Имидазо(1,2-а) пиридиновые соединения в качестве ингибиторов vegf-r2
Номер патента: 11691
Опубликовано: 28.04.2009
Авторы: Нобелок Джон Монте, Хао Янь, Барда Дэвид Энтони, Клэйтон Джошуа Райан, Ремик Дэвид Майкл, Беркхолдер Тимоти Пол, Генри Джеймс Роберт, Чжун Боюй, Маклин Джонатан Александер, Ван Чжао-Цин, Ремпала Марк Эдвард, Хит Перри Кларк, Мендел Дэвид, Йип Ивонне Йи Май




















Формула / Реферат
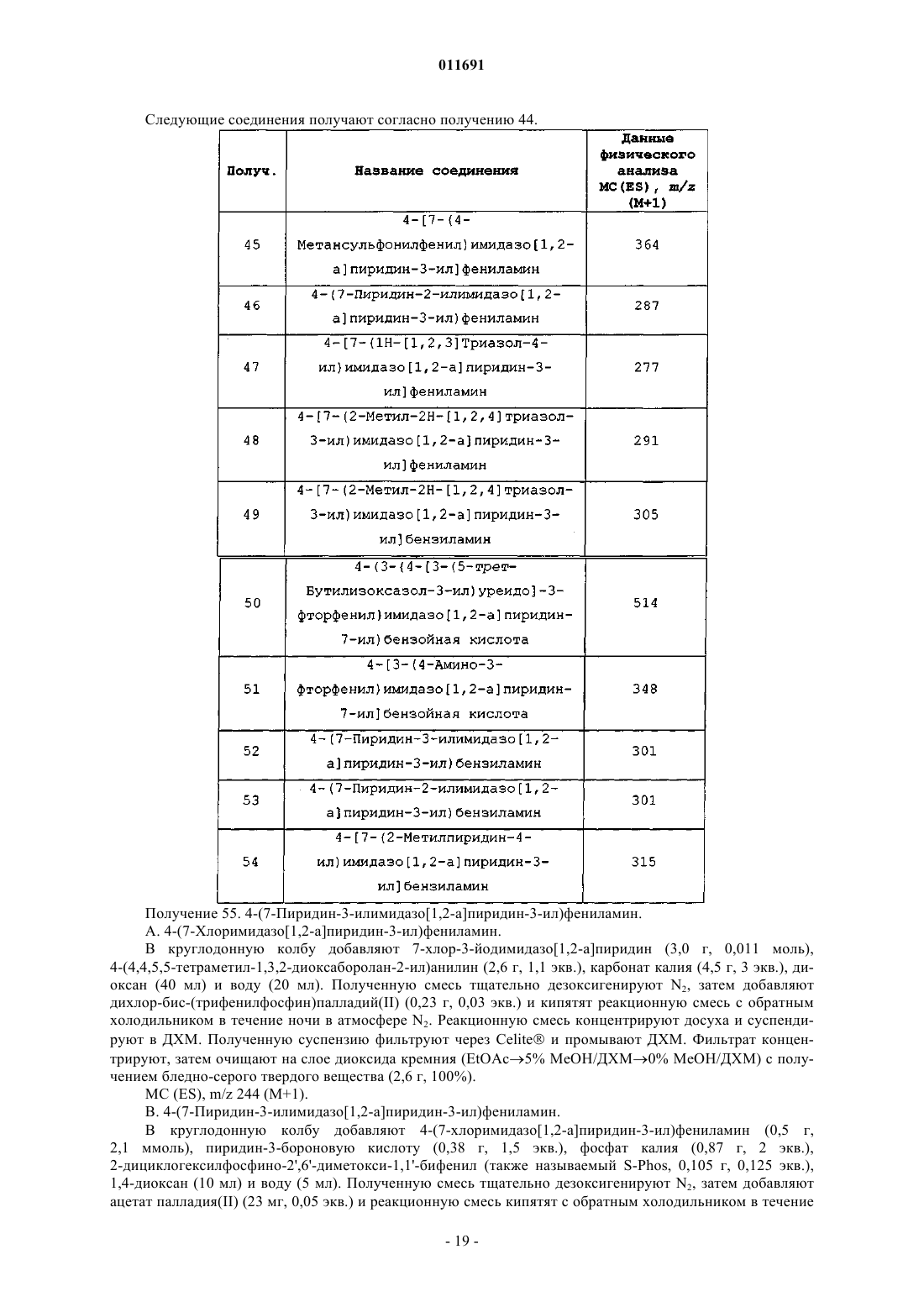
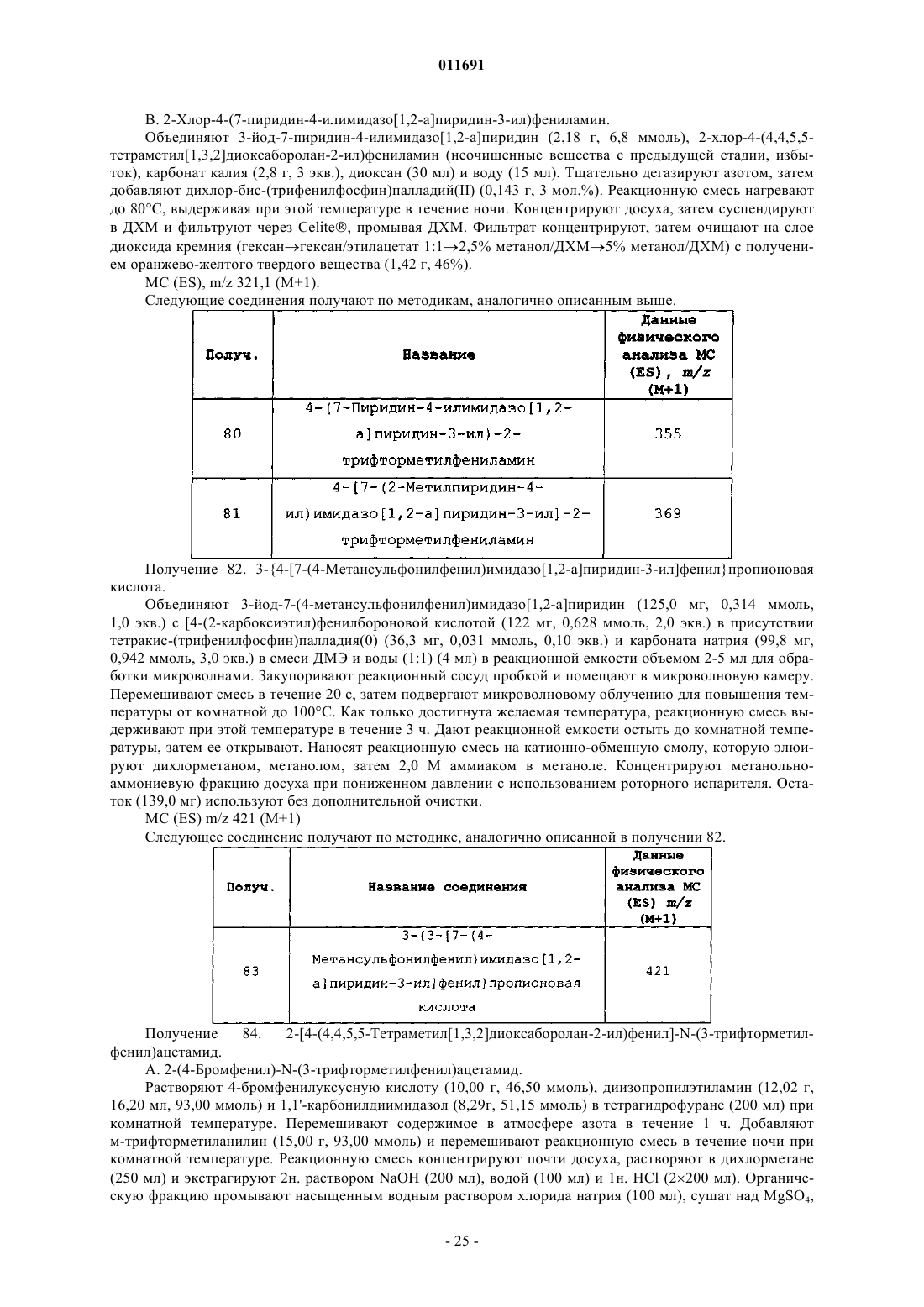
1. Соединение формулы (I)

где R1 представляет собой:
(а) 2-пиридонил, необязательно замещенный -(СН2)1-4NR2R3; или
(b) фенил, тиенил, тиазолил, имидазолил, пиразолил, триазолил, оксазолил, пиридинил,
N-оксопиридинил или пиримидинил, все из которых необязательно замещены -(СН2)0-4NR2R3,
C1-C6алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4алкокси, галогена,
(C1-C6алкил)сульфонила, нитро, -сульфонил(СН2)0-4NR2R3 и -карбонил(СН2)0-4NR2R3;
R2 представляет собой водород или C1-C6алкил, необязательно замещенный гидрокси;
R3 представляет собой водород или C1-C6алкил, необязательно замещенный гидрокси, трифторметилом или пирролидинилом; или R2, R3 и азот, к которому они присоединены, образуют пиперидинил, пиперазинил, необязательно замещенный C1-C6алкилом, или морфолинил;
R4 представляет собой тиазолил, пиридинил или фенил, необязательно замещенный 1-3 заместителями, выбранными из группы, состоящей из галогена, амино, метила, трифторметила и нитро;
R5 представляет собой C(O)NHR6, OC(O)NHR6, NHC(O)CH2R6, NHC(O)NHR6 или C(S)NHR6;
n равно 0-4 для OC(O)NHR6, NHC(O)CH2R6, NHC(O)NHR6 и n равно 1-4 для С(О)NHR6 и C(S)NHR6 и
R6 представляет собой:
(а) незамещенный тетрагидробензотиазолил или
(b) фенил, пиридинил, пиримидинил, пиразолил, тиазолил, изотиазолил, тиадиазолил, изоксазолил, все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом; C2-C6алкенила, необязательно замещенного диметиламинокарбонилом, C1-C6алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано; C3-C6циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5-иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, C2-C6алкилкарбонила, морфолинилкарбонила и пиперазинилкарбонила;
или его фармацевтически приемлемые соли.
2. Соединение по п.1, где R5 представляет собой C(O)NHR6.
3. Соединение по п.1 или 2, где R1 представляет собой фенил, тиенил, тиазолил или пиридинил, все из которых необязательно замещены -(CH2)0-4NR2R3, C1-C6алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4алкокси, галогена, (C1-C6алкил)сульфонила, нитро, -сульфонил(СН2)0-4NR2R3 и
-карбонил(СН2)0-4NR2R3.
4. Соединение по п.1, представляющее собой 2-{2-фтор-4-[7-(2-метилпиридин-4-ил)имидазо[1,2-а]пиридин-3-ил]фенил}-N-[3-трифторметилфенил)ацетамид, или его фармацевтически приемлемая соль.
5. Соединение соединение по п.1, представляющее собой N-(5-трет-бутилизоксазол-3-ил)-2-[2-фтор-4-(7-пиридин-2-илимидазо[1,2-а]пиридин-3-ил)фенил]ацетамид, или его фармацевтически приемлемая соль.
6. Фармацевтическая композиция, содержащая соединение по любому из пп.1-5 в комбинации с фармацевтически приемлемым носителем, разбавителем или сольватом.
7. Применение соединения по любому из пп.1-5 в качестве лекарственного средства.
8. Применение соединения по любому из пп.1-5 для производства лекарственного средства для блокирования ангиогенеза.
9. Применение соединения по любому из пп.1-5 для производства лекарственного средства для лечения чувствительных новообразований.
Текст
011691 Нежелательный ангиогенез представляет собой отличительную черту некоторых заболеваний, таких как ретинопатии, псориаз, ревматоидный артрит, возрастная дегенерация желтого пятна и рак (солидные опухоли), (Folkman, Nature Med., 1, 27-31 (1995. Поскольку для сохранения жизнеспособности опухоли необходимо кровоснабжение, ангиогенез представляет собой критический компонент, способствующий развитию ракового заболевания (Е. Ruoslahti, Nature Rev. Cancer, 2, 83-90 (2002). Таким образом, создание новых средств для ингибирования ангиогенеза является перспективным подходом в лечении рака (R. Kerbel and J. Folkman, Nature Rev. Cancer, 2, 727-739 (2002). Другим потенциальным преимуществом ингибирования опухолевого ангиогенеза является то, что с помощью такого подхода можно избежать токсичных побочных эффектов или резистентности к лекарственным средствам, которую вызывают традиционные средства химиотерапии (Judah Folkman, Endogenous Inhibitors of Angiogenesis, TheHarvey Lectures, Series 92, p. 65-82, Wiley-Liss Inc., (1998. Одна из протеинкиназ, для которых продемонстрировано участие в процессе ангиогенеза, входит в состав семейства тирозинкиназ рецептора фактора роста под названием VEGF-R2 (рецептор 2 фактора роста эндотелия сосудов, также известный как KDR (рецептор домена вставки киназы. VEGF-R2, который экспрессируется прежде всего на эндотелиальных клетках, связывается с мощным ангиогенным фактором роста VEGF и опосредует последующее преобразование сигнала путем активации его внутриклеточной киназной активности. Поэтому ожидается, что прямое ингибирование киназной активностиVEGF-R2 будет приводить к уменьшению ангиогенеза даже в присутствии экзогенного VEGF (см.Strawn et al., Cancer Research, 56, 3540-3545 (1996, что было продемонстрировано на мутантахVEGF-R2, которые не способны опосредовать преобразование сигнала (Millauer et al., Cancer Research,56, 1615-1620 (1996. Кроме того, на важность VEGF-R2 в качестве мишени для фармакотерапии рака указывают недавние исследования (S. Rafii et al., Nature Rev. Cancer, 2, 826-835 (2002) and Y. Snaked etal., Cancer Cell, 7, 101-111 (2004, которые продемонстрировали, что VEGF-R2 экспрессируется на циркулирующих клетках-прекурсорах эндотелия из костного мозга, которые также могут способствовать ангиогенезу и росту опухолей. Кроме того, похоже, что VEGF-R2 не выполняет функций у взрослого,кроме опосредования ангиогенной активности VEGF. Было предложено лечить ангиогенез путем применения соединений, ингибирующих киназную активность VEGF-R2. Например, в международной публикации WIPO WO 97/34876 раскрыты некоторые производные циннолина, которые являются ингибиторами VEGF-R2 и заявлены как пригодные для лечения патологических состояний, связанных с аномальным ангиогенезом и/или повышенной проницаемостью сосудов, включая диабет (гипергликемию), псориаз, ревматоидный артрит, саркому Капоши,гемангиомы, острые и хронические нефропатии, атеромы, рестеноз артерий, аутоиммунные заболевания,острые воспаления, заболевания глаз с пролиферацией сосудов сетчатки и рак. Далее, в WO 03/092595 предложены некоторые имидазо[1,2-а]пиридинильные соединения, которые ингибируют, регулируют и/или модулируют преобразование сигнала тирозинкиназой. Однако по-прежнему существует потребность в эффективных ингибиторах протеинкиназ. Настоящее изобретение предлагает новые имидазо[1,2-а]пиридинильные соединения, которые ингибируют VEGF-R2 и, таким образом, полезны для лечения опосредованных или зависимых от VEGF-R2 заболеваний, например для лечения различных форм рака. Настоящее изобретение предлагает соединения формулы (I) Соединение формулы (I):(b) фенил, тиенил, тиазолил, имидазолил, пиразолил, триазолил, оксазолил, пиридинил,N-оксопиридинил или пиримидинил, все из которых необязательно замещены -(СН 2)0-4NR2R3,C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3;R2 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси;R3 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси, трифторметилом или пирролидинилом; или(b) фенил, пиридинил, пиримидинил, пиразолил, тиазолил, изотиазолил, тиадиазолил, изоксазолил,все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом; C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом,C1-C6 алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано; C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом;, 3,4-диметилизоксазол-5 иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, С 2-Сбалкилкарбонила,морфолинилкарбонила и пиперазинилкарбонила; или его фармацевтически приемлемые соли. Настоящее изобретение дополнительно предлагает способ ингибирования VEGF-R2 у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, ингибирующегоVEGF-R2 количества соединения формулы (I) или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно предлагает способ блокирования ангиогенеза, включающий введение млекопитающему, нуждающемуся в таком лечении, ингибирующего VEGF-R2 количества соединения формулы (I) или его фармацевтически приемлемой соли. Настоящее изобретение дополнительно предлагает способ лечения чувствительных новообразований у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, ингибирующего VEGF-R2 количества соединения формулы (I) или его фармацевтически приемлемой соли. Настоящее изобретение также предлагает способ лечения различных патологических состояний,включая гипергликемию, псориаз, ревматоидный артрит, саркому Капоши, гемангиомы, острые и хронические нефропатии, атеромы, рестеноз артерий, аутоиммунные заболевания, острые воспаления и заболевания глаз с пролиферацией сосудов сетчатки у млекопитающего, включающий введение млекопитающему, нуждающемуся в таком лечении, ингибирующего VEGF-R2 количества соединения формулы(I) или его фармацевтически приемлемой соли. Настоящее изобретение также предлагает фармацевтический препарат, содержащий соединение формулы (I) или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых эксципиентов. Настоящее изобретение также предлагает применение соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного средства для ингибирования VEGF-R2. Настоящее изобретение также предлагает применение соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного средства для блокирования ангиогенеза. Настоящее изобретение также предлагает применение соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного средства для лечения чувствительных новообразований. Настоящее изобретение также предлагает применение соединения формулы (I) или его фармацевтически приемлемой соли для производства лекарственного средства для лечения различных патологических состояний, выбранных из группы, состоящей из гипергликемии, псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острых и хронических нефропатий, атеромы, рестеноза артерий, аутоиммунных заболеваний, острого воспаления и заболеваний глаз с пролиферацией сосудов сетчатки. Общие химические термины, которые используются в формулах выше, имеют их обычные значения. Например, термин "C1-C6 алкил" включает алкилы с неразветвленной или разветвленной цепью и включает радикалы метил, этил, пропил, изопропил, бутил изобутил, втор-бутил, трет-бутил, пентил,изопентил и гексил. Термин "C2-C6 алкенил" включает алкиленовые группы с неразветвленной или разветвленной цепью и включает радикалы этиленил, пропиленил, изопропиленил, бутиленил, изобутиленил, втор-бутиленил,трет-бутиленил, пентиленил, изопентиленил и гексиленил. Термин "C3-C6 циклоалкил" включает радикалы циклопропил, циклобутил, циклопентил и циклогексил. Термин "C1-C6 алкокси" включает метокси, этокси, пропокси, изопропокси, бутокси, изобутокси,втор-бутокси, трет-бутокси, пентокси, изопентокси и гексокси. Термин "галоген" означает хлор, бром, фтор или йод. Термин "(C1-C6 алкил)сульфонил" означает сульфонильную группу, замещенную C1-C6 алкильной группой. Термин "млекопитающее" означает различные теплокровные позвоночные животные класса млекопитающих (Mammalia), наиболее предпочтительно человека, характеризующихся наличием волосяного покрова на коже и, у особей женского пола, вырабатывающих молоко молочных желез для вскармлива-2 011691 ния потомства. Термин "чувствительное новообразование" означает новообразование, для которого выживание,рост или метастазирование зависит от VEGF-R2. Хотя все соединения формулы (I) представляют собой пригодные ингибиторы VEGF-R2, некоторые классы соединений являются предпочтительными. В следующих абзацах описаны такие предпочтительные классы:d) R1 представляет собой замещенный пиридинил;e) R1 представляет собой замещенный пиридин-2-ил;f) R1 представляет собой замещенный пиридин-4-ил;n) R4 представляет собой замещенный фенил; о) R4 представляет собой галогенфенил; р) R4 представляет собой хлорфенил;x) R6 представляет собой замещенный пиразолил; у) R6 представляет собой пиразолил, замещенный C1-C6 алкилом;z) R6 представляет собой 5-трет-бутилпиразол-3-ил; аа) R6 представляет собой замещенный фенил;dd) R6 представляет собой замещенный тиадиазолил; ее) R6 представляет собой тиадиазолил, замещенный C1-C6 алкилом;gg) R6 представляет собой замещенный изоксазолил;kk) R6 представляет собой замещенный тиазолил;qq) R6 представляет собой замещенный пиридинил;zz) R1 представляет собой пиридинил, R4 представляет собой фторфенил, n равно 1, R5 представляет собой CONHR6 и R6 представляет собой 4-морфолинилметил-5-трет-бутилтиазол-2-ил; ааа) соединение формулы (I), которое представляет собой 2-[4-(7-пиридин-4-илимидазо[1,2 а]пиридин-3-ил)фенил]-N-(3-трифторметилфенил)ацетамид;bbb) соединение формулы (I), которое представляет собой N-(5-трет-бутил[1,3,4]тиадиазол-2-ил)-2[2-фтор-4-(7-пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фенил]ацетамид; ссс) соединение формулы (I), которое представляет собой 2-[2-фтор-4-(7-пиридин-4-илимидазо[1,2 а]пиридин-3-ил)фенил]-N-(5-изопропил-4-метилтиазол-2-ил)ацетамид;ddd) соединение формулы (I), которое представляет собой N-(4-трет-бутил-6-морфолин-4 илметилпиридин-2-ил)-2-[2-фтор-4-(7-пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фенил]ацетамид; еее) соединение формулы (I), которое представляет собой 2-(2-фтор-4-7-[2-(2-морфолин-4 илпропил)пиридин-4-ил]имидазо[1,2-а]пиридин-3-илфенил)-N-(4-трифторметилпиридин-2-ил)ацетамид;fff) соединение формулы (I), которое представляет собой N-[4-диметиламинометил-5-(1 метилциклопропил)тиазол-2-ил]-2-[2-фтор-4-(7-пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фенил]ацетамид;ggg) соединение формулы (I), которое представляет собой 2-[2-фтор-4-(7-пиридин-4-илимидазо[1,2 а]пиридин-3-ил)фенил]-N-[5-трет-бутил-4-морфолин-4-илметилтиазол-2-ил]ацетамид;hhh) соединение по п.1, где R1 представляет собой фенил, тиенил, тиазолил или пиридинил, все из которых необязательно замещены -(СН 2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино,пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена, (C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и-(СН 2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом,или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил) сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3; и R6 представляет собой фенил, пиридинил, пиримидинил, пиразолил, тиазолил, изотиазолил, тиадиазолил, изоксазолил,все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом, C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом,C1-C6 алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано, C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5 иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, C2-C6 алкилкарбонила,морфолинилкарбонила и пиперазинилкарбонила;jjj) соединение по п.1, где R1 представляет собой 2-пиридинил, который необязательно замещен (СН 2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом,или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил) сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3; и R6 представляет собой фенил, пиридинил, тиазолил, изотиазолил, тиадиазолил или изоксазолил, все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила,необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом, C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом, C1-C6 алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано, C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5-иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, C2-C6 алкилкарбонила, морфолинилкарбонила и пиперазинилкарбонила;-(СН 2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом,или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3; и R6 представляет собой фенил, пиридинил, пиразолил, тиазолил, изотиазолил, тиадиазолил или изоксазолил, все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей изlll) соединение по п.1, где R1 представляет собой тиенил или тиазолил, который необязательно замещен -(СН 2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена, (C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(CH2)0-4NR2R3; и R6 представляет собой фенил, пиразолил, тиазолил, изотиазолил, тиадиазолил или изоксазолил, все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из-(CH2)0-4NR2R3, C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом,или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3; и R6 представляет собой фенил, тиазолил, изотиазолил, тиадиазолил или изоксазолил, все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом,C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом, C1-C6 алкокси, трифторметилом,дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано, C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5-иламиносульфонила, тетрагидропиранила,тетрагидропираниламинокарбонила, C2-C6 алкилкарбонила, морфолинилкарбонила и пиперазинилкарбонила;nnn) соединение формулы (I), которое выбрано из группы, состоящей из гидрохлоридной, дигидрохлоридной, сукцинатной, L-тартратной и гидробромидной соли; ооо) соединение формулы (I), которое выбрано из группы, состоящей из гидрохлоридной и гидробромидной соли; и ррр) соединение формулы (I), которое представляет собой соль сукцинат. Следует понимать, что приведенные выше классы могут быть комбинированы с образованием дополнительных предпочтительных классов. Другой предпочтительный класс по настоящему изобретению представляет собой соединения формулы (II)(b) фенил, тиенил, тиазолил, имидазолил, пиразолил, триазолил, оксазолил, пиридинил,N-оксопиридинил или пиримидинил, все из которых необязательно замещены -(СН 2)0-4NR2R3,C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3;R2 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси;R3 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси, трифторметилом или пирролидинилом; или(b) фенил, пиридинил, пиримидинил, пиразолил, тиазолил, изотиазолил, тиадиазолил, изоксазолил,все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пипе-5 011691 ридинилом или морфолинилом, C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом,C1-C6 алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано, C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5 иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, C2-C6 алкилкарбонила,морфолинилкарбонила и пиперазинилкарбонила; или их фармацевтически приемлемые соли. Другой предпочтительный класс по настоящему изобретению представляет собой соединения формулы (III)(b) фенил, тиенил, тиазолил, имидазолил, пиразолил, триазолил, оксазолил, пиридинил,N-оксопиридинил или пиримидинил, все из которых необязательно замещены -(СН 2)0-4NR2R3,C1-C6 алкилом, необязательно замещенным амино, пирролидинилом или морфолинилом, или 1-2 заместителями, независимо выбранными из группы, состоящей из C1-C4 алкокси, галогена,(C1-C6 алкил)сульфонила, нитро, -сульфонил(СН 2)0-4NR2R3 и -карбонил(СН 2)0-4NR2R3;R2 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси;R3 представляет собой водород или C1-C6 алкил, необязательно замещенный гидрокси, трифторметилом или пирролидинилом; или(b) фенил, пиридинил, пиримидинил, пиразолил, тиазолил, изотиазолил, тиадиазолил, изоксазолил,все из которых необязательно замещены 1-3 заместителями, независимо выбранными из группы, состоящей из C1-C6 алкила, необязательно замещенного гидрокси, диметиламино, пирролидинилом, пиперидинилом или морфолинилом, C2-C6 алкенила, необязательно замещенного диметиламинокарбонилом,C1-C6 алкокси, трифторметилом, дифторметокси, трифторметокси, диметиламиноэтокси, фенокси, толилом, галогеном, метилсульфонилом, диметиламино, диэтиламино, циано, C3-C6 циклоалкила, необязательно замещенного гидрокси, метокси, метоксиэтокси или метилом, 3,4-диметилизоксазол-5 иламиносульфонила, тетрагидропиранила, тетрагидропираниламинокарбонила, C2-C6 алкилкарбонила,морфолинилкарбонила и пиперазинилкарбонила; или их фармацевтически приемлемые соли. Другой вариант осуществления настоящего изобретения представляет собой соединение формулы(b) фенил, тиенил, тиазолил, тиадиазолил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изоксазолил, пиридинил или пиримидинил, все из которых необязательно могут быть замещены-(СН 2)0-4NR2R3 или 1-2 заместителями, независимо выбранными из группы, состоящей изR2 представляет собой водород или C1-C6 алкил;R3 представляет собой водород, C1-C6 алкил, необязательно замещенный пирролидинилом, пиперидинилом, пиперазинилом, который необязательно замещен C1-C4 алкилом, или морфолинилом; илиR4 представляет собой тиазолил, тиенил, пиридинил или фенил, необязательно замещенный одним или двумя атомами галогена;R6 представляет собой фенил, необязательно замещенный 1-3 заместителями, независимо выбранными из группы, состоящей из галогена, C1-C6 алкила, C1-C4 алкокси, циано, диметиламино, дифторметокси, трифторметила, трифторметокси, метилсульфонила, диметилизоксазолилсульфонамида и-O-фенила, пиридинила, необязательно замещенного трифторметилом, пиразолила, необязательно замещенного 1-2 заместителями, независимо выбранными из группы, состоящей из толила и C1-C6 алкила,который необязательно замещен гидрокси, пиперидинилом, пирролидинилом или морфолинилом,(C1-C6 алкил)изоксазолила, (C1-C6 алкил)тиазолила, (C1-C6 алкил)изотиазолила, (C1-C6 алкил)тиадиазолила;R7 представляет собой Н или C1-C6 алкил или его фармацевтически приемлемые соли. Соединения по настоящему изобретению представляют собой ингибиторы VEGF-R2, протеинкиназа которого показана как принимающая участие в процессе ангиогенеза. VEGF-R2, который экспрессируется прежде всего на эндотелиальных клетках, связывается с мощным ангиогенным фактором ростаVEGF и опосредует последующее преобразование сигнала путем активации его внутриклеточной киназной активности. Таким образом, специалисту в данной области будет понятно, что прямое ингибирование киназной активности VEGF-R2 будет приводить к уменьшению ангиогенеза и, таким образом, будет воздействовать на лечение состояний, характерным признаком которых является аномальный ангиогенез и/или повышенная проницаемость сосудов, например диабета (гипергликемии), псориаза, ревматоидного артрита, саркомы Капоши, гемангиомы, острых и хронических нефропатий, атеромы, рестеноза артерий,аутоиммунных заболеваний, острого воспаления, заболеваний глаз с пролиферацией сосудов сетчатки и рака. Применимость ингибиторов ангиогенеза при лечении рака известна из литературы, см., например,Rak et al. Cancer Research, 55:4575-4580, 1995. Роль ангиогенеза в патогенезе рака продемонстрирована для многочисленных видов рака и тканей: карцинома молочной железы (G. Gasparinin and A.L. Harris, J.Clin. Oncol., 1995, 13:765-782; M. Toi et al. Japan. J. Cancer Res., 1994, 85:1045-1049); карциномы мочевого пузыря (A.J. Dickinson et al., Br. J. Urol., 1994, 74:762-766), карциномы ободочной кишки (L.M. Ellis et al.,Surgery, 1996, 120(5): 871-878); а также опухоли ротовой полости (J.K. Williams et al., Am J. Surg., 1994,168:373-380). Предпочтительные новообразования, которые, как ожидается, будут чувствительны к лечению соединениями по настоящему изобретению, включают следующие виды рака: рак мозга, мочеполовой системы, лимфатической системы, желудка, гортани, легкого (включая мелкоклеточный и немелкоклеточный), поджелудочной железы, молочной железы, предстательной железы, гистиоцитарный,лимфома, гепатоцелюлярный рак, рак женских половых органов (например, яичника), саркома Капоши,опухоли ЦНС (включая астроцитомы, медуллобластомы и менингиомы), рак толстого кишечника и прямой кишки, рак головы и шеи, меланому, хроническую лимфоцитную лейкемию, множественную миелому, острую миелоидную лейкемию (ОМЛ), хроническую миелоидную лейкемию (ХМЛ), злокачественную мезотелиому, миелодиспластический синдром (например, истинную полицитемию, тромбоцитемию), рак желудка и почечно-клеточную карциному. Специалисту в данной области будет понятно, что введение некоторых заместителей будет создавать асимметрию в соединениях формулы (I). Настоящее изобретение охватывает все энантиомеры и смеси энантиомеров и стереоизомеров, как в стереомерно чистой форме (например, в виде чистых геометрических изомеров, чистых энантиомеров или чистых диастереомеров), так и в виде смесей энантиомеров и стереоизомеров. Смеси энантиомеров и стереоизомеров могут быть разделены на отдельные компоненты: энантиомеры или стереоизомеры с помощью хорошо известных методов, например высокоэффективной жидкостной хроматографией с хиральной фазой или кристаллизацией соединения в виде комплекса хиральной соли. Энантиомеры и стереоизомеры также могут быть получены из стереомерно или энантиомерно чистых промежуточных продуктов, реагентов и катализаторов хорошо известными способами асимметричного синтеза. Предусматривается, что фармацевтически приемлемые соли входят в объем настоящего изобретения. Соединения по настоящему изобретению представляют собой основания, и могут быть образованы соли таких соединений с неорганическими или органическими кислотами, например, гидрохлорид, дигидрохлорид, сукцинат, L-тартрат и гидробромид, причем гидрохлорид, дигидрохлорид и сукцинат являются предпочтительными. Соединения по настоящему изобретению могут быть получены с использованием реакций, показанных на следующих схемах, в дополнение к другим стандартным методам, которые известны из литературы или примеры которых приведены в экспериментальных методиках. Таким образом, приведенные схемы не ограничиваются перечисленными соединениями или какими-либо конкретными заместителями, которые использованы для целей иллюстрации.-7 011691 Следующие схемы используются для получения соединений формулы (I), где R5 представляет собой NHC(O)CH2R6 или NHC(O)NHR6. Если не указано иное, все остальные переменные являются такими, как определено выше. Схема I Соединения мочевины (АЕ) формулы (I) обычно могут быть получены реакцией амина (АА) с карбаматом (АВ), карбамоилхлоридом (АС) или арилизоцианатом (AD), все из которых являются коммерчески доступными или непосредственно получены по способам, известным специалистам в данной области. Амидные (AG) соединения формулы (I) обычно могут быть получены реакцией амина (АА) с ацилхлоридом (AF). Требуемый промежуточный амин (АА) может быть получен из известного 7-хлоримидазо[1,2 а]пиридина (АН), как показано на схеме II. X представляет собой Cl, Br или I, и Y представляет собой бороновую кислоту, бороновый эфир или триалкилстаннан. Если не указано иное, все другие переменные являются такими, как определено выше. Схема II Арильную группу вводят в положение 7 имидазо[1,2-а]пиридиновой группы путем катализируемого Pd перекрестного сочетания или через образование боронового эфира с получением биарильного соединения (AI). Данное промежуточное соединение (AI) селективно галогенируют с помощью NIS илиNBS в положении 3 с последующей второй реакцией перекрестного сочетания в этом положении. Амино- или аминоалкильная группа или скрытая амино или аминоалкильная группа представляет собой заместитель арилборонатного партнера для сочетания. Альтернативно, реакции сочетания могут быть осуществлены сначала в положении 3, и затем в положении 7 посредством (AK) и (AL), как показано на-8 011691 схеме II. Также следует отметить, что реакция перекрестного сочетания, которую используют для введения арильной группы в положение 7 имидазо[1,2-а]пиридина, может происходить в конце цепи реакций,после образования мочевины (не показано). Амиды формулы (I) могут быть получены, как показано на схеме III. Если не указано иное, все переменные являются такими, как определено выше. Схема III Катализируемое Pd(0) перекрестное сочетание (AJ) или (AK) с фенилбороновой кислотой, которое осуществляют со связанной с алкилом карбоновой кислотой (или защищенной кислотой), дает (AM). Карбоксилатную группу сочетают с амином с использованием стандартных способов, хорошо известных специалистам в данной области, с образованием (AN). Альтернативно, амид (АР) может быть получен из галогенфенильной кислоты (АО) с использованием стандартных способов, перед сочетанием арильной группы с имидазо[1,2-а]пиридиновым кольцом. В этом случае галогеновый заместитель на арильной группе (АР) превращают в бороновый эфир (AQ) для сочетания с (AJ) или (AK), как показано на схемеIII. В случаях, когда галоген занимает положение 7 имидазо[1,2-а]пиридинового кольца в (AN), осуществляют второе катализируемое Pd(0) сочетание с получением заявляемого соединения, как описано выше на схеме I. Карбаматы формулы (I) могут быть получены, как показано на схеме IV. Если не указано иное, все переменные являются такими, как определено выше. Схема IV Карбаматы (AR) образуют путем объединения спирта (AM') с изоцианатом. Синтез такого исходного вещества (AM') осуществляют путем катализируемого Pd(0) перекрестного сочетания в положении 3 имидазо[1,2-а]пиридина (AK или AJ) со спиртом, таким как гидроксилметилфенилбороновая кислота. Арильная группа в положении 7 имидазо[1,2-а]пиридина может быть введена путем второго катализируемого Pd(0) сочетания в любой точке, как показано для производных мочевины. Соединения формулы (I), где R1 представляет собой пиридонил, замещенный метоксипиридинил или ди(замещенный)аминопиридинил, могут быть получены, как показано на схеме V. Если не указано иное, все другие переменные являются такими, как определено выше. Схема V Диалкиламиногруппа может быть введена путем нуклеофильного ароматического замещения 2 фторпиридильной группы (AS) с получением (AT) в конце синтеза. Подобная стратегия с использованием кислотного метанолиза дает (AU), и расщепление метилового эфира с помощью HBr обеспечивает пиридонильную группу (AV). Арилбромиды на схеме II, R1X могут быть получены, как описано на схеме VI. R7 является таким,как определено выше. Схема VI Алкил- или диалкиламинометилпиридильная группа может быть введена в качестве заместителя на арильной группе в положении 7 имидазо[1,2-а]пиридина с помощью алкилирования 5-бром-2-бромметилпиридина (AW) с использованием вторичного амина, как показано на схеме VI. Далее арилбромид(ы) могут непосредственно принимать участие в описанных выше реакциях. Схема VII Соединения класса амидов также можно получить, как показано на схеме VII, с помощью катализируемой Pd(0) реакции арилирования промежуточных соединений 3-Н имидазо[1,2-а]пиридина (АН) или(AI) с защищенным (PG) эфиром бромфенилуксусной кислоты (АХ) с образованием связанной кислоты(AY). трет-Бутильная группа представляет собой подходящую защитную группу для сложного эфира,показанную как PG, которую впоследствии отщепляют в кислых условиях. Далее может быть образован амид (AN) с использованием оксалилхлорида/ДМФА с образованием ацилхлорида in situ или с использованием реагентов для сочетания DMTMM/NMM или HATU/диизопропилэтиламин. Альтернативно, амиды (ВА) получают из бромарилкислот (AZ) с использованием описанных выше методик образования амида и осуществляют сочетание ВА с помощью катализируемой Pd(0) реакции арилирования с (АН) или (AI). В случаях, когда катализируемую Pd(0) реакцию арилирования осуществляют с помощью (АН), последовательную обработку осуществляют в замещенном Cl положении, как описано раньше. Специалисту в данной области будет понятно, что не все заместители в соединении формулы (I) способны выдержать определенные условия реакции, которые применяются для синтеза соединений. Указанные фрагменты могут быть введены в традиционной точке синтеза или могут быть защищены с последующим снятием защиты при необходимости или по желанию. Специалисту в данной области будет понятно, что защитные группы могут быть удалены в любой традиционной точке в ходе синтеза соединений по настоящему изобретению. Способы введения и удаления защитных групп для азота и кислорода хорошо известны в данной области; см., например, Greene and Wuts, Protective Groups in OrganicSynthesis, 3rd Ed., John WileySons, New York, Chapter 7 (1999). Кроме того, специалисту будет понятно,что во многих обстоятельствах порядок введения фрагментов не является критическим. Конкретный порядок стадий, необходимый для получения соединений формулы (I), может зависеть от конкретного соединения, которое должно быть синтезировано, исходного соединения и относительной лабильности замещенных фрагментов. СокращенияISCO - высокоэффективная жидкостная хроматография ISCO;A. 2-(5-Амино-3-трет-бутилпиразол-1-ил)этанол. К раствору 4,4-диметил-3-оксопентаннитрила (5,0 г, 0,04 моль) в абсолютном этаноле (50 мл) добавляют 2-гидразиноэтанол (3 мл, 1,1 экв.) и концентрированную HCl (0,5 мл). Реакционную смесь кипятят с обратным холодильником в течение 4 ч, затем охлаждают до комнатной температуры и разбавляют водой и этилацетатом. Органические фракции промывают водой, затем насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Растирание с гексаном и дихлорметаном дает белое твердое вещество (4,8 г, 66%). МС (ES), m/z 184 (М+1).B. 5-трет-Бутил-2-[2-(трет-бутилдиметилсиланилокси)этил]-2 Н-пиразол-3-иламин. К 2-(5-амино-3-трет-бутилпиразол-1-ил)этанолу (3,42 г, 0,019 моль) добавляют TBDMCCl (3,38 г,1,2 экв.) и имидазол (3,18 г, 2,5 экв.) в ДМФА (7 мл) и перемешивают в течение ночи при комнатной температуре в атмосфере N2. Реакционную смесь разбавляют этилацетатом и водой. Промывают органический слой водой, затем насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением твердого вещества (5,5 г, 99%), которое используют без дополнительной очистки. МС (ES), m/z 298 (М+1). Следующие промежуточные соединения получают в соответствии с общей методикой, описанной в заявке РСТ WO 200026202 от 11 мая 2000 г., поданной 27 октября 1999 г. (стр. 52). Получение 6. 2,2,2-Трихлорэтиловый эфир 2-[2-(трет-бутилдиметилсиланилокси)этил]-(5-третбутил-2 Н-пиразол-3-ил)карбаминовой кислоты. Растворяют 5-трет-бутил-2-[2-(трет-бутилдиметилсиланилокси)этил]-2 Н-пиразол-3-иламин (5,5 г,0,018 моль) в ТГФ (100 мл) в атмосфере N2 и охлаждают до 0 С. С помощью шприца добавляют пиридин(1,6 мл, 1,1 экв.) с последующим добавлением по каплям 2,2,2-трихлорэтилхлорформиата (2,7 мл,1,1 экв.). Реакционную смесь перемешивают при 0 С, выдерживая при этой температуре в течение 1 ч,затем охлаждающую баню удаляют и реакционную смесь перемешивают в течение в общей сложности 5 ч. Реакционную смесь разбавляют этилацетатом и водой. Промывают органический слой водой, затем- 12011691 насыщенным водным раствором хлорида натрия, затем сушат над сульфатом магния, фильтруют и концентрируют в вакууме с получением остатка (8,7 г, 100%), который используют без дополнительной очистки. МС (ES), m/z 474 (М+1).- 13011691 С использованием методики, аналогично описанной в получении 6, получают следующие промежуточные соединения из коммерчески доступных исходных соединений или соединений, описанных в получениях 1-5.N-йодсукцинамид. Перемешивают в течение 30 мин. Осадок отфильтровывают и затем промывают ацетонитрилом. Перекристаллизовывают осадок из ацетонитрила с получением белого твердого вещества. Фильтрат концентрируют, разбавляют этилацетатом, промывают 10% раствором гидросульфита натрия (NaHSO3),насыщенным водным раствором бикарбоната натрия, насыщенным водным раствором NaCl, сушат надMgSO4, фильтруют и выпаривают. Объединенные твердые вещества используют без дополнительной очистки (8,0 г, 73%). 1 Н ЯМР (ДМСО) : 8,33 (д, 1 Н, J=7,3 Гц), 7,79 (д, 1 Н, J=2,0 Гц), 7,72 (с, 1 Н), 7,07 (дд, 1 Н, J=7,3 и 2,0 Гц). Получение 23. Диметиламид 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиразол-1 сульфоновой кислоты. А. Диметиламид пиразол-1-сульфоновой кислоты. Растворяют 4-йодпиразол (9,24 г, 48,0 ммоль) в ТГФ (100 мл), порциями добавляют NaH (2,26 г,63,6 ммоль, 60% в масле) и перемешивают в течение 30 мин при 0 С. По каплям добавляют диметилсульфамоилхлорид (6,18 мл, 57,6 ммоль) и перемешивают в течение 1 ч при 0 С и 1 ч при комнатной температуре. Гасят реакцию насыщенным раствором NaHCO3 и экстрагируют CH2Cl2, сушат над безводным MgSO4, фильтруют и концентрируют. Хроматографируют остаток смесью гексан/этилацетат от 1:0 до 2:1 с получением прозрачного масла (выход 11,8 г, 82%). МС (ES), m/z 176 (M+1). В. Диметиламид 4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)пиразол-1-сульфоновой кислоты. Объединяют диметиламид пиразол-1-сульфоновой кислоты (7,0 г, 23,2 ммоль), ацетат калия (6,3 г,69,8 ммоль), бис-(пинаколят)дибор (6,5 г, 25,5 ммоль) и ДМСО (140 мл) и дегазируют струей азота. Добавляют PdCl2(dppf)CH2Cl2 (0,51 г, 0,70 ммоль) и нагревают реакционную смесь до 80 С, выдерживая при этой температуре в течение ночи. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. Объединенные органические фракции промывают водой и сушат над безводным MgSO4, фильтруют и концентрируют. Используют указанное в заголовке соединение в виде смеси с примесью 33% бипиразола. Получение 24. 4-Бром-2-этилпиридин. Указанное в заголовке соединение получают в соответствии с общей методикой, описанной вJournal Organic Chemistry, vol. 50, No. 22, 1985, p. 4410-4411. Суспендируют гидрохлорид 4-бромпиридина (4,17 г, 0,021 моль) в ТГФ (100 мл) и охлаждают до -78 С в атмосфере азота. С помощью шприца по каплям добавляют этилмагнийбромид (15,7 мл 3,0 М раствора в диэтиловом эфире,2,2 экв.). Реакционную смесь перемешивают при -78 С, выдерживая при этой температуре в течение 10 мин, затем ледяную баню удаляют и дают реакционной смеси нагреться до комнатной температуры. Гасят реакцию 20% водным раствором хлорида аммония, затем разбавляют диэтиловым эфиром. Органические фракции промывают водой, 1 н. HCl (водн.), затем насыщенным водным раствором хлорида натрия. Органические фракции сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Снова растворяют остаток в толуоле (100 мл) и помещают в атмосферу азота. Растворяют тетрахлор-1,2 бензохинон (5,8 г, 1,1 экв.) в уксусной кислоте (50 мл) и по каплям добавляют к реакционной смеси. Реакционную смесь перемешивают в течение ночи при комнатной температуре. Подщелачивают добавлением 1 н. NaOH (водн.), затем экстрагируют этилацетатом. Подкисляют органическую фракцию 1 н. HCl(водн.) и экстрагируют этилацетатом. Органические фракции отбрасывают. Подщелачивают водный слой 1 н. NaOH (водн.) и экстрагируют ДХМ. Промывают полученную органическую фракцию водным насыщенным раствором хлорида натрия, затем сушат над сульфатом магния. Фильтруют и концентрируют в вакууме с получением коричневого остатка (2,64 г, 66%). ЖХМС (ES), m/z 188 (М+1, характерный для бромида). Следующее соединение получают согласно методике, аналогично описанной в получении 24. Получение 26. 1-(4-Хлорпиридин-2-ил)пропан-2-он. Указанное в заголовке соединение получают с помощью методики, аналогично описанной в получении 162 А ниже, с использованием 4-хлор-2-метилпиридина.- 15011691 Получение 27. 3-Йод-7-пиридин-2-илимидазо[1,2-а]пиридин. А. 7-Пиридин-2-илимидазо[1,2-а]пиридин. В круглодонную колбу добавляют 7-хлоримидазо[1,2-а]пиридин (0,25 г, 1,6 ммоль), трициклогексилфосфин (55 мг, 0,12 экв.), ацетат калия (0,24 г, 1,5 экв.), бис-(пинаколят)дибор (0,46 г, 1,1 экв.) и диоксан (10 мл). Полученную смесь деоксигенируют N2, затем добавляют трис-(дибензилиденацетон)дипалладий(0) (75 мг, 0,05 экв.) и нагревают реакционную смесь до 80 С, выдерживая при этой температуре в течение ночи в атмосфере N2. Реакционную смесь фильтруют через Celite и промывают ДХМ, затем концентрируют досуха. К полученному остатку добавляют 2-бромпиридин (0,14 мл,1,5 ммоль), S-Phos (75 мг, 0,125 экв.), фосфат калия (0,62 г, 2 экв.), диоксан (10 мл) и воду (5 мл). Полученную смесь тщательно деоксигенируют N2, добавляют ацетат палладия(II) (16 мг, 0,05 экв.) и кипятят реакционную смесь с обратным холодильником в течение ночи. Реакционную смесь концентрируют досуха и суспендируют в ДХМ. Полученную суспензию фильтруют через Celite и промывают ДХМ. Фильтрат концентрируют, затем очищают на колонке с диоксидом кремния (градиент от EtOAc до 5% МеОН/ДХМ) с получением остатка (0,325 г, выход 100%). МС (ES), m/z. 196 (М+1). В. 3-Йод-7-пиридин-2-илимидазо[1,2-а]пиридин. Растворяют 7-пиридин-2-илимидазо[1,2-а]пиридин (0,3 г, 1,5 ммоль) в абсолютном этаноле (10 мл) и добавляют NIS (0,35 г, 1 экв.). Нагревают реакционную смесь до 50 С, выдерживая при этой температуре в течение 30 мин в атмосфере N2, затем разбавляют этилацетатом. Промывают органическую фракцию 1 н. NaOH с последующим промыванием насыщенным раствором NaCl. Сушат органическую фракцию над сульфатом магния, фильтруют и концентрируют с получением светло-рыжевато-коричневого твердого вещества (0,4 г, 82%). МС (ES), m/z 322 (М+1). Следующие соединения получают по методике, аналогично описанной в получении 27.A. 4-Имидазо[1,2-а]пиридин-7-ил-1 Н-пиридин-2-он. Данное промежуточное соединение получают по методикам аналогично получению 27, за исключением использования 4-бром-2-фторпиридина в качестве партнера для сочетания. МС (ES), m/z 214 (M+1). Промежуточное соединение 2-фторпиридин нагревают в круглодонной колбе с 5 н. HCl в атмосфере N2 до 80 С, выдерживая при этой температуре в течение 2 ч. Охлаждают,добавляют 50 мл 2 М NH3 в МеОН и ДХМ, переносят в делительную воронку и экстрагируют ДХМ(10). Фильтруют водную фракцию и объединяют ДХМ экстракты, отгоняя ДХМ при пониженном давлении с получением 1,26 г продукта (общий выход 26%) МС (ES), m/z 212 (M+1).B. 4-Имидазо[1,2-а]пиридин-7-ил-1-(3-пиперидин-1-илпропил)-1 Н-пиридин-2-он В круглодонную колбу в атмосфере азота помещают (850 мг, 4,0 ммоль), 1-(3-хлорпропил)пиперидин HCl (1,18 г, 6 ммоль), ДМФА (40 мл) и Cs2CO3 (2,8 г, 8,8 ммоль), NaI (4 50 мг, 3 ммоль) и нагревают до 78 С, выдерживая при этой температуре в течение 24 ч. Реакционную смесь фильтруют и промывают твердые вещества ДХМ. Объединяют ДХМ и твердые вещества и концентрируют при пониженном давлении, затем очищают на колонке SCW VARIAN (интенсивного катионного обмена) (10 г),предварительно промытую водой и метанолом, продукт элюируют смесью (20%) 2 н. NH3 в метаноле/(80%) ДХМ. Выпаривают растворитель из фракций, содержащих продукт, при пониженном давлении. Хроматографируют с использованием (40 г ISCO) SiO2, элюируя градиентом от 0 до 10% 2 М NH3 в МеОН, уравновешивая ДХМ. Выпаривают растворители с получением твердого вещества цвета слоновой кости (410 мг, 30%). МС (ES), m/z 337 (M+1). В. 4-(3-Бромимидазо[1,2-а]пиридин-7-ил)-1-(3-пиперидин-1-илпропил)-1 Н-пиридин-2-он. Указанное в заголовке соединение получают по методике, аналогично описанной в получении 78 В,за исключением того, что в качестве растворителей используют EtOH и ацетонитрил. МС (ES), m/z 415, 417 изотопы Br (M+1). Получение 39. 7-[2-(3,3-Диэтоксипропил)пиридин-4-ил]имидазо[1,2-а]пиридин. В атмосфере азота в круглодонную колбу помещают 9-BBN (0,5 М в ТГФ, 42,2 мл, 2 экв.). С помощью шприца добавляют акролеиндиэтилацеталь (3,4 мл, 2,1 экв.) и перемешивают при комнатной температуре в течение ночи. На второй день в отдельной колбе объединяют 7-(2-хлорпиридин-4 ил)имидазо[1,2-а]пиридин (2,42 г, 11 ммоль), фосфат калия (4,47 г, 2 экв.), S-Phos (0,54 г, 12,5 мол.%),диоксан (90 мл) и воду (45 мл). Дегазируют азотом, затем добавляют ацетат палладия(II) (0,118 г,5 мол.%) в атмосфере азота. К полученному раствору через канюлю добавляют вещество из первой колбы. Реакционную смесь нагревают до 80 С, выдерживая при этой температуре в течение 5,5 ч. Концентрируют досуха. Суспендируют в ДХМ, затем фильтруют для удаления нерастворимых веществ. Промывают ДХМ. Фильтрат концентрируют и очищают на слое диоксида кремния (гексан/этилацетат 1:1 этилацетат 5% метанол/ДХМ 10% метанол/ДХМ) с получением остатка (4,15 г, 100%), который используют, как таковой, на следующей стадии. МС (ES), m/z 326 (М+1). Получение 40. 7-[2-(2-Морфолин-4-илпропил)пиридин-4-ил]имидазо[1,2-а]пиридин. Растворяют 1-(4-имидазо[1,2-а]пиридин-7-илпиридин-2-ил)пропан-2-он (3,9 г, 15,5 ммоль) в метаноле (400 мл) в атмосфере азота. Добавляют гидрохлорид морфолина (38,4 г, 20 экв.) с последующим добавлением порошка молекулярных сит 3 (7,8 г, высушенные в вакуумной печи при температуре 100 С в течение ночи). Перемешивают в течение 5 мин, затем с помощью шприца добавляют цианоборогидгид натрия (1,0 М в ТГФ, 28 мл, 1,8 экв.) и перемешивают при комнатной температуре в течение 5 дней. Фильтруют для удаления нерастворимых веществ и промывают метанолом. Концентрируют досуха, затем подщелачивают 20% NaOH (водн.) и экстрагируют этилацетатом. Органические фракции промывают насыщенным водным раствором хлорида натрия, затем сушат над MgSO4. Фильтруют и концентрируют, затем очищают на силикагеле (этилацетат 5% метанол/ДХМ 10% метанол/ДХМ 5% 2 МNH3 в метаноле/ДХМ 10% 2 М NH3 в метаноле/ДХМ) с получением продукта в виде рыжеватокоричневого твердого вещества (3,7 г, 74%). МС (ES), m/z 323 (М+1).- 17011691 Следующее соединение получают с использованием методик, аналогично описанным в получении 40. Получение 42. трет-Бутиловый эфир [2-(4-имидазо[1,2-а]пиридин-7-илпиридин-2-ил)-1-метилэтил]карбаминовой кислоты. Растворяют 2-(4-имидазо[1,2-а]пиридин-7-илпиридин-2-ил)-1-метилэтиламин (0,1 г, 0,4 ммоль) в смеси ТГФ (10 мл) и ДХМ (10 мл) в атмосфере азота. Добавляют ди-трет-бутилдикарбонат (0,095 г,1,1 экв.). Перемешивают в течение 45 мин при комнатной температуре, затем разбавляют этилацетатом. Органические фракции экстрагируют водой, 1 н. NaOH (водн.), затем водным раствором хлорида натрия. Органические фракции сушат над MgSO4, затем фильтруют и концентрируют. Очищают на силикагеле(10% метанол/ДХМ) с получением продукта (0,125 г, 89%). МС (ES), m/z 353,2 (М+1). Получение 42 А. 3-Йод-7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин. А. 7-(4-Метансульфонилфенил)имидазо[1,2-а]пиридин. Нагревают смесь 7-хлоримидазо[1,2-а]пиридина (100 г, 152,5 ммоль, 1 экв.), 4-метилсульфонилфенилбороновой кислоты (157,3 г, 786,8 ммоль, 1,2 экв.), Pd(PPh3)4 (19 г, 16,3 ммоль, 0,025 экв.) и карбоната цезия (472,4 г, 1,44 моль, 2,2 экв.) в смеси безводного ДМЭ (2000 мл) и EtOH (1000 мл) при 80 С в атмосфере N2 в течение 24 ч. Охлаждают смесь до комнатной температуры и фильтруют через Celite для удаления Pd катализатора. Добавляют воду (5000 мл) и экстрагируют полученный раствор CH2Cl2(32000 мл). Органическую фракцию сушат над MgSO4 и выпаривают. Добавляют к неочищенному веществу 2000 мл CH2Cl2 и нагревают до кипения с обратным холодильником. Удаляют нерастворимые вещества фильтрованием и выпаривают растворитель с получением желтого твердого вещества. Промывают твердое вещество этиловым эфиром с получением 150 г целевого соединения в виде светло-желтого твердого вещества. Выход: 85%. МС (ES), m/z 273 (М+1). В. 3-Йод-7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин. Обрабатывают раствор 7-(4-метансульфонилфенил)имидазо[1,2-а]пиридина (101 г, 371,3 ммоль,1 экв.) в 2000 мл CH3CN при 0 С N-йодсукцикимида (83,5 г, 371,3 ммоль, 1 экв.). Смесь перемешивают при комнатной температуре в течение 1 ч. Удаляют растворитель и остаток растворяют в 5000 мл CH2Cl2,промывают 10% раствором NaOH, насыщенным раствором NaHSO3, водой и насыщенным водным раствором хлорида натрия. Сушат над MgSO4 и концентрируют. Растирают полученное твердое вещество с гексаном, фильтруют и сушат в вакууме с получением 110 г указанного в заголовке соединения в виде желтого твердого вещества. Выход: 75%. МС (ES), m/z 399 (М+1). Получение 43. Метиловый эфир 4-(3-йодимидазо[1,2-а]пиридин-7-ил)бензойной кислоты. Указанное в заголовке соединение получают с использованием методики, аналогично получению 3-йод-7-(4-метансульфонилфенил)имидазо[1,2-а]пиридина. МС (ES), m/z 379 (М+1). Получение 44. 4-[7-(4-Метансульфонилфенил)имидазо[1,2-а]пиридин-3-ил]бензиламин. Объединяют 3-йод-7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин (2,50 г, 6,28 ммоль), гидрохлорид (4-аминометилфенил)бороновой кислоты (1,29 г, 6,91 ммоль) и K2CO3 (3,47 г, 25,11 ммоль) в смеси 1,4-диоксана (30 мл) и воды (15 мл). Через смесь в течение 5 мин барботируют азот. Добавляют дихлор-бис-(трифенилфосфин)палладий(II) (0,132 г, 0,188 ммоль). Присоединяют обратный холодильник и нагревают смесь до 110 С. Перемешивают в течение ночи (15 ч) и охлаждают до комнатной температуры. Концентрируют смесь досуха в вакууме. Суспендируют полученное твердое вещество в смеси дихлорметан/метанол и фильтруют через Celite 521. Концентрируют раствор в вакууме до получения желтого твердого вещества. Очищают колоночной хроматографией (этилацетат 5% метанол в дихлорметане 10% метанол в дихлорметане 10% 2 М NH3 в метаноле в дихлорметане) с получением продукта (1,57 г, 66%). МС (ES), m/z 378 (M+1).- 18011691 Следующие соединения получают согласно получению 44.A. 4-(7-Хлоримидазо[1,2-а]пиридин-3-ил)фениламин. В круглодонную колбу добавляют 7-хлор-3-йодимидазо[1,2-а]пиридин (3,0 г, 0,011 моль),4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (2,6 г, 1,1 экв.), карбонат калия (4,5 г, 3 экв.), диоксан (40 мл) и воду (20 мл). Полученную смесь тщательно дезоксигенируют N2, затем добавляют дихлор-бис-(трифенилфосфин)палладий(II) (0,23 г, 0,03 экв.) и кипятят реакционную смесь с обратным холодильником в течение ночи в атмосфере N2. Реакционную смесь концентрируют досуха и суспендируют в ДХМ. Полученную суспензию фильтруют через Celite и промывают ДХМ. Фильтрат концентрируют, затем очищают на слое диоксида кремния (EtOAc5% МеОН/ДХМ 0% МеОН/ДХМ) с получением бледно-серого твердого вещества (2,6 г, 100%). МС (ES), m/z 244 (М+1).B. 4-(7-Пиридин-3-илимидазо[1,2-а]пиридин-3-ил)фениламин. В круглодонную колбу добавляют 4-(7-хлоримидазо[1,2-а]пиридин-3-ил)фениламин (0,5 г,2,1 ммоль), пиридин-3-бороновую кислоту (0,38 г, 1,5 экв.), фосфат калия (0,87 г, 2 экв.),2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил (также называемый S-Phos, 0,105 г, 0,125 экв.),1,4-диоксан (10 мл) и воду (5 мл). Полученную смесь тщательно дезоксигенируют N2, затем добавляют ацетат палладия(II) (23 мг, 0,05 экв.) и реакционную смесь кипятят с обратным холодильником в течение- 19011691 ночи. Реакционную смесь концентрируют досуха и суспендируют в ДХМ. Полученную суспензию фильтруют через Celite и промывают ДХМ. Фильтрат концентрируют, затем очищают на слое диоксида кремния (EtOAc5% МеОН/ДХМ 10% МеОН/ДХМ) с получением желтого твердого вещества(0,39 г, 66%). МС (ES), m/z 287 (М+1). Следующие соединения получают по методике, аналогично описанной в получении 55.(8 мл). К раствору добавляют раствор BH3Me2S (2 М, 1,0 мл, 2,0 ммоль) при комнатной температуре. Перемешивают раствор в течение 15 мин и затем нагревают до 50 С, выдерживая при этой температуре в течение 3,5 ч. Охлаждают смесь до 0 С, медленно подкисляют до рН 1 и перемешивают в течение 30 мин. Подщелачивают реакционную смесь твердым NaOH до рН 12-14 с последующей экстракцией этилацетатом. Промывают экстракты насыщенным водным раствором хлорида натрия, сушат надMgSO4, фильтруют и выпаривают с получением 0,070 г. МС (ES), m/z 306 (M+1). Следующее соединение получают по методике, аналогично описанной в получении 62. Получение 64. 4-(7-Пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фениламин. А. 7-Пиридин-4-илимидазо[1,2-а]пиридин. Способ А. В круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой, нагревательным кожухом, который дает возможность контролировать температуру, и холодильником, в атмосфере N2 помещают 7-хлоримидазо[1,2-а]пиридин (4,0 г, 26,2 ммоль), 4-пиридилбороновую кислоту (3,54 г,28,8 ммоль 1,1 экв.), 2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил (600 мг) [в данной реакции в качестве альтернативного лиганда можно использовать X-Phos], Pd(OAc)2 (145 мг), K3PO4 (11,1 г,52,6 ммоль) и смесь диоксан/H2O (2:1, 170 мл). Слегка нагревают реакционную смесь при продувании N2 с помощью иглы, затем нагревают до 65 С, выдерживая при этой температуре в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры, переносят в делительную воронку и сливают нижний слой (25 мл). Добавляют EtOAc и растворители выпаривают при пониженном давлении. Твердые вещества снова переносят в EtOAc и выпаривают при пониженном давлении для азеотропной отгонки следовых количеств воды. Растворяют коричневое твердое вещество в CH2Cl2 и 5% МеОН, затем хроматогра- 20011691 фируют с использованием SiO2 при медленном элюировании градиентом 2 М NH3 в МеОН от 0 до 10%,уравновешивая CH2Cl2. Концентрируют фракции, содержащие продукт, при пониженном давлении с получением светло-желтого/рыжевато-коричневого твердого вещества (4,5 г, 89%). МС (ES), m/z 196 (М+1). А. 7-Пиридин-4-илимидазо[1,2-а]пиридин. Способ В. 1) В круглодонную колбу объемом 250 мл в атмосфере азота помещают 7-хлоримидазо[1,2-а]пиридин (5,07 г, 33,2 ммоль, 1 экв.), бис-(пинаколят)дибор (10,18 г, 40,1 ммоль, 1,2 экв.), карбонат калия(6,86 г, 49,6 ммоль, 1,5 экв.), Pd(OAc)2 (370 мг, 1,6 ммоль, 0,05 экв.), трициклогексилфосфин (914 мг,3,3 ммоль, 0,10 экв.), диглим (50 мл) и воду (68 мкл). Нагревают реакционную смесь до 100 С, выдерживая при этой температуре в течение 24 ч, затем перемешивают в течение двух дней при комнатной температуре. Смесь фильтруют и остаток промывают 210 мл диглима. Суспендируют твердые вещества в 50 мл воды в течение 1 ч, затем фильтруют и промывают 210 мл воды. Сушат влажный остаток (10 г) в вакууме при 60 С, выдерживая при этой температуре в течение ночи, с получением указанного в заголовке соединения в виде серого твердого вещества (выход 6,54 г, 26,8 ммоль, 81%) с чистотой 99,3% по данным ВЭЖХ. 2) В атмосфере азота в круглодонную колбу объемом 100 мл с мешалкой и холодильником помещают гидрохлорид 4-бромпиридина (3,98 г, 20,5 ммоль, 1 экв.), 7-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)имидазо[1,2-а]пиридин (5,47 г, 22,4 ммоль, 1,1 экв.), Pd(OAc)2 (90 мг, 401 ммоль, 0,02 экв.),трифенилфосфин (217 мг, 827 ммоль, 0,04 экв.), K3PO4 (8,6 г, 40,5 ммоль, 2 экв.), 1-пропанол (36 мл) и воду (12 мл). Полученную реакционную смесь нагревают до кипения с обратным холодильником (90 С) в течение ночи, затем охлаждают до комнатной температуры. Разделяют фракции и добавляют 40 мл метил-трет-бутилового эфира и 40 мл 1 М HCl к фракции PrOH. Промывают водную фракцию 40 мл МТВЕ и дважды промывают порциями по 6 мл 1-пропанола. Добавляют метанол (4 мл) к водной фракции, которую нагревают до 45 С, перед добавлением 9 мл 5 М NaOH. Реакционную смесь охлаждают и вносят затравку при 25 С. Спустя 1,5 ч реакционную смесь фильтруют и промывают 24 мл воды с 10% МеОН, затем 8 мл МТВЕ (для облегчения выхода воды из остатка и сушки). Сушат твердые вещества в вакууме при температуре 60 С. Указанное в заголовке соединение выделяют в виде желтого твердого вещества (выход 2,90 г, 73%) с чистотой 98% по данным ВЭЖХ (на длине волны 215 нм).B. 3-Йод-7-пиридин-4-илимидазо[1,2-а]пиридин. В круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой, нагревательным кожухом, который дает возможность контролировать температуру, и холодильником, в атмосфере N2 помещают 7-пиридин-4-илимидазо[1,2-а]пиридин (3,35 г, 17,2 ммоль), N-йодсукцинимид (3,8 г, 16,8 ммоль) и EtOH (3A). Реакционную смесь нагревают до 65 С, выдерживая при этой температуре в течение 1 ч. Далее может следовать ТСХ на SiO2 (100% EtOAc). Добавляют еще NIS (3,8 г, 16,8 ммоль) и нагревают реакционную смесь до 65 С, выдерживая при этой температуре в течение 1 ч. Реакционную смесь перемешивают при охлаждении на ледяной бане в течение 30 мин, затем фильтруют и промывают твердые вещества метанолом. Сушат твердые вещества на воздухе и в вакуумной печи при температуре 40 С с получением 4,59 г (83%). Дополнительное количество продукта может быть получено из фильтрата. МС (ES), m/z 322 (М+1).C. 4-(7-Пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фениламин. В круглодонную колбу объемом 250 мл, оборудованную магнитной мешалкой, нагревательным кожухом, который дает возможность контролировать температуру, и холодильником, в атмосфере N2 помещают 3-йод-7-пиридин-4-илимидазо[1,2-а]пиридин (4,95 г, 15,4 ммоль), 4-аминофенилбороновую кислоту (1,07 г), 4-(4,4,5,5-тетраметил-1,3,2-диоксанборолан-2-ил)анилин (2,69 г) [общее количество бороновой кислоты составляет (18,5 ммоль 1,2 экв.)], диметоксиэтан (140 мл), 2 М раствор K2CO3 (40 мл,28,8 ммоль, 1,1 экв.) и Pd(PPh3)4 (836 мг). Слегка нагревают реакционную смесь при продувании N2 с помощью иглы, затем нагревают до 65 С, выдерживая при этой температуре в течение 18 ч. Реакционную смесь охлаждают до комнатной температуры и переносят в делительную воронку и сливают нижний слой (25-30 мл). ДМЭ выпаривают при пониженном давлении и остаток переносят в МеОН (1 л) и 2 МNH3 в МеОН (10 мл). Фильтруют раствор для удаления Pd(0) и выпаривают при пониженном давлении для перенесения на силикагель. Сушат силикагель в вакууме и хроматографируют с использованиемSiO2, при градиентном элюировании 2 М NH3 в МеОН от 0% до 10%, уравновешивая CH2Cl2. Выпаривают растворители при пониженном давлении с получением ярко-желтого твердого вещества 2,6 г (60%). МС (ES), m/z 286 (М+1).- 21011691 Следующие соединения получают согласно получению 64.(0,480 г, 1,52 ммоль, 1,0 экв.) и 5-бромпиридин-2-иламин (0,263 г, 1,52 ммоль, 1,0 экв.) в ДМСО (2,0 мл). Добавляют триэтиламин (0,22 мл, 1,52 ммоль, 1,0 экв.). Реакционную смесь перемешивают при температуре 80 С, выдерживая при этой температуре в течение приблизительно 16 ч, охлаждают, затем разбавляют диэтиловым эфиром (100 мл), промывают насыщенным водным раствором хлорида натрия(220 мл), водой (220 мл), сушат, фильтруют и концентрируют с образованием коричневого масла. Очищают масло флэш-хроматографией на силикагеле с использованием градиента этилацетата в дихлорметане 0-10% с получением 0,302 г (58%) указанного в заголовке соединения в виде коричневого твердого вещества. МС (ES), m/z 339, 341 (М+1). Следующее соединение получают по методике, аналогично описанной в получении 71.- 22011691 Следующие промежуточные соединения получают по методике, аналогично описанной в примере 64, ниже. Получение 75. трет-Бутиловый эфир 4-[7-(6-фторпиридин-3-ил)имидазо[1,2-а]пиридин-3 ил]бензилкарбаминовой кислоты. А. трет-Бутиловый эфир [4-(7-хлоримидазо[1,2-а]пиридин-3-ил) бензил]карбаминовой кислоты. К суспензии 3-йод-7-хлоримидазо[1,2-а]пиридина (4,80 г, 17,2 ммоль, 1,0 экв.) в диоксане (90 мл) добавляют 2 М Na2CO3 (30 мл) и (4-аминометилфенил)-бороновой кислоты (3,88 г, 20,7 ммоль, 1,2 экв.). Дезоксигенируют смесь и заполняют азотом. Добавляют тетракис-(трифенилфосфин)палладий (0,50 г, 0,43 ммоль, 0,025 экв.). Дезоксигенируют реакционную смесь и заполняют азотом. Реакционную смесь перемешивают при 85 С, выдерживая при этой температуре в течение 3 дней. Добавляют (Boc)2 О (4,51 г, 20,7 ммоль, 1,2 экв.) и перемешивают смесь при 60 С, выдерживая при этой температуре в течение 20 мин. Концентрируют смесь досуха в вакууме. Суспендируют полученное твердое вещество в смеси дихлорметан/метанол и фильтруют черезCelite 521. Концентрируют раствор в вакууме до получения желтого твердого вещества (5,60 г). Очищают флэш-хроматографией на силикагеле с использованием градиента 0-4% МеОН/ДХМ, с получением 4,50 г (12,6 ммоль, 73%) указанного в заголовке соединения в виде слабо-желтого твердого вещества в качестве продукта. МС (ES), m/z 358 (M+1). В. трет-Бутиловый эфир 4-[7-(6-фторпиридин-3-ил)имидазо[1,2-а]пиридин-3-ил]бензилкарбаминовой кислоты. Суспендируют трет-бутиловый эфир [4-(7-хлоримидазо[1,2-а]пиридин-3-ил)бензил]карбаминовой кислоты (1,90 г, 5,30 ммоль, 1,0 экв.) в смеси диоксан/вода (2:1, 36 мл). Добавляют 2-фтор-5-пиридинбороновую кислоту (0,75 г, 5,30 ммоль, 1,0 экв.), K3PO4 (2,25 г, 10,6 ммоль, 2,0 экв.) и S-phos (0,272 г,0,66 ммоль, 0,125 экв.). Дезоксигенируют смесь и заполняют азотом. Добавляют Pd(OAc)2 (0,059 г,0,265 ммоль, 0,05 экв.). Дезоксигенируют реакционную смесь и заполняют азотом. Реакционную смесь перемешивают при температуре 80 С в атмосфере азота в течение 16 ч. После охлаждения раствора образуется белое твердое вещество. Белое твердое вещество отфильтровывают, промывают водой(315 мл), EtOAc (315 мл). Собирают твердое вещество (1,50 г). Очищают фильтрат флэшхроматографией на силикагеле с градиентом 0-5% МеОН/ДХМ, с получением указанного в заголовке соединения (0,52 г) в виде слабо-желтого твердого вещества. Объединяют продукт хроматографии и отфильтрованное твердое вещество с получением слабо-желтого твердого вещества (2,02 г, 4,83 ммоль). МС (ES), m/z 419 (M+1). Следующие соединения получают по методике, аналогично описанной в получении 75.- 23011691 Получение 78. 4-(7-Тиазол-2-илимидазо[1,2-а]пиридин-3-ил)фениламин. А. 7-Тиазол-2-илимидазо[1,2-а]пиридин. В круглодонной колбе объемом 500 мл перемешивают суспензию 7-хлоримидазо[1,2-а]пиридина(7,5 г, 49,2 ммоль) в 250 мл 1,4-диоксана с бис-(пинаколят)дибором (13,74 г, 54,1 ммоль, 1,1 экв.), ацетатом калия (7,24 г, 73,8 ммоль, 1,5 экв.) и трициклогексилфосфином (1,66 г, 5,9 ммоль, 0,12 экв.). Дезоксигенируют полученную суспензию двумя циклами удаления и барботирования азота через суспензию,каждый цикл длительностью 10 мин. К колбе присоединяют обратный холодильник, добавляют трис-(дибензилидинацетон)дипалладий(0) (2,25 г, 2,45 ммоль, 0,05 экв.) и перемешивают смесь в атмосфере азота при 80 С, выдерживая при этой температуре в течение ночи. Фильтруют горячую смесь через слой целита 1 см, промывают 50 мл 1,4-диоксана, затем концентрируют объединенный фильтрат и промывают с получением коричневого пастообразного твердого вещества. Суспендируют часть твердого вещества (32,8 ммоль) в 80 мл диоксана, объединяют с 41,25 мл 2 М водного раствора карбоната натрия(82,5 ммоль, 2,5 экв.) и 2-бромтиазола (4,38 мл, 8,07 г, 49,2 ммоль, 1,5 экв.), затем дважды дезоксигенируют путем удаления и барботирования азота через суспензию. Добавляют тетракис(трифенилфосфин)палладий (1,89 г, 1,64 ммоль, 0,05 экв.), к колбе присоединяют обратный холодильник и перемешивают смесь в атмосфере азота при 100 С, выдерживая при этой температуре в течение ночи. Фильтруют горячую темно-коричневую/черную смесь через слой Celite толщиной 1 см, промывают 50 мл диоксана, затем наносят объединенный фильтрат на картриджи SCX Mega Bond-Elut 25 г(VARIAN) и промывают количеством, эквивалентным трехкратному избытку, каждый картридж предварительно промывают 200 мл смеси CH2Cl2/МеОН (1:1). После загрузки с помощью вакуума каждый картридж промывают 300 мл смеси CH2Cl2/МеОН (1:1), затем элюируют 160 мл смеси CH2Cl2/2 MNH3-МеОН (1:1). Концентрируют объединенные элюаты в вакууме и очищают на картридже с силикагелем 330 г с использованием градиента метанола в дихлорметане (0-5%). Концентрируют объединенные чистые фракции и сушат с получением 3 г (45%) 7-тиазол-2-илимидазо[1,2-a]пиридина в виде рыжеватокоричневого твердого вещества. МС (ES) m/z 202, (М+1).B. 3-Бром-7-тиазол-2-илимидазо[1,2-а]пиридин. Растворяют 7-тиазол-2-илимидазо[1,2-а]пиридин (1,14 г, 5,66 ммоль) в 25 мл абсолютного этанола,затем добавляют NBS (1,0 г, 5,66 ммоль), перемешивают смесь при комнатной температуре в течение 15 мин, разбавляют 35 мл дихлорметана и наносят на картридж SCX Mega Bond-Elut (VARIAN) 10 г,предварительно промытый 150 мл смеси CH2Cl2/МеОН (1:1). Промывают картридж 300 мл смесиC. 4-(7-Тиазол-2-илимидазо[1,2-а]пиридин-3-ил)фениламин. 3-Бром-7-тиазол-2-илимидазо[1,2-а]пиридин (1,07 г, 3,82 ммоль) и гидрохлорид 4-аминофенилбороновой кислоты (793 мг, 4,58 ммоль, 1,2 экв.) переносят в 15 мл диоксана и 7 мл 2 н. водного раствора карбоната натрия, затем дезоксигенируют с помощью вакуума/барботирования азота, как описано выше. Добавляют тетракис-(трифенилфосфин)палладий(0) (221 мг, 0,19 1 ммоль, 0,05 экв.), к реакционной колбе присоединяют обратный холодильник и нагревают смесь до 95 С, перемешивая в атмосфере азота в течение приблизительно 16 ч. Охлаждают темно-коричневую реакционную смесь, разбавляют этилацетатом (40 мл) и разделяют фракции в делительной воронке. Органическую фракцию сушат над твердым сульфатом магния, фильтруют, затем наносят на картридж SCX Mega Bond-Elut (VARIAN) 10 г, предварительно промытый 100 мл смеси CH2Cl2/МеОН (1:1). После загрузки промывают картридж еще 200-250 мл смеси CH2Cl2/МеОН, затем элюируют продукт 100 мл CH2Cl2/2 М NH3-MeOH, концентрируют до образования оранжево-коричневого масла и очищают флэш-хроматографией с градиентом от 0 до 5% метанола в дихлорметане. Чистые фракции объединяют и концентрируют с получением после сушки 750 мг (67%) указанного в заголовке соединения в виде желто-коричневого твердого вещества. МС (ES) m/z 293 (М+1). Получение 79. 2-Хлор-4-(7-пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фениламин.A. 2-Хлор-4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фениламин. В круглодонной колбе в атмосфере азота растворяют 4-бром-2-хлорфениламин (4,2 г, 20 ммоль) в диоксане (80 мл). Добавляют триэтиламин (10 мл, 3,6 экв.), затем продувают азотом для дегазации. По каплям с помощью шприца добавляют 4,4,5,5-тетраметил[1,3,2]диоксаборолан (8,5 мл, 2,9 экв.) в течение приблизительно 5 мин. Еще в течение 5 мин барботируют азот, затем добавляют[1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,603 г, 3,7 мол.%, комплекс с дихлорметаном 1:1). Помещают в атмосферу азота и нагревают до 80 С, выдерживая при этой температуре в течение ночи. После охлаждения реакционную смесь фильтруют через Celite, промывая гексаном. Фильтрат концентрируют досуха и остаток используют неочищенным.B. 2-Хлор-4-(7-пиридин-4-илимидазо[1,2-а]пиридин-3-ил)фениламин. Объединяют 3-йод-7-пиридин-4-илимидазо[1,2-а]пиридин (2,18 г, 6,8 ммоль), 2-хлор-4-(4,4,5,5 тетраметил[1,3,2]диоксаборолан-2-ил)фениламин (неочищенные вещества с предыдущей стадии, избыток), карбонат калия (2,8 г, 3 экв.), диоксан (30 мл) и воду (15 мл). Тщательно дегазируют азотом, затем добавляют дихлор-бис-(трифенилфосфин)палладий(II) (0,143 г, 3 мол.%). Реакционную смесь нагревают до 80 С, выдерживая при этой температуре в течение ночи. Концентрируют досуха, затем суспендируют в ДХМ и фильтруют через Celite, промывая ДХМ. Фильтрат концентрируют, затем очищают на слое диоксида кремния (гексангексан/этилацетат 1:12,5% метанол/ДХМ 5% метанол/ДХМ) с получением оранжево-желтого твердого вещества (1,42 г, 46%). МС (ES), m/z 321,1 (М+1). Следующие соединения получают по методикам, аналогично описанным выше. Получение 82. 3-4-[7-(4-Метансульфонилфенил)имидазо[1,2-а]пиридин-3-ил]фенилпропионовая кислота. Объединяют 3-йод-7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин (125,0 мг, 0,314 ммоль,1,0 экв.) с [4-(2-карбоксиэтил)фенилбороновой кислотой (122 мг, 0,628 ммоль, 2,0 экв.) в присутствии тетракис-(трифенилфосфин)палладия(0) (36,3 мг, 0,031 ммоль, 0,10 экв.) и карбоната натрия (99,8 мг,0,942 ммоль, 3,0 экв.) в смеси ДМЭ и воды (1:1) (4 мл) в реакционной емкости объемом 2-5 мл для обработки микроволнами. Закупоривают реакционный сосуд пробкой и помещают в микроволновую камеру. Перемешивают смесь в течение 20 с, затем подвергают микроволновому облучению для повышения температуры от комнатной до 100 С. Как только достигнута желаемая температура, реакционную смесь выдерживают при этой температуре в течение 3 ч. Дают реакционной емкости остыть до комнатной температуры, затем ее открывают. Наносят реакционную смесь на катионно-обменную смолу, которую элюируют дихлорметаном, метанолом, затем 2,0 М аммиаком в метаноле. Концентрируют метанольноаммониевую фракцию досуха при пониженном давлении с использованием роторного испарителя. Остаток (139,0 мг) используют без дополнительной очистки. МС (ES) m/z 421 (М+1) Следующее соединение получают по методике, аналогично описанной в получении 82.A. 2-(4-Бромфенил)-N-(3-трифторметилфенил)ацетамид. Растворяют 4-бромфенилуксусную кислоту (10,00 г, 46,50 ммоль), диизопропилэтиламин (12,02 г,16,20 мл, 93,00 ммоль) и 1,1'-карбонилдиимидазол (8,29 г, 51,15 ммоль) в тетрагидрофуране (200 мл) при комнатной температуре. Перемешивают содержимое в атмосфере азота в течение 1 ч. Добавляют м-трифторметиланилин (15,00 г, 93,00 ммоль) и перемешивают реакционную смесь в течение ночи при комнатной температуре. Реакционную смесь концентрируют почти досуха, растворяют в дихлорметане(250 мл) и экстрагируют 2 н. раствором NaOH (200 мл), водой (100 мл) и 1 н. HCl (2200 мл). Органическую фракцию промывают насыщенным водным раствором хлорида натрия (100 мл), сушат над MgSO4,- 25011691 фильтруют и концентрируют. Сухой продукт загружают на диоксид кремния (100 г) и хроматографируют на диоксиде кремния, используя дихлорметан в качестве элюента, с получением продукта (13,00 г,78,1%). МС (ES), m/z 356/358 (М+1).(295 мг, 1,05 ммоль) в безводном диоксане (95 мл). Дезоксигенируют реакционную смесь азотом в течение 10 мин при комнатной температуре. К реакционной смеси добавляют ацетат палладия(II) (95 мг,0,42 ммоль). Присоединяют обратный холодильник и нагревают реакционную смесь до 80 С, выдерживая при этой температуре в течение 15 ч. Реакционную смесь охлаждают и разбавляют этилацетатом(200 мл). Экстрагируют/промывают этилацетатный раствор водой (3100 мл), сушат над MgSO4, фильтруют и концентрируют досуха. Растворяют неочищенное твердое вещество в теплом дихлорметане и медленно добавляют гексан. При охлаждении образуются кристаллы; добавляют дополнительное количество гексана к органическим фракциям и после осаждения в течение 2 ч фильтруют суспензию с получением продукта (3,00 г, 88,5%). Промывают гексаном. МС (ES), m/z 404 (M-1). Следующие соединения получают по методике, аналогично описанной в получении 49.- 26011691 Получение 92. N-(4-Хлор-3-трифторметилфенил)-2-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2 ил)фенил]ацетамид. А. 2-(4-Бромфенил)-N-(4-хлор-3-трифторметилфенил)ацетамид. Растворяют 4-бромфенилуксусную кислоту (25,39; 118,07 ммоль) в эфире (200 мл). Добавляют безводный пиридин (1 мл) и охлаждают реакционную смесь до 0 С, затем добавляют оксалилхлорид(18,44 г, 12,86 мл, 145,32 ммоль). Спустя 15 мин дают реакционной смеси нагреться до комнатной температуры. Через 1,5 ч добавляют ДМФА (0,4 мл) и перемешивают в течение еще 1 ч. Концентрируют до получения густого янтарного масла. Разбавляют дихлорметаном (118 мл) с получением 1 н. запасного раствора хлорангидрида (4-бромфенил)уксусной кислоты (118 ммоль). Растворяют 4-хлор-3-трифторметиланилин (4,13 г, 21,1 ммоль) в безводном дихлорметане (21 мл)N,N-диизопропилэтиламина (13,57 г, 18,29 мл, 105 ммоль) и охлаждают до 0 С. По каплям добавляют к анилину в растворе, охлажденном льдом, запасной раствор хлорангидрида кислоты в дихлорметане(21,1 мл). Перемешивают в течение ночи при комнатной температуре. Дополнительно разбавляют дихлорметаном (100 мл) и промывают 1 н. HCl (150 мл), водой (150 мл), 1 н. NaOH. (250 мл) и насыщенным водным раствором хлорида натрия (50 мл). Органические фракции сушат над MgSO4, фильтруют и затем концентрируют с получением неочищенного амида. Хроматографируют на диоксиде кремния с использованием смеси (15% этилацетат в дихлорметане)/гексан (1:1). Перекристаллизовывают из смеси дихлорметан/гексан. (2,78 г, 7,08 ммоль, 42,2%). МС (ES), m/z 390/392 (M+1). В.N-(4-Хлор-3-трифторметилфенил)-2-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]ацетамид. Используют методику, аналогично описанной в получении 84. МС (ES), m/z 438 (M-1). Следующие соединения получают по методике, аналогично описанной в получении 92. Получение 97. 4-[4-(7-Хлоримидазо[1,2-а]пиридин-3-ил)фенил]-N-(3-трифторметилфенил)бутирамид. Суспендируют 4-[4-(4,4,5,5-тетраметил[1,3,2]диоксаборолан-2-ил)фенил]-N-(3-трифторметилфенил)бутирамид (0,47 г, 1,15 ммоль), 7-хлор-3-йодимидазо[1,2-а]пиридин (0,29 г, 1,05 ммоль) и твердый карбонат калия (0,48 г, 3,45 ммоль) в диоксане (4 мл) и воде (2 мл). Дезоксигенируют реакционную смесь азотом в течение 10 мин при комнатной температуре. К реакционной смеси добавляют транс-дихлорбис-(трифенилфосфин)палладий(II) (22 мг, 0,03 ммоль). Присоединяют обратный холодильник и нагревают реакционную смесь до 105 С, выдерживая при этой температуре в течение 2 дней. Реакционную смесь охлаждают, разбавляют дихлорметаном и фильтруют через Celite. Фильтрат концентрируют и хроматографируют на диоксиде кремния с использованием смеси метанол/этилацетат (0-3%), с получением продукта (301 мг, 57%). МС (ES), m/z 458 (M+1).- 27011691 Следующее соединение получают по методике, аналогично описанной в получении 97.(0,480 г, 1,27 ммоль, 1,0 экв.) [получен согласно заявке РСТ WO2004046101, 3 июня 2004 г., подана 10 ноября 2003 г.] в безводном диоксане (4 мл) добавляют 7-хлор-3-йодимидазо[1,2-а]пиридин (0,354 г,1,27 ммоль, 1,0 экв.) и LiCl (0,162 г, 3,0 экв.). Добавляют Pd(PPh3)4 (0,102 г, 0,07 экв.). Дезоксигенируют реакционную смесь и заполняют азотом. Перемешивают при температуре 90 С, выдерживая при этой температуре в течение ночи, охлаждают, затем разбавляют смесью 5% МеОН/ДХМ (100 мл), промывают насыщенным водным раствором хлорида натрия (220 мл), водой (220 мл), сушат, фильтруют и концентрируют. Хроматографируют на силикагеле, сначала с градиентом 50%-100% EtOAc/ДХМ, затем с градиентом 5% МеОН/ДХМ, с получением 0,180 г указанного в заголовке соединения, которое используют на следующей стадии без дополнительной очистки. МС (ES), m/z 365 (M+1).B. трет-Бутиловый эфир 4-[7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин-3-ил]тиазол-2 илметилкарбаминовой кислоты. К суспензии трет-бутилового эфира [4-(7-хлоримидазо[1,2-а]пиридин-3-ил)тиазол-2-илметил]карбаминовой кислоты (0,180 г, 0,49 ммоль, 1,0 экв.) в смеси диоксан/вода (2:1, 6 мл) добавляют 4-(метилсульфонил)бензолбороновую кислоту (0,108 г, 1,1 экв.), K3PO4 (0,208 г, 2,0 экв.), S-phos (0,025 г,12,5% экв.) и Pd(OAc)2 (0,006 г, 0,05 экв.). Дезоксигенируют реакционную смесь, заполняют азотом и перемешивают при температуре 100 С, выдерживая при этой температуре в течение 4 ч. Охлаждают смесь до комнатной температуры и разбавляют 5:95 МеОН/ДХМ (100 мл). Промывают смесь насыщенным водным раствором хлорида натрия (220 мл), водой (220 мл), сушат, фильтруют и концентрируют. Очищают остаток на силикагеле с градиентом 0-80% EtOAc/гексан, затем смесью МеОН/ДХМ (5:95) с получением 0,160 г указанного в заголовке соединения в виде слабо-желтого твердого вещества.C. 4-[7-(4-Метансульфонилфенил)имидазо[1,2-а]пиридин-3-ил]тиазол-2-илметиламин. К раствору трет-бутилового эфира 4-[7-(4-метансульфонилфенил)имидазо[1,2-а]пиридин-3 ил]тиазол-2-илметилкарбаминовой кислоты (0,160 г, 0,33 ммоль, 1,0 экв.) в смеси МеОН/ДХМ (1:1,20 мл), добавляют HCl (4 М в диоксане, 6 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч, затем при температуре 60 С, выдерживая при этой температуре в течение 2 ч. Концентрируют реакционную смесь в вакууме и используют без дополнительной очистки. Получение 100. 2-[4-(7-Хлоримидазо[1,2-а]пиридин-3-ил)фенил]-N-(3-трифторметилфенил)ацетамид. 3-Йод-7-хлоримидазо[1,2-а]пиридин (655 мг, 2,35 ммоль) подвергают сочетанию с 2-[4-(4,4,5,5 тетраметил[1,3,2]диоксаборолан-2-ил)фенил]-N-(3-трифторметилфенил)ацетамидом (1,00 г, 2,47 ммоль) с использованием методики, аналогично получению 99 В. МС (ES), m/z 430 (M+1). Следующее соединение получают по методике, аналогично описанной в получении 100. Получение 102. 7-Йодимидазо[1,2-а]пиридин. Объединяют 4-йодпиридин-2-иламин (4,00 г, 18,18 ммоль) и хлорацетальдегид (2,77 мл,21,82 ммоль) в этаноле (40 мл). Присоединяют обратный холодильник и нагревают смесь до 83 С, перемешивают в течение ночи(15 ч) и охлаждают до комнатной температуры. Фильтруют полученный раствор с получением продукта(50 мл). Через смесь в течение 5 мин барботируют азот. Добавляют [1,1'-бис-(дифенилфосфино)ферроцен]дихлорпалладий(II) (0,442 г, 0,541 ммоль, комплекс с дихлорметаном 1:1). Присоединяют обратный холодильник, нагревают смесь до 85 С, перемешивают в течение ночи (15 ч) и охлаждают до комнатной температуры. Фильтруют смесь через Celite 521. Концентрируют раствор в вакууме до образования темно-коричневого масла, которое используют как таковое. МС (ES), m/z 299 (M+1).B. 7-Этинилимидазо[1,2-а]пиридин. Объединяют 7-[(триизопропилсиланил)этинил]имидазо[1,2-а]пиридин (3,23 г, 10,82 ммоль) и тетрабутиламмонийфторид (1,19 мл, 1,19 ммоль, 1,0 М в ТГФ) в ТГФ (5 мл). Перемешивают смесь при комнатной температуре в течение 40 мин, затем концентрируют в вакууме с получением черного масла. Очищают колоночной хроматографией (этилацетат) с получением продукта (1,10 г, 71%). МС (ES), m/z 143,1 (M+1). Получение 104. 7-(1 Н-[1,2,3]Триазол-4-ил)имидазо[1,2-а]пиридин. Объединяют 7-этинилимидазо[1,2-а]пиридин (1,10 г, 7,74 ммоль), йодид меди(I) (0,074 г,0,387 ммоль) и триметилсилилазид (1,53 мл, 11,61 ммоль) в смеси ДМФА/метанол (9:1, 13,8 мл) в колбе для обработки под давлением. Закупоривают колбу навинчивающейся тефлоновой крышкой и нагревают реакционную смесь до 100 С, перемешивают в течение ночи (15 ч) и охлаждают до комнатной температуры. Концентрируют смесь досуха в вакууме. Суспендируют полученное твердое вещество в дихлорметане и фильтруют через Celite 521. Концентрируют раствор в вакууме с получением твердого вещества оранжевого цвета. Очищают колоночной хроматографией (этилацетат 5% метанол в дихлорметане 8% метанол в дихлорметане) с получением продукта (0,82 г, 57%). МС (ES), m/z 186 (M+1). Получение 105. 3-Йод-7-(1 Н-[1,2,3]триазол-4-ил)имидазо[1,2-а]пиридин. По методике, аналогично получению 27 В, с использованием 7-(1 Н-[1,2,3]триазол-4-ил)имидазо[1,2 а]пиридина получают указанное в заголовке соединение. МС (ES), m/z 312 (M+1). Получение 106. 7-(2,6-Диметилпиридин-4-ил)имидазо[1,2-а]пиридин. Получают из 7-йодимидазо[1,2-а]пиридина и 4-бром-2,6-диметилпиридина (Acta. Chemica. Scandinavica. В 42, (1988) pages 373-377) с использованием методики, аналогично получению 27 А. МС (ES), m/z 224 (M+1). Получение 107. 5-(1-Метилциклопропил)-[1,3,4]тиадиазол-2-иламин. Объединяют 1-метилциклопропан-1-карбоновую кислоту (10,00 г, 99,88 ммоль) и тиосемикарбазид(9,10 г, 99,88 ммоль) в диоксане (110 мл). Нагревают смесь до 90 С в атмосфере N2, затем в течение 25 мин по каплям добавляют оксихлорид фосфора(III) (9,14 мл, 99,88 ммоль). Реакционную смесь перемешивают в течение 6 ч при температуре 90 С, затем в течение 8 ч при комнатной температуре. Декантируют на 200 г льда и добавляют гидроксид аммония до щелочной реакции. Фильтруют для удаления твердых веществ; фильтрат экстрагируют этилацетатом. Органическую фракцию промывают водой. Сушат полученные органические фракции над сульфатом магния, фильтруют и концентрируют с получением продукта (2,54 г, 16%). МС (ES), m/z 156 (М+1). Получение 108. Дигидрохлорид 4-диметиламинометил-5-(1-метилциклопропил)тиазол-2-иламина. А. Метиловый эфир 2-амино-5-(1-метилциклопропил)тиазол-4-карбоновой кислоты. Объединяют (1-метилциклопропил)метанол (5,00 г, 58,05 ммоль), N-оксид 4-метилморфолина(10,20 г, 87,08 ммоль) и сита 4 (5,6 г) в CH2Cl2 (200 мл). Реакционную смесь перемешивают в течение 20 мин при комнатной температуре в атмосфере N2. Добавляют перрутенат тетрапропиламмония (1,02 г,2,90 ммоль) и перемешивают в течение 5 ч. Очищают на слое силикагеля; элюируют CH2Cl2. Объединяют фракции, содержащие продукт, и концентрируют в вакууме; остается некоторое количество CH2Cl2. Полученное вещество переносят непосредственно на следующую стадию реакции. Объединяют 1-метилциклопропанкарбальдегид (4,88 г, 58,01 ммоль) и метилдихлорацетат (5,46 мл,52,74 ммоль) в диэтиловом эфире (20 мл); охлаждают до 0 С. Добавляют по каплям раствор натрия(1,21 г, 52,74 ммоль) в метаноле (20 мл). Реакционную смесь перемешивают в течение 4 ч при 0 С в атмосфере N2. Экстрагируют смесь диэтиловым эфиром для отделения от воды. Сушат полученные органические фракции над сульфатом магния, фильтруют и концентрируют до получения прозрачной жидкости. Объединяют жидкость с тиомочевиной (4,42 г, 58,01 ммоль) в метаноле (25 мл). Нагревают реакционную смесь до 60 С в атмосфере N2, выдерживая при этой температуре в течение 14 ч. Концентрируют- 29011691 в вакууме и очищают на слое силикагеля, элюируя гексаном 3% метанолом в дихлорметане 5% метанолом в дихлорметане с получением продукта (5,10 г, выход 46% для двух стадий). МС (ES), m/z 213 (М+1). В. 2-трет-Бутоксикарбониламино-5-(1-метилциклопропил)тиазол-4-карбоновая кислота. Объединяют 2-амино-5-(1-метилциклопропил)тиазол-4-карбоновой кислоты метиловый эфир(5,10 г, 24,03 ммоль) и ди-трет-бутилдикарбонат (5,24 г, 24,03 ммоль) в пиридине (15 мл). Реакционную смесь перемешивают в течение 1 ч, затем добавляют раствор триметилсиланолята калия (17,12 г,120,13 ммоль) в ТГФ (100 мл). Реакционную смесь перемешивают в течение 14 ч в атмосфере N2. Экстрагируют смесь этилацетатом против 1 н. HCl. Органический слой промывают насыщенным водным раствором хлорида натрия. Полученные органические фракции сушат над сульфатом магния, фильтруют и концентрируют. Очищают на слое силикагеля, элюируя гексаном 5% метанолом в дихлорметане с получением продукта (4,55 г, 64%). МС (ES), m/z 243 (М+1, продукт - трет-бутил).(2 г, 6,7 ммоль) в ТГФ (100 мл) и охлаждают до 0 С в атмосфере азота. Добавляют триэтиламин (0,93 мл,1 экв.) с последующим добавлением по каплям изобутилхлорформиата (0,87 мл, 1 экв.). Перемешивают в течение 30 мин при 0 С, затем фильтруют, промывая ТГФ. Снова охлаждают фильтрат 0 С, затем добавляют боргидрид натрия (0,76 г, 3 экв.) в виде одной порции с последующим добавлением по каплям метанола (4,1 мл, 15 экв.). Спустя 45 мин при температуре 0 С охлаждающую баню удаляют и дают возможность реакционной смеси нагреться до комнатной температуры в течение 15 мин. Гасят реакцию(осторожно) 1 н. HCl (водн.), приблизительно 50 мл. Экстрагируют ДХМ. Органические фракции промывают насыщенным водным раствором хлорида натрия. Органические фракции сушат над MgSO4,затем фильтруют и концентрируют. Очищают на силикагеле (смесь гексан/этилацетат 2:1 гексан/этилацетат 1:1) с получением белого твердого вещества (1,11 г, 58%). ЖХМС (ES), m/z 229 (М+1, продукт - трет-бутил).D. трет-Бутиловый эфир [4-диметиламинометил-5-(1-метилциклопропил)тиазол-2-ил]карбаминовой кислоты. В атмосфере азота растворяют трет-бутиловый эфир [4-гидроксиметил-5-(1-метилциклопропил)тиазол-2-ил]карбаминовой кислоты (1,11 г, 3,9 ммоль) в ДХМ (50 мл). Добавляют трифенилфосфин(2,05 г, 2 экв.) с последующим добавлением тетрабромида углерода (2,6 г, 2 экв.). Перемешивают при комнатной температуре в течение 15 мин, затем концентрируют и очищают на силикагеле (гексангексан/этилацетат 9:1) с получением бромида (1,02 г, 75%). Снова растворяют полученное вещество в ТГФ (40 мл), добавляют диметиламин (7,3 мл 2 М раствора в ТГФ, 5. экв.) и перемешивают при комнатной температуре в течение 4 ч. Реакционную смесь фильтруют, промывая ТГФ. Фильтрат концентрируют с получением продукта (0,9 г, 99%). 1 Н ЯМР (400 МГц, ДМСО-d6) : 11,2 (шс, 1 Н), 3,34 (с, 2 Н), 2,15 (с, 6 Н), 1,43 (с, 9 Н), 1,28 (с, 3H),0,80 (м, 2 Н), 0,73 (м, 2 Н). Е. Дигидрохлорид 4-диметиламинометил-5-(1-метилциклопропил)тиазол-2-иламина. В атмосфере азота растворяют трет-бутиловый эфир[4-диметиламинометил-5-(1 метилциклопропил)тиазол-2-ил]карбаминовой кислоты (0,9 г, 2,9 ммоль) в диоксане (40 мл), затем добавляют 4 М HCl в диоксане (7,2 мл, 10 экв.) и перемешивают при комнатной температуре в течение ночи. ЖХМС показывает в основном исходное соединение. Реакционную смесь нагревают до 40 С, выдерживая при этой температуре в течение ночи. ЖХМС показывает частичное превращение в продукт. Повышают температуру до 60 С и нагревают в течение ночи. ЖХМС показывает завершение реакции. Присутствует белый осадок; смесь фильтруют для выделения твердого вещества. Твердое вещество выглядит гигроскопичным. Твердое вещество снова растворяют в метаноле и концентрируют с получением белого твердого вещества (0,784 г, 95%). МС (ES), m/z 212 (М+1). Получение 109. 5-трет-Бутил-4-диметиламинометилтиазол-2-иламин. Получают с использованием методик, аналогично описанным в получении 108. Выделяют продукт с использованием картриджа SCX (10 г VARIAN Bond-Elut), элюируя смесью метанол/дихлорметан(1:1), затем смесью 2 М NH3 в метаноле/дихлорметан. Последнюю фракцию упаривают с получением указанного в заголовке соединения (0,190 г, 1100%). МС (ES), m/z 169 (М+1, продукт - NMe2).
МПК / Метки
МПК: C07D 471/04, A61P 35/04, A61K 31/4375
Метки: пиридиновые, имидазо(1,2-а, соединения, vegf-r2, ингибиторов, качестве
Код ссылки
<a href="https://eas.patents.su/30-11691-imidazo12-a-piridinovye-soedineniya-v-kachestve-ingibitorov-vegf-r2.html" rel="bookmark" title="База патентов Евразийского Союза">Имидазо(1,2-а) пиридиновые соединения в качестве ингибиторов vegf-r2</a>
Аминокислотные соединения и их применение в качестве ингибиторов nep, ace и ece

Номер патента: 5004
Опубликовано: 28.10.2004
Авторы: Фурнье-Залуски Мари-Клод, Пора Эрве, Рок Бернар П., Ренар Пьер, Беннжан Каролина, Скальбер Элизабет, Ингимбер Никола
МПК: A61K 31/33, C07D 209/20, A61P 43/00...
Метки: ингибиторов, применение, качестве, соединения, аминокислотные
Формула / Реферат:
1. Соединения формулы (I) в которой n представляет собой коэффициент, в котором 0_n_3, m представляет собой коэффициент, в котором 0_m_6, R3 и R4 совместно образуют с двумя атомами углерода, которые их несут, фенильную группу, которая незамещена или замещена от 1 до 3 одинаковыми или различными группами, выбираемыми из алкила, алкенила, алкинила, алкокси, гидрокси, алкилтио, меркапто, циано, нитро, амино, алкиламино, диалкиламино,...
Пиридиновые соединения, способ их получения и содержащие их фармацевтические композиции

Номер патента: 4102
Опубликовано: 25.12.2003
Авторы: Пуатевен Кристоф, Дессанж Эме, Декейн Анн, Пеглион Жан-Луи, Миллан Марк
МПК: C07D 471/04, A61K 31/496, A61P 25/00...
Метки: способ, пиридиновые, композиции, содержащие, получения, фармацевтические, соединения
Формула / Реферат:
1. Соединения формулы (I) где W представляет либо нафтильную группу, необязательно замещенную одной или несколькими одинаковыми или различными группами, выбранными из галогена, линейного или разветвленного (C1-C6)алкила, гидрокси, линейного или разветвленного (C1-C6)алкокси, циано, нитро, линейного или разветвленного тригалоген(C1-C6)алкила, метилендиокси и этилендиокси, либо группу формулы Y где E, вместе с углеродными атомами фенильного...
Соединения и композиции в качестве ингибиторов катепсина

Номер патента: 7334
Опубликовано: 25.08.2006
Авторы: Паттерсон Джон В., Ципфель Шила
МПК: C07C 255/42, A61K 31/277
Метки: качестве, ингибиторов, катепсина, композиции, соединения
Формула / Реферат:
1. Соединение формулы I где X1 является -NHC(R1)(R2)2; X2 является циано, -C(R7)(R8)X3; R7 и R8 вместе образуют оксогруппу; X3 содержит гетеромоноциклическое кольцо, имеющее от 4 до 6 атомов, являющихся членами кольца или конденсированную гетеробициклическую кольцевую систему, содержащую от 8 до 14 атомов, являющихся членами кольца и любой карбоциклический кетон, иминокетон или его тиокетоновое производное; R1 является водородом и R2 является...
Тиенодибензоазуленовые соединения в качестве ингибиторов фактора некроза опухоли

Номер патента: 6069
Опубликовано: 25.08.2005
Авторы: Мерчеп Младен, Жупанович Желько, Пешич Дияна, Месич Милан, Хрвачич Бошка
МПК: A61K 31/55, C07D 495/04, A61P 35/00...
Метки: качестве, фактора, некроза, опухоли, тиенодибензоазуленовые, ингибиторов, соединения
Формула / Реферат:
1. Соединение формулы I и его фармакологически приемлемые соли и сольваты где X представляет CH2 или гетероатом, выбранный из группы, включающей O, S и NR13, в котором R13 означает водород, C7-C10арилкарбонил; R1, R5, R7, R8 и R9 независимо друг от друга представляют водород; R2, R3 и R4 независимо представляют водород, фтор, хлор, бром, C1-C7алкил, галогенметил, C1-C7 алкокси или C1-C7алкилтио; R6 представляет водород, фтор, хлор, бром; R10...
Аминогетероарильные соединения в качестве ингибиторов протеинкиназ

Номер патента: 10727
Опубликовано: 30.10.2008
Авторы: Фанк Ли А., Нелсон Кристофер Джи., Харрис Джи Дейвис Мл., Нэмбу Митчелл Дейвид, Жанг Фанг-Джи, Шен Хонг, Джиа Ли, Джонсон Джоанна, Ботрос Айрини, Уолтер Эллисон, Ли Ксяоюан, Куй Джингджонг Джин, Чу Джи Ю, Жанг Дженнифер, (US)ЛИН Джейсон, Менг Джерри Джиалун, Бумралькар Дилип, Тран-Дюб Мишель, Пэйриш Мейсон Алан, Ханау Кэтлин Элизабет, Колодзей Стивен А., Канг Пей-Пей
МПК: A61K 31/4965, C07D 401/00, C07D 211/72...
Метки: протеинкиназ, соединения, качестве, ингибиторов, аминогетероарильные
Формула / Реферат:
1. Аминогетероарильное соединение формулы 1 где Y представляет собой N или CR12; R1 выбран из C6-12арила, 5-12-членного гетероарила, C3-12циклоалкила, 3-12-членного гетероалициклила, -O(CR6R7)nR4, -C(O)R4, -C(O)OR4, -CN, -NO2, -S(O)mR4, -SO2NR4R5, -C(O)NR4R5, -NR4C(O)R5, -C(=NR6)NR4R5, C1-8алкила, C2-8алкенила и C2-8алкинила; и любой атом водорода в R1 возможно заменен одной или более чем одной группой R3; R2 представляет собой водород,...
Следующий патент: Способ ингибирования выработки микотоксина
Случайный патент: Механизм плавного закрытия крышки унитаза