Замещенные пиразолохиназолиновые производные, способ их получения и их применение в качестве ингибиторов киназы
Номер патента: 17769
Опубликовано: 29.03.2013
Авторы: Постери Элена, Карузо Микеле, Фергюсон Рон, Берия Итало, Браска Мария Габриелла, Вальсазина Барбара

















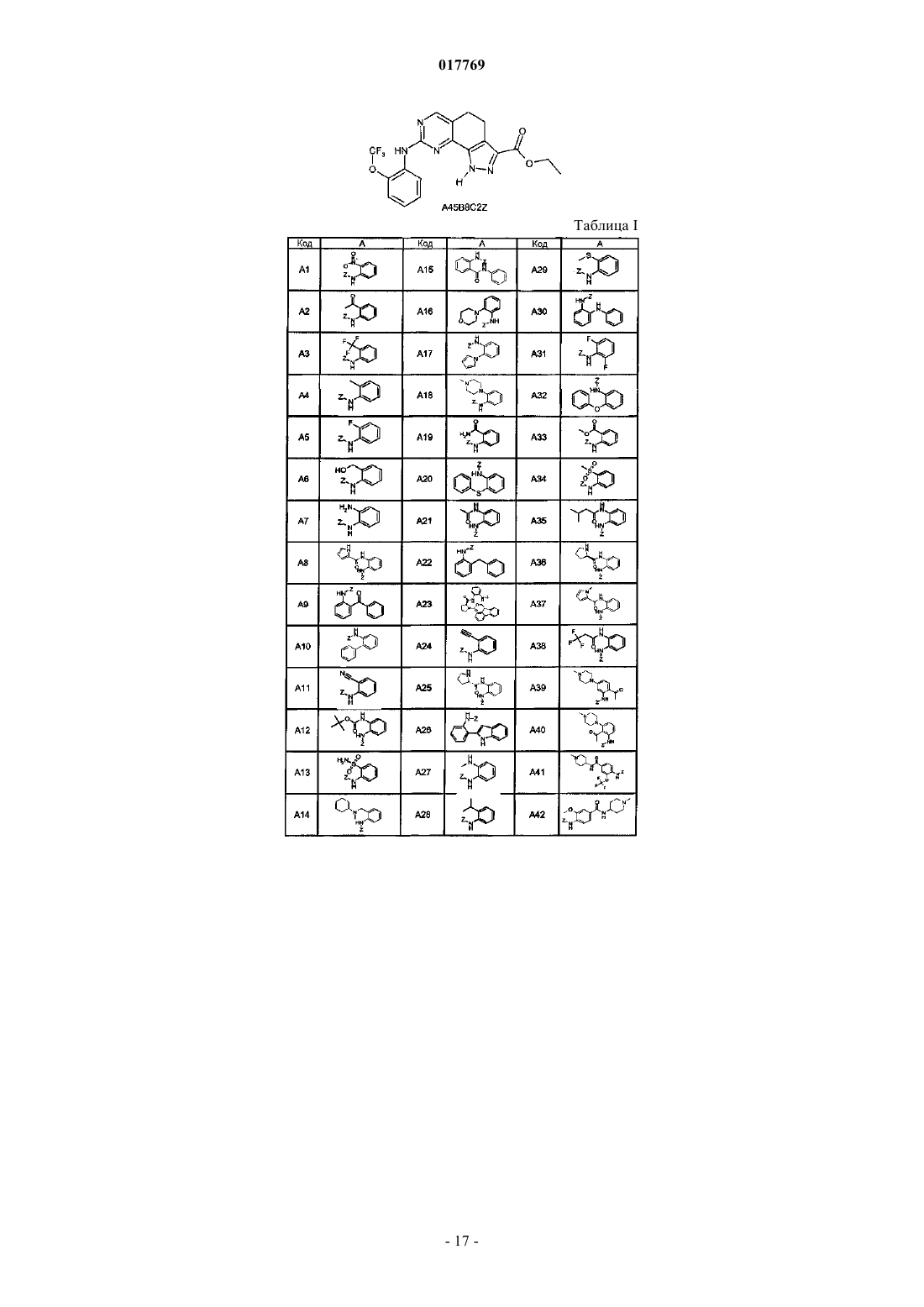
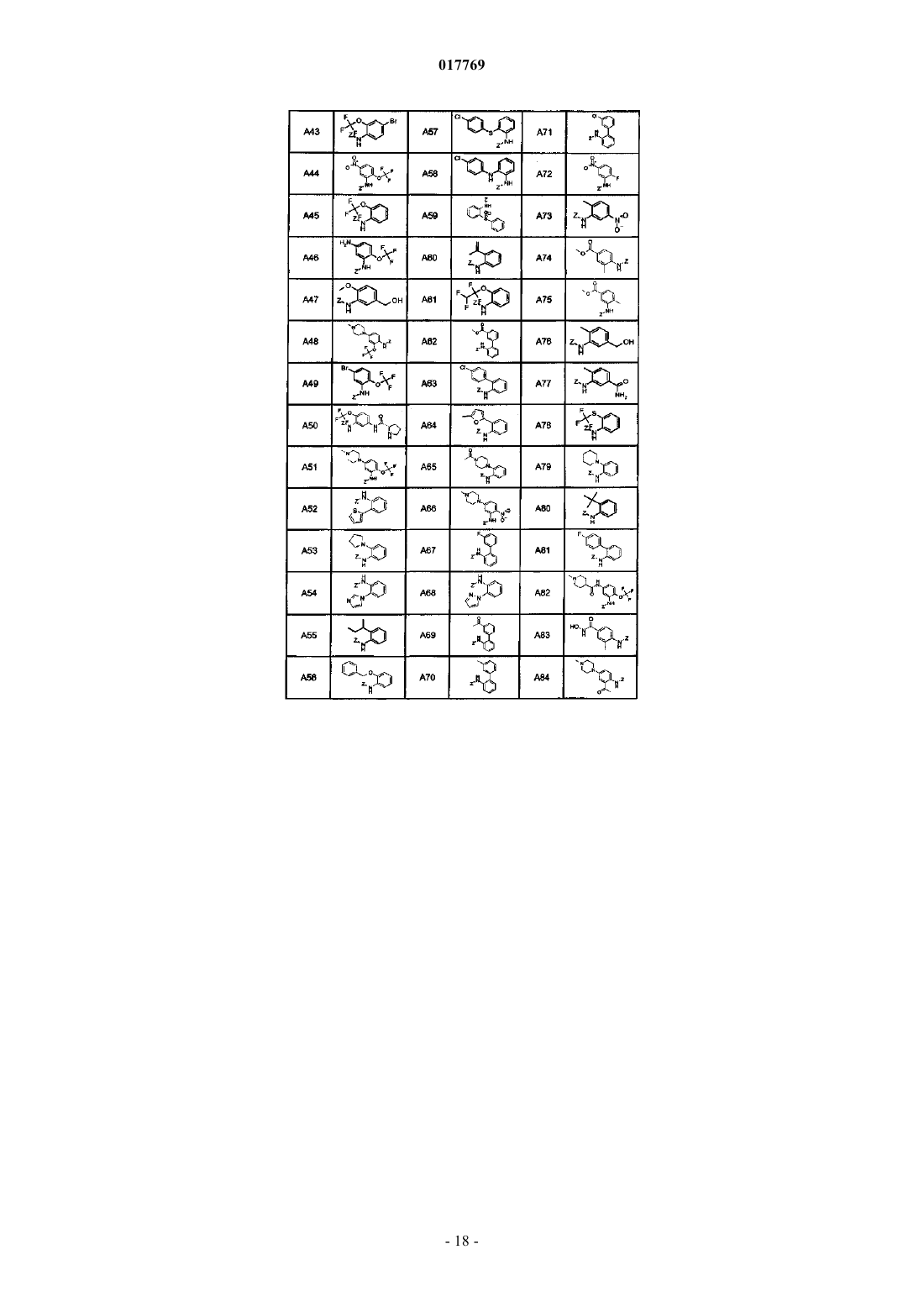
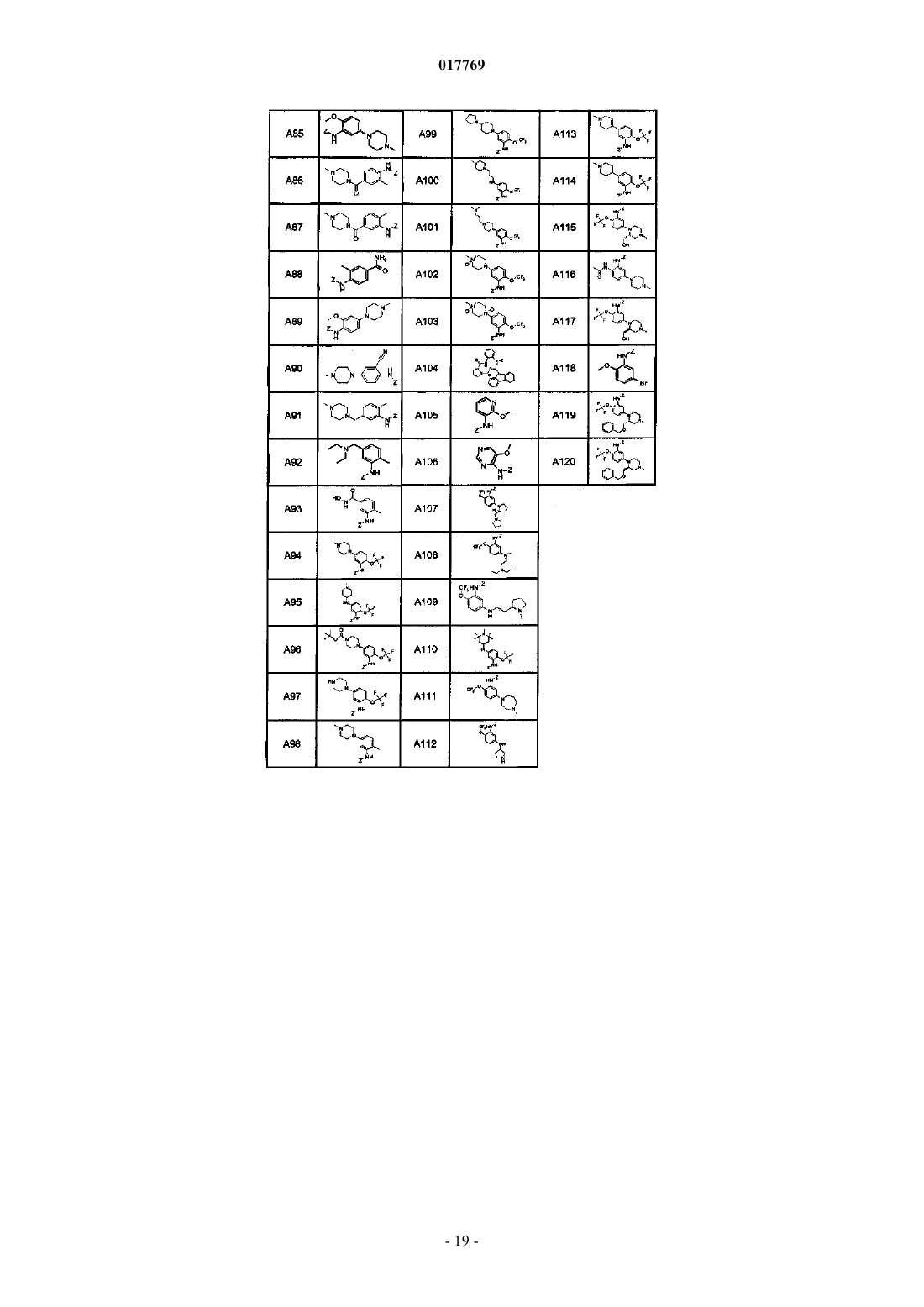
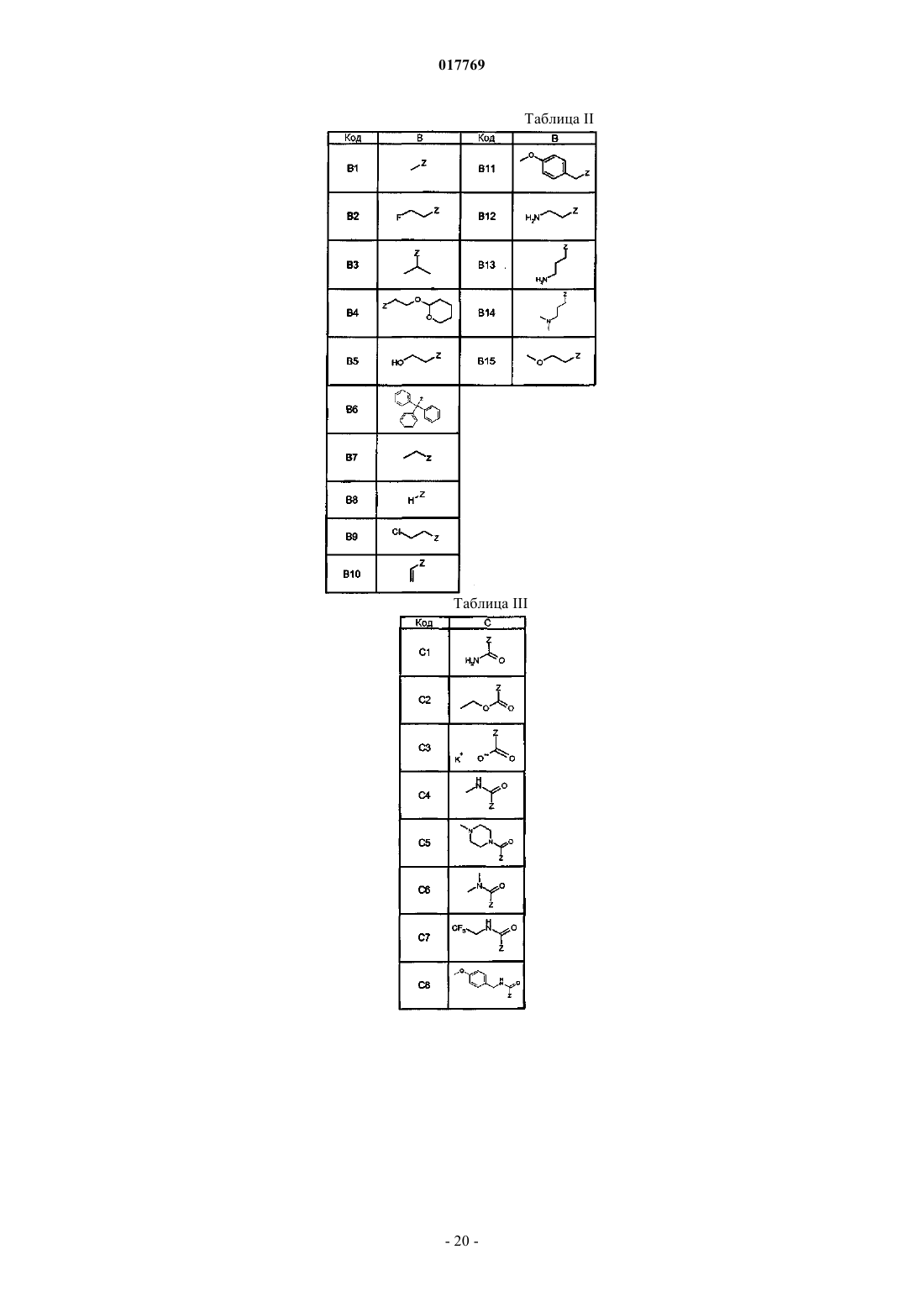







Формула / Реферат
1. Соединение формулы (I)

где R1 представляет собой ортозамещенный ариламино формулы

где R'4 и R''4 независимо выбраны из группы, включающей галоген, нитро, циано, C1-C6-алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, С3-С6-циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, алкилкарбонилокси, арилкарбонилокси, гетероциклилкарбонилокси, карбокси, алкоксикарбонил, гетероциклилоксикарбонил, амино, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино, формил, алкилкарбонил, арилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, ариламиносульфонил, арилтио и алкилтио;
R2 представляет собой водород, C1-C6-алкил с линейной или разветвленной цепью, необязательно замещенный галогеном, гетероциклилокси, гидрокси, арилом, алкоксиарилом, амино, алкиламино, диалкиламино или алкокси, или представляет собой С2-С6-алкенил с линейной или разветвленной цепью; и
R3 представляет собой CO-OR' или CO-NR'R", где R' и R" представляют собой, каждый независимо, водород, C1-C6-алкил с линейной или разветвленной цепью, необязательно замещенный галогеном или алкоксиарилом; или R' и R", взятые вместе с атомом азота, с которым они связаны, могут образовывать гетероциклильную группу, необязательно содержащую один дополнительный атом азота и необязательно замещенную алкилом;
где любая алкильная группа представляет собой C1-C6-алкил;
любая алкоксигруппа представляет собой C1-C6-алкокси;
любая алкенильная группа представляет собой С2-С6-алкенил;
любая алкинильная группа представляет собой C1-C6-алкинил;
любая арильная группа представляет собой карбоциклическую или гетероциклическую группу, содержащую от 1 до 2 кольцевых групп, либо конденсированных, либо связанных друг с другом при помощи простых связей, в которых по меньшей мере одно из колец является ароматическим; любое ароматическое гетероциклическое кольцо, в случае его присутствия, также называемое гетероарильной группой, включает 5-6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из N, NH, О или S;
любой гетероциклил представляет собой 3-7-членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, в котором один или несколько углеродных атомов замещены гетероатомами, такими как азот, кислород и сера;
любой циклоалкил представляет собой 3-6-членное моноциклическое кольцо, состоящее только из углеродных атомов, которое может содержать одну или несколько двойных связей, но не имеет полностью сопряженной p-электронной системы;
и его фармацевтически приемлемые соли.
2. Соединение формулы (I) по п.1, где R3 представляет собой СО-ОН или CO-NRR", где R' и R" имеют значения, определенные в п.1.
3. Соединение формулы (I) по п.1 или 2, где R2 представляет собой C1-C6-алкил с линейной или разветвленной цепью, необязательно замещенный галогеном, гетероциклилокси, гидрокси, арилом, алкоксиарилом, амино, алкиламино, диалкиламино или алкокси, или представляет собой С2-С6-алкенил с линейной или разветвленной цепью.
4. Соединение формулы (I) по пп.1-3, где R3 представляет собой CO-NR'R", где R' и R" имеют значения, определенные в п.1.
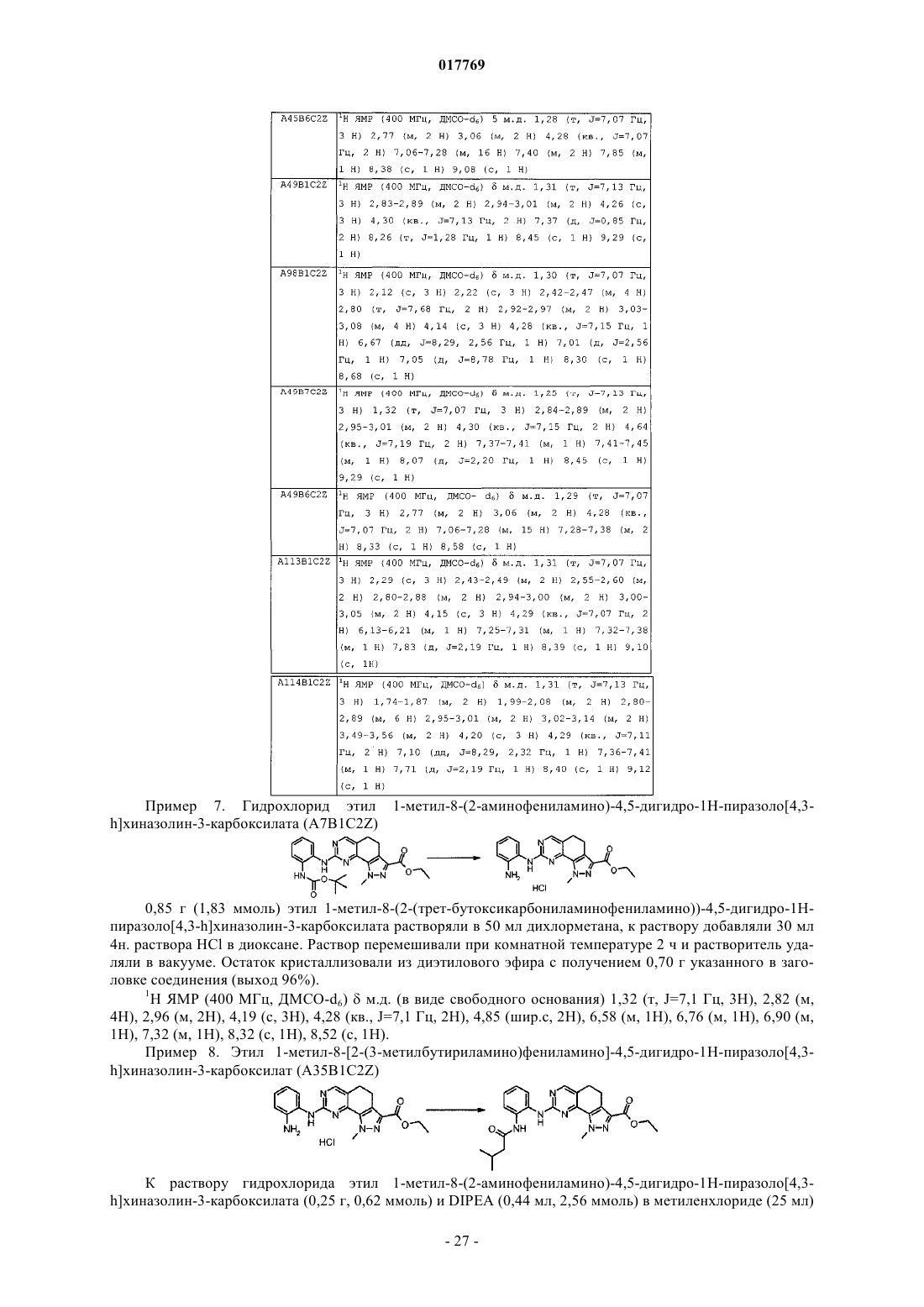
5. Соединение или его фармацевтически приемлемая соль, которое выбрано из группы, состоящей из
8-[2-ацетил-5-(4-метилпиперазин-1-ил)фениламино]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A39B1C1Z);
8-[2-ацетил-5-(4-метилпиперазин-1-ил)фениламино]-1-(2-фторэтил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A39B2C1Z);
1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B1C1Z);
этил 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксилата (A51B1C2Z);
1-метил-8-[2-метокси-5-(4-метилпиперазин-1-ил)фениламино]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A85B1C1Z);
8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-(2-фторэтил)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B2C1Z);
1-метил-8-[4-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A48B1C1Z);
1-метил-8-[2-трифторметокси-5-пиперазин-1-илфениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A97B1C1Z);
1-метил-8-[2-метил-5-(4-метилпиперазин-1-ил)фениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A98B1C1Z);
1-метил-8-[5-(4-пирролидин-1-илпиперидин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A99B1C1Z);
метиламид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A51B1C4Z);
метиламид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-метоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A85B1C4Z);
1-метил-8-[2-метил-5-(4-метилпиперазин-1-карбонил)фениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A87B1C1Z);
1-метил-8-[2-метил-4-(4-метилпиперазин-1-карбонил)фениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A86B1C1Z);
1-метил-8-{2-трифторметокси-5-[(1-метилпиперидин-4-карбонил)амино]фениламино}-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A82B1C1Z);
8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксилат калия (A51B1C3Z);
1-этил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B7C1Z);
(2,2,2-трифторэтил)амид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A51B1C7Z);
1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B5C1Z);
8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-винил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B10C1Z);
1-(2-хлорэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B9C1Z);
8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A51B8C1Z);
1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксилата калия (A51B5C3Z);
этил 1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксилата (A51B5C2Z);
1-метил-8-[5-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A113B1C1Z);
1-метил-8-[5-(1-метилпиперидин-4-ил)-2-трифторметоксифениламино]-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A114B1C1Z);
8-(5-бром-2-трифторметоксифениламино)-1-метил-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A49B1C1Z) и
8-(5-бром-2-трифторметоксифениламино)-4,5-дигидро-1Н-пиразоло[4,3-h]хиназолин-3-карбоксамида (A49B8C1Z).
6. Способ получения соединения формулы (I), включающий следующие стадии:
1) взаимодействие соединения формулы (II)

с гидразиновым производным формулы (III)
где R2 имеет значение, определенное в п.1, в присутствии уксусной кислоты с получением соединения формулы (IV)

где R2 имеет значение, определенное выше;
необязательно, алкилирование соединения формулы (IV), где R2 представляет собой водород, при помощи соединений формулы (V)
где Y представляет собой подходящую отщепляемую группу, такую как мезил, тозил, галоген, и R2 имеет значение, определенное выше, но отличное от водорода, с получением соединения формулы (IV), где R2 имеет значение, определенное выше, но отличное от водорода;
2) взаимодействие соединения формулы (IV) с диметилформамид-ди-трет-бутилацеталем или диметилформамид-диизопропилацеталем с получением соединения формулы (VI)

где R2 имеет значение, определенное выше; и
3) взаимодействие соединения формулы (VI) в соответствии с любой одной из альтернативных стадий 3а) или 3b):
3а) с гуанидином с получением соединения формулы (VII), где R2 имеет значение, определенное выше; преобразование аминогруппы полученного соединения формулы (VII) в йод и затем взаимодействие полученного йодпроизводного формулы (VIII) с ортозамещенным ариламином формулы R1-H (IX), где R1 имеет значение, определенное в п.1, с получением соединения формулы (I)

где R1 и R2 имеют значения, определенные выше;
3b) с гуанидиновым производным формулы (X)
где R1 имеет значение, определенное выше, с получением соединения формулы (I)

где R1 и R2 имеют значения, определенные выше, и, необязательно, преобразование его в другие производные формулы (I) и/или в их фармацевтически приемлемые соли.
7. Способ получения соединения формулы (I) по п.6, отличающийся тем, что соединение формулы (I) получают в соответствии со способом, который включает:
4) преобразование этоксикарбонильной группы соединения формулы (VIII), определенного в п.6, в соединение формулы (XIII) или соответствующую соль посредством кислотного или щелочного гидролиза; преобразование полученного соединения формулы (XIII) или соответствующей соли в соединение формулы (XIV) посредством реакции в щелочных условиях и в присутсвии подходящего агента конденсации с амином формулы R'R''-NH (XI), где R' и R'' имеют значения, определенные в п.1; взаимодействие соединения формулы (XIV) с ортозамещенным ариламином формулы R1-H (IX), где R1 имеет значение, определенное в п.1, с получением соединения формулы (I)

где R1, R2, R' и R'' имеют значения, определенные выше, и, необязательно, его преобразование в другие производные формулы (I) и/или в их фармацевтически приемлемые соли.
8. Способ получения соединения формулы (I) по п.6 или 7, отличающийся тем, что необязательное преобразование соединения формулы (I) в другие соединения формулы (I) осуществляют при помощи одной или нескольких из следующих реакций:
а) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой аминокарбонил, путем обработки гидроксидом аммония

b) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой группу CO-NR'R'', путем обработки амином формулы R'R''-NH (XI), где R' и R'' имеют значения, определенные в п.1

с) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой группу СО-ОН или соответствующую соль посредством кислотного или щелочного гидролиза

d) преобразование соединения формулы (I), где R3 представляет собой СО-ОН или соответствующую соль, в соединения формулы (I), где R3 представляет собой группу CO-NR'R'', посредством реакции с амином формулы R'R''-NH (XI) в щелочных условиях и в присутствии подходящего агента конденсации, где R' и R'' имеют значения, определенные выше

е) преобразование соединения формулы (I), где R2 представляет собой тритил, в соединение формулы (I), где R2 представляет собой водород, в кислотных условиях

f) преобразование соединения формулы (I), где R2 представляет собой водород, в соединение формулы (I), где R2 имеет значение, определенное в п.1, но отличное от водорода, посредством реакции со спиртом формулы R2-OH (XII), где R2 имеет значение, определенное выше, но отличное от водорода

g) преобразование соединения формулы (I), где R2 представляет собой водород, в соединение формулы (I), где R2 имеет значение, определенное в п.1, но отличное от водорода, посредством реакции с соединением формулы R2-X (XV), где R2 имеет значение, определенное выше, но отличное от водорода, и X представляет собой галоген

h) преобразование соединения формулы (I), где R2 представляет собой галогенэтил, в соединение формулы (I), где R2 представляет собой винил

i) преобразование соединения формулы (I), где R1 представляет собой ортозамещенный ариламино формулы

где R'4 или R''4 представляет собой бром, в соединение формулы (I), где R'4 или R''4 представляет собой группу -NR'R'', путем обработки амином формулы R'R''-NH (XI), где R' и R'' имеют значения, определенные в п.1.
9. Способ лечения заболевания, вызванного и/или связанного с нарушенной регуляцией активности протеинкиназы, который включает введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I) по п.1, где заболевание представляет собой рак.
10. Фармацевтическая композиция, обладающая противораковой активностью, включающая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере один фармацевтически приемлемый эксципиент, носитель и/или разбавитель.
11. Набор, включающий соединение формулы (I) или его фармацевтически приемлемую соль или фармацевтические композиции этого соединения по п.10 и одно или несколько химиотерапевтических средств, в виде комбинированного препарата для одновременного, отдельного или последовательного применения в противораковой терапии.
12. Применение соединения формулы (I) или его фармацевтически приемлемой соли в качестве лекарственного средства, обладающего противораковой активностью.
13. Применение соединения формулы (I) или его фармацевтически приемлемой соли для получения лекарственного средства, обладающего противораковой активностью.
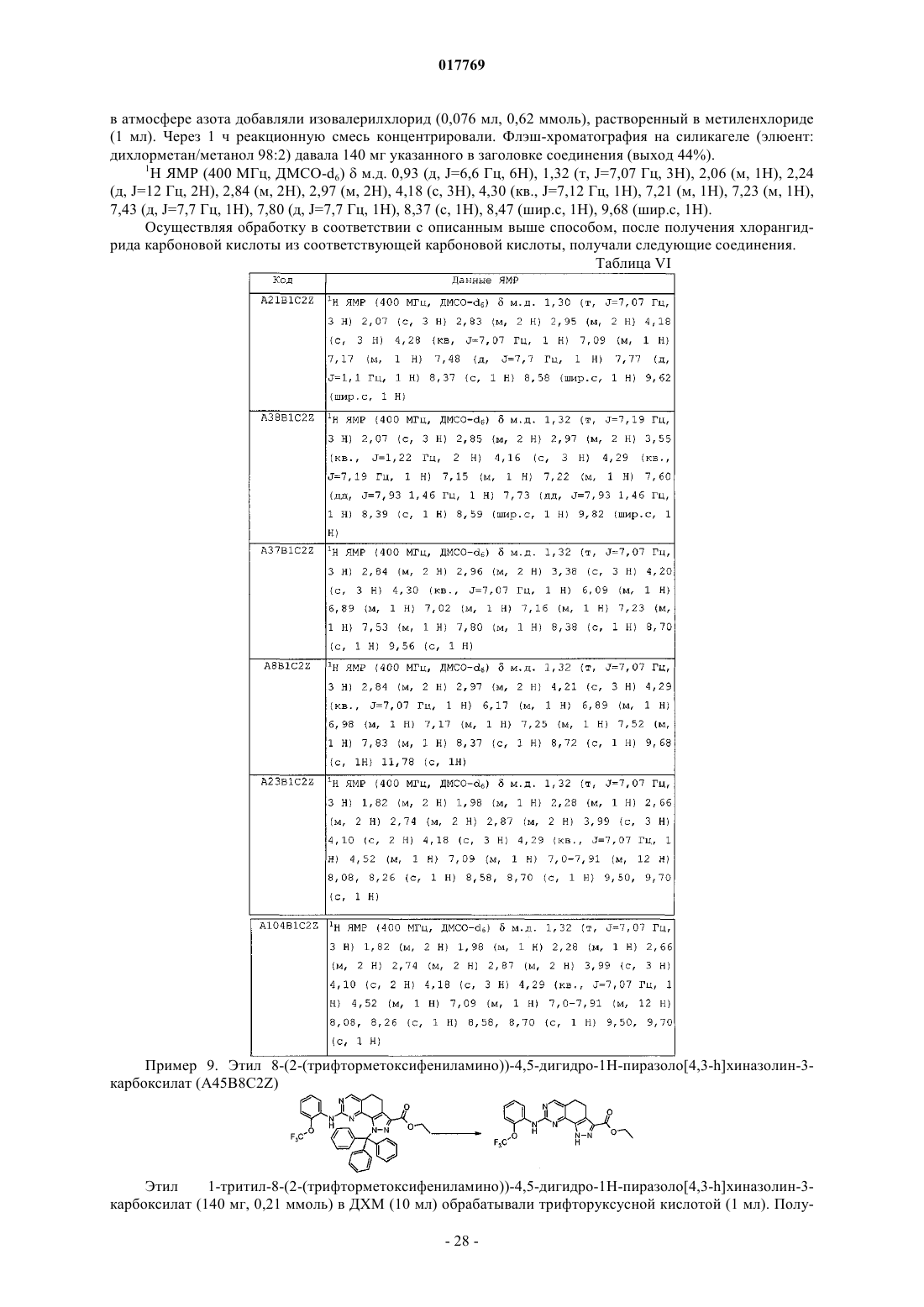
14. Промежуточное соединение формулы (IX')
где R1' представляет собой

15. Промежуточное соединение формулы (X')
где R1' представляет собой

Текст
ЗАМЕЩЕННЫЕ ПИРАЗОЛОХИНАЗОЛИНОВЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ Раскрыты пиразолохиназолиновые производные формулы (I), как определено в описании, и их фармацевтически приемлемые соли, способ их получения и содержащие их фармацевтические композиции; соединения по настоящему изобретению можно применять в терапии, при лечении заболеваний, связанных с нарушенной регуляцией активности протеинкиназы, например рака.(71)(73) Заявитель и патентовладелец: НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. (IT) 017769 Настоящее изобретение относится к конкретным замещенным пиразолохиназолиновым соединениям, которые модулируют активность протеинкиназ. Поэтому соединения по настоящему изобретению используют при лечении заболеваний, вызванных нарушенной регуляцией активности протеинкиназы. Настоящее изобретение также обеспечивает способы получения этих соединений, фармацевтические композиции, содержащие эти соединения, и способы лечения заболеваний с использованием фармацевтических композиций, содержащих эти соединения. Применение митотических ингибиторов в терапии раковых заболеваний является широко распространенной клинической стратегией для лечения широкого диапазона раковых заболеваний у человека. Таксаны (паклитаксел и доцетаксел) и алкалоиды барвинка (винкристин и винбластин) действуют либо путем стабилизации, либо дестабилизации микротрубочек с катастрофическими последствиями в клетках, проходящих через митоз. Они представляют собой первоочередную терапию для некоторых типов опухолей и вторую очередь при цисплатин-резистентном раке яичников, молочной железы, легкого, мочевого пузыря и пищевода (таксаны). Однако из-за роли микротрубочек в таких процессах, как движение клеток, фагоцитоз и аксональный транспорт, часто при использовании таких средств наблюдаются некоторые токсические эффекты, такие как периферическая невропатия. Прохождение через митоз обязательно для всех пролиферирующих клеток, и, следовательно, терапевтические методы лечения рака, которые нацелены на мишени в митозе, как правило, являются применимыми для широкого ряда различных опухолей. Некоторые протеинкиназы играют ключевые роли в гармоничном функционировании клеточного цикла, и некоторые из них уже являются объектом для прицельно действующей терапии в онкологии, включая Cdk-2 и Aurora-A. Правильность митоза имеет огромное значение, и некоторые"контрольные точки" существуют в нормальных клетках для поддержания целостности хромосом на протяжении клеточного цикла. Эти контрольные точки часто исчезают в процессе онкогенной трансформации, и это позволяет раковым клеткам выносить анэуплоидию и хромосомную нестабильность. Ингибирование митоза в опухолевых клетках с "подверженными риску контрольными точками" должно иметь катастрофические последствия, так как раковые клетки пытаются продвигать аберрантный митоз. Семейство Polo-подобных киназ, включающее 4 сериновых/треониновых киназы (Plk-1-4), преимущественно связано с вхождением в, прохождением через и выходом из митоза. Эти киназы характеризуются тем, что имеют N-концевой киназный домен и уникальный С-концевой "Polo-Box" домен. Этот домен является ответственным за нацеливание киназы на различные митотические структуры (центросомы, кинетохоры, полюсы веретен, группа гранул в плоскости экватора веретена), и временная и пространственная регуляция Plks имеют важное значение для прохождения через митоз (см. обзор van Vugt и Medema, Oncogene 2005, 24 (17):2844-59; Barr et al., Nat. Rev. Mol. Cell. Biol. 2004, 5 (6):429-40; Dai иCogswell, Prog. Cell. Cycle Res. 2003, 5:327-34; Glover et al., Genes Dev. 1998, 12(24):3777-87). Наиболее охарактеризованным членом этого семейства является Plk-1, и ее активность связана с некоторыми процессами в ходе митоза, включая G2/M переход, посредством регуляции Cdk-1 активности различными путями (активация Cdc25c, ядерная транслокация циклина В, инактивация Myt-1 и Wee-1) (Inoue et al.,EMBO J. 2005, 24(5):1057-67; van Vugt et al., J Biol. Chem. 2004, 9 (35):36841-54; Watanabe et al., Proc. Natl(15):4949-59; Qian et al., Mol. Biol. Cell. 2001, 12(6):1791-9; Roshak et al., Cell. Signal. 2000, 12(6):405-l 1); созревание и разделение центросомы; регуляцию хромосомально-плечевой когезии в профазе и отделение сестринской хроматиды при переходе метафаза/анафаза; активацию Анафаза Промотирующего Комплекса, чтобы дать начало митотическому выходу; цитокинез.Plk-1 чрезмерно экспрессируется в некоторых опухолевых клетках, включая карциному молочной железы, яичников, не-мелкоклеточную карциному легкого, карциному толстой кишки, головы и шеи,эндометриальные и эзофагеальные карциномы, и ее чрезмерную экспрессию часто соотносят с плохим прогнозом. Нарушение функции Plk-1 различными путями в опухолевых клетках (siPHK и антисмысловое удаление, доминантные негативные белки и иммуноистощение) приводит к аберрантному митозу с последующей митотической катастрофой, вызывая при этом "опосредованную контрольными точками" остановку клеточного цикла в нормальных клетках. Таким образом, фармакологическая аттенуация функцииPlk-1 может иметь терапевтическую пользу при лечении некоторых различных типов рака. Сущность изобретения Конденсированные бициклические пиримидиновые производные для лечения гиперпролиферативных заболеваний раскрыты в WO 96/40042 на имя Pfizer Inc. Конденсированные полициклические пиримидиновые производные в качестве ингибиторов протеинкиназы также раскрыты в WO 98/58926 и WO 98/28281, оба на имя Celltech Therapeutics Ltd. Конденсированные трициклические пиразольные соединения, известные из уровня техники в качестве ингибиторов протеинкиназы, раскрыты в WO 03/070236 и WO 03/070706 на имя Pharmacia Italia S.P.A. и Pharmacia Corp. соответственно. Пиразолохиназолиновые производные, обладающие активностью ингибирования киназы, также раскрыты в WO 04/104007, на имя самого заявителя. Некоторые специфические соединения указанного-1 017769 выше WO 04/104007 исключены из общей формулы, представленной в настоящей заявке. Несмотря на эти разработки, все еще остается потребность в эффективных средствах для лечения указанного заболевания. К настоящему времени авторами настоящего изобретения обнаружено, что соединения формулы (I), описанные ниже, являются ингибиторами киназ и, таким образом, являются полезными в терапии в качестве противоопухолевых средств, и они не имеют указанных выше недостатков,в том, что касается токсичности и побочных эффектов, которые связаны с существующими в настоящее время противоопухолевыми средствами. Соответственно первой целью настоящего изобретения является обеспечение замещенного пиразолохиназолинового соединения, представленного формулой (I) где R1 представляет собой ортозамещенный ариламино формулы: где R'4 и R4 независимо выбраны из группы, включающей галоген, нитро, циано, С 1-С 6-алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, С 3-С 6-циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, алкилкарбонилокси, арилкарбонилокси, гетероциклилкарбонилокси, карбокси, алкоксикарбонил, гетероциклилоксикарбонил, амино, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино,формил, алкилкарбонил, арилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, ариламиносульфонил, арилтио и алкилтио;R2 представляет собой водород, C1-C6-алкил с линейной или разветвленной цепью, необязательно замещенный галогеном, гетероциклилокси, гидрокси, арилом, алкоксиарилом, амино, алкиламино, диалкиламино или алкокси, или представляет собой С 2-С 6-алкенил с линейной или разветвленной цепью; иR3 представляет собой CO-OR' или CO-NR'R", где R' и R" представляют собой, каждый независимо,водород, C1-C6-алкил, необязательно замещенный галогеном или алкоксиарилом; или R' и R" взятые вместе с атомом азота, с которым они связаны, могут образовывать гетероциклильную группу, необязательно содержащую один дополнительный атом азота и необязательно замещенную алкилом; где любая алкильная группа представляет собой С 1-С 6-алкил; любая алкоксигруппа представляет собой C1-C6-алкокси; любая алкенильная группа представляет собой С 2-С 6-алкенил; любая алкинильная группа представляет собой C1-C6-алкинил; любая арильная группа представляет собой карбоциклическую или гетероциклическую группу, содержащую от 1 до 2 кольцевых групп, либо конденсированных, либо связанных друг с другом при помощи простых связей, в которых по меньшей мере одно из колец является ароматическим; любое ароматическое гетероциклическое кольцо, в случае его присутствия, также называемое гетероарильной группой, включает 5-6 членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из N, NH, О или S; любой гетероциклил представляет собой 3-7-членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, в котором один или несколько углеродных атомов замещены гетероатомами,такими как азот, кислород и сера, и любой циклоалкил представляет собой 3-6-членное моноциклическое кольцо, состоящее только из углеродных атомов, которое может содержать одну или несколько двойных связей, но не имеет полностью сопряженной -электронной системы; и его фармацевтически приемлемые соли. Настоящее изобретение также обеспечивает способы синтеза замещенных пиразолохиназолиновых соединений, представленных формулой (I), которые получают с использованием способа, состоящего из стандартных синтетических преобразований. Настоящее изобретение также обеспечивает способ лечения заболеваний, вызванных и/или связанных с нарушенной регуляцией активности протеинкиназы, в частности, семейства PLK, протеинкиназы С в различных изоформах, Met, PAK-4, PAK-5, ZC-I, STLK-2, DDR-2, Aurora 1, Aurora 2, Bub-1, Chk1,Chk2, HER2, raf1, MEK1, MAPK, EGF-R, PDGF-R, FGF-R, IGF-R, PBK, weel киназы, Src, Abl, Akt,-2 017769MAPK, ILK, MK-2, IKK-2, Cdc7, Nek, семейства Cdk/циклиновых киназ, более конкретно PLK-1 и PLK-3,который включает введение млекопитающему, нуждающемуся в этом, эффективного количества замещенного пиразолохиназолинового соединения, представленного формулой (I), определенного выше. Предпочтительный способ по настоящему изобретению предназначен для лечения заболевания, вызванного и/или связанного с нарушенной регуляцией активности протеинкиназы, представляющего собой рак. Другой предпочтительный способ по настоящему изобретению предназначен для лечения специфических типов рака, включая, но не ограничиваясь этим: карциному, такую как карцинома мочевого пузыря, молочной железы, толстой кишки, почки, печени, легкого, включая мелкоклеточный рак легкого, пищевода, желчного пузыря, яичника, поджелудочной железы, желудка, цервикальную, щитовидной железы, предстательной железы и кожи, включая сквамозно-клеточную карциному; гематопоэтические опухоли лимфоидной линии, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, не-ходжкинскую лимфому,волосисто-клеточную лимфому и лимфому Беркетта; гематопоэтические опухоли миелоидной линии,включая острые и хронические миелогенные лейкозы, миелодиспластический синдром и промиелоцитарный лейкоз; опухоли мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванномы; другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, xerodermapigmentosum, кератоксантому, фолликулярный рак щитовидной железы и саркому Капоши. Еще один предпочтительный способ по настоящему изобретению предназначен для лечения специфических клеточно-пролиферативных расстройств, таких как, например, доброкачественная гиперплазия предстательной железы, семейный аденоматозный полипоз, нейрофиброматоз, псориаз, пролиферация клеток гладких мышц сосудов, связанная с атеросклерозом, фиброзом легких, артритом, гломерулонефритом и послеоперационным стенозом и рестенозом. Кроме того, способ по настоящему изобретению также обеспечивает ингибирование опухолевого ангиогенеза и метастазов, а также лечение отторжения трансплантата органа и болезни "хозяин против трансплантата". Настоящее изобретение также обеспечивает фармацевтическую композицию, обладающую противораковой активностью, включающую одно или несколько соединений формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент, носитель или разбавитель. Подробное описание изобретения Если не определено иное, что касается соединений формулы (I) как таковых, а также любой содержащей их фармацевтической композиции или любого терапевтического лечения с использованием таких соединений, настоящее изобретение включает все фармацевтически приемлемые соли соединений по настоящему изобретению. В настоящем описании, если не определено иное, под термином "ортозамещенный ариламино", который представляет R1, подразумевается любая арильная группа, связанная с остальной частью молекулы через группу -(NH)-, при этом указанный ариламино является замещенным в орто-положении, а также необязательно замещенным в других свободных положениях. Под термином "арил" подразумевается карбоциклические или гетероциклические группы, содержащие от 1 до 2 кольцевых групп, либо конденсированные, либо связанные друг с другом при помощи простых связей, в которых по меньшей мере одно из колец является ароматическим; в случае его присутствия любое ароматическое гетероциклическое кольцо, также указанное как гетероарильная группа,включает 5-6 членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из N, NH, О или S. Примерами арильных групп в соответствии с настоящим изобретением являются, например, фенил, бифенил,- или -нафтил, дигидронафтил, тиенил, бензотиенил, фурил, бензофуранил, пирролил имидазолил,пиразолил, тиазолил, изотиазолил, оксазолил, изоксазолил, пиридил, пиразинил, пиримидинил, пиридазинил, индолил, изоиндолил, пуринил, хинолил, изохинолил, дигидрохинолинил, хиноксалинил, бензодиоксолил, инданил, инденил, триазолил и подобные. Термин "линейный или разветвленный C1-C6-алкил", соответственно охватывающий C1-C4-алкил,подразумевает любую из групп, таких как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, н-гексил и подобные. Под термином "линейный или разветвленный С 2-С 6-алкенил" подразумевается любая из групп, таких как, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил,1-гексенил и подобные. Под термином "линейный или разветвленный С 2-С 6-алкинил" подразумевается любая из групп, таких как, например, этинил, 2-пропинил, 4-пентинил и подобные. Под термином "С 3-С 6-циклоалкил" подразумевается, если не указано иное, 3-6-членное моноциклическое кольцо, состоящее только из углеродных атомов, которое может содержать одну или несколько двойных связей, но не имеет полностью сопряженной -электронной системы. Примерами циклоалкильных групп, без ограничения, являются циклопропан, циклобутан, циклопентан, циклопентен, циклогек-3 017769 сан, циклогексен и циклогексадиен. Под термином "гетероциклил" (также известным как "гетероциклоалкил") подразумевается 3-7 членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, в котором один или несколько углеродных атомов замещены гетероатомами, такими как азот, кислород и сера. Неограничивающими примерами гетероциклильных групп являются, например, пиран, пирролидин, пирролин, имидазолин, имидазолидин, пиразолидин, пиразолин, тиазолин, тиазолидин, дигидрофуран, тетрагидрофуран, 1,3-диоксолан, пиперидин, пиперазин, морфолин и подобные. В соответствии с настоящим изобретением, если не указано иное, любая из указанных выше группR1, R2, R3, R' и R, необязательно, может быть замещенной в любом из свободных положений одной или несколькими группами, например 1-6 группами, независимо выбранными из следующих: галоген, нитро,циано, C1-C6-алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, С 3-С 6-циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, алкилкарбонилокси, гетероциклилкарбонилокси, карбокси, алкоксикарбонил, гетероциклилоксикарбонил, амино, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино,формил, алкилкарбонил, арилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, ариламиносульфонил, арилтио и алкилтио. В свою очередь, где это является подходящим, каждый из указанных выше заместителей может быть дополнительно замещен одной или несколькими из указанных выше групп. В этой связи, под термином "атом галогена" подразумевается атом фтора, хлора, брома или йода. Под термином "циано" подразумевается остаток -CN. Под термином "нитро" подразумевается группа -NO2. Под термином "алкенил" или "алкинил" подразумевается любая из указанных выше С 2-С 6 алкильных групп с линейной или разветвленной цепью, содержащих также двойную или тройную связь. Неограничивающими примерами алкенильных или алкинильных групп по настоящему изобретению являются, например, винил, аллил, 1-пропенил, изопропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-пентенил,1-гексенил, этинил, 2-пропинил, 4-пентинил и подобные. Под термином "полифторированный алкил" или "алкокси" подразумевается любая из указанных выше C1-C6-алкильных или алкоксигрупп с линейной или разветвленной цепью, которые замещены более чем одним атомом фтора, такие как, например, трифторметил, трифторэтил, 1,1,1,3,3,3 гексафторпропил, трифторметокси и подобные. Под термином "алкокси", "арилокси", "гетероциклилокси" и их производными подразумевается любая из указанных выше C1-C6-алкильных, арильных или гетероциклильных групп, связанных с остальной частью молекулы через атом кислорода (-O-). Из всего вышесказанного специалистам в данной области должно быть понятно, что любую группу,имеющую название, состоящее из нескольких частей, такую как, например, ариламино, следует рассматривать так, что название группы стандартно составлено из частей, из которых эта группа образована,например из аминогруппы, которая затем была замещена арилом, где арил имеет значение, определенное выше. Подобным образом, любой из терминов, таких как, например, алкилтио, алкиламино, диалкиламино, алкоксикарбонил, алкокси карбониламино, гетероциклилкарбонил, гетероциклилкарбониламино,циклоалкилоксикарбонил и подобные, включает группы, где алкильные, алкокси, арильные, С 3-С 6 циклоалкильные и гетероциклильные фрагменты имеют значения, определенные выше. Фармацевтически приемлемые соли соединений формулы (I) включают кислотно-аддитивные соли с неорганическими или органическими кислотами, например азотной, хлористо-водородной, бромистоводородной, серной, перхлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой,молочной, щавелевой, малоновой, яблочной, малеиновой, винной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изетионовой и салициловой кислотой. Предпочтительно кислотноаддитивную соль соединений по настоящему изобретению выбирают из гидрохлорида или мезилата. Фармацевтически приемлемые соли соединений формулы (I) также включают соли с неорганическими или органическими основаниями, например гидроксиды, карбонаты или бикарбонаты щелочных или щелочно-земельных металлов, в частности натрия, калия, кальция, аммония или магния, ациклические или циклические амины, предпочтительно метиламин, этиламин, диэтиламин, триэтиламин, пиперидин и подобные. Предпочтительный класс соединений формулы (I) представляют собой соединения, в которых R3 представляет собой СО-ОН или CO-NR'R", где R' и R имеют значения, определенные выше. Следующий предпочтительный класс соединений формулы (I) представляют собой соединения, в которых R2 представляет собой необязательно замещенный С 1-С 6-алкил или С 2-С 6-алкенил с линейной или разветвленной цепью. Особенно предпочтительный класс соединений формулы (I) представляют собой соединения, в ко-4 017769 торых R3 представляет собой CO-NR'R", где R' и R" имеют значения, определенные выше. Конкретные предпочтительные соединения формулы (I) представляют собой соединения, перечисленные ниже (значения кодов смотри в разделе "Примеры"): 1) 8-[2-ацетил-5-(4-метилпиперазин-1-ил)фениламино]-1-метил-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксамид (A39B1C1Z); 2) 8-[2-ацетил-5-(4-метилпиперазин-1-ил)фениламино]-1-(2-фторэтил)-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A39B2C1Z); 3) 1-метил-8-(2-трифторметоксифениламино)-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A45B1C1Z); 4) 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A51B1C1Z); 5) этил 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксилат (A51B1C2Z); 6) 1-метил-8-[2-метокси-5-(4-метилпиперазин-1-ил)фениламино]-1-метил-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксамид (A85B1C1Z); 7) 8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-(2-фторэтил)-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксамид (A51B2C1Z); 8) 1-метил-8-[4-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A48B1C1Z); 9) 1-метил-8-[2-трифторметокси-5-пиперазин-1-илфениламино]-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксамид (A97B1C1Z); 10) 1-метил-8-[2-метил-5-(4-метилпиперазин-1-ил)фениламино]-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксамид (A98B1C1Z); 11) 1-метил-8-[5-(4-пирролидин-1-илпиперидин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро 1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A99B1C1Z); 12) метиламид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро 1 Н-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A51B1C4Z); 13) метиламид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-метоксифениламино]-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A85B1C4Z); 14) 1-метил-8-[2-метил-5-(4-метилпиперазин-1-карбонил)фениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A87B1C1Z); 15) 1-метил-8-[2-метил-4-(4-метилпиперазин-1-карбонил)фениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A86B1C1Z); 16) 1-метил-8-2-трифторметокси-5-[(1-метилпиперидин-4-карбонил)амино]фениламино-4,5 дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A82B1C1Z); 17) 8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-метил-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксилат калия (A51B1C3Z); 18) 1-этил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A51B7C1Z); 19) (2,2,2-трифторэтил)амид 1-метил-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоновой кислоты (A51B1C7Z); 20) 1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро 1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A51B5C1Z); 21) 8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-1-винил-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A51B10C1Z); 22) 1-(2-хлорэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксамид (A51B9C1Z); 23) 8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксамид (A51B8C1Z); 24) 1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5-дигидро 1 Н-пиразоло[4,3-h]хиназолин-3-карбоксилат калия (A51B5C3Z); 25) этил 1-(2-гидроксиэтил)-8-[5-(4-метилпиперазин-1-ил)-2-трифторметоксифениламино]-4,5 дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксилат (A51B5C2Z); 26) 1-метил-8-[5-(1-метил-1,2,3,6-тетрагидропиридин-4-ил)-2-трифторметоксифениламино]-4,5 дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A113B1C1Z); 27) 1-метил-8-[5-(1-метилпиперидин-4-ил)-2-трифторметоксифениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A114B1C1Z); 28) 8-(5-бром-2-трифторметоксифениламино)-1-метил-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3 карбоксамид (A49B1C1Z) и 29) 8-(5-бром-2-трифторметоксифениламино)-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3-карбоксамид (A49B8C1Z). Для ссылки на любое конкретное соединение формулы (I) по настоящему изобретению, необяза-5 017769 тельно в форме фармацевтически приемлемой соли, смотри экспериментальный раздел и формулу изобретения. Настоящее изобретение также обеспечивает способ получения соединений формулы (I), определенных выше, где способ отличается тем, что включает следующие стадии: 1) взаимодействие соединения формулы (II) с гидразиновым производным формулы (III) где R2 имеет значение, определенное выше, в присутствии уксусной кислоты с получением соединения формулы (IV) где R2 имеет значение, определенное выше; необязательно, алкилирование соединения формулы (IV), где R2 представляет собой водород, при помощи соединений формулы (V) где Y представляет собой подходящую отщепляемую группу, такую как мезил, тозил, галоген, и R2 имеет значение, определенное выше, но отличное от водорода, с получением соединения формулы (IV),где R2 имеет значение, определенное выше, но отличное от водорода; 2) взаимодействие соединения формулы (IV) с диметилформамид-ди-трет-бутилацеталем или диметилформамид-диизопропилацеталем с получением соединения формулы (VI) где R2 имеет значение, определенное выше; и стадия 3) взаимодействие соединения формулы (VI) в соответствии с любой одной из альтернативных стадий (стадия 3 а) или (стадия 3b): 3 а) с гуанидином с получением соединения формулы (VII), где R2 имеет значение, определенное выше; преобразование аминогруппы полученного соединения формулы (VII) в йод и затем взаимодействие полученного йодпроизводного формулы (VIII) с ортозамещенным ариламином формулы R1-H (IX),где R1 имеет значение, определенное выше, с получением соединения формулы (I) где R1 и R2 имеют значения, определенные выше; 3b) с гуанидиновым производным формулы (X) где R1 имеет значение, определенное выше, с получением соединения формулы (I) где R1 и R2 имеют значения, определенные выше, и необязательно, преобразование его в другие производные формулы (I) и/или в их фармацевтически приемлемые соли. Настоящее изобретение также обеспечивает способ получения соединения формулы (I), определенного выше, где способ отличается тем, что включает: 4) преобразование этоксикарбонильной группы соединения формулы (VIII), определенного выше, в-6 017769 соединение формулы (XIII) или соответствующую соль посредством кислотного или щелочного гидролиза; преобразование полученного соединения формулы (XIII) или соответствующей соли в соединение формулы (XIV) посредством реакции в щелочных условиях и в присутсвии подходящего агента конденсации с амином формулы R'R-NH (XI), где R' и R имеют значения, определенные выше; взаимодействие соединения формулы (XIV) с ортозамещенным ариламином формулы R1-H (IX), где R1 имеет значение, определенное выше, с получением соединения формулы (I) где R1, R2, R' и R имеют значения, определенные выше, и, необязательно, его преобразование в другие производные формулы (I) и/или в их фармацевтически приемлемые соли. Как определено выше, соединения формулы (I), которые получают в соответствии со способом, являющимся предметом настоящего изобретения, можно удобным образом преобразовать в другие соединения формулы (I), осуществляя взаимодействия в соответствии с хорошо известными условиями синтеза, при этом в качестве примеров возможных преобразований можно указать следующие: а) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой аминокарбонил, путем обработки гидроксидом аммонияb) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой группу CO-NR'R, путем обработки амином формулы R'RNH (XI), где R' и R имеют значения, определенные выше с) преобразование соединения формулы (I), где R3 представляет собой этоксикарбонил, в соединение формулы (I), где R3 представляет собой группу СО-ОН, или соответствующую соль посредством кислотного или щелочного гидролизаd) преобразование соединения формулы (I), где R3 представляет собой СО-ОН или соответствующую соль, в соединения формулы (I), где R3 представляет собой группу CO-NR'R, посредством реакции с амином формулы R'R-NH (XI) в щелочных условиях и в присутствии подходящего агента конденсации, где R' и R имеют значения, определенные выше е) преобразование соединения формулы (I), где R2 представляет собой тритил, в соединение формулы (I), где R2 представляет собой водород, в кислотных условияхf) преобразование соединения формулы (I), где R2 представляет собой водород, в соединение формулы (I), где R2 имеет значение, определенное выше, но отличное от водорода, посредством реакции со спиртом формулы R2-OH (XII), где R2 имеет значение, определенное выше, но отличное от водородаg) преобразование соединения формулы (I), где R2 представляет собой водород, в соединение формулы (I), где R2 имеет значение, определенное выше, но отличное от водорода, посредством реакции с соединением формулы R2-X (XV), где R2 имеет значение, определенное выше, но отличное от водорода,и X представляет собой галогенh) преобразование соединения формулы (I), где R2 представляет собой галогенэтил, в соединение формулы (I), где R2 представляет собой винилi) преобразование соединения формулы (I), где R1 представляет собой ортозамещенный ариламино формулы где R'4 или R4 представляет собой бром, в соединение формулы (I), где R'4 или R4 представляет собой группу -NR'R, путем обработки амином формулы R'R-NH (XI), где R' и R имеют значения, определенные выше. Описанный выше способ в любом из указанных выше вариантов является аналогичным способу,который можно осуществить в соответствии с хорошо известными способами, известными в данной области. Согласно стадии (стадия 1) способа соединение формулы (II) подвергали взаимодействию с гидразиновым производным формулы (III) в присутствии уксусной кислоты, таким образом получали соединение формулы (IV). Предпочтительно осуществление реакции при комнатной температуре. Необязательно, соединение формулы (IV), где R2 представляет собой водород, подвергали взаимодействию с подходящим соединением формулы (V) в присутствии основания, такого как гидрид натрия или триэтиламин или карбонат цезия, в подходящем растворителе, например дихлорметане, тетрагидрофуране, диоксане или диметилформамиде, при температуре в диапазоне от комнатной температуры до 100 С, так, чтобы получить соединение (IV), где R2 имеет значение, определенное выше, но отличное от водорода. Согласно стадии (стадия 2) способа соединение формулы (IV) подвергали взаимодействию с диметилформамид-ди-трет-бутилацеталем или диметилформамид-диизопропилацеталем в присутствии подходящего растворителя, такого как, например, диметилформамид, так, чтобы получить соединения формулы (VI). Предпочтительно осуществление реакции при температуре в диапазоне от комнатной температуры до около 80 С. Согласно стадии (стадия 3 а) способа соединение формулы (VI) подвергали взаимодействию с гуанидином или солями гуанидина так, чтобы получить соединение формулы (VII) посредством образования пиримидинового кольца. Соединения формулы (I), где R1 представляет ортозамещенную ариламиногруппу, можно получить с помощью соответствующих йодпроизводных формулы (VIII), которые, в свою очередь, получают при помощи соответствующих соединений формулы (VII). Получение йодпроизводных формулы (VIII) можно осуществить в подходящем растворителе, таком как тетрагидрофуран, диэтиловый эфир или диметоксиэтан, при температуре в диапазоне от комнатной-8 017769 температуры до около 80 С и в течение времени от около 2 до около 48 ч. Последующее преобразование йодпроизводного формулы (VIII) в соединения формулы (I) можно осуществить в присутствии ортозамещенного ариламина формулы R1-H (IX) в подходящем растворителе, таком как диметилформамид, диметоксиэтан или ацетонитрил, и в присутствии каталитических количеств ацетата палладия, (2,2'-бис-(дифенилфосфино)-1,1'-бинафталина (BINAP) и основания, такого как карбонат калия, фосфат калия или карбонат цезия, при температуре в диапазоне от комнатной температуры до 110 С и в течение времени от около 2 до около 24 ч. Согласно стадии (стадия 3b) способа соединение формулы (VI) подвергали взаимодействию с гуанидиновыми производными формулы (X), так, чтобы получить соответствующее соединение формулы(I) посредством образования пиримидинового кольца. Любую из описанных выше реакций проводят в соответствии с общепринятыми способами. В качестве примера реакции с гуанидином или его солями,такими как гидрохлорид, карбонат или нитрат, или с гуанидиновым производным формулы (X), как указано в стадиях (стадия 3 а) или (стадия 3b), осуществляют в диметилформамиде при температуре в диапазоне от 80 С до температуры кипения с обратным холодильником обычно в присутствии карбоната калия. Согласно стадии (стадия 4) способа соединения формулы (VIII) можно преобразовать в карбоновокислотные производные формулы (XIII) или соответствующую соль в условиях щелочного или кислотного гидролиза, хорошо известных в данной области. Соединения формулы (XIII) можно преобразовать в карбоксамидо-производные формулы (XIV),где R' и R" имеют значения, определенные выше. Реакцию осуществляют в присутствии хлорида аммония или подходящего первичного или вторичного амина формулы (XI), в щелочных условиях, предпочтительно с N,N-диизопропил-N-этиламином или триэтиламином, в подходящем растворителе, таком как дихлорметан, диметилформамид, тетрагидрофуран или диоксан, и в присутствии подходящего агента конденсации, такого как N,N'-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N'этилкарбодиимидгидрохлорид(EDCI) или тетрафторборат О-(бензотриазол-1-ил)-N,N,N',N'тетраметилизоурония (TBTU); также могут потребоваться каталитические количества гексафторфосфата(бензотриазол-1-илокси)трипирролидинофосфония (РуВОР) или N-гидроксибензотриазола. Последующее преобразование соединения формулы (XIV) в соединение формулы (I) можно осуществить в подходящем растворителе, таком как диметилформамид, диметоксиэтан или ацетонитрил, и в присутствии ортозамещенного ариламина формулы R1-H (IX), каталитических количеств ацетата палладия, (2,2'-бис-(дифенилфосфино)-1,1'-бинафталина (BINAP) и основания, такого как карбонат калия,фосфат калия или карбонат цезия, при температуре в диапазоне от комнатной температуры до 110 С и в течение времени от около 2 до около 24 ч. Как указано ранее, соединения формулы (I), полученные, как указано выше, можно легко преобразовать в некоторые другие соединения формулы (I) по настоящему изобретению. В качестве примера соединения формулы (I), содержащие R3 в качестве этоксикарбонильной группы или даже в качестве алкоксикарбонильной группы, можно преобразовать в различные производные в соответствии со способами, хорошо известными из уровня техники для преобразования карбоксиэфирной группы (-COOR) в карбоксамиды (-CONH2), N-замещенные карбоксамиды (-CONHR'), N,Nдизамещенные карбоксамиды (-CONR'R") и карбоновые кислоты (-СООН), например, как указано в преобразовании (а)-(с). Рабочие условия хорошо известны из уровня техники и могут включать, например, преобразование карбоксиэфирной группы в карбоксамидную группу, взаимодействие с аммиаком или гидроксидом аммония в присутствии подходящего растворителя, такого как низший спирт, диметилформамид или их смеси; предпочтительно реакцию осуществляют с гидроксидом аммония в смеси метанол/диметилформамид при температуре в пределах от около 50 до около 100 С. Аналогичные рабочие условия применимы для получения N-замещенных карбоксамидов или N,Nдизамещенных карбоксамидов, где подходящий первичный или вторичный амин используют вместо аммиака или гидроксида аммония. Альтернативно, карбоксиэфирные группы можно преобразовать в карбоксамид или N-замещенные карбоксамиды или N,N-дизамещенные карбоксамиды в щелочных условиях, таких как бистриметилсилиламид лития, 1 н. раствор в ТГФ, используя хлорид аммония или подходящий первичный или вторичный амин; предпочтительно реакцию осуществляют в тетрагидрофуране при температуре в пределах от 20 С до температуры кипения с обратным холодильником. Подобным образом, карбоксиэфирные группы можно преобразовать в карбоновокислотные производные с использованием условий щелочного или кислотного гидролиза, хорошо известных из уровня техники. В соответствии с преобразованием (d) способа соединения формулы (I), где R3 представляет собой карбоновую кислоту (-СООН), можно преобразовать в карбоксамидо-производные (-CONR'R"), где R' иR" имеют значения, определенные выше. Реакцию осуществляют в присутствии хлорида аммония или подходящего первичного или вторичного амина формулы (XI), в щелочных условиях, предпочтительно с N,N-диизопропил-N-этиламином-9 017769 или триэтиламином, в подходящем растворителе, таком как дихлорметан, диметилформамид, тетрагидрофуран или диоксан, и в присутствии подходящего агента конденсации, такого как N,N'дициклогексилкарбодиимид (DCC), гидрохлорид N-(3-диметиламинопропил)-N'-этилкарбодиимида(EDCI) или тетрафторбората O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилизоурония (TBTU); также могут потребоваться каталитические количества гексафторфосфата(бензотриазол-1 илокси)трипирролидинофосфония (РуВОР) или N-гидроксибензотриазола. В соответствии с преобразованием (е) тритильную группу соединений формулы (I) удаляют в кислотных условиях, например, с использованием трифторуксусной кислоты и в присутствии подходящего растворителя, такого как дихлорметан, чтобы получить соответствующее соединение формулы (I), гдеR2 представляет собой водород. В соответствии с преобразованием (f) способа соединения формулы (I), где R2 представляет собой водород, подвергают взаимодействию со спиртом формулы R2-OH (XII), где R2 имеет значение, определенное выше, но отличное от водорода, в присутствии ди-трет-бутилазадикарбоксилата и трифенилфосфина или трифенилфосфина на полимерном носителе, в подходящем растворителе, таком как, например,тетрагидрофуран, чтобы получить соответствующие соединения формулы (I). В соответствии с преобразованием (g) способа соединения формулы (I), где R2 представляет собой водород, подвергают взаимодействию с соединением формулы R2-X (XV), где R2 имеет значение, определенное выше, но отличное от водорода, и X представляет собой галоген, предпочтительно хлор, бром или йод, в присутствии основания, такого как карбонат цезия, в подходящем растворителе, таком как,например, диметилформамид, чтобы получить соответствующие соединения формулы (I). В соответствии с преобразованием (h) способа соединения формулы (I), где R2 представляет собой галогенэтил, предпочтительно хлорэтил, обрабатывают основанием, предпочтительно DBU, при температуре в пределах от 20 до 80 С, чтобы получить соответствующие соединения формулы (I), где R2 представляет собой винил. В соответствии с преобразованием (i) способа преобразование соединения формулы (I), где R1 представляет собой ортозамещенный ариламино, содержащий в каком-либо положении бром, в соединение формулы (I), где R1 представляет собой ортозамещенный ариламино, содержащий в каком-либо положении группу -NRR", можно осуществить различными путями в соответствии с традиционными способами. Предпочтительно его осуществляют в подходящем растворителе, таком как тетрагидрофуран или диоксан, путем обработки амином формулы RR-NH (XI) и в присутствии каталитических количеств трис-(дибензилиденацетон)дипалладия, 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила и основания, такого как LiN (TMS)2, при температуре в диапазоне от комнатной температуры до температуры кипения с обратным холодильником и в течение времени в пределах от 1 до около 24 ч. Из вышесказанного специалистам в данной области должно быть понятно, что соединение формулы (I), содержащее функциональную группу, которую затем можно дериватизировать до другой функциональной группы, осуществляя обработку в соответствии со способами, хорошо известными из уровня техники, приводящие, таким образом, к другим соединениям формулы (I), входит в объем настоящего изобретения. В соответствии с любым вариантом способа получения соединений формулы (I), исходные вещества и любые другие реагенты являются известными, или их можно легко получить в соответствии с известными способами. В качестве примера, когда исходное вещество для получения соединений формулы(II) является коммерчески доступным, соединения формулы (II) можно получить, как описано в указанном выше WO 04/104007. Соединения формулы (III), (V), (XII) и (XV) являются коммерчески доступными. Некоторые соединения формулы (IX), (X) и (XI) являются коммерчески доступными, другие были получены, см. представленные ниже примеры 28-35 и 43-44. Из вышесказанного специалистам в данной области должно быть понятно, что при получении соединений формулы (I) в соответствии с любым из указанных выше вариантов способа, необязательные функциональные группы в исходных веществах или их промежуточных соединениях, которые могут вызвать нежелательные побочные реакции, необходимо подходящим образом защитить в соответствии с традиционными методами. Также, преобразование этих последних в свободные незащищенные соединения можно осуществить в соответствии с известными процедурами. Должно быть понятно, что, если соединения формулы (I), полученные в соответствии со способом,описанным выше, получают в виде смеси изомеров, их разделение с использованием традиционных методов на отдельные изомеры формулы (I) входит в объем настоящего изобретения. Традиционные методы разделения рацематов включают, например, фракционированную кристаллизацию диастереизомерных солевых производных или препаративную хиральную ВЭЖХ. Кроме того, соединения формулы (I) по настоящему изобретению также могут быть получены в соответствии с методами комбинаторной химии, хорошо известными из уровня техники, например путем осуществления указанных выше взаимодействий между некоторыми промежуточными соединениями параллельным и/или последовательным образом и с использованием условий твердофазного синтеза(SPS). Общие сведения по получению соединений формулы (I) по настоящему изобретению в соответст- 10017769 вии с методами комбинаторной химии смотри в экспериментальной части. Какие-либо конкретные примеры, касающиеся получения соединений формулы (I) по настоящему изобретению и их преобразования в другие соединения формулы (I) смотри в экспериментальной части. Следовательно, следующим объектом настоящего изобретения является библиотека двух или более соединений формулы (I) где R1 представляет собой ортозамещенный ариламино формулы где R'4 и R4 независимо выбраны из группы, включающей галоген, нитро, циано, C1-C6-алкил, полифторированный алкил, полифторированный алкокси, алкенил, алкинил, гидроксиалкил, арил, арилалкил, гетероциклил, С 3-С 6-циклоалкил, гидрокси, алкокси, арилокси, гетероциклилокси, алкилкарбонилокси, арилкарбонилокси, гетероциклилкарбонилокси, карбокси, алкоксикарбонил, гетероциклилоксикарбонил, амино, алкиламино, диалкиламино, ариламино, диариламино, гетероциклиламино, формиламино, алкилкарбониламино, арилкарбониламино, гетероциклилкарбониламино, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил, ариламинокарбонил, гетероциклиламинокарбонил, алкоксикарбониламино, гидроксиаминокарбонил, алкоксиимино, алкилсульфониламино, арилсульфониламино,формил, алкилкарбонил, арилкарбонил, гетероциклилкарбонил, алкилсульфонил, арилсульфонил, аминосульфонил, ариламиносульфонил, арилтио и алкилтио;R2 представляет собой водород, C1-C6-алкил с линейной или разветвленной цепью, необязательно замещенный галогеном, гетероциклилокси, гидрокси, арилом, алкоксиарилом, амино, алкиламино, диалкиламино или алкокси, или представляет собой С 2-С 6-алкенил с линейной или разветвленной цепью;R3 представляет собой CO-OR' или CO-NR'R", где R' и R представляют собой, каждый независимо,водород, C1-C6-алкил, необязательно замещенный галогеном или алкоксиарилом; или R' и R" взятые вместе с атомом азота, с которым они связаны, могут образовывать гетероциклильную группу, необязательно содержащую один дополнительный атом азота и необязательно замещенную алкилом; где любая алкильная группа представляет собой C1-C6-алкил; любая алкокси группа представляет собой C1-C6-алкокси; любая алкенильная группа представляет собой С 2-С 6-алкенил; любая алкинильная группа представляет собой C1-C6-алкинил; любая арильная группа представляет собой карбоциклическую или гетероциклическую группу, содержащую от 1 до 2 кольцевых групп, либо конденсированных, либо связанных друг с другом при помощи простых связей, в которых по меньшей мере одно из колец является ароматическим; любое ароматическое гетероциклическое кольцо, в случае его присутствия, также называемое гетероарильной группой, включает 5-6-членное кольцо, содержащее от 1 до 3 гетероатомов, выбранных из N, NH, О или S; любой гетероциклил представляет собой 3-7-членное, насыщенное или частично ненасыщенное карбоциклическое кольцо, в котором один или несколько углеродных атомов замещены гетероатомами,такими как азот, кислород и сера, и любой циклоалкил представляет собой 3-6-членное моноциклическое кольцо, состоящее только из углеродных атомов, которое может содержать одну или несколько двойных связей, но не имеет полностью сопряженной -электронной системы; и его фармацевтически приемлемые соли. Еще одной целью настоящего изобретения является обеспечение промежуточного соединения формулы (IX') где R1' представляет собой Еще одной целью настоящего изобретения является обеспечение промежуточного соединения формулы (X') Фармакология Соединения формулы (I) являются активными в качестве ингибиторов протеинкиназы и поэтому являются полезными, например, для ограничения нерегулируемой пролиферации опухолевых клеток. В терапии их можно использовать для лечения различных опухолей, таких как указанные выше, а также для лечения других клеточно-пролиферативных расстройств, таких как доброкачественная гиперплазия предстательной железы, семейный аденоматозный полипоз, нейрофиброматоз, псориаз, пролиферация клеток гладких мышц сосудов, связанная с атеросклерозом, фиброз легких, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз. Ингибирующую активность предполагаемых ингибиторов PLK-1 и активность избранных соединений определяли с использованием анализа, описанного ниже.Swiss-Prot номер доступа Р 53350) был амплифицирован с использованием ПЦР из полноразмерного человеческого PLK1 гена, закупленного у imaGenes в виде клона IRATp970A078D. Амплификацию осуществляли с использованием прямого олигонуклеотида и обратного олигонуклеотида В целях клонирования олигонуклеотиды включали attB сайты для получения attB-фланкированного ПЦР продукта, подходящего для клонирования, используя Gateway технологию (Invitrogen). Кроме того, в целях очистки прямой праймер включал TEV сайт расщепления (Amersham Biosciences). Полученный ПЦР продукт клонировали в pDONR221 плазмиду и затем переносили в бакуловирусный экспрессирующий вектор pVL1393 (Invitrogen), Gateway-модифицированный. В целях экспрессии и очистки His метку добавляли по N-концу PLK киназного домена. Клонирование осуществляли в соответствии с протоколами, описанными в руководстве Gateway. Бакуловирусы получали путем ко-трансфекции клеток насекомого Sf9 с вектором экспрессии и вирусной ДНК с использованием набора для трансфекции BaculoGold (Pharmingen). Вирусный супернатант извлекали через 5 дней и подвергали 3 циклам амплификации для увеличения титра вируса. Рекомбинантный белок получали путем инфицирования клеток насекомого High5. После инфицирования в течение 48 ч клетки извлекали, осаждали путем центрифугирования и замораживали при -80 С. Для очистки рекомбинантного белка осадок после центрифугирования оттаивали, ресуспендировали в лизисном буфере (PBS, NaCl 150 мМ, CHAPS 0,1%, DTT 20 мМ, глицерин 10%, ингибиторы протеазы) и лизировали путем обработки ультразвуком. Лизат очищали при помощи центрифугирования и загружали на аффинную колонку Nichel. После тщательной промывки рекомбинантный белок расщепляли и элюировали путем инкубации с TEV протеазой. Биохимический анализ определения активности ингибиторов PLK-1 киназы. Ингибирующую активность предполагаемых ингибиторов PLK-1 и активность избранных соедине- 12017769 ний определяли с использованием анализа транс-фосфорилирования. Специфические пептидные или белковые субстраты транс-фосфорилировали при помощи их специфической сериновой-треониновой или тирозиновой киназы, в присутствии АТФ, отслеживаемого при помощи 33 РАТФ, и в присутствии их собственного оптимального буфера и кофакторов. По окончании реакции фосфорилирования более чем 98% холодного АТФ и радиоактивного АТФ было захвачено избыточным количество ионообменной смолы dowex; смола затем осаждалась на дно реакционного планшета под действием силы тяжести. Супернатант, содержащий фосфорилированный субстрат, затем извлекали и переносили на счетную пластинку, затем оценивали при помощи -счетчика. Реагенты/условия анализа.i. Подготовка смолы Dowex. Отвешивали 500 г мокрой смолы (SIGMA, изготовленная по специальному заказу смола DOWEX 18 200-400 меш, 2,5 кг) и разбавляли до 2 л в 150 мМ формиата натрия, рН 3,00. Смоле давали осадиться (несколько часов) и затем супернатант сливали. После трех промывок, как указано выше, в течение двух дней смоле давали осадиться, супернатант сливали и добавляли два объема 150 мМ натрийформиатного буфера на объем осадка. Затем измеряли рН, который должен быть около 3,00. Промытая смола является стабильной больше чем в течение недели; подготовленную для использования смолу хранили при 4 С до использования.ii. Киназный буфер (KB). Киназный буфер состоял из 50 мМ HEPES рН 7,9, содержащего 10 мМ MgCl2, 1 мМ DTT, 3 мкМiii. Условия анализа. Киназный анализ осуществляли с конечной концентрацией фермента PLK-1 3 нМ, в присутствии 40 мкМ АТФ, 3 нМ 33 РАТФ и 85 мкМ субстрата альфа-казеина, SIGMA,С-3240. Автоматизированный анализ Dowex: 1) 3 смеси фермента (полученной в киназном буфере 3 Х), 5 мкл/лунка; 2) 3 смеси субстрата и АТФ (полученной в ddH2O), вместе с 33PАТФ, 5 мкл/лунка; 3) 3 испытываемого соединения (разбавленного в ddH2O-3% DMSO) - 5 мкл/лунка; Разведение соединений и схема анализа описаны ниже.i. Разведение соединений. 10 мМ исходных растворов испытываемых соединений в 100% DMSO распределяли в 96-луночные микротитровальные планшеты формата 128. Для исследования % ингибирования подготавливали планшеты с отдельными разведениями при 1 мМ, 100 и 10 мкМ в 100% DMSO, затем разбавляли при 3 Х концентрации (30, 3 и 0,3 мкМ) в ddH2O, 3%DMSO. Для разведений использовали Multimek 96 (Beckman) и соединение добавляли через пипетку в планшеты для испытаний. Для определения ИК 50 соединения получали в виде 1 мМ, 100% DMSO растворов, добавляли в первую колонку микротитровального планшета (A1-G1), 100 мкл.Biomek 2000 (Beckman) использовали для серийных 1:3 разведений в воде, 3% DMSO, от колонки А 1 до А 10 и для всех семи соединений в планшете. В стандартном эксперименте наивысшая концентрация всех соединений составляла 30 мкМ, и затем разбавляли с получением конечной испытываемой смеси до 10 мкМ.ii. Схема анализа. 384-луночные планшеты с V-образным дном (планшеты для испытаний) подготавливали с разведением соединения 5 мкл (3 Х) и затем помещали на роботизированную установку Plate Trak 12 (PerkinElmer; робот имел одну головку для пипетирования с 384 наконечниками для начала анализа плюс одну головку с 96 наконечниками для распределения смолы) вместе с одним резервуаром для смеси фермента(3 Х) и одним для смеси АТФ (3 Х). В начале эксперимента робот аспирировал 5 мкл смеси АТФ, делал воздушное пространство внутри наконечников (3 мкл) и аспирировал 5 мкл смеси PLK1. Последующее распределение в планшеты давало начало киназной реакции после 3 циклов смешивания, осуществляемых при помощи робота. В этот момент восстанавливали нужную концентрацию для всех реагентов. Робот осуществлял инкубацию планшетов в течение 60 мин при комнатной температуре и затем останавливал реакцию путем пипетирования 70 мкл суспензии смолы dowex в реакционную смесь. Сразу после добавления смолы осуществляли три цикла смешивания. Еще один цикл смешивания осуществляли после остановки всех планшетов, на этот раз с использованием нормальных наконечников: планшетам затем давали выстояться в течение около 1 ч для максимального захвата АТФ. В этот момент 20 мкл супернатанта переносили в 384-луночные планшеты Optiplates (Perkin-Elmer), с 70 мкл Microscint 40 (Perkin-Elmer); после 5 мин встряхивания на орбительном шейкере планшеты считывали с использованием счетчика радиоактивности Perkin-Elmer Top Count.- 13017769 Данные анализировали с использованием адаптированного для этого пакета программ "Assay Explorer", который обеспечивал либо % ингибирования для первичных анализов, либо сигмоидальные подгонки кривых десяти разведений для определения ИК 50 для вторичных анализов/рутинных методов подтверждения успеха эксперимента. Биохимический анализ определения активности ингибиторов Aurora-2 киназы.In vitro анализ ингибирования киназы осуществляли таким же образом, как описано для ферментаi. Киназный буфер (KB) для Aurora-2. Киназный буфер состоял из 50 мМ HEPES, рН 7,0, 10 мМ MgCl2, 1 мМ DTT, 3 мкМ NaVO3 и 0,2 мг/мл BSA.ii. Условия анализа для Aurora-2 (конечные концентрации). Киназный анализ осуществляли с использованием концентрации фермента 2,5 нМ, 10 мкМ АТФ, 1 нМ 33 РАТФ и 8 мкМ субстрата, состоящего из 4 LRRWSLG повторов. Анализ ингибирования активности Cdk2/циклин А. Киназная реакция: 1,5 мкМ субстрата гистон H1, 25 мкМ АТФ (0,2 мкКи Р 33-АТФ), 30 нг бакуловирус-коэкспресируемого Cdk2/циклин А, 10 мкМ ингибитора в конечном объеме 100 мкл буфера (TRISHCl 10 мМ рН 7,5, MgCl2 10 мМ, 7,5 мМ DTT) добавляли в каждую лунку 96-луночного планшета с Uобразным дном. После инкубации в течение 10 мин при 37 С реакцию останавливали при помощи 20 мкл EDTA 120 мМ. Захват: 100 мкл переносили из каждой лунки в Multiscreen планшет для обеспечения возможности связывания субстрата с фосфоцеллюлозным фильтром. Планшеты затем промывали 3 раза при помощиIn vitro анализ клеточной пролиферации. Клетки А 2780 рака яичников человека и MCF7 рака молочной железы человека (1250 клеток/лунка) высевали в белые 384-луночные планшеты в полную среду (RPMI 1640 или ЕМЕМ плюс 10% фетальной бычьей сыворотки) и обрабатывали соединениями, растворенными в 0,1% DMSO, через 24 ч после посева. Клетки инкубировали при 37 С и 5% СО 2 и через 72 ч планшеты обрабатывали с использованием набора для анализа CellTiter-Glo (Promega), следуя инструкциям изготовителя.CellTiter-Glo представляет собой гомогенный метод, основанный на определении количества присутствующего АТФ, индикатора метаболически активных клеток. Количество АТФ определяют с использованием системы на основе люциферазы и D-люциферина, которые обеспечивают генерацию света. Люминисцентный сигнал является пропорциональным количеству клеток, присутствующих в культуре. Вкратце, 25 мкл/лунка раствора реагента добавляли в каждую лунку и после 5 мин встряхивания микропланшеты считывали с использованием люминометра. Люминисцентный сигнал является пропорциональным количеству клеток, присутствующих в культуре. Принимая во внимание описанные выше анализы ингибирования, в результате было определено,что соединения формулы (I) по настоящему изобретению обладают великолепной активностью ингибирования PLK, типично с ИК 50 меньше чем 0,07 мкМ. Смотри в качестве примера представленную ниже табл. А, содержащую экспериментальные данные для некоторых репрезентативных соединений формулы (I) по настоящему изобретению (значения кодов см. в разделе "Примеры"), которые были испытаны в биохимическом анализе как ингибиторы PLK-1 и в анализе пролиферации клеток А 2780 (ИК 50 мкМ), в сравнении соединением, известным из уровня техники как ближайший аналог, которое описано в указанном выше WO 04/104007, с. 105, табл. IX, соединение В 08-Х 00-М 00(С 01)-D03. К удивлению, ингибиторная активность соединений по настоящему изобретению в отношенииPLK-1 оказалась значительно выше, чем активность ссылочного соединения. Таким образом, неожиданно оказалось, что новые соединения по настоящему изобретению обладают активностью ингибирования PLK-1, которая существенно выше активности структурно близкого соединения предшествующего уровня техники, описанного в указанном выше WO 04/104007, и, таким образом, являются особенно полезными для использования в терапии, против пролиферативных расстройств, связанных с зависимой от измененного клеточного цикла киназной активностью. Соединения по настоящему изобретению можно вводить либо как отдельное средство, либо, альтернативно, в сочетании с известными противораковыми лечениями, такими как режим радиационной терапии или химиотерапии, в сочетании с цитостатическими или цитотоксическими средствами, средствами типа антибиотиков, алкилирующими средствами, антиметаболитами, гормональными средствами,иммунологическими средствами, средствами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторами СОХ-2), ингибиторами металлопротеазы матрикса, ингибиторами теломеразы, ингибиторами тирозиновых киназ, средствами против рецептора фактора роста, анти-HER средствами, антиEGFR средствами, средствами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути сигнальной трансдукции ras-raf, ингибиторами клеточного цикла, другими ингибиторами cdk, средствами, связывающими тубулин, ингибиторами топоизомеразы I,ингибиторами топоизомеразы II и подобными средствами. Если их формулируют в виде фиксированной дозы, такие комбинированные продукты включают соединения по настоящему изобретению в дозах, находящихся в пределах, которые описаны ниже, и другое фармацевтически активное средство в дозах, находящихся в пределах, которые считаются приемлемыми. Соединения формулы (I) можно использовать последовательно с известными противораковыми средствами, когда комбинированная композиция не является приемлемой. Соединения формулы (I) по настоящему изобретению, подходящие для введения млекопитающему,например человеку, можно вводить с использованием обычных путей введения, и уровень доз зависит от возраста, массы тела, состояния пациента и пути введения. Например, подходящая доза, принятая для перорального введения соединения формулы (I), может находиться в пределах от около 10 до около 500 мг в расчете на дозу, от 1 до 5 раз в день. Соединения по настоящему изобретению можно вводить в различных лекарственных формах, например перорально, в форме таблеток, капсул, таблеток с сахарным или пленочным покрытием, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например внутримышечно или через внутривенную, и/или интратекальную, и/или интраспинальную инъекцию или инфузию. Настоящее изобретение также включает фармацевтические композиции, включающие соединение формулы (I) или его фармацевтически приемлемую соль в ассоциации с фармацевтически приемлемым эксципиентом, который может представлять собой носитель или разбавитель. Фармацевтические композиции, содержащие соединения по настоящему изобретению, обычно получают, следуя традиционным способам, и вводят в подходящей фармацевтической форме. Например,твердые пероральные формы могут содержать вместе с активным соединением, разбавителем, например,лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; смазывающие- 15017769 вещества, например диоксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; связующие, например крахмалы, аравийскую камедь, желатин, метилцеллюлозу,карбоксиметилцеллюлозу или поливинилпирролидон; разрыхлители, например крахмал, альгиновую кислоту, альгинаты или натрийкрахмалгликолят; шипучие смеси; красители; подсластители; смачивающие вещества, такие как лецитин, полисорбаты, лаурилсульфаты; и, как правило, нетоксичные и фармакологически неактивные вещества, используемые в фармацевтических композициях. Эти фармацевтические препараты могут быть получены известным способом, например путем смешивания, гранулирования, таблетирования, способами нанесения сахарного покрытия или нанесения пленочного покрытия. Жидкие дисперсии для перорального введения могут представлять собой, например, сиропы,эмульсии и суспензии. В качестве примера сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннитом и сорбитом. Суспензии и эмульсии могут содержать в качестве примеров носителей природную смолу, агар,альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензия или растворы для внутримышечных инъекций содержат вместе с активным соединением фармацевтически приемлемый носитель, например стерильную воду, оливковое масло, этилолеат, гликоли, например пропиленгликоль, и, если это желательно, подходящее количество лидокаингидрохлорида. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду или предпочтительно они могут быть в форме стерильных водных изотонических физиологических растворов либо они могут содержать пропиленгликоль в качестве носителя. Суппозитории могут содержать вместе с активным соединением фармацевтически приемлемый носитель, например масло какао, полиэтиленгликоль, поверхностно-активное вещество на основе сложного эфира полиоксиэтилена и жирных кислот сорбитана или лецитин. Для лучшей иллюстрации настоящего изобретения без какого-либо его ограничения представлены следующие примеры. Примеры Синтетическое получение некоторых соединений формулы (I) по настоящему изобретению описано в следующих примерах. Все соединения удобным образом и однозначно обозначены с использованием кодовой системы (см. табл. IV ниже), некоторые из них перечислены и указаны также в соответствии с их химическим названием, тогда как другие удобным образом и однозначно обозначены с использованием кодовой системы, вместе с их данными 1 Н-ЯМР или данными ВЭЖХ/мас. (см. табл. V-XX ниже). Каждый код, который однозначно определяет одно конкретное соединение формулы (I), состоит из четырех единиц А-B-C-Z. Код А представляет любой R1 заместитель, как в формуле (I), присоединенный к остальной части молекулы в положении 8; каждая группа А представлена посредством соответствующей химической формулы в табл. I ниже, также указывающей точку ее присоединения к остальной части молекулы. Код В представляет R2 группу, присоединенную к остальной части молекулы через атом азота пиразола, как в формуле (I). Каждая группа В представлена посредством соответствующей химической формулы в табл. II ниже, также указывающей точку ее присоединения к остальной части молекулы. Код С представляет группу R3, присоединенную к остальной части молекулы в положении 3, как в формуле (I). Каждая группа С представлена посредством соответствующей химической формулы в табл.III ниже, также указывающей точку ее присоединения к остальной части молекулы. Каждая специфическая группа А В и С представлена и последовательно пронумерована в представленных ниже табл. I-III соответственно. Наконец, код Z относится к центральному ядру молекулы (I). Из вышесказанного специалистам в данной области должно быть понятно, что Z замещен группой R1 (код A), R2 (код В) и R3 (код С), как определено в формуле (I), также указывая положения других заместителей. Поэтому систему кодов, используемую в настоящей заявке для некоторых соединений формулы (I),можно вкратце представить следующим образом: Только в качестве примера, который не предназначен для ограничения объема настоящего изобретения, соединение A45B8C2Z (см. пример) представляет собой пиразолохиназолиновое производное формулы (I), где центральное ядро представлено фрагментом Z, R1 представляет собой группу формулы А 45 из табл. I, R2 представляет собой группу формулы В 8 из табл. II, R3 представляет собой группу формулы С 2 из табл. III, имеющее формулу Соединения по настоящему изобретению, полученные в соответствии со следующими примерами,также были охарактеризованы при помощи аналитических данных 1 Н ЯМР или ВЭЖХ/МС; данные ВЭЖХ/МС получали, следуя одному из способов 1-4. ВЭЖХ/МС. Аналитический способ 1. Оборудование для ВЭЖХ состояло из системы ВЭЖХ Waters Acquity, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ, снабженным источником ионизации - электроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 45 С, при скорости потока 0,8 мл/мин, используя колонку ВЕН С 18 1,7 мкм Waters Acquity HPLC (2,150 мм). Подвижная фаза А представляла собой буфер муравьиной кислоты 0,1% рН 3,3 с ацетонитрилом (98:2) и подвижная фаза В представляла собой H2O/ацетонитрил(5:95); использовали градиент от 5 до 95% В в течение 2 мин, затем удерживание при 95% В в течение 0,1 мин. Объем вводимой пробы был 2 мкл. Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 3,5 кВ (ES+) и 28 В(ES+); температура источника составляла 120 С; напряжение на конусе было 14 В (ES+) и 2,8 кВ(ES-); был установлен режим полной развертки, массовый диапазон от 100 до 800 amu (атомных единиц массы). Аналитический способ 2. Оборудование для ВЭЖХ состояло из системы ВЭЖХ Waters 2795 Alliance HT, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ,снабженным источником ионизации - электроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 30 С при скорости потока 1,0 мл/мин, используя колонку С 18, 3 мкм Phenomenex (4,650 мМ). Подвижная фаза А представляла собой буфер ацетата аммония 5 мМ рН 5,2 с аце- 21017769 тонитрилом (95:5), и подвижная фаза В представляла собой Н 2 О/ацетонитрил (5:95); использовали градиент от 10 до 90% В в течение 8 мин, затем равномерное повышение до 100% В в течение 1,0 мин. Объем вводимой пробы был 10 мкл. Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 3,5 кВ (ES+) и 28 В (ES-); температура источника составляла 120 С; напряжение на конусе было 14 В (ES+) и 2,8 кВ (ES-); был установлен режим полной развертки, массовый диапазон от 100 до 800 amu (атомных единиц массы). Аналитический способ 3. Оборудование для ВЭЖХ состояло из системы ВЭЖХ Waters Acquity, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ, снабженным источником ионизации электроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 45 С, при скорости потока 0,8 мл/мин, используя колонку ВЕН C18 1,7 мкм Waters Acquity HPLC (2,1x50 мм). Подвижная фаза А представляла собой буфер гидроксида аммония 0,05% рН 10 с ацетонитрилом (95:5), и подвижная фаза В представляла собой H2O/ацетонитрил(5:95); использовали градиент от 5 до 95% В в течение 2 мин, затем удерживание при 95% В в течение 0.1 мин. Объем вводимой пробы был 2 мкл. Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 3,5 кВ (ES+) и 28 В(ES-); температура источника составляла 120 С; напряжение на конусе было 14 В (ES+) и 2,8 кВ (ES-); был установлен режим полной развертки, массовый диапазон от 100 до 800 amu (атомных единиц массы). Аналитический способ 4. Оборудование для ВЭЖХ состояло из системы ВЭЖХ Waters 2790, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ, снабженным источником ионизации электроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 25 С, при скорости потока 1 мл/мин, используя колонку RP 18 Waters XTerra (3,020 мм). Подвижная фаза А представляла собой буфер гидроксида аммония 0,05% рН 10 с ацетонитрилом (95:5), и подвижная фаза В представляла собой H2O/ацетонитрил (5:95); использовали градиент от 10 до 90% В в течение 4 мин, затем удерживание при 90% В в течение 1 мин. Объем вводимой пробы был 10 мкл. Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 2,5 кВ; температура источника составляла 120 С; напряжение на конусе было 10 В; был установлен режим полной развертки, массовый диапазон от 100 до 800 amu (атомных единиц массы). Некоторые соединения формулы (I) по настоящему изобретению, полученные в соответствии с представленными ниже примерами, были очищены при помощи препаративной ВЭЖХ. Рабочие условия определены ниже. ВЭЖХ/МС. Препаративный способ 1. Оборудование для ВЭЖХ состояло системы ВЭЖХ Waters 2790, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ, снабженным источником ионизации злектроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 25 С, при скорости потока 20 мл/мин, используя колонку RP 18 Waters XTerra 10 мкм (19250 мм). Подвижная фаза А представляла собой буфер гидроксида аммония 0,05% рН 10 с ацетонитрилом (95:5), и подвижная фаза В представляла собой ацетонитрил; использовали градиент от 10 до 90% В в течение 15 мин, затем удерживание при 90% В в течение 3 мин. Объем вводимой пробы был 10 мкл. Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 2,5 кВ; температура источника составляла 120 С; напряжение на конусе было 10 В; был установлен режим полной развертки, массовый диапазон от 100 до 800amu (атомных единиц массы). Препаративный способ 2. Оборудование для ВЭЖХ состояло системы ВЭЖХ Waters 2790, которая была снабжена детектором 2996 Waters PDA и одним квадрупольным масс-спектрометром Micromass мод. ZQ, снабженным источником ионизации электроспреем (ESI). Контроль оборудования, сбор данных и обработку данных осуществляли при помощи Empower и программы MassLynx 4.0. ВЭЖХ осуществляли при 25 С, при скорости потока 20 мл/мин, используя колонку RP 18 Waters XTerra 10 мкм (19250 мм). Подвижная фаза А представляла собой 0,1% трифторуксусной кислоты в воде/ацетонитриле (95:5), и подвижная фаза В представляла собой ацетонитрил; использовали градиент от 10 до 90% В в течение 15 мин, затем удерживание при 90% В в течение 3 мин. Объем вводимой пробы был 10 мкл.- 22017769 Работу масс-спектрометра осуществляли в режиме положительной и отрицательной ионизации, капиллярное напряжение было установлено при 2,5 кВ; температура источника составляла 120 С; напряжение на конусе было 10 В; был установлен режим полной развертки, массовый диапазон от 100 до 800 30 г (0,125 моль) этил (3-этокси-2-оксоциклогекс-3-ен-1-ил)(оксо)ацетата растворяли в 150 мл ледяной уксусной кислоты и добавляли 6,5 мл метилгидразина (0,125 моль). Смесь перемешивали при комнатной температуре в течение 6 ч. Растворитель затем выпаривали и неочищенное вещество повторно растворяли в воде, подщелачивали при помощи 30% раствора NH4OH и экстрагировали дихлорметаном. Органический слой затем сушили над Na2SO4 и концентрировали. Остаток кристаллизовали из диэтилового эфира с получением 19,2 г (выход 68%) указанного в заголовке соединения. 1H ЯМР (400 МГц, ДМСО-d6)м.д. 1,12 (т, J=6,89 Гц, 3 Н), 1,51 (т, J=6,94 Гц, 3 Н), 2,06-2,58 (м, 4 Н),3,57 (м, 1 Н), 3,86 (кв., J=6,83 Гц, 2 Н), 4,38 (кв., J=6,94 Гц, 2 Н), 6,09 (м, 1 Н). Согласно тому же способу, но используя подходящим образом замещенное гидразиновое производное, получали следующие соединения: этил 1-(2-гидроксиэтил)-7-оксо-4,5,6,7-тетрагидро-1 Н-индазол-3-карбоксилат; 1 10,0 г (42 ммоль) этил (3-этокси-2-оксоциклогекс-3-ен-1-ил)(оксо)ацетата растворяли в 100 мл этанола, добавляли 2,1 мл гидразингидрата и раствор перемешивали при кипячении с обратным холодильником в течение одного дня. Растворитель затем выпаривали и остаток повторно растворяли с дихлорметаном. Органический слой промывали водой, сушили над Na2SO4 и концентрировали. Неочищенное вещество растирали в порошок с диэтиловым эфиром и фильтровали с получением 6,0 г указанного в заголовке соединения (выход 70%). 1H ЯМР (400 МГц, ДМСО-d6)м.д. 1,28 (т, J=7,07 Гц, 3 Н), 2,04 (м, 2 Н), 2,51 (м, 2 Н), 2,87 (т, J=6,10 Гц, 2 Н), 4,27 (кв., J=7,11 Гц, 2 Н), 14,39 (с, 1 Н). Стадия 2. Этил 7-оксо-1-тритил-4,5,6,7-тетрагидро-1 Н-индазол-3-карбоксилат. 2,08 г (10,0 ммоль) этил 7-оксо-4,5,6,7-тетрагидро-1 Н-индазол-3-карбоксилата растворяли в 100 мл дихлорметана и 0,76 мл триэтиламина и добавляли 3,07 г (11,0 ммоль) трифенилметилхлорида. Раствор перемешивали при комнатной температуре в течение 6 ч. Затем раствор далее разбавляли дихлорметаном и промывали водой. Органический слой обрабатывали с помощью безводного Na2SO4 и упаривали досуха. Конечный продукт получали при помощи кристаллизации из диэтилового эфира (3,24 г, выход 72%). 1 16 г (72 ммоль) этил 1-метил-7-оксо-4,5,6,7-тетрагидро-1 Н-индазол-3-карбоксилата растворяли в 100 мл диметилформамида и добавляли 32 мл диметилформамид-ди-трет-бутилацеталя. Смесь перемешивали при 60 С в течение 8 ч. Растворитель затем выпаривали в вакууме и продукт кристаллизовали из этанола с получением указанного в заголовке соединения (17,96 г, выход 90%). 1 К раствору 16,62 г (60 ммоль) этил 6-[(диметиламино)метилен]-7-оксо-1-метил-4,5,6,7-тетрагидро 1 Н-индазол-3-карбоксилата в 0,5 л ДМФА, добавляли 27 г (150 ммоль) гуанидинкарбоната. Смесь перемешивали при 110 С в течение ночи. После охлаждения смесь вливали в воду (2,5 л) и перемешивали в течение 30 мин. Осадок фильтровали, промывали водой и сушили с получением 26,83 г указанного в заголовке соединения (91%). 1 К тщательно перемешиваемой суспензии этил 8-амино-1-метил-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксилата (9,0 г, 33 ммоль) в диметоксиэтане (0,7 л), в атмосфере N2, последовательно добавляли йодид цезия (8,6 г, 33 ммоль), бисублимированный йод (4,19 г, 16,5 ммоль), йодид меди (2,0 г,10 ммоль) и изопентилнитрит (6,62 мл, 49,5 ммоль). Реакционную смесь тщательно перемешивали при 65-70 С в течение 3 ч. После охлаждения в бане лед-вода твердое вещество отфильтровывали. Фильтрат- 24017769 разбавляли дихлорметаном (2,0 л), промывали 30% раствором гидроксида аммония (150 мл), тиосульфатом натрия (300 мл), насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали с получением 5,69 г указанного в заголовке соединения (выход 46%). 1 Ацетат палладия [Pd(OAc)2] (101 мг, 0,45 ммоль), (+)-BINAP (280 мг, 0,45 ммоль) и диметилформамид (65 мл) загружали в круглодонную колбу после ее продувки аргоном. Из колбы откачивали газ и снова заполняли аргоном. Смесь перемешивали в атмосфере аргона в течение 30 мин и добавляли к смеси 2-(трет-бутоксикарбониламино)анилина (2,6 г, 12,5 ммоль), этил 8-йод-1-метил-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксилата (1,6 г, 4,16 ммоль) и карбоната калия (5,74 г, 41,6 ммоль) в диметилформамиде (50 мл). Полученную смесь перемешивали при 70 С в течение 6 ч в атмосфере аргона. После охлаждения до комнатной температуры реакционную смесь фильтровали на слое целита. Растворитель концентрировали, неочищенное твердое вещество очищали при помощи флэш-хроматографии на силикагеле (элюент: гексан/этилацетат 60/40) с получением 1,18 г (61% выход) указанного в заголовке соединения. 1 0,85 г (1,83 ммоль) этил 1-метил-8-(2-(трет-бутоксикарбониламинофениламино-4,5-дигидро-1 Нпиразоло[4,3-h]хиназолин-3-карбоксилата растворяли в 50 мл дихлорметана, к раствору добавляли 30 мл 4 н. раствора HCl в диоксане. Раствор перемешивали при комнатной температуре 2 ч и растворитель удаляли в вакууме. Остаток кристаллизовали из диэтилового эфира с получением 0,70 г указанного в заголовке соединения (выход 96%). 1 Н ЯМР (400 МГц, ДМСО-d6)м.д. (в виде свободного основания) 1,32 (т, J=7,1 Гц, 3 Н), 2,82 (м,4 Н), 2,96 (м, 2 Н), 4,19 (с, 3 Н), 4,28 (кв., J=7,1 Гц, 2 Н), 4,85 (шир.с, 2 Н), 6,58 (м, 1 Н), 6,76 (м, 1 Н), 6,90 (м,1 Н), 7,32 (м, 1 Н), 8,32 (с, 1 Н), 8,52 (с, 1 Н). Пример 8. Этил 1-метил-8-[2-(3-метилбутириламино)фениламино]-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксилат (A35B1C2Z) К раствору гидрохлорида этил 1-метил-8-(2-аминофениламино)-4,5-дигидро-1 Н-пиразоло[4,3h]хиназолин-3-карбоксилата (0,25 г, 0,62 ммоль) и DIPEA (0,44 мл, 2,56 ммоль) в метиленхлориде (25 мл)- 27017769 в атмосфере азота добавляли изовалерилхлорид (0,076 мл, 0,62 ммоль), растворенный в метиленхлориде(д, J=12 Гц, 2 Н), 2,84 (м, 2 Н), 2,97 (м, 2 Н), 4,18 (с, 3 Н), 4,30 (кв., J=7,12 Гц, 1 Н), 7,21 (м, 1 Н), 7,23 (м, 1 Н),7,43 (д, J=7,7 Гц, 1 Н), 7,80 (д, J=7,7 Гц, 1 Н), 8,37 (с, 1 Н), 8,47 (шир.с, 1 Н), 9,68 (шир.с, 1 Н). Осуществляя обработку в соответствии с описанным выше способом, после получения хлорангидрида карбоновой кислоты из соответствующей карбоновой кислоты, получали следующие соединения. Таблица VI Этил 1-тритил-8-(2-(трифторметоксифениламино-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин-3 карбоксилат (140 мг, 0,21 ммоль) в ДХМ (10 мл) обрабатывали трифторуксусной кислотой (1 мл). Полу- 28017769 ченную смесь перемешивали при комнатной температуре в течение 1 ч и растворитель удаляли в вакууме. Остаток снова растворяли в дихлорметане и промывали насыщенным раствором NaHCO3. Органический слой затем сушили над Na2SO4 и растворитель выпаривали досуха. Неочищенное твердое вещество очищали при помощи флэш-хроматографии на силикагеле (элюент: гексан/этилацетат 60/40) с получением продукта с количественным выходом, 88 мг указанного в заголовке соединения. 1 Этил 8-2-[S)-N-FMOC-пирролидин-2-карбонил)амино]фениламино-1-метил-1 Н-пиразоло[4,3h]хиназолин-3-карбоксилат (200 мг, 0,29 ммоль) суспедировали в смеси 20 мл этанола и 20 мл 30% раствора NH4OH. Температуру смеси поддерживали при 65 С при перемешивании в течение 12 ч в закрытом сосуде. Растворитель затем выпаривали досуха, остаток повторно растворяли при помощи дихлорметана и промывали водой. Органический слой сушили над Na2SO4 и упаривали. Неочищенный продукт очищали при помощи флэш-хроматографии на силикагеле (элюент: дихлорметан/метанол 94/6) с получением 60 мг (выход 47%) указанного в заголовке соединения. 1 Этил 1-метил-8-(2-трифторметокси-4-бромфениламино]-4,5-дигидро-1 Н-пиразоло[4,3-h]хиназолин 3-карбоксилат (330 мг, 0,64 ммоль) суспедировали в 10 мл тетрагидрофурана. Добавляли хлорид аммония (106 мг, 2,0 ммоль) и 1 н. раствор LiN(TMS)2 в ТГФ (4,0 мл, 4,0 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 ч. Растворитель затем выпаривали досуха, остаток суспендировали в воде и фильтровали с получением 288 мг (выход 93%) указанного в заголовке соединения. 1
МПК / Метки
МПК: C40B 40/04, A61P 35/00, C07D 487/04, A61K 31/519, C07D 295/135, C07D 295/155
Метки: производные, качестве, ингибиторов, киназы, замещенные, применение, способ, пиразолохиназолиновые, получения
Код ссылки
<a href="https://eas.patents.su/30-17769-zameshhennye-pirazolohinazolinovye-proizvodnye-sposob-ih-polucheniya-i-ih-primenenie-v-kachestve-ingibitorov-kinazy.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные пиразолохиназолиновые производные, способ их получения и их применение в качестве ингибиторов киназы</a>
Замещённые пирролопиразольные производные в качестве ингибиторов киназы

Номер патента: 10596
Опубликовано: 30.10.2008
Авторы: Орци Фабрицио, Певарелло Паоло, Руссель Патрик, Браска Мария Габриелла, Фанчелли Даниеле, Вульпетти Анна, Нези Марчелла, Орзини Паоло, Амичи Раффаэлла
МПК: A61P 25/28, A61K 31/4162, A61P 17/00...
Метки: производные, качестве, пирролопиразольные, ингибиторов, киназы, замещённые
Формула / Реферат:
1. Способ лечения заболеваний, связанных с пролиферацией клеток, которые вызваны и/или связаны с измененной киназной активностью, зависящей от клеточного цикла, путем введения нуждающемуся в лечении млекопитающему эффективного количества соединения, представленного формулами (Ia) и (Ib) в которых R означает группу -CORa, -CONHRa или -CONRaRb, где Ra и Rb, каждый независимо, означают водород или необязательно замещенную группу, выбранную из...
Замещённые бензазолы и их применение в качестве ингибиторов киназы raf

Номер патента: 7987
Опубликовано: 27.02.2007
Авторы: Ренхауэ Пол А., Рамуртхи Савитхри, Левайн Берри Хаскелл, Сунг Леонард, Эмайри Пэйман, Субраманиан Шарадха, Фэнтл Уэнди, Пун Дэниел Дж.
МПК: C07D 401/14, C07D 401/12, A61K 31/41...
Метки: применение, киназы, замещённые, бензазолы, качестве, ингибиторов
Формула / Реферат:
1. Соединение формулы (I) где X1 и Х2 независимо выбраны из =N-, -NR4-, -О- или -S- при условии, что если X1 представляет собой -NR4-, -О- или -S-, тогда Х2 представляет собой =N-, или если Х2 представляет -NR4-, -О- или -S-, тогда X1 представляет собой =N-, и как X1, так и Х2 не являются =N-; Y представляет О или S; Y представляет О или S; А1 представляет замещенный или незамещенный С1-С12алкил, С3-С8циклоалкил, С3-С8гетероциклоалкил,...
Новые аминоциклические производные мочевины, их получение и фармацевтическое применение в качестве ингибиторов киназы

Номер патента: 11088
Опубликовано: 30.12.2008
Авторы: Бушар Эрве, Бенар Дидье, Гюссреген Штефан, Лебрэн Анн, Штробель Хартмут, Иттэнжер Огюстэн, Риттер Курт, Немесек Консепсьон, Эль Амад Юссеф, Лесюисс Доминик, Руф Свен
МПК: C07D 401/06, A61K 31/4166, C07D 401/14...
Метки: получение, фармацевтическое, новые, ингибиторов, аминоциклические, мочевины, применение, производные, киназы, качестве
Формула / Реферат:
1. Соединение формулы (I) в которой р представляет собой целые числа 0, 1 и 2; A представляет собой арильный, гетероарильный или моноциклический или бициклический конденсированный карбоциклический или гетероциклический 5-11-членный радикал, все указанные радикалы необязательно замещены одним или несколькими заместителями, которые могут быть одинаковыми или различными, выбранными из значений R3; X представляет собой прямую связь или следующие...
Производные пиримидина и их применение в терапии в качестве ингибиторов киназы csbp/rk/p38

Номер патента: 12875
Опубликовано: 30.12.2009
Авторы: Боэм Джеффри С., Вань Цзехун, Нортон Бет А., Невинс Нейса, Ливия Стефано, Купер Энтони Уилльям Джеймс, Каллахан Джеймс Фрэнсис
МПК: A61K 31/5365, A61P 29/00, A61K 31/519...
Метки: производные, применение, терапии, киназы, пиримидина, ингибиторов, качестве
Формула / Реферат:
1. Соединение формул в которых G3 представляет CH2; G4 представляет CH; R1 представляет C(Z)N(R10')(CR10R20)vRb, C(Z)O(CR10R20)vRb, N(R10')C(Z)(CR10R20)vRb, N(R10')C(Z)N(R10')(CR10R20)vRb или N(R10')OC(Z)(CR10R20)vRb; R1' независимо выбран в каждом случае из атома галогена, C1-4алкила, замещенного атомом галогена C1-4алкила, циано, нитро, (CR10R20)v'NRdRd', (CR10R20)v'C(О)R12, SR5, S(O)R5, S(O)2R5 или (CR10R20)v'OR13; Rb представляет атом...
Замещённые бензазолы и их применение в качестве ингибиторов raf-киназы

Номер патента: 11890
Опубликовано: 30.06.2009
Авторы: Ренхауэ Пол А., Пун Дэниел Дж., Фантл Уэнди, Левайн Бэрри Хэскелл, Рамуртхи Савинтхри, Амири Пэйман, Сунг Леонард, Субраманиан Шарадха
МПК: A61K 31/41, C07D 401/14, C07D 401/12...
Метки: бензазолы, замещённые, ингибиторов, применение, raf-киназы, качестве
Формула / Реферат:
1. Соединение формулы (I) где пунктирная линия представляет собой простую или двойную связь; X1 и Х2 независимо выбраны из =N-, -NR4-, -О- или -S- при условии, что если X1 представляет собой -NR4-, -О- или -S-, то Х2 представляет собой =N-, или если Х2 представляет собой -NR4-, -O- или -S-, то X1 представляет собой =N-, и оба X1 и Х2 не представляют собой =N-; Y представляет собой O; A1 представляет собой замещенный или незамещенный С3-С14арил;...
Предыдущий патент: Устройство регулирования мощности транспортной системы
Следующий патент: Способы лечения с использованием гликопэгилированного g-csf
Случайный патент: Игровой автомат