Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения
Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Толлон Катрин, Вийенов Николь, Шименти Стефано, Кеньяр Паскаль, Вилен Жан-Поль, Дессинже Эмея, Пеглион Жан-Луи, Гумен Бертран




























Формула / Реферат
1. Соединение формулы (I)

где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,
R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или гидроксигруппы; метиловой группы; группы -OSO2R10; группы -OCOR10 или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, необязательно замещенной метокси или -(CO)-NR12R'12 группой; или R2 и R3, или R3 и R4, или R4 и R5, образующих вместе -O-(CH2)q-O-, -O-CH=CH-O- или -О-СН=СН-,
R6, R7, R8 и R9могут быть одинаковыми или различаться, и каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6алкоксигруппы, или R6 и R7, или R7 и R8, или R8и R9, образующих вместе -O-(CH2)q-O-,
R10 выбирают из группы, представленной линейной или разветвленной C1-C6алкоксигруппой, NR11R'11 и линейным или разветвленным C1-C6 алкилом, который необязательно может быть замещенным одним или несколькими атомами галогена,
R11 и R'11 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной C1-C6алкильной группы, или R11 и R'11, образующих вместе с атомом азота моноциклический или бициклический 5- или 8-членный гетероцикл, содержащий азот и необязательно содержащий другой гетероатом, выбранный из О и N, причем этот гетероцикл необязательно может быть замещенным одним или несколькими атомами галогена,
R12 и R'12 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной С1-C6 алкильной группы,
X является О, NH или CH2,
m и p могут быть одинаковыми или различаться и являются 0 или 1,
n и q могут быть одинаковыми или различаться и являются 1 или 2,
его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
2. Соединение формулы (I) согласно п.1, где R1 выбирают из атома водорода или линейной или разветвленной C1-C6алкильной группы, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
3. Соединение формулы (I) согласно п.1, где R1 выбирают из C3-C7 циклоалкильной группы или циклоалкильной группы, где циклоалкильная часть содержит от 3 до 7 атомов углерода, а алкильная часть содержит от 1 до 6 атомов углерода и является линейной или разветвленной, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
4. Соединение формулы (I) согласно любому из пп.1-3, где R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6алкоксигруппы или -OCOR10 группы, где R10 является группой NR11R'11, как описывается выше, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
5. Соединение формулы (I) согласно любому из пп.1-4, где R6, R7, R8и R9 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С1-C6алкоксигруппы, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
6. Соединение формулы (I) согласно любому из пп.1-5, где m является 0, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
7. Соединение формулы (I) согласно любому из пп.1-5, где m является 1, а X - CH2, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
8. Соединение формулы (I) согласно любому из пп.1-7, где р является 0, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
9. Соединение формулы (I) согласно любому из пп.1-7, где р является 1, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
10. Соединение формулы (I) согласно п.1, где R1 выбирают из атома водорода или линейной или разветвленной C1-C6алкильной группы, R2, R3, R4 и R5, которые могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С1-C6 алкоксигруппы, R6, R7, R8и R9, которые могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С1-C6алкоксигруппы, m является 0, n является 1 и р является 0, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой.
11. Соединение формулы (I) согласно п.1, которое выбирают из
N-{[3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амина, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой;
N-{[3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амина, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой;
N-[2-(5,6-диметокси-2,3-дигидро-1Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амина, а также его солей с фармацевтически приемлемой кислотой;
N-{[3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-4-оксобутан-1-амина, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой;
N-{[3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил}-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-4-оксобутан-1-амина, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой;
7-{[[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил)амино]метил}бицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамата, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой.
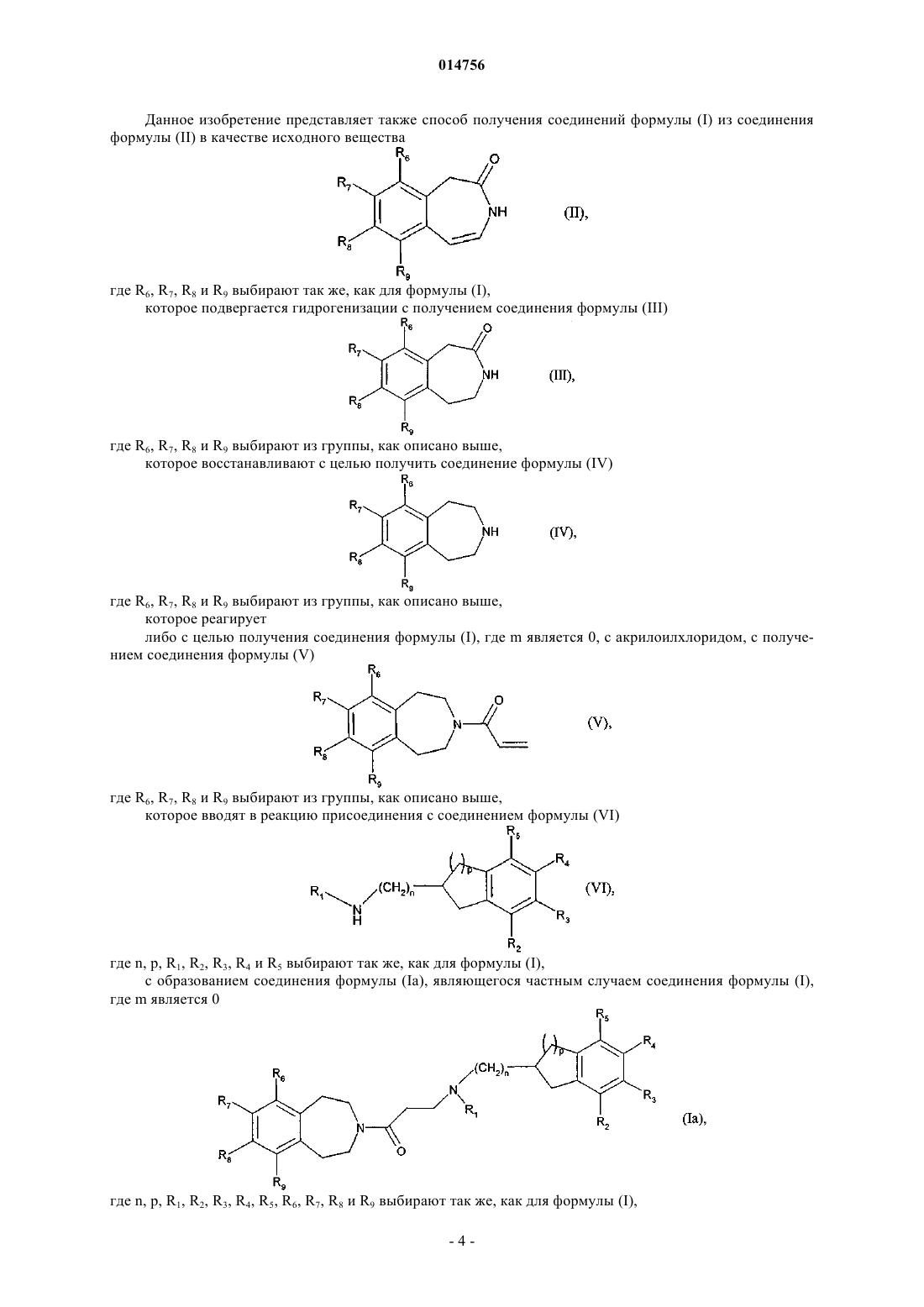
12. Способ получения соединений формулы (I) согласно п.1 с использованием в качестве исходного соединения формулы (II)

где R6, R7, R8 и R9определяются согласно п.1,
которое гидрогенируют с образованием соединения формулы (III)

где R6, R7, R8и R9 определяются согласно п.1,
которое восстанавливают с образованием соединения формулы (IV)

где R6, R7, R8и R9 определяются согласно п.1,
которое подвергают реакции для получения соединения формулы (I), где m является 0, с акрилоилхлоридом, с получением соединения формулы (V)

где R6, R7, R8и R9 определяются согласно п.1,
которое подвергают реакции присоединения с соединением формулы (VI)

где n, p, R1, R2, R3, R4 и R5определяются согласно п.1,
получая соединения формулы (Ia), которые являются частным случаем соединений формулы (I), где m является 0

где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9определяются согласно п.1.
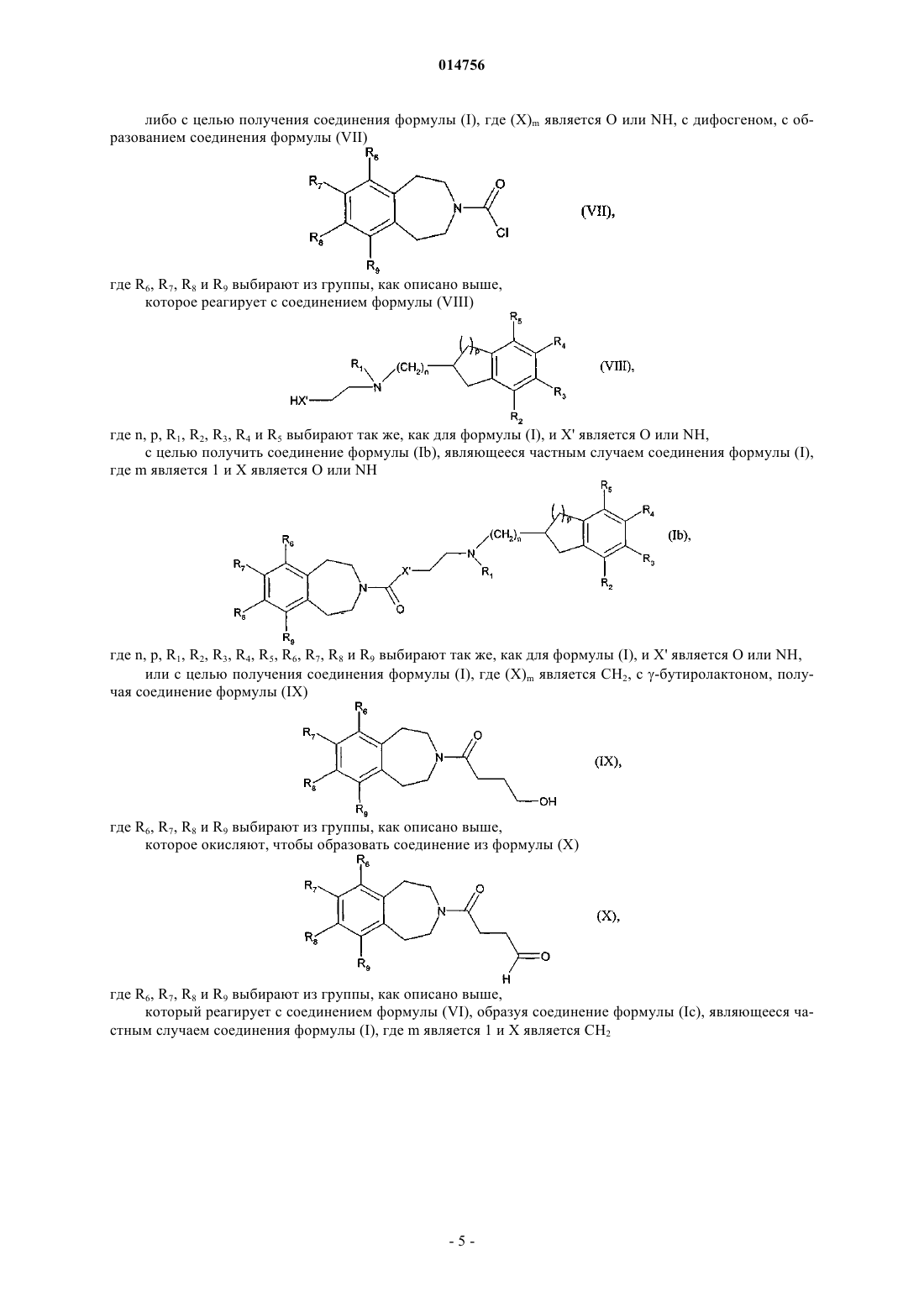
13. Способ получения соединений формулы (I) согласно п.1 с использованием в качестве исходного соединения формулы (II)

где R6, R7, R8и R9 определяются согласно п.1,
которое гидрогенируют с образованием соединения формулы (III)

где R6, R7, R8и R9 определяются согласно п.1,
которое восстанавливают с образованием соединения формулы (IV)

где R6, R7, R8и R9 определяются согласно п.1,
которое подвергают реакции для получения соединения формулы (I), где (Х)m является О или NH, с дифосгеном, с образованием соединения формулы (VII)

где R6, R7, R8и R9 определяются согласно п.1,
которое реагирует с соединением формулы (VIII)

где n, р, R1, R2, R3, R4 и R5определяются согласно п.1 и X' является О или NH,
с образованием соединений формулы (Ib), которые являются частным случаем соединений формулы (I), где m является 1 и X является О или NH

где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9определяются согласно п.1 и X' является О или NH.
14. Способ получения соединений формулы (I) согласно п.1 с использованием в качестве исходного соединения формулы (II)

где R6, R7, R8и R9 определяются согласно п.1,
которое гидрогенируют с образованием соединения формулы (III)

где R6, R7, R8и R9 определяются согласно п.1,
которое восстанавливают с образованием соединения формулы (IV)

где R6, R7, R8и R9 определяются согласно п.1,
которое подвергают реакции для получения соединения формулы (I), где (Х)m является СН2, с гамма-бутиролактоном, с образованием соединения формулы (IX)

где R6, R7, R8и R9 определяются согласно п.1,
которое окисляют для образования соединения формулы (X)

где R6, R7, R8и R9 определяются согласно п.1,
которое взаимодействует с соединением (VI) с образованием соединения (Ic), которое является частным случаем соединения формулы (I), где m является 1 и X - CH2

где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9определяются согласно п.1.
15. Фармацевтическая композиция, включающая как активный ингредиент соединение формулы (I) согласно любому из пп.1-11 в комбинации с одним или несколькими фармацевтически приемлемыми инертными нетоксичными наполнителями или носителями.
16. Фармацевтическая композиция по п.15 для лечения или профилактики патологий, при которых учащенное сердцебиение является инициатором или играет роль раздражителя.
17. Фармацевтическая композиция по п.16 для лечения или профилактики ишемических кардиопатологий, систолических или диастолических сердечных недостаточностей в хронической или острой форме, вентрикулярных или суправентрикулярных нарушений ритма или патологий, являющихся васкулярным фактором риска.
18. Фармацевтическая композиция по п.17 для лечения или профилактики стабильной стенокардии, нестабильной стенокардии, угрожающего симптомокомплекса, миокардиального инфаркта или постинфарктного состояния.
19. Фармацевтическая композиция по п.15 для лечения или профилактики артериальной гипертензии, диабета или гиперхолестеринемии.
20. Фармацевтическая композиция по п.15 для лечения или профилактики боли, повышенной деятельности мочевого пузыря или ощущения сухости глаз.
Текст
СОЕДИНЕНИЯ 1,2,4,5-ТЕТРАГИДРО-3 Н-БЕНЗАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ где R1 представляет собой атом водорода или группу, выбранную из циклоалкильной, бензильной и необязательно замещнной алкильной группы, R2, R3, R4 и R5 выбираются из атома водорода или гидроксигруппы; метиловой группы; группы -OSO2R10; группы -OCOR10; алкоксигруппы или R2 и R3, или R3 и R4, или R4 и R5, образующих вместе -O-(CH2)q-O-, -О-СН=СН-О- или -OСН=СН-, R6, R7, R8 и R9 выбираются из атома водорода или алкоксигруппы или R6 и R7, или R7 иR8, или R8 и R9, образующих вместе -O-(CH2)q-O-, R10 выбирают из группы, представленной линейной или разветвленной C1-C6 алкоксигруппой, NR11R'11 и алкилом, который необязательно может быть замещенным, R11 и R'11 выбираются из атома водорода или алкильной группы илиR11 и R'11, образующих вместе с атомом азота моноциклический или бициклический гетероцикл,который может быть дополнительно замещенным, X является О, NH или CH2, m и p являются 0 или 1, n и q являются 1 или 2, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. Фармацевтические композиции, которые содержат соединения, представленные в данном изобретении в качестве активных ингредиентов.(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ (FR) 014756 Данное изобретение относится к новым соединениям 1,2,4,5-тетрагидро-3H-бензазепина, к способу их получения и к фармацевтическим композициям, которые их содержат. Соединения, представленные в данном изобретении, блокируют HCN (гиперполяризованноактивированный циклический нуклеотидуправляемый) канал. Блокирование каналов HCN соединениями данного изобретения позволяет использовать их для лечения и профилактики какой-либо патологии, при которой активность или экспрессия каналов HCN уменьшается и становится анормальной. Соединения, представленные в данном изобретении, можно использовать для лечения и профилактики боли, в частности невропатической и воспалительной (Chaplan S.R., Guo H.Q., Lee D.H., Luo L., Liuin Neuropathic Pain. Neurochem Res. 2008 May 7), повышенной деятельности мочевого пузыря (StamatiouNephrol. 2008 Feb 1), ощущения сухости глаз (Ingram S.L., Williams J.T. Modulation of the hyperpolarization-activated current (Ih) by cikloic nucleotides in guinea-pig primary afferent neurons. J Physiol. 1996; 492:97-106; Belmonte C. Eye dryness sensations after refractive surgery: impaired tear secretion or "phantom"cornea J Refract Surg 2007; 23(6):598-602). Соединения, представленные в данном изобретении, восстанавливают работу пейсмекера прямым и селективным способами. Селективное восстановление сердечного ритма соединениями данного изобретения дает возможность использовать их для лечения и профилактики различных патологий, при которых учащенное сердцебиение имеет провоцирующее действие или играет роль раздражителя. Использование данных соединения делает более эффективным лечение и профилактику ишемических кардиопатологий (Dyer A.R.,Persky V., Stamler J., et al. Heart rate as a prognostic factor for coronary heart disease and mortality: findings inand sinus rhythm. Am J Cardiol. 1996; 78: 1175-1176) при их различных клинических проявлениях: стабильная стенокардия (Borer J.S., Fox K., Jaillon P., et al. Antianginal and antiischemic effects of ivabradine,an If inhibitor, in stable angina. A randomized, double-blind, multicentered, placebo-controlled trial. Circulation 2003; 107:817-23; Diaz A., Bourassa M.G., Guertin M.C., Tardif J.C.: Long-term prognostic value of restingacute myocardial infarction: prospective evaluation of the use of heart rate and left ventricular function. J Electrocardiol 2005; 38:106-112), постинфарктных состояний; систолической или диастолической сердечной недостаточности (Aaronson K.D., Schwartz J.S., Chen T.M., Wong K.L., Goin J.E., Mancini D.M.: Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiacsufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet 1999; 353:9-13; Mulder P., Barbier S.,Chagraoui A., et al. Long-term heart rate reduction induced by the selective If current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation 2004; 109: 1674-9); вентрикулярного или суправентрикулярного нарушения ритма (James R., Arnold J., Allen J.,et al. The effects of heart rate, myocardial ischemia and vagal stimulation on the threshold of ventricular fibrillation. Circulation. 1977; 55: 311-7; Bernier M., Curtis J.M., Hearse D.J. Ischemia-induced and reperfusioninduced arrhythmias: importance of heart rate. Am J Physiol 1989; 256: H21-H31), патологий, связанных с сосудистым фактором риска: артериальная гипертензия, диабет, гиперхолестеринемия, путем замедления главным образом развития атеросклерозных патологических изменений и осложнений (Beere P.A.,Glagov S., Zarins C.K. Retarding effect of lowered heart rate on coronary atherosclerosis. Science 1984; 226: 180-2; Kaplan J.R., Manuck S.B., Clarkson Т.В. The influence of heart rate on coronary atherosclerosis. J Cardiovasc Pharmacol 1987; 10: S100-S102; Beere P.A., Glagov S., Zarins C.K. Experimental atherosclerosis at theelevated heart rate are associated with coronary plaque disruption. Circulation. 2001; 104: 1477-82). Соединения, непосредственно или селективно замедляющие сердечный ритм, известны и особенно широко представлены в патенте ЕР 0534859. Задачей данного изобретения было получение новых соединений, которые замедляют сердечный ритм, являющихся одновременно сильнодействующими, селективными и безопасными для использования. При такой постановке задачи имеет значение риск при возможных взаимодействиях различных соединений. Данное изобретение представляет соединения формулы (I) где R1 представляет собой атом водорода или группу, выбранную из С 3-С 7 циклоалкила, бензила и линейного или разветвленного C1-C6 алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещнной гидроксигруппой или С 3-С 7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или гидроксигруппы; метиловой группы; группы -OSO2R10; группы -OCOR10 или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, необязательно замещнной метокси или-(CO)-NR12R'12 группой; или R2 и R3, или R3 и R4, или R4 и R5, образующих вместе -O-(CH2)q-O-, -OCH=CH-O- или -O-СН=СН-,R6, R7, R8 и R9 могут быть одинаковыми или различаться, и каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, или R6 и R7,или R7 и R8, или R8 и R9, образующих вместе -O-(CH2)q-O-,R10 выбирают из группы, представленной линейной или разветвленной C1-C6 алкоксигруппой,NR11R'11 и линейным или разветвленным C1-C6 алкилом, который необязательно может быть замещенным одним или несколькими атомами галогена,R11 и R'11 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной C1-C6 алкильной группы, или R11 и R'11, образующих вместе с атомом азота моноциклический или бициклический 5- или 8-членный гетероцикл, содержащий азот и необязательно содержащий другой гетероатом, выбранный из О и N, причем этот гетероцикл необязательно может быть замещенным одним или несколькими атомами галогена,R12 и R'12 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной C1-C6 алкильной группы,X является О, NH или CH2,m и p могут быть одинаковыми или различаться и являются 0 или 1,n и q могут быть одинаковыми или различаться и являются 1 или 2,-2 014756 их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Среди фармацевтически приемлемых кислот, не ограничивая изобретении приведенными примерами, можно отметить соляную кислоту, бромисто-водородную кислоту, серную кислоту, фосфорную кислоту, уксусную кислоту, трифторуксусную кислоту, молочную кислоту, пировиноградную кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, винную кислоту, малеиновую кислоту, лимонную кислоту, аскорбиновую кислоту, щавелевую кислоту, метансульфоновую кислоту, бензолсульфоновую кислоту, камфорную кислоту, памовую кислоту, 1,5-нафталиндисульфоновую кислоту. Одним из воплощений данного изобретения являются соединения формулы (I), где R1 выбирают из атома водорода или линейной или разветвленной C1-C6 алкильной группы, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где R1 выбирают из C3-C7 циклоалкильной группы или циклоалкильной группы, где циклоалкильная часть содержит от 3 до 7 атомов углерода, а алкильная часть содержит от 1 до 6 атомов углерода и является линейной или разветвленной, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где R2, R3, R4 и R5 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы или -OCOR10 группы, где R10 является группой NR11R'11, как описывается выше, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Предпочтительным является воплощение, где R11 и R'11 являются линейной или разветвленной C1C6 алкильной группой. Другое воплощение изобретения представляет соединения формулы (I), где R6, R7, R8 и R9 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где m является 0, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где m является 1, а X - CH2,их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где р является 0, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения формулы (I), где р является 1, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения, где R1 выбирают из атома водорода или линейной или разветвленной C1-C6 алкильной группы, R2, R3, R4 и R5 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, R6, R7, R8 и R9 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, m является 0, n является 1 и р является 0, их оптические изомеры, если таковые существуют, а также их соли с фармацевтически приемлемой кислотой. Другое воплощение изобретения представляет соединения-3 014756 Данное изобретение представляет также способ получения соединений формулы (I) из соединения формулы (II) в качестве исходного вещества где R6, R7, R8 и R9 выбирают так же, как для формулы (I),которое подвергается гидрогенизации с получением соединения формулы (III) где R6, R7, R8 и R9 выбирают из группы, как описано выше,которое восстанавливают с целью получить соединение формулы (IV) где R6, R7, R8 и R9 выбирают из группы, как описано выше,которое реагирует либо с целью получения соединения формулы (I), где m является 0, с акрилоилхлоридом, с получением соединения формулы (V) где n, p, R1, R2, R3, R4 и R5 выбирают так же, как для формулы (I),с образованием соединения формулы (Ia), являющегося частным случаем соединения формулы (I),где m является 0 где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9 выбирают так же, как для формулы (I),-4 014756 либо с целью получения соединения формулы (I), где (Х)m является О или NH, с дифосгеном, с образованием соединения формулы (VII) где n, р, R1, R2, R3, R4 и R5 выбирают так же, как для формулы (I), и X' является О или NH,с целью получить соединение формулы (Ib), являющееся частным случаем соединения формулы (I),где m является 1 и X является О или NH где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9 выбирают так же, как для формулы (I), и X' является О или NH,или с целью получения соединения формулы (I), где (Х)m является СН 2, с -бутиролактоном, получая соединение формулы (IX) где R6, R7, R8 и R9 выбирают из группы, как описано выше,которое окисляют, чтобы образовать соединение из формулы (X) где R6, R7, R8 и R9 выбирают из группы, как описано выше,который реагирует с соединением формулы (VI), образуя соединение формулы (Ic), являющееся частным случаем соединения формулы (I), где m является 1 и X является CH2 где n, p, R1, R2, R3, R4, R5, R6, R7, R8 и R9 выбирают так же, как для формулы (I). Оптически активные формы соединений формулы (I) получены как с использованием оптически активных форм промежуточных соединений формул (VI) и (VIII), так и разделением рацемической смеси соединений формулы (I) методами, известными из литературы. Соединения, представленные в данном изобретении, блокируют HCN (гиперполяризованноактивированный циклический нуклеотидуправляемый) канал. Блокирование каналов HCN соединениями данного изобретения позволяет использовать их для лечения и профилактики какой-либо патологии, при которой активность или экспрессия каналов HCN уменьшается и становится анормальной. Соединения, представленные в данном изобретении, можно использовать для лечения и профилактики боли, в частности невропатической и воспалительной, повышенной деятельности мочевого пузыря,ощущения сухости глаз. Соединения, представленные в данном изобретении, восстанавливают работу пейсмейкера прямым и селективным способами. Селективное восстановление сердечного ритма соединениями данного изобретения дает возможность использовать их для лечения и профилактики различных патологий, при которых учащенное сердцебиение имеет провоцирующее действие или играет роль раздражителя. Использование данных соединений делает более эффективным лечение и профилактику при ишемических кардиопатологиях в различных клинических проявлениях: стабильная стенокардия, острый коронарный синдром: нестабильная стенокардия, угрожающий симптомокомплекс и инфаркт миокарда, постинфарктное состояние; систолических или диастолических сердечных недостаточностей в хронической или острой форме; вентрикулярных или суправентрикулярных нарушениях ритма или патологий, являющихся васкулярным фактором риска: повышенное артериальное кровяное давление, диабет, гиперхолестеринемия, особенно эндотелиальная дисфункция, развитие атеросклерозных патологических изменений и их осложнений. Данные соединения могут обеспечивать миокардиальную защиту больных в зоне риска, во время хирургического вмешательства или септического шока. Замедление сердцебиения может также лечить заболевания, как,например, гипертиреоз, сопровождаемые тахикардией. Данное изобретение также описывает фармацевтические композиции, включающие как активный ингредиент соединение формулы I или его соль с фармацевтически приемлемой кислотой в комбинации с одним или несколькими приемлемыми фармацевтическими, инертными, нетоксичными наполнителями или носителями. Согласно данному изобретению, не ограничивая изобретения приведенными примерами, можно выделить формы фармацевтических композиций, которые применяются орально, парентерально (внутривенно, внутримышечно или подкожно), подкожно или чрескожно, назально, ректально, под язык, офтальмологическим или респираторным введением, а конкретней пилюли или драже, подязыковые таблетки, твердые желатиновые капсулы, капсулы, суппозитории, кремы, мази, кожные гели, ампулы для внутримышечного введения или питья, аэрозоли, глазные капли и капли для носа. Согласно изобретению, кроме соединения формулы (I), фармацевтические композиции содержат один или несколько наполнителей или носителей, как, например, растворители, смазки, связующие вещества, распадающиеся вспомогательные соединения, абсорбенты, красители, подсластители. Например, в качестве наполнителей или носителей можно использовать (приведенные примеры не ограничивают изобретение) в качестве растворителя - лактозу, глюкозу, сахарозу, маннитол, сорбит, целлюлозу, глицерин,в качестве смазки - силикагель, тальк, стеариновую кислоту и е магниевые и кальцевые соли, полиэтиленгликоль,в качестве связующего вещества - силикат алюминия, силикат магния, крахмал, желатин, трагант,метилцеллюлозу, натрийкарбоксиметилцеллюлозу и поливинилпирролидон,в качестве распадающихся вспомогательных соединений - агар, альгиновую кислоту и е натриевую соль, шипучие смеси. Содержание активного ингредиента формулы (I) в фармацевтической композиции желательно должно составлять от 5 до 50% от общей массы.-6 014756 Дозировка зависит от возраста и веса больного, способа введения, природы и степени нарушения и введения каких-либо других препаратов и составляет от 0,5 до 500 мг в день за одно или несколько введений. Приведенные ниже примеры иллюстрируют данное изобретение. Структуры соединений, описанные в примерах, были определены стандартными методами спектрометрии (инфракрасной, ЯМР и массспектрометрии). В способах получения описывается методика получения промежуточных соединений. СокращенияBINAP - 2,2-бис(дифенилфосфино)-1,1-динафтил,TLC - тонкослойная хроматография,DMF - N,N-диметилформамид,ДМСО - диметилсульфоксид,экв. - эквивалент,IR - инфракрасный,PEG 300 - полиэтиленгликоль, имеющий средний молекулярный вес 300 г/моль,THF - тетрагидрофуран. Способ получения 1. [(4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 8,8-Диэтокси-2,3-диметоксибицикло[4.2.0]окта-1,3,5-триен. 24 г (107 ммоль) бромвератрола приливают к смеси 8,4 г (215 ммоль)/2 экв.) амида натрия в 10 мл толуола и 25 г (215 ммоль/2 экв.) 1,1-диэтоксиэтилена. Реакционную массу кипятят при 80 С в течение 4 ч 30 мин; охлаждают до -10 С, после чего добавляют 50 мл воды, а затем 50 мл толуола. Отделяют органическую фазу, а водную опять экстрагируют толуолом. Объединенные экстракты толуола промывают водой до нейтрального рН и затем выпаривают. Полученный остаток разделяют хроматографически на 600 г силикагеля (элюент: дихлорметан), получая нужный продукт. Стадия 2. 4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-он. К 7,5 г (29,7 ммоль) полученного на предыдущей стадии вещества, растворенного в 120 мл THF,добавляют 30 мл 1 М раствор соляной кислоты. Перемешивают при комнатной температуре в течение 19 ч, после чего максимально выпаривают THF. Полученный белый осадок смешивают с 150 мл воды и перемешивают в течение 1 ч при 5 С. После фильтрования осадок 3 раза промывают 15 мл воды и сушат под вакуумом с получением необходимого продукта. Стадия 3. 4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ол. К 4,73 г (26,5 ммоль) полученного на предыдущей стадии вещества, суспензированого в 138 мл метанола, дважды добавляют 1,21 г (31,8 ммоль) боргидрида натрия при 0 С в течение 5 мин. После этого смесь перемешивают в течение 1 ч при 0 С, приливают к 350 мл воды, а затем экстрагируют дихлорметаном 3 раза по 100 мл. Объединенные органические фазы промывают дважды 50 мл воды, а затем сушат над сульфатом магния и выпаривают с получением желаемого продукта. Стадия 4. 8-Бромо-2,3-диметоксибицикло[4.2.0]окта-1,3,5-триен. Раствор 4,55 г (25,3 ммоль) полученного на предыдущей стадии вещества, 11,9 г (45,5 ммоль/1,8 экв.) трифенилфосфина и 10,05 г (30,3 ммоль/1,2 экв.) тетрабромметана в 150 мл эфира кипятят с обратным холодильником в течение 2 ч 30 мин. После охлаждения нерастворимый осадок фильтруют через фритту и промывают 5 раз 50 мл эфира. После выпаривания фильтратов остаток разделяют хроматографически на 550 г силикагеля (элюент: дихлорметан) с получением желаемого продукта. Стадия 5. 4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 6,0 г (24,8 ммоль) полученного на предыдущей стадии вещества и 8,64 г (32,2 ммоль/1,3 экв.) тетрабутиламмоний цианида перемешивают с 125 мл THF при комнатной температуре в течение 21 ч. После выпаривания реакционной смеси остаток разделяют хроматографически на 350 г силикагеля (элюент: дихлорметан) с получением желаемого продукта. Стадия 6. [(4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. К 4,01 г (21,2 ммоль) полученного на предыдущей стадии вещества, растворенного в 60 мл метанола и 60 мл 7N аммиачного метанола, добавляют 2 мл никель Ренея. Гидрогенирование проводят при комнатной температуре и нормальном давлении в течение 6 ч 30 мин. Смесь фильтруют на целите, остаток промывают метанолом и выпаривают с получением желаемого продукта. Способ получения 2. [(2-Метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 3-(2,6-Дибромфенил)-2-хлорпропаннитрил. К 62,4 г (464 ммоль/1,2 экв.) хлорида меди, 76 мл (580 ммоль/1,5 экв.) трет-бутилнитрита и 483 мл(7,34 моль/19 экв.) акрилонитрила в 475 мл ацетонитрила при комнатной температуре прикапывают в течение 1 ч 100 г (386 ммоль) 2,6-диброманилина, растворенного в 380 мл ацетонитрила. Смесь перемешивают в течение 1 ч при комнатной температуре, а затем приливают к 1 л 20% раствора соляной кислоты. Водную фазу экстрагируют толуолом и объединенные толуольные фазы промываются насыщенным раствором хлорида натрия. После выпаривания получают необходимый продукт, который используют в качестве исходного соединения на следующей стадии. Стадия 2. 3-(2,6-Дибромфенил)пропаннитрил.-7 014756 Вещество, полученное на предыдущей стадии, растворяют в 1400 мл уксусной кислоты. Быстро добавляют 68 г (1,04 моль/2,7 экв.) порошкообразного цинка. Медленно нагревают смесь до 58 С. Через 1 ч после прохождения реакции реакционную смесь выливают на 3 кг льда. Полученную водную фазу экстрагируют 1 л, а затем 500 мл толуола. Объединенные толуольные фазы промываются насыщенным раствором хлорида натрия. После выпаривания остаток разделяют хроматографически на силикагеле с получением необходимого продукта. Стадия 3. 2-Бромбицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 138 г (477 ммоль) полученного на предыдущей стадии вещества смешивают с амидом натрия (1,91 моль/4 экв.) в 3 л нашатырного спирта. Смесь перемешивают в течение 2 ч с обратным холодильником,после чего реакцию прекращают путем добавления 102 г (4 экв.) кристаллического хлористого аммония,а затем аммиак выпаривают. Остаток помещают в 1 л эфира и перемешивают; осадок фильтруют и промывают эфиром. После выпаривания объединенных фильтратов остаток разделяют хроматографически на 2 кг силикагеля (элюент: дихлорметан) с получением необходимого продукта. Стадия 4. 2-Бромбицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. К 75 г (360 ммоль) полученного на предыдущей стадии вещества в 375 мл этанола при комнатной температуре добавляют раствор 38 г (612 ммоль/1,7 экв.) гидроксида калия, растворенного в 125 мл воды. Кипятят с обратным холодильником в течение 16 ч, после чего выпаривают этанол. Добавляют 1 л воды, водная фаза промывается дважды 250 мл эфира, а затем подкисляется концентрированной соляной кислотой до рН 1. Образованный осадок отфильтровывают, промывают водой и сушат с получением необходимого продукта. Стадия 5. 2-(2-Бромбицикло[4.2.0]окта-1,3,5-триен-7-ил)-4,4-диметил-4,5-дигидрооксазол. 79 г (348 ммоль) полученного на предыдущей стадии вещества и 39 г (417 ммоль/1,2 экв.) 1,1 диметил-2-гидроксиэтиламина перемешивают с 800 мл ксилола. Смесь кипятят при 140 С в течение 6 ч,проводя азеотропную перегонку воды. После этого реакционную смесь охлаждают и перемешивают при комнатной температуре в течение ночи. Образованный осадок, в основном исходная кислота, отфильтровывают и промывают толуолом. Объединенные фильтраты выпаривают с получением необходимого продукта. Стадия 6.[7-(4,4-Диметил-4,5-дигидро-1,3-оксазол-2-ил)бицикло[4.2.0]окта-1,3,5-триен-2-ил(дифенилметилен)амин. При комнатной температуре 48 г (171 ммоль) полученного на предыдущей стадии вещества, 37,3 г(205 ммоль/1,2 экв.) бензофенонимина, 1,57 г (1,71 ммоль/0,01 экв.) дипалладий трис(дибензилиденацетона), 3,2 г (5,14 ммоль/0,03 экв.) BINAP и 23 г (240 ммоль/1,4 экв.) трет-бутилата натрия перемешивают с 800 мл толуола. Смесь греют при 85 С в течение 24 ч, через 4 ч добавляют 9,3 г (0,3 экв.) бензофенонимина. После нагревания осадок отфильтровывают при 50 С, после чего фильтрат выпаривают,получая необходимый продукт, который используется в качестве исходного соединения на следующей стадии. Стадия 7. Этиловый эфир 2-аминобицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты. 117 г (около 171 ммоль) полученного на предыдущей стадии вещества в 2400 мл этанола, содержащего 200 мл концентрированной серной кислоты, кипятят с обратным холодильником в течение 1 ч 30 мин. Максимально выпаривают растворитель, получая необходимое вещество в 1,5 л воды. Водную фазу экстрагируют эфиром, подщелачивая ее смешанным углекислым натрий-калием, а затем экстрагируют эфиром. После сушки над сульфатом магния и выпаривания объединенных эфирных экстрактов получают необходимый продукт. Стадия 8. 2-Гидроксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновая кислота. На холоду 18,6 мл (345 ммоль/6 экв.) концентрированной серной кислоты приливают к 11 г (57,5 ммоль) полученного на предыдущей стадии вещества, растворенного в 220 мл воды, поддерживая температуру ниже 15 С. Затем смесь охлаждают до температуры от 0 до 5 С и через 15 мин прикапывают раствор 4,36 г (63,3 ммоль/1,1 экв.) нитрита натрия в 25 мл воды; перемешивают в течение 5 мин при 0 С, а затем в течение 10 мин при температуре ниже 10 С прикапывают раствор 144 мг (0,57 ммоль/0,01 экв.) пентагидрата сульфата меди в 166 мл 1 М водного раствора серной кислоты. Греют при 80 С в течение 1 ч, а затем перемешивают при комнатной температуре ночь. Водную фазу экстрагируют эфиром, объединенные эфирные экстракты промывают насыщенным раствором хлорида натрия, сушат над сульфатом магния и выпаривают. Остаток разделяют хроматографически на 500 г силикагеля (элюент: дихлорметан/этанол: 90/10) с получением необходимого продукта. Стадия 9. Этиловый эфир 2-метоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты. 6 г (33 ммоль) полученного на предыдущей стадии вещества и 2,7 г (8,3 ммоль/0,25 экв.) карбоната цезия растворяют в 330 мл диметилкарбоната, греют при 130 С в течение 45 ч. После фильтрования и выпаривания реакционной смеси получают необходимый продукт. Стадия 10. Амид 2-метоксибицикло[4.2.0]окта-1,3,5-триен-7-карбоновой кислоты. К раствору 6 г (31 ммоль) полученного на предыдущей стадии вещества в 50 мл THF при 0 С прикапывают 120 мл 28% гидроокиси аммония. Перемешивают полученную смесь при комнатной температуре в течение 2 дней, после чего максимально выпаривают THF. Водную фазу экстрагируют этилацета-8 014756 том. Объединенные органические экстракты сушат над сульфатом магния и выпаривают, получая необходимый продукт. Стадия 11. 1-(2-Метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метанамин. 1,39 мл (14,7 ммоль/3 экв.) борандиметилсульфида приливают по каплям при комнатной температуре в течение 10 мин к 0,87 г (4,9 ммоль) полученного на предыдущей стадии вещества, растворенного в 20 мл THF, а затем нагревают до 70 С и греют в течение 24 ч. Смесь охлаждают до комнатной температуры, подвергают сольволизу 7,5 мл безводного метанола и опять нагревают с обратным холодильником в течение 1 ч. После выпаривания остаток (0,99 г) разделяют хроматографически на колонке с 50 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), чтобы отделить первую фракцию необходимого продукта и вторую фракцию этого же продукта в форме комплекса с бором. Вторая фракция после взаимодействия с этанольным раствором HCl, а затем кислотно-основной экстракции восстанавливается до необходимого продукта. Способ получения 3. 7-(Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. Стадия 1. 3-Гидроксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 68,7 г (431,6 ммоль) 3-метоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила растворяют в 1500 мл дихлорметана и охлаждают до 0 С. В течение 1 ч 15 мин по каплям добавляют 863 мл 1 М раствора трибромида бора в дихлорметане (863 ммоль/2 экв.). Реакционную массу доводят до комнатной температуры и оставляют на ночь. После этого ее опять охлаждают до 0 С и приливают к 1 кг льда. Смесь перемешивают в течение 30 мин при комнатной температуре, водную фазу экстрагируют дважды 500 мл дихлорметана. Объединенные органические экстракты промывают 600 мл воды, а затем сушат над сульфатом магния. После фильтрования и выпаривания получают нужный продукт. Стадия 2. 7-(Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. К 8,0 г (55,1 ммоль) полученного нитрила, растворенного в 170 мл метанола и 170 мл 7N аммиачного метанола, добавляют 8 мл никеля Ренея. Гидрогенирование проводят при комнатной температуре и нормальном давлении, пока теоретический объем водорода не будет поглощен реакционной массой. Смесь отфильтровывают на целите, промывают метанолом и выпаривают, получая необходимый амин. Способ получения 4. 8-(Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. Способ идентичен способу 3, в качестве исходного вещества используют 4-метоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. Способ получения 5. [(3-Этоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 3-Этоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 1,5 г (10,19 ммоль) продукта, полученного на стадии 1 способа 3, растворяют в 15 мл диметилформамида (DMF). К смеси добавляют 2,8 г (2 экв.) карбоната калия, а затем 2,3 мл (3 экв.) этилбромида и 100 мг (0,06 экв.) йодида калия. Реакционную массу кипятят при 80 С в течение 3 ч. Затем ее охлаждают и выпаривают DMF, используя роторный испаритель. Полученное вещество из воды извлекается методами экстракции дихлорметаном (CH2Cl2). Органическую фазу сушат над MgSO4, отфильтровывают, а затем выпаривают с получением 1,8 г масла. Масло очищают методами хроматографии на 100 г силикагеля (элюент: CH2Cl2 100%), получая необходимый продукт, который кристаллизуют при комнатной температуре. Точка плавления 50-52 С. Стадия 2. [(3-Этоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. 1,5 г (8,65 ммоль) продукта со стадии 1 растворяют в 30 мл метанола (МеОН), после чего добавляют 30 мл (7N) аммиачного раствора метанола и 0,5 г никеля Ренея. Реакционную смесь гидрогенизируют под давлением 1 бар при комнатной температуре в течение 12 ч. Катализатор отфильтровывают, а фильтрат выпаривают досуха. Необходимый продукт получают в виде масла. Способ получения 6. [(3-трет-Бутоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 3-трет-Бутоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 2,0 г (13,59 ммоль) продукта со стадии 1 способа 3 растворяют в 100 мл дихлорметана (CH2Cl2). Охлаждают до 0 С и пропускают через раствор изобутен, пока он не станет насыщенным. После чего к реакционной массе добавляют 0,2 мл концентрированной серной кислоты и перемешивают при комнатной температуре в течение 24 ч. Раствор нейтрализуют раствором NaOH (1N), экстрагируют CH2Cl2, промывают водой и сушат над MgSO4, после чего отфильтровывают и выпаривают фильтрат досуха. Полученное вещество очищают методами хроматографии на 150 г силикагеля (элюент: CH2Cl2 100%), продукт получают в форме масла. Стадия 2. [(3-трет-Бутоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. 1,1 г (5,46 ммоль) продукта со стадии 1 растворяют в 30 мл метанола (МеОН), добавляют 30 мл (7N) аммиачного раствора метанола и 0,5 г никеля Ренея. Реакционную смесь гидрогенизируют под давлением 1 бар при комнатной температуре в течение 12 ч. Катализатор отфильтровывают, а фильтрат выпаривают досуха. Необходимый продукт получают в виде масла. Способ получения 7. 5,6-Дигидроциклобута[f][1,3]бензодиоксол-5-илметиламин. Стадия 1. Этил (5,6-дигидроциклобута[4,5]бензо[1,2-d][1,3]диоксол-5-илметил)карбамат. 7,9 г (44,6 ммоль) (5,6-дигидроциклобута[4,5]бензо[1,2-d][1,3]диоксол-5-илметил)амина растворяют в 80 мл дихлорметана. К смеси добавляют 80 мл воды и 6 мл (1,6 экв.) (12N) водного раствора гидрокси-9 014756 да натрия. В течение 15 мин добавляют 3,95 мл (1,1 экв.) этилхлорформиата и перемешивают при 25 С в течение 2 ч. Смесь экстрагируют, органический слой промывают насыщенным водным растворомNaHCO3, сушат над MgSO4, фильтруют и выпаривают досуха. Необходимый продукт получают в виде коричневого кристаллического вещества. Стадия 2. 5,6-Дигидроциклобута[f][1,3]бензодиоксол-5-илметиламин. 7,5 г (30 ммоль) продукта со стадии 1 растворяют в 15 мл безводного тетрагидрофурана. Раствор прикапывают к суспензии 1,48 г (1,3 экв.) LiAlH4, растворенного в 38 мл безводного тетрагидрофурана. Смесь перемешивают при 25 С в течение 2 ч. Смесь гидролизуют при добавлении 2,7 мл холодной воды,2,7 мл NaOH (20%), а затем 2,7 мл воды, после чего раствор фильтруют, а фильтрат выпаривают досуха. Получают 2,4 г вещества, которое очищают на хроматографической колонке с силикагелем (элюент:CH2Cl2/этанол/NH4OH: 95/5/0,5). Необходимый продукт был получен в форме коричневого масла. Способ получения 8. [2-(5,6-Диметокси-2,3-дигидро-1 Н-инден-2-ил)этил]амин. 5 г (2,3 ммоль) (5,6-диметокси-2,3-дигидро-1 Н-инден-2-ил)ацетонитрила растворяют в 50 мл метанола, после чего добавляют 50 мл (7N) раствора МеОН-NH3 и 0,5 г никеля Ренея. Водород пропускают через реакционную смесь в течение 4 дней под давлением 5 бар, при 25 С. Фильтруют на целите и выпаривают фильтрат досуха. Остаток смешивают с 50 мл HCl (1N) и промывают этилацетатом; водную фазу доводят до рН 10, добавляя NaOH (20%). Экстрагируют Et2O, промывают водой, сушат над MgSO4, отфильтровывают и выпаривают досуха. Необходимый продукт получен в форме масла. Способ получения 9. 2-(5-Метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метиламин. 3 г (19 ммоль) 5-метоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила растворяют в 45 мл метанола и, добавляя 45 мл 7 М аммиачного раствора метанола, гидрогенируют в присутствии 1,5 г никеля Ренея 50% в воде при атмосферном давлении. Через 2 ч после прохождения реакции реакционную смесь фильтруют и выпаривают, получая необходимый продукт. Способ получения 10. 2-(2,3-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метиламин. Получение идентично получению продукта по способу 9 с использованием 2,3 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила в качестве исходного соединения. Способ получения 11. 2-(2,3,4-Триметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метиламин. Получение идентично получению продукта по способу 9 с использованием 2,3,4 триметоксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила в качестве исходного соединения. Способ получения 12. (Циклопропилметил)[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метиламин. Стадия 1. [(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилциклопропан-карбоксамид. К 4,6 г (20 ммоль) гидрохлорида[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метиламина, растворенного в 60 мл дихлорметана, при комнатной температуре быстро добавляют 6,1 мл (44 ммоль/2,2 экв.) триэтиламина и перемешивают в течение около 30 мин. После этого в течение 40 мин по каплям добавляют 2 мл (22 ммоль/1,1 экв.) хлорида циклопропанкарбоновой кислоты. Смесь перемешивают при комнатной температуре в течение 1 ч 30 мин; экстрагируют 60 мл дихлорметана,промывают органический слой 40 мл воды, два раза по 40 мл 1N соляной кислоты, два раза по 40 мл насыщенным водным раствором NaHCO3 и 40 мл воды. После чего раствор сушат над сульфатом магния,отфильтровывают и выпаривают, получая необходимый продукт. Стадия 2. (Циклопропилметил)[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламин. К 1,2 г (30 ммоль) LiAlH4, растворенного в 40 мл THF, при комнатной температуре прикапывают раствор 5,2 г (20 ммоль) исходного соединения, растворенного в 80 мл THF, после чего смесь кипятят с обратным холодильником в течение 4 ч 30 мин, охлаждают до 0 С и добавляют 0,79 мл воды, 0,63 мл 20% водного раствора гидроокиси натрия и 2,9 мл воды. Перемешивают смесь при комнатной температуре, затем фильтруют на фритте, промывают THF. Объединенные фильтраты выпаривают, получая нужный продукт. Способ получения 13. [(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилэтанамин. Получение идентично получению продукта по способу 12 с использованием в качестве исходного соединения ацетилхлорида вместо хлорида циклопропанкарбоновой кислоты на стадии 1. Способ получения 14.N-Бензил-1-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метанамин. Получение идентично получению продукта по способу 12 с использованием в качестве исходного соединения бензоила хлорида вместо хлорида циклопропанкарбоновой кислоты на стадии 2. Способ получения 15. (Циклопентилметил)[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метиламин. Получение идентично получению продукта по способу 12 с использованием в качестве исходного соединения хлорида циклопентанкарбоновой кислоты вместо хлорида циклопропанкарбоновой кислоты на стадии 2. Способ получения 16. (Циклобутилметил)[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метиламин. Получение идентично получению продукта по способу 12 с использованием в качестве исходного- 10014756 соединения хлорида циклобутанкарбоновой кислоты вместо хлорида циклопропанкарбоновой кислоты на стадии 2. Способ получения 17. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-2,2,2 трифторметанамин. Стадия 1. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-2,2,2-трифторацетамид. К 2,9 г (15 ммоль) [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламина в 7,5 мл метанола при комнатной температуре быстро добавляют 2,1 мл (15 ммоль/1 экв.) триэтиламина, а затем по каплям в течение 20 мин приливают 2,3 мл (19 ммоль/1,25 экв.) этилтрифторацетата. Перемешивают реакционную массу при комнатной температуре в течение 2 ч, после чего выпаривают досуха. Остаток растворяют в 100 мл дихлорметана, промывают 50 мл 1N соляной кислоты, а затем 50 мл воды и сушат над сульфатом магния. После фильтрования и выпаривания получают нужный продукт. Стадия 2. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-2,2,2-трифторэтанамин. К 4,07 г (14,0 ммоль) соединения со стадии 1 в 28 мл THF при около 0 С в течение 10 мин прикапывают 21 мл (21 ммоль/1,5 экв.) 1 М раствора борана в THF, а затем кипятят с обратным холодильником в течение 18 ч. Смесь охлаждают и по каплям добавляют 14 мл 2,6N этанольного раствора HCl; затем соединение снова кипятят с обратным холодильником в течение 2 ч, охлаждают, отфильтровывают на фритте, промывают эфиром и сушат над вакуумом с получением гидрохлорида и после обработки в щелочной среде получают необходимый продукт. Способ получения 18. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилпроп-2 ен-1-амин. К 3,9 г (20 ммоль) [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламина в 40 мл ацетона при комнатной температуре быстро добавляют 4,1 г (30 ммоль/1,5 экв.) карбоната калия, а затем быстро прикапывают 1,9 мл (22 ммоль/1,1 экв.) аллилбромида. Смесь перемешивают при комнатной температуре в течение 60 ч; соль отфильтровывают, фильтрат выпаривают. Остаток (4,8 г) разделяют хроматографически на 300 г силикагеля (элюент: дихлорметан/этанол/аммиак: 98/2/0,2), получая нужный продукт. Способ получения 19. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилциклопентанамин. При комнатной температуре перемешивают 2,9 г (15 ммоль) [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламина и 1,3 мл (15 ммоль/1 экв.) циклопентанона в 60 мл дихлорметана. Добавляют 4,8 г (22,5 ммоль/1,5 экв.) триацетоксиборгидрида натрия, а затем 0,86 мл (15 ммоль/1 экв.) уксусной кислоты и перемешивают при комнатной температуре в течение 4 ч. Смесь выливают в 90 мл 1N раствора гидроокиси натрия; экстрагируют эфиром два раза по 120 мл. Объединенные органические фазы промываются 90 мл воды, а затем 90 мл насыщенного водного раствора хлорида натрия и сушат над сульфатом магния. После фильтрования и выпаривания получают нужный продукт. Способ получения 20. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилциклобутанамин. Способ идентичен способу 19, где в качестве исходного соединения используют циклобутанон вместо циклопентанона. Способ получения 21. (7-Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил трифторметилсульфонат. Стадия 1. 7-Цианобицикло[4.2.0]окта-1,3,5-триен-3-ил трифторметансульфонат. 2,4 мл ангидрида трифлатной кислоты (1,2 экв.) приливаются при 0 С в течение 1 ч к раствору 2 г(13,8 ммоль) 3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила в 40 мл дихлорметана и 3,9 мл (2 экв.) триэтиламина. Перемешивают смесь в течение 3 дней при 25 С. Реакционную массу выливают в воду, экстрагируют дихлорметаном, сушат над MgSO4, отфильтровывают и выпаривают, чтобы получить 3,8 г масла. Это вещество очищают методами хроматографии на 150 г силикагеля; элюент 100% толуол. Необходимый продукт получают в форме масла. Стадия 2. (7-Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил трифторметилсульфонат. 2,7 г (9,74 ммоль) продукта со стадии 1 растворяют в 40 мл метанола. Добавляют 40 мл (7N) раствора МеОН-NH3 и 1 г никеля Ренея. Гидрогенирование проводят под давлением 5 бар при 25 С, в течение 4 дней. Отфильтровывают на целите и выпаривают фильтрат досуха. Необходимый продукт получают в форме масла. Способ получения 22. (7-Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил диметилсульфамат. Стадия 1. 7-Цианобицикло[4.2.0]окта-1,3,5-триен-3-ил диметилсульфамат. 2 г (13,8 ммоль) 3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-карбонитрила смешивают с 40 мл дихлорметана, 3,9 г (2 экв.) триэтиламина и 1,63 мл (1,1 экв.) диметилсульфамоил хлорида. Перемешивают смесь при 25 С в течение 4 дней. Реакционную массу выливают в воду, экстрагируют дихлорметаном,сушат над MgSO4, фильтруют и выпаривают, получая масло, которое очищают методами хроматографии на 70 г силикагеля (элюент: дихлорметан/толуол: 50/50). Необходимый продукт получают в виде белого кристаллического соединения. Точка плавления (М.K.) 78-79 С. Стадия 2. (7-Аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил диметилсульфомат. 1,8 г (7,1 ммоль) продукта со стадии 1 растворяют в 40 мл метанола. Добавляют 40 мл (7N) раствораMeOH-NH3 и 1 г никеля Ренея. Гидрогенирование проводят под давлением 5 бар при 25 С в течение 4 дней. Отфильтровывают смесь на целите и выпаривают фильтрат досуха. Получается масло, которое очищают методами хроматографии на 50 г силикагеля (элюент: дихлорметан/этанол: 90/10). Необходимый продукт получают в виде масла. Способ получения 23. (5,6-Дигидроциклобута[4,5]бензо[1,2-b]фуран-6-илметил)амин. 7 г (41,3 ммоль) 5,6-дигидроциклобута[4,5]бензо[1,2-b]фуран-6-карбонитрила растворяют в 490 мл этанола и 70 мл 28% NH4OH. Добавляют 1 г никеля Ренея. Гидрогенирование проводят под давлением 5 бар при 25 С в течение 3 дней. Отфильтровывают полученную смесь на целите и выпаривают фильтрат досуха. Необходимый продукт получают в форме желтого масла. Способ получения 24. 2-(Бромметил)-5,6-диметоксииндан. 1 г (4,8 ммоль) (5,6-диметокси-2,3-дигидро-1 Н-инден-2-ил)метанола растворяют в 20 мл дихлорметана и 1 мл триэтиламина. Охлаждают смесь до 0 С и приливают 0,41 мл (1,1 экв.) мезилхлорида. Перемешивают реакционную массу при 25 С в течение 1 ч, гидролизуют ее, выливая на 30 г льда. Экстрагируют, промывают HCl (1N), а затем с H2O. Сушат над MgSO4, отфильтровывают и выпаривают. Получают 1,2 г бежевых кристаллов, которые растворяют в 30 мл ацетона. 0,83 г LiBr (2 экв.) добавляют к полученному раствору и перемешивают при 25 С в течение 12 ч. Выпаривают ацетон, вещество из воды извлекают экстракцией диэтиловым эфиром. Отделяют органическую фазу, сушат над MgSO4, фильтруют и выпаривают. Необходимый продукт получают в виде масла. Способ получения 25. 2-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил) амино]этанола. При комнатной температуре перемешивают 1,04 г (5 ммоль) [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина, 0,44 мл (6 ммоль/1,2 экв.) 2-бромэтанола и 2,07 г(15 ммоль/ 3 экв.) карбоната калия и 10 мл ацетонитрила. Смесь кипятят с обратным холодильником в течение 12 ч, отгоняют растворители. Продукт растворяют в дихлорметане, промывают водой и сушат над сульфатом магния, после выпаривания получают желаемый продукт. Способ получения 26. 2-(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил) амино]этанол. Получение идентично способу 25, а в качестве исходного соединения используют вместо [(7S)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина[(7R)-3,4-диметоксибицикло[4.2.0] окта-1,3,5-триен-7-ил]метилметиламин. Способ получения 27. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метилэтан-1,2-диамин. Стадия 1. (7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]ацетонитрил. При комнатной температуре перемешивают 1,04 г (5 ммоль) [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина, 0,35 мл (5 ммоль/1 экв.) бромацетонитрила и 2,16 г (20 ммоль/4 экв.) карбоната натрия и 16 мл метилизобутил кетона. Кипячение с обратным холодильником длится 12 ч, после чего отгоняют растворители. Остаток выливают в воду и экстрагируют дихлорметаном. Объединенные органические фазы сушат над сульфатом магния, после выпаривания получают необходимый продукт. Стадия 2. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил)-N-метилэтан-1,2 диамин. Раствор 1,1 г (4,5 ммоль) соединения со стадии 1 в 10 мл THF прикапывают при комнатной температуре к 0,21 г литийаллюмогидрида (5,4 ммоль), растворенного в 5 мл THF, и перемешивают в течение 5 ч. Охлаждают полученную смесь до 0 С и добавляют сначала 0,14 мл воды, 0,11 мл 20% водного раствора гидроокиси натрия и 0,51 мл воды. Перемешивают в течение ночи при комнатной температуре, а затем отфильтровывают на фритте и промывают THF. Объединенные фильтраты выпаривают, получая нужный продукт. Способ получения 28. N-[(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метилэтан-1,2-диамин. Получение идентично способу 27, а в качестве исходного соединения используют вместо [(7S)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина[(7R)-3,4-диметоксибицикло[4.2.0] окта-1,3,5-триен-7-ил]метилметиламин. Способ получения 29. [(4-Метокси-3-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 3-(3-Бром-4-метокси-5-метилфенил)пропаннитрил. 62,8 г (358,4 ммоль) 3-(4-метокси-5-метилфенил)пропаннитрила растворяют в 390 мл уксусной кислоты. К раствору быстро добавляют 58,8 г (716,8 ммоль/2 экв.) ацетата натрия, а затем прикапывают 20,2 мл (394,2 ммоль/1,1 экв.) брома и перемешивают при комнатной температуре в течение ночи. Реакционную смесь выливают в 2 л воды, водную фазу экстрагируют 3 раза 500 мл дихлорметана. Объединенные органические экстракты промывают 500 мл воды, 500 мл насыщенного водного раствора гидрокарбоната натрия и 500 мл воды, сушат над сульфатом магния. После выпаривания получают необходимый продукт.- 12014756 Стадия 2. 4-Метокси-3-метилбицикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. 94 г (370 ммоль) полученного на предыдущей стадии вещества растворяют в 230 мл эфира и приливают амид натрия (925 ммоль/2,5 экв.) в 1850 мл нашатырного спирта. После перемешивания в течение 3 ч при кипячении с обратным холодильником реакцию останавливают, добавляя 99 г (5 экв.) кристаллического хлористого аммония, после чего аммиак отгоняют. Остаток растворяют в 500 мл воды и 500 мл дихлорметана, перемешивают, разделяют фазы, водную фазу экстрагируют дважды 500 мл дихлорметана. Объединенные органические экстракты сушат над сульфатом магния и выпаривают. Остаток (68 г) разделяют хроматографически на 800 г силикагеля (элюент: дихлорметан/циклогексан: 50/50), после перекристаллизации из 150 мл изопропилового эфира получают необходимый продукт. Стадия 3. [(4-Метокси-3-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. 4,0 г (23,1 ммоль) полученного на предыдущей стадии вещества растворяют в 75 мл метанола и 75 мл 7N аммиачного метанола, после чего добавляют 3,3 мл никеля Ренея. Гидрогенирование проводят при комнатной температуре и нормальном давлении в течение 2 ч. Отфильтровывают на целите, промывают метанолом и выпаривают, получая нужный продукт. Способ получения 30. [(3-Метокси-4-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Стадия 1. 5-Метокси-4-метилбензальдегид. 36,35 г (238,8 ммоль) (3-метокси-4-метилфенил)метанола и 207,6 г (2,4 моль/10 экв.) окиси марганца перемешивают с 730 мл дихлорметана при комнатной температуре в течение ночи, отфильтровывают на целите и промывают дихлорметаном. После выпаривания объединенных фильтратов получают нужный продукт. Стадия 2. 2-Бром-5-метокси-4-метилбензальдегид. При комнатной температуре растворяют 35,5 г (238,7 ммоль) полученного на предыдущей стадии вещества в 290 мл дихлорметана, быстро добавляют к полученному раствору 29,4 г (358 ммоль/1,5 экв.) ацетата натрия, а затем прикапывают раствор 15 мл (286,4 ммоль/1,2 экв.) брома в 130 мл дихлорметана. Перемешивают реакционную смесь при комнатной температуре в течение 4 ч и отфильтровывают; фильтрат промывают 1N раствором тиосульфата натрия и сушат над сульфатом магния. После выпаривания остаток перекристаллизовывают из 130 мл гептана, получая на выходе нужный продукт. Стадия 3. 3-(2-Бром-5-метокси-4-метилфенил)акрилонитрил. 4,22 г (183,6 ммоль/1 экв.) натрия растворяют в 180 мл безводного этанола и охлаждают полученный раствор до 0 С. Добавляют по каплям 29,7 мл (183,6 ммоль/1 экв.) диэтоксифософоноацетонитрила. Перемешивают смесь при 0 С в течение 30 мин; по частям добавляют 42,05 г (183,6 ммоль) полученного на предыдущей стадии вещества и снова перемешивают при 0 С в течение 30 мин, а затем при комнатной температуре в течение 1 ч. Реакционную смесь приливают к 1800 мл воды и перемешивают в течение 30 мин; твердое вещество отфильтровывают, промывают водой и сушат под вакуумом с получением нужного продукта. Стадия 4. 3-(2-Бром-5-метокси-4-метилфенил)пропаннитрил. При комнатной температуре перемешивают 46 г (182,5 ммоль) полученного на предыдущей стадии вещества и 27,7 г (730 ммоль/4 экв.) боргидрида натрия в 380 мл изопропанола, после чего смесь кипятят с обратным холодильником в течение 2 дней (1 экв. боргидрида натрия добавляют через 1 день прохождения реакции). Смесь охлаждают, а затем выпаривают. Остаток растворяют в 630 мл воды и льда и подкисляют до рН 1, добавляя 90 мл концентрированной соляной кислоты. Водный раствор экстрагируют дважды этилацетатом. Объединенные органические фазы промывают насыщенным водным раствором хлорида натрия, а затем сушат над сульфатом магния. После выпаривания остаток перекристаллизовывают из 100 мл гептана, получая нужный продукт. Стадия 5. 3-Метокси-4-метилбщикло[4.2.0]окта-1,3,5-триен-7-карбонитрил. Способ получения аналогичен стадии 2 способа 29. Стадия 6. [(3-Метокси-4-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]амин. Способ получения аналогичен стадии 3 способа 29. Пример 1. N-[(3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Стадия 1. 7,8-Диметокси-1,3,4,5-тетрагидро-2 Н-3-бензазепин-2-он. При комнатной температуре растворяют 35 г (159,6 ммоль) 7,8-диметокси-1,3-дигидро-2 Н-3 бензазепин-2-она в 525 мл этанола и 175 мл этилацетата. После этого добавляют 7 г (20 вес.%) 10% палладий на углероде и проводят гидрогенирование при 65 С и 3,5 бар в течение 22 ч. Катализатор отфильтровывают на целите; промывают этанол/этилацетатом и выпаривают досуха, получая на выходе нужный продукт. Стадия 2. 7,8-Диметокси-2,3,4,5-тетрагидро-1 Н-3-бензазепина гидрохлорид. 22,13 г (100 ммоль) продукта, полученного на стадии 1, растворяют в 250 мл THF, охлаждают раствор до 0 С. Прикапывают в течение 15 мин 250 мл борана в THF (1M в THF) и кипятят при 80 С в течение 24 ч. Смесь охлаждают, после чего к ней прикапывают 250 мл этанола, а затем 2,6N раствора HCl в этаноле. Реакционную массу кипятят с обратным холодильником в течение 40 мин; отфильтровывают и выпаривают досуха. Таким образом получают остаток, который перекристаллизовывают из 150 мл изо- 13014756 пропанола и 20 мл воды, получая нужный продукт. Стадия 3. 3-Акрилоил-7,8-диметокси-2,3,4,5-тетрагидро-1 Н-3-бензазепин. При комнатной температуре растворяют 70 г (287,2 ммоль) продукта, полученного на стадии 2 в 1200 мл дихлорметана, после чего быстро добавляют 100 мл (718 ммоль/2,5 экв.) триэтиламина. Смесь охлаждают до 0 С и по каплям добавляют раствор 25,8 мл (315,9 ммоль/1,1 экв.) акрилоил хлорида в 270 мл дихлорметана, поддерживая температуру близко 0 С. Через 2 ч при 0 С смесь перемешивают при комнатной температуре в течение ночи. Экстрагируют 750 мл воды, 750 мл 1N соляной кислоты и 750 мл воды, а затем сушат над сульфатом магния. Полученный после фильтрования и выпаривания остаток разделяют хроматографически на 3,2 кг силикагеля (элюент: дихлорметан/этанол: 95/5), перекристаллизовывают из изопропанола. После фильтрования на фритте сушат вещество под вакуумом при 50 С, получая нужный продукт. Стадия 4. N-[(3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. 1,05 г (4,0 ммоль) продукта, полученного на стадии 3, и 0,82 г (4,0 ммоль) [(7R,S)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина растворяют в 5 мл THF. Полученный раствор кипятят с обратным холодильником (масляная баня 90 С) в течение ночи, продувая азот таким образом, чтобы растворитель постепенно выпаривался. Полученный остаток разделяют хроматографически на силикагеле (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), после чего вещество растворяют в изопропаноле и обрабатывают 1,1 экв. HCl в эфире и продолжают перемешивать, пока не образуется осадок. После сушки полученное вещество уплотняют, растворяя в этилацетате (10 мл) и ацетонитриле(20 мл), отфильтровывают и сушат, получая необходимый продукт. Точка плавления (М.K.): 176-179 С. Пример 2. N-[(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо рацемического [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина[(7R)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин. Точка плавления (M.K.): 155-158 С. Оптическое вращение: растворитель: ДМСО, С=0,01 г/см 3, Т=20 С, L=589 нм, D=-1,46. Пример 3 а. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо рацемического [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина [(7S)3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин. Точка плавления (M.K.): 157-160 С. Оптическое вращение: растворитель: ДМСО, С=0,01 г/см 3, Т=20 С, L=589 нм, D =+1,6.IR (cm-1): 2438 (NH+), 1625 (C=O), 1234-1203-1179 (С-О-С), 865-846 (СН-Ar). Пример 3b. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин фумарат. Необходимый продукт получают преобразованием N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5 триен-7-ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1 амина, полученного реакцией гидрохлорида в примере 3a, с основанием с образованием соли фумаровой кислоты. Элементарный микроанализ: Пример 3c. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гемипамоат. Необходимый продукт получают преобразованием N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5 триен-7-ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1 амина, полученного реакцией гидрохлорида в примере 3a, с основанием с образованием соли памовой кислоты. Элементарный микроанализ: Пример 3d. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин геминападисилат. Необходимый продукт получают преобразованием N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5- 14014756 триен-7-ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1 амина, полученного реакцией гидрохлорида в примере 3a, с основанием с образованием соли 1,5 нафталиндисульфоновой кислоты. ЯМР 1H: Пример 4. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо рацемического [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина [(7S)3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламин и вместо THF диоксан. Точка плавления (M.K.): 177-180 С. Оптическое вращение: растворитель: МеОН, С=0,011 г/см 3, Т=20 С, L=589 нм, D=-4,15.IR (cm-1): 2723 (NH+), 1645 (С=O). Пример 5. N-[(4,5-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина продукт, полученный по способу 1, и заменяя THF на диоксан. Точка плавления (M.K.): 199-201 С.N-[2-(5,6-Диметокси-2,3-дигидро-1H-инден-2-ил)этил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина [2-(5,6-диметокси-2,3 дигидро-1 Н-инден-2-ил)этил]метиламин. Точка плавления (М.K.): 193-199 С. Пример 7. N-[(5,6-Диметокси-2,3-дигидро-1 Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина [(5,6-диметокси-2,3 дигидро-1 Н-инден-2-ил)метил]метиламин. Точка плавления (M.K.): 194-197 С. Пример 8. N-[(2,3-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина продукт, полученный в способе 10. Точка плавления (M.K.): 220-222 С. Пример 9.N-[(2,3,4-Триметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гемифумарат. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина продукт, полученный способом 11. Конверсия соли проходит при наличии фумаровой кислоты. Точка плавления (M.K.): 155-157 С. Пример 10. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[(5-метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 4 в качестве исходного соединения вместо [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламина продукт, полученныйN-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7-метокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают аналогично соединению примера 1, используя на стадии 1 в качестве исходного соединения вместо 7,8-диметокси-1,3-дигидро-2 Н-3-бензазепин-2-она 8-метокси-1,3-дигидро-2 Н-3-бензазепин-2 он, а на стадии 4 заменяя рацемический (3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил)метил]метиламин на [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метиламин и THF на диоксан. Точка плавления (M.K.) 171-173 С.N-[2-(5,6-Диметокси-2,3-дигидро-1 Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Стадия 1. N-Бензил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1 амин. Получают аналогично соединению в примере 1 стадия 4, заменяя [(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]метиламин на бензиламин. Точка плавления (M.K.) 100-102 С. Стадия 2. N-Бензил-N-[(5,6-диметокси-2,3-дигидро-1 Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин. 1,7 г (4,6 ммоль) продукта, полученного на стадии 1, и 1,.25 г (1 экв.) продукта, полученного в способе 24, растворяют в 20 мл ацетонитрила. 5,1 г (8 экв.) K2CO3 добавляют к полученной смеси и кипятят с обратным холодильником в течение 24 ч. Смесь охлаждают и фильтруют, а полученный фильтрат выпаривают. Остаток очищают хроматографически на силикагеле (элюент: CH2Cl2/этанол: 95/5), получая необходимый продукт в виде масла. Стадия 3.N-[2-(5,6-Диметокси-2,3-дигидро-1 Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. 2,3 г (4,1 ммоль) продукта, полученного на стадии 2, растворяют в 46 мл этанола и добавляют 230 мг Pd/C 5%. Гидрогенирование проводят под давлением 1 бар и при комнатной температуре в течение 24 ч. Смесь фильтруют на целите, промывают этанолом, а фильтрат выпаривают досуха. Получают 1,95 г вещества, которое растворяют в 15 мл этанола и добавляют 2,5 мл (2 М) раствора Et2O-HCl. Продукт выкристаллизовывается, отфильтровывается и сушится. Необходимое вещество получают в форме белых кристаллов. Точка плавления (M.K.) 198-204 С. Пример 13. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[(2-метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-оксопропан-1-амин гидрохлорид. При комнатной температуре перемешивают 0,93 г (3,6 ммоль/1 экв.) продукта, полученного на стадии 3 примера 1, 0,58 г (3,6 ммоль) продукта, полученного способом 2, и 0,11 г (0,18 ммоль/0,05 экв.) трифлата иттербия в 10 мл толуола. Кипятят при 100 С, пока не пройдет реакция (определяют методами тонкослойной хроматографии), а затем выпаривают досуха. Полученный остаток разделяют хроматографически на силикагеле (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в виде основания. Затем растворяют его в 10 мл ацетонитрила и 0,56 мл (1,1 экв.) 3,5N этанольного раствора HCl, фильтруют и сушат, получая необходимый продукт. Точка плавления (M.K.) 202-204 С.[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный в способе 2, продуктом,полученным в способе 5. Точка плавления (M.K.) 221-225 С. Пример 15. N-[(3-трет-Бутоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гемифумарат. Получают так же, как продукт примера 13, заменяя продукт, полученный в способе 2, продуктом,полученным в способе 6, и используя при солеобразовании фумаровую кислоту вместо соляной. Точка плавления (M.K.) 189-191 С. Пример 16. N-(5,6-Дигидроциклобута[f][1,3]бензодиоксол-5-илметил)-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный в способе 2, продуктом,полученным в способе 7. Точка плавления 180-184 С. Пример 17. N-(5,6-Дигидроциклобута[f][1,3]бензодиоксол-5-илметил)-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид.- 16014756 Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, на (5,6 дигидроциклобута[4,5]бензо[1,2-d][1,3]диоксол-5-илметил)амин. Точка плавления (M.K.) 201-207 С. Пример 18.N-[2-(5,6-Диметокси-2,3-дигидро-1 Н-инден-2-ил)этил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 8. Точка плавления (M.K.) 186-189 С. Пример 19. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил трифторметансульфонат гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 21. Точка плавления (M.K.) 162-168 С. Пример 20. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил диметилсульфамат гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 22. Точка плавления (M.K.) 228-235 С. Пример 21. N-(5,6-Дигидроциклобута[f][1]бензофуран-6-илметил)-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 23. Точка плавления (M.K.) 245-255 С. Пример 22. N-(Циклопропилметил)-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил] метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 12, и толуол на диоксан. Точка плавления (M.K.) 88-92 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -13,25.IR (cm-1): 1632 (C=O), 3420 (H2O), 3000 2200 (NH+). Пример 23. N-Аллил-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8 диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 18, и толуол на диоксан. Точка плавления (M.K.) 74-80 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -4,6.IR (cm-1): 1631 (C=O), 3400 (H2O), 3000 2000 (NH+). Пример 24. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-этил-3-оксопропан-1-амин гемифумарат. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 13, и толуол на диоксан. Соль получают, используя фумаровую кислоту. Точка плавления (M.K.) 71-73 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -6,67.IR (cm-1): 2200 2700 (NH+/OH), 1701-1631 (C=O), 1276-1203 (С-О-С). Пример 25. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-(2,2,2-трифторэтил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 17, и толуол на диоксан. Точка плавления (M.K.) 68-70 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -8,04.IR (cm-1): 1633 (С=O), 2800 1900 (-NH+), 1107 1206 (-CF3). Пример 26. N-Бензил-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8 диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин фумарат. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 14, и толуол на диоксан. Соль получают, используя фумаровую кислоту. Точка плавления (M.K.) 146-148 С. Оптическое вращение: растворитель: МеОН, С=0,011 г/см 3, Т=20 С, L=589 нм, D = +2,07.IR (cm-1): 1708 и 1646 (C=O), 3000 2500 (-ОН). Пример 27. N-Циклопентил-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин фумарат. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,- 17014756 полученным способом 19, и толуол на диоксан. Точка плавления (M.K.) 79-82 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -10,54.N-(Циклопентилметил)-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 15, и толуол на диоксан. Точка плавления (M.K.) 87-90 С. Оптическое вращение: растворитель: МеОН, С=0,008 г/см 3, Т=20 С, L=589 нм, D = -19,07.N-(Циклобутилметил)-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7 ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин фумарат. Получают так же, как продукт примера 13, заменяя продукт, полученный способом 2, продуктом,полученным способом 16, и толуол на диоксан. Соль получают реакцией с фумаровой кислотой. Точка плавления (M.K.) 68-71 С. Оптическое вращение: растворитель: МеОН, C=0,01 г/см 3, Т=20 С, L=589 нм, D = -12,04.IR (cm-1): 1703 и 1634 (C=O), 1279-1072 (С-О-С). Пример 30. N-(Циклобутил)-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. При комнатной температуре перемешивают 1 г (2,2 ммоль) N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро-3 Н-3-бензазепин-3-ил)3-оксопропан-1-амина (полученного восстановлением соединения примера 4 до основания) и 0,17 мл (2,2 ммоль/1 экв.) циклобутанона и 7 мл дихлорметана. Затем к смеси добавляют 0,7 г (3,3 ммоль/1,5 экв.) триацетоксиборгидрида натрия, потом 0,13 мл (2,2 ммоль/1 экв.) уксусной кислоты и перемешивают при комнатной температуре в течение 5 ч. Если реакция не завершается, добавляют 0,08 мл (0,5 экв.) циклобутанона и 350 мг (0,75 экв.) триацетоксиборгидрид натрия и перемешивают при комнатной температуре в течение 1 ч 45 мин и больше. После этого к реакционной массе добавляют 1N водный раствор гидроокиси натрия и экстрагируют дважды 15 мл эфира. Объединенные органические экстракты промывают 10 мл воды, а потом 10 мл насыщенного водного раствора хлорида натрия и сушат над сульфатом магния. После фильтрования и выпаривания остаток (1 г) разделяют хроматографически на колонке с 50 г силикагеля (элюент: дихлорметан/этанол/аммиак: 98/2/0,2), получая нужный продукт в виде основания. Полученное вещество растворяют в 9 мл этилацетата и 12 мл эфира и превращают в соль реакцией с 0,63 мл 2N HCl в эфире, получая необходимый продукт. Точка плавления (M.K.) 93-95 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -5,63.IR (cm-1): 2424 (-NH+), 1633 (С=O), 1205-1107 (С-О-С). Пример 31. N-[(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-(6,7,8 триметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт со стадии 3 примера 1 на 3-акрилоил 6,7,8-триметокси-2,3,4,5-тетрагидро-1H-3-бензазепин и продукт по способу 2 на [(7R)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин, а также толуол на THF. Точка плавления (M.K.) 83-86 С. Пример 32. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-(6,7,8 триметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт стадии 3 примера 1 на 3-акрилоил 6,7,8-триметокси-2,3,4,5-тетрагидро-1 Н-3-бензазепин и продукт способа 2 на [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин, а также толуол на THF. Точка плавления (M.K.) 83-87 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -3,00.N-[(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7-метокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт стадии 3 примера 1 на 3-акрилоил 6,7,8-триметокси-2,3,4,5-тетрагидро-1 Н-3-бензазепин и продукт способа 2 на [(7R)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин, а также толуол на THF. Точка плавления (M.K.) 126-129 С. Оптическое вращение: растворитель: МеОН, С=0,0126 г/см 3, Т=20 С, L=589 нм, D = +2,67. Пример 34.N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7-метокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 13, заменяя продукт стадии 3 примера 1 на 3-акрилоил- 18014756 6,7,8-триметокси-2,3,4,5-тетрагидро-1H-3-бензазепин и продукт способа 2 на [(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин, а также толуол на THF. Точка плавления (M.K.) 127-129 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -2,7.IR (cm-1): 1629 (С=O), 3000 1800 (-NH+), 3500 (-ОН) (слабый). Пример 35. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ол гидрохлорид. 10 г (38,3 ммоль/0,95 экв.) продукта, полученного на стадии 3 примера 1, 6 г (40,2 ммоль) продукта,полученного способом 3 (0,2 ммоль/0,05 экв.), и 42 г трихлорида рутения растворяют в 19 г ПЭГ 300. Раствор кипятят при 40 С в течение 4 ч. Реакционную смесь выливают в 600 мл дихлорметана и экстрагируют дважды 500 мл, а затем дважды 800 мл воды. Органические экстракты сушат над сульфатом магния. После выпаривания остаток разделяют хроматографически на 500 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в виде основания. 1 г чистого основания уплотняют в 9 мл этанола и затем превращают в соль, используя 1,1 экв. 2N HCl в эфире, в 9 мл ацетонитрила и затем перекристаллизовывают из этанола (27 мл) и воды (4 мл), фильтруют и сушат, получая необходимый продукт в форме гидрохлорида. Точка плавления (M.K.) 212-215 С.IR (cm-1): 1633 (C=O), 3400 2400 (-NH2+/OH). Пример 36. 8-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. Получают так же, как продукт примера 35, заменяя продукт способа 3 продуктом способа 4. Точка плавления (M.K.) 193-195 С.IR (cm-1): 1627 (С=O), 3700 2200 (-NH2+/OH). Пример 37. N-[(Бицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5-тетрагидро 3H-3-бензазепин-3-ил)-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 35, заменяя продукт способа 5 на (бицикло[4.2.0]окта-1,3,5 триен-7-илметил)амин. Точка плавления (M.K.)189-193 С. Пример 38. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[4-метокси-3-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 35, заменяя продукт способа 3 продуктом способа 29. Точка плавления (M.K.) 166-168 С. Пример 39. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-метокси-4-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-оксопропан-1-амин гидрохлорид. Получают так же, как продукт примера 35, заменяя продукт способа 3 продуктом способа 30. Точка плавления (M.K.) 211-214 С. Пример 40. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-4-оксобутан-1-амин гидрохлорид. Стадия 1. 4-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-4-оксобутан-1-ол. К 6,0 г (29 ммоль) 7,8-диметокси-2,3,4,5-тетрагидро-1H-3-бензазепина в 30 мл THF при 0 С по каплям добавляют в течение 15 мин 11,6 мл (29 ммоль/1 экв.) 2,6 М раствор н-бутиллития в гексане. Смесь охлаждают до около -78 С и прикапывают в течение 10 мин к раствору 2,7 мл (34,8 ммоль/1,2 экв.) гамма-бутиролактона в 15 мл THF. Перемешивают реакционную массу при -78 С в течение 30 мин, после чего температуру доводят до -30 С и удерживают в течение 1 ч; после чего температуру поднимают до комнатной и перемешивают в течение 48 ч. Смесь приливают к 75 мл насыщенного водного раствора хлористого аммония и экстрагируют дважды 120 мл этилацетата. Объединенные органические экстракты промываются 150 мл насыщенного водного раствора хлорида натрия, сушат над сульфатом магния и выпаривают. Остаток растворяют в 300 мл этилацетата и экстрагируют дважды 75 мл водного 1 М раствора соляной кислоты. Объединенные водные фазы повторно экстрагируют дважды 100 мл дихлорметана. Органические фазы объединяют, сушат над сульфатом магния и выпаривают, получая нужный продукт. Стадия 2. 4-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-ил)-4-оксобутаналъ. 3,3 мл (46 ммоль/6 экв.) ДМСО прикапывают при -60 С к 2,0 мл (23 ммоль/3 экв.) оксалилхлорида в 45 мл дихлорметана. Смесь нагревают до около -25 С и добавляют по каплям раствор 2,25 г (7,7 ммоль) соединения, полученного на стадии 1, в 22 мл дихлорметана. Перемешивают полученную реакционную массу при -25 С в течение 15 мин; после чего прикапывают 6,5 мл (46 ммоль/6 экв.) триэтиламина и доводят температуру до комнатной. Затем к смеси добавляют 75 мл дихлорметана и 75 мл воды; перемешивают, разделяют фазы и промывают органическую фазу 75 мл 1N водного раствора соляной кислоты,75 мл воды, 75 мл 1N водного раствора гидроокиси натрия и 75 мл воды. После сушки над сульфатом магния и выпаривания получают нужный продукт. Стадия 3. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-4-оксобутан-1-амин гидрохлорид. При комнатной температуре смешивают 2,53 г (12,2 ммоль) [(7S)-3,4-диметоксиби- 19014756 цикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламина, 3,95 г (12,2 ммоль/1 экв.) соединения, полученного на стадии 2, и 90 мл дихлорметана. К смеси добавляют 3,88 г (18,3 ммоль/1,5 экв.) триацетоксиборгидрид натрия и перемешивают при комнатной температуре в течение 4 ч. Затем добавляют 60 мл 1N водного раствора гидроокиси натрия, экстрагируют дважды 150 мл этилацетата. Объединенные органические фазы промывают 100 мл воды, а затем 100 мл насыщенного водного раствора хлорида натрия и сушат над сульфатом магния. После фильтрования и выпаривания полученный остаток разделяют хроматографически на 300 г силикагеля (элюент: дихлорметан/этанол/аммиак: 97/3/0,3), получая нужный продукт в форме основания. Продукт растворяют в 13 мл изопропанола и превращают в соль реакцией с 0,67 мл 4,8N HCl в этаноле, а затем перекристаллизовывают из 25 мл этилацетата, получая необходимый продукт. Точка плавления (M.K.) 132-136 С. Оптическое вращение: растворитель: МеОН, С=0,012 г/см 3, Т=20 С, L=578 нм, D = +4,28.IR (cm-1): 3500 (ОН), 2441 (NH+), 1610 (C=O), 1278-1234-1204 (C-O-C), 865-845 (CH-Ar). Пример 41. N-[(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-4-оксобутан-1-амин гидрохлорид. Получают так же, как и продукт, полученный в примере 40, заменяя на стадии 3 [(7S)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин на [(7R)-3,4-диметоксибицикло[4.2.0] окта-1,3,5-триен-7-ил]метилметиламин (2 экв.). Точка плавления (M.K.) 106-137 С. Пример 42. N-[(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-4-оксобутан-1-амин гидрохлорид. Получают так же, как и продукт, полученный в примере 40, заменяя на стадии 3 [(7S)-3,4 диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метилметиламин на [(7R)-3,4-диметоксибицикло[4.2.0] окта-1,3,5-триен-7-ил]метилметиламин. Точка плавления (M.K.) 169-171 С. Оптическое вращение: растворитель: МеОН, С=0,009 г/см 3, Т=20 С, L=589 нм, D = -0,9.IR (cm-1): 2719 (NH2+), 1630 (C=O), 1276-1202-1177 (С-О-С), 862-832-781 (СН-Ar). Пример 43. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. При комнатной температуре смешивают 3,2 г (7,8 ммоль) продукта, полученного в примере 35, с 6,4 мл (78 ммоль/10 экв.) 37% водного раствора формальдегида и 0,98 г (15,6 ммоль) цианоборгидрида натрия и 145 мл ацетонитрила. Перемешивают полученную смесь в течение 1 ч 40 мин при комнатной температуре, затем выпаривают досуха. К остатку добавляют 250 мл воды, водную фазу экстрагируют дважды 150 мл дихлорметана. Объединенные органические фазы промывают водой и сушат над сульфатом магния. После фильтрования, выпаривания и хроматографического разделения на 200 г силикагеля (элюент: этанол/аммиак/дихлорметан: 95/5/0,5) получают необходимый продукт в форме основания. В результате преобразования основания, растворенного в ацетонитриле, путем реакции с HCl в эфире получают гидрохлорид необходимого продукта. Точка плавления (M.K.) 167-170 С.IR (cm-1): 3600 2000 (OH/NH+), 1616 (С=O). Пример 44. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил метилкарбамат гидрохлорид. К 0,7 г (1,65 ммоль) соединения, полученного в примере 43, в 30 мл толуола при комнатной температуре в автоклаве добавляют 0,2 мл (3,3 ммоль/2 экв.) метилизоцианата и кипятят при 90 С в течение 48 ч. После выпаривания остаток разделяют хроматографически на 130 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в форме основания. Его растворяют в 6 мл этанола и превращают в соль реакцией с 0,5 мл 3N HCl в этаноле, получая необходимый продукт. Точка плавления (M.K.) 185-188 С. Пример 45. 8-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ол. Получают так же, как продукт, полученный в примере 43, заменяя продукт, полученный в примере 35, на продукт примера 36. Точка плавления (M.K.) 201-205 С.IR (cm-1): 3071 (ОН), 2672 (NH+), 1634 (C=O), 1250-1228-1196 (С-О-С), 885-749-741 (СН-Ar). Пример 46. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил метилкарбамат гидрохлорид. Получают так же, как продукт, полученный в примере 44, заменяя продукт, полученный в примере 45, на продукт примера 43. Точка плавления (M.K.) 107-110 С. Пример 47. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид.- 20014756 При комнатной температуре к раствору 0,7 г (1,65 ммоль) соединения, полученного в примере 43, в 5 мл пиридина быстро добавляют 0,3 мл (2,06 ммоль/1,25 экв.) триэтиламина, а затем прикапывают 0,2 мл (2,06 ммоль/1,25 экв.) диметилкарбомоил хлорида. Перемешивают полученную реакционную массу при комнатной температуре в течение ночи, после чего смесь выпаривают. Остаток разделяют хроматографически на 130 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в форме основания. Его растворяют в 7 мл этанола и подвергают реакции с 0,5 мл 3N HCl в этаноле, получая гидрохлорид. Точка плавления (M.K.) 176-180 С. Пример 48. (-) Энантиомер 7-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3 оксопропил](метил)амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. 1,9 г соединения, полученного в примере 47, разделяют хроматографически на хиральной фазе; подвижной фазой является MeOH/DEA: 1000/1. Первый вымываемый продукт соответствовал ожидаемому продукту, который превращали в соль, используя 3N HCl в этаноле. Точка плавления (M.K.) 132-137 С. Оптическое вращение: растворитель: МеОН, С=0,012 г/см 3, Т=20 С, L=589 нм, D = -6,72. Пример 49. (+) Энантиомер 7-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3 оксопропил](метил)амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. Второй продукт, вымываемый в примере 48, соответствовал ожидаемому продукту, который превращали в соль при тех же условиях, что и продукт в примере 48. Точка плавления (M.K.) 164-167 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = +7,15. Пример 50. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. Получают так же, как и соединение в примере 47, используя соединение, полученное в примере 45,вместо соединения примера 43. Точка плавления (M.K.) 108-112 С. Пример 51. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат гидрохлорид. К раствору 0,95 г (2,24 ммоль) соединения, полученного в примере 43, в 20 мл дихлорметана при комнатной температуре быстро добавляют 0,33 мл (2,35 ммоль/1,05 экв.) триэтиламина. Реакционную массу охлаждают до 0 С и прикапывают к ней 0,23 мл (2,46 ммоль/1,1 экв.) этилхлорформиата. Перемешивают смесь при 0 С в течение 1 ч 30 мин, затем температуру доводят до комнатной. Органическую фазу дважды промывают 20 мл воды и сушат над сульфатом магния. После выпаривания остаток (0,96 г) разделяют хроматографически на 130 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в форме основания, который, когда растворяют в 9 мл этанола и подвергают реакции с 0,65 мл 3N HCl в этаноле, дает гидрохлорид. Точка плавления (M.K.) 149-152 С. Пример 52. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил метилкарбамат гидрохлорид. Стадия 1. трет-Бутил[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил-3-оксопропил]-[(4 гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат. При комнатной температуре смешивают 1,93 г (4,7 ммоль) продукта, полученного в примере 36, и 1,03 г (4,7 ммоль/1 экв.) ди-трет-бутила дикарбоната с 20 мл дихлорметана. После перемешивания в течение 2 ч при комнатной температуре к реакционной смеси добавляют 50 мл дихлорметана; промывают дважды 40 мл воды и сушат над сульфатом магния. После выпаривания получают необходимый продукт. Точка плавления (M.K.) 150-154 С. Стадия 2. 8-трет-Бутоксикарбонил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил)метилкарбамат. Способ получения аналогичен описанному в примере 44, используя вместо продукта, полученного в примере 43, продукт, полученный на стадии 1, описанной выше. Стадия 3. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил метилкарбамат гидрохлорид. 2,8 мл раствора (8,4 ммоль/10 экв.) 3N HCl в этилацетате добавляют к раствору 0,48 г (0,84 ммоль) соединения, полученного на стадии 2, в 8 мл этилацетата. Перемешивают смесь при комнатной температуре в течение ночи; образованный осадок отфильтровывают, промывают этилацетатом и сушат под вакуумом, получая необходимый продукт. Точка плавления (M.K.) 150-154 С. Пример 53. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. Стадия 1. 8-трет-Бутоксикарбонил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил)диметилкарбамат.- 21014756 Способ получения аналогичен описанному в примере 47, используя вместо продукта, полученного в примере 43, продукт, полученный на стадии 1 примера 52. Стадия 2. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. К 0,93 г (1,6 ммоль) соединения, полученного на стадии 1 и растворенного в 9 мл этанола, добавляют 2,4 мл (7,2 ммоль/4,5 экв.) 3N HCl в этаноле. Перемешивают смесь при комнатной температуре в течение ночи; полученный осадок отфильтровывают и сушат под вакуумом, получая необходимый продукт. Точка плавления (M.K.) 220-222 С. Пример 54. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. Стадия 1. 7-трет-Бутоксикарбонил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил)диметилкарбамат. 3,6 г (8,8 ммоль) трет-бутил[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3 оксопропил][(3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат растворяют в 50 мл дихлорметана. При 0 С к раствору прибавляют 2,1 г (1,1 экв.) хлорида N,N-диметилкарбомоила, перемешивают при 25 С в течение 3 дней. Смесь выливают в воду, экстрагируют дихлорметаном, сушат надMgSO4, фильтруют и выпаривают досуха. Остаток очищают методами хроматографии на 70 г силикагеля(элюент: CH2Cl2/этанол: 98/2 90/10), получая необходимый продукт. Стадия 2. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамат гидрохлорид. 0,8 г (1,37 ммоль) продукта, полученного на стадии 1, растворяют в 4 мл этанола и 2 мл 3,2N HCl в этаноле. Кипятят реакционную массу при 60 С в течение 2 ч, затем охлаждают до комнатной температуры и перемешивают в течение 2 ч. Кристаллы отфильтровывают, перекристаллизовывают из 6 мл этанола. Реакционная масса кристаллизуется при 25 С, фильтруется и сушится. Необходимый продукт получают в виде белого кристаллического вещества. Точка плавления (M.K.) 217-230 С. Пример 55. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил 4-морфолинкарбоксилат гидрохлорид. Способ аналогичен получению соединения в примере 54 с использованием на стадии 1 вместо N,Nдиметилкарбамоилхлорида морфолинил-4-карбонил хлорид. Точка плавления (M.K.) 217-220 С. Пример 56. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диэтилкарбамат гидрохлорид. Способ аналогичен получению соединения в примере 54 с использованием на стадии 1 вместо N,Nдиметилкарбамоилхлорида N,N-диэтилкарбамоилхлорид. Точка плавления (M.K.) 197-200 С. Пример 57. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил 1-пирролидинкарбоксилат гидрохлорид. Способ аналогичен получению соединения в примере 54 с использованием на стадии 1 вместо N,Nдиметилкарбамоилхлорида пирролидинил-1-карбонилхлорид. Точка плавления (M.K.) 220-222 С. Пример 58. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат гидрохлорид. Стадия 1. 8-трет-Бутоксикарбонил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат. Способ аналогичен получению соединения в примере 51, заменяя продукт, полученный в примере 43, на трет-бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил][(4-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат, полученный на стадии 1 примера 52. Стадия 2. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат гидрохлорид. К 2,55 г ( 4,4 ммоль) соединения, полученного на стадии 1 и растворенного в 50 мл этилацетата, добавляют 14,5 мл (44 ммоль/10 экв.) 3N HCl в этилацетате. Перемешивают реакционную массу при комнатной температуре в течение 4 дней; полученный осадок фильтруют, промывают этилацетатом и сушат под вакуумом, получая 2,15 г остатка, который очищают на силикагеле (элюент: этанол/аммиак/дихлорметан: 95/5/0,5). После получения соли, используя HCl в этаноле, необходимый продукт получен. Точка плавления (M.K.) 164-167 С. Пример 59. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат гидрохлорид. Способ аналогичен получению соединения в примере 58, заменяя на стадии 1 продукт, полученный на стадии 1 примера 52, на трет-бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3- 22014756 оксопропил][3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат. Точка плавления (M.K.) 208-212 С. Пример 60. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил ацетат гидрохлорид. Стадия 1. 8-трет-Бутоксикарбонил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]аминметил)бицикло[4.2.0]окта-1,3,5-триен-3-ил) ацетат. К раствору 3,25 г (6,4 ммоль) трет-бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3 ил)-3-оксопропил][4-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамата. полученного на стадии 1 примера 52, в 20 мл дихлорметана при комнатной температуре быстро добавляют 0,33 мл (6,7 ммоль/1,05 экв.) триэтиламина. Смесь охлаждают до 0 С и прикапывают 0,45 мл (6,4 ммоль/1 экв.) ацетил хлорида. Перемешивают реакционную массу при 0 С в течение 1 ч и нагревают до комнатной температуры. Органическая фаза промывается дважды 20 мл воды и сушится над сульфатом магния. После выпаривания остаток разделяют хроматографически на 200 г силикагеля (элюент: этилацетат/дихлорметан: 97/3), получая нужный продукт. Стадия 2. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино] метилбицикло[4.2.0]окта-1,3,5-триен-3-ил ацетат гидрохлорид. 2,2 г (4,0 ммоль) соединения, полученного на стадии 1, растворяют в 45 мл этилацетата, добавляют 13,5 мл (40 ммоль/10 экв.) 3N HCl в этилацетате. Перемешивают смесь при комнатной температуре в течение 4 дней, после чего реакционную массу выливают в 300мл эфира; полученный осадок фильтруют, промывают эфиром и сушат под вакуумом, получая остаток, который разделяют хроматографически на силикагеле (элюент: дихлорметан/этанол/аммиак: 95/5/0,5). После получения соли реакцией с этанольным HCl в этаноле получается необходимый продукт в форме гидрохлорида. Точка плавления (M.K.) 176-179 С. Пример 61. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил пента]пиррол-2(1H)-карбоксилат (цис-соединение). Стадия 1. 7-трет-Бутоксикарбонил)-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил гексагидроциклопента[с]пиррол-2(1 Н)карбоксилат. 1,5 г (2,94 ммоль) трет-бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3 оксопропил][3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамата, 4,1 мл триэтиламина и 2,37 г (11,74 ммоль) 4-нитрофенилхлоркарбоната растворяют в 210 мл THF. Перемешивают смесь при комнатной температуре в течение 1 ч, а затем добавляют 0,65 г (5,87 ммоль) октагидропента[с]пиррол и 2,9 мл триэтиламина. Через 2 ч выдержки при комнатной температуре опять добавляют 0,65 г октагидропента[с]пиррол и доводят до комнатной температуры в течение 1 ч. Реакционную смесь разбавляют 700 мл этилацетата и промывают водой, сушат над MgSO4 и выпаривают. Остаток разделяют хроматографически на силикагеле (элюент: CH2Cl2/этанол/NH4OH: 99/1/0,1), получая необходимый продукт. Стадия 2. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил гексагидроциклопента[с]пиррол-2(1H)-карбоксилат (циссоединение). Продукт, полученный на стадии 1, растворяют в 10 объемах этанола и 10 объемах 3N раствора HCl в этаноле. Перемешивают смесь при комнатной температуре в течение 24 ч и полученный осадок, который является необходимым продуктом, отфильтровывают. Точка плавления (M.K.) 214-217 С. Пример 62. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино метил)бицикло[4.2.0]окта-1,3,5-триен-3-ил-4,4-дифтор-1-пиперидинкарбоксилат. Способ аналогичен получению соединения в примере 61 с использованием на стадии 1 4,4 дифторпипередин вместо октагидропента[с]пиррола. Точка плавления (M.K.) 212-216 С. Пример 63. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[(3-изопропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-оксопропан-1-амин гемифумарат. Стадия 1. трет-Бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3 Н-3-бензазепин-3-ил)-3-оксопропил][(3-гидроксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат. 3,6 г (8,8 ммоль) продукта, полученного в примере 35, растворяют в 60 мл дихлорметана. Добавляют 2,1 г (1,1 экв.) ди-трет-бутилдикарбоната и перемешивают при комнатной температуре в течение 3 ч(пока не перестанет выделяться газ). Дихлорметан выпаривают, сушат вещество, используя вакуумнасос, получают необходимый продукт. Стадия 2. трет-Бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил][(3-изопропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]карбамат. 1,4 г (2,7 ммоль) продукта, полученного на стадии 1, растворяют в 15 мл DMF, добавляют 740 мг (2 экв.) карбоната калия, 160 мг (0,18 экв.) карбоната цезия и 0,54 мл (2 экв.) изопропилйодида. Смесь кипятят при 40 С в течение 24 ч. DMF выпаривают при помощи роторного испарителя. Полученный остаток разбавляют водой и экстрагируют CH2Cl2. Органическую фазу сушат над MgSO4, фильтруют и выпари- 23014756 вают досуха. Полученное масло очищают методами хроматографии на 100 г силикагеля (элюент:CH2Cl2/этилацетат: 80/20), получая необходимый продукт в маслянистой форме. Стадия 3. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[(3-изопропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-оксопропан-1-амин гемифумарат. 1,1 г (1,99 ммоль) продукта, полученного на стадии 2, растворяют в 10 мл этанола и добавляют 14 мл (18 экв.) раствора (2,6N) HCl в этаноле. Перемешивают смесь при комнатной температуре в течение ночи. Продукт кристаллизуется, его отфильтровывают и сушат, получая 800 мг продукта. Это твердое вещество растворяют в воде; добавляют NaOH (1N), доводят рН до 9; экстрагируют CH2Cl2, сушат надMgSO4, отфильтровывают и выпаривают досуха, получая 650 мг масла. Масло очищают методами хроматографии на 100 г силикагеля (элюент: CH2Cl2/этанол/NH4OH: 95/5/0,5). Необходимый продукт получают в форме основания, которое растворяют в 10 мл этанола и добавляют 8,1 мл (1 экв.) 2% раствор фумаровой кислоты в этаноле (М = 0,172). Перемешивают смесь при комнатной температуре в течение 30 мин, а затем выпаривают досуха. Полученный остаток кристаллизуют из ацетонитрила при 25 С. Кристаллы отфильтровывают получая на выходе 314 мг, который перекристаллизуют кипячением с обратным холодильником из ацетонитрила. Отфильтровывают, не охлаждая смеси, а затем кристаллизуют в течение 1 ч при комнатной температуре. Осадок фильтруют и сушат, чтобы получить необходимый продукт в форме белых кристаллов. Точка плавления (M.K.) 208-211 С. Пример 64. 2-[7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил](метил)амино. Стадия 1. 7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепине-3-карбонилхлорид. К 1,2 мл (10 ммоль/1 экв.) дифосгена, растворенного в 20 мл дихлорметана при температуре 0 С,добавляют по каплям раствор 3,55 г (20 ммоль) 7,8-диметокси-2,3,4,5-тетрагидро-1 Н-3-бензазепина в 22 мл дихлорметана, а затем 4,2 мл (30 ммоль/1,5 экв.) триэтиламина. Перемешивают в течение ночи при комнатной температуре, а затем выпаривают. Остаток растворяют в дихлорметане, промывают водой и сушат над сульфатом магния, после выпаривания получают желаемый продукт. Стадия 2. 2-[(3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил](метил)аминоэтиловый 7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепине-3-карбоксилат. При комнатной температуре 0,23 г (5,7 ммоль/1,25 экв.) гидрида натрия (60%) промывают пентаном, а затем добавляют 7 мл THF и по каплям раствор 1,14 г (4,5 ммоль/1 экв.) соединения, полученного способом 25, в 7 мл THF; перемешивают в течение 1 ч, после чего порционно добавляют 1,02 г (3,8 ммоль/0,85 экв.) соединения, полученного на стадии 1, перемешивают в течение ночи при комнатной температуре. Реакционную смесь приливают к 50 мл 1N водного раствора соляной кислоты. Проводят экстракцию дихлорметаном, после чего объединенные органические фазы промывают 1N раствором гидроокиси натрия и сушат над сульфатом магния. После фильтрования и выпаривания остаток разделяют хроматографически на 150 г силикагеля (элюент: дихлорметан/этанол/аммиак: 98/2/0,2). Полученное основание растворяют в изопропаноле и превращают в соль реакцией с HCl в этаноле, получая необходимый продукт. Точка плавления (M.K.) 147-150 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = +7,1. Пример 65. 2-[7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил](метил)амино. Способ аналогичен получению соединения в примере 64, используя на стадии 2 продукт, полученный способом 26, вместо продукта, полученного способом 25. Точка плавления (M.K.) 144-147 С. Оптическое вращение: растворитель: МеОН, С=0,01 г/см 3, Т=20 С, L=589 нм, D = -6,94. Пример 66. N-2-(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино] этил-7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепине-3-карбоксамид метансульфонат. При комнатной температуре к 1,06 г (4,5 ммоль) соединения, полученного способом 27, растворенного в 40 мл дихлорметана, быстро добавляют 2,34 мл (13,4 ммоль/3 экв.) диизопропилэтиламина, а затем по частям 1,02 г (3,8 ммоль/0,85 экв.) соединения, полученного на стадии 1 примера 64, перемешивают в течение ночи при комнатной температуре. Реакционную смесь промывают водой и выпаривают. Остаток разделяют хроматографически на 140 г силикагеля (элюент: дихлорметан/этанол/аммиак: 95/5/0,5). Очищенный продукт превращают в соль метансульфоновой кислоты реакцией в этилацетате и после выпаривания уплотняют в эфире, получая необходимый продукт. Точка плавления (M.K.) 65-85 С. Пример 67. N-2-(7R)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино] этил-7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-карбоксамид метансульфонат. Способ аналогичен получению соединения в примере 66 с использованием продукта, полученного способом 28, вместо продукта, полученного способом 27. Точка плавления (M.K.) 65-85 С. Пример 68. N-[(2,3-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид.- 24014756 Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 8. Точка плавления (M.K.) 185-186 С. Пример 69. N-[(2,3,4-Триметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин фумарат. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 9. Соль получают, используя фумаровую кислоту. Точка плавления (M.K.) 158-160 С. Пример 70. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[(5-метоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил-N-метил-3-оксопропан-1-амин гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 10. Точка плавления (M.K.) 130-132 С. Пример 71. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диэтилкарбамат гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 56. Точка плавления (M.K.) 175-178 С. Пример 72. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил 4-морфолинкарбоксилат гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 55. Точка плавления (M.K.) 196-198 С. Пример 73. 7-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил-1-пирролидинкарбоксилат гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 57. Точка плавления (M.K.) 174-177 С. Пример 74. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-метокси-4-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-оксопропан-1-амин гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 39. Точка плавления (M.K.) 172-175 С. Пример 75. 8-3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил этилкарбонат гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 58. Точка плавления (M.K.) 156-159 С. Пример 76. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[4-метокси-3-метилбицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-оксопропан-1-амин гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 38. Точка плавления (M.K.) 191-193 С. Пример 77. N-[(Бицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3-(7,8-диметокси-1,2,4,5-тетрагидро 3H-3-бензазепин-3-ил)-N-метил-3-оксопропан-1-амин гидрохлорид. Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 37. Точка плавления (M.K.) 110-113 С. Пример 78. 7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил) аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил гексагидроциклопента[с]пиррол-2(1 Н)-карбоксилат(цис-соединение). Способ аналогичен получению соединения в примере 43 с использованием продукта, полученного в примере 35, вместо продукта, полученного в примере 61. Точка плавления (M.K.) 110-113 С. Пример 79. 2-([(7S)-3,4-Диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил[3-(7,8-диметокси 1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]амино)этанол гидрохлорид. Перемешивают 1 г (2,2 ммоль) (75)-N-[(3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил)метил]-3(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропан-1-амина (полученный восстановлением соединения, полученного в примере 4, до основания), 0,4 г (3,3 ммоль) 1,4-диоксан-2,5-диола, 0,7 г (3,3 ммоль) триацетоксиборгидрид натрия и 8 мл хлорида метилена. Через 45 мин при комнатной температуре реакционную смесь смешивают с 10 мл 1N раствора гидроокиси натрия и перемешивают в течение получаса. Разделяют фазы, водную фазу экстрагируют дважды этилацетатом, органические экс- 25014756 тракты объединяют, промывают водой, а затем насыщенным раствором NaCl и сушат над MgSO4. Полученный после выпаривания растворителя остаток разделяют хроматографически на силикагеле (элюент:CH2Cl2/EtOH/NH4OH: 95/5/0,5), получая продукт, который затем превращают в гидрохлорид, используя 2N эфирный раствор HCl. Точка плавления (M.K.) 75-79 С. Пример 80. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-(3-метоксипропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-оксопропан-1-амин гидрохлорид. Стадия А. трет-Бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил][3 гидроксибицикло[4.2.0]окта-1,3,5-триен 7-ил)метил]карбамат. К 10 г (24,4 ммоль) 7-([3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил] аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ола, полученного восстановлением соединения, полученного в примере 35, до основания и растворенного в 120 мл дихлорметана, при комнатной температуре добавляют 20 мл этанола, чтобы частично растворить осадок, а затем 6,4 г (29,3 ммоль) ди-третбутилдикарбоната. Перемешивают смесь при комнатной температуре в течение 2 ч, после чего добавляют 250 мл дихлорметана и экстрагируют дважды 200 мл воды. После сушки органической фазы над сульфатом магния, а затем выпаривания получают остаток, который очищают на силикагеле (элюент: дихлорметан/метанол: 97/3). Стадия В. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-(3-метоксипропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил)-3-оксопропан-1-амин гидрохлорид. При комнатной температуре смешивают 1,33 г (2,6 ммоль) соединения, полученного на стадии А,43 мг (0,26 ммоль) йодида калия, 1,27 г (3,9 ммоль) карбоната цезия и 0,40 мл (3,7 ммоль) метоксипропилхлорида с 2,6 мл DMF, а затем кипятят при 60 С в течение 24 ч. Смесь приливают к 50 мл ледяной воды и экстрагируют дважды 50 мл этилацетата. Объединенные органические фазы промывают 30 мл воды, сушат над сульфатом магния и выпаривают. Остаток разделяют хроматографически на 100 г аллюмогеле (элюент: циклогексан/этилацетат: 75/25), получая соединение, которое растворяют в 8 мл безводного этанола и 1,55 мл (5,0 ммоль) 3,7N HCl в этаноле. Перемешивают смесь при комнатной температуре в течение 45 ч. Отфильтровывают полученные кристаллы, получая продукт в форме гидрохлорида. Точка плавления (M.K.) 155-159 С. Пример 81. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-(3-метоксипропоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-оксопропан-1-амин гидрохлорид. При комнатной температуре смешивают 1,06 г (2,5 ммоль) 7-([3-(7,8-диметокси-1,2,4,5 тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил)аминометил)бицикло[4.2.0]окта-1,3,5-триен-3 ола (полученного восстановлением соединения, полученного в примере 43, до основания), 42 мг (0,25 ммоль) калий йодида, 0,98 г (3,0 ммоль) карбоната цезия и 0,35 мл (3,25 ммоль) метоксипропилхлорида и 2,5 мл DMF, а затем кипятят при 50 С в течение 3 дней. Затем смесь выливают в 50 мл ледяной воды и экстрагируют дважды 50 мл этилацетата. Объединенные органические экстракты промывают с 30 мл воды, сушат над сульфатом магния и выпаривают. Остаток (1,21 г) разделяют хроматографически на силикагеле (элюент: дихлорметан/метанол: 95/5), получая 1,07 г продукта в форме основы. Его растворяют в 5 мл ацетонитрила и 1,2 мл 2N эфирного раствора HCl. Перемешивают смесь при комнатной температуре в течение 2 ч. Кристаллы отфильтровывают, получая 0,91 г желаемого продукта в форме гидрохлорида. Точка плавления (M.K.) 142-147 С. Пример 82. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-(3-метоксиэтоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-оксопропан-1-амин гидрохлорид. Получение аналогично получению в примере 80, используя на стадии В метоксиэтилхлорид вместо метоксипропилхлорида. Точка плавления (M.K.) 159-170 С. Пример 83. 3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-N-[3-(3-метоксиэтоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-3-оксопропан-1-амин гидрохлорид. Получение аналогично получению в примере 81, используя метоксиэтилхлорид вместо метоксипропилхлорида. Точка плавления (M.K.) 129-131 С. Пример 84. 2-[7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил] аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил]окси-N-метилацетамид гидрохлорид. Стадия А. Этиловый эфир [7-трет-бутоксикарбонил)[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3 бензазепин-3-ил)-3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил)окси]карбоновой кислоты. 6,8 г (13,3 ммоль) соединения, полученного на стадии А примера 80, 2,3 г (16,6 ммоль) карбоната калия и 2,21 мл (20,0 ммоль) этилбромацетата смешивают с 13,3 мл DMF. Реакционную смесь перемешивают при комнатной температуре в течение 25 ч, выливают в 250 мл ледяной воды и экстрагируют дважды 150 мл этилацетата. Объединенные органические экстракты промывают 100 мл воды, сушат над сульфатом магния и выпаривают, получая необходимый продукт.- 26014756 Стадия В. трет-Бутил-[3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](3-[2-(метиламино)-2-оксоэтокси]бицикло[4.2.0]окта-1,3,5-триен-7-илметил)карбамат. При комнатной температуре смешивают раствор 2,6 г (4,4 ммоль) соединения, полученного на описанной выше стадии, в 20 мл этанола с 20 мл 40% водного раствора монометиламина. Перемешивают смесь при комнатной температуре в течение 3 дней, а затем выпаривают. Остаток промывают толуолом,этанолом, а затем опять толуолом и выпаривают, получая нужный продукт. Стадия С. 2-[7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил]аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил]окси-N-метилацетамид гидрохлорид. При комнатной температуре смешивают 2,63 г (4,5 ммоль) соединения, полученного на стадии В, с 120 мл этанола и 25 мл 3,7N HCl в этаноле. После перемешивания в течение 1 дня при комнатной температуре отфильтровывают полученные кристаллы, промывают этанолом и эфиром и сушат под вакуумом для получения продукта в форме гидрохлорида. Точка плавления (M.K.) 145-146 С. Пример 85. 2-[7-([3-(7,8-Диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил)аминометил)бицикло[4.2.0]окта-1,3,5-триен-3-ил]окси-N-метилацетамид гидрохлорид. При комнатной температуре к раствору 0,83 г (1,72 ммоль) соединения, полученного на стадии С примера 84, в 32 мл ацетонитрила быстро добавляют 1,4 мл (17,2 ммоль) 37% водного раствора формальдегида, а затем 0,22 г (3,44 ммоль) цианоборгидрид натрия. После перемешивания смеси в течение 45 мин при комнатной температуре выпаривают, а полученный остаток смешивают с 30 мл воды; водную фазу экстрагируют дважды 30 мл дихлорметана; органические экстракты объединяют, промывают 30 мл воды и сушат над сульфатом магния. После выпаривания остаток разделяют хроматографически на силикагеле (элюент: дихлорметан/этанол/аммиак: 95/5/0,5), получая необходимый продукт в форме основания. Основание растворяют в 4 мл этанола и 0,3 мл 3,7N HCl в этаноле. После перемешивания реакционной массы в течение 1 ч при комнатной температуре отфильтровывают полученный осадок, промывают этанолом и сушат под вакуумом, получая необходимый продукт в форме гидрохлорида. Точка плавления (M.K.) 147-153 С. Фармакологическое исследование Пример 86. Влияние соединений на спонтанную скорость сокращения правого предсердия у крыс. Крыс мужского пола WISTAR весом от 325 до 375 г анестезировали инъекцией пентобарбитала натрия (30 мг/кг). После чего сердце быстро вынимают и размещают в физиологическом растворе при 4 С,которое содержит (в М): NaCl 120,3, KCl 4,8, CaCl2 2,5, KH2PO4 1,0, MgSO4 1,3, NaHCO3 24,2, глюкоза 11,1, Ca-EDTA 0,016, и подкисляют (карбоген 95% O2 + 5% CO2), рН 7,4. Правое предсердье (ПП), которое спонтанно сокращается, изолировали, размещали в резервуаре, который термостатически контролирует температуру 35 С, содержит 20 мл физиологического раствора и соединен с изометрическим датчиком напряжения (model IT-25, EMKA Technologies, Paris, France). Начальное напряжение устанавливали на 0,4 г. Скорость сокращения измеряли программой IOX (EMKA Technologies, Paris, France). Образцы со скоростью сокращения, не попадающие в интервал 200-300 ударов в минуту, исключались. Растворы соединений с концентрацией 10-2 M приготавливались ежедневно. Последующие разбавления проводили во льду во время эксперимента. После стабилизации в течение 30 мин соединение добавляли в средство кумулятивным способом каждые 15 мин (4 концентрации). Уменьшение скорости сокращения выражалось в процентах относительно начального параметра. Концентрация, которая сокращает начальное сокращение на 30% (IC30), вычислялалсь и выражалась молярностью (М). Исходя из данных таблицы видно, что соединения изобретения непосредственно уменьшают скорость сердцебиения. Пример 87. Фармацевтическая композиция. Формула для получения 1000 таблеток по 10 мг каждая, содержащих активное вещество: где R1 представляет собой атом водорода или группу, выбранную из С 3-С 7 циклоалкила, бензила и линейного или разветвленного C1-C6 алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С 3-С 7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или гидроксигруппы; метиловой группы; группы -OSO2R10; группы -OCOR10 или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, необязательно замещенной метокси или (CO)-NR12R'12 группой; или R2 и R3, или R3 и R4, или R4 и R5, образующих вместе -O-(CH2)q-O-, -OCH=CH-O- или -О-СН=СН-,R6, R7, R8 и R9 могут быть одинаковыми или различаться, и каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы, или R6 и R7,или R7 и R8, или R8 и R9, образующих вместе -O-(CH2)q-O-,R10 выбирают из группы, представленной линейной или разветвленной C1-C6 алкоксигруппой,NR11R'11 и линейным или разветвленным C1-C6 алкилом, который необязательно может быть замещенным одним или несколькими атомами галогена,R11 и R'11 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной C1-C6 алкильной группы, или R11 и R'11, образующих вместе с атомом азота моноциклический или бициклический 5- или 8-членный гетероцикл, содержащий азот и необязательно- 28014756 содержащий другой гетероатом, выбранный из О и N, причем этот гетероцикл необязательно может быть замещенным одним или несколькими атомами галогена,R12 и R'12 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной С 1-C6 алкильной группы,X является О, NH или CH2,m и p могут быть одинаковыми или различаться и являются 0 или 1,n и q могут быть одинаковыми или различаться и являются 1 или 2,его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 2. Соединение формулы (I) согласно п.1, где R1 выбирают из атома водорода или линейной или разветвленной C1-C6 алкильной группы, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 3. Соединение формулы (I) согласно п.1, где R1 выбирают из C3-C7 циклоалкильной группы или циклоалкильной группы, где циклоалкильная часть содержит от 3 до 7 атомов углерода, а алкильная часть содержит от 1 до 6 атомов углерода и является линейной или разветвленной, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 4. Соединение формулы (I) согласно любому из пп.1-3, где R2, R3, R4 и R5 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной C1-C6 алкоксигруппы или -OCOR10 группы, где R10 является группой NR11R'11, как описывается выше, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 5. Соединение формулы (I) согласно любому из пп.1-4, где R6, R7, R8 и R9 могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С 1-C6 алкоксигруппы, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 6. Соединение формулы (I) согласно любому из пп.1-5, где m является 0, его оптические изомеры,если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 7. Соединение формулы (I) согласно любому из пп.1-5, где m является 1, а X - CH2, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 8. Соединение формулы (I) согласно любому из пп.1-7, где р является 0, его оптические изомеры,если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 9. Соединение формулы (I) согласно любому из пп.1-7, где р является 1, его оптические изомеры,если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 10. Соединение формулы (I) согласно п.1, где R1 выбирают из атома водорода или линейной или разветвленной C1-C6 алкильной группы, R2, R3, R4 и R5, которые могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С 1-C6 алкоксигруппы, R6, R7, R8 и R9, которые могут быть одинаковыми или различаться, каждый выбирается из атома водорода или линейной или разветвленной, насыщенной или ненасыщенной С 1-C6 алкоксигруппы, m является 0, n является 1 и р является 0, его оптические изомеры, если таковые существуют, а также его соли с фармацевтически приемлемой кислотой. 11. Соединение формулы (I) согласно п.1, которое выбирают изN-[2-(5,6-диметокси-2,3-дигидро-1 Н-инден-2-ил)метил]-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3 бензазепин-3-ил)-3-оксопропан-1-амина, а также его солей с фармацевтически приемлемой кислотой;N-[3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-3-(7,8-диметокси-1,2,4,5-тетрагидро 3H-3-бензазепин-3-ил)-4-оксобутан-1-амина, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой; 7-3-(7,8-диметокси-1,2,4,5-тетрагидро-3H-3-бензазепин-3-ил)-3-оксопропил](метил)амино]метилбицикло[4.2.0]окта-1,3,5-триен-3-ил диметилкарбамата, его оптических изомеров, а также солей с фармацевтически приемлемой кислотой. 12. Способ получения соединений формулы (I) согласно п.1 с использованием в качестве исходного соединения формулы (II)
МПК / Метки
МПК: C07D 403/12, C07D 405/12, A61K 31/55, A61P 9/00, C07D 413/12, C07D 223/16, C07D 401/12
Метки: 1,2,4,5-тетрагидро-3н-бензазепина, получения, соединения, эти, композиции, фармацевтические, способ, содержащие
Код ссылки
<a href="https://eas.patents.su/30-14756-soedineniya-1245-tetragidro-3n-benzazepina-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-soderzhashhie-eti-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения</a>
Новые соединения 7-оксо-2,3,7,14-тетрагидро-1h-бензо[b]-пирано [3,2-h]акридинкарбоксилата и содержащие их фармацевтические композиции

Номер патента: 3361
Опубликовано: 24.04.2003
Авторы: Кош Мишель, Мишель Сильви, Ренар Пьер, Тиллекэн Франсуа, Пьер Ален, Пфеффер Брюно, Атасси Ганем
МПК: C07D 491/052, A61K 31/436, A61P 35/00...
Метки: композиции, фармацевтические, содержащие, 3,2-h]акридинкарбоксилата, соединения, 7-оксо-2,3,7,14-тетрагидро-1h-бензо[b]-пирано, новые
Формула / Реферат:
1. Соединения формулы (I) где X и Y, которые могут быть одинаковыми или различными, каждый независимо от другого, представляет группу, выбранную из атомов водорода, атома галогена, гидроксигруппы, линейной или разветвленной (C1-C6)алкильной группы, линейной или разветвленной (C1-C6)алкоксигруппы, аминогруппы (необязательно замещенной одной или двумя одинаковыми или различными линейными или разветвленными (C1-C6)алкильными группами, которые сами...
17α-фторалкилстероиды, способ их получения и содержащие эти соединения фармацевтические композиции

Номер патента: 5772
Опубликовано: 30.06.2005
Авторы: Вирва Ральф, Кауфманн Гюнтер, Элгер Вальтер, Ринг Свен
МПК: A61P 5/26, C07J 1/00, A61K 31/565...
Метки: способ, эти, 17α-фторалкилстероиды, композиции, содержащие, фармацевтические, получения, соединения
Формула / Реферат:
1. 17a-фторалкилстероиды общей формулы I в которой R1 представляет собой C1-C4алкильную группу; R2 представляет собой гидроксигруппу, группу OC(O)-R20 или OR21, где R20 и R21 обозначают C1-C12алкильную группу, C3-C8циклоалкильную группу, арильную группу или арил-C1-C4алкильную группу; R3 представляет собой остаток формулы CnF2n+1, где n обозначает 1, 2, 3, 4, 5 или 6; R4 и R5 каждый представляет собой атом водорода или оба вместе образуют...
Новые соединения инденоиндолона, способ их получения и фармацевтические композиции их содержащие

Номер патента: 4571
Опубликовано: 24.06.2004
Авторы: Кро-Бертье Лоренс, Буссар Мари-Франсуа, Леонс Стефан, Хикман Джон, Гийбо Николя, Пьерре Ален, Вержбицкий Мишель, Руссо Анн, Атасси Ганем
МПК: A61P 35/00, A61K 31/403, C07D 209/58...
Метки: композиции, получения, содержащие, инденоиндолона, фармацевтические, соединения, новые, способ
Формула / Реферат:
1. Соединения формулы (I) в которой R представляет собой атом водорода, линейную или разветвленную (C1-C6)алкильную группу, необязательно замещенную карбоксигруппой, линейной или разветвленной (C1-C6)алкоксикарбонильной группой или группой NR10R11 (в которой R10 и R11, которые могут быть одинаковы или различны, каждый представляет собой линейную или разветвленную (C1-C6)алкильную группу или совместно с азотом, который их несет, образуют...
Соединения пиримидин-4-она способ их получения и содержащие их фармацевтические композиции

Номер патента: 5402
Опубликовано: 24.02.2005
Авторы: Мюлле Оивье, Брокко Морисетт, Дюбюффе Тьерри, Лавие Жильбер, Декен Анн, Миллан Марк
МПК: C07D 471/00, A61K 31/519, A61P 25/00...
Метки: способ, соединения, получения, фармацевтические, содержащие, пиримидин-4-она, композиции
Формула / Реферат:
1. Соединения формулы (I) в которой R1, R2, R3 и R4, которые могут быть одинаковыми или различными, каждый представляет собой атом водорода, атом галогена или группу, выбираемую из линейного или разветвленного (C1-C6)алкила, линейного или разветвленного (C1-C6)алкокси, линейного или разветвленного (C1-C6)полигалоалкил, гидрокси, циано, нитро и амино, или R1 и R2, R2 и R3 или R3 и R4 совместно с атомами углерода, их несущими, образуют...
4-галогенированные 17-метиленстероиды, способ их получения и содержащие эти соединения фармацевтические композиции

Номер патента: 6387
Опубликовано: 29.12.2005
Авторы: Дрёшер Петер, Швайкерт Ханс-Удо, Элгер Вальтер, Менценбах Бернд, Хиллиш Александр, Кауфманн Гюнтер, Мюллер Герд
МПК: C07J 13/00, A61P 5/28, A61K 31/56...
Метки: получения, содержащие, соединения, композиции, 17-метиленстероиды, способ, фармацевтические, 4-галогенированные, эти
Формула / Реферат:
1. 17-Метиленстероиды общей формулы I в которой R4 представляет собой атом галогена или псевдогалоген, R10 представляет собой атом водорода или прямоцепочечную либо разветвленную C1-C4алкильную группу, R20 и R20a, каждый независимо друг от друга, представляет собой атом водорода, прямоцепочечную либо разветвленную C1-C4алкильную или гидрокси-C1-C4алкильную группу или один из радикалов R20 и R20a обозначает атом водорода, прямоцепочечную либо...
Предыдущий патент: Применение фенофибрата или его производного для предотвращения диабетической ретинопатии
Следующий патент: Химерный аденовирусный вектор, иммуногенная композиция на его основе, способ индукции иммуного ответа с его помощью и выделенная нуклеиновая кислота
Случайный патент: Антагонисты хемокинного рецептора и способы их применения