Хинуклидиновые производные в качестве антагонистов мускаринового м3 рецептора
Номер патента: 17627
Опубликовано: 30.01.2013
Авторы: Скидмор Элизабет Энн, Булл Ричард Джеймс, Мэтер Эндрю Найджел, Форд Ронан Ли, Метэ Антонио







Формула / Реферат
1. Соединение, выбранное из группы, состоящей из
(R)-1-[(6-метилпиридин-3-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(6-метилпиразин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-[(6-трифторметилпиридазин-3-ил-карбамоил)метил]-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(бензо[d]изоксазол-3-ил-карбамоилметил)-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(пиридазин-3-ил-карбамоилметил)-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-метилизоксазол-3-ил-карбамоил)метил]-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(3-метилизоксазол-5-ил-карбамоил)метил]-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(3-фторфенилкарбамоил)метил]-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-метилпиразин-2-ил-карбамоил)метил]-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(бензо[d]изоксазол-3-ил-карбамоилметил)-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(пиразин-2-ил-карбамоилметил)-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-[1-(3-фторфенил)циклогептанкарбонилокси]-1-(пиразин-2-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-[1-(3-фторфенил)циклогептанкарбонилокси]-1-(изоксазол-3-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиридин-2-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиридин-4-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-фторпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-метилпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиридин-3-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2-метилпиридин-4-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-фенилкарбамоилметил-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиримидин-4-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2-фторфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2,3-дифторфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[2-(2,3-дигидробензофуран-5-ил)этил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[2-(4-фторфенокси)этил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиридазин-4-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-фторпиридин-3-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-[2-(пиридин-3-ил-окси)этил]-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(6-метилпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(орто-толилкарбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(2-пиразин-2-ил-этил)-1-азониа-бицикло[2.2.2]октана Х;
(S)-1-(3-феноксипропил)-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-{[2-(3-фторфенокси)этилкарбамоил]метил}-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(3,5-дифторфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[2-(4-метоксибензилокси)этил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(2-фенилэтилоксиэтил)-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2,6-дифторфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(метилфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[3-(4-цианофенокси)пропил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2,5-дифторфенилкарбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[2-(4-цианобензилокси)этил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-[(6-трифторметилпиридин-2-ил-карбамоил)метил]-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(4-метилпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-хлорпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(паратолилкарбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(метатолилкарбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-(оксазол-2-ил-карбамоилметил)-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(6-метилпиридазин-3-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиримидин-2-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(5-цианопиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(пиримидин-5-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(3-фторпиридин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(3-фторпиридин-4-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-{2-[(пиразин-2-карбонил)амино]этил}-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-([1,2,4]тиадиазол-5-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-{3-[(пиридин-2-карбонил)амино]пропил}-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(2-метилпиримидин-4-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-1-[(6-метилпиримидин-4-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1-азониа-бицикло[2.2.2]октана Х;
(R)-3-(1-фенилциклогептанкарбонилокси)-1-{2-[(пиридин-2-карбонил)амино]этил}-1-азониа-бицикло[2.2.2]октана Х; и
(R)-3-(1-фенилциклогептанкарбонилокси)-1-(3-пиридин-4-ил-пропил)-1-азониа-бицикло[2.2.2]октана Х;
где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты.
2. Соединение по п.1, выбранное из группы, состоящей из



где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты.
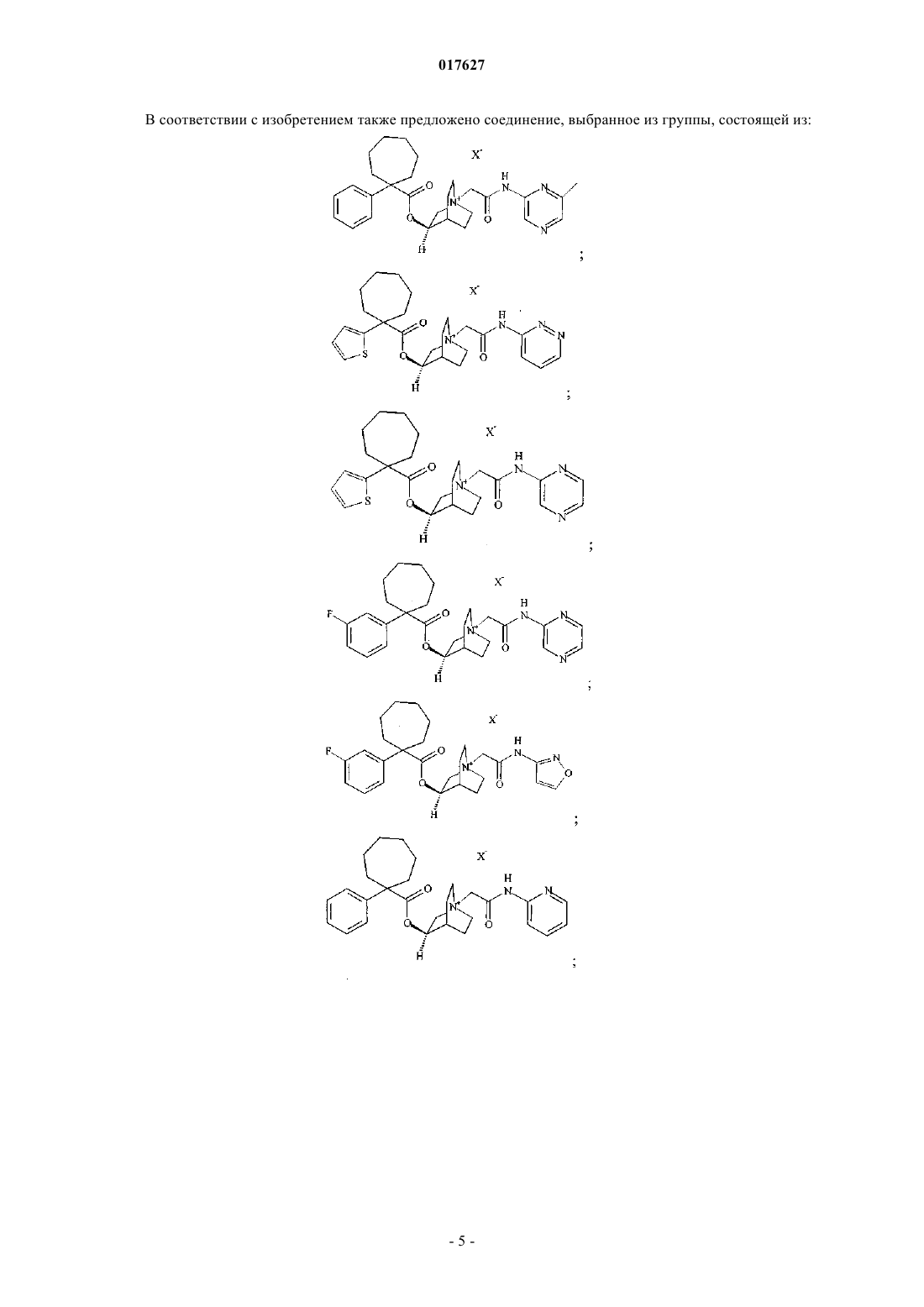
3. Соединение по п.2, представляющее собой

где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты.
4. Соединение по п.3, представляющее собой

5. Фармацевтическая композиция, содержащая соединение по любому из пп.1-4 вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
6. Фармацевтическая композиция по п.5, подходящая для легочного введения.
7. Способ изготовления фармацевтической композиции по п.5, при котором соединение по любому из пп.1-4 смешивают с фармацевтически приемлемым адъювантом, разбавителем или носителем.
8. Применение соединения по любому из пп.1-4 в изготовлении лекарственного средства для использования в лечении хронической обструктивной болезни легких или астмы.
9. Способ лечения хронической обструктивной болезни легких или астмы у теплокровного животного, такого как человек, при котором млекопитающему, нуждающемуся в таком лечении, вводят эффективное количество соединения по любому из пп.1-4.
10. Фармацевтический продукт, содержащий в комбинации первый активный ингредиент, который представляет собой соединение по любому из пп.1-4, и по меньшей мере один дополнительный активный ингредиент, выбранный из
ингибитора фосфодиэстеразы,
агониста β2-адренорецептора,
модулятора функции хемокинового рецептора,
ингибитора киназной функции,
ингибитора протеазы,
стероидного агониста глюкокортикоидного рецептора и
нестероидного агониста глюкокортикоидного рецептора.
11. Фармацевтический продукт по п.10, где по меньшей мере один дополнительный активный ингредиент представляет собой ингибитор киназной функции, выбранный из ингибитора р38 киназы и ингибитора IKK (IkB киназный комплекс).
12. Фармацевтический продукт, содержащий в комбинации первый активный ингредиент, который представляет собой соединение по любому из пп.1-4, и по меньшей мере один дополнительный активный ингредиент, который представляет собой агент, модулирующий ядерный рецептор гормона.
13. Фармацевтический продукт по п.12, где агент, модулирующий ядерный рецептор гормона, представляет собой PPAR (рецептор, активирующий пролиферацию пероксисом).
Текст
ХИНУКЛИДИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АНТАГОНИСТОВ МУСКАРИНОВОГО М 3 РЕЦЕПТОРА В изобретении предложены конкретные соединения общей структурной формулы, приведенной ниже, в которой R3 представляет собой тиен-2-ил или фенил, возможно замещенный атомом фтора,R4 представляет собой замещенный 1-азабицикло[2.2.2]октан, содержащие их фармацевтические композиции и способ изготовления фармацевтических композиций. Также раскрыто их применение в терапии для лечения состояний, опосредованных М 3 мускариновыми рецепторами, таких как хроническая обструктивная болезнь легких. Форд Ронан Ли, Мэтер Эндрю Найджел, Метэ Антонио, Булл Ричард Джеймс, Скидмор Элизабет Энн (GB) Поликарпов А.В., Борисова Е.Н. (RU) 017627 Настоящее изобретение относится к циклоалкилзамещенным алкиловым эфирам полициклических аминоспиртов, способу их получения, содержащим им фармацевтическим композициям, способу изготовления фармацевтических композиций, их применению в терапии и промежуточным соединениям,используемым при их получении. Предшествующий уровень техники Мускариновые рецепторы представляют собой семейство рецепторов, связанных с G-белком(GPCR), имеющее пять членов семейства M1, M2, M3, M4 и M5. Из пяти мускариновых подтипов три (M1,M2 и М 3), как известно, демонстрируют физиологические действия в отношении человеческой легочной ткани. Парасимпатические нервы представляют собой основной путь для рефлекторной бронхоконстрикции в человеческих дыхательных путях и опосредуют тонус дыхательных путей путем высвобождения ацетилхолина на мускариновые рецепторы. Тонус дыхательных путей повышен у пациентов, страдающих респираторными расстройствами, такими как астма и хроническая обструктивная болезнь легких(COPD), и по этой причине антагонисты мускариновых рецепторов разработаны для применения в лечении заболеваний дыхательных путей. Антагонисты мускариновых рецепторов, часто называемые в клинической практике антихолинергическими средствами, получили широкое признание в качестве терапии первой линии людей, страдающих COPD, и их применение подробно рассмотрено в литературе (например, Lee et al., Current Opinion в Pharmacology 2001, 1, 223-229). При использовании для лечения респираторных расстройств антагонисты мускариновых рецепторов типично вводят путем ингаляции. Тем не менее, при введении путем ингаляции значительная доля антагониста мускариновых рецепторов часто абсорбируется в системный кровоток, приводя в результате к побочным эффектам, таким как сухость во рту. Дополнительно, большая часть мускариновых антагонистов обладает относительно короткой длительностью действия, приводя к необходимости их введения несколько раз в течение суток. Такая схема введения несколько раз в течение суток не только не удобна для пациента, но также представляет значительный риск неправильного лечения ввиду не соблюдения пациентом схемы лечения, связанной с частым повторяющимся приемом дозы. Таким образом, сохраняется потребность в новых соединениях, которые способны блокировать мускариновые рецепторы. В частности, существует потребность в новых мускариновых антагонистах,которые обладают высокой эффективностью и уменьшенными системными побочными действиями при введении путем ингаляции. Кроме того, существует потребность в новых мускариновых антагонистах,которые демонстрируют длительное действие при дозировании путем ингаляции и которые подлежат введению один раз или дважды в сутки. В WO 98/04517 описаны сложные эфиры арилциклопропан-, арилциклобутан-, арилциклопентан- и арилциклогексан-карбоновых кислот, обладающие антимускариновой активностью в отношении гладкой мускулатуры мочевого пузыря. Находящаяся на одновременном рассмотрении заявка на изобретение авторов данного изобретения где R1 и R2 вместе с атомом углерода, к которому они оба непосредственно присоединены, образуют 7-членное алифатическое карбоциклическое кольцо, которое может быть возможно замещено одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, C1-6 алкокси, NH2,групп NH(C1-6 алкил), N(C1-6 алкил)2 и C1-6 алкила, где С 1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена и гидроксила;R3 представляет собой фенил или 5-6-членное гетероарильное кольцо, каждое из которых может быть возможно замещено одним или более чем одним заместителем, независимо выбранным из галогена,циано, нитро, SH, S(O)0-2R9, NR10R11, S(O)2NR12R13, C(O)NR14R15, C(O)2R16, NR17S(O)2R18, NR19C(O)R20,NR21C(O)2R22, NR23C(O)NR24R25, OR26 и С 1-6 алкила, где С 1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, C1-6 алкокси, NH2,групп NH(C1-6 алкил) и N(C1-6 алкил)2;R4 представляет собой группу формулы (II), или (IIIa), или (IIIb) где Y представляет собой -СН 2-, -СН 2 СН 2- или -CH2CH2CH2-, и замещение по кольцу в группе (II)-1 017627 может происходить по 3 или 4 положениям; а равен 1 или 2;R5 представляет собой группу формулы (IV)R6 представляет собой С 1-4 алкилен, возможно замещенный одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, С 1-6 алкокси, NH2, групп NH(C1-6 алкил) иR7 представляет собой циклическую группу Cyc1 или C1-4 алкильную группу, где С 1-4 алкильная группа может быть возможно замещена одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, С 1-4 алкокси, NH2, групп NH(C1-4 алкил), N(C1-4 алкил)2, циклической группы Сус 2 и -ОСус 2; иR7 дополнительно может представлять собой атом водорода, когда Q представляет собой О, NR8,-CONR8-, -SO2NR8-, -C(O)O-, -НС=СН- или этинилен; каждый из Сус 1 и Сус 2 независимо представляет собой арил, гетероарил, 3-8-членное алифатическое карбоциклическое кольцо или 4-8-членное алифатическое гетероциклическое кольцо, каждый из которых может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена, циано, нитро, SH, S(O)0-2R9, NR10R11, S(O)2NR12R13, C(O)NR14R15, C(O)2R16,NR17S(O)2R18, NR19C(O)R20, NR21C(O)2R22, NR23C(O)NR24R25, OR26, фенила и С 1-6 алкила, где фенил илиC1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, С 1-6 алкокси, NH2, групп NH(С 1-6 алкил) и N(C1-6 алкил)2;R8 представляет собой водород или С 1-6 алкил; каждый из R9 и R18 независимо представляет собой C1-6 алкил, где С 1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила,C1-6 алкокси, NH2, групп NH(C1-6 алкил) и N(C1-6 алкил)2; и каждый из R10, R11, R12, R13, R14, R15, R16, R17, R19, R20, R21, R22, R23, R24, R25 и R26 независимо представляет собой водород или С 1-6 алкил, где С 1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила, C1-6 алкокси, NH2, группNH(C1-6 алкил) и N(C1-6 алкил)2; или любой из R10 и R11, R12 и R13, R14 и R15 или R24 и R25 вместе с атомом азота, к которому они оба присоединены, могут образовывать 4-8-членное алифатическое гетероциклическое кольцо, где гетероциклическое кольцо может быть возможно замещено одним или более чем одним заместителем, независимо выбранным из галогена, гидроксила и C1-6 алкила, где C1-6 алкил может быть возможно замещен одним или более чем одним заместителем, независимо выбранным из галогена и гидроксила;X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты. Краткое изложение сущности изобретения В настоящем изобретении предложены соединения, находящиеся в объеме находящейся на одновременном рассмотрении заявки на изобретение авторов данного изобретения PCT/GB2007/004350, на которую ссылались выше, но не раскрытые там специально. Таким образом, в настоящем изобретении предложено соединение, которое имеет ион четвертичного аммония, выбранное из группы, состоящей из:(R)-3-(1-фенилциклогептанкарбонилокси)-1-(3-пиридин-4-ил-пропил)-1-азониабицикло[2.2.2]октана Х; где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты. Соединения формулы (I), на которые ссылались выше, и соединения по настоящему изобретению включают анион X, связанный с положительным зарядом на четвертичном атоме азота. Анион X может представлять собой любой фармацевтически приемлемый анион моно- или поливалентной (например,бивалентной) кислоты. В воплощении изобретения X может представлять собой анион неорганической кислоты, например хлорид, бромид, йодид, сульфат, нитрат или фосфат; или анион подходящей органической кислоты, например ацетат, малеат, фумарат, цитрат, оксалат, сукцинат, тартрат, метансульфонат,пара-толуолсульфонат, бензолсульфонат, нападизилат (нафталин-1,5-дисульфонат) (например, геминападизилат), 2,5-дихлорбензолсульфонат, 1-гидроксинафталин-2-сульфонат или ксинафоат (1-гидрокси-2 нафтоат).-4 017627 В соответствии с изобретением также предложено соединение, выбранное из группы, состоящей из: где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты. В одном воплощении соединение представляет собой где X представляет собой фармацевтически приемлемый анион моно- или поливалентной кислоты. В еще одном воплощении соединение представляет собой В одном аспекте изобретения предложена фармацевтическая композиция, содержащая соединение по настоящему изобретению, как определено ранее, вместе с фармацевтически приемлемым адъювантом,разбавителем или носителем. В одном воплощении фармацевтическая композиция подходит для легочного введения. В еще одном аспекте изобретения предложен способ изготовления фармацевтической композиции,при котором соединение по настоящему изобретению, как определено ранее, смешивают с фармацевтически приемлемым адъювантом, разбавителем или носителем. В еще одном аспекте изобретения предложено применение соединения по настоящему изобретению, как определено ранее, в изготовлении лекарственного средства для использования в лечении хронической обструктивной болезни легких или астмы. В еще одном аспекте изобретения предложен способ лечения хронической обструктивной болезни легких или астмы у теплокровного животного, такого как человек, при котором млекопитающему, нуждающемуся в таком лечении, вводят эффективное количество соединения по настоящему изобретению,как определено ранее. В еще одном аспекте изобретения предложен фармацевтический продукт, содержащий в комбинации первый активный ингредиент, который представляет собой соединение по настоящему изобретению,как определено ранее, и по меньшей мере один дополнительный активный ингредиент, выбранный из ингибитора фосфодиэстеразы,агониста 2-адренорецептора,модулятора функции хемокинового рецептора,ингибитора киназной функции,-6 017627 ингибитора протеазы,стероидного агониста глюкокортикоидного рецептора и нестероидного агониста глюкокортикоидного рецептора. В одном воплощении по меньшей мере один дополнительный активный ингредиент представляет собой ингибитор киназной функции, выбранный из ингибитора р 38 киназы и ингибитора IKK (IkB киназный комплекс). В еще одном аспекте изобретения предложен фармацевтический продукт, содержащий в комбинации первый активный ингредиент, который представляет собой соединение по настоящему изобретению,как определено ранее, и по меньшей мере один дополнительный активный ингредиент, который представляет собой агент, модулирующий ядерный рецептор гормона. В одном воплощении агент, модулирующий ядерный рецептор гормона, представляет собой PPAR(рецептор, активирующий пролиферацию пероксисом). Понятно, что некоторые соединения по настоящему изобретению могут существовать в сольватированной, например гидратированной, а также несольватированной формах. Понятно, что настоящее изобретение охватывает все такие сольватированные формы. Некоторые соединения по настоящему изобретению могут существовать в виде таутомеров. Таутомеры и их смеси также образуют аспект настоящего изобретения. Соединения по настоящему изобретению демонстрируют благоприятные фармацевтические свойства. Например, соединения по изобретению демонстрируют активность в качестве антагонистов мускариновых рецепторов, в частности мускариновых M3 рецепторов. Кроме того, соединения также демонстрируют желаемые свойства связывания белков плазмы крови. Связывание белков плазмы крови может представлять собой благоприятное свойство соединений, вводимых путем ингаляции, поскольку оно может ослаблять влияние любого системного действия, которое соединение может оказывать. Соединения по изобретению обладают активностью в качестве фармацевтических агентов, в частности в качестве антихолинергических агентов, включая антагонистов мускариновых рецепторов (M1, М 2 и М 3), в частности антагонистов М 3. Заболевания и состояния, которые можно лечить указанными соединениями, включают: 1) дыхательные пути: обструктивные заболевания дыхательных путей, включающие астму, включая бронхиальную, аллергическую, наследственную, приобретенную, астму, вызванную физическими нагрузками, астму, вызванную приемом лекарств (включая астму, вызванную аспирином и NSAID (нестероидные противовоспалительные средства, и астму, вызванную пылью, как перемежающуюся, так и персистирующую, и всех тяжестей, и другие случаи гиперчувствительности дыхательных путей; хроническое обструктивное заболевание легких (COPD); бронхит, включая инфекционный и эозинофильный бронхит; эмфизему; бронхоэктаз; муковисцидоз; саркоидоз; экзогенный аллергический альвеолит и родственные заболевания; аллергический пневмонит (hypersensitivity pneumonitis); легочный фиброз, включая криптогенный фиброзирующий альвеолит, идиопатические интерстициальные пневмонии, фиброз,осложняющий противоопухолевую терапию, и хроническую инфекцию, включая туберкулез и аспергиллез и другие грибковые инфекции; осложнения трансплантации легких; сосудистые и тромботические расстройства сосудистой сети легких и легочную гипертензию; противокашлевую активность, включая лечение хронического кашля, ассоциированного с воспалительными и секреторными состояниями дыхательных путей, и ятрогенного кашля; острый и хронический ринит, включая лекарственный ринит, и вазомоторный ринит; круглогодичный и сезонный аллергический ринит, включая нервный ринит (сенная лихорадка); полипоз носа; острую вирусную инфекцию, включая простуду, и инфекцию, обусловленную респираторно-синцитиальным вирусом, вирусом гриппа, коронавирусом (включая SARS (тяжелый острый респираторный синдром и аденовирусом; 2) кости и суставы: артриты, ассоциированные с или включающие остеоартрит/остеоартроз, первичный и вторичный относительно, например, врожденной дисплазии тазобедренного сустава; шейный и поясничный спондилит, и боль в нижней части спины и шее; ревматоидный артрит и юношеский ревматоидный артрит; серонегативные спондилоартропатии, включая анкилозирующий спондилоартрит, псориатический артрит, реактивный артрит и недифференцированную спондилоартропатию; септический артрит и другие связанные с инфекциями артропатии и костные расстройства, такие как туберкулез,включая болезнь Потта и полиартрит Понсе; острый и хронический вызванный кристаллами синовит,включая подагру, болезнь отложения пирофосфата кальция и связанное с отложением апатита кальция воспаление сухожилий, синовиальной сумки и синовиальной полости; болезнь Бехчета; первичный и вторичный синдром Шегрена; системный склероз и ограниченную склеродерму; системную красную волчанку, смешанное заболевание соединительной ткани и недифференцированное заболевание соединительной ткани; воспалительные миопатии, включающие дерматомиозит и полимиозит; ревматическую полимиалгию; юношеский артрит, включая идиопатические воспалительные артриты совместного распределения и ассоциированные синдромы, и ревматическую атаку и ее системные осложнения; васкулиты, включая гигантоклеточный артериит, синдром Такаясу, синдром Чарга-Стросса, нодозный полиартериит, микроскопический полиартериит и васкулитиды, ассоциированные свирусной инфекцией, реакцией гиперчувствительности, криоглобулинами и парапротеинами; боль в нижней части спины; семейную-7 017627 средиземноморскую лихорадку, синдром Макла-Уэлса и семейную ирландскую лихорадку, болезнь Кукучи; вызванные лекарствами артралгии, тендониты и миопатии; 3) боль и ремоделирование соединительной ткани при скелетно-мышечных расстройствах, вызванных повреждением [например, спортивные травмы] или заболеванием: артриты (например, ревматоидный артрит, остеоартрит, подагра или кристаллическая артропатия), другое заболевание суставов (такое как дегенерация межпозвоночного диска или дегенерация височно-нижнечелюстного сустава), ремоделирование кости (такое как остеопороз, болезнь Педжета или остеонекроз), полихондрит, склеродерма,смешанное расстройство соединительной ткани, спондилоартропатии или болезнь периодонта (такая как парадонтоз); 4) кожа: псориаз, атопический дерматит, контактный дерматит или другие экзематозные дерматозы и реакции гиперчувствительности замедленного типа; фито- и фотодерматит; себорейный дерматит, герпетиформный дерматит, красный плоский лишай, склероатрофический лишай, гангренозная пиодермия,кожный саркоид, дискоидная красная волчанка, пузырчатка, пемфигоид, буллезный эпидермоз, крапивница, болезнь Квинке, васкулит, токсические эритемы, кожные эозинофилии, очаговая алопеция, алопеция по мужскому типу, синдром Свита, синдром Вебера-Кристиана, полиморфная эритема; целлюлит как инфекционный, так и не инфекционный; панникулит; кожные лимфомы, немеланомный рак кожи и другие диспластические поражения; вызванные лекарствами расстройства, включая локальные лекарственные сыпи; 5) глаза: блефарит; конъюнктивит, включая хронический и весенний аллергический конъюнктивит; воспаление радужной оболочки глаза; передний и задний увеит; хориоидит; аутоиммунные; дегенеративные или воспалительные расстройства, поражающие сетчатку глаза; офтальмит, включая симпатический офтальмит; саркоидоз; инфекции, включая вирусные, грибковые и бактериальные; 6) желудочно-кишечный тракт: глоссит, гингивит, периодонтит; эзофагит, включая рефлюксэзофагит; эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, колит, включая неспецифический язвенный колит, проктит, анальный зуд; целиакия, синдром разраженной толстой кишки и пищевые аллергии, которые могут оказывать действия на расстоянии от кишечника (например, мигрень, ринит или экзема); 7) брюшная полость: гепатит, включая аутоиммунный, алкогольный и вирусный; фиброз и цирроз печени; холецистит; панкреатит как острый, так и хронический; 8) мочеполовой тракт: нефрит, включая интерстициальный и гломерулонефрит; нефротический синдром; цистит, включая острый и хронический (интерстициальный) цистит и язву Ханнера; острый и хронический уретрит, простатит, эпидидимит, оофорит и сальпингит; вульвовагинит; болезнь Пейрони; эректильная дисфункция (у мужчин и женщин); 9) отторжение аллотрансплантата: острое и хроническое после, например, трансплантации почки,сердца, печени, легкого, костного мозга, кожи или роговицы или после переливания крови; или хроническое заболевание трансплантат против хозяина; 10) ЦНС (центральная нервная система): болезнь Альцгеймера и другие дементные расстройства,включая CJD (болезнь Крейтцфельда-Якоба) и nvCJD (новый вариант CJD); амилоидоз; рассеянный склероз и другие демиелинизирующие синдромы; церебральный атеросклероз и васкулит; височный артрит; миастения гравис; острая и хроническая боль (острая, перемежающаяся или персистирующая, центральная или периферическая), включая висцеральную боль, головную боль, мигрень, невралгию тройничного нерва, атипичную лицевую боль, боль суставов и костей, боль, возникающую в результате инвазии рака и опухоли, нейропатические болевые синдромы, включая диабетическую, постгерпетическую и ассоциированную с HIV (ВИЧ, вирус иммунодефицита человека) нейропатии; нейросаркоидоз; осложнения центральной и периферической нервной системы после злокачественных, инфекционных или аутоиммунных процессов; 11) другие аутоиммунные и аллергические расстройства, включая тиреоидит Хашимото, диффузный токсический зоб, болезнь Аддисона, сахарный диабет, идиопатическую тромбоцитопеническую пурпуру, эозинофильный фасцит, гипер-IgE синдром, антифосфолипидный синдром; 12) другие расстройства с воспалительным или иммунологическим компонентом, включая синдром приобретенного иммунодефицита (AIDS), лепру, синдром Сезари и паранеопластические синдромы; 13) сердечно-сосудистая система: атеросклероз, поражающий коронарное и периферическое кровообращение; перикардит; миокардит, воспалительные и аутоиммунные кардиомиопатии, включая миокардиальный саркоид; ишемические реперфузионные повреждения; эндокардит, вальвулит и аортит,включая инфекционный (например, сифилитический); васкулиты; расстройства проксимальных и периферических вен, включая флебит и тромбоз, включая тромбоз глубоких вен и осложнения варикозного расширения вен. 14) онкология: лечении обычных видов рака, включая опухоли предстательной железы, молочной железы, легкого, яичника, поджелудочной железы, кишечника и толстой кишки, желудка, кожи и головного мозга, и злокачественные образования, поражающие костный мозг (включая лейкемии) и лимфопролиферативную систему, такие как ходжкинская и неходжкинская лимфома; включая профилактику и лечение метастатического заболевания и рецидивов опухолей, и паранеопластические синдромы;-8 017627 15) желудочно-кишечный тракт: целиакия, проктит, эозинофильный гастроэнтерит, мастоцитоз, болезнь Крона, неспецифический язвенный колит, микроскопический колит, недетерминантный колит, болезнь раздраженной толстой кишки, синдром раздраженной толстой кишки, невоспалительная диарея,пищевые аллергии, который оказывают действие на расстоянии от кишки, например мигрень, ринит и экзема. В контексте настоящего описания, если не указано иное, термин "терапия" также включает "профилактику". Термины "терапевтический" и "терапевтически" следует рассматривать соответствующим образом. Для применения соединения по изобретению для терапевтического лечения теплокровного животного, такого как человек, указанный ингредиент обычно готовят в соответствии со стандартной фармацевтической практикой в виде фармацевтической композиции. Таким образом, в еще одном аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение по изобретению, как определено ранее, и фармацевтически приемлемый адъювант, разбавитель или носитель. В еще одном аспекте настоящего изобретения предложен способ изготовления указанной композиции, включающий смешивание активного ингредиента с фармацевтически приемлемым адъювантом, разбавителем или носителем. В зависимости от способа введения фармацевтическая композиция, например, содержит от 0,05 до 99 мас.%, например от 0,05 до 80 мас.%,например от 0,10 до 70 мас.%, например от 0,10 до 50 мас.% активного ингредиента, где все проценты по массе основаны на массе всей композиции. Фармацевтические композиции по изобретению могут быть введены стандартным образом при болезненном состоянии, которое желательно лечить, например, при помощи местного (такого как в легкое и/или дыхательные пути, или на кожу), перорального, ректального или парентерального введения. Для этих задач соединения по изобретению могут быть приготовлены при помощи способов, известных в области техники, в форме, например, аэрозолей, сухих порошковых препаратов, таблеток, капсул, сиропов, порошков, гранул, водных или маслянистых растворов или суспензий, (жидких) эмульсий, диспергируемых порошков, суппозиториев, мазей, кремов, капель и стерильных инъецируемых водных или маслянистых растворов или суспензий. Подходящая фармацевтическая композиция по изобретению представляет собой фармацевтическую композицию, подходящую для перорального введения в стандартной лекарственной форме, например в виде таблетки или капсулы, которая содержит от 0,1 мг до 1 г активного ингредиента. В еще одном аспекте фармацевтическая композиция по изобретению представляет собой композицию, подходящую для внутривенной, подкожной или внутримышечной инъекции. Каждый пациент может получать, например, внутривенную, подкожную или внутримышечную дозу от 0,01 до 100 мг/кг соединения, предпочтительно в диапазоне от 0,1 до 20 мг/кг по изобретению, где композицию вводят от 1 до 4 раз в сутки. Внутривенная, подкожная и внутримышечная доза может быть введена путем болюсного введения. Альтернативно, внутривенная доза может быть введена путем непрерывной инфузии в течение периода времени. Альтернативно, каждый пациент получает суточную дозу для перорального введения, которая приблизительно эквивалентна суточной дозе для парентерального введения, где композицию вводили от 1 до 4 раз в сутки. Еще одна подходящая фармацевтическая композиция по изобретению представляет собой композицию, подходящую для ингалируемого введения, где ингаляция представляет собой особенно полезный способ для введения соединения по изобретению при лечении респираторных заболеваний, таких как хроническая обструктивная болезнь легких (COPD) или астма. При введении путем ингаляции соединения по настоящему изобретению могут быть использованы эффективно в дозах, находящихся в мкг диапазоне, например, от 0,1 до 500 мкг, от 0,1 до 50 мкг, от 0,1 до 40 мкг, от 0,1 до 30 мкг, от 0,1 до 20 мкг,от 0,1 до 10 мкг, от 5 до 10 мкг, от 5 до 50 мкг, от 5 до 40 мкг, от 5 до 30 мкг, от 5 до 20 мкг, от 5 до 10 мкг, от 10 до 50 мкг, от 10 до 40 мкг, от 10 до 30 мкг или от 10 до 20 мкг активного ингредиента. В одном воплощении изобретения предложена фармацевтическая композиция, содержащая соединение по изобретению, как определено ранее, вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем, которая изготовлена для ингалируемого введения. При введении путем ингаляции устройства дозированных ингаляторов могут быть использованы для введения активного ингредиента, диспергируемого в подходящем пропелленте, и с добавлением или без дополнительных эксципиентов, таких как этанол, поверхностно-активные вещества, смазывающие вещества или стабилизаторы. Подходящие пропелленты включают углеводородные, хлорфторуглеродные и гидрофторалкановые (например, гептафторалкан) пропелленты или смеси любых таких пропеллентов. Предпочтительные пропелленты представляют собой Р 134 а и Р 227, каждый из которых может быть использован самостоятельно или в комбинации с другими пропеллентами и/или поверхностноактивным веществом, и/или другими экципиентами. Распыляемые водные суспензии или, предпочтительно, растворы, также могут быть использованы, с регулированием рН и/или тоничности или без него,в виде композиций со стандартной дозой или множеством доз. Сухие порошкообразные ингаляторы могут быть использованы для введения активного ингредиента, самостоятельно или в комбинации с фармацевтически приемлемым носителем, в последнем случае в-9 017627 виде тонкоизмельченного порошка или в виде упорядоченной смеси. Сухой порошковый ингалятор может представлять собой однодозовый или содержащий множество доз ингалятор и может использовать сухой порошок или капсулу, содержащую порошок. Дозированный ингалятор, небулайзер и сухой порошковый ингалятор хорошо известны и имеется большое разнообразие таких устройств. Таким образом, изобретение также относится к комбинированным способам терапии, при которых соединение по изобретению или фармацевтическую композицию или препарат, содержащий соединение по изобретению, вводят одновременно или последовательно, или в виде комбинированного препарата с другим терапевтическим агентом или агентами для лечения одного или более чем одного из перечисленных состояний. В частности, для лечения воспалительных заболеваний, таких как (но не ограничиваясь ими) ревматоидный артрит, остеоартрит, астма, аллергический ринит, хроническая обструктивная болезнь легких(COPD), псориаз и воспалительное заболевание кишечника, соединения по изобретению могут быть комбинированы с агентами, перечисленными ниже. Нестероидные противовоспалительные агенты (далее NSAID), включающие неселективные ингибиторы циклооксигеназы СОХ-1/СОХ-2 при местном или системном применении (такие как пироксикам,диклофенак, пропионовые кислоты, такие как напроксен, флурбипрофен, фенопрофен, кетопрофен и ибупрофен, фенаматы, такие как мефенамовая кислота, индометацин, сулиндак, азапропазон, пиразолоны, такие как фенилбутазон, салицилаты, такие как аспирин); избирательные ингибиторы СОХ-2 (такие как мелоксикам, целекоксиб, рофекоксиб, валдекоксиб, лумарококсиб, парекоксиб и эторикоксиб); ингибирующие циклооксигеназу доноры окиси азота (CINOD); глюкокортикостероиды (вводимые местным,пероральным, внутримышечным, внутривенным или внутрисуставным путями); метотрексат; лефлуномид; гидроксихлорхин; d-пеницилламин; ауранофин или другие препараты на основе золота для парентерального или перорального введения; обезболивающие средства; диацереин; средства внутрисуставной терапии, такие как производные гиалуроновой кислоты; и пищевые добавки, такие как глюкозамин. Настоящее изобретение также относится к комбинации соединений по изобретению с цитокином,или агонистом, или антагонистом цитокиновой функции (включая агенты, которые действуют на сигнальные цитокиновые пути, такие как модуляторы системы SOCS), включая альфа-, бета- и гаммаинтерфероны; инсулиноподобный фактор роста I (IGF-1); интерлейкины (IL), включая IL1-17, и интерлейкиновые антагонисты или ингибиторы, такие как анакинра; ингибиторы фактора некроза опухоли альфа (TNF-), такие как моноклональные антитела против TNF (например, инфликсимаб; адалимумаб иCDP-870) и антагонисты рецептора TNF, включая молекулы иммуноглобулина (такие как этанерцепт) и низкомолекулярные агенты, такие как пентоксифиллин. Дополнительно, изобретение относится к комбинации соединения по изобретению с моноклональным антителом, направленным против В-лимфоцитов (таким как CD20 (ритуксимаб), MRA-aIL16R и Тлимфоцитов, CTLA4-Ig, HuMax Il-15). Настоящее изобретение также относится к комбинации соединения по изобретению с модулятором хемокиновой рецепторной функции, таким как антагонист CCR1, CCR2, CCR2A, CCR2B, CCR3, CCR4,CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 и CCR11 (для семейства С-С); CXCR1, CXCR2, CXCR3,CXCR4 и CXCR5 (для семейства С-Х-С) и CX3CR1 для семейства С-Х 3-С. Настоящее изобретение также относится к комбинации соединения по изобретению с ингибитором матриксных металлопротеиназ (ММР), т.е. стромелизинами, коллагеназами и желатиназами, а также аггреканазой; в частности коллагеназой-1 (ММР-1), коллагеназой-2 (ММР-8), коллагеназой-3 (ММР-13),стромелизином-1 (ММР-3), стромелизином-2 (ММР-10) и стромелизином-3 (ММР-11), и ММР-9, и ММР-12, включая агенты, такие как доксоциклин. Настоящее изобретение также относится к комбинации соединения по изобретению с ингибитором биосинтеза лейкотриенов, ингибитором 5-липоксигеназы (5-LO) или антагонистом белка-активатора 5 липоксигеназы (FLAP), таким как зилеутон; АВТ-761; фенфлеутон; тепоксалин; Abbott-79175; Abbott85761; a N-(5-замещеный)-тиофен-2-алкилсульфонамид; 2,6-ди-трет-бутилфеноолгидразоны; метокситетрагидропираны, такие как Zeneca ZD-2138; соединение SB-210661; пиридинилзамещенное 2 цианонафталиновое соединение, такое как L-739,010; 2-цианохинолиновое соединение, такое как L746,530; или индольное или хинолиновое соединение, такое как MK-591, MK-886 и BAY х 1005. Настоящее изобретение также относится к комбинации соединения по изобретению с антагонистом рецептора лейкотриенов (LT) B4, LTC4, LTD4 и LTE4, выбранным из группы, состоящей из фенотиазин 3-1-ов, таких как L-651,392; амидиносоединений, таких как CGS-25019c; бензоксаламинов, таких как онтазоласт; бензолкарбоксимидамидов, таких как BIIL 284/260; и соединений, таких как зафирлукаст,аблукаст, монтелукаст, пранлукаст, верлукаст (МК-679), RG-12525, Ro-245913, иралукаст (CGP 45715A) и BAY х 7195. Настоящее изобретение также относится к комбинации соединения по изобретению с ингибитором фосфодиэстеразы (PDE), таким как метилксантанин, включая теофиллин и аминофиллин; селективный ингибитор изофермента PDE, включая ингибитор PDE4, ингибитор изоформы PDE4D или ингибитор- 10017627 Настоящее изобретение также относится к комбинации соединения по изобретению с антагонистом гистаминового рецептора 1 типа, таким как цетиризин, лоратадин, деслоратадин, фексофенадин, акривастин, терфенадин, астемизол, азеластин, левокабастин, хлорфенирамин, прометазин, циклизин или мизоластин; применяемые перорально, местно или парентерально. Настоящее изобретение также относится к комбинации соединения по изобретению с ингибитором протонного насоса (таким как омепразол) или гастропротективным антагонистом гистаминового рецептора 2 типа. Настоящее изобретение также относится к комбинации соединения по изобретению с антагонистом гистаминового рецептора 4 типа. Настоящее изобретение также относится к комбинации соединения по изобретению с сосудосуживающим симпатомиметиком, представляющим собой агонист альфа-1/альфа-2 адренорецептора, таким как пропилгекседрин, фенилэфрин, фенилпропаноламин, эфедрин, псевдоэфедрин, нафазолина гидрохлорид, оксиметазолина гидрохлорид, тетрагидрозолина гидрохлорид, ксилометазолина гидрохлорид,трамазолина гидрохлорид или этилнорэпинефрина гидрохлорид. Настоящее изобретение также относится к комбинации соединения по изобретению с агонистом бета-адренорецептора (включая бета-рецептор подтипов 1-4), таким как изопреналин, сальбутамол, формотерол, сальметерол, тербуталин, орципреналин, битолтерола мезилат или пирбутерол, или индакатерол,или их хиральный энантиомер. Настоящее изобретение также относится к комбинации соединения по изобретению с хромоном,таким как натрия кромогликат или недокромил натрия. Настоящее изобретение также относится к комбинации соединения по изобретению с глюкокортикоидом, таким как флунизолид, триамцинолона ацетонид, беклометазона дипропионат, будесонид, флутиказона пропионат, циклезонид или мометазона фуроат. Настоящее изобретение также относится к комбинации соединения по изобретению с агентом, модулирующим ядерный рецептор гормона, такой как PPAR (рецепторы, активирующие пролиферацию пероксисом). Настоящее изобретение также относится к комбинации соединения по изобретению с иммуноглобулином (Ig) или препаратом Ig, или антагонистом, или антителом, модулирующим функцию Ig, таким как антитело против IgE (например, омализумаб). Настоящее изобретение также относится к комбинации соединения по изобретению с другим противовоспалительным агентом, применяемым системно или местно, таким как талидомид или его производное, ретиноид, дитранол или кальципотриол. Настоящее изобретение также относится к комбинации соединения по изобретению с комбинацией аминосалицилатов и сульфапиридина, такого как сульфасалазин, месалазин, балсалазид и олсалазин; и иммуномодулирующих агентов, таких как тиопурины, и кортикостероидов, таких как будесонид. Настоящее изобретение также относится к комбинации соединения по изобретению с антибактериальным агентом, таким как производное пенициллина, тетрациклин, макролид, бета-лактам, фторхинолон, метронидазол, ингалируемый аминогликозид; противовирусный агент, включая ацикловир, фамцикловир, валацикловир, ганцикловир, цидофовир, амантадин, римантадин, рибавирин, занамавир и оселтамавир; ингибитором протеазы, таким как индинавир, нелфинавир, ритонавир и саквинавир; нуклеозидным ингибитором обратной транскриптазы, таким как диданозин, ламивудин, ставудин, залцитабин или зидовудин; или ненуклеозидным ингибитором обратной транскриптазы, таким как невирапин или эфавиренз. Настоящее изобретение также относится к комбинации соединения по изобретению с сердечнососудистым агентом, таким как блокатор кальциевых каналов, блокатор бета-адренорецептора, ингибитор ангиотензин-конвертирующего фермента (АСЕ), антагонист рецептора ангиотензина-2; агентом,снижающим уровень липидов, таким как статин или фибрат; модулятором морфологии клеток крови,таким как пентоксифиллин; тромболитическим агентом или антикоагулянтом, таким как ингибитор агрегации тромбоцитов. Настоящее изобретение также относится к комбинации соединения по изобретению с агентом, действующим в отношении CNS (центральной нервной системы), таким как антидепрессант (такой как сертралин), лекарство против болезни Паркинсона [такое как депренил, L-допа, ропинирол, прамипексол,ингибитор МАОВ (моноаминоксидазы В), такой как селегин и разагилин, ингибитор comP, такой как тасмар, ингибитор А-2, ингибитор обратного захвата дофамина, антагонист NMDA (N-метил-Dаспартатный рецептор), никотиновый агонист, дофаминовый агонист или ингибитор нейрональной синтазы окиси азота] или лекарство против болезни Альцгеймера, такое как донепезил, ривастигмин, такрин, ингибитор СОХ-2, пропентофиллин или метрифонат. Настоящее изобретение также относится к комбинации соединения по изобретению с агентом для лечения острой или хронической боли, таким как обезболивающее средство центрального или периферического действия (например, опиоид или его производное), карбамазепин, фенитоин, натрия вальпроат, амитриптилин или другой(ие) антидепрессант(ы), парацетамол или нестероидный противовоспалительный агент.- 11017627 Настоящее изобретение также относится к комбинации соединения по изобретению с используемым парентерально или местно (включая ингаляцию) обезболивающим агентом, таким как лигнокаин или его производное. Соединение по настоящему изобретению также можно использовать в комбинации с агентом против остеопороза, включая гормональный агент, такой как ралоксифен, или бифосфонат, такой как алендронат. Настоящее изобретение также относится к комбинации соединения по изобретению с: (1) ингибитором триптазы; (2) антагонистом фактора активации тромбоцитов (PAF); (3) ингибитором интерлейкинконвертазы (ICE); (4) ингибитором IMPDH (инозинмонофосфатдегидрогеназы); (5) ингибиторами молекул адгезии, включая антагонист VLA-4; (6) катепсином; (7) киназным ингибитором, таким как ингибитор тирозинкиназы (таких как Btk, Itk, Jak3 или MAP, например гефитиниб или иматиниба мезилат), ингибитором сериновой/треониновой киназы (таким как ингибитор MAP киназы, такой как р 38, JNK, протеинкиназы А, В или С, или IKK), или киназы, вовлеченной в регуляцию клеточного цикла (такой как циклинзависимая киназа); (8) ингибитором глюкозо-6 фосфатдегидрогеназы; (9) антагонистом кининового-В 1- или В 2-рецептора; (10) противоподагрическим средством, например колхицином; (11) ингибитором ксантиноксидазы, например аллопуринолом; (12) урикозурическим агентом, например пробенецидом, сульфинпиразоном или бензбромароном; (13) агентом, увеличивающим секрецию гормона роста;(14) трансформирующим фактором роста (TGF); (15) тромбоцитарным фактором роста (PDGF);(16) фактором роста фибробластов, например основным фактором роста фибробластов (bFGF); (17) колониестимулирующим фактором гранулоцитов-макрофагов (GM-CSF); (18) капсаициновым кремом;(19) антагонистом тахикининового NK1 или NK3 рецептора, таким как NKP-608C, SB-233412 (талнетант) или D-4418; (20) ингибитором эластазы, таким как UT-77 или ZD-0892; (21) ингибитором TNFальфа конвертазы (ТАСЕ); (22) ингибитором индуцируемой синтазы окиси азота (iNOS); (23) молекулой,гомологичной хемоаттрактантному рецептору, экспрессирующемуся на клетках ТН 2 (такой как антагонист CRTH2); (24) ингибитором Р 38; (25) агентом, модулирующим функцию Toll-подобных рецепторов(TLR), (26) агентом, модулирующим активность пуринергических рецепторов, таким как Р 2 Х 7; или(27) ингибитором активации фактора транскрипции, таким как NFkB, API или STATS. Соединение по изобретению также может быть использовано в комбинации с существующим терапевтическим агентом для лечения рака, например, подходящие агенты включают:(1) антипролиферативное/антинеопластическое лекарственное средство или их комбинация, которая используется в медицинской онкологии, такое как алкилирующий агент (например, цисплатин, карбоплатин, циклофосфамид, азотистый иприт, мелфалан, хлорамбуцил, бусульфан или нитрозомочевина); антиметаболит (например, антифолат, такой как фторпиримидин, такой как 5-фторурацил или тегафур,ралтитрексед, метотрексат, цитозинарабинозид, гидроксимочевина, гемцитабин или паклитаксел); противоопухолевый антибиотик (например, антрациклин, такой как адриамицин, блеомицин, доксорубицин,дауномицин, эпирубицин, идарубицин, митомицин-С, дактиномицин или митрамицин); антимитотический агент (например, алкалоид барвинка, такой как винкристин, винбластин, виндесин или винорелбин,или таксоид, такой как таксол или таксотере); или ингибитор топоизомеразы (например, эпиподофиллотоксин, такой как этопозид, тенипозид, амсакрин, топотекан или камптотецин);(2) цитостатический агент, такой как антиэстроген (например, тамоксифен, торемифен, ралоксифен,дролоксифен или йодоксифен), понижающий регулятор эстрогенового рецептора (например, фулвестрант), антиандроген (например, бикалутамид, флутамид, нилутамид или ципротерона ацетат), антагонист(3) агент, ингибирующий инвазию раковых клеток (например, ингибитор металлопротеиназы, такой как маримастат, или ингибитор рецепторной функции урокиназного активатора плазминогена);(4) ингибитор функции фактора роста, например антитело против фактора роста (например, антитело против erbb2 трастузумаб, или антитело против erbb1 цетуксимаб [С 225]), ингибитор фарнезилтрансферазы, ингибитор тирозинкиназы или ингибитор сериновой/треониновой киназы, ингибитор семейства фактора роста эпидермиса (например, семейство EGFR тирозинкиназного ингибитора, такого как N-(3 хлор-4-фторфенил)-7-метокси-6-(3-морфолинопропокси)хиназолин-4-амин (гефитиниб, AZD1839), N-(3 этинилфенил)-6,7-бис-(2-метоксиэтокси)хиназолин-4-амин (эрлотиниб, OSI-774) или 6-акриламидо-N-(3 хлор-4-фторфенил)-7-(3-морфолинопропокси)хиназолин-4-амин (CI 1033, ингибитор семейства тромбоцитарных факторов роста или ингибитор семейства фактора роста гепатоцитов;(5) антиангиогенный агент, такой как агент, ингибирующий действия фактора роста эндотелия сосудов (например, антитело против фактора роста эндотелия сосудов бевацизумаб, соединение, раскрытое в WO 97/22596, WO 97/30035, WO 97/32856 или WO 98/13354), или соединение, действующее посредством другого механизма (например, линомид, ингибитор интегриновой v3 функции или ангиостатин);(6) агент, повреждающий сосуды, такой как комбретастатин А 4, или соединение, раскрытое в(7) агент, используемый в антисмысловой терапии, например агент, направленный на одну из перечисленных выше мишеней, такой как ISIS 2503, антисмысловой агент против ras;(8) агент, используемый в способах генной терапии, например в способах по замене аберрантных генов, таких как способы с использованием аберрантного р 53 или аберрантного BRCA1 или BRCA2,GDEPT (ген-направленная терапия пролекарством фермента), такие как способы с использованием цитозиндеаминазы, тимидинкиназы или бактериального фермента нитроредуктазы, и способы, усиливающие толерантность пациента к химиотерапии или радиотерапии, такие как генная терапия, обеспечивающая мультилекарственную резистентность; или(9) агент, используемый в иммунотерапевтическом способе, например способах ex vivo и in vivo для увеличения иммуногенности опухолевых клеток пациента, таких как трансфекция цитокинами, такими как интерлейкин 2, интерлейкин 4 или колониестимулирующий фактор гранулоцитов и макрофагов, способы по уменьшению Т-клеточной толерантности, способы с использованием трансфицированных иммунных клеток, таких как трансфицированных цитокинами дендритные клетки, способы с использованием трансфицированных цитокинами линий опухолевых клеток и способы с использованием антиидиотипических антител. Фармацевтический продукт может представлять собой, например, фармацевтическую композицию,содержащую первый и дополнительный активный ингредиент в смеси. Альтернативно, фармацевтический продукт может, например, содержать первый и дополнительный активный ингредиент в отдельных фармацевтических препаратах, подходящих для одновременного, последовательного или раздельного введения пациенту, нуждающемуся в таком введении. Фармацевтический продукт особенно полезен в лечении респираторных заболеваний, таких как астма, COPD или ринит. Примеры ингибитора фосфодиэстеразы, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают ингибитор PDE4, такой как ингибитор изоформы PDE4D, ингибитор PDE3 и ингибитор PDE5. Примеры включают соединения(V11294A). Примеры агонистов 2-адренорецепторов, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают метапротеренол, изопротеренол, изопреналин,альбутерол, сальбутамол (например, в виде сульфата), формотерол (например, в виде фумарата), салметерол (например, в виде ксинафоата), тербуталин, орципреналин, битолтерол (например, в виде мезилата), пирбутерол или индакатерол. Агонист 2-адренорецептора по изобретению может представлять собой 2-агонисты длительного действия, например салметерол (например, в виде ксинафоата), формотерол (например, в виде фумарата), бамбутерол (например, в виде гидрохлорида), кармотерол (ТА 2005,химически идентифицированный как 8-гидрокси-5-[1-гидрокси-2-2-(4-метоксифенил)-1-метилэтил]- 13017627 амино]этил]-2(1 Н)-хинолон моногидрохлорид, [R-(R,R)], также идентифицированный под ChemicalUS 2005/222144, и соединения GSK 159797, GSK 159802, GSK 597901, GSK 642444 и GSK 678007. Примеры модулятора функции хемокинового рецептора, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают антагонист CCR1 рецептора. Примеры ингибитора киназной функции, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают ингибитор киназы р 38 и ингибитор IKK. Примеры ингибитора протеазы, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают ингибитор эластазы нейтрофилов или ингибитор MMP12. Примеры стероидных агонистов глюкокортикоидных рецепторов, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают будесонид, флутиказонWO 2002/00679, WO 2005/041980 или стероиды GSK 870086, GSK 685698 и GSK 799943. Примеры модулятора нестероидного агониста глюкокортикоидного рецептора, которые могут быть использованы в фармацевтическом продукте в соответствии с этим воплощением, включают примеры,описанные в WO 2006/046916. Изобретение проиллюстрировано примерами. В примерах представлены следующие фигуры: фиг. 1: картина дифракции рентгеновских лучей на порошке для формы А примера 14; фиг. 2: картина дифракции рентгеновских лучей на порошке для формы А примера 15. В примерах спектры NMR (ядерного магнитного резонанса) измеряли на спектрометре Varian UnityInova с частотой протонов 300, или 400, или 500 МГц, или на спектрометре Bruker DRX с частотой протонов 400 или 500 МГц, или на спектрометре Bruker Avance с частотой протонов 600 МГц, или на спектрометре Bruker Avance DPX 300 с частотой протонов 300 МГц. Спектры MS (масс-спектрометрия) измеряли на спектрометре Agilent 1100 MSD G1946D, или спектрометре Hewlett Packard HP1100 MSDG1946A, или спектрометре Waters Micromass ZQ2000. Названия получены с использованием программного обеспечения Autonom 2000 (версия 4.01.305), поставляемого MDL. Данные XRPD (дифракции рентгеновских лучей на порошке) собирали с использованием инструмента PANalytical CubiX PRO или инструмента PANalytical X-Pert.XRPD - PANalytical CubiX PRO. Данные собирали с использованием инструмента PANalytical CubiX PRO в конфигурации - в диапазоне сканирования 2-40 2 с 100-секундной экспозицией на прирост 0,02. Рентгеновские лучи генерировали в медной длиннофокусной трубке, функционирующей при 45 кВ и 40 мА. Длина волны рентгеновских лучей с трубки составляла 1,5418 . Данные собирали с подложек с нулевым фоном, на которые помещали приблизительно 2 мг соединения. Подложку готвили из монокристаллического диоксида кремния, который разрезали вдоль недиффракционирующей плоскости и затем полировали на оптически плоском конце. Проникновение рентгеновских лучей через эту поверхность устранялось в соответствии с экстинкцией Брэгга.PANalvtical X-Pert. Данные собирали с использованием инструмента PANalytical X-Pert в конфигурации 2- в диапазоне сканирования 2-49 2 с 100-секундной экспозицией на прирост 0,02. Рентгеновские лучи генерировали в медной длиннофокусной трубке, функционирующей при 45 кВ и 40 мА. Длина волны рентгеновских лучей с трубки составляла 1,5418 . Данные собирали с подложек с нулевым фоном, на которые помещали приблизительно 2 мг соединения. Подложку готовили из монокристаллического диоксида кремния, который разрезали вдоль недиффракционирующей плоскости и затем полировали на оптически плоском конце. Проникновение рентгеновских лучей через эту поверхность устранялось в соответствии с экстинкцией Брэгга.- 14017627 Термограммы DSC (дифференциальной сканирующей калориметрии) измеряли с использованием дифференциального сканирующего калориметра ТА Q1000 с алюминиевыми стаканчиками и крышками с отверстиями. Массы образцов варьировали от 0,5 до 5 мг. Процедуру осуществляли в потоке газообразного азота (50 мл/мин) и исследуемой температуре от 25 до 300 С при постоянной скорости увеличения температуры 10 С/мин. Термограммы TGA (термогравиметрический анализ) измеряли с использованием термогравиметрического анализатора ТА Q500 с платиновыми стаканчиками. Масса образца варьировала от 1 до 5 мг. Процедуру осуществляли в потоке газообразного азота (60 мл/мин) и исследуемой температуре от 25 до 300 С при постоянной скорости увеличения температуры 10 С/мин. Профили GVS (гравиметрическая сорбция пара) измеряли с использованием аппарата для динамической сорбции пара DVS-1. Образец твердого вещества приблизительно 1-5 мг помещали в стеклянный сосуд и массу образца регистрировали в способе с двойными циклами (от 40 до 90, до 0, до 90, до 0% относительной влажности (ОВ), в шагах по 10% ОВ). Сокращения, используемые в экспериментальном разделе: водн. = водный;XRPD = дифракция рентгеновских лучей на порошке. Пример 1. К магнию (1,2 г) в безводном тетрагидрофуране (60 мл) в среде азота добавляли кристалл йода, а затем бромбензол (7,85 г) с такой скоростью, что реакционная смесь поддерживалась при постоянной дефлегмации. Реакционную смесь перемешивали в течение 20 мин, затем осторожно добавляли циклогептанон (4,48 г). После перемешивания в течение 10 мин добавляли насыщенный водный хлорид аммония (10 мл) и реакционная смесь распределялась между водой (100 мл) и изогексаном (100 мл). Органический слой сушили (MgSO4) и упаривали с получением указанного в подзаголовке соединения (7,6 г) в виде масла. 1 Н NMR (300 МГц, CDCl3):7.53-7.47 (m, 2H), 7.36-7.29 (m, 2H), 7.26-7.19 (m, 1H), 2.07 (ddd, 2H),1.97-1.50 (m, 11 Н). 1-Фенилциклогептанол (пример 1 а) (7,6 г) растворяли в тетрагидрофуране (100 мл) и добавляли гидрид натрия (60% в масле, 2,0 г). Реакционную смесь перемешивали при 60 С в течение 5 мин и добавляли йодметан (7,1 г). Смесь поддерживали при 60 С в течение ночи и затем добавляли дополнительные количества гидрида натрия (60% в масле, 2,0 г) и йодметана (7,1 г), реакционную смесь кипятили с- 15017627 обратным холодильником в течение 70 ч. Реакционную смесь распределяли между водой (100 мл) и изогексаном (100 мл), органический слой отделяли, сушили (MgSO4) и упаривали с получением указанного в подзаголовке соединения (11,31 г). 1 Н NMR (300 МГц, CDCl3):7.43-7.37 (m, 2H), 7.37-7.30 (m, 2H), 7.24-7.19 (m, 1H), 2.98 (s, 3 Н),2.12-1.88 (m, 4H), 1.88-1.45 (m, 8H). с) 1-Фенилциклогептанкарбоновая кислота Калий (2,62 г) и натрий (0,52 г) нагревали вместе при 120 С в минеральном масле в среде азота в течение 30 мин и затем охлаждали до комнатной температуры. Масло удаляли и замещали простым эфиром (100 мл), добавляли 1-метокси-1-фенилциклогептан (пример 1b) (4,9 г) и реакционную смесь перемешивали в атмосфере азота в течение ночи при комнатной температуре. Реакционную смесь охлаждали до -78 С и твердый диоксид углерода (приблизительно 20 г) добавляли при перемешивании. Реакционную смесь оставляли нагреваться до комнатной температуры и осторожно добавляли воду (150 мл) в среде азота. Водный слой отделяли, нейтрализовали концентрированной соляной кислотой и экстрагировали диэтиловым эфиром (150 мл). Органический слой сушили (MgSO4) и упаривали с получением указанного в подзаголовке соединения (4,15 г) в виде масла. 1 Н NMR (300 МГц, CDCl3):7.40-7.20 (m, 5H), 2.49-2.35 (m, 2H), 2.16-2.03 (m, 2H), 1.76-1.47 (m,8H). 1-Фенилциклогептанкарбоновую кислоту (пример 1 с) (4,15 г) кипятили с обратным холодильником в метаноле (150 мл) и концентрированной соляной кислоте (5 мл) в течение 24 ч. Растворитель упаривали и остаток растворяли в простом эфире (100 мл), который промывали водой (100 мл), насыщенным бикарбонатом натрия (50 мл) и водой (100 мл), сушили (MgSO4) и упаривали с получением указанного в подзаголовке соединения (3,5 г) в виде масла. 1 Н NMR (300 МГц, CDCl3):7.37-7.18 (m, 5H), 3.63 (s, 3 Н), 2.47-2.35 (m, 2H), 2.08-1.97 (m, 2H),1.70-1.48 (m, 8H). е) (R)-(1-Азабицикло[2.2.2]окт-3-иловый) эфир 1-фенилциклогептанкарбоновой кислоты Метиловый эфир 1-фенилциклогептанкарбоновой кислоты (пример 1d) (1,0 г) и (R)-хинуклидин-3 ол (0,39 г) кипятили с обратным холодильником в гептане (50 мл), содержащем натрий (приблизительно 5 мг) в аппарате Дин-Старка в течение 24 ч. Гептан (20 мл) замещали толуолом (20 мл) и кипячение с обратным холодильником продолжали в течение 3 суток. Реакционную смесь распределяли между водой(50 мл) и простым эфиром (50 мл) и эфирный слой отделяли, сушили (MgSO4) и упаривали. Неочищенный продукт очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/триэтиламином (99/1) с получением указанного в заголовке соединения в виде масла (0,83 г). Смесь 6-метилпиридин-3-амина (1 г) и триэтиламина (2,2 мл) в безводном THF (20 мл) перемешивали и охлаждали до -60 С. 2-Хлорацетил хлорид (1,567 г) добавляли через шприц к перемешиваемой смеси с образованием желтой суспензии. Смесь перемешивали при -60 С до тех пор, пока анализ не про- 16017627 демонстрировал полное исчезновение исходного вещества. Реакционную суспензию выливали в воду и продукты экстрагировали этилацетатом (2150 мл). Объединенные органические экстракты сушили над сульфатом магния и концентрировали досуха. Неочищенное коричневое твердое вещество перекристаллизовывали из простого эфира с получением указанного в подзаголовке соединения (700 мг). 1 Н NMR (400 МГц, DMSO-D6):10.40 (1 Н, s), 8.60 (1 Н, d), 7.91 (1H, dd), 7.22 (1 Н, d), 4.27 (2 Н, s),2.42 (3 Н, s). Пример 1. (R)-1-[(6-Метилпиридин-3-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)1-азониа-бицикло[2.2.2]октан хлорид(52 мг) растворяли в ацетонитриле (2 мл) и добавляли 2-хлор-N-(6-метилпиридин-3-ил)ацетамид (пример 1f) (29 мг). Реакционную смесь перемешивали в течение 10 суток и разбавляли этилацетатом (4 мл) и изогексаном (14 мл). Смесь оставляли на 5 суток, после чего образующиеся в результате кристаллы отделяли и промывали диэтиловым эфиром (0,5 мл) с получением указанного в заголовке соединения в виде твердого вещества (36 мг). 6-Метилпиразин-2-ил-амин (150 мг) и карбонат калия (571 мг) добавляли к дихлорметану (25 мл). 2-Бромацетил бромид (0,120 мл) добавляли к суспензии при перемешивании. Реакционную смесь перемешивали в течение ночи, затем добавляли воду (0,1 мл) при дополнительном перемешивании. Дополнительные количества карбоната калия (571 мг), 2-бромацетил бромида (0,120 мл) и воды (0,1 мл) добавляли в течение 2 ч до тех пор, пока реакция не придет к завершению. Реакционную смесь разбавляли водой(100 мл), осторожно подкисляли соляной кислотой и экстрагировали дихлорметаном (250 мл), который сушили и упаривали с получением указанного в подзаголовке соединения, которое использовали неочищенным (365 мг). 1 Н NMR (400 МГц, CDCl3):9.45 (s, 1H), 9.37 (s, 1H), 8.33 (s, 1H), 4.05 (s, 2H), 2.51 (s, 3H). Пример 2. (R)-1-[(6-Метилпиразин-2-ил-карбамоил)метил]-3-(1-фенилциклогептанкарбонилокси)-1 азониа-бицикло[2.2.2]октан бромид(1 мл) и оставляли на ночь. Кристаллы отделяли при стоянии и фильтровали и промывали ацетонитрилом (21 мл), этилацетатом (23 мл) и диэтиловым эфиром (23 мл) и сушили с получением указанного в заголовке соединения (24 мг).(0,214 г). Добавляли 2-бромацетил бромид (0,12 мл) и перемешивание продолжали в течение 1,5 ч. Добавляли воду (0,24 мл) и реакционную смесь перемешивали в течение 1,5 ч, после чего добавляли воду(40 мл) и реакционную смесь перемешивали в течение еще 1,5 ч. Дихлорметан отделяли, сушили(MgSO4) и упаривали с получением указанного в подзаголовке соединения в виде белого твердого вещества (0,053 г).(61,1 мг) и 2-бром-N-(6-трифторметилпиридазин-3-ил)ацетамид (пример 3 а) (53,0 мг) растворяли в ацетонитриле (2 мл) и оставляли на ночь. Растворитель упаривали и продукт очищали путем колоночной хроматографии на диоксиде кремния с элюцией с 10% метанолом в дихлорметане с получением указанного в заголовке соединения (107 мг). К смеси бензо[d]изоксазол-3-ил-амина (1 г) и карбоната цезия (2,42 г) в безводном DMF (20 мл), перемешиваемой при к.т., добавляли бромацетил хлорид (0,62 мл) путем добавления по каплям. После перемешивания смеси в течение 8 ч реакционную смесь выливали в воду (100 мл) и продукты экстрагировали в простом эфире (2200 мл). Объединенные экстракты сушили над сульфатом магния и концентрировали досуха. Неочищенный продукт очищали на силикагеле с использованием простого эфира/изогексана (4/6) с получением указанного в подзаголовке соединения в виде бесцветного твердого вещества (0,5 г).(10 мл) и оставляли на одну неделю. Образующиеся в результате кристаллы отфильтровывали и промывали диэтиловым эфиром (310 мл) с получением указанного в заголовке соединения в виде твердого вещества (120 мг). Этил-2-(тиофен-2-ил)ацетат (2,35 г) растворяли в тетрагидрофуране (30 мл) и охлаждали до -78 С. Добавляли литий бис-(триметилсилил)амид (2,31 г) в THF (1 M раствор, 13,8 мл) и раствор перемешивали в течение 30 мин. Добавляли 4-бром-бут-1-ен (1,4 мл), реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь повторно охлаждали до-78 С и добавляли литий бис-(триметилсилил)амид (2,31 г) в THF (1 M раствор, 13,8 мл) и раствор перемешивали в течение 30 мин. Добавляли 4-бром-бут-1-ен (1,4 мл), реакционную смесь оставляли нагреваться до комнатной температуры и оставляли на ночь. Анализ при помощи HPLC-MS (высокоэффективная жидкостная хроматография/масс-спектрометрия) указывал на то, что реакция не завершена, таким образом реакционную смесь повторно охлаждали до -78 С и добавляли дополнительные аликвоты литий бис-(триметилсилил)амида (1 М раствор, 10 мл) и 4-бром-бут-1-ена (1,0 мл) в соответствии со способом,изложенным выше. После перемешивания в течение еще 2 ч добавляли воду (30 мл) и реакционную смесь экстрагировали диэтиловым эфиром (260 мл). Объединенные органические экстракты сушили(MgSO4) и упаривали. Получающееся в результате масло очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/изогексаном (1/99) с получением указанного в подзаголовке соединения (3,18 г). 1 Н NMR (400 МГц, DMSO-D6):7.21 (dd, 1 Н), 6.97-6.94 (m, 2H), 5.79 (ddt, 2H), 5.01 (dq, 2H), 4.95 К этиловому эфиру 2-бут-3-енил-2-тиофен-2-ил-гекс-5-еновой кислоты (пример 5 а) (3,18 г) в дихлорметане (100 мл) добавляли катализатор Граббса (2-е поколение, Sigma-Aldrich Company Ltd)(0,100 г). Смесь кипятили с обратным холодильником в атмосфере азота. Через 20 ч смесь оставляли охлаждаться до комнатной температуры и упаривали до масла. Очистка путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/изогексаном (10:90) позволила получить указанное в подзаголовке соединение (2,60 г) в виде окрашенного масла. 1 Этиловый эфир 1-тиофен-2-ил-циклогепт-4-енкарбоновой кислоты (пример 5b) (2,86 г) растворяли в этаноле (30 мл) и добавляли трис-(трифенилфосфин)родий(I) хлорид (0,100 г). Реакционную смесь быстро перемешивали под давлением 5 атм (506625 Па) водорода в течение ночи. Добавляли дополнительное количество трис-(трифенилфосфин)родий(I) хлорида (0,050 г) и реакционную смесь перемешивали под давлением 5 атм (506625 Па) водорода в течение 3 суток. Осуществляли третье добавление трис(трифенилфосфин)родий(I) хлорида (0,050 г) и реакционную смесь перемешивали под давлением 3 атм(303975 Па) водорода в течение ночи. Содержимое упаривали досуха и очищали на диоксиде кремния с элюцией этилацетатом/изогексаном (5/95) с получением указанного в подзаголовке соединения (2,500 г) в виде прозрачного почти бесцветного масла. Этиловый эфир 1-тиофен-2-ил-циклогептанкарбоновой кислоты (пример 5 с) (2,5 г) и (R)хинуклидин-3-ол (2,08 г) растворяли в толуоле (350 мл) и добавляли гидрид натрия (0,1 г) в атмосфере азота. Смесь кипятили с обратным холодильником в течение 20 ч, после чего толуол осторожно отгоняли, оставляя приблизительно 100 мл, которые охлаждали и промывали водой (100 мл), сушили (MgSO4) и упаривали. Неочищенный продукт очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/триэтиламином (99/1) с получением указанного в подзаголовке соединения (2,84 г). К суспензии пиридазин-3-ил-амина (2,7 г) и диизопропилэтиламина (6,3 мл) в дихлорметане (100 мл) при 0 С добавляли бромацетангидрид (9,0 г) в дихлорметане (10 мл) путем добавления по каплям. Смесь перемешивали при 0 С в течение 0,5 ч и затем оставляли нагреваться до к.т. Получающуюся в результате суспензию фильтровали, промывали дихлорметаном и сушили с получением указанного в подзаголовке соединения в виде твердого вещества (2,0 г). 1 Н NMR (400 МГц, DMSO-D6):11.51 (s, 1H), 9.00 (dd, 1H), 8.28 (dd, 1H), 7.74-7.68 (m, 1H), 4.15 (s,2H). Пример 5. (R)-1-(Пиридазин-3-ил-карбамоилметил)-3-(1-тиофен-2-ил-циклогептанкарбонилокси)-1 азониа-бицикло[2.2.2]октан бромид(3 мл) и перемешивали в течение ночи. Добавляли этилацетат (9 мл) и изогексан (4 мл) и перемешивали в течение ночи. Образующиеся в результате кристаллы отфильтровывали и затем растирали сэтилацетатом с получением указанного в заголовке соединения (14 мг). К перемешиваемой суспензии бикарбоната натрия (1,242 г) и 5-метилизоксазол-3-ил-амина (1,45 г) в дихлорметане (50 мл) добавляли 2-бромацетил бромид (1,28 мл) путем добавления по каплям. Реакционную смесь перемешивали в течение ночи и затем промывали водой (20 мл). Органическую фракцию отделяли, сушили над сульфатом магния и упаривали с получением указанного в подзаголовке соединения (279 мг). 1(R)-(1-Азабицикло[2.2.2]окт-3-иловый) эфир 1-тиофен-2-ил-циклогептанкарбоновой кислоты (пример 5d) (68 мг) и 2-бром-N-(5-метилизоксазол-3-ил)ацетамид (пример 6 а) (45 мг) растворяли в ацетонитриле (2 мл) и перемешивали в течение ночи. Добавляли этилацетат (10 мл) и изогексан (9 мл) и перемешивали в течение ночи. Образующиеся в результате кристаллы отфильтровывали и промывали этилацетатом с получением указанного в заголовке соединения (82 мг).(100 мл) при комнатной температуре и по каплям добавляли 2-бромацетил бромид (6 г). Смесь оставляли перемешиваться в течение ночи. Воду (0,3 мл) добавляли вместе с дополнительным количеством карбоната калия (3 г) и реакционную смесь перемешивали в течение еще 30 мин. Реакционную смесь выливали в воду (100 мл) и экстрагировали дихлорметаном (250 мл). Объединенные органические экстракты сушили над сульфатом магния и затем упаривали в вакууме. Неочищенный продукт очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/изогексаном (50:50) с получением указанного в подзаголовке соединения (4,8 г). К суспензии бикарбоната натрия (1 г) и 3-фторанилина (0,46 г) в дихлорметане (100 мл) добавляли 2-бромацетил бромид (0,36 мл) путем добавления по каплям. После перемешивания в течение ночи реакционную смесь промывали водой, сушили над сульфатом магния и упаривали с получением указанного в подзаголовке соединения (1,07 г).R)-(1-Азабицикло[2.2.2]окт-3-иловый) эфир 1-тиофен-2-ил-циклогептанкарбоновой кислоты (пример 5d) (96 мг) и 2-бром-N-(фторфенил)ацетамид (пример 8 а) (67 мг) растворяли в ацетонитриле (2 мл) и оставляли на ночь. Добавляли диэтиловый эфир (10 мл) и изогексан (8 мл) и смесь оставляли на ночь. Образующиеся в результате кристаллы отфильтровывали и промывали диэтиловым эфиром с получением указанного в заголовке соединения (90 мг).(30 мл), добавляли по каплям бромацетилбромид (2,89 г) и смесь перемешивали при к.т. в течение 2 ч. Добавляли воду (200 мл), смесь экстрагировали этилацетатом (2100 мл) и сушили над сульфатом магния. Концентрирование экстракта до приблизительно 50 мл и добавление изогексана (100 мл) позволило получить указанное в подзаголовке соединение в виде твердого вещества (1,64 г). 1(пример 9 а) (33 мг). После перемешивания в течение 1 недели добавляли диэтиловый эфир (8 мл) и изогексан (5 мл). Кристаллы собирали путем фильтрования, промывали этилацетатом (24 мл) и сушили с получением указанного в заголовке соединения (26 мг).(R)-(1-Азабицикло[2.2.2]окт-3-иловый) эфир 1-тиофен-2-ил-циклогептанкарбоновой кислоты (пример 5d) (71 мг) и N-бензо[d]изоксазол-3-ил-2-хлорацетамид (пример 4 а) (54 мг) растворяли в ацетонитриле (10 мл) и оставляли на 6 суток. Образующиеся в результате кристаллы отфильтровывали и промывали диэтиловым эфиром (310 мл) с получением указанного в заголовке соединения (82 мг). К перемешиваемой суспензии пиразин-2-ил-амина (1,87 г) и карбоната калия (8,19 г) в дихлорметане (25 мл) добавляли по каплям 2-бромацетила бромид (1,72 мл). Реакционную смесь перемешивали в течение ночи и затем промывали водой (250 мл). Органическую фазу отделяли, сушили над сульфатом магния и упаривали с получением указанного в подзаголовке соединения (0,70 г). 1(2 мл) и оставляли на ночь. Добавляли диэтиловый эфир (10 мл) и изогексан (8 мл) и смесь оставляли на ночь. Образующиеся в результате кристаллы отфильтровывали и промывали диэтиловым эфиром с получением указанного в заголовке соединения (117 мг). Метиловый эфир (3-фторфенил)уксусной кислоты (4,30 г) растворяли в тетрагидрофуране (20 мл) и охлаждали до -78 С. Добавляли литий бис-(триметилсилил)амид (25,6 мл, 1 М раствор THF) и раствор перемешивали в течение 30 мин. Добавляли 4-бром-бут-1-ен (2,60 мл) и содержимое реакционной смеси оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Реакционную смесь вновь охлаждали до -78 С. Добавляли литий бис-(триметилсилил)амид (25,6 мл, 1 М раствор THF) и раствор перемешивали в течение 30 мин. Добавляли 4-бром-1-бутен (2,60 мл) и реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение 1 ч. Содержимое вновь охлаждали до -78 С и добавляли дополнительные аликвоты литий бис-(триметилсилил)амида (25,6 мл, 1 М раствор THF) и 4-бром-1-бутена (2,60 мл) в соответствии со способом, изложенным выше. После перемешивания в течение ночи добавляли воду (20 мл) и реакционную смесь экстрагировали диэтиловым эфиром(260 мл). Объединенные органические экстракты сушили над сульфатом магния и упаривали. Получающуюся в результате жидкость очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/изогексаном (1/99) с получением указанного в подзаголовке соединения (5,0 г). К метиловому эфиру 2-бут-3-енил-2-(3-фторфенил)гекс-5-еновой кислоты (пример 12 а) (5,0 г) в дихлорметане (100 мл) добавляли катализатор Граббса (2-е поколение, Sigma-Aldrich Company Ltd) (0,05 г). Смесь кипятили с обратным холодильником в атмосфере азота. Через 20 ч реакционную смесь охлаждали до комнатной температуры, упаривали до масла и очищали путем колоночной хроматографии на диоксиде кремния с элюцией этилацетатом/изогексаном (5/95) с получением масла. Анализ продукта продемонстрировал, что значительные количества исходного вещества присутствуют в смеси, таким образом, смесь повторно подвергали реакционным условиям и очистке, как указано выше, с получением указанного в подзаголовке соединения в виде окрашенного масла (3,60 г). Метиловый эфир 1-(3-фторфенил)циклогепт-4-енкарбоновой кислоты (пример 12b) (1,09 г) растворяли в метаноле (20 мл), добавляли палладий на углероде (50 мг) и смесь перемешивали под давлением 4 атм (405300 Па) водорода в течение ночи. Раствор фильтровали и упаривали с получением указанного в подзаголовке соединения (1,09 г). Метиловый эфир 1-(3-фторфенил)циклогептанкарбоновой кислоты (пример 12 с) (0,280 г) растворяли в толуоле (100 мл) и добавляли (R)-хинуклидин-3-ол (0,320 г). Толуол (10 мл) отгоняли в аппарате Дин-Старка и после охлаждения добавляли гидрид натрия (10 мг). Реакционную смесь кипятили с обратным холодильником в аппарате Дин-Старка в течение 4 ч, после чего добавляли дополнительное количество гидрида натрия (10 мг) и реакционную смесь кипятили с обратным холодильником в течение еще 4 ч. После охлаждения до комнатной температуры толуол промывали водой, сушили (MgSO4) и упаривали. Остаток очищали путем колоночной хроматографии с элюцией этилацетатом/изогексаном/триэтиламин(пример 11 а) (0,05 г). Реакционную смесь перемешивали в течение 3 суток, разбавляли диэтиловым эфиром (8 мл), перемешивали в течение еще 10 мин, получающееся в результате твердое вещество фильтровали и промывали диэтиловым эфиром (38 мл) с получением твердого вещества, которое перекристаллизовывали из горячего бутанона (8 мл) с получением указанного в заголовке соединения в виде твердого вещества (0,081 г).(3,74 г). Бромацетил хлорид (1,12 мл) медленно добавляли при перемешивании и суспензию перемешивали в течение ночи. Реакционную смесь промывали водой (250 мл), сушили и упаривали. Продукт перекристаллизовывали из дихлорметана/изогексана с получением указанного в подзаголовке соединения(пример 12d) (50 мг) и 2-бром-N-изоксазол-3-ил-ацетамид (пример 13 а) (30 мг) растворяли в ацетонитриле (4 мл) и перемешивали в течение ночи. Раствор разбавляли диэтиловым эфиром (12 мл) и перемешивали в течение ночи. Образующиеся в результате кристаллы отфильтровывали, промывали простым эфиром (310 мл) и сушили с получением указанного в заголовке соединения в виде твердого вещества Фенилмагния бромид (3,0 М раствор в диэтиловом эфире) (271 мл) добавляли по каплям к перемешиваемому (верхняя мешалка) раствору циклогептанкарбонитрила (50 г) в 229 мл диэтилового эфира в атмосфере азота с такой скоростью, чтобы поддерживать мягкое кипячение с обратным холодильником. Реакционную смесь затем кипятили с обратным холодильником в течение 3 ч. TLC (тонкослойная хроматография) указывала на то, что в реакционной смеси больше не присутствует исходное вещество. Реакционную смесь оставляли охлаждаться до комнатной температуры и оставляли в атмосфере азота на ночь. Реакционную смесь охлаждали до 0 С и обрабатывали по каплям 102 мл 4 н. HCl (водн.), поддер- 26017627 живая температуру ниже 20 С. 4 н. серную кислоту (203 мл) быстро добавляли по каплям вначале и затем медленнее ближе к концу. Ледяную баню удаляли и диэтиловый эфир отгоняли. Реакционную смесь нагревали при 80-90 С в течение 3,5 ч, затем оставляли охлаждаться до комнатной температуры и оставляли на ночь. Смесь разбавляли простым эфиром (прибл. 450 мл) и водой (100 мл). Слои разделяли и водный слой экстрагировали простым эфиром (2400 мл). Органические слои объединяли и промывали насыщенным водным гидрокарбонатом натрия (600 мл) и рассолом (600 мл), сушили над сульфатом магния, фильтровали и упаривали с получением указанного в подзаголовке соединения в виде оранжевой жидкости (86,5 г). 1 Сульфурил хлорид (210 мл) добавляли по каплям к чистому циклогептилфенилметанону (пример 14 а) (86,5 г) при 0 С в течение приблизительно 1 ч. Обнаружили выделение газа и экзотермию. Внутреннюю температуру поддерживали ниже 15 С во время добавления и выделенный газ поглощали путем пропускания через 10,2 М водный раствор NaOH. Реакционную смесь кипятили с обратным холодильником в течение ночи. Реакционную смесь охлаждали до 0 С и медленно выливали в лед (1 л) при перемешивании. Слои разделяли и водный слой экстрагировали простым эфиром (2400 мл). Объединенные органические слои промывали водой (600 мл), насыщенным водным гидрокарбонатом натрия (600 мл) и рассолом (600 мл), сушили над сульфатом магния, фильтровали и упаривали с получением указанного в подзаголовке соединения в виде коричневого масла (100 г). 1 Н NMR (400 МГц, CDCl3):8.10-8.06 (d, 2H), 7.52-7.46 (t, 1H), 7.44-7.36 (t, 2H), 2.50 (ddd, 2H), 2.29 Раствор (1-хлорциклогептил)фенилметанона (пример 14b) (100 г) в 750 мл диоксана по каплям быстро обрабатывали мутным раствором нитрата серебра (137 г) в воде (85 мл), вызывая образование осадка. Реакционную смесь нагревали до 75 С в течение 4,5 ч. Реакционную смесь охлаждали до комнатной температуры, затем фильтровали и концентрировали до приблизительно 200 мл. Добавляли воду(200 мл) и простой эфир (300 мл) и слои отделяли. Водный слой экстрагировали простым эфиром(2250 мл). Объединенные органические слои экстрагировали 10% водным карбонатом натрия(3250 мл). Объединенные основные экстракты нагревали до 90 С в течение 40 мин и затем охлаждали до комнатной температуры и подкисляли концентрированной HCl (водн.). Получающееся в результате коричневое твердое вещество отфильтровывали, промывали водой (2) и сушили в вакууме при 50 С. Кристаллизация из горячего этанола (40 мл) позволила получить указанное в подзаголовке соединение в виде бледно-коричневых кристаллов (9,83 г). 1 Н NMR (400 МГц, CD3OD):7.36-7.26 (m, 4H), 7.21-7.15 (m, 1H), 2.43-2.35 (m, 2H), 2.07-1.98 (m,2H), 1.70-1.53 (m, 8H). 2,0 М раствор триметилсилил диазометана (29,2 мл) добавляли по каплям к раствору 1 фенилциклогептанкарбоновой кислоты (пример 14 с) (9,8 г) в метаноле (85 мл) и толуоле (300 мл) в атмосфере азота. Через 45 мин реакционную смесь концентрировали в вакууме и неочищенный продукт очищали путем колоночной хроматографии с элюцией 0-10% этилацетатом/циклогексаном с получением продукта в Раствор (R)-(3)-хинуклидинола (10,13 г) и метилового эфира 1-фенилциклогептанкарбоновой кислоты (пример 14d) (9,25 г) в толуоле (90 мл) кипятили с обратным холодильником с ловушкой ДинСтарка в течение 30 мин. Реакционную смесь оставляли охлаждаться до комнатной температуры и ловушку удаляли. Гидрид натрия (60% дисперсия в минеральном масле) (3,19 г) порциями добавляли в атмосфере азота и реакционную смесь кипятили с обратным холодильником в течение ночи в атмосфере азота. Реакционную смесь охлаждали в ледяной бане и разбавляли этилацетатом (200 мл) и водой(200 мл). Смесь фильтровали и слои разделяли. Водный слой экстрагировали этилацетатом (2250 мл) и объединенные органические слои промывали рассолом, сушили над сульфатом магния и упаривали с получением неочищенного продукта, который очищали путем хроматографии на силикагеле с элюциейEtOAc, содержащим 1% триэтиламин, с получением указанного в подзаголовке соединения в виде бесцветного масла (7,63 г). 1 Н NMR (400 МГц, CD3OD):7.34-7.28 (m, 4H), 7.23-7.17 (m, 1H), 4.80-4.75 (m, 1 Н), 3.12 (ddd, 1H),2.75-2.65 (m, 3 Н), 2.53-2.37 (m, 4H), 2.14-2.06 (m, 2H), 1.88-1.85 (m, 1H), 1.69-1.54 (m, 10H), 1.54-1.42 (m,1H), 1.35-1.24 (m, 1H). Раствор 2-аминопиридина (1,0 г) в безводном дихлорметане (10,6 мл) в атмосфере азота при 0 С обрабатывали триэтиламином (1,63 мл), а затем медленно добавляли хлорацетила хлорид (0,93 мл). Реакционную смесь оставляли нагреваться до комнатной температуры. Через 2 ч смесь распределялась между дихлорметаном и водой. Фазы разделяли и водный слой экстрагировали дихлорметаном (2). Объединенные органические слои промывали рассолом, сушили над сульфатом магния, фильтровали и концентрировали с получением неочищенного продукта, который очищали путем хроматографии на силикагеле с элюцией 0-30% этилацетатом/циклогексаном с получением указанного в заголовке соединения(1,43 г) в виде розового твердого вещества. Дополнительной очистки достигали путем растирания с 40-60 петролейным эфиром с получением 1,15 г желаемого продукта. Кристаллизация 0,94 г порции материала из дефлегмируемого ацетонитрила (2,4 мл) позволила получить указанное в подзаголовке соединение в виде розового твердого вещества (0,73 г). 1(пример 14 е) (254 мг) в ацетонитриле (5 мл) обрабатывали 2-хлор-N-пиридин-2-ил-ацетамидом (пример 14f) (146 мг) и получающийся в результате желтый раствор перемешивали при комнатной температуре в течение ночи, в течение которой твердое вещество осаждалось. Реакционную смесь обрабатывали приблизительно 2 мл простого эфира, твердое вещество отфильтровывали, промывали простым эфиром и сушили в вакууме с получением указанного в заголовке соединения в виде беловатого твердого вещества(217 мг). Кристаллизация из дефлегмируемого ацетонитрила (20 мл) позволила получить 98 мг указанного в заголовке соединения в виде белого кристаллического твердого вещества.(пример 14 е) (254 мг, 0,78 ммоль) в ацетонитриле (5 мл) обрабатывали 2-хлор-N-пиридин-2-илацетамидом (пример 14f) (146 мг), получающийся в результате желтый раствор перемешивали при комнатной температуре в течение ночи, при этом твердое вещество осаждалось. Реакционную смесь обрабатывали парой миллилитров простого эфира, твердое вещество отфильтровывали, промывали простым эфиром и сушили в вакууме с получением указанного в заголовке соединения (217 мг) в виде беловаго твердого вещества. Кристаллизация из дефлегмируемого ацетонитрила (20 мл) позволила получить 98 мг указанного в заголовке соединения в виде белого кристаллического твердого вещества.m/e 462 [М]+. 1 Н NMR (400 МГц, DMSO-D6):11.09 (s, 1H), 8.34-8.32 (d, 1H), 7.97 (d, 1H), 7.85-7.79 (t, 1H), 7.337.25 (m, 4H), 7.21-7.13 (m, 2H), 5.07 (m, 1H), 4.29 (s, 2H), 4.07 (ddd, 1H), 3.65-3.51 (m, 4H), 3.41-3.29 (m,1H), 2.36-2.23 (m, 2H), 2.17-2.04 (m, 2H), 1.99-1.81 (m, 3H), 1.78-1.66 (m, 1H), 1.77-1.19 (m, 9H). Анализ примера 14. Кристаллическая форма А хлорида (R)-3-(1-фенилциклогептанкарбонилокси)1-(пиридин-2-ил-карбамоилметил)-1-азониа-бицикло[2.2.2]октана. Образец кристаллического примера 14 кристаллической формы А, полученной при помощи способа, описанного выше, анализировали при помощи XRPD (PANalytical X'Pert system), DSC и TGA. Температура плавления для примера 14 хлорида формы А, определенная при помощи DSC, составила 239 С (начало) (2 С). Потеря массы, обнаруженная перед плавлением при помощи TGA, оказалась незначительной. Определение GVS позволило получить незначительное увеличение массы (% мас./мас.) при 80% ОВ (относительной влажности) (0,2%). Спектр XRPD для примера 14 хлорида формы А представлен на фиг. 1. Пример 15. К раствору 2-аминопиридина (48,8 ммоль) в безводном THF (98 мл) при комнатной температуре по каплям добавляли Et3N (58,6 ммоль) и бромацетил бромид (58,6 ммоль). Смесь перемешивали в течение ночи и гасили насыщ. NaHCO3 (водн.). EtOAc добавляли к смеси и слои разделяли. Водную фазу экстрагировали EtOAc, объединенные органические слои сушили (MgSO4) и концентрировали в вакууме до коричневого твердого вещества. Очистка путем флэш-хроматографии на силикагеле с элюцией 1-2% МеОН/дихлорметан позволила получить указанное в подзаголовке соединение в виде желтого твердого вещества (1,14 г). 1 Н NMR (400 МГц, CDCl3):8.75 (s, 1H), 8.26 (ddd, 1H), 8.10 (d, 1H), 7.67 (ddd, 1H), 7.03 (ddd, 1H),3.94 (s, 2H). Пример 15. (R)-3-(1-Фенилциклогептанкарбонилокси)-1-(пиридин-2-ил-карбамоилметил)-1-азониабицикло[2.2.2]октан бромид(0,79 ммоль) и 2-бром-N-пиридин-2-ил-ацетамид (пример 15 а) (0,87 ммоль) перемешивали вместе в безводном MeCN при комнатной температуре в течение 2,5 суток. Реакционную смесь концентрировали в вакууме и желтое твердое вещество очищали путем колоночной флэш-хроматографии на силикагеле с элюцией 2-8% МеОН/дихлорметан с получением рыжевато-коричневого твердого вещества, которое кристаллизовали из кипящего MeCN с получением указанного в заголовке соединения в виде белого твердого вещества (211 мг).
МПК / Метки
МПК: A61P 11/00, A61K 31/439, C07D 453/02
Метки: мускаринового, качестве, антагонистов, рецептора, хинуклидиновые, производные
Код ссылки
<a href="https://eas.patents.su/30-17627-hinuklidinovye-proizvodnye-v-kachestve-antagonistov-muskarinovogo-m3-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Хинуклидиновые производные в качестве антагонистов мускаринового м3 рецептора</a>
Производные карбамата 1-алкил-1-азониабицикло [2.2.2] октана и их применение в качестве антагонистов мускаринового рецептора

Номер патента: 6505
Опубликовано: 29.12.2005
Авторы: Катена Руис Хуан Лоренсо, Фернандес Серрат Анна, Сальседо Рока Каролина, Бальса Лопес Долорс, Микель Боно Игнасио-Хосе, Фарреронс Гальеми Карлес, Лагунас Арналь Кармен, Толедо Меса Нативидад, Фернандес Гарсия Андрес
МПК: A61K 31/435, A61P 43/00, C07D 453/06...
Метки: применение, октана, рецептора, карбамата, антагонистов, мускаринового, производные, качестве, 1-алкил-1-азониабицикло, 2.2.2
Формула / Реферат:
1. Соединение общей формулы (I) где R1, R2 и R3 означают радикалы, независимо выбранные из группы, состоящей из H, OH, NO2, SH, CN, F, Cl, Br, I, COOH, CONH2, (C1-C4)-алкоксикарбонила, (C1-C4)-алкилсульфанила, (C1-C4)-алкилсульфинила, (C1-C4)-алкилсульфонила, (C1-C4)-алкоксила, необязательно замещенного одним или несколькими атомами F, и (C1-C4)-алкила, необязательно замещенного одним или несколькими атомами F или OH; альтернативно, или R1 и...
Азабициклические производные в качестве антагонистов мускаринового рецептора

Номер патента: 9387
Опубликовано: 28.12.2007
Авторы: Дхармараджан Санкаранарайанан, Кхугх Анита, Сарма Пакала Кумара Савитхру, Кумар Нареш, Салман Мохаммад
МПК: A61P 1/00, A61P 11/00, A61K 31/403...
Метки: рецептора, мускаринового, антагонистов, качестве, азабициклические, производные
Формула / Реферат:
1. 6-Замещенные азабицикло[3,1,0]гексаны, выбранные из следующих соединений: N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамид (соединение 1); N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-фенил-2-гидрокси-2-(N-метил)фенилацетамида тартрат (соединение 2); (2R,2S)-N-[(1a,5a,6a)-3-азабицикло[3,1,0]гекс-6-ил-метил]-2-изопропил-2-гидрокси-2-фенилацетамид (соединение 3);...
Азабициклические производные в качестве антагонистов мускаринового рецептора

Номер патента: 9942
Опубликовано: 28.04.2008
Авторы: Салман Мохаммад, Кумар Нареш, Каур Кирандип, Сарма Пакала Кумара Савитхру, Кхугх Анита, Дхармараджан Санкаранарайанан, Мехта Анита
МПК: A61K 31/403, A61P 1/00, A61P 11/00...
Метки: антагонистов, рецептора, мускаринового, производные, качестве, азабициклические
Формула / Реферат:
1. Азабициклические производные в качестве антагонистов мускаринового рецептора, выбранные из следующих соединений: (2R,2S)(1a,5a,6a)-N-{[4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-бутил]-3-азабицикло[3.1.0]гекс-6-ил-метил}-2-гидрокси-2-циклопентил-2-фенилацетамид (соединение 1), (2R)(1a,5a,6a)-N-(3-бензил-3-азабицикло[3.1.0]гекс-6-ил-метил)-2-гидрокси-2-циклопент-1-енил-2-фенилацетамид (соединение 2),...
Замещенные производные азабициклогексана в качестве антагонистов мускаринового рецептора

Номер патента: 9059
Опубликовано: 26.10.2007
Авторы: Гупта Джанг Бахадур, Силамкоти Арундутт Висванатхам, Мехта Анита
МПК: A61P 11/00, A61P 1/00, A61K 31/403...
Метки: производные, азабициклогексана, замещенные, антагонистов, мускаринового, качестве, рецептора
Формула / Реферат:
1. Соединения структурной формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереомеры, N-оксиды, полиморфные формы, где Ar представляет собой арильное или гетероарильное кольцо, имеющее 1-2 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота, арильное или гетероарильное кольца могут быть незамещенными или замещенными от одного до трех заместителями, независимо...
Производные циклопропила в качестве антагонистов рецептора nk3

Номер патента: 9477
Опубликовано: 28.02.2008
Авторы: Хансен Туре, Руланд Томас, Нергор Мортен Банг, Поульсен Андерс, Бьернхольм Берит, Нильсен Серен Меллер, Келер Ян
МПК: A61K 31/495, A61P 25/00, A61K 31/435...
Метки: рецептора, циклопропила, качестве, производные, антагонистов
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль; где R1-R5 независимо выбирают из водорода и галогена; R6 выбирают из водорода и C1-6алкила; R7 является группой фенил-CR8R9-, R8 и R9 независимо выбирают из водорода и C1-6алкила; n равно 0, 1 или 2; Q выбирают из (ii), (iii), стрелки указывают на место присоединения: где R11 выбирают из фенила или бензила; R12 является фенилом; R13 является водородом, или гидрокси, или одной...
Предыдущий патент: Фармацевтические композиции, содержащие антагонисты брадикинина и гиалуроновую кислоту, и их применения
Следующий патент: Соединения хинуклидина в качестве лигандов никотинового ацетилхолинового рецептора α7
Случайный патент: Способ получения энтакапона