Циклические пептиды – антагонисты cxcr4





























Формула / Реферат
1. Пептид - антагонист CXCR4, включающий лактамный цикл и соответствующий формуле I

причем:
a) указанный лактам образован амидной связью между аминогруппой боковой цепи Х1 и карбоксильной группой боковой цепи Х7, и при этом X1 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из (D/L)Agl/Glu, Dab/Glu и Dap/Glu, и R1 представляет собой Ас или н-гексаноил; или
b) указанный лактам образован амидной связью между карбоксильной группой боковой цепи фрагмента Х1, и аминогруппой боковой цепи фрагмента Х7, и при этом Х1 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из Asp/(D/L)Agl, Asp/Dab, Asp/Dap, Glu/(D/L)Agl, Glu/Dab, Glu/Dap, Glu/DDap и Glu/Lys, и R1 представляет собой Ас или Bz, или при этом Х1 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из сукцинила/(D/L)Agl, сукцинила/Dab, сукцинила/Dap, сукцинила/Lys и сукцинила/Orn, и R1 отсутствует; или
c) указанный лактам образован амидной связью, между α-аминогруппой фрагмента X1 и карбоксильной группой боковой цепи Х7, при этом X1 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из Ala/Glu, Ala/DGlu, DAla/Glu, DAla/DGlu, Dap(Ac)/Glu, Gly/Asp, Gly/Glu, Gly/DGlu, Leu/Glu, Leu/DGlu, Lys/DGlu, Lys(Ac)/Glu, 2Nal/Glu, Phe/Glu, Phe/DGlu, DPhe/Glu и DPhe/DGlu, и R1 отсутствует; или
d) указанный лактам образован амидной связью между аминогруппой, не являющейся аминогруппой боковой цепи Х1 или α-аминогруппой, и карбоксильной группой боковой цепи Х7, и при этом Х1 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из β-Ala/Asp, β-Ala/Glu, 5-аминовалерила/Asp, 5-аминовалерила/Glu, 4-AMB/Glu, 4-AMPA/Asp и 4-AMPA/Glu, и R1 отсутствует; или
е) указанный лактам образован амидной связью между α-аминогруппой Х2 и карбоксильной группой боковой цепи Х7, и при этом Х2 и Х7, соответственно, представляют собой пару, выбранную из группы, состоящей из Tyr/Asp, Tyr/Glu и Tyr/DGlu, и как R1, так и X1 отсутствует;
R1 представляет собой заместитель на α-аминогруппе Х1, в случае если Х1 содержит α-аминогруппу, и указанная α-аминогруппа не участвует в образовании амидной связи указанного лактама, и при этом R1 выбран из группы, состоящей из Ac, Bz и н-гексаноила, или отсутствует, и при этом Х1 выбран из группы, состоящей из (D/L)Agl, Asp, Dab, Dap и Glu;
X1 выбран из группы, состоящей из (D/L)Agl, Ala, β-Ala, DAla, 5-аминовалерила, 4-AMB, 4-AMPA, Asp, Dab, Dap, Dap(Ac), Glu, Gly, Leu, Lys, Lys(Ac), 2Nal, Phe, DPhe и сукцинила, или отсутствует;
Х3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys(Me2);
X7 выбран из группы, состоящей из (D/L)Agl, Asp, Dab, Dap, DDap, Glu, DGlu, Lys и Orn;
X8 выбран из группы, состоящей из β-Ala, Arg, DArg, Gly, Lys, Lys(iPr) и Orn, или отсутствует;
X9 выбран из группы, состоящей из Gly, 2Nal, D2Nal и DPhe, или отсутствует;
Х10 представляет собой 2Nal или отсутствует;
при этом, если Х8 отсутствует, то как Х9, так и Х10 отсутствует, и, если Х9 отсутствует, то Х10 отсутствует, и
R2 выбран из группы, состоящей из NH2 и NHEt, или фармацевтически приемлемая соль указанного пептида.
2. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по п.1, отличающийся тем, что
R1 выбран из группы, состоящей из Ас и Bz, или отсутствует;
Х1 выбран из группы, состоящей из β-Ala, 4-АМВ, 4-АМРА, Asp, Dab, Dap, Dap(Ac), Glu, 2Nal, Phe и сукцинила, или отсутствует;
Х3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys (Me2);
Х7 выбран из группы, состоящей из Asp, Dab, Dap, Glu, DGlu, Lys и Orn;
X8 выбран из группы, состоящей из Arg и Lys, или отсутствует;
Х9 отсутствует;
Х10 отсутствует и
R2 выбран из группы, состоящей из NH2 и NHEt.
3. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по п.1, отличающийся тем, что
R1 выбран из группы, состоящей из Ас и Bz, или отсутствует;
Х1 выбран из группы, состоящей из DAla, 5-аминовалерила, 4-АМРА, Asp, Glu, Leu, Lys(Ac), Phe, DPhe, и сукцинила;
X3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys(Me2);
X7 выбран из группы, состоящей из (D/L)Agl, Asp, Dab, Dap, DDap, Glu и DGlu;
X8 выбран из группы, состоящей из Arg, DArg и Lys, или отсутствует;
Х9 отсутствует;
Х10 отсутствует и
R2 выбран из группы, состоящей из NH2 и NHEt.
4. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по п.1, отличающийся тем, что
R1 выбран из группы, состоящей из Ac, Bz, и н-гексаноила, или R1 отсутствует;
X1 выбран из группы, состоящей из (D/L)Agl, Ala, β-Ala, Asp, Dap, Glu, Gly, Lys и Phe;
X3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys(Me2);
X7 выбран из группы, состоящей из (D/L)Agl, Asp, Dap, Glu и DGlu;
X8 выбран из группы, состоящей из β-Ala, Arg, Gly, Lys, Lys(iPr) и Orn, или отсутствует;
X9 выбран из группы, состоящей из Gly, 2Nal, D2Nal и DPhe, или отсутствует;
Х10 представляет собой 2Nal или отсутствует и
R2 выбран из группы, состоящей из NH2 и NHEt.
5. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по п.1, отличающийся тем, что
R1 выбран из группы, состоящей из Ас и Bz, или отсутствует;
X1 выбран из группы, состоящей из Ala, 5-аминовалерила, Asp, Glu, Gly, Phe, DPhe и сукцинила;
X3 выбран из группы, состоящей из Arg, Lys(iPr) и Lys(Me2);
Х7 выбран из группы, состоящей из (D/L)Agl, Asp, Dap, Glu и DGlu;
X8 выбран из группы, состоящей из β-Ala, Arg, Gly, Lys, Lys(iPr) и Orn, или отсутствует;
Х9 выбран из группы, состоящей из Gly, D2Nal и DPhe, или отсутствует;
Х10 представляет собой 2Nal или отсутствует; и
R2 выбран из группы, состоящей из NH2 и NHEt.
6. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по пп.1, 4 или 5, при этом
Х1 выбран из группы, состоящей из Gly и Phe; Х3 представляет собой Lys(iPr) и Х7 представляет собой DGlu.
7. Пептид, включающий лактамный цикл, или фармацевтически приемлемая соль указанного пептида по п.5, отличающийся тем, что
R1 отсутствует;
X1 выбран из группы, состоящей из Gly и Phe;
Х3 представляет собой Lys(iPr);
Х7 представляет собой DGlu;
Х8 выбран из группы, состоящей из Arg и Lys(iPr), или отсутствует;
Х9 отсутствует;
Х10 отсутствует и
R2 выбран из группы, состоящей из NH2 и NHEt.
8. Пептид - антагонист CXCR4, включающий лактамный цикл, соответствующий формуле

или фармацевтически приемлемая соль указанного пептида.
9. Пептид, включающий лактамный цикл, по п.8, отличающийся тем, что указанная фармацевтически приемлемая соль представляет собой соль уксусной кислоты.
10. Фармацевтическая композиция, содержащая пептид, включающий лактамный цикл, или фармацевтически приемлемую соль указанного пептида по любому из пп.1-9, и фармацевтически приемлемый носитель, разбавитель или эксципиент.
11. Применение пептида, включающего лактамный цикл, или его фармацевтически приемлемой соли по любому из пп.1-9 в лечении заболеваний, в патогенезе которых участвуют CXCR4 и SDF-1.
12. Применение пептида, включающего лактамный цикл, или его фармацевтически приемлемой соли по любому из пп.1-9 в лечении ревматоидного артрита, фиброза легких, ВИЧ-инфекции или рака, выбранного из группы, состоящей из рака молочной железы; рака поджелудочной железы; меланомы; рака предстательной железы; рака почек; нейробластомы; неходжкинской лимфомы; рака легких; рака яичников; рака прямой и ободочной кишки; множественной миеломы; мультиформной глиобластомы и хронического лимфолейкоза.
13. Применение пептида, включающего лактамный цикл, или фармацевтически приемлемой соли указанного пептида по любому из пп.1-9 для получения лекарственного средства для лечения ревматоидного артрита, фиброза легких, ВИЧ-инфекции или рака, выбранного из группы, состоящей из рака молочной железы; рака поджелудочной железы; меланомы; рака предстательной железы; рака почек; нейробластомы; заболевания, отличного от лимфомы Ходжкина; рака легких; рака яичников; рака прямой и ободочной кишки; множественной миеломы; мультиформной глиобластомы и хронического лимфолейкоза.
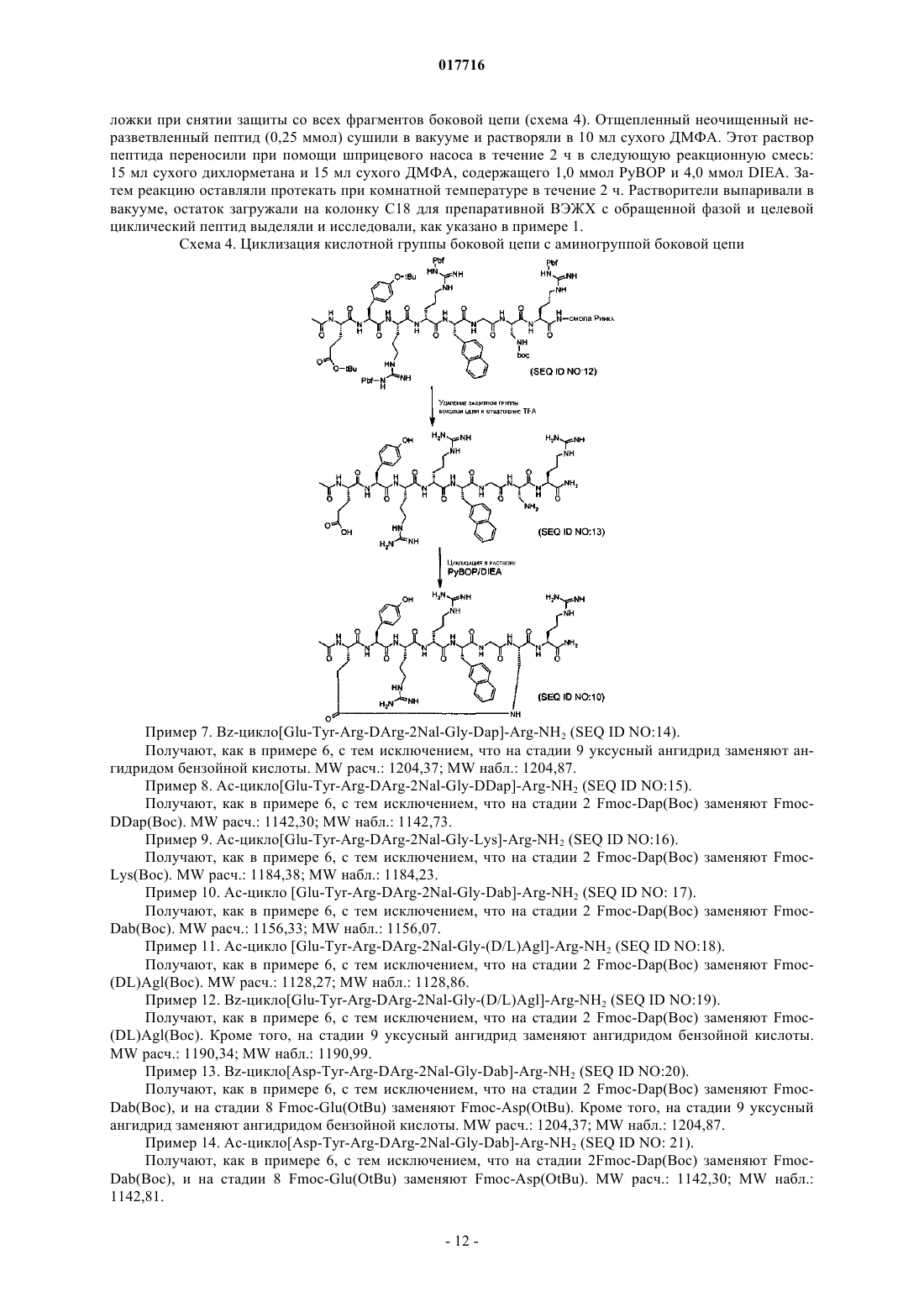
Текст
Изобретение относится к антагонистам CXCR4, представляющим собой пептиды, включающие лактамный цикл, применяемым для лечения раковых заболеваний, ревматоидного артрита, фиброза легких и ВИЧ-инфекции. Кон Вэйн Дэвид, Пэн Шэн-бинь, Янь Лян Цзэн (US) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 017716 Изобретение относится к новым соединениям - циклическим пептидам - антагонистам CXCR4 и их применению в лечении заболеваний, в патогенезе которых участвуют CXCR4 и SDF-1.CXCR4, рецептор, конъюгированный с G-белком, и его природный лиганд - фактор-1 клеток стромы (SDF-1; CXCL12) представляют собой пару хемокиновый рецептор-лиганд. CXCR4 конститутивно или в избытке экспрессируется во многих видах раковых опухолей человека. SDF-1, единственный известный лиганд CXCR4, экспрессируется на высоком уровне в микроокружении опухолей, а также в костном мозге, легких, печени и лимфатических узлах, т.е. органах-мишенях, наиболее часто вовлеченных в образование метастазов. Взаимодействие CXCR4/SDF-1 играет важную роль на многих стадиях развития опухолей, включая рост опухоли, инвазию, ангиогенез и образование метастазов, а также в развитии ревматоидного артрита, фиброза легких и ВИЧ-инфекции (Tsutsumi et al. (2006) Peptide Science 88(2):279289). Ввиду участия CXCR4/SDF-1 в развитии этих серьезных заболеваний, CXCR4 является привлекательной мишенью терапевтических воздействий.AMD3100, бицикламный антагонист CXCR4, в настоящее время проходит III фазу клинических испытаний, в отношении его применения для мобилизации стволовых клеток при трансплантации или стволовых клеток у пациентов с множественной миеломой и неходжкинской лимфомой. AMD070 - другой низкомолекулярный антагонист CXCR4, в настоящее время проходит II фазу клинических испытаний в отношении его применения при ВИЧ-инфекции. СТСЕ 9908, двухвалентный (димерный) пептидный антагонист CXCR4 в настоящее время проходит фазу Ib/II клинических испытаний в отношении его применения при раке. FC131 - циклический пентапептидный антагонист CXCR4 ингибирует связывание 125Araki et al. (2003) Peptide Science. The Japanese Peptide Society (2004):207-210). Существует необходимость в улучшенных антагонистах CXCR4, которые являлись бы эффективными и селективными, проявляли низкую активность в отношении рецепторов хемокинов или вообще проявляли такую активность. Соединения, предложенные в настоящем изобретении, являются такими эффективными и селективными антагонистами CXCR4. Высокая эффективность позволяет применять низкие дозы при режимах терапии, тогда как высокая селективность минимизирует нежелательные побочные эффектов, связанные с органами - не мишенями. Кроме того, соединения, предложенные в настоящем раскрытии, обладают другими особенно благоприятными фармакологическими свойствами,такими как высокая биодоступность при подкожном введении, хорошая метаболическая стабильность invivo и фармакокинетические/фармакодинамические свойства, которые дают возможность удобного введения. Соответственно, согласно первому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, формулы Ia) указанный лактам образован амидной связью между аминогруппой боковой цепи X1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из (D/L)Agl/Glu, Dab/Glu и Dap/Glu, и R1 представляет собой Ас или nгексаноил; илиb) указанный лактам образован амидной связью между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7, и при этом Х 1 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из (D/L)Agl/Glu, Dab/Glu и Dap/Glu, a R1 представляет собой Ас или Bz, или где X1 и X7, соответственно, представляют собой пару, выбранную из группы, состоящей из сукцинил/(D/L) Agl, сукцинил/Dab, сукцинил/Dap, сукцинил/Lys и сукцинил/Orn, a R1 отсутствует; илиc) указанный лактам образован амидной связью между -аминогруппой Х 1 и карбоксильной группой боковой цепи X7, где X1 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из Ala/Glu, Ala/DGlu, DAla/Glu, DAla/DGlu, Dap(Ac)/Glu, Gly/Asp, Gly/Glu, Gly/DGlu, Leu/Glu,Leu/DGlu, Lys/DGlu, Lys(Ac)/Glu, 2Nal/Glu, Phe/Glu, Phe/DGlu, DPhe/Glu и DPhe/DGlu, a R1 отсутствует; илиd) указанный лактам образован амидной связью между аминогруппой, не являющейся аминогруппой или аминогруппой боковой цепи X1, и карбоксильной группой боковой цепи X1, где X1 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из -Ala/Asp, -Ala/Glu,5-аминовалерил/Asp, 5-аминовалерил/Glu, 4-AMB/Glu, 4-AMPA/Asp и 4-AMPA/Glu, и R1 отсутствует; илиe) указанный лактам образован амидной связью между -аминогруппой Х 2 и карбоксильной группой боковой цепи Х 7, где Х 2 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из Tyr/Asp, Tyr/Glu и Tyr/DGlu, a R1 и X1 оба отсутствуют.R1 представляет собой заместитель -аминогруппы X1, где X1 содержит -аминогруппу, и указанная -аминогруппа не участвует в образовании амидной связи указанного лактама, выбранного из груп-1 017716 пы, состоящей из Ac, Bz и n-гексаноил, или отсутствует, где X1 выбран из группы, состоящей из D/L)Agl,Asp, Dab, Dap и Glu;X9 выбран из группы, состоящей из Gly, 2Nal, D2Nal и DPhe, или отсутствует; Х 10 представляет собой 2Nal, или отсутствует; где при отсутствии Х 8 отсутствуют и Х 9 и Х 10, при отсутствии X9, отсутствует и Х 10, иR2 выбран из группы, состоящей из NH2 и NHEt или их фармацевтически приемлемые соли. Другими словами, это эквивалентно лактам-циклизованному пептиду, соответствующему формуле где R1 представляет собой заместитель -аминогруппы X1, где X1 содержит -аминогруппу, и указанная -аминогруппа не является составляющей частью амидной связи указанного лактама, выбранного из группы, состоящей из Ac, Bz и n-гексаноил, или отсутствует,где X1 выбран из группы, состоящей из D/L)Agl, Asp, Dab, Dap и Glu;X9 выбран из группы, состоящей из Gly, 2Nal, D2Nal и DPhe, или отсутствует; Х 10 представляет собой 2Nal или отсутствует; где при отсутствии Х 8 отсутствуют и Х 9 и Х 10, при отсутствии Х 9, отсутствует и X10, иR2 выбран из группы, состоящей из NH2 и NHEt или их фармацевтически приемлемые соли,причем:a) указанный лактам образован амидной связью между аминогруппой боковой цепи X1 и карбоксильной группой боковой цепи Х 7, и при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из (D/L) Agl/Glu, Dab/Glu и Dap/Glu, a R1 представляет собой Ас или nгексаноил; илиb) указанный лактам образован амидной связью между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7, и при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Asp/(D/L)Agl, Asp/Dab, Asp/Dap, Glu/(D/L) Agl, Glu/Dab, Glu/Dap,Glu/DDap и Glu/Lys, a R1 представляет собой Ас или Bz, или где X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из сукцинил/(D/L)Agl, сукцинил/Dab, сукцинил/Dap, сукцинил/Lys и сукцинил/Orn, и R1 отсутствует; илиc) указанный лактам образован амидной связью между -аминогруппой Х 1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Ala/Glu, Ala/DGlu, DAla/Glu, DAla/DGlu, Dap(Ac)/Glu, Gly/Asp, Gly/Glu, Gly/DGlu,Leu/Glu, Leu/DGlu, Lys/DGlu, Lys(Ac)/Glu, 2Nal/Glu, Phe/Glu, Phe/DGlu, DPhe/Glu и DPhe/DGlu, и R1 отсутствует; илиd) указанный лактам образован амидной связью между аминогруппой, не являющейся -аминогруппой или аминогруппой боковой цепи X1, и карбоксильной группой боковой цепи Х 7, и при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из -Ala/Asp,-Ala/Glu, 5-аминовалерил/Asp, 5-аминовалерил/Glu, 4-AMB/Glu, 4-AMPA/Asp и 4-AMPA/Glu, и R1 отсутствует; илиe) указанный лактам образован амидной связью между -аминогруппой X1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Tyr/Asp, Tyr/Glu и Tyr/DGlu, a R1 и X1 оба отсутствуют, или фармацевтически приемлемую соль указанного пептида. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, отвечающему формуле I: или его фармацевтически приемлемую соль, в котором:X1 выбран из группы, состоящей из (D/L)Agl, Ala, -Ala, DAla, 5-аминовалерил, 4-АМВ, 4-АМРА,Asp, Dab, Dap, Dap(Ac), Glu, Gly, Leu, Lys, Lys (Ac), 2Nal, Phe, DPhe и сукцинил, или отсутствует; причем если X1 представляет собой (D/L)Agl, Dab или Dap, a -аминогруппа X1 не участвует в об-2 017716 разовании амидной связи лактама, указанная -аминогруппа содержит в качестве заместителя R1, который выбран из группы, состоящей из Fc и n-гексоноиал; причем если X1 представляет собой Asp или Glu, и -аминогруппа X1 не участвует в образовании амидной связи лактама, указанная -аминогруппа содержит в качестве заместителя R1, который выбран из группы, состоящей из Ас и Bz; и причем если X1 представляет собой Ala, -Ala, DAla, 5-аминовалерил, 4-АМВ, 4-АМРА, Dap(Ac),Gly, Leu, Lys, Lys(Ac), 2Nal, Phe, DPhe или сукцинил, R1 отсутствует; Х 3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys(Me2); Х 7 выбран из группы, состоящей из (D/L)Agl, Asp, Dab, Dap, DDap, Glu, DGlu, Lys и Orn;X9 выбран из группы, состоящей из Gly, 2Nal, D2Nal и DPhe,или отсутствует; и Х 10 представляет собой 2Nal или отсутствует; в котором при отсутствии Х 8 отсутствуют и Х 9 и Х 10, отсутствии в случае отсутствия Х 9, отсутствует и Х 10, иR2 выбран из группы, состоящей из NH2 и NHEt,в котором также указанный лактам образован амидной связью между аминогруппой боковой цепи X1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из (D/L)Agl/Glu, Dab/Glu и Dap/Glu, и R1 представляет собой Ас или n-гексаноил; или указанный лактам образован амидной связью между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7, где X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Asp/(D/L)Agl, Asp/Dab, Asp/Dap, Glu/(D/L)Agl, Glu/Dab, Glu/Dap, Glu/DDap иGlu/Lys, a R1 представляет собой Ас или Bz; или, или в котором X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из сукцинил/(D/L)Agl, сукцинил/Dab, сукцинил/Dap, сукцинил/Lys и сукцинил/ Orn, a R1 отсутствует; или указанный лактам образован амидной связью между -аминогруппой Х 1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно представляют собой пару, выбранную из группы, состоящей из Ala/Glu, Ala/DGlu, DAla/Glu, DAla/DGlu, Dap(Ac)/Glu, Gly/Asp, Gly/Glu, Gly/DGlu, Leu/Glu,Leu/DGlu, Lys/DGlu, Lys(Ac)/Glu, 2Nal/Glu, Phe/Glu, Phe/DGlu, DPhe/Glu и DPhe/DGlu, a R1 отсутствует; или указанный лактам образован амидной связью между аминогруппой, не являющейся аминогруппой или аминогруппой боковой цепи X1, и карбоксильной группой боковой цепи Х 7, при этомX1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из -Ala/Asp, Ala/Glu, 5-аминовалерил/Asp, 5-аминовалерил/Glu, 4-AMB/Glu, 4-AMPA/Asp и 4-AMPA/Glu, и R1 отсутствует; или указанный лактам образован амидной связью между -аминогруппой Tyr на Х 2 и карбоксильной группой боковой цепи Х 7, и Х 7 выбран из группы, состоящей из Asp, Glu и DGlu, a R1 и Х 1 оба отсутствуют. Повторяющимся мотивом последовательности во всех соединениях, отвечающих формуле I, является наличие Tyr в положении Х 2, DArg в положении Х 4, 2Nal в положении Х 5 и Gly в положении Х 6. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которыхR1 выбран из группы, состоящей из Ас и Bz, или отсутствует;R2 выбран из группы, состоящей из NH2 и NHEt. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которыхR1 выбран из группы, состоящей из Ас и Bz, или отсутствует; Х 1 выбран из группы, состоящей из DAla, 5-аминовалерила, 4-АМРА, Asp, Glu, Leu, Lys(Ac), Phe,DPhe и сукцинила; Х 3 выбран из группы, состоящей из Arg, Lys, Lys(iPr) и Lys(Me2);R2 выбран из группы, состоящей из NH2 и NHEt. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых:R2 выбран из группы, состоящей из NH2 и NHEt. Согласно дополнительному аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которыхR1 выбран из группы, состоящей из Ас и Bz, или отсутствует;R2 выбран из группы, состоящей из NH2 и NHEt. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которыхX1 выбран из группы, состоящей из Gly и Phe; Х 3 представляет собой Lys(iPr); и Х 7 представляет собой DGLu. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которыхR2 выбран из группы, состоящей из NH2 и NHEt. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между аминогруппой боковой цепи X1 и карбоксильной группой боковой цепи Х 7;R1 выбран из группы, состоящей из Ас и n-гексаноил;R2 представляет собой NH2. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, X1 представляет собой (D/L)Agl или Dap. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7;R1 выбран из группы, состоящей из Ас и Bz;R2 представляет собой NH2. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 7 представляет собой (D/L)Agl, Dab, Dap или DDap. Согласно более предпочтительному варианту осуществления Х 7 (D/L)Agl или Dap. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7;R2 представляет собой NH2. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 7 представляет собой (D/L)Agl или Dap. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между -аминогруппой X1 и карбоксильной группой боковой цепи Х 7;R2 выбран из группы, состоящей из NH2 и NHEt. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, X1 представляет собой Ala, DAla, Gly, Leu, Lys, Lys (Ac), Phe или DPhe. Согласно более предпочтительному варианту осуществления, X1 представляет собой Ala, Gly, Lys или Phe. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 3 представляет собой Arg, Lys, Lys(iPr) или Lys(Ме 2). Согласно более предпочтительному варианту осуществления, Х 3 представляет собой Arg. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 7 представляет собой Asp, Glu или DGlu. Согласно более предпочтительному варианту осуществления, Х 7 представляет собой Asp. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 8 представляет собой -Ala, Arg, Gly, Lys, Lys(iPr), Orn, или отсутствует. Согласно более предпочтительному варианту осуществления, Х 8 представляет собой -Ala, Gly, Lys, Lys(iPr), Orn или отсутствует. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 9 представляет собой Gly, 2Nal, D2Nal, DPhe или отсутствует. Согласно более предпочтительному варианту осуществления, Х 9 представляет собой Gly, 2Nal, D2Nal или DPhe. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 10 представляет собой 2Nal или отсутствует. Согласно более предпочтительному варианту осуществления Х 10 представляет собой 2Nal. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, R2 представляет собой NHEt. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между неаминогруппой не из боковой цепи X1 и карбоксильной группой боковой цепи Х 7;R2 представляет собой NH2. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, X1 представляет собой -Ala, 5-аминовалерил или 4-АМРА. Согласно более предпочтительному варианту осуществления, X1 представляет собой -Ala. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, Х 7 представляет собой Asp. Согласно предпочтительному варианту осуществления этого аспекта настоящего изобретения, X8 представляет собой Arg. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, отвечающим формуле I (SEQ ID NO:1), в которых указанный лактам образован амидной связью между -аминогруппой Х 2 и карбоксильной группой боковой цепи Х 7;X9 отсутствует; Х 10 отсутствует; и R2 представляет собой NH2. Согласно другому аспекту настоящего изобретения предложен пептид, включающий лактамный цикл, отвечающий формуле: или его фармацевтически приемлемой соли. Этот лактам образован амидной связью между аминогруппой Phe и карбоксильной группой боковой цепи DGlu. Фармацевтически приемлемой солью может быть соль уксусной кислоты. Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей пептид, включающий лактамный цикл, или фармацевтически приемлемую соль такого пептида, которые описаны выше в разных вариантах, и фармацевтически приемлемый носитель, разбавитель или эксципиент. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, как описано выше в разных вариантах, для применения в терапии. Согласно другому аспекту настоящее изобретение относится к пептиду, включающему лактамный цикл, или его фармацевтически приемлемой соли, которые описаны выше в разных вариантах, для лечения ревматоидного артрита, фиброза легких, инфекции ВИЧ или рака, выбранного из группы, состоящей из рака молочной железы, рака поджелудочной железы, меланомы, рака предстательной железы, рака почек, нейробластомы, не-ходжкинской лимфомы, рака легких, рака яичников, рака прямой и ободочной кишки, множественной миеломы, мультиформной глиобластомы и хронического лимфолейкоза. Согласно другому аспекту настоящее изобретение относится к применению пептида, включающего лактамный цикл, или его фармацевтически приемлемой соли, которые описаны выше в разных вариантах, для получения лекарственного средства для лечения ревматоидного артрита, фиброза легких, инфекции ВИЧ или рака, выбранного из группы, состоящей из рака молочной железы, рака поджелудочной железы, меланомы, рак предстательной железы, рака почек, нейробластомы, неходжкинской лимфомы,рака легких, рак яичников, рака прямой и ободочной кишки, множественной миеломы, мультиформной глиобластомы и хронического лимфолейкоза. Согласно другому аспекту настоящее изобретение относится к способу лечения ревматоидного артрита, фиброза легких, ВИЧ-инфекции или рака, выбранного из группы, состоящей из рака молочной железы, рака поджелудочной железы, меланомы, рака предстательной железы, рака почек, нейробластомы,неходжкинской лимфомы, рака легких, рака яичников, рака прямой и ободочной кишки, множественной-6 017716 миеломы, мультиформной глиобластомы и хронического лимфолейкоза, включающему введение пациенту эффективного количества пептида, содержащего лактамный цикл, или его фармацевтически приемлемой соли, которые описаны выше в разных вариантах. В соединениях макроциклических пептидов согласно настоящему изобретению (SEQ ID NO:1),аминокислоты с Х 1 по Х 10 обозначают стандартно используемыми для этого трехбуквенными символами,показываемыми слева направо от амино-конца к карбокси-концу. D- и L- (малые прописные буквы) означают абсолютную стереохимию. В случае, когда конкретная формула не содержит никакого обозначения, имеет место L-форма аминокислоты. Также X1 может представлять собой остаток дикарбоновой кислоты, т.е., группу сукцинила. Остаток аминокислоты или карбоновой кислоты в скобках "[ ]" входит в циклическую структуру; группы за пределами скобок находятся вне циклического кольца. Во всех случаях циклизация происходит посредством образования лактамной (амидной) связи между X1 (или Х 2, т.е.Tyr) и Х 7, механизмы образования которой могут несколько различаться в зависимости от структуры Х 1,Х 2 и Х 7. В случае образования лактамной связи между аминогруппой X1 боковой цепи и карбоксильной группой боковой цепи Х 7 (схемы 1 и 2; примеры 1-5) -аминогруппу Xi блокируют Ас или пгексаноилом. В случае образования лактамной связи между карбоксильной группой боковой цепи X1 и аминогруппой боковой цепи Х 7, -аминогруппу X1 блокируют Ас или Bz (схема 3 и 4; примеры 6-19). ТакжеX1 может представлять собой двуфункциональный остаток, отличный от -аминокислоты, например,остаток с двумя карбоксильными группами, т.е., остаток сукцинила. В таком случае одна карбоксильная группа образует амидную связь с -аминогруппой Tyr, а другая карбоксильная группа образует структуру циклического лактама посредством амидной связи с аминогруппой боковой цели Х 7 (схема 3 и 4; примеры 20-24). В большинстве пептидов, содержащих лактамный цикл, раскрытых в настоящем документе (схемы 5-15; примеры 25-28, 32-66 и 75-89), -аминогруппа X1 образует структуру лактама посредством амидной связи с карбоксильной группой боковой цепи Х 7, a R1 отсутствует. Также схемы синтеза данной категории включают циклические пептиды, содержащие остатки Х 1, т.е., -Ala, 4-АМВ, 5-аминовалерил и 4-АМРА, в которых аминогруппа находится в не боковой цепи, и это не -аминогруппа (схемы 5 и 6; примеры 67-74). R1 также в этих случаях отсутствует. Когда и R1 и X1 отсутствуют, структура лактама образуется посредством амидной связи между аминогруппой Tyr(Х 2) и карбоксильной группой Х 7 (схемы 5 и 6; примеры 29-31). Структуры распространенных аминокислот, т.е. аланина, глицина и др., хорошо известны в технике. Структуры нестандартных и замещенных аминокислот, присутствующих в настоящем изобретении,показаны ниже. Пептиды, включающие лактамный цикл, согласно настоящему изобретению можно получать в форме их фармацевтически приемлемых солей. Такие соли и общепринятая методология их получения хорошо известны в технике. См., например, P. Stahl et al. (2002) Handbook of Pharmaceutical Salts: Properties, Selection and Use, VCHA/Wiley-VCH; Berge et al. (1977) "Pharmaceutical Salts," Journal of Pharmaceuti-7 017716cal Sciences 66(1):1-19. Соединения, предложенные в настоящем изобретении, являются эффективными антагонистами взаимодействия CXCR4/SDF-1. Соединения, отвечающие формуле (I), и фармацевтически приемлемые соли таких соединений,конкретно указанные в настоящей заявке в качестве примеров, демонстрируют среднее значение Ki, приблизительно равное 7,5 нМ или менее, которое определяли путем анализа связывания CXCR4/125I-SDF1, описанного ниже. Более предпочтительные соединения, отвечающие формуле (I), и фармацевтически приемлемые соли таких соединений демонстрируют в этом тесте среднее значение Ki в диапазоне приблизительно 0,2-1 нМ. Самые предпочтительные соединения, отвечающие формуле (I), и их фармацевтически приемлемые соли проявляют в данном анализе среднее значение Ki приблизительно 0,2 нМ. Кроме того, соединения и их фармацевтически приемлемые соли, предложенные в настоящем изобретении, предпочтительно высоко селективны в отношении рецептора CXCR4, и при этом проявляют низкую активность в отношении других хемокиновых рецепторов CCR1, CCR2, CXCR2, CXCR3 и других рецепторов, сопряженных с G-белком, или вообще не проявляя такой активности в исследованных концентрациях, и не проявляют значительной активности в отношении рецепторов серотонина, дофамина и опиоидов. Также они предпочтительно проявляют хорошую стабильность в крови и плазме, хорошую биодоступность при подкожном введении, желаемые фармакокинетические/фармакодинамические свойства и высокую эффективность in vivo в отношении подавления роста опухолей, с широким диапазоном безопасности. Благодаря таким фармакологическим свойствам соединения, предложенные в настоящем изобретении, показаны для лечения нарушений, включающих взаимодействие CXCR4/SDF-1 или активность рецепторов CXCR4, таких как инфекция ВИЧ. В частности, настоящие соединения полезны в лечении злокачественного роста, в патогенез которого задействованы механизмы ангиогенеза, роста, выживания и метастазирования, опосредуемые CXCR4 и SDF-1, включая рак молочной железы, рак поджелудочной железы, меланому, рак предстательной железы, рак почек, нейробластому, не-ходжкинскую лимфому,рак легких, рак яичников, рак прямой и ободочной кишки, множественную миелому, мультиформную глиобластому и хронический лимфолейкоз, а также ревматоидный артрит, фиброз легких и инфекцию ВИЧ (Tsutsumi et al. (2006) Peptide Science 88 (2):279-289).Agl (аминоглицин) является прохиральным структурным элементом. Когда его остаток присутствует в формуле пептида в настоящем документе, -атом углерода становится хиральным центром, в котором каждая из двух ассоциированных -аминогруппы отдельно связана с разными компонентами. В этом случае, конечный пептидный продукт содержит два диастереоизомера, которые не разделяются на отдельные части, и которые могут присутствовать в соотношении, отличном от 1:1. "(D/L)Agl" в формуле пептида означает такую смесь диастереоизомеров. "(DL)Agl" означает производное Agl, которое является рацемическим, например, Fmoc-(DL)Agl(Boc)."Значения Ki" вычисляют с применением значений IC50, определенных при анализе связыванияStructure, Mechanism, and Data Analysis, Robert A. Copeland, Wiley-VCH, New York, 1996, стр. 207. Термин "SDF-1" включает две изоформы - SDF-1 и SDF-1, которые, как сейчас предполагают,проявляют сходную функциональность. Термин "лечение" в настоящем документе означает терапевтическое лечение нарушений, ассоциированных с взаимодействием CXCR4/SDF-1 или активностью рецепторов CXCR4. Терапевтическое лечение означает процессы, подразумевающие замедление, прерывание, приостановку или прекращение прогрессирования заболевания, но не обязательно подразумевает полное устранение симптомов, патологических состояний или нарушений. Соединения, предложенные в настоящем изобретении можно применять в качестве лекарственных средств, вводимых разными путями, у людей или в ветеринарии. Наиболее предпочтительны композиции для парентерального введения. Такие фармацевтические композиции можно получать при помощи методов, хорошо известных в данной области. См., например, Remington: The Science and Practice of Pharmacy, 19th ed. (1995), A. Gennaro et al., Mack Publishing Co., и они состоят из соединений, отвечающих формуле (I) или их фармацевтически приемлемых солей, и фармацевтически приемлемых носителя, растворителя или наполнителя. Эффективное количество соединений согласно настоящему изобретению лежит в диапазоне приблизительно от 1 мг до приблизительно 300 мг, более предпочтительно от приблизительно 1 мг до приблизительно 200 мг, еще более предпочтительно от приблизительно 1 мг до приблизительно 100 мг и даже более предпочтительно от приблизительно 1 мг до приблизительно 50 мг, которое вводят раз в день. Все пептиды, содержащие лактамный цикл, предложенные в настоящем изобретении, можно синтезировать либо путем твердофазного синтеза, либо путем синтеза в растворе, или при помощи комбинации указанных способов, при которой сборка пептидной цепи будет происходить на твердой фазе, а циклизация или другие модификации - на смоле или в растворе. Такие методы хорошо известны в технике.-8 017716 Следующие сокращения имеют указанные значения: Ас: ацетил;Dap: 2,3-диаминопропионовая кислота; DCC: дициклогексилкарбодиимид; DCM: дихлорметан; DIC: диизопропилкарбодиимид; DIEA: диизопропилэтиламин; DMF: N,N-диметилформамид; DMSO: диметилсульфоксид; EDT: 1, 2-этандитиол; Et: этил; Fm: 9-фтороэнилметил; Emoc: 9-фтороэнилметоксикарбонил; HATU: N-[(диметиламино)-1 Н-1,2,3-триазоло[4,5-b]пиридин-1-илметилен]-Nметилметанаммоний гексафторофосфат N-оксид; HBTU: O-бензотриазолил-N,N,N',N'-тетраметилуроний гексафторофосфат; HCTU: 1 Н-бензотриазолиум 1-[бис(диметиламино)метилен]-5-хлор-3-оксид гексафторофосфат; HF: фтороводород; HOBt: гидроксибензотриазол; IBCF: изобутил хлорформат; iPr: изопропил; IPA: изопропиловый спирт; Me: метил; 2Nal: 2-нафтилаланин; NMM: N-метилморфолин; NMP: Nметил-пирролидон; OtBu: тетробутиловый эфир; Pbf: 2,2,4,6,7-пентаметилдигидробензофуран-5 сульфонил; PBS: физиологический раствор с фосфатном буфером; РуВОР: (бензотриазол-1-илокси)трис(пирролидино)-фосфония гексафторофосфат; PyBrOP: бромотрис (пирролидино)фосфония гексафторофосфат; tBu: тетрабутил; TFA: трифторуксусная кислота; THF: тетрагидрофуран; TIS: триизопропиловый солевой раствор; Tos: р-толуолсульфонил; Z: бензилоксикарбонил; ZOSu: N-(бензилоксикарбонилокси сукцинимид. Получение соединений, предложенных в настоящем изобретении, описанное в следующих примерах, приведено в целях иллюстрации, а не для ограничения. В каждом из этих примеров наблюдаемую молекулярную массу приводят как обращенное значение. Обращенное значение получают по формуле:MW (наблюдаемая)=n(m/z)-n, где m/z относится к заряженному иону (положительному), а n число зарядов конкретной молекулы. Когда в масс-спектре присутствует много заряженных молекул, наблюдаемую молекулярную массу(вес) приводят как среднее. Способы синтеза, раскрываемые в примерах 85-87, и процедуры присоединения изотопных меток,раскрываемые в примере 89, для циклических лактамных пептидов, содержащих изопропиловые боковые цепи лизина, в равной степени применимы к другим пептидам, раскрытым в настоящем документе, содержащим алкильные боковые цепи лизина, при соответствующих модификациях. Способы синтеза в примерах 86-88, которые исключают необходимость в применении токсических катализаторов на основе палладия, также в равной степени применимы к другим пептидам, раскрываемым в настоящем документе, при соответствующих модификациях. Пример 1 Ac-цикло[Dap-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:2) Последовательность Ac-Dap(Alloc)-Tyr(tBu)-Arg(Pbf) DArg(Pbf)-2Kal-Gly-Clu(O-аллил)-Arg(Pbf) (SEQ ID NO:3) собирают при помощи стандартной процедуры с применением Fmoc, используя устройство для синтеза пептидов ABI 431 (Applied Biosystems),как показано на Схеме 1, приведенной ниже. Автоматизированный синтез осуществляли с использованием стандартного протоколаDCC/HOBt Applied Biosystems или протокол FastMoc HBTU/DIEA в соответствии с указаниями поставщика (РЕ Applied Biosystems Inc., Foster City, CA). Для амидов, содержащих С-концевую группу,твердая подложка представляла собой амидную смолу Ринка (4-(2',4'-диметоксифенил-Fmocаминометил)феноксидную смолу), а для этиламидов, содержащих С-концевую группу - индольную смолу (3-(этил-Fmoc-аминометил)индол-1-ил]ацетильную AM смолу) (NovaBiochem, EMD Biosciences,Inc., San Diego, CA). Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и осуществляли за 9 основных стадий. При выполнении стадии 1, четыре эквивалента защищенной аминокислоты Fmoc-Arg(Pbf) подвергали активации под действием DCC/HOBt (или HBTU/DIEA для протоколаFastMoc) в NMP, и присоединяли к амидной смоле Ринка со снятой защитой, используя 20% пиперидин. На стадии 2 четыре эквивалента Fmoc-Glu(О-аллил) подвергали активации и присоединяли к пептидной смоле со снятой защитой, полученной при проведении стадии 1. Соответствующие стадии выполняли до проведения стадии 8, взаимодействия Fmoc-Dap(Alloc). Затем Fmoc на N-конце удаляли, используя 20% пиперидина в ДМФА, и отдельно выполняли ацетилирование -аминогруппы, используя 5 эквивалентов уксусного ангидрида, 10 эквивалентов DIEA в сухом ДМФА или NMP, в течение 1 ч при комнатной температуре. Защитные группы боковой цепи, аллильную и Alloc удаляли в присутствии 0,1 эквивалентаPd(Ph3P)4 и в присутствии 24 эквивалентов фенилсилана в дихлорметане (схема 2). Эту операцию повторяли один раз для завершения снятия защиты с боковой цепи. Фрагмент карбоновой кислоты Glu со снятой защитой подвергали активации в присутствии PyBOP/DIEA и подвергали циклизации с аминогруппой боковой цепи фрагмента Dap на смоле. С циклизованного пептида одновременно снимали защиту и отщепляли от смолы, промывая поглотительной смесью, состоящей из ТФУ/Н 2 О/TIS/EDT (95/2/1/2,-9 017716 об./об./об./об.), или ТФУ/H2O/TIS/анизола (92/2/4/2, об./об./об./об.) в течение 2 ч при комнатной температуре. Затем растворители испаряли в вакууме, и пептид осаждали и три раза промывали холодным диэтиловым эфиром с удалением поглотителей. Расчетная молекулярная масса (MW расч.): 1142,30, наблюдаемая MW(MW набл.): 1142,50. Схема 1. Синтез цепочки пептида от аминогруппы боковой цепи до карбоксильной группы боковой цепи Очистку пептида выполняли, используя стандартные методики препаративной ВЭЖХ. Сразу после проведения циклизации, раствор пептида разбавляли водой, содержащей 0,1% (об./об.) ТФУ, загружали на колонку С 18 для ВЭЖХ с обращенной фазой и элюировали градиентом водной смеси 0,1% трифторуксусная кислота/ацетонитрил (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали. Другие характеристики конечного продукта получали при помощи аналитической ВЭЖХ и масс-спектрометрического анализа в соответствии с традиционными методиками. Для пептидов, содержащих основную группу в боковой цепи, конечный лиофилизованый продукт представляет собой соль ТФУ. Схема 2. Циклизация аминогруппы боковой цепи с карбоксильной группой боковой цепи и отщепление Пример 2. Ac-цикло[Dab-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:6). Получают, как в примере 1, с тем исключением, что при выполнении стадии 8 Fmoc-Dap(Alloc) заменяют Fmoc-Dab(Alloc). MW расч.: 1156,33; MW набл.: 1156,10.- 10017716 Пример 3. Ас-цикло[Dap-Tyr-Lys (iPr)-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:7). Получают, как в примере 1, с тем исключением, что при выполнении стадии 6 Fmoc-Arg(Pbf) заменяют Fmoc-Lys(iPr)(Boc). MW расч.: 1156,37; MW набл.: 1156,78. Пример 4. н-гексаноил-цикло[Dap-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:8). Получают, как в примере 1, с тем исключением, что при выполнении стадии 6 Fmoc-Arg(Pbf) заменяют Fmoc-Lys(iPr)(Boc). Кроме того, при выполнении стадии 9 уксусный ангидрид заменяют гексановой кислотой, активированной PyBOP/DIEA. MW расч.: 1212,47; MW набл.: 1212,92. Пример 5. Ас-цикло[(D/L)Agl-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:9). Получают, как в примере 1, с тем исключением, что при выполнении стадии 2 Fmoc-Glu(O-аллил) заменяют Fmoc-Glu(OtBu) и при выполнении стадии 8 Fmoc-Dap(Alloc) заменяют Fmoc-(DL)Agl(Boc). После получения цепи, обработку Pd(Ph3P)4 не производят, поскольку отсутствует аллильная защита. Вместо этого, циклизацию выполняли в растворе после отщепления неразветвленного пептида от твердой подложки и снятия защиты. Неочищенный неразветвленный пептид (0,25 ммол), полученный при отщеплении, сушили в вакууме и растворяли в 10 мл сухого ДМФА. Этот раствор пептида переносили при помощи шприцевого насоса в течение 2 ч в следующий раствор: 15 мл сухого дихлорметана и 15 мл сухого ДМФА, содержащего 1,0 ммол РуВОР и 4,0 ммол DIEA. Затем реакцию оставляли протекать при комнатной температуре в течение 2 ч. Растворители затем испаряли в вакууме, остаток загружали на колонку С 18 для препаративной ВЭЖХ с обращенной фазой, и целевой циклический пептид выделяли и исследовали, как описано в примере 1. MW расч.: 1128,27; MW набл.: 1128,26. Пример 6. Ас-цикло[Glu-Tyr-Arg-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:10). Последовательность Glu(OtBu)-Tyr(tBu)-Arg(Pbf)-DArg(Pbf)-2Nal-Gly-Dap(Boc)-Arg(Pbf) (SEQ IDNO:13) собирают при помощи стандартной процедуры с применением Fmoc, используя устройство ABI 431, как показано на схеме 3, приведенной ниже. Автоматизированный синтез выполняли с использованием стандартного протокола DCC/HOBt Applied Bxosystems или протокола FastMoc (HBTU/DIEA) в соответствии с указаниями поставщика (РЕ Applied Biosystems Inc., Foster City, CA). Для амидов, содержащих С-концевую группу, твердая подложка представляла собой амидную смолу Ринка, а для этиламидов, содержащих С-концевую группу - индольную смолу (3-(этил-Fmoc-аминометил)индол-1-ил]ацетил AM смолу). Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и осуществляли путем проведения 9 основных стадий. На стадии 1 четыре эквивалента защищенной аминокислоты Fmoc-Arg(Pbf) подвергали активации в присутствии DCC/HOBt (или HBTU/DIEA для протоколаFastMoc) в NMP и закрепляли на амидной смоле Ринка со снятой защитой. На стадии 2 четыре эквивалента Fmoc-Dap(Boc) подвергали активации и закрепляли на смоле со снятой защитой, полученной на стадии 1. Соответствующие стадии выполняли до проведения стадии 8, взаимодействия Fmoc-Glu(OtBu). На стадии 9 Fmoc на N-концевом конце удаляли, используя 20% пиперидина в ДМФА, и автономно выполняли ацетилирование -аминогруппы 5 эквивалентами уксусного ангидрида, 10 эквивалентами DIEA в сухом ДМФА или NMP, в течение 1 ч при комнатной температуре. С готового пептида одновременно снимали защиту и отщепляли его от смолы, используя поглотительный коктейль, состоящий из ТФУ/Н 2 О/TIS/EDT (95/2/1/2, об./об./об./об.), или ТФУ/H2O/TIS/анизола (92/2/4/2, об./об./об./об.) в течение 2 ч при комнатной температуре (схема 4). Затем растворители испаряли в вакууме, и пептид осаждали и три раза промывали холодным диэтиловым эфиром для удаления поглотителей. Неочищенный продукт непосредственно использовали в реакции циклизации. MW расч.: 1142,30; MW набл.: 1142,83. Схема 3. Синтез цепочки пептида от кислотной группы боковой цепи до аминогруппы боковой цепи Циклизацию выполняли в растворе после отщепления неразветвленного пептида от твердой под- 11017716 ложки при снятии защиты со всех фрагментов боковой цепи (схема 4). Отщепленный неочищенный неразветвленный пептид (0,25 ммол) сушили в вакууме и растворяли в 10 мл сухого ДМФА. Этот раствор пептида переносили при помощи шприцевого насоса в течение 2 ч в следующую реакционную смесь: 15 мл сухого дихлорметана и 15 мл сухого ДМФА, содержащего 1,0 ммол РуВОР и 4,0 ммол DIEA. Затем реакцию оставляли протекать при комнатной температуре в течение 2 ч. Растворители выпаривали в вакууме, остаток загружали на колонку С 18 для препаративной ВЭЖХ с обращенной фазой и целевой циклический пептид выделяли и исследовали, как указано в примере 1. Схема 4. Циклизация кислотной группы боковой цепи с аминогруппой боковой цепи Пример 7. Bz-цикло[Glu-Tyr-Arg-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:14). Получают, как в примере 6, с тем исключением, что на стадии 9 уксусный ангидрид заменяют ангидридом бензойной кислоты. MW расч.: 1204,37; MW набл.: 1204,87. Пример 8. Ac-цикло[Glu-Tyr-Arg-DArg-2Nal-Gly-DDap]-Arg-NH2 (SEQ ID NO:15). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocDDap(Boc). MW расч.: 1142,30; MW набл.: 1142,73. Пример 9. Ас-цикло[Glu-Tyr-Arg-DArg-2Nal-Gly-Lys]-Arg-NH2 (SEQ ID NO:16). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocLys(Boc). MW расч.: 1184,38; MW набл.: 1184,23. Пример 10. Ас-цикло [Glu-Tyr-Arg-DArg-2Nal-Gly-Dab]-Arg-NH2 (SEQ ID NO: 17). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocDab(Boc). MW расч.: 1156,33; MW набл.: 1156,07. Пример 11. Ас-цикло [Glu-Tyr-Arg-DArg-2Nal-Gly-(D/L)Agl]-Arg-NH2 (SEQ ID NO:18). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют Fmoc(DL)Agl(Boc). MW расч.: 1128,27; MW набл.: 1128,86. Пример 12. Bz-цикло[Glu-Tyr-Arg-DArg-2Nal-Gly-(D/L)Agl]-Arg-NH2 (SEQ ID NO:19). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют Fmoc(DL)Agl(Boc). Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом бензойной кислоты.MW расч.: 1190,34; MW набл.: 1190,99. Пример 13. Bz-цикло[Asp-Tyr-Arg-DArg-2Nal-Gly-Dab]-Arg-NH2 (SEQ ID NO:20). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocDab(Boc), и на стадии 8 Fmoc-Glu(OtBu) заменяют Fmoc-Asp(OtBu). Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом бензойной кислоты. MW расч.: 1204,37; MW набл.: 1204,87. Пример 14. Ас-цикло[Asp-Tyr-Arg-DArg-2Nal-Gly-Dab]-Arg-NH2 (SEQ ID NO: 21). Получают, как в примере 6, с тем исключением, что на стадии 2Fmoc-Dap(Boc) заменяют FmocDab(Boc), и на стадии 8 Fmoc-Glu(OtBu) заменяют Fmoc-Asp(OtBu). MW расч.: 1142,30; MW набл.: 1142,81.- 12017716 Пример 15. Ac-цикло[Asp-Tyr-Arg-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:22). Получают, как в примере 6, с тем исключением, что на стадии 8 Fmoc-Glu(OtBu) заменяют FmocAsp(OtBu). MW расч.: 1128,27; MW набл.: 1128,78. Пример 16. Bz-цикло[Asp-Tyr-Arg-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:23). Получают, как в примере 6, с тем исключением, что на стадии 8 Fmoc-Glu(OtBu) заменяют FmocAsp(OtBu). Кроме того, уксусный ангидрид на стадии 9 заменяют ангидридом бензойной кислоты. MW расч.: 1190,34; MW набл.: 1190,69. Пример 17. Ас-цикло[Asp-Tyr-Arg-DArg-2Nal-Gly-(D/L)Agl]-Arg-NH2 (SEQ ID NO:24). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют Fmoc(DL) Agl(Boc) , и на стадии 8 Fmoc-Glu(OtBu) заменяют Fmoc-Asp(OtBu). MW расч.: 1114 24; MW набл.: 1114,85. Пример 18. Ас-цикло [Asp-Tyr-Lys(Me2)-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:25). Получают, как в примере 6, с тем исключением, что на стадии 6 Fmoc-Arg(Pbf) заменяют FmocLys(Me2), и на стадии 8 Fmoc-Glu(OtBu) заменяют Fmoc-Asp(OtBu). MW расч.: 1128,31; MW набл.: 1128,92. Пример 19. Bz-цикло [Asp-Tyr-Lys(Me2)-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:26). Получают, как в примере 6, с тем исключением, что на стадии 6 Fmoc-Arg(Pbf) заменяют FmocLys(Me2), и на стадии 8Fmoc-Glu(OtBu) заменяют Fmoc-Asp(OtBu). Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом бензойной кислоты. MW расч.: 1190,38; MW набл.: 1191,14. Пример 20. Цикло[сукцинил-Tyr-Arg-DArg-2Nal-Gly-(D/L)Agl]-Arg-NH2 (SEQ ID NO:27). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют Fmoc(DL)Agl(Boc), на стадии 8 Fmoc-Glu(OtBu) не используют, и эту стадию не проводят. Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом янтарной кислоты. MW расч.: 1057,19; MW набл.: 1057,87. Пример 21. Цикло[сукцинилин-Tyr-Arg-DArg-2Nal-Gly-Dap]-Arg-NH2 (SEQ ID NO:28). Получают, как в примере 6, с тем исключением, что на стадии 8 Fmoc-Glu(OtBu) не используют, и эту стадию не проводят. Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом янтарной кислоты. MW расч.; 1071,22; MW набл.: 1071,85. Пример 22. Цикло[сукцилин-Tyr-Arg-DArg-2Nal-Gly-Dab]-Arg-NH2 (SEQ ID NO:29). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocDab(Boc), на стадии 8 Fmoc-Glu(OtBu) не используют, и эту стадию не проводят. Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом янтарной кислоты. MW расч.: 1085,25; MW набл.: 1085,87. Пример 23. Цикло[сукцилин-Tyr-Arg-DArg-2Nal-Gly-Orn]-Arg-NH2 (SEQ ID NO:30). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocOrn(Boc), на стадии 8 Fmoc-Glu(OtBu) не используют, и эту стадию не проводят. Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом янтарной кислоты. MW расч.: 1099,27; MW набл.: 1100,23. Пример 24. Цикло[сукцилин-Tyr-Arg-DArg-2Nal-Gly-Lys]-Arg-NH2 (SEQ ID NO:31). Получают, как в примере 6, с тем исключением, что на стадии 2 Fmoc-Dap(Boc) заменяют FmocLys(Boc), на стадии 8 Fmoc-Glu(OtBu) не используют, и эту стадию не проводят. Кроме того, на стадии 9 уксусный ангидрид заменяют ангидридом янтарной кислоты. MW расч.: 1113,30; MW набл.: 1114,25. Пример 25 Цикло [Gly-Tyr-Lys (iPr) -DArg-2Nal-Gly-DGlu] -Arg-NH2 (SEQ ID NO:32). Последовательность Gly-Tyr(tBu)-Lys(iPr) (Boc)-DArg(Pbf)-2Nal-Gly-DGlu(Оаллил)-Arg(Pbf) (SEQID NO:33) собирают при помощи стандартной процедуры с применением стандартных реагентов Fmoc,используя устройство ABI 431, как представлено на схеме 5, приведенной ниже. Автоматизированный синтез выполняли с использованием стандартного протокола DCC/HOBt Applied Biosystems или протокола FastMoc HBTU/DIEA в соответствии с указаниями поставщика (РЕ Applied Biosystems Inc., FosterCity, CA). Для амидов твердая подложка представляла собой амидную смолу Ринка, а для содержащих Сконцевую группу этиламидов - индольную смолу [3-(этил-Fmoc-аминометил)индол-1-ил]ацетил AM смолу. Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и завершали при выполнении 8 основных стадий (схема 5). На стадии 1 четыре эквивалента защищенной аминокислотыFmoc-Arg(Pbf) подвергали активации в присутствии DCC/HOBt (или HBTU/DIEA для протокола FastMoc) в NMP и закрепляли на амидной смоле Ринка со снятой защитой. На стадии 2, четыре эквивалентаof Fmoc-DGlu(O-аллил) подвергали активации и присоединяли к пептидной смоле со снятой защитой,полученной на стадии 1. Соответствующие стадии выполняли до проведения стадии 8, взаимодействияFmoc-Gly. Аллилэфирную защитную группу боковой цепи удаляли 0,1 эквивалентом Pd(Ph3P)4 в присутствии 24 эквивалентов фенилсилана в дихлорметане (схема 6). Эту операцию повторяли один раз для полного снятия защиты с боковой цепи. Затем удаляли Fmoc, находящуюся на N-конце, используя 20% пиперидина в ДМФА. Фрагмент карбоновой кислоты DGlu со снятой защитой подвергали активации в присутствии PyBOP/DIEA и циклизации с -аминогруппой глицина на смоле. С циклизованного пептида одновременно снимали защиту и отщепляли его от смолы, используя поглотительный коктейль, состоящий из ТФУ/Н 2 О/TIS/EDT(95/2/1/2, об./об./об./об.) или ТФУ/H2O/TIS/анизола (92/2/4/2, об./об./об./об.) в течение 2 ч при комнатной температуре. Затем растворители выпаривали в вакууме, и пептид осаждали и три раза промывали холодным диэтиловым эфиром для удаления поглотителей. MW расч.: 1085,29; MW набл.; 1085,32. Пример 25 а. Цикло [Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Arg-NH2 уксусной кислоты соль (SKQID NO:34). Соль трифторуксусной кислоты пептида, полученного в примере 25, превращали в соль уксусной кислоты адсорбцией материала на препаративной колонке С 18 подходящего размера, уравновешенной смесью 2% уксусной кислоты/H2O (об./об.). Затем колонку промывали, используя от трех до пяти объемов колонки, 2% водной уксусной кислотой (об./об.). Пептид элюировали, используя 1:1 смесь воды/ацетонитрила (об./об.), содержащую 2% об. уксусной кислоты, и лиофилизировали. MW расч.: 1085,29; MW набл.: 1085,32. Схема 5. Синтез цепочки пептида от -аминогруппы до карбоксильной группы боковой цепи Очистку пептида выполняли, используя стандартную методику препаративной ВЭЖХ. Сразу после циклизации раствор пептида разбавляли водой, содержащей 0,1% (об./об.) ТФУ, загружали на колонку С 18 для ВЭЖХ с обращенной фазой и элюировали градиентом водной смеси 0,1% трифторуксусной кислоты/ацетонитрила (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали. Другие характеристики конечного продукта получали при помощи аналитической ВЭЖХ и масс-спектрометрического анализа, выполняемого традиционными способами. Для пептидов, содержащих основные группы в боковой цепи, конечный лиофилизированный продукт представляет собой соль ТФУ. Схема 6. Циклизация -аминогруппы с карбоксильной группой боковой цепи и отщепление Пример 26. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:37). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят. MW расч.: 929,10; MW набл.: 929,39. Пример 26 а. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 уксусной кислоты соль (SEQ IDNO:38). Соль трифторуксусной кислоты пептида, полученного в примере 26, превращали в соль уксусной кислоты адсорбцией материала на препаративной колонке С 18 подходящего размера, уравновешенной смесью 2% уксусной кислоты/Н 2 О (об./об.). Затем колонку промывали, используя от трех до пяти объемов колонки, 2% водной уксусной кислотой (об./об.). Пептид элюировали, используя 1:1 смесь воды/ацетонитрила (об./об.), содержащую 2% об. уксусной кислоты, и лиофилизировали. MW расч.: 929,10; MW набл.: 929,39. Пример 27. Цикло[Gly-Tyr-Arg-DArg-2Nal-Gly-DGlu]-Arg-NH2 (SEQ ID NO:39). Получают, как в примере 25, с тем исключением, что на стадии 6 Fmoc-Lys(iPr)(Boc) заменяютFmoc-Arg(Pbf). MW расч.: 1071,22; MW набл.: 1071,02. Пример 28. Цикло [Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys (iPr)-NH2 (SEQ ID NO:40). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc). MW расч.: 1099,35; MW набл.: 1099,91. Пример 29. цикло[Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:41). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Glu(Оаллил), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), на стадии 8 Fmoc-Gly не используют, и стадию 8 не проводят. MW расч.: 1014,17; MW набл.: 1014,78. Пример 30. Цикло[Tyr-Arg-DArg-2Nal-Gly-DGlu]-Arg-NH2 (SEQ ID NO:42). Получают, как в примере 25, с тем исключением, что на стадии 6 Fmoc-Lys(iPr)(Boc) заменяютFmoc-Arg(Pbf), на стадии 8 Fmoc-Gly не используют, и стадию 8 не проводят. MW расч.: 1014,17; MW набл.: 1014,65. Пример 31. Цикло[Tyr-Arg-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:43). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Asp(аллил), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), на стадии 8Fmoc-Gly не используют, и стадию 8 не проводят. MW расч.: 1000,14; MW набл.: 1000,63. Пример 32. Цикло[Gly-Tyr-Arg-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:44). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Asp(аллил), и на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf). MW расч.: 1057,19; MW набл.: 1057,35. Пример 33. Цикло[Gly-Tyr-Lys(Me2)-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:45). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Asp(аллил), и на стадии 6 Fmoc-Lys(iPr) (Boc) заменяют Fmoc-Lys(Me2). MW расч.: 1057,23; MW набл.: 1057,86. Пример 34. Цикло[Gly-Tyr-Lys(iPr) -DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:46). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Asp(аллил). MW расч.: 1071,26; MW набл.: 1071,76. Пример 35. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Asp]-NH2 (SEQ ID NO:47). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят и на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Asp (аллил). MW расч.: 915,07; MW набл.: 915,38. Пример 36. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Asp]-Lys(iPr)-NH2 (SEQ ID NO:48). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc) и на стадии 2 Fmoc-DGlu(Оаллил) заменяют Fmoc-Asp(аллил). MW расч.: 1085,33; MW набл.: 1085,78. Пример 37. Цикло[Gly-Tyr-Lys(Me2)-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:49). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Glu(О-аллил) и на стадии 6 Fmoc-Lys(iPr) (Boc) заменяют Fmoc-Lys(Me2). MW расч.: 1071,26; MW набл.: 1071,05. Пример 38. Цикло[Gly-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:50). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Glu(О-аллил) и на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf). MW расч.: 1071,22; MW набл.: 1071,50. Пример 39. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:51) Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяют Fmoc-Glu(О-аллил). MW расч.: 1085,29; MW набл.: 1085,91. Пример 40. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:52). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят и на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(O-аллил). MW расч.: 929,10; MW набл.: 929,39. Пример 41. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Asp]-NHEt (SEQ ID NO:53). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят и на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Asp(аллилом). MW расч.: 943,13; MW набл.: 943,36. Пример 42. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NHEt (SEQ ID NO:54). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят и на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(О-аллилом). MW расч.: 957,15; MW набл.: 957,50. Пример 43. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NHEt (SEQ ID NO: 55). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, и стадию 1 не проводят. MW расч.: 957,15; MW набл.: 957,46. Пример 44. Цикло[Gly-Tyr-Lys (iPr)-DArg-2Nal-Gly-DGlu]-Arg-NHEt (SEQ ID NO:56). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой. MW расч.: 1113,34; MW набл.: 1113,81. Пример 44 а. Соль уксусной кислоты цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Arg-NHEt (SEQID NO:57). Соль трифторуксусной кислоты пептида, полученного в примере 44, превращали в соль уксусной кислоты адсорбцией материала на препаративной колонке С 18 подходящего размера, уравновешенной смесью 2% уксусной кислоты/Н 2 О (об./об.). Затем колонку промывали, используя от трех до пяти объемов колонки, 2% водной уксусной кислотой (об./об.). Пептид элюировали, используя 1:1 смесь воды/ацетонитрила (об./об.), содержащую 2% об. уксусной кислоты, и лиофилизировали. MW расч.: 1113,34; MW набл.: 1113,81. Пример 45. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NHEt (SEQ ID NO:58). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, и Fmoc-Arg(Pbf) на стадии 1 заменяют FmocLys(iPr)(Boc). MW расч.: 1127,41; MW набл.: 1127,35. Пример 46. цикло[Lys(Ac)-Tyr-Lys(Me2)-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:59). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Glu(OtBu), на стадии 6 Fmoc-Lys(iPr) (Boc) заменяют Fmoc-Lys(Me2), и при выполнении стадии 8Fmoc-Gly заменяют Boc-Lys(Fmoc). После получения цепи Fmoc на боковой цепи N-концевого Lys удаляли в присутствии 20% пиперидина в ДМФА, и боковую цепь Lys затем ацетилировали, используя 10 эквивалентов уксусного ангидрида/DIEA при комнатной температуре в течение одного часа. Все защитные группы боковой цепи удаляли и неразветвленный пептид отщепляли от твердой подложки, используя смесь ТФУ/вода/TIS/анизол (90/5/2,5/2,5, об./об./об./об.) в течение 2 ч при комнатной температуре. Циклизацию неочищенного неразветвленного пептида выполняли в растворе. Отщепленный неочищенный неразветвленный пептид (0,25 ммоль) сушили в вакууме и растворяли в 10 мл сухого ДМФА. Этот раствор пептида переносили при помощи шприцевого насоса в течение 2 ч в следующую реакционную смесь: 15 мл сухого дихлорметана и 15 мл сухого ДМФА, содержащего 1,0 ммол РуВОР и 4,0 ммолDIEA. Затем смесь оставляли для протекания реакции при комнатной температуре в течение 2 ч. Растворители затем испаряли в вакууме, остаток загружали на колонку С 18 для препаративной ВЭЖХ с обращенной фазой, и целевой циклический пептид выделяли и исследовали, как указано в примере 1. MW расч.: 118 4,42; MW набл.: 1184,06. Пример 47. Цикло[Dap(Ac)-Tyr-Lys(Me2)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO: 60). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Glu(OtBu), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Lys(Me2), и на стадии 8 Fmoc-Gly заменяют Boc-Dap(Fmoc). После получения цепи, Fmoc на фрагменте боковой цепиN-концевого Dap удаляли, используя 20% пиперидина в ДМФА, и Dap боковой цепи затем ацетилировали 10 эквивалентами уксусного ангидрида/DIEA при комнатной температуре в течение одного часа. Все защитные группы боковой цепи затем удаляли и неразветвленный пептид отщепляли от твердой подложки, используя смесь ТФУ/воды/TIS/анизола (90/5/2,5/2,5, об./об./об./об.) в течение 2 ч при комнатной температуре. Циклизацию сырого неразветвленного пептида выполняли в растворе. Отщепленный неочищенный неразветвленный пептид (0,25 ммол) сушили в вакууме и растворяли в 10 мл сухого ДМФА. Этот раствор пептида переносили при помощи шприцевого насоса в течение 2 ч в следующую- 16017716 реакционную смесь: 15 мл сухого дихлорметана и 15 мл сухого ДМФА, содержащего 1,0 ммол РуВОР и 4.0 ммол DIEA. Реакцию оставляли протекать при комнатной температуре в течение 2 ч. Растворители затем испаряли в вакууме, остаток загружали на колонку для препаративной ВЭЖХ, и целевой циклический пептид выделяли и исследовали, как указано в примере 1. MW расч.: 986,15; MW набл.: 985,97. Пример 48. Цикло[Ala-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:61). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(О-аллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-Ala. MW расч.: 943,13; MW набл.: 942,92. Пример 49. Цикло[DAla-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:62). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Glu(Оаллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-DAla. MW расч.: 943,13; MW набл.: 943,44. Пример 50. Цикло[DAla-Tyr-Lys(iPr) -DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:63). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, и на стадии 8 Fmoc-Gly заменяют Fmoc-DAla. MW расч.: 943,13; MW набл.: 943,42. Пример 51. Цикло[Ala-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:64). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, и на стадии 8 Fmoc-Gly заменяют Fmoc-Ala. MW расч.: 943,13; MW набл.: 943,48. Пример 52. Цикло[Leu-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:65). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, и на стадии 8 Fmoc-Gly заменяют Fmoc-Leu. MW расч.: 985,21; MW набл.: 985,56. Пример 53. Цикло[Leu-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:66). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Glu(Оаллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-Leu. MW расч.: 985,21; MW набл.: 985,49. Пример 54. Цикло[DPhe-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:67). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(О-аллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-DPhe. MW расч.: 1019,22; MW набл.: 1019,52. Пример 55. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NH2 (SEQ ID NO:68). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(Оаллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1019,22; MW набл.: 1019,53. Пример 56. Цикло[DPhe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:69). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, и на стадии 8 Fmoc-Gly заменяют Fmoc-DPhe. MW расч.: 1019,22; MW набл.: 1019,50. Пример 57. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2 (SEQ ID NO:70). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1189,48; MW набл.: 1189,92. Пример 57 а. Соль уксусной кислоты цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2(SEQ ID NO:71). Соль трифторуксусной кислоты пептида, полученного в примере 57, превращали в соль уксусной кислоты адсорбцией материала на препаративной колонке С 18 подходящего размера, уравновешенной смесью 2% уксусной "кислоты/H2O (об./об.). Затем колонку промывали, используя от трех до пяти объемов колонки, 2% водной уксусной кислотой (об./об.). Пептид элюировали, используя 1:1 смесь воды/ацетонитрила (об./об.), содержащую 2 об.% уксусной кислоты, и лиофилизировали. MW расч.: 1189,48; MW набл.: 1189,92. Пример 58. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Arg-NH2 (SEQ ID NO:72). Получают, как в примере 25, с тем исключением, что на стадии 8 Fmoc-Gly заменяют Fmoc-Phe.MW расч.: 1175,41; MW набл.: 1175,81. Пример 59. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-NH2 (SEQ ID NO:73). Получают, как в примере 25, с тем исключением, что стадию 1 не проводят, и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1019,22; MW набл.: 1019,56. Пример 60. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NHEt (SEQ ID NO:74). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1217,53; MW набл.: 1217,97. Пример 60 а. Соль уксусной кислоты цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NHEt(SEQ ID NO:75). Соль трифторуксусной кислоты пептида, полученного в примере 60, превращали в соль уксусной кислоты адсорбцией материала на препаративной колонке С 18 подходящего размера, уравновешенной смесью 2% уксусной кислоты/Н 2 О (об./об.). Затем колонку промывали, используя от трех до пяти объе- 17017716 мов колонки, 2% водной уксусной кислотой (об./об.). Пептид элюировали, используя 1:1 смесь воды/ацетонитрила (об./об.), содержащую 2% об. уксусной кислоты, и лиофилизировали. MW расч.: 1217,53; MW набл.: 1217,97. Пример 61. Цикло[DAla-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NHEt (SEQ ID NO:76). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят, на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Glu(Оаллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-DAla. MW расч.: 971,18; MW набл.: 971,49. Пример 62. Цикло[2Nal-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NHEt (SEQ ID NO:77). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят, на стадии 2 FmocDGlu(О-аллил) заменяют Fmoc-Glu(Оаллил), и на стадии 8 Fmoc-Gly заменяют Fmoc-2Nal. MW расч.: 1097,34; MW набл.: 1097,53. Пример 63. Цикло [DPhe-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NHEt (SEQ ID NO:78). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят, на стадии 2-FmocDGlu(О-аллил) заменяют Fmoc-Glu(О-аллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc-DPhe. MW расч.: 1047,28; MW набл.: 1047,51. Пример 64. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-NHEt (SEQ ID NO:79). Получают, как в примере 25, с тем исключением, что амидную смолу Ринка заменяют 3-(этилFmoc-аминометил)индол-1-ил]ацетильной AM смолой, стадию 1 не проводят, на стадии 2 FmocDGlu(Оаллил) заменяют Fmoc-Glu(О-аллилом), и на стадии 8 Fmoc-Gly заменяют Fmoc- Phe. MW расч.: 1047,28; MW набл.: 1047,57. Пример 65. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Gly-2Nal-NH2 (SEQ ID NO:80). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют Fmoc2Nal, и между стадиями 1 и 2 добавляют одну стадию, в которой используют Fmoc-Gly. MW расч.: 1183,39; MW набл.: 1183,26. Пример 66. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-Glu]-(3-Ala-D2Nal-NH2 (SEQ ID NO:81). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocD2Nal, между стадиями 1 и 2 добавляют одну стадию, на которой используют FmocAla, и на стадии 2Fmoc-DGlu(Оаллил) заменяют Fmoc-Glu(О-аллилом). MW расч.: 1197,40; MW набл.: 1196,70. Пример 67. Цикло[-Ala-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO: 82). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Glu(Оаллил), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют FmocAla. MW расч.: 1085,25; MW набл.: 1085,05. Пример 68. Цикло[-Ala-Tyr-Arg-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:83). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Asp(О-аллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-p-Ala. MW расч.: 1071,22; MW набл.: 1071,15. Пример 69. Цикло[5-аминовалерил-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:84). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Glu(О-аллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-5-аминовалериановой кислотой. MW расч.: 1113,30; MW набл.: 1113,40. Пример 70. Цикло[5-аминовалерил-Tyr-Arg-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:85). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(О-аллил) заменяютFmoc-Asp(О-аллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-5-аминовалериановой кислотой. MW расч.: 1099,27; MW набл.: 1100,25. Пример 71. Цикло[(4-AMPA)-Tyr-Arg-DArg-2Nal-Gly-Asp]-Arg-NH2 (SEQ ID NO:86). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Asp(Оаллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-4-аминометилфенилуксусной кислотой (4-АМРА). MW расч.: 1147,32; MW набл.: 1148,20. Пример 72. Цикло[(4-AMPA)-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:87). Получают, как в Примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Glu(Оаллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-4-аминометилфенилуксусную кислоту (4-АМРА). MW расч.: 1161,34; MW набл.: 1161,99. Пример 73. Цикло[(4-АМРА)-Tyr-Arg-DArg-2Nal-Gly-Glu]-DArg-NH2 (SEQ ID NO:88). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocDArg(Pbf), на стадии 2 Fmoc-DGlu(Оаллил) заменяют Fmoc-Glu(Оаллил), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-4-аминометилфенилуксусной кислотой(4-АМРА). MW расч.: 1161,34; MW набл.: 1161,83. Пример 74. Цикло[(4-АМВ)-Tyr-Arg-DArg-2Nal-Gly-Glu]-Arg-NH2 (SEQ ID NO:89). Получают, как в примере 25, с тем исключением, что на стадии 2 Fmoc-DGlu(Оаллил) заменяютFmoc-Glu(Оаллилом), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Arg(Pbf), и на стадии 8 Fmoc-Gly заменяют Fmoc-4-аминометилбензойной кислотой (4-АМВ). MW pacч.: 1147,32; MW набл.: 1147,66. Пример 75. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-Gly-2Nal-NH2 (SEQ ID NO:90). Получают, как в примере 25, с тем исключением, что стадию 1 заменяют тремя последовательными присоединениями следующих остатков: сначала, Fmoc-2Nal, затем Fmoc-Gly и затем Fmoc-Lys(iPr)(Boc).MW расч.: 1353,69; MW набл.: 1354.03. Пример 76. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-Gly-2Nal-NH2 (SEQ ID NO: 91). Получают, как в примере 25, с тем исключением, что стадию 1 заменяют тремя последовательными присоединениями следующих остатков: сначала, Fmoc-2Nal, затем Fmoc-Gly и затем Fmoc-Lys(iPr)(Boc). Кроме того, на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 14 4 3,82; MW набл.: 1444.13. Пример 77. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Gly-DPhe-NH2 (SEQ ID NO:92). Получают, как в примере 25, с тем исключением, что стадию 1 заменяют двумя последовательными присоединениями следующих остатков: сначала, Fmoc-DPhe, затем Fmoc-Gly. MW расч.: 1133,36; MW набл.: 1133,73. Пример 78. Цикло[Gly-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-DPhe-NH2 (SEQ ID NO:93). Получают, как в Примере 25, с тем исключением, что стадию 1 заменяют двумя последовательными присоединениями следующих остатков: сначала, Fmoc-DPhe, затем Fmoc-Lys(iPr)(Boc). MW расч.: 1246,58; MW набл.: 1246,88. Пример 79. Цикло[Lys-Tyr-Lys (iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2 (SEQ ID NO:94). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Lys(Boc). MW расч.: 1170,50; MW набл.: 1169,80. Пример 80. Цикло[Phe-Tyr-Lys-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2 (SEQ ID NO:95). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(iPr)(Boc), на стадии 6 Fmoc-Lys(iPr)(Boc) заменяют Fmoc-Lys(Boc), и на стадии 8 Fmoc-Gly заменяютFmoc-Phe. MW расч.: 1147,40; MW набл.: 1146,70. Пример 81. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys-NH2 (SEQ ID NO:96). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocLys(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1147,40; MW набл.: 1146,70. Пример 82. Цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Orn-NH2 (SEQ ID NO:97). Получают, как в примере 25, с тем исключением, что на стадии 1 Fmoc-Arg(Pbf) заменяют FmocOrn(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1133,40; MW набл.: 1132,70. Пример 83. Цикло[Phe-Tyr-Lys-DArg-2Nal-Gly-DGlu]-Lys-NH2 (SEQ ID NO:98). Получают, как в примере 25, с тем исключением, что как Fmoc-Arg(Pbf) на стадии 1, так и FmocLys(iPr)(Boc) на стадии 6 заменяют Fmoc-Lys(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1105,37; MW набл.: 1105,40. Пример 84. Цикло[Phe-Tyr-Lys-DArg-2Nal-Gly-DGlu]-Lys-NHEt (SEQ ID NO:99). Получают, как в примере 25, с тем исключением, что смолу Ринка заменяют 3-(этил-Fmocаминометил)индол-1-ил]ацетильной AM смолой, и как Fmoc-Arg(Pbf) на стадии 1, так и FmocLys(iPr)(Boc) на стадии 6 заменяют Fmoc-Lys(Boc), и на стадии 8 Fmoc-Gly заменяют Fmoc-Phe. MW расч.: 1133,36; MW набл.: 1133,82. Пример 85. Альтернативный синтез I цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2(SEQ ID NO:70). В примере 57 описан синтез SEQ ID NO:70 методом твердофазного синтеза пептидов по протоколуFmoc, с использованием коммерчески доступного элемента Fmoc-Lys(iPr)Boc, который представляет собой дорогостоящий материал, который сложно получить в больших количествах. Способ, описываемый в настоящем примере, позволяет осуществлять синтез SEQ ID NO:70, используя менее дорогостоящийFmoc-Lys(Boc), циклизацию в растворе, и алкилирование лизина посредством восстановительного аминирования с использованием цианоборгидрида натрия, что экономически представляет собой более оправданный подход к указанному конечному продукту. Другие преимущества состоят в том, что реакционные среды (уксусная кислота, ацетон и метанол) относительно недороги, условия реакции легко контролируются, соотношение растворителей может быть значительно изменено и при этом не влиять на реакцию алкилирования, и получаемый выход составляет 90% или более. Последовательность Phe-Tyr(tBu)-Lys(Boc)-DArg(Pbf)-2Nal-Gly-DGlu(Оаллил)-Lys(Boc) (SEQ IDNO:100) собирают на амидной смоле Ринка при помощи стандартных реагентов Fmoc, используя устройство ABI 431 для синтеза пептидов, как показано на схеме 7. Автоматизированный синтез выполняли,используя стандартный протокол DCC/HOBt Applied Biosystems или протокол FastMoc (HBTU/DIEA) в соответствии с указаниями поставщика (РЕ Applied Biosystems Inc., Foster City, CA). Схема защитных групп фрагмента боковой цепи следующая: Lys(Boc), DGlu(Оаллил), DArg(Pbf), Tyr(tBu). Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и завершали при выполнении 8 стадий. На- 19017716 стадии 1, четыре эквивалента защищенной аминокислоты Fmoc-Lys(Boc) подвергали активации в присутствии DCC/HOBt (или HBTU/DIEA для протокола FastMoc) в NMP, и закрепляли на амидной смоле Ринка со снятой защитой. На стадии 2 четыре эквивалента Fmoc-DGlu(О-аллил) подвергали активации и присоединяли к пептидной смоле со снятой защитой, полученной на стадии 1. Указанные стадии повторяли соответствующим образом до проведения стадии 8, т.е. взаимодействия с Fmoc-Phe. Сложноэфирную аллильную защитную группу боковой цепи удаляли при помощи 0,1 эквивалентаPd(Ph3P)4 в присутствии 24 эквивалентов фенилсилана в дихлорметане (схема 8). Эту операцию повторяли один раз для завершения снятия защиты с боковой цепи. Затем удаляли фрагмент Fmoc на N-конце,используя 20% пиперидина в ДМФА. Фрагмент карбоновой кислоты DGlu со снятой защитой подвергали активации в присутствии PyBOP/DIEA и подвергали циклизации с -аминогруппой Phe на смоле. С циклизованного пептида одновременно снимали защиту и отщепляли от смолы, используя поглотительный коктейль, состоящий из ТФУ/Н 2 О/TIS/EDT (95/2/1/2, об./об./об./об.) или ТФУ/H2O/TIS/анизола (92/2/4/2, об./об./об./об.) в течение 2 ч при комнатной температуре. Затем растворители выпаривали в вакууме, и пептид осаждали и три раза промывали холодным диэтиловым эфиром для удаления поглотителей. Схема 7. Сборка пептидной цепи при помощи твердофазного синтеза Схема 8. Получение циклического пептидного предшественника- 20017716 Очистку циклического пептида-предшественника выполняли, используя методики стандартной препаративной ВЭЖХ. Неочищенный отщепленный продукт растворяли минимальном количествеDMSO, загружали на колонку С 18 для ВЭЖХ с обращенной фазой и элюировали градиентом водной смеси 0,1% трифторуксусной кислоты/ацетонитрила (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали. Другие характеристики промежуточного предшественника циклического пептида получали, используя аналитическую ВЭЖХ и масс-спектрометрический анализ в соответствии с традиционными методиками. Лиофилизированный циклический пептид-предшественник затем алкилировали в растворе уксусной кислоты/ацетона/метанола (1:1:4, об./об./об.) посредством восстановительного аминирования, используя цианоборгидрид натрия (cхема 9). Концентрация пептида составляла приблизительно 10 мг/мл; она может варьировать в широком диапазоне, не влияя на результаты. Использовали от трех до пяти эквивалентов восстановителя, цианоборгидрида натрия, и реакцию обычно завершали в течение 2 ч при комнатной температуре. Получаемый выход составлял 90% или более. Например, 20 мг предшественника циклического пептида растворяли в 2 мл метанола, к которому добавляли 0,5 мл уксусной кислоты и 0,5 мл ацетона, хорошо перемешивали, и затем добавляли при перемешивании 5,6 мг цианоборгидрида натрия (2,5 эквивалента в метаноле). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего добавляли еще 5,6 мг цианоборгидрида натрия. Протекание реакции отслеживали при помощи ВЭЖХ и масс-спектрометрического анализа. После завершения реакции проводили высаливание и лиофилизацию реакционной смеси, получая 18,9 мг конечного продукта (SEQ ID NO:70),чистота которого составляла 97,5%. MW расч.: 1189,48; MW набл.: 1189,6. Схема 9. Восстановительное аминирование, превращающее Lys в Lys(iPr) Соединение, полученное в примере 60 (SEQ ID NO:74) также может быть получено аналогичным способом, в который вносят подходящие модификации, соответствующие аминокислотному составу, и применяя [3-(этил-Fmoc-аминометил)индол-1-ил]ацетил AM смолу. Сначала получают предшественник пептида (SEQ ID NO:99), как указано в примере 84, и затем выполняют восстановительное аминирование, как показано на схеме 9. Эта последовательность реакций позволяет получить конечный циклический пептид, включающий С-концевую этиламидную группу, чистота которого составляет 99,16%. MW расч.: 1217,53; MW набл.: 1217,84. Пример 86. Альтернативный синтез IX цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2(SEQ ID NO:70). В примере 85 описан экономически эффективный синтез SEQ ID NO:70 посредством проведенияFmoc твердофазного пептидного синтеза с использованием относительно недорогого Fmoc-Lys(Boc) и осуществлением алкилирования лизина посредством восстановительного аминирования в присутствии цианоборгидрида натрия в относительно недорогих растворителях (уксусной кислоте, ацетоне и метаноле). Тем не менее, эта операция включает использование катализатора на основе тяжелого металла палладия, применяемого для удаления аллильной сложноэфирной защитной группы боковой цепи. Палладий высоко токсичен, и качественный контроль, обеспечивающий полное удаление этого элемента, чрезвычайно сложен. Твердофазный синтез пептида с использованием Boc и циклизации на смоле, описанный в этом примере, позволяет получать SEQ ID NO:70, используя относительно недорогой реактив BocLys(2-Cl-Z) в отсутствие токсичного и дорогостоящего палладиевого катализатора, что обеспечивает бо- 21017716 лее экономически выгодный синтез, масштаб которого легко увеличить, и менее токсичный подход к получению конечного продукта, включающий менее сложный процесс контроля качества. Последовательность Fmoc-Phe-Tyr(2-Br-Z)-Lys(2-Cl-Z)-DArg(Tos)-2Nal-Gly-DGlu(OFm)-Lys(2-Cl-Z)(SEQ ID NO:102) собирали вручную на смоле МВНА (4-метилбензгидриламиновая смола) (Cat. No. D2095, BaChem California Inc., Torrance, CA), используя известный твердофазный синтез пептида с использованием Boc (Schnolzer et al. (1992) Int. J. Pept. Protein Res. 40:180-193), как показано на схеме 10. Сборку цепи выполняли, используя процедуру нейтрализации in situ/активации HBTU/DIEA, как указано в цитируемой статье. Схема защитных групп фрагмента боковой цепи следующая: Lys(2-Cl-Z),DGlu(OFm), DArg(Tos), и Tyr(2-Br-Z). Альфа-аминогруппы всех аминокислотных элементов защищали трет-бутоксикарбонильной группой (Boc), за исключением N-концевого остатка Phe, который защищалиFmoc для повышения эффективности синтеза. Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и завершали при выполнении 8 стадий. На стадии 1 пять эквивалентов защищенной аминокислоты Boc-Lys(2-Cl-Z) подвергали активации в присутствии HBTU/DIEA в ДМФА и закрепляли на МВНА смоле. На стадии 2 пять эквивалентов BOC-DGlu(OFm) подвергали активации и присоединяли к пептидной смоле со снятой защитой, полученной на стадии 1, используя чистую ТФУ. Указанные стадии повторяли соответствующим образом до проведения стадии 8, т.е. взаимодействия Fmoc-Phe. Защитную Fm группу боковой цепи DGlu удаляли вместе с Fmoc на N-конце, используя 20% пиперидина в ДМФА. Фрагмент карбоновой DGlu кислоты со снятой защитой подвергали активации в присутствии PyBOP/DIEA, HCTU/DIEA, или других подходящих активирующих реагентов, и подвергали циклизации с -аминогруппой Phe на смоле. С циклического пептида одновременно снимали защиту и отщепляли его от смолы, используя в качестве поглотителя HF с добавлением 5% м-крезола или пкрезола в течение 1 ч при 0 С. Затем растворители выпаривали, и неочищенный пептид осаждали и три раза промывали холодным диэтиловым эфиром. Схема 10. Сборка пептидной цепи на твердой фазе с использованием протокола Boc Очистку циклического пептидного предшественника (SEQ ID NO:98, как показано на схеме 10),выполняли, используя методики стандартной препаративной ВЭЖХ. Неочищенный продукт отщепления растворяли в минимальном количестве DMSO, загружали на колонку С 18 для ВЭЖХ с обращенной фазой, и элюировали градиентом водной смеси 0,1% трифторуксусной кислоты/ацетонитрила (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали. Другие характеристики промежуточного циклического пептида-предшественника получали, используя аналитическую ВЭЖХ и масс-спектрометрический анализ в соответствии со стандартными методиками. Для SEQ ID- 22017716 Лиофилизированный циклический пептид-предшественник (SEQ ID NO:98) затем алкилировали в растворе уксусной кислоты/ацетона/метанола (1:1:4, об./об./об.) посредством восстановительного аминирования, используя цианоборгидрид натрия, как указано на схеме 9. Концентрация пептида составляла приблизительно 10 мг/мл; она может варьировать в широком диапазоне без влияния на результаты. Использовали от пяти эквивалентов восстановителя, цианоборгидрида натрия, и реакция обычно завершалась в течение 2 ч при комнатной температуре. Получаемый выход составлял 90% или более. Например,6,6 мг предшественника циклического пептида растворяли в 0,8 мл метанола, к которому добавляли 0,2 мл уксусной кислоты и 0,2 мл ацетона, хорошо перемешивали, и затем двумя равными порциями при перемешивании добавляли 1,9 мг цианоборгидрида натрия (2,5 эквивалента в метаноле). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин, после чего добавляли еще 1,9 мг цианоборгидрида натрия. Протекание реакции отслеживали при помощи ВЭЖХ и массспектрометрического анализа. После завершения реакции проводили высаливание и лиофилизацию реакционной смеси, получая конечный продукт (SEQ ID NO:70), чистота которого составляла 96,5%. MW расч.: 1189,45; MW набл.: 1189,6. Пример 87. Альтернативный синтез III цикло[Phe-Tyr-Lys(iPr)-DArg-2Nal-Gly-DGlu]-Lys(iPr)-NH2(SEQ ID NO:70). Соединение, полученное в примере 57 (SEQ ID NO:70), также может быть получено без использования палладиевого катализатора при проведении циклизации в растворе, с возможным увеличением масштаба синтеза, следующим образом. Последовательность(SEQ ID NO:103) собирали вручную на смоле МВНА, используя твердофазный синтез пептида с использованием Boc (Schnolzer et al. (1992) Int. J. Pept. Protein Res. 40:180-193), как показано на схеме 11. Сборку цепи выполняли, используя процедуру нейтрализации in situ/активации HBTU/DIEA, как указано уSchnolzer et al. Схема защитных групп боковой цепи следующая: Lys(Fmoc), DGlu(OBzl), DArg(Tos),Tyr(2-Br-Z). Альфа-аминогруппы всех аминокислотных элементов защищали трет-бутоксикарбонильной группой (Boc). Пошаговый синтез цепочки начинали с С-конца неразветвленного пептида и осуществляли в 8 стадий, как показано на схеме 11. На стадии 1, пять эквивалентов защищенной аминокислоты BocLys(2-C1-Z) подвергали активации в присутствии HBTU (4 экв.) /DIEA (10 экв.) в ДМФА, и закрепляли на МВНА смоле. На стадии 2, пять эквивалентов BOC-DGlu(OBzl) подвергали активации и присоединяли к пептидной смоле со снятой защитой, полученной на стадии 1, используя чистую ТФУ. Указанные стадии повторяли соответствующим образом до стадии 8, т.е. взаимодействия Вос-Phe. Защитную группуBoc удаляли в присутствии чистой ТФУ, смолу нейтрализовали DIEA, и промывали ДМФА и метанолом,и сушили на воздухе, а затем проводили отщепление в присутствии HF. С неразветвленного пептида одновременно снимали защиту и отщепляли его от смолы, используя в качестве поглотителя HF с добавлением 5% м-крезола или п-крезола в течение 1 ч при 0 С. Затем пастворители выпаривали, и неочищенный пептид осаждали и три раза промывали холодным диэтиловым эфиром. Схема 11. Сборка цепи с использованием протокола Boc- 23017716 Очистку неразветвленного предшественника пептида (SEQ ID NO:104) выполняли, используя методики стандартной препаративной ВЭЖХ. Неочищенный продукт отщепления растворяли минимальном количестве DMSO, загружали на колонку С 18 для ВЭЖХ с обращенной фазой и элюировали градиентом водной смеси 0,1% трифторуксусной кислоты/ацетонитрила (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали. Другие характеристики промежуточного циклического пептида-предшественника получали, используя аналитическую ВЭЖХ и масс-спектрометрический анализ в соответствии с традиционными методиками. Для SEQ ID NO:104, MW расч.: 1567,78;MW набл.: 1567,6. Циклизацию лиофилизированного неразветвленного пептида-предшественника (SEQ ID NO:104) выполняли в растворе (схема 12). Неразветвленный пептид растворяли в небольшом количестве сухого ДМФА (10 мг/мл). Этот раствор пептида медленно переносили при помощи шприцевого насоса в реакционную смесь, состоящую из РуВОР (2 экв., или других подходящих активирующих реагентов, например, HCTU, ВОР, HBTU, и т.д.) и DIEA (10 экв.) в сухом ДМФА при перемешивании магнитной мешалкой. Затем смесь оставляли для протекания реакции при комнатной температуре в течение 2 ч. Затем к реакционной смеси добавляли чистый пиперидин до получения конечной концентрации, равной 25%(об./об.). Реакционную смесь выдерживали при перемешивании в течение еще 20 мин до полного удаления защиты Fmoc. Растворители выпаривали в вакууме, и остаток загружали на колонку С 18 для препаративной ВЭЖХ с обращенной фазой, и элюировали градиентом водной смеси 0,1% трифторуксусной кислоты/ацетонитрила (об./об.) с мониторингом при 214 нм. Соответствующие фракции объединяли и лиофилизировали, получая циклический пептидный предшественник (SEQ ID NO:98). Другие характеристики промежуточного предшественника циклического пептида получали, используя аналитическую ВЭЖХ и масс-спектрометрический анализ в соответствии с традиционными методиками. Для SEQ IDNO:98, MW расч.: 1105,29; MW набл.: 1105,4. Алкилирование циклического пептида SEQ ID NO:98 выполняли в растворе уксусной кислоты/ацетона/метанола (1:1:4, об./об./об.) посредством восстановительного аминирования, используя цианоборгидрид натрия, как указано на схеме 9, получая конечный продукт (SEQ ID NO:70). MW расч.: 1189,45; MW набл.: 118 9,6. Схема 12. Циклизация и удаление Fmoc(SEQ ID NO:70). Соединение, полученное в примере 57 (SEQ ID NO:70), также может быть получено без использования палладиевого катализатора в соответствии со схемой синтеза, показанной на схеме 13, приведенной ниже. Сначала получают в растворе дипептид Fmoc-DGlu-Lys(iPr,Z)-NH2, содержащий незащищенную Dглютаминовую кислоту в боковой цепи. Дипептид фиксировали на СТС смоле, гиперчувствительной к действию кислот (hyper-acid labile) (2-хлортритилхлоридной PS смоле, 1% DVB (ДВБ) (100-200 меш)(Senn Chemicals USA Inc., San Diego, CA; номер по каталогу 40207), и конечный пептидный продукт затем синтезировали на этой смоле посредством стандартного синтеза с использованием Fmoc, как указано выше. При селективном удалении пептида с СТС смолы только боковая цепь D-Глютаминовой кислоты- 24017716 реагирует с N-концевой группой пептида в растворе с образованием циклического пептидного продукта. Затем оставшиеся боковые цепи отщепляли в присутствии 95% ТФУ или другой сильной кислоты. Схема 13. Общая схема синтеза из Fmoc-DGlu-Lys(iPr, Z)-NH2 Сначала получают дипептид Fmoc-DGlu-Lys(iPr, Z)-NH2, как показано на приведенной ниже схемеBoc-Lys(iPr,Z)-ОН вводили в реакцию с NMM и IBCF в ТГФ. После добавления NH4OH, растворители удаляли на роторном испарителе, и продукт растворяли в этилацетате. Этилацетатную фазу тщательно промывали 5% NaHCO3 и затем 0,1N HCl, и затем сушили над безводным сульфатом натрия. Сульфат натрия удаляли фильтрованием, и этилацетат испаряли при уменьшенном давлении. Полученный Boc-Lys(iPr,Z)-NH2 растворяли в ДХМ и добавляли ТФУ. По завершении реакции растворители удаляли на роторном испарителе. Затем H-Lys(iPr,Z)-NH2 растворяли в ДМФА. Показатель рН доводили до 8 добавлением DIEA. В отдельном сосуде растворяли в ДМФА Fmoc-DGlu(OtBu)-ОН, HBTU и HOBt; добавляли DIEA для получения рН, равного 8. Два раствора перемешивали и протекание реакции отслеживали при помощи ВЭЖХ С 18 с обращенной фазой. Отслеживали рН, и при необходимости регулировали его добавлением DIEA. Растворители удаляли на роторном испарителе, и продукт растворяли в этилацетате. Этилацетатную фазу тщательно промывали 5% NaHCO3 и затем 0,1N HCl, и затем сушили над безводным сульфатом натрия. Сульфат натрия удаляли фильтрованием, и этилацетат испаряли при уменьшенном давлении. Роторное испарение продолжали до получения сухого остатка. ПолученныйFmoc-DGlu(OtBu)-Lys(iPr, Z)-NH2 растворяли в ДХМ и добавляли ТФУ. По завершении реакции растворители удаляли на роторном испарителе. Твердый продукт получали размешиванием в диэтиловом эфире. После промывки осадка эфиром продукт сушили в вакуумной печи. Твердофазный синтез пептида конечного продукта выполняли следующим образом. FmocDGlu(OtBu)-Lys(iPr,Z)-NH2 растворяли в ДХМ и вводили в реакцию с СТС смолой в присутствии DIEA в реакционном сосуде. Спустя 3 ч пептид-смолу промывали ДХМ до исчезновения реагентов в промывных водах и добавляли Z-OSu. Отслеживали рН и при необходимости регулировали его до значения рН 8-9 добавлением DIEA. Спустя 8 ч пептид-смолу промывали ДХМ до исчезновения реагентов в промывных водах, переносили в полипропиленовый контейнер и сушили в вакуумной печи. Защищенный пептид-смолу собирают, используя протокол Fmoc следующим образом. Применяют следующий цикл взаимодействий:- 25017716 1) Разблокировка: обработка 25% пиперидина в ДМФА; 2) Циклы промывки: ДМФА, IPA и снова ДМФА; 3) Нингидриновый тест (качественный: если положительный, то переходят к стадии 4 взаимодействия); 4) Взаимодействие с 2 эквивалентами Fmoc-аминокислоты в присутствии HOBt /DIC в ДМФА; 5) Циклы промывки ДМФА; 6) Нингидриновый тест (качественный: если отрицательный, то переходят к следующему циклу разблокировки/взаимодействия; если положительный, то переходят к стадии 7 повторного взаимодействия; если слегка положительный, то переходят к стадии 10 ацетилирования); 7) Повторное взаимодействие (при необходимости), с 1 эквивалентом Fmoc-аминокислоты в присутствии HOBt, HBTU/DIEA в ДМФА; 8) Циклы промывки ДМФА; 9) Нингидриновый тест (качественный: если отрицательный, то переходят к следующей разблокировки/взаимодействие стадии; если положительный, то переходят к стадии 10 ацетилирования); 10) Ацетилирование (при необходимости) в присутствии 2% уксусного ангидрида в 4% DIEA в ДМФА; 11) Циклы промывки (ДМФА, IPA, и вновь ДМФА); 12) Нингидриновый тест (качественный: если положительный, то переходят к следующему циклу разблокировки/взаимодействия). После проведения конечного цикла взаимодействия пептид-смолу (SEQ ID NO:105) промывали эфиром и сушили в вакууме. Защищенный пептид-смолу промывали ДХМ. Отщепление полностью защищенного неразветвленного пептида от смолы проводили в присутствии 2% ТФУ в ДХМ с последующим фильтрованием. Растворители удаляли на роторном испарителе и полностью защищенный неразветвленный пептид (SEQ ID NO:106) осаждали размешиванием в эфире. Полностью защищенный неразветвленный пептид переносили в полипропиленовый контейнер и сушили в вакуумной печи. Полностью защищенный неразветвленный пептид подвергали циклизации в присутствии РуВОР,HOBt и DIEA в ДМФА. При необходимости доводили рН до значения рН 7-8 добавлением DIEA. По завершении реакции, растворители удаляли на роторном испарителе, и продукт растворяли в этилацетате. Этилацетатную фазу тщательно промывали 5% NaHCO3 и затем 0,1N HCl и насыщенным растворомNaCl. Затем эту фазу сушили над безводным сульфатом натрия. Сульфат натрия удаляли фильтрованием,и этилацетат удаляли на роторном испарителе при уменьшенном давлении. Защищенный циклический пептид (SEQ ID NO:107) осаждали размешиванием в эфир и сушили в вакуумной печи. Если реакция завершена, снятие защиты выполняли в смеси ТФУ:H2O:TIS, растворители удаляли на роторном испарителе и циклический пептид (SEQ ID NO:70) осаждали размешиванием в эфире и сушили в вакуумной печи. MW расч.: 1189,48; MW набл.: 1189,50. Пример 89. Введение изотопных меток: Синтез цикло[Phe-Tyr-Lys(iPr-d6)-DArg-2Nal-Gly-DGlu]Lys(iPr-d6) -NH2 (SEQ ID NO:108). Применение ацетона, содержащего изотопную метку, например, ацетона, содержащего 13 С-, 14 С-,дейтерий или тритий, как показано на схеме, приведенной ниже, позволяет вводить в соответствии с прописями примеров 85-87 сайт-специфическую изотопную метку в циклические пептидные антагонисты CXCR4, что позволяет использовать их в различных фармакологических исследованиях и для получения изображений. Молекулы ацетона, содержащего изотопную метку, коммерчески доступны; их поставляют многие организации. Ниже приведен пример использования ацетона-d6 для получения пептида,содержащего 12 атомов дейтерия. Полученное соединение, молекулярная масса которого отличается на 12 Da по сравнению с немеченым ацетоном, легко распознается по масс-спектрам и при этом проявляет идентичное сродство к целевому рецептору. Циклические пептиды-предшественники (SEQ ID NO:98, SEQ ID NO:99, и т.д.) могут быть получены и очищены любым из способов, рассмотренных в примерах 85-87. Алкилирование выполняют в растворе уксусной кислоты/ацетона/метанола (1:1:4, об./об./об.) посредством восстановительного аминирования, используя цианоборгидрид натрия, как указано на схеме 9, за исключением того, что стандартный ацетон заменяют соответствующим ацетоном, содержащим изотопную метку. В рассматриваемом примере применяют дейтерированный ацетон-d6.- 26017716 Пептид (97 мг) растворяли в 15 мл уксусной кислоты/ацетона-d6/метанола (1:1:4, об./об./об.). Концентрация пептида может в значительной степени варьироваться, не влияя на результаты. Использовали пять эквивалентов цианоборгидрида натрия, и реакция обычно завершается в течение 2 ч при комнатной температуре. Протекание реакции отслеживали при помощи ВЭЖХ и масс-спектрометрического анализа. После завершения реакции проводили высаливание реакционной смеси и лиофилизацию, получая 90,5 мг конечного продукта (SEQ ID NO:108), чистота которого составляла 99,9%. MW расч.: 1201,48;MW набл.: 12 01,7. Схема 15. Сайт-специфичное введение дейтериевой метки в антагонист CXCR4 Применение содержащего изотопную метку цианоборгидрида натрия (например, NaBD3CN,NaBT3CN и т.д.) позволяет осуществлять и другие варианты сайт-специфичного введения радиоактивных меток. Фармакологические свойства предложенных соединений могут быть определены в соответствии с описанными ниже испытаниями. Тест на ингибирование связывания CXCR4/125I-SDF-1 человека Связывание SDF-1 с CXCR4 представляет собой первую стадию в активации внутриклеточного пути передачи сигнала CXCR4. Для определения способности соединения блокировать взаимодействиеSDF-1 и CXCR4, проводили тест на связывание SDF-1, меченого 125I, с использованием клеток лейкемии человека CCRF-CEM (АТСС CCL 119), экспрессирующих эндогенный CXCR4. Анализ проводили в необработанных 96-луночных полистирольных планшетах с полукруглым дном (Corning Incorporated,Costar,3632). Буферный раствор для анализа связывания готовят из среды RPMI 1640 (Gibco, GrandElmer) и 240 пикомоль холодного SDF-la (RD Systems, различные концентрации испытуемых соединений в буфере для анализа, 100000 клеток CCRF-CEM человека и 0,5 мг гранул SPA (Wheatgerm agglutinin beads; Amersham) инкубировали при комнатной температуре в течение 2 ч. Затем планшеты считывали на считывающем устройстве жидкостного сцинтилляционного и люминесцентного анализа Microbeta 1450 (Wallac) в режиме SPA. В этом тесте антагонисты CXCR4 снижают связанную радиоактивность дозозависимым образом. Эффективность ингибирования (Ki или IC50) тестируемым соединением рассчитывали, используя программное обеспечение GraphPad Prism, по дозозависимому снижению связанной радиоактивности. В данном тесте средние значения Ki всех соединений, примеры которых приведены выше, составляет приблизительно 7,5 нМ или менее. Например, при проведении этого анализа среднее значение Ki соединения, полученного в примере 1, составляет 3,45 нМ. Среднее значение Ki многих из указанных соединений составляют приблизительно от 0,2 нМ до 1 нМ. Например, в данном тесте среднее значениеKi соединения, полученного в примере 50, составляет 0,285 нМ. Среднее значение Ki других соединений составляет приблизительно менее 0,2 нМ. Например, в этом тесте среднее значение Ki соединения, полученного в примере 75, составляет 0,096 нМ. Анализ хемотаксической активности Взаимодействие CXCR4/SDF-1 регулирует миграцию (хемотаксис) клеток, на поверхности которых присутствует CXCR4.- 27017716 Для определения антагонистической и клеточной активности тестируемого соединения использовали анализ хемотаксической активности с применением клеток U937 пистиоцитарной лимфомы человека(АТСС CRL 1593), которые экспрессируют эндогенный CXCR4. Вкратце, клетки U937, выращенные в среде DMEM (модифицированная по способу Дульбекко среда Игла) (Gibco, Grand Island, NY), содержащей 10% ЭБС (эмбриональной бычьей сыворотки), 1% раствор натриевой соли пировиноградной кислоты в MEM (модифицированная среда Игла) (Gibco), 1% заменимых аминокислот в MEM (Gibco), и 1% GlutaMAX 1 (Gibco), собирали и один раз промывали буферным раствором для теста на хемотаксическую активность, полученным из 1 среды RPMI (Gibco), содержащей 10 мМ HEPES, рН 7,5, и 0,3% БСА. После промывки, клетки вновь суспендировали в буфере для теста при концентрации, составляющей 5106 клеток/мл. Анализ проводили в 96-луночных планшетах ChemoTx (NeuroProbe) в соответствии с указаниями изготовителя. В общем случае, 50 мкл смеси клеток с тестируемым соединением или без него, помещали на планшет в верхнюю камеру, а в нижнюю камеру добавляли 30 мкл SDF-1 (RDSystems, 10 нг/мл), полученного в 1 буфере для теста на хемотаксическую активность. После сборки,планшет инкубировали в течение 2,5 часов при 37 С в атмосфере 5% CO2. После инкубации в нижнюю камеру добавляли 5 мкл CellTiter 96 AQ (Promega, Madison, WI). Затем планшет инкубировали в течение 60 мин при 37 С, и мигрировавшие клетки детектировали, измеряя поглощение при 492 нм на считывающем устройстве для микропланшетов Тесап Spectrafluor Plus (Salzburg, Austria). Антагонисты CXCR4 ингибируют миграцию клеток, снижая значения поглощения. В данном тесте эффективность ингибирования (IC50) тестируемого соединения рассчитывали, используя программное обеспечение GraphPadPrism, по дозозависимому снижению поглощения при 492 нм. В этом тесте среднее значение IC50 для большинства соединений, примеры которых даны выше, составляло приблизительно 60 нМ или менее. В этом тесте среднее значение IC50 для многих из указанных соединений составляло приблизительно 6 нМ или менее, например, среднее значение IC50 для соединения, полученного в Примере 19, составляло 2,05 нМ. В этом тесте среднее значение IC50 для многих из указанных соединений составляло приблизительно 0,6 нМ или менее, например, среднее значение IC50 Для соединения, полученного в Примере 50, составляло 0,171 нМ. Анализ селективности связывания рецептора хемокина Можно сравнить селективность связывания предлагаемых соединений с рецептором CXCR4 с селективностью по отношению к другим рецепторам хемокина, например, рецепторам человека CCR1,CCR2, CXCR2 или CXCR3, и другим рецепторам, сопряженным с G-белком, в клетках, трансфицированных нуклеиновой кислотой, кодирующей и экспрессирующей такие рецепторы, или в клетках, эндогенно экспресирующих такие рецепторы. Для оценки конкурирования тестируемых соединений и соответствующих лигандов за эти рецепторы может быть проведен конкурентный тест с использованием целых клеток или фрагментов мембран, аналогичный тесту, описанному выше для анализа ингибирования связывания CXCR4/125I-SDF-1. Например, значение Ki, полученное в анализе связывания лигандов с рецептором хемокина человека CXCR2 для соединения, полученного в примере 57 а, превышает 73000 нМ. Мобилизация лейкоцитов и нейтрофилов у мышей C57BL/6 под действием соединений Стволовые клетки костного мозга активно поддерживают непрерывное образование всех видов зрелых клеток крови на протяжении жизни. Костный мозг является основным местом образования лейкоцитов/ нейтрофилов, откуда они поступают в кровяное русло. Ось CXCR4/SDF-1, по-видимому, является важным элементом для сохранения и высвобождения лейкоцитов, нейтрофилов и клетокпредшественников гемопоэза в костном мозге, и нарушение взаимодействия CXCR4/SDF-1 в костном мозге приводит к повышению концентрации этих клеток в периферической крови. Для определения мишень-модулирующей активности тестируемых соединений in vivo использовали модель краткосрочной мобилизации лейкоцитов/нейтрофилов у мышей. Вкратце, здоровых (без патогенов) самок мышейC57BL/6 в возрасте 5-6 недель (Taconic) содержали в течение одной недели перед тестом. Животные имели свободный доступ к стерилизованному корму для грызунов и подкисленной воде. Мышам, разделенным на группы по 5 животных, вводили тестируемые соединения подкожно в солевом растворе; контрольной группе вводили солевой раствор, а затем умерщвляли животных путем удушения в атмосфереCO2 и смещения шейных позвонков в различные моменты времени после введения соединения. Образцы периферической крови собирали путем пункции сердца с использованием шприцев и пробирок с покрытием ЭДТУ. Полный анализ кровяных клеток проводили на гематологическом анализаторе Hemavet Mascot (Drew Scientific Group, Dallas, TX). Регистрировали общее содержание лейкоцитов, нейтрофилов и лимфоцитов в периферической крови. Эффективные антагонисты CXCR4, вводимые мышам подкожно,повышали содержание нейтрофилов и лейкоцитов в периферической крови по сравнению с аналогичными показателями, полученными для мышей контрольной группы, получавших солевой раствор. Для многих из предложенных соединений, использованных при проведении этого теста, демонстрировали среднее отношение нейтрофилов (отношение увеличения количества нейтрофилов в группе,получавшей соединения, к увеличению количества нейтрофилов в группе, получавшей солевой раствор),измеренное спустя 3 ч после введения соединения, превышало 2. Например, среднее отношение нейтро- 28017716 филов, измеренное после введения соединения, полученного в примере 39, составляло 4,6 при дозировке,равной 5 мг/кг. Противоопухолевое действие при модели ксенотрансплантата SCID/Namalwa По-видимому, взаимодействие SDF-1/CXCR4 играет важную роль на различных стадиях развития опухолей, включая рост опухоли, инвазию, развитие кровеносных сосудов и возникновение метастазов. Для оценки противоопухолевого действия тестируемых соединений in vivo применяли ксенотрансплантат опухоли у мышей NOD/SCID (Jackson Laboratories) с использованием клеток Namalwa лимфомы человека, неходжкинской лимфомы (АТСС CRL 1432). Вкратце, 200000 клеток Namalwa перемешивали с матригелем (1:1) и имплантировали подкожно в боковую заднюю часть тела животных. Имплантированные клетки опухоли вырастали в твердую опухоль, размеры которой постоянно измеряли при помощи циркуля. Для определения эффективности тестируемых соединений in vivo в этом опыте, животным (10 животных в группе) вводили различные дозы тестируемых соединений, растворенных в солевом растворе или фосфатно-солевом буфере; введение соединений начинали через 48 ч после имплантации клеток опухоли. Соединения вводили подкожно, и затем определяли объем опухоли и массу тела каждые 2 или 3 суток. Исследования обычно проводили в течение 3-4 недель в зависимости от роста опухоли. Противоопухолевую эффективность тестируемых соединений определяли по процентному уменьшению опухоли в сравнении с группой, которой вводили только носитель, но не вводили соединения. Некоторые из описанных выше соединений, например, соединение, полученное в примере 26, значительно замедляли рост опухоли в указанном тесте при введении в количестве, равном 1 мг/кг два раза в сутки. Фармакологические свойства, такие как биодоступность соединения, метаболическая стабильностьin vivo и фармакокинетические/фармакодинамические свойства могут быть определены методами, хорошо известными в области разработки лекарственных средств. Предпочтительные соединения согласно настоящему изобретению имеют высокую биодоступность при подкожном введении. Биодоступность некоторых из приведенных выше соединений, например, соединения, полученного в примере 44, для крыс составляет приблизительно 100%. Предпочтительные соединения также имеют хорошую метаболическую стабильность in vivo. Например, после введения соединения, полученного в примере 57 а, собакам и обезьянам в плазме крови и моче животных измеряемые количества метаболитов указанного соединения не обнаруживались до 24 ч с момента введения. Предпочтительные соединения также имеют хорошие фармакокинетические/фармакодинамические характеристики, которые позволяют использовать традиционные режимы дозирования. Например, период полувыведения (Т 1/2) соединения, полученного в примере 58, у мышей составляет приблизительно 3 ч. Что касается фармакодинамических свойств, то предпочтительные соединения вызывают продолжительную мобилизацию нейтрофилов и лейкоцитов в организме мышей. Например, соединение, полученное в примере 25, вызывает значительное увеличение концентрации нейтрофилов и белых клеток крови в периферической крови мышей в течение по меньшей мере 6 ч после однократного подкожного введения соединения в количестве, составляющем 5 мг/кг. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Пептид-антагонист CXCR4, включающий лактамный цикл и соответствующий формуле Ia) указанный лактам образован амидной связью между аминогруппой боковой цепи Х 1 и карбоксильной группой боковой цепи Х 7, и при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из (D/L)Agl/Glu, Dab/Glu и Dap/Glu, и R1 представляет собой Ас или нгексаноил; илиb) указанный лактам образован амидной связью между карбоксильной группой боковой цепи фрагмента Х 1, и аминогруппой боковой цепи фрагмента Х 7, и при этом Х 1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Asp/(D/L)Agl, Asp/Dab, Asp/Dap, Glu/(D/L)Agl, Glu/Dab,Glu/Dap, Glu/DDap и Glu/Lys, и R1 представляет собой Ас или Bz, или при этом Х 1 и Х 7, соответственно,представляют собой пару, выбранную из группы, состоящей из сукцинила/(D/L)Agl, сукцинила/Dab, сукцинила/Dap, сукцинила/Lys и сукцинила/Orn, и R1 отсутствует; илиc) указанный лактам образован амидной связью, между -аминогруппой фрагмента X1 и карбоксильной группой боковой цепи Х 7, при этом X1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из Ala/Glu, Ala/DGlu, DAla/Glu, DAla/DGlu, Dap(Ac)/Glu, Gly/Asp, Gly/Glu,Gly/DGlu, Leu/Glu, Leu/DGlu, Lys/DGlu, Lys(Ac)/Glu, 2Nal/Glu, Phe/Glu, Phe/DGlu, DPhe/Glu иd) указанный лактам образован амидной связью между аминогруппой, не являющейся аминогруппой боковой цепи Х 1 или -аминогруппой, и карбоксильной группой боковой цепи Х 7, и при этом Х 1 и Х 7, соответственно, представляют собой пару, выбранную из группы, состоящей из -Ala/Asp, -Ala/Glu,- 29
МПК / Метки
МПК: A61P 31/18, C07K 7/54, A61K 38/12
Метки: циклические, cxcr4, пептиды, антагонисты
Код ссылки
<a href="https://eas.patents.su/30-17716-ciklicheskie-peptidy-antagonisty-cxcr4.html" rel="bookmark" title="База патентов Евразийского Союза">Циклические пептиды – антагонисты cxcr4</a>
Иммуногенные композиции, содержащие циклические пептиды, полученные из бета-амилоидного пептида

Номер патента: 10586
Опубликовано: 30.10.2008
Автор: Цурбригген Ринальдо
МПК: G01N 33/68, C07K 14/47, C07K 7/64...
Метки: иммуногенные, пептиды, beta;-амилоидного, композиции, пептида, полученные, содержащие, циклические
Формула / Реферат:
1. Антигенный пептид формулы где Ab представляет собой пептид из 12-20 аминокислотных остатков, производный от b-амилоидного пептида (Ab1-26; SEQ ID NO:2), предпочтительно Ab1-16 (SEQ ID NO:3) или Ab11-26 (SEQ ID NO:4), или его аналога, содержащего консервативную аминокислотную замену; m равно от 1 до 6; А и B ковалентно связаны с образованием циклического пептида; A и B представляют собой прямую связь, линкерный фрагмент или обозначают...
Связанные с матрицей пептидомиметики β-шпилечной структуры с активностью антагонистов cxcr4

Номер патента: 10163
Опубликовано: 30.06.2008
Авторы: Димарко Стивен Дж., Цумбрунн Юрг, Обрехт Даниель, Романьоли Барбара, Лудин Кристиан, Робинсон Джон Энтони, Мукерджи Решми, Гомберт Франк, Хенце Хайко, Меле Керстин, Фрийблуд Ян Вим, Лочиуро Серджио
МПК: A61K 38/12, C07K 1/04, A61P 31/18...
Метки: активностью, beta;-шпилечной, связанные, матрицей, антагонистов, пептидомиметики, структуры, cxcr4
Формула / Реферат:
1. Соединения общей формулы где представляет собой группу, характеризуемую одной из формул A-CO означает DPro, DGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-DPro или Gly; B-CO означает LPro, LGln, 4-{2-[2-(метоксиэтокси)этокси]ацетиламино}-LPro или Gly; но A-CO и B-CO не могут оба быть Gly; Z означает цепь из n a-аминокислотных остатков, причем n означает целое число 12, 14 или 18, а положения указанных аминокислотных остатков в цепи...
Циклические конструкты натрийуретических пептидов

Номер патента: 16804
Опубликовано: 30.07.2012
Авторы: Цай Хуэй-Чжи, Шарама Шабх Д., Бастос Маргарита, Ян Вэй
МПК: A61K 38/12
Метки: циклические, натрийуретических, пептидов, конструкты
Формула / Реферат:
1. Циклический конструкт формулы Vгде Ааа2 и Ааа13 одинаковы или различны и каждый из них представляет собой аминокислотный остаток, образующий циклический мостик посредством боковых цепей каждого из Ааа2 и Ааа13, где R1 образует часть боковой цепи каждого из фрагментов Ааа2 и Ааа13;Ааа3 - L- или D-изомер α-аминокислоты или β-аминокислоты, являющейся аминокислотой или производным аминокислоты, выбранной из группы, включающей His, Ala,...
Алкоксиалкилзамещенные циклические кетоенолы

Номер патента: 15590
Опубликовано: 31.10.2011
Авторы: Лер Штефан, Мальзам Ольга, Розингер Крис, Гергенс Ульрих, Фишер Райнер, Хиллс Мартин Джеффри, Диттген Ян, Кене Хайнц, Фойхт Дитер
МПК: C07D 207/36, A01N 43/36
Метки: алкоксиалкилзамещенные, кетоенолы, циклические
Формула / Реферат:
1. Соединения формулы (I)где W означает водород или (С1-С6)-алкил,X означает галоид, (С1-С6)-алкил или (С1-С6)-алкокси,Y означает галоид, (С1-С6)-алкил или (С2-С6)-алкинил,при условии, что когда Y означает галоид, алкильный остаток заместителя X содержит по меньшей мере два атома углерода,А означает -СН2- или -СН2-СН2-,В означает (С1-С6)-алкил,D означает (С1-С6)-алкокси, (С5-С6)-гетероциклил с одним атомом кислорода, замещенный галогеном, или...
Циклические пептидные антигрибковые агенты

Номер патента: 464
Опубликовано: 26.08.1999
Авторы: Васудеван Венкатрагхаван, Родригез Майкл Дж., Тернер Уильям В.Мл., Джемисон Джеймс Э., Борромео Питер С.
МПК: C07K 7/56, A61K 38/12
Метки: агенты, антигрибковые, циклические, пептидные
Формула / Реферат:
1. Соединение формулы I: где R' обозначает водород, метил или NH2C(O)CH2-; R" и R'" обозначают, независимо, метил или водород; Rx1, Rx2, Ry1, Ry2, Ry3, Ry4 обозначают, независимо, гидрокси или водород; R0 обозначает группу формулы: R1 обозначает С1-С6 алкил, C1-C6 алкокси, фенил, п-галогенфенил, п-нитрофенил, фенокси, бензил, п-галогенбензил или п-нитробензил; I) R2 обозначает группу формулы где A) R3 обозначает...
Предыдущий патент: Способ лечения рака
Следующий патент: Высокоэффективный котел
Случайный патент: Двуслойная таблетка, содержащая телмисартан и амлодипин