Производные &beta -d-нуклеозида и их применение





















Формула / Реферат
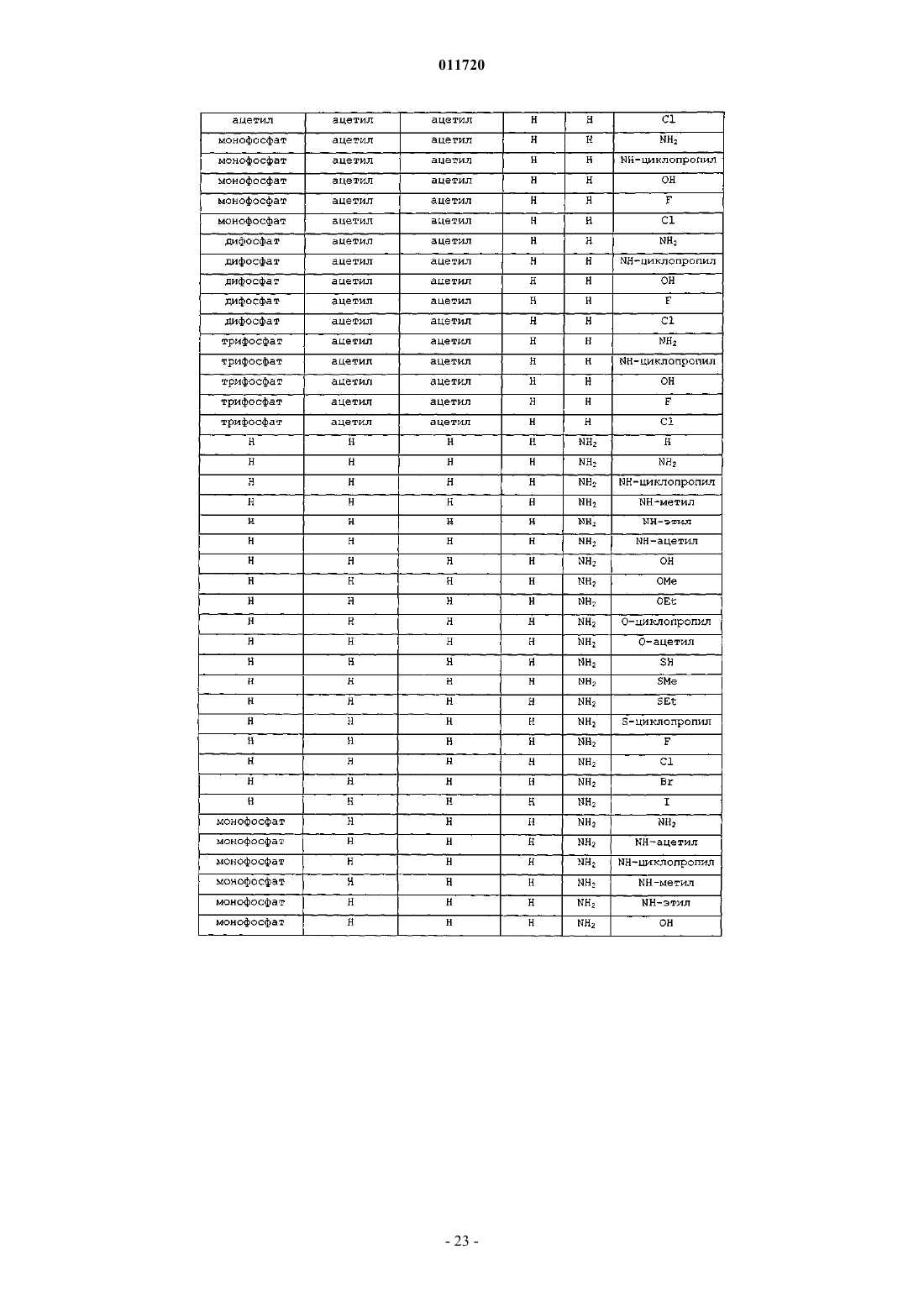
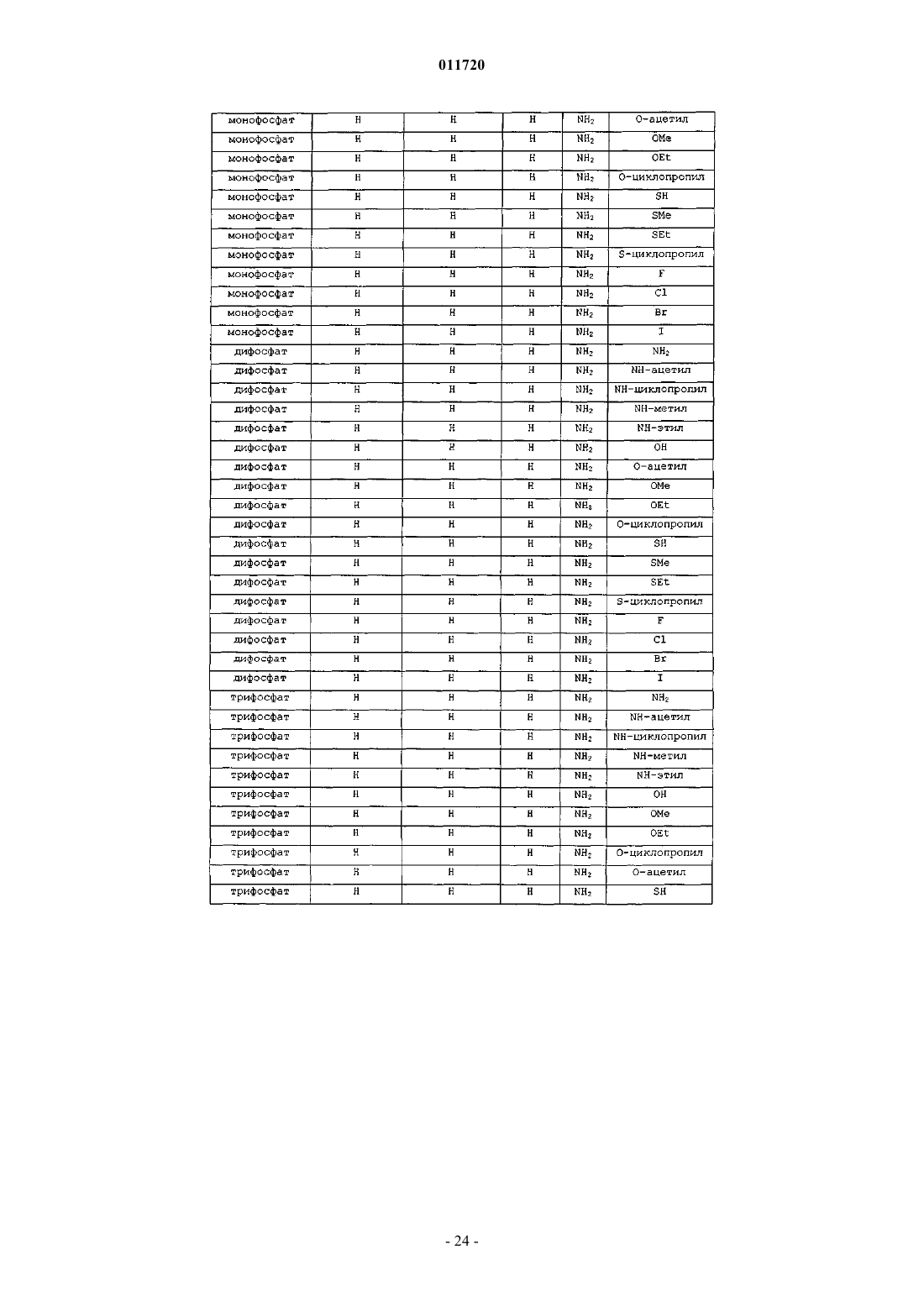
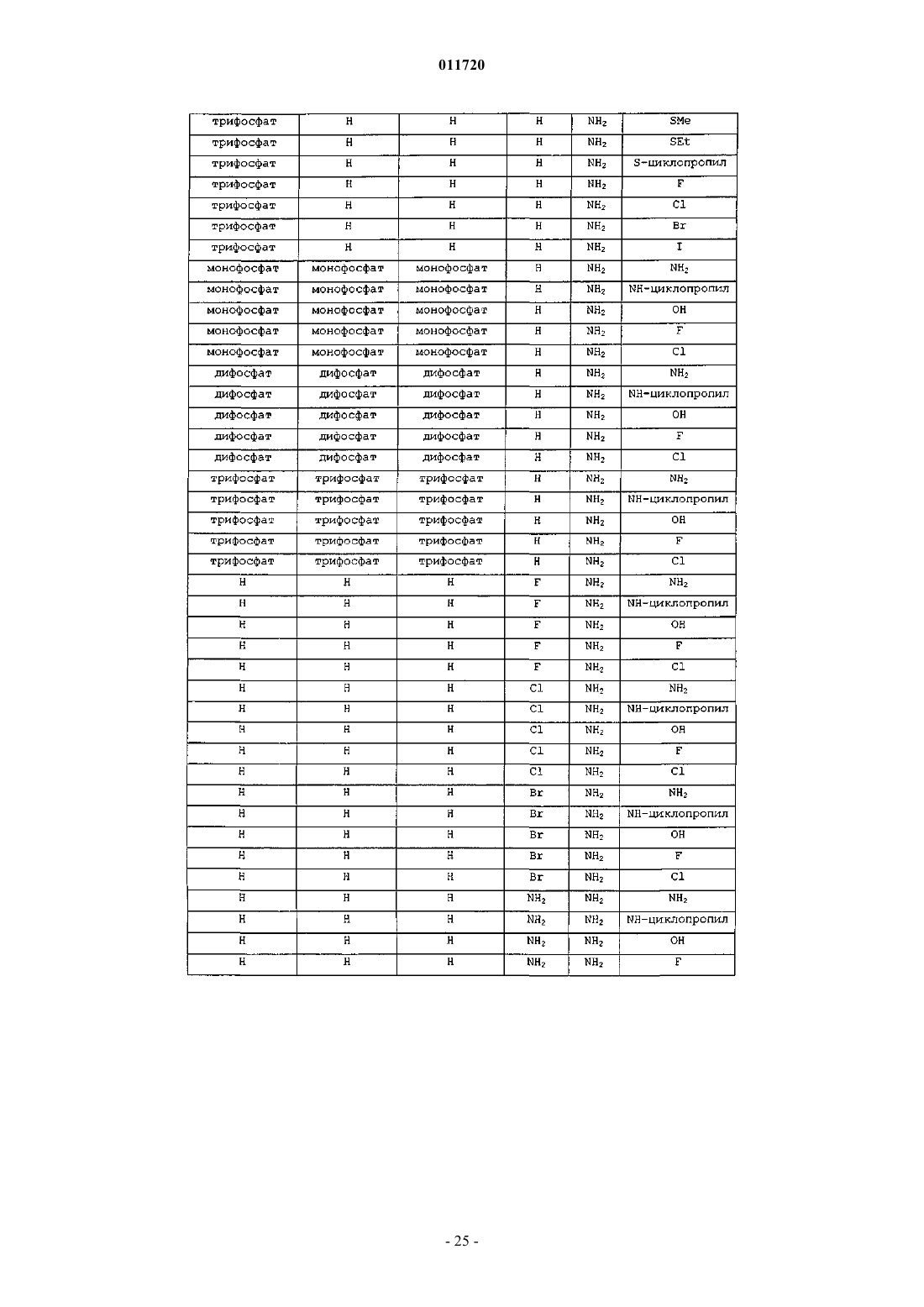
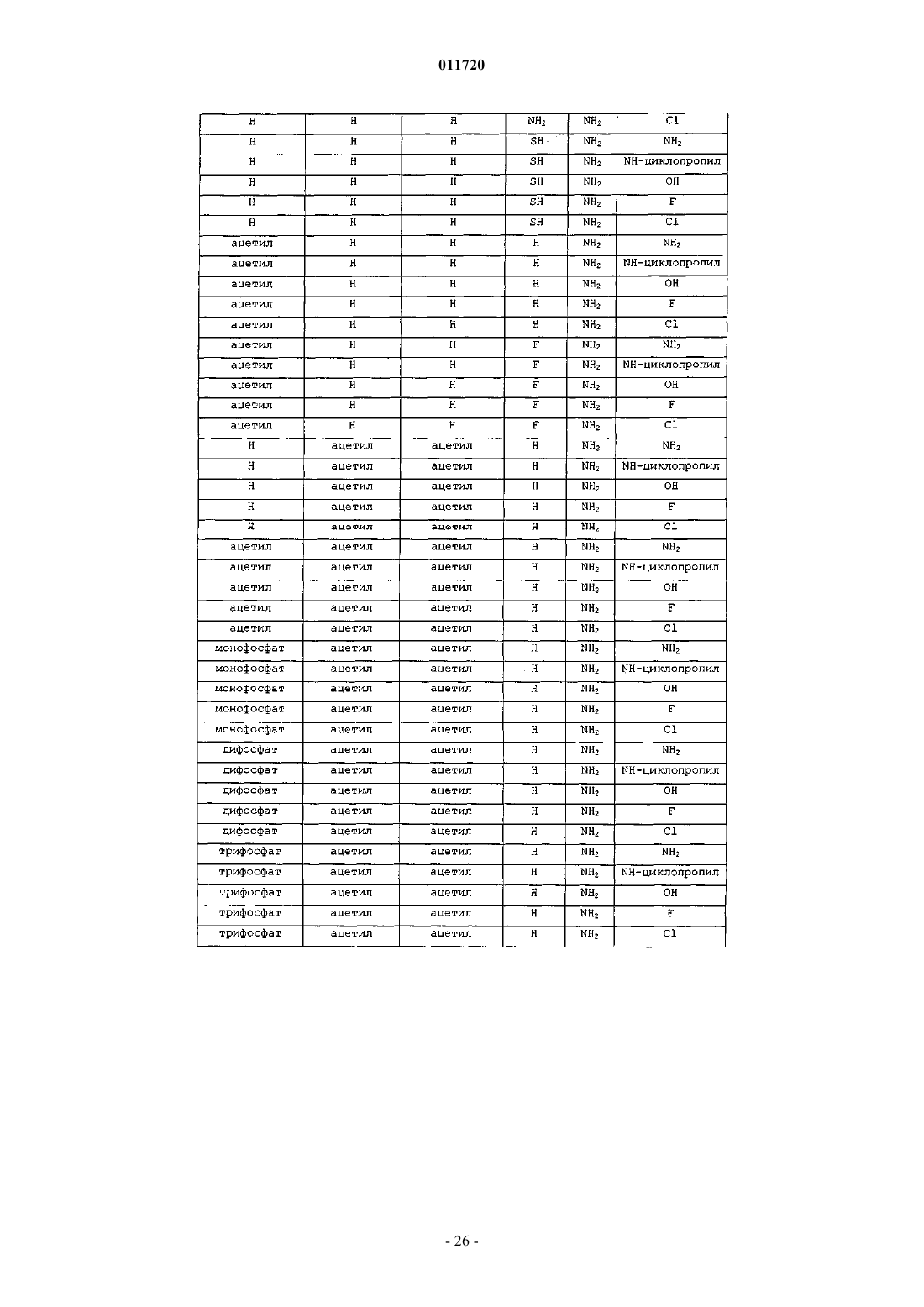
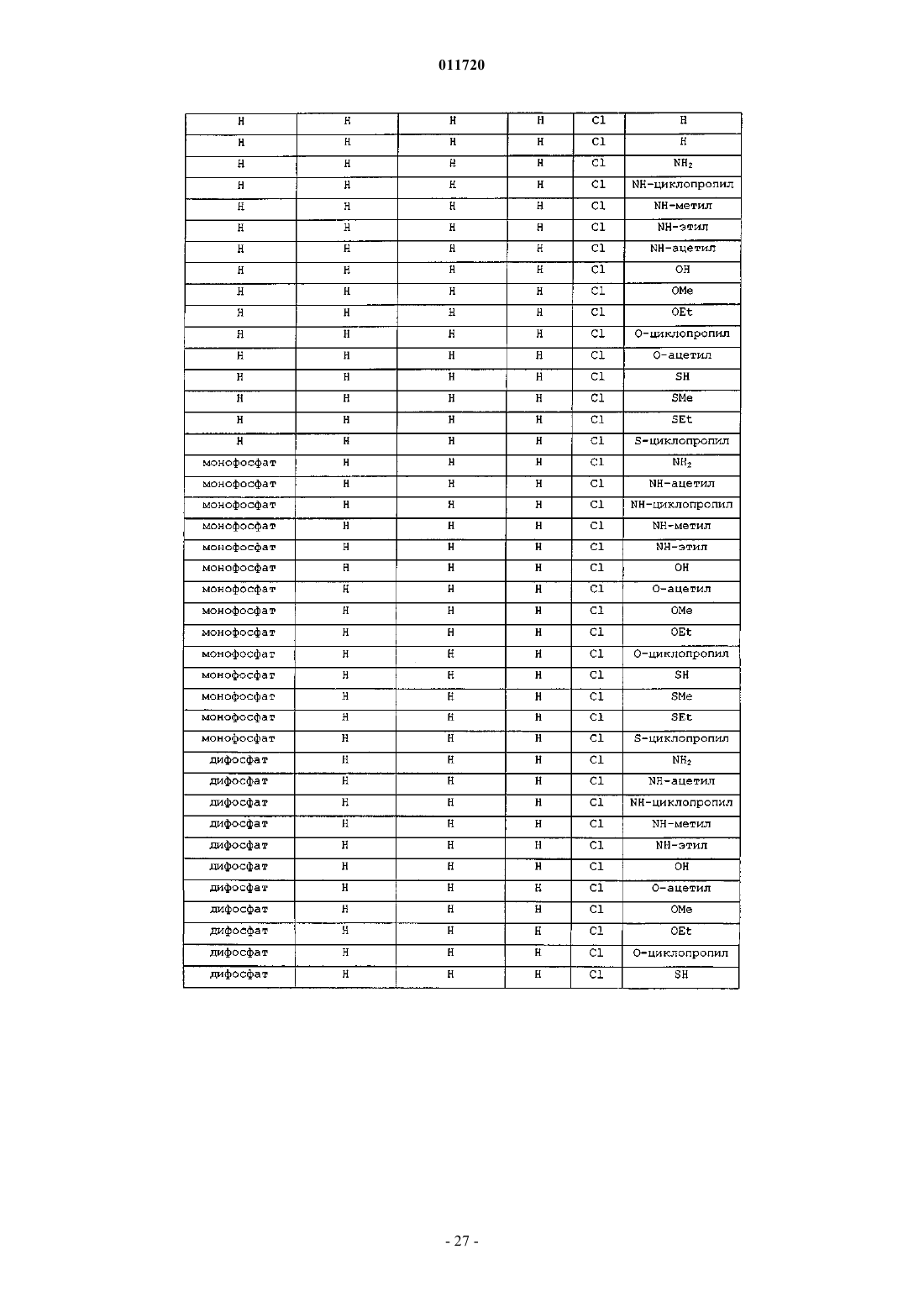
1. Соединение формулы XVII:

или его фармацевтически приемлемая соль, сложный эфир или фосфат,
где "основание" представляет собой пирролпиримидин;
R1 представляет собой Н, фосфат или С1-С10ацил;
R6 представляет собой C1-С10алкил;
R7 и R9 независимо представляют водород, OR1, гидрокси или -NH(C1-С10ацил), -N(С1-С4алкил)2, -N(C1-С10ацил)2;
R10 представляет собой Н, C1-С10алкил, хлор, бром или иод и
X представляет собой О.
2. Соединение по п.1 или его фармацевтически приемлемая соль, сложный эфир или фосфат,
где R1 представляет собой Н или фосфат;
R6 представляет собой C1-С10алкил;
R7 и R9 независимо представляют собой OR1;
R10 представляет собой Н;
X представляет О.
3. Соединение по п.2 или его фармацевтически приемлемая соль, сложный эфир или фосфат, где X означает О.
4. Соединение по п.2 или 3 или его фармацевтически приемлемая соль, сложный эфир или фосфат, где R1 означает водород.
5. Соединение по любому из пп.2-4 или его фармацевтически приемлемая соль, сложный эфир или фосфат, где R6 означает метил.
6. Лекарственное средство, содержащее соединение по любому из пп.1-5 или его фармацевтически приемлемую соль, сложный эфир или фосфат.
7. Лекарственное средство по п.6, где указанное соединение или его фармацевтически приемлемая соль, сложный эфир или фосфат представлены в виде стандартной лекарственной формы.
8. Лекарственное средство по п.7, где указанная стандартная лекарственная форма содержит 10-1500 мг указанного соединения.
9. Лекарственное средство по п.7, где указанной стандартной лекарственной формой является таблетка или капсула.
10. Лекарственное средство по любому из пп.6-8, дополнительно содержащее один или более других антивирусных агентов.
11. Лекарственное средство по п.10, где другой антивирусный агент представляет собой агент против вируса гепатита С.
12. Лекарственное средство по п.11, где агент против вируса гепатита С выбран из группы, включающей интерферон, рибавирин, ингибитор протеазы, производное тиазолидина, ингибитор полимеразы и ингибитор геликазы.
13. Лекарственное средство по п.11, где агент против вируса гепатита С представляет собой интерферон.
14. Лекарственное средство по п.12, где агент против вируса гепатита С представляет собой рибавирин.
15. Применение соединения по любому из пп.1-5, или его фармацевтически приемлемой соли, или фосфата для изготовления лекарственного средства для лечения хозяина, инфицированного вирусом гепатита С.
16. Применение по п.15, где хозяин представляет собой человека.
Текст
011720 Область, к которой относится изобретение Настоящее изобретение относится к области фармацевтической химии, а в частности к соединению,способу и композиции для лечения вирусного гепатита С. В настоящей заявке испрашивается приоритет предварительной заявки США 60/206585, поданной 23 мая 2000. Предпосылки создания изобретения Вирус гепатита С (HCV) является главной причиной хронических заболеваний печени во всем мире.(Boyer, N. et al., J.Hepatol. 32:98-112, 2000). HCV вызывает медленно развивающуюся вирусную инфекцию и является главной причиной цирроза и гепатоцеллюлярной карциномы (Di Besceglie, A.M.Bacon, B.R.,Scientific American, Oct.: 80-85, (1999); Boyer, N. et al., J.Hepatol. 32:98-112, 2000). По оценкам специалистов, вирусом HCV инфицировано 170 миллионов человек во всем мире. (Воуег, N. et al. J.Hepatol. 32:98112, 2000). В США от цирроза печени, вызываемого хронической инфекцией гепатита С, ежегодно умирает 8000-12000 человек и HCV-инфекция является главным показанием к пересадке печени. Известно, что вирус HCV по крайней мере в 80% случаях вызывает посттрансфузионный гепатит, а в большинстве случаев, он вызывает спорадический острый гепатит. По предварительной оценке, HCV во многих случаях также является причиной "идиопатического" хронического гепатита, "криптогенного" цирроза и, вероятно, гепатоцеллюлярной карциномы, не ассоциированных с другими вирусами гепатита,такими как вирус гепатита В (HBV). Очевидно, что незначительное количество здоровых людей является носителями хронического HCV, и число этих людей варьируется в зависимости от географических и других эпидемиологических факторов. Это число может значительно превышать число людей с HBV, хотя эти данные являются пока предварительными, и еще неизвестно, сколько людей имеют субклиническое хроническое заболевание печени. (The Merck Manual, ch.69, p.901, 16th ed., (1992.HCV был классифицирован как член семейства Flaviviridae, которое включает вирусы рода флавивирусов, пестивирусов и гепацивирусов, которые включают вирусы гепатита С (Rice, C.M., Flaviviridae:The viruses and their replication. In: Fields Virology, Editors: Fields, B.N., Knipe, D.M.,Howley, P.M., Lippincott-Raven Publishers, Philadelphia, PA, Chapter 30, 931-959, 1996). HCV представляет собой вирус с оболочкой, содержащий позитивный геном одноцепочечной РНК, составляющий приблизительно 9,4 т.п.н. Вирусный геном состоит из 5'-нетранслируемой области (UTR), длинной открытой рамки считывания, кодирующей полипротеин-предшественник приблизительно из 3011 аминокислот, и короткой 3'UTR. 5'UTR представляет собой наиболее высококонсервативную часть генома HCV и играет важную роль для инициации и регуляции трансляции полипротеина. Трансляция генома HCV инициируется кэпнезависимым механизмом, известным как внутреннее включение рибосомы. Этот механизм заключается в связывании рибосомы с РНК-последовательностью, известной как внутренний сайт связывания с рибосомой (IRES). Недавно было установлено, что псевдоузловая структура РНК-последовательности является главным структурным элементом IRES HCV. Вирусные структурные белки включают нуклеокапсидный коровый белок (С) и два оболочечных гликопротеина, Е 1 и Е 2. HCV также кодирует две протеиназы, цинкзависимую металлопротеиназу, кодируемую NS2-NS3-областью и сериновую протеиназу, кодируемую в NS3-области. Эти протеиназы необходимы для расщепления специфических областей полипротеина-предшественника с образованием зрелых пептидов. Карбоксильная половина неструктурного белка 5, NS5B, содержит РНК-зависимую РНК-полимеразу. Функции остальных неструктурных белков,NS4A и NS4B и белка NS5A (аминоконцевой половины неструктурного белка 5) пока не известны. Современные исследования по противовирусной терапии направлены, главным образом, на разработку улучшенных способов лечения хронических HCV-инфекций у человека (Di Besceglie, A.M.Bacon, B.R., Scientific American, Oct.: 80-85, (1999. В настоящее время имеются два главных протививирусных соединения, рибавирин и интерферон-альфа, которые используются для лечения хроническихHCV-инфекций у человека. Лечение HCV-инфекций рибавирином Рибавирин (1 рибофуранозил-1-1,2,4-триазол-3-карбоксамид) представляет собой синтетический не индуцирующий интерферон противовирусный нуклеозидный аналог широкого спектра, имеющийся в продаже под торговым знаком "виразол" (The Merck Index, 11th edition, Editor: Budavari, S., MerckCo.,Inc., Rahway, NJ, pl304, 1989). В патенте США 3798209 и в RE29835 описан и заявлен рибавирин. По своей структуре, рибавирин аналогичен гуанозину и обладает in vitro активностью против нескольких ДНК- и РНК-вирусов, включая Flaviviridae (Gary L. Davis. Gastroenterology 118:S104-S114, 2000). У 40% пациентов рибавирин снижает сывороточные уровни аминотрансферазы до нормальных, но не снижает сывороточные уровни HCV-PHK (Gary L. Davis. Gastroenterology 118:S104-S114, 2000). Таким образом, рибавирин, взятый отдельно, не эффективен в снижении уровней вирусной РНК. Кроме того, рибавирин обладает значительной токсичностью, и известно, что он вызывает анемию. Лечение HCV-инфекций интерфероном Интерфероны (IFN) представляют собой соединения, которые являются коммерчески доступными и уже почти 10 лет используются для лечения хронического гепатита. IFN представляют собой гликопротеины, продуцируемые иммунными клетками в ответ на вирусную инфекцию. IFN ингибируют репликацию множества вирусов, включая HCV, и при их использовании как единственного средства для лечения инфекций гепатита С, IFN способствуют снижению сывороточной HCl-РНК до недетектируемых уров-1 011720 ней. Кроме того, IFN нормализует уровни аминотрансферазы в сыворотке. К сожалению, действие IFN является кратковременным, и продолжительный ответ наблюдается лишь у 8-9% пациентов с хронической HCV-инфекцией (Gary L. Davis. Gastroenterology 118:S104-S114, 2000). Описано HCV-лечение с использованием интерфероновой терапии у ряда пациентов. Так, например, в патенте США 5980884, Blatt et al., описаны методы повторного лечения HCV-инфицированных пациентов с использованием "канонического" интерферона. В патенте США 5942223, Bazer et al., описана анти-HCV-терапия с использованием овечьего или бычьего интерферона-тау. В патенте США 5928636, Alber et al., описана комбинированная терапия с использованием интерлейкина-12 и интерферона-альфа для лечения инфекционных заболеваний, включая HCV. В патенте США 5908621, Glue etal., описано использование модифицированного полиэтиленгликолем интерферона для лечения HCVинфекций. В патенте США 5849696, Chreitien et al., описано использование тимозинов, взятых отдельно или в комбинации с интерфероном, для лечения HCV-инфекций. В патенте США 5830455, Valtuena et al., описана комбинированная HCV-терапия с использованием интерферона и акцептора свободных радикалов. В патенте США 5738845, Imakawa, описано использование белков человеческого интерферона-тау для лечения HCV-инфекций. Другие методы лечения HCV-инфекций с использованием интерферона описаны в патенте США 5676942, Testa et al., в патенте США 5372808, Blatt et al., и в патенте США 5849696. Комбинация интерферона и рибавирина Сообщалось, что комбинация IFN и рибавирина для лечения HCV-инфекции эффективна для лечения пациентов, не подвергавшихся ранее лечению IFN (Battaglia, A.M. et al., Ann. Pharmacother. 34:487494, 2000). Лечение этой комбинацией дало многообещающие результаты в случае, когда она использовалась до развития гепатита или когда имелось гистологическое подтверждение заболевания (Berenguer,M. et al. Antivir. Ther. 3(Suppl. 3):125-136, 1998). Побочными эффектами комбинированной терапии являются гемолиз, симптомы, подобные гриппу, анемия и усталость.(Gary L. Davis. Gastroenterology 118:S104-S114, 2000). Дополнительное описание методов лечения HCV-инфекций Обзор ряда методов HCV-терапии дан в работе Bymock et al., Antiviral ChemistryChemotherapy,11:2; 79-95 (2000). Описание нескольких субстратных ингибиторов протеаз NS3, в которых расщепляемая амидная связь гидролизованного субстрата заменена электрофилом, который взаимодействует с каталитическим серином, можно найти в литературе. Attwood et al. (1998) Antiviral peptide derivatives, 98/22496; Attwoodamino acid derivatives as antiviral agents, публикация патента Германии DE 19914474; Tung et al. (1998)Inhibitors of serine proteases, particularly hepatitis С virus NS3 protease, WO 98/17679. Описанные ингибиторы в своем концевом положении имеют электрофил, такой как бороновая кислота или фосфонат. Llinas-Brunet et al. (1999) Hepatitis C inhibitor peptide analogues, WO 99/07734. Было описано два класса ингибиторов на основе электрофилов, таких как альфакетоамиды и гидразономочевины. В литературе также описан ряд несубстратных ингибиторов. Так, например, была проведена оценка ингибирующего действия 2,4,6-тригидрокси-3-нитробензамидных производных на протеазу HCV и другие сериновые протеазы. Sudo K. et al., (1997) Biochemical and Biophysical Research Communications,238:643-647; Sudo K. et al. (1998) Antiviral Chemistry and Chemotherapy, 9:186. В анализе с использованием обращенно-фазовой ВЭЖХ были идентифицированы два наиболее эффективных соединения RD34082 и RD3-4078, где первое соединение замещено у амида с цепью из 14 атомов углерода, а второе имеет парафеноксифенильную группу. В анализе, проводимом посредством обращенно-фазовой ВЭЖХ с использованием гибридного белка NS3/4A и субстрата NS5A/5B, были идентифицированы триазолидиновые производные как микромолярные ингибиторы. Sudo, K. et al. (1996) Antiviral Research 32:9-18. Соединение RD-1-6250, имеющее конденсированную циннамоильную группу, замещенную длинной алкильной цепью, обладало наиболее сильным действием против выделенного фермента. Двумя другими примерами активных соединений являются RD4-6205 и RD4-6193. В других работах описан скрининг относительно небольшой библиотеки с использованием ELISAанализа и идентификация трех соединений как сильных ингибиторов, тиазолидина и двух бензанилидов.Kakiuchi N. et al. J. EBS Letters 421:217-220; Takeshita N. et al., Analytical Biochemistry 247:242-246, 1997. В нескольких патентах США описаны ингибиторы, используемые для лечения HCV-инфекций. Так, например, в патенте США 6004933, Spruce et al., описан класс ингибиторов цистеиновых протеаз, ингибирующих HCV-эндопептидазу 2. В патенте США 5990276, Zhang et al., описаны синтетические ингибиторы протеазы NS3 вируса гепатита С. Этот ингибитор представляет собой подпоследовательность субстрата протеазы NS3 или субстрата кофактора NS4A. Использование рестриктирующих ферментов для лечения HCV описано в патенте США 5538865, Reyes et al. Выделенный из бульона ферментационной культуры Streptomyces sp., Sch 68631, фенан-тренехинон обладал микромолекулярной активностью против протеазы HCV в анализе посредством электрофореза в ДСН-ПААГ и авторадиографии. Chu M. et al., Tetrahedron Letters 37:1229-7232, 1996. В другом примере-2 011720 тех же авторов, Sch 351633, выделенный из грибов Penicillium griscofuluum обнаруживал микромолярную активность в сцинтилляционном проксимальном анализе. Chu M. et al., Bioorganic and MedicinalChemistry Letters 9:1949-1952. Наномолярная активность против фермента протеазы NS3 HCV была достигнута путем конструирования селективных ингибиторов на основе макромолекулярного эглина с. Эглин с, выделенный из пиявок, является сильным ингибитором нескольких сериновых протеаз, таких как протеазы S. griseus А и В, -химотрипсин, химаза и субтилизин. Qasim M.A. et al., Biochemistry 36:15981607, 1997. Были также описаны ингибиторы геликазы HCV. Патент США 5633358, Diana G.D. et al.; публикация PCTWO 97/36554, Diana G.D. et al. Имеется несколько описаний ингибиторов полимеразыal., Journal of Virology 73:1649-1654, 1999; Lohmann V. et al., Virology 249:108-118, 1998. Сообщалось, что антисмысловые фосфортиоат-олигодезоксинуклеотиды, которые комплементарны последовательности, находящейся в 5'некодирующей области HCV, являются эффективными ингибиторами экспрессии гена HCV при in vitro-трансляции и в системах клеточных культур с IICV-люциферазой IIcpG2.Alt M. et al., Hepatology 22:707-717, 1995. В недавно опубликованной работе было продемонстрировано, что нуклеотиды 326-348, содержащие 3'-конец NCR, и нуклеотиды 371-388, локализованные в коровой кодирующей области РНК HCV, являются эффективными мишенями для опосредованного антисмысловым олигонуклеотидом ингибирования трансляции вируса. Alt M. et al., Archives of Virology 142: 589-599, 1997. В патенте США 6001990, Wands et al., описаны олигонуклеотиды, ингибирующие репликацию HCV. В публикации РСТWO 99/29350 описаны композиции и способы лечения инфекционного гепатита С, предусматривающие введение антисмысловых олигонуклеотидов, которые являются комплементарными HCV-PHK и гибридизуются с нею. В патенте США 5922857, Han et al., описаны нуклеиновые кислоты, соответствующие последовательности области гомологии пестивируса, т.е. боксу IV, регулирующей трансляцию HCV. Недавно были также описаны антисмысловые олигонуклеотиды, используемые в качестве терапевтических агентов(Galderisi U. et al., Journal of Cellular Physiology 181:251-257, 1999). Были также описаны другие соединения, являющиеся ингибиторами IRES-зависимой трансляции вHCV. Публикация японского патента JP-08268890, Ikeda N. et al.; публикация Японского патента JP10101591, Kai Y. et al Нуклеазо-резистентные рибозимы нацелены на IRES и, как недавно сообщалось,действуют как ингибиторы в анализе на образование бляшек химерой "HCV-полиовирус". Maccjak D.J. etal., Hepatology 30 abstract 995, 1999. Использование рибозимов для лечения HCV также описано в патенте США 6043077, Barber et al. и в патенте США 5869253 и 5610054, Draper et al. В других патентах описано использование соединений, стимулирующих иммунную систему, для лечения HCV-инфекций. Так, например, в патенте США 6001799, Chretien et al., описан метод лечения гепатита С у не восприимчивых к интерферону пациентов путем введения этим пациентам дозы тимозина или его фрагмента, стимулирующей иммунную систему. В патенте США 5972347, Eder et al., и в патенте США 5969109, Bona et al., описано лечение HCV-инфекции с использованием антител. В патенте США 6034134, Gold et al., описаны некоторые агонисты рецепторов NMDA, обладающих иммуномодулирующей противомалярийной активностью, направленной против вируса Борна и против гепатита С. Описанные агонисты рецепторов NMDA принадлежат к семейству 1-аминоалкилциклогексанов. В патенте США 6030960, Morris-Natschke et al., описано использование некоторых алкиллипидов для ингибирования продуцирования индуцируемых гепатитом антигенов, включая антигены, продуцируемые вирусом HCV. В патенте США 5922757, Chojkier et al., описано использование витамина Е и других антиоксидантов для лечения заболеваний печени, включая HCV-инфекции. В патенте США 5858389, Elsherbi et al., описано использование сквалена для лечения гепатита С. В патенте США 5849800, Smith et al., описано использование амантадина для лечения гепатита С. В патенте США 5846964, Ozeki et al., описано использование желчных кислот для лечения HCV-инфекций. В патенте США 5491135, Blough et al., описано использование N-(фосфоноацетил)-L-аспарагиновой кислоты для лечения инфекций, вызываемых флавивирусами, такими как HCV. Другими соединениями, предлагаемыми для лечения HCV-инфекций, являются растительные экстракты(патент США 5837257, Tsai et al., патент США 5725859, Omer et al., и патент США 6056961), пиперидины (патент США 5830905, Diana et al.), бензолдикарбоксамиды (патент США 5633388, Diana et al.),производные полиадениловых кислот (патент США 5496546, Wang et al.), 2',3'-дидезоксиинозин (патент США 5026687, Yarchoan et al.) и бензимидазолы (патент США 5891874, Colacino et al). Если принять во внимание тот факт, что вирус гепатита С вызывает инфекции, достигающие уровня эпидемий во всем мире, и приводит к трагическим последствиям для инфицированных пациентов, то крайняя необходимость в получении новых эффективных фармацевтических средств для лечения гепатита С, не обладающих токсичностью по отношению к хозяину, остается актуальной. Поэтому, целью настоящего изобретения является получение соединения, способа и композиции для лечения хозяина, инфицированного вирусом гепатита С. Краткое описание изобретения Описаны соединения и композиции для лечения инфекционного гепатита С, где указанное лечение-3 011720 предусматривает использование эффективного для лечения гепатита С количества -D-нуклеозида формул (I)-(XVIII) или его фармацевтически приемлемой соли или пролекарства. Настоящее изобретение относится к соединению формулы XVII или его фармацевтически приемлемой соли, сложному эфиру или фосфату где "основание" представляет собой пирролпиримидин;R10 представляет собой Н, C1-С 10 алкил, хлор, бром или иод иX представляет собой О. В предпочтительном варианте R1 представляет собой Н или фосфат, предпочтительно Н; R6 представляет собой C1-С 10 алкил, предпочтительно метил; R7 и R9 независимо представляют собой OR1; R10 представляет собой Н и X представляет O. В одном из вариантов предложенного изобретения соединение формулы XVII или его фармацевтически приемлемую соль или фосфат используют для изготовления лекарственного средства для лечения хозяина, инфицированного вирусом гепатита С, где хозяин предпочтительно представляет собой человека. Таким образом, настоящее изобретение также относится к лекарственному средству, содержащему соединение формулы XVII или его фармацевтически приемлемую соль, сложный эфир или фосфат. В предпочтительном варианте соединение формулы XVII или его фармацевтически приемлемая соль, сложный эфир или фосфат представлены в виде стандартной лекарственной формы, которая предпочтительно содержит 10-1500 мг указанного соединения. Указанная стандартная лекарственная форма может представлять собой таблетку или капсулу. В предпочтительном варианте лекарственное средство может дополнительно содержать один или более других антивирусных агентов. Предпочтительно другой антивирусный агент представляет собой агент против вируса гепатита С, где указанный агент против вируса гепатита С выбирают из группы,включающей интерферон, рибавирин, ингибитор протеазы, производное тиазолидина, ингибитор полимеразы и ингибитор геликазы. Наиболее предпочтительно агент против вируса гепатита С представляет собой интерферон или рибавирин.-D-нуклеозиды настоящего изобретения могут ингибировать HCV-полимеразную активность. Нуклеозиды могут быть скринированы на их способность ингибировать HCV-полимеразную активностьin vitro в соответствии с методами скрининга, которые более конкретно описаны ниже. Спектр активности может быть легко определен путем оценки указанного соединения в анализах, описанных ниже, или в других подтверждающих анализах. В одном из вариантов осуществления изобретения эффективность анти-HCV соединения измеряли по концентрации соединения, необходимой для снижения числа вирусных бляшек in vitro на 50% (т.е., ES50 данного соединения), методами, более конкретно описанными ниже. В предпочтительных вариантах осуществления изобретения, ЕС 50 указанного соединения составляет менее чем 25, 15, 10, 5 или 1 мкмоль. В другом варианте осуществления изобретения активное соединение может быть введено в комбинации или поочередно с другим анти-HCV агентом. При комбинированной терапии эффективную дозу двух или нескольких агентов вводят вместе, а при альтернативной терапии эффективную дозу каждого агента вводят поочередно. Указанные дозы зависят от абсорбции, инактивации и экскреции лекарственного средства, а также от других известных факторов. Следует отметить, что величины доз могут также варьироваться в зависимости от тяжести состояния, которое подвергается лечению. Кроме того, следует отметить, что для любого конкретного индивидуума конкретные схемы приема и введения лекарственного средства должны быть скорректированы в зависимости от времени в соответствии с потребностью индивидуума и в соответствии с назначением врача, осуществляющего введение или наблюдение за введением данных композиций. Неограничивающими примерами противовирусных агентов, которые могут быть использованы в комбинации с соединениями, описанными в настоящей заявке, являются:(2) ингибиторы протеазы NS3 на основе субстрата (Attwood et al. (1998) Antiviral peptide derivatives,PCT WO 98/22496, 1998; Attwood et al. (1999), Antiviral Chemistry and Chemotherapy 10.259-273, 1999;Attwood et al. (1999) Preparation and use of amino acid derivatives as anti-viral agents, публикация патента Германии DE 19914474; Tung et al. (1998) Inhibitors of serine proteases, particularly hepatitis С virus NS3protease, PCT WO 98/17679), включая альфакетоамиды и гидразиномочевины, и ингибиторы, которые в своем концевом положении имеют электрофил, такой как бороновая кислота или фосфонат. Llinas-Brunet(3) несубстратные ингибиторы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (Sudo K.et al., (1997) Biochemical and Biophysical Research Communications, 238:643-647; Sudo K. et al. (1998) Antiviral Chemistry and Chemotherapy, 9:186, 1998), включая RD3-4082 и RD3-4078, где первое соединение было замещено у амида с цепью из 14 атомов углерода, а второе имеет парафеноксифенильную группу.(4) тиазолидиновые производные, которые обладают соответствующим ингибирующим действием в анализе, проводимом обращенно-фазовой ВЭЖХ с использованием гибридного белка NS3/4A и субстрата NS5A/5B (Sudo, K. et al. (1996) Antiviral Research 32:9-18, 1996), а в частности, соединение RD-16250, имеющее конденсированную циннамоильную группу, замещенную длинной алкильной цепью,RD4-6205 и RD4-6193;(6) фенан-тренехинон, обладающий активностью против HCV-протеазы в анализе с использованием электрофореза в ДСН-ПААГ и авторадиографии и выделенный из бульона ферментационной культурыStreptomyces sp., Sch 68631 (Chu M. et al., Tetrahedron Letters 37:7229-7232, 1996), и Sch 351633, выделенный из грибов Penicillium griscofuluum, который обнаруживал активность в сцинтилляционном проксимальном анализе (Chu M. et al., Bioorganic and Medicinal Chemistry Letters 9:1949-1952);(7) селективные ингибиторы протеазы NS3 на основе макромолекулярного эглина с, выделенного из пиявок (Qasim M.A. et al., Biochemistry 36:1598-1607, 1997);(9) ингибиторы HCV-полимеразы, такие как нуклеотидные аналоги, глиотоксин (Ferrari R. et al.,Journal of Virology 73:1649-1654, 1999) и природный продукт церуленин (Lohmann V. et al., Virology 249:108-118, 1998);(10) антисмысловые фосфортиоат-олигодезоксинуклеотиды (S-ODN), которые комплементарны последовательности, находящейся в 5'некодирующей области (NCR) HCV (Alt M. et al., Hepatology 22:707717, 1995), или нуклеотиды 326-348, содержащие 3'-конец NCR, и нуклеотиды 371-388, локализованные в коровой кодирующей области РНК HCV (Alt M. et al., Archives of Virology 142:589-599, 1997, Galderisihepatitis C; публикация Японского патента JP-08268890, Kai Y. et al. Prevention and treatment of viral diseases, публикация японского патента JP-10101591);(патент США 5922757, Chojkier et al), сквален, амантадин, желчные кислоты (патент США 5846964,Ozeki et al.), N-(фосфоноацетил)-L-аспарагиновая кислота (патент США 5830905, Diana et al.), бензолдикарбоксамиды (патент США 5633388, Diana et al.), производные полиадениловой кислоты (патент США 5496546, Wang et al.), 2',3'-дидезоксиинозин (патент США 5026687, Yarchoan et al.) и бензимидазолы (патент США 5891874, Colacino et al). Краткое описание графического материала На фиг. 1 представлены различные неограничивающие примеры структуры нуклеозидов настоящего изобретения, а также других известных нуклеозидов, FIAU и рибавирина, которые используются в настоящем описании в качестве сравнительных примеров. На фиг. 2 представлены линейные кривые фармакокинетики (концентрации в плазме) -D-2'-CH3 рибоG, введенного шести собакоподобным обезьянам, в зависимости от времени введения. На фиг. 3 а и 3b представлены линейные кривые фармакокинетики (концентраций в плазме) -D-2'CH3-рибоG, введенного внутривенно (3 а) или перорально (3b) собакоподобным обезьянам, в зависимости от времени введения. Подробное описание изобретения Описанное здесь настоящее изобретение относится к соединениям и к композициям для лечения гепатита С у человека или других животных-хозяев, где указанное лечение предусматривает введение эффективного для лечения HCV-инфекций количества -D-нуклеозида, описанного в настоящей заявке,или его фармацевтически приемлемой соли, или пролекарства, необязательно в фармацевтически прием-5 011720 лемом носителе. Соединения настоящего изобретения обладают либо противовирусной (т.е. анти-HCV) активностью, либо они подвергаются метаболизму с образованием соединения, обладающего указанной активностью.I. Активное соединение и его физиологически приемлемые соли и пролекарства. Настоящее изобретение относится к соединению формулы XVII или его фармацевтически приемлемой соли, сложному эфиру или фосфату где "основание" представляет собой пирролпиримидин;R10 представляет собой Н, C1-С 10 алкил, хлор, бром или иод иX представляет собой О. В предпочтительном варианте R1 представляет собой Н или фосфат, предпочтительно Н; R6 представляет собой C1-С 10 алкил, предпочтительно метил; R7 и R9 независимо представляют собой OR1; R10 представляет собой Н и X представляет О.-D-нуклеозиды настоящего изобретения ингибируют HCV-полимеразную активность. Нуклеозиды могут быть скринированы на их способность ингибировать HCV-полимеразную активность in vitro в соответствии с методами скрининга, которые более конкретно описаны ниже. Спектр активности может быть легко определен путем оценки указанного соединения в анализах, описанных ниже, или в других подтверждающих анализах. В одном из вариантов осуществления изобретения эффективность анти-HCV соединения определяли путем измерения концентрации соединения, необходимой для 50%-ного снижения числа вирусных бляшек in vitro (т.е. EC50 данного соединения), методами, более конкретно описанными ниже. В предпочтительных вариантах осуществления изобретения, ЕС 50 указанного соединения составляет менее чем 25,15 или 10 мкмоль, как было измерено в анализе на полимеразную активность, описанном Ferrari R. et al.,Jnl of Vir. 73:1649-1654, 1999; Ishii et al., Hepatology, 29:1227-1235, 1999; Lohmann et al., Jnl. of Bio.-D-нуклеозиды настоящего изобретения могут ингибировать HCV-полимеразную активность. Нуклеозиды могут быть скринированы на их способность ингибировать HCV-полимеразную активностьin vitro в соответствии с методами скрининга, которые более конкретно описаны ниже. Спектр активности может быть легко определен путем оценки указанного соединения в анализах, описанных ниже, или в других подтверждающих анализах. В одном из вариантов осуществления изобретения эффективность анти-HCV соединения измеряли по концентрации соединения, необходимой для снижения числа вирусных бляшек in vitro на 50% (т.е., ЕС 50 данного соединения), методами, более конкретно описанными ниже. В предпочтительных вариантах осуществления изобретения EC50 указанного соединения составляет менее чем 25, 15, 10, 5 или 1 мкмоль. В другом варианте осуществления изобретения активное соединение может быть введено в комбинации или поочередно с другим анти-HCV агентом. При комбинированной терапии эффективную дозу двух или нескольких агентов вводят вместе, а при альтернативной терапии эффективную дозу каждого агента вводят поочередно. Указанные дозы зависят от абсорбции, инактивации и экскреции лекарственного средства, а также от других известных факторов. Следует отметить, что величины доз могут также варьироваться в зависимости от тяжести состояния, которое подвергается лечению. Кроме того, следует отметить, что для любого конкретного индивидуума конкретные схемы приема и введения лекарственного средства должны быть скорректированы в зависимости от времени в соответствии с потребностью индивидуума и в соответствии с назначением врача, осуществляющего введение или наблюдение за введением данных композиций. Неограничивающими примерами противовирусных агентов, которые могут быть использованы в комбинации с соединениями, описанными в настоящей заявке, являются(2) ингибиторы протеазы NS3 на основе субстрата (Attwood et al. (1998) Antiviral peptide derivatives,PCT WO 98/22496, 1998; Attwood et al. (1999), Antiviral Chemistry and Chemotherapy 10.259-273, 1999;Attwood et al. (1999) Preparation and use of amino acid derivatives as anti-viral agents, публикация патента Германии DE 19914474; Tung et al. (1998) Inhibitors of serine proteases, particularly hepatitis С virus NS3protease, PCT WO 98/17679), включая альфакетоамиды и гидразиномочевины, и ингибиторы, которые в своем концевом положении имеют электрофил, такой как бороновая кислота или фосфонат. Llinas-Brunet(3) несубстратные ингибиторы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (Sudo K.et al., (1997) Biochemical and Biophysical Research Communications, 238:643-647; Sudo K. et al. (1998) Antiviral Chemistry and Chemotherapy, 9:186, 1998), включая RD3-4082 и RD3-4078, где первое соединение было замещено у амида с цепью из 14 атомов углерода, а второе имеет парафеноксифенильную группу;(4) тиазолидиновые производные, которые обладают соответствующим ингибирующим действием в анализе, проводимом обращенно-фазовой ВЭЖХ с использованием гибридного белка NS3/4A и субстрата NS5A/5B (Sudo, K. et al. (1996) Antiviral Research 32:9-18, 1996), а в частности, соединение RD-16250, имеющее конденсированную циннамоильную группу, замещенную длинной алкильной цепью,RD4-6205 и RD4-6193;(6) фенан-тренехинон, обладающий активностью против HCV-протеазы в анализе с использованием электрофореза в ДСН-ПААГ и авторадиографии и выделенный из бульона ферментационной культурыStreptomyces sp., Sch 68631 (Chu M. et al., Tetrahedron Letters 37:7229-7232, 1996), и Sch 351633, выделенный из грибов Penicillium griscofuluum, который обнаруживал активность в сцинтилляционном проксимальном анализе (Chu M. et al., Bioorganic and Medicinal Chemistry Letters 9:1949-1952);(7) селективные ингибиторы протеазы NS3 на основе макромолекулярного эглина с, выделенного из пиявок (Qasim M.A. et al., Biochemistry 36:1598-1607, 1997);(9) ингибиторы HCV-полимеразы, такие как нуклеотидные аналоги, глиотоксин (Ferrari R. et al.,Journal of Virology 73:1649-1654, 1999) и природный продукт церуленин (Lohmann V. et al., Virology 249:108-118, 1998);(10) антисмысловые фосфортиоат-олигодезоксинуклеотиды (S-ODN), которые комплементарны последовательности, находящейся в 5'некодирующей области (NCR) HCV (Alt M. et al., Hepatology 22:707717, 1995), или нуклеотиды 326-348, содержащие 3'-конец NCR и нуклеотиды 371-388, локализованные в коровой кодирующей области РНК HCV (Alt M. et al., Archives of Virology 142:589-599, 1997, Galderisi U.hepatitis C; публикация Японского патента JP-08268890, Kai Y. et al. Prevention and treatment of viral diseases, публикация японского патента JP-10101591);(патент США 5922757, Chojkier et al), сквален, амантадин, желчные кислоты (патент США 5846964,Ozeki et al.), N-(фосфоноацетил)-L-аспарагиновая кислота (патент США 5830905, Diana et al.), бензолдикарбоксамиды (патент США 5633388, Diana et al.), производные полиадениловой кислоты (патент США 5496546, Wang et al.), 2',3'-дидезоксиинозин (патент США 5026687, Yarchoan et al.) и бензимидазолы (патент США 5891874, Colacino et al). Активное соединение может быть введено в виде любой соли или пролекарства, которые после введения реципиенту могут прямо или опосредованно превращаться в исходное соединение либо сами обладают нужной активностью. Неограничивающими примерами таких соединений являются фармацевтически приемлемые соли (иначе называемые "физиологически приемлемыми солями") и соединения, которые были алкилированы или ацилированы в 5'-положении, либо у пуринового или пиримидинового основания (типа "фармацевтически приемлемого пролекарства"). Кроме того, указанные модификации могут влиять на биологическую активность данного соединения, а в некоторых случаях эта активность может быть увеличена по сравнению с исходным соединением. Это может быть легко проанализировано путем получения соли или пролекарства и тестирования их противовирусной активности методами, описанными в настоящей заявке, или другими методами, известными специалистам.II. Определения. Используемый здесь термин "алкил", если это не оговорено особо, означает насыщенный прямой,разветвленный или циклический первичный, вторичный или третичный углеводород, обычно C1-С 10 углеводород, а в частности метил, этил, пропил, изопропил, циклопропил, бутил, изобутил, трет-бутил,пентил, циклопентил, изопентил, неопентил, гексил, изогексил, циклогексил, циклогексилметил, 3 метилпентил, 2,2-диметилбутил и 2,3-диметилбутил. Этот термин включает как замещенные, так и незамещенные алкильные группы. Группы, которые могут замещать алкильную группу, выбирают из груп-7 011720 пы, состоящей из гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, и могут быть, незащищенными или защищенными, если это необходимо, как очевидно каждому специалисту и как описано, например, в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, которая вводится в настоящее описание посредством ссылки. Используемый здесь термин "низший алкил", если это не оговорено особо, означает насыщенную,прямую, разветвленную или, если это необходимо, циклическую (например, циклопропильную) С 1 С 4 алкильную группу, включая как замещенные, так и незамещенные формы. В настоящем описании, если это не оговорено особо, в случае, когда подходящей группой является алкил, то предпочтительным является низший алкил. Аналогичным образом, если подходящей группой является алкил или низший алкил, то предпочтительным является незамещенный алкил или низший алкил. Термин "алкиламино" или "ариламино" означает аминогруппу, которая имеет один или два алкильных или арильных заместителя, соответственно. Используемый здесь термин "защищенный" относится, если это не оговорено особо, к группе, которая присоединяется к атомам кислорода, азота или фосфора для предотвращения их последующих реакций или в каких-либо других целях. Специалистам в области органического синтеза известен широкий ряд кислород- и азотзащитных групп. Используемый здесь термин "арил" означает, если это не оговорено особо, фенил, бифенил или нафтил, а предпочтительно, фенил. Этот термин включает как замещенные, так и незамещенные группы. Арильная группа может быть замещена одним или несколькими радикалами, выбранными из группы,состоящей из гидроксила, амино, алкиламино, ариламино, алкокси, арилокси, нитро, циано, сульфоновой кислоты, сульфата, фосфоновой кислоты, фосфата или фосфоната, которые могут быть, если это необходимо, незащищенными или защищенными, как очевидно каждому специалисту и как описано, например,в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991. Термин "алкарил" или "алкиларил" означает алкильную группу с арильным заместителем. Термин"аралкил" или "арилалкил" означает арильную группу с алкильным заместителем. Используемый здесь термин "галоген" означает хлор, бром, иод и фтор. Термин "пуриновое или пиримидиновое основание" означает, но не ограничивается ими, аденин,N6-алкилпурины, N6-ацилпурины (где ацил представляет С(О) алкил, арил, алкиларил или арилалкил),N6-бензилпурин,N6-галогенпурин,N6-винилпурин,N6-ацетиленпурин,N6-ацилпурин,N 66 2 2 гидроксиалкилпурин, N -тиоалкилпурин, N -алкилпурины, N -алкил-6-тиопурины, тимин, цитозин, 5 фторцитозин, 5-метилцитозин, 6-азапиримидин, включая 6-азацитозин, 2- и/или 4-меркаптопиримидин,урацил, 5-галогенурацил, включая 5-фторурацил, С 5-алкилпиримидины, С 5-бензилпиримидины, С 5 галогенпиримидины,С 5-винилпиримидин,С 5-ацетиленпиримидин,С 5-ацилпиримидин,С 55 5 5 гидроксиалкилпурин,С -амидопиримидин,С -цианопиримидин,С -нитропиримидин,С 52 2 аминопиримидин, N -алкилпурины, N -алкил-6-тиопурины, 5-азацитидин, 5-азаурацил, тиазолпиримидин, имидазолпиридинил, пирролпиримидинил и пиразолпиримидинил. Пуриновыми основаниями являются, но не ограничиваются ими, гуанин, аденин, гипоксантин, 2,6-диаминопурин и 6-хлорпурин. При необходимости или по желанию, функциональные кислородные и азотные группы на этом основании могут быть защищены. Подходящие защитные группы хорошо известны специалистам, и такими группами являются триметилсилил,диметилгексилсилил,трет-бутилдиметилсилил,третбутилдифенилсилил, тритил, алкильные группы и ацильные группы, такие как ацетил, пропионил, метансульфонил и п-толуолсульфонил. Термин "ацил" означает эфир карбоновой кислоты, в котором некарбонильная часть сложноэфирной группы выбрана из прямого, разветвленного или циклического алкила или низшего алкила, алкоксиалкила, включая метоксиметил, аралкила, включая бензил, арилоксиалкила, такого как феноксиметил,арила, включая фенил, необязательно замещенный хлором, бромом, фтором, иодом, C1-4 алкилом или С 1-4 алкокси; эфиры сульфоновой кислоты, такие как алкил- или аралкилсульфонил, включая метансульфонил; эфир моно-, ди- или трифосфорной кислоты, тритил или монометокситритил, замещенный бензил,триалкилсилил (например, диметил-трет-бутилсилил) или дифенилметилсилил. Арильные группы в указанных сложных эфирах могут, но необязательно, содержать фенильную группу. Термин "низший ацил" относится к ацильной группе, в которой некарбонильной частью является низший алкил. Используемый здесь термин "по существу, не содержащий" или "по существу, в отсутствии" относится к нуклеозидной композиции, которая включает по крайней мере 85-90 мас.%, предпочтительно 9598 мас.% и даже более предпочтительно 99-100 мас.%, определенного энантиомера данного нуклеозида. В предпочтительном варианте способов и соединений настоящего изобретения, указанные соединения,по существу, не содержат энантиомеров. Аналогичным образом, термин "выделенный" относится к нуклеозидной композиции, которая включает по крайней мере 85-90 мас.%, предпочтительно 95-98 мас.% и даже более предпочтительно, 99100 мас.% указанного нуклеозида, где остальное количество композиции составляют другие химические молекулы или энантиомеры. Используемый здесь термин "независимо" указывает на то, что данный независимо используемый-8 011720 радикал независимо варьируется для каждого применения. Таким образом, в таком соединении, какR"XYR", где R" независимо представляет углерод или азот, оба R" могут представлять углерод, оба R" могут представлять азот или один R" может представлять углерод, а другой R" может представлять азот. Используемый здесь термин "хозяин" относится к одноклеточному или многоклеточному организму, в котором может реплицироваться данный вирус, включая клеточные линии и животных, а предпочтительно человека. Альтернативно, указанный хозяин может содержать часть генома вируса гепатита С,репликация или функция которого может быть изменена под действием соединений настоящего изобретения. В частности, термин "хозяин" относится к неинфицированным клеткам; клеткам, трансфецированным целым геномом HCV или его частью, и к животным, в частности к приматам (включая шимпанзе) и к человеку. В большинстве применений настоящего изобретения хозяином является человек. Однако очевидно, что при некоторых показаниях настоящее изобретение может быть использовано в ветеринарии (для таких животных, как шимпанзе). Используемый в настоящем описании термин "фармацевтически приемлемая соль или пролекарство" означает любую фармацевтически приемлемую форму (такую как сложный эфир, эфир фосфорной кислоты, соль сложного эфира или родственную группу) нуклеозидного соединения, которая при ее введении пациенту превращается в нуклеозидное соединение. Фармацевтически приемлемыми солями являются соли, полученные из фармацевтически приемлемых неорганических или органических оснований и кислот. Подходящими солями, образованными из большого числа других кислот, применяемых в фармацевтической практике, являются соли щелочных металлов, таких как калий и натрий, и щелочноземельных металлов, таких как кальций и магний. Термин "фармацевтически приемлемое пролекарство" означает соединение, которое подвергается метаболизму, например гидролизуется или окисляется в организме хозяина, с образованием соединения настоящего изобретения. Типичными примерами пролекарств являются соединения, которые имеют биологически лабильные защитные группы на функциональной части активного соединения. Пролекарствами являются соединения, которые могут быть окислены, восстановлены, аминированы, дезаминированы, гидроксилированы, дегидроксилированы, гидролизованы, дегидролизованы, алкилированы, дезалкилированы, ацилированы, деацилированы, фосфорилированы и дефосфорилированы с образованием активного соединения. Соединения настоящего изобретения обладают противовирусной активностью, направленной против HCV, либо подвергаются метаболизму с образованием соединения, обладающего такой активностью.III. Композиции, включающие соль нуклеотидов или их пролекарство. В случаях, когда соединения являются достаточно основными или кислотными для образования стабильных нетоксических солей кислот или оснований, может оказаться необходимым введение указанного соединения в виде фармацевтически приемлемой соли. Примерами фармацевтически приемлемых солей являются кислотно-аддитивные соли, образованные органическими кислотами, образующими физиологически приемлемый анион, например тозилат, метансульфонат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, -кетоглутарат и -глицерофосфат. Могут быть также образованы подходящие соли неорганических кислот, включая сульфат, нитрат, бикарбонат и карбонат. Фармацевтически приемлемые соли могут быть получены в соответствии со стандартными процедурами, хорошо известными специалистам, например посредством взаимодействия достаточно основного соединения, такого как амин, с подходящей кислотой с образованием физиологически приемлемого аниона. Могут быть также получены соли карбоновых кислот и щелочных металлов (например, натрия,калия или лития) или щелочно-земельных металлов (например, кальция). Для повышения активности, биологической доступности, стабильности или улучшения каких-либо других свойств указанного нуклеозида все описанные здесь нуклеозиды могут быть введены в качестве нуклеотидного пролекарства. Хорошо известен ряд лигандов нуклеотидного пролекарства. Вообще говоря, алкилирование, ацилирование или другая липофильная модификация моно-, ди- или трифосфата нуклеозида может повышать стабильность нуклеотида. Примерами групп-заместителей, которые могут замещать водород на фосфатной части, являются алкил, арил, стероиды, углеводы, включая сахара, 1,2 диацилглицерин и спирты. Многие из этих групп описаны в работе R. JonesN. Bischofberger, AntiviralResearch, 27 (1995) 1-17. Для достижения нужного эффекта многие из них могут быть использованы в комбинации с описанными нуклеозидами. Активный нуклеозид может также представлять собой 5'-фосфоэфирный липид или 5'-эфирный липид, описанные в нижеследующих работах, которые вводятся в настоящее описание посредством ссылки: Kucera, L.S., N. Iyer, Е. Leake, A. Raben, Modest E.K, D.L.W., и С. Piantadosi. 1990. "Novel membraneinteractive ether lipid analogs that inhibit infectious HIV-1 production and induce defective virus formation."Bosch, and D.D. Richman, 1990. "Synthesis and antiretroviral activity of phospho lipid analogs of azidothymidine and other antiviral nucleosides." J. Biol. Chem. 265:61127. Неограничивающими примерами патентов США, где описаны подходящие липофильные заместители, которые могут быть введены в нуклеозид, предпочтительно в 5'-ОН-положении нуклеозидного или липофильного препаратов, являются патенты США 5149794 (Sep.22, 1992, Yatvin et al.);5194654al.); 5411947 (May 2, 1995, Hostetler et al.); 5463092 (Oct. 31, 1995, Hostetler et al.); 5543389 (Aug. 6, 1996,Yatvin et al.); 5543390 (Aug. 6, 1996, Yatvin et al.); 5543391 (Aug. 6, 1996, Yatvin et al.); и 5554728 (Sep. 10,1996; Basava et al), каждый из которых вводится в настоящее описание посредством ссылки. Иностранными патентными заявками, где описаны липофильные заместители, которые могут быть присоединены к нуклеозидам настоящего изобретения, или липофильные препараты, являются WO 89/02733, WO 90/00555, WO 91/16920, WO 91/18914, WO 93/00910, WO 94/26273, WO 96/15132, ЕР 0350287, ЕР 93917054.4 и WO 91/19271.IV. Комбинированная и альтернативная терапия. Следует отметить, что длительное лечение противовирусным агентом может приводить к появлению резистентности вариантов HCV к лекарственному средству. Резистентность к лекарственному средству наиболее часто возникает в результате мутации в гене, кодирующем фермент, используемый при репликации вируса. Действие лекарственного средства против HCV-инфекции может быть продлено,усилено или восстановлено путем введения соединения в комбинации или поочередно со вторым, а вероятно, и с третьим противовирусным соединением, которое индуцирует другую мутацию, вызываемую главным лекарственным средством. Альтернативно, фармакокинетика, биораспределение или другие параметры лекарственного средства могут быть изменены путем применения такой комбинированной или альтернативной терапии. В основном, комбинированная терапия является предпочтительной по сравнению с альтернативной терапией, поскольку она индуцирует множественные одновременные стрессовые воздействия на вирус. Неограничивающими примерами противовирусных агентов, которые могут быть использованы в комбинации с описанными здесь соединениями, являются:(2) ингибиторы протеазы NS3 на основе субстрата (Attwood et al. (1998) Antiviral peptide derivatives,PCT WO 98/22496, 1998; Attwood et al. (1999), Antiviral Chemistry and Chemotherapy 10.259-273, 1999;Attwood et al. (1999) Preparation and use of amino acid derivatives as anti-viral agents, публикация патента Германии DE 19914474; Tung et al. (1998) Inhibitors of serine proteases, particularly hepatitis С virus N53protease, PCT WO 98/17679), включая альфакетоамиды и гидразиномочевины, и ингибиторы, которые в своем концевом положении имеют электрофил, такой как бороновая кислота или фосфонат. Llinas-Brunet(3) нeсубстратные ингибиторы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (SudoAntiviral Chemistry and Chemotherapy, 9:186, 1998), включая RD3-4082 и RD3-4078, где первое соединение замещено у амида с цепью из 14 атомов углерода, а второе имеет парафеноксифенильную группу;(4) тиазолидиновые производные, которые обнаруживали соответствующее ингибирующее действие в анализе, проводимом посредством обращенно-фазовой ВЭЖХ с использованием гибридного белкаNS3/4A и субстрата NS5A/5B (Sudo, K. et al. (1996) Antiviral Research 32:9-18, 1996), а в частности соединение RD-1-6250, имеющее конденсированную циннамоильную группу, замещенную длинной алкильной цепью, RD4-6205 и RD4-6193;(6) фенан-тренехинон, обладающий активностью против протеазы HCV в анализе с использованием электрофореза в ДСН-ПААГ и авторадиографии и выделенный из бульона ферментационной культурыStreptomyces sp., Sch 68631 (Chu M. et al., Tetrahedron Letters 37:7229-7232, 1996), и Sch 351633, выделенный из грибов Penicillium griscofuluum, который обнаруживал активность в сцинтилляционном проксимальном анализе (Chu M. et al., Bioorganic and Medicinal Chemistry Letters 9:1949-1952);(7) селективные ингибиторы протеазы NS3 на основе макромолекулярного элгина с, выделенного из пиявок (Qasim M.A. et al., Biochemistry 36:1598-1607, 1997);(9) ингибиторы HCV-полимеразы, такие как нуклеотидные аналоги, глиотоксин (Ferrari R. et al.,Journal of Virology 73:1649-1654, 1999) и природный продукт церуленин (Lohmann V. et al., Virology 249:108-118, 1998);(10) антисмысловые фосфортиоат-олигодезоксинуклеотиды (S-ODN), которые комплементарны последовательности, находящейся в 5' некодирующей области (NCR) HCV (Alt M. et al., Hepatology 22:707- 10011720 717, 1995), или нуклеотиды 326-348, содержащие 3'-конец NCR, и нуклеотиды 371-388, локализованные в коровой кодирующей области РНК HCV (Alt M. et al., Archives of Virology 142:589-599, 1997, Galderisihepatitis C; публикация Японского патента JP-08268890, Kai Y. et al. Prevention and treatment of viral diseases, публикация японского патента JP-10101591);(патент США 5922757, Chojkier et al), сквален, амантадин, желчные кислоты (патент США 5846964,Ozeki et al.), N-(фосфоноацетил)-L-аспарагиновая кислота (патент США 5830905, Diana et al.), бензолдикарбоксамиды (патент США 5633388, Diana et al.), производные полиадениловых кислот (патент США 5496546, Wang et al.), 2',3'-дидезоксиинозин (патент США 5026687, Yarchoan et al.) и бензимидазолы (патент США 5891874, Colacino et al).V. Фармацевтические композиции. Хозяева, включая человека, инфицированные HCV или его генным фрагментом, могут быть подвергнуты лечению путем введения пациенту эффективного количества активного соединения или его фармацевтически приемлемого пролекарства или соли в присутствии фармацевтически приемлемого носителя или разбавителя. Активные материалы могут быть введены любым подходящим способом, например перорально, парентерально, внутривенно, внутрикожно, подкожно или местно в виде жидкой или твердой формы. Предпочтительная доза соединения против HCV составляет в пределах примерно от 1 до 50 мг/кг,предпочтительно 1-20 мг/кг массы тела в день, а обычно от 0,1 до около 100 мг на килограмм массы тела реципиента в день. Диапазон эффективных доз фармацевтически приемлемых солей и пролекарств может быть вычислен исходя из массы исходного нуклеозида, необходимого для доставки. Если соль или пролекарство сами обладают нужной активностью, то эффективная доза может быть определена как описано выше по массе соли или пролекарства, или другими способами, известными специалистам. Данное соединение обычно вводят в виде подходящей разовой лекарственной формы, включая, но не ограничиваясь ею, форму, содержащую 7-3000 мг, предпочтительно 70-1400 мг активного ингредиента на одну разовую лекарственную форму. Подходящая пероральная доза обычно составляет 50-1000 мг. В идеальном случае активный ингредиент должен быть введен так, чтобы максимальная концентрация активного соединения в плазме составляла примерно 0,2-70 мкМ, а предпочтительно примерно 1,0-10 мкМ. Это может быть достигнуто, например, путем внутривенной инъекции 0,1-5% раствора активного ингредиента, необязательно в физиологическом растворе, или путем введения болюса, содержащего активный ингредиент. Концентрация активного соединения в лекарственной композиции зависит от абсорбции, инактивации и скорости экскреции лекарственного средства, а также от других известных факторов. Следует отметить, что величины доз могут также варьироваться в зависимости от тяжести состояния, на которое направлено лечение. Кроме того, следует отметить, что для любого отдельного индивидуума, конкретные схемы введения лекарственного средства должны быть скорректированы в зависимости от времени,в соответствии с потребностью индивидуума и назначением врача, осуществляющего введение или наблюдение за введением данных композиций, и интервалы концентраций, указанные выше, приводятся лишь в целях иллюстрации и не ограничивают объема или практического применения заявленной композиции. Активный ингредиент может быть введен один раз или несколько раз в виде более мелких дробных доз, вводимых через разные интервалы времени. Предпочтительным способом введения активного соединения является пероральное введение. Пероральные композиции обычно включают инертный разбавитель или пищевой носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для перорального терапевтического введения активное соединение может быть введено вместе с наполнителем и использовано в форме таблеток, пастилок или капсул. Фармацевтически совместимые связывающие агенты и/или адъюванты могут включены как часть композиции. Указанные таблетки, драже, капсулы, пастилки и т.п. могут содержать любой из нижеследующих ингредиентов или соединения аналогичной природы: связующие вещества, такие как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; наполнитель, такой как крахмал или лактоза; дезинтегрирующий агент, такой как альгиновая кислота, примогель или кукурузный крахмал; замасливатель,такой как стеарат магния или стероты; агент, увеличивающий скольжение, такой как коллоидальный диоксид кремния; подслащивающий агент, такой как сахароза или сахарин; или ароматизирующий агент,такой как перечная мята, метилсалицилат или апельсиновая отдушка. Если разовая лекарственная форма представляет собой капсулу, то она, помимо ингредиентов вышеуказанного типа, может содержать жидкий носитель, такой как жирное масло. Кроме того, разовые лекарственные формы могут содержать другие различные ингредиенты, которые модифицируют физическую форму разовой лекарственной формы,например покрытия сахаром, шеллаком или другими энтеросолюбильными веществами.- 11011720 Указанное соединение может быть введено как компонент эликсира, суспензии, сиропа, облатки,жевательной резинки или т.п. Сироп, помимо активных соединений, может содержать сахарозу в качестве подсластителя и некоторые консерванты, красители и подкрашивающие вещества и отдушки. Соединение или его фармацевтически приемлемые пролекарство или соли могут быть также смешаны с другими активными веществами, которые не влияют на желаемый эффект, или с веществами,которые дополняют желаемый эффект, такими как антибиотики, противогрибковые средства, противовоспалительные средства или другие противовирусные средства, включая другие нуклеозидные соединения. Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного введения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, физиологический раствор, жирные масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатообразующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для корректировки тоничности, такие как хлорид натрия или декстроза. Препарат для парентерального введения может быть заключен в ампулы, в одноразовые шприцы или во флаконы для многократных доз, изготовленные из стекла или пластика. При внутривенном введении предпочтительными носителями являются физиологический раствор или забуференный фосфатом физиологический раствор (PBS). В предпочтительном варианте осуществления изобретения активные соединения смешивают вместе с носителями, которые защищают указанное соединение от быстрого выведения из организма, например приготавливают в виде препарата с регулируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. При этом могут быть использованы биологически разлагаемые и биологически совместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Методы получения указанных композиций известны каждому специалисту. Указанные материалы могут быть также закуплены у Alza Corporation. В качестве фармацевтически приемлемых носителей также предпочтительными являются липосомные суспензии (включая липосомы, нацеленные на инфицированные клетки с помощью моноклональных антител против вирусных антигенов). Они могут быть получены методами, известными специалистам,например, как описано в патенте США 4522811 (который во всей своей полноте вводятся в настоящее описание посредством ссылки). Так, например, липосомные композиции могут быть получены путем растворения соответствующего липида(ов) (такого как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в неорганическом растворителе, который затем выпаривают, после чего на поверхности контейнера остается тонкая пленка осушенного липида. Затем в этот контейнер вводят водный раствор активного соединения или его монофосфатное, дифосфатное и/или трифосфатное производное. После этого контейнер встряхивают вручную для удаления со стенок контейнера липидного материала и диспергирования липидных агрегатов, в результате чего получают липосомную суспензию.VI. Способ получения активных соединений. Нуклеозиды настоящего изобретения могут быть синтезированы любым способом, известным специалистам. В частности, синтез нуклеозидов настоящего изобретения может быть достигнут путем алкилирования соответствующим образом модифицированного сахара с последующим гликозилированием, или путем гликозилирования с последующим алкилированием нуклеозида. Нижеследующие неограничивающие варианты иллюстрируют некоторые общие способы получения нуклеозидов настоящего изобретения. А. Общий синтез 1'-С-разветвленных нуклеозидов. 1'-С-разветвленные рибонуклеозиды следующей структуры: где "основание" представляет пуриновое или пиримидиновое основание, определенное выше;R1 и R2 независимо представляют Н, фосфат (включая монофосфат, дифосфат, трифосфат или стабилизированное фосфатное пролекарство); ацил (включая низший ацил); алкил (включая низший алкил); эфир сульфоновой кислоты, включая алкил- или арилалкилсульфонил (включая метансульфонил и бензил), где фенильная группа необязательно замещена одним или несколькими заместителями, описанны- 12011720 ми выше при определении арила; липид, включая фосфолипид; аминокислоту; углевод; пептид; холестерин или другую фармацевтически приемлемую уходящую группу, которая, при введении in vivo, способна обеспечивать образование соединения, где R1 или R2 независимо представляют Н или фосфат;R6 представляет алкил, хлор-, бром-, фтор- или иодалкил (т.е. CF3), алкенил или алкинил (т.е. аллил) и X представляет О, S, SO2 или СН 2,могут быть получены одним из нижеследующих общих методов. 1) Модификация лактона. Ключевым исходным материалом, используемым в данном способе, является соответствующим образом замещенный лактон. Этот лактон может быть закуплен, либо он может быть получен любым известным методом, включая стандартные методы эпимеризации, замещения и циклизации. Указанный лактон может быть, но необязательно, защищен подходящей защитной группой, предпочтительно ацильной или силильной группой, методами, хорошо известными специалистам и описанными в работе Greeneet al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991. Затем, защищенный лактон может быть подвергнут реакции взаимодействия с подходящим агентом сочетания, таким как металлорганический С-нуклеофил, такой как реактив Гриньяра, литийорганическое соединение, литийдиалкилмедь или R6-SiMe3 в TBAF в соответствующем апротонном растворителе при подходящей температуре, с получением 1'-алкилированного сахара. Необязательно активированный сахар может быть затем подвергнут реакции взаимодействия с основанием методами, хорошо известными специалистам и описанными в работе Townsend, Chemistry ofNucleosides and Nucleotides, Plenum Press, 1994. Так, например, ацилированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии кислоты Льюиса, такой как тетрахлорид олова, тетрахлорид титана или триметилсилилтрифлат в соответствующем растворителе при подходящей температуре. Затем, указанный нуклеозид может быть подвергнут реакции снятия защиты методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, JohnWileySons, Second Edition, 1991. В конкретном варианте осуществления изобретения предпочтительным является 1'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 1. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов, имеющийся рибонуклеозид может быть, но необязательно, защищен методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, то есть,реакции восстановления Бартона. Схема 1 2. Альтернативный метод получения 1'-С-разветвленных нуклеозидов. Ключевым исходным материалом, используемым в данном способе, является соответствующим образом замещенная гексоза. Эта гексоза может быть закуплена либо она может быть получена любым известным методом, включая стандартный метод эпимеризации, такой как щелочная обработка, замещение и реакция сочетания. Указанная гексоза может быть селективно защищена с получением соответствующей гексафуранозы, как описано в работе Townsend, Chemistry of Nucleosides and Nucleotides, Plenum- 13011720 1'-Гидрокси может быть, но необязательно, активирован с присоединением подходящей уходящей группы, такой как ацильная группа или хлор, бром, фтор, иод, посредством ацилирования или галогенирования, соответственно. Указанный необязательно активированный сахар может быть затем подвергнут реакции взаимодействия с основанием методами, хорошо известными специалистам и описанными в работе Townsend, Chemistry of Nucleosides and Nucleotides, Plenum Press, 1994. Так, например, ацилированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии кислоты Льюиса, такой как тетрахлорид олова, тетрахлорид титана или триметилсилилтрифлат, в соответствующем растворителе при подходящей температуре. Альтернативно, галогенированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии триметилсилилтрифлата. 1'-СН 2-ОН, если он защищен, может быть подвергнут реакции селективного снятия защиты методами, хорошо известными специалистам. Полученный первичный гидроксил может быть функционализирован с получением различных С-разветвленных нуклеозидов. Так, например, первичный гидроксил может быть восстановлен до метила с использованием подходящего восстановителя. Альтернативно,гидроксил может быть активирован для облегчения реакции восстановления, т.е. восстановления Бартона. В альтернативном варианте осуществления изобретения, первичный гидроксил может быть окислен до альдегида, а затем подвергнут реакции взаимодействия с С-нуклеофилом, таким как реактив Гриньяра, литийорганическим соединением, литийдиалкилмедью или R6-SiMe3 в TBAF, с соответствующим апротонным растворителем при подходящей температуре. В конкретном варианте осуществления изобретения предпочтительным является 1'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 2. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов имеющийся рибонуклеозид может быть, но необязательно, защищен методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'Гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, т.е. реакции восстановления Бартона. Схема 2 Кроме того, L-энантиомеры, соответствующие соединению настоящего изобретения, могут быть получены теми же самыми общими методами (1 или 2), описанными ниже, с использованием соответствующего L-caxapa или нуклеозидного L-энантиомера в качестве исходного материала. В. Общий синтез 2'-С-разветвленных нуклеозидов. 2'-С-разветвленные рибонуклеозиды следующей структуры: где "основание" представляет пуриновое или пиримидиновое основание, определенное выше;R1 и R2 независимо представляют Н, фосфат (включая монофосфат, дифосфат, трифосфат или стабилизированное фосфатное пролекарство); ацил (включая низший ацил); алкил (включая низший алкил); эфир сульфоновой кислоты, включая алкил- или арилалкилсульфонил (включая метансульфонил и бензил), где фенильная группа необязательно замещена одним или несколькими заместителями, описанными выше при определении арила; липид, включая фосфолипид; аминокислоту; углевод; пептид; холестерин или другую фармацевтически приемлемую уходящую группу, которая, при введении in vivo, спо- 14011720 собна обеспечивать образование соединения, где R1 или R2 независимо представляют Н или фосфат;R6 представляет алкил, хлор-, бром-, фтор- или иодалкил (т.е. CF3), алкенил или алкинил (т.е. аллил) иX представляет О, S, SO2 или СН 2,могут быть получены одним из нижеследующих общих методов. 1. Гликозилирование нуклеооснования с использованием соответствующим образом модифицированного сахара. Ключевым исходным материалом, используемым в данном способе, является сахар, имеющий группы 2'-ОН и 2'-Н и соответствующим образом замещенный подходящей уходящей группой (LG), например ацильной группой или хлором, бромом, фтором или иодом. Этот сахар может быть закуплен либо он может быть получен любыми известными методами, включая стандартный метод эпимеризации,замещения, окисления и восстановления. Замещенный сахар может быть затем окислен соответствующим окислителем в совместимом растворителе при подходящей температуре с получением 2'модифицированного сахара. Возможными окислителями являются реактив Джонса (смесь хромовой кислоты и серной кислоты), реактив Коллинза (оксид Cr(VI)-дипиридина), реактив Корри (хлорхромат пиридиния), дихромат пиридиния, дихромат кислоты, перманганат калия, MnO2, тетроксид рутения, межфазные катализаторы, такие как хромовая кислота или перманганат, присоединенный к полимеру, Cl2 пиридин, молибдат Н 2 О 2-аммония, NaBrO2-CAN, NaOCl в HOAc, хромит меди, оксид меди, никелевый катализатор Ренея, ацетат палладия, реактив Мейервейна-Понндорфа-Верлея (трет-бутоксид алюминия с другим кетоном) и N-бромсукцинимид. Затем, в результате реакции взаимодействия металлоорганического С-нуклеофила, такого как реактив Гриньяра, литийорганическое соединение, литий-диалкилмедь или R6-SiMe3 в TBAF, с кетоном в соответствующем апротонном растворителе при подходящей температуре получают 2'-алкилированный сахар. Этот алкилированный сахар может быть, но необязательно, защищен подходящей защитной группой, предпочтительно ацильной или силильной группой, методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, SecondEdition, 1991. Необязательно защищенный сахар может быть затем подвергнут реакции взаимодействия с основанием методами, хорошо известными специалистам и описанными в работе Townsend, Chemistry of Nucleosides and Nucleotides, Plenum Press, 1994. Так, например, ацилированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии кислоты Льюиса, такой как тетрахлорид олова, тетрахлорид титана или триметилсилилтрифлат, в соответствующем растворителе при подходящей температуре. Альтернативно, галогенированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии триметилсилилтрифлата. Затем указанный нуклеозид может быть подвергнут реакции снятия защиты методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, JohnWileySons, Second Edition, 1991. В конкретном варианте осуществления изобретения предпочтительным является 2'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 3. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов имеющийся рибонуклеозид может быть, но необязательно, защищен методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'Гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, то есть реакции восстановления Бартона. Схема 3- 15011720 2. Модификация предварительно полученного нуклеозида. Ключевым исходным материалом, используемым в данном способе, является соответствующим образом замещенный нуклеозид с группами 2'-ОН и 2'-Н. Этот нуклеозид может быть закуплен либо он может быть получен любыми известными методами, включая стандартные методы проведения реакций сочетания. Этот нуклеозид может быть, но необязательно, защищен подходящими защитными группами,предпочтительно ацильной или силильной группами, методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, SecondEdition, 1991. Соответствующим образом защищенный нуклеозид может быть затем окислен соответствующим окислителем в совместимом растворителе при подходящей температуре с получением 2'модифицированного сахара. Возможными окислителями являются реактив Джонса (смесь хромовой кислоты и серной кислоты), реактив Коллинза (оксид Cr(VI)-дипиридина), реактив Корри (хлорхромат пиридиния), дихромат пиридиния, дихромат кислоты, перманганат калия, MnO2, тетроксид рутения, межфазные катализаторы, такие как хромовая кислота или перманганат, присоединенный к полимеру, Cl2 пиридин, молибдат H2O2-аммония, NaBrO2-CAN, NaOCl в HOAc, хромит меди, оксид меди, никелевый катализатор Ренея, ацетат палладия, реактив Мейервейна-Понндорфа-Верлея (трет-бутоксид алюминия с другим кетоном) и N-бромсукцинимид. Затем указанный нуклеозид может быть подвергнут реакции снятия защиты методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, JohnWileySons, Second Edition, 1991. В конкретном варианте осуществления изобретения предпочтительным является 2'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 4. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов имеющийся рибонуклеозид может быть, но необязательно, защищен методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'Гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, то есть реакции восстановления Бартона. Схема 4 В другом варианте осуществления изобретения предпочтительными являются L-энантиомеры. Поэтому указанные L-энантиомеры, соответствующие соединению настоящего изобретения, могут быть получены теми же самыми общими методами, описанными выше, с использованием соответствующего где "основание" представляет пуриновое или пиримидиновое основание, определенное выше;R1 и R2 независимо представляют Н, фосфат (включая монофосфат, дифосфат, трифосфат или стабилизированное фосфатное пролекарство); ацил (включая низший ацил); алкил (включая низший алкил); эфир сульфоновой кислоты, включая алкил- или арилалкилсульфонил (включая метансульфонил и бензил), где фенильная группа необязательно замещена одним или несколькими заместителями, описанными выше при определении арила; липид, включая фосфолипид; аминокислоту; углевод; пептид; холестерин или другую фармацевтически приемлемую уходящую группу, которая при введении in vivo способна обеспечивать образование соединения, где R1 или R2 независимо представляют Н или фосфат;R6 представляет алкил, хлор-, фтор-, бром-, иодалкил (т.е. CF3), алкенил или алкинил (т.е. аллил) иX представляет О, S, SO2 или СН 2,могут быть получены одним из нижеследующих общих методов. 1. Гликозилирование нуклеооснования с использованием соответствующим образом модифицированного сахара. Ключевым исходным материалом, используемым в данном способе, является сахар, имеющий группы 2'-ОН и 2'-Н и соответствующим образом замещенный подходящей уходящей группой (LG), например ацильной группой или хлором, бромом, фтором или иодом. Этот сахар может быть закуплен либо он может быть получен любыми известными методами, включая стандартный метод эпимеризации,замещения, окисления и восстановления. Замещенный сахар может быть затем окислен соответствующим окислителем в совместимом растворителе при подходящей температуре с получением 3'модифицированного сахара. Возможными окислителями являются реактив Джонса (смесь хромовой кислоты и серной кислоты), реактив Коллинза (оксид Cr(VI)-дипиридина), реактив Корри (хлорхромат пиридиния), дихромат пиридиния, дихромат кислоты, перманганат калия, MnO2, тетроксид рутения, межфазные катализаторы, такие как хромовая кислота или перманганат, присоединенный к полимеру, Cl2 пиридин, молибдат Н 2 О 2-аммония, NaBrO2-CAN, NaOCl в HOAc, хромит меди, оксид меди, никелевый катализатор Ренея, ацетат палладия, реактив Мейервейна-Понндорфа-Верлея (трет-бутоксид алюминия с другим кетоном) и N-бромсукцинимид. Затем посредством реакции взаимодействия металлоорганического С-нуклеофила, такого как реактив Гриньяра, литийорганическое соединение, литий-диалкилмедь или R6-SiMe3 в TBAF, с кетоном в соответствующем апротонном растворителе при подходящей температуре получают 3'-С-разветвленный сахар. Этот 3'-С-разветвленный сахар может быть, но необязательно, защищен подходящей защитной группой, предпочтительно ацильной или силильной группой, методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, SecondEdition, 1991. Необязательно защищенный сахар может быть затем подвергнут реакции взаимодействия с основанием в соответствии с процедурами, хорошо известными специалистам и описанными в работе Townsend, Chemistry of Nucleosides and Nucleotides, Plenum Press, 1994. Так, например, ацилированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии кислоты Льюиса, такой как тетрахлорид олова, тетрахлорид титана или триметилсилилтрифлат в соответствующем растворителе при подходящей температуре. Альтернативно, галогенированный сахар может быть подвергнут реакции взаимодействия с силилированным основанием в присутствии триметилсилилтрифлата. Затем, указанный нуклеозид может быть подвергнут реакции снятия защиты способами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, JohnWileySons, Second Edition, 1991. В конкретном варианте осуществления изобретения предпочтительным является 3'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 5. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов образованный рибонуклеозид может быть, но необязательно, защищен способами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'Гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, то есть реакции восстановления Бартона. 2. Модификация предварительно полученного нуклеозида. Ключевым исходным материалом, используемым в данном способе, является соответствующим образом замещенный нуклеозид с группами 3'-ОН и 3'-Н. Этот нуклеозид может быть закуплен либо он может быть получен любыми известными методами, включая стандартные методы сочетания. Этот нуклеозид может быть, но необязательно, защищен подходящими защитными группами, предпочтительно ацильной или силильной группами, методами, хорошо известными специалистам и описанными в работеGreene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991. Соответствующим образом защищенный нуклеозид может быть затем окислен соответствующим окислителем в совместимом растворителе при подходящей температуре с получением 2'-модифицированного сахара. Возможными окислителями являются реактив Джонса (смесь хромовой кислоты и серной кислоты), реактив Коллинза (оксид Cr(VI)-дипиридина), реактив Корри (хлорхромат пиридиния),дихромат пиридиния, дихромат кислоты, перманганат калия, MnO2, тетроксид рутения, межфазные катализаторы, такие как хромовая кислота или перманганат, присоединенный к полимеру, Cl2-пиридин, молибдат Н 2 О 2-аммония, NaBrO2-CAN, NaOCl в HOAc, хромит меди, оксид меди, никелевый катализатор Ренея, ацетат палладия, реактив Мейервейна-Понндорфа-Верлея (трет-бутоксид алюминия с другим кетоном) и N-бромсукцинимид. Затем указанный нуклеозид может быть подвергнут реакции снятия защиты методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, JohnWileySons, Second Edition, 1991. В конкретном варианте осуществления изобретения предпочтительным является 3'-Сразветвленный рибонуклеозид. Синтез рибонуклеозида описан в схеме 6. Альтернативно, предпочтительным является дезоксирибонуклеозид. Для получения этих нуклеозидов образованный рибонуклеозид может быть, но необязательно, защищен методами, хорошо известными специалистам и описанными в работе Greene et al., Protective Groups in Organic Syntesis, John WileySons, Second Edition, 1991, после чего группа 2'-ОН может быть восстановлена с использованием подходящего восстановителя. 2'гидроксил может быть, но необязательно, активирован для облегчения реакции восстановления, то есть реакции восстановления Бартона. В другом варианте осуществления изобретения предпочтительными являются L-энантиомеры. Поэтому указанные L-энантиомеры, соответствующие соединению настоящего изобретения, могут быть получены теми же самыми общими методами, которые были описаны выше, с использованием соответствующего L-caxapa или нуклеозидного L-энантиомера в качестве исходного материала. Примеры Пример 1. Получение 1'-С-метилрибоаденина с использованием 6-амино-9-(1-дезоксиD-псикофуранозил)пурина. Указанное в заголовке соединение может быть также получено другим альтернативными методом в соответствии с процедурой, описанной в литературе (J. FarkasF.Sorm, "Nucleic acid components and Аналогичным способом, но с использованием соответствующего сахара и соответствующих пиримидиновых или пуриновых оснований были получены нижеследующие нуклеозиды формулы I Альтернативно, с использованием соответствующего сахара и соответствующих пиримидиновых или пуриновых оснований получали нижеследующие нуклеозиды формулы IV
МПК / Метки
МПК: C07H 19/16, C07H 19/06, C07H 19/10, A61K 31/7076, A61P 31/14, A61K 31/7068, C07H 19/20
Метки: beta, производные, применение, d-нуклеозида
Код ссылки
<a href="https://eas.patents.su/30-11720-proizvodnye-beta-d-nukleozida-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Производные &beta -d-нуклеозида и их применение</a>
Производные &beta-d-нуклеозида в качестве лекарственного средства для лечения инфекции вируса гепатита c у хозяина

Номер патента: 7178
Опубликовано: 25.08.2006
Авторы: Соммадосси Жан-Пьер, Лаколла Пауло
МПК: A61P 31/14, A61K 31/7076, A61K 31/7068...
Метки: вируса, лечения, beta-d-нуклеозида, инфекции, хозяина, гепатита, качестве, средства, производные, лекарственного
Формула / Реферат:
1. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или пролекарства, необязательно, в фармацевтически приемлемом носителе или разбавителе при получении лекарственного средства для лечения инфекции вируса гепатита С у хозяина. 2. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или...
Способ получения дроспиренона (6&beta, 7β 15&beta, 16&beta – диметилен -3 – оксо -17α-прегн- 4 -ен-21,17-карболактона, drsp) и 6&beta,7β15&beta,16&beta – диметилен -5&beta-гидрокси -3- оксо – 17&alpha – андростан – 21,17 – карболактон (zk 90965) в качестве промежуточного продукта

Номер патента: 1947
Опубликовано: 22.10.2001
Авторы: Мор Йорг-Торштен, Никкиш Клаус
МПК: C07J 53/00
Метки: 6&beta,7β15&beta,16&beta, 15&beta, промежуточного, 16&beta, drsp, 6&beta, оксо, 17α-прегн, дроспиренона, ен-21,17-карболактона, 21,17, андростан, 5&beta-гидрокси, качестве, 7&beta, продукта, 90965, способ, диметилен, карболактон, 17&alpha, получения
Формула / Реферат:
1. Способ получения дроспиренона (6b,7b;15b,16b-диметилен-3-оксо-17a-прегн-4-ен-21,17-карболактон DRSP) осуществляемый каталитическим гидрированием 17a-(3-гидрокси-1-пропинил)-6b,7b;15b,16b-диметилен-5b-андростан-3b,5,17b-триола (ZK 34506) с получением 17a-(3-гидрокси-1-пропил)-6b,7b;15b,16b-диметилен-5b-андростан-3b,5,17b-триола (ZK 92836) окислением в присутствии соли рутения до...
Гемисульфат карбоциклического нуклеозида и его применение при лечении вирусных инфекций

Номер патента: 1809
Опубликовано: 27.08.2001
Авторы: Джоунз Мартин Фрэнсис, Уоллис Кристофер Джон, Броуди Аластер Купер, Сигер Джон Фредерик
МПК: C07D 473/16, A61K 31/52, A61P 31/12...
Метки: нуклеозида, вирусных, инфекций, лечении, применение, карбоциклического, гемисульфат
Формула / Реферат:
1. Гемисульфат (1S,4R)-цис-4-[2-амино-6-(циклопропиламино)-9Н-пурин-9-ил]-2-цикло-пентен-1-метанола или его сольват. 2. Способ получения соединения по п.1, при котором смешивают серную кислоту и (1S,4R)-цис-4-[2-амино-6-(циклопропиламино)-9Н-пурин-9-ил]-2-циклопентен-1-метанол в стехиометрическом соотношении приблизительно 1:2. 3. Способ получения соединения по п.1, при котором смешивают сульфат...
Аналоги нуклеозида (варианты) и их применение, комбинация и способ лечения вирусных инфекций, фармацевтическая композиция

Номер патента: 4767
Опубликовано: 26.08.2004
Автор: Нгуйен-Ба Нгхе
МПК: C07D 473/16, A61K 31/52, A61P 31/12...
Метки: нуклеозида, комбинация, лечения, способ, вирусных, аналоги, фармацевтическая, инфекций, применение, варианты, композиция
Формула / Реферат:
1. цис-Нуклеозид формулы I и его фармацевтически приемлемые соли, где n равно 1 или 2, R4 выбирают из следующих заместителей: H, COOH, CONH2, OH, SH, NH2, NO2, C1-6алкил, C2-6алкенил, C2-6алкинил, галоген, CORa, где Ra означает C1-6алкил, C2-6алкенил, C2-6алкинил или COORb, где Rb означает C1-6алкил, C2-6алкенил или C2-6алкинил; R3 означает H или C1-6алкил, C2-6алкенил, C2-6алкинил; X выбирают из H, монофосфата, дифосфата, трифосфата,...
Производные пиримидона в качестве промежуточных соединений для получения ингибиторов gsk3&beta или gsk3&beta и cdk5/p25

Номер патента: 9456
Опубликовано: 28.12.2007
Авторы: Локхид Алистер, Саади Мурад, Словински Франк, Неделек Ален, Йеш Филипп, Ларденуа Патрик, Маргери Северен, Галле Тьерри
МПК: A61P 25/28, A61K 31/519, C07D 235/00...
Метки: производные, gsk3&beta, пиримидона, соединений, получения, качестве, ингибиторов, промежуточных
Формула / Реферат:
1. Производное пиримидона, представленное формулой (III) где R1 представляет собой пиримидиновое кольцо, возможно замещенное С3-6циклоалкильной группой, С1-4алкильной группой, С1-4алкоксигруппой, бензильной группой или атомом галогена; R3 и R4 представляют собой, каждый независимо, атом водорода, С1-6алкильную группу, гидроксигруппу, С1-4алкоксигруппу или атом галогена; R5 представляет собой атом водорода, С1-6алкильную группу или атом...
Предыдущий патент: Ингибитор высвобождения воспалительного цитокина
Следующий патент: Композиция, для сдерживания деградации алкоголя в организме человека
Случайный патент: Применение акриловых водорастворимых сополимеров в качестве загустителя, водная композиция, возможно содержащая пигменты, и способ ее получения