Производные 3-аминопирроло[3,4-c]пиразол-5(1h,4h,6h)-карбальдегида в качестве ингибиторов ркс
Номер патента: 17937
Опубликовано: 30.04.2013
Авторы: Инь Чуньфын, Тэн Минь, Ли Хой, Скейлс Стефани Энн, Нукуи Сейдзи





























Формула / Реферат
1. Соединение формулы (А)

где X представляет собой C-R11 или N, где R11 представляет собой Н или F;
А и В выбирают из C-R2 или N при условии, что А и В одновременно не являются N;
R1 выбирают из (С1-С4)алкиламино, диметиламино, циклопропилметиламино, циклопропиламино, 1-морфолино, (C1-С3)алкила, (C1-C2)алкокси, циано, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С2-С3)алкокси;
R2 представляет собой метил, F или Н;
R3 представляет собой Н или метил;
R4 представляет собой Н, метил или изопропил;
R5 представляет собой Н;
R6 и R7, каждый независимо, представляют собой Н, (C1-С2)алкил или НО(С2-С3)алкил; или R6 и R7, взятые вместе, могут быть (С3-С7)циклоалкилом;
R8 выбирают из тетрагидропиран-4-илметила, 4-гидрокситетрагидропиран-4-илметила, тетрагидропиран-4-ила, (С1-С2)алкила, НО(С2-С3)алкила, МеО(С2-С3)алкила или 3,3,3-трифторпропила; и
R9 и R10 представляют собой метил;
при условии, что если X представляет собой N, то оба R5 и R7 не являются Н, а если X представляет собой C-R11, то оба R6 и R7 представляют собой Н;
или его фармацевтически приемлемая соль.
2. Соединение по п.1 или его фармацевтически приемлемая соль, где X представляет собой N, а каждый из R6 и R7 независимо представляет собой Н или С1-С2алкил, но оба они не являются Н.
3. Соединение по п.1 или его фармацевтически приемлемая соль, где А представляет собой N, а В представляет собой C-R2.
4. Соединение по п.1 или его фармацевтически приемлемая соль, где А представляет собой C-R2, а В представляет собой N.
5. Соединение по п.1 или его фармацевтически приемлемая соль, где оба R6 и R7 представляют собой метил.
6. Соединение по п.1 или его фармацевтически приемлемая соль, где R6 представляет собой Н, a R7 представляет собой метил.
7. Соединение по п.1 или его фармацевтически приемлемая соль, где R4 представляет собой метил или изопропил.
8. Соединение по п.1 или его фармацевтически приемлемая соль, где R4 представляет собой метил.
9. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 выбирают из (C1-C4)алкиламино, диметиламино, циклопропилметиламино, циклопропиламино, морфолино, (C1-С3)алкила, (C1-C2)алкокси, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С2-С3)алкокси.
10. Соединение по п.1 или его фармацевтически приемлемая соль, где R8 выбирают из тетрагидропиран-4-илметила, 4-гидрокситетрагидропиран-4-илметила, тетрагидропиран-4-ила, (C1-С2)алкила, НО(С2-С3)алкила или МеО(С2-С3)алкила.
11. Соединение по п.1 или его фармацевтически приемлемая соль, где R1 представляет собой (С1-С3)алкил; R4 представляет собой метил; R6 представляет собой Н; R7 представляет собой метил и R8 представляет собой тетрагидропиран-4-илметил.
12. Соединение формулы (В)

где X представляет собой C-R11 или N, где R11 представляет собой Н или F;
А и В выбирают из C-R2 или N при условии, что А и В одновременно не являются N;
R1 выбирают из (C1-C4)алкиламино, диметиламино, циклопропилметиламино, циклопропиламино, 1-морфолино, (C1-С3)алкила, (C1-C2)алкокси, циано, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С2-С3)алкокси;
R2 представляет собой метил, F или Н;
R3 представляет собой Н или метил;
R4 представляет собой Н, метил или изопропил;
R5 представляет собой Н или метил;
R6 и R7, каждый независимо, представляют собой Н, (C1-С2)алкил или НО(С2-С3) алкил; или R6 и R7, взятые вместе, могут быть (С3-С7)циклоалкилом;
R8 выбирают из тетрагидропиран-4-илметила, 4-гидрокситетрагидропиран-4-илметила, тетрагидропиран-4-ила, (C1-С2)алкила, НО(С2-С3)алкила, МеО(С2-С3)алкила или 3,3,3-трифторпропила;
R9 и R10 представляют собой метил;
или его фармацевтически приемлемая соль.
13. Соединение по п.12 или его фармацевтически приемлемая соль, где А представляет собой N, а В представляет собой C-R2.
14. Соединение по п.12 или 13 или его фармацевтически приемлемая соль, где R4 представляет собой метил или изопропил.
15. Соединение по любому из пп.12, 13 или 14 или его фармацевтически приемлемая соль, где R4 представляет собой метил.
16. Соединение по любому из пп.12, 13, 14 или 15 или его фармацевтически приемлемая соль, где R1 выбирают из (C1-С4)алкиламино, диметиламино, циклопропилметиламино, циклопропиламино, морфолино, (C1-C3)алкила, (C1-C2)алкокси, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С2-С3)алкокси.
17. Соединение, выбранное из группы, состоящей из
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-5-фтор-N2,N2-диметилпиримидин-2,4-диамина,
N2-циклопропил-N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-5-фторпиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-5-фтор-N2-метилпиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-5-фтор-N2-изопропилпиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этилпиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2,N2-диметилпиримидин-2,4-диамина,
5-{[(8S)-6,8-диметил-6,9-диазаспиро[4.5]дек-9-ил]карбонил}-N-(5-фтор-2-метилпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N4-(5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4-диамина,
N4-(5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4-диамина,
ацетатной соли N2-этил-5-фтор-N4-(5-{[(2S,5R)-4-(3-метоксипропил)-2,5-диметилпиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)пиримидин-2,4-диамина,
N4-(6,6-диметил-5-{[(2S,5R)-2,4,5-триметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4-диамина,
4-[(6,6-диметил-5-{[(2S,5R)-2,4,5-триметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)амино]пиримидин-2-карбонитрила,
N-(2-этил-5-фторпиримидин-4-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этил-5-фторпиримидин-4-ил)-5-{[(2S,5R)-4-(3-метоксипропил)-2,5-диметилпиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
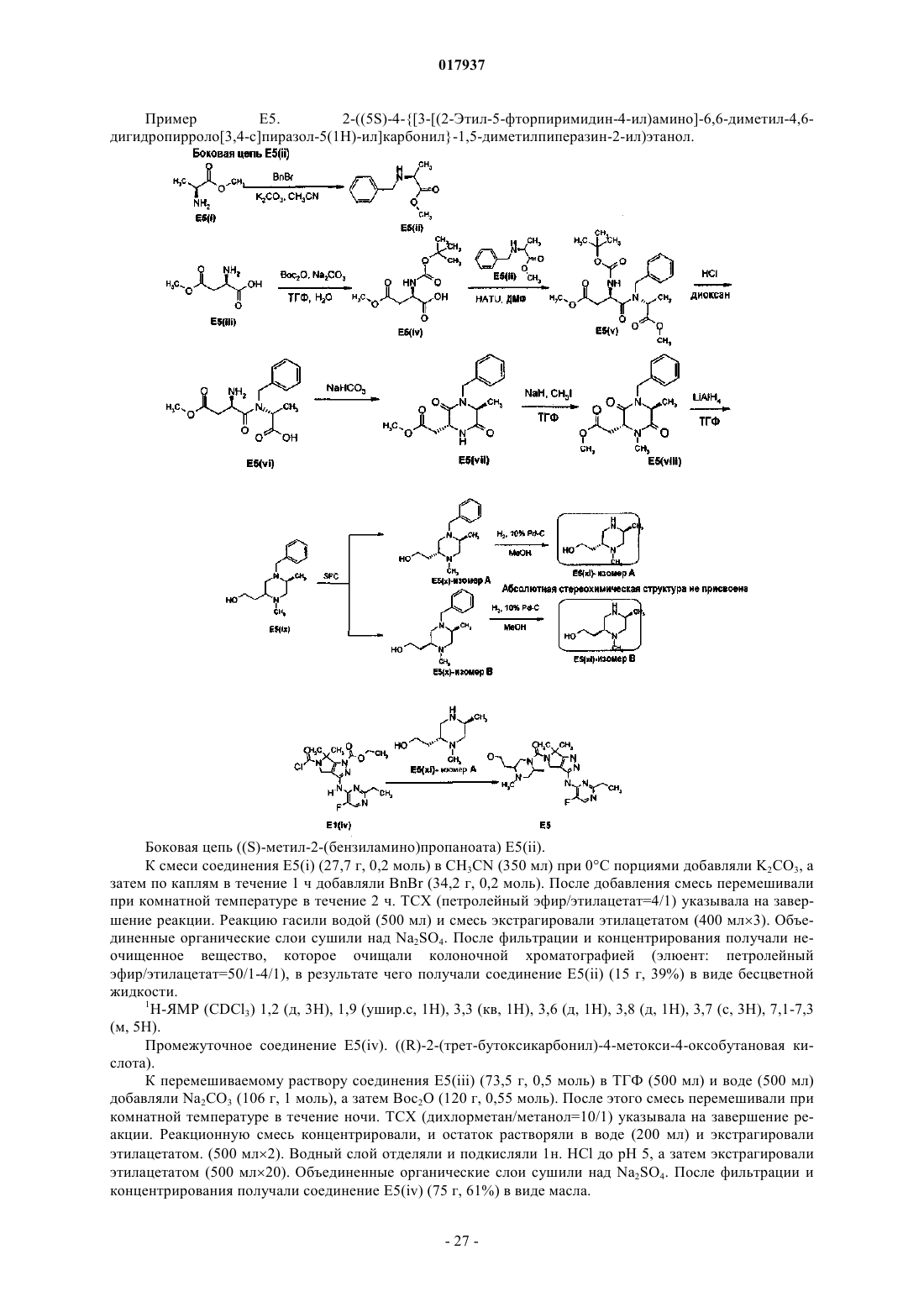
2-((5S)-4-{[3-[(2-этил-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1H)-ил]карбонил}-1,5-диметилпиперазин-2-ил)этанола,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил}-N-(5-фтор-2-метилпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(5-фтор-2-метилпиримидин-4-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(5-фтор-2-пропилпиримидин-4-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(5-фтор-2-изопропилпиримидин-4-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(3-фтор-6-метилпиридин-2-ил)-6,6-диметил-5-{[(2S,5R)-2,4,5-триметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(3S,8aS)-3,8а-диметилгексагидропирроло[1,2-а]пиразин-2(1H)-ил]карбонил}-N-(3-фтор-6-метилпиридин-2-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(3-фтор-6-метилпиридин-2-ил)-6,6-диметил-5-{[(3S,8aS)-3-метилгексагидропирроло[1,2-а]пиразин-2(1H)-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(3-фтор-6-метилпиридин-2-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-[5-фтор-2-(метоксиметил)пиримидин-4-ил]-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2Н-пиран-4-илметил)пиперазин-1-ил]карбонил}-N-(2-этил-5-фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2Н-пиран-4-ил)пиперазин-1-ил]карбонил}-N-(4-метоксипиримидин-2-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]карбонил}-6,6-диметил-N-(4-метилпиримидин-2-ил)-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]карбонил}-6,6-диметил-N-[4-(трифторметил)пиримидин-2-ил]-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил}-6,6-диметил-N-(4-метилпиримидин-2-ил)-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-5-{[(2S,5R)-2,4,5-триметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-5-{[4-этил(2S,5R)-2,5-диметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этокси-5-фторпиримидин-4-ил)-5-{[(2S,5R)-4-(2-метоксиэтил)-2,5-диметилпиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-(2-этокси-5-фторпиримидин-4-ил)-5-{[(2S,5R)-4-(3-метоксипропил)-2,5-диметилпиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-5-{[(2S,5R)-4-(3-метоксипропил)-2,5-диметилпиперазин-1-ил]карбонил}-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-6,6-диметил-5-{[(2S,5R)-2,4,5-триметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-6,6-диметил-5-{[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил}-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил}-N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]карбонил}-N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
2-((5S)-4-{[3-[(2-этокси-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1H)-ил]карбонил}-1,5-диметилпиперазин-2-ил)этанола,
2-((5S)-4-{[3-[(2-этокси-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1H)-ил]карбонил}-1,5-диметилпиперазин-2-ил)этанола,
5-[(4-фтор-1-метилпиперидин-4-ил)карбонил]-N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
5-{[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил}-N-[5-фтор-2-(метоксиметил)пиримидин-4-ил]-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,
2-((5S)-4-{[3-{[5-фтор-2-(метоксиметил)пиримидин-4-ил]амино}-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1H)-ил]карбонил}-1,5-диметилпиперазин-2-ил)этанола
или их фармацевтически приемлемой соли.
18. Фармацевтическая композиция, содержащая эффективное количество соединения по любому из предыдущих пунктов или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
19. Способ лечения сахарного диабета и его осложнений, рака, ишемии, воспалений, расстройств центральной нервной системы, сердечно-сосудистых заболеваний, болезни Альцгеймера, кожных болезней, депрессий, вирусных заболеваний, воспалительных расстройств или заболеваний, при которых органом-мишенью является печень, включающий введение млекопитающему эффективного количества соединения по любому из предыдущих пунктов или его фармацевтически приемлемой соли.
20. Способ лечения сахарного диабета и его осложнений, включающий введение млекопитающему эффективного количества соединения или его фармацевтически приемлемой соли по любому из предыдущих пунктов.
21. Способ по п.20, где указанными осложнениями являются диабетическая ретинопатия, нефропатия и невропатия.
Текст
Настоящее изобретение относится к соединениям формул (А) и (В) и к их фармацевтически приемлемым солям, где в формулах (А) и (В) А, В, R1, R2, R3, R4, R5, R6, R7, R8, R9 и R10 являются такими, как они были определены выше. Настоящее изобретение также относится к фармацевтическим композициям, содержащим указанные соединения и их фармацевтически приемлемые соли, и к способам лечения сахарного диабета и его осложнений, рака, ишемии,воспалений, расстройств центральной нервной системы, сердечно-сосудистых заболеваний,болезни Альцгеймера, кожных болезней, депрессий, вирусных заболеваний, воспалительных расстройств или заболеваний, при которых органом-мишенью является печень. В настоящем изобретении заявляется приоритет предварительной заявки на патент США 60/888749, поданной 7 февраля 2007 г., предварительной заявки на патент США 60/989086, поданной 19 ноября 2007 г., и предварительной заявки на патент США 61/020965, поданной 14 января 2008 г.,содержание которых во всей своей полноте вводятся в настоящее описание посредством ссылки. Область техники, к которой относится изобретение Настоящее изобретение относится к новым соединениям, к фармацевтическим композициям, содержащим такие соединения, а также к применению указанных соединений в медицине и для приготовления лекарственного препарата, воздействующего на человеческий фермент протеинкиназу С, в частности его изоформу бета II (PKCII). Предшествующий уровень техники Протеинкиназа С (PKC) принадлежит к суперсемейству активируемых липидом Ser/Thr-киназ, участвующих во множестве путей передачи сигналов. Существует тринадцать PKC-изоформ, которые были идентифицированы и классифицированы по их регуляции передачи клеточных сигналов, опосредуемых такими молекулами, как диацилглицерин, фосфолипиды и кальций. Для полной активации изоферментов протеинкиназы С, таких как альфа, бета (двух сплайсированных вариантов PKCI и PKCII) и гамма,требуются мембранные фосфолипиды, кальций и сложные эфиры диацилглицеринфорбола. По своему механизму активации такие формы PKC, как дельта, эпсилон, эта и тета, являются кальцийнезависимыми. Дзета- и лямбда-формы PKC являются кальций- и диацилглицерин-зависимыми, и для осуществления их механизмов активации, очевидно, требуются только мембранные фосфолипиды. Исходя из тканеспецифической экспрессии и активации PKC-изоформ, предполагается, что отдельные PKC-изоформы могут быть потенциальными терапевтическими мишенями. Было продемонстрировано, что в тканях у животных с диабетом наблюдается активация изоформы PKC-бета, и такая активация приводит к развитию микрососудистых патологий, ассоциированных с гипергликемией. В Японии у пациентов с диабетом типа II был идентифицирован генетический полиморфизм в 5'-фланкирующей области, расположенной выше гена PKC. Такая генетическая вариабельность PKC ассоциируется со значительным повышением восприимчивости к развитию сосудистых осложнений при диабете и макроваскулярных заболеваний, таких как ишемическая болезнь сердца. В крупномасштабных исследованиях методом "случай-контроль", проводимых специалистами Центра диабета Джослина (Joslin), был идентифицирован дополнительный полиморфизм в промоторной области PKC, который ассоциируется с сахарным диабетом типа I (с длительностью 24 лет) и с повышенным риском развития диабетической нефропатии. Было показано, что введение ингибиторов PKC,таких как мезилат рибоксистаурина (LY333531, Lilly), животным с моделью диабета приводит к предотвращению или к снижению уровня гемодинамических изменений и сосудистых поражений, ассоциированных с диабетической нефропатией, диабетической периферической невропатией и с диабетической ретинопатией (Way, K.J. et al., Diabet. Med. 18:945-959 (2001); Vinik, A., Expert Opin. Investig. Drugs 14:1547-1559 (2005. Наряду с дополнительными данными, полученными в клинических испытаниях фазы II и фазы III по применению мезилата рибоксистаурина для лечения диабета и микроваскулярных осложнений при диабете, было собрано много данных, которые явно свидетельствуют о том, что PKC может служить молекулярной мишенью при диабетических осложнениях и может быть использована для разработки селективных ингибиторов PKC, как потенциальных терапевтических средств. Соединениями согласно изобретению являются ингибиторы протеинкиназы С бета II, а поэтому очевидно, что они могут быть использованы для лечения состояний, ассоциированных с сахарным диабетом и его осложнениями, а также с раком, ишемией, воспалениями, расстройствами центральной нервной системы, сердечно-сосудистыми заболеваниями и кожными болезнями. Описание сущности изобретения Настоящее изобретение относится к соединению формулы (А) или к его фармацевтически приемлемой соли где X представляет собой C-R11 или N, где R11 представляет собой Н или F; А и В выбирают из C-R2 или N при условии, что А и В одновременно не являются N;R9 и R10 представляют собой метил; при условии, что если X представляет собой N, то оба R6 и R7 не являются Н, а если X представляет собой C-R11, то оба R6 и R7 представляют собой Н. В одном варианте изобретения X представляет собой N, а каждый из R6 и R7 независимо представляет собой Н или С 1-С 2 алкил, но оба они не являются Н. В одном из вариантов изобретения А представляет собой N, а В представляет собой C-R2. В альтернативном варианте А представляет собой C-R2, а В представляет собой N. В другом варианте изобретения R6 и R7 представляют собой метил. В альтернативном варианте изобретения R6 представляет собой Н, a R7 представляет собой метил. В одном из вариантов изобретения R4 представляет собой метил или изопропил. В другом варианте изобретения R4 представляет собой метил. В другом варианте изобретения R1 выбирают из (C1-С 4)алкиламино, диметиламино, циклопропилметиламино, циклопропиламино, морфолино, (C1-С 3)алкила, (C1-C2)алкокси, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С 2-С 3)алкокси. В другом варианте изобретения R1 представляет собой (C1-С 3)алкил; R4 представляет собой метил; 6R представляет собой Н; R7 представляет собой метил и R8 представляет собой тетрагидропиран-4 илметил. Настоящее изобретение также относится к соединению или его фармацевтически приемлемой соли формулы (В) где X представляет собой C-R11 или N, где R11 представляет собой Н или F; А и В выбирают из C-R2 или N при условии, что А и В одновременно не являются N;R9 и R10 представляют собой метил. В одном из вариантов соединения формулы (В) А представляет собой N, а В представляет собой CR2. В другом варианте соединения формулы (В) R4 представляет собой метил или изопропил. В другом варианте соединения формулы (В) R4 представляет собой метил. В другом варианте соединения формулы (В) R1 выбирают из (С 1-С 4)алкиламино, диметиламино,циклопропилметиламино, циклопропиламино, морфолино, (C1-C3)алкила, (C1-C2)алкокси, метоксиметила, трифторметила, 2,2,2-трифторэтокси или МеО(С 2-С 3)алкокси. Настоящее изобретение относится к соединениям или к их фармацевтически приемлемым солям,-2 017937 ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-5-[4-этил(2S,5R)-2,5-диметилпиперазин-1 ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-(2-этокси-5-фторпиримидин-4-ил)-6,6-диметил-5-[(2S)-2,4,5,5-тетраметилпиперазин-1 ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-(2-этокси-5-фторпиримидин-4-ил)-5-[2S,5R)-4-(2-метоксиэтил)-2,5-диметилпиперазин-1 ил]карбонил-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-4-амина,N-(2-этокси-5-фторпиримидин-4-ил)-5-[(2S,5R)-4-(3-метоксипропил)-2,5-диметилпиперазин-1 ил]карбонил-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-5-[(2S,5R)-4-(3-метоксипропил)-2,5 диметилпиперазин-1-ил]карбонил-б,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-6,6-диметил-5-[(2S,5R)-2,4,5 триметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]-6,6-диметил-5-[(2S)-2,4,5,5 тетраметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,5-[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил-N-(2 этокси-5-фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,5-[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-ил)пиперазин-1-ил]карбонил-N-(2-этокси-5 фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,2-5S)-4-[3-[(2-этокси-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4 с]пиразол-5(1H)-ил]карбонил-1,5-диметилпиперазин-2-ил)этанола,2-5S)-4-[3-[(2-этокси-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4 с]пиразол-5(1H)-ил]карбонил-1,5-диметилпиперазин-2-ил)этанола,5-[(4-фтор-1-метилпиперидин-4-ил)карбонил]-N-[5-фтор-2-(2,2,2-трифторэтокси)пиримидин-4-ил]6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,5-[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1-ил]карбонил-N-[5-фтор 2-(метоксиметил)пиримидин-4-ил]-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина,2-5S)-4-[3-[5-фтор-2-(метоксиметил)пиримидин-4-ил]амино-6,6-диметил-4,6 дигидропирроло[3,4-c]пиразол-5(1H)-ил]карбонил-1,5-диметилпиперазин-2-ил)этанола. Настоящее изобретение также относится к фармацевтической композиции, содержащей эффективное количество соединения по любому из предыдущих выриантов или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу лечения сахарного диабета и его осложнений,рака, ишемии, воспалений, расстройств центральной нервной системы, сердечно-сосудистых заболеваний, болезни Альцгеймера, кожных болезней, депрессий, вирусных заболеваний, воспалительных расстройств или заболеваний, при которых органом-мишенью является печень, указанный способ включает введение млекопитающему эффективного количества соединения по любому из предыдущих вариантов или его фармацевтически приемлемой соли. Настоящее изобретение также относится к способу лечения сахарного диабета и его осложнений,указанный способ включает введение млекопитающему эффективного количества соединения по любому из предыдущих пунктов. В другом варианте осуществления изобретения указанными осложнениями являются диабетическая ретинопатия, нефропатия и невропатия. Определения. Используемые здесь термины "содержащий" и "включающий" употребляются в их обычном неограничивающем смысле. Используемые здесь термины "C1-C8" или "С 2-С 8" и т.п. означают группы,имеющие 1-8 или 2-8 атомов углерода соответственно. Используемый здесь термин "алкил", если это не оговорено особо, включает насыщенные одновалентные углеводородные радикалы, имеющие прямые или разветвленные группы. Репрезентативные алкильные группы имеют 1-8, 1-6 или 1-4 атомов углерода. Используемый здесь термин "алкенил", если это не оговорено особо, включает алкильные группы,имеющие по меньшей мере одну углерод-углеродную двойную связь, где указанный алкил определен выше, и содержащие Е- и Z-изомеры указанной алкенильной группы. Используемый здесь термин "алкинил", если это не оговорено особо, включает алкильные группы,имеющие по меньшей мере одну углерод-углеродную тройную связь, где указанный алкил определен выше. Используемый здесь термин "алкоксил", если это не оговорено особо, включает О-алкильные группы, где алкил определен выше. Используемый здесь термин "гидроксил", если это не оговорено особо, включает -ОН. Используемый здесь термин "амино", если это не оговорено особо, включает радикал -NH2 и любые заместители у атома N. Используемые здесь термины "галоген" и "галогено", если это не оговорено особо, означают хлор,фтор, бром или йод. Используемый здесь термин "трифторметил", если это не оговорено особо, означает группу -CF3. Используемый здесь термин "перфторалкил" означает алкильную группу, в которой все атомы водорода, связанные с атомами углерода, заменены фтором, таким как CF3, CF2-CF3, C(CF2)(CF2) и т.п. Используемый здесь термин "трифторметокси", если это не оговорено особо, означает группу-OCF3. Используемый здесь термин "циано", если это не оговорено особо, означает группу -CN. Используемый здесь термин "CH2Cl2", если это не оговорено особо, означает дихлорметан. Используемые здесь термины "С 3-С 12-циклоалкил" или "С 5-С 8-циклоалкил", если это не оговорено особо, означают неароматический, насыщенный или частично насыщенный моноциклический или конденсированный, спиро или неконденсированный бициклический или трициклический углеводород, описанный в настоящем патенте и содержащий всего от 3 до 12 атомов углерода или 5-8 атомов углерода на кольце соответственно. Репрезентативными циклоалкилами являются кольца, имеющие 3-10 атомов углерода, такие как циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и адамантил. Иллюстративными примерами циклоалкилов являются циклоалкилы, происходящие от нижеследующих групп, не ограничиваясь ими: Используемый здесь термин "арил", если это не оговорено особо, включает органический радикал,образующийся от ароматического углеводорода в результате удаления одного атома водорода, такой как фенил или нафтил. Используемые здесь термины "(3-15)-членный гетероциклил", "(3-7)-членный гетероциклил", "(610)-членный гетероциклил" или "(4-10)-членный гетероциклил", если это не оговорено особо, включают ароматические и неароматические гетероциклические группы, содержащие от одного до четырех гетероатомов, выбранных из О, S и N, где каждая гетероциклическая группа имеет 3-15, 3-7, 6-10 или 4-10 атомов, соответственно, в своей циклической системе, при условии, что кольцо указанной группы не содержит два смежных атома О или S. Неароматическими гетероциклическими группами являются группы, имеющие только 3 атома в своей циклической системе, тогда как ароматические гетероциклические группы должны иметь по меньшей мере 5 атомов в своей циклической системе. Гетероциклическими группами являются бензо-конденсированные циклические системы. Примером 3-членной гетероциклической группы является азиридин, а примером 4-членной гетероциклической группы является азетидинил (производное азетидина). Примером 5-членной гетероциклической группы является тиазолил, примером 7-членного кольца является азепинил, а примером 10-членной гетероциклической группы является хинолинил. Примерами неароматических гетероциклических групп являются пирролидинил, тетрагидрофуранил, дигидрофуранил, тетрагидротиенил, тетрагидропиранил, дигидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, азетидинил, оксетанил,тиэтанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 1,2,3,6 тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2 Н-пиранил, 4 Н-пиранил, диоксанил,1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил,пиразолидинил,имидазолинил,имидазолидинил,3-азабицикло[3.1.0]гексанил,3 азабицикло[4.1.0]гептанил, 3 Н-индолил и хинолизинил. Гетероциклы включают моноциклические и полициклические ароматические кольцевые структуры,а термин "(5-12)-членные гетероарилы" означает гетероциклы, имеющие в своей(их) циклической(их) системе(ах) 5-12 атомов. Примерами "(5-12)-членных гетероарилов" являются пиридинил, имидазолил,пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил,циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил,пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил. Вышеупомянутые группы,происходящие от вышеперечисленных групп, могут быть связаны, если это возможно, с атомами С или(N-связанный) или имидазол-3-ил (С-связанный). Вышеупомянутые гетероциклические группы могут быть необязательно замещены у любого(ых) атома(ов) углерода, серы или азота на кольце одной или двумя оксогруппами на одно кольцо. Примером гетероциклической группы, в которой 2 атома углерода на кольце замещены оксогруппами, является 1,1-диоксотиоморфолинил. Другими иллюстративными примерами 4-10-членных гетероциклических групп являются группы, происходящие от нижеследующих групп, не ограничиваясь ими: Используемый здесь термин "(12-15)-членный гетероциклил", если это не оговорено особо, включает ароматические и неароматические гетероциклические группы, которые имеют частично конденсированную или спироциклическую конфигурацию и которые содержат по меньшей мере один N и необязательно дополнительные 1-5 гетероатомов, каждый из которых выбран из О, S и N, где указанная гетероциклическая группа имеет в своей системе от 12 до 15 атомов соответственно, при условии, что любое кольцо указанной группы не содержит двух смежных атомов О или S. Гетероциклическими группами являются трициклические конденсированные системы и спироциклические системы. Примером 13 членной трициклической гетероциклической группы является 3,4-дигидропиразино[1,2-а]бензимидазол,а примером 15-членной спироциклической гетероциклической группы является 3,4-дигидро-1'Hспирохромен. Термин "оксо", если это не оговорено особо, означает =O. Термин "сольват" означает фармацевтически приемлемую сольватную форму конкретного соединения, которая сохраняет биологическую эффективность такого соединения. Примерами сольватов являются соединения согласно изобретению, представляющие собой комбинацию с водой, изопропанолом,этанолом, метанолом, ДМСО (диметилсульфоксидом), этилацетатом, уксусной кислотой или этаноламином. Используемый здесь термин "фармацевтически приемлемая(ые) соль(соли)", включает, если это не оговорено особо, соли кислотных или основных групп, которые могут присутствовать в соединениях формулы (А) или формулы (В). Соединения формулы (А) или формулы (В), которые по своей природе являются основными, способны образовывать соли широкого ряда с различными неорганическими и органическими кислотами. Кислотами, которые могут быть использованы для получения фармацевтически приемлемых кислотно-аддитивных солей указанных основных соединений формулы (А) или формулы(В), являются кислоты, способные образовывать нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как ацетат, бензолсульфонат, бензоат, бикарбонат, бисульфат, битартрат, борат, бромид, эдетат кальция, камзилат, карбонат, хлорид, клавуланат, цитрат, дигидрохлорид, эдетат, эдисилат, эстолат, эзилат, этилсукцинат, фумарат, глюцептат, глюконат, глутамат, гликолиларсанилат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, йодид, изотионат,лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилсульфат, мукат, напсилат, нитрат,олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, салицилат, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид и валерат. Используемый здесь термин "лечение", если это не оговорено особо, означает реверсию, ослабление и ингибирование прогрессирования расстройства или состояния, которые определяются данным термином, или одного или нескольких симптомов такого расстройства или состояния, или их предупреждение. Используемый здесь термин "терапия", если это не оговорено особо, означает действие, направленное на "лечение", где указанный термин определен непосредственно выше. Используемый здесь термин "терапевтически эффективное количество" означает количество лекарственного средства или фармацевтического средства, которое способствует вырабатыванию биологического или клинического ответа в ткани, в системе, у животного или человека, где указанный ответ может быть определен исследователем, ветеринаром, практикующим врачом или т.п. Термин "замещенный" означает, что конкретная группа или часть содержит один или несколько заместителей. Термин "незамещенный" означает, что конкретная группа не содержит заместителей. Термин "необязательно замещенный" означает, что конкретная группа либо является незамещенной, либо замещена одним или несколькими заместителями. В соответствии с общепринятым соглашением в некоторых представленных здесь структурных формулах атомы углерода и связанные с ними атомы водорода конкретно не указываются, например означает метильную группу,означает этильную группу,означает циклопентильную группу и т.п. Кроме того, если любая циклическая группа (арильная, гетероциклическая или циклоалкильная) указана вместе со связью, которая непосредственно не присоединена к атому на кольце, например то это означает, что атом на кольце циклической группы может быть присоединен в любом доступном положении. Некоторые соединения формулы (А) или формулы (В) могут иметь асимметрические центры, поэтому они присутствуют в различных энантиомерных формах. Следует отметить, что все оптические изомеры и стереоизомеры формулы (А) или формулы (В) и их смеси входят в объем настоящего изобретения. Настоящее изобретение, если оно относится к соединениям формулы (А) или формулы (В), включает применение рацемата, одной или нескольких энантиомерных форм, одной или нескольких диастереомерных форм или их смесей. Соединения формулы (А) или формулы (В) могут также существовать в виде таутомеров. Настоящее изобретение относится к применению всех указанных таутомеров и их смесей. Некоторые функциональные группы, присутствующие в соединениях согласно изобретению, могут быть замещены биоизостерическими группами, т.е. группами, которые удовлетворяют таким же пространственным и электронным потребностям, как и родительская группа, но которые обладают отличающимися или улучшенными физико-химическими или другими свойствами. Подходящие примеры таких групп хорошо известны специалистам и такими группами являются, но не ограничиваются ими,группы, описанные в публикации Patini et al., Chem. Rev, 1996, 96, 3147-3176 и в цитируемых там работах. Настоящее изобретение также включает меченные изотопами соединения, которые идентичны соединениям, представленным формулой (А) или формулой (В), но в которых фактически один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или массового числа, обычно имеющихся в природе. Примерами изотопов, которые могут быть включены в соединения согласно изобретению, являются изотопы водорода, углерода, азота,кислорода, фосфора, фтора и хлора, такие как 2 Н, 3 Н, 13 С, 14 С, 15N, 18O, 17O, 31P, 32P, 35S, 18F и 36Cl соответственно. Соединения согласно изобретению и фармацевтически приемлемые соли или сольваты указанных соединений, которые содержат вышеупомянутые изотопы и/или другие изотопы других атомов,входят в объем настоящего изобретения. Некоторые меченные изотопами соединения согласно изобретению, например соединения, в которые включены радиоактивные изотопы, такие как 3 Н и 14 С, могут быть использованы в анализах на распределение лекарственных средств и/или субстратов в ткани. Тритий, т.е. 3 Н, и углерод-14, т.е. 14 С, представляют собой изотопы, которые являются особенно предпочтительными благодаря легкости их получения и детектирования. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2 Н, может иметь некоторые терапевтические преимущества, заключающиеся в их большей метаболической стабильности, например, в более продолжительном времени полужизни in vivo или в возможности введения в более низких дозах, и поэтому в некоторых случаях такое замещение может оказаться предпочтительным. Меченные изотопами соединения формулы (А) или формулы (В) согласно изобретению могут быть, в основном, получены путем проведения процедур,описанных в представленных ниже схемах и/или в примерах, в результате замены реагентов, не меченных изотопами, легкодоступными реагентами, меченными изотопами. Используемый здесь термин "ммоль", если это не оговорено особо, означает миллимоль. Используемый здесь термин "экв.", если это не оговорено особо, означает эквивалент. Используемый здесь термин "мл", если это не оговорено особо, означает миллилитр. Используемый здесь термин "ед.", если это не оговорено особо, означает единицы. Используемый здесь термин "мм", если это не оговорено особо,означает миллиметр. Используемый здесь термин "г", если это не оговорено особо, означает граммы. Используемый здесь термин "кг", если это не оговорено особо, означает килограммы. Используемый здесь термин "ч", если это не оговорено особо, означает часы. Используемый здесь термин "мин", если это не оговорено особо, означает минуты. Используемый здесь термин "мкл", если это не оговорено осо-7 017937 бо, означает микролитры. Используемый здесь термин "мкМ", если это не оговорено особо, означает микромоли. Используемый здесь термин "мкм", если это не оговорено особо, означает микрометры. Используемый здесь термин "М", если это не оговорено особо, означает молярную концентрацию. Используемый здесь термин "н.", если это не оговорено особо, означает нормальный. Используемый здесь термин "нм", если это не оговорено особо, означает нанометры. Используемый здесь термин "нМ", если это не оговорено особо, означает наномолярную концентрацию. Используемый здесь термин "е.а.м.", если это не оговорено особо, означает единицу атомной массы. Используемый здесь термин "С", если это не оговорено особо, означает градусы Цельсия. Используемый здесь термин "m/z", если это не оговорено особо, означает среднее отношение масса/заряд. Используемый здесь термин "мас./мас." если это не оговорено особо, означает масса/масса. Используемый здесь термин "об./об.", если это не оговорено особо,означает объем/объем. Используемый здесь термин "мл/мин", если это не оговорено особо, означает миллилитр в минуту. Используемый здесь термин "УФ", если это не оговорено особо, означает ультрафиолетовое излучение. Используемый здесь термин "APCI-MS", если это не оговорено особо, означает масс-спектроскопию методом химической ионизации при атмосферном давлении. Используемый здесь термин "ВЭЖХ", если это не оговорено особо, означает высокоэффективную жидкостную хроматографию. Хроматографию осуществляют, если это не оговорено особо, при температуре примерно 20 С. Используемый здесь термин "ЖХ", если это не оговорено особо, означает жидкостную хроматографию. Используемый здесь термин "ЖХМС", если это не оговорено особо, означает масс-спектроскопию методом жидкостной хроматографии. Используемый здесь термин "ТСХ", если это не оговорено особо, означает тонкослойную хроматографию. Используемый здесь термин "SFC", если это не оговорено особо,означает хроматографию в надкритической жидкости. Используемый здесь термин "нас.", если это не оговорено особо, означает насыщенный. Используемый здесь термин "вод.", если это не оговорено особо, означает водный. Используемый здесь термин "ELSD", если это не оговорено особо, означает детектирование методом рассеяния света с выпариванием. Используемый здесь термин "МС", если это не оговорено особо, означает масс-спектроскопию. Используемый здесь термин "ВРМС (ИЭР)", если это не оговорено особо, означает масс-спектрометрию высокого разрешения (с ионизацией электрораспылением). Используемый здесь термин "анал.", если это не оговорено особо, означает "аналитический". Используемый здесь термин "выч.", если это не оговорено особо, означает вычислено. Используемый здесь термин "н.т.", если это не оговорено особо, означает "не тестировали". Используемый здесь термин"к.т.", если это не оговорено особо, означает комнатную температуру. Используемый здесь термин"Mth", если это не оговорено особо, означает метод. Используемый здесь термин "целит", если это не оговорено особо, означает фильтрующий агент в виде белого твердого диатомита, поставляемого компанией World Minerals в Лос-Анджелесе, Калифорния, США. Используемый здесь термин "прим.", если это не оговорено особо, означает "пример". В используемых здесь терминах -(CR3R4)t или -(CR10R11)v, например, R3, R4, R10 и R11 могут варьироваться, где каждое число t или v превышает 1. Так, например, если t или v равно 2, то термины-(CR3R4)v или -(CR10R11)t могут означать -СН 2 СН 2- или -СН(СН 3)С(СН 2 СН 3)(СН 2 СН 2 СН 3)-, или любое количество подобных молекул, входящих в объем определений R3, R4, R10 и R11. Используемый здесь термин "Ki", если это не оговорено особо, означает величины константы ингибирования ферментов. Используемый здесь термин "Ki app", если это не оговорено особо, означает кажущуюся величину Ki. Используемый здесь термин "IC50", если это не оговорено особо, означает концентрации, необходимые для по меньшей мере 50% ингибирования фермента. Другие аспекты, преимущества и отличительные признаки настоящего изобретения будут более очевидными из представленного ниже подробного описания. Подробное описание и варианты настоящего изобретения В нижеследующих реакционных схемах проиллюстрировано получение соединений согласно изобретению. Если это не оговорено особо, то в указанных реакционных схемах значения R1-R12 и Ra-Re и их обсуждение приводятся выше. Подробное описание изобретения Соединения формул I-III могут быть получены методами синтеза, проиллюстрированными на схеме 1 и схеме 2. На схеме 1 и схеме 2 и в нижеследующем описании "ВОС", "Boc" или "boc" означают Nтрет-бутоксикарбонил, ДХМ означает CH2Cl2, DIPEA (также известный как основание Хьюнига) означает диизопропилэтиламин, DMA означает диметиламин, "ДМФ" означает диметилформамид, "ДМСО" означает диметилсульфоксид, Et означает -СН 2 СН 3, "МТВЕ" означает метил-трет-бутиловый эфир,"NMP" означает 1-метил-2-пирролидинон, "TEA" означает триэтиламин, "TFA" означает трифторуксусную кислоту, "ТГФ" означает тетрагидрофуран. Хотя схемы 1 и 2 и их описание относятся к соединениюI, однако эти схемы и их описание в равной степени относятся и к соединениям II и III. На схеме 1 проиллюстрирован синтез промежуточного соединения I(А), используемого для получения соединений формулы I. Аминогруппу замещенной аминокислоты I(А) алкилируют с получением соединения I(В). Обычно эту процедуру осуществляют путем обработки соединения I(А) алкилирующим агентом в присутствии основания. Наиболее часто используемым алкилирующим реагентом является активированная электрофильная молекула с двойной связью. Типичными условиями реакции алкилирования соединения I(А) активированной электрофильной молекулой с двойной связью является обработка соединения I(А) активированной молекулой с двойной связью в присутствии сильного основания. Последующая водная обработка приводит к образованию соединения I(В). Затем аминогруппу соединенияI(В) защищают boc-группой с получением соединения I(С). Обычно такую реакцию проводят путем обработки соединения I(В) Вос-агентом в присутствии основания. Типичными условиями является обработка соединения I(В) реагентом (Вос)2O в присутствии Me4NOH в MeCN, используемого в качестве растворителя. Затем карбоксильную группу соединения I(С) превращают в метиловый эфир соединенияI(D). Типичными условиями реакции превращения карбоксильной группы в метиловый эфир является обработка соединения I(С) метилйодидом в ДМФ в присутствии основания. После этого соединение I(D) подвергают внутримолекулярной альдольной конденсации с получением соединения I(Е). Такую реакцию обычно проводят путем обработки соединения I(D) сильным основанием в апротонном растворителе. Типичными условиями является обработка соединения I(D) трет-бутоксидом калия (t-BuOK) в толуоле. Последующая водная обработка приводит к образованию соединения I(Е). Затем, соединение I(Е) подвергают реакции 2+3-циклизации с гидразиновой молекулой, в результате чего получают соединениеI(F). Типичными условиями реакции циклизации является обработка соединения I(E) гидразином и уксусной кислотой в EtOH при кипячении с обратным холодильником. Затем, азот пиразолового свободного основания соединения I(F) ацилируют с получением соединения I(G). Типичными условиями ацилирования является обработка соединения I(F) хлорэтилкарбонатом в ТГФ. Более подробное описание условий синтеза соединения I(G) схемы 1 можно найти в публикации заявки на патент США 2003/0171357 и в публикации РСТ WO 02/12242, которые вводятся в настоящее описание посредством ссылки. На схеме 2 проиллюстрированы два способа получения соединений формулы I из промежуточного соединения I(G). В первом способе схемы 2 соединение I(G) подвергают нуклеофильной реакции с электрофильной группой Ra. Такими нуклеофильными реакциями могут быть ацилирование, алкилирование,сульфонирование, восстановительное аминирование или одна из многих других реакций, в результате которой образуется функциональная аминогруппа. Типичным условием реакции ацилирования является обработка соединения I(G) ацилирующим агентом, таким как Ra-COCl, в присутствии основания, такого как 2 экв. DIPEA, в растворителе, таком как дихлорметан. Реакционную смесь перемешивают при температуре от 0 С до комнатной температуры в течение 12 ч. Последующая водная обработка приводит к образованию соединения II(А). Затем Вос-группу у азота пирролового соединения II(А) удаляют с получением соединения II(В). Такую реакцию обычно проводят путем обработки соединения II(А) сильной кислотой. Типичным условием реакции является обработка соединения II(А) 4 н. HCl в диоксане и ДХМ. Последующая водная обработка приводит к образованию соединения II(В). Затем группу NH пирролового соединения II(В) превращают в хлорформиат II(С). Такая реакция может быть осуществлена с использованием фосгена, трифосгена или некоторых их эквивалентов. Типичным условием реакции является обработка соединения II(В) 2 экв. трифосгена в ДХМ при 0 С в течение 4 ч. Последующая обработка слабым основанием в присутствии насыщенного NaHCO3 и очистка приводят к образованию соединенияII(С). Затем соединение II(С) обрабатывают нуклеофильной молекулой Ra. Нуклеофилом может быть спирт, амин или одна из любых других функциональных групп, которая может взаимодействовать с электрофильным соединением II(С). Типичной реакцией является обработка соединения II(С) нуклеофилом, таким как 1,5 экв. спирта в присутствии 2 экв. основания, такого как K2CO3 в растворителе, таком как DME. Затем реакционную смесь нагревают до 80 С в течение 8 ч и растворитель удаляют. Альтернативно соединение II(С) может быть обработано 1,5 экв. амина в присутствии 1 экв. основания, такого какDIPEA, в растворителе, таком как ТГФ. Дальнейшая обработка в протонном растворителе, таком как метанол, в присутствии основания, такого как TEA, с последующей очисткой приводит к образованию соединения формулы I. Альтернативно соединение II(В) может быть затем подвергнуто нуклеофильной реакции с электрофилом (CORb) с получением соединения II(D). Нуклеофильной реакцией, проводимой для такой трансформации, может быть алкилирование, ацилирование, сульфонирование, восстановительное аминирование. Реакцию ацилирования соединения II(В) с образованием соединения II(D) осуществляют путем обработки соединения II(В) ацилирующим реагентом в присутствии основания. Типичным условием реакции является смешивание соединения II(В) с избытком основания, такого как DIPEA, в ДХМ и добавление полученного раствора к изоцианату при 0 С. Реакционную смесь перемешивают в течение 2 ч, а затем проводят водную обработку с получением соединения II(D). Защитную группу в виде этилового эфира у азота пиразолового соединения II(D) удаляют с получением соединения формулы I. Такую реакцию обычно проводят путем обработки соединения II(D) основанием. Типичным условием реакции яв- 10017937 ляется обработка соединения II(D) в диоксане и ДХМ в присутствии 2-3 экв. LiOH при кипячении с обратным холодильником. Последующая водная обработка приводит к образованию соединения формулы I. Во втором способе, проиллюстрированном на схеме 2, Вос-группу у азота пиррола удаляют с получением соединения III(А). Такую реакцию обычно осуществляют путем обработки соединения I(G) сильной кислотой. Типичным условием реакции является обработка соединения I(G) 4 н. HCl в диоксане и ДХМ. Последующая водная обработка приводит к образованию соединения III(А). Соединение III(А) может быть затем подвергнуто нуклеофильной реакции с электрофилом (CORb) с получением соединения III(В). Поскольку группа -NH2, присоединенная к пиразолу соединения III(А), является менее реакционноспособной, чем азот пиррола соединения III(А), то превращение соединения III(А) в соединениеIII(В) может быть осуществлено без проведения реакции защиты группы -NH2 пиразола соединенияIII(А). Нуклеофильной реакцией, осуществляемой для такого превращения, может быть алкилирование,ацилирование, сульфонирование и восстановительное аминирование. Для придания реакции селективности, такую реакцию предпочтительно осуществлять в относительно слабых условиях проведения реакций. Реакцию ацилирования соединения III(А) с получением соединения III(В) осуществляют путем обработки соединения III(А) ацилирующим реагентом в присутствии основания. Типичным условием проведения реакции является смешивание соединения III(А) с избытком основания, такого как DIPEA, в ДХМ и добавление полученного раствора к изоцианату при 0 С. Реакционную смесь выдерживают при 0 С в течение примерно 2 ч, а затем проводят водную обработку с получением соединения III(В). После этого соединение III(В) подвергают нуклеофильной реакции с электрофильной группой Ra. Такой нуклеофильной реакцией может быть ацилирование, алкилирование, сульфонирование, восстановительное аминирование или одна из многих других реакций, в результате которой образуется функциональная аминогруппа. Типичным условием проведения реакции ацилирования является обработка соединения III(В) ацилирующим агентом, таким как Ra-NCO, в присутствии основания, такого как 2 экв.DIPEA, в растворителе, таком как дихлорметан, в течение 2 ч. Альтернативно соединение III(В) может быть обработано ацилирующим агентом, таким как Ra-COOR, где R представляет собой активирующую группу, такую как п-нитрофенил, в присутствии основания, такого как 2 экв. DIPEA, в растворителе, таком как 1,2-дихлорэтан. Последующая водная обработка приводит к образованию соединения III(С). Защитную группу в виде этилового эфира у атома азота пиразола соединения III(С) обычно удаляют путем обработки основанием, в результате чего получают соединение I в виде свободного основания. Типичным условием проведения реакции является смешивание соединения III(С) с TEA в протонном растворителе, таком как метанол, с последующей очисткой с получением соединения формулы I. Альтернативно защитную группу в виде этилового эфира у атома азота пиразола соединения III(В) удаляют с получением соединения III(D) в виде свободного основания. Эту реакцию обычно проводят путем обработки соединения III(В) основанием. Типичным условием проведения реакции является кипячение соединения III(В) с обратным холодильником в диоксане и ДХМ в присутствии 2-3 экв. LiOH. Последующая водная обработка приводит к образованию соединения III(D). Затем соединение III(D) подвергают нуклеофильной реакции с электрофильной группой Ra. Такой нуклеофильной реакцией может быть ацилирование, алкилирование, сульфонирование, восстановительное аминирование или одна из многих других реакций, в результате которой образуется функциональная аминогруппа. Типичным условием проведения реакции ацилирования является обработка соединения III(D) ацилирующим агентом,таким как Ra-COCl, в присутствии основания, такого как 2 экв. DIPEA, в растворителе, таком как дихлорметан. Реакционную смесь перемешивают в течение 4 ч, а затем проводят водную обработку и очистку, в результате чего получают соединение формулы I. Любое из вышеуказанных соединений формулы (А) и (В), определенных выше, может быть превращено в другое аналогичное соединение стандартными методами химического синтеза. Все исходные вещества, реагенты и растворители являются коммерчески доступными и известны специалистам, если это не оговорено особо. Такие методы химического синтеза известны специалистам и включают (а) удаление защитной группы методами, описанными в публикациях Т.W. Greene и P.G.M. Wuts, Protective(галогенида, мезилата, тозилата и т.п.) первичным или вторичным амином, тиолом или спиртом с образованием вторичного или третичного амина, тиоэфира или простого эфира соответственно; (с) обработку первичного и вторичного аминов изоцианатом, хлорангидридом (или другим активированным производным карбоновой кислоты), алкил/арилхлорформиатом или сульфонилхлоридом с получением соответствующего соединения мочевины, амида, карбамата или сульфонамида; (d) восстановительное аминирование первичного или вторичного амина с использованием альдегида. Соединения согласно изобретению могут иметь асимметрические атомы углерода. Диастереомерные смеси могут быть разделены на отдельные диастереомеры, исходя из различий их физикохимических свойств, методами, известными специалистам, например с помощью хроматографии или фракционированной кристаллизации. Энантиомеры могут быть разделены путем превращения энантиомерных смесей в диастереомерную смесь в результате проведения реакции взаимодействия с соответствующим оптически активным соединением (например, спиртом), разделения диастереомеров и превращения (например, гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Все указанные изомеры, включая диастереомерные смеси и чистые энантиомеры, рассматриваются как часть настоящего изобретения. Соединения формулы (А) или формулы (В), которые являются основными по своей природе, могут образовывать различные соли с различными неорганическими и органическими кислотами широкого ряда. Хотя такие соли должны быть фармацевтически приемлемыми для их введения человеку, однако на практике, в большинстве случаев может оказаться желательным сначала выделение соединения формулы(А) или формулы (В) из реакционной смеси в виде фармацевтически неприемлемой соли, а затем простое превращение данного соединения снова в свободное основание путем обработки щелочным реагентом и последующее превращение полученного свободного основания в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений согласно изобретению могут быть легко получены путем обработки основного соединения, по существу, эквивалентным количеством выбранной минеральной или органической кислоты в водном растворителе или в подходящем органическом растворителе, таком как метанол или этанол. После тщательного выпаривания растворителя может быть легко получена нужная твердая соль. Нужная соль кислоты может быть также подвергнута осаждению из раствора свободного основания в органическом растворителе путем добавления к этому раствору соответствующей минеральной или органической кислоты. Соединения формулы (А) или формулы (В), которые являются кислотными по своей природе, могут образовывать соли оснований с различными фармакологически приемлемыми катионами. Примерами таких солей являются соли щелочных или щелочно-земельных металлов, в частности соли натрия и калия. Все эти соли получают стандартными методами. Химическими основаниями, которые используются в качестве реагентов для получения фармацевтически приемлемых солей оснований согласно изобретению, являются основания, образующие нетоксичные соли оснований с кислотными соединениями формулы (А) или формулы (В). Такими нетоксичными солями оснований являются соли, происходящие от фармакологически приемлемых катионов, таких как натрий, калий, кальций, магний и т.п. Эти соли могут быть легко получены путем обработки соответствующих кислотных соединений водным раствором, содержащим нужные фармакологически приемлемые катионы, с последующим выпариванием полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно они могут быть также получены путем смешивания низших алканоловых растворов кислотных соединений и нужного алкоксида щелочного металла с последующим выпариванием полученного раствора досуха методом,аналогичным методу, описанному выше. В любом случае, для гарантии завершения реакции и для достижения максимальных выходов желаемого конечного продукта предпочтительно использовать реагенты в стехиометрических количествах. Соединения согласно изобретению представляют собой ингибиторы протеинкиназы С и предпочтительно селективно ингибируют такие изоферменты протеинкиназы С, как бета-1-, бета-2- и необязательно альфа. В частности, что касается изофермента бета-2, то соединения согласно изобретению имеют величины Ki менее чем 100 нМ. В качестве ингибиторов протеинкиназы С указанные соединения могут быть использованы для лечения состояний, в патологии которых определенную роль играет протеинкиназа С. Такие состояния известны специалистам, и ими являются сахарный диабет и его осложнения, рак, ишемия, воспаления,расстройства центральной нервной системы, сердечно-сосудистые заболевания, болезнь Альцгеймера и кожные болезни. Протеинкиназа С ассоциируется с диабетом нескольких типов. Избыточная активность протеинкиназы С ассоциируется с нарушением передачи сигнала инсулина, а следовательно, и с инсулинорезистентностью, наблюдаемой при диабете типа II. Karasik, A. et al. J. Biol. Chem. 265:10226-10231 (1990);Chen, K.S. et al. Trans. Assoc. Am. Physicians 104:206-212 (1991); Chin, J.E. et al. J. Biol. Chem. 268:63386347 (1993). Кроме того, исследования продемонстрировали заметное увеличение активности протеинкиназы С в тканях, которые, как известно, являются восприимчивыми к осложнениям диабета, возникающим при гипергликемии. Lee, T.S. et al., J. Clin. Invest. 83:90-94 (1989); Lee, T.S. et al. Proc. Natl. Acad.Sci USA 86:5141-5145 (1989); Craven, P.A. and DeRubertis, F.R. J. Clin. Invest. 83:1667-1675 (1989); Wolf,B.A. J. Clin. Invest. 87:1643-1648 (1991). Давно известно, что активность протеинкиназы С ассоциируется с клеточным ростом, с развитием опухолей и рака. Rotenberg, S.A.Weinstein, I.В. Biochem. Mol. Aspects Sel. Cancer 1:25-73 (1991).Ahamd et al., Molecular Pharmacology: 43, 858-862 (1993). Известно, что ингибиторы протеинкиназы С являются эффективными для предупреждения роста опухолей у животных. Meyer, Т. et al. Int. J. Cancer 43:851-856 (1989); Akinagaka, S. et al. Cancer Res. 51:4888-4892 (1991). Совсем недавно было обнаружено,что ингибитор протеинкиназы С, энзастаурин (LY317615-HCl), оказывает прямое воздействие на опухоль в результате индуцирования апоптоза и подавления пролиферации культивируемых опухолевых клеток, а в частности на человеческую глиобластому и карциному толстой кишки. Graff et al., Cancer Res. 16:7462-7469 (2005). Соединения согласно изобретению также действуют как агенты, которые подавляют действие многих лекарственных средств (MDR), что делает их эффективными при введении в комбинации с другими химиотерапевтическими средствами. Было показано, что ингибиторы протеинкиназы С блокируют воспалительные ответы, такие как окислительная вспышка нейтрофилов, ингибирование CD3 в Т-лимфоцитах и индуцированный форболом отек лапы. Towemy, В. et al. Biochem. Biophys. Res. Commun. 171:1087-1092 (199; Mulqueen, M.J. etal. Agents Actions 37:85-89 (1992). В соответствии с этим соединения согласно изобретению, используемые в качестве ингибиторов PKC, могут быть эффективными для лечения воспалений. Активность протеинкиназы С играет главную роль в функционировании центральной нервной системы. Huang, K.P. Trends Neurosci. 12:425-432 (1989). Кроме того, было показано, что ингибиторы протеинкиназы С предупреждают поражения, наблюдаемые при очаговом и центральном ишемическом повреждении мозга и при отеке головного мозга. Hara, Н. et al. J. Cereb. Blood Flow Metab. 10:646653(1990); Shibata, S. et al. Brain Res. 594:290-294 (1992). Было также установлено, что протеинкиназа С вызывает болезнь Альцгеймера. Shimohama, S. et al. Neurology 43:1407-1413 (1993). В соответствии с этим соединения согласно изобретению могут быть использованы для лечения болезни Альцгеймера и ишемического повреждения головного мозга. Активность протеинкиназы С также играет важную роль в развитии сердечно-сосудистых заболеваний. Было показано, что повышенная активность протеинкиназы С в сосудистой системе приводит к усилению сужения кровеносных сосудов и к гипертензии. Такое усиление предотвращает известный ингибитор протеинкиназы С. Bilder, G.E. et al. J. Pharmacol. Exp. Ter. 252:526-430 (1990). Поскольку ингибиторы протеинкиназы С подавляют окислительную вспышку нейтрофилов, то такие ингибиторы протеинкиназы С могут быть также использованы для лечения сердечно-сосудистой ишемии и для улучшения сердечной функции после ишемии. Muid, R.E. et al. FEBS Lett. 293:169-172 (1990); Sonoki, H. et al. KokyuTo Junkan 37:669-674 (1989). Была также исследована роль протеинкиназы С в функции тромбоцитов, и было показано, что повышение уровней протеинкиназы С коррелирует с усилением ответа на действие агонистов. Bastyr III, E.J.Lu, J. Diabetes 42: (Suppl. 1) 97A (1993). PKC участвует в биохимическом пути модуляции проницаемости капилляров под действием фактора активации тромбоцитов. Kobayashi etal., Amer. Pjus. Soc. H1214-H1220 (1994). Было продемонстрировано, что сильные ингибиторы протеинкиназы С влияют на индуцированную агонистом агрегацию тромбоцитов. Toullec, D. et al. J. Biol. Chem. 266:15771-15781 (1991). Ингибиторы протеинкиназы С также блокируют индуцированную агонистом пролиферацию клеток гладких мышц. Matsumoto, H.Sasaki, Y. Biochem. Biophys, Res. Commun. 158:105-109 (1989). Поэтому соединения согласно изобретению могут быть использованы для лечения сердечно-сосудистых заболеваний, атеросклероза и рестеноза. Аномальная активность протеинкиназы С также ассоциируется с кожными заболеваниями, такими как псориаз. Horn, F. et al. J. Invest. Dermatol. 88:220-222 (1987); Raynaud, F. and Evain-Brion, D. Br. J.Dermatol. 124:542-546 (1991). Псориаз характеризуется аномальной пролиферацией кератиноцитов. Было показано, что известные ингибиторы протеинкиназы С ингибируют пролиферацию кератиноцитов с активностью, аналогичной их активности как ингибиторов PKC. Hegemann, L. et al. Aarch. Dermatol. Res. 283:456-460 (1991); Bollag, W.B. et al. J. Invest. Dermatol. 100:240-246 (1993). В соответствии с этим ингибиторы PKC могут быть использованы для лечения псориаза. Соединения согласно изобретению также являются селективными по отношению к изоферменту. Указанные соединения ингибируют преимущественно изоферменты протеинкиназы С бета-1 и бета-2 и необязательно изофермент альфа, но не остальные изоферменты протеинкиназы С, т.е. изоферменты гамма, дельта, эпсилон, дзета и эта. В соответствии с этим соединения согласно изобретению ингибируют изоферменты протеинкиназы С бета-1 и бета-2 и необязательно изофермент альфа в значительно меньших концентрациях, и в минимальной степени ингибируют другие изоферменты PKC. Соединения согласно изобретению являются особенно подходящими для лечения патологических состояний, ассоциированных с действием изоферментов протеинкиназы С бета-1, бета-2 и необязательно альфа. Так, например, увеличение уровней глюкозы в крови, наблюдаемых при диабете, приводит к изофермент-специфическому повышению уровня изофермента бета-2 в тканях сосудов. Proc. Natl Acad. Sci.USA 89:11059-11065 (1992). Ассоциированное с диабетом увеличение уровней изофермента бета в человеческих тромбоцитах коррелирует с изменением ответа на действие агонистов. Bastyr III, E.J.Lu, J.Diabetes 42: (Suppl. 1), 97A (1993). Было показано, что человеческий рецептор витамина D селективно фосфорилируется протеинкиназой С бета. Такое фосфорилирование ассоциируется с изменениями функции рецептора. Hsieh et al., Proc. Natl. Acad. Sci. USA 88:9315-9319 (1991); Hsieh et al. J. Biol. Chem. 268:15118-15126 (1993). Кроме того, недавно проведенные исследования показали, что изофермент бета 2 ответственен за пролиферацию клеток эритролейкоза, а изофермент альфа участвует в дифференцировке мегакариоцитов, образующихся из этих же клеток. Murray et al., J. Biol. Chem. 268:15847-15853(1993). Было обнаружено, что изофермент протеинкиназы С альфа, помимо изоферментов бета-1 и бета-2,обсуждаемых выше, может быть использован для лечения нефропатии, т.е. у PKC-альфа-дефицитных мышей, страдающих STZ-индуцированным диабетом, наблюдалось улучшение состояния при нефропатии. Menne et al., Diabetes 53:2101-2109 (2005). PKC альфа участвует в сердечных сокращениях, Braz et al. Анализ. Протеинкиназа С бета 2 (PKCII) катализирует продуцирование АДФ из АТФ, которое сопровождается переносом фосфорила на пептид-псевдосубстрат PKC (АS, RFARKGSLRQKNV). Такой перенос происходит одновременно с окислением -NADH благодаря активации пируваткиназы (РК) и лактат-дегидрогеназы (LDH). Мониторинг превращения -NADH в NAD+ осуществляют по снижению оптической плотности при 340 нм (е=6,22 см-1 мМ-1) на спектрофотометре Molecular DevicesSPECTRA max PLUS. Такой анализ обычно осуществляют в 96-луночном прозрачном микротитрационном планшете на спектрофотометре Molecular Devices в течение 20 мин при 30 С в 0,1 мл аналитического буфера, содержащего 50 мМ HEPES, рН 7,4, 5 нМ PKC, 23 единицы пируваткиназы, 33 единицы лактат-дегидрогеназы,0,15 мМ пептида, 0,1 мМ АТФ, 1 мМ DTT, 4 мМ PEP, 8 мМ MgCl2, 0,3 мМ NADH, 60 мМ CaCl2, 10 мг/млPS, 50 нг/мл ФМА, 7,5% ДМСО и примерно 10000 нМ-0,169 нМ соединения-ингибитора. Маточные растворы 3-sn-фосфатидил-L-серина (ФС) и форбол-12-миристат-13-ацетата (ФМА) обрабатывают ультразвуком в течение 30 с, после чего сразу добавляют аналитический буфер и анализы инициируют путем добавления 100 мкМ АТФ. Стационарные кинетические параметры для bi-bi-киназной реакции определяют при насыщающей концентрации фосфоакцепторного пептидного субстрата (0,15 мМ) путем построения кривой по данным начальной скорости в соответствии с уравнением Михаэлиса-Ментона: где v представляет собой измеренную начальную скорость;Vmax представляет собой максимальную скорость ферментативной реакции;[S] представляет собой концентрацию АТФ-субстрата;KM представляет собой константу Михаэлиса для АТФ. Величины кататаболизма ферментов (kcat) вычисляли по уравнению kcat=Vmax[Е], где [Е] представляет собой общую концентрацию ферментов. Константы ингибирования ферменов (кажущиеся величиныKi) определяют путем построения кривой зависимости начальных скоростей от различных концентраций ингибитора в соответствии с моделью конкурентного ингибирования АТФ по уравнению Моррисона(Morrison, J.F., Biochim. Biophys Acta 185:269-286 (1969. Фармацевтические композиции/препараты, дозы и способы введения. Методы получения различных фармацевтических композиций с конкретным количеством активного соединения известны специалистам или являются очевидными для специалистов. Кроме того, среднему специалисту в данной области известны методы приготовления композиций и их введения. Такие методы в основном обсуждаются, например, в публикациях GoodmanGilman's. The Pharmaceutical Basisof Therapeutics, current ed., Pergamon Press; и Remington's Pharmaceutical Sciences, current ed., Mack Publishing, Co., Easton, PA. Эти методы могут быть применены в соответствии с аспектами и вариантами описанных здесь методов и композиций. Нижеследующие примеры приводятся лишь в целях иллюстрации и не должны рассматриваться как ограничение настоящего изобретения. Соединения формул (А) и (В) могут быть получены в виде фармацевтических препаратов, подходящих для местного, перорального и парентерального введения, которые могут быть использованы для лечения PKCII-опосредуемых заболеваний. Соединения согласно изобретению могут быть введены перорально в виде таблеток или капсул, маслянистых или водных суспензий, пастилок, лепешек, порошков,гранул, эмульсий, сиропов или эликсиров. Композиции для перорального введения могут включать одну или несколько добавок, таких как отдушки, подсластители, красители и консерванты, что будет придавать фармацевтическим препаратам эстетический вид и приятный вкус. Таблетки могут содержать фармацевтически приемлемые наполнители, облегчающие приготовление таких таблеток. Как известно специалистам, такие таблетки могут иметь фармацевтически приемлемые энтеросолюбильные покрытия,такие как глицерилмоностеаратные или глицерилдистеаратные покрытия, что будет приводить к замедлению дезинтеграции и абсорбции в желудочно-кишечном тракте и обеспечивать пролонгированное действие в течение длительного периода времени. Препараты для перорального введения могут быть приготовлены в виде жестких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, таким как, например,карбонат кальция, фосфат кальция или каолин. Такие препараты могут быть приготовлены в виде мягких желатиновых капсул, в которых активный ингредиент смешан с водной или с масляной средой, такой как арахисовое масло, жидкое вазелиновое масло или оливковое масло. Водные суспензии обычно содержат активные ингредиенты в смеси с наполнителями, подходящими для приготовления водной суспензии. Такими наполнителями могут быть суспендирующий агент,такой как натрийсодержащая карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующий или смачивающий агент, который может представлять собой природный фосфатид, такой как лецитин, продукт конденсации этиленоксида и длинноцепочечной жирной кислоты, например, полиоксиэтиленстеарат, продукт конденсации этиленоксида и длинноцепочечного алифатического спирта, такой как гептадекаэтиленоксицетанол, продукт конденсации этиленоксида и неполного сложного эфира, происходящего от жирной кислоты и гексита, такой как моноолеат полиоксиэтиленсорбита, или ангидриды гексита и жирной кислоты, такие как моноолеат полиоксиэтиленсорбитана. Фармацевтические композиции могут быть приготовлены в форме стерильной водной или масляной суспензии для инъекций. Такая суспензия может быть приготовлена известными методами с использованием подходящих диспергирующих или смачивающих агентов и суспендирующих агентов, упомянутых выше. Стерильный препарат для инъекций может быть также приготовлен в виде суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3 бутандиоле. Приемлемыми наполнителями и растворителями, которые могут быть использованы, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Для этих целей может быть использовано любое мягкое жирное масло, включающее синтетические моно- или диглицериды. Кроме того, в целях приготовления препаратов для инъекций могут быть использованы жирные кислоты, такие как олеиновая кислота. Соединения формул (А) и (В) могут быть также получены в форме суппозиториев для ректального введения лекарственного средства. Эти композиции могут быть получены путем смешивания данного лекарственного средства с подходящим нераздражающим наполнителем, который является твердым примерно при 25 С и становится жидким при температуре прямой кишки, т.е. расплавляется в прямой кишке с высвобождением лекарственного средства. Такими наполнителями являются масло какао и другие глицериды. Для местного применения могут быть приготовлены такие препараты, как, например, кремы, мази,желеобразные растворы или суспензии, содержащие соединения согласно изобретению. Соединения формул (А) и (В) могут быть также введены в виде систем доставки типа липосом, таких как небольшие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть приготовлены из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Дозы соединений согласно изобретению составляют примерно 0,5 мг/кг массы тела-100 мг/кг массы тела. Предпочтительная доза составляет от примерно 30 мг/кг массы тела до примерно 100 мг/кг массы тела. Однако следует отметить, что конкретный уровень дозы для любого конкретного пациента зависит от ряда факторов, включая активность конкретно вводимого соединения, возраст, массу тела, общее состояние здоровья, пол и режим питания пациента, время введения, способ введения, скорость экскреции,комбинации лекарственных средств и тяжесть конкретного заболевания, подвергаемого лечению. Для повышения терапевтической активности соединений согласно изобретению они могут быть введены одновременно с другими перорально вводимыми активными противодиабетическими соединениями, такими как сульфонилмочевины, например толбутамид и т.п. Для закапывания в глаза соединения согласно изобретению могут быть включены в фармацевтически приемлемый офтальмический носитель, так чтобы указанное соединение контактировало с поверхностью глаза в течение периода времени, достаточного для проникновения данного соединения в роговицу и/или склеру и во внутренние области глаза, включая, например, переднюю камеру глаза, заднюю камеру глаза, стекловидное тело, внутриглазную жидкость, жидкую часть стекловидного тела, роговицу,радужку/ресничное тело, хрусталик, сосудистую оболочку глаза/сетчатку и склеру. Фармацевтически приемлемыми офтальмическими носителями могут быть мазь, растительное масло или инкапсулирующий материал. Соединение согласно изобретению может быть также введено непосредственно в жидкую часть стекловидного тела или во внутриглазную жидкость. Кроме того, соединение может быть также введено хорошо известными приемлемыми методами,такими как инъекции под тенонову капсулу и/или субконъюнктивальные инъекции. Как хорошо известно специалистам-офтальмологам, желтое пятно состоит главным образом из колбочек сетчатки и является областью максимальной остроты зрения в сетчатке. Тенонова капсула или тенонова оболочка находится на склере. Конъюнктива покрывает небольшую область глазного яблока позади лимба (луковичная конъюнктива) и верхних складок (верхнего слепого мешка) или нижних складок (нижнего слепого мешка), и тем самым она покрывает внутренние области верхнего века и нижнего века соответственно. Конъюнктива находится в верхней части теноновой капсулы. Склера и тенонова капсула определяют внешнюю поверхность глазного яблока. Для лечения возрастной дегенерации желтого пятна (ВДЖП),хороидальной неоваскуляризации, ретинопатии (такой как диабетическая ретинопатия (включая отек желтого пятна), ретинопатия недоношенных), ретинита, увеита, кистоидного отека желтого пятна(КОЖП), глаукомы и других заболеваний или патологий заднего сегмента глаза может оказаться предпочтительным размещение депо-препарата, содержащего конкретное количество офтальмически приемлемого фармацевтически активного средства, непосредственно на внешнюю поверхность склеры и на поверхность, расположенную ниже теноновой капсулы. Кроме того, в случае ВДЖП и КОЖП наиболее предпочтительно размещать депо-препарат непосредственно на внешнюю поверхность склеры, на поверхность, расположенную ниже теноновой капсулы, и в основном выше желтого пятна. Указанные соединения могут быть приготовлены в виде депо-препарата. Такие препараты пролонгированного действия могут быть введены путем имплантации (например, подкожно или внутримышеч- 15017937 но), путем внутримышечной инъекции или вышеупомянутой инъекции в область, расположенную под теноновой капсулой или внутри стекловидного тела. Альтернативно активный ингредиент может быть приготовлен в виде порошка, который перед его применением может быть разведен подходящим носителем, например стерильной апирогенной водой. В особенно предпочтительных вариантах изобретения указанные соединения могут быть приготовлены для местного введения в физиологическом растворе (в комбинации с любыми консервантами и противомикробными средствами, обычно используемыми для получения офтальмических препаратов) и для введения в виде глазных капель. Раствор или суспензия могут быть приготовлены в чистом виде и введены ежедневно несколько раз в день. Альтернативно композиции согласно изобретению, полученные как описано выше, могут быть также введены непосредственно в роговицу. В предпочтительных вариантах изобретения композицию получают с использованием мукоадгезивного полимера, который связывается с роговицей. Так, например, указанные соединения могут быть получены с использованием подходящих полимерных или гидрофобных веществ (например, в виде эмульсии в подходящем масле) или ионообменных смол, или в виде слаборастворимых производных,например слаборастворимой соли. Фармацевтический носитель для гидрофобных соединений представляет собой систему сорастворителей, включающую бензиловый спирт, неполярное поверхностно-активное вещество, смешиваемый с водой органический полимер и водную фазу. Указанной системой сорастворителей может быть также система сорастворителей VPD. VPD представляет собой раствор, содержащий 3% мас./об. бензилового спирта, 8% мас./об. неполярного поверхностно-активного вещества полисорбата 80 и 65% мас./об. полиэтиленгликоля 300, и доведенный до нужного объема добавлением абсолютного этанола. Система сорастворителей VPD (VPD:5W) содержит VPD, разведенный в отношении 1:1 5% раствором декстрозы в воде. Эта система сорастворителей хорошо растворяет гидрофобные соединения и обладает низкой токсичностью после системного введения. Разумеется, что состав системы сорастворителей может значительно варьироваться, но при этом ее растворимость и токсичность не изменяются. Кроме того, могут варьироваться свойства компонентов сорастворителей, так, например, вместо полисорбата 80 могут быть использованы другие низкотоксичные неполярные поверхностно-активные вещества; может варьироваться размер фракций полиэтиленгликоля; вместо полиэтиленгликоля могут быть использованы другие биологически совместимые полимеры, например поливинилпирролидон, а вместо декстрозы могут быть использованы другие сахара или полисахариды. Альтернативно могут быть использованы и другие системы доставки гидрофобных фармацевтических соединений. Известными примерами носителей для доставки или носителей для гидрофобных лекарственных средств являются липосомы и эмульсии. Также могут быть использованы некоторые органические растворители, такие как диметилсульфоксид, хотя обычно он является более токсичным. Кроме того, указанные соединения могут быть доставлены с помощью системы пролонгированного высвобождения, такой как полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие терапевтическое средство. Существуют различные материалы, применяемые для пролонгированного высвобождения, и такие материалы известны специалистам. Капсулы пролонгированного высвобождения, в зависимости от их химической природы, могут высвобождать соединения за период времени от нескольких недель до 100 дней и выше. В зависимости от химической природы и биологической стабильности терапевтического реагента могут быть применены и другие стратегии стабилизации белка. Фармацевтические композиции могут также содержать подходящие носители или наполнители в виде твердого вещества или геля. Примерами таких носителей или наполнителей являются карбонат кальция, фосфат кальция, сахара, крахмалы, производные целлюлозы, желатин и полимеры, такие как полиэтиленгликоли. Некоторые соединения согласно изобретению могут быть получены в виде солей с фармацевтически совместимыми противоионами. Фармацевтически совместимые соли могут быть образованы многими кислотами, включая хлористоводородную, серную, уксусную, молочную, винную, яблочную, янтарную кислоты и т.п. Соли в отличие от соответствующих свободных оснований могут лучше растворяться в водных или других протонных растворителях. Получение предпочтительных соединений согласно изобретению более подробно описано в нижеследующих примерах, хотя для специалиста в данной области очевидно, что описанные химические реакции могут быть легко адаптированы для получения различных других соединений согласно изобретению. Так, например, синтез нерепрезентативных соединений согласно изобретению может быть с успехом осуществлен путем модификаций, которые являются очевидными для специалиста в данной области,например путем соответствующей защиты интерферирующих групп, путем замены одних реагентов другими подходящими реагентами, известными специалистам, или путем проведения рутинных изменений условий реакции. Альтернативно могут быть проведены и другие реакции, описанные в настоящем патенте или известные специалистам, которые являются подходящими для получения других соединений согласно изобретению. Примеры Примеры и препаративные примеры, представленные ниже, служат для более подробной иллюстрации и описания соединений согласно изобретению и методов их получения. Очевидно, что объем настоящего изобретения не ограничивается объемом представленных ниже примеров и препаративных примеров. В нижеследующих примерах молекулы, имеющие один хиральный центр, если это не оговорено особо, присутствуют в виде рацемической смеси. Молекулы, имеющие два или более хиральных центра, если это не оговорено особо, присутствуют в виде рацемической смеси диастереомеров. Отдельные энантиомеры/диастереомеры могут быть получены методами, известными специалистам. Структуры соединений были подтверждены с помощью элементного анализа или ЯМР, где для указанного в заголовке соединения, если это необходимо, представлены пики характеристических протонов. 1 Н-ЯМР-сдвиги (H) в сторону нижнего поля приводятся в миллионных долях (м.д.) по отношению к внутреннему стандарту. Представленное ниже описание изобретения приводится со ссылками на нижеследующие примеры. Эти примеры не должны рассматриваться как ограничение объема настоящего изобретения и служат лишь для иллюстрации. В таблице приводится полный список соединений согласно изобретению и в эту таблицу включены релевантные данные 1 Н-ЯМР и вычисленные величины Ki. Пример А 1. Промежуточное соединение A1(i). К раствору 5-трет-бутил 1-этил 3-амино-6,6-диметил-4,6 дигидропирроло[3,4-с]пиразол-1,5-дикарбоксилата (16,2 г, 49,9 ммоль) в ТГФ (100 мл) при 0 С 3 порциями добавляли NaH (2,4 г, 59,9 ммоль). Реакционную смесь перемешивали в течение 15 мин на ледяной бане, а затем в течение 10 мин добавляли этилхлорформиат (6,5 г, 59,9 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч, а затем реакцию гасили NH4Cl(насыщенным) и смесь экстрагировали EtOAc (250 мл). Объединенные экстракты промывали насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали с получением нужного соединения A1(i) (19,8 г, 99%). Масс-спектр: вычислено для C18H29N4O6 (M+H): 397. Найдено 397. Промежуточное соединение A1(ii). Этил 5-(хлоркарбонил)-3-[(этоксикарбонил)амино]-6,6-диметил 5,6-дигидропирроло[3,4-с]пиразол-1(4 Н)-карбоксилат. К раствору соединения A1(i) (19,8 г, 49,9 ммоль) в диоксане (20 мл) добавляли HCl (60 мл, 4 М в диоксане). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, а затем концентрировали и сушили в вакууме. HCl-соль этил-3-[(этоксикарбонил)амино]-6,6-диметил-5,6 дигидропирроло[3,4-с]пиразол-1(4H)-карбоксилата растворяли в CH2Cl2 (60 мл). Затем добавляли DIPEA(16,1 г, 125 ммоль) и реакционную смесь охлаждали на ледяной бане. После этого медленно добавляли фосген (30 мл, 20% в толуоле), а затем реакционную смесь нагревали до комнатной температуры и подвергали перегонке в течение ночи. Затем концентрированную реакционную смесь растворяли в EtOAc(100 мл) и воде (100 мл). Водную фазу экстрагировали EtOAc (225 мл), а затем объединенные органические экстракты промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованиемCH2Cl2-2% 7 н. NH3/MeOH в CH2Cl2, в результате чего получали указанное в заголовке соединение A1(ii) в виде твердого вещества белого цвета (9,58 г, 54%). Масс-спектр: вычислено для C14H20ClN4O5 (M+H): 359. Найдено: 359. Промежуточное соединение А 1(iii). Этил-3-[(этоксикарбонил)амино]-6,6-диметил-5-[(2S)-2,4,5,5 тетраметилпиперазин-1-ил]карбонил-5,6-дигидропирроло[3,4-с]пиразол-1(4 Н)-карбоксилат. В герметично закрывающуюся пробирку добавляли (55)-1,2,2,5-тетраметилпиперазин (2,8 г,20 ммоль), DIPEA (7,63 г, 59,1 ммоль) и ТГФ (40 мл), а затем соединение A1(ii) (7,1 г, 20 ммоль). Затем пробирку герметично закрывали, помещали на масляную баню при 80 С и нагревали в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, после чего концентрировали и очищали колоночной хроматографией на силикагеле с использованием CH2Cl2-3% (7 н. NH3/MeOH) в CH2Cl2, в результате чего получали указанное в заголовке соединение A1(iii) (4,62 г, 51%). Масс-спектр: вычислено для C22H37N6O5 (М+Н): 465. Найдено: 465. Промежуточное соединениеA1(iv). 6,6-Диметил-5-[(2S)-2,4,5,5-тетраметилпиперазин-1 ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. В микроволновый сосуд добавляли соединение A1(iii) (4,6 г, 10 ммоль), МеОН (45 мл) и LiOH(6,4 г, 40 ммоль). Реакционную смесь нагревали при 110 С в микроволновой печи в течение 20 мин. Неочищенную реакционную смесь концентрировали и растворяли в ТГФ (75 мл). Нерастворимое вещество отфильтровывали и фильтрат концентрировали с получением указанного в заголовке соединения A1(iv)(2,3 г, 72%). 1 Н-ЯМР (300 МГц, CDCl3) м.д. 1,02 (с, 3 Н), 1,08-1,18 (м, 6 Н), 1,66 (с, 3 Н), 1,76 (с, 3 Н), 2,22 (с, 3 Н),2,25-2,41 (м, 1 Н), 2,59-2,70 (м, 1 Н), 2,70-2,91 (м, 2 Н), 3,42-3,57 (м, 1 Н), 4,28-4,51 (м, 2 Н). Масс-спектр: вычислено для C16H29N6O (М+Н): 321. Найдено: 321. Промежуточное соединение A1(v). N-(2-Хлор-5-фторпиримидин-4-ил)-6,6-диметил-5-[(2S)-2,4,5,5 тетраметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. К раствору соединения A1(iv) (1,92 г, 5,9 ммоль) и 2,4-дихлор-5-фторпиримидина (1,0 г, 5,9 ммоль) в ДМСО (8 мл) добавляли дигидрофосфат калия (0,82 г, 5,9 ммоль), а затем Н 3 РО 4 (0,12 г, 1,2 ммоль). Реакционную смесь помещали на масляную баню при 90 С и нагревали в течение 20 ч. Неочищенную реакционную смесь охлаждали до комнатной температуры, а затем выливали в охлажденный льдомNaHCO3 (30 мл) и экстрагировали EtOAc (230 мл). Объединенные органические экстракты промывали насыщенным раствором соли, а затем сушили (MgSO4), фильтровали и концентрировали. Неочищенное твердое вещество растирали с EtOAc, а затем фильтрат очищали колоночной хроматографией на силикагеле с использованием 1-4% 7 н. NH3/MeOH в CH2Cl2, в результате чего получали указанное в заголовке соединение A1(v) в виде твердого вещества белого цвета (1,4 г, 53%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 0,90 (с, 3 Н), 1,00 (с, 3 Н), 1,06 (д, J=6,4 Гц, 3 Н), 1,58 (с, 3 Н),1,69 (с, 3 Н), 2,08 (с, 3 Н), 2,14-2,25 (м, 1 Н), 2,55-2,64 (м, 2 Н), 2,73-2,89 (м, 1 Н), 3,38-3,51 (м, 1 Н), 4,51-4,72N4-(6,6-Диметил-5-[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил-1,4,5,6 тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4-диамин. В микроволновый сосуд добавляли соединение A1(v) (200 мг, 0,44 ммоль) и этиламин (5 мл, 2 М в МеОН). Сосуд нагревали до 150 С в микроволновой печи в течение 1,5 ч. Неочищенный образец концентрировали, а затем очищали колоночной хроматографией на силикагеле с использованием 1-4% 7 н.NH3/MeOH в CH2Cl2, в результате чего получали нужный продукт А 1 в виде твердого вещества желтого цвета (161 мг, 79%). ЯМР-данные представлены далее в таблице. Примеры А 2-А 9. Соединения примеров A2-A9 получали методами, аналогичными методам, описанным выше в примере А 1. Названия соединений и их ЯМР-данные приводятся ниже в таблице. Пример А 10. 5-[(8S)-6,8-Диметил-6,9-диазаспиро[4.5]дек-9-ил]карбонил-N-(5-фтор-2 метилпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин Промежуточное соединение A10(i). 5-[(8S)-6,8-Диметил-6,9-диазаспиро[4.5]дек-9-ил]карбонил 6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. В соответствии с процедурой, описанной выше для получения промежуточного соединения A1(iv),указанное в заголовке промежуточное соединение очищали флэш-хроматографией, элюируя метиленхлоридом, содержащим 5% 7 н. NH3 в метаноле, в результате чего получали твердое вещество желтого цвета (506 мг, 90%). Синтез спироциклопентилпиперазина описан в разделе "Общий синтез пиперазинов". 1 Н-ЯМР (400 МГц, хлороформ-d):м.д. 1,13 (3 Н, д, J=6,32 Гц), 1,36-1,41 (1 Н, м), 1,52-1,64 (8 Н, м),1,66 (3 Н, с), 1,77 (3 Н, с), 2,20-2,22 (3 Н, м), 2,23-2,29 (1 Н, м), 2,60 (1 Н, дд, J=11,75, 3,66 Гц), 2,78-2,89 (2 Н,м), 3,49 (2 Н, с), 3,52-3,56 (1 Н, м), 3,64 (1 Н, с), 4,33-4,44 (2 Н, м). Промежуточное соединение A10(ii). 4-Хлор-5-фтор-2-метилпиримидин. Гидрид натрия (60%, 5,0 г, 125 ммоль) промывали гексаном для удаления минерального масла и сушили, а затем суспендировали в ТГФ (50 мл) и охлаждали до 0 С. Этилфторацетат (13,30 г, 125 ммоль) и этилформиат (15,14 мл, 187 ммоль) смешивали и добавляли к перемешиваемой суспензии. Реакционную смесь медленно нагревали до температуры окружающей среды и перемешивали в течение 3 дней. Растворитель удаляли. К реакционной смеси добавляли смесь гидрохлорида ацетамидина (11,81 г,125 ммоль), этоксида натрия (8,86 г, 125 ммоль) и этанола (60 мл), после чего смесь кипятили с обратным холодильником в течение ночи. Этанол удаляли при пониженном давлении. Остаток растворяли в минимальном количестве воды и подкисляли до рН 6 путем добавления концентрированной HCl. Затем неочищенные продукты экстрагировали путем высаливания из водной фазы и истощающей промывки смесью 4:1 CHCl3/изопропанола. Объединенные органические фазы сушили (MgSO4) и выпаривали. Неочищенное твердое вещество очищали хроматографией на силикагеле, элюируя смесью 5-90%EtOAc/гексан, в результате чего получали твердое вещество белого цвета (0,95 г, 6%).H-ЯМР (400 МГц, ДМСО-d6):2,25 (д, J=1,0 Гц, 3 Н), 7,93 (д, J=3,8 Гц, 1 Н), 12,95 (ушир, 1 Н). ЖХМС: 129. 2-Метил-5-фторурацил (1,04 г, 7,21 ммоль) и N,N-диметиланилин (1,80 мл) нагревали в POCl3 при 110 С в течение 90 мин. После охлаждения реакционную смесь осторожно помещали на лед. Продукт экстрагировали диэтиловым эфиром. Эфирный слой последовательно промывали 2 н. HCl, водой и насыщенным раствором соли, а затем сушили (MgSO4). Эфир осторожно удаляли при пониженном давлении,в результате чего получали летучую жидкость (0,39 г, 34%), которую использовали без дополнительной очистки.H-ЯМР (400 МГц, ДМСО-d6):3,91 (с, 3 Н), 8,79 (с, 1 Н). Пример А 10. 5-[(8S)-6,8-Диметил-6,9-диазаспиро[4.5]дек-9-ил]карбонил-N-(5-фтор-2 метилпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. Указанное в заголовке соединение А 10 очищали в соответствии с процедурой, описанной выше для промежуточного соединения A1(v), в результате чего получали порошок белого цвета (49,7 мг, 24%). ЯМР-данные приводятся ниже в таблице. Пример Промежуточные соединения A11(ii) (2S,5R)-1-бензил-2,5-диметилпиперазин и A11(iii) (2R,5S)-1 бензил-2,5-диметилпиперазин. Соединение A11(i) (800 г, 7 моль) растворяли в 12,8 л EtOH. Раствор бензилбромида (1 кг, 5,8 моль) в 1,6 л EtOH добавляли по каплям при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. ТСХ (CH2Cl2:MeOH=10:1) указывала на завершение реакции. Растворитель удаляли в вакууме. Остаток разбавляли 32 л воды и полученную смесь перемешивали при комнатной температуре в течение 30 мин, а затем фильтровали. Фильтрат экстрагировали CH2Cl2 (8 л 3). Органическую фазу сушили над Na2SO4 и концентрировали досуха, в результате чего получали смесь соединений A11(ii) и A11(iii) (800 г, 56%) в виде масла коричневого цвета. Промежуточное соединение A11(iv). (2S,5R)-1-Бензил-2,5-диметилпиперазин. К перемешиваемому раствору соединений A11(ii) и A11(iii) (300 г, 1,48 моль) в МеОН (1 л) добавляли раствор L-(+)-винной кислоты (444 г, 2,96 моль) в МеОН (2 л). После этого смесь перемешивали при комнатной температуре в течение 20 мин, а затем оставляли на 24 ч при 0 С. Образовавшееся твердое вещество собирали путем фильтрации, а затем перекристаллизовывали из МеОН (2 л) с получением твердого вещества. Полученное твердое вещество растворяли в 1,6 л воды. Полученную смесь подщелачивали до рН 9-10 насыщенным водным раствором Na2CO3, Затем смесь экстрагировали CH2Cl2(1,5 л 2). Органическую фазу сушили над Na2SO4 и концентрировали досуха с получением соединенияA11(iv) (122 г, 81%) в виде жидкости желтого цвета. 1H-ЯМР (400 МГц, CDCl3):7,21-7,13 (м, 5 Н), 4,02 (д, 1 Н), 3,00 (д, 1H), 2,82-2,51 (м, 4 Н), 2,15 (м,1 Н), 1,57 (м, 2 Н), 1,05 (д, 3 Н), 0,85 (д, 3 Н). Промежуточное соединение A11(v). (2S,5R)-1-Бензил-2,5-диметил-4-(тетрагидро-2H-пиран-4 илметил)пиперазин. В микроволновый сосуд добавляли соединение A11(iv) (7,50 г, 36,7 ммоль), (бромметил)тетрагидропиран (6,57 г, 36,7 ммоль), триэтиламин (12,8 мл, 91,8 ммоль) и МеОН (9 мл). Реакционную смесь нагревали до 150 С в течение 2 ч в микроволновой печи, а затем полученное твердое вещество растирали с МеОН, фильтровали и сушили с получением нужного продукта в виде твердого вещества белого цвета A11(v) (6,1 г, 55%). Масс-спектр: вычислено для C19H31N2O (M+H): 303. Найдено: 303. Промежуточное соединение(2R,5S)-2,5-Диметил-1-(тетрагидро-2H-пиран-4 илметил)пиперазин. К раствору соединения A11(v) (6,10 г, 20,0 ммоль) в MeOH (200 мл) добавляли 10% палладий на угле (0,600 г, 0,570 ммоль). Из суспензии откачивали воздух и снова заполняли водородом (3), а затем смесь перемешивали в течение 15 ч в атмосфере водорода. Суспензию фильтровали через целит, осадок на фильтре промывали CH2Cl2, и фильтрат концентрировали с получением нужного продукта в виде бесцветного масла A11(vi) (4,3 г, 88%). Масс-спектр: вычислено для C12H25N2O (M+H): 213. Найдено: 213. ПримерN4-(5-[(2S,5R)-2,5-диметил-4-(тетрагидро-2H-пиран-4-илметил)пиперазин-1 ил]карбонил-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-ил)-N2-этил-5-фторпиримидин-2,4 диамин. Указанное в заголовке соединение получали методами, аналогичными методам, описанным в примере А 1, за исключением того, что вместо (55)-1,2,2,5-тетраметилпиперазин использовали A11(vi). ЯМРданные представлены ниже в таблице. Пример АА 1. 5-[(2S,5R)-4-Этил-2,5-диметилпиперазин-1-ил]карбонил-N-(5-фтор-2 метоксипиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин(257 мг, 2 экв.) в 5 мл 50% уксусной кислоты в воде нагревали в микроволновой печи в течение 30 мин при 80 С. Очистку проводили, как описано в примере A1, в результате чего получали указанное в заголовке соединение AA1 в виде порошка белого цвета (13,1 мг, 3%). ЯМР-данные представлены ниже в таблице. Примеры АА 2-АА 5. Соединения примеров АА 2-АА 5 получали методами, аналогичными методам, описанным в примере АА 1. Названия и ЯМР-данные представлены ниже в таблице. Промежуточное соединение B1(i). трет-Бутил 3-амино-6,6-диметил-4,6-дигидропирроло[3,4 с]пиразол-5(1H)-карбоксилат. К взвеси 5-трет-бутил 1-этил 3-амино-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-1,5 дикарбоксилата (25,00 г, 77,1 ммоль) в МеОН (50 мл) добавляли LiOH (1,92 г, 77,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали. Неочищенную реакционную смесь растворяли в EtOAc (50 мл), а затем промывали NaHCO3 (20 мл) и водой (20 мл). Органический слой сушили (MgSO4), фильтровали и концентрировали с получением твердого вещества оранжевого цвета, которое растирали с ACN, а затем фильтровали и промывали ацетонитрилом (ACN)(50 мл) с получением указанного в заголовке соединения B1(i) в виде твердого вещества белого цвета(ушир.с, 1 Н), 11,15 (с, 1 Н). Масс-спектр: вычислено для C12H21N4O2 (M+H): 253. Найдено 253. Промежуточное соединение B1(ii). трет-Бутил 3-[(2-хлор-5-фторпиримидин-4-ил)амино]-6,6 диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1 Н)-карбоксилат. К раствору соединения B1(i) (8,01 г, 31,7 ммоль) и 2,4-дихлор-5-фтор пиримидина (5,30 г,31,7 ммоль) в ДМСО (40 мл) добавляли дигидрофосфат калия (4,32 г, 31,7 ммоль), а затем Н 3 РО 4 (0,62 г,6,4 ммоль). Реакционную смесь помещали на масляную баню при 95 С и нагревали в течение 20 ч. Неочищенную реакционную смесь охлаждали до комнатной температуры, а затем выливали в охлажденный льдом NaHCO3 (насыщенный, 100 мл) и добавляли EtOAC (75 мл). Полученную смесь фильтровали с получением нужного соединения B1(ii) в виде твердого вещества белого цвета (6,1 г, 50%). 1(м, 1 Н), 10,70 (ушир.с, 1 Н), 12,53 (ушир.с, 1 Н). Масс-спектр: вычислено для C16H21ClFN6O2 (M+H): 383. Найдено 383. Промежуточное соединение B1(iii). трет-Бутил 3-[2-(этиламино)-5-фторпиримидин-4-ил]амино 6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1 Н)-карбоксилат. В герметично закрывающуюся пробирку добавляли соединение B1(ii) (7,64 г, 20,0 ммоль) и этиламин (40,0 мл, 2 М в МеОН). Герметично закрытую пробирку нагревали на масляной бане при 150 С в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, и полученное твердое вещество фильтровали и промывали холодным МеОН, в результате чего получали указанное в заголовке соединение B1(iii) в виде твердого вещества белого цвета (6,1 г, 78%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 1,10 (т, J=7,2 Гц, 3 Н), 1,39-1,48 (м, 9 Н), 1,53-1,61 (м, 6 Н), 3,183,27 (м, 2 Н), 4,23-4,37 (м, 2 Н), 7,06 (ушир.с, 1 Н), 7,93 (с, 1 Н), 10,10 (ушир.с, 1 Н), 12,45 (ушир.с, 1 Н). Масс-спектр: вычислено для C18H27FN7O2 (M+H): 392. Найдено: 392. Промежуточное соединение B1(iv). 5-трет-Бутил 1-этил 3-[2-(этиламино)-5-фторпиримидин-4 ил]амино-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-1,5-дикарбоксилат. Раствор соединения B1(iii) (5,14 г, 13,1 ммоль) в ТГФ (60 мл) и DIPEA (4,24 г, 13,1 ммоль) охлаждали на ледяной бане, а затем по каплям добавляли этилхлорформиат (1,42 г, 13,1 ммоль). Реакционную смесь медленно нагревали до комнатной температуры и перемешивали в течение 5 ч, а затем реакцию гасили водой (50 мл) и смесь экстрагировали EtOAc (2100 мл). Объединенные экстракты промывали насыщенным раствором соли (50 мл), а затем сушили (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием 10-40%EtOAC в CH2Cl2, в результате чего получали указанное в заголовке соединение B1(iv) в виде пены белого цвета (5,8 г, 95%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 1,01-1,11 (м, 3 Н), 1,33 (т, J=7,2 Гц, 3 Н), 1,38-1,49 (м, 9 Н), 1,701,83 (м, 6 Н), 3,13-3,27 (м, 2 Н), 4,28-4,50 (м, 4 Н), 6,54-6,86 (м, 1 Н), 7,94 (м, 1 Н), 10,06-10,41 (м, 1 Н). Масс-спектр: вычислено для C21H31FN7O4 (M+H): 464. Найдено: 464. Промежуточное соединение В 1(v). Этил 5-(хлоркарбонил)-3-[2-(этиламино)-5-фторпиримидин-4 ил]амино-6,6-диметил-5,6-дигидропирроло[3,4-с]пиразол-1(4 Н)-карбоксилат. К суспензии соединения B1(iv) (4,43 г, 9,6 ммоль) в диоксане (25 мл) добавляли HCl (20 мл, 4 М в диоксане). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали. К раствору НС 1-соли этил 3-[2-(этиламино)-5-фторпиримидин-4-ил]амино-6,6-диметил 5,6-дигидропирроло[3,4-с]пиразол-1(4H)-карбоксилата (1,27 г, 2,7 ммоль) в CH2Cl2 (40 мл) добавлялиDIPEA (1,57 г, 12,1). Реакционную смесь охлаждали до -78 С и с помощью капельной воронки в течение 15 мин добавляли трифосген (0,48 г, 0,6 ммоль) в растворе CH2Cl2 (10 мл). Реакцию гасили водой при-78 С, а затем смесь нагревали до комнатной температуры. Смесь доводили до рН 8-9 добавлением NaHCO3 и экстрагировали CH2Cl2 (215 мл). Объединенные экстракты промывали насыщенным раствором соли (15 мл), а затем сушили, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием 0-3% 7 н. NH3/МеОН в CH2Cl2, в результате чего получали нужное соединение B1(v) в виде пены белого цвета (489 мг, 36%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 1,07 (т, J=7,2 Гц, 3 Н), 1,33 (т, J=7,2 Гц, 3 Н), 1,78-1,84 (м, 6 Н),3,12-3,28 (м, 2 Н), 4,42 (кв, J=7,2 Гц, 2 Н), 4,78 (с, 2 Н), 6,69-6,97 (м,1 Н), 7,77-8,08 (м, 1 Н), 10,41 (с, 1 Н). Масс-спектр: вычислено для C17H22CIFN7O3 (M+H): 426. Найдено 426. Получение соединения В 1(с) с боковой цепью:(2S,5R)-1-Бензил-4-трет-бутоксисарбонил-2,5 диметилпиперазин. К раствору исходного вещества (3,00 г, 9,85 ммоль) в CH2Cl2 (100 мл) добавляли 4 н. HCl в 1,4 диоксане (20 мл). Раствор перемешивали в течение 1 ч, а затем летучие вещества удаляли в вакууме, в результате чего получали нужный продукт в виде твердого вещества белого цвета с количественным выходом. Масс-спектр: вычислено для C13H21N2 (M+H): 205. Найдено: 205. Промежуточное соединение B1(b). (2S,5R)-1-Бензил-2,5-диметилпиперазин. В микроволновый сосуд добавляли исходное вещество (1,60 г, 5,77 ммоль), 1-бром-3 метоксипропан (3,09 г, 20,2 ммоль), триэтиламин (6,03 мл, 43,3 ммоль), ТГФ (6 мл) и МеОН (6 мл). Суспензию нагревали при 150 С в микроволновой печи в течение 2 ч. Охлажденный раствор разбавлялиEtOAc (20 мл), после чего органический слой промывали NaHCO3 (насыщенным, водным) (325 мл) и насыщенным раствором соли (125 мл). Органический слой сушили над MgSO4 и концентрировали с получением нужного продукта в виде масла коричневого цвета (1,5 г, 92%). Масс-спектр: вычислено для C17H29N2O (M+H): 277. Найдено: 277. Промежуточное соединение В 1(с). (2R,5S)-1-Бензил-4-(3-метоксипропил)-2,5-диметилпиперазин. К раствору исходного вещества (1,50 г, 5,43 ммоль) в МеОН (50 мл) добавляли 10% палладий на угле (0,150 г, 1,41 ммоль). Из суспензии откачивали воздух, а затем ее снова заполняли водородом (3) и оставляли на 15 ч для перемешивания в атмосфере водорода. Суспензию фильтровали через целит, а за- 22017937 тем осадок на фильтре промывали CH2Cl2 и фильтрат концентрировали с получением нужного продукта в виде пены коричневого цвета (0,83 г, 82%). Масс-спектр: вычислено для C10H23N2O (M+H): 187, Найдено: 187. Пример В 1. Ацетатная сольN2-этил-5-фтор-N4-(5-[(2S,5R)-4-(3-метоксипропил)-2,5 диметилпиперазин-1-ил]карбонил-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3 ил)пиримидин-2,4-диамина. В герметично закрывающуюся пробирку добавляли соединение В 1(v) (393 мг, 0,92 ммоль) в растворе ТГФ (10 мл), а затем добавляли DIPEA (537 мг, 4,2 ммоль) и соединение В 1(с) (172 мг, 0,92 ммоль). Реакционную смесь помещали на масляную баню при 85 С и нагревали в течение 16 ч. Неочищенную реакционную смесь концентрировали, растворяли в МеОН (5 мл) и Et3N (5 мл), а затем перемешивали еще 16 ч. После проведения ВЭЖХ с использованием 5-50% ACN/H2O (0,1% AcOH) получали указанное в заголовке соединение B1 в виде твердого вещества белого цвета (185 мг, 36%). ЯМР-данные представлены ниже в таблице. Примеры В 2-В 5. Соединения примеров В 2-В 5 получали методами, аналогичными методам, описанным выше в примере В 1. Названия соединений и ЯМР-данные представлены ниже в таблице. ПримерB1, за исключением того, что в процессе получения промежуточного соединения B1(ii) вместо промежуточного соединения B1(b) использовали гидрохлорид (2R,5S)-1,2,5-триметилпиперазина, а вместо 2,4 дихлор-5-фторпиримидина использовали 4-хлорпиримидин-2-карбонитрил. ЯМР-данные представлены ниже в таблице. Пример В 7.N-(5-Фтор-2-морфолин-4-илпиримидин-4-ил)-6,6-диметил-5-[(2S,5R)-2,4,5 триметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. Соединение примера В 7 получали методами, аналогичными методам, описанным выше в примере В 1, за исключением того, что в процессе получения промежуточного соединения B1(iii) вместо промежуточного соединения B1(b) использовали гидрохлорид (2R,5S)-1,2,5-триметилпиперазина, а вместо этиламина использовали морфолин. ЯМР-данные представлены ниже в таблице. Пример С 1. N2-Этил-5-фтор-N4-5-[(4-фтор-1-метилпиперидин-4-ил)карбонил]-6,6-диметил-1,4,5,6 тетрагидропирроло [3,4-с]пиразол-3-илпиримидин-2,4-диамин.HCl (20 мл, 4 M в диоксане). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали с получением указанного в заголовке соединения C1(i) в виде три-HCl-соли(3,8 г, 84%). Масс-спектр: вычислено для C16H23FN7O2 (M+H): 364. Найдено 364. Пример С 1. N2-Этил-5-фтор-N4-5-[(4-фтор-1-метилпиперидин-4-ил)карбонил]-6,6-диметил-1,4,5,6 тетрагидропирроло[3,4-с]пиразол-3-илпиримидин-2,4-диамин. В герметично закрывающуюся пробирку добавляли C1(i) (400 мг, 0,85 ммоль) в растворе ТГФ(10 мл), а затем DIPEA (492 мг, 3,8 ммоль) и 4-фтор-1-метилпиперидине-4-карбонилхлорид (228 мг,1,3 ммоль). Реакционную смесь нагревали на масляной бане при 80 С в течение 16 ч, а затем концентрировали, растворяли в МеОН (3 мл) и Et3N (5 мл) и перемешивали еще 10 ч. Неочищенную реакционную смесь концентрировали, а затем очищали путем колоночной хроматографии на силикагеле с использованием 1-3% 7 н. NH3/МеОН в CH2Cl2, в результате чего получали указанное в заголовке соединение С 1 в виде твердого вещества желтого цвета (175 мг, 47%). ЯМР-данные представлены ниже в таблице. Промежуточное соединение D1(i). N-(5-Фтор-2-винилпиримидин-4-ил)-6,6-диметил-5-[(2S)2,4,5,5-тетраметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. В снабженную перегородкой герметично закрывающуюся пробирку добавляли N-(2-хлор-5 фторпиримидин-4-ил)-6,6-диметил-5-[(2S)-2,4,5,5-тетраметилпиперазин-1-ил]карбонил-1,4,5 тетрагидропирроло[3,4-с]пиразол-3-амин (109 мг, 0,242 ммоль), 4,4,5,5-тетраметил-2-винил-1,3,2 диоксаборолан (122 мг, 0,725 ммоль), Na2CO3 (77 мг, 0,73 ммоль) и продукт присоединения дихлор[1,1'бис-(дифенилфосфино)ферроцен]палладия(II) дихлорметана (40 мг, 0,048 ммоль), а затем DME (2,5 мл) и воду (0,5 мл). Реакционную смесь продували аргоном в течение 2 мин, а затем помещали на предварительно нагретую до 100 С масляную баню и перемешивали в течение 16 ч. Неочищенную реакционную смесь концентрировали, растворяли в EtOAc (15 мл) и промывали водой (10 мл). Раствор EtOAc сушили(MgSO4), фильтровали и концентрировали. После очистки колоночной хроматографией на силикагеле с использованием 1-3% 7 н. NH3/MeOH в CH2Cl2 получали указанное в заголовке соединение D1(i) в виде твердого вещества желтого цвета (45 мг, 42%). 1 Н-ЯМР (300 МГц, CDCl3):м.д. 0,99 (с, 3 Н), 1,05 (с, 3 Н), 1,18 (д, J=6,4 Гц, 3 Н), 1,72 (с, 3 Н), 1,83 (с,3 Н), 2,19 (с, 3 Н), 2,25-2,36 (м, 1 Н), 2,63-2,75 (м, 1 Н), 2,76-2,95 (м, 2 Н), 3,52-3,73 (м, 1 Н), 4,57-4,81 (м, 2 Н), 5,65 (м, 1 Н), 6,40 (м, 1 Н), 6,76 (м, 1 Н), 8,13-8,31 (м, 1 Н), 8,53 (с, 1 Н). Масс-спектр: вычислено для C22H32FN8O (M+H): 443. Найдено: 443. Пример D1. N-(2-Этил-5-фторпиримидин-4-ил)-6,6-диметил-5-[(2S)-2,4,5,5-тетраметилпиперазин 1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. В продутую азотом колбу, содержащую соединение D1(i) (54 мг, 0,12 ммоль), добавляли Pd/C(6,5 мг, 0,006 ммоль) и МеОН (3 мл). Затем колбу накачивали баллонным Н 2 и реакционную смесь перемешивали в течение 16 ч. Неочищенную смесь выливали на слой целита и промывали МеОН (30 мл), а затем концентрировали и очищали колоночной хроматографией с использованием 1-3% 7 н. NH3/MeOH вCH2Cl2 с получением указанного в заголовке соединения D1 в виде твердого вещества бледно-желтого цвета (33 мг, 61%). ЯМР-данные представлены ниже в таблице. Пример Е 1. Ацетатная соль N-(2-этил-5-фторпиримидин-4-ил)-6,6-диметил-5-[(2S,5R)-2,4,5 триметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина Промежуточное соединение E1(i). трет-Бутил 3-[(5-фтор-2-винилпиримидин-4-ил)амино]-6,6 диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1H)-карбоксилат. В снабженную перегородкой герметично закрывающуюся пробирку добавляли трет-бутил 3-[(2 хлор-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-5(1 Н)-карбоксилат(2,00 г, 5,22 ммоль), 4,4,5,5-тетраметил-2-винил-1,3,2-диоксаборолан (2,41 г, 15,7 ммоль), Na2CO3 (1,66 г,15,7 ммоль) и продукт присоединения дихлор[1,1'-бис-(дифенилфосфино)ферроцен]палладия(II) дихлорметана (850 мг, 1,04 ммоль), а затем DME (50 мл) и воду (12 мл). Реакционную смесь продували аргоном в течение 2 мин, а затем помещали на предварительно нагретую до 100 С масляную баню и перемешивали в течение 16 ч. Неочищенную реакционную смесь концентрировали, а затем растворяли в EtOAc(100 мл) и промывали водой (50 мл). Раствор EtOAc сушили (MgSO4), фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле с использованием 1-10% 7 н.NH3/МеОН в CH2Cl2, в результате чего получали нужное соединение E1(i) в виде твердого вещества оранжевого цвета (1,8 г, 91%). 1(241 мг, 0,277 ммоль) и МеОН (30 мл). Колбу накачивали баллонным Н 2 и реакционную смесь перемешивали в течение 16 ч. Неочищенную смесь выливали на слой целита и промывали МеОН (75 мл), а затем концентрировали. Твердое вещество темно-коричневого цвета растирали с Et2O (35 мл) и получали указанное в заголовке соединение E1(ii) в виде твердого вещества не совсем белого цвета (1,02 г, 60%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 1,21 (т, J=7,5 Гц, 3 Н), 1,40-1,49 (м, 9 Н), 1,52-1,65 (м, 6 Н), 2,602,87 (м, 2 Н), 4,37-4,68 (м, 2 Н), 8,01-8,55 (м, 1 Н), 9,75-10,42 (м, 1 Н), 11,60-12,54 (м, 1 Н). Масс-спектр: вычислено для C18H26FN6O2 (M+H): 377. Найдено: 377. Промежуточное соединение E1(iii). 5-трет-Бутил 1-этил-3-[(2-этил-5-фторпиримидин-4-ил)амино]6,6-диметил-4,6-дигидропирроло[3,4-с]пиразол-1,5-дикарбоксилат. Раствор соединения E1(ii) (1,00 г, 2,66 ммоль) в ТГФ (40 мл) и DIPEA (858 мг, 6,64 ммоль) охлаждали на ледяной бане. Затем добавляли этилхлорформиат (317 мг, 2,92 ммоль), и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Реакцию гасили водой (50 мл), и смесь экстрагировали EtOAc (2100 мл). Объединенные экстракты промывали насыщенным раствором соли (50 мл), а затем сушили (MgSO4), фильтровали и концентрировали. Очистку проводили с помощью колоночной хроматографии на силикагеле с использованием 0-40% EtOAc в CH2Cl2, в результате чего получали указанное в заголовке соединение E1(iii) в виде твердого вещества желтого цвета (879 мг,74%). 1 Н-ЯМР (300 МГц, CDCl3):м.д. 1,28-1,34 (м, 3 Н), 1,47 (т, J=7,2 Гц, 3 Н), 1,50-1,56 (м, 9 Н), 1,79-1,90(м, 6 Н), 2,75-2,87 (м, 2 Н), 4,45-4,56 (м, 2 Н), 4,74-4,77 (м, 2 Н), 7,65 (с, 1 Н), 8,06-8,31 (м, 1 Н). Масс-спектр: вычислено для C21H30FN6O4 (M+H): 449. Найдено: 449. Промежуточное соединение E1(iv). Этил 5-(хлоркарбонил)-3-[(2-этил-5-фторпиримидин-4 ил)амино]-6,6-диметил-5,6-дигидропирроло[3,4-с]пиразол-1(4 Н)-карбоксилат. К суспензии соединения E1(iii) (879 мг, 1,96 ммоль) в диоксане (5 мл) добавляли HCl (10 мл, 4 М в диоксане). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, а затем концентрировали. Дигидрохлоридную соль растворяли в CH2Cl2 (40 мл), и добавляли DIPEA (1,27 г, 9,8 ммоль),а затем реакционную смесь охлаждали до -78 С и через капельную воронку в течение 15 мин медленно добавляли трифосген (0,41 г, 1,37 ммоль) в растворе CH2Cl2 (10 мл). Реакцию гасили водой при -78 С, а затем реакционную смесь нагревали до комнатной температуры. Смесь доводили до рН 8-9 добавлениемNaHCO3 и экстрагировали CH2Cl2 (215 мл). Объединенные экстракты промывали насыщенным раствором соли (15 мл), а затем сушили, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле с использованием 0-3% 7 н. NH3/MeOH в CH2Cl2, в результате чего получали указанное в заголовке соединение E1(iv) в виде твердого вещества белого цвета (244 мг,31%). 1 Н-ЯМР (300 МГц, ДМСО-d6):м.д. 1,19-1,30 (м, 3 Н), 1,32-1,42 (м, 3 Н), 1,69 (с, 6 Н), 2,67-2,95 (м,2 Н), 4,47 (кв, J=7,0 Гц, 2 Н), 4,66-5,51 (м, 2 Н), 8,27-8,76 (м, 1 Н), 9,73-10,36 (м, 1 Н). Масс-спектр: вычислено для C17H21ClFN6O3 (M+H): 411. Найдено: 411. Пример E1. Ацетатная соль N-(2-этил-5-фторпиримидин-4-ил)-6,6-диметил-5-[(2S,5R)-2,4,5 триметилпиперазин-1-ил]карбонил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амина. В герметично закрывающуюся пробирку добавляли соединение E1(iv) (200 мг, 0,487 ммоль), DIPEA(283 мг, 2,19 ммоль) и ТГФ (10 мл). Пробирку помещали на масляную баню при 90 С и нагревали в течение 16 ч. Реакционную смесь концентрировали, а затем растворяли в МеОН (5 мл) и Et3N (5 мл) и перемешивали при комнатной температуре в течение 16 ч. Затем проводили препаративную ВЭЖХ с использованием 5-50% ACN (0,1% АсОН), в результате чего получали указанное в заголовке соединение Е 1 в виде ацетатной соли (61 мг, 26%). ЯМР-данные представлены ниже в таблице. Примеры Е 2 и Е 3. Соединения примеров Е 2 и Е 3 получали методами, аналогичными методам, описанным выше в примере Е 1. Названия соединений и их ЯМР-данные представлены ниже в таблице.(10 мл) и TEA (1,0 мл) добавляли 3,3,3-трифторпропиональдегид (1,62 г, 14,4 ммоль) и АсОН (0,826 мл,14,4 ммоль), а затем цианоборгидрид натрия (0,907 г, 14,4 ммоль). Реакционную смесь перемешивали в течение 2 ч, а затем реакцию гасили водой (15 мл), после чего смесь подщелачивали NaHCO3 и экстрагировали EtOAc. Органический слой промывали насыщенным раствором соли, сушили и концентрировали в вакууме с получением порошка белого цвета (2,0 г, 90%). Масс-спектр: вычислено для C16H24N2F3 (M+H): 301. Найдено: 301. Промежуточное соединение E4(ii). (2R,5S)-2,5-Диметил-1-(3,3,3-трифторпропил)пиперазин. К раствору соединения E4(i) (1,90 г, 6,30 ммоль) в МеОН (20 мл) добавляли Pd(OH)2 (1,00 г,7,10 ммоль). Из суспензии откачивали воздух, а затем ее снова заполняли водородом (3), после чего оставляли на 15 ч для перемешивания в атмосфере водорода. Суспензию фильтровали через целит, а затем осадок на фильтре промывали CH2Cl2 и фильтрат концентрировали с получением твердого вещества белого цвета (1,3 г, 96%). Масс-спектр: вычислено для C9H18N2F3 (М+Н): 211. Найдено: 211. Пример Е 4. 5-[(2S,5R)-2,5-Диметил-4-(3,3,3-трифторпропил)пиперазин-1-ил]карбонил-N-(2-этил 5-фторпиримидин-4-ил)-6,6-диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. Соединение примера E4 получали методами, аналогичными методам, описанным выше в примереE1, за исключением того, что вместо гидрохлорида (2R,5S)-1,2,5-триметилпиперазина использовали промежуточное соединение E4(ii). ЯМР-данные представлены ниже в таблице. Боковая цепь S)-метил-2-(бензиламино)пропаноата) E5(ii). К смеси соединения Е 5(i) (27,7 г, 0,2 моль) в CH3CN (350 мл) при 0 С порциями добавляли K2CO3, а затем по каплям в течение 1 ч добавляли BnBr (34,2 г, 0,2 моль). После добавления смесь перемешивали при комнатной температуре в течение 2 ч. ТСХ (петролейный эфир/этилацетат=4/1) указывала на завершение реакции. Реакцию гасили водой (500 мл) и смесь экстрагировали этилацетатом (400 мл 3). Объединенные органические слои сушили над Na2SO4. После фильтрации и концентрирования получали неочищенное вещество, которое очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат=50/1-4/1), в результате чего получали соединение Е 5(ii) (15 г, 39%) в виде бесцветной жидкости. 1(м, 5 Н). Промежуточное соединение Е 5(iv). R)-2-(трет-бутоксикарбонил)-4-метокси-4-оксобутановая кислота). К перемешиваемому раствору соединения Е 5(iii) (73,5 г, 0,5 моль) в ТГФ (500 мл) и воде (500 мл) добавляли Na2CO3 (106 г, 1 моль), а затем Boc2O (120 г, 0,55 моль). После этого смесь перемешивали при комнатной температуре в течение ночи. ТСХ (дихлорметан/метанол=10/1) указывала на завершение реакции. Реакционную смесь концентрировали, и остаток растворяли в воде (200 мл) и экстрагировали этилацетатом. (500 мл 2). Водный слой отделяли и подкисляли 1 н. HCl до рН 5, а затем экстрагировали этилацетатом (500 мл 20). Объединенные органические слои сушили над Na2SO4. После фильтрации и концентрирования получали соединение Е 5(iv) (75 г, 61%) в виде масла.H-ЯМР (CDCl3) 1,4 (с, 9 Н), 2,8 (дд, 1H), 3,0 (дд, 1 Н), 3,6 (с, 3 Н), 4,6 (м, 1 Н), 5,5 (д, 1 Н), 9,3 (ушир, 1 Н). Промежуточное соединение Е 5(v). (R)-Метил 4-(бензилR)-1-метокси-1-оксопропан-2-ил)амино)-3(трет-бутоксикарбонил)-4-оксобутаноат. К размешиваемому раствору соединения Е 5(iv) (117,5 г, 0,475 моль) в ДМФ (1,5 л) последовательно добавляли NMM (80,1 г, 0,792 моль) и HATU (150,6 г, 0,396 моль). Смесь перемешивали при 0 С в течение 30 мин, а затем по каплям добавляли соединение 9 (76,5 г, 0,396 моль). Полученный раствор перемешивали при комнатной температуре в течение ночи. ТСХ (петролейный эфир/этилацетат=1/1) показала, что реакция была завершена. Реакционный раствор выливали в воду (1 л) и экстрагировали этилацетатом (1 л 3). Объединенные органические слои промывали 1 н. HCl (200 мл 2), водным NaHCO3(200 мл 2) и насыщенным раствором соли (400 мл), а затем сушили над Na2SO4 и концентрировали с получением соединения Е 5(v) (152 г, 91%) в виде масла, которое использовали в следующей стадии без дополнительной очистки. Промежуточное соединение(R)-2-R)-2-Амино-N-бензил-4-метокси-4 оксобутанамидо)пропановая кислота. К перемешиваемому раствору соединения Е 5(v) (152 г, 0,362 моль) в диоксане (200 мл) добавлялиHCl (г) в диоксане (1 л). Раствор перемешивали при комнатной температуре в течение ночи. Затем раствор концентрировали с получением соединения Е 5(vi) (100 г, 94%) в виде масла, которое использовали в следующей стадии без дополнительной очистки. Промежуточное соединение E5(vii). Метил-2 2R,5S)-4-бензил-5-метил-3,6-диоксопиперазин-2 ил)ацетат). Соединение Е 5(vi) (110 г, 0,307 моль) растворяли в смеси дихлорметан/вода (1 л/500 мл). Затем при перемешивании по каплям добавляли водный NaHCO3 до рН 9, после чего раствор перемешивали при комнатной температуре в течение 1 ч. Дихлорметановый слой отделяли, а водный слой экстрагировали дихлорметаном (300 мл 2). Объединенные органические слои сушили над Na2SO4. После фильтрации и выпаривания получали соединение Е 5(vii) (95 г, 99%) в виде масла. 1(0,24 г, 5 ммоль). Смесь перемешивали в течение 20 мин, а затем по каплям добавляли CH3I (0,85 г,6 ммоль) в ТГФ (10 мл). Затем смесь перемешивали при комнатной температуре в течение 5 ч. ТСХ (петролейный эфир/этилацетат=1/1) указывала на завершение реакции. Реакцию гасили водой (20 мл), и смесь экстрагировали этилацетатом (30 мл 2). Объединенные органические слои сушили над Na2SO4. После фильтрации и концентрирования получали неочищенное вещество, которое очищали колоночной хроматографией (элюент: петролейный эфир/этилацетат=5/1) и получали соединение Е 5(viii) (0,7 г, 46%) в виде масла. 1LiAlH4 (7,6 г, 0,2 моль). После добавления реакционную смесь кипятили с обратным холодильником в течение 36 ч. ТСХ (дихлорметан/метанол=10/1) указывала на завершение реакции. Реакцию гасили водой(5 мл), а затем смесь фильтровали, и осадок на фильтре несколько раз промывали этилацетатом. Фильтрат промывали Na2SO4. После фильтрации и концентрирования получали неочищенное вещество, которое очищали колоночной хроматографией (элюент: дихлорметан/метанол=100/1-10/1) с получением рацемического соединения Е 5(ix) (8,3 г, 84%) в виде масла. Промежуточное соединение Е 5(х) - изомеры А и В. 2-2R,5S)-4-Бензил-1,5-диметилпиперазин-2 ил)этанол и 2-2S,5S)-4-бензил-1,5-диметилпиперазин-2-ил)этанол. Рацемическое соединение Е 5(ix) разделяли с помощью SFC на колонке AS-H (0,462,5 см 5 мкм) с 5% МеОН (0,025% DEA) и 95% СО 2, используемого в качестве подвижной фазы, в результате чего получали соединения Е 5(х) - изомеры А и В (5,2 г и 1,9 г соответственно). Е 5(х) - изомер А: 1 Промежуточные соединения E5(xi) - изомеры А и В: 2-2R,5S)-1,5-диметилпиперазин-2-ил)этанол и 2-2S,5S)-1,5-диметилпиперазин-2-ил)этанол. Смесь соединения Е 5(х) - изомера А (5,2 г, 0,021 моль) и Pd/C (0,5 г) в МеОН (40 мл) или смесь соединения Е 5(х) - изомера В (1,9 г, 7,7 ммоль) и Pd/C (0,2 г) в МеОН (40 мл) в атмосфере Н 2 под давлением 50 фунт/кв.дюйм (35150 кг/м 2) перемешивали при комнатной температуре в течение ночи. ТСХ (дихлорметан/метанол=10/1) указывала на завершение реакции. Реакционную смесь фильтровали и фильтрат концентрировали с получением Е 5(xi) - изомера А (3,1 г, 94%) в виде твердого вещества не совсем белого цвета или Е 5(xi) - изомера В (1,2 г, 93%) в виде твердого вещества не совсем белого цвета. Е 5(xi) - изомер А: 1(м, 1 Н), 3,9 (м, 1 Н). Пример Е 5. 2-5S)-4-[3-[(2-этил-5-фторпиримидин-4-ил)амино]-6,6-диметил-4,6 дигидропирроло[3,4-с]пиразол-5(1H)-ил]карбонил-1,5-диметилпиперазин-2-ил)этанол. Указанное в заголовке соединение получали методом, аналогичным методу, описанному выше в примере Е 1, за исключением того, что вместо 2R,5S)-1,2,5-триметилпиперазина использовали Е 5(xi)изомер А. ЯМР-данные представлены ниже в таблице. Пример F1. 5-[(4-Фтор-1-метилпиперидин-4-ил)карбонил]-N-(5-фтор-2-метилпиримидин-4-ил)-6,6 диметил-1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-аминHCl в 1,4-диоксане (20 мл). Раствор перемешивали в течение 1 ч, а затем летучие вещества удаляли в вакууме с получением нужного продукта F1(i) в виде твердого вещества белого цвета (4,8 г, 91%). Масс-спектр: вычислено для C13H21N4O4 (M+H): 297. Найдено: 297. Промежуточное соединение F1(ii). Этил 3-[(этоксикарбонил)амино]-5-[(4-фтор-1-метилпиперидин 4-ил)карбонил]-6,6-диметил-5,6-дигидропирроло[3,4-с]пиразол-1(4 Н)-карбоксилат. В сосуд под давлением добавляли соединение F1(i) (4,80 г, 16,2 ммоль), 4-фтор-1-метилпиперидин 4-карбонилхлорид (2,45 г, 13,6 ммоль), диизопропилэтиламин (9,06 мл, 52,0 ммоль) и ТГФ (300 мл). Суспензию нагревали при 80 С в течение 15 ч. Летучие вещества удаляли в вакууме и получали нужный продукт F11(ii) в виде пены коричневого цвета (5,6 г, 98%). Масс-спектр: вычислено для C20H31N5O5F (M+H): 440. Найдено: 440. Промежуточное соединение F1(iii). 5-[(4-Фтор-1-метилпиперидин-4-ил)карбонил]-6,6-диметил 1,4,5,6-тетрагидропирроло[3,4-с]пиразол-3-амин. В микроволновый сосуд добавляли соединение F1(ii) (9,80 г, 22,0 ммоль) в растворе МеОН (35 мл) и LiOH (2,14 г, 89,2 ммоль). Реакционную смесь нагревали при 110 С в микроволновой печи в течение 2 ч. Реакционную смесь концентрировали в вакууме и получали нужный продукт F1(iii) в виде пены желтовато-коричневого цвета (4,4 г, 67%). Масс-спектр: вычислено для C14H23N5OF (M+H): 296. Найдено: 296.
МПК / Метки
МПК: A61P 3/00, C07D 487/04, A61K 31/4162
Метки: ркс, производные, качестве, ингибиторов, 3-аминопирроло[3,4-c]пиразол-5(1h,4h,6h)-карбальдегида
Код ссылки
<a href="https://eas.patents.su/30-17937-proizvodnye-3-aminopirrolo34-cpirazol-51h4h6h-karbaldegida-v-kachestve-ingibitorov-rks.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 3-аминопирроло[3,4-c]пиразол-5(1h,4h,6h)-карбальдегида в качестве ингибиторов ркс</a>
Производные пиразол-4-илбензоила и их применение в качестве гербицидов.

Номер патента: 753
Опубликовано: 24.04.2000
Авторы: Вестфален Карл-Отто, Фоссен Маркус, Плат Петер, Гётц Норберт, Фон Дейн Вольфганг, Хилл Регина Луиза, Вальтер Хельмут, Кардорфф Увэ, Энгель Штефан, Оттен Мартина
МПК: C07D 409/06, A01N 43/56
Метки: пиразол-4-илбензоила, качестве, гербицидов, производные, применение
Формула / Реферат:
1. Производные пиразол-4-илбензоила формулы I в которой заместители имеют следующие значения: L, М представляют собой водород, С1-С6алкил, С2-С6алкенил, С2-С6алкинил, С1-C4алкокси, причем эти группы необязательно могут быть замещены одним-пятью атомами галогена или C1-C4алкоксигруппой; галоген, циано, нитро, группу -(А)m-S(O)nR1 или группу -(А)m-СО-R2; Y представляет собой группу, включающую С=O, C=N-R3, CR7-NR5R6, CR7-OR8, CR10R11,...
Производные пиразол-4-илгетароила в качестве гербицидов.

Номер патента: 1515
Опубликовано: 23.04.2001
Авторы: Гётц Норберт, Фон Деин Вольфганг, Вальтер Хельмут, Плат Петер, Мисслитц Ульф, Энгель Штефан, Вестфален Карл-Отто, Фоссен Маркус, Оттен Мартина, Кардорфф Увэ, Хилл Регина Луизе
МПК: A01N 43/56, C07D 411/06
Метки: производные, качестве, пиразол-4-илгетароила, гербицидов
Формула / Реферат:
1. Производные пиразол-4-илгетароила формулы I в которой заместители имеют следующие значения: L, М обозначают водород, С1-С6алкил, С2-С6алкенил, С2-С6алкинил, С1-С4алкокси, причем эти группы необязательно могут быть замещены одним-пятью атомами галогена или С1-С4алкоксигруппой; галоген, циано, нитро; X обозначает кислород или серу, которая может быть замещена одним либо двумя кислородами; n обозначает 0, 1, 2; R1 обозначает водород,...
Производные пиразол-4-илбензоила и их применение в качестве гербицидов

Номер патента: 1228
Опубликовано: 25.12.2000
Авторы: Мисслитц Ульф, Хилл Регина Луизе, Фоссен Маркус, Оттен Мартина, Кениг Хартманн, Плат Петер, Вальтер Хельмут, Ранг Харальд, Вестфален Карл-Отто, Энгель Штефан, Фон Дайн Вольфганг, Харреус Альбрехт, Кардорфф Увэ
МПК: A01N 43/56, C07D 409/10
Метки: пиразол-4-илбензоила, производные, гербицидов, качестве, применение
Формула / Реферат:
1. Производные пиразол-4-илбензоила формулы I в которой заместители имеют следующие значения: L обозначает водород, галоген, нитрогруппу, С1-С4алкилсульфонил; М обозначает водород, С1-С4алкил, галоген, С1-С4алкилтиогруппу; Z обозначает 5-членный гетероциклический остаток, содержащий от одного до двух гетероатомов, выбранных из группы, включающей кислород, серу или азот, который необязательно замещен С1-С4алкилом; Q обозначает присоединенное в...
Производные n-(пиразол-3-ил)бензамида в качестве активаторов глюкокиназы

Номер патента: 17114
Опубликовано: 28.09.2012
Авторы: Байер Норберт, Глайтц Иоганнес, Карниато Денис, Эмде Ульрих, Бургдорф Ларс Торе, Шарон Кристин
МПК: A61P 3/10, A61P 3/04, A61K 31/4155...
Метки: активаторов, качестве, производные, глюкокиназы, n-(пиразол-3-ил)бензамида
Формула / Реферат:
1. Соединения, выбранные из группыи их фармацевтически пригодные соли и стереоизомеры, включая их смеси во всех соотношениях.2. Лекарственное средство, обладающее активностью активаторов глюкокиназы, содержащее по меньшей мере одно соединение в соответствии с п.1 и/или его фармацевтически пригодные соли и стереоизомеры, включая их смеси во всех соотношениях, и необязательно наполнители и/или вспомогательные...
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Гадек Томас, Карниато Дени, Кнолле Йохен, Макдауэлл Роберт, Шойнеманн Карлхайнц, Вилл Дэвид Вильям, Бодари Сара Кэтрин, Пейман Ануширван, Гурвест Жан-Франсуа, Катбертсон Роберт Эндрю
МПК: A61P 19/10, A61K 31/505, C07D 239/42...
Метки: ингибиторов, клеток,способ, адгезии, композиция, применение, сульфонамидные, производные, костной, фармацевтическая, ткани, рассасывания, получения, качестве
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...