Стабильный при хранении инфузионный раствор дигидроптеридинонов







Формула / Реферат
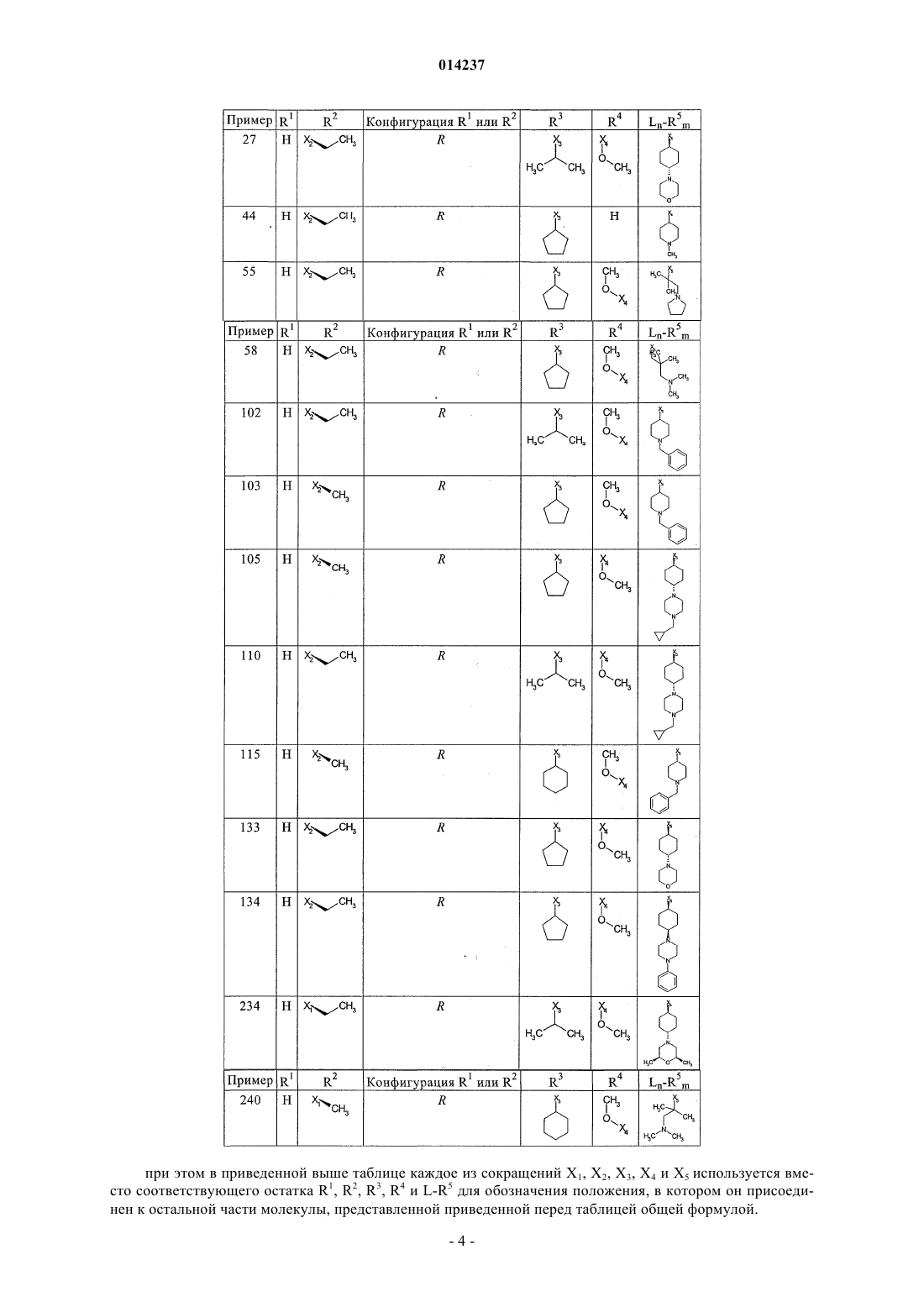
1. Стабильные при хранении водные инфузионные растворы или растворы для инъекций, содержащие действующее вещество общей формулы (I)

где































где каждое из сокращений Х1, Х2, Х3, X4и Х5 используется вместо соответствующего остатка R1, R2, R3, R4и Ln-Rm5 для обозначения положения, в котором он присоединен к остальной части молекулы, представленной приведенной перед таблицей общей формулой, или его таутомеры, рацематы, энантиомеры, диастереомеры или необязательно физиологически совместимую кислоту или смесь физиологически совместимых кислот в достаточном для растворения действующего вещества и для стабилизации количестве, а также необязательно содержащие другие пригодные для парентерального введения в организм вспомогательные вещества.
2. Водные инфузионные растворы или растворы для инъекций по п.1, отличающиеся тем, что они дополнительно содержат EDTA.
3. Водные инфузионные растворы или растворы для инъекций по п.1 или 2, отличающиеся тем, что содержание растворенного действующего вещества в 1 мл инфузионного раствора или раствора для инъекций составляет от 0,1 до 10,0 мг.
4. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-3, отличающиеся тем, что одна или несколько используемых для обеспечения стабильности при хранении и стабильности в разведенном состоянии кислот выбрана(ы) из группы, включающей соляную кислоту, уксусную кислоту, гидроксиуксусную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, фосфорную кислоту, азотную кислоту, серную кислоту, лимонную кислоту, винную кислоту, фумаровую кислоту, янтарную кислоту, глутаровую кислоту, адипиновую кислоту, пропионовую кислоту, аскорбиновую кислоту, малеиновую кислоту, яблочную кислоту, глутаминовую кислоту, глюконовую кислоту, глюкуроновую кислоту, галактуроновую кислоту и молочную кислоту.
5. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-4, отличающиеся тем, что молярное соотношение между физиологически совместимой кислотой или смесью физиологически совместимых кислот и действующим веществом составляет максимум 3:1.
6. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-5, отличающиеся тем, что они содержат одно или несколько дополнительных вспомогательных веществ, используемых в технологии приготовления лекарственных средств и выбранных из группы, включающей комплексообразователи, светостабилизаторы, ингибиторы кристаллизации, загустители, придающие изотоничность агенты, антиокислители и средства для поддержания нормального водного баланса организма.
7. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-6, отличающиеся тем, что их осмомолярность составляет от 200 до 600 мосмомоль/кг.
8. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-7, отличающиеся тем, что их значение рН составляет от 2,4 до 5,3.
9. Водные инфузионные растворы или растворы для инъекций по одному из пп.1-8, отличающиеся тем, что они содержат в пересчете на 100 мл своего объема от 1,25 до 3,0 моль соляной кислоты на моль действующего вещества и от 0,75 до 1,2 г NaCl и имеют осмомолярность от 260 до 350 мосмомоль/кг, а также значение рН от 3,5 до 5,0.
10. Лиофилизаты, концентраты или суспензии, отличающиеся тем, что из них добавлением к ним воды возможно получение водного инфузионного раствора или раствора для инъекций по одному из пп.1-9.
11. Инфузионные растворы или растворы для инъекций по одному из пп.1-9 для применения в качестве лекарственных средств с антипролиферативным действием.
12. Применение инфузионного раствора или раствора для инъекций по одному из пп.1-9 для приготовления лекарственного средства, предназначенного для лечения опухолевых, инфекционных, воспалительных и аутоиммунных заболеваний.
13. Применение инфузионного раствора или раствора для инъекций по одному из пп.1-10 в количестве, которое соответствует интервалу доз от 0,1 до 50 мг действующего вещества на 1 кг веса тела.
14. Пригодный для хранения парентеральных лекарственных форм контейнер, выбранный из стеклянной емкости, мягкого пластикового пакета и мешка, содержащий инфузионные растворы или растворы для инъекций по одному из пп.1-9 соответственно, лиофилизаты, концентраты или суспензии по п.10.
Текст
СТАБИЛЬНЫЙ ПРИ ХРАНЕНИИ ИНФУЗИОННЫЙ РАСТВОР ДИГИДРОПТЕРИДИНОНОВ(71)(73) Заявитель и патентовладелец: БРИНГЕР ИНГЕЛЬХАЙМ ИНТЕРНАЦИОНАЛЬ ГМБХ (DE) Предложены стабильные при хранении водные инфузионные растворы или растворы для инъекций, содержащие действующее вещество общей формулы (I) в которой остатки L, R1, R2, R3, R4 и R5 имеют указанные в описании значения, или его таутомеры, рацематы, энантиомеры, диастереомеры или необязательно физиологически совместимую кислоту или смесь физиологически совместимых кислот в достаточном для растворения действующего вещества и для стабилизации количестве, а также необязательно содержащие другие пригодные для парентерального введения в организм вспомогательные вещества. Также предложены лиофилизаты, концентраты или суспензии, а также инфузионные растворы или растворы для инъекций и их применение в качестве лекарственных средств с антипролиферативным действием. Предложен контейнер, пригодный для хранения парентеральных лекарственных форм. 014237 Настоящее изобретение относится к стабильным при хранении водным инфузионным растворам,соответственно растворам для инъекций, содержащим действующее вещество формулы (I) в которой остатки L, R1, R2, R3, R4 и R5 имеют указанные в формуле изобретения и последующем описании значения, и физиологически совместимую кислоту или смесь физиологически совместимых кислот в достаточном для растворения действующего вещества и для стабилизации количестве, а также необязательно содержащим другие пригодные для парентерального введения в организм вспомогательные вещества, используемые в технологии приготовления лекарственных средств, а также относится к способу приготовления предлагаемых в изобретении инфузионных растворов, соответственно растворов для инъекций. Предпосылки создания изобретения Предлагаемые в изобретении дигидроптеридиноны формулы (I) представляют собой соединения,которые обладают инновационным, новым цитостатическим механизмом действия и благодаря этому эффективно используются в онкологии для лечения быстро растущих раковых опухолей различных типов. Обычно цитостатические средства применяются в составе парентеральных лекарственных форм,хотя могут обладать вполне достаточной биодоступностью и при пероральном применении. Причина, по которой цитостатические средства приходится вводить в организм парентерально, заключается в том,что лечение цитостатическими средствами обычно сопровождается побочными действиями на желудочно-кишечный тракт, которые часто проявляются в виде тошноты, рвоты и/или диареи и которые поэтому при пероральном применении цитостатических средств могли бы снизить эффективность лечения. Сказанное в полной мере относится и к дигидроптеридинонам формулы (I), чем обусловлена необходимость их парентерального применения в составе инфузионного раствора, соответственно раствора для инъекций. В ЕР 0219784 и WO 01/78732 описаны способы приготовления и меры по стабилизации содержащих ципрофлоксаксин инфузионных растворов за счет применения одной или нескольких физиологически совместимых органических или неорганических кислот. В ЕР 0287926 говорится, что избежать опасности образования дисперсной твердой фазы (частиц) в растворе ципрофлоксаксина позволяет его применение в высокочистом виде. В ЕР 0143478 А 1 описан способ приготовления стабильного солянокислого раствора цисплатина, пригодного для инъекций и прежде всего не содержащего никаких дополнительных добавок. В DE 19703023 говорится, что стабильность инфузионных растворов касательно выделения в них частиц загрязняющих примесей можно в решающей степени улучшить за счет хранения таких растворов в стеклянных емкостях с силиконизированными поверхностями. В основу настоящего изобретения была положена задача предложить стабильный инфузионный раствор, соответственно раствор для инъекций с дигидроптеридинонами формулы (I) для требуемого терапевтически оптимизированного интервала доз. В соответствии еще с одной задачей изобретения такой стабильный инфузионный раствор, соответственно раствор для инъекций для возможности гибкого регулирования вводимой в организм дозы содержащегося в нем действующего вещества должен быть пригоден и в виде готового к применению раствора, и в виде концентрата для дополнительного разбавления или разведения обычно используемыми для парентерального введения в организм растворами,такими, например, как изотонический раствор NaCl, изотонический раствор декстрозы или содержащий лактат раствор Рингера. Подробное описание изобретения При создании изобретения неожиданно было установлено, что получение остающихся стабильными при длительном хранении и не проявляющих склонность к выделению частиц водных инфузионных растворов, соответственно растворов для инъекций, содержащих действующее вещество общей формулы(I), физиологически совместимую кислоту или смесь физиологически совместимых кислот в достаточном для растворения действующего вещества и для стабилизации количестве, а также необязательно содержащих другие пригодные для парентерального введения в организм вспомогательные вещества, используемые в технологии приготовления лекарственных средств, возможно вне зависимости от конкретного качества действующего вещества и прежде всего вне зависимости от характера и особенностей содержащихся в нем примесей. В соответствии с этим в изобретении предлагаются стабильные при хранении водные инфузионные-1 014237 растворы, соответственно растворы для инъекций, содержащие действующее вещество общей формулыR1 , R2 имеют идентичные или различные значения и обозначают водород или необязательно замещенный C1-С 6 алкил либоR1 и R2 совместно обозначают 2-5-членный алкильный мостик, который может содержать 1 или 2 гетероатома,R3 обозначает водород либо остаток, выбранный из группы, включающей необязательно замещенный С 1-С 12 алкил, С 2-С 12 алкенил, С 2-С 12 алкинил и С 6-С 14 арил, или остаток, выбранный из группы, включающей необязательно замещенный и/или соединенный мостиком С 3-С 12 циклоалкил, С 3-С 12 циклоалкенил, С 7-С 12 полициклоалкил, С 7-С 12 полициклоалкенил, С 5-С 12 спироциклоалкил, С 3-С 12 гетероциклоалкил, содержащий 1 или 2 гетероатома, и С 3-С 12 гетероциклоалкенил, содержащий 1 или 2 гетероатома,либоR1 и R3 или R2 и R3 совместно образуют насыщенный или ненасыщенный С 3-С 4 алкильный мостик,который может содержать 1 гетероатом,R4 обозначает остаток, выбранный из группы, включающей водород, -CN, гидроксигруппу, -NR6R7 и галоген, или остаток, выбранный из группы, включающей необязательно замещенный C1-С 6 алкил, С 2 С 6 алкенил, С 2-С 6 алкинил, С 1-С 6 алкилоксигруппу, С 2-С 5 алкенилоксигруппу, С 2-С 5 алкинилоксигруппу, С 1 С 6 алкилтиогруппу, C1-С 6 алкилсульфоксогруппу и C1-С 6 алкилсульфонил,L обозначает линкер, выбранный из группы, включающей необязательно замещенный C2-С 10 алкил,C2-С 10 алкенил, С 6-С 10 арил, -С 2-С 4 алкил-С 6-С 14 арил, -С 6-С 14 арил-С 1-С 4 алкил, необязательно соединенный мостиком С 3-С 12 циклоалкил и гетероарил, содержащий 1 или 2 атома азота,n обозначает 0 или 1,m обозначает 1 или 2,R5 обозначает остаток, выбранный из группы, включающей необязательно замещенный морфолинил, пиперидинил, пиперазинил, пиперазинилкарбонил, пирролидинил, тропенил, R8-дикетометилпиперазинил, сульфоксоморфолинил, сульфонилморфолинил, тиоморфолинил, -NR8R9 и азациклогептил,R6, R7 имеют идентичные или различные значения и обозначают водород или С 1-С 4 алкил иR8, R9 представляют собой идентичные или различные незамещенные заместители азота при R5 и обозначают либо водород, либо остаток, выбранный из группы, включающей С 1-С 6 алкил, -С 1-С 4 алкилС 3-С 10 циклоалкил, С 3-С 10 циклоалкил, С 6-С 14 арил, -С 1-С 4 алкил-С 6-С 14 арил, пиранил, пиридинил, пиримидинил, С 1-С 4 алкилоксикарбонил, С 6-С 14 арилкарбонил, С 1-С 4 алкилкарбонил, С 6-С 14 арилметилоксикарбонил, С 6-С 14 арилсульфонил, С 1-С 4 алкилсульфонил и С 6-С 14 арил-С 1-С 4 алкилсульфонил,или его таутомеры, рацематы, энантиомеры, диастереомеры или необязательно физиологически совместимую кислоту или смесь физиологически совместимых кислот в достаточном для растворения действующего вещества и для стабилизации количестве, а также необязательно содержащие другие пригодные для парентерального введения в организм вспомогательные вещества, используемые в технологии приготовления лекарственных средств. Предпочтительны стабильные при хранении растворы, содержащие соединения формулы (I), в которойR1-R4, R6 и R7 имеют указанные выше значения, аL обозначает линкер, выбранный из группы, включающей необязательно замещенный C2-С 10 алкил,C2-С 10 алкенил, С 6-С 14 арил, -С 2-С 4 алкил-С 6-С 14 арил, -С 6-С 14 арил-С 1-С 4 алкил, необязательно соединенный мостиком С 3-С 12 циклоалкил и гетероарил, содержащий 1 или 2 атома азота,n обозначает 1,m обозначает 1 или 2,R5 обозначает остаток, присоединенный к L через атом азота и выбранный из группы, включающей необязательно замещенный морфолинил, пиперидинил, R8-пиперазинил, пирролидинил, тропенил, R8 дикетометилпиперазинил, сульфоксоморфолинил, сульфонилморфолинил, тиоморфолинил, -NR8R9 иR8, R9 представляют собой идентичные или различные незамещенные заместители азота при R5 и обозначают водород или остаток, выбранный из группы, включающей C1-С 6 алкил, -С 1-С 4 алкил-С 3 С 10 циклоалкил, С 3-С 10 циклоалкил, С 6-С 14 арил, -С 1-С 4 алкил-С 6-С 14 арил, пиранил, пиридинил, пиримидинил, С 1-С 4 алкилоксикарбонил, С 6-С 14 арилкарбонил, С 1-С 4 алкилкарбонил, С 6-С 14 арилметилоксикарбонил,С 6-С 14 арилсульфонил, С 1-С 4 алкилсульфонил и С 6-С 14 арил-С 1-С 4 алкилсульфонил,необязательно в виде их таутомеров, их рацематов, их энантиомеров, их диастереомеров и их смесей, а также необязательно их фармакологически приемлемых кислотно-аддитивных солей. Предпочтительны далее стабильные при хранении растворы, содержащие соединения формулы (I),в которойR1-R4, R6 и R7 имеют указанные выше значения, аL обозначает линкер, выбранный из группы, включающей необязательно замещенный C2-С 10 алкил,C2-С 10 алкенил, С 6-С 14 арил, -С 2-С 4 алкил-С 6-С 14 арил, -С 6-С 14 арил-С 1-С 4 алкил, необязательно соединенный мостиком С 3-С 12 циклоалкил и гетероарил, содержащий 1 или 2 атома азота,n обозначает 0 или 1,m обозначает 1 или 2,R5 обозначает остаток, присоединенный к L через атом углерода и выбранный из группы, включающей R8-пиперидинил-, R8R9-пиперазинил, R8-пирролидинил, R8-пиперазинилкарбонил, R8-тропенил,R8-морфолинил и R8-азациклогептил, иR8, R9 представляют собой идентичные или различные незамещенные заместители азота при R5 и обозначают водород или остаток, выбранный из группы, включающей C1-С 6 алкил, -С 1-С 4 алкил-С 3-С 10 циклоалкил, С 3-С 10 циклоалкил, С 6-С 14 арил, -С 1-С 4 алкил-С 6-С 14 арил, пиранил, пиридинил, пиримидинил,С 1-С 4 алкилоксикарбонил, С 6-С 14 арилкарбонил, С 1-С 4 алкилкарбонил, С 6-С 14 арилметилоксикарбонил, С 6 С 14 арилсульфонил, С 1-С 4 алкилсульфонил и С 6-С 14 арил-С 1-С 4 алкилсульфонил,необязательно в виде их таутомеров, их рацематов, их энантиомеров, их диастереомеров и их смесей, а также необязательно их фармакологически приемлемых кислотно-аддитивных солей. Особенно предпочтительны стабильные при хранении растворы, содержащие соединения формулыL, m, n и R3-R9 имеют указанные выше значения, аR1, R2 имеют идентичные или различные значения и обозначают остаток, выбранный из группы,включающей водород, Me, Et и Pr, либоR1 и R2 совместно образуют С 2-С 4 алкильный мостик, необязательно в виде их таутомеров, их рацематов, их энантиомеров, их диастереомеров и их смесей, а также необязательно их фармакологически приемлемых кислотно-аддитивных солей. Наиболее предпочтительны стабильные при хранении растворы, содержащие соединения формулыR1, R2, m, n и R5-R8 имеют указанные выше значения, аR3 обозначает остаток, выбранный из группы, включающей необязательно замещенный C1 С 10 алкил, С 3-С 7 циклоалкил, С 3-С 6 гетероциклоалкил и С 6-С 14 арил, либоR1 и R3 или R2 и R3 совместно образуют насыщенный или ненасыщенный С 3-С 4 алкильный мостик,который может содержать 1 или 2 гетероатома,R4 обозначает остаток, выбранный из группы, включающей водород, ОМе, ОН, Me, Et, Pr, OEt,NHMe, NH2, F, Cl, Br, О-пропаргил, О-бутинил, CN, SMe, NMe2, CONH2, этинил, пропинил, бутинил и аллил, иL обозначает линкер, выбранный из группы, включающей необязательно замещенный фенил, фенилметил, циклогексил и разветвленный C1-С 6 алкил, необязательно в виде их таутомеров, их рацематов,их энантиомеров, их диастереомеров и их смесей, а также необязательно их фармакологически приемлемых кислотно-аддитивных солей. Еще одним объектом изобретения является стабильный при хранении раствор, содержащий один из дигидроптеридинонов общей формулы (I), выбранный из группы, включающей следующие соединения общей формулы (I) при этом в приведенной выше таблице каждое из сокращений X1, Х 2, Х 3, Х 4 и Х 5 используется вместо соответствующего остатка R1, R2, R3, R4 и L-R5 для обозначения положения, в котором он присоединен к остальной части молекулы, представленной приведенной перед таблицей общей формулой.-4 014237 Под долговременной стабильностью согласно изобретению подразумевается стабильность раствора при хранении в течение по меньшей мере 12 месяцев при 25 С/60%-ной относительной влажности и 30 С/70%-ной относительной влажности, предпочтительно в течение по меньшей мере 36 месяцев при 25 С/60%-ной относительной влажности и 30 С/70%-ной относительной влажности. Предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций помимо физиологически совместимой кислоты или смеси физиологически совместимых кислот могут не содержать никаких иных гидротропных солюбилизаторов или органических сорастворителей, прежде всего органических сорастворителей. Предпочтительны водные инфузионные растворы, соответственно растворы для инъекций, в 1 мл которых содержание растворенного действующего вещества формулы (I) составляет от 0,1 до 10,0 мг,прежде всего от 0,5 до 5 мг. Предпочтительны далее водные инфузионные растворы, соответственно растворы для инъекций, в которых одна или несколько используемых для обеспечения стабильности при хранении и стабильности разбавления кислот выбраны из группы, включающей соляную кислоту, уксусную кислоту, гидроксиуксусную кислоту, метансульфоновую кислоту, этансульфоновую кислоту, фосфорную кислоту, азотную кислоту, серную кислоту, лимонную кислоту, винную кислоту, фумаровую кислоту, янтарную кислоту,глутаровую кислоту, адипиновую кислоту, пропионовую кислоту, аскорбиновую кислоту, малеиновую кислоту, яблочную кислоту, глутаминовую кислоту, глюконовую кислоту, глюкуроновую кислоту, галактуроновую кислоту и молочную кислоту, предпочтительно из группы, включающей уксусную кислоту, соляную кислоту, фосфорную кислоту, винную кислоту, лимонную кислоту и фумаровую кислоту,наиболее предпочтительно из группы, включающей соляную кислоту, лимонную кислоту и уксусную кислоту. С учетом обеспечения рН-совместимости предпочтительны, как следует из прилагаемого к описанию чертежа, водные инфузионные растворы, соответственно растворы для инъекций, в которых для установления их рН на значение выше 2,4 молярное соотношение между физиологически совместимой кислотой или смесью физиологически совместимых кислот и действующим веществом должно составлять максимум 3:1, предпочтительно от 1,25:1 до 3:1, наиболее предпочтительно от 1,5:1 до 3:1. В предпочтительном варианте изобретение относится также к инфузионным растворам, соответственно растворам для инъекций, которые содержат от 0,1 до 10,0 мг действующего вещества на миллилитр водного раствора и до 3,0 молей соляной кислоты в пересчете на моль действующего вещества. Предпочтительное содержание соляной кислоты при этом составляет от 1,25 до 3,0 молей, прежде всего от 1,5 до 2,4 моля. Еще одним объектом изобретения являются инфузионные растворы, соответственно растворы для инъекций, содержащие 4-(7R)-8-циклопентил-7-этил-5,6,7,8-тетрагидро-5-метил-6-оксо-2-птеридинил] амино]-3-метокси-N-(1-метил-4-пиперидинил)бензамид и от 1,6 до 2,0 молей соляной кислоты на моль действующего вещества. В соответствии с модифицированным вариантом предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций могут также содержать действующее вещество в количестве до 10 мг/мл и соляную кислоту в количестве до 1 моля на моль действующего вещества, а также одну или несколько других физиологически совместимых кислот, при условии, что суммарное количество кислот должно быть не меньше 1,25 моля и не больше 3,0 молей на моль действующего вещества. Минимально необходимое в пересчете на моль действующего вещества количество кислоты зависит от концентрации действующего вещества и конкретно применяемой(-ых) кислоты(кислот) и в поэтому не является постоянной величиной. Однако содержание конкретной кислоты или кислот, ограниченное предлагаемыми в изобретении пределами, можно определить проведением простых опытов, как это описано, например, в ЕР 0219784 и WO 01/78732. Особенно предпочтительны водные инфузионные растворы, соответственно растворы для инъекций, которые содержат одно или несколько дополнительных вспомогательных веществ, используемых в технологии приготовления лекарственных средств и выбранных из группы, включающей комплексообразователи, ингибиторы кристаллизации, загустители, придающие изотоничность агенты, консерванты,светостабилизаторы и антиокислители. В качестве примера пригодных для применения в предлагаемых в изобретении растворах комплексообразователей можно назвать истинные (незамещенные) и замещенные циклодекстрины, ЭДТК, альбумины, а также лимонную кислоту, ее соли и производные. В качестве примера пригодных для применения в предлагаемых в изобретении растворах ингибиторов кристаллизации можно назвать поливинилпирролидон, производные целлюлозы, альгинаты, полоксамеры и полисорбаты. В качестве примера пригодных для применения в предлагаемых в изобретении растворах загустителей можно назвать декстраны, глицерин и растворимые производные целлюлозы, прежде всего карбоксиметилцеллюлозу и ее соли, а также гидроксиалкилцеллюлозы. В качестве примера пригодных для применения в предлагаемых в изобретении растворах придающих изотоничность агентов можно назвать NaCl, маннит, сорбит, ксилит, сахарозу, лактозу, глюкозу и-5 014237 глицерин, предпочтительны среди которых NaCl, маннит, глюкоза, сахароза и глицерин, а осбенно предпочтительны NaCl, маннит и глюкоза. В качестве примера пригодных для применения в предлагаемых в изобретении растворах консервантов можно назвать эфиры n-гидроксибензойной кислоты, бензиловый спирт, сорбиновую кислоту и бензойную кислоту. В качестве примера пригодных для применения в предлагаемых в изобретении растворах светостабилизаторов можно назвать производные n-гидроксибензойной кислоты, а также коричную кислоту и ее производные. В качестве примера пригодных для применения в предлагаемых в изобретении растворах антиокислителей можно назвать аскорбиновую кислоту и ее соли. Особенно предпочтительны также водные инфузионные растворы, соответственно растворы для инъекций, осмомолярность которых составляет от 200 до 600 мосмомолей/кг, предпочтительно от 260 до 350 мосмомолей/кг. Осмомолярность предлагаемых в изобретении растворов можно устанавливать на указанные значения добавлением придающих изотоничность агентов, таких как NaCl, маннит, сорбит,глюкоза, сахароза, ксилит, фруктоза и глицерин или смеси указанных веществ. Предпочтительны инфузионные растворы, соответственно растворы для инъекций, которые помимо действующего вещества,воды, кислоты(кислот) и прочих вспомогательных веществ, используемых в технологии приготовления лекарственных средств, содержат NaCl или иной придающий изотоничность агент в количестве, при котором раствор является изотоническим либо слегка гипо- или гипертоническим по отношению к жидкости тканей организма человека или животного. Предпочтительны, кроме того, водные инфузионные растворы, соответственно растворы для инъекций, значение рН которых лежит в интервале от 2,4 до 5,3, предпочтительно от 3,5 до 5,0, наиболее предпочтительно от 3,9 до 4,5. Предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций можно, кроме того, в целях снабжения их электролитами без добавления углеводов разбавлять или разводить имеющимися в продаже инфузионными физиологическими растворами, соответственно физиологическими растворами для инъекций, такими как изотонический раствор NaCl, изотонический раствор глюкозы, содержащий лактат раствор Рингера и аналогичные растворы (Rote Liste 2004, Verzeichnis der Fertigarzneimittel der Mitglieder des Bundesverbandes der Pharmazeutischen Industrie e.V., Editio Cantor, Aulendorf/Wrtt., основные группы 52.1 und 52.2.1), с доведением таким путем концентрации, соответственно дозы содержащегося в них действующего вещества до требуемого уровня без опасности возникновения физической или химической несовместимости. Равным образом предпочтительны водные инфузионные растворы, соответственно растворы для инъекций, которые в пересчете на 100 мл своего объема содержат соляную кислоту в количестве от 1,25 до 3,0 молей, предпочтительно от 1,5 до 2,4 моля, на моль действующего вещества и NaCl в количестве от 0,75 до 1,2 г, предпочтительно от 0,85 до 0,95 г и которые имеют осмомолярность от 260 до 350 мосмомолей/кг и значение рН от 3,5 до 5,0. Еще одним объектом изобретения являются лиофилизаты, концентраты и суспензии, из которых добавлением к ним воды получают один из предлагаемых в изобретении водных инфузионных растворов, соответственно растворов для инъекций. Еще одним объектом изобретения являются предлагаемые в нем инфузионные растворы, соответственно растворы для инъекций для применения в качестве лекарственных средств с антипролиферативным действием. Еще одним объектом изобретения является применение предлагаемых в нем инфузионных растворов, соответственно растворов для инъекций для приготовления лекарственного средства, предназначенного для лечения опухолевых, инфекционных, воспалительных и аутоиммунных заболеваний. Еще одним объектом изобретения является способ лечения и/или профилактики опухолевых, инфекционных, воспалительных и аутоиммунных заболеваний, предпочтительно опухолевых заболеваний,заключающийся во введении в организм пациента предлагаемого в изобретении инфузионного раствора,соответственно раствора для инъекций в эффективном количестве. Еще одним объектом изобретения является применение предлагаемых в нем инфузионных растворов, соответственно растворов для инъекций в количестве, которое соответствует интервалу доз от 0,1 до 50 мг действующего вещества на кг веса тела, предпочтительно от 0,5 до 25 мг действующего вещества на кг веса тела. Предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций можно расфасовывать в пригодные для этой цели стеклянные емкости для парентеральных лекарственных форм или в мягкие пластиковые пакеты или мешки, предпочтительно из материалов, отличных от поливинилхлорида, например, из материалов на полиолефиновой основе, с вместимостью от 20 до 1000 мл,предпочтительно от 50 до 500 мл. Такие емкости могут иметь исполнение, при котором они обеспечивают специальную защиту находящихся в них предлагаемых в изобретении инфузионных растворов, соответственно растворов для инъекций, например, от воздействия света или контакта с кислородом. Особая обработка поверхностей первичных упаковок (например, силиконизация поверхностей стеклянных емко-6 014237 стей (путем обжига для повышения стабильности хранящихся в них предлагаемых в изобретении инфузионных растворов, соответственно растворов для инъекций в принципе не требуется, но и в то же время не оказывает никакого отрицательного влияния. Мягкие (гибкие) пластиковые пакеты или мешки могут быть снабжены дополнительной защитой, например, в виде алюминиевой упаковки. Предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций пригодны для их заключительной стерилизации, например, острым водяным паром, что тем самым позволяет приготавливать их с особо высокой экономичностью и с высокой безопасностью (с малым риском бактериального загрязнения) в стерильном виде без пирогенных продуктов. Предлагаемые в изобретении инфузионные растворы, соответственно растворы для инъекций можно приготавливать известными из литературы методами приготовления водных жидких лекарственных форм. В соответствии с этим настоящее изобретение относится также к способу приготовления предлагаемых в нем инфузионных растворов, соответственно растворов для инъекций, содержащих на миллилитр их объема от 0,1 до 10 мг действующего вещества формулы (I). Такой способ отличается тем, что взятое в соответствующем количестве действующего вещество, необязательно в виде его соли с анионным противоионом, гидрата либо гидрата соли, или же взятые в соответствующем количестве смеси этих солей/гидратов смешивают с физиологически совместимой кислотой или смесью физиологически совместимых кислот, взятой в количестве, избыточном по отношению к количеству, которое требуется непосредственно для растворения действующего вещества, соответственно его солей или гидратов, а также для предупреждения физической нестабильности, необязательно добавляют другие вспомогательные вещества, используемые в технологии приготовления лекарственных средств, и добавлением воды (для инъекций) объем инфузионного раствора, соответственно раствора для инъекций доводят до величины,при которой концентрация в нем действующего вещества лежит в интервале от 0,1 до 10 мг на миллилитр раствора. При приготовлении инфузионных растворов, соответственно растворов для инъекций необходимо также учитывать, что по своим свойствам касательно значения рН, содержания кислоты(кислот) и осмомолярности они должны удовлетворять уже рассмотренным выше требованиям. При использовании соли действующего вещества может оказаться целесообразным использовать кислоту, анион которой соответствует аниону соли действующего вещества, соответственно аниону гидрата такой соли. При необходимости действующее вещество или его соль либо гидрат суспендируют в воде и добавляют физиологически совместимую кислоту или смесь физиологически совместимых кислот, предпочтительно соляную кислоту, в количестве до 3,0 молей на моль действующего вещества. В завершение добавляют другие вспомогательные вещества, используемые в технологии приготовления лекарственных средств, прежде всего придающие изотоничность агенты, предпочтительно NaCl,который в некоторых случаях может также образовываться в результате реакции нейтрализации в перерабатываемой в инфузионный раствор, соответственно раствор для инъекций смеси, после чего объем раствора добавлением воды доводят до величины, при которой концентрация в нем действующего вещества соответствует требуемой. Значение рН предлагаемых в изобретении инфузионных растворов, соответственно растворов для инъекций можно устанавливать на указанные выше значения с помощью (физиологически) совместимых кислот и/или оснований, прежде всего NaOH. Для ускорения процесса приготовления растворов, прежде всего для ускорения процесса растворения твердых компонентов, растворы можно по частям или целиком слегка нагревать, предпочтительно до температуры в пределах от 20 до 80 С. Особо экономичный способ приготовления предлагаемых в изобретении растворов состоит в предварительном или промежуточном приготовлении концентрированных растворов. Для этого действующее вещество в необходимом для приготовления одной порции смеси количестве смешивают с физиологически совместимой кислотой или смесью физиологически совместимых кислот, взятой в основном количестве (90%), и растворяют в ней, при необходимости при слабом нагреве и/или при добавлении небольшого количества воды. Полученный таким путем концентрат затем разбавляют водой, после чего добавляют другие вспомогательные вещества, используемые в технологии приготовления лекарственных средств, и в завершение остальное количество кислоты(кислот), соответственно воды, доводя объем раствора до номинального. После приготовления раствора его несмотря на заключительную стерилизацию острым паром обычно фильтруют через мембранный фильтр или фильтр для глубокой очистки с размером пор 0,2 мкм для отделения возможно присутствующих частиц и/или пирогенных продуктов. Соответствующие методы фильтрации известны из уровня техники (M.J. Groves, Parenteral Technology Manual, изд-во InterpharmPress Inc., 2-е изд., 1988). Количество частиц ограничивают при этом регламентированно необходимой и экономически целесообразной величиной, например, 6000 частицами размером 10 мкм и 600 частицами размером 25 мкм в одной порции (одна порция соответствует 100 мл), соответственно 25 частицами размером 10 мкм и 3 частицами размером 25 мкм на миллилитр (при объеме одной порции более 100 мл),USP 27 788.-7 014237 Предлагаемые в изобретении растворы обладают высокой стабильностью при хранении, которая обусловлена присутствием в них видимых и не видимых невооруженным глазом частиц лишь в количестве, не превышающем допустимые пределы, и отсутствием реакций, приводящих к существенному разложению действующего вещества. Предлагаемые в изобретении растворы обладают с точки зрения фармакодинамических свойств действующего вещества достаточной локальной переносимостью и не являются гемолитическими. Ниже на примерах рассмотрены некоторые возможные составы предлагаемых в изобретении инфузионных растворов, соответственно растворов для инъекций. Эти примеры носят исключительно иллюстративный характер и не ограничивают объем изобретения. На прилагаемом к описанию чертеже показан график зависимости значения рН готового к применению раствора от молярного соотношения в нем между кислотой/смесью кислот и действующим веществом. В данном случае действующим веществом является 4-(7R)-8-циклопентил-7-этил-5,6,7,8 тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)бензамид (пример 46 в табл. 1). Для улучшения локальной переносимости при внутривенной инфузии или инъекции максимальное молярное соотношение между кислотой/смесью кислот и действующим веществом в предлагаемом в изобретении инфузионном растворе, соответственно растворе для инъекций ограничено величиной в 3:1 с целью установления рН раствора на значение выше 2,4. Примеры парентеральных инфузионных растворов, соответственно растворов для инъекций. В приведенных ниже примерах сокращение "ВДИ" означает "вода для инъекций". В представленном ниже общем примере 1 действующим веществом является один из дигидроптеридинонов приведенной выше общей формулы (I). Общий пример 1 В последующих примерах действующим веществом является 4-(7R)-8-циклопентил-7-этил-5,6,7,8 тетрагидро-5-метил-6-оксо-2-птеридинил]амино]-3-метокси-N-(1-метил-4-пиперидинил)бензамид (пример 46 в табл. 1). Пример 2 Пример 3 В последующих примерах действующим веществом является -N-[транс-4-[4-(циклопропилметил)-1 пиперазинил]циклогексил]-4-(7R)-7-этил-5,6,7,8-тетрагидро-5-метил-8-(1-метилэтил)-6-оксо-2 птеридинил]амино]-3-метокси-бензамид (пример 110 в табл. 1). Пример 12 Пример 13 Предлагаемые в изобретении соединения можно получать описанным ниже методом синтеза А, при этом заместители в общих формулах (А 1)-(А 9) имеют указанные выше значения. Этот метод лишь иллюстрирует настоящее изобретение и не ограничивает его объем.-9 014237 Метод А. Стадия 1 А. Соединение формулы (А 1) подвергают взаимодействию с соединением формулы (А 2) с получением соединения формулы (A3) (схема 1 А). Эту реакцию можно проводить в соответствии с методом, описанным в WO 00/43369 или WO 00/43372. Соединение (А 1) представляет собой коммерчески доступный продукт, выпускаемый, например, фирмой City Chemical LLC, расположенной по адресу: 139 AllingsCrossing Road, West Haven, CT, 06516, USA. Соединение формулы (А 2) можно получать известными из литературы методами, описанными, например, в следующих публикациях: (a) F. Effenberger, U. Burkhart,J. Willfahrt, Liebigs Ann. Chem. 1986, cc. 314-333, (б) Т. Fukuyama, C.-K. Jow, M. Cheung, Tetrahedron Lett. 36, 1995, cc. 6373-6374, (в) R.K. Olsen, J. Org. Chem. 35, 1970, cc. 1912-1915, (г) F.E. Dutton, B.H. Byung,Tetrahedron Lett. 30, 1998, cc. 5313-5316, или (д) J.M. Ranajuhi, M.M. Joullie, Synth. Commun. 26, 1996, cc. 1379-1384. Схема 1 А На стадии 1 А 1 эквивалент соединения (А 1) и 1-1,5 эквивалента, предпочтительно 1,1 эквивалента,основания, предпочтительно карбоната калия, гидрокарбоната калия, карбоната натрия, гидрокарбоната натрия или карбоната кальция, наиболее предпочтительно карбоната калия, перемешивают в разбавителе, смешанном в некоторых случаях с водой, например, в ацетоне, тетрагидрофуране, диэтиловом эфире,циклогексане, петролейном эфире или диоксане, предпочтительно циклогексане или диэтиловом эфире. Далее при температуре 0-15 С, предпочтительно 5-10 С, по каплям добавляют 1 эквивалент аминокислоты формулы (А 2), растворенной в органическом растворителе, например, ацетоне, тетрагидрофуране, диэтиловом эфире, циклогексане или диоксане. После этого реакционную смесь при перемешивании нагревают до температуры 18-30 С, предпочтительно примерно 22 С, и затем перемешивают еще в течение 10-24 ч, предпочтительно примерно 12 ч. После этого отгоняют разбавитель, остаток смешивают с водой и смесь два-три раза экстрагируют органическим растворителем, например, диэтиловым эфиром или этилацетатом, предпочтительно этилацетатом. Объединенные органические экстракты сушат и отгоняют растворитель. Остаток (соединение формулы (A3 можно без предварительной очистки использовать на стадии 2. Стадия 2 А. Полученное на стадии 1 А соединение (A3) восстанавливают по нитрогруппе и циклизуют до соединения формулы (А 4) (схема 2 А). Схема 2 А На стадии 2 А 1 эквивалент нитросоединения формулы (A3) растворяют в кислоте, предпочтительно ледяной уксусной кислоте, муравьиной кислоте или водной соляной кислоте, предпочтительно ледяной уксусной кислоте, и нагревают до 50-70 С, предпочтительно до примерно 60 С. Затем до завершения экзотермической реакции добавляют восстановитель, например, цинк, олово или железо, предпочтительно железный порошок, и перемешивают в течение 0,2-2 ч, предпочтительно 0,5 ч, при 100-125 С, предпочтительно при примерно 117 С. После охлаждения до комнатной температуры железную соль отфильтровывают и отгоняют растворитель. Остаток растворяют в растворителе или смеси растворителей,например, в этилацетате или смеси дихлорметан/метанол в соотношении 9:1, и полунасыщенном растворе NaCl и фильтруют, например, через кизельгур. Органическую фазу сушат и концентрируют. Остаток(соединение формулы (А 4 можно очищать путем хроматографии или кристаллизации либо использовать для синтеза на стадии 3 А в виде сырого продукта. Стадия 3 А. Полученное на стадии 2 А соединение формулы (А 4) можно путем электрофильного замещения в соответствии со схемой 3 А превращать в соединение формулы (А 5). Схема 3 А На стадии 3 А 1 эквивалент амида формулы (А 4) растворяют в органическом растворителе, например, диметилформамиде или диметилацетамиде, предпочтительно диметилацетамиде, и охлаждают до температуры в интервале примерно от -5 до 5 С, предпочтительно до температуры 0 С. Затем добавляют 0,9-1,3 эквивалента гидрида натрия и 0,9-1,3 эквивалента агента метилирования,например, метилиодида. Далее реакционную смесь перемешивают в течение 0,1-3 ч, предпочтительно в течение примерно 1 ч, при примерно 0-10 С, предпочтительно при примерно 5 С, и затем ее при необходимости можно оставлять стоять еще на 12 ч при температуре в указанном интервале. После этого реакционную смесь сливают в смесь воды со льдом и отделяют осадок. Остаток (соединение формулы (А 5 можно очищать путем хроматографии, предпочтительно на силикагеле, или путем кристаллизации либо использовать для синтеза на стадии 4 А в виде сырого продукта. Стадия 4 А. Полученное на стадии 3 А соединение формулы (А 5) можно аминировать до соединения формулы(А 9) (схема 4 А) известными из литературы методами, которые для варианта 4.1 А, описаны, например, в следующих публикациях: (а) M.P.V. Boarland, J.F.W. McOmie, J. Chem. Soc. 1951, cc., 1218-1221, или (б) Согласно варианту 4.1 А, например, 1 эквивалент соединения формулы (А 5) и 1-3 эквивалента,предпочтительно примерно 2 эквивалента, соединения формулы (А 6) без растворителя или в органическом растворителе, таком, например, как сульфолан, диметилформамид, диметилацетамид, толуол, Nметилпирролидон, диметилсульфоксид или диоксан, предпочтительно сульфолан, нагревают до 100220 С, предпочтительно до примерно 160 С, с выдержкой при этой температуре в течение 0,1-4 ч, предпочтительно 1 ч. После охлаждения продукт формулы (А 9) кристаллизуют добавлением органических растворителей или смеси растворителей, например, смеси диэтиловый эфир/метанол, этилацетата, метиленхлорида или диэтилового эфира, предпочтительно смеси диэтиловый эфир/метанол в соотношении 9:1, или очищают путем хроматографии. Согласно варианту 4.2 А, например, 1 эквивалент соединения формулы (А 5) и 1-3 эквивалента соединения формулы (А 6) при нагревании с обратным холодильником перемешивают в течение 1-48 ч,- 11014237 предпочтительно в течение примерно 5 ч, с кислотой, например, 1-10 эквивалентами 10-38%-ной соляной кислоты, и/или спиртом, например, этанолом, пропанолом или бутанолом, предпочтительно этанолом. Выпавший в осадок продукт формулы (А 9) отфильтровывают, при необходимости промывают водой, сушат и кристаллизуют из приемлемого органического растворителя. Согласно варианту 4.3 А, например, 1 эквивалент соединения формулы (А 5) и 1-3 эквивалента соединения формулы (А 7) растворяют в растворителе, например, толуоле или диоксане, смешивают с фосфиновыми лигандами, например, 2,2'-бис-(дифенилфосфино)-1,1'-бинафтилом, палладиевым катализатором, например, трис-(дибензилиденацетон)дипалладием(0), и основанием, например, карбонатом цезия,и кипятят с обратным холодильником в течение 1-24 ч, предпочтительно в течение 17 ч. Затем реакционную смесь очищают, например, на силикагеле и продукт формулы (А 8) выделяют из раствора либо получают путем соответствующей кристаллизации. Продукт формулы (А 8) растворяют в приемлемом растворителе, например, диоксане, и смешивают с кислотой, например, полуконцентрированной соляной кислотой, например, при соотношении между растворителем и кислотой 3:1. После этого нагревают с обратным холодильником в течение 1-48 ч, например, в течение 12 ч, и отделяют выпавший осадок. При необходимости продукт формулы (А 9) очищают путем кристаллизации. Стадия 5 А. Схема 5 А На этой стадии, например, 1 эквивалент соединения формулы (А 9) растворяют с 1 эквивалентом активатора, например, тетрафторбората О-бензотриазолил-N,N,N',N'-тетраметилурония (ТБТУ), и основанием, взятым, например, в количестве примерно 1,5 эквивалента, в частности диизопропилэтиламином(ДИПЭА), в органическом разбавителе, например, дихлорметане, тетрагидрофуране, диметилформамиде,N-метилпирролидоне, диметилацетамиде, предпочтительно дихлорметане или диметилформамиде. После добавления 1 эквивалента амина формулы (А 10) реакционную смесь перемешивают в течение 0,1-24 ч, предпочтительно в течение примерно 2 ч, при температуре в интервале от 20 до 100 С. Продукт формулы (A11) получают, например, кристаллизацией или путем хроматографической очистки. Соединения общей формулы (I) можно синтезировать аналогично описанным ниже примерам. Эти примеры, однако, служат для более детального пояснения настоящего изобретения и не ограничивают его объем. Ниже рассмотрено также получение некоторых промежуточных соединений, используемых для синтеза соединений из соответствующих примеров. Получение кислот Для синтеза соединений из примера 94 и примера 95 сначала описанным ниже методом получают промежуточное соединение Z1 50,0 г (0,48 моля) гидрохлорида метилового эфира D-аланина и 49,1 г (0,50 моля) циклогексанона добавляют в 300 мл дихлорметана и затем смешивают с 41,0 г (0,50 моля) ацетата натрия и 159,0 г (0,75 моля) триацетоксиборгидрида натрия. Далее смесь оставляют перемешиваться на ночь и затем добавляют 300 мл 10%-ного раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы промывают 10%-ным раствором гидрокарбоната натрия, сушат над Na2SO4 и концентрируют. Выход: 72,5 г соединения Z1a (прозрачная жидкость). 72,5 г соединения Z1a добавляют в 500 мл воды и добавляют 76,6 г (0,39 моля) 2,4-дихлор-5 нитропиримидина в 500 мл диэтилового эфира. Далее при температуре -5 С по каплям добавляют 100 мл- 12014237 10%-ного раствора гидрокарбоната калия. После этого сначала перемешивают в течение 3 ч при -5 С, а затем еще в течение 12 ч при комнатной температуре. Органическую фазу отделяют и сушат над Na2SO4. При концентрировании выкристаллизовывается продукт. Выход: 48,0 г соединения Z1b (желтые кристаллы). 48,0 г соединения Z1b растворяют в 350 мл ледяной уксусной кислоты и нагревают до 60 С. Далее порциями добавляют 47,5 г железного порошка, что сопровождается повышением температуры до 105 С. Затем реакционную смесь перемешивают в течение трех часов при 80 С, после чего в горячем состоянии фильтруют через целлюлозу и концентрируют. Остаток размешивают в воде и этилацетате,получая светло-серый осадок, который отделяют вакуум-фильтрацией и промывают этилацетатом. Фильтрат промывают разбавленным аммиаком и водой, органические фазы сушат над Na2SO4, фильтруют через активированный уголь и концентрируют. Таким путем получают дополнительное количество светло-серого твердого вещества. Выход: 29,5 г соединения Z1c (светло-серые кристаллы). 32,1 г соединения Z1c добавляют в 300 мл диметилацетамида и смешивают с 13 мл (0,2 моля) метилиодида. Далее при -5 С порциями добавляют 6,4 г (0,16 моля) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Через 2 ч реакционную смесь сливают в 800 мл смеси воды со льдом. Выпавший осадок отделяют вакуум-фильтрацией и промывают петролейным эфиром. Выход: 33,0 г соединения Z1d (бежевые кристаллы). 4,0 г соединения Z1d и 2,3 г (15 ммолей) 4-амино-3-метилбензойной кислоты суспендируют в 50 мл этанола и 120 мл воды, смешивают с 2 мл конц. соляной кислоты и в течение 48 ч кипятят с обратным холодильником. Выпавший при охлаждении осадок отделяют вакуум-фильтрацией и промывают водой,этанолом и диэтиловым эфиром. Выход: 2,9 г соединения Z1 (бесцветные кристаллы). Для синтеза соединений из примера 188 и примера 203 сначала описанным ниже методом получают промежуточное соединение Z2 Раствор 128,2 г (0,83 моля) гидрохлорида этилового эфира D-аланина и 71,5 г (0,85 моля) циклопентанона в 1500 мл дихлорметана смешивают с 70,1 (0,85 моля) ацетата натрия и 265,6 г (1,25 моля) триацетоксиборгидрида натрия. Далее реакционную смесь перемешивают в течение 12 ч и затем сливают в 1,5 л 10%-ного раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и концентрируют. Выход: 143,4 г соединения Z2a (бесцветное масло). 66,0 г соединения Z2a добавляют в 500 мл воды и смешивают с 85,0 г (0,44 моля) 2,4-дихлор-5 нитропиримидина в 500 мл диэтилового эфира. Далее при -5 С по каплям добавляют 100 мл 10%-ного раствора гидрокарбоната калия и реакционную смесь перемешивают в течение 48 ч при комнатной температуре. Водную фазу экстрагируют диэтиловым эфиром, объединенные органические фазы сушат надNa2SO4 и концентрируют. Размешиванием с петролейным эфиром осаждают темно-красное твердое вещество, которое затем отделяют вакуум-фильтрацией. Выход: 88,0 г соединения Z2b (желтые кристаллы). 88,0 г соединения Z2b растворяют в 1000 мл ледяной уксусной кислоты и при 60 С порциями смешивают с 85 г железного порошка, что сопровождается повышением температуры до 110 С. Далее перемешивают в течение 1 ч при 60 С, после чего в горячем состоянии подвергают вакуум-фильтрации через целлюлозу и концентрируют. Размешиванием с 700 мл воды осаждают коричневое твердое вещество,которое отделяют вакуум-фильтрацией. Выход: 53,3 г соединения Z2c (коричневатые кристаллы). 53,3 г соединения Z2c растворяют в 300 мл диметилацетамида и смешивают с 13 мл (0,21 моля) метилиодида. Далее при -5 С порциями добавляют 5,0 г (0,21 моля) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Через 12 ч реакционную смесь сливают в 1000 мл смеси воды со льдом и образовавшийся осадок отделяют вакуум-фильтрацией. Выход: 40,0 г соединения Z2d (бесцветные кристаллы). 4,0 г соединения Z2d и 2,8 г (16 ммолей) 4-амино-3-хлорбензойной кислоты суспендируют в 25 мл этанола и 60 мл воды, смешивают с 3 мл конц. соляной кислоты и кипятят с обратным холодильником в течение 43 ч. Выпавший при охлаждении осадок отделяют вакуум-фильтрацией и промывают его водой,этанолом и диэтиловым эфиром. Выход: 0,9 г соединения Z2 (бесцветные кристаллы).- 13014237 Для синтеза соединений из примеров 19, 21, 22, 23, 45, 55, 58, 116, 128, 131, 133, 134, 136, 138, 177,217, 231, 239, 46, 184, 166 и 187 сначала описанным ниже методом получают промежуточное соединение 54,0 г (0,52 моля) D-2-аминомасляной кислоты суспендируют в 540 мл метанола и при охлаждении льдом медленно смешивают с 132 г (1,1 моля) тионилхлорида. Далее в течение 1,5 ч кипятят с обратным холодильником и затем концентрируют. Оставшееся масло смешивают с 540 мл трет-бутилметилового эфира и образовавшиеся бесцветные кристаллы отделяют вакуум-фильтрацией. Выход: 78,8 г соединения Z3a (бесцветные кристаллы). 74,2 г соединения Z3a и 43,5 мл (0,49 моля) циклопентанона растворяют в 800 мл дихлорметана. После добавления 40,0 г (0,49 моля) ацетата натрия и 150,0 г (0,71 моля) триацетоксиборгидрида натрия при 0 С перемешивают в течение 12 ч при комнатной температуре и затем добавляют 500 мл 20%-ного раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы промывают водой, сушат над MgSO4 и концентрируют. Выход: 85,8 г соединения Z3b (желтоватое масло). 40,0 г соединения Z3b и 30,0 г (0,22 моля) карбоната калия суспендируют в 600 мл ацетона и при охлаждении льдом смешивают с 45,0 г (0,23 моля) 2,4-дихлор-5-нитропиримидина в 200 мл ацетона. Через 12 ч добавляют еще 5,0 г 2,4-дихлор-5-нитропиримидина и перемешивают в течение 3 ч. Далее реакционную смесь концентрируют, растворяют в 800 мл этилацетата и 600 мл воды и водную фазу экстрагируют этилацетатом. Объединенные органические фазы промывают водой, сушат над MgSO4 и концентрируют. Выход: 75,0 г соединения Z3c (коричневое масло). 100 г соединения Z3c растворяют в 650 мл ледяной уксусной кислоты и при 70 С порциями смешивают с 20 г железного порошка. Далее сначала перемешивают в течение 1 ч при 70 С, а затем в течение 1,5 ч при 100 С, после чего в горячем состоянии фильтруют через кизельгур. Затем реакционную смесь концентрируют, растворяют в смеси метанол/дихлорметан, подают на силикагель и очищают путем экстракции этилацетатом в приборе Сокслета. Растворитель удаляют и остаток осаждают размешиванием с метанолом. Выход: 30,0 г соединения Z3d (светло-коричневые кристаллы). 25,0 г соединения Z3d и 6,5 мл (0,1 моля) метилиодида добавляют в 250 мл диметилацетамида и при-10 С смешивают с 3,8 г (0,95 моля) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Далее сначала перемешивают в течение 20 мин при 0 С, а затем в течение 30 мин при комнатной температуре и в завершение добавляют лед. После этого реакционную смесь концентрируют и смешивают с 300 мл воды. Образовавшийся осадок отделяют вакуум-фильтрацией и промывают петролейным эфиром. Выход: 23,0 г соединения Z3e (бесцветное твердое вещество). 6,0 г соединения Z3e и 5,1 г (31 ммоль) 4-амино-3-метоксибензойной кислоты суспендируют в 90 мл этанола и 350 мл воды, смешивают с 3,5 мл конц. соляной кислоты и кипятят с обратным холодильником в течение 48 ч. Далее реакционную смесь концентрируют, остаток размешивают со смесью метанол/диэтиловый эфир и образовавшийся осадок отделяют вакуум-фильтрацией. Выход: 6,3 г соединения Z3 (светло-бежевые кристаллы). Для синтеза соединений из примеров 81, 82, 93, 137 сначала описанным ниже методом получают промежуточное соединение Z4 25,0 г (0,19 моля) гидрохлорида этилового эфира 1-аминоциклопропан-1-карбоновой кислоты и 16,8 г (0,20 моля) циклопентанона растворяют в 300 мл дихлорметана и смешивают с 16,4 г (0,20 моля) ацетата натрия и 61,7 г (0,29 моля) триацетоксиборгидрида натрия. После этого реакционную смесь оставляют перемешиваться на ночь и затем сливают в 400 мл 10%-ного раствора гидрокарбоната натрия. Водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и концентри- 14014237 руют. Выход: 34,5 г соединения Z4a (бесцветное масло). К смеси 34,5 г соединения Z4a в 350 мл воды добавляют 42,5 г (0,22 моля) 2,4-дихлор-5 нитропиримидина в 350 мл диэтилового эфира. Далее при -5 С смешивают с 80 мл 10%-ного раствора гидрокарбоната калия и оставляют перемешиваться на ночь при комнатной температуре. Водную фазу экстрагируют диэтиловым эфиром. Объединенные органические фазы сушат над Na2SO4 и концентрируют. Выход: 53,8 г соединения Z4b (коричневое масло). 20,1 г соединения Z4b растворяют в 200 мл ледяной уксусной кислоты и при 60 С порциями смешивают с 19,1 г железного порошка, что сопровождается повышением температуры до 100 С. Затем смесь перемешивают в течение 3 ч при 60 С, после чего подвергают вакуум-фильтрации через целлюлозу и концентрируют. Остаток размешивают в воде и этилацетате, получая желтый осадок, который отделяют вакуум-фильтрацией. Фильтрат промывают разбавленным аммиаком и водой, органические фазы сушат над Na2SO4 и концентрируют. Продукт кристаллизуют после добавления диэтилового эфира. Выход: 4,0 г соединения Z4c (желтые кристаллы). 7,8 г соединения Z4c и 2,6 мл (0,04 моля) метилиодида растворяют в 100 мл диметилацетамида и при -5 С порциями смешивают с 1,5 г (0,04 моля) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Через 2 ч реакционную смесь сливают в смесь воды со льдом и образовавшийся осадок отделяют вакуум-фильтрацией. Выход: 7,5 г соединения Z4d (коричневатые кристаллы). 3,0 г соединения Z4d и 1,9 г (11 ммолей) 4-амино-3-метоксибензойной кислоты суспендируют в 40 мл этанола и 80 мл воды, смешивают с 2 мл конц. соляной кислоты и в течение 20 ч кипятят с обратным холодильником. После этого добавляют еще 0,5 г 4-амино-3-метоксибензойной кислоты и кипятят с обратным холодильником в течение 48 ч. Выпавший при охлаждении осадок отделяют вакуумфильтрацией и промывают водой, этанолом и диэтиловым эфиром. Выход: 2,1 г соединения Z4 (бесцветные кристаллы). Для синтеза соединений из примеров 162, 43, 53, 161, 202, 211, 215 и 212 сначала описанным ниже методом получают промежуточное соединение Z5 Смесь из 73,4 мл (0,5 моля) этилового эфира 2-бромизомасляной кислоты, 87,1 мл (0,75 моля) 3 метил-1-бутиламина, 82,5 г (0,6 моля) иодида натрия и 76,0 г (0,6 моля) карбоната калия в течение 3 дней кипятят с обратным холодильником в 1000 мл этилацетата. Имеющиеся соли отфильтровывают и фильтрат концентрируют. Выход: 97,0 г соединения Z5a (красное масло). 49,0 г (0,25 моля) 2,4-дихлор-5-нитропиримидина и 38,3 г (0,28 моля) карбоната калия суспендируют в 500 мл ацетона и при 0 С смешивают с 93,0 г соединения Z5a в 375 мл ацетона. Реакционную смесь оставлют перемешиваться на ночь при комнатной температуре, фильтруют и концентрируют. Растворенный в этилацетате остаток промывают водой и органические фазы сушат над MgSO4 и концентрируют. Выход: 102,7 г соединения Z5b (коричневое масло). 22,7 г соединения Z5b растворяют в 350 мл ледяной уксусной кислоты и при 60 С порциями смешивают с 17,4 г железного порошка. По завершении этой операции добавления в течение 0,5 ч кипятят с обратным холодильником, после чего фильтруют в горячем состоянии и концентрируют. Остаток растворяют в 200 мл смеси дихлорметан/метанол (в соотношении 9:1) и промывают раствором хлорида натрия. Органическую фазу отделяют вакуум-фильтрацией через кизельгур, сушат над MgSO4, концентрируют и очищают колоночной хроматографией (элюент: этилацетат/циклогексан в соотношении 1:1). Выход: 1,9 г соединения Z5c (бесцветные кристаллы). 1,9 г соединения Z5c растворяют в 32 мл диметилацетамида и при охлаждении льдом смешивают с 0,3 г (7 ммолей) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Через 10 мин добавляют 0,5 мл (7 ммолей) метилиодида и в течение 3 ч перемешивают при комнатной температуре. Далее реакционную смесь концентрируют и смешивают с водой. Образовашийся осадок отделяют вакуумфильтрацией и промывают петролейным эфиром. Выход: 1,6 г соединения Z5d (бесцветные кристаллы). 14,0 г соединения Z5d и 10,0 г (0,06 моля) 4-амино-3-метоксибензойной кислоты суспендируют в 200 мл диоксана и 80 мл воды, смешивают с 10 мл конц. соляной кислоты и кипятят с обратным холодильником в течение 40 ч. Выпавший при охлаждении осадок отделяют вакуум-фильтрацией и промы- 15014237 вают водой, диоксаном и диэтиловым эфиром. Выход: 13,9 г соединения Z5 (бесцветные кристаллы). Для синтеза соединений из примеров 88, 194, 229 и 89 сначала описанным ниже методом получают промежуточное соединение Z6 6,0 г (0,06 моля) L-2-аминомасляной кислоты добавляют в 80 мл 0,5-молярной серной кислоты и при 0 С смешивают с 5,5 г (0,08 моля) нитрита натрия в 15 мл воды. Далее реакционную смесь в течение 22 ч перемешивают при 0 С, смешивают с сульфатом аммония и фильтруют. Фильтрат экстрагируют диэтиловым эфиром и объединенные органические фазы сушат над MgSO4 и концентрируют. Выход: 6,0 г соединения Z6a (желтое масло). 200 мл метанола при охлаждении льдом последовательно смешивают с 65,0 мл (0,89 моля) тионилхлорида и 76,0 г соединения Z6a в 50 мл метанола. Далее смесь сначала перемешивают в течение 1 ч при 0 С, а затем в течение 2 ч при комнатной температуре и после этого метанол и остаточный тионилхлорид удаляют в вакууме при 0 С. Выход: 40,0 г соединения Z6b (желтое масло). 30,0 мл (0,17 моля) трифторметансульфонового ангидрида добавляют в 150 мл дихлорметана и при охлаждении льдом смешивают в течение часа с раствором 20,0 г соединения Z6b и 14,0 мл (0,17 моля) пиридина в 50 мл дихлорметана. После этого смесь в течение 2 ч перемешивают при комнатной температуре, образовавшиеся соли отделяют вакуум-фильтрацией и затем промывают 100 мл воды. Органическую фазу сушат над MgSO4 и концентрируют. Выход: 42,0 г соединения Z6c (светло-желтое масло). К раствору 15,5 мл (0,17 моля) анилина и 24,0 мл (0,17 моля) триэтиламина в 400 мл дихлорметана при охлаждении льдом в течение часа по каплям добавляют 42,0 г соединения Z6c в 200 мл дихлорметана. После этого смесь перемешивают сначала в течение 1 ч при комнатной температуре, а затем в течение 2 ч при 35 С. Далее реакционную смесь промывают водой, сушат над MgSO4 и концентрируют. Полученный остаток очищают перегонкой (95-100 С, 110-3 мбар). Выход: 14,0 г соединения Z6d (бесцветное масло). 14,0 г соединения Z6d и 16,0 г (0,1 моля) карбоната калия суспендируют в 100 мл ацетона и при 10 С смешивают с 16,0 г (0,08 моля) 2,4-дихлор-5-нитропиримидина. Затем смесь перемешивают в течение 4 ч при 40 С, образовавшиеся соли отделяют вакуум-фильтрацией и фильтрат концентрируют. Остаток растворяют в 300 мл этилацетата и промывают водой. Органическую фазу сушат над MgSO4 и концентрируют. Выход: 31,0 г соединения Z6e (коричневое масло). 31,0 г соединения Z6e растворяют в 200 мл ледяной уксусной кислоты и при 60 С порциями смешивают с 10 г железного порошка, что сопровождается повышением температуры до 85 С. Затем смесь перемешивают в течение последующего часа при 60 С, фильтруют через кизельгур и концентрируют. Остаток размешивают с метанолом, осаждая продукт. Выход: 4,5 г соединения Z6f (коричневые кристаллы). К смеси 4,5 г соединения Z6f и 1,0 мл (16 ммолей) метилиодида в 100 мл диметилацетамида при-20 С порциями добавляют 0,6 г (16 ммолей) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Через 1 ч реакционную смесь смешивают с 50 мл воды и концентрируют. Остаток размешивают с 200 мл воды, осадок отделяют вакуум-фильтрацией и промывают петролейным эфиром. Выход: 4,5 г соединения Z6g (бесцветные кристаллы). Суспензию 1,5 г соединения Z6g и 1,4 г (8 ммолей) метилового эфира 4-амино-3-метоксибензойной кислоты в 30 мл толуола смешивают с 0,4 г (0,6 ммоля) 2,2'-бис-(дифенилфосфино)-1,1'-бинафтила, 0,23 г(0,3 ммоля) трис-(дибензилиденацетон)дипалладия(0) и 7,0 г (21 ммоль) карбоната цезия и кипятят с обратным холодильником в течение 17 ч. Далее реакционную смесь подают на силикагель и очищают колоночной хроматографией (элюент: смесь дихлорметан/метанол в соотношении 9:1). Выход: 1,7 г соединения Z6h (желтые кристаллы). 1,7 г соединения Z6h растворяют в 50 мл диоксана, смешивают с 15 мл полуконцентрированной соляной кислоты и в течение 12 ч кипятят с обратным холодильником. Образовавшийся после охлаждения осадок отделяют вакуум-фильтрацией. Выход: 1,1 г соединения Z6 (бесцветное твердое вещество). Для синтеза соединений из примеров 26, 20, 32, 56, 101, 112, 209 сначала описанным ниже методом получают промежуточное соединение Z7 50,0 г (0,36 моля) гидрохлорида метилового эфира D-аланина суспендируют в 500 мл дихлорметана и 35 мл ацетона и смешивают с 30,0 г (0,37 моля) ацетата натрия и 80,0 г (0,38 моля) триацетоксиборгидрида натрия. Затем смесь перемешивают в течение 12 ч, после чего сливают в 400 мл 10%-ного раствора гидрокарбоната натрия. Органическую фазу сушат над Na2SO4 и концентрируют. Выход: 51,0 г соединения Z7a (желтое масло). Суспензию 51,0 г соединения Z7a в 450 мл воды смешивают с 80,0 г (0,41 моля) 2,4-дихлор-5 нитропиридина в 450 мл диэтилового эфира. Далее при -5 С по каплям добавляют 100 мл 10%-ного раствора гидрокарбоната калия. После этого реакционную смесь перемешивают в течение 3 ч, органические фазы сушат над Na2SO4 и концентрируют. Выход: 74 г соединения Z7b (желтое масло). 18,6 г соединения Z7b растворяют в 200 мл ледяной уксусной кислоты и при 60 С порциями смешивают с 20,0 г железного порошка. Далее смесь в течение 2 ч перемешивают при 60 С, а затем подвергают вакуум-фильтрации через целлюлозу. Остаток растворяют в этилацетате и промывают водой и конц. аммиаком. Органическую фазу сушат над Na2SO4 и концентрируют. Остаток кристаллизуют из диэтилового эфира. Выход: 9,8 г соединения Z7c (бесцветные кристаллы). 17,0 г соединения Z7c и 7 мл (0,1 моля) метилиодида растворяют в 200 мл диметилацетамида и при-5 С смешивают с 4,0 г (0,1 моля) гидрида натрия в виде 60%-ной дисперсии в минеральном масле. Далее реакционную смесь перемешивают в течение 30 мин и затем сливают в 300 мл смеси воды со льдом. Образовашийся осадок отделяют вакуум-фильтрацией и выделяют, размешивая с петролейным эфиром. Выход: 14,8 г соединения Z7d (бежевые кристаллы). 0,9 г соединения Z7d и 1,5 г (9 ммолей) 4-амино-3-метоксибензойной кислоты нагревают до 210 С с выдержкой при этой температуре в течение 30 мин. После охлаждения остаток размешивают с этилацетатом, получая осадок, который отделяют вакуум-фильтрацией. Выход: 1,2 г соединения Z7 (серые кристаллы). Аналогично рассмотренным выше методам синтеза получают, например, следующие кислоты: Синтез аминовых фрагментов L-R5 Представленные ниже амины получают следующими методами. 1,1-Диметил-2-диметиламино-1-илэтиламин и 1,1-диметил-2-пиперидин-1-илэтиламин- 17014237 Указанные соединения получают методами, описанными в следующих литературных источниках:(a) S. Schuetz и др., Arzneimittel-Forschung 21, 1971, cc. 739-763, (б) V.M. Belikov и др., Tetrahedron 26,1970, cc. 1199-1216, и (в) Е.В. Butler и McMillan, J. Amer. Chem. Soc. 72, 1950, с. 2978. Другие амины получают модификациями описанных в вышеуказанных публикациях методов следующим путем. 1,1-Диметил-2-морфолин-1-илэтиламин К 8,7 мл морфолина и 9,3 мл 2-нитропропана при охлаждении льдом медленно по каплям добавляют (10 С) 7,5 мл формальдегида (37%-ного) и 4 мл раствора NaOH в количестве 0,5 моля/л. После этого перемешивают сначала в течение 1 ч при 25 С, а затем в течение 1 ч при 50 С. Далее раствор обрабатывают водой и диэтиловым эфиром и водную фазу трижды экстрагируют диэтиловым эфиром. Объединенные органические фазы сушат над NaSO4, смешивают с HCl в диоксане (4 моля/л) и образовавшийся осадок отделяют вакуум-фильтрацией. Выход: 21,7 г белого порошка. 5 г этого белого порошка растворяют в 80 мл метанола и при добавлении 2 г никеля Ренея в течение 40 мин обрабатывают водородом при темературе 35 С и давлении 50 фунтов/кв. дюйм. Таким путем получают 3,6 г 1,1-диметил-2 морфолин-1-илэтиламина. Аналогично этому методу получают следующие амины. 1,1-диметил-N-метилпиперазин-1-илэтиламин 5 г 1,3-диморфолин-2-нитропропана фирмы Aldrich растворяют в 80 мл метанола и при добавлении 2 г никеля Ренея в течение 5,5 ч обрабатывают водородом при температуре 30 С и давлении 50 фунтов/кв.дюйм. Таким путем получают 4,2 г 1,3-диморфолин-2-аминопропана. 4-Аминобензилморфолин Получение этого амина описано в следующей публикации: S. Mitsuru и др., J. Med. Chem. 43, 2000,cc. 2049-2063. 4-Амино-1-тетрагидро-4 Н-пиран-4-илпиперидин 20 г (100 ммолей) 4-трет-бутилоксикарбониламинопиперидина растворяют в 250 мл CH2Cl2 и в течение 12 ч перемешивают при комнатной температуре с 10 г (100 ммолей) тетрагидро-4 Н-пиран-4-она и 42 г (200 ммолей) NaBH(OAc)3. После этого смешивают с водой и карбонатом калия, органическую фазу отделяют, сушат и в вакууме отгоняют растворитель. Остаток растворяют в 200 мл CH2Cl2 и в течение 12 ч перемешивают при комнатной температуре с 100 мл трифторуксусной кислоты. После этого растворитель отгоняют в вакууме, остаток растворяют в CHCl3 и вновь упаривают, после чего растворяют в ацетоне и обработкой раствором HCl в диэтиловом эфире осаждают гидрохлорид. Выход: 14,3 г (56%). цис- и транс-4-морфолиноциклогексиламин Дибензил-4-морфолиноциклогексиламин 3,9 г (30 ммолей) 4-дибензилциклогексанона растворяют в 100 мл CH2Cl2 и в течение 12 ч перемешивают при комнатной температуре с 3,9 г (45 ммолей) морфолина и 9,5 г (45 ммолей) NaBH(OAc)3. После этого смешивают с водой и карбонатом калия, органическую фазу отделяют, сушат и в вакууме отгоняют растворитель. Остаток очищают на силикагелевой колонке (около 20 мл силикагеля, около 500 мл смеси этилацетат/метанол в соотношении 90:10 + 1% конц. аммиака). Объединеные фракции концентрируют в вакууме. Выход 6,6 г (60%) цис-изомера и 2 г (18%) транс-изомера. В другом варианте транс-дибензил-4-морфолиноциклогексиламин можно поучать следующим путем. 33 г (112 ммолей) 4-дибензилциклогексанона растворяют в 300 мл МеОН, смешивают с 17,4 г (250 ммолей) гидрохлорида гидроксиламина и в течение 4 ч перемешивают при 60 С. Затем в вакууме удаляют растворитель, смешивают с 500 мл воды и 50 г карбоната калия и дважды экстрагируют дихлорметаном порциями по 300 мл. Органическую фазу сушат, концентрируют в вакууме, остаток кристаллизуют из петролейного эфира, растворяют в 1,5 л EtOH и нагревают до 70 С. После этого порциями добавляют 166 г натрия и кипятят с обратным холодильником до растворения натрия. Затем растворитель отгоняют в вакууме, остаток смешивают с 100 мл воды и дважды экстрагируют диэтиловым эфиром порциями по 400 мл. Органическую фазу промывают водой, сушат, концентрируют в вакууме и пропусканием через колонку выделяют транс-изомер (около 1,5 л силикагеля, около 2 л смеси этилацетат/метанол в соотношении 80:20 + 2% конц. аммиака). Выход: 12,6 г (41,2%). 6,8 г (23 ммоля) транс-1-амино-4-дибензиламиноциклогексана растворяют в 90 мл ДМФ и в течение 8 ч перемешивают при 100 С с 5 мл (42 ммоля) 2,2'-дихлорэтилового эфира и 5 г карбоната калия. После охлаждения смешивают с 30 мл воды, выпавшие кристаллы отделяют вакуум-фильтрацией и очищают на короткой колонке (около 20 мл силикагеля, около 100 мл этилацетата). Остаток кристаллизуют из метанола и конц. HCl в виде дигидрохлорида. Выход: 7,3 г (72,4%). транс-4-Морфолиноциклогексиламин 7,2 г (16,4 ммоля) транс-дибензил-4-морфолиноциклогексиламина растворяют в 100 мл МеОН и гидрируют на 1,4 г Pd/C (10%-ного) при 30-50 С. Затем растворитель отгоняют в вакууме и остаток кристаллизуют из этанола и конц. HCl. Выход: 3,9 г (93%). цис-Изомер можно получить аналогичным путем. цис- и транс -4-пиперидиноциклогексиламины транс-Дибензил-4-пиперидиноциклогексиламин.2,0 г (6,8 ммоля) транс-1-амино-4-дибензиламиноциклогексана (см. пример 2) растворяют в 50 мл ДМФ и в течение 48 ч перемешивают при комнатной температуре с 1,6 г (7 ммолей) 1,5-дибромпентана и 2 г карбоната калия. Затем смесь охлаждают, смешивают с водой, дважды экстрагируют дихлорметаном порциями по 100 мл, сушат и растворитель отгоняют в вакууме. Остаток очищают на колонке (около 100 мл силикагеля, около 500 мл смеси этилацетат/метанол в соотношении 80:20 + 1% конц. аммиака). Объединенные фракции концентрируют в вакууме и кристаллизуют из петролейного эфира. Выход: 1,2 г (49%). транс-4-Пиперидиноциклогексиламин. 1,7 г (4,8 ммоля) транс-дибензил-4-пиперидиноциклогексиламина растворяют в 35 мл МеОН и гидрируют на 350 мг Pd/C (10%-ного) при 20 С. Затем растворитель отгоняют в вакууме и остаток кристаллизуют из этанола и конц. HCl. Выход: 1,1 г (78%). цис-Изомер можно получить аналогичным путем. цис- и транс-4-(4-фенилпиперазин-1-ил)циклогексиламины 4,1 г (25,3 ммоля) 4-дибензилциклогексанона растворяют в 50 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 7,4 г (25,3 ммоля) N-фенилпиперазина и 7,4 г (35 ммолей)NaBH(OAc)3. После этого смешивают с водой и карбонатом калия, органическую фазу отделяют, сушат и растворитель отгоняют в вакууме. Остаток очищают на силикагелевой колонке (смесь этилаце- 19014237 тат/метанол в соотношении 80:20 + 0,5% конц. аммиака). Выход: 1,7 г (15,8%) цис-изомера и 0,27 (2,5%) транс-изомера. транс-4-(4-Фенилпиперазин-1-ил)циклогексиламин. 270 мг (0,61 ммоля) транс-дибензил-[4-(4-фенилпиперазин-1-ил)циклогексил]амина растворяют в 5 мл МеОН и гидрируют на 40 мг Pd/C (10%-ного) при 20-30 С. Затем растворитель отгоняют в вакууме и остаток кристаллизуют из этанола и конц. HCl. Выход: 110 мг (69%). цис-Изомер можно получить аналогичным путем. цис- и транс-4-(4-циклопропилметилпиперазин-1-ил)циклогексиламины 9,8 г (33,4 ммоля) 4-дибензилциклогексанона растворяют в 100 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 5,6 г (40 ммолей) N-циклопропилметилпиперазина и 8,5 г(40 ммолей) NaBH(OAc)3. После этого смешивают с водой и карбонатом калия, органическую фазу отделяют, сушат и растворитель отгоняют в вакууме. Остаток очищают на силикагелевой колонке (около 50 мл силикагеля, около 3 л смеси этилацетат/метанол в соотношении 95:5 + 0,25% конц. аммиака). Объединенные фракции концентрируют в вакууме. Быстрее элюирующееся цис-соединение кристаллизуют из этилацетата. транс-Соединение кристаллизуют из этанола и конц. HCl. Выход: 8,5 г (61%) цис-изомера и 2,2 г (13%) транс-изомера. цис-4-(4-Циклопропилметилпиперазин-1 -ил)циклогексиламин 8,5 г (20 ммолей) цис-дибензил[4-(4-циклопропилметилпиперазин-1-ил)циклогексил]амина растворяют в 170 мл МеОН и гидрируют на 1,7 г Pd/C (10%-ного) при 30-50 С. Затем растворитель отгоняют в вакууме и остаток кристаллизуют из этанола и конц. HCl. Выход: 4,4 г (91%). транс-Изомер можно получить аналогичным путем. Синтез соединений из примеров Пример 152. 0,15 г соединения Z10, 0,14 г ТБТУ и 0,13 мл ДИПЭА растворяют в дихлорметане и в течение 20 мин перемешивают при 25 С. Затем добавляют 90 мкл 1-(3-аминопропил)-4-метилпиперазина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют дихлорметаном и экстрагируют водой. Добавлением петролейного эфира, диэтилового эфира и этилацетата к органической фазе осаждают продукт. Выход: 0,16 г бежевого твердого вещества. Пример 164. 0,10 г соединения Z10, 0,1 г ТБТУ и 0,08 мл ДИПЭА растворяют в 4 мл дихлорметана и в течение 20 мин перемешивают при 25 С. Затем добавляют 44 мкл диметиламинопропиламина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют дихлорметаном и экстрагируют водой. Добавлением петролейного эфира, диэтилового эфира и ацетона к органической фазе осаждают продукт. Выход: 0,08 г желтого твердого вещества. Пример 242. 0,15 г соединения Z10, 0,14 г ТБТУ и 0,13 мл ДИПЭА растворяют в 5 мл дихлорметана и в течение 20 мин перемешивают при 25 С. Затем добавляют 75 мкл 1-(2-аминоэтил)пиперидина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют дихлорметаном и экстрагируют водой. Добавлением петролейного эфира, диэтилового эфира и этилацетата к органической фазе осаждают продукт. Выход: 0,14 г желтого твердого вещества. Пример 188. 0,1 г соединения Z2, 0,09 г ТБТУ и 0,05 мл ДИПЭА растворяют в 15 мл дихлорметана и в течение 20 мин перемешивают при 25 С. Затем добавляют 33 мг 1-метил-4-аминопиперидина и перемешивают еще в течение 3 ч при 25 С. Далее раствор экстрагируют 20 мл воды и после этого концентрируют в вакууме. Продукт кристаллизуют с помощью диэтилового эфира. Выход: 0,047 г белых кристаллов. Пример 203. 0,1 г соединения Z2, 0,09 г ТБТУ и 0,5 мл ДИПЭА растворяют в 15 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 50 мг 4-амино-1-бензилпиперидина и перемешивают еще в течение 3 ч при 25 С. Далее раствор экстрагируют 20 мл воды и после этого концентрируют в вакууме. Затем хроматографируют на силикагеле и выделенный продукт кристаллизуют с помощью диэтилового эфира. Выход: 0,015 г белых кристаллов. Пример 94.- 20014237 0,17 г соединения Z1, 0,19 г ТБТУ и 0,11 мл ДИПЭА растворяют в 50 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 63 мг 1-метил-4-аминопиперидина и перемешивают еще в течение 17 ч при 25 С. Далее к раствору добавляют 50 мл воды и 1 г карбоната калия и органические фазы отделяют с помощью фазоразделительного патрона, после чего концентрируют в вакууме. Затем продукт очищают хроматографией на силикагеле и очищенный продукт кристаллизуют с помощью диэтилового эфира. Выход: 0,1 г белых кристаллов. Пример 95. 0,17 г соединения Z1, 0,19 г ТБТУ и 0,11 мл ДИПЭА растворяют в 50 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 77 мг экзо-3 аминотропана и перемешивают еще в течение 17 ч при 25 С. Далее к раствору добавляют 50 мл воды и 1 г карбоната калия и органические фазы отделяют с помощью фазоразделительного патрона, после чего концентрируют в вакууме. Затем продукт очищают хроматографией на силикагеле и очищенный продукт кристаллизуют с помощью диэтилового эфира. Выход: 0,03 г белых кристаллов. Пример 46. 0,15 г соединения Z3, 0,12 г ТБТУ и 0,12 мл ДИПЭА растворяют в 5 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 50 мг 1-метил-4-аминопиперидина и перемешивают еще в течение 2,5 ч при 25 С. После этого раствор экстрагируют водой и затем концентрируют. Остаток растворяют в теплом этилацетате и кристаллизуют с помощью диэтилового эфира и петролейного эфира. Выход: 0,025 г белых кристаллов. Пример 80. 0,2 г соединения Z8, 0,2 г ТБТУ и 0,1 мл ДИПЭА растворяют в 10 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 100 мг 1-метил-4-аминопиперидина и перемешивают еще в течение 17 ч при 25 С. После этого раствор экстрагируют разбавленным раствором карбоната калия и концентрируют. Остаток кристаллизуют с помощью диэтилового эфира. Выход: 0,12 г белых кристаллов. Пример 190. 0,2 г соединения Z8, 0,2 г ТБТУ и 0,3 мл ДИПЭА растворяют в 5 мл дихлорметана и в течение 1 ч перемешивают при 25 С. Затем добавляют 0,13 г 4-амино-1-бензилпиперидина и перемешивают еще в течение часа при 25 С. После этого раствор разбавляют 10 мл метиленхлорида и экстрагируют 20 мл воды. Затем продукт очищают на силикагеле и кристаллизуют с помощью этилацетата и диэтилового эфира. Выход: 0,23 г соединения Z8. 0,23 г бензиламина Z8 растворяют в 10 мл метанола, смешивают с 50 мг Pd/C и в течение 3 ч гидрируют при давлении 3 бара и температуре 25 С. Далее добавлением петролейного эфира и этилацетата получают белые кристаллы. Их хроматографируют на силикагеле и кристаллизуют с помощью этилацетата и диэтилового эфира. Выход: 0,075 г белых кристаллов. Пример 196. 0,1 г соединения Z10, 0,09 г ТБТУ и 0,3 мл ДИПЭА растворяют в 4 мл дихлорметана и в течение 20 мин перемешивают при 25 С. Далее добавляют 67 мг 1,1-диметил-N-метилпиперазин-1-илэтиламина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют дихлорметаном и экстрагируют водой. Затем хроматографируют на силикагеле, остаток растворяют в ацетоне, смешивают с раствором HCl в диэтиловом эфире и отделяют образовавшийся осадок. Выход: 0,09 г светло-желтого твердого вещества. Пример 166. 0,1 г соединения Z10, 0,11 г ТБТУ и 0,14 мл ДИПЭА растворяют в 2 мл диметилформамида и в течение 3 ч перемешивают при 50 С. Далее добавляют 55 мг 4-морфолинометилфениламина. После этого реакционную смесь в течение 17 ч охлаждают до комнатной температуры. Затем диметилформамид удаляют в вакууме, остаток растворяют в дихлорметане и экстрагируют водой. После этого хроматографируют на силикагеле и продукт кристаллизуют из этилацетата и диэтилового эфира. Выход: 0,06 г желтоватых кристаллов. Пример 81. 0,2 г соединения Z4, 0,2 г ТБТУ и 0,1 мл ДИПЭА растворяют в 10 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 0,1 г 1-метил-4-аминопиперидина и перемешивают еще в течение 17 ч при 25 С. После этого раствор экстрагируют водным раствором карбоната калия и затем концентрируют. Продукт кристаллизуют с помощью диэтилового эфира. Выход: 0,16 г белых кристаллов. Пример 162. 0,1 г соединения Z5, 0,07 г ТБТУ и 0,15 мл ДИПЭА растворяют в 5 мл дихлорметана и в течение 20 мин перемешивают при 25 С. Затем добавляют 0,04 г 1-метил-4-аминопиперидина и перемешивают еще- 21014237 в течение 2 ч при 25 С. После этого раствор разбавляют 15 мл дихлорметана и экстрагируют 20 мл воды. Остаток растворяют в МеОН и ацетоне, смешивают с 1 мл раствора HCl в диэтиловом эфире и концентрируют. С помощью диэтилового эфира, этилацетата и небольшого количества МеОН получают кристаллический продукт. Выход: 0,1 г белых кристаллов. Пример 88. 0,1 г соединения Z6, 0,12 г ТБТУ и 0,12 мл ДИПЭА растворяют в 10 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 0,04 г 1-метил-4-аминопиперидина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют 10 мл дихлорметана и экстрагируют 10 мл воды. С помощью этилацетата, диэтилового эфира и петролейного эфира получают кристаллический продукт. Выход: 0,6 г белых кристаллов. Пример 89. 0,1 г соединения Z6, 0,08 г ТБТУ и 0,08 мл ДИПЭА растворяют в 10 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 37 мкл N,N-диметилнеопентандиамина и перемешивают еще в течение 2 ч при 25 С. После этого раствор разбавляют 10 мл дихлорметана и экстрагируют 10 мл воды. Затем продукт хроматографируют на силикагеле и кристаллизуют с помощью этилацетата, диэтилового эфира и петролейного эфира. Выход: 0,005 г белых кристаллов. Пример 26. 0,15 г соединения Z7, 0,16 г ТБТУ и 1 мл ДИПЭА растворяют в 5 мл дихлорметана и в течение 30 мин перемешивают при 25 С. Затем добавляют 0,1 г 4-морфолиноциклогексиламина и перемешивают еще в течение 17 ч при 25 С. Затем остаток смешивают с 10 мл 10%-ного раствора карбоната калия, отделяют осадок и промывают его водой. После этого его растворяют в дихлорметане и вновь концентрируют. Продукт кристаллизуют с помощью этилацетата. Выход: 0,1 г белых кристаллов. Пример 9. 150 мг соединения Z9 и 93 мг цис-4-морфолиноциклогексиламина растворяют в 5 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 160 мг ТБТУ и 1 мл ДИПЭА. Затем растворитель отгоняют в вакууме и остаток смешивают с 10 мл 10%-ного раствора карбоната калия. Осадок отделяют вакуум-фильтрацией, промывают водой, растворяют в дихлорметане, сушат и растворитель отгоняют в вакууме. Остаток кристаллизуют из этилацетата. Выход: 82,0 мг. Пример 16. 150 мг соединения Z8 и 73 мг транс-4-пиперидиноциклогексиламина растворяют в 5 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 160 мг (0,50 ммоля) ТБТУ и 1 мл ДИПЭА. Затем растворитель отгоняют в вакууме и остаток смешивают с 10 мл 10%-ного раствора карбоната калия. Осадок отделяют вакуум-фильтрацией, промывают водой, растворяют в дихлорметане, сушат и растворитель отгоняют в вакууме. Остаток кристаллизуют из этилацетата. Выход: 87,0 мг. Пример 37. 100 мг соединения Z9 и 42 мг 3-амино-1-этилпирролидина растворяют в 10 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 90 мг ТБТУ и 0,5 мл ДИПЭА. Затем растворитель отгоняют в вакууме и остаток смешивают с 10 мл 10%-ного раствора карбоната калия. Осадок отделяют вакуум-фильтрацией, промывают водой, растворяют в дихлорметане, сушат и растворитель отгоняют в вакууме. Остаток кристаллизуют из смеси этилацетат/петролейный эфир. Выход: 24,0 мг. Пример 120. 100 мг соединения Z11 и 73 мг 4-амино-1-тетрагидро-4 Н-пиран-4-илпиперидина растворяют в 10 мл дихлорметана и в течение 1 ч перемешивают при комнатной температуре с 90 мг ТБТУ и 0,5 мл ДИПЭА. Затем растворитель отгоняют в вакууме и остаток смешивают с 10 мл 10%-ного раствора карбоната калия. Осадок отделяют вакуум-фильтрацией, промывают водой, растворяют в дихлорметане, сушат и растворитель отгоняют в вакууме. Остаток кристаллизуют из смеси этилацетат/петролейный эфир. Выход: 89 мг. Пример 212. 150 мг соединения Z5 и 150 мг транс-4-(4-циклопропилметилпиперазин-1-ил)циклогексиламина (в виде гидрохлорида) растворяют в 5 мл дихлорметана и в течение 2 ч перемешивают при комнатной температуре с 160 мг ТБТУ и 2 мл ДИПЭА. Затем растворитель отгоняют в вакууме и остаток смешивают с 10 мл 10%-ного раствора карбоната калия. Осадок отделяют вакуум-фильтрацией, промывают водой,растворяют в дихлорметане, сушат и растворитель отгоняют в вакууме. Остаток очищают на колонке (20 мл силикагеля, 300 мл смеси этилацетат/метанол в соотношении 90:10 + 2% конц. аммиака). Объединеные фракции концентрируют в вакууме и кристаллизуют из этилацетата.- 22014237 Выход: 140 мг. Пример 232. 390 мг соединения Z11 и 240 мг транс-4-(4-трет-бутилоксикарбонилпиперазин-1-ил)циклогексиламина растворяют в 2,5 мл N-МП и в течение 2 ч перемешивают при комнатной температуре с 482 мг ТБТУ и 1 мл триэтиламина. Затем смешивают с 100 мл воды и 200 мг карбоната калия, осадок отделяют вакуум-фильтрацией, промывают водой и очищают на силикагелевой колонке. Объединенные фракции концентрируют в вакууме, растворяют в 2 мл дихлорметана, смешивают с 2 мл трифторуксусной кислоты и в течение 2 ч перемешивают при комнатной температуре, после чего вновь смешивают с 100 мл воды и 200 мг карбоната калия, осадок отделяют вакуум-фильтрацией и промывают его водой. После этого осадок очищают на силикагелевой колонке. Объединенные фракции концентрируют в вакууме и остаток кристаллизуют из этанола и конц. соляной кислоты. Выход: 95 мг. Пример 213. 60 мг соединения из примера 232 растворяют в 10 мл этилацетата и в течение 30 мин перемешивают при комнатной температуре с 1 мл уксусного ангидрида и 1 мл триэтиламина. Затем растворитель отгоняют в вакууме, остаток смешивают с водой и аммиаком, выпавшие кристаллы отделяют вакуумфильтрацией и промывают водой и небольшим количеством холодного ацетона. Выход: 40 мг. Пример 218. 1,2 г соединения Z9 и 0,5 г 1,4-диоксаспиро[4.5]дец-8-иламина растворяют в 20 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 1,28 г ТБТУ и 4 мл триэтиламина. Далее смешивают с 50 мл воды и 0,5 г карбоната калия, органическую фазу отделяют, сушат и концентрируют в вакууме. Остаток кристаллизуют из этилацетата, смешивают с 25 мл 1 н. соляной кислоты и 20 мл метанола и в течение 30 мин перемешивают при 50 С. После этого метанол отгоняют в вакууме, осадок отделяют вакуум-фильтрацией, промывают водой и сушат. Остаток растворяют в 20 мл дихлорметана и в течение 12 ч перемешивают при комнатной температуре с 0,5 г тиоморфолина и 0,5 г NaBH(OAc)3. После этого смешивают с водой и карбонатом калия, органическую фазу отделяют, сушат и растворитель отгоняют в вакууме. Остаток очищают на силикагелевой колонке. Объединеные фракции концентрируют в вакууме и обработкой раствором HCl в диэтиловом эфире осаждают гидрохлорид. Выход: 86 мг транс-изомера в виде аморфного порошка. Пример 187. 200 мг соединения Z3 в 5 мл дихлорметана смешивают с 0,1 мл диизопропилэтиламина и 180 мг ТБТУ и перемешивают в течение 30 мин. Затем добавляют 191 мг 4-(4-метилпиперазин-1-ил)фениламина и оставляют перемешиваться на ночь. Далее реакционную смесь смешивают с водой и водную фазу экстрагируют дихлорметаном. Объединенные органические фазы сушат над Na2SO4 и концентрируют. Остаток очищают колоночной хроматографией (элюент: смесь дихлорметан/метанол в соотношении 100:7). Выход: 128 мг (желтоватых кристаллов). Аналогично описанным выше методам получают, в частности, представленные ниже в табл.1 соединения формулы (I). В приведенной ниже табл. 1 каждое из сокращений Х 1, Х 2, Х 3, Х 4 и Х 5 используется вместо соответствующего остатка R1 , R2 , R3 , R4 и L-R5 для обозначения положения, в котором он присоединен к остальной части молекулы, представленной приведенной перед табл. 1 общей формулой. Таблица 1
МПК / Метки
МПК: A61K 31/4985, A61P 35/00, A61K 31/519
Метки: хранении, инфузионный, дигидроптеридинонов, раствор, стабильный
Код ссылки
<a href="https://eas.patents.su/30-14237-stabilnyjj-pri-hranenii-infuzionnyjj-rastvor-digidropteridinonov.html" rel="bookmark" title="База патентов Евразийского Союза">Стабильный при хранении инфузионный раствор дигидроптеридинонов</a>
Дезинтоксикационный инфузионный раствор

Номер патента: 7865
Опубликовано: 27.02.2007
Авторы: Коваленко Алексей Леонидович, Алексеева Людмила Евгеньевна
МПК: A61K 33/14, A61K 31/19, A61K 33/06...
Метки: раствор, дезинтоксикационный, инфузионный
Формула / Реферат:
Дезинтоксикационный инфузионный раствор, содержащий хлориды натрия, магния и калия, биологически активный компонент - натрия N-метилглюкамина сукцинат и растворитель - воду для инъекций, отличающийся тем, что он дополнительно содержит в качестве биологически активных компонентов - рибоксин, метионин и никотинамид, при следующем соотношении компонентов, мас.%: Хлорид натрия 0,4-0,6 Хлорид магния 0,011-0,013 Хлорид калия 0,027-0,033 ...
Инфузионный раствор “реамберин”

Номер патента: 879
Опубликовано: 26.06.2000
Авторы: Алексеева Людмила Евгеньевна, Коваленко Алексей Леонидович
МПК: A61K 33/14, A61K 31/205, A61K 31/194...
Метки: реамберин, раствор, инфузионный
Формула / Реферат:
Детоксицирующий инфузионный раствор, содержащий хлориды натрия, калия и магния, растворитель и биологически активный компонент, отличающийся тем, что в качестве растворителя содержит воду для инъекций, а в качестве биологически активного компонента содержит натрия глюкамина сукцинат общей формулы: Na+ [НОСН2(СНОН)4СН2NH2СН3]+[ООССН2СН2СОО]2- при следующем соотношении компонентов, маc.%: Хлорид натрия 0,4-0,6 Хлорид...
Инфузионный и инъекционный раствор леводопа

Номер патента: 12415
Опубликовано: 30.10.2009
Автор: Диздар Сегрелл Нил
МПК: A61K 9/08, A61P 25/16, A61K 31/198...
Метки: инфузионный, леводопа, раствор, инъекционный
Формула / Реферат:
1. Инфузионный или инъекционный раствор леводопа, содержащий: а1) по меньшей мере 10 мг/мл леводопа или а2) по меньшей мере 5 мг/мл леводопа вместе по меньшей мере с 0,5 мг/мл по меньшей мере одного ингибитора фермента, метаболизирующего леводопа, выбранного из группы, состоящей из ингибиторов дофадекарбоксилазы (DDC), ингибиторов катехол-о-метилтрансферазы (СОМТ) и ингибиторов моноаминооксидазы (МАО-В), б) буфер, в) физиологически приемлемый...
Стабильный раствор ксилометазолина и оксиметазолина

Номер патента: 3329
Опубликовано: 24.04.2003
Автор: Мэрц Фридер Ульрих
МПК: A61K 31/4174, A61K 31/415
Метки: стабильный, оксиметазолина, ксилометазолина, раствор
Формула / Реферат:
1. Композиция в виде стабильного изотонического раствора со значением pH от 4,5 до 7,5, содержащая гидрохлорид ксилометазолина и/или гидрохлорид оксиметазолина в качестве действующего вещества, фармакологически приемлемый для назального введения растворитель, адъювант из группы, включающей сорбит и/или глицерин, буфер из группы, включающей неорганический pH-буфер или органический буфер, которым является трометамол, и необязательно соляную...
Новый стабильный кристалл производного тиазолидиндиона и способ его получения

Номер патента: 4244
Опубликовано: 26.02.2004
Авторы: Орита Казуо, Охнота Михиро
МПК: A61K 31/426, A61P 3/10, C07D 277/34...
Метки: способ, тиазолидиндиона, новый, кристалл, получения, производного, стабильный
Формула / Реферат:
1. Новые кристаллы 5-[(2,4-диоксотиазолидин-5-ил)метил]-2-метокси-N-{[4-(трифторметил)фенил]метил}бензамида, характеризующиеся при порошковой дифракции рентгеновских лучей наличием дифракционных максимумов при значениях дифракционных углов (2Q) при по меньшей мере 9,7, 15,0 и 22,5ш. 2. Способ приготовления новых кристаллов по п.1 путем перекристаллизации из приемлемого растворителя. 3. Способ приготовления новых кристаллов по п.2, в котором...
Предыдущий патент: Новые пиперазиновые производные диалкилоксиндолов
Следующий патент: Производные n-гидроксиамида и их применение
Случайный патент: Гуманизированные антитела с противоопухолевой активностью