Соединения тиадиазола и их применение
Номер патента: 12181
Опубликовано: 28.08.2009
Авторы: Таскер Эндрю, Райдер Джеймс Томас, Волхитер Джордж Эрих, Бурбо Мэттью Пол, Моненшайн Хольгер, Висванадхан Велларкад Н., Зенг Кингпинг, Домингэз Селиа, Йао Гуомин
























Формула / Реферат
1. Соединение тиадиазола формулы I

где Y представляет собой -N(R4)R5;
X представляет собой N(R6);
R1 представляет собой арил или гетероарил;
R2 представляет собой Н;
R3 выбран из группы, состоящей из -(CR7R8)t(арила) и -(CR7R8)t(гетероарила);
R4 представляет собой -Н или C1-C8-алкил;
R5 и R6 независимо выбраны из -Н или C1-C8-алкила;
R7 и R8 независимо выбраны из -Н и С1-С6-алкила;
n, m и t равны 1;
каждый из указанных выше алкильных, арильных и гетероарильных остатков необязательно и независимо замещены с помощью 1-3 заместителей, выбранных из амино, арила, гетероарила, циклоалкила или гетероциклила, необязательно замещенного с помощью 1-5 заместителей, выбранных из С1-С6-алкокси, C1-С6-алкила, необязательно замещенного с помощью галогена, арила, галогена, гетероарила, С1-С6-гидроксила и -NHS(O)2-(C1-C6-алкила);
C1-C6-алкил, C1-C6-галогеналкил, C1-C6-гидроксиалкил, C1-C6-алкокси, C1-C6-алкиламино, С2-С6-алкенил или С2-С6-алкинил, каждый из которых может быть прерван с помощью одного или большего количества гетероатомов, циано, галогена, гидрокси, нитро или -О-арила;
или его фармацевтически приемлемая соль, гидрат или стереоизомер.
2. Соединение по п.1 или его фармацевтически приемлемая соль, гидрат или стереоизомер, где R4 и R5 представляют собой Н.
3. Соединение по п.1 или его фармацевтически приемлемая соль, гидрат или стереоизомер, где R6 представляет собой Н.
4. Соединение по п.1 или его фармацевтически приемлемая соль, гидрат или стереоизомер, где R3 представляет собой -(CR7R8)t(арил).
5. Соединение по п.1 или его фармацевтически приемлемая соль, гидрат или стереоизомер, где R3 представляет собой -(CR7R8)t(гетероарил).
6. Соединение по п.1, которое выбрано из
(S)-3-(3-фторфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-3-(3,4-дифторфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-3-(3,4-дихлорфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
N-((S)-2-амино-3-(4-(трифторметил)фенил)пропил)-5-(3-метил-1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(2-бромфенил)пропил)-5-(3-метил-1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-этилфенил)пропил)-5-(3-метил-1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(3,5-дифторфенил)пропил)-5-(3-метил-1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(2-метоксифенил)пропил)-5-(3-метил-1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
(S)-3-(4-метоксифенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-3-(2-хлорфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]-3-о-толилпропан-1,2-диамина;
(S)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]-3-(3-трифторметилфенил)пропан-1,2-диамина;
(S)-3-(4-фторфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-3-(4-хлорфенил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2-диамина;
(S)-N-[5-(3-метил-1Н-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]-3-m-толилпропан-1,2-диамина;
N-((S)-2-амино-3-(3-бромфенил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(3-хлорфенил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-изопропилфенил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(2,4-дихлорфенил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-р-толилпропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(нафталин-2-ил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(бензо[b]тиофен-3-ил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-хлорфенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-(трифторметил)фенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(нафталин-2-ил)пропил)-5-(1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-изопропилфенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(3,4-дихлорфенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(2,4-дихлорфенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-метоксифенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-бромфенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-((3-трифторметил)фенил)пропил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-((3-трифторметил)фенил)пропил)-5-(3-метил-1H-пиразоло[3,4-b]пиридин-5-ил)-1,3,4-тиадиазол-2-амина;
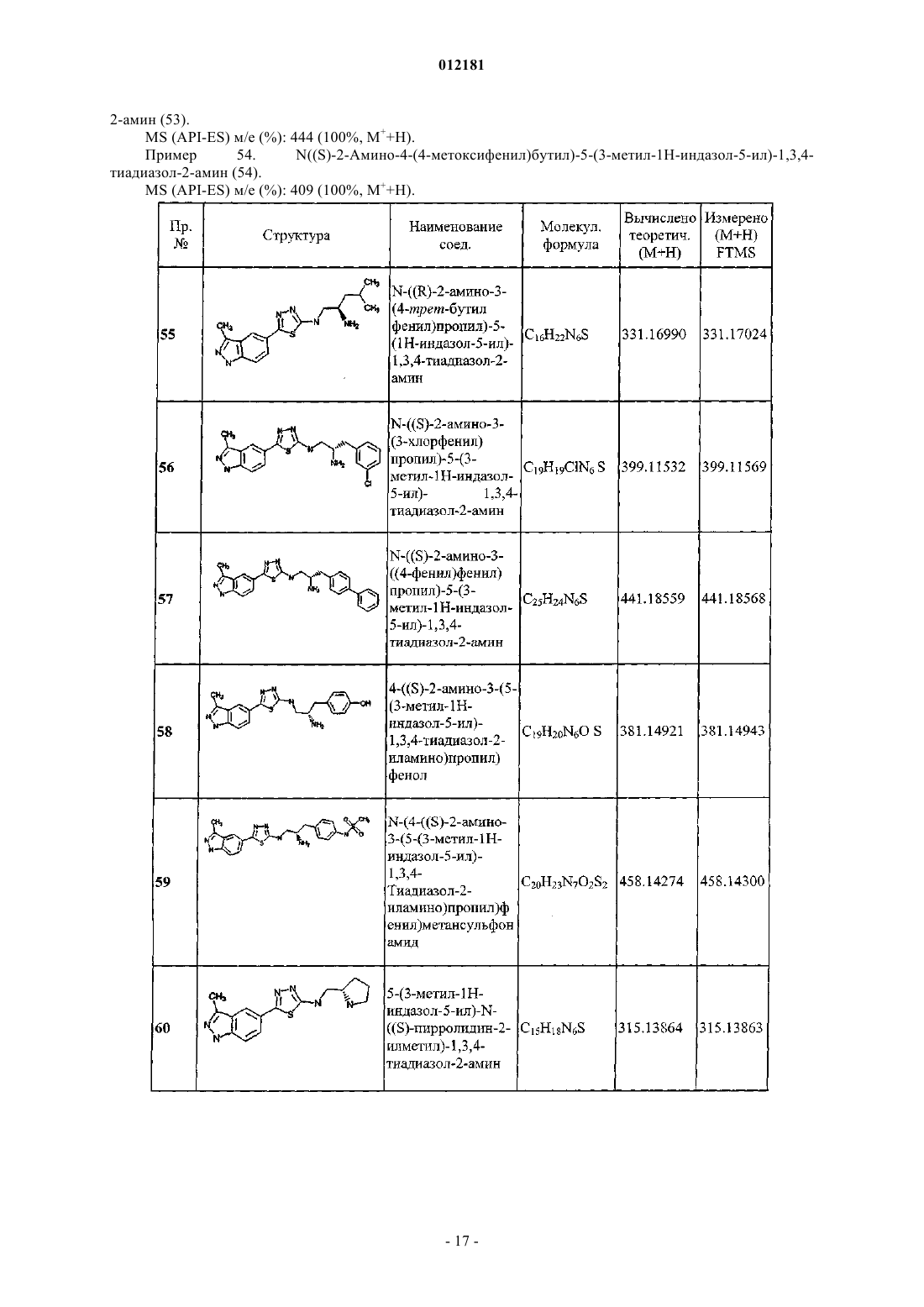
N-((S)-3-(4-хлорфенил)-2-(метиламино)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
5-(3-метил-1H-индазол-5-ил)-N-((S)-2-(метиламино)-3-(3-(трифторметил)фенил)пропил)-1,3,4-тиадиазол-2-амина;
5-(1H-индазол-5-ил)-N-((S)-2-(метиламино)-3-(4-(трифторметил)фенил)пропил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-метоксифенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(3-(трифторметил)фенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-(трифторметил)фенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
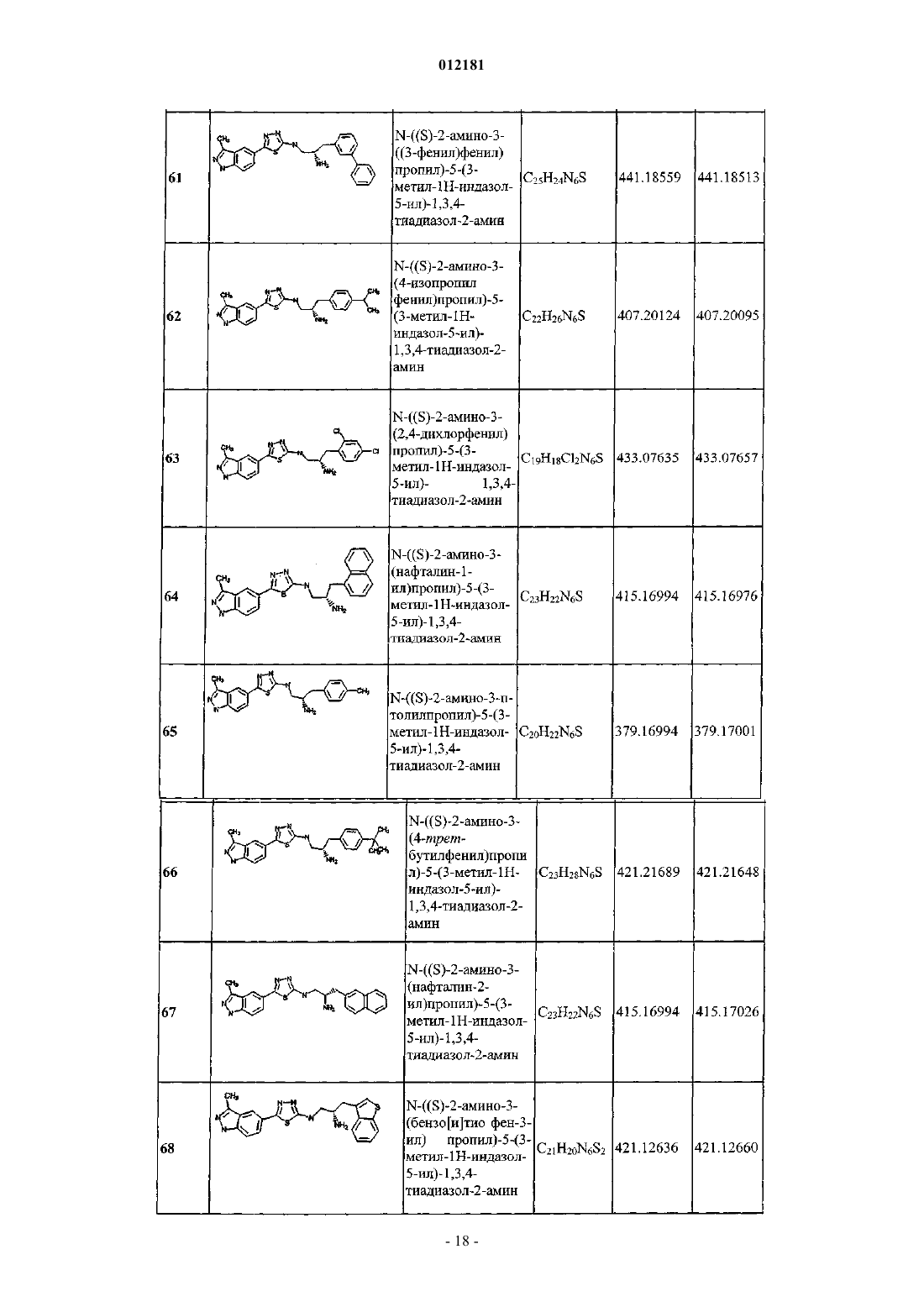
N-((S)-2-амино-3-(3-метоксифенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(2,4-дихлорфенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-хлорфенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(3,5-дифторфенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-(S)-2-амино-3-(м-толилпропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-(трифторметил)фенил)пропил)-5-(1,6-нафтиридин-2-ил)-1,3,4-тиадиазол-2-амина;
N-((S)-2-амино-3-(4-бромфенил)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина;
N-((2S,3S)-2-амино-3-(4-(трифторметил)фенил)бутил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амина и
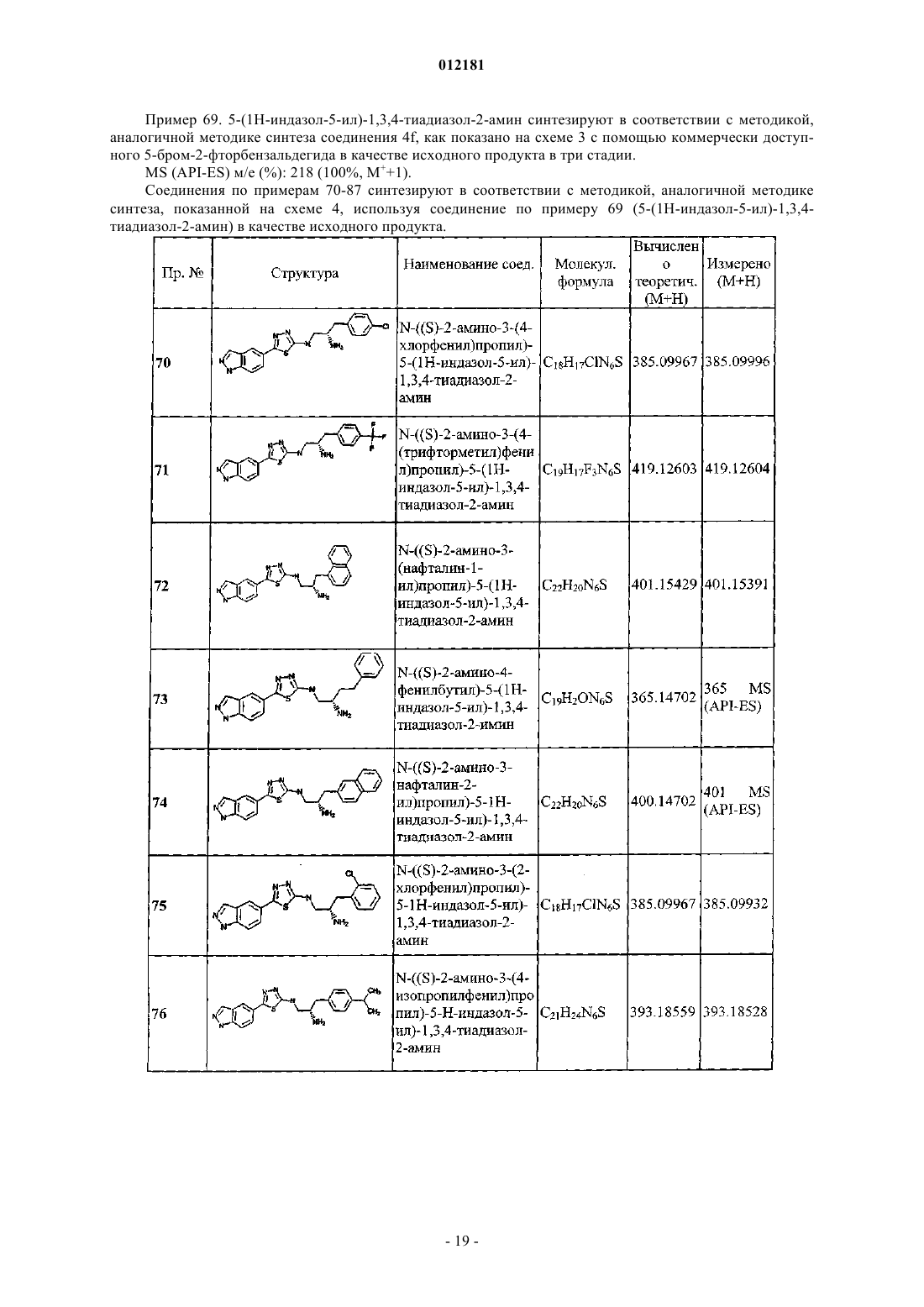
N-((2S,3S)-2-амино-3-(4-(трифторметил)фенил)бутил)-5-(1Н-индазол-5-ил)-1,3,4-тиадиазол-2-амина.
7. Фармацевтическая композиция для лечения опосредованной киназой болезни у млекопитающих, содержащая соединение по п.1 и фармацевтически приемлемый носитель.
8. Композиция по п.7, дополнительно содержащая по крайней мере один дополнительный терапевтический агент.
9. Способ лечения опосредованной киназой болезни у млекопитающих, нуждающихся в таком лечении, включающий введение млекопитающему терапевтически эффективного количества соединения по п.1.
10. Способ по п.9, отличающийся тем, что болезнь опосредована с помощью PKB.
11. Способ по п.9, отличающийся тем, что лечение включает селективное ингибирование PKB.
12. Способ по п.9, отличающийся тем, что болезнь представляет собой рак.
13. Способ лечения пролиферативно связанной болезни у млекопитающих, нуждающихся в таком лечении, включающий введение млекопитающему терапевтически эффективного количества соединения по п.1.
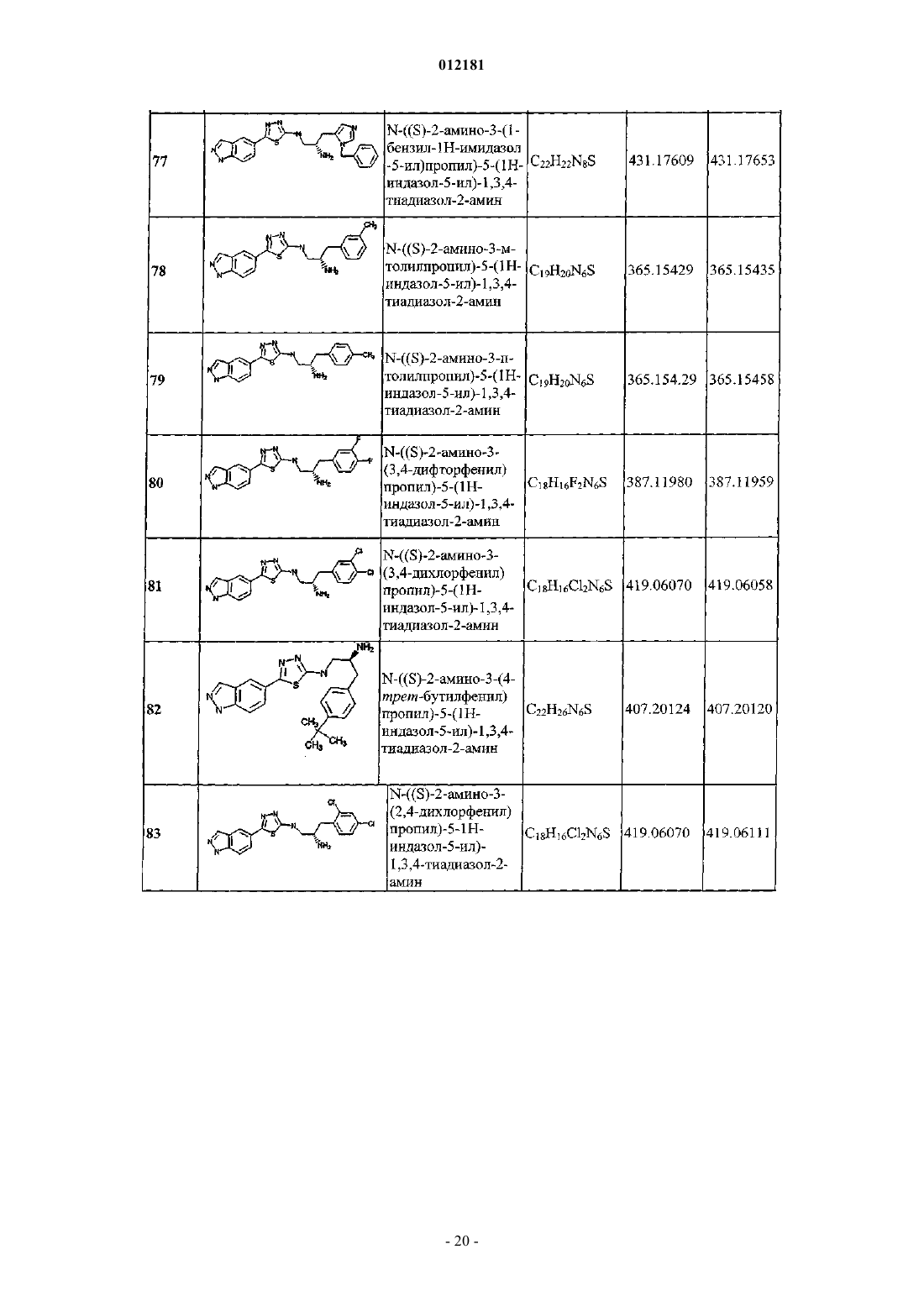
14. Способ по п.13, отличающийся тем, что болезнь представляет сосющ аномальный рост клетки.
Текст
012181 Область техники Изобретение относится к соединениям тиадиазола, применимых при лечении заболеваний, опосредованных протеинкиназой В (PKB). Изобретение также относится к терапевтическому применению таких соединений тиадиазола и их композиций при лечении патологических состояний, связанных с патологическим клеточным ростом, раком, воспалением и метаболическими расстройствами. Уровень техники Протеинкиназы представляют собой большое семейство белков, которые играют центральную роль в регуляции большого разнообразия клеточных процессов, обеспечивая контроль клеточной функции. Неполный список таких киназ включает abl, AKT, bcr-ab1, Blk, Brk, Btk, c-kit, c-met, c-src, c-fms, CDK1,CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, cRafl, CSFlR, CSK, EGFR, ErbB2,ErbB3, ErbB4, Erk, Fak, fes, FGFR1, FGFR2, FGFR3, FGFR4, FGFR5, Fgr, flt-1, Fps, Frk, Fyn, GSKSo;GSK3/3, Hck, IGF-IR, INS-R, Jak, KDR, Lck, Lyn, MEK, MK2, MSK1, p38, PDGFR, PIK, PKB, PKA, PRAK,PRK2, PKC, PYK2, P70S6, ROCK2, ros, tie, tie2, TRK, Yes и Zap70. Ингибирование таких киназ становится важной мишенью для терапии. Фермент AKT (также известный как протеинкиназа В (PKB) или Rac-PK-beta), и продукты его семейства генов были идентифицированы как серин/треонин протеинкиназы. Testa et al., Proc. Natl. Acad.Sci, 2001, 98,10983-10985; Lawlor et al., J. Cell Sci, 2001, 114, 2903-2910; Duan, Circ. Res., 2000, 86,15-23. Три изоформы PKB известны в настоящий момент, PKB (AKT1), PKB (AKT2) и PKB (AKT3). Cheng,Proc. Natl. Acad. Sci. USA, 1992, 89, 9267-9271; Brodbeck et al., J. Biol. Chem. 1999, 274, 9133-9136. PKB опосредует множество эффектов IGF-1 и других факторов роста на опухолевый рост и ингибирование апоптоза. Nicholson, et al., Cell. Signal, 2002, 14, 381-395. PKB играет важную роль в пролиферации клеток, апоптозе и ответе на инсулин. По этим причинам регулирование ферментов PKB представляет интерес в лечении канцерогенеза, патологической пролиферации клеток и диабетов. Молекулярная структура ферментов PKB включает регуляторный сайт вблизи от С-конца полипептида, каталитический домен с активируемым петлевым фрагментом, содержащим треонин, а также Nконцевой плекстрин-гомологичный домен. Плекстрин-гомологичный домен обеспечивает фиксацию фермента к клеточной мембране путем взаимодействия с фосфолипидами, которое запускает активацию ферментов PKB. Работа плекстрин-гомологичного домена требует фосфорилирования фосфатидилинозитола в позиции D-3 с помощью фосфатидилинозитол-3-киназы PI3K, содержащего SH2 домен белка,который связан с активированными рецепторными киназами, в частности, IGF-1R. В частности, фосфоинозитол-3-киназа, когда активируется рецепторной тирозинкиназой, катализирует синтез фосфоинозитол-3,4-дифосфата и фосфатидилинозитол 3,4,5-трифосфата. Плекстрингомологичный домен связывает 3-фосфоинозитиды, которые синтезируются ферментом PI3K под действием факторов роста, таких как фактор роста тромбоцитов (PDGF), фактор роста нервов (NGF) и инсулиноподобный фактор роста (IGF-I). Kulik et al., Mol. Cell. Biol, 1997, 17, 1595-1606; Hemmings, Science,1997, 275, 628-630; Datta, et al. Genes Dev., 1999, 13, 2905-2927. Связывание липидов с плекстрингомологичным доменом обеспечивает транслокацию PKB на плазматическую мембрану. Последующая активация PKB происходит за счет фосфорилирования с помощью другой протеинкиназы, PDK1 остатков Thr308, Thr309 и Thr305 в изоформах 1, 2 и 3 фермента PKB, соответственно. Третий этап активации катализируется киназой, которая фосфорилирует Ser473, Ser474 или Ser472 в С-концевых областяхPKB/AKT-1, -2 и -3 соответственно. Активность Ser473 киназы, как было установлено, связана с плазматической мембраной и не относится к активности киназ PKB и PDK1. Hill et al., Current Biology, 2002, 12,1251-1255; Hresko et al., J. Biol. Chem., 2003, 278, 21615-21622. Этот процесс приводит к образованию полностью активированной формы PKB. Активация PKB может также происходить путем ингибирования D-3 фосфоинозитидспецифической фосфатазы, PTEN, которая является мембранно-ассоциированной FYVE пальцеобразной фосфатазой, обычно инактивированной во многих раковых опухолях, включая рак предстательной железы. Besson, et al., Eur. J. Biochem., 1999, 263, 605-611; Li, et al., Cancer Res., 1997, 57, 2124-2129. Каталитический домен PKB отвечает за фосфорилирование серина или треонина в белке-мишени. Однократно активированная PKB опосредует несколько клеточных функций, включая пролиферацию, клеточный рост и обеспечение выживания. Интракоронарно, опосредованный аденовирусом перенос в сердце гена akt ограничивает объем инфаркта в результате ишемическо-реперфузионного повреждения in vivo. Miao et al., J. Mol. Cell. Cardiol, 2000, 32, 2397-2402. Противоапоптотическая функция PKB,как сообщается, опосредуется за счет ее способности фосфорилировать молекулы, регулирующие апоптоз, включая BAD, каспазу 9, DCK- и forkhead транскрипционный фактор FKHRL1. Datta et al., в 2905. Сигнализация PKB также участвует в физиологической регуляции размера органа (Verdu, et al., Nat. CellBiol, 1999, 1, 500-506), обмена глюкозы (Czech, et al., J. Biol Chem., 1999, 274, 1865-1868), вазомоторного тонуса (Luo, et al. J. Clin. Invest. 1999, 106, 493-499) и ангиогенеза (Kureishi, et al., Nat. Med., 2000, 6,1004-1010). Проявления нарушенной регуляции PKB проявляется как в виде повреждения, так и в виде болезни,играя наиболее важную роль в случае рака. Активность киназы PKB значительно активируется в опухолях с мутацией PTEN, мутацией и сверхсинтезом PI3-киназы, а также сверхсинтезом рецептора тирозин-1 012181 киназы. PKB является также медиатором нормальных клеточных функций в ответ на передачу сигналов фактора роста. Экспрессия гена AKT, как было установлено, увеличивается в 15% случаев рака яичников человека. Cheng, et al., Proc. Natl. Acad. Sci. U.S.A., 1992, 89, 9267-9271. Также было установлено, чтоU.S.A., 1996, 93, 3636-3641. В частности, AKT-2 сверхсинтезируется в 12% карцином яичников и в 50% недифференцированных опухолей, что указывает на то, что PKB может быть связана с агрессивностью опухоли. Bellacosa, et al., Int. J. Cancer, 1995, 64, 280-285. PKB также является медиатором нормальных клеточных функций. Khwaja, Nature, 1999, 401, 33-34; Yuan, et al., Oncogene, 2000, 19, 2324-2330; Namikawa, et al., J Neurosci., 2000, 20, 2875-2886. Объяснение роли PKB в усилении роста и ингибировании апоптоза осложняется большим количеством белковых субстратов PKB, включая BAD, Forkhead (семейство FOXO), GSK3, Tuberin (TSC2), р 27Rev. Cancer, 2002, 2, 489-501. Содержание различных киназ PKB различается в разных типах клеток млекопитающих. Например,PKB особенно много содержится в тканях, высокочувствительных к инсулину, включая бурый жир. Модуляция PKB посредством малых молекул может быть достигнута путем идентификации соединений, которые связываются и активируют или ингибируют одну или более PKB. Cao et al. в заявке США 2004/0122016, опубликованной 24 июня 2004 г., раскрывает некоторые производные тиофена и аналоги тиофена в качестве ингибиторов протеинкиназ. В частности, раскрытие относится к композициям,эффективным в качестве ингибиторов Rho-ассоциированных образующих биспираль протеинкиназ серина/треонина (ROCK), внеклеточной сигнал-регулируемой киназы (ERK), киназы гликогенсинтазы (GSK) и членов AGC подсемейства протеинкиназ. Id. в 4.AGC подсемейство киназ включает протеинкиназу А (PKA), PDK, р 70S6K -1, р 70S6K -2 и PKB. Id. Трицирибин, как сообщается, ингибирует клеточный рост в клетках, сверхсинтезирующих PBK,трансформированных клетках и эффективен при концентрации 50 нМ. Yang et al., Cancer Res., 2004, 64,4394-4399.US 390456, опубликованный 9 сентября 1975 г., раскрывает замещенные нитроимидазолилтиадиазолы и оксадиазолы в качестве антибактериальных агентов и соединений, способствующих росту. Патент не относится к модуляции PKB.US 5086053, опубликованный 4 февраля 1992 г., раскрывает некоторые производные 1,3,4 тиадиазола, способ их получения и фармацевтические композиции, содержащие их. Агенты описаны как мускариновые холинергические агонисты Id. в col. 2, 11. 6-7. Патент '053, тем не менее, не раскрывает модуляторы PKB. Синтезированы производные 1,3,4-окса(тиа)диазолопиримидин-5-оны и родственные соединения.Yadav et al., Synthesis, 2003, 1, 63-66. Некоторые производные тиазолопиридопиримидинов и тиазолотиадиазолопиримидинов синтезированы Singh и коллегами и протестированы на противогрибковую активность. Singh et al., Indian J. Chem., 1994, 33B, 350-354. Производные 2-амино-1,3,4-тиадиазола и родственные соединения синтезированы и протестированы на анестетическую активность. Mazzone et al.,IlFarmaco, 1993, 48, 1207-1224. Некоторые производные тиадиазолов синтезированы и протестированы на антимикробную активность. Pachhamia et al., J. Inst. Chemists (India), 1989, 61, 54-56. Более того, сообщается о синтезе ацетамидных производных 1,3,4-тиадиазолов и родственных соединений. Shah et al.,J. Indian Chem. Soc., 1982, LIX, 678-680. Ни один из вышеуказанных источников не раскрывает модуляцию PKB. Сообщается о противоопухолевых эффектах некоторых 1,3,4-тиадиазоловых производных. Platonova, Akad Med Nauk, SSSR 2, 167, цитировано Shah et al. в 678. Сущность изобретения Настоящее изобретение охватывает новые соединения, применимые для лечения заболеваний и патологических состояний, опосредованных PKB. Изобретение также охватывает терапевтическое применение таких соединений и их композиций в лечении патологических состояний, связанных с патологическим клеточным ростом, таких как рак, или патологических состояний обмена веществ, таких как диабет или воспаление. Настоящее изобретение охватывает новые соединения, пригодные для лечения заболеваний или состояний, опосредованных PKB. Изобретение также охватывает терапевтическое применение таких соединений и их композиций при лечении болезненных состояний, связанных с аномальным ростом клетки, таких как рак, или нарушение обмена веществ состояния, такие как диабет или воспаление. В одном аспекте изобретение включает в себя соединение тиадиазола формулы IR1 представляет собой арил или гетероарил;R5 и R6 независимо выбраны из -Н или C1-C8-алкила;R7 и R8 независимо выбраны из -Н и C1-С 6-алкила;n, m и t равны 1; каждый из указанных выше алкильных, арильных и гетероарильных остатков необязательно и независимо замещены с помощью 1-3 заместителей, выбранных из амино, арила, гетероарила, циклоалкила,или гетероциклила, необязательно замещенного с помощью 1-5 заместителей, выбранных из C1-С 6 алкокси, C1-С 6-алкила, необязательно замещенного с помощью галогена, арила, галогена, гетероарила,С 1-С 6-гидроксила и -NHS(O)2-(C1-C6-алкила);C1-С 6-алкил, C1-С 6-галогеналкил, C1-С 6-гидроксиалкил, С 1-С 6-алкокси, С 1-С 6-алкиламино, С 2-С 6 алкенил или С 2-С 6-алкинил, каждый из которых может быть прерван с помощью одного или большего количества гетероатомов, циано, галогена, гидрокси, нитро или -О-арила; или его фармацевтически приемлемая соль, гидрат или стереоизомер. В одном воплощении изобретение - это соединение формулы (I), где R4 и R5 представляют собой Н. В другом воплощении изобретение - это соединение формулы (I), где R6 представляет собой Н. В другом воплощении изобретение - это соединение формулы (I), где R3 представляет собой 7 8-(CR R )t(арил). В другом воплощении изобретение - это соединение формулы (I), где R3 представляет собой 7 8-(CR R )t(гетероарил). В другом воплощении, изобретение - это соединение формулы (I), выбранное изN-2S,3S)-2-амино-3-(4-(трифторметил)фенил)бутил)-5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2 амина. В другом аспекте изобретение охватывает фармацевтически приемлемую соль, гидрат или сольват соединения формулы (I). Фармацевтически приемлемые соли формулы (I) соединений могут быть выбраны из трифторацетата аммония и хлорида аммония. Другой аспект изобретения включает фармацевтическую композицию, состоящую из фармацевтически приемлемого носителя и соединения формулы (I). В другом аспекте изобретение включает способ лечения опосредованного киназой расстройства у млекопитающего, включающий введение млекопитающему терапевтически эффективного количества соединения формулы (I). Расстройство может быть расстройством, опосредованным PKA, PKB, PKC,FKHR, SGK, LCK, BTK, Tie2, KDR, Erk, MSK, MK2, MSK, p38, P70S6, ROCK2, GSK3 или комплексомCDK. В другом воплощении изобретение охватывает соединения формулы (I), которые обладают избирательной киназной активностью, т.е. они обладают значительной активностью в отношении одной конкретной киназы, одновременно обладая меньшей или минимальной активностью в отношении других киназ. Другое воплощение изобретения включает лечение патологического клеточного роста путем введения терапевтически эффективного количества соединения по изобретению субъекту, который в этом нуждается. Патологический клеточный рост может быть доброкачественным или злокачественным ростом. В частности, патологический клеточный рост может быть при карциноме, саркоме, лимфоме или лейкозе. В одном воплощении этого способа патологический клеточный рост представлен раком, включая помимо прочего рак легкого, рак костей, рак поджелудочной железы, рак кожи, рак головы и шеи, кожную или внутриглазную меланому, рак матки, рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак ободочной кишки, рак молочной железы, карциному фаллопиевых труб, карциному эндометрия, карциному шейки матки, карциному влагалища, карциному вульвы, болезнь Ходжкина, рак пищевода, рак тонкой кишки, рак эндокринной системы, рак щитовидной железы, рак паращитовидной железы, рак надпочечников, саркому мягких тканей, рак мочеиспускательного канала, рак полового члена, рак предстательной железы, хронический и острый лейкоз, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки и мочеточника, почечно-клеточную карциному, карциному почечной лоханки, новообразования центральной нервной системы (ЦНС), первичную лимфому ЦНС, опухоли позвоночника,глиому ствола мозга, питуитарную аденому или комбинацию одного или более вышеперечисленных раковых опухолей. Способ также включает лечение пациента, страдающего раком, у которого рак выбран из группы, состоящей из мелкоклеточного рака легкого, немелкоклеточного рака легкого, рака пищевода, рака почки, рака поджелудочной железы, меланомы, рака мочевого пузыря, рака молочной железы,рака ободочной кишки, рака печени, рака легкого, саркомы, рака желудка, холангиокарциномы, мезотелиомы или рака предстательной железы. В другом воплощении указанного способа указанный патологи-4 012181 ческий клеточный рост является доброкачественным пролиферативным заболеванием, включающим помимо прочего псориаз, доброкачественную гиперплазию предстательной железы или рестеноз. В другом воплощении данное изобретение охватывает способ введения терапевтически эффективного количества соединения формулы (I) млекопитающему для лечения патологических состояний или болезней, выбранных из следующих: диабет, воспаление и метаболические расстройства. В другом воплощении изобретение охватывает способ лечения или профилактики рака у пациентов,которые нуждаются в подобном лечении, включающий введение пациенту терапевтически или профилактически эффективного количества соединения, согласно формуле I и фармацевтически приемлемого наполнителя или носителя. В другом аспекте изобретение охватывает способ лечения или профилактики рака у пациентов, которые нуждаются в подобном лечении, включающий введение пациенту терапевтически или профилактически эффективного количества соединения формулы (I) и по меньшей мере одного вспомогательного терапевтического агента. Подробное описание изобретения Определения. Когда следующие термины используются в этих обозначениях, они используются, как определено ниже. Термины "содержащий" и "включающий" используют в настоящем изобретении, их широком, неограниченном понимании. Как используется в настоящем изобретении, если иное не определено, термин "алкил" означает насыщенный с нормальной цепью или разветвленный нециклический углеводород, имеющий от 1 до 20 атомов углерода, предпочтительно 1-10 атомов углерода и более предпочтительно 1-4 атомов углерода. Представители насыщенных алкилов с нормальной цепью включают -метил, -этил, -н-пропил, -н-бутил,-н-пентил, -н-гексил, -н-гептил, -н-октил, -н-нонил и -н-децил; тогда как насыщенные разветвленные алкилы включают -изопропил, -втор-бутил, -изобутил, -трет-бутил, -изопентил, 2-метилбутил, 3 метилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2-метилгексил, 3-метилгексил, 4 метилгексил, 5-метилгексил, 2,3-диметилбутил, 2,3-диметилпентил, 2,4-диметилпентил, 2,3 диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилпентил, 2,2-диметилгексил, 3,3 диметилпентил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилпентил, 3-этилпентил, 2-этилгексил, 3 этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, 2-метил-4-этилпентил, 2-метил 2-этилгексил, 2-метил-3-этилгексил, 2-метил-4-этилгексил, 2,2-диэтилпентил, 3,3-диэтилгексил, 2,2 диэтилгексил, 3,3-диэтилгексил и им подобные. Алкильная группа может быть незамещенной или замещенной. Как используют в настоящем изобретении, если иное не определено, термин "алкенил" означает ненасыщенный с нормальной цепью или разветвленный нециклический углеводород, имеющий от 2 до 20 атомов углерода и по крайней мере одну углерод-углеродную двойную связь. Предпочтительно алкенил имеет от 2 до 10 атомов углерода и наиболее предпочтительно от 2 до 4 атомов углерода. Примерная нормальная цепь алкенилов включает -бут-3-ен, -гекс-4-ен и -окт-1-ен. Примерная разветвленная цепочка алкенилов включает -2-метил-бут-2-ен, -1-метил-гекс-4-ен и-4-этил-окт-1-ен. Алкенильная группа может быть замещенной или незамещенной. Как используется в настоящем изобретении, и если иначе не определено, термин "алкинил" означает алкильную группу, в которой одна или более углерод-углеродных простых связей замещены эквивалентным количеством углерод-углеродных тройных связей. Алкинильная группа должна включать по крайней мере два атома углерода и может быть замещена или незамещена. Как используется в настоящем изобретении, если иначе не определено, термин "галгеноалкил" означает алкильную группу, в которой один или более водородов были замещены атомом галогена. Атом галогена - это атом фтора, хлора, брома или йода. Как используется в настоящем изобретении, если иначе не определено, термин "гидроксиалкил" означает алкильную группу, в которой один или более водородов были замещены гидроксильной группой. Термин "алкокси" означает структуру формулы -О-алкил. Термин "алкилсульфонил" означает структуру формулы -S(О)2-алкил. Термины "алкиламин" и "диалкиламино" означают структуру формулы-N-алкил и -NH(алкил)алкил, соответственно, где алкил является таким, как определено выше. Термин "алканоил", один или в комбинации с другим термином, означает радикал типа "R-C(O)-",где "R" алкильный радикал, как определено выше, и "-С(О)-" является карбонильным радикалом. Примеры таких алканоильных радикалов включают ацетил, трифторацетил, гидроксиацетил, пропионил, бутирил, валерил, 4-метилвалерил и т.п. Термины "алканоиламино" и "алканоилокси" означают-NH-алканоил и -О-алканоил соответственно. Термин "алкоксикарбониламино" означает структуру формулы -NHC(О)О-алкил. Термин "алкилсульфониламино" означает структуру общей формулы -NHS(O)2-алкил. Как используется в настоящем изобретении, если иначе не определено, термин "арил" означает карбоциклическую кольцевую или кольцевую систему, содержащую от 5 до 14 кольцевых атомов, где по-5 012181 крайней мере одно кольцо является ароматическим. Кольцевые атомы карбоциклической арильной группы - все атомы углерода. Арильные группы включают моно-, би- или трициклические группы, а также бензоконденсированные карбоциклические группы, такие как 5,6,7,8-тетрагидронафтил и т.п. Предпочтительно арильная группа представляет собой моноциклическое кольцо или бициклическое кольцо. Примерные арильные группы включают фенил, толил, антраценил, флуоренил, инденил, азуленил, фенантренил и нафтил. Арильная группа может быть незамещенной или замещенной. Термин гетероарил означает арильную группу, в которой один или более, но не все, кольцевых атомов углерода замещены гетероатомом. Примерные гетероатомы - это N, О, S и Si. Гетероарильная группа может быть незамещенной или замещенной. Термин "циклоалкил" означает ненасыщенный или насыщенный углеводород, который образует по крайней мере одно кольцо, имеющее от 3 до 20 кольцевых атомов углерода, предпочтительно от 3 до 10 кольцевых атомов углерода. Кольца в циклоалкильной группе не являются ароматическими. Циклоалкильная группа может быть незамещенной или замещенной. Термин "гетероциклил" означает циклоалкил, в котором по крайней мере один, но не все, кольцевые атомы углерода замещены гетероатомом. Примерные гетероатомы - это NH, О и S. Как описано в настоящем изобретении, соединения по изобретению могут произвольно быть замещенными одним или более заместителями, такими как проиллюстрированы главным образом выше, или как иллюстрируются конкретными классами, подклассами и разновидностями изобретения. Будет понятно, что фраза "необязательно замещенный" используется попеременно с фразой "замещенный или незамещенный". В общем, термин "замещенный", которому предшествует термин "необязательно" или нет,относится к замене водородных радикалов в данной структуре радикалом указанного заместителя. Если не обозначено иначе, необязательно замещенная группа может иметь заместитель в каждом приемлемом положении группы, и когда более чем одно положение в любой данной структуре может быть замещено более чем одним заместителем, выбранным из указанной группы, заместитель может быть или тем же самым или другим в каждом положении. Комбинации заместителей, предполагаемых в соответствии с настоящим изобретением - предпочтительно те, которые приводят к образованию стабильных или химически возможных соединений. Термин "PKB" относится к протеинкиназе В, также известной, как AKT. Термин "лечение" относится к:(i) профилактике болезни, нарушения или патологического состояния, наблюдаемого у млекопитающего, которое может быть предрасположено к болезни, нарушению и/или патологическому состоянию, но еще не было диагностировано, как имеющееся в наличии;(ii) ингибированию заболевания, нарушения или патологического состояния, т.е. подавлению его развития; и(iii) ингибированию заболевания, нарушения или патологического состояния, т.е. вызывая регрессию заболевания, нарушения и/или патологического состояния, или одного или большего количества его симптомов. Термин "профилактика" относится к способности соединения или композиции изобретения к предотвращению заболевания, идентифицированного здесь у млекопитающих, диагностированных как имеющих заболевание, или у того, кто подвергается риску получить такое заболевание. Термин также охватывает профилактику дальнейшего прогрессирования заболевания у млекопитающих, которые уже страдают от или имеют симптомы такого заболевания. Термин "млекопитающее" относится к животным, не относящимся к человеку, или к людям. Как используется в настоящем изобретении, термин "пациент" или "испытуемый" означает животное (например, корова, лошадь, овцы, свиньи, цыплята, индейки, перепела, кот, собака, мышь, крыса,кролик, гвинейская свинка и т.д.) или млекопитающее, включая химерных и трансгенных животных и млекопитающих. В лечении или профилактике рака термин "пациент" или "испытуемый" предпочтительно означает обезьяну или человека, наиболее предпочтительно человека. В конкретном воплощении пациент или испытуемый поражены раком. Как используется в настоящем изобретении, "терапевтически эффективное количество" относится к количеству соединения формулы (I) изобретения, или его пролекарству, достаточному для обеспечения эффекта в лечении или профилактике патологического состояния или болезни, такой как рак, замедлению или снижению симптомов, связанных с патологическим состоянием или болезнью, или выздоровлению или улучшению состояния при болезни или причине этого. В частности, терапевтически эффективное количество означает количество, достаточное обеспечивать терапевтический эффект in vivo. Используемый совместно с количеством соединения по изобретению, термин предпочтительно охватывает нетоксичное количество, которое улучшает полную терапию, восстанавливает или избегает симптомов или причин болезни, или усиливает терапевтическую эффективность или синергизм с другим терапевтическим агентом. Как используется в настоящем изобретении, "профилактически эффективное количество" относится к количеству соединения по изобретению или другого активного ингредиента, достаточного приводить к профилактике патологического состояния или заболевания, такого как рак, или рецидива или метастаза-6 012181 рака. Профилактически эффективное количество может относиться к количеству, достаточному, чтобы предотвратить начальное заболевание или рецидив или распространение заболевания. Термин предпочтительно охватывает нетоксичное количество, которое улучшает полную профилактику или усиливает эффективность профилактического средства или синергизм с другим профилактическим или терапевтическим агентом. Как используется в настоящем изобретении, "в комбинации" относится к использованию более чем одного профилактического и/или терапевтического агентов одновременно или последовательно. Агенты могут быть выбраны и введены таким образом, что их соответствующие эффекты являются аддитивными или синергическими. Как используется в настоящем изобретении, термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных кислот или оснований, включая неорганические и органические кислоты и основания. Если соединение формулы (I) представляет собой основание, то необходимая фармацевтически приемлемая соль может быть получена любым подходящим методом, доступным в технологии, например, обработкой свободного основания с минеральной кислотой, такой как соляная кислота, бромисто-водородная кислота, серная кислота, азотная кислота,фосфорная кислота и т.п., или с органической кислотой, такой как уксусная кислота, малеиновая кислота, янтарная кислота, миндальная кислота, фумаровая кислота, малоновая кислота, пировиноградная кислота, щавелевая кислота, гликолевая кислота, салициловая кислота, пиранозидиловая кислота, такая,как глюкуроновая кислота или галактуроновая кислота, альфа-гидроксикислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глутаминовая кислота,ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая кислота, такая как п-толуолсульфоновая кислота или этансульфоновая кислота или т.п. Если соединение формулы (I) представляет собой кислоту, то необходимая фармацевтически приемлемая соль может быть получена любым подходящим методом, например, обработкой свободной кислоты с неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидрооксидом щелочного металла или гидрооксидом щелочно-земельного металла или т.п. Иллюстративные примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиака,первичных, вторичных и третичных аминов и циклических аминов, таких как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития. Термин "пролекарство" предназначен для обозначения любой химической структуры, которая после введения преобразуется в другую терапевтически эффективную химическую структуру. Соединения по настоящему изобретению могут содержать один или более асимметричных центров,и таким образом встречаются как рацематы и рацемические смеси, энантиообогащенные смеси, отдельные энантиомеры, индивидуальные диастереомеры и диастереомерные смеси. Все такие изомерные формы этих соединений специально включены в настоящее изобретение. Как используется в настоящем изобретении, и если не обозначено иначе, термин "оптически чистый" или "стереоизомерно чистый" означает композицию, которая включает один стереоизомер соединения и в основном не содержит других стереоизомеров этого соединения. Например, стереоизомерно чистое соединение, имеющее один хиральный центр, будет в основном не содержать противоположного энантиомера соединения. Типичное стереоизомерно чистое соединение включает больше чем приблизительно 80% по весу одного стереоизомера соединения и меньше чем приблизительно 20% по весу других стереоизомеров соединения, более предпочтительно больше чем приблизительно 90% по весу одного стереоизомера соединения и меньше чем приблизительно 10% по весу других стереоизомеров соединения, еще более предпочтительно больше чем приблизительно 95% по весу одного стереоизомера соединения и меньше чем приблизительно 5% по весу других стереоизомеров соединения и наиболее предпочтительно больше чем приблизительно 97% по весу одного стереоизомера соединения и меньше чем приблизительно 3% по весу других стереоизомеров соединения. Соединения по изобретению могут обнаруживать явление таутомерии. В то время как формула (I) не может явно изобразить все возможные таутомерные формы, должно быть понятно, что формула (I) предназначена, чтобы представить любую таутомерную форму изображенного соединения и не для ограничения только конкретной формой соединения, показанной изображениями формулы. Способы лечения и профилактики патологических состояний, опосредованных активностью PKB Настоящее изобретение охватывает способы лечения или профилактики PKB-опосредованных патологических состояний, таких как рак. Дозы. Величина профилактической или терапевтической дозы соединения данного изобретения формулы(I) или его фармацевтически приемлемых солей, сольвата, гидрата или стереоизомера, в случае кратковременного или длительного лечения или профилактики заболевания или состояния, такого как патологический клеточный рост или рак, будет зависеть от природы и тяжести заболевания, а также от пути,посредством которого вводится активный ингредиент. Доза и в некоторых случаях частота дозирования будут также варьироваться в соответствии с патологическим клеточным ростом, по поводу которого-7 012181 проводится лечение, возрастом, массой тела и реакциями конкретного пациента. Подходящие режимы дозирования могут быть без труда подобраны средним специалистом в данной области при надлежащем рассмотрении этих факторов. Величина профилактической или терапевтической дозы соединения данного изобретения формулы(I) или его фармацевтически приемлемых соли, сольвата, гидрата или стереоизомера, в случае кратковременного или длительного лечения или профилактики рака или патологического состояния, будет варьировать в зависимости от природы и тяжести заболевания, а также от пути, посредством которого вводится активный ингредиент. Доза и в некоторых случаях частота дозирования будут также варьировать в соответствии с патологическим состоянием, по поводу которого проводится лечение, возрастом,массой тела и реакциями конкретного пациента. Подходящие режимы дозирования могут быть без труда подобраны средним специалистом в данной области при надлежащем рассмотрении этих факторов. В одном воплощении вводимая доза зависит от конкретного применяемого соединения, массы тела и общего состояния пациента. В общем случае, дневная доза находится в пределах от около 0,001 до 100 мг/кг, предпочтительно около 1 до 25 мг/кг, более предпочтительно от около 1 до около 5 мг/кг. Для лечения людей, страдающих раком, назначают от около 0,1 мг до около 15 г в день, разделенные на 1-4 приема, предпочтительно от 10 мг до 12 г в день, более предпочтительно от 40 до 500 мг в день. В одном воплощении соединения данного изобретения назначают в дозе от 40 до 500 мг в день, разделенной на 14 приема. Дополнительно, рекомендуемая дневная доза может быть назначена курсами в виде монотерапии или в комбинации с другими терапевтическими агентами, в одном воплощении, дневная доза может быть назначена в виде разовой дозы или в равных разделенных дозах. В родственном воплощении рекомендуемая дневная доза может быть назначена несколько раз в неделю, три раза в неделю, четыре раза в неделю или пять раз в неделю. Соединения по настоящему изобретению могут быть назначены для обеспечения системного распределения соединения в организме пациента. В родственном воплощении соединения по изобретению назначают для получения системного действия на организм. В другом воплощении соединения данного изобретения вводят непосредственно в место, пораженное патологическим процессом, например, доступная кожа или рак пищевода. В другом воплощении соединения данного изобретения применяют путем перорального, чресслизистого (включая подъязычное, защечное, ректальное, интраназальное или вагинальное), парентерального (включая подкожное, внутримышечное, болюсные инъекции, внутриартериальное или внутривенное),чрескожного или местного введения. В отдельном воплощении соединения по настоящему изобретению применяют посредством чресслизистого (включая подъязычное, защечное, ректальное, интраназальное или вагинальное), парентерального (включая подкожное, внутримышечное, болюсные инъекции, внутриартериальное или внутривенное), чрескожного или местного введения. В более конкретном воплощении, соединения данного изобретения применяют путем перорального введения. В альтернативном отдельном воплощении соединения данного изобретения не применяют путем перорального введения. Различные терапевтически эффективные количества могут быть применимыми при различных патологических состояниях, что хорошо известно среднему специалисту в данной области. Аналогично,количества, достаточные для лечения или профилактики таких патологических состояний, но недостаточные вызывать (или достаточные, чтобы уменьшить) побочные эффекты, связанные с традиционным лечением, также включены в вышеописанные дозы и режим дозирования. Комбинированная терапия. Отдельные способы данного изобретения также включают введение дополнительного терапевтического агента (т.е. другого терапевтического агента, иного нежели соединение данного изобретения). В некоторых воплощениях настоящего изобретения соединения по изобретению могут быть применены в комбинации, по меньшей мере, с одним другим терапевтическим агентом. Терапевтические агенты включают помимо прочего антибиотики, противорвотные агенты, антидепрессанты и противогрибковые агенты, противовоспалительные агенты, противовирусные агенты, другие противоопухолевые агенты,иммуномодуляторы, -интерфероны, -интерфероны, алкилирующие агенты, гормоны и цитокины. В предпочтительном воплощении изобретение охватывает назначение дополнительного терапевтического агента, который проявляет противоопухолевую активность. Соединения данного изобретения и другой терапевтический агент могут действовать аддитивно или, предпочтительно, синергично. В предпочтительном воплощении композиция, содержащая соединение изобретения, вводится одновременно с введением другого терапевтического агента, который может быть частью той же композиции или в другой композиции, нежели та, что содержит соединения по изобретению. В другом воплощении соединение изобретения вводят перед введением или после введения другого терапевтического агента. В отдельном воплощении соединение изобретения вводят пациенту,который ранее не проходил или в настоящее время не проходит лечение другим терапевтическим агентом. В одном воплощении способы изобретения включают введение одного или более соединений формулы (I) данного изобретения без вспомогательного терапевтического агента.-8 012181 Фармацевтические композиции и лекарственные формы Фармацевтические композиции и отдельные лекарственные формы, содержащие соединение формулы (I) данного изобретения или его фармацевтически приемлемую соль, гидрат, метаболит или стереоизомер, также охвачены этим изобретением. Конкретные лекарственные формы изобретения могут быть пригодны для перорального, чресслизистого (включая, подъязычное, защечное, ректальное, интраназальное или внутривенное), чрескожного или местного применения. Фармацевтические композиции и лекарственные формы изобретения обычно также содержат один или более фармацевтически приемлемых наполнителей. Стерильные лекарственные формы также предусмотрены. В альтернативном воплощении фармацевтическая композиция включает соединение формулы (I) изобретения или его фармацевтически приемлемую соль, гидрат или стереоизомер и по меньшей мере один дополнительный терапевтический агент. Примеры дополнительных терапевтических агентов включают помимо прочего те, которые перечислены выше. Композиция, форма и тип лекарственных форм изобретения, как правило, варьируют в зависимости от их применения. Например, лекарственная форма, применяемая при кратковременном лечении заболевания или сопутствующего заболевания может содержать большие количества одного или более активных ингредиентов, чем лекарственная форма, применяемая при длительном лечении того же заболевания. Аналогично, парентеральная лекарственная форма может содержать меньшие количества одного или более активных ингредиентов, чем пероральная форма для лечения того же заболевания или расстройства. Эти и другие пути, при которых конкретные лекарственные формы, охваченные данным изобретением, будут варьировать между собой, будут очевидны среднему специалисту в данной области. См., например, Remington's Pharmaceutical Sciences, 20th ed., Mack Publishing, Easton PA 2000. Примеры лекарственных форм включают, помимо прочего, таблетки, каплеты; капсулы, такие как мягкие эластичные желатиновые капсулы; крахмальные капсулы; пастилки; леденцы; дисперсии; суппозитории; мази; припарки (примочки); пасты; порошки; повязки; кремы; гипс; растворы; пластыри; аэрозоли (например,интраназальные спреи или ингаляторы); гели; жидкие лекарственные формы, подходящие для перорального или слизистого введения пациенту, включая суспензии (например, водные и неводные жидкие суспензии, эмульсии масло-в-воде или эмульсии вода-в-масле), растворы и эликсиры; жидкие лекарственные формы, особенно подходящие для парентерального введения пациенту; и стерильные твердые вещества (например, кристаллические или аморфные твердые вещества), которые могут быть восстановлены для получения жидких лекарственных форм, подходящих для парентерального введения пациенту. Подобно количеству и видам наполнителей, количество и конкретные виды активных ингредиентов в лекарственной форме могут различаться в зависимости от факторов, включающих помимо прочего путь, которым они будут вводиться пациентам. Тем не менее, типичные лекарственные формы изобретения включают соединения формулы (I) изобретения или их фармацевтически приемлемые соли, гидраты или стереоизомеры, в количестве от 0,1 мг до 1500 мг на одну форму для обеспечения дозы около 0,01 до 200 мг/кг в день. Вышесказанное демонстрирует существенные и важные свойства настоящего изобретения. Средний специалист в данной области должен понимать, что многочисленные модификации и воплощения изобретения могут быть разработаны. Тем не менее, предполагается, что прилагаемая формула изобретения охватывает все такие модификации и воплощения. Примеры Соединения формулы (I) получают в соответствии со следующими схемами синтеза и отдельными примерами, детализированными в настоящем описании. Соединения именуют, используя Chemdraw Ultra, v.8.07. Указанные схемы и примеры обеспечены только для иллюстративных целей и не предназначены для ограничения области изобретения. Если иное не отмечено, все продукты получают от коммерческих поставщиков и используют без дополнительной очистки. Безводные растворители, такие как ДМФА, ТГФ, CH2Cl2 и толуол получают отAldrich Chemical Company. Все реакции, касающиеся соединений, чувствительных к воздушной среде или влажности, выполняют в атмосфере азота. Флэш-хроматографию выполняют, используя силикагель от Aldrich Chemical Company (200-400 меш, 60 А) или Biotage предварительно загруженную колонку. Тонкослойную хроматографию (ТСХ) выполняют с помощью Analtech геля ТСХ плат (250 мкм). Препаративную ТСХ выполняют с помощью Analtech силикагельных плат (1000-2000 mu). ПрепаративнуюHPLC проводят на Beckman или Waters HPLC системе со смесью 0,1% TFA/H2O и 0,1% TFA/CH3CN в качестве мобильной фазы. Используют скорость потока 20 мл/мин и градиентный способ. 1H ЯМР спектр определяют с помощью super conducting FT ЯМР спектрометров operating при 400 МГц или прибора Varian 300 МГц. Химические сдвиги выражают в част. на млн, по отношению к внутреннему стандарту тетраметилсилану. Все соединения, имеющие ЯМР спектр, совместимы с их заданными структурами. Масс-спектр (MS) определяют на электроспрейном масс-спектрометре Perkin Elmer-SCIEX API 165 (положительный и/или отрицательный) или HP 1100 MSD LC-MS с помощью электроспрейной ионизации и квадропольного определения. Все части определяют в расчете на массу и значения температур определяют в градусах по Цельсию, если иное не отмечено. Используют следующие сокращения: АсОН или НОАс (уксусная кислота), Ac2O (уксусный ангид-9 012181 рид), Al2O3 (оксид алюминия), AIBN (2,2'-азобисизобутиронитрил), Ar (аргон), AgSO4 (сульфат серебра),АТР (аденозинтрифосфат), 9-BBN (9-борабицикло[3,3,1]нонан), ВН 3 (боран), BINAP (2,2'бис(дифенилфосфино)-1,1'-бинафтил), Boc (трет-бутилоксикарбонил), ВоС 2 О (Вос ангидрид), ВОР-С 1(12,2 г, 133 ммоль) смешивают в 150 мл оксихлорида фосфора. Смесь нагревают при температуре 80 С в течение 60 ч. После удаления избытка оксихлорида фосфора через роторный испаритель при пониженном давлении, оставшийся остаток смешивают с водой со льдом и значение pH повышают до pH 12 с помощью KOH. После фильтрования и промывания водой получают аморфное твердое вещество желтого цвета 1 а в виде сырого продукта (17 г, у (выход) = 85%). Его используют непосредственно на следующей стадии. Чистый образец продукта получают путем воздействия сырого продукта хроматографии на колонке с силикагелем с градиентом 1-5% 2 М NH3 раствор метанола в дихлорметане в виде элюента. 1- 10012181 ацетонитрила до температуры 60 С в круглодонной колбе. 5-Изохинолин-6-ил-[1,3,4]тиадиазол-2-иламин(Ia) тонко суспендируют в 100 мл ацетонитрила. Суспензию по каплям добавляют в нагретую круглодонную колбу и полученную смесь нагревают при температуре 70 С в течение полутора часов. Реакционную смесь концентрируют до 30 мл при пониженном давлении и смешивают с 100 мл 20% водным раствором HBr. Полученной смеси дают возможность отстояться в холодильнике (-20 С) в течение 12 ч. После фильтрования промывают фильтратную лепешку 10% водным раствором HBr, затем водой, сушат в вакууме, получают зеленоватого цвета твердое вещество (3,7 г, выход = 79%) в виде HBr соли желаемого продукта.MS (API-ES) м/е (%): 292 (100%, М 1), 294(100%, М 3). Сырой продукт используют непосредственно на следующей стадии. Пример 1. 6-(5-S)-3-(4-Хлорбензил)пиперазин-1-ил)-1,3,4-тиадиазол-2-ил)изохинолин (1). Соединение 1b (0,31 г, 0,83 ммоль), S-2-(4-хлорбензил)пиперазин (0,23 г, 1,10 ммоль), диизопропилэтиламин (0,58 мл, 3,33 ммоль) и 1,5 мл N-метилпирролидинон смешивают в пробирке объемом 2 мл для нагревания при микроволновом излучении. Смесь нагревают при микроволновом излучении при температуре 180 С в течение 40 мин. Реакционную смесь распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. После удаления этилацетата сырой продукт подвергаютHPLC хроматографии на колонке с силикагелем, чтобы получить на выходе аморфное твердое вещество желтого цвета в виде чистого продукта (0,16 г, выход = 46%). 1MS (API-ES) м/е (%): 422 (100%, М 1). Пример 2. N-S)-2-Амино-3-фенилпропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-амин (2). Соединение синтезируют в соответствии с методикой, аналогичной методике синтеза соединения по примеру 1, и очищают с помощью HPLC с обратимой фазой в виде TFA соли. 1 Н ЯМР (400 МГц, Метанол-d4):3,08 (м, 2 Н), 3,71 (м, 1H), 3,83 (м, 1H), 3,91 (м, 1H), 7,38 (м, 5 Н),8,50-8,60 (м, 5 Н), 9,75 (с, 1H).(S)-2-(1-Гидрокси-3-(1 Н-индол-3-ил)пропан-2-ил)изоиндолин-1,3-дион (150 мг, 0,47 ммоль) и 60% раствор гидрида натрия в неорганическом масле (37,6 мг, 0,94 ммоль) смешивают в 2 мл Nметилпирролидинона. После перемешивания в течение 10 мин добавляют соединение 1b, суспендированное в 2 мл N-метилпирролидинона, и реакционную смесь перемешивают при температуре 20 С в течение часа. Реакционную смесь распределяют между этилацетатом и насыщенным водным раствором хлорида аммония. Этилацетатную фазу промывают более насыщенным водным раствором хлорида аммония и сушат над безводным сульфатом натрия. После удаления этилацетата, оставшийся остаток наносят на хроматографическую колонку с силикагелем, чтобы получить на выходе промежуточное соединение 2-S)-3-(1H-индол-3-ил)-1-(5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-илокси)пропан-2-ил)изоиндолин 1,3-дион (3 а). Соединение 3 а растворяют в 4 мл этилового спирта, 0,5 мл воды и 0,5 мл моногидрата гидразина в колбе для нагревания при микроволновом излучении. Колбу нагревают при температуре 100 С при микроволновом излучении в течение пяти мин. После удаления полностью растворителя остаток подвергают разделению с помощью HPLC хроматографии на С 18 колонке с обратимой фазой, чтобы получить на выходе (2S)-1-(1 Н-индол-3-ил)-3-(5-(изохинолин-6-ил)-1,3,4-тиадиазол-2-илокси)пропан-2 амин (3) (2,4 мг) в виде TFA соли. 1 5-Бром-2-фторфенилэтанол (4b). Коммерчески доступное соединение 4 а (150,0 г, 739 ммоль) загружают в круглодонную колбу объемом 2 л. Реакционную смесь в колбе погружают на баню воды со льдом по каплям добавляют через делительную воронку метилмагнийбромид (270 мл, 812 ммоль). После добавления реакционную смесь непрерывно перемешивают в течение более часа. После завершения реакции смесь медленно выливают в 500 мл воды со льдом и 250 мл насыщенного хлорида аммония. Полученный водный раствор экстрагируют эфиром (800 мл 2) в делительной воронке. Объединенный эфирный слой промывают рассолом и сушат над сульфатом натрия. Удаление растворителя дает продукт 4b (150 г, выход = 93%). Продукт используют непосредственно на следующей стадии без дополнительной очистки. 1-(5-Бром-2-фторфенил)этанон (4 с). Соединение 4b (50,0 г, 228 ммоль) вместе с 300 мл дихлорметана загружают в круглодонную колбу объемом 2 л. В колбу добавляют размельченный дихромат пиридиния (171,0 г, 456 ммоль) и порошкообразные молекулярные сита (10 г). Гетерогенную реакционную смесь перемешивают в течение 16 ч при температуре 20 С. Полученную реакционную смесь фильтруют через целит и промывают эфиром(500 мл 3). Объединенный фильтрат концентрируют при пониженном давлении. Сырой продукт элюируют через небольшую таблетку из силикагеля (3 дюйма в длину) с помощью 10% EtOAc в гексане. Полученный продукт (42,0 г, выход=84%) используют на следующей стадии. 5-Бром-3-метил-1H-индазол (4d). Соединение 4 с (66,0 г, 304 ммоль) и 350 мл безводного гидразина загружают в круглодонную колбу объемом 1 л. Полученную реакционную смесь нагревают при температуре кипения с обратным холодильником при температуре 117 С в течение 5 ч. Затем реакционной смеси дают возможность остыть до комнатной температуры, избыток гидразина выпаривают при пониженном давлении, чтобы получить на выходе твердое вещество белого цвета. Затем 400 мл воды выливают в полученное твердое вещество и воду отфильтровывают. Твердое вещество промывают 400 мл воды дважды, чтобы удалить следы гидразина, твердое вещество белого цвета помещают в 600 мл EtOAc и промывают дважды 300 мл воды и насыщенным раствором рассола. Затем слой EtOAc сушат над сульфатом натрия. Удаление растворителя дает требуемый продукт в виде аморфного твердого вещества белого цвета (60,0 г, выход = 94%). Продукт используют непосредственно на следующей стадии без дополнительной очистки. 3-Метил-1H-индазол-5-карбоновая кислота (4 е). В трехгорлую круглодонную колбу, оборудованную внутренним термометром и подвесным мотором для перемешивания, загружают 600 мл ТГФ и охлаждают до температуры -78 С. В колбу добавляют трет-BuLi (1,7 М в ТГФ, 200 мл, 0,340 моль), и смесь перемешивают в течение 15 мин. Затем по каплям добавляют через делительную воронку 5-бром-3-метил-1H-индазол (4d) (22,4 г, 0,106 моль) в 200 мл ТГФ. Скорость добавления тщательно контролируют для обеспечения того, чтобы внутренняя температура оставалась ниже -70 С. Полученный раствор оранжевого цвета перемешивают в течение 30 мин, в течение которых СО 2 пробулькивают через смесь. Наблюдают осадок белого цвета. Через 20 мин баню со льдом удаляют и температуре дают возможность подняться до комнатной температуры (rt), после чего перемешивание продолжают в течение еще 30 мин. Затем добавляют воду,- 12012181 вначале 40 мл, а затем еще 200 мл. Двухфазную смесь частично концентрируют при пониженном давлении, удаляя 75% исходной органической части. Двухфазный раствор затем переносят в делительную воронку, и органическую фазу экстрагируют 100 мл 2 М NaOH. Объединенный водные экстракты промывают эфиром и затем подкисляют до значения pH 2,0 с помощью концентрированной HCl. Осадок начинает образовываться, и смесь охлаждают до температуры 0 С, чтобы завершить осаждение. Полученное твердое вещество фильтруют, промывают 1 М HCl и сушат при пониженном давлении при температуре 160 С над пентоксидом фосфора, получая на выходе 3-метил-1H-индазол-5-карбоновую кислоту (4 е)(18,1 г, 96% выход) в виде твердого вещества розово-бежевого цвета. 1 Н ЯМР 400 МГц (d4 MeOH) 2,61 (3 H, с), 3,33 (2 Н, с), 7,52 (1 Н, д, J = 6,0 Гц), 8,05 (1 Н, т, J = 5,2 Гц), 8,50 (1 Н, с).MS (API-ES) м/е (%): 177 (100%, МН). Используют два способа для получения 5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-иламина(4f). Способ 1 использует методику, аналогичную методике получения соединения 1 а. Способ 2. В круглодонную колбу, оборудованную подвесным мотором для перемешивания, добавляют 80 г полифосфорной кислоты. Колбу нагревают до температуры 90 С и медленно добавляют в течение 30 мин мелкоизмельченную смесь 3-метил-1H-индазол-5-карбоновой кислоты 4 е (8,0 г, 45,5 ммоль) и тиосемикарбазида (4,1 г, 45,4 ммоль). Полученную смесь перемешивают в течение 24 ч. На этой стадии к раствору добавляют 200 мл воды со льдом, и значение pH полученной смеси доводят до 7,0, используя твердый КОН. В процессе реакции образуется осадок. Осадок отделяют фильтрованием,промывают последовательно водой и эфиром и сушат при пониженном давлении, получая на выходе 5(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-иламин (4f) (5,50 г, 53% выход) в виде твердого вещества,окрашенного в желто-коричневый цвет. 1MS (API-ES) м/е (%): 232 (100%, М'+Н). 5-(5-Бром-[1,3,4]тиадиазол-2-ил)-3-метил-1H-индазол (4g). Суспензию 5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-иламина (4f) (1,10 г, 4,76 ммоль) в 20 мл ацетонитрила добавляют в смесь трет-бутилнитрита (0,74 г, 7,14 ммоль) и бромида меди(II) (1,27 г,5,71 ммоль), которую предварительно нагревают до температуры 60 С. Полученную смесь нагревают при температуре 60 С в течение 2 ч. После удаления полностью растворителя путем выпаривания при пониженном давлении, оставшийся остаток распределяют между этилацетатом и насыщенным рассолом. Раствор этилацетата промывают рассолом и сушат над сульфатом натрия. После удаления растворителя получают твердое вещество оранжевого цвета в виде сырого продукта (1,10 г). Указанное вещество используют непосредственно на следующей стадии.MS (API-ES) м/е (%): 295 (100%, М 1), 297 (97%, М 3). Пример 4. S-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]-3-фенилпропан-1,2-диамин (4). Соединение по примеру 4 синтезируют в соответствии с методикой, аналогичной методике синтеза соединения по примеру 1, используя соединение 4g и S-3-фенилпропан-1,2-диамин в виде исходных продуктов. Полученное соединение очищают с помощью методики HPLC с обратимой фазой в виде TFA соли. 1[M+1]: 365,1543; Найдено: 365,1542. Пример 5. S-3-(1H-Индол-3-ил)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол-2-ил]пропан-1,2 диамин (5). Соединение по примеру 5 синтезируют в соответствии с методикой, аналогичной методике синтеза соединения по примеру 1, используя соединение 4g и S-3-(1H-индол-3-ил)пропан-1,2-диамин в виде исходных продуктов. Полученное соединение очищают с помощью методики HPLC с обратимой фазой в виде TFA соли. 1[M+H]: 404,1652; Найдено: 404,1651. Соединения по примерам 6-69 синтезируют, используя следующую общую методику, показанную на схеме 4. Общая методика реакции образования аминотиадиазоламидной связи. 150 мг аминотиадиазола (0,65 ммоль, 1 экв.) растворяют в 6 мл ДМФА и PS-EDC (1083 мг,1,95 ммоль, 3 экв.), HOBt (263 мг, 1,95 ммоль, 3 экв.) и добавляют соответствующую кислоту (1,95 ммоль, 3 экв.). Реакционную смесь перемешивают в течение ночи при комнатной температуре и фильтруют. Смолу промывают трижды 20 мл ДМФА (каждый раз) и объединенные ДМФА фазы выпаривают. Сырого желтого цвета масло подвергают затем стадии восстановления без дополнительной очистки. Общая методика для (LAH), индуцированного литийалюминийгидридом восстановления амида. Сырой продукт из предыдущей стадии растворяют в 5 мл ТГФ и охлаждают до температуры 0 С. Добавляют 6 мл LAH (1M в ТГФ) и охлажденную баню удаляют. Перемешивание продолжают в течение 2 ч при комнатной температуре. Добавляют 50 мл сухого ТГФ и реакционную смесь выливают в перемешиваемую смесь 10 г Na2SO4 10 Н 2 О в 50 мл ТГФ. Перемешивание продолжают при комнатной температуре в течение 30 мин и реакционную смесь фильтруют. После промывания (трижды каждый раз 80 мл CH2Cl2) и высушивания объединенных органических слоев над MgSO4, смесь выпаривают. Сырой продукт частично очищают на 2 препаративных TLC, что дает восстановленное промежуточное соединение со средним значением чистоты между 75-85%. Этот продукт используют без дополнительной очистки на конечной стадии ВОС-снятие защиты. Общая методика для индуцированного TFA снятия ВОС защиты. Получистый продукт из предыдущей реакции растворяют в 10 мл CH2Cl2 и добавляют 3 мл TFA при комнатной температуре. Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем добавляют 50 мл толуола. Реакционную смесь выпаривают и повторно растворяют в 2 мл метанола. Значение pH смеси повышают с помощью 5 М NaOH(aq) (3-10 капель из пипетки Пастера) и помещают на препаративную ТСХ пластину для очистки (10% МеОН в CH2Cl2), получая свободные амины. Для получения соответствующей HCl соли соединения, препаративную ТСХ на силикагеле промывают раствором, подкисленным 1 мл HCl (1 М в Et2O) до упаривания. Соответствующие TFA соли получают на основе необходимой дополнительной стадии очистки с помощью препаративной HPLC (TFA буфер). Пример 6. Трифторацетат 2-[5-(3-метил-1 Н-индазол-5-ил)-[1,3,4]тиадиазол-2-иламино]-1(тетрагидропиран-4-илметил)этиламмония (6).- 18012181 Пример 69. 5-(1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин синтезируют в соответствии с методикой,аналогичной методике синтеза соединения 4f, как показано на схеме 3 с помощью коммерчески доступного 5-бром-2-фторбензальдегида в качестве исходного продукта в три стадии.MS (API-ES) м/е (%): 218 (100%, М 1). Соединения по примерам 70-87 синтезируют в соответствии с методикой, аналогичной методике синтеза, показанной на схеме 4, используя соединение по примеру 69 (5-(1H-индазол-5-ил)-1,3,4 тиадиазол-2-амин) в качестве исходного продукта. Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 3, используя 5-бром-6-фтор-1H-индазол (88d), который обрабатывают трет-бутиллитием и диоксидом углерода, чтобы получить на выходе 6-фтор-1H-индазол-5-карбоновую кислоту. Указанное соединение в свою очередь обрабатывают тиосемикарбазидом и полифосфорной кислотой для образования 5-(6-фтор-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амина.MS (API-ES) м/е (%): 236 (100%, М 1). Методики получения промежуточных соединений по примеру 88 показаны ниже на схеме 5. Схема 5 Исходный продукт, 5-фтор-2-метилбензоламин (12,37 г, 99 ммоль), растворяют в толуоле (120 мл) и добавляют уксусный ангидрид (12,5 мл, 113 ммоль). Смесь нагревают до температуры 100 С в течение часа. Растворитель удаляют в вакууме, и полученное твердое вещество (88 а) растворяют в уксусной кислоте (70 мл) и затем по каплям добавляют бром (4,82 мл, 94 ммоль). Раствору темного цвета дают возможность перемешиваться при комнатной температуре в течение 12 ч, в течение которых образуется осадок рыжего цвета. Осадок размельчают и помещают в воду (50 мл), фильтруют и промывают водой (50 мл), что даетN-(4-бром-5-фтор-2-метилфенил)ацетамид (88b) с 94% выходом. Ацетамид (88b) (10 г, 41 ммоль) суспендируют в хлороформе (90 мл) и последовательно добавляют уксусный ангидрид (11,5 мл, 122 ммоль),ацетат калия (KOAc, 8,0 г, 81 ммоль), 18-Краун эфир-6 (0,54 г, 2 ммоль) и изоамилонитрит (12,3 мл,92 ммоль). Смесь нагревают при температуре 65 С в течение 24 ч, охлаждают до комнатной температуры и промывают бикарбонатом натрия (Na2COs насыщенный, 70 мл 3), сушат над сульфатом натрия(Na2SO4) и помещают непосредственно на силикагель. Хроматография на колонке (0-20% EtOAc в гексан) дает 1-(5-бром-6-фтор-1H-индазол-1-ил)этанон (89 с) с 55% выходом. Полученный индазол суспендируют в 10% HCl (70 мл) и добавляют метанол (приблизительно 20 мл). Суспензию нагревают при температуре кипения с обратным холодильником (приблизительно в течение часа), в течение которого она становится прозрачной, затем ее охлаждают и значение pH повышают путем добавления раствора гидроксида натрия (NaOH, 5 N), что приводит к выпадению твердого вещества в виде осадка грязно-белого цвета. Раствор фильтруют и полученное твердое вещество сушат в вакууме, что дает требуемый продукт (88d) с 84% выходом. LCMS (М+Н) 215,1 Вычислено для C7H5BrFN2 214,96. 1H ЯМР (400 МГц) MeOD: 8,06 (д, J = 6,8 Гц, 1H), 8,03 (с, 1H), 7,37 (д, J = 8,8 Гц, 1H). Соединения по примерам 89-93 получают, применяя методику, аналогичную методике получения соединения, показанной на схеме 4, используя соединение 88 в качестве исходного продукта. Пример 94. 5-(6-Метил-1 Н-индазол-5-ил)-1,3,4-тиадиазол-2-амин (94). Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 3, используя 5-бром-6-метил-1H-индазол в качестве исходного продукта,который получают в соответствии с методикой, аналогичной методике получения 5-бром-6-фтор-1Hиндазола. Исходный продукт обрабатывают трет-бутиллитием и диоксидом углерода, чтобы получить на выходе 6-метил-1 Н-индазол-5-карбоновую кислоту, которую в свою очередь обрабатывают тиосемикарбазидом и полифосфорной кислотой для образования соединения 94. Соединения по примерам 95-100 получают, применяя методику, аналогичную методике получения соединения, показанной на схеме 4, используя соединение 94 в качестве исходного продукта. Пример 101. NS)-2-Амино-3-метил-3-фенилбутил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол 2-амин (101). Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 4. Пример 102. 5-(7-Метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин (102). Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения по примеру 88, используя коммерчески доступный 4-бром-2,5-диметиланилин в качестве исходного продукта. После стадии ацилирования, N-(4-бром-2,6-диметилфенил)ацетамид обрабатывают изоамилонитритом и затем 10% водным раствором HCl, чтобы получить на выходе 5-бром-7-метил-1 Ниндазол. 5-Бром-7-метил-1 Н-индазол обрабатывают трет-бутиллитием и диоксидом углерода, чтобы получить на выходе 6-метил-1 Н-индазол-5-карбоновую кислоту, которую в свою очередь обрабатывают тиосемикарбазидом и полифосфорной кислотой для образования 5-(7-метил-1H-индазол-5-ил)-1,3,4 тиадиазол-2-амина.MS (API-ES) м/е (%): 232 (100%, М 1). Соединения по примерам 103-105 получают в соответствии с методикой, аналогичной методике,показанной на схеме 4, используя соединение 102 в качестве исходного продукта. Пример 103. N-S)-2-Амино-3-(4-(трифторметил)фенил)пропил)-5-(7-метил-1H-индазол-5-ил)1,3,4-тиадиазол-2-амин (103).FTMS Вычислено (М+Н) 433,07635; Найдено 433,07629. Пример 106. 5-(7-Хлор-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения 102, используя коммерчески доступный 4-бром-2-хлор-6-метилбензоламин в качестве исходного продукта.MS (API-ES) м/е (%): 232 (100%, М 1). Пример 107. N-S)-2-Амино-3-(4-хлорфенил)пропил)-5-(7-хлор-1H-индазол-5-ил)-1,3,4-тиадиазол 2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике, показанной на схеме 4, используя соединение 106 в качестве исходного продукта.FTMS Вычислено (М+Н) 419,06070; Найдено 419,06044. Пример 108. 5-(1H-Пиразоло[3,4-с]пиридин-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение по примеру получают, как показано на схеме 6. Схема 6 5-Бром-1H-пиразоло[3,4-с]пиридин получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 5, используя коммерчески доступный 6-бром-4 метилпиридин-3-амин в качестве исходного продукта. После стадии ацилирования N-(6-бром-4 метилпиридин-3-ил)ацетамид обрабатывают изоамилонитритом, затем 10% водным раствором HCl, чтобы получить на выходе 5-бром-1 Н-пиразоло [3,4-с]пиридин. трет-BuLi (27 мл, 1,7 М в пентане) добавляют к 100 мл ТГФ при температуре -78 С. К раствору через делительную воронку по каплям добавляют 5-бром-1H-пиразоло[3,4-с]пиридин (3,0 г, 15 ммоль) в 50 мл ТГФ. Полученную смесь перемешивают в течение часа, в течение которого по каплям добавляют ДМФА (6,0 мл, 76 ммоль). Реакционной смеси дают возможность нагреться до комнатной температуры и перемешивают 2 ч. Реакционную смесь затем осторожно гасят водным раствором NH4Cl и разбавляютEtOAc. Полученную двухфазную смесь разделяют в делительной колбе. Водную часть экстрагируют трижды EtOAc, и объединенные органические экстракты промывают рассолом и сушат над MgSO4. Фильтрование и концентрация при пониженном давлении и последующая флэш-хроматография на силикагеле (от 100% CH2Cl2 до 7,5% МеОН/CH2Cl2), дает требуемый 1H-пиразоло[3,4-с]пиридин-5 карбальдегид (1,1 г, 50% выход) в виде твердого вещества белого цвета. 1 Н ЯМР (MeOD, 400 МГц) кето таутомер: 10,17 (с, 1 Н), 9,19 (с, 1H), 8,56 (с, 1H), 8,46 (с, 1 Н); энольный таутомер: 9,00 (с, 1H), 8,25 (с, 1H), 8,05 (с, 2H), 5,73 (с, 1H).NaOAc (0,44 г) и тиосемикарбазид (1,1 г) помещают в 30 мл EtOH и нагревают до температуры кипения. Добавляют воду, пока смесь не станет гомогенной. Нагревающую баню удаляют и добавляют одну часть 1 Н-пиразоло[3,4-с]приридин-5-карбальдегида (1,1 г, 7,5 ммоль). Реакционную смесь возвращают к температуре кипения и перемешивают в течение 3 ч. Во время реакции начинает образовываться осадок. Реакционную смесь затем охлаждают до комнатной температуры и осадок собирают фильтрованием, затем промывают МеОН и Et2O и сушат под высоким вакуумом, получая на выходе 1,4 г сырого продукта, который переносят непосредственно на следующую стадию. Продукт предыдущей реакции помещают в 15 мл А.с 2 О и нагревают до температуры кипения в течение 30 мин. Концентрация при пониженном давлении дает вязкое твердое вещество. Вязкое твердое вещество помещают в 17 мл уксусной кислоты. Добавляют перуксусную кислоту (4,2 мл, 25% веса в уксусной кислоте), и смесь нагревают до температуры 65 С. Через 90 мин реакционную смесь охлаждают до комнатной температуры и образуется осадок. Осадок собирают фильтрованием, промывают Н 2 О иEt2O и сушат под высоким вакуумом. Высушенный осадок помещают в 30 мл 10% HCl и нагревают до температуры кипения для завершения растворения. Реакционную смесь затем охлаждают до комнатной температуры и значение pH раствора доводят до 7,0 с помощью 20% водного раствора NaOH. Образуется твердое вещество белого цвета, которое собирают фильтрованием, промывают Et2O, и сушат под высоким вакуумом, получая на выходе требуемый 5-(1H-пиразоло[3,4-с]пиридин-5-ил)-1,3,4-тиадиазол-2- 24012181 амин 108 (0,88 г, 72% выход) в виде в значительной степени нерастворимого твердого вещества белого цвета.MS (API-ES) м/е (%): 219 (100%, М 1). Соединения по примерам 109-111 получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 4, используя соединение 108 в качестве исходного продукта. Пример 109. N-S)-2-Амино-3-(2,4-дихлорфенил)пропил)-5-(1H-пиразоло[3,4-с]пиридин-5-ил)1,3,4-тиадиазол-2-амин.N-S)-2-Амино-3-(3-(трифторметил)фенил)пропил)-5-(3-метил-1 Н-пиразоло[3,4b]пиридин-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, показанной на схеме 7. Схема 7 5-Бром-2-хлор-N-метокси-N-метилникотинамид. Коммерчески доступную 5-бром-2-хлорникотиновую кислоту (1 а) (15,0 г, 63,44 ммоль) растворяют в 50 мл ДМФА и добавляют порциями N,N'-карбонилдиимидазол (11,30 г, 69,78 ммоль). Полученный раствор перемешивают в течение 10 мин и затем добавляют гидрохлорид N,O-диметилгидроксиламина. Через час ДМФА удаляют через роторный испаритель при пониженном давлении и полученный остаток разбавляют насыщенным раствором бикарбоната натрия и экстрагируют дихлорметаном в делительной воронке. Объединенный органический слой промывают рассолом и сушат над сульфатом натрия. Удаление растворителя дает продукт с количественным выходом. Продукт используют непосредственно на следующей стадии без дополнительной очистки. 1-(5-Бром-2-хлорпиридин-3-ил)этанон. Исходный продукт 5-бром-2-хлор-N-метокси-N-метилникотинамид (5,0 г, 17,89 ммоль) загружают в круглодонную колбу объемом 250 мл, и колбу охлаждают до температуры -78 С. По каплям добавляют через делительную воронку метилмагнийбромид (96,5 мл, 19,68 ммоль) в 5 мл ТГФ. Полученной смеси- 25012181 дают возможность перемешиваться в течение 3 ч при температуре -78 С. После удаления ТГФ растворителя через роторный испаритель при пониженном давлении, реакционную смесь распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. После удаления этилацетата, полученный сырой продукт переносят на колонку с силикагелем, элюируя 20% этилацетатом в гексане. Полученный продукт (2,28 г, выход = 54,3%) используют на следующей стадии. 5-Бром-3-метил-1 Н-пиразоло[3,4-b]пиридин. Исходный продукт 1-(5-бром-2-хлорпиридин-3-ил)этанон (5,8 г, 24,7 ммоль) и 100 мл безводного гидразина загружают в круглодонную колбу, объемом 500 мл, и полученной смеси дают возможность перемешиваться при комнатной температуре в течение ночи. После удаления избытка гидразина через роторный испаритель при пониженном давлении оставшийся остаток разбавляют, отгоняя воду и получают твердое вещество. После фильтрования воды полученное твердое вещество помещают в этилацетат и насыщенный водный раствор бикарбоната натрия и экстрагируют дважды этилацетатом. Объединенные органический слой промывают водой и рассолом и сушат над сульфатом натрия. Сырой продукт элюируют через короткую колонку с силикагелем (3 дюйма в длину, 1 дюйм = 2,5 см) с этилацетатом в виде твердого вещества белого цвета (4,0 г, выход = 77%). 3-Метил-1H-пиразоло[3,4-b]пиридин-5-карбальдегид. 80 мл безводного ТГФ загружают в круглодонную колбу объемом 250 мл и охлаждают до температуры -78 С. В колбу добавляют через пипетку трет-BuLi (1,70 М в ТГФ, 17,6 мл, 30 ммоль). После перемешивания в течение 5 мин по каплям добавляют через пипетку 5-бром-3-метил-1H-пиразоло[3,4b]пиридин (2,12 г,10 ммоль). Через 30 мин по каплям добавляют ДМФА (3,65 г, 50 ммоль) в 20 мл ТГФ и смесь перемешивают в течение ночи. Реакционную смесь гасят насыщенным водным раствором хлорида аммония. После выпаривания избытка ТГФ, реакционную смесь экстрагируют дважды этилацетатом. Объединенный органический слой промывают рассолом один раз и сушат над сульфатом натрия. Сырой продукт хроматографируют с помощью 50% этилацетата в гексане, что дает требуемый продукт (0,23 г,выход = 20,5%). 1-3-Метил-1 Н-пиразоло[3,4-b]пиридин-5-ил)метилен)тиосемикарбазид. Ацетат натрия (166 мг, 2,02 ммоль), тиосемикарбазид (333 мг, 3,66 ммоль), 100 мл этанола и 2 мл дистиллированной воды загружают в круглодонную колбу объемом 250 мл. Полученную суспензию нагревают до температуры 80 С, пока она не станет прозрачным раствором. Реакционную смесь затем охлаждают до комнатной температуры и полученную смесь добавляют в круглодонную колбу объемом 250 мл, содержащую 3-метил-1 Н-пиразоло[3,4-b]пиридин-5-карбальдегид (333 мг, 2,1 ммоль). После завершения добавления полученную смесь нагревают до температуры 80 С в течение 16 ч. Смесь охлаждают до комнатной температуры и твердое вещество осаждают. Твердое вещество отфильтровывают и промывают эфиром (50 мл). Продукт (500 мг, выход = 95%) используют непосредственно на следующей стадии без дополнительной очистки. 1-Ацетил-3-Е)-(3-метил-1H-пиразоло[3,4-b]пиридин-5-ил)метиленамино)изотиомочевина. Исходный продукт 1-3-метил-1H-пиразоло[3,4-b]пиридин-5-ил)метилен)тиосемикарбазид (500 мг,2,0 ммоль) и уксусный ангидрид (20 мл) смешивают и нагревают до температуры 80 С в течение часа. Реакционную смесь затем охлаждают до комнатной температуры. Твердое вещество желтого цвета осаждают и отфильтровывают. Твердое вещество желтого цвета промывают эфиром (50 мл). Продукт получают с количественным выходом и используют на следующей стадии без дополнительной очистки.(80 мг, 0,22 ммоль) помещают в 5 мл уксусной кислоты и 1,2 мл перуксусной кислоты в круглодонную колбу. Смесь нагревают до температуры 60 С. Через 15 мин продукт осаждают. Твердое вещество отфильтровывают и промывают эфиром (50 мл). Выход в течение указанной реакции является количественным и полученный продукт используют на следующей стадии без дополнительной очистки. 5-(3-Метил-1 Н-пиразоло[3,4-b]пиридин-5-ил)-1,3,4-тиадиазол-2-амин. Исходный продукт N-(5-(3-метил-1H-пиразоло[3,4-b]пиридин-5-ил)-1,3,4-тиадиазол-2-ил)ацетамид(60 мг, 0,18 ммоль) и 20 мл 10% раствора HCl смешивают в круглодонной колбе, объемом 100 мл. Полученную смесь нагревают в течение часа. Реакционную смесь охлаждают и нейтрализуют с помощью 20% раствора гидроксида натрия, пока раствор не станет щелочным. Раствор распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой промывают раствором рассола и сушат с помощью сульфата натрия. Полученный продукт (50 мг, выход = 90%) используют на следующей стадии без дополнительной очистки. И наконец, 5-(3-метил-1H-пиразоло[3,4-b]пиридин-5-ил)-1,3,4-тиадиазол-2-амин превращают в соединение 112 в соответствии с методикой, аналогичной методике, показанной на схеме 4. Пример 113. 5-(4-Метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения 88, используя коммерчески доступный 4-бром-2,3-диметиланилин в качестве исходного продукта. После стадии ацилирования N-(4-бром-2,3-диметилфенил)ацетамид обрабатывают изоамилонитритом, затем 10% водным раствором HCl, чтобы получить на выходе 5-бром-4-метил-1H-индазол. 5 Бром-4-метил-1 Н-индазол обрабатывают трет-бутиллитием и диоксидом углерода, чтобы получить на выходе 4-метил-1 Н-индазол-5-карбоновую кислоту, которую в свою очередь обрабатывают тиосемикарбазидом и полифосфорной кислотой для образования 5-(4-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2 амина.MS (API-ES) м/е (%): 232 (100%, М 1). Соединения по примерам 114-116 получают в соответствии с методикой аналогичной методике, показанной на схеме 4, используя соединение 113 в качестве исходного продукта. Пример 114. N-S)-2-Амино-3-(4-(трифторметил)фенил)пропил)-5-(4-метил-1 Н-индазол-5-ил)1,3,4-тиадиазол-2-амин. Пример 117. 5-(3-Циклопропил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике, показанной на схеме 3. В начальной стадии используют циклопропилмагнийбромид вместо метилмагнийбромида. Соединения по примерам 118-120 получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 4, используя соединение 117 в качестве исходного продукта. Пример 118. N-S)-2-Амино-3-фенилпропил)-5-(3-циклопропил-1H-индазол-5-ил)-1,3,4-тиадиазол 2-амин. Пример 121. 5-(3-Фенил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 3, за исключением того, что на начальной стадии используют фенилмагнийбромид вместо метилмагнийбромида.MS (API-ES) м/е (%): 294 (100%, М 1). Соединения по примерам 122-124 получают в соответствии с методикой, аналогичной методике получения, показанной на схеме 4, используя соединение 117 в качестве исходного продукта. Пример 122. N-S)-2-Амино-4-метилпентил)-5-(3-фенил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Синтез соединения 125 представлен на схеме 8. Схема 8 4-Фтор-3-пропионилбензойную кислоту синтезируют путем обработки коммерчески доступной 3 бром-4-фторбензойной кислоты 1-этоксипроп-1-еном (6 экв.), ацетатом палладия (0,03 экв.), 1,3-бис(дифенилфосфино)пропаном (0,06 экв.) и карбонатом калия (1,2 экв.) в ДМФА и воде при микроволновом излучении при температуре 130 С в течение 3 ч с последующей обработкой кислотой. 3-Этил-1Hиндазол-5-карбоновую кислоту получают путем обработки 4-фтор-3-пропионилбензойной кислоты гидразином при микроволновом излучении при температуре 160 С в течение 30 мин. 5-(3-Этил-1H-индазол 5-ил)-1,3,4-тиадиазол-2-амин получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 3 с помощью POCl3 и тиосемикарбазида.MS (API-ES) м/е (%): 246 (100%, М 1). Пример 126. N-S)-2-Амино-3-фенилпропил)-5-(3-этил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Указанное соединение получают в соответствии с методикой, аналогичной методике, показанной на схеме 4, за исключением того, что используют соединение 125 в качестве исходного продукта. Соединения по примерам 127-128 синтезируют в соответствии с методикой, аналогичной методике получения, показанной на схеме 4. Пример 127. N-S)-2-амино-5-фенилпентил)-5-(3-метил-1H-индазол-5-ил)-1,3,4-тиадиазол-2-амин. Соединения по примерам 129-134 получают в соответствии с общей методикой, в которой используют одновременно LAH для восстановления амидной связи и Вос группу при температуре 60 С вместо температуры 0 С, как показано на схеме 4. Сырой продукт (1 ммоль), образующийся при формировании амидной связи, как показано на схеме 4, растворяют в 5 мл ТГФ и добавляют 6 мл LAH (1M в ТГФ). Реакционную смесь нагревают в герметичной колбе до температуры 60 С в течение 2 ч и после этого, охлаждают до комнатной температуры. Реакционную смесь разбавляют 20 мл ТГФ и выливают в смесь 10 гNa2SO4 10 Н 2 О в 30 мл ТГФ, после чего перемешивание продолжают в течение 20 мин. Реакционную смесь фильтруют, промывают, сушат над MgSO4 и концентрируют. Продукт реакции очищают с помощью препаративной ТСХ (12% МеОН в CH2Cl2). Пример 129. N-S)-3-(4-Хлорфенил)-2-(метиламино)пропил)-5-(3-метил-1H-индазол-5-ил)-1,3,4 тиадиазол-2-амин.FTMS Вычислено (М+Н) 433,1417; Найдено 433,1411. Соединения по примерам 135-144 получают, используя соединение 37 в качестве исходного продукта через упрощенную методику алкилирования. (S)-N-[5-(3-метил-1H-индазол-5-ил)-[1,3,4]тиадиазол 2-ил]-3-(3-трифторметилфенил)пропан-1,2-диамин (15 мг) растворяют в 0,1 мл МеОН и добавляют 5 капель (из Пастеровкой пипетки) АсОН. Затем добавляют 6 экв. карбонильного соединения и перемешивание продолжают в течение 30 мин. Добавляют 3 экв. Na(OAc)3BH и перемешивание продолжают в течение ночи. Реакционную смесь или загружают без дополнительной очистки на препаративную ТСХ пластину или используют препаративную HPLC для очистки. Пример 135. Хлорид N-изопропил-1-(5-(3-метил-1 Н-индазол-5-ил)-1,3,4-тиадиазол-2-иламино)-3-(3(трифторметил)фенил)пропан-2-аминия.MS (API-ES) м/е (%): 513 (100%, М 1). Соединения по примерам 145-155 получают в соответствии с методикой, аналогичной методике получения соединения, показанной на схеме 4 за исключением того, что 5-изохинолин-6-ил[1,3,4]тиадиазол-2-ил-амин (см. схему 1) используют в качестве исходного продукта. Эти соединения получают после связывания 5-изохинолин-6-ил-[1,3,4]тиадиазол-2-иламина с соответствующими Вос защищенными аминокислотами, с последующим LAH восстановлением, а затем обработкой кислотой,чтобы удалить Вос группу. Пример 145. N-S)-2-Амино-3-(4-метоксифенил)пропил)-5-(изохинолин-6-ил)-1,3,4-тиадиазол-2 амин.
МПК / Метки
МПК: C07D 285/12, A61K 31/433
Метки: применение, тиадиазола, соединения
Код ссылки
<a href="https://eas.patents.su/30-12181-soedineniya-tiadiazola-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения тиадиазола и их применение</a>
Фармкомпозиция, содержащая соединения с анти-ха активностью и соединения антагониста агрегации тромбоцитов, их применение, набор, содержащий эти соединения, способ лечения или профилактики заболеваний, сопутствующих тромбообразованию

Номер патента: 2475
Опубликовано: 27.06.2002
Авторы: Данвидди Кристофер Т., Юзан Андре, Перроне Марк Х., Лидли Роберт Дж., Кюродо Алан Х.
МПК: A61K 31/715, A61P 7/02
Метки: набор, профилактики, содержащая, сопутствующих, фармкомпозиция, соединения, тромбоцитов, анти-ха, тромбообразованию, активностью, заболеваний, применение, содержащий, антагониста, эти, способ, лечения, агрегации
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически пригодный носитель и фармацевтически эффективные количества соединения, обладающего анти-Ха активностью, и соединения антагониста агрегации тромбоцитов. 2. Фармацевтическая композиция по п.1, отличающаяся тем, что соединение, обладающее анти-Ха активностью, является низкомолекулярно весовым гепарином. 3. Фармацевтическая композиция по п.2, отличающаяся тем, что низкомолекулярно весовой...
Диароматические соединения пропинила или диенила, промежуточные соединения, фармацевтические и косметические композиции на основе указанных соединений и применение косметических композиций.

Номер патента: 1060
Опубликовано: 30.10.2000
Автор: Бернардон Жан-Мишель
МПК: A61P 17/00, A61K 31/192, C07C 63/66...
Метки: пропинила, основе, фармацевтические, диенила, композиций, композиции, промежуточные, косметические, применение, соединений, соединения, косметических, указанных, диароматические
Формула / Реферат:
1. Диароматические соединения пропинила или диенила общей формулы (I) где R1 является (I) радикалом -СН3, (II) радикалом -СН2-O-R6, (III) радикалом -О-R6, (IV) радикалом -CO-R7, где R6 и R7 имеют значения, приведенные ниже, Аr является радикалом, отвечающим следующей формуле (а): где R5 имеет значения, приведенные ниже, Х является радикалом формулы или или где R8 и R9 имеют значения, приведенные ниже, R2 и R3,...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Кроуелл Томас А., Брайант Генри У., Джонс Чарльз Д., Палковиц Алан Д.
МПК: A61K 31/33, A61P 19/10, C07C 47/546...
Метки: способ, соединения, снижения, соединений, нафтильные, холестерина, промежуточные, получения, нафтильных, применение
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Аминокислотные соединения и их применение в качестве ингибиторов nep, ace и ece

Номер патента: 5004
Опубликовано: 28.10.2004
Авторы: Скальбер Элизабет, Беннжан Каролина, Фурнье-Залуски Мари-Клод, Ренар Пьер, Ингимбер Никола, Рок Бернар П., Пора Эрве
МПК: A61K 31/33, C07D 209/20, A61P 43/00...
Метки: применение, соединения, качестве, аминокислотные, ингибиторов
Формула / Реферат:
1. Соединения формулы (I) в которой n представляет собой коэффициент, в котором 0_n_3, m представляет собой коэффициент, в котором 0_m_6, R3 и R4 совместно образуют с двумя атомами углерода, которые их несут, фенильную группу, которая незамещена или замещена от 1 до 3 одинаковыми или различными группами, выбираемыми из алкила, алкенила, алкинила, алкокси, гидрокси, алкилтио, меркапто, циано, нитро, амино, алкиламино, диалкиламино,...
Арилсульфонамидные соединения и их применение для лечения ожирения, диабета типа ii

Номер патента: 9647
Опубликовано: 28.02.2008
Авторы: Тейбрант Ян, Сутин Лори, Йоханссон Гари, Кальдирола Патриция, Йенсен Анника, Мотт Эндрю, Бремберг Ульф
МПК: C07D 211/46, A61K 31/495, A61K 31/551...
Метки: соединения, применение, арилсульфонамидные, диабета, лечения, ожирения, типа
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, где X означает R1 и R3 независимо означают: (a) Н; (b) C1-6алкил; (c) C1-6алкокси; (d) C1-6гидроксиалкил с прямой или разветвленной цепью; (e) C1-6алкилгалогениды с прямой или разветвленной цепью или (f) группу Ar; Ar означает: (a) фенил; (b) 1-нафтил; (c) 2-нафтил; (d) 5-7-членное, частично или полностью насыщенное, гетероциклическое кольцо, содержащее 1-4 гетероатома,...
Предыдущий патент: Способ получения арипипразола и соответствующие промежуточные продукты и их получение
Следующий патент: Электропорация микобактерии и сверхэкспрессия антигенов микобактерий
Случайный патент: Фармацевтическая композиция для пероральной доставки дииндолилметана