Диокса-бицикло[3.2.1]октан-2,3,4-триольные производные






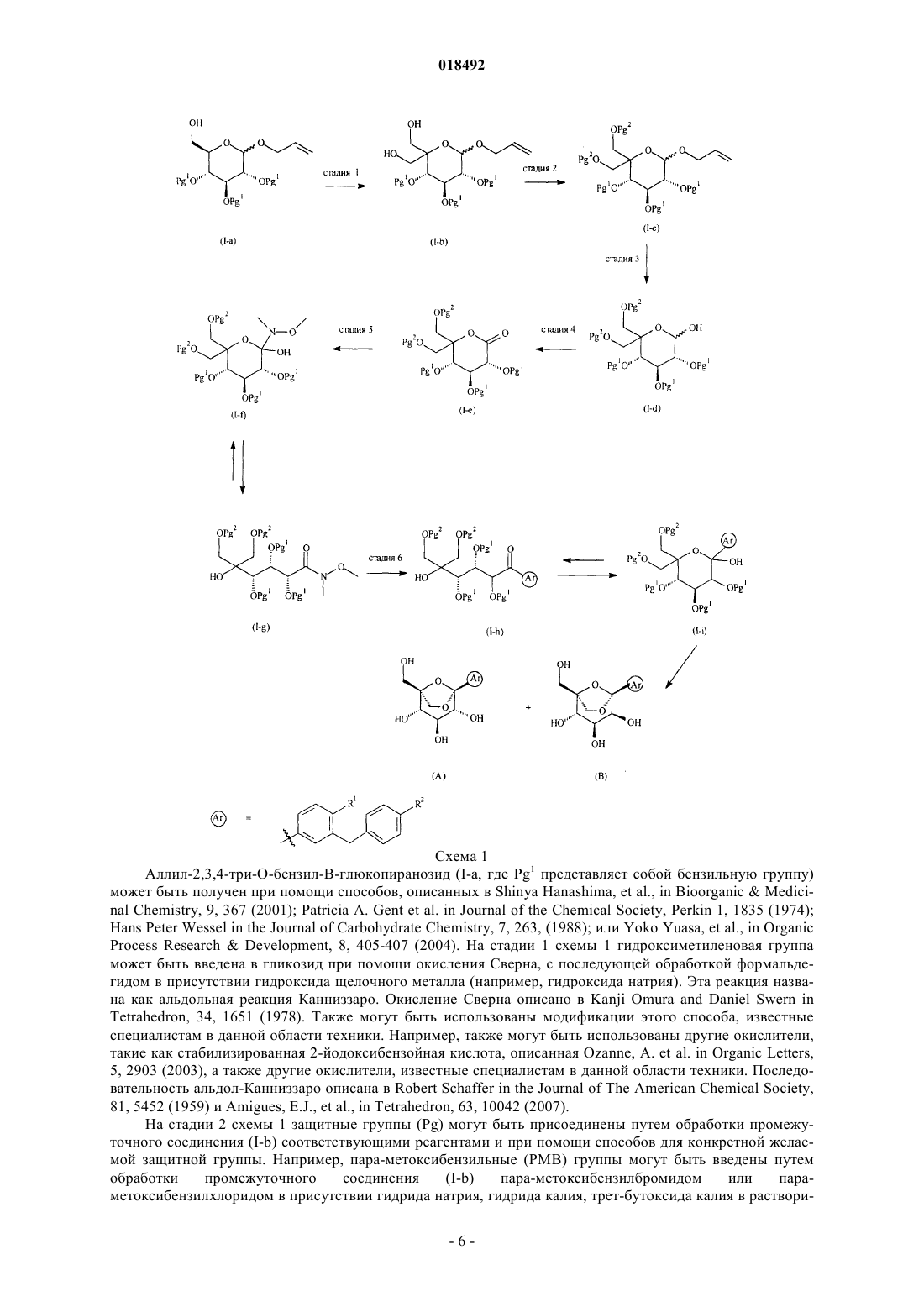

Формула / Реферат
1. Соединение формулы (А) или формулы (В)

где R1 представляет собой Н, (С1-С4)алкил, (С1-С4)алкокси, Cl, F, циано, замещенный фторо (С1-С2)алкил, (С1-С4)алкил-SO2- или (С3-С6)циклоалкил; и
R2 представляет собой (С1-С4)алкил, (С1-С4)алкокси, (С2-С4)алкинил, 3-оксетанилокси, 3-тетрагидрофуранилокси, Cl, F, циано, замещенный фторо (С1-С2)алкил, (С1-С4)алкил-SO2-, (С3-С6)циклоалкил или (С5-С6)гетероцикл, имеющий 1 или 2 гетероатома, каждый из которых независимо выбран из N, О или S.
2. Соединение по п.1, где указанное соединение представляет собой соединение формулы (А).
3. Соединение по п.1 или 2, где
R1 представляет собой Н, метил, этил, пропил, изопропил, метокси, этокси, F, Cl, циано, -CF3, циклопропил или циклобутил и
R2 представляет собой метил, этил, пропил, изопропил, метокси, этокси, F, Cl, циано, -CF3, -CF2CH3, этинил, 3-оксетанилокси, 3-тетрагидрофуранилокси или циклопропил.
4. Соединение по п.3, где
R1 представляет собой Н, метил, этил, изопропил, метокси, этокси, F, Cl, циано, -CF3 или циклопропил и
R2 представляет собой метил, этил, изопропил, метокси, этокси, F, Cl, циано, -CF3, -CF2CH3, этинил, 3-оксетанилокси, 3-тетрагидрофуранилокси или циклопропил.
5. Соединение по п.4, где
R1 представляет собой Н, метил, этил, метокси, этокси, F, Cl, циано, -CF3 или циклопропил и
R2 представляет собой метил, этил, метокси, этокси, F, Cl, циано, -CF3, -CF2CH3, этинил, 3-оксетанилокси, 3-тетрагидрофуранилокси или циклопропил.
6. Соединение по п.5, где
R1 представляет собой метил, этил, F, Cl, циано, CF3 или циклопропил и
R2 представляет собой метокси или этокси.
7. Соединение, выбранное из группы, состоящей из
(1S,2S,3S,4R,5S)-1-гидроксиметил-5-[3-(4-метоксибензил)-4-метилфенил]-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-[3-(4-этоксибензил)-4-метилфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-[4-хлор-3-(4-метоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-[4-хлор-3-(4-этоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-[4-фтор-3-(4-метоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
2-(4-метоксибензил)-4-((1S,2S,3S,4R,5S)-2,3,4-тригидрокси-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]окт-5-ил)бензонитрила;
2-(4-этоксибензил)-4-((1S,2S,3S,4R,5S)-2,3,4-тригидрокси-1-(гидроксиметил)-6,8-диоксабицикло[3.2.1]окт-5-ил)бензонитрила;
(1S,2S,3S,4R,5S)-5-[3-(4-этоксибензил)-4-фторфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-{4-фтор-3-[4-(тетрагидрофуран-3-илокси)бензил]фенил}-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-[3-(4-хлорбензил)-4-фторфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4R,5S)-5-{4-фтор-3-[4-(оксетан-3-илокси)бензил]фенил}-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола и
(1S,2S,3S,4R,5S)-5-{4-хлор-3-[4-(оксетан-3-илокси)бензил]фенил}-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола.
8. Соединение, представляющее собой (1S,2S,3S,4R,5S)-5-[4-хлор-3-(4-этоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол.
9. Соединение, выбранное из группы, состоящей из
(1S,2S,3S,4S,5S)-1-гидроксиметил-5-[3-(4-метоксибензил)-4-метилфенил]-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4S,5S)-5-[3-(4-этоксибензил)-4-метилфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4S,5S)-5-[4-хлор-3-(4-метоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4S,5S)-5-[4-хлор-3-(4-этоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4S,5S)-5-[4-фтор-3-(4-метоксибензил)фенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола;
(1S,2S,3S,4S,5S)-5-[3-(4-этоксибензил)-4-фторфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола и
(1S,2S,3S,4S,5S)-5-[3-(4-хлорбензил)-4-фторфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триола.
10. Кристаллическое соединение формулы (4А)

11. Сокристалл, содержащий соединение формулы (4А)

и L-пролин или L-пироглутаминовую кислоту.
12. Сокристалл по п.11, содержащий соединение формулы (4А) и L-пироглутаминовую кислоту в стехиометрическом отношении 1:1.
13. Сокристалл по п.11, содержащий L-пироглутаминовую кислоту и имеющий:
а) картину дифракции рентгеновских лучей на порошке, содержащую 2-тэта величины (излучение CuKα, длина волны 1,54056Å) 6,4 ± 0,2; и
б) спектр 13С ЯМР твердого состояния, содержащий пики в положениях 16,5 ± 0,2, 158,7 ±0,2 и 181,5 ± 0,2 млн-1, определенные на спектрометре при 500 МГц относительно кристалла адамантина с 29,5 млн-1.
14. Сокристалл, содержащий соединение формулы (4А)

и L-пироглутаминовую кислоту и имеющий одну или более чем одну из следующих характеристик:
а) пространственную группу Р2(1)2(1)2(1) и параметры элементарной ячейки:

б) картину дифракции рентгеновских лучей на порошке, содержащую 2-тэта величины (излучение CuKα, длина волны 1,54056Å) 6,4 ± 0,2, 16,7 ± 0,2, 17,4 ± 0,2 и 21,1 ± 0,2; или
в) спектр 13С ЯМР твердого состояния, содержащий пики в положениях 16,5 ± 0,2, 131,1 ± 0,2, 158,7 ± 0,2 и 181,5 ± 0,2 млн-1, определенные на спектрометре при 500 МГц относительно кристалла адамантина с 29,5 млн-1.
15. Сокристалл, содержащий соединение формулы (4А)

и L-пролин и имеющий одну или более чем одну из следующих характеристик:
а) пространственную группу С2 и параметры элементарной ячейки:

б) картину дифракции рентгеновских лучей на порошке, содержащую 2-тэта величины (излучение CuKα, длина волны 1,54056Å) 7,6 ± 0,2, 12,1 ± 0,2, 20,3 ± 0,2 и 28,8 ± 0,2.
16. Фармацевтическая композиция, содержащая:
(1) соединение по любому из пп.1-10 или сокристалл по любому из пп.11-15 и
(2) фармацевтически приемлемый эксципиент, разбавитель или носитель.
17. Способ лечения ожирения и связанных с ожирением расстройств у животных, при котором животному, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-10 или терапевтически эффективное количество сокристалла по любому из пп.11-15.
18. Способ лечения или замедления прогрессирования или начала диабета 2 типа и связанных с диабетом расстройств у животных, при котором животному, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-10 или терапевтически эффективное количество сокристалла по любому из пп.11-15.
19. Способ лечения ожирения и связанных с ожирением расстройств у животных, при котором животному, нуждающемуся в таком лечении, вводят фармацевтическую композицию по п.16.
20. Способ лечения или замедления прогрессирования или начала диабета 2 типа и связанных с диабетом расстройств у животных, при котором животному, нуждающемуся в таком лечении, вводят фармацевтическую композицию по п.16.
Текст
Маситти Винсент, Коллман Бенджамин Миках (US) Поликарпов А.В., Борисова Е.Н. (RU) Здесь раскрыты соединения формулы (I) и их применения для лечения заболеваний, состояний и/или расстройств, опосредованных ингибиторами натрий-глюкозного транспортера (в частности ингибиторами SGLT2). Раскрытые здесь соединения полезны для предупреждения и лечения ожирения и ассоциированных с ним сопутствующих заболеваний, в частности диабета II типа (2 типа). Область изобретения Настоящее изобретение относится к диоксабицикло[3.2.1]октан-2,3,4-триольным производным,кристаллическим структурам, фармацевтическим композициям и их применению в качестве ингибиторов натрий-глюкозного котранспортера (SGLT). Предшествующий уровень техники Ожирение представляет собой значительную проблему для здоровья вследствие тяжелых медицинских осложнений, которые включают сопутствующие заболевания, такие как гипертензия, инсулинорезистентность, сахарный диабет, ишемическая болезнь сердца и сердечная недостаточность (в совокупности называемые метаболическим синдромом). Ожирение и связанные с ним сопутствующие заболевания продолжают вызывать растущие проблемы здоровья в развитых странах и начинают поражать также и развивающиеся страны. Отрицательные последствия ожирения для здоровья привели к тому, что оно является второй лидирующей причиной преждевременных смертей в Соединенных Штатах Америки и оказывает значительное экономическое и психологическое влияние на общество. См. McGinnis M, FoegeWH., "Actual Causes of Death in the United States," JAMA, 270, 2207-12 (1993). Существует потребность в идентификации и разработке новых лекарственных препаратов, которые позволили бы лечить и/или предупреждать ожирение и ассоциированные с ним сопутствующие заболевания, в частности диабет II типа(тип 2). Недавно продемонстрировали, что ингибиторы натрий-глюкозного котранспорта (SGLT), в частности ингибиторы SGLT2 блокируют реабсорбцию глюкозы из почечного фильтрата в клубочек, таким образом вызывая экскрецию глюкозы в мочу. Поскольку экскретируется избыток глюкозы, уровень глюкозы в крови уменьшается, уменьшается запас глюкозы в печени, уменьшается секреция инсулина и,наконец, уменьшается превращение углеводов в жир и, в конечном итоге, уменьшается накопленный жир. Полагают, что избирательное ингибирование SGLT2 нормализует уровень глюкозы в плазме крови путем усиления экскреции глюкозы. Следовательно, ингибиторы SGLT2 предлагают привлекательные способы улучшения диабетических состояний без увеличения массы организма или риска гипогликемии. См., Isaji M., Current Opinion Investigational Drugs, 8(4), 285-292 (2007). Общий обзор SGLT в качестве терапевтического агента см. также в Asano, Т., et al., Drugs of the Future, 29(5),461-466(2004). Типичные примеры гликозидов, которые, как показано, полезны для лечения NIDDM (инсулиннезависимого сахарного диабета) и ожирения можно найти в следующих описаниях: патенты США 6515117; 6414126; 7101856; 7169761 и 7202350; публикации СШАUS 2002/0111315; US 2002/0137903; US 2004/0138439; US 2005/0233988; US 2006/0025349; US 2006/0035841 и US 2006/0632722; и публикациях РСТWO 01/027128; WO 02/044192; WO 02/088157; WO 03/099836;WO 04/087727; WO 05/021566; WO 05/085267; WO 06/008038; WO 06/002912; WO 06/062224; WO 07/000445; WO 07/093610 и WO 08/002824. Некоторые гликозиды являются генотоксическими и влияют на клеточный генетический материал,таким образом, они могут быть потенциально мутагенными или канцерогенными. Генотоксические вещества можно обнаружить с использованием стандартных анализов, таких как микроядерный тест на клетках млекопитающих in vitro (MNvit), Organization for Economic Co-Operation and Development(OECD) Draft Test Guideline (Draft TG) 487 (2007); тест хромосомальной аберрации на клетках млекопитающих in vitro, OECD TG 473 (1997); тест бактериальной обратной мутации, OECD TG 471 (1997); микроядерный тест на эритроцитах млекопитающих, OECD TG 474 (1997) и т.п. Следовательно, все еще сохраняется потребность в более эффективном и безопасном терапевтическом лечении и/или предупреждении ожирения и ассоциированных с ним сопутствующих заболеваний, в частности диабета II типа. Краткое изложение сущности изобретения Обнаружено, что соединения формулы (А) и формулы (В) действуют в качестве ингибиторов натрий-глюкозного котранспортера (SGLT), в частности, ингибиторов SGLT2; таким образом, они могут быть использованы в лечении заболеваний, опосредованных таким ингибированием (например, заболеваний, связанных с ожирением, диабетом 2 типа и связанных с ожирением и связанных с диабетом сопутствующих заболеваний). Эти соединения могут быть представлены формулами (А) и (В), как изображено нижеR2 представляет собой (С 1-С 4)алкил, (С 1-С 4)алкокси, (С 2-С 4)алкинил, 3-оксетанилокси, 3 тетрагидрофуранилокси, Cl, F, циано, замещенный фторо (C1-С 2)алкил, (С 1-С 4)алкил-SO2-, (С 3 С 6)циклоалкил или (С 5-С 6)гетероцикл, имеющий 1 или 2 гетероатома, каждый из которых независимо выбран из N, О или S. Специалистам в данной области техники в общем понятно, что различные заместители могут быть присоединены к соединениям формулы (А) или формулы (В), при условии, что выбранный(е) заместитель(и) не оказывает(ют) отрицательного влияния на фармакологические свойства соединения или не оказывает(ют) неблагоприятного влияния на применение лекарственного средства. Конкретные соединения формулы (А) включают(1S,2S,3S,4R,5S)-5-4-хлор-3-[4-(оксетан-3-илокси)бензил]фенил-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол. Конкретные соединения формулы (В) включают(1S,2S,3S,4S,5S)-5-[3-(4-хлорбензил)-4-фторфенил]-1-гидроксиметил-6,8-диоксабицикло[3.2.1]октан-2,3,4-триол. Еще один аспект настоящего изобретения представляет собой кристалл, содержащий соединение,имеющее формулу (4 А) Еще один аспект настоящего изобретения представляет собой фармацевтическую композицию, со-2 018492 держащую (1) соединение по настоящему изобретению и (2) фармацевтически приемлемый эксципиент,разбавитель или носитель. Предпочтительно композиция содержит терапевтически эффективное количество соединения по настоящему изобретению. Композиция также может содержать по меньшей мере один дополнительный фармацевтический агент (описанный здесь). Предпочтительные агенты включают агенты против ожирения и/или антидиабетические агенты (описанные здесь ниже). В еще одном аспекте настоящего изобретения предложен способ лечения заболевания, расстройства или состояния, модулируемого путем ингибирования SGLT2 у животных, при котором осуществляют стадию введения животному (предпочтительно человеку), нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению (или его фармацевтической композиции). Заболевания, состояния и/или расстройства, модулируемые путем ингибирования SGLT2,включают, например, диабет II типа, диабетическую нефропатию, синдром инсулинорезистентности,гипергликемию, гиперинсулинемию, гипердипидемию, нарушенную толерантность к глюкозе, ожирение(включая контроль массы организма или поддержание массы организма), гипертензию и понижение уровня глюкозы в крови. Соединения по настоящему изобретению могут быть введены в комбинации с другими фармацевтическими агентами (в частности, агентами против ожирения и антидиабетическими агентами, описанными здесь ниже). Комбинированное терапевтическое средство может быть введено в виде: (а) единой фармацевтической композиции, содержащей соединение по настоящему изобретению, по меньшей мере один описанный здесь дополнительный фармацевтический агент и фармацевтически приемлемый эксципиент, разбавитель или носитель или (б) двух отдельных фармацевтических композиций, включая (1) первую композицию, содержащую соединение по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель, и (2) вторую композицию, содержащую по меньшей мере один дополнительный фармацевтический агент, описанный здесь, и фармацевтически приемлемый эксципиент, разбавитель или носитель. Фармацевтические композиции могут быть введены одновременно или последовательно и в любом порядке. Понятно, что приведенное выше общее описание и следующее подробное описание представляют собой только примеры и объяснение, но не ограничивают заявленное изобретение. Краткое описание графических материалов На фиг. 1 представлена уточненная кристаллическая структура для соединения примера 8 А, построенная с использованием программного пакета для графического изображения SHELXTL. На фиг. 2 - уточненная кристаллическая структура для соединения примера 9 А, построенная с использованием программного пакета для графического изображения SHELXTL. На фиг. 3 - наблюдаемая картина дифракции рентгеновских лучей на порошке для примера 22: сокристалл примера 18 соединения примера 4 А и L-пролина. На фиг. 4 - наблюдаемая картина дифракции рентгеновских лучей на порошке для примера 22: сокристалл примера 20 соединения примера 4 А и L-пироглутаминовой кислоты. На фиг. 5 - наблюдаемая термограмма дифференциальной сканирующей калориметрии для примера 23: сокристалл примера 18 соединения примера 4 А и L-пролина На фиг. 6 - наблюдаемая термограмма дифференциальной сканирующей калориметрии для примера 23: сокристалл примера 20 соединения примера 4 А и L-пироглутаминовой кислоты. На фиг. 7 - уточненная кристаллическая структура для примера 24: сокристалл соединения примера 4 А и L-пролина, построенная с использованием программного пакета для графического изображенияSHELXTL. На фиг. 8 - уточненная кристаллическая структура для примера 25: сокристалл соединения примера 4 А и L-пироглутаминовой кислоты, построенная с использованием программного пакета для графического изображения SHELXTL. На фиг. 9 - наблюдаемый спектр ядерного магнитного резонанса 13 С твердого состояния для примера 26: сокристалл соединения примера 4 А и L-пироглутаминовой кислоты. Пики, отмеченные звездочками, представляют собой вращающиеся боковые полосы. Подробное описания изобретения Настоящее изобретение может быть понято еще легче со ссылкой на следующее подробное описание приведенных для примера воплощений изобретения и включенных сюда примеров. Перед тем, как раскрыть и описать соединения, композиции и способы по настоящему изобретению, следует понимать, что данное изобретение не ограничено конкретными способами синтеза для получения, которые безусловно могут варьировать. Также следует понимать, что используемая здесь терминология приведена только для задачи описания конкретных воплощений и не предполагается, что она ограничивает объем изобретения. Формы множественного и единственного числа являются взаимозаменяемыми, кроме указания на количество. Используемый здесь термин "алкил" относится к углеводородному радикалу общей формулыCnH2n+1. Алкановый радикал может быть прямоцепочечным или разветвленным. Например, термин "(С 1 С 6)алкил" относится к одновалентной, прямоцепочечной или разветвленной алифатической группе, содержащей от 1 до 6 атомов углерода (например, метил, этил, n-пропил, изопропил, н-бутил, изобутил,-3 018492 втор-бутил, трет-бутил, н-пентил, 1-метилбутил, 2-метилбутил, 3-метилбутил, неопентил, 3,3 диметилпропил, гексил, 2-метилпентил и т.п.). Аналогично, алкильный фрагмент (т.е. алкильная группировка) группы алкокси, ацила (например, алканоила), алкиламино, диалкиламино, алкилсульфонила и алкилтио имеет то же самое определение, как указано выше. Когда указано, что алкановый радикал или алкильная группировка является "возможно замещенной", тогда она может быть незамещенной или замещенной одним или более чем одним заместителем (обычно от одного до трех заместителей, за исключением случая галогеновых заместителей, таких как перхлор или перфторалкилы), независимо выбранным из группы заместителей, перечисленных ниже в определении "замещенного". "Замещенный группой галогеноалкил" относится к алкильной группе, замещенной одним или более чем одним атомом галогена(например, фторметил, дифторметил, трифторметил, перфторэтил, 1,1-дифторэтил и т.п). Термин "циклоалкил" относится к неароматическим кольцам, которые полностью гидрированы и могут существовать в виде единичного кольца, бициклического кольца или спирокольца. Если не указано иное, тогда карбоциклическое кольцо, как правило, представляет собой 3-8-членное кольцо. Например, циклоалкил включает группы, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогексенил, норборнил (бицикло[2.2.1]гептил), бицикло[2.2.2]октил и т.п. Термин "гетероцикл" относится к неароматическим кольцам, которые полностью гидрированы и могут существовать в виде единичного кольца, бициклического кольца или спирального кольца. Если не указано иное, то гетероциклическое кольцо обычно представляет собой 3-6-членное кольцо, содержащее 1-3 гетероатома (предпочтительно 1 или 2 гетероатома), независимо выбранные из серы, кислорода и/или азота. Гетероциклические кольца включают группы, такие как эпокси, азиридинил, тетрагидрофуранил,пирролидинил, N-метилпирролидинил, пиперидинил, пиперазинил, пиразолидинил, 4 Н-пиранил, морфолино, тиоморфолино, тетрагидротиенил, тетрагидротиенил 1,1-диоксид и т.п. Фраза "терапевтически эффективное количество" обозначает количество соединения по настоящему изобретению, которое (1) лечит конкретное заболевание, состояние или расстройство, (2) ослабляет,уменьшает интенсивность или устраняет один или более чем один симптом конкретного заболевания,состояния или расстройства, или (3) предотвращает или замедляет начало одного или более чем одного симптома конкретного описанного здесь заболевания, состояния или расстройства. Термин "животное" относится к людям (мужчинам или женщинам), животным-компаньонам (например, собакам, кошкам и лошадям), животным, представляющим собой источник пищи, животным в зоопарках, морским животным, птицам и другим похожим видам животных. "Пригодные в пищу животные" относится к животным, представляющим собой источник пищи, таким как коровы, свиньи, овцы и домашняя птица. Фраза "фармацевтически приемлемый" указывает на то, что вещество или композиция должны быть совместимы химически и/или токсикологически с другими ингредиентами, составляющими композицию, и/или млекопитающим, которое лечат ими. Термины "процесс лечения", "лечить" или "лечение" охватывает предупредительное, т.е. профилактическое, и паллиативное лечение. Если не указано иное, то используемые здесь термины "модулированный" или "модуляция", или"модулируют(ет)" относятся к ингибированию натрийглюкозного транспортера (в частности SGLT2) при помощи соединений по настоящему изобретению, таким образом частично или полностью предотвращая транспорт глюкозы через транспортер. Термин "соединения по настоящему изобретению" (если конкретно не определено иное) относится к соединениям формулы (А), формулы (В) и всем очищенным и смешанным стереоизомерам (включая диастереоизомеры и энантиомеры), таутомерам и изотопно-меченым соединениям. Гидраты и сольваты соединений по настоящему изобретению рассматривают как композиции по настоящему изобретению,где соединение находится соответственно в комбинации с водой или растворителем. Соединения также могут существовать в одном или более чем одном кристаллическом состоянии, т.е. в виде сокристаллов,полиморфов, или они могут существовать в виде аморфных твердых веществ. Все такие формы входят в объем формулы изобретения. В одном из воплощений R1 представляет собой Н, метил, этил, пропил, изопропил, метокси, этокси,F, Cl, циано, -CF3, циклопропил или циклобутил. В еще одном воплощении R1 представляет собой Н,метил, этил, изопропил, метокси, этокси, F, Cl циано, -CF3 или циклопропил. В еще одном воплощенииR1 представляет собой Н, метил, этил, метокси, этокси, F, C1, циано, -CF3 или циклопропил. В еще одном воплощении R1 представляет собой метил, этил, F, C1, циано, CF3 или циклопропил. В одном из воплощений R2 представляет собой метил, этил, пропил, изопропил, метокси, этокси, F,Cl, циано, -CF3, -CF2CH3, этинил, 3-оксетанилокси, 3-тетрагидрофуранилокси или циклопропил. В еще одном воплощении R представляет собой метил, этил, изопропил, метокси, этокси, F, Cl, циано, -CF3,-CF2CH3, этинил, 3-оксетанилокси, 3-тетрагидрофуранилокси или циклопропил. В еще одном воплощении R2 представляет собой метил, этил, метокси, этокси, F, Cl, циано, -CF3, -CF2CH3, этинил, 3 оксетанилокси, 3-тетрагидрофуранилокси или циклопропил. В еще одном воплощении R2 представляет собой метокси или этокси. В одном из воплощений кристалл содержит соединение 4 А и L-пролин или L-пироглутаминовую кислоту. В еще одном воплощении кристалл имеет одно или более чем одно из следующих: а) пространственную группу Р 2(1)2(1)2(1) и параметры элементарной ячейки, по существу, равные следующим: б) картину дифракции рентгеновских лучей на порошке, содержащую 2-тэта величины (излучениеCuK, длина волны 1,54056) 6,40,2, 16,70,2, 17,40,2 и 21,10,2; в) спектр 13 С NMR (ядерный магнитный резонанс) твердого состояния, содержащий пики в положениях 16,50,2, 131,10,2, 158,70,2 и 181,50,2 млн-1 ,определенные на спектрометре при 500 МГц относительно кристалла адамантина с 29,5 млн-11; или г) термограмму дифференциальной сканирующей калориметрии, имеющую эндотерму при приблизительно 142,52 С. В еще одном воплощении кристалл представляет собой сокристалл, содержащий соединение формулы (4 А) и L-пироглутаминовую кислоту в стехиометрическом отношении 1:1. Соединения по настоящему изобретению могут быть синтезированы при помощи путей синтеза,которые включают способы, аналогичные хорошо известным в химической области, в частности в свете содержащегося здесь описания. Исходные вещества в общем доступны из коммерческих источников,таких как Aldrich Chemicals (Milwaukee, WI), или их можно легко получить с использованием способов,хорошо известных специалистам в данной области техники (например, получают при помощи способов,в общем описанных в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, NewYork (1967-1999 ed.), или Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin,включая дополнения (также доступные через базу данных Beilstein в Интернете. Для иллюстративных задач изображенные ниже схемы реакций представляют потенциальные пути для синтеза соединений по настоящему изобретению, а также ключевых промежуточных соединений. Более подробное описание отдельных реакционных стадий см. в нижеприведенном разделе Примеры. Специалистам в данной области техники понятно, что другие синтетические пути могут быть использованы для синтеза заявленных соединений. Хотя конкретные исходные вещества и реагенты изображены на схемах и обсуждаются ниже, другие исходные вещества и реагенты легко могут быть замещены с получением различных производных и/или реакционных условий. Дополнительно, множество соединений,полученных при помощи описанных ниже способов, может быть дополнительно модифицировано в свете данного описания с использованием обычной химии, хорошо известной специалистам в данной области техники. При получении соединений по настоящему изобретению может быть необходима защита удаленной функциональной группы промежуточных соединений. Потребность в такой защите варьирует в зависимости от природы удаленной функциональной группы и условий в способах получения. "Защитная группа гидрокси" относится к заместителю группы гидрокси, который блокирует или защищает функциональную группу гидрокси. Подходящие защитные группы гидрокси (O-Pg) включают, например, аллил,ацетил (Ас), силил (такой как триметилсилил (TMS) или трет-бутилдиметилсилил (TBS, бензил (Вn),пара-метоксибензил (РМВ), тритил (Tr), пара-бромбензоил, пара-нитробензоил и т.п (бензилиден для защиты 1,3-диолов). Потребность в такой защите легко может определить специалист в данной области техники. Общее описание защитных групп и их применение см. в Т. W. Greene, Protective Groups in Organic Synthesis, John WileySons, New York, 1991. На схеме 1 изложены общие способы, которые можно использовать для получения соединений по настоящему изобретению. Схема 1 Аллил-2,3,4-три-O-бензил-В-глюкопиранозид (I-а, где Pg1 представляет собой бензильную группу) может быть получен при помощи способов, описанных в Shinya Hanashima, et al., in BioorganicMedicinal Chemistry, 9, 367 (2001); Patricia A. Gent et al. in Journal of the Chemical Society, Perkin 1, 1835 (1974);Process ResearchDevelopment, 8, 405-407 (2004). На стадии 1 схемы 1 гидроксиметиленовая группа может быть введена в гликозид при помощи окисления Сверна, с последующей обработкой формальдегидом в присутствии гидроксида щелочного металла (например, гидроксида натрия). Эта реакция названа как альдольная реакция Канниззаро. Окисление Сверна описано в Kanji Omura and Daniel Swern inTetrahedron, 34, 1651 (1978). Также могут быть использованы модификации этого способа, известные специалистам в данной области техники. Например, также могут быть использованы другие окислители,такие как стабилизированная 2-йодоксибензойная кислота, описанная Ozanne, A. et al. in Organic Letters,5, 2903 (2003), а также другие окислители, известные специалистам в данной области техники. Последовательность альдол-Канниззаро описана в Robert Schaffer in the Journal of The American Chemical Society,81, 5452 (1959) и Amigues, E.J., et al., in Tetrahedron, 63, 10042 (2007). На стадии 2 схемы 1 защитные группы (Pg) могут быть присоединены путем обработки промежуточного соединения (I-b) соответствующими реагентами и при помощи способов для конкретной желаемой защитной группы. Например, пара-метоксибензильные (РМВ) группы могут быть введены путем обработки промежуточного соединения теле, таком как тетрагидрофуран, 1,2-диметоксиэтан или N,N-диметилформамид (DMF). Также могут быть использованы условия, охватывающие пара-метоксибензилтрихлорацетимидат, в присутствии каталитического количества кислоты (например, трифторметансульфоновой кислоты, метансульфоновой кислоты или камфорсульфоновой кислоты) в растворителе, таком как дихлорметан, гептан или гексаны. Бензильные (Bn) группы могут быть введены путем обработки промежуточного соединения (I-b) бензилбромидом или бензилхлоридом в присутствии гидрида натрия, гидрида калия, трет-бутоксида калия в растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан или N,N-диметилформамид. Также могут быть использованы условия, охватывающие бензилтрихлорацетимидат в присутствии каталитического количества кислоты (например, трифторметансульфоновой кислоты, метансульфоновой кислоты или камфорсульфоновой кислоты) в растворителе, таком как дихлорметан, гептан или гексаны. На стадии 3 схемы 1 аллильную защитную группу удаляют (например, путем обработки хлоридом палладия в метаноле также может быть использован сорастворитель, такой как дихлорметан; также могут быть использованы другие условия, известные специалистам в данной области техники, см. Т. W.(I-d). На стадии 4 схемы 1 окисление незащищенной гидроксильной группы до оксогруппы (например,окисление Сверна) затем позволяет получить лактон (1-е). На стадии 5 схемы 1 лактон (I-е) приводят во взаимодействие с N,O-диметилгидроксиламина гидрохлоридом с образованием соответствующего амида Вайнреба, который может существовать в равновесии в закрытой/открытой форме (I-f/I-g). "Амид Вайнреба" (I-g) может быть получен с использованием способов, хорошо известных специалистам в данной области техники. См. Nahm, S. and S.M. Weinreb,Tetrahedron Letters, 22 (39), 3815-1818 (1981). Например, промежуточное соединение (I-f/I-g) может быть получено из имеющегося в продаже N,O-диметилгидроксиламина гидрохлорида и активирующего агента(например, триметилалюминия). На стадии 6 схемы 1 арилбензильную группу (Ar) вводят с использованием желаемого металлоорганического реагента (например литийорганического соединения (ArLi) или магнийорганического соединения (ArMgX в тетрагидрофуране (THF) при температуре, находящейся в диапазоне от приблизительно -78 С до приблизительно 20 С, с последующим гидролизом (при нахождении в протонных условиях) до соответствующего лактола (I-i), который может находиться в равновесии с соответствующим кетоном (I-h). Связанный мостиковой связью кетальный мотив, обнаруженный в (А) и (В), может быть получен путем удаления защитных групп (Pg ) с использованием соответствующих реагентов для используемых защитных групп. Например, РМВ защитные группы могут быть удалены путем обработки трифторуксусной кислотой в присутствии анизола и дихлорметана (DCM) при приблизительно от 0 С до приблизительно 23 С (комнатная температура). Оставшиеся защитные группы (Pg1) затем могут быть удалены с использованием соответствующей химии для конкретных защитных групп. Например, бензильные защитные группы могут быть удалены путем обработки муравьиной кислотой в присутствии палладия (Pd чернь) в протонном растворителе (например, этаноле/THF) при приблизительно комнатной температуре с получением конечных продуктов (А) и (В). Когда R1 представляет собой CN, тогда кислота Льюиса, такая как трихлорид бора, при температуре, находящейся в диапазоне от приблизительно -78 С до приблизительно комнатной температуры в растворителе, таком как дихлорметан или 1,2-дихлорэтан,также может быть использована для удаления бензильной и/или пара-метоксибензильной защитных групп. Когда R1 представляет собой CN и R2 представляет собой (С 1-С 4)алкокси в промежуточном соединении (I-i) или в продуктах (А) или (В), тогда после обработки кислотой Льюиса, такой как трихлорид бора или трибромид бора, может возникнуть частичное-полное деалкилирование до соответствующего фенола, приводящее к соответствующему соединению (А) или (В), где R1 представляет собой CN, и R2 представляет собой ОН. Если это происходит, то группа (С 1-С 4)алкокси может быть повторно введена путем избирательного алкилирования с использованием (С 1-С 4)алкилйодида в умеренно основных условиях, например с карбонатом калия в ацетоне, при температуре, находящейся в диапазоне от приблизительно комнатной температуры до приблизительно 56C. Когда R1 и/или R2 представляет собой (С 1-С 4)алкил-SO2-, тогда специалисту в данной области техники понятно, что стадию 6 (Схема 1) добавления металлоорганического соединения осуществляют с использованием соответствующего металлоорганического реагента, содержащего (С 1-С 4)алкил-S-. Тиоалкил затем окисляют на более поздней стадии до соответствующего сульфона с использованием обычных способов, известных специалистам в данной области техники. Соединения по настоящему изобретению могут быть получены в виде сокристаллов с использованием любого подходящего способа. Типичная схема получения таких сокристаллов описана на схеме 2. На схеме 2, где Me представляет собой метил и Et представляет собой этил, на стадии 1 1-(5-бром 2-хлорбензил)-4-этоксибензол растворяют в смеси 3:1 толуол:тетрагидрофуран, а затем получающийся в результате раствор охлаждают до температуры меньше -70 С. К этому раствору добавляют гексиллитий при поддержании температуры реакционной среды не более -65 С, а затем перемешивают в течение 1 ч.(3R,4S,5R,6R)-3,4,5-трис(триметилсилилокси)-6-триметилсилилокси)метил)тетрагидропиран-2-он (II-а) растворяют в толуоле и получающийся в результате раствор охлаждают до -15 С. Этот раствор затем добавляют к раствору ариллития при -70 С, а затем перемешивают в течение 1 ч. Затем добавляют раствор метансульфоновой кислоты в метаноле, а затем нагревают до комнатной температуры и перемешивают в течение 16-24 ч. Реакция считается завершенной, когда уровень -аномера составляет не более 3%. Реакционную смесь затем подщелачивают путем добавления 5 М водного раствора гидроксида натрия. Получающиеся в результате соли отфильтровывают, а затем раствор неочищенного продукта концентрируют. 2-Метилтетрагидрофуран добавляют в качестве сорастворителя и органическую фазу дважды экстрагируют водой. Органическую фазу затем концентрируют до 4 объемов в толуоле. Этот концентрат затем добавляют к раствору 5:1 гептан:толуол, вызывая образование осадка. Твердые вещества собирают и сушат в вакууме с получением твердого вещества. На стадии 2 схемы 2 к (II-b) в метиленхлориде добавляют имидазол, а затем охлаждают до 0 С и затем добавляют триметилсилилхлорид с получением персилилированного продукта. Реакционную смесь нагревают до комнатной температуры и гасят путем добавления воды, и органическую фазу промывают водой. Этот неочищенный раствор (II-с) в метиленхлориде сушат над сульфатом натрия и затем используют неочищенным на следующей стадии. На стадии 3 схемы 2 раствор неочищенного вещества (II-с) в метиленхлориде концентрируют до небольшого объема и затем растворитель меняют на метанол. Метанольный раствор (II-с) охлаждают до 0 С, затем добавляют 1 моль.% карбоната калия в виде раствора в метаноле, а затем перемешивают в течение 5 ч. Реакционную смесь затем гасят путем добавления 1 мол.% уксусной кислоты в метаноле, а затем нагревают до комнатной температуры, растворитель меняют на этилацетат и затем фильтруют небольшое количество неорганических твердых веществ. Этилацетатный раствор неочищенного вещества(II-d) используют непосредственно на следующей стадии. На стадии 4 схемы 2 раствор неочищенного вещества (II-d) концентрируют до небольшого объема,-8 018492 затем разбавляют метиленхлоридом и диметилсульфоксидом. Добавляют триэтиламин, а затем охлаждают до 10 С и затем комплекс триоксида серы с пиридином добавляют 3 порциями в виде твердого вещества с 10-минутными интервалами. Реакционную смесь перемешивают в течение еще 3 ч при 10 С, а затем гасят водой и нагревают до комнатной температуры. Фазы разделяют, а затем слой в метиленхлориде промывают водным хлоридом аммония. Раствор неочищенного вещества (II-е) в метиленхлориде используют непосредственно на следующей стадии. На стадии 5 схемы 2 раствор неочищенного вещества (II-е) концентрируют до небольшого объема,и затем растворитель меняют на этанол. Добавляют тридцать эквивалентов водного формальдегида, а затем нагревают до 55 С. Добавляют водный раствор 2 эквивалентов фосфата калия трехосновного, а затем перемешивают в течение 24 ч при 55 С. Температуру реакционной смеси затем увеличивают до 70 С в течение еще 12 ч. Реакционную смесь охлаждают до комнатной температуры, разбавляют третбутилметиловым эфиром и рассолом. Фазы разделяют, а затем органическую фазу меняют на этилацетат. Фазу в этилацетате промывают рассолом и концентрируют до небольшого объема. Концентрат неочищенного вещества затем очищают путем флэш-хроматографии на силикагеле, элюируя 5% метанолом,95% толуолом. Фракции, содержащие продукт, комбинируют и концентрируют до небольшого объема. Добавляют метанол, а затем перемешивают до осаждения. Суспензию охлаждают, и твердые вещества собирают и промывают гептаном, а затем сушат. Продукт (II-f) выделяют в виде твердого вещества. На стадии 6 схемы 2 соединение (II-f) растворяют в 5 объемах метиленхлорида, а затем добавляют 1 мол.% SiliaBond тозиновой кислоты и перемешивают в течение 18 ч при комнатной температуре. Кислотный катализатор отфильтровывают и раствор (II-g) в метиленхлориде используют непосредственно на следующей стадии способа сокристаллизации. На стадии 7 схемы 2 раствор (II-g) в метиленхлориде концентрируют и затем растворитель меняют на 2-пропанол. Добавляют воду, а затем нагревают до 55 С. Добавляют водный раствор Lпироглутаминовой кислоты, а затем получающийся в результате раствор охлаждают до комнатной температуры. В раствор затем вводят затравку и гранулируют в течение 18 ч. После охлаждения твердые вещества собирают и промывают гептаном, а затем сушат. Продукт (II-h) выделяют в виде твердого вещества. Альтернативный путь синтеза соединения (А) по настоящему изобретению изображен на схеме 3 и описан ниже. Схема 3. Синтез (III-а), где R3 представляет собой алкил или фторзамещенный алкил (за исключением атома углерода, располагающегося рядом с атомом кислорода) может быть осуществлен по аналогии с описанным для стадии 1 схемы 2. На стадии 1 схемы 3 первичную гидроксильную группу избирательно защищают при помощи подходящей защитной группы. Например, тритильная группа (Pg3 = Tr) может быть введена путем обработки промежуточного соединения (III-а) хлортрифенилметаном в присутствии основания, такого как пиридин, в растворителе, таком как толуол, тетрагидрофуран или дихлорметан, при температуре, находящейся в диапазоне от приблизительно 0C до приблизительно комнатной температуры. Дополнительные примеры таких защитных групп и экспериментальных условий известны специали-9 018492 стам в данной области техники и могут быть найдены в Т. W. Greene, Protective Groups in Organic Synthesis, John WileySons, New York, 1991. На стадии 2 схемы 3 вторичные гидроксильные группы могут быть защищены при помощи подходящих защитных групп. Например, бензильные группы (Pg4 представляет собой Bn) могут быть введены путем обработки промежуточного соединения (III-b) бензилбромидом или бензилхлоридом в присутствии гидрида натрия, гидрида калия, трет-бутоксида калия в растворителе, таком как тетрагидрофуран,1,2-диметоксиэтан или N,N-диметилформамид, при температуре, находящейся в диапазоне от приблизительно 0C до приблизительно 80C. Ацетильные или бензоильные группы (Pg4 = Ac или Bz) могут быть введены путем обработки промежуточного соединения (III-b) ацетилхлоридом, ацетилбромидом или уксусным ангидридом или бензоилхлоридом, или бензойным ангидридом в присутствии основания, такого как триэтиламин, N,N-диизопропилэтиламин или 4-(диметиламино)пиридин, в растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан или дихлорметан, при температуре находящейся в диапазоне от приблизительно 0C до приблизительно 80C. На стадии 3 схемы 3 защиту с первичной гидроксильной группы удаляют, получая промежуточное соединение (III-d). Когда Pg3 представляет собой Tr, тогда промежуточное соединение (III-с) обрабатывают в присутствии кислоты, такой как трет-толуолсульфоновая кислота, в спиртовом растворителе, таком как метанол, при температуре, находящейся в диапазоне от приблизительно -20C до приблизительно комнатной температуры, с получением промежуточного соединения (III-d). Могут быть использованы сорастворители, такие как хлороформ. На стадии 4 схемы 3 гидроксиметиленовую группу вводят посредством способа, аналогичного уже описанному на схеме 1 (стадия 1) и схеме 2 (стадии 4 и 5). Другие источники формальдегида, такие как параформальдегид, в растворителе, таком как этанол, при температуре, находящейся в диапазоне от приблизительно комнатной температуры до приблизительно 70C, в присутствии алкоксида щелочного металла, также могут быть использованы на этой стадии. Когда Pg4 представляет собой Bn, тогда эта стадия позволяет получить промежуточное соединение (III-е), и когда Pg4 представляет собой Ас или Bz, тогда эта стадия позволяет получить промежуточное соединение (III-f). На стадии 5 схемы 3 промежуточное соединение (III-е) обрабатывают кислотой, такой как трифторуксусная кислота, или кислотной смолой в растворителе, таком как дихлорметан, при температуре, находящейся в диапазоне от приблизительно -10C до приблизительно комнатной температуры, с получением промежуточного соединения (III-g). На стадии 6 схемы 3 оставшиеся защитные группы (Pg4) затем могут быть удалены с использованием химии, подходящей для конкретных защитных групп. Например, бензильные защитные группы могут быть удалены путем обработки муравьиной кислотой в присутствии палладия (Pd чернь) в протонном растворителе (например, этанол/THF) при приблизительно комнатной температуре с получением конечного продукта (А). На стадии 7 схемы 3 промежуточное соединение (III-f) обрабатывают кислотой, такой как трифторуксусная кислота, или кислотной смолой в растворителе, таком как дихлорметан, при температуре, находящейся в диапазоне от приблизительно -10C до приблизительно комнатной температуры, с получением конечного продукта (А). Еще одна альтернативная схема синтеза продукта (А) изображена на схеме 4 и описана ниже. Схема 4 На стадии 1 схемы 4 промежуточное соединение (III-а) обрабатывают подходящим арилсульфонилхлоридом R4SO2Cl или арилсульфоновым ангидридом R4S(O)2OS(O)2R4 (где R4 представляет собой возможно замещенную арильную группу, такую как обнаруживаемая в арилсульфонилхлоридах: 4-метилбензолсульфонилхлориде, 4-нитробензолсульфонилхлориде, 4-фторбензолсульфонилхлориде, 2,6 дихлорбензолсульфонилхлориде,4-фтор-2-метилбензолсульфонилхлориде и 2,4,6-трихлорбензолсульфонилхлориде, и в арилсульфоновом ангидриде, пара-толуолсульфоновом ангидриде) в присутствии основания, такого как пиридин, триэтиламин, N,N-диизопропилэтиламин, в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, при температуре, находящейся в диапазоне от при- 10018492 близительно -20C до приблизительно комнатной температуры. Некоторые кислоты Льюиса, такие как бромид цинка(II), могут быть использованы в качестве добавок. На стадии 2 схемы 4 промежуточное соединение (IV-a) подвергают окислению по типу Корнблума(см. Kornblum, N., et al., Journal of The American Chemical Society, 81, 4113 (1959 с получением соответствующего альдегида, который может существовать в равновесии с соответствующей формой гидрата и/или полуацеталя. Например, промежуточное соединение (IV-a) обрабатывают в присутствии основания,такого как пиридин,2,6-лутидин,2,4,6-коллидин,N,N-диизопропилэтиламин,4(диметиламино)пиридин, в растворителе, таком как диметилсульфоксид, при температуре, находящейся в диапазоне от приблизительно комнатной температуры до приблизительно 150C. Полученное альдегидное промежуточное соединение затем подвергают альдольным условиям/условиям Канниззаро, описанным для стадии 1 (схема 1) и стадии 5 (схема 2) с получением промежуточного соединения (IV-b). На стадии 3 схемы 4 промежуточное соединение (IV-b) обрабатывают кислотой, такой как трифторуксусная кислота, или кислотной смолой в растворителе, таком как дихлорметан, при температуре, находящейся в диапазоне от приблизительно -10C до приблизительно комнатной температуры, с получением конечного продукта (А). Когда R2 представляет собой (С 1-С 4)алкинил, то способ может быть осуществлен с использованием схемы 5, где R6 представляет собой Н или (С 1-С 2)алкил. Схема 5 На стадии 1 схемы 5, которая позволяет получить промежуточное соединение (V-i), стадию добавления металлоорганического соединения осуществляют по аналогии с описанным на схеме 1, стадия 6, с использованием металлоорганического реагента, являющегося производным (V-a), где Pg5 представляет собой подходящую защитную группу для гидроксильной группы. Например, Pg5 может представлять собой трет-бутилдиметилсилильную группу (TBS) (см. US 2007/0054867 в отношении получения, например, 4-[(5-бром-2-хлорфенил)метил]фенокси-трет-бутилдиметилсилана). На стадии 2 схемы 5, когда Pg2 = РМВ, промежуточное соединение (V-i) обрабатывают кислотой, такой как трифторуксусная кислота, метансульфоновая кислота или кислотная смола, в присутствии анизола в растворителе, таком как дихлорметан, при температуре, находящейся в диапазоне от приблизительно -10C до приблизительно комнатной температуры, с получением промежуточного соединения (V-j). На стадии 3 схемы 5 защитные группы (Pg5) и (Pg1) могут быть удалены с получением (V-k). Типично (Pg5) представляет собой TBS и Pg1 редставляет собой Bn. В этом случае защитные группы удаляют путем последовательной обработки (V-j) (1) фторидом тетрабутиламмония в растворителе, таком как тетрагидрофуран или 2-метилтетрагидрофуран, при температуре, находящейся в диапазоне от 0C до приблизительно 40C и (2) муравьиной кислотой в присутствии палладия (Pd чернь) в протонном растворителе (например, этанол/THF) при приблизительно комнатной температуре. В этой последовательности порядок 2 реакций взаимозаменяем. На стадии 4 схемы 5 промежуточное соединение (V-k) обрабатывают N,N-бис(трифторметансульфонил)анилином в присутствии основания, такого как триэтиламин или 4 диметиламинопиридин, в растворителе, таком как дихлорметан или 1,2-дихлорэтан, при температуре,находящейся в диапазоне от 0C до приблизительно 40C, с получением промежуточного соединения (V1). На стадии 5 схемы 5 промежуточное соединение (V-1) подвергают реакции типа Соногашира (см.(eds. Trost, В. М., Fleming, I.), 3, 521-549, (Pergamon, Oxford, 1991. Например (V-1) обрабатывают соответствующим терминальным алкином HCCR6 в присутствии йодида меди (I), катализатора, такого как бис-(трифенилфосфин)палладия дихлорид или тетракис(трифенилфосфин)палладий(0), в присутствии основания, такого как триэтиламин или N,N-диизопропилэтиламин, в растворителе, таком как N,Nдиметилформамид, при температуре, находящейся в диапазоне от приблизительно комнатной температуры до приблизительно 120C, с получением желаемого продукта (А) и (В). Когда R6 представляет собой Н, то удобней использовать триметилсилилацетилен. В этом случае неочищенное вещество, полученное в результате вышеописанной реакции, обрабатывают основанием, таким как карбонат калия, в спиртовом растворителе, таком как МеОН, при приблизительно комнатной температуре, с получением после классической обработки, известной специалистам в данной области техники, желаемого продукта(А) и (В), где R2 представляет собой -ССН. Специалисту в данной области техники понятно, что химия, описанная выше на схемах 1-5, представляет собой различные пути получения промежуточного соединения (V-k). В свою очередь, в частности, когда R1 представляет собой Cl, (V-k) может быть обработан алкилирующим агентом, выбираемым в классических условиях, для избирательного алкилирования фенольной группы с получением (А) и (В) на схемах 1 и 5, где R2 представляет собой (С 1-С 4)алкокси. Соединения по настоящему изобретению содержат асимметрические или хиральные центры, и, таким образом, существуют в различных стереоизомерных формах. Если не указано иное, то предполагают, что все стереоизомерные формы соединений по настоящему изобретению, а также их смеси, включающие рацемические смеси, образуют часть настоящего изобретения. Дополнительно настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение по настоящему изобретению включает двойную связь или конденсированное кольцо, то цис- и транс-формы, а также смеси охвачены в объеме изобретения. Диастереоизомерные смеси могут быть разделены на индивидуальные диастереоизомеры на основе их физико-химических различий при использовании способов, хорошо известных специалистам в данной области техники, таких как хроматография и/или фракционная кристаллизация, дистилляция, возгонка. Энантиомеры могут быть разделены путем превращения энантиомерной смеси в диастереоизомерную смесь путем взаимодействия с соответствующим оптически активным соединением (например, хиральным дополнительным веществом, таким как хиральный спирт или хлорангидрид Мошера), разделения диастереоизомеров и превращения (например, гидролизом) индивидуальных диастереоизомеров в соответствующие чистые энантиомеры. Кроме того, некоторые из соединений по настоящему изобретению могут представлять собой атропизомеры (например, замещенные биарилы), и их рассматривают как часть данного изобретения. Энантиомеры также могут быть разделены путем использования колонки для хиральной HPLC(жидкостной хроматографии высокого давления). Также возможно, чтобы промежуточные соединения и соединения по настоящему изобретению существовали в различных таутомерных формах, и все такие формы входят в объем изобретения. Термин"таутомер" или "таутомерная форма" относится к структурным изомерам, обладающим отличающимися энергиями, которые являются взаимопревращаемыми через низкоэнергетический барьер. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения по- 12018492 средством миграции протона, такие как изомеризации кетоенол и иминэнамин. Конкретный пример протонного таутомера представляет собой имидазольную группировку, где протон может мигрировать между двумя кольцевыми атомами азота. Валентные таутомеры включают взаимопревращения путем реорганизации некоторых из связывающих электронов. Равновесие между закрытой и открытой формой некоторых промежуточных соединений (и/или смесей промежуточных соединений) напоминает процесс мутаротации, захватывающий альдозы, известные специалистам в данной области техники. Настоящее изобретение также охватывает изотопно-меченые соединения по настоящему изобретению, которые идентичны приведенным здесь, за исключением того, что один или более чем один атом заменен на атом, имеющий атомную массу или атомное число, отличающиеся от атомной массы или массового числа, обычно обнаруживаемого в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора,серы, фтора, йода и хлора, такие как, соответственно 2 Н, 3 Н, 11 С, 13 С, 14 С, 13N, 15N, 15O, 17O, 18O,31 Р, 32 Р,35S, 18F, 123I, 125I и 36Cl. Некоторые изотопно-меченые соединения по настоящему изобретению (например, меченые 3 Н и 14 С) полезны в анализах тканевого распределения соединения и/или субстрата. Тритиевые (т.е. 3 Н) и углерод-14 (т.е. 14 С) изотопы особенно предпочтительны ввиду легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий (т.е. 2 Н) может обеспечить некоторые терапевтические преимущества, являющиеся результатом большей метаболической стабильности (например, увеличенного периода полувыведения in vivo или уменьшенной потребностью в дозах) и следовательно в некоторых случаях может быть предпочтительным. Позитронно-активные изотопы, такие как 15O, l3N, 11 С и l8F, полезны для исследований при помощи позитронно-эмиссионной томографии(PET) для проверки субстратной оккупации. Изотопно-меченые соединения по настоящему изобретению в общем могут быть получены при помощи следующих способов, аналогичных описанным на схемах и/или в приведенных ниже примерах, путем замещения изотопно-меченого реагента на не меченый изотопно реагент. Соединения по настоящему изобретению полезны для лечения заболеваний, состояний и/или расстройств, модулируемых путем ингибирования натрий-глюкозных транспортеров (в частности SGLT2); таким образом, еще одно воплощение настоящего изобретения представляет собой фармацевтическую композицию, содержащую терапевтически эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Соединения по настоящему изобретению (включая композиции и используемые здесь способы) также могут быть использованы в изготовлении лекарственного средства для описанных здесь терапевтических применений. Типичную композицию готовят путем смешивания соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Подходящие носители, разбавители и эксципиенты хорошо известны специалистам в данной области техники и включают вещества, такие как углеводы, воски, водорастворимые и/или набухаемые полимеры, гидрофильные или гидрофобные вещества, желатин, масла,растворители, вода и т.п. Конкретный используемый носитель, разбавитель или эксципиент зависит от средств и задачи, для которой применяется соединение по настоящему изобретению. Растворители в общем выбирают на основе растворителей, известных специалистам в данной области техники как безопасные (GRAS) при введении млекопитающему. В общем, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые растворимы в воде или смешиваются с ней. Подходящие водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG400, PEG300) и т.д. и их смеси. Композиции также могут включать один или более чем один буфер, стабилизатор, поверхностно-активное вещество, увлажнитель,смазывающий агент, эмульгатор, суспендирующий агент, консервант, антиоксидант, агент, придающий непрозрачность, скользящее вещество, агент, используемый при обработке, краситель, подсластитель,ароматизатор, корригент и другие известные добавки, придающие лучший внешний вид лекарству (т.е. соединению по настоящему изобретению или его фармацевтической композиции) или служащие для изготовления фармацевтического продукта (т.е. лекарственного средства). Композиции могут быть приготовлены с использованием обычных способов растворения и смешивания. Например, нефасованное лекарственное вещество (т.е. соединение по настоящему изобретению или стабилизированная форма соединения (например, комплекс с циклодекстриновым производным или другим известным комплексообразующим агентом растворяют в подходящем растворителе в присутствии одного или более чем одного из описанных выше эксципиентов. Соединение по настоящему изобретению типично готовят в фармацевтических лекарственных формах для обеспечения легко контролируемой дозировки лекарства и для обеспечения пациента приятным и легким в обращении продуктом. Фармацевтические композиции также включают сольваты и гидраты соединений формулы (I). Термин "сольват" относится к молекулярному комплексу соединения, представленного формулой (I) (включая их фармацевтически приемлемые соли), с одной или более чем одной молекулой растворителя. Такие молекулы растворителя представляют собой молекулы, обычно используемые в области фармации, которые известны как безвредные для реципиента, например воду, этанол, этиленгликоль и т.п. Термин"гидрат" относится к комплексу, в котором молекула растворителя представляет собой воду. Сольваты и/или гидраты предпочтительно существуют в кристаллической форме. Другие растворители могут быть использованы в качестве промежуточных сольватов при получении более желательных сольватов, такие как метанол, метил-трет-бутиловый эфир, этилацетат, метилацетат, (S)-пропиленгликоль, (R)пропиленгликоль, 1,4-бутин-диол и т.п. Кристаллические формы также могут существовать в виде комплексов с другими безвредными небольшими молекулами, такими как L-фенилаланин, L-пролин, Lпироглутаминовая кислота и т.п, в виде сокристаллов или сольватов, или гидратов сокристаллического вещества. Сольваты, гидраты и сокристаллические соединения могут быть получены с использованием способов, описанных в публикации РСТWO 08/002824, включенной сюда путем ссылки, или других способов, хорошо известных специалистам в данной области техники. Фармацевтическая композиция (или препарат) для применения может быть упакована различными путями в зависимости от способа, используемого для введения лекарства. Как правило, изделие для распределения включает контейнер, внутри которого находится фармацевтическая композиция в подходящей форме. Подходящие контейнеры хорошо известны специалистам в данной области техники и включают материалы, такие как бутыли (пластиковые и стеклянные), саше, ампулы, пластиковые контейнеры,металлические цилиндры и т.п. Контейнер также может включать защищающий от неумелого обращения механизм для предотвращения неосторожного доступа к содержимому контейнера. Дополнительно, контейнер несет на себе этикетку, которая описывает содержимое контейнера. Этикетка также может включать соответствующие предупреждения. В настоящем изобретении также предложен способ лечения заболеваний, состояний и/или расстройств, модулируемых путем ингибирования натрий-глюкозных транспортеров, у животного, при котором животному, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по настоящему изобретению или фармацевтической композиции, содержащей эффективное количество соединения по настоящему изобретению и фармацевтически приемлемый эксципиент, разбавитель или носитель. Способ особенно полезен для лечения заболеваний, состояний и/или расстройств,для которых благоприятно ингибирование SGLT2. Один из аспектов настоящего изобретения представляет собой лечение ожирения и связанных с ожирением расстройств (например, избыточная масса тела, увеличение массы тела или поддержание массы тела). Ожирение и избыточная масса тела в общем определяются индексом массы тела (BMI), который коррелирует с общим содержанием в организме жира и оценивает относительный риск заболевания. BMI вычисляют по массе в килограммах, деленной на рост в квадратных метрах (кг/м 2). Избыточная масса тела как правило определяется как BMI 25-29,9 кг/м 2, и ожирение как правило определяется как BMI 30 кг/м 2. См., например National Heart, Lung and Blood Institute, Clinical Guidelines on the Identification,Evaluation, and Treatment of Overweight and Obesity in Adults, The Evidence Report, Washington, DC: U.S.Department of Health and Human Services, NIH publication no. 98-4083(1998). Еще один аспект настоящего изобретения заключается в лечении или замедлении прогрессирования или начала диабета или связанных с диабетом расстройств, включая диабет 1 типа (инсулинзависимый сахарный диабет, также называемый как "IDDM") и 2 типа (инсулиннезависимый сахарный диабет, также называемый как "NIDDM"), нарушенную толерантность к глюкозе, замедленное заживление ран, гиперинсулинемию, повышенный уровень жирных кислот в крови, гиперлипидемию, гипертриглицеридемию, синдром X, повышенный уровень липопротеинов высокой плотности, резистентность к инсулину,гипергликемию и осложнения сахарного диабета (такие как атеросклероз, ишемическая болезнь сердца,инсульт, болезнь периферических сосудов, нефропатия, гипертензия, нейропатия и ретинопатия). Еще один аспект настоящего изобретения представляет собой лечение сопутствующих ожирению заболеваний, таких как метаболический синдром. Метаболический синдром включает заболевания, состояния или расстройства, такие как дислипидемия, гипертензия, резистентность к инсулину, диабет (например, диабет 2 типа), заболевание коронарных артерий и сердечная недостаточность. Более подробную информацию о метаболическом синдроме см., например в Zimmet, P.Z. et al., "The Metabolic Syndrome:Definition," Lancet, 366, 1059-62 (2005). Предпочтительно введение соединений по настоящему изобретению обеспечивает статистически значимое (р 0,05) уменьшение по меньшей мере одного фактора риска сердечно-сосудистого заболевания, такого как уменьшение в плазме крови лептина, С-реактивного белка (CRP) и/или холестерина, по сравнению с контролем, представляющем собой носитель без лекарства. Введение соединений по настоящему изобретению также может обеспечить статистически значимое (р 0,05) уменьшение уровней глюкозы в крови. Для нормального взрослого человека, имеющего массу тела приблизительно 100 кг, как правило,достаточна доза, находящаяся в диапазоне от приблизительно 0,001 до приблизительно 10 мг на килограмм массы тела, предпочтительно от приблизительно 0,01 до приблизительно 5,0 мг/кг, более предпочтительно от приблизительно 0,01 до приблизительно 1 мг/кг. Тем не менее, некоторое изменение общего диапазона дозы может потребоваться в зависимости от возраста и массы субъекта, которого лечат, предполагаемого пути введения, конкретного соединения, которое вводят, и т.п. Определение диапазонов доз и оптимальных доз для конкретного пациента находится в пределах знаний специалиста в данной области техники, получающего пользу от настоящего описания. Также отмечают, что соединения по настоящему изобретению могут быть использованы в препаратах с длительным высвобождением, контролируемым высвобождением и замедленным высвобождением, причем эти формы также хорошо известны специалистам в данной области техники. Соединения по изобретению также могут быть использованы в сочетании с другими фармацевтическими агентами для лечения описанных здесь заболеваний, состояний и/или расстройств. Таким образом,также предложены способы лечения, при которых вводят соединения по настоящему изобретению в комбинации с другими фармацевтическими агентами. Подходящие фармацевтические агенты, которые могут быть использованы в комбинации с соединениями по настоящему изобретению, включают агенты против ожирения (включая агенты, подавляющие аппетит), антидиабетические агенты, антигипергликемические агенты, агенты, снижающие уровень липидов, противовоспалительные агенты и антигипертензивные агенты. Подходящие агенты против ожирения включают антагонисты каннабиноидных рецепторов-1 (СВ 1) (такие как римонабант), ингибиторы 11-гидроксистероиддегидрогеназы-1 (11-HSD тип 1), ингибиторы стеароил-СоА десатуразы-1 (SCD-1), агонисты меланокортиновых рецепторов MCR-4, агонисты холецистокинина-А (ССК-А), ингибиторы обратного захвата моноамина (такие как сибутрамин), симпатомиметики, (3 адренергические агонисты, дофаминовые агонисты (такие как бромкриптин), аналоги меланоцитстимулирующего гормона, агонисты 5 НТ 2 с, антагонисты меланинконцентрирующего гормона, лептин (белок ОВ), лептиновые аналоги, лептиновые агонисты, галаниновые антагонисты, ингибиторы липазы (такие как тетрагидролипстатин, т.е. орлистат), анорексические агенты (такие как бомбезиновый агонист), антагонисты нейропептида-Y (например, антагонисты NPY Y5), пептид PYY3-36 (включая его аналоги), тиреомиметики, дегидроэпиандростерон или его аналоги, глюкокортикоидные агонисты или антагонисты, орексиновые антагонисты, агонисты глюкагонподобного пептида-1, цилиарные нейротрофические факторы (такие как Axokine, доступный от Regeneron Pharmaceuticals, Inc., Tarrytown, NY и ProcterGamble Company, Cincinnati, ОН), ингибиторы человеческого агути-связанного белка (AGRP),грелиновые антагонисты, антагонисты или обратные агонисты гистамина 3, агонисты нейромедина U,ингибиторы МТР/АроВ (например, селективные для кишечника ингибиторы МТР, такие как дирлотапид), опиоидные антагонисты, орексиновые антагонисты и т.п. Предпочтительные агенты против ожирения для применения в комбинированных аспектах настоящего изобретения включают антагонисты СВ-1 (например, римонабант, таранабант, суринабант, отенабант, SLV319 (CAS464213-10-3) и AVE1625 (CAS358970-97-5, селективные для кишечника ингибиторы МТР (например, дирлотапид, митратапид и имплитапид, R56918 (CAS403987) и CAS913541-47-6), агонисты ССКа (например, N-бензил-2-[4-(1 Н-индол-3-илметил)-5-оксо-1-фенил-4,5 дигидро-2,3,6,10b-тетраазабензо[е]азулен-6-ил]-N-изопропил-ацетамид, описанный в публикации РСТWO 2005/116034 или публикации США 2005-0267100 А 1), агонисты 5 НТ 2 с (например, лоркасерин),агонисты MCR4 (например, соединения, описанные в US 6818658), ингибитор липазы (например, Cetilistat), PYY3.36 (используемый здесь "PYY3-36" включает аналоги, такие как ПЭГилированный PYY3-36,например описанный в публикации заявки на патент США 2006/0178501), опиоидные антагонисты (например, налтрексон), олеоил-эстрон (CAS180003-17-2), обинепитид (ТМ 30338), прамлинтид (Symlin), тезофензин (NS2330), лептин, лираглутид, бромкриптин, орлистат, эксенатид (Byetta), AOD-9604(CAS221231-10-3) и сибутрамин. Предпочтительно соединения по настоящему изобретению и комбинированные способы лечения применяют в сочетании с физической нагрузкой и целесообразной диетой. Подходящие антидиабетические агенты включают ингибитор ацетил-СоА карбоксилазы-2 (АСС-2),ингибитор фосфодиэстеразы (PDE)-10, ингибитор диацилглицеролацилтрансферазы (DGAT) 1 или 2,сульфонилмочевину (например, ацетогексамид, хлорпропамид, диабинез, глибенкламид, глипизид, глибурид, глимепирид, гликлазид, глипентид, глихидон, глизоламид, толазамид и толбутамид), меглитинид,ингибитор -амилазы (например, тендамистат, трестатин и AL-3688), ингибитор -глюкозидгидролазы(например, акарбоза), ингибитор -глюкозидазы (например, адипозин, камиглибоза, эмиглитат, миглитол, воглибоза, прадимицин-Q и салбостатин), агонист PPAR (например, балаглитазон, циглитазон,дарглитазон, энглитазон, изаглитазон, пиоглитазон, розиглитазон и троглитазон), агонист PPAR / (например, CLX-0940, GW-1536, GW-1929, GW-2433, KRP-297, L-796449, LR-90, МК-0767 и SB-219994),бигуанид (например, метформин), агонист глюкагонподобного пептида 1 (GLP-1) (например, эксендин-3 и эксендин-4), ингибитор протеин тирозинфосфатазы-1 В (РТР-1 В) (например, тродусквемин, экстрактhyrtiosal и соединения, раскрытые Zhang, S., et al., Drug Discovery Today, 12(9/10), 373-381 (2007, ингибитор SIRT-1 (например, резерватрол), ингибитор дипептидилпептидазы IV (DPP-IV) (например, ситаглиптин, вилдаглиптин, алоглиптин и саксаглиптин), агент, усиливающий секрецию инсулина, ингибитор окисления жирных кислот, антагонист А 2, ингибитор c-jun аминоконцевой киназы (JNK), инсулин, ми- 15018492 метик инсулина, ингибитор гликогенфосфорилазы, агонист рецептора VPAC2 и активатор глюкокиназы. Предпочтительные антидиабетические агенты представляют собой метформин и ингибиторы DPP-IV(например, ситаглиптин, вилдаглиптин, алоглиптин и саксаглиптин). Подходящие противовоспалительные агенты включают средства для предупреждения и лечения инфекции полового тракта/мочевого тракта. Примеры агентов включают клюкву (т.е. Vaccinium macrocarpon) и производные клюквы, такие как клюквенный сок, клюквенные экстракты или флавонолы клюквы. Клюквенные экстракты могут включать один или более чем один флавонол (т.е. антоцианины и проантоцианидины) или очищенное флавонольное соединение из клюквы, включающее мирицетин-3-ксилопиранозид, кверцетин-3 глюкозид, кверцетин-3 арабинопиранозид, 3'-метоксикверцетин-3-ксилопиранозид,кверцетин-3-O-(6"-пара-кумароил)галактозид,кверцетин-3-O-(6"-бензоил)-галактозид и/или кверцетин-3 арабинофуранозид. Воплощения настоящего изобретения проиллюстрированы следующими примерами. Тем не менее,понятно, что воплощения изобретения не ограничиваются конкретными деталями этих примеров, поскольку другие их вариации известны или понятны в свете настоящего описания специалистам в данной области техники. Примеры Если не указано иное, то исходные вещества в общем доступны из коммерческих источников, таких как Aldrich Chemicals Co. (Milwaukee, WI), Lancaster Synthesis, Inc. (Windham, NH), Acros Organics (Fairlawn, NJ), Maybridge Chemical Company, Ltd. (Cornwall, England), Tyger Scientific (Princeton, NJ), AstraZeneca Pharmaceuticals (London, England) и Accela ChemBio (San Diego, CA). Общие экспериментальные процедуры Спектры ядерного магнитного резонанса (NMR) регистрировали на Varian Unity 400 (доступном от Varian Inc., Palo Alto, CA) при комнатной температуре при 400 МГц для протона. Химические сдвиги выражены в миллионных долях (дельта) относительно остаточного растворителя в качестве внутреннего стандарта. Формы пиков обозначены следующим образом: s, синглет; d, дублет; dd, дублет дублетов; t,триплет; q, квартет; m, мультиплет; bs или br.s., широкий синглет; 2s, два синглета; br.d., широкий дублет. Масс-спектры с ионизацией путем распыления электронов (ES) получали на приборе Waters ZMD(газ-носитель: азот; растворитель А: вода/0,01% муравьиная кислота, растворитель В: ацетонитрил/0,005% муравьиная кислота; доступном от Waters Corp., Milford, MA). Масс-спектры высокого разрешения (HRMS) получали на времяпролетном масс-спектрометре Agilen Model 6210. Когда описана интенсивность ионов, содержащих один атом хлора или один атом брома, тогда обнаруживали ожидаемое отношение интенсивности (приблизительно 3:1 для 35Cl/37Cl-содержащих ионов и 1:1 для 79Br/81Brсодержащих ионов) и приведена интенсивность только малой массы. В некоторых случаях приведены только типичные пики 1 Н NMR. Колоночную хроматографию осуществляли на силикагеле Baker (40 мкм; J.T. Baker, Phillipsburg,NJ) или Silica Gel 50 (EM Sciences, Gibbstown, NJ) в стеклянных колонках или в колонках Flash 40 Biotagetttt (ISC, Inc., Shelton, CT). MPLC (жидкостную хроматографию среднего давления) осуществляли с использованием системы очистки Biotage SP или Combiflash Companion от Teledyne Isco; использовали картридж Biotage SNAP KPsil или диоксид кремния Redisep Rf (от Teledyne Isco) при низком давлении азота.HPLC (жидкостную хроматографию высокого давления) осуществляли с использованием Shimadzu 10A LC-UV (жидкостной хроматографии-УФ) или препаративной HPLC Agilent 1100. За исключением того, когда указано иное, все взаимодействия осуществляли в инертной атмосфере газообразного азота с использованием безводных растворителей. Также за исключением того, когда указано иное, все взаимодействия осуществляли при комнатной температуре (приблизительно 23 С). При проведении TLC (тонкослойная хроматография) Rf определяют как отношение расстояния,пройденного соединением, к расстоянию, пройденному растворителем.Rt (время удерживания). Исходные вещества Как правило, любое из следующих исходных веществ может быть получено с использованием способов, описанных на схемах 7 или 8 публикации на патент США 2008/0132563 или альтернативно схемах 2, 3 или 8 публикации на патент США 2007/0259821. Конкретней, следующие исходные вещества, используемые в следующих примерах, могут быть получены с использованием способов, описанных в соответствующих ссылках, или приобретены у соответствующего поставщика. 4-Бром-2-(4-метоксибензил)-1-метилбензол может быть получен посредством способов, описанных в примере 8 публикации РСТWO 01/027128. 4-Бром-2-(4-этоксибензил)-1-метилбензол может быть получен посредством способов, описанных в подготовительном примере 17 в US 2008/0132563. 4-Бром-1-хлор-2-(4-метоксибензил)бензол может быть получен посредством способов, описанных в подготовительном примере 19 в US 2008/0132563 или примере V в US 2007/0259821. 4-Бром-1-хлор-2-(4-этоксибензил)бензол может быть приобретен в Shanghai Haoyuan ChemexpressCo., Ltd., Shanghai, People's Republic of China. 4-Бром-2-(4-метоксибензил)бензонитрил может быть получен посредством способов, описанных в примере XXII в US 2007/0259821. Следующие исходные вещества получали, как описано ниже. Получение 4-бром-1-фтор-2-(4-метоксибензил)бензола: Оксалилхлорид (11,0 мл, 126 ммоль) по каплям добавляли к хорошо перемешиваемой суспензии 5 бром-2-фторбензойной кислоты (25,0 г, 114 ммоль) в дихлорметане (150 мл) и N,N-диметилформамиде(1,5 мл) при 0 С. Получающуюся в результате смесь оставляли постепенно нагреваться до комнатной температуры. Через 18 ч твердое вещество превращалось в раствор. Получающийся в результате светлооранжевый раствор концентрировали при пониженном давлении и дважды очищали диэтиловым эфиром с получением 5-бром-2-фторбензоилхлорида (27,0 г, количественный выход) в виде бледно-оранжевого масла. К раствору 5-бром-2-фторбензоилхлорида (27,0 г, 114 ммоль) и анизолу (12,9 г, 13,0 мл, 119 ммоль) в дихлорметане (150 мл) при 0 С порциями добавляли трихлорид алюминия (16,2 г, 119 ммоль) таким образом, что внутренняя температура оставалась ниже 10 С. После перемешивания в течение 4 ч при 0 С раствор выливали в колотый лед и получающуюся в результате смесь перемешивали. Через 30 мин органическую фазу удаляли и водную фазу экстрагировали дважды дихлорметаном. Объединенные органические фазы однократно промывали водным 1 М раствором соляной кислоты, однократно водным 1 М раствором гидроксида натрия и однократно рассолом. Органическую фазу сушили над сульфатом натрия, фильтровали, и концентрировали при пониженном давлении. Получающийся в результате остаток перекристаллизовывали из этанола с получением (5-бром-2-фторфенил)-(4-метоксифенил)метанона (22,5 г, 64%) в виде белого твердого вещества. К хорошо перемешиваемому раствору (5-бром-2-фторфенил)-(4-метоксифенил)метанона (22,5 г,72,80 ммоль) и триэтилсилана (27,9 мл, 20,3 г, 175,0 ммоль) в дихлорметане (20 мл) и ацетонитриле (60 мл) при 0 С по каплям добавляли эфират трифторида бора (32,0 мл, 36,2 г, 255,0 ммоль). Эфират трифторида бора добавляли со скоростью такой, что внутренняя температура не превышала 20 С. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение ночи. После в общей сложности 18 ч добавляли раствор гидроксида калия (5,0 г) в воде (15,0 мл) и получающуюся в результате смесь перемешивали в течение 2 ч. Органическую фазу отделяли и водную фазу дважды экстрагировали диэтиловым эфиром. Объединенные органические фазы однократно промывали водным 1 М раствором гидроксида натрия и однократно рассолом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. После добавления этанола к получающемуся в результате остатку образовывалось белое твердое вещество. Твердое вещество собирали и сушили в высоком вакууме с получением 4-бром-1-фтор-2-(4-метоксибензил)бензола (20,1 г, выход 93%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, хлороформ-d) дельта млн-1 3.79 (s, 3 Н), 3.89 (s, 2 Н), 6.85 (d, J=8.6 Hz, 2 Н), 6.91 (t,1 Н), 7.12 (d, J=8.8 Hz, 2 Н), 7.21-7.31 (m, 2 Н). Получение исходного вещества 4-бром-2-(4-этоксибензил)бензонитрила: Раствор этил-(4-этоксифенил)ацетата (2,68 г, 12,87 ммоль), 4-бром-2-фторбензонитрила (2,74 г,13,70 ммоль) в N-метилпирролидоне (4 мл) медленно добавляли к суспензии трет-бутоксида калия (3,14 г, 27,98 ммоль) в N-метилпирролидоне (13 мл) при 0 С. После добавления раствор становился темнокрасным. Темно-красную смесь перемешивали при 0 С в течение 30 мин и затем при комнатной температуре в течение 1 ч. Добавляли метанол (10 мл) и водный 1 М раствор гидроксида натрия (13,7 мл) и смесь перемешивали в течение ночи при комнатной температуре. рН доводили до приблизительно 4 соляной кислотой (1 М водный раствор) и смесь экстрагировали этилацетатом (50 мл 4). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и упаривали досуха. ДобавлялиN,N-диметилформамид (5 мл) и карбонат калия (7 г), смесь нагревали до 100 С в течение 1 ч и охлаждали до комнатной температуры. Добавляли воду и смесь экстрагировали этилацетатом (60 мл 3). Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и упаривали досуха. Неочищенное вещество очищали путем флэш-хроматографии на силикагеле (элюируя градиентом от 0 до 14% этилацетат в гептане) с получением 2,26 г неочищенного продукта (содержащего желаемый продукт и еще один продукт). Неочищенный продукт осаждали метанолом с получением 4-бром-2-(4 этоксибензил)бензонитрила (1,2 г, содержащего 5% еще одного соединения, имеющего пики NMR при 4,15 млн-1 квартет, и 1,5 млн-1 триплет). 1 Н NMR (400 МГц, хлороформ-d) дельта 7.48-7.38 (m, 3 Н), 7.13 (d, J = 8,4 Гц, 2 Н), 6.85 (d, J=8,4 Гц,2 Н), 4.08 (s, 2 Н), 4.03 (q, J= 7,2 Гц, 2 Н), 1.41 (t, J = 7,2 Гц, 3 Н). Получение исходного вещества 4-бром-2-(4-этоксибензил)-1-фторбензола К раствору 4-бром-1-фтор-2-(4-метоксибензил)бензола (4,2 г, 14,2 ммоль) в дихлорметане (20 мл) при 0 С по каплям в течение 10 мин медленно добавляли 1 М раствор трибромида бора в дихлорметане(15,7 мл, 16,0 ммоль). После завершения добавления трибромида бора реакционную смесь постепенно нагревали до комнатной температуры. Через 4 ч реакционную смесь охлаждали до 0 С и гасили путем медленного добавления 1 н. водного раствора соляной кислоты (20 мл). Реакционную смесь перемешивали в течение 30 мин и дважды экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом магния, фильтровали, и концентрировали при пониженном давлении с получением светло-розового твердого вещества (3,83 г, 96%). Неочищенный продукт 4-(5-бром-2-фторбензил)фенол использовали на следующей стадии без дополнительной очистки. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 3.88 (s, 2 Н), 4.76 (br. s., 1 Н), 6.77 (d, J=8,2 Гц, 2 Н),6.91 (t, J=9,1 Гц, 1 Н), 7.07 (d, J=8,6 Гц, 2 Н), 7.23 (dd, J=6.8, 2,3 Гц, 1 Н), 7.26-7.31 (m, 1 Н). К раствору 4-(5-бром-2-фторбензил)фенола (6,0 г, 21,0 ммоль) в безводном N,N-диметилформамиде(20 мл), охлажденном до 0 С, добавляли гидрид натрия (60% дисперсия с минеральном масле, 1,02 г,25,6 ммоль). После перемешивания при 0 С в течение 45 мин по каплям добавляли йодэтан (2,08 мл, 25,6 ммоль) и получающуюся в результате смесь оставляли нагреваться до комнатной температуры. Через 18 ч реакционную смесь гасили водой и дважды экстрагировали этилацетатом. Объединенные органические слои дважды промывали водой и однократно рассолом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэшхроматографии на силикагеле, элюируя градиентом от 0 до 10% этилацетата в гептане, с получением 4,6 г (выход 58%) желаемого продукта в виде желтого масла. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 1.40 (t, J=7,0 Гц, 3 Н), 3.89 (s, 2 Н), 4.01 (q, J=6,9 Гц,2 Н), 6.83 (d, J=8,4 Гц, 2 Н), 6.91 (t, J=9,0 Гц, 1 Н), 7.10 (d, J=8,8 Гц, 2 Н), 7.20-7.30 (m, 2 Н). Получение тетрагидрофуран-3-илового эфира толуол-4-сульфоновой кислоты К раствору 3-гидрокситетрагидрофурана (2,5 г, 28,0 ммоль) в безводном пиридине (60 мл) при комнатной температуре добавляли 4-толуолсульфонилхлорид (6,49 г, 34,0 ммоль). После перемешивания реакционной смеси в течение 18 ч при комнатной температуре реакционную смесь концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем флэш-хроматографии на силикагеле, элюируя градиентом от 0 до 30% этилацетата в гептане, с получением 3,5 г (выход 51%) желаемого продукта в виде бесцветного масла. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 2.05-2.12 (m, 2 Н), 2.45 (s, 3 Н), 3.77-3.92 (m, 4 Н), 5.095.14 (m, 1 Н), 7.35 (d, J=8,00 Гц, 2 Н), 7.79 (d, 2 Н). Получение оксетан-3-илового эфира толуол-4-сулъфоновой кислоты К раствору оксетан-3-ола (1,0 г, 13,0 ммоль) в безводном пиридине (25 мл) при комнатной температуре добавляли 4-толуолсульфонилхлорид (3,09 г, 16,2 ммоль). После перемешивания реакционной смеси в течение 18 ч при комнатной температуре реакционную смесь концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем флэш-хроматографии на силикагеле,элюируя градиентом от 0 до 30% этилацетата в гептане, с получением 1,9 г (выход 62%) желаемого продукта в виде белого твердого вещества. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 2.46 (s, 3 Н), 4.63-4.75 (m, 4 Н), 5.26-5.34 (m, 1 Н), 7.36(d, J=8,00 Гц, 2 Н), 7.78 (d, J=8,40 Гц, 2 Н). Получение исходного вещества 3-[4-(5-бром-2-фторбензил)фенокси]тетрагидрофурана К раствору 4-(5-бром-2-фторбензил)фенола (1,5 г, 5,3 ммоль) и карбоната цезия (2,61 г, 8,0 ммоль) вN-диметилформамиде (15,0 мл) при комнатной температуре добавляли раствор тетрагидрофуран-3 илового эфира толуол-4-сульфоновой кислоты (1,94 г, 8,0 ммоль) в N,N-диметилформамиде (10,0 мл). Реакционную смесь затем перемешивали в течение ночи при 50 С. После в общей сложности 18 ч реакционную смесь охлаждали до комнатной температуры, разбавляли рассолом и трижды экстрагировали этилацетатом. Объединенные органические слои дважды промывали водой и однократно рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате неочищенный остаток очищали путем флэш-хроматографии на силикагеле, элюируя градиентом от 0 до 30% этилацетата в гептане, с получением 1,66 г (выход 89%) желаемого продукта в виде бесцветного масла. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 2.09-2.24 (m, 2 Н), 3.86-4.01 (m, 6 Н), 4.86-4.91 (m,1 Н), 6.80 (d, J=8,6 Гц, 2 Н), 6.91 (t, J=9 Гц, 1 Н), 7.10 (d, J=8,6 Гц, 2 Н), 7.23 (dd, J=6,8, 2,5 Гц, 1 Н), 7.26-7.31(m, 1 Н). Получение исходного вещества 3-[4-(5-бром-2-фторбензил)фенокси]оксетана К раствору 4-(5-бром-2-фторбензил)фенола (1,1 г, 3,9 ммоль) и карбоната цезия (1,91 г, 5,87 ммоль) в N,N-диметилформамиде (15,0 мл) при комнатной температуре добавляли раствор оксетан-3-илового эфира толуол-4-сульфоновой кислоты (1,34 г, 8,0 ммоль) в N,N-диметилформамиде (10,0 мл). Реакционную смесь затем перемешивали в течение ночи при 65 С. После в общей сложности 18 ч реакционную смесь охлаждали до комнатной температуры, разбавляли рассолом и трижды экстрагировали этилацетатом. Объединенные органические слои дважды промывали водой и однократно рассолом, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле, элюируя градиентом от 0 до 30% этилацетата в гептане, с получением 0,948 г (выход 72%) желаемого продукта в виде белого твердого вещества. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 3.88 (s, 2 Н), 4.76 (dd, J=7,22, 5,3 Гц, 2 Н), 4.95 (t, J=6,6 Гц, 2 Н), 5.14-5.21 (m, 1 Н), 6.63 (d, J=8,4 Гц, 2 Н), 6.92 (dd, 1 Н), 7.10 (d, J=8,6 Гц, 2 Н), 7.23 (dd, J=6,6, 2,15 Гц, 1 Н), 7.26-7.31 (m, 1 Н). Получение 3-(4-(5-бром-2-хлорбензил)фенокси)оксетана: 4-Бром-1-хлор-2-(4-метоксибензил)бензол (10 г, 32 ммоль) растворяли в дихлорметане (32 мл) и охлаждали до 0 С в атмосфере азота. 1,0 М раствор трибромида бора в дихлорметане (35,3 мл, 34,3 ммоль) по каплям добавляли в течение 10 мин. После добавления ледяную баню удаляли и раствор перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0 С и гасили путем добавления 1 н. водного раствора соляной кислоты (45 мл). Смесь перемешивали в течение 30 мин, переносили в делительную воронку, органический слой отбирали и водный слой экстрагировали дихлорметаном (45 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением 4-(5-бром-2-хлорбензил)фенола (9,5 г, выход 99%) в виде белого твердого вещества. К раствору неочищенного 4-(5-бром-2-хлорбензил)фенола (3,0 г, 10 ммоль) и карбоната цезия (4,9 г,15 ммоль) в N,N-диметилформамиде (77,5 мл) при комнатной температуре добавляли раствор оксетан-3 илового эфира толуол-4-сульфоновой кислоты (3,5 г, 15 ммоль) в N,N-диметилформамиде (8 мл). Смесь нагревали до 65 С в течение 22 ч, после чего добавляли дополнительную аликвоту карбоната цезия (3,3 г, 10 ммоль). Реакционную смесь перемешивали в течение еще 12 ч при 120 С, охлаждали до комнатной температуры, после чего добавляли воду и этилацетат, и смесь постепенно подкисляли 1 н. водным раствором соляной кислоты. Органический слой отделяли, промывали рассолом (трижды) и концентрировали в вакууме. Очистка путем Biotage MPLC (силикагель, элюируя градиентом от 0 до 25% этилацетата в гептане) позволила получить 3-(4-(5-бром-2-хлорбензил)фенокси)оксетан (2,5 г, выход 70%) в виде белого твердого вещества. 1 Н NMR (400 МГц, дихлорметан-d2) дельта млн-1 7.34-7.28 (m, 2 Н), 7.26 (d, J=8,4 Гц, 1 Н), 7.14-7.09(m, 2 Н), 6.69-6.35 (m, 2 Н), 5.22-5.16 (m, 1 Н), 4.96-4.91 (m, 2 Н), 4.72-4.68 (m, 2 Н), 4.01 (s, 2H). Получение 4-бром-2-(4-хлорбензил)-1-фторбензола Раствор 5-бром-2-фторбензальдегида (10,2 г, 50 ммоль) в безводном тетрагидрофуране (200 мл) охлаждали до -78 С. Раствор 4-хлорфенилмагния бромида (1M B диэтиловом эфире, 60 мл, 60 ммоль) добавляли через шприц в течение 8 мин. Перемешивание продолжали при низкой температуре в течение 5 мин и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч при этой температуре. Раствор охлаждали в бане лед-вода и гасили путем добавления насыщенного водного раствор хлорида аммония (40 мл). Органическую фазу декантировали и водный остаток концентрировали при пониженном давлении для удаления какого-либо оставшегося органического растворителя. Водную фазу экстрагировали этилацетатом (200 мл 2) и экстракты объединяли с декантированным раствором тетрагидрофурана. Этот раствор промывали рассолом (25 мл) и сушили (сульфат натрия), фильтровали и концентрировали при пониженном давлении с получением неочищенного (5-бром-2-фторфенил)-(4 хлорфенил)метанола (15,2 г, выход 96%) в виде желтого твердого вещества. К раствору вышеприведенного (5-бром-2-фторфенил)-(4-хлорфенил)метанола (15,0 г, 48 ммоль) и триэтилсилана (18,5 мл, 116 ммоль) в дихлорметане (40 мл) и ацетонитриле (20 мл) при 0 С в атмосфере азота медленно добавляли диэтилэфират трифторида бора (22,7 мл, 181 ммоль). Получающийся в результате раствор перемешивали в течение 18 ч, медленно нагревая до комнатной температуры. Реакционную смесь охлаждали в бане лед-вода, гасили путем медленного добавления 7 М водного раствора гидроксида калия (30 мл) и экстрагировали метил-трет-бутиловым эфиром (200 мл 2). Объединенный органический раствор промывали водой (25 мл 2), рассолом (25 мл 2), сушили (сульфат натрия), фильтровали и концентрировали при пониженном давлении. Очистка путем колоночной флэш-хроматографии на силикагеле, элюируя градиентом от этилацетата в гептане, позволила получить 2-(4-хлорбензил)-4-бром-1 фторбензол (5,0 г, выход 35%) в виде бесцветного масла. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 7.33-7.22 (m, 4H), 7.13 (d, J = 8,4 Гц, 2 Н), 6.93 (dd, J= 9,2, 9,2 Гц, 1 Н), 3.92 (s, 2H). Получение промежуточных соединений Получение промежуточного соединения 2R,3R,4S,5R)-6-аллилокси-3,4,5-трис-бензилокситетрагидропиран-2-ил)метанола (I-1 а) Суспензию D-глюкозы (1,2 кг, 6,6 моль), трифторметансульфоновой кислоты (12 мл) и аллилового спирта (5 л) нагревали при 80 С в течение 3 суток. Смесь охлаждали до комнатной температуры, летучие вещества удаляли в вакууме и остаток растворяли в N,N-диметилформамиде (8 л). Его разделяли на четыре равные реакционные смеси и к каждой добавляли тритил хлорид (463 г, 1,67 моль) и триэтиламин(231 мл, 1,67 моль). Наблюдали небольшую экзотермическую реакцию при добавлении триэтиламина. Реакционную смесь перемешивали в течение 2 суток при 30 С и затем каждую реакционную смесь разделяли на две половины, получая восемь равных реакционных смесей. К каждой из этих реакционных смесей добавляли бензилхлорид (300 мл, 2,60 моль), а затем порциями добавляли гидрид натрия (102,5 г,2,60 моль), поддерживая температуру реакционной смеси от 40 до 50 С. После завершения добавления реакционные смеси перемешивали при комнатной температуре в течение 20 ч. Каждую реакционную смесь затем выливали в смесь лед/вода (2 л) и экстрагировали этилацетатом (2,5 л). Каждую из органических фаз промывали насыщенным рассолом/водой (1:1, 22 л), комбинировали и сушили над сульфатом магния (продукт Rf 0,85 в 3:1 гексаны/этилацетат). После фильтрования и упаривания остаток растворяли в смеси дихлорметана (16 л) и метанола (4 л). Смесь разделяли на 5 равных частей и к каждой добавляли серную кислоту (32 мл). Реакционные смеси перемешивали в течение 3 ч, промывали рассолом/2 М водным раствором гидроксида натрия (1:1, 22 л), комбинировали и сушили над сульфатом магния. После фильтрования и концентрирования в вакууме остаток дополнительно очищали на силикагеле, элюируя 30% этилацетатом в толуоле, с получением промежуточного соединения (I-1 а) в виде смеси аномеров(1,77 кг, выход 54% из D-глюкозы). Rf 0,15 в 3:1 смеси гексаны/этилацетат. Получение промежуточного соединения 3S,4S,5R)-6-аллилокси-3,4,5-трис-бензилокси-2 гидроксиметилтетрагидропиран-2-ил)метанола (I-1b) Раствор диметилсульфоксида (87 мл, 1,22 моль) в дихлорметане (160 мл) по каплям добавляли к раствору оксалилхлорида (64,7 мл, 0,76 моль) в дихлорметане (2,5 л) при -78 С. После завершения добавления по каплям при -78 С добавляли раствор промежуточного соединения (I-1 а) (287 г, 0,59 моль) в дихлорметане (500 мл). После завершения добавления реакционную смесь перемешивали в течение 30 мин и по каплям добавляли триэтиламин (417 мл, 2,9 моль). После завершения добавления реакционную смесь оставляли нагреваться до комнатной температуры. Реакционную смесь затем промывали 1 М водным раствором соляной кислоты (2 л) и воды (2 л) и затем сушили над сульфатом магния. Эту процедуру реакции повторяли для шести эквивалентных реакционных смесей и после сушки их комбинировали и упаривали с получением альдегида в виде желтого масла (1,71 кг). Это масло растворяли в изопропаноле(2,57 л) и разделяли на семь равных реакционных смесей. К каждой из них добавляли 37% водный раствор формальдегида (0,79 л, 10 моль), а затем по каплям добавляли раствор гидроксида натрия (32 г, 0,8 моль) в воде (130 мл). После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение 2 суток. Реакционную смесь разбавляли рассолом (2 л) и экстрагировали этилацетатом (2 л). Органическую фазу дополнительно промывали насыщенным водным раствором бикарбоната натрия (2 л), рассолом (2 л) и затем сушили над сульфатом магния. Органические фазы после семи реакций комбинировали, упаривали и остаток очищали на силикагеле (элюируя от 4 к 1 до 1 к 1 гексаны в этилацетате) с получением промежуточного соединения (I-1b) в виде смеси аномеров (980 г, выход 53% после двух стадий). Rf 0,57 и 0,60 в 1:1 смеси гексаны/этилацетат. Исходный диол [3S,4S,5R)-6-аллилокси-3,4,5-трис-бензилокси-2-гидроксиметилтетрагидропиран 2-ил)метанол (I-1b: 10 г, 19,208 ммоль) растворяли в Т-диметилформамиде (70 мл) и охлаждали до 0 С. Добавляли гидрид натрия (60% дисперсия в минеральном масле, 1,69 г, 42,3 ммоль) и реакционную смесь оставляли перемешиваться при 0 С в течение 1 ч, а затем добавляли 1-бромметил-4-метоксибензол (5,96 мл, 40,3 ммоль). Реакционную смесь затем нагревали до 60 С в течение ночи. Смесь охлаждали до комнатной температуры и реакционную смесь гасили водой и экстрагировали этилацетатом (2 раза). Объединенные органические слои промывали водой, рассолом, сушили над сульфатом натрия,фильтровали и концентрировали при пониженном давлении. Реакционную смесь затем подвергали хроматографии на силикагеле (элюируя градиентом от 0 до 80% этилацетата в гептане) с получением 7,55 г К раствору исходного вещества 3S,4S,5R)-6-аллилокси-3,4,5-трис-бензилокси-2,2-бис-(4-метоксибензилоксиметил)тетрагидропирана (I-1 с: 7,55 г, 9,92 ммоль) в метаноле (60 мл) и дихлорметане (20 мл) при комнатной температуре добавляли хлорид палладия (II) (528 мг, 2,98 ммоль) и получающуюся в результате смесь перемешивали при этой температуре в течение 4 ч. TLC указывала на чистое образование более полярного продукта. Реакционную смесь фильтровали через Celite и концентрировали при пониженном давлении. Неочищенное вещество подвергали хроматографии на силикагеле, элюируя градиентом от 0 до 80% этилацетата в гептане, с получением 5,6 г (выход 78%) продукта (I-1d). MS 738,8 (М + К раствору оксалилдихлорида (1,9 мл, 23 ммоль) в дихлорметане (65 мл) при -78 С добавляли раствор диметилсульфоксида (3,3 мл, 47 ммоль) в дихлорметане (5 мл) и получающийся в результате раствор перемешивали при этой температуре в течение 30 мин. Затем по каплям добавляли раствор исходного вещества 3R,4S,5S)-3,4,5-трис-бензилокси-6,6-бис-(4-метоксибензилоксиметил)тетрагидропиран 2-ола (I-1d, 5,6 г, 7,7 ммоль) в дихлорметане (15,0 мл), и получающуюся в результате смесь перемешивали в течение 30 мин, давая возможность температуре вырасти до -60 С. По каплям добавляли триэтиламин (9,7 мл, 69,5 ммоль), и смесь оставляли нагреваться до 0 С в течение 1 ч. Реакционную смесь гасили путем добавления насыщенного водного раствора хлорида аммония и органическую фазу сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали путем флэш-хроматографии на силикагеле, элюируя градиентом от 0 до 60% этилацетата в гептане, с получением продукта (I-1 е) (4 г, выход 72%). 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 3.24 (d, J=10 Гц, 1 Н), 3.40-3.47 (m, 2 Н), 3.74 (s, 3 Н),3.77 (s, 3 Н), 3.86 (d, J=10 Гц, 1 Н), 4.07 (d, J=8,6 Гц, 1 Н), 4.15 (d, J=9,6 Гц, 1 Н), 4.35-4.55 (m, 6 Н), 4.654.72 (m, 2 Н), 4.82 (d, J=11 Гц, 1 Н), 4.87 (d, J=11,2 Гц, 1 Н), 5.10 (d, J= 11,1 Гц, 1 Н), 6.74-6.79 (m, 2 Н), 6.816.85 (m, 2 Н), 7.11 (dd, J=7,0, 2,5 Гц, 2 Н), 7.17-7.41 (m, 17 Н). Метоксиметиламид К раствору лактона (3R,4S,5S)-3,4,5-трис-бензилокси-6,6-бис-(4-метокси-бензилоксиметил)тетрагидропиран-2-она (Me: 10,4 г, 14,5 ммоль) и N,О-диметилгидроксиламина гидрохлорида (1,77 г,29,0 ммоль) в дихлорметане (100 мл) при 0 С по каплям добавляли 2,0 М раствор триметилалюминия в гексанах (14,5 мл, 29,0 ммоль) и получающийся в результате раствор перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь охлаждали до 0 С и гасили путем медленного добавления водного 1 н. раствора соляной кислоты. Получающуюся в результате смесь оставляли перемешиваться в течение 1 ч. Органическую фазу отделяли и промывали водным 1 н. раствором соляной кислоты, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенное вещество очищали путем хроматографии среднего давления (градиент от 5 до 40% этилацетата в гептане) с получением 6,5 г (58%) продукта. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 2.62 (br. s, 1 Н), 2.94 (br. s., 3 Н), 3.23 (br. s., 3 Н), 3.42 Это соединение получали, начиная с [3S,4S,5R)-6-аллилокси-3,4,5-трис-бензилокси-2 гидроксиметилтетрагидропиран-2-ил)метанола (I-1b), с использованием способа, аналогичного описанному для синтеза метоксиметиламида 2R,3S,4S)-2,3,4-трис-бензилокси-5-гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) и/или (3R,4S,5S)-3,4,5-трисбензилокси-6,6-бис-(4-метоксибензилоксиметил)-2-(метоксиметиламино)тетрагидропиран-2-ола (I-1f), за исключением того, что алкилирующий агент, используемый в экспериментальном разделе, описывающем превращение из (I-1b) в (I-1 с), представлял собой бензилбромид вместо параметоксибензилбромида. 1 Н NMR (400 МГц, хлороформ-d) дельта млн-1 2.66 (br. s, 1 Н), 2.94 (br. s., 3 Н), 3.23 (br. s., 3 Н), 3.48 н-Бутиллитий (0,97 мл, 2,5 М/гексаны, 3,15 эквивалентов) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон для микроволнового реактора Biotage 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-2-(4-метоксибензил)-1-метилбензола (690 мг, 3 экв) в безводном тетрагидрофуране (2,7 мл) при -78 С и получающийся в результате раствор перемешивали при этой температуре в течение еще 1 ч. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трис-бензилокси-5 гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) (608 мг) в безводном тетрагидрофуране (1,35 мл) затем добавляли по каплям в течение 1,5 ч с использованием шприцевого насоса и получающуюся в результате смесь перемешивали при -78 С в течение 1 ч, а затем оставляли нагреваться до -20 С в течение 14 ч (помещали в глубокий сосуд Дьюара (Сосуд Дьюара), закрытый алюминиевой фольгой для поддержания низкой температуры; размер сосуда: внешний диаметр 10 см, внутренний диаметр 8 см, высота 9 см). Добавляли диэтиловый эфир, и реакционную смесь гасили путем добавления по каплям 1 М водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Хроматография на силикагеле с использованием градиента от 20 до 50% этилацетата в гептане позволила получить продукт в виде смеси изомеров (440 мг, выход 61%). К раствору промежуточного соединения I-1i (150 мг) в дихлорметане (3 мл) добавляли анизол (90 мкл, 5 экв), а затем 3 мл 20% раствора трифторуксусной кислоты в дихлорметане и получающуюся в результате смесь перемешивали при комнатной температуре в течение приблизительно 1 ч. Смесь концентрировали и неочищенное вещество подвергали хроматографии на силикагеле (с использованием градиента от 10 до 30% этилацетата в гептане) с получением желаемого продукта в виде смеси изомеров (66 мг, выход 61%). MS (LCMS (жидкостная хроматография/масс-спектрометрия 673,9 (М+Н+; режим положительных ионов). н-Бутиллитий (0,312 мл, 2,5 М/гексаны, 3,05 эквивалентов) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон Biotage для микроволнового реактора 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-2-(4-этоксибензил)-1-метилбензола (238 мг, 3,05 эквивалентов) в безводном тетрагидрофуране (0,9 мл) при -78 С и получающийся в результате раствор перемешивали при этой температуре в течение еще одного часа. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трисбензилокси-5-гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I1g) (200 мг) в безводном тетрагидрофуране (0,6 мл) затем добавляли по каплям в течение 1,5 ч с использованием шприцевого насоса и получающуюся в результате смесь перемешивали при -78 С в течение 1 ч, а затем оставляли нагреваться до комнатной температуры в течение 16 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры; размер сосуда: внешний диаметр 10 см, внутренний диаметр 8 см, высота 9 см). Добавляли диэтиловый эфир и реакционную смесь гасили путем добавления по каплям 1 М водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Неочищенное вещество подвергали хроматографии с использованием автоматизированного хроматографического модуля Biotage (две расположенные друг над другом колонки силикагеля 10 г; элюируя градиентом от 0 до 60% этилацетата в гептане) с получением продукта в виде смеси изомеров (136 мг, выход 56%). MS (LCMS) 968 (M+Na+; режим положительных ионов). К раствору промежуточного соединения I-2i (136 мг, 0,145 ммоль) в дихлорметане (4 мл) добавляли анизол (310 мкл, приблизительно 5 экв), а затем 4 мл 20% раствора трифторуксусной кислоты в дихлорметане и получающуюся в результате смесь перемешивали при комнатной температуре в течение 1,5 ч. Смесь концентрировали и неочищенное вещество подвергали хроматографии с использованием автоматизированного хроматографического модуля ISCO combiflash companion (4 г колонка силикагеля) и элюируя градиентом от 0 до 70% этилацетата в гептане, с получением желаемого продукта в виде смеси изомеров (85 мг, выход 85%). MS (LCMS) 687,7 (М+Н+; режим положительных ионов). н-Бутиллитий (0,97 мл, 2,5 М/гексаны, 3,15 экв) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон Biotage для микроволнового реактора 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-1-хлор-2-(4-метоксибензил)бензола (725 мг, 2,95 экв) в безводном тетрагидрофуране (2,7 мл) при -78 С, и получающийся в результате раствор перемешивали при этой температуре в течение еще одного часа. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трис-бензилокси-5 гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) (616 мг) в безводном тетрагидрофуране (1,35 мл) затем добавляли по каплям в течение 1,5 ч с использованием шприцевого насоса и получающуюся в результате смесь перемешивали при -78 С в течение 1 ч, а затем оставляли нагреваться до -20 С в течение 14 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры; размер сосуда: внешний диаметр 10 см, внутренний диаметр 8 см, высота 9 см). Добавляли диэтиловый эфир и реакционную смесь гасили путем добавления по каплям 1 М водного раствора соляной кислоты. Получившуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Хроматография на силикагеле с использованием градиента от 10 до 40% этилацетата в гептане позволила получить продукт в виде смеси изомеров (530 мг, выход 71%). К раствору промежуточного соединения I-3i (530 мг) в дихлорметане (11 мл) добавляли анизол (300 мкл, 5 экв), а затем 11 мл 20% раствора трифторуксусной кислоты в дихлорметане и получающуюся в результате смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали и неочищенное вещество подвергали хроматографии на силикагеле с использованием градиента от 10 до 40% этилацетата в гептане с получением продукта в виде смеси изомеров (229 мг, выход 59%). н-Бутиллитий (1,0 мл, 2,5 М/гексаны, 3,25 экв) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон Biotageдля микроволнового реактора 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-1-хлор-2-(4-этоксибензил)бензола (815 мг, 3,25 экв) в безводном тетрагидрофуране (2,9 мл) при -78 С, и получающийся в результате раствор перемешивали при этой температуре в течение еще одного часа. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трис-бензилокси-5 гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) (600 мг) в безводном тетрагидрофуране (1,45 мл) затем добавляли по каплям в течение 1,3 ч с использованием шприцевого насоса и получающуюся в результате смесь перемешивали при -78 С в течение 1 ч, а затем оставляли нагреваться до -25 С в течение 14 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры; размер сосуда: внешний диаметр 10 см, внутренний диаметр 8 см, высота 9 см). Добавляли диэтиловый эфир, и реакционную смесь гасили путем добавления по каплям 1 М водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали. Хроматография на силикагеле с использованием градиента от 10 до 40% этилацетата в гептане позволила получить продукт в виде смеси изомеров (280 мг, выход 38%). К раствору промежуточного соединения Mi (1,46 г) в дихлорметане (31 мл) добавляли анизол (900 мкл, приблизительно 5 экв), а затем 31 мл 20% раствора трифторуксусной кислоты в дихлорметане, и получающуюся в результате смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали, и неочищенное вещество подвергали хроматографии на силикагеле с использованием градиента от 10 до 30% этилацетата в гептане с получением продукта в виде смеси изомеров (670 мг, выход 63%). н-Бутиллитий (462 мкл, 2,5 М/гексаны, 3,0 экв) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон Biotage для микроволнового реактора 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-1-фтор-2-(4-метоксибензил)бензола (341 мг, 3 экв) в безводном тетрагидрофуране (1,4 мл) при -78 С в атмосфере азота. Получающийся в результате раствор перемешивали при этой температуре в течение 1 ч. Затем раствор метоксиметиламида (2R,3S,4S)-2,3,4-трис-бензилокси 5-гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) (300 мг,0,385 ммоль) в безводном тетрагидрофуране (0,70 мл) добавляли по каплям очень медленно (1 капля каждые 5 с) и получающуюся в результате смесь перемешивали при -78 С в течение еще 1 ч, а затем нагревали до 10 С в течение 12 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры; размер сосуда: внешний диаметр 10 см, внутренний диаметр 8 см,высота 9 см). Реакционную смесь разбавляли диэтиловым эфиром и гасили путем добавления по каплям 1 н. водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле (элюируя градиентом от 10 до 40% этилацетата в гептане) с получением продукта в виде смеси изомеров (199 мг, выход 55%). К раствору (4S,5S)-3,4,5-трис-бензилокси-2-[4-фтор-3-(4-метоксибензил)фенил]-6,6-бис-(4-метоксибензилоксиметил)тетрагидропиран-2-ола (I-5i; 191 мг, 0,204 ммоль) в дихлорметане (3,75 мл) добавляли анизол (0,178 мл, 1,63 ммоль), а затем 20% раствор трифторуксусной кислоты в дихлорметане (3,75 мл) при комнатной температуре в атмосфере азота. После перемешивания в течение 1 ч при комнатной температуре реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле (элюируя градиентом от 10 до 30% этилацетата в гептане) с получением продукта в виде смеси изомеров (115 мг, выход 83%). MS (LCMS) 677,7 (М+Н+; режим положительных ионов). н-Бутиллитий (508 мкл, 2,5 М/гексаны, 3,0 экв) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору 4-бром-2-(4-этоксибензил)-1-фторбензола (392,0 мг, 1,27 ммоль) в безводном тетрагидрофуране (1,5 мл) при -78 С в атмосфере азота. Получающийся в результате раствор перемешивали при этой температуре в течение 1 ч. Затем раствор метоксиметиламида (2R,3S,4S)-2,3,4 трис-бензилокси-5-гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты I-1g (330,0 мг, 0,423 ммоль) в безводном тетрагидрофуране (0,75 мл) добавляли по каплям очень медленно (1 капля каждые 5 с) и получающуюся в результате смесь перемешивали при -78 С в течение еще 1 ч, а затем нагревали до 10 С в течение 12 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры). Реакционную смесь разбавляли диэтиловым эфиром и гасили путем добавления по каплям 1 н. водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле К раствору промежуточного соединения I-10i (180,0 мг, 0,19 ммоль) в дихлорметане (2,0 мл) добавляли анизол (0,175 мл, 1,60 ммоль), а затем 20% раствор трифторуксусной кислоты в дихлорметане (2,0 мл) при комнатной температуре в атмосфере азота. После перемешивания в течение 1 ч реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэшхроматографии на силикагеле (элюируя градиентом от 10 до 30% этилацетата в гептане) с получением продукта в виде смеси изомеров (85,0 мг, выход 64%). н-Бутиллитий (1,0 мл, 2,5 М/гексаны, 3,0 эквивалента) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору 3-[4-(5-бром-2-фторбензил)фенокси]тетрагидрофурана (878 мг,2,50 ммоль) в безводном тетрагидрофуране (3,0 мл) при -78 С и получающийся в результате раствор перемешивали при этой температуре в течение 1 ч. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трисбензилокси-5-гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты I-1g(650 мг, 0,833 ммоль) в безводном тетрагидрофуране (1,5 мл) затем очень медленно добавляли по каплям(0,9 мл/ч) и получающуюся в результате смесь перемешивали при -78 С в течение еще 1 ч, а затем нагревали до 10 С в течение 12 ч (помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой температуры). Реакционную смесь разбавляли диэтиловым эфиром и гасили путем добавления по каплям 1 н. водного раствора соляной кислоты. Получающуюся в результате двухфазную смесь перемешивали при комнатной температуре в течение 15 мин. Органическую фазу отделяли, промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле (элюируя градиентом от 10 до 40% этилацетата в гептане) с получением продукта в виде смеси изомеров (287 мг, выход 34%). 2S,3S)-2,3,4-трис-бензилокси-5-4-фтор-3-[4-(тетрагидрофуран-3-илокси)бензил]фенил-6,8 диоксабицикло[3.2.1]окт-1-ил)метанол (I-11k)(4S,5S)-3,4,5-трис-бензилокси-2-4-фтор-3-[4-(тетрагидрофуран-3-илокси)бензил]фенил-6,6-бис-(4-метоксибензилоксиметил)тетрагидропиран-2-ола I-11i (275 мг, 0,28 ммоль) в дихлорметане (2,0 мл) добавляли анизол (0,250 мл, 2,29 ммоль), а затем 20% раствор трифторуксусной кислоты в дихлорметане (8,0 мл) при комнатной температуре в атмосфере азота. После перемешивания в течение 1 ч реакционную смесь концентрировали при пониженном давлении. Неочищенный остаток очищали путем флэш-хроматографии на силикагеле (элюируя градиентом от 10 до 30% этилацетата в гептане) с получением продукта в виде смеси изомеров (168 мг, выход 83%). м-Бутиллитий (1,0 мл, 2,5 М/гексаны, 3,1 экв) по каплям добавляли (1 капля каждые 5 с) к дегазированному по кислороду раствору (помещенному в предварительно высушенный флакон Biotage для микроволнового реактора 10-20 мл, закрытый крышкой и помещенный в поток газообразного азота при положительном давлении) 4-бром-2-(4-хлорбензил)-1-фторбензола (702 мг, 2,9 эквивалентов) в безводном тетрагидрофуране (3,0 мл) при -78 С, и получающийся в результате раствор перемешивали при этой температуре в течение 25 мин. Раствор метоксиметиламида (2R,3S,4S)-2,3,4-трис-бензилокси-5 гидрокси-6-(4-метоксибензилокси)-5-(4-метоксибензилоксиметил)капроновой кислоты (I-1g) (621 мг) в безводном тетрагидрофуране (1,5 мл) затем добавляли по каплям с использованием шприцевого насоса(0,9 мл/ч) и получающуюся в результате смесь перемешивали при низкой температуре в течение еще 17 ч(помещали в глубокий сосуд Дьюара, закрытый алюминиевой фольгой для поддержания низкой темпе- 29
МПК / Метки
МПК: A61P 3/10, C07H 15/18, A61K 31/357, C07D 493/10
Метки: производные, диокса-бицикло[3.2.1]октан-2,3,4-триольные
Код ссылки
<a href="https://eas.patents.su/30-18492-dioksa-biciklo321oktan-234-triolnye-proizvodnye.html" rel="bookmark" title="База патентов Евразийского Союза">Диокса-бицикло[3.2.1]октан-2,3,4-триольные производные</a>
Бицикло [2.2.1] гептаны и родственные соединения

Номер патента: 3946
Опубликовано: 30.10.2003
Автор: Ченард Бертранд Лео
МПК: A61P 25/00, C07C 229/50, A61K 31/198...
Метки: гептаны, родственные, бицикло, соединения, 2.2.1
Формула / Реферат:
1. Соединение формулы где n принимает значения от 0 до 6, X обозначает CH2, CH2CH2 или кислород, Z обозначает CHR2 или NR2 и R1 и R2 выбирают независимо из водорода, (C1-C6)алкила, арила и гетероарила, при этом указанный арил выбирают из фенила и нафтила и указанный гетероарил выбирают из 5- и 6-членных ароматических гетероциклических колец, которые содержат от одного до четырех гетероатомов, выбранных независимо из азота, кислорода и серы, и...
Применение производных бицикло [2.2.1] гептана для приготовления нейропротекторных фармацевтических композиций

Номер патента: 10868
Опубликовано: 30.12.2008
Авторы: Сенаши Габор, Харшин Ласло Габор, Леваи Дьёрдь, Гиглер Габор, Гачальи Иштван, Шимо Аннамария, Мориц Кристина
МПК: A61K 31/135, A61P 25/28
Метки: применение, фармацевтических, приготовления, 2.2.1, нейропротекторных, композиций, производных, бицикло, гептана
Формула / Реферат:
1. Применение соединения формулы и его фармацевтически приемлемых кислотно-аддитивных солей для приготовления лекарственных препаратов для уменьшения последствий острых ишемических или травматических повреждений головного и спинного мозга, особенно различных типов инсульта или церебрального вазоспазма, тяжелой окклюзии сосудов головного мозга, потери нервных клеток и при повреждении головного и спинного мозга в результате несчастного случая, ее...
Стабильная полиморфная форма кристаллического моногидрата цитрата (2s,3s)-n-(2-метокси-5-трет-бутилфенил)метил-2-дифенилметил-1-азабицикло[2.2.2]октан-3-амина и фармацевтическая композиция на её основе

Номер патента: 3731
Опубликовано: 28.08.2003
Авторы: Куоллич Джордж Джозеф, Уинт Льюин Теофилус, Касталди Майкл Джеймс
МПК: A61K 31/439, A61P 1/08, C07D 453/02...
Метки: форма, 2s,3s)-n-(2-метокси-5-трет-бутилфенил)метил-2-дифенилметил-1-азабицикло[2.2.2]октан-3-амина, стабильная, полиморфная, композиция, фармацевтическая, моногидрата, цитрата, основе, кристаллического
Формула / Реферат:
1. Стабильная полиморфная форма A кристаллического моногидрата цитрата (2S,3S)-N-(2-метокси-5-трет-бутилфенил)метил-2-дифенилметил-1-азабицикло[2.2.2]октан-3-амина формулы характеризующаяся следующей картиной порошковой дифракции рентгеновских лучей: Пик ь Расстояние d 1 13,28 2 7,70 3 7,45 4 6,34 5 5,33 6 5,06 7 4,40 2. Полиморфная форма моногидрата цитрата по п.1, где габитусы ее кристалла представляют собой...
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Гиглер Габор, Сабо Геза, Леваи Дьердь, Вег Миклош, Грефф Зольтан, Мартонне Марко Бернадетт, Баркоци Йожеф, Харшинг Ласло Габор, Раткаи Зольтан, Шимиг Дьюла, Сенаши Габор, Линг Иштван
МПК: C07D 243/02, A61K 31/551, A61P 25/00...
Метки: ингредиента, качестве, 2,3-бензодиазепина, композиции, активного, фармацевтические, производные, содержащие, эти
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Кларк Робин Дуглас, Чжао Шухай, Бергер Якоб
МПК: A61K 31/536, A61P 25/18, C07D 265/36...
Метки: качестве, 5-нт-6, бензоксазина, производные, содержащие, композиции, эти, получения, фармацевтические, применение, модуляторов, способ
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Предыдущий патент: Способ получения сахаров из биомассы
Следующий патент: Способ получения p4o6 с высоким выходом
Случайный патент: Производные 2-(4-замещенного)-бензиламино-2-метилпропанамида.