Пролекарства возбуждающих аминокислот
Номер патента: 14979
Опубликовано: 29.04.2011
Авторы: Монн Джеймс Аллен, Колладо Кано Иван, Мохер Эрик Дэвид, Бланко -Ургоити Хаиме Гонзало, Педрегал-Терсеро Консепсьон





























Формула / Реферат
1. Соединение формулы (I)

где А представляет собой H-(Q)p-,
Q каждый раз независимо выбран из группы аминоацила, полученного из аминокислоты, выбранной из группы, состоящей из природных и синтетических аминокислот, выбранных из D-изомеров природных a-аминокислот; Aib (аминомасляной кислоты), bAib (3-аминоизомасляной кислоты), Nva (норвалина), b-Ala, Aad (2-аминоадипиновой кислоты), bAad (3-аминоадипиновой кислоты), Abu (2-аминомасляной кислоты), Gaba (g-аминомасляной кислоты), Acp (6-аминокапроновой кислоты), Dbu (2,4-диаминомасляной кислоты), a-аминопимелиновой кислоты, TMSA (триметилсилил-Ala), aIle (алло-изолейцина), Nle (норлейцина), трет-Leu, Cit (цитруллина), Orn, Dpm (2,2'-диаминопимелиновой кислоты), Dpr (2,3-диаминопропионовой кислоты), a- или b-Nal, Cha (циклогексил-Ala), гидроксипролина, Sar (саркозина), О-метилтирозина, фенилглицина; циклических аминокислот; Na-алкилированных аминокислот, где Na-алкилированная аминокислота представляет собой Na-(1-10C)алкиламинокислоту, такую как MeGly (Na-метилглицин), EtGly (Na-этилглицин) и EtAsn (Na-этиласпарагин);
р является целым числом от 1 до 10;
X представляет собой SO2;
R10 представляет собой водород или фтор и
R11 представляет собой водород, фтор или гидрокси;
при условии, что, когда p равно 1, Q не является метионилом;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где p является целым числом от 1 до 3.
3. Соединение по п.1, где p равно 1.
4. Соединение по любому из пп.1-3, где Q является аминоацилом, полученным из природной аминокислоты.
5. Соединение формулы (I)

где А представляет собой H-(Q)p-,
Q представляет собой глицил, аланил, валил, лейцил, изолейцил, пролил, фенилаланил, тирозил, триптофил, метионил, лизил или серинил;
p является целым числом от 1 до 10;
X представляет собой SO2;
R10 представляет собой водород или фтор и
R11 представляет собой водород, фтор или гидрокси;
при условии, что, когда p равно 1, Q не является метионилом;
или его фармацевтически приемлемая соль.
6. Соединение по п.5, где p является целым числом от 1 до 3.
7. Соединение по п.5, где p равно 1.
8. Соединение по любому из пп.1-6, где Q является метионилом и p не равно 1.
9. Соединение по любому из пп.1-8, где R10 представляет собой водород.
10. Соединение по любому из пп.1-9, где R11 представляет собой водород.
11. Фармацевтически приемлемая соль по п.1 или 5, которая является кислотно-аддитивной солью, полученной из кислоты, которая дает фармацевтически приемлемый анион, основно-аддитивной солью, полученной из основания, которое дает фармацевтически приемлемый анион для соединения, которое содержит кислотную группу, или цвиттерионным соединением, которое содержит противоположно заряженные группы.
12. Соединение по п.1 или 5,
где А представляет собой H-(Q)P-;
Q представляет собой L-аланил;
р равно 1;
X представляет собой SO2;
R10 представляет собой водород и
R11 представляет собой водород;
или его гидрохлоридная соль, толуолсульфонатная соль, метансульфонатная соль, этансульфонатная соль, бензолсульфонатная соль или однозамещенная натриевая соль.
13. Соединение по п.12, которое представляет собой гидрохлорид (1R,4S,5S,6S)-4-(2'S-аминопропионил)амино-2,2-диоксо-2l6-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты или толуолсульфонат (1R,4S,5S,6S)-4-(2'S-2'-аминопропионил)амино-2,2-диоксо-2l6-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты.
14. Способ получения соединения формулы (I) или его фармацевтически приемлемой соли по любому из пп.1-13, включающий
ацилирование соединения формулы (ii)

соответствующим аминоацилом формулы (III)
![]() ,
,
где PgN представляет собой защитную группу для атома азота,
затем, для любой из вышеуказанных процедур, когда функциональная группа защищена с использованием защитной группы, удаление защитной группы,
после этого, для любой из вышеуказанных процедур, когда требуется фармацевтически приемлемая соль соединения формулы (I), взаимодействие основной формы такого соединения формулы (I) с кислотой, дающей фармацевтически приемлемый противоион, или для соединения формулы (I), имеющего кислотную группу, взаимодействие кислотной формы такого соединения формулы (I) с основанием, которое дает фармацевтически приемлемый катион, или для цвиттерионного соединения формулы (I), нейтрализацию кислотно-аддитивной солевой формы или основно-аддитивной солевой формы такого соединения формулы (I), или любой другой традиционной процедурой.
15. Применение соединения по любому из пп.1-13 в качестве пролекарства соединения формулы (II)

где X и R10 являются такими, как определено в п.1.
16. Применение соединения по любому из пп.1-13 для лечения неврологических расстройств у пациента.
17. Применение по п.16, где указанное неврологическое расстройство представляет собой церебральную недостаточность, являющуюся результатом операции шунтирования или имплантации на сердце, ишемию головного мозга, травму спинного мозга, травму головы, болезнь Альцгеймера, хорею Гентингтона, боковой амиотрофический склероз, СПИД-ассоциированную деменцию, перинатальную гипоксию, гипогликемическое повреждение нейронов, поражение зрения и ретинопатию, расстройства познавательной способности, идиопатическую и обусловленную действием лекарственного средства болезнь Паркинсона, мышечные спазмы, мигрень, непроизвольное мочеиспускание, толерантность к лекарственному средству, абстиненцию, прекращение приема и пристрастие к наркотикам, прекращение курения, рвоту, отек головного мозга, хроническую боль, нарушения сна, конвульсии, синдром Туретта, синдром нарушения внимания и позднюю дискинезию.
18. Применение по п.17, где указанное неврологическое расстройство представляет собой толерантность к лекарству, абстиненцию, прекращение приема и пристрастие к наркотикам или прекращение курения.
19. Применение соединения по любому из пп.1-13 для лечения психиатрического расстройства у пациента.
20. Применение по п.19, где указанное психиатрическое расстройство представляет собой шизофрению, тревожное расстройство и родственные расстройства, депрессию, биполярные расстройства, психоз и обсессивно-компульсивные расстройства.
21. Применение по п.20, где указанное психиатрическое расстройство представляет собой тревожное расстройство и родственные расстройства.
22. Применение по п.20, где указанное психиатрическое расстройство представляет собой шизофрению.
23. Фармацевтическая композиция, включающая в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем соединение по любому из пп.1-13.
Текст
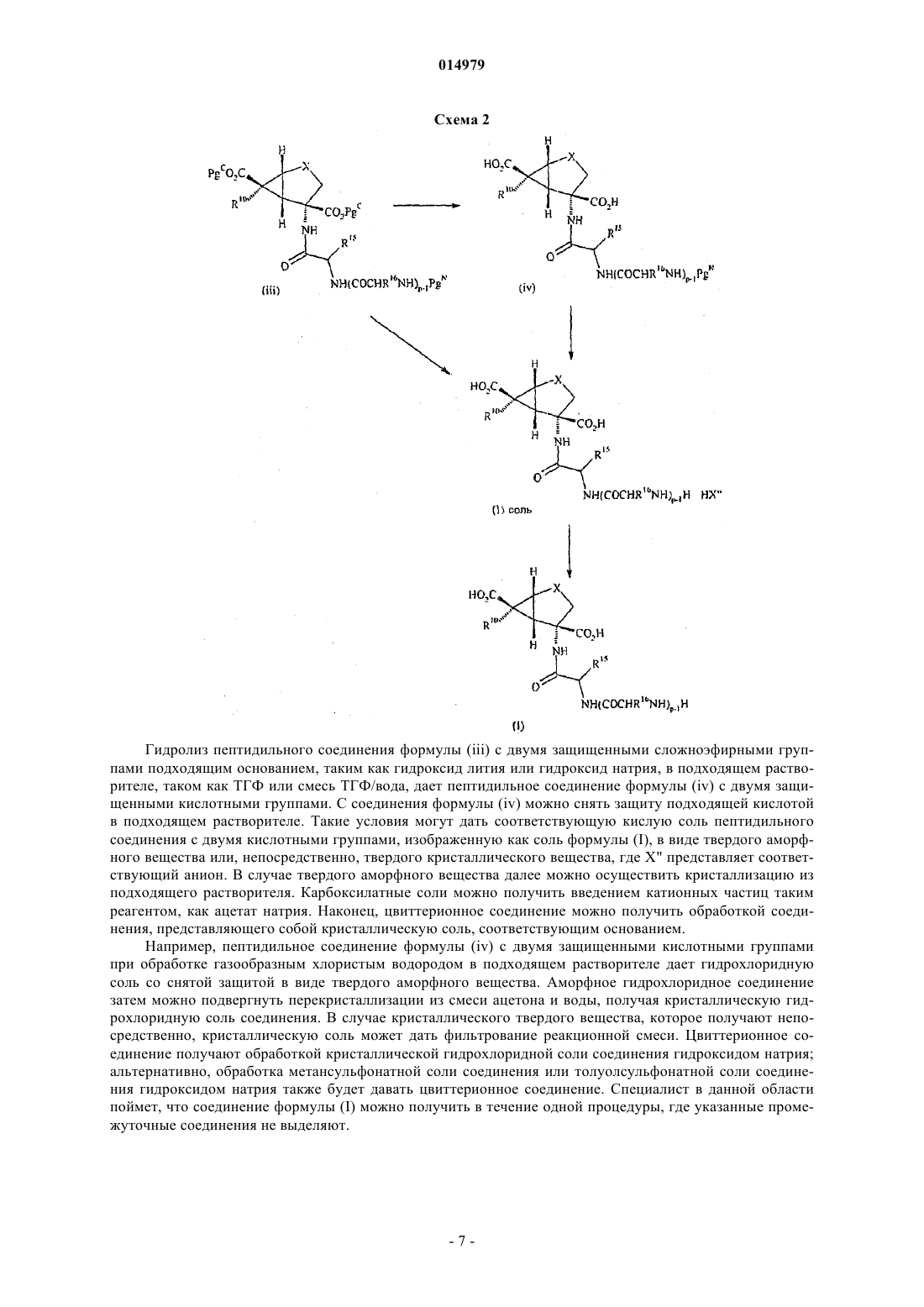
Данное изобретение относится к синтетическим пролекарствам возбуждающих аминокислот формулы (I) и к способам их получения. Далее изобретение относится к применению данных соединений и фармацевтическим композициям, включающим данные соединения, для лечения неврологических расстройств и психиатрических расстройств.(US), Педрегал-Терсеро Консепсьон, Бланко-Ургоити Хаиме Гонзало, Колладо Кано Иван (ES)(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 014979 Данное изобретение относится к синтетическим пролекарствам возбуждающих аминокислот (соединения формулы (I и способам их получения. Далее изобретение относится к способам применения соединений формулы (I) и фармацевтическим композициям, включающим соединения формулы (I), для лечения неврологических и психиатрических нарушений. Предпосылки изобретения Лечение неврологических или психиатрических нарушений, таких как тревожные расстройства,было связано с селективной активацией метаботропных рецепторов возбуждающих аминокислот. Например, (+)-4-амино-2-сульфонилбицикло[3.1.0]гексан-4,6-дикарбоновая кислота описана в качестве активного агониста mGluR2 рецептора в патенте США 5688826 (патент '826), выданном 18 ноября 1997 г. Кроме того, (+)-2-амино-4-фторбицикло[3.1.0]гексан-2,6-дикарбоновая кислота описана в качестве активного агониста mGluR2 рецептора в патенте США 5958960 (патент '960), выданном 28 сентября 1999 г. Настоящее изобретение относится к пролекарственным формам соединений агонистов mGluR2 рецептора, которые усиливают активность in vivo соответствующего исходного соединения и оказывают более сильное воздействие исходного соединения при пероральном применении. Соединения по настоящему изобретению представляют наилучший подход для сохранения безопасности и эффективности описанных ранее агонистов mGluR2 рецептора при увеличенной биологической доступности при пероральном введении. Пролекарства синтетических возбуждающих аминокислот и способы их получения описываются в заявках РСТ, порядковыеPCT/US01/45866 и PCT/US02/00488. Сущность изобретения Настоящее изобретение относится к соединению формулы (I) где А представляет собой H-(Q)P-,Q каждый раз независимо выбран из группы аминоацила, полученного из аминокислоты, выбранной из группы, состоящей из природных и синтетических аминокислот, выбранных из D-изомеров природных -аминокислот; Aib (аминомасляной кислоты), Aib (3-аминоизомасляной кислоты), Nva (норвалина), -Ala, Aad (2-аминоадипиновой кислоты), Aad (3-аминоадипиновой кислоты), Abu (2 аминомасляной кислоты), Gaba (-аминомасляной кислоты), Аср (6-аминокапроновой кислоты), Dbu(саркозина), О-метилтирозина, фенилглицина; циклических аминокислот; N-алкилированных аминокислот, где N-алкилированная аминокислота представляет собой N-(1-10C)алкиламинокислоту, такую как MeGly (N-метилглицин), EtGly (N-этилглицин) и EtAsn (N-этиласпарагин); р является целым числом от 1 до 10;R10 представляет собой водород или фтор;R11 представляет собой водород, фтор или гидрокси; при условии, что, когда р равно 1, Q не является метионилом; или его фармацевтически приемлемой соли. В частности, настоящее изобретение относится к соединению формулы (I), где р является целым числом от 1 до 3, предпочтительно р равно 1. Еще один вариант изобретения относится к соединению формулы (I), где А представляет собойH-(Q)P- и Q является аминоацилом, полученным из природной аминокислоты. Еще один вариант изобретения относится к соединению формулы (I), где А представляет собойH-(Q)P-, Q представляет собой глицил, аланил, валил, лейцил, изолейцил, пролил, фенилаланил, тирозил,триптофил, метионил, лизил или серинил; р является целым числом от 1 до 10; X представляет собойSO2; R10 представляет собой водород или фтор; R11 представляет собой водород, фтор или гидрокси; при условии, что, когда р равно 1, Q не является метионилом; или его фармацевтически приемлемой соли. Предпочтительными являются варианты соединения формулы (I), в которых р является целым числом от 1 до 3, более предпочтительно р равно 1;R11 представляет собой водород. Необходимо принять во внимание, что соединения формулы (I) содержат по меньшей мере четыре асимметричных атома углерода. Настоящее изобретение включает все стереоизомерные формы соединений формулы (I), включая каждый индивидуальный энантиомер и их смеси, такие как пролекарственные формы соединений, описанных в '826 патенте, например 1SR,4RS,5RS,6RS-4-амино-(2 сульфонилбицикло[3.1.0]гексан)-4,6-дикарбоновой кислоты. Еще один предпочтительный вариант изобретения относится к фармацевтически приемлемой соли соединения формулы (I), которая является кислотно-аддитивной солью, полученной из кислоты, которая дает фармацевтически приемлемый анион, основно-аддитивной солью, полученной из основания, которое дает фармацевтически приемлемый анион для соединения, которое содержит кислотную группу, или цвиттерионным соединением, которое содержит противоположно заряженные группы. Следующий предпочтительный вариант изобретения относится к соединению формулы (I), где А представляет собой H-(Q)P-; Q представляет собой L-аланил; р равно 1; X представляет собой SO2; R10 представляет собой водород; R11 представляет собой водород; или его гидрохлоридной соли, толуолсульфонатной соли, метансульфонатной соли, этансульфонатной соли, бензолсульфонатной соли или однозамещенной натриевой соли. Еще один предпочтительный вариант изобретения относится к соединению, которое представляет собой гидрохлорид (1R,4S,5S,6S)-4-(2'S-аминопропионил)амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан 4,6-дикарбоновой кислоты или толуолсульфонат (1R,4S,5S,6S)-4-(2'S-2'-аминопропионил)амино-2,2 диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты. Следующий аспект изобретения относится к способу получения соединения формулы (I) или его фармацевтически приемлемой соли, включающему ацилирование соединения формулы (ii) соответствующим аминоацилом формулы (III) где PgN представляет собой защитную группу для атома азота,затем, для любой из вышеуказанных процедур, когда функциональная группа защищена с использованием защитной группы, удаление защитной группы,после этого, для любой из вышеуказанных процедур, когда требуется фармацевтически приемлемая соль соединения формулы (I), взаимодействие основной формы такого соединения формулы (I) с кислотой, дающей фармацевтически приемлемый противоион, или для соединения формулы (I), имеющего кислотную группу, взаимодействие кислотной формы такого соединения формулы (I) с основанием, которое дает фармацевтически приемлемый катион, или для цвиттерионного соединения формулы (I), нейтрализацию кислотно-аддитивной солевой формы или основно-аддитивной солевой формы такого соединения формулы (I), или любой другой традиционной процедурой. Еще один аспект изобретения относится к применению соединения формулы (I) в качестве пролекарства соединения формулы (II) где X и R10 являются такими, как определено для соединения формулы (I). Еще один аспект изобретения относится к применению соединения формулы (I) для лечения неврологических расстройств у пациента. В одном из вариантов изобретения указанное неврологическое расстройство представляет собой церебральную недостаточность, являющуюся результатом операции шунтирования или имплантации на сердце, ишемию головного мозга, травму спинного мозга, травму головы, болезнь Альцгеймера, хорею Гентингтона, боковой амиотрофический склероз, СПИД-ассоциированную деменцию, перинатальную гипоксию, гипогликемическое повреждение нейронов, поражение зрения и ретинопатию, расстройства познавательной способности, идиопатическую и обусловленную действием лекарственного средства бо-2 014979 лезнь Паркинсона, мышечные спазмы, мигрень, непроизвольное мочеиспускание, толерантность к лекарственному средству, абстиненцию, прекращение приема и пристрастие к наркотикам, прекращение курения, рвоту, отек головного мозга, хроническую боль, нарушения сна, конвульсии, синдром Туретта, синдром нарушения внимания и позднюю дискинезию. Еще в одном варианте изобретения указанное неврологическое расстройство представляет собой толерантность к лекарству, абстиненцию, прекращение приема и пристрастие к наркотикам или прекращение курения. Следующий аспект изобретения относится к применению соединения формулы (I) для лечения психиатрического расстройства у пациента. В одном из вариантов изобретения указанное психиатрическое расстройство представляет собой шизофрению, тревожное расстройство и родственные расстройства, депрессию, биполярные расстройства, психоз и обсессивно-компульсивные расстройства. Предпочтительно указанное психиатрическое расстройство представляет собой тревожное расстройство и родственные расстройства. Еще в одном предпочтительном варианте изобретения указанное психиатрическое расстройство представляет собой шизофрению. В дальнейшем аспекте настоящее изобретение относится к фармацевтической композиции, включающей в сочетании с фармацевтически приемлемым носителем, разбавителем или наполнителем соединение формулы (I) или его фармацевтически приемлемую соль. Подробное описание изобретения Соединения по изобретению, как было обнаружено, представляют собой применимые пролекарства соединений, которые являются селективными агонистами метаботропных глутаматных рецепторов и поэтому применимы для лечения заболеваний центральной нервной системы, таких как неврологические расстройства, например нейродегенеративных заболеваний, и в качестве антипсихотического, анксиолитического, антиабстинентного, антидепрессивного, противосудорожного, обезболивающего и противорвотного средства. Необходимо принимать во внимание, что соединения формулы (I) содержат по меньшей мере четыре асимметричных атома углерода, три из которых находятся в кольце циклопропана и один у -атома углерода аминокислотной группы. Соответственно соединения по изобретению могут существовать и быть выделены в энантиомерно чистой форме, в рацемической форме или в виде смеси диастереоизомеров. Аминокислотная часть молекулы предпочтительно имеет конфигурацию природной аминокислоты,т.е. L-конфигурацию относительно D-глицеролальдегида. Настоящее изобретение включает фармацевтически приемлемые соли соединения формулы (I). Данные соли могут существовать в сочетании с кислотной или основной частью молекулы и могут существовать в виде кислотно-аддитивных солей первичного, вторичного, третичного или четвертичного аммония, щелочных металлов или щелочно-земельных металлов. Обычно кислотно-аддитивные соли можно получить реакцией кислоты с соединением формулы (I). Альтернативно, кислотно-аддитивные соли можно приготовить реакцией предпоследнего соединения (блокированного промежуточного соединения) с соответствующими эквивалентами кислоты с получением соответствующей солевой формы, которая, в свою очередь, может прореагировать с получением соединения формулы (I) или других солей. Соли щелочных металлов и щелочно-земельных металлов обычно получают взаимодействием гидроксидной формы соли желаемого металла с соединением формулы (I). Некоторые конкретные соли дают определенные рецептурные преимущества благодаря своей кристаллической форме. Некристаллические аморфные формы соединений могут быть гигроскопичными. Кристаллические формы фармацевтических соединений иногда являются более желательными, поскольку они показывают благоприятные свойства в твердом состоянии. Кислоты, обычно используемые для получения таких солей, включают неорганические кислоты,например хлористо-водородную, бромисто-водородную, азотную, серную или фосфорную кислоту, или органические кислоты, такие как органические карбоновые кислоты, например гликолевую, малеиновую,гидроксималеиновую, фумаровую, яблочную, винную, лимонную, салициловую, о-ацетоксибензойную,или органические сульфоновые, например 2-гидроксиэтансульфоновую, толуол-п-сульфоновую, метансульфоновую, нафталин-2-сульфоновую, бензолсульфоновую или этансульфоновую кислоту. Кроме фармацевтически приемлемых солей в настоящее изобретение включены другие соли. Они могут служить в качестве промежуточных соединений при очистке соединений или при получении других фармацевтически приемлемых кислотно-аддитивных солей или являются применимыми для идентификации, характеризации или очистки. Было показано, что различные физиологические функции являются объектом воздействия чрезмерной или несоответствующей стимуляцией передачи возбуждающей аминокислотой. Считается, что соединения формулы (I) по настоящему изобретению обладают способностью лечить различные неврологические расстройства у млекопитающих, связанные с данным состоянием, включая острое неврологическое расстройство, такое как церебральная недостаточность, являющаяся результатом операции шунтирования или имплантации на сердце, инсульт, ишемия головного мозга, травма спинного мозга, травма-3 014979 головы, перинатальная гипоксия, остановка сердца и гипогликемическое повреждение нейронов. Считается, что соединения формулы (I) обладают способностью лечить различные хронические неврологические расстройства, такие как болезнь Альцгеймера, хорея Гентингтона, боковой амиотрофический склероз, СПИД-ассоциированная деменция, поражение зрения и ретинопатия, расстройства познавательной способности и идиопатическая и обусловленная действием лекарственного средства болезнь Паркинсона. Соединения формулы (I) по настоящему изобретению лечат множество других неврологических расстройств у пациентов, которые связаны с глутаматной дисфункцией, включая мышечные спазмы, конвульсии, мигрень, непроизвольное мочеиспускание, боль, предменструальное дисфорическое расстройство (PDD), психоз (такой как шизофрения), толерантность к лекарству, абстиненцию, прекращение приема и пристрастие (например, к никотину, опиатам, кокаину, бензодиазепинам и этанолу), тревожные и родственные расстройства, рвоту, отек головного мозга, хроническую боль и позднюю дискинезию. Соединения формулы (I) также применимы в качестве антидепрессантов и анальгетиков. Следующие ниже определения объясняют смысл и рамки различных используемых в настоящем описании терминов. Общие используемые здесь термины имеют свои обычные значения. Термин "воздействие" относится к соединению формулы (II), действующему в качестве агониста на рецептор возбуждающей аминокислоты. Термин "рецептор возбуждающей аминокислоты" относится к метаботропному глутаматному рецептору, рецептору, который соединен с клеточными нервными окончаниями посредством GTP-связывающих белков. Термин "связанный с цАМФ метаботропный глутаматный рецептор" относится к метаботропному рецептору, который связан, чтобы ингибировать аденилатциклазную активность. Термин "неврологическое расстройство" относится как к острому, так и хроническому нейродегенеративному состоянию, включая церебральную недостаточность, являющуюся результатом операции шунтирования или имплантации на сердце, ишемию головного мозга (например, инсульт, в результате остановки сердца), травму спинного мозга, травму головы, болезнь Альцгеймера, хорею Гентингтона,боковой амиотрофический склероз, СПИД-ассоциированную деменцию, перинатальную гипоксию, гипогликемическое повреждение нейронов, поражение зрения и ретинопатию, расстройства познавательной способности и идиопатическую и обусловленную действием лекарственного средства болезнь Паркинсона. Данный термин также включает другие неврологические состояния, которые вызваны глутаматной дисфункцией, включая мышечные спазмы, мигрень, непроизвольное мочеиспускание, толерантность к лекарству, абстиненцию, прекращение приема и пристрастие (например, к опиатам, бензодиазепинам, никотину, кокаину или этанолу), прекращение курения, рвоту, отек головного мозга, хроническую боль, нарушения сна, конвульсии, синдром Туретта, синдром нарушения внимания и позднюю дискинезию. Термин "психиатрическое расстройство" относится как к острым, так и хроническим психиатрическим состояниям, включая шизофрению, тревожное расстройство и родственные расстройства (например, острое тревожное состояние с реакцией паники и связанные со стрессом сердечно-сосудистые заболевания), депрессию, биполярный психоз, психоз, обсессивно-компульсивный синдром, генерализированное тревожное расстройство, острое стрессовое расстройство и расстройства панического типа. Используемый в настоящем описании термин "эффективное количество" относится к количеству или дозе соединения, которое при однократной или многократной дозе введения пациенту дает желаемый эффект у пациента, которому поставлен данный диагноз или проводится лечение. Эффективное количество может быть легко определено лечащим врачом, специалистом в данной области, с использованием известных методов и при наблюдении за результатами, полученными в аналогичных обстоятельствах. При определении эффективного количества или дозы вводимого соединения лечащим врачом рассматривается ряд факторов, включая, но не ограничиваясь этим, вид млекопитающего; его размер, возраст и общее здоровье; конкретное заболевание; степень или запущенность или серьезность заболевания; реакцию конкретного пациента; конкретное вводимое соединение; режим введения; характеристики биодоступности вводимого препарата; выбранный режим введения; использование сопутствующего лекарственного лечения и другие существенные обстоятельства. Например, типичная суточная доза может содержать примерно от 5 до 300 мг активного ингредиента. Соединения можно вводить разнообразными маршрутами, включая пероральный, ректальный, трансдермальный, подкожный,внутривенный, внутримышечный, трансбуккальный или интраназальный пути. Альтернативно, соединение можно вводить непрерывной инфузией. Используемый здесь термин "пациент" относится к млекопитающему, такому как мышь, морская свинка, крыса, собака или человек. Понятно, что предпочтительным пациентом является человек. Используемый здесь термин "лечение" (или "терапия") включает свои общепринятые значения, которые охватывают предотвращение, профилактику, сдерживание и замедление, остановку или изменение направления на обратное развития проистекающих симптомов. По существу, способы по данному изобретению охватывают как терапевтическое, так и профилактическое введение. Общие химические термины, используемые в настоящем описании, имеют свои обычные значения. Используемый здесь термин "защитная группа для азота", представленный в виде "PgN", относится-4 014979 к группам, предназначенным для защиты или блокирования азота от нежелательных реакций в течение процедур синтеза. Выбор используемой подходящей защитной группы для азота зависит от условий, которые будут использоваться в последующих реакционных стадиях, где требуется защита, как хорошо известно специалисту в данной области. Обычно используемые защитные группы для азота описаны в(1999. Предпочтительной защитной группой для азота является трет-бутоксикарбонил. Используемый здесь термин "защитная группа для карбоксила", представленный в виде "Pgc", относится к одному из сложноэфирных производных карбоксильной группы, обычно применяемому для защиты или блокирования карбоксильной группы, когда осуществляют реакции на других функциональных группах соединения. Конкретные примеры включают, например, метил, этил, трет-бутил, бензил,метоксиметил, триметилсилил и аналогичные группы. Дополнительные примеры таких групп можно найти в T.W. Greene и P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed. (John WileySons, NewYork (1999. Предпочтительными защитными группами для карбоксила являются метил и этил. Сложный эфир разлагают, используя традиционную процедуру, которая не влияет на другую часть молекулы. Специалисту в области органической химии понятно, что термин "защитная группа для гидроксила" обозначает группу такого типа, который описан в главе 2 книги Грина и Вутса. Типичные представители защитных групп для гидроксила включают, например, группы простых эфиров, группы замещенного этилового эфира, группы изопропилового эфира, группы фенилового и замещенного фенилового эфира, группы бензилового и замещенного бензилового эфира, группы алкилсилильного эфира, сложноэфирные защитные группы и аналогичные. Тип используемой защитной группы для гидроксила не является критичным при условии, что полученное производное гидроксильной группы является стабильным к условиям последующей(их) реакции(й) в других положениях молекулы промежуточного соединения и ее можно селективно удалить в соответствующий момент без разрушения оставшейся части молекулы,включая любую другую защитную(ые) группу(ы) для гидроксила. Термин "аминоацил" обозначает аминоацил, полученный из аминокислоты, выбранной из группы,состоящей из природных и синтетических аминокислот, как определено в настоящем описании. Природные аминокислоты могут быть нейтральными, положительно заряженными или отрицательно заряженными в зависимости от заместителей боковой цепи. Термин "нейтральные аминокислоты" обозначает аминокислоты, содержащие незаряженные заместители боковой цепи. Примеры нейтральных аминокислот включают аланин, валин, лейцин, изолейцин, пролин, фенилаланин, триптофан, метионин, глицин,серин, треонин, цистеин, глутамин и аспарагин. Термин "положительно заряженные аминокислоты" обозначает аминокислоты, в которых заместители боковой цепи являются положительно заряженными при физиологическом рН. Примеры положительно заряженных аминокислот включают лизин, аргинин и гистидин. Термин "отрицательно заряженные аминокислоты" обозначает аминокислоты, в которых заместители боковой цепи несут отрицательный заряд при физиологическом рН. Примеры отрицательно заряженных аминокислот включают аспарагиновую кислоту и глутаминовую кислоту. Предпочтительными аминокислотами являются -аминокислоты. Наиболее предпочтительными аминокислотами являются аминокислоты, имеющие L-стереохимию на -углероде. Примерами природных -аминокислот являются валин, изолейцин, пролин, фенилаланин, триптофан, метионин, глицин, серин, треонин, цистеин, тирозин, аспарагин, глутамин, лизин, аргинин, гистидин, аспарагиновая кислота и глутаминовая кислота. Термин "синтетическая аминокислота" обозначает аминокислоту, для которой нет кодона нуклеиновой кислоты. Примеры синтетических аминокислот включают, например, D-изомеры природных аминокислот, как указано выше; Aib (аминомасляная кислота), Aib (3-аминоизомасляная кислота), Nva(норвалин), -Ala, Aad (2-аминоадипиновая кислота), Aad (3-аминоадипиновая кислота), Abu (2 аминомасляная кислота), Gaba (-аминомасляная кислота), Аср (6-аминокапроновая кислота), Dbu (2,4 диаминомасляная кислота), -аминопимелиновая кислота, TMSA (триметилсилил-Ala), aIle (аллоизолейцин), Nle (норлейцин), трет-Leu, Cit (цитруллин), Orn, Dpm (2,2'-диаминопимелиновая кислота),Dpr (2,3-диаминопропионовая кислота), - или -Nal, Cha (циклогексил-Ala), гидроксипролин, Sar (саркозин), О-метилтирозин, фенилглицин и аналогичные; циклические аминокислоты; N-алкилированные аминокислоты, где N-алкилированная аминокислота представляет собой N-(1-10 С)алкиламинокислоту,такую как MeGly (N-метилглицин), EtGly (N-этилглицин) и EtAsn (N-этиласпарагин). Примеры синтетических -аминокислот включают D-аланин, D-лейцин и фенилглицин. Названия природных и синтетических аминокислот и их остатков, используемые в настоящем описании, следуют конвенции обозначений, предложенной Совместной комиссией по биохимической номенклатуре (JCBN) ИЮПАК, как сформулировано в "Nomenclature and Symbolism for Amino Acids and Peptides (Recommendations, 1983)",European Journal of Biochemistry, 138, 9-37 (1984). Поскольку используемые в данном описании и прилагаемой формуле изобретения названия и сокращения аминокислот отличаются от указанных названий,отличающиеся названия и сокращения будут разъяснены. Хотя все соединения формулы (I) являются применимыми активными агонистами mGluR2 рецептора, определенные соединения являются предпочтительными. Следующие далее параграфы определяют предпочтительные классы.C) Q представляет собой метионил при условии, что р не равно 1.I) Соединение является свободным основанием.K) Соединение является гидрохлоридной солью.L) Соединение является метансульфонатной солью.M) Соединение является этансульфонатной солью.N) Соединение является толуолсульфонатной солью. Предшествующие параграфы могут быть объединены, чтобы характеризовать дополнительные предпочтительные классы соединений. Соединения формулы (I) применимы для лечения расстройств у млекопитающих и предпочтительным млекопитающим является человек. Соединения по настоящему изобретению можно получить различными процедурами, некоторые из которых иллюстрируются на приведенных ниже схемах. Конкретный порядок стадий, требующихся для получения соединений формулы (I), зависит от конкретных соединений, которые синтезируются, от исходного соединения и относительной лабильности замещенных групп. Некоторые заместители могут быть исключены из следующих ниже схем ради понятности изложения, и они не имеют намерения каким-либо образом ограничивать описание схем. Специалист в данной области поймет, что заместителиR15 и R16 представляют соответствующие боковые цепи для получения желаемого аминоацила. Если необходимые исходные вещества для приведенных ниже схем отсутствуют в продаже, то данные вещества можно изготовить процедурами, которые выбраны из стандартных методов органической и гетероциклической химии, методами, которые аналогичны синтезам известных, структурно-сходных соединений, и процедурами, описанными в методах получения и примерах, включая новые процедуры. Схема 1 Соединения формулы (I) конвертируют посредством ферментных или гидролитических процессовin vivo с получением соединений формулы (II), как показано на приведенной выше схеме 1. В частности,кристаллическую форму соединения формулы (I) можно получить маршрутом, показанным в общих чертах на приведенной ниже схеме 2. Гидролиз пептидильного соединения формулы (iii) с двумя защищенными сложноэфирными группами подходящим основанием, таким как гидроксид лития или гидроксид натрия, в подходящем растворителе, таком как ТГФ или смесь ТГФ/вода, дает пептидильное соединение формулы (iv) с двумя защищенными кислотными группами. С соединения формулы (iv) можно снять защиту подходящей кислотой в подходящем растворителе. Такие условия могут дать соответствующую кислую соль пептидильного соединения с двумя кислотными группами, изображенную как соль формулы (I), в виде твердого аморфного вещества или, непосредственно, твердого кристаллического вещества, где X" представляет соответствующий анион. В случае твердого аморфного вещества далее можно осуществить кристаллизацию из подходящего растворителя. Карбоксилатные соли можно получить введением катионных частиц таким реагентом, как ацетат натрия. Наконец, цвиттерионное соединение можно получить обработкой соединения, представляющего собой кристаллическую соль, соответствующим основанием. Например, пептидильное соединение формулы (iv) с двумя защищенными кислотными группами при обработке газообразным хлористым водородом в подходящем растворителе дает гидрохлоридную соль со снятой защитой в виде твердого аморфного вещества. Аморфное гидрохлоридное соединение затем можно подвергнуть перекристаллизации из смеси ацетона и воды, получая кристаллическую гидрохлоридную соль соединения. В случае кристаллического твердого вещества, которое получают непосредственно, кристаллическую соль может дать фильтрование реакционной смеси. Цвиттерионное соединение получают обработкой кристаллической гидрохлоридной соли соединения гидроксидом натрия; альтернативно, обработка метансульфонатной соли соединения или толуолсульфонатной соли соединения гидроксидом натрия также будет давать цвиттерионное соединение. Специалист в данной области поймет, что соединение формулы (I) можно получить в течение одной процедуры, где указанные промежуточные соединения не выделяют. Сложный диэфир формулы (ii) ацилируют соединением формулы (III), используя подходящее связующее вещество, получая пептидильное соединение формулы (iii) с двумя защищенными сложноэфирными группами. Альтернативно, данное превращение можно осуществить с использованием хлорангидрида соединения формулы (III). Подходящие связующие вещества для пептида включают дициклогексилкарбодиимид (DCC), 1-(3 диметиламинопропил)-3-этилкарбодиимид (EDC), изобутилхлорформиат, дифенилхлорфосфат, 2-хлор 4,6-диметокси-1,3,5-триазин (CDMT), хлорангидрид бис-(2-оксо-3-оксазолидинил)фосфиновой кислоты и гексафторфосфат бензотриазол-1-илокси-трис-(диметиламино)фосфония. Схема 4 На вышеприведенной схеме 4 соединение формулы (II), дикислоту, обрабатывают подходящим защищающим карбоксигруппу реагентом, таким как смесь каталитического количества хлористоводородной кислоты или тионилхлорида и метанола или этанола, получая соответствующий сложный диэфир формулы (ii). Альтернативно, соединение формулы (II) вначале можно обработать реагентом,защищающим атом азота, таким как BOC2O, получая соединение формулы (i) с защитной группой на атоме азота. Затем соединение формулы (i) можно обработать реагентом, защищающим карбоксигруппу,таким как метилйодид, в присутствии основания, такого как карбонат калия, после чего следует обработка реагентом для снятия защитной группы с азота, таким как хлористо-водородная кислота или трифторуксусная кислота, получая соединение формулы (ii). Кроме того, специалист в данной области поймет, что в зависимости от X может требоваться соответствующий защищающий реагент. Например, если X представляет собой CR3R4, R3 представляет собой гидроксил и R4 представляет собой водород, то специалист в данной области поймет, что перед осуществлением любой из вышеуказанных схем может потребоваться подходящая защитная группа для гидроксила. Соединения формулы (II) известны из уровня техники. Например, способы получения данных соединений можно найти в патентах США 5688826 (патент '826) и 5958960 (патент '960). По сравнению с ранее описанными способами были сделаны различные улучшения маршрутов синтеза соединений формулы (II). Улучшения включают окисление серы или спирта, а также оптическое разрешение различных промежуточных соединений, как описываетсяниже. Первое улучшение относится к превращению, описанному в патенте '826, столбец 8, строки 22-34 и столбец 7, начиная со строки 33 (формула V), включающему окисление соединения формулы VII патента с получением соединения формулы V патента '826 Было обнаружено, что комплекс триоксид серы/пиридин или трифторуксусный ангидрид в сочетании с ДМСО являются предпочтительнее многих методов окисления, известных из уровня техники. Во-вторых, что касается разделения соединения формулы (III) патента '826 где R2 представляет собой карбоксильную группу, ссылаясь на столбец 8, строки 3-7 и столбец 6, начиная со строки 1 (формула III), было обнаружено, что предпочтительными являются (R)-метилбензиламин и хинин. (R)метилбензиламин является особенно предпочтительным. Кроме того, было обнаружено, что при окислении сульфида соединения формулы (III) патента '826,где X представляет собой серу, с получением соединения формулы (III) патента '826, где X представляет собой сульфонил, как ссылаются в патенте '826, столбец 8, строки 39-53, предпочтительной является основная водная система и пероксид водорода, используемые в комбинации с катализатором. Следующие ниже примеры дополнительно иллюстрируют соединения по настоящему изобретению и способы их синтеза. Примеры не имеют намерения каким-либо образом ограничивать объем изобретения, и их не следует так истолковывать. Все эксперименты проводят при избыточном давлении сухого азота или аргона. Все растворители и реагенты получают из коммерческих источников и используют без дополнительной очистки, если это не указано иным образом. Сухой тетрагидрофуран (ТГФ) можно получить перегонкой с натрием или натрий бензофенонкетилом перед использованием. Спектры протонного ядерного магнитного резонанса (1 Н ЯМР) получают на Bruker Avance II bay-500 при 500 МГц, BrukerAvance I bay-200 при 200 МГц или Varian Inova/Varian 300/Varian 400 при 500 МГц. Масс-спектрометрию с электрораспылительной ионизацией (ЭРИ) осуществляют на приборе Agilent MSD/B, используя в качестве подвижной фазы смесь ацетонитрил/водный ацетат аммония. Масс-спектроскопию с ионизацией бомбардировкой быстрыми атомами (МС ББА) проводят на приборе VG ZAB-2SE. Масс-спектрометрию с ионизацией полевой десорбцией (МС ПД) осуществляют, используя либо спектрометр VG 70SE, либоVarian MAT 731. Оптическое вращение измеряют на поляриметре Perkin-Elmer 241. Хроматографическое разделение на Waters Prep 500 LC обычно проводят, используя линейный градиент растворителей, указанных в тексте. Завершенность протекания реакций контролируют, используя тонкослойную хроматографию (ТСХ). Тонкослойную хроматографию проводят с использованием пластин Е. Merck Kieselgel 60F254, 5 см 10 см, толщина 0,25 мм. Пятна детектируют с использованием комбинации УФ и химического определения (пластины погружают в раствор, содержащий соль церия и молибдат аммония [75 г молибдата аммония и 4 г сульфата церия (IV) в 500 мл 10% водного раствора серной кислоты], и затем нагревают на горячей пластине). Флэш-хроматографию осуществляют, как описано в работе Still, et al. Still,Kahn, Mitra, J. Org. Chem., 43, 2923 (1978). Элементный анализ на углерод, водород и азот осуществляют на Control Equipment Corporation 440 Elemental Analyzer или проводят в Universidad ComplutenseAnalytical Centre (Facultad de Farmacia, Madrid, Spain). Температуры плавления определяют в открытых стеклянных капиллярах на приборе для определения температуры плавления с горячевоздушной банейGallenkamp или на приборе для определения температуры плавления Bchi, и они являются приблизительными. Сокращения, символы и термины, используемые в примерах, имеют следующие значения. Ас=ацетил;EDC сочетание между аминами и N-Boc-(L)-аминокислотами. Исходный сложный аминодиалкильный эфир (соединение формулы (ii), схема 3) (1,0 экв.) суспендируют в сухом дихлорметане под атмосферой азота. Последовательно добавляют соответствующую NBoc-(L)-аминокислоту (1,5-2,0 экв.), EDC (1,5-2,0 экв.), HOBt (1,5-2,0 экв.) и диметиламинопиридин(ДМАП, 0,1-0,2 экв.). Реакционную смесь перемешивают при комнатной температуре до завершения реакции, о чем судят по данным ТСХ, если это не указано иным образом. Реакционную смесь разбавляют этилацетатом и последовательно промывают насыщенным водным раствором NaHCO3 и/или водным раствором NaHSO4 и насыщенным раствором соли. После сушки над сульфатом натрия и выпаривания под вакуумом неочищенный остаток (соединение формулы (iii очищают хроматографией на силикагеле, используя соответствующий элюент (типично смесь этилацетат/гексан). Общая процедура В. Ангидридное сочетание между амином и изобутильными ангидридами N-Boc-(L)-аминокислоты. К раствору соответствующей N-Boc-(L)-аминокислоты (1,5 экв.) в сухом дихлорметане (10 мл) при-20 С под атмосферой азота добавляют N-метилморфолин (NMM, 1,5 экв., в 1 мл CH2Cl2), после чего по каплям добавляют изобутилхлорформиат (ИБХФ, 1,5 экв., в 5 мл CH2Cl2) с такой скоростью, чтобы- 10014979 внутренняя температура реакционной смеси не превышала -15 С. Полученную в результате реакционную смесь перемешивают при -20 С в течение 30 мин, затем добавляют раствор гидрохлорида диэтилового эфира(1S,2S,4S,5R,6R)-2-(2'-аминопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (1,0 экв.) в дихлорметане (10 мл) при -20 С с такой скоростью, чтобы внутренняя температура реакционной смеси не превышала -15 С. После завершения добавления убирают охлаждающую баню и реакционную смесь перемешивают при комнатной температуре до завершения реакции по данным ТСХ. Реакционную смесь разбавляют этилацетатом и последовательно промывают насыщенным водным раствором NaHCO3, водным раствором NaHSO4 и насыщенным раствором соли. После сушки над сульфатом магния и выпариванием под вакуумом неочищенный остаток очищают хроматографией на силикагеле, используя соответствующий элюент (типично смесь гексан/этилацетат). Общая процедура С. Последовательное удаление N-Boc защитной группы и защищающей сложноэфирной группы. Соответствующее N-Boc диэфирное пептидное производное (соединение формулы (iii), схема 2)(1,0 экв.) перемешивают в смеси 1:1 ТГФ/2,5 н LiOH (10-20 экв.) при комнатной температуре в течение 4 ч. Реакционную смесь разбавляют H2O и промывают этилацетатом. Органический слой сливают. рН водной фазы доводят до 2, используя 1 н. HCl (к водной фазе добавляют NaCl для увеличения способности к экстракции до необходимого уровня) и полностью экстрагируют продукт, представляющий собойN-Boc дикарбоновую кислоту (соединение формулы (iv этилацетатом. Все органические слои объединяют, промывают насыщенным раствором соли, сушат над MgSO4 и концентрируют до сухого состояния под вакуумом, получая желаемый карбоксилатный продукт в виде твердого пенообразного вещества. Растворяют в этилацетате и охлаждают до 0 С. Реакционную смесь продувают безводным газообразнымHCl до насыщения HCl. Полученную в результате реакционную смесь перемешивают при 0 С в течение 4 ч. Выделяют пептидное производное с полностью снятой защитой (соединение формулы (I в виде его гидрохлоридной соли фильтрованием под атмосферой N2 или концентрированием реакционной смеси до сухого состояния, после чего следует растирание в этилацетате или Et2O и концентрирование до получения белого порошка. Необязательно, для удаления остаточного количества растворителя и избытка HCl,повторно разводят продукты в H2O, замораживают и затем подвергают лиофилизации, получая желаемые гидрохлоридные продукты. Метод получения 1. Диэтиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты К суспензии (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты (10 г, 42,5 ммоль, патент США 5688826) в 100 мл 2 В этанола при комнатной температуре по каплям в течение 20 мин добавляют тионилхлорид (15,5 мл, 212,6 ммоль), после чего следует промывка 40 мл этанола. Суспензию кипятят с обратным холодильником и перемешивают в течение ночи. Анализ 500 МГц 1 Н ЯМР (CD3OD) концентрированной аликвоты показывает расходование исходного вещества и промежуточного моноэфира. Полученному в результате раствору дают охладиться до комнатной температуры, затем концентрируют до желатинообразного остатка. К желатинообразному веществу добавляют EtOAc (50 мл) и далее концентрируют до твердого вещества, затем разбавляют дополнительными 94 мл EtOAc. К смеси медленно добавляют 1,5% водный раствор карбоната натрия (70 мл) при вращении вручную, чтобы постепенно способствовать растворению, получая конечное значение рН 7,95. Отфильтровывают образовавшийся в результате осадок карбоната натрия, после чего экстрагируют слои. Осуществляют обратную экстракцию водного слоя EtOAc (2100 мл). Объединенные органические экстракты промывают насыщенным раствором соли (1100 мл), сушат (MgSO4), фильтруют и концентрируют под вакуумом, получая бледно-желтое маслянистое вещество, которое затвердевает, давая указанное в заголовке соединение в виде твердого, не совсем белого вещества (11,71 г, выход 95%). Перекристаллизация. Смесь указанного в заголовке соединения (200 мг) в EtOAc (800 мкл) нагревают до 56 С, и в данный момент происходит растворение. После перемешивания в течение 15 мин при 56 С к раствору по каплям добавляют гептан (1 мл). Нагрев выключают. Раствору дают охладиться до 52 С, и в данный момент происходит выпадение осадка. После охлаждения и дополнительного разбавления гептаном(600 мкл) образуется суспензия. Полученную в результате суспензию перемешивают при комнатной температуре в течение 1 ч, после чего фильтруют, промывают гептаном (2500 мкл) и сушат при 45 С в течение ночи, получая 145 мг (выход 73%) указанного в заголовке соединения в виде твердого белого вещества. Тпл.=80-83 С. К раствору N-Boc-L-аланина (43,52 г, 230 ммоль) и N-метилморфолина (25,5 мл, 232 ммоль) в 457 мл метиленхлорида при -30 С под атмосферой азота по каплям в течение 10 мин добавляют изобутилхлорформиат (30,4 мл, 234 ммоль). Полученную в результате жидкую суспензию перемешивают при температуре от -25 до -30 С в течение 30 мин, после чего в течение 25 мин добавляют раствор диэтилового эфира (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты(63,90 г, 219 ммоль, метод получения 1) в 213 мл метиленхлорида так, чтобы температура реакционной смеси не превышала -25 С. После завершения добавления убирают охлаждающую баню и перемешивают при комнатной температуре в течение 60 мин, после чего температура реакционной смеси достигает 19 С и цвет становится бледно-оранжевым. Реакционную смесь обрабатывают 350 мл 1 н. HCl и разделяют слои. Органический слой промывают насыщенным водным раствором NaHCO3 (1350 мл) и насыщенным раствором соли (1350 мл), сушат (Na2SO4), фильтруют и концентрируют под вакуумом, получая белое пенообразное вещество (105,2 г, 104%). 1 Н ЯМР (300 МГц, CDCl3)7,62 (ушир.с, 1 Н), 4,90 (ушир.д, 1 Н, J=7,1 Гц), 4,34-4,10 (м, 6 Н), 3,39 К раствору диэтилового эфира (1R,4S,5S,6S)-4-(2'S-трет-бутоксикарбониламинопропиониламино)2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты (181,4 г, 392 ммоль теоретических,метод получения 2) в 292 мл ТГФ при комнатной температуре добавляют 490 мл (980 ммоль) 2 н. раствора гидроксида натрия. Двухфазную смесь энергично перемешивают при комнатной температуре в течение 1,25 ч, после чего реакционная смесь становится гомогенной. Смесь разбавляют 490 мл этилацетата и слои разделяют. Водный слой разбавляют 490 мл этилацетата и рН смеси понижают до 1,5 концентрированной HCl. Разделяют слои и проводят обратную экстракцию водного слоя 245 мл этилацетата. Объединенные органические слои сушат (Na2SO4), фильтруют и концентрируют, получая 167,9 г (105%) указанного в заголовке соединения в виде белого пенообразного вещества. Данное вещество используют без характеризации в примерах 1 и 2.- 12014979 Метод получения 4. Диметиловый эфир дикарбоновой кислоты(1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6 К быстро перемешиваемой суспензии (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты (10,0 г, 42,5 ммоль, патент США 5688826) в МеОН (170 мл, 5 С) по каплям добавляют тионилхлорид (6,2 мл, 85,0 ммоль). После завершения добавления реакционной смеси дают медленно нагреться до комнатной температуры, затем кипятят с обратным холодильником в течение 48 ч. Удаляют летучие компоненты при пониженном давлении и остаток распределяют между насыщенным раствором NaHCO3 (200 мл) и этилацетатом (400 мл). Слои разделяют и водный слой экстрагируют этилацетатом (2400 мл, каждый раз). Объединенные органические слои сушат над K2CO3 и концентрируют при пониженном давлении, получая 8,10 г (30,8 ммоль) указанного в заголовке соединения с выходом 72%. Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-Boc-(L)фенилаланин и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6 дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Выход 0,85 г (88%, 1,67 ммоль) белого пенообразного вещества. Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-Boc-(L)изолейцин и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6 дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-Boc-(L)-валин и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Выход 0,41 г (47%, 0,89 ммоль) белого пенообразного вещества. Получают в соответствии с общей процедурой А, используя имеющийся в продаже моногидрат NBoc-(L)-лейцина и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан 4,6-дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Выход 0,85 г (94%, 1,78 ммоль) белого пенообразного вещества. Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-BocLys(Boc)-ОН и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6 дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Выход 1,04 г (93%, 1,76 ммоль) белого пенообразного вещества. Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-Boc-(L)глутамин(Trt)-ОН и диметиловый эфир (1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан 4,6-дикарбоновой кислоты (метод получения 4). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают ПЦ-ТСХ, 4 мм SiO2 ротор (от смеси 10% этилацетат/гексан до 100% этилацетата). Выход 0,53 г (48%, 0,72 ммоль) белого пенообразного вещества.N 5,57. МС(ЭР) m/z 731,9 [М-Н]-. МСВР вычислено для C38H43N3O10S [M+Na]+, 756,2567. Найдено, 756,2585.[]23D=-19,6 (c=0,51, CHCl3). Элементный анализ. Вычислено для C30H39N3O11S1,0 С 4 Н 8 О 2: С 55,35; Н 6,42; N 5,70. Найдено: С 54,98; Н 6,09; N 6,07. МСВР вычислено для C30H39N3O11NaS, 672,2203. Найдено, 672,2180. Метод получения 14. Диметиловый эфир (1R,4S,5S,6S)-4-[2'S-трет-бутоксикарбониламино-3'-(4-трет-бутоксикарбонилоксифенил)пропиониламино]-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты(1 Н, д, J=14,3 Гц), 4,22-4,29 (1 Н, каж. кв., J=7,3 Гц), 4,92 (1 Н, ушир.д, J=7,7 Гц), 7,07 (1 Н, ушир.с), 7,17,26 (4 Н, м). МСВР вычислено для C28H38N2O12SNa, 649,2043. Найдено, 649,2001. Метод получения 15. Диметиловый эфир (1R,4S,5S,65)-4-(3'-ацетокси-2'S-трет-бутоксикарбониламинопропионил)амино 2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой кислоты(0,2 г,(1R,4S,5S,6S)-4-амино-2,2-диоксо-26-тиабицикло[3.1.0]гексан-4,6-дикарбоновой 0,8 ммоль, метод получения 4), за исключением того, что ДМАП не используют. Очищают, используя ПЦ-ТСХ (этилацетат/гексан), получая 0,19 г (48,2%) указанного в заголовке соединения.(1 Н, дд, J=4,0, 7,3 Гц), 3,77 (3 Н, с), 3,86 (3 Н, с), 4,14-4,40 (4 Н, м), 5,29 (1 Н, ушир.д, J=7,3 Гц), 7,64 (1 Н,ушир.с). МСВР вычислено для C19H28N2O11SNa, 515,1312. Найдено, 515,1305. Метод получения 16. Диэтиловый эфир (1R,2S,4R,5R,6R)-2-амино-4-фторбицикло[3.1.0]гексан-2,6-дикарбоновой кислоты(14,45 г, 71,12 ммоль, патент США 5958960) в 202 мл абсолютного этанола при комнатной температуре в течение 20 мин по каплям добавляют тионилхлорид (26 мл, 356 ммоль). Суспензию кипятят с обратным холодильником при перемешивании в течение 3 ч. Охлаждают до комнатной температуры и пере- 17014979 мешивают в течение ночи. Концентрируют полученный в результате раствор под вакуумом до получения остатка, затем разбавляют 136 мл этилацетата и обрабатывают 306 мл 10% водного раствора карбоната натрия в течение 15 мин при вращении вручную, так что конечная рН равна 10. Слои разделяют и водный слой промывают этилацетатом (1136 мл). Объединенные органические экстракты промывают насыщенным раствором соли (1136 мл), сушат (MgSO4), фильтруют и концентрируют под вакуумом, получая 17,07 г (93%) указанного в заголовке соединения в виде твердого белого вещества. К раствору N-Boc-L-аланина (38,62 г, 204 ммоль) в 396 мл метиленхлорида при -22 С под атмосферой азота добавляют N-метилморфолин (22,44 мл, 204 ммоль), после чего по каплям в течение 15 мин добавляют изобутилхлорформиат (26,48 мл, 204 ммоль) так, чтобы температура реакционной смеси не превышала -18 С. Полученную в результате жидкую суспензию перемешивают при -20 С в течение 30 мин, после чего в течение 40 мин добавляют раствор диэтилового эфира (1R,2S,4R,5R,6R)-2-амино-4 фторбицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (49,46 г, 191 ммоль, метод получения 16) в 247 мл метиленхлорида так, чтобы температура реакционной смеси не превышала -16 С. После завершения добавления реакционную смесь удаляют из охлаждающей бани и перемешивают при комнатной температуре в течение 70 мин, после чего температура реакционной смеси достигает 15 С и цвет становится бледно-оранжевым. Реакционную смесь обрабатывают 408 мл 1 н. HCl и перемешивают в течение 5 мин,затем слои разделяют. Органический слой промывают насыщенным водным раствором бикарбоната натрия (1408 мл), сушат (Na2SO4), фильтруют и концентрируют под вакуумом, получая белое пенообразное вещество (88,16 г). Элементный анализ. Вычислено для C20H31FN2O70,1CH2Cl2: С 55,00; Н 7,16; N 6,38. Найдено: С 55,18; Н 7,18; N 6,49. МС(ЭР) m/z 431,3 [М+Н]+, 331,2 [М+Н-Вос]+. Метод получения 18.(1R,2S,4R,5R,6R)-2-[2'S-(трет-бутоксикарбониламино)пропионил]амино-4-фторбицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (88,16 г, 191 ммоль, метод получения 17) в 238 мл ТГФ при комнатной температуре добавляют 238 мл (477 ммоль) 2 н. гидроксида натрия. Двухфазную смесь энергично перемешивают при комнатной температуре в течение 2,5 ч, после чего реакционная смесь становится гомогенной. Смесь разбавляют 238 мл трет-бутилметилового эфира,после чего следует перемешивание и разделение слоев. Водный слой дополнительно разбавляют 238 мл воды и фильтруют, удаляя порошкообразное вещество. Раствор обрабатывают концентрированной HCl(42,9 мл, 515 ммоль) в течение 30 мин, после чего необязательно следует внесение затравки указанного в заголовке соединения и перемешивание в течение 1 ч. Полученную в результате суспензию фильтруют,промывают водой (2100 мл) и подвергают вакуумной сушке при 45 С в течение 40 ч, получая 72,2 г- 18014979 указанного в заголовке соединения в виде твердого белого вещества. Перемешивают часть твердого вещества (69,5 г) с 490 мл ацетона в течение 1 ч, получая мутный раствор, фильтруют и промывают ацетоном (2100 мл). Фильтрат концентрируют под вакуумом до получения белого пенообразного вещества,которое далее сушат под вакуумом при 45 С в течение 16 ч, получая 61,8 г (86% скорректированных с учетом 12 мас.%/мас. ацетона) указанного в заголовке соединения. Данное вещество используют в примерах 14-18 без характеризации. Метод получения 19. Диэтиловый эфир (1S,2S,4S,5R,6R)-4-ацетилокси-2-(трет-бутоксикарбонил)аминобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты К раствору диэтилового эфира (1S,2S,4S,5R,6R)-2-(трет-бутоксикарбонил)амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (1,50 г, 4,20 ммоль, патент США 5958960), пиридина(0,365 мл, 4,62 ммоль) и ДМАП (0,513 г, 4,20 ммоль) в дихлорметане (40 мл) под атмосферой азота добавляют уксусный ангидрид (0,514 мл, 5,0 ммоль). Перемешивают при комнатной температуре в течение 16 ч, разбавляют дихлорметаном и выливают в 10% водный раствор лимонной кислоты (50 мл). Органический слой промывают водой (50 мл) и насыщенным раствором соли (50 мл). Сушат над MgSO4, фильтруют и концентрируют под вакуумом, получая указанное в заголовке соединение в виде твердого белого вещества (1,295 г, 75%). ЖХМС: m/z 400 [М+Н]+ и m/z 300 [М+Н-CO2-трет-Bu]+RT 1,39 мин. Метод получения 20. Диэтиловый эфир (1S,2S,4S,5R,6R)-4-ацетилокси-2-аминобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Растворяют диэтиловый эфир (1S,2S,4S,5R,6R)-4-ацетилокси-2-(трет-бутоксикарбонил)аминобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (1,25 г, 3,13 ммоль, метод получения 19) в растворе 95% ТФУ в дихлорметане (60 мл) и перемешивают под атмосферой азота при комнатной температуре в течение 5 мин. После этого удаляют смесь ТФУ/дихлорметан под вакуумом. Неочищенный продукт растворяют в суспензии NaHCO3 (1,00 г) в дихлорметане (50 мл) и перемешивают в течение 30 мин. Суспензию фильтруют, промывают дихлорметаном (325 мл) и концентрируют под вакуумом, получая 916 мг (98%) продукта в виде желтого маслянистого вещества. ЖХМС: m/z 300 [М+Н]+RT 0,92 мин. Метод получения 21. Диэтиловый эфир (1S,2S,4S,5R,6R)-4-ацетилокси-2-[2'S-(трет-бутокси)карбониламинопропионил]аминобицикло[3.1.0]гексан-2,6-дикарбоновой кислоты К суспензии диэтилового эфира (1S,2S,4S,5R,6R)-4-ацетилокси-2-аминобицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (0,80 г, 1,94 ммоль, метод получения 20) и Boc-L-Ala (0,477 г, 2,52 ммоль) в безводном дихлорметане (50 мл) под атмосферой азота последовательно добавляют EDC (0,519 г,2,72 ммоль), HOBt (0,314 г, 2,32 ммоль), каталитическое количество ДМАП (0,024 г, 0,19 ммоль) и триэтиламин (1,08 мл, 7,76 ммоль). После перемешивания при комнатной температуре в течение примерно 15 мин исходная белая суспензия полностью растворяется. Реакционную смесь перемешивают в течение 16 ч, разбавляют дихлорметаном и промывают насыщенным водным раствором NaHCO3 (50 мл), 1,0 н. водным раствором HCl (320 мл) и насыщенным раствором соли (40 мл). Органический слой сушат надMgSO4, фильтруют и концентрируют под вакуумом. Полученный в результате неочищенный амид очищают колоночной хроматографией, используя смесь (4:1) этилацетата и гексана в качестве элюента, получая 638 мг (75%) продукта в виде твердого белого вещества. Раствор диэтилового эфира (1S,2S,4S,5R,6R)-2-трет-бутоксикарбониламино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (5,0 г, 14,0 ммоль, патент США 5958960) в этилацетате охлаждают до 0 С. Через раствор барботируют безводный HCl (газ.) до насыщения и перемешивают в течение 0,5 ч. Реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 1 ч. Реакционную смесь концентрируют и распределяют между этилацетатом и H2O. Водный слой обрабатывают NaHCO3 (водн.) и экстрагируют этилацетатом. Органические слои сушат над K2CO3 и концентрируют, получая 2,2 г (59,7%) твердого белого вещества.- 20014979 Получают в соответствии с общей процедурой А, используя Boc-L-валин (569 мг, 2,62 ммоль,Sigma) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (450 мг, 1,75 ммоль, метод получения 22). Неочищенное вещество очищают на 35 г силикагеля; элюируют смесью гексан/этилацетат с градиентом от 70/30 до 20/80. Выход: 694 мг (87%) белого пенообразного вещества. 1 Н ЯМР (400 МГц, CDCl3)0,95 (3 Н, д, J=6,8 Гц), 1,00 (3 Н, д, J=6,8 Гц), 1,29 (6 Н, т, J=7,3 Гц), 1,45(1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (450 мг, 1,75 ммоль, метод получения 22). Неочищенное вещество очищают на 35 г силикагеля; элюируют смесью гексан/этилацетат с градиентом от 70/30 до 20/80. Выход: 689 мг (84%) белого пенообразного вещества. 1 Н ЯМР (400 МГц, CDCl3)0,96 (6 Н, м), 1,28 (3 Н, т, J=7,3 Гц), 1,29 (3 Н, т, J=7,3 Гц), 1,46 (9 Н, с),1,45-1,73 (5 Н, м), 2,20 (1 Н, м), 2,42 (1 Н, м), 2,80 (1 Н, д, J=16,1 Гц), 4,07-4,35 (7 Н, м), 4,81 (1 Н, ушир.д),6,86 (1 Н, ушир.с). Элементный анализ. Вычислено для C23H38N2O80,1H2O: С 58,48; Н 8,15; N 5,93. Найдено: С 58,22; Н 7,94; N 5,92. МС(ЭР) m/z 471,2 [М+Н]+, 493,2 [M+Na]+. Метод получения 26. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-(2'S-трет-бутоксикарбониламино-3'S-метилпентаноиламино)4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Получают в соответствии с общей процедурой А, используя Boc-L-изолейцин (606 мг, 2,62 ммоль,Aldrich) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (450 мг, 1,75 ммоль, метод получения 22). Неочищенное вещество очищают на 35 г силикагеля,элюируя смесью гексан/этилацетат с градиентом от 70/30 до 20/80. Выход: 731 мг (89%). 1 Н ЯМР (400 МГц, CDCl3)0,92 (3 Н, т, J=7,3 Гц), 0,97 (3 Н, д, J=6,8 Гц), 1,17 (1 Н, м), 1,29 (6 Н, т,J=6,8 Гц), 1,46 (9 Н, с), 1,53 (1 Н, дд, J=5,8, 15,6 Гц), 1,58 (1 Н, т, J=2,4 Гц), 1,74 (1 Н, м), 1,83 (1 Н, м), 2,20(1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (400 мг, 1,36 ммоль, метод получения 22 без обработки основанием) за исключением нижеследующего. ДМАП не используют. Добавляют один эквивалент триэтиламина. Неочищенное вещество очищают на 35 г силикагеля; элюируют этилацетатом. Выход: 517 мг (81%).(9 Н, с), 1,59 (1 Н, т, J=3,4 Гц), 1,62 (1 Н, дд, J=6,3, 16,1 Гц), 2,21 (1 Н, дд, J=2,9, 5,8 Гц), 2,46 (1 Н, дд, J=2,4,5,8 Гц), 2,73 (1 Н, д, J=15,6 Гц), 3,78-3,91 (3 Н, м), 4,01-4,15 (3 Н, м), 4,24 (2 Н, кв., J=6,8 Гц), 4,32 (1 Н, д,J=5,8 Гц), 5,28 (1 Н, ушир.), 6,87 (1 Н, ушир.т, J=4,9 Гц), 7,39 (1 Н, ушир.с). МСВР вычислено для C21H34N3O9, 472,2295. Найдено, 472,2303. ВЭЖХ: 16,755 мин. Колонка: Symmetry C18, 3,5 мкм, 4,6150 мм. =230 нМ. Скорость потока: 1 мл/мин. Градиент: от 10 до 50% акрилонитрил/вода, содержащая 0,1% ТФУ в течение 25 мин. Метод получения 28. Диэтиловый эфир(1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (400 мг, 1,36 ммоль, метод получения 22 без обработки основанием) за исключением нижеследующего. ДМАП не используют. Добавляют один эквивалент триэтиламина. Неочищенное вещество очищают на 35 г силикагеля, элюируя этилацетатом. Выход: 500 мг (76%).(1 Н, м), 4,07-4,16 (3 Н, м), 4,24 (2 Н, кв., J=7,3 Гц), 4,32 (1 Н, ушир.), 5,08 (1 Н, ушир.), 6,84 (1 Н, ушир.т,J=4,88 Гц), 7,14 (1 Н, ушир.с). МСВР вычислено для C22H36M3O9, 486,2452. Найдено, 486,2451. Метод получения 29. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-(2'-трет-бутоксикарбониламино-3'-фенилпропиониламино)-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Получают в соответствии с общей процедурой А, используя Boc-L-фенилаланин (772 мг,2,91 ммоль) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (480 мг, 1,94 ммоль, метод получения 22). Неочищенную реакционную смесь концентрируют, растворяют в дихлорметане, подвергают флэш-хроматографии через короткий слой силикагеля с использованием смеси этилацетат/ДХМ (1:1) и концентрируют под вакуумом, получая желтое маслянистое вещество. Дополнительно очищают вещество хроматографией на силикагеле, элюируя смесью с градиентом от 30% этилацетат/гексан до 80% этилацетат/гексан, получая 852 мг (87%) указанного в заголовке соединения.[]23D=-23,66 (с=0,93, МеОН). 1 Н ЯМР (400 МГц, CD3OD)1,22-1,30 (6 Н, м), 1,35 (9 Н, с), 1,59 (1 Н, т, J=2,9 Гц), 1,70 (1 Н, дд,J=5,5, 15,4 Гц), 2,06 (1 Н, м), 2,40 (1 Н, д, J=15,4 Гц), 2,63 (1 Н, дд, J=2,9, 5,9 Гц), 2,76 (1 Н, дд, J=9,2, 13,9 Гц), 4,08-4,33 (6 Н, м), 7,20-7,29 (5 Н, м). МС найдено 505,0 [М+Н]+. МСВР вычислено для C26H36N2O8, 505,2550. Найдено, 505,2559. Метод получения 30. Диэтиловый эфир Получают в соответствии с общей процедурой А, используя Boc-L-глутамин(тритил)-ОН (1,40 г,2,86 ммоль) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (490 мг, 1,90 ммоль, метод получения 22) за исключением, что ДМАП не используют. Неочищенную реакционную смесь концентрируют, растворяют в дихлорметане, подвергают флэшхроматографии через короткий слой силикагеля с использованием этилацетата и концентрируют под вакуумом, получая желтое маслянистое вещество. Дополнительно очищают вещество хроматографией на силикагеле, элюируя градиентом от 30% этилацетат/гексан до 100% этилацетат/гексан, получая 1,3 г(1 Н, м), 2,38-2,48 (3 Н, м), 2,54 (1 Н, м), 4,00-4,27 (6 Н, м), 7,18-7,30 (15 Н, м). МС найдено 728,2 [М+Н]+. МСВР вычислено для C41H49N3O9, 728,3547. Найдено, 728,3533. Метод получения 31. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-(2'S,6'-бис-трет-бутоксикарбониламиногексаноиламино)-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Получают в соответствии с общей процедурой А, используя Boc-L-лизин(Boc)-ОН (910 мг,- 23014979 2,63 ммоль) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (450 мг, 1,75 ммоль, метод получения 22) за исключением того, что к реакционной смеси не добавляют ДМАП. Неочищенную реакционную смесь концентрируют, растворяют в дихлорметане, подвергают флэш-хроматографии через короткий слой силикагеля с использованием этилацетата и концентрируют под вакуумом, получая желтое маслянистое вещество. Дополнительно очищают вещество хроматографией на силикагеле, элюируя смесью этилацетат/гексан (1:1), получая 800 мг[]23D=-30,19 (c=0,53, МеОН). 1 Н ЯМР (400 МГц, CD3OD)1,21-1,24 (6 Н, м), 1,26-1,77 (27 Н, м), 2,07 (1 Н, м), 2,42 (1 Н, д,J=15,8 Гц), 2,59 (1 Н, дд, J=2,6, 5,5 Гц), 3,03 (1 Н, т, J=6,2 Гц), 3,99 (1 Н, м), 4,07-4,28 (5 Н, м). МС найдено 586,1 [М+Н]+. МСВР вычислено для C28H47N3O10, 586,3340. Найдено, 586,3348. Метод получения 32. Диэтиловый эфир Получают в соответствии с общей процедурой А, используя Boc-L-аспарагин(тритил)-ОН (1,35 г,2,84 ммоль) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (476 мг, 1,85 ммоль, метод получения 22) за исключением того, что к реакционной смеси не добавляют ДМАП. Неочищенную реакционную смесь концентрируют, растворяют в дихлорметане, подвергают флэш-хроматографии через короткий слой силикагеля с использованием этилацетата и концентрируют под вакуумом, получая желтое маслянистое вещество. Дополнительно очищают вещество хроматографией на силикагеле, элюируя градиентом от 20% этилацетат/гексан до 50% этилацетат/гексан, получая 1,07 г (81%) указанного в заголовке соединения.[]23D=-25,45 (c=0,55, МеОН). 1 Н ЯМР (400 МГц, CD3OD)1,19-1,26 (6 Н, м), 1,45 (9 Н, с), 1,61-1,69 (2 Н, м), 2,05 (1 Н, м), 2,44 (1 Н,д, J=15,4 Гц), 2,51-2,72 (3 Н, м), 4,02-4,21 (4 Н, м), 4,25 (1 Н, д, J=5,5 Гц), 4,36 (1 Н, дд, J=4,8, 8,8 Гц), 7,187,30 (15 Н, м). МС найдено 714,1 [М+Н]+. МСВР вычислено для C40H47N3O9, 714,3391. Найдено, 714,3380. Метод получения 33. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-[2'-трет-бутоксикарбониламино-3'-(1'-трет-бутоксикарбонил 1 Н-индол-3'-ил)пропиониламино]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Получают в соответствии с общей процедурой А, используя Boc-L-триптофан(Boc)-ОН (1,18 г,2,91 ммоль) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (520 мг, 1,90 ммоль, метод получения 22). Неочищенную реакционную смесь концентрируют, растворяют в дихлорметане, подвергают флэш-хроматографии через короткий слой силикагеля с использованием этилацетата и концентрируют под вакуумом, получая желтое маслянистое вещество. Дополнительно очищают вещество хроматографией на силикагеле, элюируя смесью этилацетат/ДХМ (1:1), получая 1,2 г (92%) указанного в заголовке соединения.(1 Н, дд, J=5,9, 15,2 Гц), 2,07 (1 Н, м), 2,46 (1 Н, д, J=15,7 Гц), 2,60 (1 Н, д, J=2,9, 5,9 Гц), 2,89 (1 Н, дд, J=9,3,15,0 Гц), 3,16 (1 Н, дд, J=4,9, 15,2 Гц), 4,04-4,12 (2 Н, м), 4,18-4,24 (2 Н, м), 4,27 (1 Н, д, J=5,4 Гц), 4,43 (1 Н,дд, J=5,4, 9,3 Гц), 7,26 (2 Н, м), 7,52 (1 Н, с), 7,64 (1 Н, д, J=7,3 Гц), 8,10 (1 Н, д, J=8, 3 Гц). МС найдено 644,8 [М+Н]+, 666,8 [M+Na]+. МСВР вычислено для C33H44N3O10, 666,3002. Найдено, 666,2988.(1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (метод получения 22). Неочищенное вещество очищают на 110 г силикагеля на системе ISCO, элюируя градиентом от 80% этилацетат/гексан до 100% этилацетата, получая 699 мг (84%) указанного в заголовке соединения.(1 Н, м), 1,97 (1 Н, м), 2,07 (4 Н, м), 2,41 (1 Н, д, J=15,3 Гц), 2,50 (1 Н, м), 2,59 (1 Н, м), 4,12 (2 Н, м), 4,20 (1 Н,кв., J=7,4 Гц), 4,27 (1 Н, д, J=5,4 Гц). МС найдено 454,9 [М+Н]+, 476,8 [M+Na]+. МСВР вычислено для C22H34N2O8Na, 477,2213. Найдено, 477,2210. Метод получения 35. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-[2'S-трет-бутоксикарбониламино-3'-(4-трет-бутоксикарбонилоксифенил)пропиониламино]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Указанное в заголовке соединение получают в соответствии с общей процедурой А, используя 2Sтрет-бутоксикарбониламино-3-(4-трет-бутоксикарбонилоксифенил)пропионовую кислоту и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (метод получения 22). Неочищенное вещество очищают на 110 г силикагеля на системе ISCO, элюируя градиентом от 60% этилацетат/гексан до 90% этилацетат/гексан, получая 1,09 г (91%) указанного в заголовке соединения.(6 Н, м), 7,04 (2 Н, д, J=8,4 Гц), 7,26 (2 Н, д, J=8,4 Гц). МС найдено 621,8 [М+Н]+, 643,8 [M+Na]+. МСВР вычислено для C31H44N2O11Na, 643,2843. Найдено, 643,2845. Метод получения 36. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-(2'S-трет-бутоксикарбониламино-4'-метилсульфанилбутириламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты- 26014979 Указанное в заголовке соединение получают в соответствии с общей процедурой А, используя(Boc-L-метионин) и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6 дикарбоновой кислоты (метод получения 22). Неочищенное вещество очищают на 110 г силикагеля на системе ISCO, элюируя градиентом от 80% этилацетат/гексан до 100% этилацетата, получая 1,0 г (92%) указанного в заголовке соединения.(1 Н, м), 2,61 (1 Н, м), 3,37 (1 Н, м), 2,49 (1 Н, м), 4,2 (6 Н, м). МС найдено 488,8 [М+Н]+, 510,8 [M+Na]+. МСВР вычислено для C22H36N2O8SNa, 511,2090. Найдено, 511,2071. Метод получения 37. Диэтиловый эфир Получают в соответствии с общей процедурой А, используя имеющийся в продаже N-Boc-(L)аланин и диэтиловый эфир (1S,2S,4S,5R,6R)-2-амино-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (метод получения 22). Реакционную смесь кипятят с обратным холодильником в течение ночи. Очищают препаративной ВЭЖХ, 1500 г SiO2 колонка (от 10% этилацетат/гексан до 100% этилацетата). Выход 3,0 г (90%, 7,00 ммоль) белого пенообразного вещества.(6 Н, м), 4,92 (1 Н, ушир.с), 6, 94 (1 Н, ушир.с). Элементный анализ. Вычислено для C2OH32N2O80,1H2O: С 55,83; Н 7,54; N 6,51. Найдено: С 55,57; Н 7,64; N 6,44. МС(ЭР) m/z 429,2 [М+Н]+, 329,1 [М-Вос]+. Метод получения 38. Гидрохлорид диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'S-аминопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Через раствор диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'S-трет-бутоксикарбониламинопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (2,95 г, 6,88 ммоль, метод получения 37) в этилацетате (30 мл) при 0 С пропускают безводный газообразный HCl, пока раствор не будет насыщен HCl. Полученную в результате реакционную смесь перемешивают при 0 С, пока не будет сделан вывод о завершении реакции по данным ТСХ. Через реакционную смесь в течение 30 мин продувают N2 для удаления газообразного HCl. Полученную в результате суспензию концентрируют под вакуумом до сухого состояния, получая 2,47 г (98%, 6,78 ммоль) желаемую гидрохлоридную соль амина. Никакая дальнейшая очистка не требуется. Получают в соответствии с общей процедурой А, используя гидрохлорид диэтилового эфира(3 Н, д, J=7, 0 Гц), 1,44 (9 Н, с), 1,58-1,65 (2 Н, м), 2,19 (1 Н, дд, J=3,0, 5,9 Гц), 2,46 (1 Н, дд, J=2,6, 5,9 Гц),2,70 (1H, д, J=15,4 Гц), 4,09-4,33 (7H, м), 4,48 (1 Н, каж. р, J=7, 0 Гц), 5,05 (1 Н, ушир.д, J=6,6 Гц), 6,79 (1 Н,ушир.д, J=7,7 Гц), 7,26 (1 Н, с). МСВР вычислено для С 23 Н 38 Н 3 О 9, 500,2608. Найдено, 500,2598. Метод получения 40. Диэтиловый эфир Получают в соответствии с общей процедурой А, используя Вос-глицин (0,29 г, 1,6 ммоль) и гидрохлорид диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'S-аминопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,4 г, 1,1 ммоль, метод получения 38) за исключением того, что не используют ДМАП. Очищают, используя ПЦ-ТСХ (смесь этилацетат/гексан), получая 0,14 г (26,2%) указанного в заголовке соединения.(9 Н, с), 1,57-1,65 (2 Н, м), 2,19 (1 Н, дд, J=3,3, 5,9 Гц), 2,44 (1 Н, дд, J=2,9, 5,9 Гц), 2,74 (1 Н, д, J=15,8 Гц),3,70-3,86 (2 Н, м), 4,08-4,34 (6 Н, м), 4,56 (1 Н, каж. р, J=7,0 Гц), 5,31 (1 Н, ушир.с), 6,88 (1 Н, ушир.д, J=7,0 Гц), 7,50 (1 Н, с). МСВР вычислено для C22H36N3O9, 486,2452. Найдено, 486,2444. Метод получения 41. Диэтиловый эфир Получают в соответствии с общей процедурой В, используя имеющийся в продаже моногидрат NBoc-(L)-лейцина и гидрохлорид диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'S-аминопропиониламино)-4 гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,2 г, 0,55 ммоль, метод получения 38). Очи- 28014979 щают ПЦ-ТСХ, 4 мм SiO2 ротор (от 10% этилацетат/гексан до 100% этилацетата). Выход 0,54 г (62%, 1,00 ммоль) белого пенообразного вещества.(3 Н, т, J=7, 3 Гц), 1,36 (3 Н, д, J=7,0 Гц), 1,43 (9 Н, с), 1,53 (1 Н, д, J=9,5 Гц), 1,55-1,69 (5 Н, м), 2,18 (1 Н, дд,J=2,9, 5,9 Гц), 2,44 (1 Н, дд, J=2,9, 6,2 Гц), 2,70 (1 Н, д, J=15,8 Гц), 4,06 (1 Н, ушир.с), 4,13 (4 Н, кв.,J=7,3 Гц), 4,18-4,28 (2 Н, м), 4,31 (1 Н, д, J=5,9 Гц), 4,41-4,45 (1 Н, м), 4,85 (1 Н, ушир.с), 6,57 (1 Н, д,J=7,3 Гц), 6,97 (1 Н, ушир.с). Элементный анализ. Вычислено для C26H43N3O90,1H2O: С 57,46; Н 8,01; N 7,73. Найдено: С 57,18; Н 8,00; N 7,64. МСВР вычислено для C26H43N3O9 [M+Na]+, 564,2897. Найдено, 564,2922. Метод получения 42. Диэтиловый эфир (1S,2S,4S,5R,6R)-2-[2'-(2S-трет-бутоксикарбониламино-3-метилбутириламино)пропиониламино]-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты Получают в соответствии с общей процедурой В, используя имеющийся в продаже N-Boc-(L)-валин и гидрохлорид диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'S-аминопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,2 г, 0,55 ммоль, метод получения 38). Очищают ПЦТСХ, 4 мм SiO2 ротор (от 10% этилацетат/гексан до 67% этилацетата). Выход 0,36 г (43%, 0,68 ммоль) белого пенообразного вещества. К перемешиваемому при 0 С раствору гидрохлорида диэтилового эфира (1S,2S,4S,5R,6R)-2-(2'Sаминопропиониламино)-4-гидроксибицикло[3.1.0]гексан-2,6-дикарбоновой кислоты (0,30 г, 0,82 ммоль,метод получения 38) в CH2Cl2 (30 мл) последовательно добавляют триэтиламин (0,16 г, 1,6 ммоль) и затем ацетилхлорид (0,10 г, 1,18 ммоль). Реакционной смеси дают нагреться, перемешивая ее в течение ночи. Реакционную смесь разбавляют этилацетатом (700 мл) и промывают водным раствором NaHSO4 и насыщенным раствором соли. Органический слой сушат над сульфатом магния. Очищают ПЦ-ТСХ, 4 мм
МПК / Метки
МПК: A61K 38/05, C07K 5/06
Метки: возбуждающих, пролекарства, аминокислот
Код ссылки
<a href="https://eas.patents.su/30-14979-prolekarstva-vozbuzhdayushhih-aminokislot.html" rel="bookmark" title="База патентов Евразийского Союза">Пролекарства возбуждающих аминокислот</a>
Производные индола в качестве антагонистов возбуждающих аминокислот

Номер патента: 308
Опубликовано: 29.04.1999
Авторы: Де Маджистрис Элизабетта, Конти Надия, Ди Фабио Романо, Ферьяни Альдо
МПК: C07D 209/42, A61K 31/40
Метки: аминокислот, антагонистов, качестве, возбуждающих, производные, индола
Формула / Реферат:
1. Соединение формулы (I) или его соль, или метаболически лабильный сложный эфир, где m равно 2, a R представляет собой хлор в положении 4 и 6, А представляет собой незамещенную этенильную группу в трансконфигурации; R1 представляет собой водород, С1-4алкил, возможно замещенный карбоксилом, С3-6циклоалкил, фенил, возможно замещенный метоксилом, 3-пиридил, 4-тетрагидропиранил, R2 представляет собой водород или метил, R3 представляет...
Замещенные бициклогексанкарбоновые кислоты и их производные в качестве антагонистов рецептора возбуждающих аминокислот, способ их получения и применение.

Номер патента: 894
Опубликовано: 26.06.2000
Авторы: Мэсси Стивен М., Монн Джеймс Э., Хелтон Дэвид Р., Домингес-Фернандес Кармен
МПК: A61K 31/19, C07D 233/58, C07C 211/38...
Метки: применение, замещенные, бициклогексанкарбоновые, качестве, получения, аминокислот, кислоты, рецептора, возбуждающих, способ, производные, антагонистов
Формула / Реферат:
1. Соединение формулы где Х представляет собой связь, S, О или NRa; R представляет собой группу (1-4С)алкил или группу фенил(1-4С)алкил, либо дифенил(1-4С)алкил, в которой фенильное кольцо является незамещенным или замещенным одним, двумя или тремя заместителями, выбранными независимо из галогена, (1-4С)алкила, (1-4С)алкокси, (1-4С)фторалкила, (1-4С)фторалкокси, фенила, фенокси, 3-трифторметилфенокси и 4-хлорфенокси; Ra представляет водород...
Пролекарства

Номер патента: 2647
Опубликовано: 29.08.2002
Авторы: Линдстрем Стефан, Жоу Ксиао-Ксионг, Велинг Хорст, Йоханссон Нильс Гуннар, Сальвадор Лурдес, Сунд Кристиан, Валльберг Ханс
МПК: C07H 19/16, C07K 5/00, C07D 473/32...
Метки: пролекарства
Формула / Реферат:
1. Производное гуанозина, выбранное из 2',3'-дидезокси-3'-фтор-5'-О-[2-(L-валилокси)пропионил]гуанозина или 2',3'-дидезокси-3'-фтор-5'-О-[2,3-бис(L-валилокси)пропаноил]гуанозина, или его фармацевтически приемлемая соль. 2. Соединение по п.1, где 2-(L-валилокси)пропионильный фрагмент молекулы определяет производное L-молочной кислоты. 3. Соединение по п.1, обозначенное как 2',3'-дидезокси-3'-фтор-5'-О-[(R)-2,3-бис(L-валилокси)пропаноил]гуанозин. ...
Окулоселективные лекарственные средства и пролекарства

Номер патента: 12668
Опубликовано: 30.12.2009
Авторы: Патил Гханшиам, Матьер Уилльям Л.
МПК: A61K 31/24
Метки: средства, окулоселективные, пролекарства, лекарственные
Формула / Реферат:
1. Соединение формулы I где каждый из R1 и R2 независимо представляет собой Н или бензилзащитную группу; R3 представляет собой Н или линейный или разветвленный C1-С5алкил; R4 представляет собой Н или W; при условии, что по крайней мере один из R1, R2 и R4 представляет собой W; R5 представляет собой линейный или разветвленный С1-С5алкил, циклоалкил, С1-С5алкоксиалкил, тетрагидрофуранил, тетрагидрооксазолил, диоксоланил и диоксанил; W...
Водорастворимые пролекарства противоракового действия

Номер патента: 2400
Опубликовано: 25.04.2002
Авторы: Ли Чун, Янг Дэвид Дж., Ю Донг-Фанг, Уоллейс Сидни
МПК: A61M 36/00, A01N 43/02, A61K 35/00...
Метки: действия, пролекарства, противоракового, водорастворимые
Формула / Реферат:
1. Композиция, включающая противоопухолевое лекарственное средство, конъюгированное с водорастворимым полимером или сополимером, причем указанный водорастворимый полимер или сополимер выбирают из группы, включающей поли-(d-глутаминовую кислоту), поли-(l-глутаминовую кислоту), поли- (dl-глутаминовую кислоту), поли-(d-аспарагиновую кислоту), поли-(l-аспарагиновую кислоту) и поли(dl-аспарагиновую кислоту) и их сополимеры, где используемое...
Предыдущий патент: Крутильный инструмент и способ затягивания или ослабления соединения с использованием этого инструмента
Следующий патент: Энзиматический метод разделения рацемической 3-арил-4-аминомасляной кислоты
Случайный патент: Способ получения однородной фильтрующей структуры для каталитического применения