Агонисты fxr
Номер патента: 15632
Опубликовано: 31.10.2011
Авторы: Стельзер Линдсэй Скотт, Доти Роберт Энтони, Варшавски Алан М., Цюй Фучэн, Ститс Райан Эдвард, Ма Тяньвэй, Маннинен Питер Рудольф, Ландер Питер Амброз, Белл Майкл Грегори, Очоада Джейсон Мэттью, Генин Майкл Джеймс






























Формула / Реферат
1. Соединение формулы

где р равен 0, или 1, или 2;
X1 представляет собой С или N и Х2 представляет собой С или N; при условии, что X1 и Х2 одновременно не являются N;
R1 и R2независимо выбраны из группы, состоящей из атома водорода, C1-С6алкила, С1-С6галогеналкила, C1-С6алкокси, C1-С6галогеналкокси, атома галогена, -SC1-С6алкила и -S-C1-С3галогеналкила;
каждый R3независимо выбран из группы, состоящей из атома водорода, С1-С6алкила, C1-С6галогеналкила, С1-С6алкокси, С1-С6галогеналкокси и атома галогена;
R4a выбран из группы, состоящей из атома водорода, C1-С6алкила, С1-С6галогеналкила, С3-С8циклоалкила, С4-С8алкилциклоалкила, C1-С6алкокси и С1-С6галогеналкокси;
R4b выбран из группы, состоящей из атома водорода, C1-С6алкила, C1-С6галогеналкила, С3-С8циклоалкила, С4-С8алкилциклоалкила, С1-С6алкокси и C1-С6галогеналкокси;
R5 и R5a независимо выбраны из группы, состоящей из атома водорода и С1-С3алкила;
R6 выбран из группы, состоящей из атома водорода, C1-С6алкила, С1-С6галогеналкила, атома галогена, С1-С6алкокси, C1-С6галогеналкокси, NO2, С3-С8циклоалкила и С4-С8алкилциклоалкила;
L1 выбран из группы, состоящей из связи, C1-С6алкила, CRa=CRb, этинила, С1-С5алкилена, С1-С5алкил-S-, С1-С5алкил-O-, N(Rc)-С1-С5алкила и -С1-С5алкил-N(Rc)-, где Ra и Rb независимо выбраны из группы, состоящей из атома водорода и C1-С3алкила, и Rc независимо выбран из группы, состоящей из Н, С1-С5алкила, С1-С3алкилфенила и С4-С8алкилциклоалкила;
Ar1 выбран из группы, состоящей из индолила, тиенила, бензотиенила, нафтила, фенила, пиридинила, пиразолила, оксазолила, бензоизоксазолила, бензофуранила, пирролила, тиазолила, бензоизотиазолила, индазолила и фуранила, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, С1-С6алкила, С1-С6галогеналкила, атома галогена, С2-С6алкенила, С2-С6алкинила, C1-С6алкокси, -OC1-С2алкилфенила, N(RC)SO2 С1-С6алкила, -C(O)R10 и NHC(O)R10;
R7 выбран из группы, состоящей из СООН, С1-С6алкилСООН, -О-С1-С5алкилСООН, С2-С4алкенилСООН, С3-С8циклоалкилСООН и CONR11R11;
каждый R10 независимо выбран из группы, состоящей из атома водорода, C1-С6алкила и фенила;
каждый R11 независимо представляет собой атом водорода или C1-С6алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где
р равен 0, или 1, или 2;
X1 представляет собой С или N и Х2 представляет собой С или N, при условии, что X1 и Х2 одновременно не являются N;
R1 и R2независимо выбраны из группы, состоящей из атома водорода, C1-С3алкила, С1-С3галогеналкила, C1-С3тиогалогеналкила, С1-С3алкокси, C1-С3галогеналкокси и атома галогена;
R3 отсутствует или независимо выбран из группы, состоящей из C1-С3алкила, С1-С3галогеналкила, C1-С3алкокси, C1-С3галогеналкокси и атома галогена;
R4a выбран из группы, состоящей из атома водорода, С1-С3алкила, C1-С3галогеналкила, С3-С6циклоалкила и С4-С5алкилциклоалкила;
R4b выбран из группы, состоящей из атома водорода, C1-С3алкила, C1-С3галогеналкила, С3-С6циклоалкила и С4-С5алкилциклоалкила;
R5 и R5a независимо выбраны из группы, состоящей из атома водорода и C1-С3алкила;
R6 выбран из группы, состоящей из атома водорода, С1-С3алкила, С1-С3галогеналкила, атома галогена и
-NO2;
L1 выбран из группы, состоящей из связи, CRa=CRb, этинила, С1-С3алкил-S-, С1-С3алкил-O-, N(Rc)-С1-С3алкила и -C1-С3алкил-N(Rc)-, где Ra и Rb независимо выбраны из группы, состоящей из атома водорода и С1-С3алкила, и Rc независимо выбран из группы, состоящей из Н, С1-С5алкила, С1-С3алкилфенила и С4-С8алкилциклоалкила;
Ar1 выбран из группы, состоящей из индолила, бензотиенила, бензоизотиазолила, индазолила, нафтила, фенила, пиридинила, пиразолила, пирролила, тиенила, тиазолила и фуранила, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, C1-С3алкила, C1-С3галогеналкила, атома галогена, С2-С4алкенила, С2-С4алкинила, С1-С4алкокси,
-OC1-С2алкилфенила, -NHC(О)R10;
R7 выбран из группы, состоящей из -СООН, -С1-С3алкилСООН, -O-C1-С3алкилСООН и -CONR11R11;
каждый R10 независимо выбран из группы, состоящей из атома водорода, C1-С3алкила и фенила;
каждый R11 независимо представляет собой атом водорода или С1-С5алкил;
или его фармацевтически приемлемая соль.
3. Соединение по п.1, где
р равен 0 или 1;
X1 и Х2 оба являются С или X1 представляет собой N и Х2 представляет собой С;
R1 и R2независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора, CF3, SCF3, OCF3;
R3 представляет собой атом фтора, атом хлора, C1-С3алкил, CF3, SCF3 или OCF3;
R4a представляет собой атом водорода, метил, этил, изопропил или циклопропил;
R4b представляет собой Н, С1-С3алкил, С1-С3галогеналкил, C1-С3галогеналкокси или С3-С4циклоалкил;
R5 и R5a, каждый независимо, выбран из Н или C1-С3алкила;
группа Ar1представляет собой фенил, индолил, пиридинил, пирролил, тиенил, нафтил, тиазолил, фуранил, пиразолил, индазолил, бензоизотиазолил и бензотиенил, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из С1-С5алкила, С1-С3алкокси, С1-С2галогеналкокси и C1-С3галогеналкила;
R6 представляет собой атом водорода, метил, этил или атом хлора;
L1 представляет собой связь, этенил, -CH(CH3)-S-, С(СН3)2-S-, -CH2O-, -CH2CH2O-, -СН(СН3)-O-,
-СН(СН3)СН2-O-, -СН(СН2СН3)-O-, -CH2NH-, -CH2CH2NH-, -N(Rc)CH2-, N(Rc)CH2CH2- или N(Rc)CH2CH2CH2-, где Rc представляет собой атом водорода, С1-С2алкил, бензил или -СН2СН2-O-СН2-;
R7 представляет собой СООН, -СН2СООН, -СН(СН3)СООН, -циклопропилСООН, -С(СН3)2СООН, CONH2, C(O)NHCH3или С(О)NHCH2CH3;
R10 представляет собой атом водорода или С1-С2алкил и
R11 представляет собой атом водорода или С1-С2алкил.
4. Соединение по п.1 или 3, где X1 и Х2 оба являются С; р равен 0; R1 и R2независимо выбраны из группы, состоящей из атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой этенил, этинил, -N(СН3)СН2- или -N(СН3)СН2СН2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, атом хлора или брома; Ar1 представляет собой фенил, индолил, индазолил, бензотиенил или бензоизотиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН.
5. Соединение, выбранное из группы, состоящей из
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-метоксибензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-метилбензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-изопропоксибензойной кислоты,
2-бутокси-4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
2-бензилокси-4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]нафталин-1-карбоновой кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-пентилбензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-5-метилбензойной кислоты,
2-бутириламино-4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-трифторметилбензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-5-трифторметилбензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-3-гидроксибензойной кислоты,
5-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-фторбензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-4-фторбензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-4-метоксибензойной кислоты,
2-бутокси-5-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
3-бутокси-5-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
3-[({4-[4-циклопропил-2-(2,6-дихлорфенил)-2Н-пиразол-3-илметокси]фенил}метиламино)метил]бензойной кислоты,
4-[({4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
4-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
3-[({4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]бензойной кислоты,
4-[({6-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилпиридин-3-ил}метиламино)метил]бензойной кислоты,
3-[({6-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилпиридин-3-ил}метиламино)метил]бензойной кислоты,
5-[({6-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилпиридин-3-ил}метиламино)метил]-2-метоксибензойной кислоты,
4-[({6-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилпиридин-3-ил}метиламино)метил]-2-пентилбензойной кислоты,
4-[({6-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилпиридин-3-ил}метиламино)метил]-2-метилбензойной кислоты,
6-{4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}-1-метил-1Н-индол-3-карбоновой кислоты,
6-{4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил]бензо[b]тиофен-3-карбоновой кислоты,
5-{4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}тиофен-2-карбоновой кислоты,
5-{4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}тиофен-2-карбоновой кислоты,
2-{4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}-4-метилтиазол-5-карбоновой кислоты,
6-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]никотиновой кислоты,
4-[({4-[4-изопропил-2-(2-трифторметоксифенил)-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-2-метилбензойной кислоты,
3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}этиламино)метил]бензойной кислоты,
и их фармацевтически приемлемой соли, сольвата, энантиомера или диастереоизомера.
6. Соединение, представляющее собой 2-[2-({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)этил]-5-метилбензойную кислоту.
7. Соединение, представляющее собой 3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}метиламино)метил]-5-метилбензойную кислоту.
8. Соединение, представляющее собой 3-[({4-[2-(2,6-дихлорфенил)-4-изопропил-2Н-пиразол-3-илметокси]-2-метилфенил}этиламино)метил]бензойную кислоту.
9. Фармацевтическая композиция, содержащая соединение по любому из пп.1-8 и носитель, разбавитель или эксципиент.
10. Применение соединения по любому из пп.1-8 для получения лекарственного препарата для лечения заболеваний, опосредуемых FXR.
Текст
ИСПРАВЛЕННОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ЕВРАЗИЙСКОМУ ПАТЕНТУ В изобретении предложены соединения формулы Белл Майкл Грегори, Доти Роберт Энтони, Генин Майкл Джеймс, где переменные являются такими, как определено в данном описании, и фармацевтические композиции, содержащие данные соединения, а также способы их применения, при этом указанные соединения и фармацевтические композиции полезны в лечении дислипидемии и родственных заболеваний. Примечание: библиография отражает состояние при переиздании(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 015632 Изобретение относится к области медицинской органической химии, фармакологии и медицины. В частности, настоящее изобретение относится к новым соединениям, полезным при лечении заболеваний,связанных с дислипидемией. Дислипидемия и заболевания, связанные с дислипидемией, например атеросклероз, ишемическая болезнь сердца, инсульт и так далее, являются основными причинами смерти, возникновения других заболеваний и экономических потерь. Общепризнанно, что липиды плазмы, в частности фракции холестерина, играют значительную роль в нормальном функционировании сердечно-сосудистой системы. Поэтому желательна модуляция липидов плазмы, таких как триглицериды, ЛПВП-холестерин (холестерин липопротеинов высокой плотности) и ЛПНП-холестерин (холестерин липопротеинов низкой плотности),оказывающая благоприятное действие. В настоящее время прилагаются значительные усилия для получения безопасных и эффективных соединений для лечения заболеваний, связанных с дислипидемией. Например, в международной заявкеWO 2004/048349 А 1 описаны соединения, полезные в качестве агонистов фарнезоидного Х-рецептора(FXR). Агонисты FXR являются лигандами ядерного рецептора, который регулирует транскрипцию генов,контролирующих метаболизм триглицеридов, холестерина и углеводов. Ввиду того, что усилия, направленные на получение таких соединений, не привели к желаемому результату, существует необходимость в обнаружении и разработке соединений, которые будут (1) высокоактивными, (2) эффективными (согласно экспериментам на in vivo моделях) и/или (3) селективными агонистами FXR. Такие соединения могут оказаться полезными при лечении расстройств, которые характеризуются нежелательным липидным профилем или являются следствием такого профиля, включая дислипидемию, атеросклероз, диабет и сходные заболевания. В настоящем изобретении предложены соединения, которые, как считается, должны являться (1) высокоактивными, (2) эффективными (согласно экспериментам на in vivo моделях) и/или (3) селективными агонистами FXR. В настоящем изобретении предложено соединение формулыX1 представляет собой С или N, и Х 2 представляет собой С или N; при условии, что X1 и Х 2 одновременно не являются N;R1 и R2 независимо выбраны из группы, состоящей из атома водорода, C1-С 6 алкила, C1-С 6 галогеналкила, С 1-С 6 алкокси, С 1-С 6 галогеналкокси, атома галогена, -SC1-С 6 алкила и -S-C1-С 3 галогеналкила; каждый R3 независимо выбран из группы, состоящей из атома водорода, C1-С 6 алкила, С 1-С 6 галогеналкила, С 1-С 6 алкокси, C1-С 6 галогеналкокси и атома галогена;R4a выбран из группы, состоящей из атома водорода, C1-С 6 алкила, C1-С 6 галогеналкила, С 3-С 8 циклоалкила, С 4-С 8 алкилциклоалкила, С 1-С 6 алкокси и C1-С 6 галогеналкокси;R4b выбран из группы, состоящей из атома водорода, C1-С 5 алкила, C1-С 6 галогеналкила, С 3-С 8 циклоалкила, С 4-С 8 алкилциклоалкила, С 1-С 6 алкокси и C1-С 6 галогеналкокси;R5 и R5a независимо выбраны из группы, состоящей из атома водорода и C1-С 3 алкила;R6 выбран из группы, состоящей из атома водорода, C1-С 6 алкила, С 1-С 6 галогеналкила, атома галогена, С 1-С 6 алкокси, C1-С 6 галогеналкокси, NO2, С 3-С 8 циклоалкила и С 4-С 8 алкилциклоалкила;L1 выбран из группы, состоящей из связи, С 1-С 6 алкила, CRa=CRb, этинила, С 1-С 5 алкилена, С 1-С 3 алкил-S-, С 1-С 5 алкил-О-, N(Rc)-С 1-С 5 алкила и -С 1-С 5 алкил-N(Rc)-, где Ra и Rb независимо выбраны из группы, состоящей из атома водорода и C1-С 3 алкила, иRc независимо выбран из группы, состоящей из Н, С 1-С 5 алкила, С 1-С 3 алкилфенила и С 4-С 8 алкилциклоалкила;Ar1 выбран из группы, состоящей из индолила, тиенила, бензотиенила, нафтила, фенила, пиридинила, пиразолила, оксазолила, бензоизоксазолила, бензофуранила, пирролила, тиазолила, бензоизотиазолила, индазолила и фуранила, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, С 1-С 6 алкила, С 1-С 6 галогеналкила, атома галогена, С 2-С 6 алкенила, С 2-С 6 алкинила, С 1-С 6 алкокси, -ОС 1-С 2 алкилфенила, N(Rc)SO2C1-С 6 алкила, -C(O)R10 иR7 выбран из группы, состоящей из СООН, С 1-С 5 алкилСООН, -О-С 1-С 5 алкилСООН, С 2-С 4 алкенил СООН, С 3-С 8 циклоалкилСООН и CONR11R11; каждый R10 независимо выбран из группы, состоящей из атома водорода, С 1-С 6 алкила и фенила; каждый R11 независимо представляет собой атом водорода или С 1-С 6 алкил;-1 015632 или его фармацевтически приемлемая соль. Соединения настоящего изобретения являются агонистами рецепторов FXR. Соединения настоящего изобретения полезны для изменения липидного профиля в требуемом направлении, включая снижение уровня общего холестерина, снижение уровня ЛПНП-холестерина, снижение уровня ЛПОНПхолестерина (холестерина липопротеинов очень низкой плотности), повышение уровня ЛПВП (липопротеинов высокой плотности) и снижение уровня триглицеридов. Соответственно, в настоящем изобретении предложен способ лечения состояний, опосредуемых FXR, таких как дислипидемия и заболевания,связанные с дислипидемией, включающий введение терапевтически эффективного количества соединения настоящего изобретения пациенту, нуждающемуся в таком лечении. В настоящем изобретении предложена фармацевтическая композиция, содержащая соединение настоящего изобретения и фармацевтически приемлемый носитель. Настоящее изобретение относится к применению соединения настоящего изобретения для снижения уровня общего холестерина, снижения уровня ЛПНП-холестерина, снижения уровня ЛПОНПхолестерина, повышения уровня ЛПВП и/или снижения уровня триглицеридов, включающему введение терапевтически эффективного количества соединения настоящего изобретения пациенту, нуждающемуся в таком лечении. Настоящее изобретение относится к применению соединения настоящего изобретения в лечении состояний, опосредуемых FRX, таких как дислипидемия и заболевания, связанные с дислипидемией,включающему введение терапевтически эффективного количества соединения настоящего изобретения пациенту, нуждающемуся в таком лечении. Настоящее изобретение также относится к применению соединения настоящего изобретения для изготовления фармацевтического препарата. Термин "дислипидемия", как использовано в данном описании, относится к нарушению уровня или аномальному количеству липидов и липопротеинов в крови и к болезненным состояниям, приводящим к такому нарушению, вызванным или усугубленным таким нарушением или сопутствующим такому нарушению (см. Dorland's Illustrated Medical Dictionary, 29th edition, W.B Saunders publishing Company, NewYork, NY). Болезненные состояния, входящие в определение дислипидемии, как использовано в данном описании, включают гиперлипидемию, гипертриглицеридемию (hypertriglyceremia), низкий уровень ЛПВП в плазме, высокий уровень ЛПНП (липопротеинов низкой плотности) в плазме, высокий уровень ЛПОНП (липопротеинов очень низкой плотности) в плазме, холестаз печени и гиперхолестеринемию. Фраза "заболевания, связанные с дислипидемией", как использовано в данном описании, относится к заболеваниям, включающим, но, не ограничиваясь ими, атеросклероз, тромбоз, ишемическую болезнь сердца, инсульт и гипертензию. Заболевания, связанные с дислипидемией, также включают нарушения обмена веществ, такие как ожирение, диабет, устойчивость к инсулину, и их осложнения. Осложнения диабета включают, например, диабетическую ретинопатию. Как использовано в данном описании, термин "пациент" относится к человеку, домашним животным (например, собакам и кошкам и т.п.) и домашнему скоту. Термины "лечение" и "лечить" включают снижение выраженности (ослабление), остановку, сдерживание, замедление и обращение прогрессирования или уменьшение тяжести патологических симптомов дислипидемии и заболеваний, связанных с дислипидемией. Как использовано в данном описании, термин "терапевтически эффективное количество" означает количество соединения настоящего изобретения, которое является частью одобренной схемы лечения или определено квалифицированным медицинским работником как достаточное при приеме в соответствии с предписанием, для лечения состояния или его неблагоприятных действий, описанных в данном описании. Термин "фармацевтически приемлемый" используется в данном описании в качестве характеристики и означает, по существу, безвредный для принимающего пациента. Термин "С 1-С 6 алкил" означает линейный или разветвленный углеводородный фрагмент, содержащий от 1 до 6 атомов углерода, и включает, но, не ограничиваясь ими, метил, этил, н-пропил, изопропил,н-бутил, изобутил, втор-бутил, трет-бутил и т.п. Аналогично, термин "С 1-С 5 алкил" означает линейный или разветвленный углеводородный фрагмент, содержащий от 1 до 5 атомов углерода, и включает, но не ограничиваясь ими, метил, этил, н-пропил и изопропил. Специалисту в данной области техники будет понятно, что термин "С 1-С 6 алкил" или близкие термины являются синонимами термина "С 1-С 6 алкилен" или подобных терминов, означающих бирадикал в случае, когда группа "С 1-С 6 алкил" расположена между двумя группами таким образом, что она становится бирадикалом. Термин "C1-С 3 алкил" означает линейный или разветвленный углеводородный фрагмент, содержащий от 1 до 3 атомов углерода, и включает метил, этил, н-пропил и изопропил. Специалисту в данной области техники будет понятно, что термин "C1-С 3 алкил" является синонимом термина "C1-С 3 алкилен",означающего бирадикал в случае, когда группа "C1-С 3 алкил" расположена между двумя группами таким образом, что она становится бирадикалом. Термин "С 2-С 6 алкенил" означает линейный или разветвленный углеводородный фрагмент, включающий по меньшей мере одну двойную связь и содержащий от 2 до 6 атомов углерода. Примеры вклю-2 015632 чают, но не ограничиваясь ими, винил, пропенил и 2-бутенил. Аналогично, термин "С 2-С 4 алкенил" означает линейный или разветвленный углеводородный фрагмент, включающий по меньшей мере одну двойную связь и содержащий от 2 до 4 атомов углерода. Термин "С 2-С 6 алкинил" или близкие термины означают линейный или разветвленный углеводородный фрагмент, включающий по меньшей мере одну тройную связь и содержащий от 2 до 6 атомов углерода. Примеры включают, но, не ограничиваясь ими, этинил, пропинил, 2-бутинил и т.п. Аналогично,термин "С 2-С 4 алкинил" или близкие термины означают линейный или разветвленный углеводородный фрагмент, имеющий по меньшей мере одну тройную связь и содержащий от 2 до 4 атомов углерода. Термин "С 3-С 8 циклоалкил" относится к насыщенному карбоциклическому кольцу, содержащему от 3 до 8 атомов углерода, и включает, но не ограничиваясь ими, циклопропил, циклопентил и циклогексил. Аналогично, термин "С 3-С 6 циклоалкил" относится к насыщенному карбоциклическому кольцу, содержащему от 3 до 6 атомов углерода, включая циклопропил, циклобутил, циклопентил и циклогексил. Термин "С 4-С 8 алкилциклоалкил", как использовано в данном описании, относится к такой комбинации алкильной и циклоалкильной групп (алкила и циклоалкила), в которой общее количество атомов углерода составляет 4-8 атомов углерода или указанное количество атомов углерода. Например, С 4-С 8 алкилциклоалкил включает циклоалкильные кольца, соединенные по меньшей мере с одним атомом углерода, так что общее количество атомов углерода составляет от 4 до 8 атомом углерода. Аналогично,термин "С 4-С 5 алкилциклоалкил", как использовано в данном описании, относится к такой комбинации алкильной и циклоалкильной групп, что общее количество атомов углерода составляет 4-5 атомов углерода. Например, С 4-С 5 алкилциклоалкил включает -СН 2-циклопропил, то есть метиленциклопропил, который представляет собой С 4 алкилциклоалкил. Термин "C1-С 6 галогеналкил" относится к С 1-С 6 алкильной группе, замещенной одним, двумя, тремя или несколькими атомами галогена, как указано или химически возможно. Примеры C1-С 6 галогеналкила включают, но не ограничиваясь ими, трифторметил, хлорэтил и 2-хлорпентил. Аналогично, термин "C1 С 3 галогеналкил" относится к C1-С 3 алкильной группе, замещенной одним, двумя, тремя или несколькими атомами галогена, как указано или химически возможно. Примеры C1-С 3 галогеналкила включают, но не ограничиваясь ими, трифторметил, хлорэтил и 2-хлорпропил. Группа "C1-С 6 алкокси" представляет собой C1-С 6 алкильный фрагмент, присоединенный через атом кислорода. Примеры алкоксигрупп включают, но не ограничиваясь ими, метокси (-ОМе), этокси (-OEt), пропокси (-OPr), изопропокси (-OiPr) и бутокси (-OBu). Аналогично, термин "C1-С 3 алкокси" означает C1-С 3 алкильный фрагмент, присоединенный через атом кислорода. C1-С 3 алкоксигруппы включают метокси (-ОМе(С 1-С 5 алкил или указанную алкильную группу), заканчивающуюся атомом кислорода, в отличие от алкокси (-O-С 1-С 5 алкил), при чтении слева направо. Например, такие радикалы или группы, как -CH2O-,-CH2CH2O- и -СН(СН 3)O-, в данном описании классифицированы как алкилоксигруппы. Аналогично, термин "-C1-С 3 алкил-О-" относится к C1-С 3 алкилоксигруппе, означающей C1-С 3 алкильную группу, заканчивающуюся атомом кислорода, в отличие от алкокси (-O-C1-С 3 алкил), при чтении слева направо. Например, такие радикалы или группы, как -CH2O-, -CH2CH2O- и -СН(СН 3)O-, в данном описании классифицированы как алкилоксигруппы. Термин "С 1-С 6 галогеналкокси" или близкие термины включают алкоксигруппы, в которых один или более чем один атом водорода в алкильной части группы заменены атомами галогена. Примеры галогеналкоксигрупп включают дифторметокси, трифторметокси, 2-галогеноэтокси, 2,2,2-трифторэтокси,4,4,4-трифторбутокси и так далее, включая все подобные группы, имеющие указанное количество атомов углерода. Аналогично, термин "C1-С 3 галогеналкокси" относится к C1-С 3 алкоксигруппе, в которой один или более чем один атом водорода в алкильной части группы заменен атомами галогена. Примеры C1-С 3 галогеналкоксигрупп включают дифторметокси, трифторметокси, 2-галогеноэтокси, 2,2,2-трифторэтокси и так далее, включая все подобные группы, имеющие указанное количество атомов углерода. Термин "S-C1-С 6 алкил" означает линейный или разветвленный тиоалкильный или S-алкильный фрагмент, содержащий от 1 до 6 атомов углерода, и включает, но не ограничиваясь ими, S-метил, S-этил,S-н-пропил, S-изопропил, S-н-бутил, S-изобутил и т.п. Аналогично, термин "S-C1-С 3 алкил" означает линейный или разветвленный тиоалкильный или S-алкильный фрагмент, содержащий от 1 до 3 атомов углерода, включая S-метил, S-этил, S-н-пропил и S-изопропил. Термин "C1-С 6 тиогалогеналкил" или "-S-C1-С 6 галогеналкил" включает -S-алкильные группы, где один или несколько атомов водорода в алкильной части группы заменены атомами галогена. ПримерыC1-С 6 тиогалогеналкильных групп включают тиодифторметил, тиотрифторметил, 2-галогенотиоэтил,2,2,2-трифтортиоэтил, 4,4,4-трифторбутил и так далее, включая все подобные группы, имеющие указанное количество атомов углерода. Аналогично, термин "C1-С 3 тиогалогеналкил" относится к S-C1 С 3 алкилу, где один или несколько атомов водорода в алкильной части группы заменены атомами галогена. Примеры C1-С 3 тиогалогеналкильных групп включают дифтортиометил, трифтортиометил, 2-галогенотиоэтил, 2,2,2-трифтортиоэтил и так далее, включая все подобные группы, имеющие указанное количество атомов углерода.-3 015632 Термин "-O-С 1-С 2 алкилфенил" относится к С 1-С 2 алкоксигруппе, присоединенной к фенильной группе или являющейся заместителем у фенильной группы. Подразумевается, что R6 может быть заместителем у X1 или X2, если X1 или X2 представляет собой углерод, но не в случае, когда X1 или X2 представляет собой азот. Термин "галоген" (гало) означает атомы галогена, включая йод, хлор, бром и фтор. Если не указано иное, то подразумевается, что в случае, когда Ar1 представляет собой бициклическое кольцо, присоединение Ar1 к кольцу, содержащему R6, может происходить по любому (химически) подходящему атому углерода или азота бициклического кольца. Соединение настоящего изобретения может быть представлено любым его изомером, каждый из которых является объектом настоящего изобретения. Некоторые соединения настоящего изобретения могут содержать один или более хиральных центров и, соответственно, могут существовать в виде оптически активных форм. Подобным образом, соединения настоящего изобретения могут содержать алкенильные группы и, соответственно, могут существовать в виде геометрических изомеров. Все такие изомеры, а также их смеси входят в объем настоящего изобретения. Если необходим конкретный стереоизомер, его можно получить с помощью методик, хорошо известных в данной области. Специалисту в данной области известно, что в случаях, когда в соединениях настоящего изобретения присутствуют аминогруппы (например, когда L1 представляет собой N(RC)CH2CH2-), получение кислотно-аддитивной соли данного соединения может привести к образованию соли четырехвалентного аммония данного соединения. Например, взаимодействие кислоты, такой как трифторуксусная кислота, с соединением настоящего изобретения, где L1 представляет собой аминогруппу, может приводить к образованию соли четырехвалентного аммония данного соединения. Предполагается, что все такие соли входят в объем настоящего изобретения. Предпочтительно каждый из X1 и Х 2 представляет собой С. Также предпочтительным является соединение настоящего изобретения, где X1 представляет собой N. Предпочтительно р равен 0 или 1. Более предпочтительно р равен 0. Предпочтительно группы R1 и R2, каждая независимо, выбрана из группы, состоящей из атома водорода, C1-С 3 алкила, C1-С 3 галогеналкила, C1-С 3 алкокси, C1-С 3 галогеналкокси, -SC1-С 3 алкила, -SC1-С 3 галогеналкила и атома галогена. Более предпочтительно группы R1 и R2 независимо выбраны из группы,состоящей из атома водород, атома хлора, атома фтора, CF3, OCF3 и SCF3. Предпочтительно группа R3 отсутствует или выбрана из группы, состоящей из атома водорода, C1 С 3 алкила, C1-С 3 галогеналкила, C1-С 3 алкокси, C1-С 3 галогеналкокси и атома галогена. Более предпочтительно группа R3 выбрана из группы, состоящей из атома водорода, атома хлора, атома фтора, CF3, OCF3 и SCF3. Наиболее предпочтительно R3 отсутствует. Предпочтительно R4a независимо выбран из Н, метила, этила, пропила, изопропила, циклопропила,CF3 и метилциклопропила. Более предпочтительно R4a представляет собой атом водорода. Предпочтительно R4b независимо выбран из Н, метила, этила, пропила, изопропила, циклопропила,CF3 и метилциклопропила. Более предпочтительно R4b представляет собой CF3, изопропил или циклопропил. Предпочтительно R5 и R5a, каждый независимо, выбран из группы, состоящей из атома водорода,метила и этила. Более предпочтительно R5 и R5a, каждый, представляет собой водород. Предпочтительно группа R6 выбрана из группы, состоящей из атома водорода, атома галогена, C1 С 3 алкила, С 2-С 3 алкенила, гидрокси, -NO2 и -ОС 1-С 2 алкила. Более предпочтительно группа R6 выбрана из группы, состоящей из атома водорода, атома галогена, метила и метокси. Наиболее предпочтительно R6 представляет собой атом водорода, атом хлора или метил. Предпочтительные L1. Предпочтительно L1 выбран из группы, состоящей из связи, СН=СН, этинила, -CH2S-, -C(CH3)2-S-,-CH(CH2CH3)S-, -CH(CH3)S-, -CH(CH3)CH2-S-, -CH(CH3)CH2O-, -С(СН 3)2O-, -СН(СН 3)O-, СН (СН 2 СН 3)O, -N (Rc) (CH2)m- и -(СН 2) m-N (Rc)-, где Rc представляет собой атом водорода или C1-С 3 алкил, m равен 1, 2 или 3. Более предпочтительно L1 представляет собой связь, СН=СН, -N(CH3)CH2, -N (СН 3) СН 2 СН 2 или N(СН 3)СН 2 СН 2 СН 2-. Особенно предпочтительно L1 представляет собой связь, -N(CH3)CH2 или N(CH3)CH2. Наиболее предпочтительно L1 представляет собой -N(CH3)CH2 или N(CH3)CH2CH2. Предпочтительно группа Ar1 выбрана из группы, состоящей из необязательно замещенных индолила, индазолила, тиенила, бензотиенила, бензоизотиазолила, фенила, пиридинила, пирролила, тиазолила и фуранила. Более предпочтительно группа Ar1 выбрана из группы, состоящей из необязательно замещенных бензотиенила, индолила, индазолила, бензоизотиазолила и фенила. Особенно предпочтительно Ar1 представляет собой фенил, индолил, бензотиенил или бензоизотиазолил. Предпочтительно Ar1 необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из атома галогена, C1-С 3 алкила, С 1-С 3 алкокси, С 1-С 3 галогеналкокси и C1-С 3 галогеналкила. Предпочтительно заместитель R7 представляет собой СООН или CONHR11. Более предпочтительно 7R представляет собой СООН или CONH2, -CONHCH3 или CONHC2H5. Наиболее предпочтительно R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где р равен 0, или 1, или 2;X1 представляет собой С или N, и Х 2 представляет собой С или N, при условии, что X1 и Х 2 одновременно не являются N;R1 и R2 независимо выбраны из группы, состоящей из атома водорода, С 1-С 3 алкила, С 1-С 3 галогеналкила, С 1-С 3 тиогалогеналкила, C1-С 3 алкокси, С 1-С 3 галогеналкокси и атома галогена;R3 отсутствует или независимо выбран из группы, состоящей из C1-С 3 алкила, С 1-С 3 галогеналкила,С 1-С 3 алкокси, C1-С 3 галогеналкокси и атома галогена;R4a выбран из группы, состоящей из атома водорода, C1-С 3 алкила, C1-С 3 галогеналкила, С 3-С 6 циклоалкила и С 4-С 5 алкилциклоалкила;R4b выбран из группы, состоящей из атома водорода, C1-С 3 алкила, С 1-С 3 галогеналкила, С 3-С 6 циклоалкила и С 4-С 5 алкилциклоалкила;R5 и R5a независимо выбраны из группы, состоящей из атома водорода и C1-С 3 алкила;R6 выбран из группы, состоящей из атома водорода, С 1-С 3 алкила, C1-С 3 галогеналкила, атома галогена и -NO2;L1 выбран из группы, состоящей из связи, CRa=CRb, этинила, С 1-С 3 алкил-S-, C1-С 3 алкил-О-, N (Rc)C1-С 3 алкила и -C1-С 3 алкил-N(Rc)-, где Ra и Rb независимо выбраны из группы, состоящей из атома водорода и С 1-С 3 алкила, и Rc независимо выбран из группы, состоящей из Н, С 1-С 5 алкила, С 1-С 3 алкилфенила и С 4-С 8 алкилциклоалкила;Ar1 выбран из группы, состоящей из индолила, бензотиенила, бензоизотиазолила, индазолила, нафтила, фенила, пиридинила, пиразолила, пирролила, тиенила, тиазолила и фуранила, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из гидрокси, С 1-С 3 алкила, C1-С 3 галогеналкила, атома галогена, С 2-С 4 алкенила, С 2-С 4 алкинила, С 1-С 4 алкокси, -ОС 1-С 2 алкилфенила, -NHC(О)R10;R7 выбран из группы, состоящей из -СООН, -C1-С 3 алкилСООН, -O-C1-С 3 алкилСООН и -CONR11R11; каждый R10 независимо выбран из группы, состоящей из атома водорода, C1-С 3 алкила и фенила; каждый R11 независимо представляет собой атом водорода или С 1-С 5 алкил; или его фармацевтически приемлемая соль. Предпочтительным является также соединение настоящего изобретения, где р равен 0 или 1;R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома фтора, атома хлора,CF3, SCF3, OCF3;R3 представляет собой атом фтора, атом хлора, C1-С 3 алкил, CF3, SCF3 или OCF3;R4a представляет собой атом водорода, метил, этил или изопропил;R5 и R5a, каждый независимо, выбран из Н или С 1-С 3 алкила; группа Ar1 представляет собой фенил, индолил, пиридинил, пирролил, тиенил, нафтил, тиазолил,фуранил, пиразолил, индазолил, бензоизотиазолил и бензотиенил, каждый из которых необязательно замещен одной или двумя группами, независимо выбранными из С 1-С 5 алкила, С 1-С 4 алкокси, С 1-С 2 галогеналкокси и C1-С 3 галогеналкила;R6 представляет собой атом водорода, метил, этил или атом хлора;CH2CH2CH2-, где RC представляет собой атом водорода, С 1-С 2 алкил или бензил;R10 представляет собой атом водорода или С 1-С 2 алкил; иR11 представляет собой атом водорода или С 1-С 2 алкил,или его фармацевтически приемлемая соль. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; R5 и R5a оба являются атомом водорода;L1 представляет собой этенил, этинил, -N(CH3)CH2- или -N (CH3) CH2CH2-; R6 представляет собой атом водорода, метил, атом хлора или атом брома; Ar1 представляет собой фенил, индолил, индазолил, бензотиенил или бензоизотиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и X2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой -N(CH3)CH2- или-N(CH3)CH2CH2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил,-5 015632 этил или атом хлора; Ar1 представляет собой фенил, бензоизотиазолил, индазолил, индолил или бензотиенил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 1; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R3 представляет собой атом водорода; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил;L1 представляет собой -N(CH3)CH2- или -N(СН 3)СН 2 СН 2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, необязательно замещенный группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси,этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой связь,-N(CH3)CH2- или -N(CH3)CH2CH2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атома хлора; Ar представляет собой фенил, бензоизотиазолил, индазолил, индолил или бензотиенил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой этенил,-N(CH3)CH2- или -N(CH3)CH2CH2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, тиенил, пирролил, фуранил или тиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила,изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой связь,-СН(СН 3)O, -СН(CH3)CH2O, -CH(CH3)S, -C(CH3)2S, -CH2-NH- и -CH2N(CH3)-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, бензоизотиазолил, индазолил, индолил или бензотиенил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой этенил, -СН(СН 3)O, -СН(CH3)CH2O, -CH(CH3)S, -C(CH3)2S, -CH2-NH- и -CH2N(CH3)-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, тиенил, пирролил, фуранил или тиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой -N(CH3)CH2- или-N(CH3)CH2CH2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил,этил или атом хлора; Ar1 представляет собой бензоизотиазолил, индазолил, индолил или бензотиенил,каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила,циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой связь; R5 и R5a,оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, бензоизотиазолил, индазолил, индолил или бензотиенил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, ме-6 015632 токси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома водорода, атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой связь; R5 и R5a,оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, необязательно замещенный группой, выбранной из метила, этила, пропила,изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой этенил; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, тиенил, пирролил, фуранил, или тиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пролила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома хлора, атома фтора, трифторметила, тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой этенил, -N(CH3)CH2- или-N(СН 3)СН 2 СН 2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил,этил или атом хлора; Ar1 представляет собой фенил, тиенил, пирролил, фуранил или тиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси, изопропокси и циклопропокси; и R7 представляет собой СООН. Предпочтительным является также соединение настоящего изобретения, где X1 и Х 2 оба являются С; р равен 0; R1 и R2 независимо выбраны из группы, состоящей из атома хлора, атома фтора, трифторметила,тиотрифторметила и трифторметокси; R4a представляет собой атом водорода; R4b представляет собой трифторметил, изопропил или циклопропил; L1 представляет собой -N(CH3)CH2- или -N(CH3)CH2CH2-; R5 и R5a оба являются атомом водорода; R6 представляет собой атом водорода, метил, этил или атом хлора; Ar1 представляет собой фенил, тиенил, пирролил, фуранил или тиазолил, каждый из которых необязательно замещен группой, выбранной из метила, этила, пропила, изопропила, циклопропила, метокси, этокси,изопропокси и циклопропокси; и R7 представляет собой СООН. Соединения настоящего изобретения (то есть соединения формулы I) могут быть получены с помощью ряда методик, известных в данной области, а также способов, описанных ниже. Продукты, получаемые на каждой стадии приведенной ниже схемы, можно выделить стандартными методами, включающими экстракцию, упаривание, осаждение, хроматографию, фильтрование, растирание, кристаллизацию и т.п. На приведенной ниже схеме все заместители, если не указано иное, имеют определенные выше значения, и подходящие реагенты представляют собой реагенты, хорошо известные и используемые в данной области. На схеме 1 показано взаимодействие подходящего соединения формулы (1) с подходящим соединением формулы (2) с образованием соединения формулы (I). Реакцию, приведенную на схеме 1, можно осуществить с использованием по меньшей мере двух вариантов синтеза, описанных ниже. Согласно первому варианту синтеза подходящее соединение формулы (1) представляет собой соединение, где R1, R2, R3, р, R4a, R4b, R5 и R5a являются такими, как определено для формулы I, и Y представляет собой -ОН, и подходящее соединение формулы (2) представляет собой соединение, где R6, R7,Х 1, Х 2, L1 и Ar1 являются такими, как определено формулы (I), или содержит, группу, которую можно преобразовать в R7, как определено для формулы (I), например, путем образования сложного эфира, амида, сульфонамида или кислоты. Например, соединение формулы (1) подвергают взаимодействию с соединением формулы (2) в условиях реакции Мицунобу с использованием подходящего диазореагента, такого как DEAD (диэтилазодикарбоксилат) или ADDP и т.п., и подходящего фосфинового реагента, такого как трифенилфосфин или трибутилфосфин, и т.п. Такие реакции проводят в подходящем растворителе, таком как толуол, тетрагидрофуран и т.п. Как правило, данные реакции проводят при температуре приблизительно от 0 С до-7 015632 50 С. Типичное стехиометрическое соотношение компонентов в данной реакции составляет, в расчете на один эквивалент соединения формулы (1), приблизительно 1-2 эквивалента соединения формулы (2) и приблизительно 1-2 эквивалента диазореагента, и 1-2 эквивалента фосфинового реагента. Согласно второму варианту синтеза подходящее соединение формулы (1) представляет собой соединение, где R1, R2, R3, р, R4a, R4b, R5 и R5a являются такими, как определено для формулы I, и Y представляет собой уходящую группу, и подходящее соединение формулы (2) определено выше. Подходящие уходящие группы хорошо известны в данной области и включают галогениды, конкретно галогениды,содержащие хлор, бром и йод; и сложные сульфонатные эфиры, такие как брозил, тозил, метансульфонил и трифторметансульфонил. Например, соединение формулы (1) подвергают взаимодействию с соединением формулы (2) в подходящем растворителе, таком как ацетонитрил, диметилформамид, тетрагидрофуран, пиридин, метилэтилкетон и т.п. Легко понять, что в данной реакции обычно используется избыток подходящего основания, включающего гидрид натрия, карбонат калия, карбонат натрия, карбонат цезия, бикарбонат натрия, триэтиламин, диизопропилэтиламин. Такие реакции обычно проводят в температурном интервале приблизительно от комнатной температуры до приблизительно температуры кипения с обратным холодильником выбранного растворителя и обычно используют приблизительно 1-2 эквивалента соединения формулы (2). На необязательной стадии может быть образована фармацевтически приемлемая соль соединения формулы (I). Получение таких солей хорошо известно и апробировано в данной области. Будет очевидно, что соединения формулы (1) и (2) можно легко получить с помощью способов,аналогично приведенным в данном описании методик, которые хорошо известны и апробированы в данной области. Например, соединения формулы (1) получают путем взаимодействия необязательно замещенного фенилгидразина с 1,3-дикетоэфиром (или с эквивалентным соединением), с последующим восстановлением и необязательным преобразованием в соединение, содержащее уходящую группу, и соединения формулы (2) получают путем образования углерод-углеродной связи, восстановительного аминирования, реакции сочетания и т.д. Также показано, что стадии, необходимые для получения соединения формулы (2), могут быть выполнены в любом порядке, например, после взаимодействия частично полученного соединения формулы (2) с соединением формулы (1), таким образом, что последние стадии образования углерод-углеродной связи, восстановительного аминирования, реакции сочетания и т.д. приводят к получению соединения формулы I. Следует понимать, что стадии получения соединения формулы I зависят от конкретного синтезируемого соединения, исходного соединения и относительной лабильности замещенных групп. Также предполагается, что при выполнении описанных выше реакций могут оказаться необходимыми или полезными стадии введения и снятия различных защитных групп. Методы выбора и использования подходящих защитных групп хорошо известны и широко используются в данной области (см., например, Protecting Groups in Organic Synthesis, Theodora Greene (WileyInterscience. Настоящее изобретение дополнительно проиллюстрировано приведенными ниже примерами и примерами получения. Данные примеры и примеры получения являются только иллюстрацией изобретения,и предполагается, что они никоим образом не ограничивают объем изобретения. Если не указано иное,термины, используемые в разделах Примеры и Примеры получения, имеют их обычно употребляемые значения. Анализ Протоколы и результаты описанных ниже исследований, демонстрирующие полезность, in vitro и invivo эффективность соединений и/или способов настоящего изобретения, приведены с целью иллюстрации, и предполагается, что они никоим образом не ограничивают объем изобретения. Аббревиатуры, используемые в данном описании, определены следующим образом. "ЛПНП" обозначает липопротеин низкой плотности; "ЛПВП" обозначает липопротеин высокой плотности; "ЛПОНП" обозначает липопротеин очень низкой плотности; "LDLR-/-" обозначает дефицит рецепторов липопротеинов низкой плотности; "DMEM" обозначает модифицированную по способу Дульбекко среду Игла;"GAPDH" обозначает глицеральдегид-3-фосфатдегидрогеназу; "Na-CMC" обозначает натрийкарбоксиметилцеллюлозу; "SLS" обозначает лаурилсульфат натрия; "FPLC" обозначает жидкостную экспрессхроматографию белков; "PBS" обозначает забуференный фосфатом солевой раствор; "ЛПОНП-Х" обозначает холестерин липопротеинов очень низкой плотности; "ЛПВП-Х" обозначает холестерин липопротеинов высокой плотности. Анализ с рДНК для мРНК SHPFXR является ключевым прямым регулятором транскрипции гена SHP (Small Heterodimer Partner),регистрационный номер NM021969, атипичного представителя семейства ядерных рецепторов, у которого отсутствует ДНК-связывающий домен. SHP взаимодействует с несколькими обычными и сиротскими рецепторами суперсемейства ядерных рецепторов, включающего ретиноидные рецепторы и рецептор тиреоидного гормона. SHP ингибирует потенциал трансактивации членов суперсемейства, с которыми он взаимодействует. Было обнаружено, что FXR и SHP оба контролируют гены, ответственные за катаболизм холестерина в печени, синтез триглицеридов и транспорт желчных кислот. Поскольку FXR-8 015632 напрямую трансактивирует транскрипцию гена SHP, активацию самого FXR лигандами можно оценить количественно с помощью метода, основанного на использовании разветвленной рДНК SHP. Таким образом, увеличение экспрессии мРНК SHP, определяемое по увеличению сигнала рДНК, указывает на связывание FXR с агонистом. Клетки гепатокарциномы человека Huh7, выращенные в DMEM:F12 с 10% эмбриональной телячьей сывороткой высевали в 96-луночные планшеты с плотностью 1105/лунка. После инкубации в течение ночи клетки обрабатывали в течение 24 ч исследуемыми соединениями в различных концентрациях. Выполняли анализ рДНК в соответствии с протоколом производителя (Panomics, Fremont, CA) для набора QuantiGene High Volume. После стимуляции клеток соединением настоящего изобретения клетки лизировали с использованием буфера для лизиса QuantiGene, содержащего олигонуклеотиды мРНКSHP, описанные ниже. Олигонуклеотидные реагенты (усилители захвата (capture extender (СЕ, усилители метки (label extender (LE и блокаторы (blocker (BL, подходящие для рДНК-анализа мРНК SHP человека, могут быть сконструированы и синтезированы в Panomics (Fremont, CA). После добавления буфера для лизиса пробы инкубировали в течение 15 мин при 37 С, затем из каждой лунки 100 мкл лизата переносили в соответствующие лунки планшета для захвата. Планшет для захвата инкубировали в течение ночи при 53 С. Планшет для захвата дважды промывали буфером для промывки QuantiGene, затем добавляли 100 мкл/лунка реагента-амплификатора QuantiGene. Планшет инкубировали в течение 60 мин при 46 С, затем дважды промывали. Анализируемую мРНК метили добавлением 100 мкл буфера QuantiGene, содержащего меченый зонд, затем инкубировали в течение 60 минут при 46 С. Планшет для захвата дважды промывали и добавляли 100 мкл/лунка субстрата QuantiGene и усиливающего реагента QuantiGene. Планшеты инкубировали при 37 С в течение 30 мин и затем считывали с помощью люменметра (Packard Fusion Alpha, время регистрации 1 с) для регистрации люминесцентного сигнала. Рассчитывали величину ЕС 50, то есть отношение фактического ответа к максимальному ответу. Согласно результатам описанного выше анализа примеры соединений являются эффективными модуляторами FXR и имеют ЕС 50 2 мкМ или менее. Например, соединение примера 90, вызывающее активацию гена SHP, имеет ЕС 50 приблизительно 53 нМ. Модуляции липидов LDLR-/-сыворотки За две недели до исследования начинали подготовку животных. Мышей помещали в поликарбонатные камеры с крышками-светофильтрами (по одному животному в каждую камеру) и выдерживали с циклом свет-темнота 12:12 ч (включение света в 6:00 утра) при температуре 21 С. Животные получали без ограничения деионизованную воду и в течение двух недель получали без ограничения корм в соответствии с диетой TD 88137 ("western diet") (42% жиров, 0,15% холестерина, Harlan Teklad). СамцовLDLR-/- мышей в возрасте десяти недель делили на группы (по пять животных в каждой группе) на основе уровней триглицеридов и холестерина в сыворотке. Животным в группах дозирования ежедневно путем перорального зондового кормления вводили различные дозы исследуемого соединения, растворенного в смеси 5% EtOH/5% раствор, содержащий Na-CMC (1%), SLS (0,5%), антивспенивающий агент(0,05%), повидон (0,085%). В конце семидневного периода животных после эвтаназии в камере с СО 2 собирали кровь путем пункции сердца. Измеряли уровень триглицеридов, глюкозы и общего холестерина в сыворотке с использованием стандартного биохимического оборудования и реагентов [Hitachi 912instrument с наборами реагентов (Roche, Indianapolis, IN)]. Пробы пула сыворотки анализировали методом FPLC, определяя содержание холестерина липопротеинов (ЛПОНП, ЛПНП, ЛПВП) во фракциях путем разделения на колонке для эксклюзионной хроматографии со встроенной системой для определения холестерина. Липопротеины разделяли методом жидкостной экспресс-хроматографии белков, и содержание холестерина определяли с помощью встроенной системы регистрации. Кратко, пробы плазмы объемом 35 мкл/50 мкл пробы пула наносили на колонку для эксклюзионной хроматографии Superose 6HR 10/30 (Amersham Pharmacia Biotech, Piscataway, NJ) и элюировали PBS, pH 7,4 (разбавленный 1:10),содержащим 5 мМ EDTA, при 0,5 мл/мин. Реагент для регистрации холестерина от Roche Diagnostics(Indianapolis, IN) при 0,16 мл/мин смешивали с элюатом через Т-образное соединение; затем данную смесь пропускали через сетчатый трубчатый химический реактор (knitted tubing reactor) 15 м 0,5 мм(Aura Industries, New York, NY), погруженный в водяную баню, имеющую температуру 37 С. Окрашенный продукт, получаемый в присутствии холестерина, регистрировали в потоке при 505 нм, и аналоговое напряжение регистрирующего устройства преобразовывали в цифровой сигнал, который регистрировали и анализировали. Строили график изменения напряжения, соответствующего изменению концентрации холестерина, со временем и вычисляли площадь под кривой, соответствующую элюированию ЛПОНП-Х и ЛПВП-Х, с использованием программы Turbochrome (версия 4,12F12) (PerkinElmer, Norwalk, СТ). Согласно результатам данного анализа при введении исследуемых соединений настоящего изобретения в дозе 10 мг/кг снижение уровня общего холестерина достигало 84% и триглицеридов - 86%. Более конкретно, при введении дозы 10 мг/кг соединение примера 199 снижает уровень общего холестерина на 60% и триглицеридов на 63%. Конечно, конкретная доза вводимого соединения настоящего изобретения зависит от конкретных-9 015632 обстоятельств, связанных с данным случаем, например от вводимого соединения, пути введения, состояния здоровья пациента и патологического состояния, которое подвергают лечению. Типичная суточная доза обычно содержит нетоксичное количество соединения настоящего изобретения и составляет приблизительно от 0,1 мг до приблизительно 1000 мг/сутки. Соединения данного изобретения можно вводить разными путями, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. Предпочтительно данные соединения готовят в виде препарата перед введением, выбор которого обычно находится в компетенции лечащего врача. Таким образом, другим аспектом настоящего изобретения является фармацевтическая композиция, содержащая эффективное количество соединения настоящего изобретения или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель, разбавитель или эксципиент. Специалист в данной области может легко выбрать нужную форму и путь введения в зависимости от конкретных характеристик выбранного соединения, расстройства или состояния, которые подвергают лечению, стадии расстройства или состояния и других имеющих отношение к лечению обстоятельств(Remington's Pharmaceutical Sciences, 18th Edition, Mack Publishing Co. (1990. Фармацевтические композиции настоящего изобретения можно адаптировать для разных путей введения и можно вводить пациенту, например, в форме таблеток, капсул, облаток, пастилок (paper), лепешек, вафель (wafer), эликсиров,мазей, чрескожных пластырей, аэрозолей, ингаляций, суппозиториев, растворов и суспензий. Соединения настоящего изобретения можно изготовить в виде препарата в виде эликсиров или растворов для традиционного перорального введения или в виде растворов, подходящих для парентерального введения, например, для внутримышечного, подкожного или внутривенного введения. Кроме того,данные соединения можно приготовить в виде дозированных форм с замедленным высвобождением и подобных. Данные препараты можно приготовить таким образом, что высвобождение активного ингредиента будет иметь исключительно или преимущественно конкретную физиологическую локализацию и,возможно, происходить в течение определенного периода времени. Покрытия, оболочки и защитные матриксы могут быть изготовлены, например, из полимерных веществ или восков. Примеры получения и примеры Изобретение дополнительно проиллюстрировано приведенными ниже примерами получения и примерами. Аббревиатуры, используемые в данном описании, определены согласно Aldrichimica Acta, Vol 17, 1, 1984. Другие аббревиатуры определены следующим образом. "ACN" обозначает ацетонитрил;"РСу 3" обозначает трициклогексилфосфин. Все соединения названы с использованием программы ChemDraw Ultra 7,0 от CambridgeSoft Corporation, Cambridge, MA, USA. Пример получения 1. Метиловый эфир 4-метил-2-оксопентановой кислоты. К раствору 4-метил-2-оксопентановой кислоты (3,4 г, 26 ммоль) в метаноле (12 мл) и 2,2 диметоксипропане (48 мл) добавляли хлортриметилсилан (0,38 мл). Данную реакционную смесь перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде масла (3,8 г, количественный выход). 1 Н ЯМР (400 МГц, CDCl3)3,86 (с, 3 Н), 2,72 (д, 2 Н), 2,16 (м, 1 Н), 0,96 (д, 6 Н). Пример получения 2. Этиловый эфир 2-метоксиимино-4-оксопентановой кислоты. Смесь этилового эфира 2,4-диоксопентановой кислоты (5,4 мл, 50 ммоль), гидрохлорида метоксиамина (4,4 г, 52,5 ммоль), молекулярных сит (3, 35 г) и сульфата натрия (15 г) в этаноле (50 мл) перемешивали при температуре окружающей среды в течение 5 ч. Реакционную смесь фильтровали и промывали этанолом. Объединенный фильтрат концентрировали при пониженном давлении и распределяли между этиловым эфиром и насыщенным раствором бикарбоната натрия. Органический слой сушили(MgSO4), фильтровали и концентрировали. Остаток очищали колоночной хроматографией (0-15% EtOAc в гексанах) с получением указанного в заголовке соединения в виде бесцветного масла (3 г, 32%). 1 Н ЯМР (400 МГц, CDCl3)4,33 (кв, 2 Н), 4,06 (с, 3 Н), 3,70 (с, 3 Н), 2,20 (с, 3 Н), 1,34 (т, 3 Н). Пример получения 3. Этиловый эфир 3-ацетил-2-метоксиимино-5-метилгексановой кислоты. К раствору этилового эфира 2-метоксиимино-4-оксопентановой кислоты (2,62 г, 14 ммоль) в ДМФА(N,N-диметилформамид) (30 мл) добавляли карбонат калия (2,5 г, 18,2 ммоль), затем 1-йод-2 метилпропан (1,62 мл, 14 ммоль) и перемешивали данную смесь при температуре окружающей среды в течение ночи. Добавляли дополнительную порцию 1-йод-2-метилпропана (0,8 мл, 7 ммоль) и перемеши- 10015632 вали смесь при 60 С в течение 1 ч. Реакционную смесь разбавляли EtOAc и доводили до рН 3 с помощью 1 н. HCl. Органический слой промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали при пониженном давлении. Остаток очищали колоночной хроматографией (0-20%EtOAc в гексанах) с получением указанного в заголовке соединения в виде бесцветного масла (1 г, 29%). 1H ЯМР (400 МГц, CDCl3)4,33 (кв, 2 Н), 4,06 (с, 3 Н), 4,03 (дд, 1 Н), 2,07 (с, 3 Н), 1,88 (м, 1 Н), 1,58 (м,1 Н), 1,43 (м, 1 Н), 1,37 (т, 3 Н), 0,89 (д, 6 Н). Пример получения 4. Этиловый эфир 2-циклопропилметил[1,3]дитиан-2-карбоновой кислоты. В высушенную пламенем колбу добавляли сухой толуол (80 мл) и гидрид натрия (60%, 33,5 ммоль,1,34 г). Реакционную смесь охлаждали на ледяной бане и добавляли по каплям в течение 10 мин раствор этилового эфира 1,3-дитианкарбоновой кислоты (52 ммоль, 10 г) и бромметилциклопропана (62,4 ммоль,8,42 г) в ДМФА (24 мл). Ледяную баню удаляли и перемешивали реакционную смесь в течение 18 ч. Добавляли воду (50 мл) и отделяли органический слой. Органический слой промывали насыщенным раствором соли, сушили (Na2SO4) и концентрировали с получением желтого масла (12 г, 92%). ЖХ/МС(жидкостная хроматография/масс-спектрометрия): 247,0 (М+1). Пример получения 5. Этиловый эфир 3-циклопропил-2-оксопропионовой кислоты. К суспензии NBS (N-бромсукцинимид) (439 ммоль, 79 г) в смеси ацетонитрила (400 мл) и воды (100 мл), имеющей температуру 0 С, добавляли в течение 15 мин раствор этилового эфира 2 циклопропилметил[1,3]дитиан-2-карбоновой кислоты (73,2 ммоль, 18,05 г) в ацетонитриле (50 мл). Реакционную смесь нагревали и перемешивали при комнатной температуре. Через 45 мин добавляли 500 мл смеси гексан/ДХМ (1:1). Слои разделяли. Органический слой промывали насыщенным раствором Na2SO3(2225 мл) и насыщенным раствором соли (2225 мл), сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении, с получением остатка. Остаток разбавляли CCl4 и фильтровали. Полученный фильтрат концентрировали с получением указанного в заголовке соединения (7 г, 61%). ЖХ/МС: 157,0 (М+1). Пример получения 6. Этиловый эфир 3-циклобутил-2-оксопропионовой кислоты. К суспензии 5 мг/мл Rieke магния в ТГФ (тетрагидрофуран) (37,9 ммоль, 40 мл) при комнатной температуре добавляли по каплям бромметилциклобутан (37,9 ммоль, 4,25 мл). После добавления реакционную смесь нагревали до 60 С в течение 1 ч. Смесь с помощью шприца переносили в раствор диэтилоксалата (37,9 ммоль, 5,14 г) в ТГФ (20 мл), имеющий температуру -78 С. Реакционную смесь оставляли нагреваться до 0 С и гасили добавлением 1 н. HCl. Добавляли диэтиловый эфир (20 мл) и полученные слои разделяли. Органический слой сушили (Na2SO4), адсорбировали на силикагеле и очищали, используя градиент 0-10% EtOAc/гексаны, с получением указанного в заголовке соединения (2,3 г, 37%). ЖХ/МС: 171,0 (М+1). Пример получения 7. Этиловый эфир 3-циклопропил-4-диметиламино-2-оксобут-3-еновой кислоты. Объединяли этиловый эфир 3-циклопропил-2-оксопропионовой кислоты (6,4 ммоль, 1,0 г) и диметилформамиддиметилацеталь (12,8 ммоль, 2,0 мл) и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,38 г, 100%). ЖХ/МС: 212,0 (М+1). Пример получения 8. Этиловый эфир 3-циклобутил-4-диметиламино-2-оксобут-3-еновой кислоты. Указанное в заголовке соединение (0,663, 51%) получали, по существу, как описано в примере получения этилового эфира 3-циклопропил-4-диметиламино-2-оксобут-3-еновой кислоты, с использованием этилового эфира 3-циклобутил-2-оксопропионовой кислоты. ЖХ-ES/MC (жидкостная хроматография/масс-спектрометрия с электрораспылением): m/е 226,0 (М+1). Пример получения 9. Гидрохлорид 2-хлор-6-трифторметилфенилгидразина. К раствору 2-хлор-6-трифторметилфениламина (35,7 ммоль, 7,0 г) в ТГФ (100 мл), имеющему температуру 0 С, добавляли 48% BF3OEt (143 ммоль, 36 мл), с последующим добавлением изоамилнитрита(143 ммоль, 19 мл). Данную реакционную смесь перемешивали в течение 1 ч и фильтровали, с получением тетрафторборатной диазониевой соли (48,7 ммоль, 9,0 г). Данную соль растворяли в смеси концентрированной HCl (30 мл) и воды (10 мл) при 0 С. К полученной смеси добавляли аскорбиновую кислоту(48,7 ммоль, 8,5 г). Реакционную смесь нагревали до 50 С в течение 3 ч и охлаждали до комнатной температуры. Полученное твердое вещество отфильтровывали и промывали ледяной водой. Влажное твердое вещество растворяли в смеси концентрированной HCl (30 мл) и воды (20 мл) и нагревали до 90 С в течение 2 ч. Реакционную смесь охлаждали до 0 С и фильтровали с получением указанного в заголовке соединения (5,0 г, 60%). ЖХ-ES/MC: m/е 157,0 (М+1). Следующий список соединений получали, по существу, как описано в примере получения гидрохлорида 2-хлор-6-трифторметилфенилгидразина с использованием подходящего исходного вещества. Пример получения 9 А. Гидрохлорид 3,5-дифтор-2-трифторметилфенилгидразина (3,0 г, 25%), ЖХES/MC: m/е 159,0 (М+1). Пример получения 9 В. Гидрохлорид 2-фтор-6-трифторметилфенилгидразина (5,2 г, 63%), ЖХES/MC: m/е 195,0 (М+1). Пример получения 9 С. Гидрохлорид 2-трифторметилсульфанилфенилгидразина (0,414 г, 68%),- 11015632ES/MC: m/е 209,0 (М+1). Пример получения 10. Гидрохлорид 2,6-дихлор-4-фторфенилгидразина. К раствору 2,6-дихлор-4-фторфениламина (3,0 г, 16,6 ммоль) в 12 М HCl (30 мл) и ТФУК (20 мл),имеющему температуру 0 С, медленно, по каплям, добавляли NaNO2 (20 ммоль, 1,37 мл) в воде (6 мл). Реакционную смесь перемешивали при 0 С в течение 1 ч. В течение 15 мин добавляли раствор SnCl2(5,74 г, 25,6 ммоль) в 12 М HCl (16 мл). Ледяную баню удаляли и перемешивали реакционную смесь в течение 18 ч. Реакционную смесь фильтровали и полученное твердое вещество промывали изопропиловым спиртом. Твердое вещество сушили с получением указанного в заголовке соединения (3,0 г, 96%). ЖХ-ES/MC: m/е 194,0 (М+1). Пример получения 11. Гидрохлорид (2-трифторметоксифенил)гидразина. К перемешиваемому раствору хлористо-водородной кислоты (37%, 1,6 л) при 0 С добавляли 2 трифторметоксифениламин (200 г, 113 ммоль), затем воду (160 мл) и еще одну порцию хлористоводородной кислоты (160 мл). Данную смесь нагревали до комнатной температуры, перемешивали в течение 20 мин и охлаждали до -5 С. Поддерживая внутреннюю температуру ниже 0 С, добавляли по каплям раствор нитрита натрия (82 г, 1,19 ммоль) в воде (400 мл). Смесь охлаждали до -5 С и, поддерживая внутреннюю температуру ниже 0 С, добавляли по каплям раствор дигидрата хлорида олова (II) (1020 г,4520 ммоль) в HCl (37%, 3,2 л). Смесь нагревали до комнатной температуры, перемешивали в течение 3 ч, фильтровали и промывали 6 н. HCl (3 л), с получением желтого твердого вещества, которое сушили в вакууме в течение ночи. Указанное в заголовке соединение (115,8 г, 54%) получали в виде розовокоричневого твердого вещества. Пример получения 12. Гидрохлорид трифторметоксифенилгидразина. Указанное в заголовке соединение (115,8 г, 54%) получали, по существу, согласно примеру получения гидрохлорида (2-трифторметоксифенил)гидразина с использованием 2-трифторметоксифениламина. Пример получения 13. Этиловый эфир 2-(2,6-дихлорфенил)-4-изобутил-5-метил-2 Н-пиразол-3 карбоновой кислоты. Смесь этилового эфира 3-ацетил-2-метоксиимино-5-метилгексановой кислоты (1 г, 4,1 ммоль), гидрохлорида 2,6-дихлорфенилгидразина (1,76 г, 8,2 ммоль) в ледяной уксусной кислоте (10 мл) и 2 метоксиэтанола (5 мл) перемешивали при 105 С в течение 3 ч. Реакционную смесь концентрировали и остаток распределяли между EtOAc и 1 н. HCl. Органический слой концентрировали и очищали колоночной хроматографией (0-20% EtOAc в гексанах) с получением указанного в заголовке продукта в виде масла, 1,1 г (76%). 1 Н ЯМР (400 МГц, CDCl3)7,41 (д, 2 Н), 7,31 (дд, 1 Н), 4,16 (кв, 2 Н), 2,65 (д, 2 Н), 2,33(с, 3 Н), 1,92 (м, 1 Н), 1,13 (т, 3 Н), 0,95 (д, 6 Н). Пример получения 14. Этиловый эфир 4-циклопропил-2-(2,6-дихлорфенил)-2 Н-пиразол-3 карбоновой кислоты. К раствору этилового эфира 3-циклопропил-4-диметиламино-2-оксобут-3-еновой кислоты (6,5 ммоль, 1,4 г) в этаноле (25 мл) добавляли гидрохлорид 2,6-дихлорфенилгидразина (7,2 ммоль, 1,5 г), затем концентрированную HCl (100 мкл). Перемешивали реакционную смесь в течение 4 ч при комнатной температуре, затем нагревали при 85 С в течение 18 ч. Реакционную смесь адсорбировали на силикагеле и очищали, используя градиент 0-20% EtOAc/гексаны, с получением указанного в заголовке соединения(0,8 г, 34%). ЖХ-ES/MC: m/е 325,0 (М+1). Следующий список соединений получали, по существу, согласно примеру получения этилового эфира 4-циклопропил-2-(2,6-дихлорфенил)-2 Н-пиразол-3-карбоновой кислоты с использованием подходящего исходного вещества. Пример получения 14 А. Этиловый эфир 4-циклобутил-2-(2,6-дихлорфенил)-2 Н-пиразол-3 карбоновой кислоты (0,56 г, 57%), ЖХ-ES/MC: m/e 339,0 (М+1). Пример получения 14 В. Метиловый эфир 2-(2-хлор-6-трифторметилфенил-4-изопропил-2 Нпиразол-3-карбоновой кислоты (0,56 г, 10%), ЖХ-ES/MC: m/e 293,0 (М+1). Пример получения 14 С. Метиловый эфир 2-(3,5-дифтор-2-трифторметилфенил)-4-изопропил-2 Нпиразол-3-карбоновой кислоты (1,6 г, 60%), ЖХ-ES/MC: m/e 295,0 (М+1). Пример получения 14D. Этиловый эфир 2-(2,6-дихлорфенил)-4-изобутил-5-метил-2 Н-пиразол-3 карбоновой кислоты (1,1 г, 16%), 1 Н ЯМР (CDCl3)7,41 (д, 2 Н), 7,31 (дд, 1 Н), 4,16 (кв, 2 Н), 2,65 (д, 2 Н),2,33 (с, 3 Н), 1,92 (м, 1 Н), 1,13 (т, 3 Н), 0,95 (д, 6 Н). Пример получения 14 Е. Метиловый эфир 2-(2-Фтор-6-трифторметилфенил)-4-изопропил-2 Нпиразол-3-карбоновой кислоты (0,063 г, 67%), ЖХ-ES/MC: m/e 277,0 (М+1). Пример получения 14F. Метиловый эфир 2-(2,6-дихлор-4-фторфенил)-4-изопропил-2 Н-пиразол-3 карбоновой кислоты (0,28 г, 39%), ЖХ-ES/MC: m/e 331,0 (М+1). Пример получения 14G. Метиловый эфир 4-изопропил-2-(2-трифторметилсульфанилфенил)-2 Нпиразол-3-карбоновой кислоты (0,25 г, 76%), ЖХ-ES/MC: m/e 345,0 (М+1). Пример получения 14 Н. Метиловый эфир 4-изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3 карбоновой кислоты (82 г, 22%). Пример получения 14I. Этиловый эфир 4-циклопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3- 12015632 карбоновой кислоты (0,65 г, 19%), ES/MC: m/e 341,0 (М+1). Пример получения 14J. Этиловый эфир 2-(2-хлор-6-трифторметилфенил)-4-циклопропил-2 Нпиразол-3-карбоновой кислоты (0,82 г, 38%), ES/MC: m/e 359,0 (М+1). Пример получения 15. Метиловый эфир 2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3 карбоновой кислоты. К раствору метилового эфира 4-метил-2-оксопентановой кислоты (3,8 г, 26 ммоль) в N,Nдиметилформамиддиметилацетале (7 мл, 52 ммоль) добавляли моногидрат паратолуолсульфоновой кислоты (30 мг) и перемешивали данную смесь при 80 С в течение ночи. Реакционную смесь концентрировали при пониженном давлении, с получением этилового эфира 3-изопропил-4-диметиламино-2-оксобут 3-еновой кислоты в виде оранжевого масла. К раствору этилового эфира 3-изопропил-4-диметиламино-2 оксобут-3-еновой кислоты и гидрохлорида 2,6-дихлорфенилгидразина (2,8 г, 13 ммоль) в EtOH (40 мл) добавляли концентрированную HCl (0,5 мл). Перемешивали данную смесь при температуре окружающей среды в течение 2 ч, затем кипятили в обратным холодильником в течение ночи. Реакционную смесь концентрировали и остаток распределяли между EtOAc и 1 н. HCl. Органическую фазу сушили (Na2SO4) и концентрировали, с получением остатка. Остаток очищали колоночной хроматографией (0-15% EtOAc в гексанах), с получением указанного в заголовке соединения в виде масла (2,2 г, 52%). 1 Н ЯМР (CDCl3):7,76 (с, 1 Н), 7,43 (д, 2 Н), 7,3,4 (дд, 1 Н), 3,75 (с, 3 Н), 3,48 (м, 1 Н), 1,32 (д, 6 Н). Пример получения 16. Метиловый эфир 4-изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3 карбоновой кислоты. Указанное в заголовке соединение (82 г, 22%) получали, по существу, согласно примеру получения метилового эфира 2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-карбоновой кислоты с использованием подходящего исходного вещества. Пример получения 17. [2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-ил]метанол. К раствору метилового эфира 2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-карбоновой кислоты(2,2 г, 6,8 ммоль) в ТГФ (30 мл), имеющему температуру 0 С, добавляли DIBAL-H (диизобутилалюминийгидрид) (33,7 мл, 1 М в толуоле). Перемешивали реакционную смесь при температуре окружающей среды в течение ночи. Реакционную смесь гасили добавлением метанола и концентрировали при пониженном давлении. Остаток распределяли между 5 н. NaOH и EtOAc. Слои разделяли и органический слой фильтровали через слой кизельгура. Полученный фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения в виде светло-желтого твердого вещества, (1,6 г, 80%). 1 Н ЯМР (400 МГц, CDCl3):7,67 (с, 1 Н), 7,44 (д, 2 Н), 7,34 (дд, 1 Н), 4,45 (с, 2 Н), 3,00 (м, 1 Н), 1,32 (д, 6 Н). Следующий список соединений получали, по существу, согласно примеру получения [2-(2,6 дихлорфенил)-4-изопропил-2 Н-пиразол-3-ил]метанола с использованием подходящих исходных веществ. Пример получения 17 А. [2-(2,6-Дихлорфенил)-4-изобутил-5-метил-2 Н-пиразол-3-ил]метанол (920 мг, 95%), 1 Н ЯМР (400 МГц, CDCl3)7,46 (д, 2 Н), 7,36 (дд, 1 Н), 4,40 (с, 2 Н), 2,39 (д, 2 Н), 2,30 (с, 3 Н),1,83 (м, 1 Н), 0,94 (д, 6 Н). Пример получения 17 В. [2-(2,6-Дихлорфенил)-4,5-диметил-2 Н-пиразол-3-ил]метанол, 1 Н ЯМР(CDCl3):7,66 (с, 1 Н), 7,42, 7,46 (м, 1 Н), 7,08 (м, 2 Н), 4,52 (с, 2 Н), 3,01 (м, 1 Н), 1,31 (д, 6 Н). Пример получения 171. [2-(2,6-Дихлорфенил)-4-метил-2 Н-пиразол-3-ил]метанол, 1 Н ЯМР (CDCl3):7,61 (с, 1 Н), 7,47 (д, 2 Н, J=8,3 Гц), 7,38 (дд, 1 Н, J=7,0, J=8,3 Гц), (м, 5 Н), 4,45 (с, 2 Н), 2,20 (с, 3 Н). Пример получения 18. [4-Циклопропил-2-(2,6-дихлорфенил)-2 Н-пиразол-3-ил]метанол. К смеси порошка LAH (алюмогидрид лития) и ТГФ (20 мл), имеющей температуру 0 С, добавляли раствор этилового эфира 4-циклопропил-2-(2,6-дихлорфенил)-2 Н-пиразол-3-карбоновой кислоты (2,4 ммоль, 0,8 г) в ТГФ (10 мл). Перемешивали реакционную смесь в течение 2 ч при 0 С. Реакцию гасили последовательным добавлением воды (0,26 мл), 5 н. NaOH (0,26 мл) и воды (0,78 мл). Перемешивали реакционную смесь в течение 1 ч при 0 С. Реакционную смесь фильтровали, полученный фильтрат адсор- 13015632 бировали на силикагеле и очищали, используя градиент 30-50% EtOAc/гексаны, с получением указанного в заголовке соединения (0,36 г, 53%). ЖХ-ES/MC: m/е 283,0 (М+1). Следующий список соединений получали, по существу, согласно примеру получения [4-циклопропил-2-(2,6-дихлорфенил)-2 Н-пиразол-3-ил]метанола, с использованием подходящих исходных веществ. Пример получения 18 А. [4-Циклобутил-2-(2,6-дихлорфенил)-2 Н-пиразол-3-ил]метанол (0,42 г,86%), ЖХ-ES/MC: m/е 297,0 (М+1). Пример получения 18 В. [2-(2-Хлор-6-трифторметилфенил)-4-изопропил-2 Н-пиразол-3-ил]метанол[2-(2-Хлор-6-трифторметилфенил)-4-циклопропил-2 Н-пиразол-3 ил]метанол (0,400, 54%), ES/MC: m/е 317,0 (М+1). Пример получения 19. 1-(2,6-Дихлорфенил)-4-изопропил-5-[3-метил-4-(4,4,5,5-тетраметил[1,3,2] диоксаборолан-2-ил)феноксиметил]-1 Н-пиразол. Смесь трициклогексилфосфина (10 мг, 0,036 ммоль) и бис(дибензилидинацетон)палладия (8,5 мг,0,015 ммоль) в диоксане (3 мл) перемешивали при комнатной температуре в течение 30 мин. К данной реакционной смеси добавляли 5-(4-бром-3-метилфеноксиметил)-1-(2,6-дихлорфенил)-4-изопропил-1 Нпиразол (227 мг, 0,500 ммоль), пинаколборан (140 мг, 0,550 ммоль) и ацетат калия (74 мг, 0,750 ммоль) и полученную смесь нагревали до 80 С в течение 20 ч. Реакционную смесь охлаждали, разбавляли водой и экстрагировали диэтиловым эфиром. Объединенные эфирные фракции сушили (MgSO4) и упаривали. Остаток очищали флэш-хроматографией (10% CH2Cl2/гептан-0% CH2Cl2/гептан), с получением указанного в заголовке соединения (119 мг, 47%). ES/MC: m/е 501,1(М+). Пример получения 20. 6-[4-Изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3-илметокси]-2 метил-3-нитропиридин. К раствору [4-изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3-ил]метанола (2,0 г, 6,66 ммоль) в дегазированном толуоле (22 мл), имеющему температуру окружающей среды, добавляли 6-хлор-2 метил-3-нитропиридин (1,15 г, 6,66 ммоль), карбонат цезия (3,25 г, 9,99 ммоль), 2-(ди-третбутилфосфино)-1,1'-бинафтил (332 мг, 0,833 ммоль, 12,5 моль%) и ацетат палладия (II) (150 мг, 0,666 ммоль, 10 мол.%). Реакционную смесь нагревали до 70 С в течение ночи. Реакционную смесь фильтровали через слой Celite, концентрировали при пониженном давлении и подвергали хроматографии (020% EtOAc/Hex), с получением указанного в заголовке соединения (2,73 г, 94%). 1 Н ЯМР (400 МГц,CDCl3)8,22 (д, 1 Н, J=8,8 Гц), 7,64 (с, 1 Н), 7,52 (дд, 1 Н, J=7,8, 1,7 Гц), 7,49-7,44 (м, 1 Н), 7,41-7,34 (м,2 Н), 6,51 (д, 1 Н, J=9,2 Гц), 5,35 (с, 2 Н), 3,05 (септ, 1 Н, J=7,0 Гц), 2,72 (с, 3 Н), 1,29 (д, 6 Н, J=7,0 Гц). Пример получения 21. 6-[4-Изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3-илметокси]-2 метилпиридин-3-иламин. К раствору 6-[4-изопропил-2-(2-трифторметоксифенил)-2 Н-пиразол-3-илметокси]-2-метил-3-нитропиридина (2,73 г, 6,25 ммоль) в EtOH/ТГФ (100/100 мл), имеющему температуру окружающей среды,добавляли оксид платины (II) (142 мг, 0,625 ммоль, 10 мол.%). Реакционную смесь помещали в атмосферу газообразного водорода. Через 3 ч реакционную смесь фильтровали через кизельгур, концентрировали и подвергали хроматографии (0-30% EtOAc/Hex), с получением указанного в заголовке соединения (2,15 г, 85%). 1 Н ЯМР (400 МГц, CDCl3)7,60 (с, 1 Н), 7,53 (дд, 1 Н, J=7,9, 1,8 Гц), 7,46-7,41 (м, 1 Н), 7,39-7,31(м, 2 Н), 7,08 (д, 1 Н, J=8,8 Гц), 6,33 (д, 1 Н, J=8,4 Гц), 5,17 (с, 2 Н), 3,06 (септ, 1 Н, J=7,0 Гц), 2,32 (с, 3 Н),1,27 (д, 6 Н, J=7,0 Гц). Пример получения 22. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилбензальдегид. К раствору 5-(4-бром-3-метилфеноксиметил)-1-(2,6-дихлорфенил)-4-изопропил-1 Н-пиразола (906 мг, 2,0 ммоль) в безводном ТГФ, имеющему температуру -78 С, добавляли 1,6 М н-бутиллитий (1,35 мл). Через 30 мин добавляли N,N-диметилформамид (0,5 мл, 6,4 ммоль). Через 30 мин добавляли насыщенный водный раствор хлорида аммония. Данную смесь экстрагировали этилацетатом (2). Объединенные этилацетатные слои сушили (MgSO4) и концентрировали при пониженном давлении. Неочищенный остаток очищали флэш-хроматографией (градиентом концентрации этилацетата в гептане) с получением- 14015632 указанного в заголовке соединения (110 мг, 14%). ЖХ-ES/MC: m/е 403,0 (М+1). Пример получения 23. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфениламин. К раствору [2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-ил]метанола (3,0 г, 10,6 ммоль), 4 амино-3-метилфенола (2,0 г, 15,8 ммоль) и три-н-бутилфосфина (3,2 г, 15,8 ммоль) в толуоле (30 мл),имеющему температуру 0 С, добавляли раствор 1,1'-(азодикарбонил)дипиперидина (4,0 г, 15,8 ммоль) в толуоле (40 мл). Данной реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 3,0 ч. Реакционную смесь распределяли между EtOAc (50 мл) и водой (60 мл). Органический слой промывали. насыщенным раствором соли (60 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, с получением остатка. Остаток очищали флэш-хроматографией(EtOAc/гексан), с получением указанного в заголовке соединения в виде твердого вещества (2,3 г, 56%). ЖХ-ES/MC: m/е 390,0 (М+1). Пример получения 24. 5-(4-Бром-3-метилфеноксиметил)-1-(2,6-дихлорфенил)-4-изопропил-1 Нпиразол. Указанное в заголовке соединение получали, по существу, согласно примеру получения 4-[2-(2,6 дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфениламина с использованием [2-(2,6 дихлорфенил)-4-изопропил-2 Н-пиразол-3-ил]метанола и 4-бром-3-метилфенола. ES/MC: m/е 454,9(М+1). Пример получения 25. трет-Бутиловый эфир 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3 илметокси]-2-метилфенилкарбаминовой кислоты. К раствору 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфениламина (2,3 г, 5,9 ммоль) и триэтиламина (0,72 г, 7,1 ммоль) в дихлорметане (20 мл), имеющему температуру 0 С,добавляли раствор ди-трет-бутилдикарбоната (1,55 г, 7,1 ммоль) в дихлорметане (3,0 мл). Перемешивали реакционную смесь при комнатной температуре в течение 16,0 ч. Реакционную смесь распределяли между дихлорметаном (50 мл) и водой (50 мл). Органическую фазу промывали насыщенным раствором соли(50 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, с получением остатка. Остаток очищали флзш-хроматографией (EtOAc/гексан) с получением указанного в заголовке соединения в виде твердого вещества (1,9 г, 66%). ЖХ-ES/MC: m/е 490,0 (М+1). Пример получения 26. трет-Бутиловый эфир 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3 илметокси]-2-метилфенилметилкарбаминовой кислоты. К раствору трет-бутилового эфира 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]2-метилфенилкарбаминовой кислоты (1,9 г, 3,9 ммоль) в ДМФА (15 мл), имеющему температуру 0 С,добавляли порциями гидрид натрия (60% дисперсия в вазелиновом масле, 0,19 г, 4,7 ммоль). Перемешивали данную смесь при 0 С в течение 30 мин. Добавляли по каплям йодметан (0,83 г, 5,9 ммоль). Затем перемешивали реакционную смесь при комнатной температуре в течение 1,0 ч. Реакцию гасили добавлением насыщенного раствора хлорида аммония (20 мл) при 0 С. Реакционную смесь распределяли междуEtOAc (30 мл) и водой (20 мл). Органическую фазу промывали насыщенным раствором соли (30 мл),сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (1,85 г, 95%) в виде пенообразного твердого вещества. ЖХ-ES/MC: m/е 526,0(М+23). Пример получения 27. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилметиламин. К раствору трет-бутилового эфира 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-лиразол-3-илметокси]2-метилфенилметилкарбаминовой кислоты (1,85 г, 3,7 ммоль) в дихлорметане (25,0 мл) при 0 С добавляли раствор 40 М HCl в 1,4-диоксане (2,8 мл, 11,0 ммоль). Затем перемешивали реакционную смесь при комнатной температуре в течение 2,0 ч. Растворители удаляли выпариванием при пониженном давлении. Остаток распределяли между EtOAc (50 мл) и 5% водным раствором бикарбоната натрия (30 мл). Органический слой промывали насыщенным раствором соли (40 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении, с получением остатка. Остаток очищали флэш-хроматографией (EtOAc/гексан) с получением указанного в заголовке соединения в виде пенообразного твердого вещества (1,25 г, 84%). ЖХ-ES/MC: m/е 404,0 (М+1). Пример получения 28. (2-Бромэтил)-4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилметиламин. К раствору 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилметиламина (1,12 г, 2,77 ммоль) в 1,2-дибромэтане (5 мл), имеющему температуру окружающей среды, добавляли триэтиламин (1,54 мл, 11,08 ммоль). Реакционную смесь нагревали до 90 С в течение ночи. К реакционной смеси добавляли EtOAc. Полученную смесь промывали насыщенным раствором соли, сушили (MgSO4) и подвергали хроматографии (0-20% EtOAc/Hex), с получением указанного в заголовке соединения (520 мг, 37%). 1 Н ЯМР (400 МГц, CDCl3)7,70 (с, 1 Н), 7,43-7,40 (м, 2 Н), 7,31 (дд, 1 Н, J=9,0,7,1 Гц), 6,93 (д, 1 Н, J=7,3 Гц), 6,61-6,54 (м, 2 Н), 4,77 (с, 2 Н), 3,34 (т, 2 Н, J=6,5 Гц), 3,26 (т, 2 Н, J=6,7 Гц),2,99 (септ, 1 Н, J=7,0 Гц), 2,65 (с, 3 Н), 2,26 (с, 3 Н), 1, 30 (д, 6 Н, J=7,0 Гц). Пример получения 29. 1-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метил- 15015632 фенилэтанон. К суспензии 4'-гидрокси-2'-метилацетофенона (716 мг, 4,77 ммоль), [2-(2,6-дихлорфенил)-4 изопропил-2 Н-пиразол-3-ил]метанола (1,36 г, 4,77 ммоль), три-н-бутилфосфина (1,78 мл, 7,15 ммоль) в толуоле (20 мл), имеющей температуру 0 С, добавляли ADDP (1,80 г, 7,15 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток подвергали хроматографии (0-30% EtOAc/Hex), с получением указанного в заголовке соединения (857 мг, 64%). 1 Н ЯМР (400 МГц, CDCl3)7,70 (с, 1 Н), 7,65 (д, 1 Н,J=8,8 Гц), 7,42-7,38 (м, 2 Н), 7,32-7,27 (м, 1 Н), 6,64-6,58 (м, 2 Н), 4,85 (с, 2 Н), 2,99 (септ, 1 Н, J=7,0 Гц), 2,50(с, 3 Н), 2,49 (с, 3 Н), 1,30 (д, 6 Н, J=7,0 Гц). Пример получения 30. 2-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилпропан-1-ол. К раствору 2-4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенил пропиональдегида (753 мг, 1,74 ммоль) в ТГФ (17 мл) и МеОН (3 мл), имеющему температуру 0 С, добавляли порциями боргидрид натрия (198 мг, 5,23 ммоль). Реакционную смесь нагревали до комнатной температуры. Через 2 ч реакционную смесь концентрировали при пониженном давлении и остаток распределяли между Et2O (100 мл) и 1 н. HCl (30 мл). Водный слой экстрагировали Et2O (100 мл), объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-30% EtOAc/Hex), с получением указанного в заголовке соединения (715 мг, 95%). 1 Н ЯМР (400 МГц, CDCl3)7,71 (с, 1 Н), 7,43-7,40 (м, 2 Н), 7,33-7,28 (м, 1 Н), 7,04 (д,1 Н, J=8,4 Гц), 6,64-6,58 (м, 2 Н), 4,79 (с, 2 Н), 3,68-3,63 (м, 2 Н), 3,14-3,16 (м, 1 Н), 2,99 (септ, 1 Н, J=7,0 Гц),2,28 (с, 3 Н), 1,30 (д, 3 Н, J=7,0 Гц), 1,21 (д, 6 Н, J=7,0 Гц). Следующий список соединений получали, по существу, согласно примеру получения 2-4-[2-(2,6 дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилпропан-1-ола с использованием подходящих исходных веществ. Пример получения 30 А. 1-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2 метилфенилэтанол, 1 Н ЯМР (400 МГц, CDCl3)7,70 (с, 1 Н), 7,41 (д, 2 Н, J=7,9 Гц), 7,36-7,27 (м, 2 Н),6,64 (дд, 1 Н, J=8,8, 2,6 Гц), 6,55 (д, 1 Н, J=2,6 Гц), 5,04 (дкв, 1 Н, J=6,6, 3,5 Гц), 4,80 (с, 2 Н), 3,00 (септ, 1 Н,J=7,0 Гц), 2,28 (с, 3 Н), 1,57 (д, 1 Н, J=3,5 Гц), 1,42 (д, 3 Н, J=6,6 Гц), 1,30 (д, 6 Н, J=7,0 Гц). Пример получения 30 В. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилметанол (322 мг, 97%), 1 Н ЯМР (400 МГц, CDCl3)7,71 (с, 1 Н), 7,43-7,39 (м, 2 Н), 7,33-7,28 (м,1 Н), 7,16 (д, 1 Н, J=7,9 Гц), 6,62-6,56 (м, 2 Н), 4,81 (с, 2 Н), 4,60 (с, 2 Н), 3,00 (септ, 1 Н, J=7,0 Гц), 2,31 (с,3 Н), 1,31 (д, 6 Н, J=7,0 Гц). Пример получения 30 С. 2-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2 метилфенилэтанол (260 мг, 87%), 1 Н ЯМР (400 МГц, CDCl3)7,71 (с, 1 Н), 7,43-7,40 (м, 2 Н), 7,31 (дд,1 Н, J=8,8, 7,5 Гц), 7,00 (д, 1 Н, J=7,9 Гц), 6,60-6,54 (м, 2 Н), 4,79 (с, 2 Н), 3,77 (т, 2 Н, J=6,8 Гц), 3,00 (септ,1 Н, J-6,6 Гц), 2,80 (т, 2 Н, J=6,8 Гц), 2,25 (с, 3 Н), 1,30 (д, 6 Н, J=6,6 Гц). Пример получения 31. 1-(2,6-Дихлорфенил)-4-изопропил-5-[4-(2-метокси-1-метилвинил)-3-метилфеноксиметил]-1 Н-пиразол. К суспензии трет-бутилата калия (1,36 г, 12,12 ммоль) в ТГФ (30 мл), имеющей комнатную температуру, добавляли (метоксиметил)трифенилфосфонийхлорид (4,14 г, 12,12 ммоль). Перемешивали реакционную смесь при комнатной температуре в течение 20 минут. Добавляли 1-4-[2-(2,6-дихлорфенил)-4 изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилэтанон (843 мг, 2,02 ммоль), в виде твердого вещества, и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl и концентрировали при пониженном давлении. Остаток распределяли между Et2O и водой. Водный слой экстрагировали Et2O, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали при пониженном давлении и подвергали хроматографии (0-30% EtOAc/Hex), с получением указанного в заголовке соединения (836 мг, 93%) в виде смеси E/Z изомеров. (Основной изомер) 1 Н ЯМР (400 МГц,CDCl3)7,70 (с, 1 Н), 7,41 (д, 2 Н, J=7,9 Гц), 7,33-7,28 (м, 1 Н), 6,93 (д, 1 Н, J=8,4 Гц), 6,64-6,52 (м, 2 Н), 5,83(кв, 1 Н, J=1,3 Гц), 4,78 (с, 2 Н), 3,63 (с, 3 Н), 3,00 (септ, 1 Н, J=7,0 Гц), 2,22 (с, 3 Н), 1,83 (д, 3 Н, J=1,3 Гц),1,30 (д, 6 Н, J=7,0 Гц). Пример получения 32. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилбензальдегид. Указанное в заголовке соединение (1,51 г, 75%) получали, по существу, согласно примеру получения 1-(2,6-дихлорфенил)-4-изопропил-5-[4-(2-метокси-1-метилвинил)-3-метилфеноксиметил]-1 Н-пиразола с использованием 5-(4-бром-3-метилфеноксиметил)-1-(2,6-дихлорфенил)-4-изопропил-1 Н-пиразола. 1 Н ЯМР (400 МГц, CDCl3)10,09 (с, 1 Н), 7,73 (с, 1 Н), 7,68 (д, 1 Н, J=8,8 Гц), 7,44-7,40 (м, 2 Н), 7,31 (дд,1 Н, J=8,6, 7,3 Гц), 6,71 (дд, 1 Н, J=8,6, 2,4 Гц), 6,61 (д, 1 Н, J=2,4 Гц), 4,89 (с, 2 Н), 3,01 (септ, 1 Н, J=6,6 Гц),2,60 (с, 3 Н), 1,32 (д, 6 Н, J=6,6 Гц). Пример получения 33. К раствору 1-(2,6-дихлорфенил)-4-изопропил-5-[4-(2-метокси-1-метилвинил)-3-метилфеноксиметил]-1 Н-пиразола (825 мг, 1,85 ммоль) в ТГФ (20 мл), имеющему температуру- 16015632 0 С, добавляли по каплям концентрированную HCl (3 мл). Через 2 ч реакционную смесь разбавляли водой и рН доводили до 7. Водный слой экстрагировали Et2O (2200 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали, с получением указанного в заголовке соединения (764 мг, 96%). 1H ЯМР (400 МГц, CDCl3)9,58 (д, 1 Н, J=1,3 Гц), 7,70 (с, 1 Н), 7,43-7,40 (м, 2 Н), 7,33-7,28 (м, 1 Н), 6,88 (д, 1 Н, J=8,4 Гц), 6,66-6,61 (м, 2 Н), 4,80 (с, 2 Н),3,73 (дкв, 1 Н, J=7,0, 1,3 Гц), 2,99 (септ, 1 Н, J=7,0 Гц), 2,27 (с, 3 Н), 1,35 (д, 3 Н, J=7, 0 Гц), 1,30 (д, 6 Н, J=7,0 Гц). Пример получения 34. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилацетальдегид. Указанное в заголовке соединение (292 мг, количественный выход) получали, по существу, согласно примеру получения 2-4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенил пропиональдегида с использованием 1-(2,6-дихлорфенил)-4-изопропил-5-[4-(2-метоксивинил)-3 метилфеноксиметил]-1 Н-пиразола. 1 Н ЯМР (400 МГц, CDCl3)9,65 (т, 1 Н, J=2,2 Гц), 7,70 (с, 1 Н), 7,41 (д,2 Н, J=8,4 Гц), 7,31 (дд, 1 Н, J=8,8, 7,4 Гц), 6,98 (д, 1 Н, J=7,9 Гц), 6,65-6,58 (м, 2 Н), 4,80 (с, 2 Н), 3,60 (д, 2 Н,J=2,2 Гц), 3,00 (септ, 1 Н, J=7,0 Гц), 2,18 (с, 3 Н), 1,31 (д, 6 Н, J=7,0 Гц). Пример получения 35. 4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилбензонитрил. К раствору [2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-ил]метанола (2,50 г, 8,76 ммоль) в ДМФА (15 мл), имеющему температуру окружающей среды, добавляли 4-фтор-2-метилбензонитрил(1,18 г, 8,76 ммоль), карбонат цезия (5,71 г, 17,53 ммоль). Реакционную смесь нагревали до 80 С в течение ночи. Реакционную смесь концентрировали и остаток распределяли между Et2O и водой. Водный слой экстрагировали Et2O, объединенные органические слои промывали насыщенным раствором соли,сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-20% EtOAc/Hex), с получением указанного в заголовке соединения (2,13 г, 61%). 1H ЯМР (400 МГц, CDCl3)7,72 (с, 1 Н),7,46-7,40 (м, 3 Н), 7,32 (дд, 1 Н, J=9,0, 7,3 Гц), 6,68-6,62 (м, 2 Н), 4,85 (с, 2 Н), 2,99 (септ, 1 Н, J=7,0 Гц), 2,46(с, 3 Н), 1,31 (д, 6 Н, J=7,0 Гц). Пример получения 36. 1-(2,6-Дихлорфенил)-4-изопропил-5-[4-(2-метоксивинил)-3-метилфеноксиметил]-1 Н-пиразол. К суспензии трет-бутилата калия (668 мг, 5,96 ммоль) в ТГФ (20 мл), имеющей температуру окружающей среды, добавляли (метоксиметил)трифенилфосфонийхлорид (2,04 г, 5,96 ммоль) и перемешивали при комнатной температуре в течение 20 мин. Добавляли 4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилбензальдегид(400 мг, 0,992 ммоль), в виде твердого вещества, и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl и концентрировали. Остаток распределяли между Et2O и водой. Водный слой экстрагировали Et2O, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали,концентрировали и подвергали хроматографии (120 г SiO2, 0-30% EtOAC/Hex), с получением указанного в заголовке соединения (302 мг, 73%) в виде смеси E/Z изомеров. 1 Н ЯМР (400 МГц, CDCl3) (основной изомер)7,70 (с, 1 Н), 7,43-7,38 (м, 2 Н), 7,33-7,26 (м, 1 Н), 7,08 (д, 1 Н, J=8,4 Гц), 6,72 (д, 1 Н, J=12,8 Гц),6,60-6,52 (м, 2 Н), 5,82 (д, 1 Н, J=12,8 Гц), 4,78 (с, 2 Н), 3,66 (с, 3 Н), 3,00 (септ, 1 Н, J=7,0 Гц), 2,22 (с, 3 Н),1,30 (д, 6 Н, J=7,0 Гц). Пример получения 37. 1-(4-Метоксифенил)бутан-1,3-дион. К суспензии гидрида натрия (853 мг, 21,8 ммоль, 60% масляная дисперсия) в ТГФ (13 мл), имеющей температуру 0 С, добавляли этилацетат (2,03 мл, 20,8 ммоль). Перемешивали реакционную смесь при комнатной температуре в течение 1 ч. Добавляли по каплям раствор 4-метоксиацетофенона (1,56 г,10,4 ммоль), дибензо-18-краун-6 (62 мг, 0,016 ммоль) и этанол (2 капли) в ТГФ (13 мл) и нагревали реакционную смесь до температуры кипения с обратным холодильником. Через 3 ч реакционную смесь охлаждали до комнатной температуры, погасили насыщенным водным раствором NH4Cl и концентрировали при пониженном давлении. Остаток распределяли между EtOAc (200 мл) и водой (50 мл). Водный слой экстрагировали EtOAc (200 мл), объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали. Остаток очищали хроматографией (0-20%EtOAc/гексаны), с получением указанного в заголовке соединения (1,62 г, 81%). ГХ-ES/MC (газовая хроматография/масс-спектрометрия с электрораспылением) m/е 192. Пример получения 38. 3-Метил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенол. Смесь трициклогексилфосфина (525 мг, 1,87 ммоль), бис(дибензилидинацетон)палладия (460 мг,0,801 ммоль) и диоксана (200 мл) перемешивали при комнатной температуре в течение 30 мин. К данной реакционной смеси добавляли 4-бром-3-метилфенол (5,00 г, 26,7 ммоль), пинаколборан (7,45 г, 40,1 ммоль) и ацетат калия (3,93 г, 40,1 ммоль). Смесь нагревали до 80 С в течение 20 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали диэтиловым эфиром. Объединенные эфирные фракции промывали насыщенным раствором соли, сушили (MgSO4) и упаривали. Остаток очищали, используя флэш-хроматографию (0-2% MeOH/CH2Cl2), с получением указанного в- 17015632 заголовке соединения (1,6 г 47%). Вторая очистка загрязненных фракций дополнительно давала 2,76 г указанного в заголовке соединения, и общий выход составлял 4,36 г (70%). ES/MC: m/e 233,3 (М-1). Пример получения 39. Метиловый эфир 6-бром-1 Н-индол-3-карбоновой кислоты. К раствору 6-броминдол-3-карбоновой кислоты (960 мг, 4,00 ммоль) в метаноле (9,5 мл) при комнатной температуре в течение двух минут добавляли (триметилсилил)диазометан (2,0 М раствор в гексанах, приблизительно 9 мл). Полученную желтую смесь концентрировали при пониженном давлении. Остаток несколько раз повторно растворяли в метаноле и концентрировали при пониженном давлении, с получением указанного в заголовке соединения в виде твердого вещества (100%). ES/MC: m/e 256,0(М+2). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 6-бром-1 Н-индол-3-карбоновой кислоты, с использованием подходящих исходных веществ. Пример получения 39 А. Метиловый эфир (5-бром-1 Н-индол-3-ил)уксусной кислоты (710 мг, 99%),ES/MC: m/e 266,2 (М-2). Пример получения 39 В. Метиловый эфир 5-бром-1 Н-индол-3-карбоновой кислоты, ES/MC: m/e 255,9 (М+2). Пример получения 40. Метиловый эфир 6-(4-гидрокси-2-метилфенил)-1 Н-индол-3-карбоновой кислоты. Смесь 3-метил-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенола (213 мг, 0,910 ммоль), метилового эфира 6-бром-1 Н-индол-3-карбоновой кислоты (193 мг, 0,759 ммоль), тетракис(трифенилфосфин) палладия(0) (57 мг, 0,046 ммоль), ДМФА (2,7 мл), этанола (1,34 мл) и 2 М водного раствора карбоната калия (1,34 мл) нагревали до 100 С в течение 60 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и подкисляли 1 н. HCl. Полученный раствор экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом магния и концентрировали. Остаток очищали флэш-хроматографией, элюируя смесью 25-40% этилацетат/гептан, с получением указанного в заголовке соединения (134 мг, 63%). ES/MC: m/e 280,3 (М-1). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 6-(4-гидрокси-2-метилфенил)-1 Н-индол-3-карбоновой кислоты с использованием подходящих исходных веществ. Пример получения 40 А. Этиловый эфир 6-(4-гидрокси-2-метилфенил)-1 Н-индол-2-карбоновой кислоты, ES/MC: m/e 296,1 (М+1). Пример получения 40 В. Метиловый эфир 5-(4-гидрокси-2-метилфенил)-1 Н-индол-3-карбоновой кислоты, ES/MC: m/e 252,1 (М-2). Пример получения 40 С. Метиловый эфир [5-(4-гидрокси-2-метилфенил)-1 Н-индол-3-ил]уксусной кислоты (180 мг, 60%), ES/MC: m/e 296,1 (М+1). Пример получения 40D. Метиловый эфир 6-(4-гидрокси-2-метилфенил)бензо[b]тиофен-2 карбоновой кислоты, ES/MC: m/e 297,3 (М-1). Пример получения 40 Е. Метиловый эфир (4'-гидрокси-2'-метилбифенил-4-ил)уксусной кислоты(400 МГц, CDCl3)7,99 (м, 2 Н), 7,70 (д, 1 Н), 7,50 (д, 1 Н), 3,92 (с, 3 Н). Пример получения 41. Метиловый эфир 6-бром-1-метил-1 Н-индол-3-карбоновой кислоты. Смесь метилового эфира 5-бром-1 Н-индол-3-карбоновой кислоты (100 мг, 0,394 ммоль), карбоната калия (163 мг, 1,18 ммоль) и ДМФА перемешивали при комнатной температуре и добавляли йодметан(30 мкл, 0,47 ммоль). Через 1,5 ч добавляли дополнительную порцию йодметана (10 мкл), перемешивали реакционную в течение 30 мин, разбавляли дихлорметаном и фильтровали. Полученный фильтрат концентрировали в условиях глубокого вакуума, разбавляли этилацетатом и концентрировали, с получением указанного в заголовке соединения (105 мг, 99%). ES/MC: m/e 270,0 (М+2). Пример получения 42. 2- и 3-Ацетил-6-бромбензотиофен. К раствору 6-бромбензотиофена (20 г, 93,8 ммоль) и ацетилхлорида (8,84 г, 112,6 ммоль) в 120 мл 1,2-дихлорэтана в атмосфере азота при комнатной температуре добавляли по каплям тетрахлорид олова(1 М раствор в дихлорметане, 112,6 ммоль, 112,6 мл). После завершения добавления перемешивали реакционную смесь при комнатной температуре в течение ночи. Смесь вливали в баню лед/вода и экстрагировали дихлорметаном. Органическую фазу промывали насыщенным раствором NaHCO3, водой и насыщенным раствором соли, сушили над MgSO4 и упаривали. Неочищенный остаток очищали флэшхроматографией с использованием в качестве элюента смеси гексан/EtOAc (6:1). Указанное в заголовке соединение (12 г, 50%) получали в виде смеси двух изомеров, 3-ацетил-6-бромбензотиофена и 2-ацетил 6-бромбензотиофена, в соотношении 7:3. ES/MC: m/e 256 (М+2). Пример получения 43. 6-Бромбензотиофен-3-карбоновая кислота и 6-бромбензотиофен-2 карбоновая кислота.- 18015632 К раствору гидроксида натрия (13,64 г, 341 ммоль) в воде (94 мл, 5,22 ммоль), имеющему температуру 0 С, медленно добавляли бром (21,92 г, 137,18 ммоль). Перемешивали реакционную смесь при 0 С в течение 15 мин. К смеси добавляли по каплям раствор 2- и 3-ацетил-6-бромбензотиофена (10,00 г, 39,19 ммоль) в диоксане (75 мл). Перемешивали реакционную смесь при комнатной температуре в течение 2 ч,затем добавляли 50 мл 40% раствора NaHSO3 и затем 10 мл HCl. Наблюдали образование оранжевого твердого вещества. Полученное твердое вещество отфильтровывали и промывали водой и гексанами, с получением указанного в заголовке соединения (7 г, 70%) в виде смеси 6-бромбензотиофен-3 карбоновой кислоты и 6-бромбензотиофен-2-карбоновой кислоты в соотношении 7:3. ES/MC: m/e 258(М+2). Пример получения 44. Метиловый эфир 6-бромбензотиофен-3-карбоновой кислоты и метиловый эфир 6-бромбензотиофен-2-карбоновой кислоты в соотношении. Раствор смеси 6-бромбензотиофен-3-карбоновой кислоты и 6-бромбензотиофен-2-карбоновой кислоты (6,5 г, 25,28 ммоль) и серной кислоты (4,65 г, 47,43 ммоль) в МеОН (100 мл) нагревали до 65 С в течение ночи. Наблюдали образование светло-коричневого твердого вещества. Раствор охлаждали до комнатной температуры. Твердое вещество отфильтровывали и промывали МеОН, с получением указанного в заголовке соединения (5,6 г, 83%) в виде смеси метилового эфира 6-бромбензотиофен-3 карбоновой кислоты и метилового эфира 6-бромбензотиофен-2-карбоновой кислоты в соотношении 7:3.ES/MC: m/e 272 (М+2). Пример получения 45. Метиловый эфир 3-(4'-гидрокси-2'-метилбифенил-4-ил)акриловой кислоты. Смесь 4-бром-3-метилфенола (460 мг, 2,01 ммоль), [4-(Е-3-метокси-3-оксо-1-пропен-1-ил)фенил] бороновой кислоты (48 6 мг, 2,21 ммоль), тетракис(трифенилфосфин)палладия(0) (232 мг, 0,201 ммоль),карбоната цезия (1,31 г, 4,02 ммоль) и ДМФА (4,8 мл) нагревали до 80 С в течение 4,5 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли водой и подкисляли 1 н. HCl. Полученный раствор экстрагировали диэтиловым эфиром. Объединенные органические слои сушили над безводным сульфатом магния и очищали флэш-хроматографией, элюируя смесью 15% этилацетат/гептан, с получением указанного в заголовке соединения (108 мг, 20%). ES/MC: m/e 267,3 (М-1). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 3-(4'-гидрокси-2'-метилбифенил-4-ил)акриловой кислоты, с использованием подходящего исходного вещества. Пример получения 45 А. Метиловый эфир 6-(4-гидрокси-2-метилфенил)-1-метил-1 Н-индол-3 карбоновой кислоты, ЖХ-ES/MC: m/e 296,0 (М+1). Пример получения 45 В. Этиловый эфир 3-(4'-гидрокси-2'-метилбифенил-4-ил)пропионовой кислоты (457 мг, 78%), ES/MC: m/e 283,3 (М-1). Пример получения 45 С. Метиловый эфир 3-(4'-гидрокси-2'-метилбифенил-3-ил)пропионовой кислоты, ES/MC: m/e 269,2 (М+1). Пример получения 45D. Метиловый эфир (4'-гидрокси-2'-метилбифенИл-3-ил)уксусной кислоты,ES/MC: m/e 255,2 (М-1). Пример получения 45 Е. Метиловый эфир 3-(4'-гидрокси-2'-метилбифенил-3-ил)акриловой кислоты,ES/MC: m/e 269,2 (М +1). Пример получения 45F. Метиловый эфир 6-(4-гидрокси-2-метилфенил)бензо[b]тиофен-3 карбоновой кислоты и метиловый эфир 6-(4-гидрокси-2-метилфенил)бензо[b]тиофен-2-карбоновой кислоты метиловый эфир получали, используя в качестве исходных веществ смесь метилового эфира 6 бромбензо[b]тиофен-3-карбоновой кислоты и метилового эфира 6-бромбензо[b]тиофен-2-карбоновой кислоты в соотношении 7:3, МС m/e: 297,0 (М-1). Пример получения 46. Стадия А. Этиловый эфир 4'-бензилоксибифенил-3-карбоновой кислоты. К раствору этил-3-йодбензоата (2,5 г, 1 экв.) в диметиловом эфире этиленгликоля (20 мл) добавляли 2 М раствор карбоната натрия (40 мл), 4-бензилоксибензолбороновую кислоту (2,5 г, 1,2 экв.) и тетракистрифенилфосфинпалладия(0) (1,05 г, 0,1 экв.). Реакционную смесь нагревали до температуры кипения с обратным холодильником в течение 2 ч. Реакционной смеси давали остыть до комнатной температуры и разбавляли 300 мл 50% раствора бикарбоната натрия. Полученный продукт три раза экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией, элюируя градиентом смеси (2:98-8:92) этилацетат/гексаны, с получением желаемого продукта (2,3 г, 76%) в виде белого твердого вещества. Стадия В. Этиловый эфир 4'-гидроксибифенил-3-карбоновой кислоты. К раствору этилового эфира 4'-бензилоксибифенил-3-карбоновой кислоты (2,3 г) в смеси этанол/этилацетат (3:1) (200 мл) добавляли 5% палладий на углероде (300 мг). Реакционную смесь помещали в атмосферу водорода (55 psi (3,79105 Па и встряхивали на аппарате Парра в течение 2 ч. К реакционной смеси добавляли концентрированную хлористо-водородную кислоту (0,3 мл). Реакцию продолжали еще в течение 10 ч. К реакционной смеси добавляли трифторуксусную кислоту (1 мл) и 20% гидроксид палладия на углероде (300 мг) и реакционную смесь оставляли перемешиваться в атмосфере водо- 19015632 рода (60 psi (4,14105 Па. Через 80 ч реакционную смесь дегазировали азотом и фильтровали через слой силикагеля, элюируя 500 мл этилацетата и 500 мл этанола. Полученный фильтрат концентрировали при пониженном давлении и остаток разбавляли 400 мл насыщенного раствора бикарбоната натрия. Данный продукт три раза экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (1,6 г, 95%) в виде белого твердого вещества. Пример получения 47. Стадия А. Метиловый эфир 2'-амино-4'-метоксибифенил-4-карбоновой кислоты. К метиловому эфиру 4'-метокси-2'-нитробифенил-4-карбоновой кислоты (4,00 г), суспендированному в этаноле (150 мл) и этилацетате (150 мл), добавляли 5% палладий на углероде (0,300 г). Реакционную смесь помещали в атмосферу водорода (50 psi (3,45105 Па и встряхивали на аппарате Парра. Через 18 ч реакционную смесь дегазировали азотом и фильтровали через слой силикагеля, элюируя 700 мл этилацетата и 600 мл метиленхлорида. Полученный фильтрат упаривали с получением указанного в заголовке соединения (3,52 г, 99%) в виде белого твердого вещества. Стадия В. Метиловый эфир 2'-бром-4'-метоксибифенил-4-карбоновой кислоты. К раствору нитрита натрия (1,40 г) в диметилсульфоксиде (50 мл) добавляли метиловый эфир 2'амино-4'-метоксибифенил-4-карбоновой кислоты (2,61 г). Через 5 мин реакционную смесь обрабатывали раствором 4,7 мл бромисто-водородной кислоты (48%) в 50 мл диметилсульфоксида. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 2 ч. Реакционную смесь разбавляли раствором 20,0 г карбоната калия в 400 мл воды. Полученный продукт пять раз экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией, элюируя градиентом смеси(99:1-86:14) гексаны/этилацетат, с получением указанного в заголовке соединения (1,00 г, 31%) в виде белого твердого вещества. Стадия С. Метиловый эфир 2'-бром-4'-гидроксибифенил-4-карбоновой кислоты. К раствору метилового эфира 2'-бром-4'-метоксибифенил-4-карбоновой кислоты (0,200 г) в метиленхлориде (5 мл), имеющему температуру 0 С, добавляли бромид бора (0,176 мл). Температуру реакционной смеси в течение 2 ч поддерживали при 0 С. Реакционную смесь медленно гасили добавлением 50 мл метанола, разбавляли 150 мл 2 н. хлористо-водородной кислоты и три раза экстрагировали метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, с получением указанного в заголовке соединения (0,180 г, 94%). Пример получения 48. Метиловый эфир 4'-гидрокси-2'-метилбифенил-4-карбоновой кислоты. К раствору 4-бром-3-метилфенола (0,300 г, 1 экв.) в ДМФА (5 мл) добавляли метиловый эфир 4 фенилбороновой кислоты (0,58 г, 2 экв.), dppf (0,27 г, 0,3 экв.), ацетат палладия (0,036 г, 0,1 зкв.) и карбонат цезия (1,04 г, 2 экв. ). Реакционную смесь нагревали до 75 С в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли водой. Полученный раствор экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали флэш-хроматографией, элюируя 3% этилацетатом в толуоле с получением желаемого продукта (0,224 г,58%). ES/MC: m/e 241,3 (М-1). Пример получения 49. Этиловый эфир 2-(4-метоксифенил)-4-метилтиазол-5-карбоновой кислоты. Смесь 4-метокситиобензамида (5 г, 30 ммоль) и этилового эфира 2-хлор-3-оксомасляной кислоты(4,6 мл, 33 ммоль) в этаноле перемешивали при температуре кипения с обратным холодильником в течение ночи. Реакционную смесь концентрировали и остаток растирали с диэтиловым эфиром с получением указанного в заголовке соединения в виде желтого твердого вещества (5,8 г, 70%). ЖХ-ES/MC: m/e 278(М+1). Пример получения 50. Этиловый эфир 2-(4-гидроксифенил)-4-метилтиазол-5-карбоновой кислоты. К раствору этилового эфира 2-(4-метоксифенил)-4-метилтиазол-5-карбоновой кислоты (550 мг, 2 ммоль) в дихлорметане (20 мл) при -80 С добавляли BBr3 (5 мл, 1 М раствор в дихлорметане). Реакционную смесь в течение ночи перемешивали при температуре окружающей среды. Реакционную смесь гасили добавлением метанола и концентрировали при пониженном давлении. Остаток распределяли междуEtOAc и 1 н. HCl. Органический слой концентрировали и остаток очищали хроматографией (0-30%EtOAc в гексанах), с получением указанного в заголовке соединения в виде желтовато-коричневого твердого вещества (500 мг, 95%). ЖХ-ES/MC: m/e 264 (М+1), 1 Н ЯМР (400 МГц, ДМСО-d6)10,22 (с,1 Н), 7,82 (д, 2 Н), 6,86 (д, 2 Н), 4,27 (кв, 2 Н), 2,64 (с, 3 Н), 1,29 (т, 3 Н). Пример получения 51. Метиловый эфир 2-(4-гидроксифенил)бензо[b]тиофен-6-карбоновой кислоты. Указанное в заголовке соединение (57 мг, 12%) получали, по существ, согласно примеру получения этилового эфира 2-(4-гидроксифенил)-4-метилтиазол-5-карбоновой кислоты с использованием метилового эфира 2-(4-метоксифенил)бензо[b]тиофен-6-карбоновой кислоты. ЖХ-ES/MC: m/е 283 (М-1).- 20015632 Пример получения 52. 2-(4-Метоксифенил)бензо[b]тиофен-6-иловый эфир трифторметансульфоновой кислоты. К раствору 2-(4-метоксифенил)бензо[b]тиофен-6-ола (512 мг, 2 ммоль) в дихлорметане (20 мл) при 0 С добавляли триэтиламин (0,58 мл, 5 ммоль) и ангидрид трифторметансульфоновой кислоты (0,67 мл,4 ммоль). Перемешивали реакционную смесь при температуре окружающей среды в течение ночи. Реакционную смесь концентрировали, остаток повторно растворяли в EtOAc, промывали 1 н. NaOH и затем 1 н. HCl. Органический слой концентрировали, с получением указанного в заголовке соединения в виде желтовато-коричневого твердого вещества (800 мг, количественный выход). Пример получения 53. Метиловый эфир 2-(4-метоксифенил)бензо[b]тиофен-6-карбоновой кислоты. Смесь 2-(4-метоксифенил)бензо[b]тиофен-6-илового эфира трифторметансульфоновой кислоты(750 мг), ацетата палладия (43 мг), 1,4-бис(дифенилфосфино)бутана (97 мг), триэтиламина (1,4 мл) в МеОН (8 мл) и ДМСО (диметилсульфоксид) (12 мл) перемешивали в атмосфере оксида углерода (100 psi(6,9105 Па при 80 С в течение 4 ч. Реакционную смесь фильтровали через слой Celite и полученный фильтрат концентрировали. Остаток очищали колоночной хроматографией (0-20% EtOAc в гексанах), с получением указанного в заголовке соединения в виде желтовато-коричневого твердого вещества (500 мг, 87%). ЖХ-ES/MC: m/е 321 (M+Na). Пример получения 54. Метиловый эфир 5-(4-гидрокси-2-метилфенил)-4-метилтиофен-2-карбоновой кислоты. Стадия А. Смесь 4-метокси-2-метилфенилбороновой кислоты (912 мг, 6 ммоль), метилового эфира 5-бром-4 метилтиофен-2-карбоновой кислоты (1,1 г, 5 ммоль) и K2CO3 (1,38 г, 10 ммоль) в толуоле (30 мл) и воде(5 мл) барботировали N2 в течение 15 мин с последующим добавлением тетракис(трифенилфосфин)палладия (28 9 мг, 0,2 5 ммоль). Перемешивали смесь при 80 С в атмосфере N2 в течение ночи. Затем реакционную смесь фильтровали через слой Celite, элюируя EtOAc. Объединенный фильтрат концентрировали при пониженном давлении и остаток очищали колоночной хроматографией (0-15% EtOAc в гексанах), с получением метилового эфира 5-(4-метокси-2-метилфенил)-4-метилтиофен-2-карбоновой кислоты(540 мг, 39%). 1H ЯМР (CDCl3):7,63 (с, 1 Н), 7,15 (д, 1 Н, J=8,4 Гц), 6,82 (д, 1 Н, J=2,8 Гц), б,78(дд, 1 Н,J=2,8, J=8,4 Гц), 4,79 (ушир.с, 1 Н), 3,88 (с, 3 Н), 3,83 (с, 3 Н), 2,17 (с, 3 Н), 2,02 (с, 3 Н). Стадия В. К раствору метилового эфира 5-(4-метокси-2-метилфенил)-4-метилтиофен-2-карбоновой кислоты(54 0 мг, 2 ммоль) в дихлорметане (30 мл) при 0 С добавляли BBr3 в дихлорметане (1 н., 5,0 мл) и перемешивали данную смесь при температуре окружающей среды в течение ночи. Затем реакционную смесь гасили добавлением метанола и упаривали. Остаток очищали колоночной хроматографией (0-20% EtOAc в гексанах), с получением метилового эфира 5-(4-гидрокси-2-метилфенил)-4-метилтиофен-2-карбоновой кислоты (420 мг, 82%). 1 Н ЯМР (CDCl3):7,62 (с, 1 Н), 7,10 (д, 1 Н, J=7,9 Гц), 6,76 (с, 1 Н), 6,70 (д, 1 Н,J=7,9 Гц), 4,79 (ушир.с, 1 Н), 3,88 (с, 3 Н), 2,15 (с, 3 Н), 2,02 (с, 3 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 5-(4-гидрокси-2-метилфенил)-4-метилтиофен-2-карбоновой кислоты с использованием подходящего исходного вещества. Пример получения 54 А. Метиловый эфир 5-(4-гидроксифенил)тиофен-2-карбоновой кислоты, 1 Н ЯМР (ДМСО-d6):9,87 (с, 1 Н), 7,74 (д, 1 Н, J=4,0 Гц), 7,57 (д, 2 Н, J=8,8 Гц), 7,40 (д, 1 Н, J=4,0 Гц), 6,83 (д,2 Н, J=8,8 Гц), 3,81 (с, 3 Н). Пример получения 54 В. Метиловый эфир 5-(4-гидрокси-2-метилфенил) тиофен-2-карбоновой кислоты, 1 Н ЯМР (ДМСО-d6):9,71 (с, 1 Н), 7,76 (д, 1 Н, J=3,5 Гц), 7,26 (д, 1 Н, J=8,4 Гц), 7,17 (д, 1 Н, J=4,0 Гц), 6,72 (д, 1 Н, J=2,6 Гц), 6,67 (дд, 1 Н, J=2,6, J=8,4 Гц), 3,81 (с, 3 Н), 2,32 (с, 3 Н). Пример получения 54 С. Метиловый эфир 5-(2-хлор-4-гидроксифенил)-тиофен-2-карбоновой кислоты, 1 Н ЯМР (ДМСО-d6):10,33 (с, 1 Н), 7,78 (д, 1 Н, J=3,8 Гц), 7,53 (д, 1 Н, J=8,6 Гц), 7,37 (д, 1 Н, J=3,8 Гц),6,96 (с, 1 Н), 6,84 (д, 1 Н, J=8,6 Гц), 3,82 (с, 3 Н). Пример получения 54D. Метиловый эфир 5-(2-хлор-4-гидроксифенил)-4-метилтиофен-2-карбоновой кислоты, 1 Н ЯМР (ДМСО-d6):10,26 (ушир.с, 1 Н), 7,68 (с, 1 Н), 7,25 (д, 1 Н, J=8,4 Гц), 6,96 (д, 1 Н,J=2,6 Гц), 6,83 (дд, 1 Н, J=2,6, 8,4 Гц), 3,82 (с, 3 Н), 2,03 (с, 3 Н). Пример получения 54 Е. Метиловый эфир 2-(4-гидрокси-2-метилфенил)-4-метилтиазол-5 карбоновой кислоты, 1 Н ЯМР (ДМСО-d6):10,0 (с, 1 Н), 7,74 (д, 1 Н, J=8,4 Гц), 6,74 (с, 1 Н), 6,73 (д, 1 Н,J=8,4 Гц), 3,81 (с, 3 Н), 2,67 (с, 1 Н), 2,50 (с, 3 Н). Пример получения 54F. Этиловый эфир 2-(4-гидрокси-2-метилфенил) тиазол-5-карбоновой кисло 1 ты, Н ЯМР (ДМСО-d6):8,44 (с, 1 Н), 7,74 (д, 1 Н, J=8,4 Гц), 6,76 (с, 1 Н), 6,75 (д, 1 Н, J=8,4 Гц), 4,33 (кв,2 Н), 2,51 (с, 3 Н), 1,30 (т, 3 Н). Пример получения 54G. Метиловый эфир 6-(4-гидрокси-2-метилфенил) никотиновой кислоты, 1 Н ЯМР (ДМСО-d6) :9, 65 (с, 1 Н), 9,10 (с, 1 Н), 8,27 (дд, 1 Н, J=2,2, J=8,4 Гц), 7,62 (дд, 1 Н, J=0,9, J=8,4 Гц),7,32 (д, 1 Н, J=8,8 Гц), 6,71 (с, 1 Н), 6,69 (т, 1 Н), 3,89 (с, 3 Н), 2,31 (с, 3 Н). Пример получения 54 Н. Метиловый эфир 4'-гидрокси-2,2'-диметилбифенил-4-карбоновой кислоты(4,6 мл, 33 ммоль) в этаноле перемешивали при температуре кипения с обратным холодильником в течение ночи. Реакционную смесь концентрировали и остаток растирали с диэтиловым эфиром, с получением этилового эфира 2-(4-метоксифенил)-4-метилтиазол-5-карбоновой кислоты в виде желтого твердого вещества, 5,8 г (70%). ЖХ/МС: 278 (М+1). Стадия В. К раствору этилового эфира 2-(4-метоксифенил)-4-метилтиазол-5-карбоновой кислоты (550 мг, 2 ммоль) в дихлорметане (20 мл) при -80 С добавляли BBr3 (5 мл, 1 М раствор в дихлорметане). Реакционную смесь в течение ночи перемешивали при температуре окружающей среды. Реакционную смесь гасили добавлением метанола и концентрировали. Остаток распределяли между EtOAc и 1 н. HCl. Органический слой концентрировали и очищали хроматографией (0-30% EtOAc в гексанах) с получением указанного в заголовке соединения в виде желтовато-коричневого твердого вещества, (500 мг, 95%). ЖХ/МС: 264 (М+1); 1 Н ЯМР (ДМСО-d6)10,22 (с, 1 Н), 7,82 (д, 2 Н), 6,86 (д, 2 Н), 4,27 (кв, 2 Н), 2,64 (с, 3 Н), 1,29 (т,3 Н). Пример получения 56. Метиловый эфир 4-[(4-гидрокси-2-метилфенил)метиламино]метилбензойной кислоты. К раствору 4-амино-3-метилфенола (1,0 г, 8,12 ммоль) в МеОН (77 мл), имеющему температуру окружающей среды, добавляли метиловый эфир 4-формилбензойной кислоты (1,47 г, 8,93 ммоль) и декаборан (329 мг, 2,68 ммоль). Перемешивали реакционную смесь при комнатной температуре. Через 2 ч добавляли формальдегид (1,23 мл, 16,93 ммоль, 37% водный раствор) и декаборан (329 мг, 2,68 ммоль) и перемешивали реакционную смесь в течение ночи. Реакционную смесь концентрировали при пониженном давлении и остаток очищали хроматографией с получением указанного в заголовке соединения (2,07 г, 90%). ЖХ-ES/MC: m/е 286,2 (М+1). Пример получения 57. Метиловый эфир 3-[(4-гидрокси-2-метилфенил)метиламино]метилбензойной кислоты. Указанное в заголовке соединение получали, по существу, как описано в примере получения метилового эфира 4-[(4-гидрокси-2-метилфенил)метиламино]метилбензойной кислоты, с использованием метилового эфира 3-формилбензойной кислоты (выход 74%). ЖХ-ES/MC: m/е 286,2 (М+1). Пример получения 58. Метиловый эфир 3-[2-(2-хлор-4-гидроксифенил)винил]бензойной кислоты. К раствору метилового эфира 3-винилбензойной кислоты (0,300 г) в диметилформамиде (3 мл) добавляли 4-бром-3-метилфенол (0,35 г), три(ортотолуил)фосфин (0,06 г), Pd(dba)2 (0,032 г) и триэтиламин(0,26 мл). Реакционную смесь нагревали до 100 С в течение ночи. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. Остаток растворяли в этилацетате. Органический слой промывали водой и насыщенным раствором соли, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на фильтре (filter chromatography), элюируя последовательно 300 мл толуола и 250 мл 10% раствора этилацетата в толуоле, с получением указанного в заголовке соединения (0,210 г). 1 Н ЯМР (400 МГц, CDCl3)8,20 (с, 1 Н), 7,95-7,93 (д, 1 Н), 7,71-7,69 (д, 1 Н), 7,53-7,51 (д, 1 Н), 7,48-7,44 (т, 1 Н), 7,38-7,34 (д, 1 Н),6,96-6,92 (д, 1 Н), 6,77-6,72 (м, 2 Н), 5,26 (ушир.с, 1 Н), 3,99 (с, 3 Н), 2,43 (с, 3 Н). Пример получения 59. Метиловый эфир 2-аллил-4-метилбензойной кислоты. К раствору метилового эфира 2-бром-4-метилбензойной кислоты (1,330 г, 5,81 ммоль) в 5 мл ДМФА и 50 мл ацетонитрила добавляли аллилтри-н-бутилолово (2,12 г, 6,39 ммоль), PdCl2(PPh3)2 (0,203 г, 0,29 ммоль) и хлорид лития (0,493 г, 11,6 ммоль). Полученную смесь нагревали до 120 С в течение 8 ч. Затем реакционную смесь охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток растворяли в дихлорметане и очищали хроматографией, элюируя 10% раствором этилацетата в гексанах, с получением 0,980 г (89%) желаемого продукта. 1 Н ЯМР (400 МГц, CDCl3)7,79-7,76 (д, 1 Н), 7,05 (с, 1 Н), 7,05-7,03 (д, 1 Н), 6,03-5,92 (м, 1 Н), 5,01 (дд, 1 Н), 4,99-4,98 (дд, 1 Н), 3,84 (с,3 Н), 3,72 (дд, 1 Н), 3,70 (дд, 1 Н), 2,33 (с, 3 Н). Пример получения 60. Метиловый эфир 4-метил-2-(2-оксоэтил)бензойной кислоты. К раствору метилового эфира 2-аллил-4-метилбензойной кислоты (0,980 г, 5,15 ммоль) в ацетоне(20 мл) и воде (2,0 мл) добавляли N-метилморфолиноксид (0,89 г, 7,57 ммоль) и OsO4 (1 мг). Полученную реакционную смесь перемешивали при комнатной температуре в течение трех часов. Реакционную смесь разбавляли этилацетатом (50 мл) и промывали Na2S2O3 (1,0 М, 20 мл). Органический слой концентрировали и остаток растворяли в ТГФ (25 мл) и воде (15 мл). К данному раствору добавляли NaIO4 (3,24 г,15,2 ммоль) и полученную смесь перемешивали при комнатной температуре в течение двух часов. Смесь фильтровали, полученный фильтрат концентрировали, разбавляли этилацетатом (60 мл) и промывалиNa2S2O3 (1,0M, 20 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный остаток очищали флэш-хроматографией, элюируя 3040% этилацетатом в гексане с получением указанного в заголовке соединения (0,260 г, 26%). 1 Н ЯМР(400 МГц, CDCl3)9,74 (с, 1 Н), 7,94-7,92 (д, 1 Н), 7,15-7,13 (д, 1 Н), 7,00 (с, 1 Н), 3,99 (с, 2 Н), 3,81 (с, 3 Н),2,35 (с, 3 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-метил-2-(2-оксоэтил)бензойной кислоты с использованием подходящего исходного вещества. Пример получения 60 А. Метиловый эфир 5-метокси-2-(2-оксоэтил)бензойной кислоты (29%), 1 Н ЯМР (400 МГц, CDCl3)9,72 (с, 1 Н), 7,54 (д, 1 Н), 7,12-7,10 (д, 1 Н), 7,14-7,00 (дд, 1 Н), 3,95 (с, 2 Н), 3,84(с, 2 Н). Пример получения 61. Метиловый эфир 2-бутокси-5-(2-оксоэтил)бензойной кислоты. Стадия А. К раствору метилового эфира 5-бром-2-гидроксибензойной кислоты (1,100 г, 4,76 ммоль) в 5 мл ДМФА добавляли 1-йодбутан (1,31 г, 7,14 ммоль) и карбонат калия (1,31 г, 9,52 ммоль). Полученную смесь нагревали до 80 С в течение 12 ч. Затем реакционную смесь охлаждали до комнатной температуры, гасили водой (30 мл) и экстрагировали этилацетатом (30 мл 2). Объединенные органические слои сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией, элюируя 15% раствором этилацетата в гексанах, с получением 1,35 г (99%) желаемого промежуточного соединения, метилового эфира 5-бром-2-бутоксибензойной кислоты. 1 Н ЯМР (400 МГц,CDCl3)7,85 (д, 1 Н), 7,50-7,47 (дд, 1 Н), 6,82-6,80 (д, 1 Н), 4,00-3,96 (т, 2 Н), 3,84 (с, 3 Н), 1,79-1,73 (м, 2 Н),1,57-1,46 (м, 2 Н), 0,96-0,93 (т, 3 Н). Стадия В. Указанное в заголовке соединение (30%) получали, по существу, согласно примеру получения метилового эфира 4-метил-2-(2-оксоэтил)бензойной кислоты с использованием метилового эфира 5-бром-2 бутоксибензойной кислоты. 1 Н ЯМР (400 МГц, CDCl3)9,70 (с, 1 Н), 7,61 (д, 1 Н), 7,26-7,23 (дд, 1 Н), 6,946,92 (д, 1 Н), 4,02-3,99 (т, 2 Н), 3,85 (с, 3 Н), 3,62 (д, 2 Н), 1,80-1,76 (м, 2 Н), 1,52-1,47 (м, 2 Н), 0,96-0,93 (т,3 Н). Пример получения 62. Метиловый эфир 4-бутокси-3-(2-оксоэтил)бензойной кислоты. Указанное в заголовке соединение получали, по существу, согласно примеру получения метилового эфира 2-бутокси-5-(2-оксоэтил)бензойной кислоты (52%) с использованием метилового эфира 3-бром-4 гидроксибензойной кислоты. 1 Н ЯМР (400 МГц, CDCl3)9,67 (д, 1 Н), 7,98-7,95 (д, 1 Н), 7,83 (с, 1 Н), 6,896,87 (д, 1 Н), 4,03-4,01 (т, 2 Н), 3,86 (с, 3 Н), 3,65 (д, 2 Н), 1,76-1,73 (м, 2 Н), 1,46-1,41 (м, 2 Н), 0,96-0,92 (т,3 Н). Пример получения 63. Метиловый эфир метилового эфира 2-аллил-5-метоксибензойной кислоты. К раствору 2-бром-5-метоксибензойной кислоты (750 мг, 3,02 ммоль) в бензоле (2 мл), имеющему температуру окружающей среды, добавляли аллилтри-н-бутилолово (1,16 мл, 3,73 ммоль) и тетракис(трифенилфосфин)палладий (0) (174 мг, 0,157 ммоль, 5 мол.%). Реакционную смесь нагревали в герметически закрытой пробирке до 100 С. Через 3 ч реакционную смесь концентрировали при пониженном давлении и остаток подвергали хроматографии (0-10% EtOAc/Hex), с получением указанного в заголовке соединения (451 мг, 73%). 1 Н ЯМР (400 МГц, CDCl3)7,40 (д, 1 Н, J=2,6 Гц), 7,18 (д, 1 Н, J=8,4 Гц),7,00 (дд, 1 Н, J=8,6, 2,9 Гц), 6,04-5,93 (м, 1 Н), 5,03-4,94 (м, 2 Н), 3,88 (с, 3 Н), 3,82 (с, 3 Н), 3,70-3,65 (м, 2 Н). Пример получения 64. Метиловый эфир 5-метокси-2-пропилбензойной кислоты. К суспензии 10% палладия на углероде (245 мг) в МеОН (5 мл), имеющей температуру окружающей среды, одной порцией добавляли раствор метилового эфира 2-аллил-5-метоксибензойной кислоты(440 мг, 2,13 ммоль) в МеОН (5 мл). Реакционную смесь помещали в атмосферу газообразного водорода и перемешивали в течение ночи. Реакционную смесь фильтровали через Celite и концентрировали с- 23015632 получением указанного в заголовке соединения (361 мг, 81%). 1 Н ЯМР (400 МГц, CDCl3)7,38 (д, 1 Н,J=3,l Гц), 7,15 (д, 1 Н, J=8,4 Гц), 6,97 (дд, 1 Н, J=8,6, 2,9 Гц), 3,89 (с, 3 Н), 3,82 (с, 3 Н), 2,88-2,82 (м, 2 Н),1,65-1,53 (м, 2 Н), 0,95 (т, 3 Н, J=7,5 Гц). Пример получения 65. Метиловый эфир 5-гидрокси-2-пропилбензойной кислоты. К раствору метилового эфира 5-метокси-2-пропилбензойной кислоты (350 мг, 1,68 ммоль) в ДХМ(18 мл), имеющему температуру -78 С, добавляли по каплям бромид бора (840 мкл, 8,40 ммоль, 1,0 М раствор в ДХМ). Реакционную смесь нагревали до комнатной температуры в течение ночи. Реакционную смесь гасили добавлением по каплям МеОН и перемешивали при комнатной температуре в течение 30 мин. Реакционную смесь концентрировали и остаток распределяли между EtOAc и водой. Водный слой экстрагировали EtOAc, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-20% EtOAc/Hex), с получением указанного в заголовке соединения (274 мг, 84%) 1 Н ЯМР (400 МГц, CDCl3)7,34 (д, 1 Н, J=2,6 Гц), 7,11 (д, 1 Н, J=8,4 Гц), 6,92 (дд, 1 Н, J=8,4, 3,1 Гц), 6,92 (дд, 1 Н, J=8,4, 3,1 Гц), 3,88 (с, 3 Н), 2,87-2,81(м, 2 Н), 1,63-1,52 (м, 2 Н), 0,94 (т, 3 Н, J=7,3 Гц). Пример получения 66. Метиловый эфир 4-гидрокси-2-метилбензойной кислоты. Указанное в заголовке соединение (77%) получали, по существу, согласно примеру получения метилового эфира 5-гидрокси-2-пропилбензойной кислоты с использованием метилового эфира 4-метокси 2-метилбензойной кислоты. 1 Н ЯМР (400 МГц, CDCl3)7,91-7,86 (м, 1 Н), 6,72-6,67 (м, 2 Н), 3,86 (с, 3 Н),2,57 (с, 3 Н). Пример получения 67. Метиловый эфир 4-метокси-2-метилбензойной кислоты. К суспензии 4-метокси-2-метилбензойной кислоты (1,0 г, 6,02 ммоль) в МеОН (10 мл), имеющей температуру окружающей среды, добавляли по каплям тионилхлорид (1,10 мл, 15,04 ммоль). Реакционную смесь нагревали до температуры кипения с обратным холодильником. Через 3 ч реакционную смесь концентрировали и остаток распределяли между EtOAc и NaHCO3. Водный слой экстрагировали EtOAc,объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и концентрировали с получением указанного в заголовке соединения (877 мг, 81%). 1 Н ЯМР (400 МГц, CDCl3)7,95-7,91 (м, 1 Н), 6,77-6,72 (м, 2 Н), 3,86 (с, 3 Н), 3,84 (с, 3 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-метокси-2-метилбензойной кислоты с использованием подходящего исходного вещества. Пример получения 67 А. Метиловый эфир 1-паратолилциклопропанкарбоновой кислоты (98%), 1 Н ЯМР (400 МГц, CDCl3)7,24 (д, 2 Н, J=7,9 Гц), 7,13 (д, 2 Н, J=8,4 Гц), 3,62 (с, 3 Н), 2,34 (с, 3 Н), 1,60-1,57(м, 2 Н), 1,19-1,15 (м, 2 Н). Пример получения 67 В. Метиловый эфир 2-метил-2-паратолилпропионовой кислоты (84%), 1 Н ЯМР (400 МГц, CDCl3)7,23 (д, 2 Н, J=8,4 Гц), 7,14 (д, 2 Н, J=8,4 Гц), 3,65 (с, 3 Н), 2,33 (с, 3 Н), 1,57 (с,6 Н). Пример получения 67 С. Метиловый эфир 2-бром-4-метилбензойной кислоты, (количественный выход), 1 Н ЯМР (400 МГц, CDCl3)7,73 (д, 1 Н, J=7,9 Гц), 7,49 (д, 1 Н, J=0,9 Гц), 7,15 (дд, 1 Н, J=7,6, 1,7 Гц),3,91 (с, 3 Н), 2,36 (с, 3 Н). Пример получения 68. Метиловый эфир 2-(4-бромметилфенил)-2-метилпропионовой кислоты. К кипящему с обратным холодильником раствору метилового эфира 2-метил-2-паратолилпропионовой кислоты (892 мг, 4,64 ммоль) и N-бромсукцинимида (826 мг, 4,64 ммоль) в CCl4 (120 мл) добавляли 2,2'-азобисизобутиронитрил (12 мг, 0,073 ммоль). Через 5 ч реакционную смесь концентрировали и остаток подвергали хроматографии (120 г SiO2, 5-20% EtOAc/Hex), с получением указанного в заголовке соединения (906 мг, 72%) 1 Н ЯМР (400 МГц, CDCl3)7,38-7,28 (м, 4 Н), 4,48 (с, 2 Н), 3,65 (с,3 Н), 1, 57 (с, 6 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 2-(4-бромметилфенил)-2-метилпропионовой кислоты с использованием подходящего исходного вещества. Пример получения 68 А. Метиловый эфир 1-(4-бромметилфенил)циклопропанкарбоновой кислоты(400 МГц, CDCl3)7,78 (д, 1 Н, J=8,4 Гц), 7,69 (д, 1 Н, J=1,3 Гц), 7,38 (дд, 1 Н, J=8,4, 1,8 Гц), 4,42 (с, 2 Н),3,93 (с, 3 Н). Пример получения 69. Метиловый эфир 2-(4-формилфенил)-2-метилпропионовой кислоты. К раствору метилового эфира 2-(4-бромметилфенил)-2-метилпропионовой кислоты (275 мг, 1,01 ммоль) в ДМСО (8 мл), имеющему температуру окружающей среды, добавляли бикарбонат натрия (127 мг, 1,52 ммоль). Реакционную смесь нагревали до 120 С. Через 3 ч реакционную смесь концентрировали и остаток распределяли между EtOAc и NaHCO3. Водный слой экстрагировали EtOAc, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-20% EtOAc/Hex) с получением указанного в заголовке соедине- 24015632 ния (179 мг, 86%). 1 Н ЯМР (400 МГц, CDCl3)10,00 (с, 1 Н), 7,85 (д, 2 Н, J=8,4 Гц), 7,50 (д, 2 Н, J=8,4 Гц),3,67 (с, 3 Н), 1,61 (с, 6 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 2-(4-формилфенил)-2-метилпропионовой кислоты с использованием подходящего исходного вещества. Пример получения 69 А. Метиловый эфир 1-(4-формилфенил)циклопропанкарбоновой кислоты(с, 3 Н). Пример получения 70. Метиловый эфир 4-(2-метоксивинил)бензойной кислоты. К суспензии трет-бутилата калия (2,05 г, 18,27 ммоль) в ТГФ (60 мл), имеющей температуру окружающей среды, добавляли (метоксиметил)трифенилфосфонийхлорид (6,26 г, 18,27 ммоль). Перемешивали реакционную смесь при комнатной температуре в течение 20 мин. Добавляли метиловый эфир 4 формилбензойной кислоты (1,0 г, 6,09 ммоль) в виде твердого вещества и перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь гасили насыщенным водным раствором NH4Cl и концентрировали. Остаток распределяли между Et2O и водой. Водный слой экстрагировали Et2O, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4),фильтровали, концентрировали и подвергали хроматографии (0-30% EtOAC/Hex) с получением указанного в заголовке соединения (939 мг, 80%) в виде смеси E/Z изомеров. 1 Н ЯМР (400 МГц, CDCl3)8,04(м, 2 Н), 7,23-7,17 (м, 1 Н), 6,99 (д, 1 Н, J=12,8 Гц), 6,74 (д, 1 Н, J=12,8 Гц), 6,22 (д, 1 Н, J=7,0 Гц), 6,06 (д, 1 Н,J=7,0 Гц), 3,89 (с, 6 Н),3,76 (с, 3 Н), 3,73 (с, 3 Н). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-(2-метоксивинил)бензойной кислоты с использованием подходящего исходного вещества. Пример получения 70 А. Метиловый эфир 3-(2-метоксивинил)бензойной кислоты (73%), 1 Н ЯМР(400 МГц, CDCl3)8,04 (д, 1 Н, J=7,9 Гц), 7,89 (д, 1 Н, J=7,9 Гц), 7,82 (дд, 1 Н, J=7,9, 1,3 Гц), 8,04 (дд, 1 Н,J=7,9, 0,9 Гц), 7,41-7,39 (м, 2 Н), 7,23-7,17 (м, 1 Н), 6,99 (д, 1 Н, J=12,8 Гц), 6,74 (д, 1 Н, J=12,8 Гц), 6,22 (д,1 Н, J=7,0 Гц), 6,06 (д, 1 Н, J=7,0 Гц), 3,89 (с, 6 Н), 3,76 (с, 3 Н), 3,73 (с, 3 Н). Пример получения 71. Метиловый эфир 4-(2-оксоэтил)бензойной кислоты. К раствору метилового эфира 4-(2-метоксивинил)бензойной кислоты (930 мг, 4,84 ммоль) в ТГФ(50 мл), имеющему температуру 0 С, добавляли по каплям концентрированную HCl (7 мл). Через 2 ч реакционную смесь разбавляли водой и рН доводили до 7. Водный слой экстрагировали Et2O (2200 мл). Объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-30% EtOAC/Hex) с получением указанного в заголовке соединения (613 мг, 71%). 1 Н ЯМР (400 МГц, CDCl3)9,77 (т, 1 Н, J=2,2 Гц), 8,04 (д, 2 Н, J=8,4 Гц), 7,30 (д, 2 Н, J=8,8 Гц), 3,92 (с, 3 Н), 3,77 (д, 2 Н, J=2,2 Гц). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-(2-оксоэтил)бензойной кислоты с использованием подходящего исходного вещества. Пример получения 71 А. Метиловый эфир 3-(2-оксоэтил)бензойной кислоты (59%), 1H ЯМР (400 МГц, CDCl3)9,78 (т, 1 Н, J=2,2 Гц), 7,98 (д, 1 Н, J=7,5 Гц), 7,91 (с, 1 Н), 7,48-7,39 (м, 2 Н), 7,48-7,39 (м,2 Н), 3,92 (с, 3 Н), 3,77 (д, 2 Н, J=2,2 Гц). Пример получения 71 В. Метиловый эфир 2-(2-оксоэтил)бензойной кислоты (62%), 1H ЯМР (400 МГц, CDCl3)9,79 (т, 1 Н, J=1,3 Гц), 8,13-8,05 (м, 2 Н), 7,58-7,49 (м, 1 Н), 7,42-7,37 (м, 1 Н), 7,31-7,23 (м,1 Н), 4,07 (д, 2 Н, J=1,3 Гц), 3,88 (с, 3 Н). Пример получения 72. Метиловый эфир (4-меркаптофенил)уксусной кислоты. К раствору 4-меркаптофенилуксусной кислоты (5,0 г, 29,72 ммоль) в МеОН (250 мл), имеющему температуру окружающей среды, добавляли серную кислоту (1,25 мл). Перемешивали реакционную смесь при комнатной температуре в течение ночи. Реакционную смесь концентрировали и остаток распределяли между Et2O и водой. Водный слой экстрагировали Et2O, объединенные органические слои промывали насыщенным раствором соли, сушили (MgSO4), фильтровали, концентрировали и подвергали хроматографии (0-30% EtOAC/Hex) с получением указанного в заголовке соединения (3,69 г, 68%). 1 Н ЯМР (400 МГц, CDCl3)7,21 (д, 2 Н, J=7,9 Гц), 7,12 (д, 2 Н, J=8,4 Гц), 3,66 (с, 3 Н), 3,54 (с, 2 Н). Пример получения 73. 1-(4-Бензилокси-2-метилфенил)этанон. К раствору гидрокси-2'-метилацетофенона (5,508 г, 0,037 моль) в ДМФА (10 мл) добавляли карбонат цезия (23,8 г, 0,073 моль). Перемешивали реакционную смесь при комнатной температуре в течение 10 мин. Добавляли бензилбромид (4,36 мл, 0,037 моль), выдерживали 1 ч при комнатной температуре и- 25015632 затем реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате и воде. Слои разделяли, органическую фазу промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения. ES/MC: m/е 241,0 (М+1). Пример получения 74. Этиловый эфир (E/Z)-3-(4-бензилокси-2-метилфенил)бут-2-еновой кислоты. К раствору триэтилфосфоноацетата (1,25 мл, 0,006 моль) в ДМФА (5 мл) добавляли гидрид натрия(0,25 г, 0,006 моль). Через 30 мин добавляли 1-(4-бензилокси-2-метилфенил)этанон (0,5 г, 0,002 моль) и реакционную смесь нагревали до 80 С в течение ночи. После охлаждения добавляли воду, затем 1 н. HCl. Водный слой два раза экстрагировали этилацетатом, органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения. 1 Н ЯМР (CDCl3)1,27 (т, 3 Н), 2,25 (с,3 Н), 2,4 (с, 3 Н), 4,17 (кв, 2 Н), 5,0 (с, 2 Н), 5,72 (с, 1 Н), 6,75 (дд, 1 Н), 6,8 (с, 1 Н), 6,98 (д, 1 Н), 7,27-7,45 (м,5 Н). Пример получения 75. Этиловый эфир (+/-)-3-(4-гидрокси-2-метилфенил)масляной кислоты. К раствору этилового эфира (E/Z)-3-(4-бензилокси-2-метилфенил)бут-2-еновой кислоты в этиловом спирте добавляли 10% палладий на углероде. Реакционную смесь помещали на 6 часов в атмосферу водорода при давлении 413,8 кПа. Катализатор отфильтровывали на кизельгуре и полученный фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения. ES/MC:m/e 221,0 (М-1). Пример получения 76. Этиловый эфир (+/-)-3-4- [2-(2,6-дихлорфенил)-4-изолропил-2 Н-пиразол-3 илметокси]-2-метилфенилмасляной кислоты. К раствору этилового эфира (+/-)-3-(4-гидрокси-2-метилфенил)масляной кислоты (0,366 г, 0,748 ммоль) в ДМФА (4 мл) добавляли карбонат цезия (1,1 г, 1,5 ммоль). После выдерживания в течение 5 мин при комнатной температуре добавляли 5-хлорметил-1-(2,6-дихлорфенил)-4-изопропил-1 Н-пиразол(0,50 г, 0,748 ммоль) и реакционную смесь нагревали до 50 С в течение ночи. После охлаждения добавляли воду, затем 1 н. HCl. Полученную смесь два раза экстрагировали этилацетатом, органические слои объединяли и промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией, элюируя смесью 3-5% этилацетат/толуол, с получением указанного в заголовке соединения. 1 Н ЯМР (CDCl3)1,113-1,163(м, 6 Н), 1,26 (д, 6 Н), 2,399-2,532 (м, 2 Н), 2,952 (м, 1 Н), 3,388 (м, 1 Н), 4,024 (кв, 2 Н), 4,741 (с, 2 Н), 6,513 (д,1 Н), 6,544 (дд, 1 Н), 6,970 (д, 1 Н), 7,264 (т, 1 Н), 7,372 (д, 2 Н), 7,661 (с, 1 Н). Пример получения 77. (+/-)-3-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2 метилфенилбутан-1-ол. Этиловый эфир (+/-)-3-4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилмасляной кислоты (0,581 г, 1,19 ммоль) растворяли в ТГФ (15 мл) и метиловом спирте (3 мл). Добавляли порциями боргидрид натрия, в избытке. После перемешивания при комнатной температуре в течение 3 суток реакцию гасили добавлением воды, затем 1 н. HCl. Полученную смесь два раза экстрагировали этилацетатом. Органические слои объединяли, промывали насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией, элюируя смесью 5-10% этилацетат/толуол с получением указанного в заголовке соединения. 1 Н ЯМР (CDCl3)1,134 (д, 3 Н), 1,262 (д, 6 Н), 1,456 (с, 2 Н), 1,764 (м, 2 Н), 2,223 (с, 3 Н),2,939-3,039 (м, 2 Н), 3,476-3,539 (м, 2 Н), 4,747 (с, 2 Н), 6,516 (д, 1 Н), 6,559 (дд, 1 Н), 6,99 (д, 1 Н), 7,263 (т,1 Н), 7,372 (д, 2 Н), 7,663 (с, 1 Н). Пример получения 78. 3-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилбутиловый эфир (+/-)-3-нитробензолсульфоновой кислоты. К раствору (+/-)-3-4-[2-(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенил бутан-1-ола (0,245 г, 0,548 ммоль) в дихлорметане (10 мл) добавляли триэтиламин (0,15 мл, 1,09 ммоль). Через 10 мин добавляли мета-нитробензолсульфонилхлорид (0,243 г, 1,09 ммоль). Перемешивали реакционную смесь при комнатной температуре в течение 4 ч и затем реакционную смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате и промывали водой и насыщенным раствором соли, сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения. 1 Н ЯМР (CDCl3)1,1 (д, 3 Н), 1,28 (д, 6 Н), 1,62 (с, 2 Н),1,85-1,95 (м, 2 Н), 2,17 (с, 3 Н), 2,91-3,039 (м, 2 Н), 3,85-3,95 (м, 1 Н), 4,04-4,12 (м, 2 Н), 4,74 (с, 2 Н), 6,45 (д,1 Н), 6,51 (дд, 1 Н), 6,86 (д, 1 Н), 7,26 (т, 1 Н), 7,38 (д, 2 Н), 7,65 (т, 3 Н), 8,1 (д, 1 Н), 8,39 (д, 1 Н), 8,63 (с, 1 Н). Пример получения 79. 3-4-[2-(2,6-Дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилбутиловый эфир(+/-)-толуол-4-сульфоновой кислоты. Указанное в заголовке соединение получали, по существу, согласно примеру получения 3-4-[2(2,6-дихлорфенил)-4-изопропил-2 Н-пиразол-3-илметокси]-2-метилфенилбутилового эфира(+/-)-3 нитробензолсульфоновой кислоты с использованием подходящего исходного вещества. 1 Н ЯМР (CDCl3)1,057 (д, 3 Н), 1,264 (д, 6 Н), 1,825 (кв, 2 Н), 2,145 (с, 3 Н), 2,394 (с, 3 Н), 2,969 (кв, 2 Н), 3,813 (м, 1 Н), 3,935- 26015632 Пример получения 80. Метиловый эфир 4-[1-(4-гидрокси-2-метилфенил)этиламино]бензойной кислоты. Смесь метил-4-аминобензоата (260 мг, 1,72 ммоль) и 4'-гидрокси-2'-метилацетофенона в ледяной уксусной кислоте (8 мл) нагревали до 50 С в течение 50 мин. Смесь охлаждали до комнатной температуры и добавляли триацетоксиборгидрид натрия (750 мг, 3,54 ммоль). Через 20 ч добавляли новую порцию триацетоксиборгидрида натрия (750 мг, 3,54 ммоль). Через 24 ч снова добавляли новую порцию триацетоксиборгидрида натрия (750 мг, 3,54 ммоль). Через семь часов смесь концентрировали, остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Слои разделяли и водный слой экстрагировали этилацетатом (2). Объединенные этилацетатные слои сушили (MgSO4) и концентрировали при пониженном давлении. Остаток очищали флэш-хроматографией с получением указанного в заголовке соединения (84 мг, 17%). 1 Н-ЯМР (ДМСО-d6, 400 МГц)9,06 (с, 1 Н), 7,55 (д, J=9,2 Гц, 2 Н),7,02 (д, 8,4 Гц, 1 Н), 6,87 (д, 6,8 Гц, 1 Н), 6,51-6,39 (м, 4 Н), 4,53 (м, 1 Н), 3,66 (с, 3 Н), 2,25 (с, 3 Н), 1,31 (д,6,4 Гц, 3 Н). Пример получения 81. Этиловый эфир 3-(4-гидрокси-2-метилбензиламино)бензойной кислоты. Стадия А. Смесь 2-метил-4-бензилоксибензальдегида (1,22 г, 5,39 ммоль) и этил-3-аминобензоата(912 мг, 5,52 ммоль) в ледяной уксусной кислоте (40 мл) перемешивали в течение 30 мин. К данной смеси добавляли триацетоксиборгидрид натрия (1,25 г, 5,90 ммоль). Через 20 ч смесь концентрировали и остаток распределяли между этилацетатом и насыщенным раствором бикарбоната натрия. Слои разделяли и водный слой экстрагировали этилацетатом (2). Объединенные этилацетатные слои сушили(MgSO4) и концентрировали. Остаток очищали флэш-хроматографией с получением бензильного промежуточного соединения (1,6 г, 80%). Стадия В. К раствору полученного на стадии А бензильного промежуточного соединения (471 мг,1,25 ммоль), в этилацетате (20 мл) в атмосфере азота добавляли 10% палладий на углероде (80 мг). Данную смесь дегазировали и перемешивали в атмосфере водорода в течение ночи. Смесь фильтровали через Celite и концентрировали при пониженном давлении с получением указанного в заголовке продукта в виде твердого вещества (300 мг, 84%). ES/MC: m/е 284,3 (М-1). Пример получения 82. Метиловый эфир 4-(4-гидрокси-2-метилбензиламино)бензойной кислоты. Указанное в заголовке соединение (546 мг, 91%) получали, по существу, согласно методике получения этилового эфира 3-(4-гидрокси-2-метилбензиламино)бензойной кислоты с использованием 2 метил-4-бензилоксибензальдегида и метил-4-аминобензоата. ES/MC: m/e 270,3 (М-1). Пример получения 83. Метиловый эфир 4-формил-2-пент-1-инилбензойной кислоты. К раствору метилового эфира 2-бром-4-формилбензойной кислоты (500 мг, 2,05 ммоль) в дегазированном ДМФА (5 мл), имеющему комнатную температуру, добавляли триэтиламин (2,0 мл, 14,35 ммоль), дихлорбис(трифенилфосфин)палладий(II) (144 мг, 0,205 ммоль, 10 мол.%), йодид меди (I) (20 мг,0,103 ммоль, 5 мол.%) и 1-пентин (406 мкл, 4,10 ммоль). Реакционную смесь нагревали до 80 С в герметически закрытой пробирке. Через 2 ч реакционную смесь концентрировали и остаток подвергали хроматографии (0-10% EtOAc/Hex) с получением указанного в заголовке соединения (279 мг, 59%). 1 Н ЯМР(400 МГц, CDCl3)10,03 (с, 1 Н), 8,02-7,98 (м, 2 Н), 7,80 (дд, 1 Н, J=7,9, 1,8 Гц), 3,95 (с, 3 Н), 2,48 (т, 2 Н,J=7,0 Гц), 1,73-1,63 (м, 2 Н), 1,09 (т, 3 Н, J=7,5 Гц). Пример получения 84. Метиловый эфир 4-формил-2-метилбензойной кислоты. В атмосфере N2 в автоклав Парра объемом 1 л загружали ацетат палладия (II) (2,19 г, 0,0097 моль) и бутил-1-диадамантилфосфин (10,42 г, 0,291 моль) в толуоле (100 мл). К данной смеси добавляли метиловый эфир 4-бром-2-метилбензойной кислоты (222 г, 0,969 моль) и тетраметилэтилендиамин (97,1 мл, 0,63 экв.) в толуоле (325 мл). Автоклав закрывали и удаляли из атмосферы N2. Автоклав заполняли газомSynGas (эквимолярная смесь СО/Н 2) и давление газа поддерживали на постоянном уровне (75 psi(5,17-105 Па. Перемешивали реакционную смесь в течение 18 ч при 85 С. Неочищенную смесь фильтровали через слой Celite и промывали CH2Cl2 до прозрачности. Растворитель удаляли при пониженном давлении с получением красного масла (86%), которое при стоянии кристаллизовалось. 1 Н ЯМР (400 МГц, CDCl3)2,6 (3 Н, с), 3,85 (3 Н, с), 7,65-8,0 (3 Н, m), 10,0 (1 Н, с). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-формил-2-метилбензойной кислоты с использованием подходящего исходного вещества. Пример получения 84 А. Метиловый эфир 2-бензилокси-4-формилбензойной кислоты (384 мг, 86%),ЖХ-ES/MC: m/е 293,0 (М+23). Пример получения 84 В. Метиловый эфир 4-формил-2-трифторметилбензойной кислоты (1,29 г,92%), ЖХ-ES/MC: m/е 233,3 (М+1). Пример получения 84 С. Метиловый эфир 3-формил-5-трифторметилбензойной кислоты (0,5 г,78%), 1 Н ЯМР (CDCl3) (м.д.): 3,95 (3 Н, с), 8,25 (1 Н, с), 8,5 (1 Н, с), 8,7 (1 Н, с), 10,1 (1 Н, с). Пример получения 85. (4-Йод-3-метилфенил)метанол. К раствору 4-йод-3-метилбензойной кислоты (5,2 г, 20 ммоль) в ТГФ (30 мл) добавляли по каплям 2,0 М боран-диметилсульфидный комплекс в ТГФ (40,0 мл, 80 ммоль). Перемешивали данную смесь в течение ночи. Реакционную смесь осторожно гасили при 0 С метанолом (20 мл) и данную смесь упари- 27015632 вали досуха при пониженном давлении. Остаток распределяли между EtOAc (80 мл) и водой (60 мл). Органическую фазу промывали насыщенным раствором соли (60 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали флэшхроматографией (градиентом EtOAc/гексан) с получением указанного в заголовке соединения в виде белого твердого вещества (4,7 г, 95%). Н ЯМР (CDCl3) (м.д.): 2,4 (3 Н, с), 4,55 (2 Н, с), 6,8-7,75 (3 Н, m). Следующий список соединений получали, по существу, согласно примеру получения (4-йод-3 метилфенил)метанола, с использованием подходящего исходного вещества. Пример получения 85 А. (4-Йод-3-трифторметилфенил)метанол (2,1 г, 92%), 1 Н ЯМР (CDCl3) (м.д.): 2,18 (1 Н, с), 4,8 (2 Н, с), 7,4-8,0 (3 Н, m). Пример получения 85 В. (3-Йод-5-трифторметилфенил)метанол (2,1 г, 92%), 1 Н ЯМР (CDCl3) (м.д.): 2,7 (1 Н, с), 4,65 (2 Н, с), 7,5-7,85 (3 Н, m). Пример получения 86. Метиловый эфир 4-гидроксиметил-2-метилбензойной кислоты. В реактор Парра из жаропрочного сплава "Хастеллой" с реакционным объемом 50 мл, предназначенный для работы под давлением, добавляли ацетат палладия (0,161 г, 0,7 ммоль), 1,4-бис(дифенилфосфино)бутан (DPPB) (0,363 г, 0,85 ммоль), (4-йод-3-метилфенил)метанол (1,80 г, 7,25 ммоль),сухой метанол (10,0 мл), сухой триэтиламин (5,25 мл, 37,7 ммоль) и сухой ацетонитрил (15,0 мл). Реакционный резервуар откачали и продували азотом (4). Затем реакционный резервуар откачали и промывали оксидом углерода (4). Затем реакционный резервуар заполняли оксидом углерода при давлении 100 psi (690 кПа), герметизировали и перемешивали реакционную смесь при 100 С в течение 4 ч, поддерживая давление оксида углерода на уровне 100 psi (690 кПа). Реакционную смесь охлаждали до температуры окружающей среды и из реакционного резервуара удаляли оксид углерода. После фильтрования фильтрат концентрировали с получением остатка. Остаток распределяли между EtOAc (50 мл) и водой (50 мл). Органическую фазу промывали насыщенным раствором соли (50 мл), сушили (Na2SO4),фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали флэш-хроматографией с получением указанного в заголовке соединения в виде сиропа (1,18 г, 90%). ЖХES/MC: m/е 181,3 (М+1). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-гидроксиметил-2-метилбензойной кислоты с использованием подходящего исходного вещества. Пример получения 86 А. Метиловый эфир 2-бензилокси-4-гидроксиметилбензойной кислоты (450 мг, 40%), ЖХ-ES/MC: m/е 273,0 (М+1). Пример получения 86 В. Метиловый эфир 4-метилнафталин-1-карбоновой кислоты (3,85 г, 85%),ЖХ-ES/MC: m/е 201,0 (М+1). Пример получения 86 С. Метиловый эфир 4-гидроксиметил-2-трифторметилбензойной кислоты(0,65 г, 24%), 1 Н ЯМР (COCl3) (м.д.): 3,05 (1 Н, ушир.с), 3,9 (3 Н, с), 4,7 (2 Н, с), 7,75 (1 Н, с), 8,1 (2 Н, с). Пример получения 86 Е. Метиловый эфир 2-бензоил-4-метилбензойной кислоты (67 г, 49%), ЖХES/MC: m/е 255,3 (М+1). Пример получения 87. Метиловый эфир 4-формил-2-метилбензойной кислоты. К раствору метилового эфира 4-гидроксиметил-2-метилбензойной кислоты (0,49 г, 2,7 ммоль) в метиленхлориде (8,0 мл), имеющему температуру 0 С, последовательно добавляли бикарбонат натрия (0,46 г, 5,4 ммоль) и перйодинан Десса-Мартина (0,14 г, 3,3 ммоль). Перемешивали данную смесь при комнатной температуре в течение 1,0 ч и гасили водой (2,0 мл). Смесь распределяли между CH2Cl2 (30 мл) и водой (30 мл). Органическую фазу промывали насыщенным раствором соли (30 мл), сушили (Na2SO4),фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали флэш-хроматографией с получением указанного в заголовке соединения в виде сиропа (0,35 г, 72%). 1 Н ЯМР (CDCl3) (м.д.): 2,6 (3 Н, с), 3,85 (3 Н, с), 7,65-8,0 (3 Н, m), 10,0 (1 Н, с). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-формил-2-метилбензойной кислоты с использованием подходящего исходного вещества. Пример получения 87 А. Метиловый эфир 2-бром-4-формилбензойной кислоты (440 мг, 61%), ЖХES/MC: m/е 261,0 (М+18). Пример получения 87 В. Метиловый эфир 2-бутокси-4-формилбензойной кислоты (240 мг, 90%),ЖХ-ES/MC: m/е 237,3 (М+1). Пример получения 87 С. Метиловый эфир 2-бутириламино-4-формилбензойной кислоты (550 мг,87%), ЖХ-ES/MC: m/е 250,3 (М+1), 248,3 (М-1). Пример получения 87D. Метиловый эфир 4-формил-2-(пропан-1-сульфониламино)бензойной кислоты (447 мг, 82%), ЖХ-ES/MC: m/е 303,3 (М+18), 284,3 (М-1). Пример получения 88. Метиловый эфир 3-бензилокси-4-йод-бензойной кислоты. Смесь метилового эфира 3-гидрокси-4-йод-бензойной кислоты (1,2 г, 4,3 ммоль), карбоната калия(1,78 г, 13 ммоль), ацетона (15,0 мл), бензилбромида (1,5 г, 8,6 ммоль) и TBAI (0,05 г) нагревали до 50 С в течение ночи. Растворитель удаляли при пониженном давлении. Остаток распределяли между EtOAc(30 мл) и водой (30 мл). Органическую фазу промывали насыщенным раствором соли (20 мл), сушили(Na2SO4), фильтровали и концентрировали при пониженном давлении, с получением остатка. Остаток очищали флэш-хроматографией с получением указанного в заголовке соединения в виде белого твердого вещества (1,58 г, 99%). ЖХ-ES/MC: m/e 386,0 (М+18). Пример получения 89. 3-Бензилокси-4-йодфенилметанол. Смесь метилового эфира 3-бензилокси-4-йод-бензойной кислоты (1,58 г, 4,3 ммоль), гидроксида лития (0,52 г, 21 ммоль), ТГФ (10 мл), МеОН (10 мл) и воды (10 мл) перемешивали при комнатной температуре в течение 2,0 ч и подкисляли 1,0 М HCl. Смесь распределяли между EtOAc (30 мл) и водой (30 мл). Органическую фазу промывали насыщенным раствором соли (20 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением промежуточного соединения. Промежуточный продукт растворяли в ТГФ (20 мл) и обрабатывали 2,0 М боран-диметилсульфидным комплексом в ТГФ (10 мл, 20 ммоль) в течение ночи. Реакционную смесь осторожно гасили при 0 С метанолом (10 мл) и упаривали досуха при пониженном давлении. Остаток распределяли между EtOAc (30 мл) и водой(30 мл). Органическую фазу промывали насыщенным раствором соли (30 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали флэшхроматографией с получением указанного в заголовке соединения в виде белого твердого вещества (1,32 г, 90%). ЖХ-ES/MC: m/e 358,0 (М+18). Пример получения 90. Метиловый эфир 2-изопропокси-4-метилбензойной кислоты. К смеси метилового эфира 2-гидрокси-4-метилбензойной кислоты (1,0 г, 6,0 ммоль), трифенилфосфина (1,9 г, 7,2 ммоль) и изопропанола (0,72 г, 12,0 ммоль) в ТГФ (10,0 мл) добавляли по каплям при 0 СDIAD (1,45 г, 7,2 ммоль). Перемешивали данную смесь при комнатной температуре в течение ночи. Смесь упаривали досуха при пониженном давлении. Остаток распределяли между EtOAc (50 мл) и водой(50 мл). Органическую фазу промывали насыщенным раствором соли (50 мл), сушили (Na2SO4), фильтровали и концентрировали при пониженном давлении с получением остатка. Остаток очищали флэшхроматографией, с получением указанного в заголовке соединения в виде белого твердого вещества (1,4 г, 96%). ЖХ-ES/MC: m/е 209,0 (М+1). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 2-изопропокси-4-метилбензойной кислоты с использованием подходящего исходного вещества. Пример получения 90 А. Метиловый эфир 2-бутокси-4-метилбензойной кислоты (1,0 г, 74%), ЖХES/MC: m/е 223,3 (М+1). Пример получения 90 В. Метиловый эфир 2-бутокси-5-метилбензойной кислоты (0,85 г, 64%), ЖХES/MC: m/е 223,3 (М+1). Пример получения 90 С. Диметиловый эфир 5-бутоксиизофталевой кислоты (2,1 г, 83%), ЖХES/MC: m/е 284,0 (М+1). Пример получения 91. Метиловый эфир 4-бромметил-2-изопропоксибензойной кислоты. Смесь метилового эфира 2-изопропокси-4-метилбензойной кислоты (1,0 г, 4,8 ммоль), дибензоилпероксида (100 мг) и NBS (0,85 г, 4,8 ммоль) в CCl4 (20 мл) нагревали при 70 С в течение ночи. Твердое вещество отфильтровывали и полученный фильтрат концентрировали, с получением остатка. Остаток очищали флэш-хроматографией с получением указанного в заголовке соединения в виде белого твердого вещества (0,7 г, 51%). ЖХ-ES/MC: m/е 287,0 (М+1). Следующий список соединений получали, по существу, согласно примеру получения метилового эфира 4-бромметил-2-изопропоксибензойной кислоты с использованием подходящего исходного вещества. Пример получения 91 А. Метиловый эфир 4-бромметил-2-бутоксибензойной кислоты (0,6 г, 52%),ЖХ-ES/MC: m/е 301,0 (М+1). Пример получения 91 В. Метиловый эфир 5-бромметил-2-бутоксибензойной кислоты (0,44 г, 65%),ЖХ-ES/MC: m/е 301,0 (М+1). Пример получения 91 С. Метиловый эфир 3-бромметил-5-метилбензойной кислоты (2,73 г, 62%),ЖХ-ES/MC: m/е 260,0 (М+18). Пример получения 91D. Метиловый эфир 6-бромметилникотиновой кислоты (575 мг, 43%), ЖХES/MC: m/е 230,0 (М+1). Пример получения 91 Е. Метиловый эфир 4-бромметил-2,3-дифторбензойной кислоты (1,95 г, 76%),ЖХ-ES/MC: m/е 282,0 (М+18). Пример получения 91F. Метиловый эфир 4-бромметил-3-трифторметилбензойной кислоты (0,7 г,89%), 1 Н ЯМР (CDCl3) (м.д.): 3,85 (3 Н, с), 4,6 (2 Н, с), 7,4-8,3 (3 Н, m). Пример получения 91G. 2-Бром-4-метилбензойная кислота (4,3 г, 63%), ЖХ-ES/MC: m/е 292,7 (М 1). Пример получения 91 Н. Метиловый эфир 4-бромметил-2-бутириламинобензойной кислоты (2,9 г,98%). ЖХ-ES/MC: m/е 314,0 (М+1). Пример получения 91I. Метиловый эфир 4-бромметилнафталин-1-карбоновой кислоты (1,44 г,86%), ЖХ-ES/MC: m/е 281,0 (М+1). Пример получения 91J. Метиловый эфир 2-бензоил-4-бромметилбензойной кислоты (450 мг, 51%),- 29
МПК / Метки
МПК: A61P 3/06, C07D 401/12, C07D 417/12, C07D 403/12, C07D 231/12
Метки: агонисты
Код ссылки
<a href="https://eas.patents.su/30-15632-agonisty-fxr.html" rel="bookmark" title="База патентов Евразийского Союза">Агонисты fxr</a>
Мускариновые агонисты

Номер патента: 6853
Опубликовано: 28.04.2006
Авторы: Хитчкок Стефен Эндрю, Джеймисон Джеймс Эндрю, Аллен Дженнифер Ребекка, Лиу Бин, Тернер Уилльям Уилсон Мл.
МПК: C07C 251/00
Метки: мускариновые, агонисты
Формула / Реферат:
1. Соединение формулы где Q, X, Y и Z независимо выбраны из группы, состоящей из CR1 и N, при условии, что не более чем два из Q, X, Y и Z представляют собой N, и по меньшей мере два из Q, X, Y и Z представляют собой СН; или Y представляет собой СН, Z представляет собой СН и группа Q=X представляет собой S, с образованием тиофенового кольца; R1, в каждом случае независимо, выбран из группы, состоящей из водорода, галогена, С1-С4-алкокси и...
Пиперидиновые агонисты gpcr
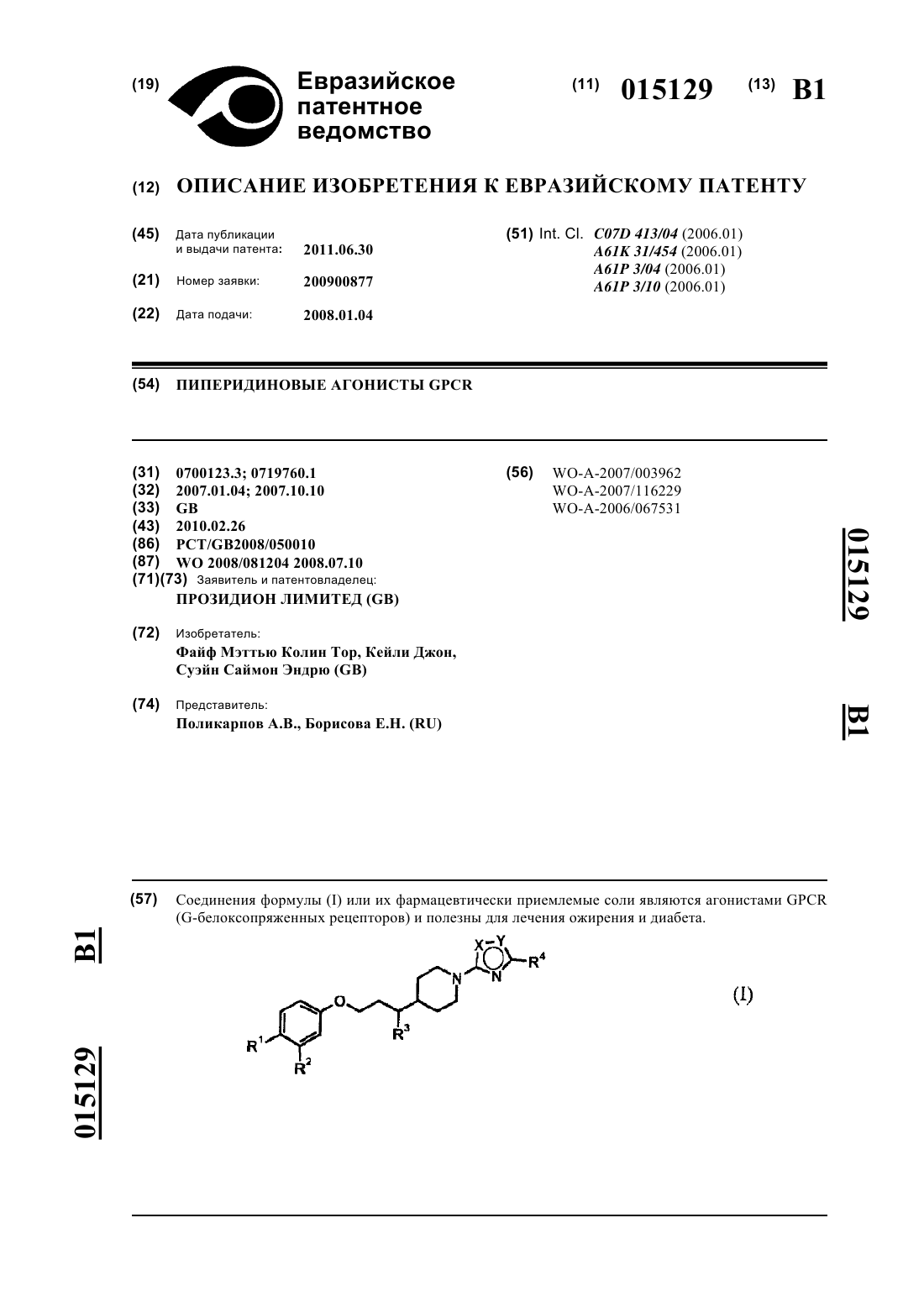
Номер патента: 15129
Опубликовано: 30.06.2011
Авторы: Файф Мэттью Колин Тор, Кейли Джон, Суэйн Саймон Эндрю
МПК: A61K 31/454, A61P 3/04, A61P 3/10...
Метки: агонисты, пиперидиновые
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая сольгде один из Х и Y представляет собой О, а другой представляет собой N;R1 представляет собой SO2R5;R2 представляет собой водород, галоген или метил;R3 представляет собой водород или метил;R4 представляет собой С2-5алкил иR5 представляет собой C1-3алкил.2. Соединение по п.1 или его фармацевтически приемлемая соль, где Х представляет собой О.3. Соединение по п.1 или его...
Пиперидиновые агонисты gpcr

Номер патента: 15130
Опубликовано: 30.06.2011
Авторы: Кейли Джон, Суэйн Саймон Эндрю, Файф Мэттью Колин Тор, Бертрам Лайза Сара, Дживаратнам Ревати Перпетуа
МПК: A61P 3/04, A61P 3/10, A61K 31/454...
Метки: агонисты, пиперидиновые
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая сольгде один из X и Y представляет собой О, а другой представляет собой N;один из Е и Q представляет собой N, а другой представляет собой СН;R1 представляет собой SO2R5или -CONHR6;R2 представляет собой водород или метил;R3 представляет собой водород или метил;R4 представляет собой С2-5алкил;R5 представляет собой С1-3алкил иR6 представляет собой водород, C1-3алкил или С2-3алкил,...
Пиридиноилпиперидины как агонисты 5-нт1f

Номер патента: 7966
Опубликовано: 27.02.2007
Авторы: Кольман Дэниел Тимоти, Лян Сидни Си, Сюй Яо-Чан, Кохен Майкл Филип, Ин Бай-Пин, Чжан Дэи, Захерл Деанна Пиатт, Манкюзо Венсан, Виктор Франтц
МПК: A61P 25/06, C07D 401/06, A61K 31/44...
Метки: пиридиноилпиперидины, 5-нт1f, агонисты
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая аддитивная соль кислоты, где R1 означает С1-С6алкил, замещенный С1-С6алкил, С3-С7циклоалкил, замещенный С3-С7циклоалкил, С3-С7циклоалкилС1-С3алкил, замещенный С3-С7циклоалкилС1-С3алкил, фенил, замещенный фенил, гетероцикл или замещенный гетероцикл; R2 означает водород, С1-С3алкил, С3-С6циклоалкилС1-С3алкил или группу формулы II R3 означает водород или С1-С3алкил; R4 означает водород,...
Агонисты и антагонисты рецепторов 5-ht2c

Номер патента: 5820
Опубликовано: 30.06.2005
Авторы: Тор Маркус, Йенссон Маттиас, Рингберг Эрик, Нильссон Бьерн, Тейбрант Ян, Пелькман Беньямин, Нильссон Йонас
МПК: A61P 25/00, A61K 31/497, C07D 241/18...
Метки: 5-ht2c, рецепторов, антагонисты, агонисты
Формула / Реферат:
1. Соединение общей формулы (Ib) где X1 представляет -O-, -S- или -N(R27)-; Y1 представляет -O-, -S-, -N(R27)- или -CH2-; Ra представляет C3-8-циклоалкил, арил или гетероарил, каждый из которых может быть, необязательно, замещен в одном или в нескольких положениях независимо друг от друга C1-6-алкилом, C1-6-алкокси, фтор-C1-6-алкокси, 2,2,2-трифторэтокси, C3-5-алкинилокси, C3-5-алкенилокси, диметиламино-C1-6-алкокси, метиламино-C1-6-алкокси,...
Предыдущий патент: Соединение, содержащее кремний в качестве осушающего агента для полиолефиновых композиций
Следующий патент: Составы сталей для специальных применений
Случайный патент: Применение 25-oh d3 для воздействия на физиологию мышц человека