенил)пиперидин-4-карбоксилат. Метил-2-(3-трет-бутилфенил)изоникотината гидрохлорид (3 г, 9,81 ммоль) растворяли в метаноле(100 мл) и добавляли оксид платины(IV) (0,111 г, 0,49 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 13 ч. К смеси добавляли оксид платины(IV) (40 мг) и смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар(0,5 МПа) в течение 1 ч. Катализатор отфильтровывали и промывали МеОН и элюат упаривали. Добавляли DCM и нас. NaHCO3 и фазы разделяли. Промывали рассолом, пропускали через фазоразделитель и упаривали с получением метил-2-(3-трет-бутилфенил)пиперидин-4-карбоксилата (2,56 г, 95%) в виде коричневого масла. Стадия 4. Диметил-2-(3-трет-бутилфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(3-трет-бутилфенил)пиперидин-4-карбоксилат (2,56 г, 9,30 ммоль) растворяли в DCM (100 мл) и добавляли DIPEA (2,429 мл, 13,94 ммоль), затем метилхлорформиат (0,9 мл, 11,43 ммоль) при 0 С. Раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь промывали 1 М водн. HCl (рН 4), затем рассолом, нас. NaHCO3. Органическую фазу пропускали через фазоразделитель и упаривали с получением диметил-2-(3-трет-бутилфенил)пиперидин-1,4-дикарбоксилата (3,17 г, колич.) в виде коричневого масла. Стадия 5. 2-(3-трет-Бутилфепил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(3-трет-бутилфенил)пиперидин-1,4-дикарбоксилат (3,17 г, 9,51 ммоль) растворяли в ацетонитриле (60 мл) и воде (1,200 мл), затем добавляли одной порцией бромид лития (6,61 г, 76,06 ммоль). Добавляли триэтиламин (5,27 мл, 38,03 ммоль) и полученную коричневую суспензию кипятили с обратным холодильником в течение 1,5 ч. Добавляли воду (120 мл) и МТВЕ (120 мл). Органическую фазу экстрагировали водой (225 мл). Объединенный водный слой подкисляли до рН 1 с 0,5 М HCl (30 мл) и экстрагировали МТВЕ (2150 мл). Объединенный органический слой промывали водой (25 мл), сушили надNa2SO4 и упаривали. 2-(3-трет-Бутилфенил)-1-(метоксикарбонил)пиперидин-4-карбоновую кислоту (2,73 г, 90%) выделяли в виде пены. Ссылочное соединение 15. 2-(4-трет-Бутилфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(4-трет-бутилфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (7 г, 40,80 ммоль), 4-трет-бутилфенилбороновую кислоту (10 г, 56,17 ммоль), карбонат калия (3,5 г, 25,32 ммоль) и PdCl2(dppf) (0,9 г, 1,24 ммоль) смешивали в метаноле (30 мл) двумя равными порциями в 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Твердые вещества удаляли фильтрацией и фильтрат упаривали с получением темно-красной суспензии. Добавляли DCM и рассол и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенные органические фазы сушили над MgSO4 и упаривали. Неочищенное вещество растворяли в МТВЕ (180 мл) и твердые вещества отфильтровывали. Во время перемешивания добавляли по каплям хлористый водород(4 M в диоксане, 10,20 мл, 40,80 ммоль) и образовывалась суспензия. Суспензию перемешивали при комнатной температуре в течение 1 ч. Твердое вещество собирали фильтрацией и промывали МТВЕ. Метил-2-(4-трет-бутилфенил)изоникотината гидрохлорид (9,6 г, 77%) выделяли в виде темнокоричневого твердого вещества. 1 Н ЯМР (400 МГц, DMSO)1.30 (s, 9H), 3.92 (s, 3H), 7.53 (d, 2H), 7.80(100 мл) добавляли оксид платины(IV) (0,713 г, 3,14 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при 5 бар (0,5 МПа). Через 8 ч реакционную смесь фильтровали через картонный фильтр с диатомовой землей, остаток промывали метанолом и фильтрат упаривали. Метил-2-(4-третбутилфенил)пиперидин-4-карбоксилата гидрохлорид (9,66 г, 99%) выделяли в виде коричневого твердого вещества. 1H ЯМР (400 МГц, DMSO)1.26 (s, 9H), 1.88-2.15 (m, 4 Н), 2.82-2.92 (m, 1H), 3.03-3.16 (m,1H), 3.27-3.38 (m, 1H), 3.61 (s, 3H), 4.22-4.29 (m, 1H), 7.42-7.53 (m, 4 Н), 9.43(br, 2 Н). MS m/z 276 (М+Н)+. Стадия 3. Диметил-2-(4-трет-бутилфенил)пиперидин-1,4-дикарбоксилат. К суспензии метил-2-(4-трет-бутилфенил)пиперидин-4-карбоксилата гидрохлорида (9,66 г, 30,98 ммоль) в дихлорметане (100 мл) добавляли DIPEA (12 мл, 68,89 ммоль). Реакционную смесь охлаждали в ледяной бане и добавляли по каплям метилхлорформиат (2,8 мл, 35,56 ммоль) в течение 5 мин. Реакционную смесь перемешивали в течение 18 ч при комнатной температуре и промывали 3,8 н. HCl. Водный слой экстрагировали DCM. Объединенные органические слои сушили над MgSO4 и упаривали. Диметил-2-(4-трет-бутилфенил)пиперидин-1,4-дикарбоксилат (10,6 г, колич.) выделяли в виде темно-коричневого масла. MS m/z 334 (М+Н). Стадия 4. 2-(4-трет-Бутилфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. К раствору диметил-2-(4-трет-бутилфенил)пиперидин-1,4-дикарбоксилата (10,937 г, 32,80 ммоль) вMeCN (100 мл) добавляли бромид лития (6,58 мл, 262,42 ммоль), триэтиламин (18,19 мл, 131,21 ммоль) и воду (2 мл). Полученную смесь кипятили с обратным холодильником в течение 3 ч, затем охлаждали до комнатной температуры. Растворители выпаривали и остаток растворяли в воде. Водный слой промывали МТВЕ, затем подкисляли путем добавления 3,8 н. HCl. Водный слой три раза экстрагировали DCM. Объединенные органические слои сушили над MgSO4 и упаривали. 2-(4-трет-Бутилфенил)-1(метоксикарбонил)пиперидин-4-карбоновую кислоту (9,5 г, 91%) выделяли в виде светло-коричневой пены. MS m/z 320 (М+Н)+. Ссылочное соединение 16. 1-(Метоксикарбонил)-2-(4-(метилсульфонил)фенил)пиперидин-4 карбоновая кислота. Стадия 1. Метил-2-(4-(метилсульфонил)фенил)изоникотинат. Метил-2-хлоризоникотинат (5 г, 29,14 ммоль), 4-(метилсульфонил)фенилбороновую кислоту (7,58 г, 37,88 ммоль), карбонат калия (2,416 г, 17,48 ммоль) и PdCl2(dppf) (0,633 г, 0,87 ммоль) растворяли в метаноле (30 мл) и распределяли в двух флаконах для микроволновой обработки. Каждый флакон нагревали до 100 С в одномодовом микроволновом реакторе в течение 35 мин. Добавляли DCM (250 мл) и воду (250 мл), встряхивали и фазы разделяли. Водную фазу экстрагировали DCM (250 мл). Объединенные органические фазы промывали рассолом (500 мл), сушили с помощью фазоразделителя и упаривали в вакууме. Остаток очищали посредством автоматизированной флэш-хроматографии на двух колонкахBiotage KP-SIL 340 г. В качестве подвижной фазы использовали градиент 20-70% EtOAc в гептане за 10 CV и затем 70% EtOAc в гептане за 5 CV. Продукт собирали, используя длину волны 253 нм. Фракции продукта собирали и упаривали в вакууме, получая метил-2-(4-(метилсульфонил)фенил)изоникотинат(5,66 г, 66,7%) в виде белого твердого вещества. 1 Н ЯМР (600 МГц, CDCl3)3.09 (s, 3H), 3.98 (s, 3H),7.85 (dd, 1H), 8.05 (d, 2H), 8.25 (d, 2 Н), 8.34 (s, 1H), 8.86 (t, 1H). Стадия 2. Метил-2-(4-(метилсульфонил)фенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(4-(метилсульфонил)фенил)изоникотинат (5,663 г, 19,44 ммоль) суспендировали в МеОН(20 мл) и добавляли хлористый водород (1,25 М в МеОН, 40 мл, 50,00 ммоль). Смесь перемешивали при комнатной температуре в течение 20 мин и затем упаривали в вакууме. HCl соль суспендировали в МеОН (100 мл) и добавляли оксид платины(IV) (0,221 г, 0,97 ммоль). Смесь гидрировали в гидрогенизатореBchi при комнатной температуре и 5 бар (0,5 МПа) в течение 2 ч. Катализатор отфильтровывали и промывали МеОН. Фильтрат упаривали в вакууме, получая метил-2-(4-(метилсульфонил)фенил)пиперидин 4-карбоксилата гидрохлорид (5,84 г, 90%) в виде белого твердого вещества. MS m/z 298 (М+Н)+. Стадия 3. Диметил-2-(4-(метилсульфонил)фенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-(метилсульфонил)фенил)пиперидин-4-карбоксилата гидрохлорид (5,2 г, 15,58 ммоль) иDIPEA (6,78 мл, 38,94 ммоль) растворяли в DCM (40 мл) и добавляли метилхлорформиат (1,472 мл, 18,69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 50 мин. ДобавлялиDCM (160 мл). Органическую фазу промывали 0,1 М HCl (2200 мл), нас. NaHCO3 (200 мл), сушили с помощью фазоразделителя и упаривали в вакууме, получая неочищенный диметил-2-(4(метилсульфонил)фенил)пиперидин-1,4-дикарбоксилат (5,95 г, 108%). MS m/z 356 (М+Н)+. Стадия 4. 1-(Метоксикарбонил)-2-(4-(метилсульфонил)фенил)пиперидин-4-карбоновая кислота. Диметил-2-(4-(метилсульфонил)фенил)пиперидин-1,4-дикарбоксилат (5,952 г, 16,75 ммоль) растворяли в ацетонитриле (50 мл) и воде (1000 мл), затем добавляли бромид лития (11,64 г, 133,98 ммоль). Добавляли триэтиламин (9,29 мл, 66,99 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 1,5 ч. Добавляли МТВЕ (150 мл) и воду (150 мл), встряхивали и фазы разделяли. Органическую фазу экстрагировали водой (2200 мл). Объединенные водные фазы подкисляли до рН 1 с помощью 3 М HCl и экстрагировали МТВЕ (2500 мл). Объединенные органические фазы сушили с помощью в метаноле (30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением оранжевого твердого вещества. Твердое вещество разбавляли МТВЕ, затем добавляли HCl (4 M в диоксане) с получением суспензии. Твердые вещества собирали фильтрацией с получением метил 6'-(трифторметил)-2,3'бипиридин-4-карбоксилата гидрохлорида (3,2 г, 57%). MS m/z 283 (М+Н)+. Стадия 2. Метил-2-(6-(трифторметил)пиридин-3-ил)пиперидин-4-карбоксилата гидрохлорид. Метил 6'-(трифторметил)-2,3'-бипиридин-4-карбоксилата гидрохлорид (2,5 г, 7,86 ммоль) растворяли в МеОН (20 мл) и добавляли оксид платины(IV) (0,036 г, 0,16 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 1 ч. Добавляли дополнительное количество катализатора и гидрирование продолжали в течение 1 ч. Катализатор отфильтровывали, промывали МеОН и элюат упаривали, получая метил-2-(6-(трифторметил)пиридин-3 ил)пиперидин-4-карбоксилата гидрохлорид (2,2 г, 86%) в виде коричневого твердого вещества. MS m/z 289 (М+Н)+. Стадия 3. Диметил-2-(6-(трифторметил)пиридин-3-ил)пиперидин-1,4-дикарбоксилат. Метил-2-(6-(трифторметил)пиридин-3-ил)пиперидин-4-карбоксилата гидрохлорид (3 г, 9,24 ммоль) растворяли в DCM (50 мл) и DIPEA (4,03 мл, 23,10 ммоль). К раствору добавляли по каплям метилхлорформиат (1,019 мл, 12,93 ммоль). Смесь перемешивали при комнатной температуре в течение 10 мин. Смесь промывали 0,1 М HCl (2100 мл) и нас. NaHCO3 (100 мл), затем сушили с помощью фазоразделителя и упаривали, получая неочищенный диметил-2-(6-(трифторметил)пиридин-3-ил)пиперидин-1,4 дикарбоксилат (3 г, 94%) в виде темно-коричневого масла. MS m/z 347 (М+Н)+. Стадия 4. 1-(Метоксикарбонил)-2-(6-(трифторметил)пиридин-3-ил)пиперидин-4-карбоновая кислота. Неочищенный диметил-2-(6-(трифторметил)пиридин-3-ил)пиперидин-1,4-дикарбоксилат (2,9 г, 8,38 ммоль) растворяли в ацетонитриле (35 мл) и воде (0,700 мл), затем добавляли бромид лития (2,91 г, 33,50 ммоль). Добавляли Et3N (2,334 мл, 16,75 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 1 ч. Добавляли воду (100 мл) и МТВЕ (150 мл). Фазы разделяли и органический слой экстрагировали водой (2 раза). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и экстрагировали в МТВЕ (2 раза). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали, получая неочищенную 1-(метоксикарбонил)-2-(6-(трифторметил)пиридин-3 ил)пиперидин-4-карбоновую кислоту (2,5 г, 90%) в виде слегка желтого твердого вещества. MS m/z 333(М+Н)+. Ссылочное соединение 18. 2-(5-трет-Бутилтиофен-2-ил)-1-(метоксикарбонил)пиперидин-4 карбоновая кислота. Стадия 1. Метил-2-(5-трет-бутилтиофен-2-ил)изоникотинат. Метил-2-хлоризоникотинат (175 мг, 1,02 ммоль), 2-(5-трет-бутилтиофен-2-ил)-4,4,5,5-тетраметил 1,3,2-диоксаборолан (300 мг, 1,13 ммоль), K2CO3(85 мг, 0,61 ммоль) и PdCl2(dppf) (37,3 мг, 0,05 ммоль) перемешивали в метаноле (2 мл) и нагревали в одномодовом микроволновом реакторе до 100 С в течение 30 мин. Добавляли DCM (50 мл) и воду (50 мл), встряхивали и фазы разделяли. Водную фазу экстрагировали DCM (50 мл). Объединенные органические фазы сушили с помощью фазоразделителя и упаривали в вакууме. Остаток очищали посредством автоматизированной флэш-хроматографии на колонкеBiotage KP-SIL 50 г. В качестве подвижной фазы использовали градиент от 5% EtOAc в гептане за 2CV, затем от 5 до 20% EtOAc в гептане за 9 CV. Продукт собирали, используя длину волны 295 нм. Фракции продукта собирали и упаривали в вакууме, получая метил-2-(5-трет-бутилтиофен-2 ил)изоникотинат (213 мг, 76%). 1H ЯМР (600 МГц, cdcl3)1.41 (s, 9H), 3.96 (s, 3H), 6.85 (d, 1H), 7.50 (s,1H), 7.61 (d, 1H), 8.13 (s, 1H), 8.64 (d, 1H). MS m/z 276 (M+H)+. Стадия 2. Метил-2-(5-трет-бутилтиофен-2-ил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(5-трет-бутилтиофен-2-ил)изоникотинат (2,02 г, 7,34 ммоль) растворяли в хлористом водороде (1,25 М в МеОН, 11,74 мл, 14,67 ммоль) и перемешивали при комнатной температуре в течение 30 мин. Растворитель выпаривали в вакууме. Остаток растворяли в МеОН (20 мл), добавляли оксид платины(IV) (0,050 г, 0,22 ммоль) и реакционную смесь гидрировали в гидрогенизаторе Bchi при 5 бар (0,5 МПа) и комнатной температуре в течение 3 ч. Добавляли дополнительно оксид платины(IV) (0,050 г, 0,22 ммоль) и гидрирование продолжали в течение 3 ч. Реакционную смесь фильтровали через целит, промывали МеОН и фильтрат упаривали в вакууме. Остаток перерастворяли в МеОН (15 мл) и добавляли оксид платины(IV) (0,167 г, 0,73 ммоль). Гидрирование продолжали в течение 3 ч. Добавляли дополнительно оксид платины(IV) (0,167 г, 0,73 ммоль) и гидрирование продолжали в течение 2 ч. Реакционную смесь фильтровали через целит, промывали МеОН и фильтрат упаривали в вакууме с получением метил-2-(5 трет-бутилтиофен-2-ил)пиперидин-4-карбоксилата гидрохлорида (2,360 г, 101%) в виде черного масла.DIPEA (3,24 мл, 18,56 ммоль) растворяли в DCM (30 мл). Добавляли метилхлорформиат (0,690 мл, 8,91 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли DCM(170 мл). Органическую фазу промывали 0,1 М HCl (2200 мл), нас. NaHCO3 (200 мл), сушили с помощью фазоразделителя и упаривали в вакууме, получая диметил-2-(5-трет-бутилтиофен-2-ил)пиперидин 1,4-дикарбоксилат (2,490 г, 99%) в виде темного масла. MS m/z 340 (М+Н)+. Стадия 4. 2-(5-трет-Бутилтиофен-2-ил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(5-трет-бутилтиофен-2-ил)пиперидин-1,4-дикарбоксилат (2,49 г, 7,34 ммоль) растворяли в ацетонитриле (20 мл). Добавляли воду (0,400 мл) и бромид лития (5,10 г, 58,68 ммоль). Через 5 мин добавляли TEA (4,09 мл, 29,34 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 1 ч. Добавляли МТВЕ (150 мл) и воду (150 мл), встряхивали и фазы разделяли. Органическую фазу экстрагировали водой (150 мл). Объединенные водные фазы подкисляли 3 М HCl до рН 1 и экстрагировали МТВЕ (2300 мл). Объединенные органические фазы сушили с помощью Na2SO4, фильтровали и упаривали в вакууме, получая 2-(5-трет-бутилтиофен-2-ил)-1-(метоксикарбонил)пиперидин-4 карбоновую кислоту (1,125 г, 47,1%) в виде белой пены. MS m/z 326 (М+Н)+. Ссылочное соединение 19. 2-(2,4-Дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(2,4-дифторфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (3 г, 17,48 ммоль), 2,4-дифторфенилбороновую кислоту (4,14 г, 26,23 ммоль), карбонат калия (1,812 г, 13,11 ммоль) и PdCl2(dppf) (0,380 г, 0,52 ммоль) смешивали в метаноле(30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Твердое вещество растворяли в МТВЕ и перемешивали в течение 15 мин, затем фильтровали. Оранжевый МТВЕ слой обрабатывали хлористым водородом (4 M в диоксане, 4,37 мл, 17,48 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Образовавшееся твердое вещество собирали фильтрацией. Метил-2-(2,4-дифторфенил)изоникотината гидрохлорид (4,0 г, 80%) выделяли в виде слегка оранжевого твердого вещества. MS m/z 250 (М+Н)+. Стадия 2. Метил-2-(2,4-дифторфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(2,4-дифторфенил)изоникотината гидрохлорид (3,93 г, 13,76 ммоль) растворяли в МеОН(40 мл) и добавляли оксид платины(IV) (0,312 г, 1,38 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 2 ч. Катализатор отфильтровывали, промывали МеОН и элюат упаривали с получением метил-2-(2,4-дифторфенил)пиперидин-4 карбоксилата гидрохлорида (2,34 г, 58%) в виде слегка желтого твердого вещества. MS m/z 256 (М+Н)+. Стадия 3. Диметил-2-(2,4-дифторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(2,4-дифторфенил)пиперидин-4-карбоксилата гидрохлорид (2,34 г, 8,02 ммоль) растворяли в DCM (50 мл), затем добавляли DIPEA (3,50 мл, 20,05 ммоль). К раствору добавляли по каплям метилхлорформиат (0,884 мл, 11,23 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч,промывали 0,1 М HCl (100 мл) и нас. NaHCO3 (100 мл), затем сушили с помощью фазоразделителя и упаривали, получая диметил-2-(2,4-дифторфенил)пиперидин-1,4-дикарбоксилат (2,38 г, 95%) в виде оранжевого масла. Стадия 4. 2-(2,4-Дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(2,4-дифторфенил)пиперидин-1,4-дикарбоксилат (2,38 г, 7,60 ммоль) растворяли в ацетонитриле (35 мл) и воде (0,700 мл), затем добавляли бромид лития (5,28 г, 60,77 ммоль). Добавляли Et3N(4,24 мл, 30,39 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 2 ч. Добавляли воду (50 мл) и МТВЕ (150 мл). Фазы разделяли и органический слой экстрагировали водой (2 раза). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и экстрагировали МТВЕ (2 раза). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали, получая 2-(2,4-дифторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновую кислоту (2,27, колич.) в виде слегка желтого полутвердого вещества. MS m/z 300 (М+Н)+. Ссылочное соединение 20. 2-(4-Хлор-2-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(4-хлор-2-фторфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (4 г, 23,31 ммоль), 4-хлор-2-фторфенилбороновую кислоту (6,10 г, 34,97 ммоль), карбонат калия (2,416 г, 17,48 ммоль) и PdCl2(dppf) (0,506 г, 0,70 ммоль) смешивали в метаноле(30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением темно-оранжевого твер- 24020825 дого вещества. Твердое вещество растворяли в МТВЕ и перемешивали в течение 15 мин, затем фильтровали. Оранжевый МТВЕ слой обрабатывали хлористым водородом (4 M в диоксане, 5,83 мл, 23,31 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Осадок собирали фильтрацией с получением метил-2-(4-хлор-2-фторфенил)изоникотината гидрохлорида (7,22 г, колич.) в виде слегка оранжевого твердого вещества. MS m/z 266 (М+Н)+. Стадия 2. Метил-2-(4-хлор-2-фторфенил)пиперидин-4-карбоксилат. Метил-2-(4-хлор-2-фторфенил)изоникотината гидрохлорид (7,26 г, 24,03 ммоль) растворяли в МеОН (100 мл) и добавляли оксид платины(IV) (0,546 г, 2,40 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 1 ч. Катализатор отфильтровывали и промывали МеОН и элюат упаривали, получая метил-2-(4-хлор-2 фторфенил)пиперидин-4-карбоксилата гидрохлорид (7,45 г, колич.) в виде коричневого твердого вещества. MS m/z 272 (М+Н)+. Стадия 3. Диметил-2-(4-хлор-2-фторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-хлор-2-фторфенил)пиперидин-4-карбоксилат (7,45 г, 27,42 ммоль) растворяли в DCM(120 мл), затем добавляли DIPEA (11,97 мл, 68,55 ммоль). К раствору добавляли по каплям метилхлорформиат (3,02 мл, 38,39 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Смесь промывали 0,1 М HCl (2100 мл) и нас. NaHCO3 (100 мл), затем сушили с помощью фазоразделителя и упаривали с получением диметил-2-(4-хлор-2-фторфенил)пиперидин-1,4-дикарбоксилата (7,77 г, 86%) в виде оранжевого масла. MS m/z 330 (М+Н)+. Стадия 4. 2-(4-Хлор-2-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(4-хлор-2-фторфенил)пиперидин-1,4-дикарбоксилат (7,77 г, 23,56 ммоль) растворяли в ацетонитриле (100 мл) и воде (2000 мл), затем добавляли бромид лития (16,37 г, 188,51 ммоль). Добавляли Et3N (13,14 мл, 94,25 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 2 ч. Добавляли воду (200 мл) и МТВЕ (300 мл). Фазы разделяли и органический слой экстрагировали водой (2 раза). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и экстрагировали МТВЕ (2 раза). Объединенный органический слой сушили над MgSO4, фильтровали и упаривали,получая 2-(4-хлор-2-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновую кислоту (7,28 г, 98%) в виде желтого полутвердого вещества. MS m/z 316 (М+Н)+. Ссылочное соединение 21. 2-(2-Хлор-4-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(2-хлор-4-фторфенил)изоникотината гидрохлорид. Метил-2-хлоризоникотинат (3,5 г, 20,40 ммоль), 2-хлор-4-фторфенилбороновую кислоту (3,91 г,22,44 ммоль), карбонат калия (2,114 г, 15,30 ммоль) и PdCl2(dppf) (0,443 г, 0,61 ммоль) смешивали в метаноле (30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением темнокоричневого твердого вещества. Твердое вещество растворяли в МТВЕ и перемешивали в течение 15 мин, затем фильтровали. МТВЕ слой обрабатывали хлористым водородом (4 М в диоксане, 5,10 мл,20,40 ммоль) и перемешивали в течение 1 ч при комнатной температуре. Осадок собирали фильтрацией. Метил-2-(2-хлор-4-фторфенил)изоникотинат (2,350 г, 38,1%) получали в виде светло-желтого твердого вещества. 1 Н ЯМР (400 МГц, cdcl3)4.13 (s, 3H), 7.24-7.33 (m, 1H), 7.36 (d, 1H), 7.82-8.03 (m, 1H), 8.34 (s,br., 1H), 8.56 (s, br., 1H), 9.08 (s, br., 1H). MS m/z 266 (M+H)+. Стадия 2. Метил-2-(2-хлор-4-фторфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(2-хлор-4-фторфенил)изоникотината гидрохлорид (2,35 г, 7,78 ммоль) растворяли в метаноле (50 мл) и добавляли оксид платины(IV) (0,177 г, 0,78 ммоль). Реакционную смесь гидрировали в гидрогенизаторе Bchi при 5 бар (0,5 МПа) и комнатной температуре в течение 9 ч. Катализатор удаляли фильтрацией и растворитель выпаривали. Остаток растворяли в метаноле (50,0 мл) и добавляли оксид платины(IV) (144 мг, 0,63 ммоль). Гидрирование продолжали при 5 бар (0,5 МПа) и комнатной температуре в течение 4 ч. Катализатор удаляли фильтрацией и растворитель выпаривали. Неочищенный гидрохлорид метил-2-(2-хлор-4-фторфенил)пиперидин-4-карбоксилата (2,4 г, 100%) получали в виде желтого твердого вещества. MS m/z 272 (М+Н)+. Стадия 3. Диметил-2-(2-хлор-4-фторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(2-хлор-4-фторфенил)пиперидин-4-карбоксилата гидрохлорид (2,4 г, 7,79 ммоль) растворяли в дихлорметане (50 мл) с получением бесцветного раствора. Добавляли DIPEA (3,39 мл, 19,47 ммоль) и метилхлорформиат (0,736 мл, 9,35 ммоль) и смесь перемешивали при комнатной температуре в течение 14 ч. Добавляли 0,1 М HCl и водную фазу экстрагировали DCM. Объединенные органические слои фильтровали через фазоразделитель и упаривали. Неочищенный 2-(2-хлор-4 фторфенил)пиперидин-1,4-дикарбоксилат (2,5 г, 97%) получали в виде желтого масла. MS m/z 330 Диметил-2-(2-хлор-4-фторфенил)пиперидин-1,4-дикарбоксилат (2,5 г, 7,58 ммоль) растворяли в ацетонитриле (50 мл) и воде (1 мл), затем добавляли бромид лития (5,27 г, 60,65 ммоль). Добавляли триэтиламин (4,20 мл, 30,33 ммоль) и полученную суспензию кипятили с обратным холодильником в течение 7 ч. Добавляли МТВЕ (170 мл) и воду (90 мл), фазы разделяли и органический слой экстрагировали водой. Объединенные водные слои подкисляли 3,8 М HCl и экстрагировали с помощью EtOAc. Объединенные органические слои сушили с помощью Na2SO4 и упаривали. 2-(2-Хлор-4-фторфенил)-1(метоксикарбонил)пиперидин-4-карбоновую кислоту (2,130 г, 89%) выделяли в виде коричневого твердого вещества. MS m/z 316 (М+Н)+. Ссылочное соединение 22. 2-(4-Хлор-3-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. 2-(4-Хлор-3-фторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан. 4-Бром-1-хлор-2-фторбензол (4 г, 19,10 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2 диоксаборолан) (5,63 г, 22,15 ммоль),PdCl2(dppf) (0,691 г, 0,95 ммоль) и ацетат калия (3,75 г, 38,20 ммоль) суспендировали в DMSO (40 мл) и нагревали до 80 С в течение 4 ч. Реакционную смесь оставляли охлаждаться до комнатной температуры. Разбавляли с помощью EtOAc (400 мл) и фильтровали через целит. Органическую фазу промывали водой (3500 мл), рассолом (500 мл), сушили с помощью фазоразделителя и упаривали в вакууме. Остаток очищали посредством автоматизированной флэшхроматографии на 2 колонках Biotage KP-SIL 100 г. В качестве подвижной фазы использовали градиент от 0 до 30% EtOAc в гептане за 15 CV. Продукт собирали, используя длину волны 258 нм. 2-(4-Хлор 3-фторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,100 г, 42,9%) выделяли в виде бесцветного масла. 1 Н ЯМР (600 МГц, cdcl3)1.32 (s, 12H), 7.37 (t, 1H), 7.48 (d, 1H), 7.52 (d, 1H). Стадия 2. Метил-2-(4-хлор-3-фторфенил)изоникотинат. Метил-2-хлоризоникотинат (1,2 г, 6,99 ммоль), карбонат калия (0,580 г, 4,20 ммоль) и PdCl2(dppf)(0,122 г, 0,17 ммоль) объединяли во флаконе для микроволновой обработки. Добавляли 2-(4-хлор-3 фторфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,1 г, 8,19 ммоль), растворенный в МеОН (15 мл). Реакционную смесь нагревали в одномодовом микроволновом реакторе при 100 С в течение 15 мин. Добавляли DCM (125 мл) и воду (125 мл), встряхивали и фазы разделяли. Водную фазу экстрагировалиDCM (125 мл). Объединенные органические фазы промывали рассолом (250 мл), сушили с помощью фазоразделителя и упаривали в вакууме. Остаток очищали посредством автоматизированной флэшхроматографии на колонке Biotage KP-SIL 100 г. В качестве подвижной фазы использовали градиент 10% EtOAc в гептане за 3 CV и затем 10 до 50% EtOAc в гептане за 12 CV. Продукт собирали, используя длину волны 253 нм. Метил-2-(4-хлор-3-фторфенил)изоникотинат (1,473 г, 79%) выделяли в виде белого твердого вещества. 1 Н ЯМР (400 МГц, cdcl3)4.00 (s, 3H), 7.48-7.54 (m, 1H), 7.76-7.83 (m, 2H), 7.88-7.94(m, 1H), 8.24-8.27 (m, 1H), 8.83 (dd, 1H). Стадия 3. Метил-2-(4-хлор-3-фторфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(4-хлор-3-фторфенил)изоникотинат (1,473 г, 5,54 ммоль) суспендировали в МеОН (30 мл) и добавляли по каплям хлористый водород (1,25 М в МеОН, 9,86 мл, 11,09 ммоль) во время перемешивания. Растворитель выпаривали через 15 мин, HCl соль перерастворяли в МеОН (10 мл) и добавляли оксид платины(IV) (0,063 г, 0,28 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 1,5 ч. Добавляли оксид платины(IV) (0,038 г, 0,17 ммоль) и гидрирование продолжали в течение 1 ч. Катализатор отфильтровывали и промывали МеОН. Растворитель выпаривали в вакууме, получая метил-2-(4-хлор-3-фторфенил)пиперидин-4-карбоксилата гидрохлорид (1,639 г, 96%) в виде белого твердого вещества. MS m/z 272 (М+Н)+. Стадия 4. Диметил-2-(4-хлор-3-фторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(4-хлор-3-фторфенил)пиперидин-4-карбоксилата гидрохлорид (1,639 г, 5,32 ммоль) и DIPEA (2,316 мл, 13,30 ммоль) растворяли в DCM (10 мл). Добавляли по каплям метилхлорформиат (0,503 мл, 6,38 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли DCM (100 мл) и промывали 0,1 М HCl (275 мл), нас. NaHCO3 (100 мл), сушили с помощью фазоразделителя и упаривали в вакууме, получая диметил-2-(4-хлор-3 фторфенил)пиперидин-1,4-дикарбоксилат (1,625 г, 93%) в виде бесцветного масла. MS m/z 330 (М+Н)+. Стадия 5. 2-(4-Хлор-3-фторфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(4-хлор-3-фторфенил)пиперидин-1,4-дикарбоксилат (1,625 г, 4,93 ммоль) растворяли в ацетонитриле (20 мл) и воде (0,400 мл). Добавляли бромид лития (3,42 г, 39,42 ммоль). Затем добавляли триэтиламин (2,73 мл, 19,71 ммоль) и реакционную смесь кипятили с обратным холодильником в течение 1,5 ч. Добавляли воду (200 мл) и МТВЕ (150 мл), встряхивали и фазы разделяли. Органическую фазу экстрагировали водой (2200 мл). Объединенные водные фазы подкисляли 3 М HCl до рН 1 и затем экстрагировали МТВЕ (2500 мл). Объединенные органические фазы сушили с помощью Na2SO4, фильтровали и упаривали в вакууме, получая 2-(4-хлор-3-фторфенил)-1-(метоксикарбонил)пиперидин-4 карбоновую кислоту (1,430 г, 92%). MS m/z 314 (M-H)-. Ссылочное соединение 23. 2-(2,4-дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(2,4-дихлорфенил)изоникотинат. Карбонат калия (1,812 г, 13,11 ммоль), метил-2-хлоризоникотинат (3 г, 17,48 ммоль), 2,4 дихлорфенилбороновую кислоту (5,00 г, 26,23 ммоль), PdCl2(dppf) (0,380 г, 0,52 ммоль) и МеОН (30,6 мл) распределяли в два 20-мл флакона для микроволновой обработки и нагревали до 100 С в течение 35 мин в одномодовом микроволновом реакторе. Реакционную смесь разбавляли водой и DCM, экстрагировали DCM и упаривали. Очищали посредством автоматизированной флэш-хроматографии с использованием 5% EtOAc в гептане 30% EtOAc за 15 CV при 280 нм. Метил-2-(2,4-дихлорфенил)изоникотинат(2,3 г, 47%) выделяли в виде белого твердого вещества. 1 Н ЯМР (400 МГц, cdcl3)3.98 (s, 3H), 7.37 (dd,1H), 7.53 (d, 1H), 7.57 (d, 1H), 7.86 (dd, 1H), 8.20 (s, 1H), 8.87 (d, 1H). MS m/z 282 (M+H)+. Стадия 2. Метил-2-(2,4-дихлорфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(2,4-дихлорфенил)изоникотината гидрохлорид (2,60 г, 8,15 ммоль) растворяли в МеОН (50 мл), добавляли оксид платины(IV) (0,019 г, 0,08 ммоль) и гидрировали в гидрогенизаторе Bchi при 5 бар(0,5 МПа) в течение 90 мин. Реакционную смесь фильтровали и упаривали с получением неочищенного метил-2-(2,4-дихлорфенил)пиперидин-4-карбоксилата гидрохлорида (2,8 г) в виде белого твердого вещества. MS m/z 288 (М+Н)+. Стадия 3. Диметил-2-(2,4-дихлорфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(2,4-дихлорфенил)пиперидин-4-карбоксилата гидрохлорид (2,8 г, 8,63 ммоль) разбавлялиDCM (24,18 мл). DIPEA (3,76 мл, 21,56 ммоль) и добавляли метилхлорформиат (0,815 мл, 10,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, затем промывали 30,1 М HCl и рассолом. Фильтровали через фазоразделитель. Упаривали с получением диметил-2-(2,4 дихлорфенил)пиперидин-1,4-дикарбоксилата (2,78 г, 93%) в виде масла. MS m/z 346 (М+Н)+. Стадия 4. 2-(2,4-Дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(2,4-дихлорфенил)пиперидин-1,4-дикарбоксилат (2,78 г, 8,03 ммоль) растворяли в ацетонитриле (27,1 мл) и воде (0,542 мл). Добавляли бромид лития (5,58 г, 64,24 ммоль) и триэтиламин (4,48 мл, 32,12 ммоль) и полученную смесь кипятили с обратным холодильником в течение 4 ч, разбавляли водой и МТВЕ и органический слой экстрагировали водой. Объединенные водные слои подкисляли 3 МHCl, экстрагировали МТВЕ, сушили над MgSO4, фильтровали и упаривали с получением 2-(2,4 дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновой кислоты (2,440 г, 91%) в виде пены. MS m/z 332 (М+Н)+. Ссылочное соединение 24. 2-(3,5-Дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(3,5-дихлорфенил)изоникотинат. Карбонат калия (2,416 г, 17,48 ммоль), метил-2-хлоризоникотинат (5 г, 29,14 ммоль), 2-(3,5 дихлорфенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (10,34 г, 37,88 ммоль), PdCl2(dppf) (0,633 г, 0,87 ммоль) и МеОН (45,7 мл) распределяли в три 20 мл флакона для микроволновой обработки и нагревали до 100 С в одномодовом микроволновом реакторе в течение 20 мин. Реакционную смесь разбавляли водой и DCM, водный слой экстрагировали DCM и объединенные органические слои упаривали. Очищали посредством автоматизированной флэш-хроматографии с использованием 5% EtOAc в гептане 30%(0,5 МПа) в течение 4 ч. Реакционную смесь фильтровали, промывали МеОН до тех пор, пока весь продукт не растворялся, и упаривали. Метил-2-(3,5-дихлорфенил)пиперидин-4-карбоксилата гидрохлорид(4,8 г, 69%) выделяли в виде желтоватого твердого вещества. MS m/z 288 (М+Н)+. Стадия 3. Диметил-2-(3,5-дихлорфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(3,5-дихлорфенил)пиперидин-4-карбоксилата гидрохлорид (4,8 г, 14,79 ммоль) растворяли в DCM (41,5 мл) и DIPEA (6,46 мл, 36,97 ммоль). Добавляли метилхлорформиат (1,374 мл, 17,74 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем разбавляли DCM и промывали 0,3 М HCl и рассолом. Органический слой фильтровали через фазоразделитель и упаривали с получением диметил-2-(3,5-дихлорфенил)пиперидин-1,4-дикарбоксилата (5,3 г) в виде масла. MS m/z 346 (М+Н)+. Стадия 4. 2-(3,5-Дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(3,5-дихлорфенил)пиперидин-1,4-дикарбоксилат (5,3 г, 15,31 ммоль) растворяли в ацетонитриле (51,7 мл). Добавляли воду (1,033 мл), бромид лития (10,64 г, 122,47 ммоль) и триэтиламин(8,53 мл, 61,24 ммоль) и полученную смесь кипятили с обратным холодильником в течение 2,5 ч. Смесь разбавляли водой и МТВЕ. Водный слой экстрагировали водой. Объединенные водные слои подкисляли 3 М HCl, экстрагировали МТВЕ, сушили над MgSO4, фильтровали и упаривали с получением 2-(3,5 дихлорфенил)-1-(метоксикарбонил)пиперидин-4-карбоновой кислоты (4,60 г, 90%) в виде зеленоватой пены. MS m/z 330 (М-Н)-. Ссылочное соединение 25. 2-(2-Фтор-4-(трифторметокси)фенил)-1-(метоксикарбонил)пиперидин-4 карбоновая кислота. Стадия 1. 2-(2-Фтор-4-(трифторметокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан. 1-Бром-2-фтор-4-(трифторметокси)бензол (7,41 г, 28,61 ммоль) растворяли в DMSO (50 мл). Добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (8,43 г, 33,19 ммоль), PdCl2(dppf) (1,035 г,1,43 ммоль) и ацетат калия (5,62 г, 57,22 ммоль) и смесь нагревали в масляной бане при 80 С в течение 15 ч. Реакционную смесь разбавляли этилацетатом и промывали водой. Органический слой сушили надNa2SO4 и упаривали. Остаток очищали посредством автоматизированной флэш-хроматографии на колонке Biotage KP-SIL 340 г. В качестве подвижной фазы использовали градиент от 0 до 25% EtOAc в гептане за 10 CV. 2-(2-Фтор-4-(трифторметокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (5,38 г,61,4%) получали в виде бесцветного твердого вещества. Стадия 2. Метил-2-(2-фтор-4-(трифторметокси)фенил)изоникотинат. Метил-2-хлоризоникотинат (3 г, 17,48 ммоль), 2-(2-фтор-4-(трифторметокси)фенил)-4,4,5,5 тетраметил-1,3,2-диоксаборолан (5,37 г, 17,55 ммоль), карбонат калия (1,812 г, 13,11 ммоль) иPdCl2(dppf) (0,380 г, 0,52 ммоль) смешивали в метаноле (30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу экстрагировали DCM и объединенные органические слои фильтровали через фазоразделитель и упаривали с получением темно-коричневого твердого вещества. Остаток очищали посредством автоматизированной флэш-хроматографии на колонке Biotage KP-SIL 340 г. В качестве подвижной фазы использовали градиент от 8:1 до 3:1 EtOAc в гептане за 10 CV. Метил-2-(2-фтор-4-(трифторметокси)фенил)изоникотинат(4,48 г, 81%) получали в виде бесцветного твердого вещества. 1H ЯМР (600 МГц, cdcl3)3.96 (s, 3H),7.07 (d, 1H), 7.14 (d, 1H), 7.79-7.84 (m, 1H), 8.03-8.09 (m, 1H), 8.31 (s, 1H), 8.82-8.88 (m, 1H). MS m/z 316(М+Н)+. Стадия 3. Метил-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(2-фтор-4-(трифторметокси)фенил)изоникотинат (4,47 г, 14,18 ммоль) растворяли в метаноле и добавляли хлористый водород (1,25 М в метаноле, 17,02 мл, 21,27 ммоль). Растворитель выпаривали и остаток перерастворяли в метаноле (50 мл). Добавляли оксид платины(IV) (0,322 г, 1,42 ммоль) и смесь гидрировали при 5 бар (0,5 МПа) при комнатной температуре в течение 5 ч. Катализатор отфильтровывали и растворитель выпаривали. Неочищенный метил-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-4-карбоксилата гидрохлорид (4,97 г, 98%) получали в виде бесцветного твердого вещества. MS m/z 322 (М+Н)+. Стадия 4. Диметил-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-1,4-дикарбоксилат. Метил-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-4-карбоксилата гидрохлорид (4,97 г, 13,89 ммоль) растворяли в дихлорметане (50 мл) и DIPEA (6,05 мл, 34,73 ммоль). Добавляли метилхлорформиат (1,313 мл, 16,67 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Добавляли 0,1 М HCl и водный слой экстрагировали DCM. Объединенные органические слои фильтровали через фазоразделитель и упаривали. Неочищенный диметил-2-(2-фтор-4(трифторметокси)фенил)пиперидин-1,4-дикарбоксилат (5,39 г, 102%) получали в виде желтого масла. MSm/z 380 (М+Н)+. Стадия 5. 2-(2-Фтор-4-(трифторметокси)фенил)-1-(метоксикарбонил)пиперидин-4-карбоновая кислота. Диметил-2-(2-фтор-4-(трифторметокси)фенил)пиперидин-1,4-дикарбоксилат (5,35 г, 14,10 ммоль) растворяли в ацетонитриле (100 мл) и воде (2000 мл), затем добавляли бромид лития (9,80 г, 112,84 ммоль). Добавляли триэтиламин (7,82 мл, 56,42 ммоль) и полученную суспензию кипятили с обратным холодильником в течение 7 ч. Добавляли МТВЕ (350 мл) и воду (180 мл), фазы разделяли и органический слой экстрагировали водой. Объединенные водные слои подкисляли 3,8 М HCl и экстрагировали с помощью EtOAc. Объединенные органические слои сушили с помощью Na2SO4 и упаривали. 2-(2-Фтор-4(трифторметокси)фенил)-1-(метоксикарбонил)пиперидин-4-карбоновую кислоту (5,09 г, 99%) получали в виде бесцветного твердого вещества. MS m/z 364 (М-Н)-. Ссылочное соединение 26. 1-(Метоксикарбонил)-2-(3,4,5-трифторфенил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(3,4,5-трифторфенил)изоникотинат. Метил-2-бромизоникотинат (4 г, 18,52 ммоль), 3,4,5-трифторфенилбороновую кислоту (4,89 г, 27,77 ммоль), карбонат калия (3,84 г, 27,77 ммоль) и PdCl2(dppf) (0,402 г, 0,56 ммоль) смешивали в метаноле(30 мл) в двух отдельных 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Добавляли воду и DCM и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу про- 28020825 мывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Остаток растворяли в DCM и очищали посредством катионообменной хроматографии SCX-2. Метил-2-(3,4,5-трифторфенил)изоникотинат (2,62 г, 52%) выделяли в виде слегка желтого твердого вещества. MS m/z 268 (М+Н)+. Стадия 2. Метил-2-(3,4,5-трифторфенил)пиперидин-4-карбоксилата гидрохлорид. Метил-2-(3,4,5-трифторфенил)изоникотинат (2,62 г) растворяли в МТВЕ, затем добавляли HCl (4 M в диоксане, 15 мл). Растворители выпаривали с получением метил-2-(3,4,5-трифторфенил)изоникотината гидрохлорида (2,94 г, 9,68 ммоль), который растворяли в МеОН (40 мл), и добавляли оксид платины(IV)(0,220 г, 0,97 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 1 ч. Катализатор отфильтровывали, промывали МеОН и элюат упаривали, получая метил-2-(3,4,5-трифторфенил)пиперидин-4-карбоксилата гидрохлорид (2,75 г, 92%) в виде коричневого твердого вещества. MS m/z 274 (М+Н)+. Стадия 3. Диметил-2-(3,4,5-трифторфенил)пиперидин-1,4-дикарбоксилат. Метил-2-(3,4,5-трифторфенил)пиперидин-4-карбоксилата гидрохлорид (2,75 г, 8,88 ммоль) растворяли в DCM (50 мл), затем добавляли DIPEA (3,88 мл, 22,20 ммоль). К раствору добавляли по каплям метилхлорформиат (0,979 мл, 12,43 ммоль). Смесь перемешивали при комнатной температуре в течение 4 ч. Смесь промывали HCl (0,1 М, 100 мл) и нас. NaHCO3(100 мл), затем сушили с помощью фазоразделителя и упаривали, получая диметил-2-(3,4,5-трифторфенил)пиперидин-1,4-дикарбоксилат (2,77 г, 94%) в виде оранжевого масла. Стадия 4. 1-(Метоксикарбонил)-2-(3,4,5-трифторфенил)пиперидин-4-карбоновая кислота. Диметил-2-(3,4,5-трифторфенил)пиперидин-1,4-дикарбоксилат (2,77 г, 8,36 ммоль) растворяли в ацетонитриле (35 мл) и воде (0,700 мл), затем добавляли бромид лития (5,81 г, 66,89 ммоль). ДобавлялиEt3N (4,66 мл, 33,45 ммоль) и полученную желтую суспензию кипятили с обратным холодильником в течение 2 ч. Добавляли воду (50 мл) и МТВЕ (150 мл). Фазы разделяли и органический слой экстрагировали водой (2 раза). Объединенные водные слои подкисляли до рН 1 с помощью 3,8 М HCl и с МТВЕ (2 раза). Объединенный органический слой сушили над Na2SO4, фильтровали и упаривали, получая 1(метоксикарбонил)-2-(3,4,5-трифторфенил)пиперидин-4-карбоновую кислоту (2,23 г, 84%) в виде слегка желтого полутвердого вещества. MS m/z 316(M-H)-. Ссылочное соединение 27. 1-(Метоксикарбонил)-2-(2,4,5-трифторфенил)пиперидин-4-карбоновая кислота. Стадия 1. Метил-2-(2,4,5-трифторфенил)изоникотинат. Метил-2-хлоризоникотинат (3,25 г, 18,94 ммоль), 2,4,5-трифторфенилбороновую кислоту (5,00 г,28,41 ммоль), карбонат калия (3,93 г, 28,41 ммоль) и PdCl2(dppf) (0,411 г, 0,57 ммоль) смешивали в метаноле (20 мл) в двух 20-мл флаконах для микроволновой обработки. Флаконы закрывали крышкой и нагревали при 100 С в течение 10 мин в одномодовом микроволновом реакторе. Реакционную смесь суспендировали в метаноле, фильтровали и фильтрат упаривали с получением темно-красной суспензии. Добавляли DCM и воду и фазы разделяли. Водную фазу (рН 9) экстрагировали DCM и объединенную органическую фазу промывали рассолом, пропускали через фазоразделитель и упаривали с получением коричневого твердого вещества. Неочищенное коричневое твердое вещество растворяли в МТВЕ (120 мл) и твердые вещества отфильтровывали. Добавляли по каплям хлористый водород (4 M в диоксане,4,74 мл, 18,94 ммоль) и образовывалась суспензия. Суспензию перемешивали при комнатной температуре в течение 45 мин. Твердое вещество собирали фильтрацией и промывали МТВЕ. Добавляли МТВЕ и нас. NaHCO3. Органическую фазу сушили над Na2SO4, фильтровали и упаривали с получением метил-2(2,4,5-трифторфенил)изоникотината (1,676 г, 33%) в виде бежевого твердого вещества. МТВЕ-фильтрат промывали нас. NaHCO3, сушили над Na2SO4, фильтровали и упаривали с получением коричневого твердого вещества. Остаток очищали с помощью Biotage (от 0 до 20% EtOAc в гептане, 6 CV; колонка Biotage KP-SIL 100 г) с получением метил-2-(2,4,5-трифторфенил)изоникотината (0,86 г, 17%) в виде белого твердого вещества. 1H ЯМР (600 МГц, cdcl3)3.97 (s, 3H), 7.00-7.07 (m, 1H), 7.81 (dd, 1H), 7.90-7.97(25 мл) и добавляли оксид платины(IV) (0,107 г, 0,47 ммоль). Полученную смесь гидрировали в гидрогенизаторе Bchi при комнатной температуре и 5 бар (0,5 МПа) в течение 6 ч. Катализатор отфильтровывали, промывали МеОН и элюат упаривали. Добавляли DCM и 10% K2CO3 и фазы разделяли. Органический слой промывали рассолом, пропускали через фазоразделитель и упаривали с получением метил-2(2,4,5-трифторфенил)пиперидин-4-карбоксилата (2,434 г, 95%) в виде коричневого масла. MS m/z 274(50 мл) и DIPEA (1,707 мл, 9,80 ммоль), затем добавляли метилхлорформиат (0,965 мл, 12,25 ммоль). Раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь промывали 0,1 М
МПК / Метки
МПК: A61P 7/04, A61K 31/4523, C07D 413/04
Метки: качестве, терапевтических, изоксазол-3(2н)-она, агентов, аналоги
Код ссылки
<a href="https://eas.patents.su/30-20825-analogi-izoksazol-32n-ona-v-kachestve-terapevticheskih-agentov.html" rel="bookmark" title="База патентов Евразийского Союза">Аналоги изоксазол-3(2н)-она в качестве терапевтических агентов</a>
Производные спиропиперидина и их использование в качестве терапевтических агентов

Номер патента: 1574
Опубликовано: 25.06.2001
Авторы: Джексон Филип Стефен, Вилльямс Брайан Джон, Свейн Кристофер Джон, Харрисон Тимоти, Холлингворт Грегори Джон, Кертис Нил Рой, Кулаговский Януш Йозеф, Эллиотт Джейсон Мэттью, Бейкер Рэймонд, Сьюард Эйлин Мэри
МПК: C07D 491/10, A61P 1/08, A61K 31/4355...
Метки: производные, качестве, спиропиперидина, агентов, терапевтических, использование
Формула / Реферат:
1. Соединение формулы (I) где R1 означает водород, гидрокси, C1-6-алкил, С2-6-алкенил, С3-7-циклоалкил, С3-7-циклоалкил-С1-4-алкил, C1-6-алкокси, фтор-С1-6-алкокси, C1-6-алкокси-С1-4-алкил, С1-б-алкокси-С1-4-алкокси, фтор-С1-6-алкокси-С1-4-алкил, С2-6-алкенилокси, С3-7-циклоалкокси, С3-7-циклоалкил-С1-4-алкокси, фенокси, бензилокси, циано, галоген, NRaRb, SRa, SORa, SO2Ra, OSO2Ra, NRaCOR14, CORa, CO2Ra или CONRaRb, где Ra и Rb независимо друг...
Бензконденсированные гетероариламидные производные тиенопиридинов, применяемые в качестве терапевтических агентов, фармацевтические композиции, включающие их, и способы их применения

Номер патента: 8255
Опубликовано: 27.04.2007
Авторы: Криппс Стефан Джеймс, Хэ Минин, Чжоу Жу, Лоу Цзихун, Палмер Синтия Луиза, Кания Роберт Стивен, Роминес Уилльям Генри III, Коллинс Майкл Рэймонд, Дил Джудит Гейл
МПК: C07D 495/04, A61P 35/00, A61K 31/4365...
Метки: гетероариламидные, агентов, качестве, терапевтических, бензконденсированные, тиенопиридинов, композиции, фармацевтические, применения, производные, способы, их, применяемые, включающие
Формула / Реферат:
1. Соединение, представленное формулой I где Y является -NH-, -O-, -S- или -СН2-; Z является -O-, -S- или -N-; R14 является C1-C6алкилом, C1-C6алкиламино, C1-C6алкилгидрокси, C3-C10циклоалкилом, С3-C10циклоалкиламино, C1-C6алкилC3-C10циклоалкилом или метилуреидогруппой; R15 и R17 независимо являются Н, галогеном или C1-C6алкилом, незамещенным или замещенным одним или более R5; R16 является Н или C1-C6алкилом, когда Z является N, и R16...
Бензонафтиридины в качестве бронхиальных терапевтических средств

Номер патента: 1551
Опубликовано: 23.04.2001
Авторы: Зандерс Карл, Хатцельманн Армин, Амшлер Германн, Хефнер Дитрих, Босс Хильдегард, Ульрих Вольф-Рюдигер, Мартин Томас, Гуттерер Биате, Беуме Рольф, Клей Ханс-Петер, Бер Томас, Флоккерци Дитер, Гёбель Карл-Йозеф
МПК: A61P 11/00, C07D 471/04, A61K 31/47...
Метки: терапевтических, бензонафтиридины, качестве, средств, бронхиальных
Формула / Реферат:
1. Соединения формулы I в которой R1 обозначает С1-С4алкил, R2 обозначает гидроксил, С1-С4алкокси, С3-С7циклоалкокси, С3-С7циклоалкилметокси или С1-С4алкокси, который полностью или большей частью замещен фтором, R3 обозначает гидроксил, С1-С4алкокси, С3-С7циклоалкокси, С3-С7циклоалкилметокси или С1-С4алкокси, который полностью или большей частью замещен фтором, или в которой R2 и R3 совместно обозначают С1-C2алкилендиоксигруппу, R4...
Трициклические азотсодержащие соединения в качестве антибактериальных агентов

Номер патента: 15821
Опубликовано: 30.12.2011
Авторы: Хеннесси Алан Джозеф, Дэвис Дэвид Томас, Джордано Илариа, Пирсон Нейл Дэвид, Дэвис Дэвид Эван
МПК: C07D 471/16, A61K 31/4985, A61P 31/00...
Метки: качестве, агентов, соединения, антибактериальных, трициклические, азотсодержащие
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль и/или N-оксидв которой Z1 и Z2независимо выбраны из CH и N;R1a и R1b независимо выбраны из водорода; галогена; циано; (C1-6)алкила; (C1-6)алкилтио; трифторметила; трифторметокси; карбокси; гидрокси, в случае необходимости замещенного (C1-6)алкилом или (C1-6)алкоксизамещенным (C1-6)алкилом; (C1-6)алкоксизамещенного (C1-6)алкила; гидрокси(C1-6)алкила; аминогруппы, в случае...
Производные индола в качестве эстрогенных агентов

Номер патента: 815
Опубликовано: 24.04.2000
Авторы: Миллер Крис П., Коллини Майкл Д., Тран Бэч Д.
МПК: A61K 31/405, C07D 209/08
Метки: агентов, качестве, эстрогенных, производные, индола
Формула / Реферат:
1. Соединение, имеющее структуру где R1 выбран из Н, ОН, -ОС(=O)(С1-С4алкил), -О(С1-С4алкил) или галогена; R2, R3, R4, R5 и R6 независимо выбраны из Н, ОН, -ОС(=O)(C1-С4алкил), -О(С1-C4алкил), галогена или C1-С6алкила, при условии, что в случае, когда R1 представляет собой Н, R2 не является группой ОН; Х выбран из Н, C1-С6алкила, нитро или галогена; Z выбран из где n равно 1, 2 или 3; Y выбран из: а) группы где R7 и R8...
Предыдущий патент: Гидравлическая приводная система для шахтной канатной дороги
Следующий патент: Самосвал
Случайный патент: Моющие составы и способ их получения