Способы образования дисульфидных связей и гликозилирования белков и реагенты, применяемые в этих способах
Номер патента: 11299
Опубликовано: 27.02.2009
Авторы: Фэйрбэнкс Энтони Джон, Дэвис Бенджамин Ги, Гэмблин Дэвид Филип, Гарньер Филипп


























Формула / Реферат
1. Способ получения соединения, содержащего дисульфидную связь, включающий взаимодействие аминокислоты, пептида или белка, содержащего по меньшей мере одну тиоловую группу, с соединением формулы I
![]()
где
X означает SO2 или Se;
R означает углеводную группу или фарнезил и
R1 означает факультативно замещенную C1-C10 алкильную группу, факультативно замещенную фенильную группу, или факультативно замещенную пиридильную группу, где факультативные заместители выбраны из -NO2, -SO3H, -CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n имеет значение от 1 до 100;
при условии, что когда X означает SO2, тогда R1 не является факультативно замещенным C1-C10 алкилом.
2. Способ химической модификации белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу, включающий взаимодействие указанного белка, пептида или аминокислоты с соединением формулы I
![]()
где
X означает SO2 или Se;
R означает углеводную группу или фарнезил и
R1 означает факультативно замещенную C1-C10 алкильную группу, факультативно замещенную фенильную группу или факультативно замещенную пиридильную группу, где факультативные заместители выбраны из -NO2, -SO3H, -CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n имеет значение от 1 до 100;
при условии, что когда X означает SO2, тогда R1 не является факультативно замещенным C1-C10 алкилом.
3. Способ по п.1 или 2, отличающийся тем, что R представляет собой олигосахарид.
4. Способ по любому из пп.1-3, отличающийся тем, что R1 представляет собой фенил.
5. Способ по любому из пп.1-4, отличающийся тем, что X представляет собой Se.
6. Способ по любому из пп.1-4, отличающийся тем, что X представляет собой SO2.
7. Соединение формулы I
![]()
где
X означает SO2 или Se;
R означает углеводную группировку или фарнезил и
R1 означает факультативно замещенную C1-C10 алкильную группу, факультативно замещенную фенильную группу или факультативно замещенную пиридильную группу, где факультативные заместители выбраны из -NO2, -SO3H, -CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n имеет значение от 1 до 100;
при условиях, что когда X означает SO2, тогда R1 не является факультативно замещенным C1-C10 алкилом, и
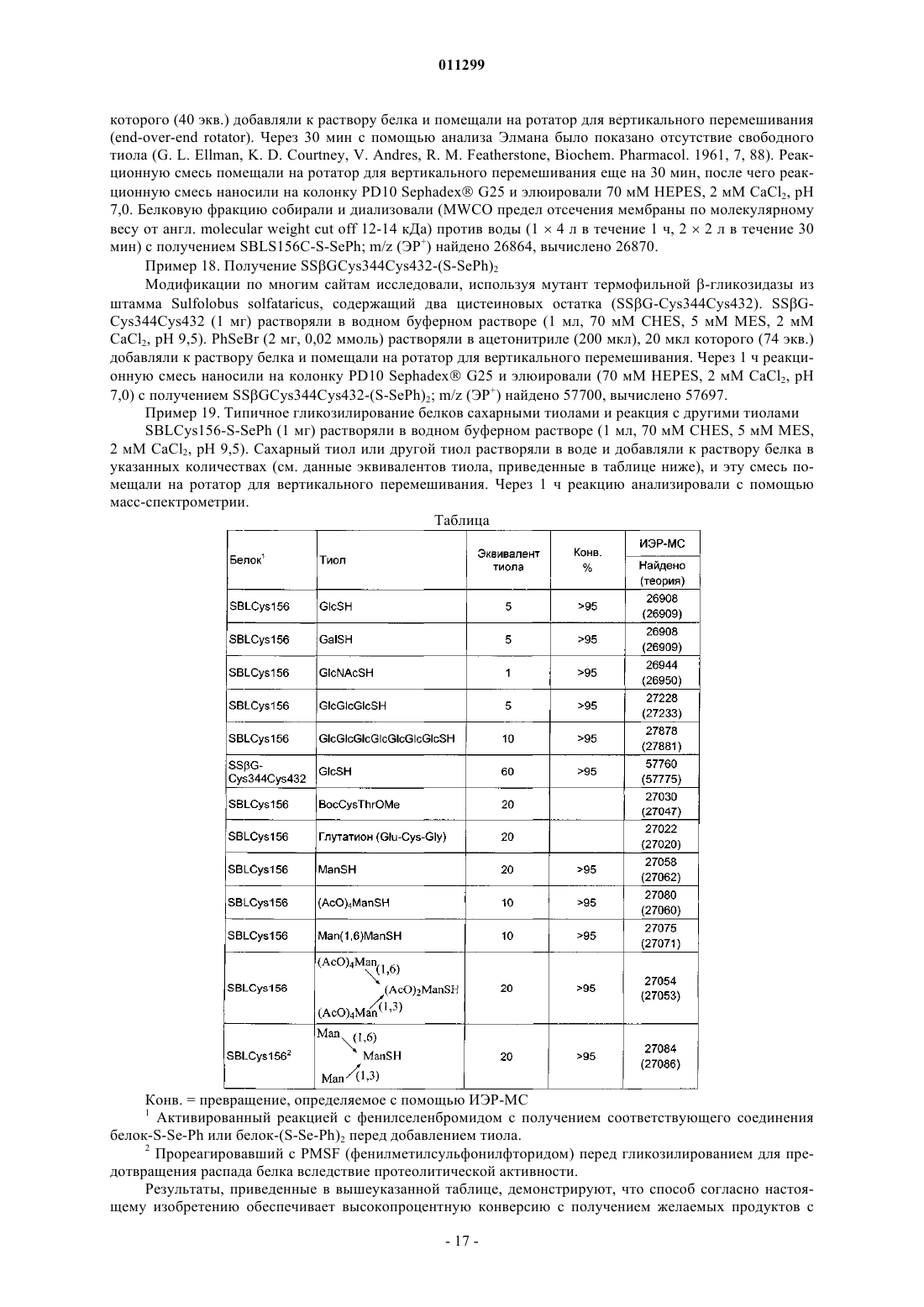
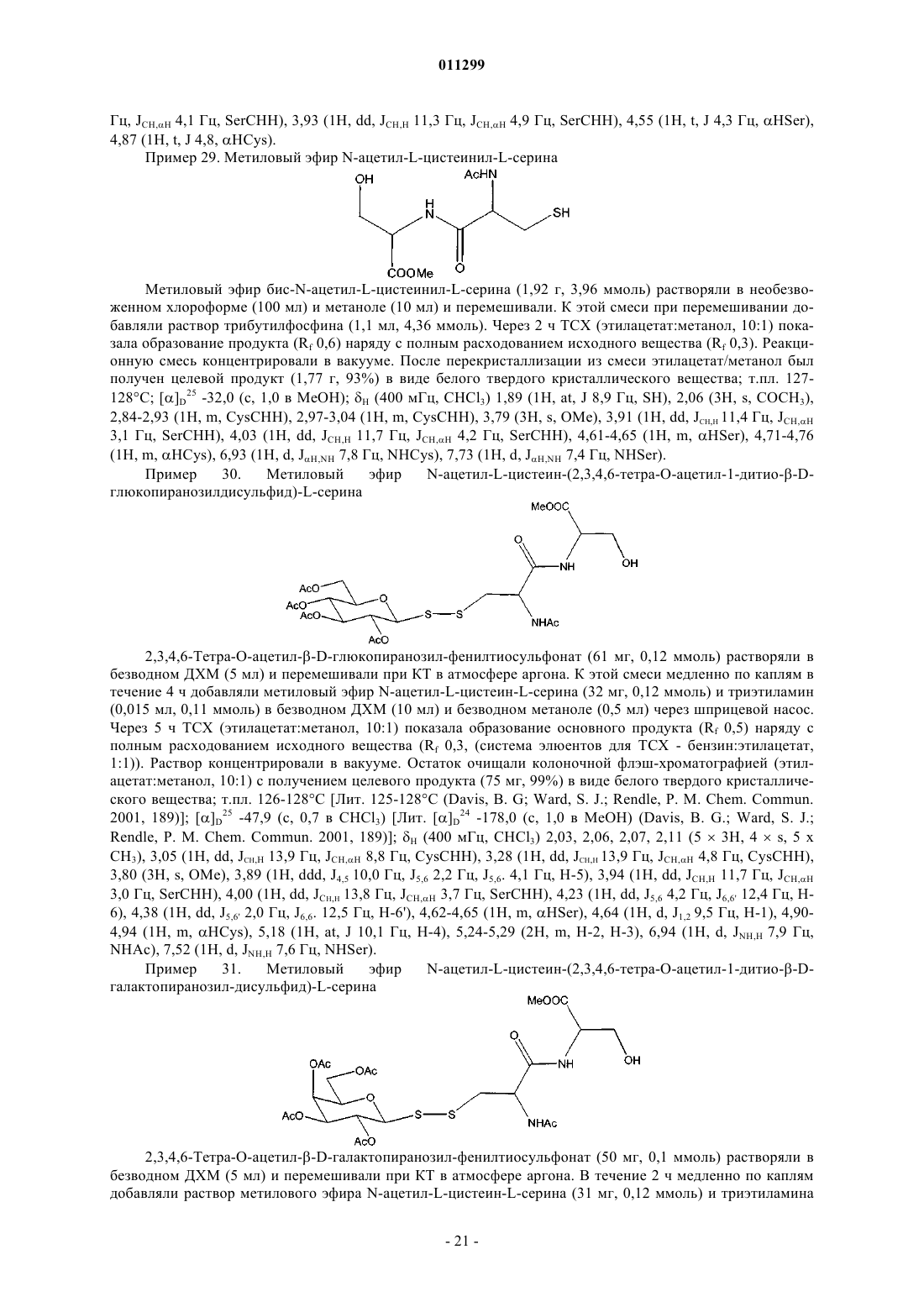
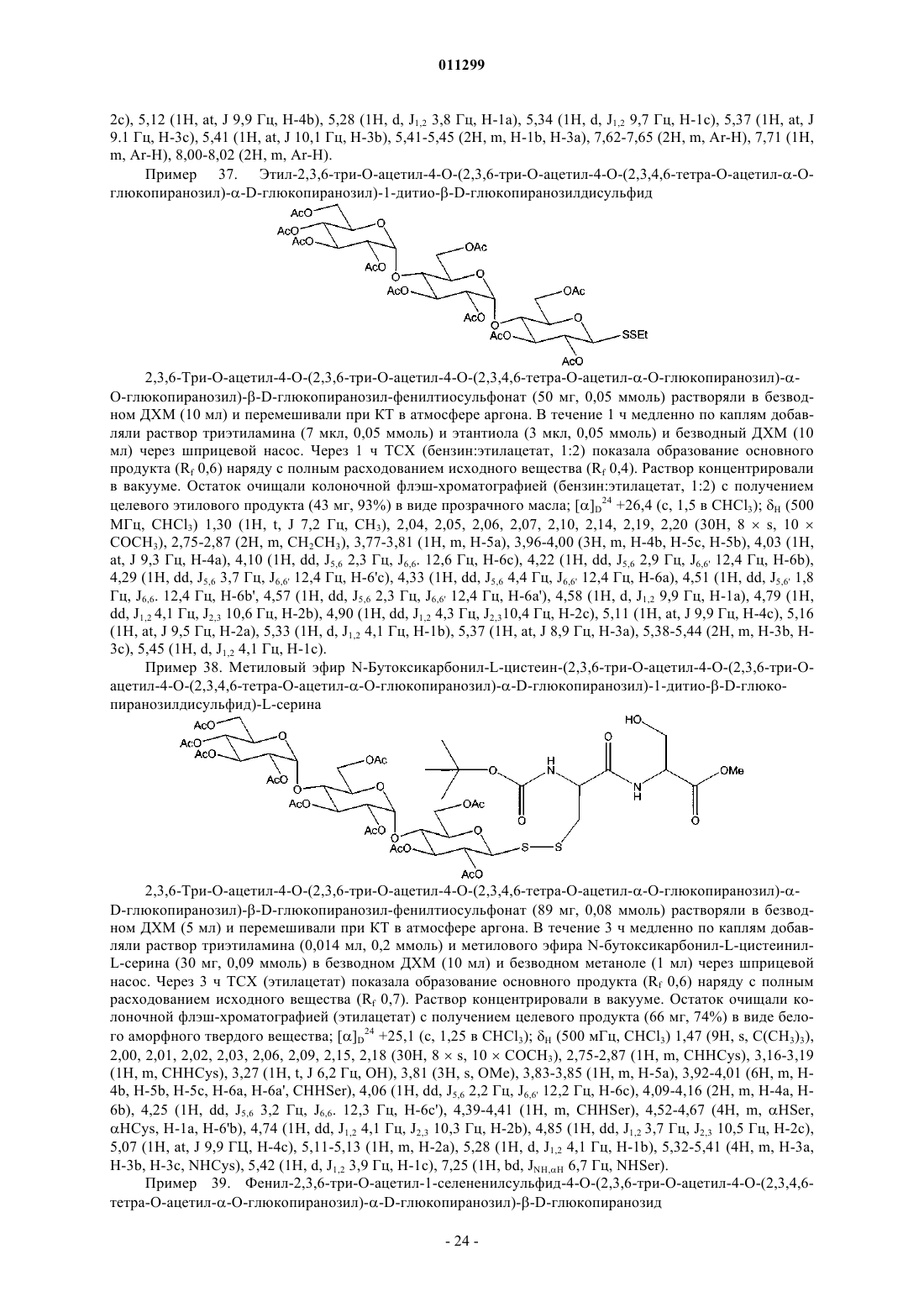
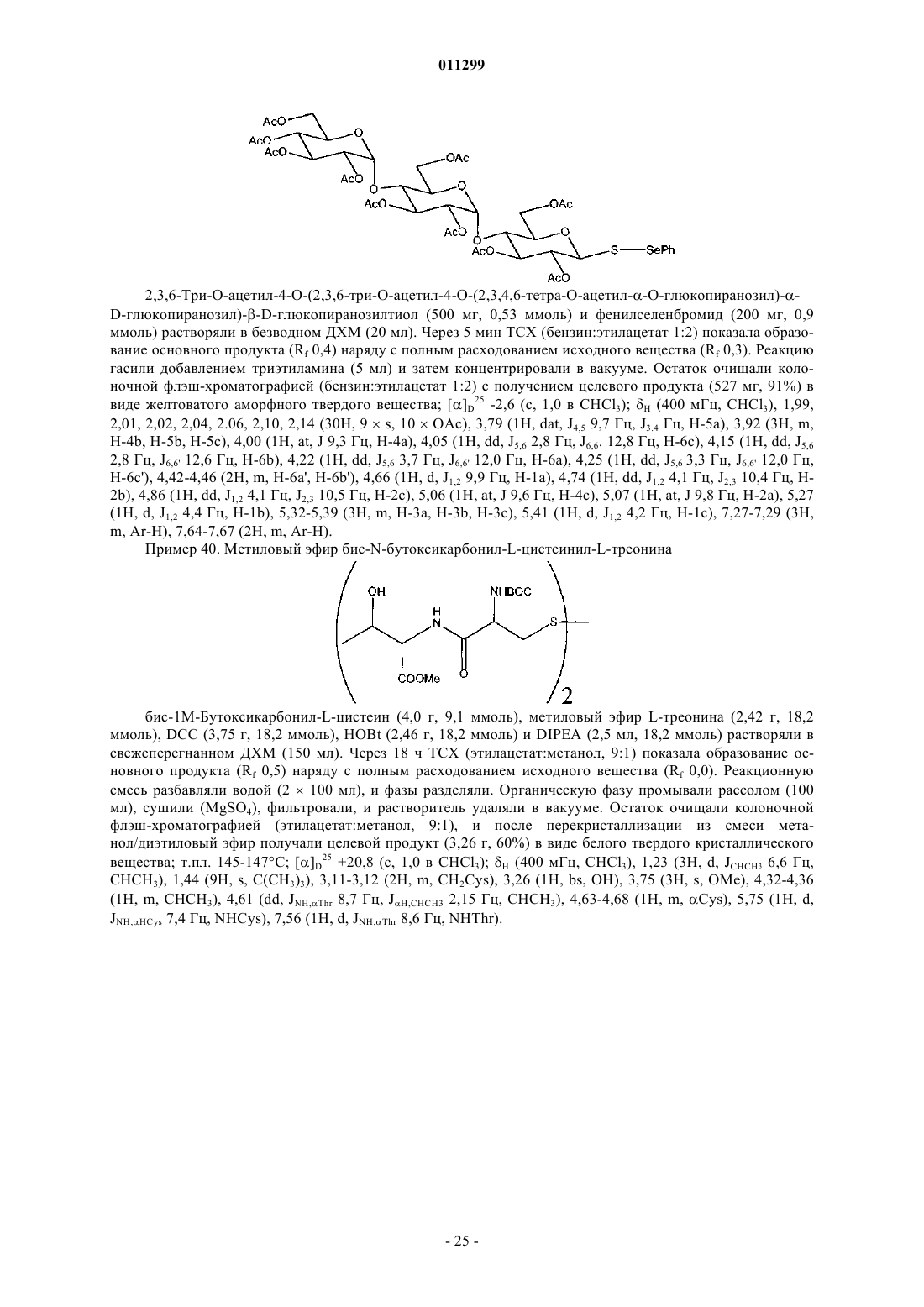
соединение не является фенилселененил 2-O-ацетил-3,4,6-три-O-бензил-1-тио-a-D-маннопиранозидом.
8. Соединение по п.7, отличающееся тем, что R1 представляет собой фенил.
9. Соединение по п.7 или 8, отличающееся тем, что R представляет собой олигосахарид.
10. Соединение по любому из пп.7-9, отличающееся тем, что X представляет собой Se.
11. Соединение по любому из пп.7-9, отличающееся тем, что X представляет собой SO2.
12. Способ получения соединения формулы I, как оно определено в п.11, включающий взаимодействие соединения формулы II
![]()
где М означает металл, например Li, Na, K, Са, Cs, Zn, Mg или Al; и k равно 1, 2 или 3; с соединением формулы III
![]()
где L означает уходящую группу.
13. Способ получения соединения формулы I, как оно определено в п.11, включающий взаимодействие дисульфидного соединения формулы VIII

с сульфинит-анионом формулы R1SO2- в присутствии ионов серебра.
14. Способ получения соединения формулы I, как оно определено в п.10, включающий взаимодействие соединения формулы V
![]()
с соединением формулы VIa или VIb

где L2 означает Br, Cl, CN или I.
15. Применение соединения формулы I, как оно определено в любом из пп.1-6, для образования дисульфидной связи.
16. Применение соединения формулы I, как оно определено в любом из пп.1-6, для модификации белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу.
17. Применение соединения формулы I, как оно определено в любом из пп.7-11, для гликозилирования белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу.
18. Способ химической модификации белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу, включающий превращение указанной тиоловой группы в селененилсульфидную группу.
19. Способ по п.18, отличающийся тем, что превращение осуществляют путем взаимодействия белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу, с соединением формулы Ха или Xb:

где
L2 означает уходящую группу; и
R2 означает факультативно замещенную C1-C10 алкильную группу, факультативно замещенную фенильную группу, факультативно замещенную пиридильную группу, или R2 образует часть твердой подложки или присоединена к ней, где факультативные заместители выбраны из -NO2, -SO3H, -CO2H,
-(CH2CH2O)nH и -(CH2CH2O)nMe, где n имеет значение от 1 до 100.
20. Способ по п.19, отличающийся тем, что R2 представляет собой фенил.
21. Способ по п.19, отличающийся тем, что соединение формулы Ха представляет собой PhSeBr.
22. Способ по любому из пп.18-21, отличающийся тем, что он дополнительно включает взаимодействие селененилсульфидной группы белка, пептида или аминокислоты с органическим соединением, содержащим тиоловую группу, отличающийся тем, что органическое соединение представляет собой углеводное соединение, белок, пептид или аминокислоту.
23. Способ химической модификации белка, пептида или аминокислоты, содержащих по меньшей мере одну селененилсульфидную группу, включающий взаимодействие белка, пептида или аминокислоты с органическим соединением, содержащим тиоловую группу, отличающийся тем, что органическое соединение представляет собой углеводное соединение, белок, пептид или аминокислоту.
24. Белок, пептид или аминокислота, содержащие по меньшей мере одну селененилсульфидную группу, где селененилсульфидная группа представляет собой группу формулы
![]()
где R2 означает факультативно замещенную C1-C10 алкильную группу, факультативно замещенную фенильную группу или факультативно замещенную пиридильную группу, где факультативные заместители выбраны из -NO2, -SO3H, -CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n имеет значение от 1 до 100.
25. Белок, пептид или аминокислота, содержащие по меньшей мере одну селененилсульфидную группу, полученную способом, как он определен в любом из пп.18-21.
26. Белок, пептид или аминокислота, содержащие по меньшей мере одну дисульфидную связь, полученную способом, как он определен в п.22 или 23.
27. Применение белка или пептида, как они определены в п.25 или 26, в определеэшш белковой структуры методами рентгеновской дифракции.
Текст
011299 Область техники Настоящая заявка относится к способам образования дисульфидных связей и/или химической модификации белков и к реагентам, применяемым в этих способах, в частности, к способам гликозилирования белков и к реагентам, применяемым в этих способах. Предшествующий уровень техники Ко- и посттрансляционное гликозилирование белков играет значительную роль в их биологическом поведении и стабильности (R. Dwek, Chem. Rev., 96:683-720 (1996. Например, гликозилирование играет главную роль в важнейших биологических процессах, таких как клеточная сигнализация и регуляция,развитие и иммунитет. Исследование этих событий осложняется тем, что гликопротеины встречаются в природе в виде смеси так называемых гликоформ, которые обладает одинаковым пептидным скелетом,но различаются как типом, так и сайтом гликозилирования. Более того, поскольку гликозилирование белков не находится под прямым генетическим контролем, экспрессия терапевтических гликопротеинов в культурах клеток млекопитающих дает в результате гетерогенную смесь гликоформ. Поэтому умение синтезировать гомогенные гликоформы гликопротеинов является не только предпосылкой для точных исследовательских целей, но и имеет вс возрастающее значение при получении терапевтических гликопротеинов, которые в настоящее время продаются в виде смеси мультигликоформ (например, эритропоэтин и интерлейкины). Другие посттрансляционные модификации белков, такие как фосфорилирование и метилирование, также имеют значение. Поэтому контролирование степени и природы такой модификации белка позволяет исследовать и контролировать его поведение в биологических системах (B.G.Davis, Science, vol. 303, р 480-482, 2004). Известен ряд способов гликозилирования белков, включая химический синтез. Химический синтез гликопротеинов дает определенные преимущества и не в последнюю очередь - возможность получать чистые гликоформы гликопротеинов. В одном известном способе синтеза используют тиол-селективные углеводные реагенты - гликозилметантиосульфонатные реагенты (глико-MTS). Такие гликозилметантиосульфонатные реагенты реагируют с тиоловыми группами белка с введением гликозильного остатка,связанного с белком через дисульфидную связь (см., например, WO00/01712). Однако глико-MTS реагенты имеют ряд недостатков, включая небольшие реакционные выходы,трудности в их получении и проблемы со стабильностью в щелочных условиях, в которых их часто используют. Поэтому существует потребность в дополнительных реагентах, применяемых при гликозилировании белков, которые могут быть легко получены, являются стабильными и дают высокие выходы гликозилированного белкового продукта. Существует также потребность в альтернативных способах гликозилирования белков, которые дают высокие выходы гликозилированного белкового продукта, являются сайт-селективными и которые обеспечивают гликозилирование как по одному, так по многим сайтам у широкого спектра различных белков. Сущность изобретения Авторы изобретения неожиданно обнаружили, что некоторые серо- и селеносодержащие реагенты гликозилирования являются относительно простыми для получения, обычно более стабильны, чем соответствующие глико-MTS реагенты, и могут быть использованы в гликозилировании широкого спектра тиол-содержащих соединений, включая белки, с высоким выходом. Поэтому в первом аспекте изобретения предусмотрен способ образования дисульфидных связей (S-S-), включающий взаимодействие органического соединения, содержащего по меньшей мере одну тиоловую группу (-SH), с соединением формулы I: гдеR означает органическую группировку, например алкильную группу, алкенильную группу, алкинильную группу или углеводную группировку; иR1 означает факультативно замещенную алкильную группу, факультативно замещенную фенильную группу, факультативно замещенную пиридильную группу или факультативно замещенную нафтильную группу; при условии, что если X означает SO2, то R1 не является факультативно замещенным алкилом. Предпочтительно, органическое соединение, содержащее по меньшей мере одну тиоловую группу,представляет собой аминокислоту, пептид или белок. Во втором аспекте изобретения, кроме того, предусмотрен способ химической модификации белка,пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу (-SH), включающий взаимодействие указанного белка, пептида или аминокислоты с соединением формулы I, как определено выше. В еще одном аспекте изобретения предусмотрены соединения формулы I, где R означает углеводную группировку. Когда R означает алкенильную или алкинильную группу, существует вероятность, что дисульфид-1 011299 ное соединение, образованное путем взаимодействия с соединением формулы I, может быть дополнительно модифицировано путем взаимодействия по С=С или С=С связи в группе R. Авторы изобретения также неожиданно обнаружили, что тиол-содержащий белок может быть превращен в соответствующий селененилсульфид и что электрофильные свойства серы в S-Se связи, образованной таким образом, делают ее чувствительной к нуклеофильному замещению тиол-содержащими соединениями, включая углеводы. Поэтому в третьем аспекте изобретения предусмотрен способ химической модификации белка, пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу (-S-H), включающий превращение указанной тиоловой группы в селененилсульфидную группу (-S-Se-R2). Таким образом, этот способ позволяет получать белок, пептид или аминокислоту, содержащие по меньшей мере одну селененилсульфидную группу. Такие белки, пептиды и аминокислоты, содержащие по меньшей мере одну селененилсульфидную группу, формируют еще один признак изобретения. Особенно предпочтительными являются белки или пептиды, содержащие по меньшей мере одну селененилсульфидную группу. Селененилсульфидная группа в белке, пептиде или аминокислоте может быть дополнительно подвергнута взаимодействию с органическим соединением, содержащим тиоловую группу, с получением дополнительных химически модифицированных белков, пептидов или аминокислот, в которых органическая группа присоединена к белку, пептиду или аминокислоте через дисульфидную связь. Предпочтительно органическое соединение, содержащее тиоловую группу, представляет собой углеводное соединение, обеспечивая таким образом способ гликозилирования аминокислоты, пептида или белка. Как использовано в данном контексте, "гликозилирование" относится к общему способу добавления гликозильной единицы к другой группировке через ковалентную связь. Поэтому в четвертом аспекте изобретения предусмотрен способ химической модификации белка,пептида или аминокислоты, содержащих по меньшей мере одну тиоловую группу (-S-H), включающий:(a) превращение указанной тиоловой группы в селененилсульфидную группу (-S-Se-R2); и(b) взаимодействие указанной селененилсульфидной группы с органическим соединением, содержащим тиоловую группу. Способ(ы) согласно первому, второму, третьему и четвертому аспектам изобретения в дальнейшем в этом документе будут называться, соответственно, первым способом, вторым способом, третьим способом и четвертым способом. Если не указано иначе, все предпочтительные признаки и определения в этом документе относятся ко всем этим способам. Более того, настоящее изобретение включает любые и все возможные комбинации любых предпочтительных признаков, о которых идет речь в данном документе, независимо от того, описаны такие комбинации конкретно или нет. Обобщенная реакционная схема образования дисульфидной связи согласно первому и второму способам показана на схеме 1: Схема 1 Предпочтительно органическая группировка, представленная на схеме 1, представляет собой белок,пептид или аминокислоту. Обобщенная реакционная схема введения селененилсульфидной группы в белок, пептид или аминокислоту согласно третьему и четвертому способам показана на схеме 2: Схема 2 Способ согласно Схеме 2 дает в результате ковалентную связь группы R2 с белком, пептидом или аминокислотой через селененилсульфидную (-S-Se-) связь. Такие белки, пептиды или аминокислоты формируют еще один признак изобретения. Белки и пептиды, содержащие одну селененилсульфидную группу, могут быть полезны в определении белковой структуры методами рентгеновской дифракции. В настоящее время методы MAD (аномальной дисперсии на различных длинах волн от англ. multiple wavelength anomalous dispersion) включают превращение любых метиониновых остатков в белке в селенометионин. Сравнение картин рентгеновской дифракции модифицированных и немодифицированных белков делает возможным последую-2 011299 щее определение структуры немодифицированного белка, с которым будут работать. Способ по изобретению обеспечивает удобный и быстрый доступ к альтернативным селеносодержащим белкам или пептидам, которые могут быть использованы в таких методах. В способах согласно изобретению предусмотрен удобный способ введения тяжелого металла в белковую структуру, который облегчает, таким образом, интерпретацию данных рентгеновской дифракции. Селененилсульфид-содержащие белки, пептиды или аминокислоты могут быть дополнительно подвергнуты взаимодействию с тиол-содержащими органическими соединениями согласно четвертому способу, как показано в обобщенной реакционной схеме 3: Схема 3 Способ согласно схеме 3 дает в результате ковалентную связь органической группировки с белком,пептидом или аминокислотой через дисульфидную связь (-S-S-). В атом способе белок, пептид или аминокислота действуют в качестве электрофила, тогда как тиол-содержащее органическое соединение действует в качестве нуклеофила. В противоположность этому, известные реакции, в которых используются глико-MTS реагенты, включают в себя реакцию нуклеофильной тиоловой группы в белке, пептиде или аминокислоте с электрофильным глико-MTS реагентом. Поэтому в способе по изобретению предусмотрена дополнительная стратегия к уже известным стратегиям модификации белков с использованием глико-MTS реагентов. Сведения, подтверждающие возможность осуществления изобретения При использовании в данном описании, термин алкил предпочтительно означает прямую или разветвленную алкильную группу, содержащую 1-10 атомов углерода, предпочтительно 1-6 атомов углерода. Предпочтительные алкильные группы включают в себя метил и этил. При использовании в данном описании, термин алкенил предпочтительно означает прямую или разветвленную углеводородную группу, включающую по меньшей мере одну двойную углерод-углеродную связь и содержащую 2-20 атомов углерода, предпочтительно 2-10 атомов углерода и более предпочтительно 2-6 атомов углерода. Предпочтительные алкенильные группы включают в себя -(СН 2)СН=СН 2, -СН 2 СН 2 СН=СН 2, пренилСН 3)2 С=СНСН 2-) и фарнезилСН 3)2 С=СН[СН 2 СН 2 С(СН 3)=СН]2 СН 2-). При использовании в данном описании, термин алкинил предпочтительно означает прямую или разветвленную углеводородную группу, включающую по меньшей мере одну тройную углерод-углеродную связь и содержащую 2-10 атомов углерода, предпочтительно 2-6 атомов углерода. Предпочтительные алкинильные группы включают в себя -СН 2 С=СН и -СН 2 СН 2 ССН. Когда R1 означает факультативно замещенную группировку, тогда подходящие заместители включают в себя любые заместители, которые не препятствуют образованию соединения формулы I или реакции образования дисульфидной связи согласно первому или второму способам, например, -NO2, -SO3H, CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n равно 1-100, предпочтительно 1-50, более предпочтительно 1-20 и еще более предпочтительно 1-10. Группа R1 может быть независимо замещена 1-5 и предпочтительно 1 или 2 заместителями. Группа R1 может также по выбору присоединяться к твердой подложке или образовывать часть твердой подложки, например, смолы, такой как полистирольная смола. Предпочтительной группой R1 является фенил. Когда группа R1 в соединениях формулы I представляет собой фенил или другую ароматическую группу, тогда существует дополнительное преимущество в том, что протекание реакции с тиол-содержащим соединением согласно первому и второму способам можно контролировать с использованием УФ спектроскопии. Так, например, PhSO2-хромофор демонстрирует максимум в УФ спектре при приблизительно 265 нм. PhSO2-группировка присутствует и в соединении формулы I, и в PhSO2-, которая является побочным продуктом реакции образования дисульфидной связи, но связанные коэффициенты экстинкции существенно различаются в ходе реакции, которую будут контролировать с использованием УФ. Аналогично, третий и четвертый способы изобретению можно контролировать с помощью УФ спектроскопии, когда группа R2 представляет собой фенил или другую ароматическую группу. В соединениях формулы I группа R может представлять собой любую органическую группировку, в частности, любую органическую группировку, которая подходит для связывания с белком, пептидом или аминокислотой. Не существует никакого специального ограничения относительно природы R. Например,-3 011299 группа R может быть первичной, вторичной или третичной. R может быть ароматической или алифатической. Группа R может быть факультативно замещена, например, фосфорильными или сульфонильными заместителями. Когда X представляет собой Se, тогда R может представлять собой также белок, пептид или аминокислоту, которые дают возможность одному белку, пептиду или аминокислоте связываться с другим белком, пептидом или аминокислотой через дисульфидную связь. Одна предпочтительная группа R представляет собой фарнезил. Фарнезилирование является природной посттрансляционной модификацией, ассоциированной со многими белками, включая онкогенный белок Ras. Поэтому способы по изобретению дают возможность получать фарнезилированные белки,пептиды и аминокислоты. Также предпочтительно, R представляет собой углеводную группировку, факультативно присоединенную через линкер к группе -S-X-. Линкер может содержать 1-10 атомов между углеводной группировкой и группой -S-X-. Например, линкер может представлять собой алкиленовую группу (например,группу -(СН 2)2, где t равно 1-10), или алкениленовую группу (например, группу -(СН 2)СН=СН- или СН 2 СН 2 СН=СН-). Предпочтительными являются соединения, в которых группа -S-X- находится в аномерном положении сахаридного остатка или присоединена к аномерному углероду через линкер. Подходящие углеводные группировки включают в себя моносахариды, олигосахариды и полисахариды, и включают в себя любую углеводную группировку, которая присутствует в природных гликопротеинах или в биологических системах. Предпочтительными являются гликозильные или гликозидные производные, которые могут быть защищены, например, глюкозильные, глюкозидные, галактозильные или галактозидные производные, которые могут быть защищены. Гликозильные и гликозидные группы включают в себя как -, так и -группы. Подходящие углеводные группировки включают в себя глюкозу, галактозу, фукозу, GlcNAc, GalNAc, сиаловую кислоту и маннозу, и олигосахариды или полисахариды, содержащие по меньшей мере один остаток глюкозы, галактозы, фукозы, GlcNAc, GalNAc, сиаловой кислоты и/или маннозы. Любые функциональные группы в углеводной группировке могут быть защищены с использованием защитных групп, известных в данной области (см., например, Greene et al., "Protective groups in organicsynthesis", 2nd Edition, Wiley, New York, 1991, раскрытие которой таким образом включено путем ссылки). Подходящие защитные группы для любой группы -ОН в углеводной группировке включают в себя ацетил (Ас), бензил (Bn), пиволил (piv), силил (например, трет-бутилдиметилсилил (TBDMSi) и третбутилдифенилсилил (TMDPSi, ацетали, кетали и метоксиметил (MOM). Любые защитные группы могут быть удалены до или после присоединения углеводной группировки к аминокислоте, пептиду или белку. Особенно предпочтительные углеводные группировки включают в себя Glc(Ac)4-, Glc(Bn)4-,Gal(Ac)4-, Gal(Bn)4-, Glc(Ac)4(1,4)Glc(Ac)3(1,4)Glc(Ac)4-, -Glc, -Gal, -Man, -Man(Ac)4,Man(1,6)Man-, Man(1-6)Man(1-3)Man-, (Ac)4Man(1-6)(Ac)4Man(1-3)(Ac)2Man-, -EtGal, -EtGlc,EtGlc, -EtMan, -Et-Lac, Glc(Ac)2, Glc(Ac)3, -EtGlc(Ac)2, -EtGlc(Ac)3, -EtGlc(Ac)4, -Et-Glc(Ac)2, -EtGlc(Ac)3, -EtGlc(Ac)4, -EtMan(Ac)3, -EtMan(Ac)4, -EtGal(Ac)3, -EtGal(Ac)4,-Et-Lac(Ac)5, -Et-Lac(Ac)6, -Et-Lac(Ac)7 и их эквиваленты после удаления защиты. Предпочтительно, любые сахаридные фрагменты, входящие в состав углеводной группировки, которые происходят из природных сахаров, будут находиться в природной энантиомерной форме, которая может представлять собой либо D-форму (например, D-глюкоза или D-галактоза), либо L-форму (например, L-рамноза или L-фукоза). Любые аномерные связи могут представлять собой - или -связи. Соединение, содержащее тиоловую группу, используемое в первом или втором способах, может представлять собой любое органическое соединение, содержащее по меньшей мере одну тиоловую группу. Тиоловая группа может быть первичной, вторичной или третичной. Соединение может быть ароматическим или алифатическим. Если в соединении присутствует более чем одна тиоловая группа, то в каждой такой тиоловой группе будет потенциально образовываться дисульфидная связь. Предпочтительно соединение представляет собой аминокислоту, пептид или белок. Как использовано в данном описании, пептид содержит минимум два аминокислотных остатка, связанных амидной связью. Любая аминокислота, содержащаяся в белке, пептиде или аминокислоте, представляет собой предпочтительно -аминокислоту. Любая аминокислота может находиться в D- или L-форме, предпочтительно в L-форме. Аминокислота, пептид или белок могут представлять собой любую природную аминокислоту, пептид или белок, содержащие тиоловую группу, например, благодаря присутствию одного или более остатков цистеина. Альтернативно, аминокислота, пептид или белок могут быть получены путем химической модификации предшественника аминокислоты, пептида или белка, не содержащих тиол. Альтернативно, тиол-содержащий пептид или белок может быть получен посредством сайтнаправленного мутагенеза с введением остатка цистеина. Сайт-направленный мутагенез является известным методом в данной области (см., например, WO00/01712 и J. Sambrook et al., Molecular Cloning: ALaboratory Manual, 3rd Edition, Cold Springs Harbour Laboratory Press, 2001, содержание которых включено в настоящую заявку путем ссылки). Предпочтительные белки включают в себя ферменты, селективность которых может быть модифи-4 011299 цирована путем контролируемого гликозилирования с использованием способов и реагентов в соответствии с изобретением, и терапевтические белки. Другие предпочтительные белки включают в себя сывороточные альбумины и другие белки крови, гормоны, интерфероны, рецепторы, антитела, интерлейкины и эритропоэтин. Было обнаружено, что соединения формулы I обычно тиол-селективны и поэтому присутствие других функциональных групп в тиол-содержащем органическом соединении обычно не препятствует реакции. Однако любые другие функциональные группы могут быть защищены с использованием любых защитных групп, известных в данной области, которые стабильны в условиях реакции. Реакцию образования дисульфидной связи согласно первому или второму способу обычно осуществляют в присутствии буфера при нейтральном или щелочном рН (рН от примерно 7 до примерно 9,5),причем предпочтительными являются слегка щелочные значения рН (рН от примерно 8 до примерно 9). Подходящие буферы включают в себя HEPES, CHES, MES и Трис. Если тиол-содержащее соединение представляет собой белок, пептид или аминокислоту, то величина рН должна быть такой, чтобы во время реакции не наблюдалось нежелательной денатурации, или она была бы незначительной. Аналогично,температура реакции должна быть выбрана такой, чтобы исключить любое значительное повреждение любых соединений, чувствительных к температуре. Например, реакцию с белком или пептидом предпочтительно осуществляют при температуре окружающей среды или ниже, чтобы избежать какой-либо денатурации. Могут быть использованы системы водных или органических растворителей, причем системы водных растворителей являются предпочтительными для реакции белков, аминокислот или пептидов для обеспечения их растворения. Реакция обычно протекает довольно быстро, например, часто в течение менее 1 ч. Обычно при проведении реакции используется избыток соединения формулы I, например, 10-20 эквивалентов по отношению к тиол-содержащему соединению. В противоположность этому, реакции с глико-MTS реагентами часто требуют применения приблизительно 30 эквивалентов, что увеличивает стоимость реагентов. Было обнаружено, что соединения формулы I, где R означает углеводную группировку, X означаетSO2, и R1 означает фенил, обычно более стабильны в щелочных условиях, чем соответствующие гликоMTS соединения. Поэтому любое непрореагировавшее соединение формулы I или его избыток часто могут быть выделены из реакционной смеси для повторного использования, что особенно выгодно, когда R означает углеводную группировку, поскольку такие соединения могут быть относительно дорогостоящими и/или трудоемкими для получения. Более того, фенилтиосульфонатные соединения формулы I обычно дешевле и легче для получения, чем соответствующие MTS соединения. Соединения формулы I могут быть получены различными способами. Соединения, где X означаетSO2, могут быть получены путем взаимодействия соединения формулы II: где М означает металл, например, Li, Na, K, Cs, Ca, Mg, Zn или Al, предпочтительно Na или K; иR такой, как определено для соединения формулы I, и L означает уходящую группу. Любая уходящая группа L может быть использована до тех пор, пока полученный анион L- не будет каким-либо образом чрезмерно препятствовать реакции, например, путем взаимодействия с продуктом. Предпочтительные уходящие группы L включают в себя галогено и сульфонаты, такие как толуолсульфонат (тозилат), метансульфонат (мезилат) и трифторметансульфонат (трифлат), в частности хлоро и бромо. Соединения формулы III имеются в продаже или могут быть получены с использованием способов,известных в данной области, например, способов образования галогеносахаров вообще и 1 галогеносахаров в частности. Предпочтительно, соединение формулы III представляет собой гликозилгалогенид. Примеры подходящих соединений формулы III на основе глюкозы и галактозы показаны в общем случае ниже: где каждый R5 независимо представляет собой Н, сахаридную группировку или подходящую защитную группу, например, Ас или Bn, предпочтительно каждый R5 представляет собой Н; один из R3 и R4 представляет собой Н, а другой представляет собой ОН, О-защитную группу или Осахаридную группировку, предпочтительно Н или О-сахаридную группировку; иt равно 1-10, предпочтительно 1-6, более предпочтительно 2 или 3. Реакция может быть осуществлена в любой системе растворителей, в которых растворимо соединение формулы III. Предпочтительно, соединение формулы II также по меньшей мере частично растворимо в системе растворителей. Подходящие растворители включают в себя алканолы, такие как этанол и метанол, N,N-диметилформамид (ДМФА) и ацетонитрил, причем ацетонитрил является особенно предпочтительным. Соединения формулы II могут быть получены путем взаимодействия соответствующей сульфинитной соли (формула VII) с серой, как показано на схеме 4: Схема 4 Соединения формулы II, являющиеся кристаллическими, предпочтительны для простоты очистки,особенно в промышленном масштабе. Сульфинитные соли формулы VII имеются в продаже (например, бензолсульфинит натрия) или могут быть получены способами, известными в данной области (см., например, JP 61205249, и М. Uchino etal., ChemicalPharmaceutical Bulletin, 1978, 26(6), 1837-45, содержание которых включено в настоящую заявку путем ссылки). Например, соответствующая тиолатная соль R1S- может быть получена путем депротонирования соответствующего тиолового соединения R1SH с использованием подходящего основания, например, метиллития. Тиолатная соль затем может быть окислена до соответствующей сульфинитной соли с использованием подходящего окисляющего агента, например, 2-(фенилсульфонил)-3 фенилоксазиридина ("реагента Дэвиса", Sandrinelli et al., Organic Letters (1999), 1(8), 1177-1180, содержание которого включено в настоящую заявку путем ссылки). Альтернативно, соединения формулы I, в которых X означает SO2, могут быть получены путем взаимодействия дисульфида формулы VIII с сульфинит-анионом R1SO2- в присутствии ионов серебра,как показано на схеме 5: Схема 5 Дисульфидные соединения формулы VIII имеются в продаже или могут быть получены с использованием способов, известных в данной области. Соединения формулы I, где X означает Se, могут быть образованы путем взаимодействия соединения формулы V: где R такой, как определено для соединения формулы I, с соединением формулы VIa или VIb: где R1 такой, как определено для соединения формулы I, и L2 означает уходящую группу, например,ОН, Br, CI, CN или I, предпочтительно Br. Реакцию можно проводить в безводном дихлорметане и затем гасить добавлением триэтиламина. Предпочтительное соединение формулы IVa представляет собойPhSeBr, a предпочтительное соединение формулы VIb представляет собой PhSe(OH)2.-6 011299 Соединения формулы VI имеются в продаже (например, PhSeBr, PhSeCl, PhSeCN, 2 нитрофенилселеноцианат) или могут быть получены способами, известными в данной области. Например, MeSeBr может быть получен согласно способу, опубликованному Hope Eric G., Kemmitt Tim и Levason William, в Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (19721999) (1987), (4), 487-90, содержание которого включено в настоящую заявку путем ссылки. Органические соединения, содержащие по меньшей мере одну тиоловую группу, включая соединения формулы V, имеются в продаже или могут быть получены с использованием способов, известных в данной области, например, способов получения тиоловых соединений вообще и тиосахаров в частности. Например, тиосахара могут быть получены из соответствующих галогеносахаров путем обработки галогеносахара тиомочевиной с получением соответствующей изотиоурониевой соли (W. A. Bonner, J. E.Kahn, J. Am. Chem. Soc. 1951, 73), с последующим мягким гидролизом метабисульфитом натрия с получением соответствующего тиола. При необходимости, в процессе синтеза любых тиосахаров могут быть использованы подходящие защитные группы. Когда R в соединении формулы V означает углеводную группировку, тогда тиоловая группа может находиться в любом положении в этой группировке. Предпочтительно, она находится в аномерном положении сахарида или присоединяется к аномерному углероду через линкер. Примеры подходящих соединений формулы V на основе глюкозы и галактозы в общем виде показаны ниже: где каждый R5 независимо представляет собой Н, сахаридную группировку или подходящую защитную группу, например, Ас или Вn, предпочтительно каждый R5 представляет собой Н; один из R3 и R4 представляет собой Н, а другой представляет собой ОН, О-защитную группу или Осахаридную группировку, предпочтительно Н или О-сахаридную группировку; иr равно 2-10, предпочтительно 2-6, более предпочтительно 2 или 3. Соединения формулы V также подходят для применения в качестве тиол-содержащего соединения в четвертом способе настоящего изобретения. При взаимодействии соединений формулы V с соединениями формулы VI любые другие функциональные группы в соединении формулы V могут быть незащищенными или могут быть защищены защитными группами, известными в данной области. Превращение по меньшей мере одной тиоловой группы в белке, пептиде или аминокислоте в одну селененилсульфидную группу согласно третьему или четвертому способу является высоко селективным. Кроме того, реакция тиол-содержащего органического соединения с селененилсульфидной группой является высоко сайт-селективной. Поэтому обычно нет необходимости в защите каких-либо других функциональных групп в белке, пептиде или аминокислоте или в тиол-содержащем органическом соединении, пока осуществляют способы согласно изобретению. Это может быть чрезвычайно полезно, поскольку исключает необходимость последующих стадий удаления защиты, которые проводятся с продуктом. Если белок, пептид или аминокислота содержат более чем одну тиоловую группу, то потенциально каждая такая тиоловая группа будет превращаться в соответствующую селененилсульфидную группу. Потенциально каждая такая селененилсульфидная группа может быть затем подвергнута взаимодействию с тиол-содержащим органическим соединением, что приводит к присоединению органического соединения через дисульфидную связь к белку, пептиду или аминокислоте по многим сайтам. Таким образом, в способах согласно настоящему изобретению предусмотрен удобный способ химической модификации белка, пептида или аминокислоты по многим сайтам. В частности, способы согласно настоящему изобретению делают возможным гликозилирование белка, пептида или аминокислоты по многим сайтам. Превращение тиоловой группы в белке, пептиде или аминокислоте в селененилсульфидную группу согласно третьему или четвертому способу удобно осуществлять путем взаимодействия указанного белка, пептида или аминокислоты с соединением формулы Ха или Xb:R2 означает факультативно замещенную алкильную группу, факультативно замещенную фенильную группу, факультативно замещенную бензильную группу, факультативно замещенную пиридильную группу или факультативно замещенную нафтильную группу. Предпочтительная группа R2 представляет собой фенил, предпочтительное соединение формулы Ха представляет собой PhSeBr, a предпочтительное соединение формулы Xb представляет собой PhSe(OH)2. Когда R2 означает факультативно замещенную группировку, тогда подходящие заместители включают в себя любые заместители, которые не препятствуют реакции с тиол-содержащим белком, пептидом или аминокислотой, и предпочтительно также не препятствуют любой последующей реакции белка,пептида или аминокислоты, например, реакции с тиол-содержащим органическим соединением. Подходящие заместители включают в себя -NO2, -SO3H, -CO2H, -(CH2CH2O)nH и -(CH2CH2O)nMe, где n равно 1100, предпочтительно 1-50, более предпочтительно 1-20 и еще более предпочтительно 1-10. Группа R2 может быть независимо замещена 1-5 и предпочтительно 1 или 2 заместителями. Группа R2 может также по выбору присоединяться к твердой подложке или образовывать часть этой твердой подложки. Например, соединение формулы Ха или Xb может быть получено из смолы, такой как полистирольная смола, как показано ниже: Соединения формулы Ха и Xb имеются в продаже или могут быть получены способами, известными в данной области, как обсуждалось выше для соединений формулы VIa и VIb. По меньшей мере один молярный эквивалент соединения формулы Ха или Xb на одну тиоловую группу белка, пептида или аминокислоты следует использовать для обеспечения превращения каждой такой тиоловой группы в соответствующую селененилсульфидную группу. Реакцию предпочтительно осуществляют в водном растворителе (таком как смесь воды и ацетонитрила) в присутствии буфера (например, MES, рН 9,5). рН и температуру реакции следует выбирать таким образом, чтобы избежать нежелательной денатурации белка или пептида. Предпочтительно, реакцию осуществляют при комнатной температуре или ниже, при слегка щелочном значении рН (например, при рН от примерно 8 до примерно 9,5). Органическое соединение, содержащее тиоловую группу, может представлять собой любое органическое соединение, которое подходит для связывания с белком, пептидом или аминокислотой, и в котором атом серы тиоловой группы может действовать в качестве нуклеофила при взаимодействии с селененилсульфидной группой. Не существует никакого специального ограничения относительно природы органического соединения. Например, тиоловая группа может быть первичной, вторичной или третичной. Соединение может быть ароматическим или алифатическим. Например, соединение может представлять собой алкил, алкенил (например, фарнезил) или алкинилтиол. Предпочтительно соединение содержит только одну тиоловую группу. Подходящие органические группировки для присоединения к белку, пептиду или аминокислоте включают в себя любую группу, которая может быть полезна при модификации физических или химических свойств белка, пептида или аминокислоты. Подходящие группировки включают в себя метки (например, флуоресцентные метки) или группы для обеспечения стабильности, процессинга или растворимости белка, пептида или аминокислоты. Органическое соединение может представлять собой также второй белок, пептид или аминокислоту, которые обеспечивают возможность связывания одного белка,пептида или аминокислоты с другим белком, пептидом или аминокислотой через дисульфидную связь с использованием способов согласно настоящему изобретению. Предпочтительно, органическое соединение, содержащее по меньшей мере одну тиоловую группу,представляет собой фарнезильное производное, или представляет собой углеводную группировку, как определено выше, и может быть присоединено через линкер к тиоловой (-S-H) группе. Линкер может содержать 1-10 атомов между углеводной группировкой и группой -SH. Например, линкер может представлять собой алкиленовую группу (например, группу -(СН 2)2, где t равно 1-10), или алкениленовую группу (например, группу -(СН 2)СН=СН- или -СН 2 СН 2 СН=СН-). Предпочтительными являются соединения, в которых тиоловая группа находится в аномерном положении сахаридного остатка или присоединена к аномерному углероду через линкер.-8 011299 Любые функциональные группы в углеводной группировке могут быть защищены с использованием защитных групп, известных в данной области, как обсуждалось выше. Любые защитные группы могут быть удалены до или после присоединения углеводной группировки к аминокислоте, пептиду или белку. Предпочтительно, их удаляют до реакции с селененилсульфидным соединением, чтобы исключить необходимость осуществления каких-либо стадий удаления защиты после стадии присоединения. Еще одно преимущество способа гликозилирования согласно изобретению состоит в том, что он обеспечивает возможность связывания незащищенных углеводных группировок с аминокислотой, пептидом или белком. Реакцию селененилсульфидной группы с органическим соединением, содержащим тиоловую группу, согласно четвертому способу (т.е. реакцию образования дисульфидной связи) обычно осуществляют в присутствии буфера при нейтральном или щелочном рН (например, при рН от примерно 7 до примерно 9,5), причем предпочтительными являются слегка щелочные значения рН (например, рН от примерно 8 до примерно 9). Подходящие буферы включают в себя HEPES, CHES, MES и Трис. Величина рН должна быть такой, чтобы во время реакции не наблюдалось нежелательной денатурации, или она была бы незначительной. Аналогично, температуру реакции следует выбирать таким образом, чтобы избежать каких-либо существенных повреждений соединений чувствительных к температуре. Например, реакцию с белком или пептидом предпочтительно осуществляют при температуре окружающей среды или ниже,чтобы избежать какой-либо денатурации. Могут быть использованы системы с водными или органическими растворителями, при этом системы с водными растворителями являются предпочтительными для обеспечения растворения белка, аминокислоты или пептида. Системы с водными растворителями также являются предпочтительными, поскольку они обеспечивают возможность использования незащищенных углеводных соединений в качестве органического соединения. Реакция обычно протекает довольно быстро, например, часто в течение менее 1 ч. В общем, при проведении реакции используется избыток органического соединения, содержащего по меньшей мере одну тиоловую группу, например, 10-20 эквивалентов по отношению к белку, аминокислоте или пептиду. Однако в некоторых случаях можно использовать только 1 молярный эквивалент. Углеводные соединения могут быть дорогостоящими и трудоемкими, что касается их получения в больших количествах. Поэтому, когда органическое соединение, содержащее по меньшей мере одну тиоловую группу, представляет собой углеводное соединение, из соображений экономии желательно использовать минимально возможное количество эквивалентов. Способы гликозилирования белков, известные из предшествующего уровня техники, часто требуют применения очень большого избытка углеводного соединения, например, часто порядка 1000 эквивалентов (В.G. Davis, Сип. Opin. Biotechnol. 2003, 14,379). Таким образом, способ согласно изобретению преимущественно обеспечивает возможность применения меньшего количества эквивалентов гликозильного соединения, чем способы известные из предшествующего уровня техники. Изобретение будет проиллюстрировано следующими не ограничивающими примерами. Описание примеров осуществления изобретения Точки плавления регистрировали на нагревающем блоке Кофлера (Kofler) и представляют собой значения без учета погрешности. Спектры протонного ядерного магнитного резонанса (H) 400 мГц определяли, используя COSY. Спектры углеродного ядерного магнитного резонанса (C) определяли, используя HMQC. Мультиплетности определяли, используя последовательность DEPT. Все химические сдвиги приведены на шкалев млн-1 с использованием остаточного растворителя в качестве внутреннего стандарта. Максимумы адсорбции в инфракрасных спектрах регистрировали в волновых числах (см-1) и классифицировали как s (интенсивный) и br (широкий). Масс-спектры низкого разрешения регистрировали,используя ионизацию электрораспылением (ИЭР) или используя методы химической ионизации (NH3,ХИ), как указано. Масс-спектры высокого разрешения (МСВР) регистрировали, используя методы химической ионизации (NH3, ХИ) или используя методы ионизации электрораспылением (NH3, ХИ), или используя ионизацию электрическим полем (ИЭП+), как указано. Значения m/z представлены в дальтонах и сопровождаются значением их процентного избытка в круглых скобках. Оптические вращения измеряли на поляриметре с длиной пробега 1 дм. Концентрации даны в г/100 мл. Тонкослойную хроматографию (ТСХ) осуществляли на предварительно покрытых Kieselgel 60F254 стеклянных пластинах от фирмы Merck. Визуализацию пластин осуществляли, используя УФ лампу (max = 254 или 365 нм), и/или молибдат аммония (5% в 2 М H2SO4) или серную кислоту (5% в EtOH). Колоночную флэшхроматографию осуществляли, используя силикагель Sorbsil C60 40/60. Дихлорметан (ДХМ) перегоняли после обезвоживания с использованием гидрида кальция. Ацетон перегоняли после обезвоживания с использованием безводного сульфата кальция. Остальные безводные растворители были получены от фирмы Fluka. 'Бензин' означает фракцию петролейного эфира, кипящую в диапазоне 40-60 С. Масс-спектрометрия белков: Жидкостную хроматографию/масс-спектрометрию осуществляли наnex Jupiter C5 (1502,1 мм 5 мкм). В качестве подвижной фазы использовали воду (растворитель А) и ацетонитрил (растворитель В), причем каждый содержал 0,5% муравьиной кислоты, при скорости потока 0,2 мл мин-1. Градиент программировали следующим образом: 95% растворителя А (3 мин изократический) до 100% растворителя В через 16 мин, затем изократический в течение 2 мин. Источник электрораспыления LCT работал с напряжением в капилляре 3 кВ и напряжением на острие 30 В. Азот использовали в качестве распылителя и десольватирующего газа при общей скорости 400 л ч-1. Миоглобин(сердце лошади) использовали в качестве калибровочного стандарта и для тестирования чувствительности системы. Пример 1. (2,3,4,6-Тетра-O-ацетилD-глюкопиранозил)-1-изотиоурония бромид 2,3,4,6-Тетра-O-ацетилD-глюкопиранозилбромид (11,0 г, 26,4 ммоль) и тиомочевину (3,10 г, 41,9 ммоль) растворяли в безводном ацетоне (30 мл) в атмосфере аргона и нагревали до 60 С. Через 20 мин осаждалось белое твердое вещество. Осадок отделяли фильтрацией, фильтрат снова нагревали до температуры дефлегмации, и этот процесс повторяли до тех пор, пока твердое вещество не прекращало осаждаться. Желтоватые кристаллы объединяли и перекристаллизовывали из смеси ацетон/бензин с получением целевого соединения (11,4 г, 76%) в виде белого твердого кристаллического вещества, т.пл. 194196 С [Лит. 191 С (Н. Beyer, U. Schuitz, Chem. Ber. 1954, 87, 78)]; []D25 -5,6 (с, 1,0 в H2O) [Лит. []D25 -7,6(4,93 г, 26,0 ммоль) добавляли при перемешивании к смеси ДХМ (150 мл) и воды (70 мл). Смесь нагревали до температуры дефлегмации в атмосфере аргона. Через 1,5 ч реакцию охлаждали до комнатной температуры (КТ) и разделяли фазы. Водный слой повторно экстрагировали ДХМ (350 мл). Объединенные органические слои промывали водой (50 мл), сушили над MgSO4, фильтровали, и растворитель удаляли в вакууме с получением целевого соединения (6,14 г, 90%) в виде белого твердого вещества, т.пл. 112-114 С [Лит. 113-114 С (R. J. Ferrier, R. Н. Furneaux, Carbohydr. Res. 1977, 57, 73)]; []D24 +6,3 (с, 1,2 в 2,3,4,6-Тетра-O-ацетил-Dгалактопиранозилбромид (5,4 г, 13,0 ммоль) и тиомочевину (1,25 г, 16,8 ммоль) растворяли в безводном ацетоне (40 мл) в атмосфере аргона и нагревали до 60 С. Через 1 ч реакцию оставляли охлаждаться до комнатной температуры, и полученный остаток фильтровали и перекристаллизовывали из смеси ацетон/бензин с получением целевого соединения (4,6 г, 70%, 2 стадии) в виде белого твердого кристаллического вещества, т.пл. 134-137 С [Лит. 170 С из изопропанола (W. A. Bonner,J. E. Kahn, J Am Chem Soc 1951, 73, 2241)]; []D25 +40,4 (c, 1,0 в H2O) [Лит. []D25 +16,0 (c, 1,6 в EtOH, (W.(2,02 г, 10,6 ммоль) добавляли при перемешивании к смеси ДХМ (60 мл) и воды (30 мл). Смесь нагревали до температуры дефлегмации в атмосфере аргона. Через 2,5 ч реакцию охлаждали до КТ и разделяли фазы. Водный слой повторно экстрагировали ДХМ (350 мл). Объединенные органические слои промывали водой (100 мл), рассолом (100 мл), сушили над MgSO4, фильтровали, и растворитель удаляли в вакууме с получением целевого соединения (2,65 г, 81%) в виде белого твердого вещества, т.пл. 83-84 С 3,4,6-Три-O-ацетил-2-ацетамидо-2-дезоксиD-глюкопиранозоилхлорид (3,0 г, 8,2 ммоль) и тиомочевину (1,21 г, 14,6 ммоль) растворяли в безводном ацетоне (25 мл) в атмосфере аргона и нагревали до 60 С. Через 2 ч осаждалось белое твердое вещество. Осадок отделяли фильтрацией, фильтрат снова нагревали до температуры дефлегмации, и этот процесс повторяли до тех пор, пока твердое вещество не прекращало осаждаться. Желтоватые кристаллы объединяли и перекристаллизовывали из смеси ацетон/бензин с получением целевого соединения (2,19 г, 61%) в виде белого твердого кристаллического вещества, т.пл. 134-137 С [Лит. 179-181 С из EtOH (D. Horton, M. L. Wolfrom, J. Org. Chem. 1962, 27,1794)]; []D25 -25,2 (с, 1,0 в H2O) [Лит. []D25 -29,3 (с, 1,1 в МеОН) (D. Horton, M. L. Wolfrom, J. Org.(3,4,6-Три-O-ацетил-2-ацетамидо-2-дезоксиD-глюкопиранозил)-1-изотиоурония хлорид (1,75 г,39,8 ммоль) и Na2S2O5 (0,91 г, 4,8 ммоль) добавляли к перемешиваемой смеси ДХМ (30 мл) и воды (15 мл). Смесь нагревали до температуры дефлегмации в атмосфере аргона. Через 2 ч реакцию охлаждали до КТ и разделяли фазы. Водный слой повторно экстрагировали ДХМ (250 мл). Объединенные органические слои промывали водой (50 мл), рассолом (50 мл), сушили над MgSO4, фильтровали, и растворитель удаляли в вакууме. После перекристаллизации из смеси ЕtOАс/бензин было получено целевое соединение (1,00 г, 68%) в виде белого твердого вещества, т.пл. 165-167 С [Лит. 167-168 С (W. M. zu Reckendorf,W. A. Bonner, J. Org. Chem. 1961, 26, 4596)]; []D25 -24,8 (c, 1,0 в CHCl3) [Лит. []D25 -14,5 (c, 0,9 в CHCl3)- 11011299 1-Тио-2,3,4,6-тетра-O-ацетилD-галактопиранозу (3,00 г, 7,3 ммоль) и NaOMe (40 мг, 0,73 ммоль) добавляли при перемешивании к раствору МеОН (40 мл). Через 2 ч ТСХ (ЕtOАс/бензин, 1:1) показала образование продукта (Rf 0,0) с полным расходованием исходного вещества (Rf 0,5). Реакционную смесь нейтрализовали добавлением ионообменной смолы Dowex-50, после чего реакционную смесь фильтровали и концентрировали в вакууме. После перекристаллизации из смеси МеОН/ЕtOАс было получено целевое соединение (1,41 г, 98%) в виде белого твердого кристаллического вещества, т.пл. 100-102 С; 3,4,6-Три-О-ацетил-2-ацетиламино-2-дезоксиD-глюкопиранозилтиол (400 мг, 0,98 ммоль) и метоксид натрия (18 мг, 0,03 ммоль) добавляли при перемешивании к раствору метанола (10 мл). Через 30 минут ТСХ (этилацетат) показала образование продукта (Rf 0,0) с полным расходованием исходного вещества (Rf 0,2). Реакционную смесь нейтрализовали добавлением ионообменной смолы Dowex-50, после чего реакционную смесь фильтровали и концентрировали в вакууме. После перекристаллизации из смеси метанол/этилацетат было получено целевой продукт (230 мг, 98%) в виде белого твердого кристаллического вещества; т.пл. 85-88 С [Лит. 86-88 С]18; []D22 -10,4 (с, 1,0 в МеОН) [Лит. []D25 +177,1 (с,1,45 в CHCl3)]18; H (400 мГц, МеОН), 2,00 (3H, s, СН 3), 3,27-3,37 (2 Н, m, Н-4, Н-5), 3,42 (1 Н, at, J 9,1 Гц,Н-3), 3,64-3,73 (2 Н, m, Н 2, Н-6), 3,87 (1 Н, dd, J5,6 2,1 Гц, J6,6, 12,0 Гц, Н-6'), 4,56 (1 Н, d, J1,2 10,0 Гц, Н-1),8,11 (1 Н, bd, JNH,2 9,1 Гц, NH). Пример 9. 1,2,3,6-Тетра-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилOглюкопиранозил)D-глюкопиранозил)-D-глюкопираноза Ацетат натрия (700 мг, 8,3 ммоль) добавляли к уксусному ангидриду (50 мл) и нагревали до температуры дефлегмации, при которой добавляли мальтотриозу (3,00 г, 6,0 ммоль) и энергично перемешивали. Через 90 мин ТСХ (бензин: этилацетат, 1:2) показала образование продукта (Rf 0,3) с полным расходованием исходного вещества (R) 0,0). Реакционную смесь оставляли охлаждаться до КТ и разбавляли ДХМ (50 мл) и распределяли с водой (100 мл). Фазы разделяли и водный слой повторно экстрагировали ДХМ (250 мл). Объединенные органические слои промывали гидрокарбонатом натрия (400 мл насыщенного водного раствора) до тех пор, пока не получали рН 8, рассолом (200 мл), сушили (MgSO4),фильтровали и концентрировали в вакууме с получением целевого продукта, представляющего собой смесь аномеров (/, 2/11) в виде белого аморфного твердого вещества; для -соединения: H (500 мГц,CHCl3) 2,05, 2,07, 2,10, 2,14, 2,15, 2,19, 2,21, 2,27 (30H, 8s, 10 ОАс), 3,92 (1 Н, , J 4,5 9,5 Гц, J5,6 2,9 Гц, J6,6 4,1 Гц, Н-5 а), 3,95-4,01 (3H, m, Н-4b, Н-5b, Н-5 с), 4,05 (1 Н, at, J 9,1 Гц, Н-4 а), 4,09 (1 Н, dd, J5,6 2,5 Гц, J6,6, 12,7 Гц, Н-6c), 4,21 (1 Н, dd, J5,6, 3,4 Гц, J6,6 12,6 Гц, H-6b), 4,29 (1 Н, dd, J5,6 3,4 Гц, J6,6, 12,4 Гц, Н 6'с), 4,35 (1 Н, dd, J5,6 4,3 Гц, J6,6. 12,3 Гц, Н-6a), 4,48-4,52 (2 Н, m, Н-6'а, H-6'b), 4,78 (1 Н, dd, J1,2 4,1 Гц, J2,3 10,3 Гц, Н-2b), 4,90 (1 Н, dd, J1,2 4,1 Гц, J2,3 10,6 Гц, Н-2 с), 5,01 (1 Н, dd, J1,2 8,0 Гц, J2,3 9,0 Гц, Н-2 а), 5,11(1 Н, at, J 10,1 Гц, Н-4 с), 5,31 (1 Н, d, J1,2 3,9 Гц, H-1b), 5,32-5,44 (3H, m, Н-3a, Н-3b, Н-3c), 5,45 (1 Н, d, J1,2 4,1 Гц, Н-1 с), 5,79 (1 Н, d, J1,2 8,2 Гц, Н-1 а); для -соединения выбрали только данные: H (500 мГц,CHCl3) 2,08, 2,09, 2,12, 2,18, 2,21, 2,23, 2,26 (30H, 8s, 10 ОАс), 5,07 (1 Н, at, J 9,9 Гц), 6,28 (1 Н, d, J1,2 3,8 Гц, Н-1 а). Оставшиеся сигналы лежат в следующих мультиплетных областях: 3,85-3,89, 3,90-3,98,3,99-4,07, 4,15-4,18, 4,23-4,27, 4,29-4,32, 4,43-4,49, 4,74-4,76, 4,84-4,87, 4,98-4,94, 5,25-5,54; m/z (ЭР+) 984 1,2,3,6-Тетра-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-D-глюкопиранозил)-D-глюкопиранозу (200 мг, 0,21 ммоль) растворяли в безводном ДХМ (5 мл). К этой смеси добавляли раствор бромоводорода (33% в уксусной кислоте, 2 мл). Смесь оставляли в атмосфере аргона при КТ. Через 30 мин ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,6) с полным расходованием исходного вещества (Rf 0,3). Реакционную смесь распределяли между ДХМ (10 мл) и водой (10 мл), и водный слой повторно экстрагировали ДХМ (310 мл). Объединенные органические слои промывали гидрокарбонатом натрия (20 мл насыщенного водного раствора) до тех пор, пока не получали рН 8, рассолом (20 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с получением целевого продукта (203 мг, 98%) в виде белой пены; []D22 +152,2 (с, 1,0 в CHCl3); H (400 мГц,CHCl3) 2,03, 2,05, 2,06, 2,08, 2,10, 2,13, 2,18, 2,21 (30H, 10 СОСН 3), 3,93-3,99 (3H, m, Н-4b, Н-5 а, Н-5b),4,05-4,10 (2 Н, m, Н-4 с, Н-6 а), 4,20 (1 Н, dd, J5,6 1-8 Гц, J6,6 12,2 Гц, Н-6b), 4,26-4,34 (2 Н, m, Н-5 с, Н-6 а'),4,35 (1 Н, dd, J5,6 3,5 Гц, J6,6. 12,7 Гц, Н-6c), 4,52 (1 Н, dd, J5,6 0,6 Гц, J6,6. 12,2 Гц, H-6b'), 4,57 (1 Н, dd, J5,6 2,1 Гц, J6,6 12,4 Гц, Н-6c), 4,74 (1 Н, dd, J1,2 4,1 Гц, J2,3 9,9 Гц, Н-2 с), 4,78 (1 Н, dd, J1,2 4,2 Гц, J2,3 10,2 Гц, H-2b),4,88 (1 Н, dd, J1,2 4,0 Гц, J2,3 10,5 Гц, Н-2 а), 5,10 (1 Н, at, J 9,7 Гц, Н-4 а), 5,32 (1 Н, d, J1,2 4,0 Гц, H-1b), 5,39(60 мл). К этой смеси добавляли безводную тиомочевину (315 мг, 4,2 ммоль) и затем нагревали до температуры дефлегмации в атмосфере аргона. Через 6,5 ч ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,0) с полным расходованием исходного вещества (Rf 0,3). Реакционную смесь концентрировали в вакууме и титровали ДХМ для удаления органических веществ из избытка тиомочевины. Фильтрат концентрировали в вакууме, и остаток очищали колоночной флэш-хроматографией (этилацетат/метанол, 9:1) с получением промежуточного соединения 2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил 4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил-1 изотиоурония бромида (1,14 г, 50%), которое использовали далее без идентификации. Это промежуточное соединение (100 мг, 0,09 ммоль) и Na2S2O5 (22 мг, 0,11 ммоль) добавляли при перемешивании к смеси ДХМ (30 мл) и воды (15 мл). Эту смесь нагревали до температуры дефлегмации в атмосфере аргона. Через 2,5 ч ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,4) с полным расходованием исходного вещества (Rf 0,0), после чего реакционную смесь охлаждали до КТ и разделяли фазы. Водный слой повторно экстрагировали ДХМ (220 мл). Объединенные органические слои промывали рассолом (20 мл), сушили (MgSO4), фильтровали, и растворитель удаляли в вакууме с получением целевого продукта (74 мг, 84%) в виде белого аморфного твердого вещества; []D22 +99,5 (с, 1,0 в CHCl3); H (400 МГц, CHCl3) 1,99, 2,00, 2,01, 2,02, 2,03, 2,05, 2,10, 2,15, 2,18 (30H, 9s,10COCH3), 3,72-3,76 (1 Н, m, Н 5 а), 3,90-4,00 (4 Н, m, Н-4 а, Н-4b, Н-5b, Н-5 с), 4,05 (1 Н, dd, J5,6 2,2 Гц, J6,6 12,3 Гц, Н-6c), 4,17 (1 Н, dd, J5,6 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-D-глюкопиранозил)D-глюкопиранозилбромид (11,2 г, 11,6 ммоль) и тиоацетат калия (3,96 г, 34,8 ммоль) суспендировали в безводном ТГФ (40 мл) и нагревали до температуры дефлегмации в атмосфере инертного аргона. Через 14 ч ТСХ (бензин/EtOAc, 1:2) показала образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,45). Реакционную смесь разбавляли водой (80 мл) и оставляли охлаждаться до КТ. Фазы разделяли, и водную фазу повторно экстрагировали ДХМ (340 мл). Объединенные органические слои промывали насыщенным раствором NaHCO3 (50 мл) до тех пор, пока не получали рН 8, рассолом (50 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин/EtOAc, 1:4) с получением целевого соединения (8,08 г, 71%) в виде белой пены; []D25 +86,4 (с, 1,0 в CHCl3); H (400 МГц, CHCl3) 2,01,2,02, 2,05, 2,08, 2,11, 2,17 (27 Н, 6s, 9 ОАс), 2,40 (3H, s, SAc), 3,88 (1 Н, , J4,5 9,8 Гц, J5,6 4,0 Гц, J5,6 2,7 Гц, Н-5 а), 3,92-4,01 (4 Н, m, Н-4 а, H-4b, H-5b, Н-5 с), 4,07 (1 Н, dd, J5,6 2,4 Гц, J6.6. 12,3 Гц, Н-6c), 4,19 (1 Н,dd, J5,6 3,5 Гц, J6,6 12,2 Гц, Н-6b), 4,27 (1 Н, dd, J5,6. 3,8 Гц, J6,6 12,3 Гц, Н-6'с), 4,30 (1 Н, dd, J5,6 4,2 Гц, J6,6. 12,4 Гц, Н-6 а), 4,46 (1 Н, dd, J5,6.2,6 Гц, J6,6 12,3 Гц, H-6'b), 4,47 (1 Н, dd, J5,6 2,2 Гц, J6,6. 12,2 Гц, Н-6'а), 4,76 1-Тиоацетил-2,3,6-три-О-ацетил-4-О-(2,3,6-три-О-ацетил-4-О-(2,3,4,6-тетра-О-ацетилOглюкопиранозил)O-глюкопиранозил)-1-тиоO-глюкопиранозу (600 мг, 0,6 ммоль) и NaOAc (18 мг,0,18 ммоль) добавляли при перемешивании к раствору МеОН (10 мл). Через 10 мин ТСХ (EtOAc/МеОН,9:1) показала образование продукта (Rf 0,0) с полным расходованием исходного вещества (Rf 0,9). Реакционную смесь нейтрализовали добавлением ионообменной смолы Dowex-50, после чего реакционную смесь фильтровали и концентрировали в вакууме с получением целевого соединения (305 мг, 98%) в виде аморфного твердого вещества; []D25 +123 (с, 1,0 в МеОН); H (400 мГц, D2O), 3,15 (1 Н, at, J 9,2 Гц, Н 2 а), 3,26 (1 Н, at, J 9,3 Гц), 3,41-3,82 (16 Н, m, H-2b, Н-2 с, Н-3a, Н-3b, Н-3c, Н-4 а, H-4b, H-4c, H-5a, H-5b, Н 5 с, Н-6 а, H-6b, H-6 с, Н-6'а, H-6'b, Н-6'с), 4,42 (1 Н, d, J1,2 9,6 Гц, Н-1 а), 5,23 (1 Н, d, J1,2 1,7 Гц, Н-1), 5,24 Ацетат натрия (420 мг, 5,2 ммоль) добавляли к уксусному ангидриду (30 мл) и нагревали до температуры дефлегмации, при которой добавляли мальтогептозу (1,00 г, 0,86 ммоль), и реакционную смесь энергично перемешивали. Через 90 мин ТСХ (бензин:этилацетат, 1:3) показала образование продукта (Rf 0,3) с полным расходованием исходного вещества (Rf 0,0). Реакционную смесь оставляли охлаждаться до КТ, разбавляли ДХМ (50 мл) и распределяли с водой (100 мл). Фазы разделяли, и водный слой повторно экстрагировали ДХМ (240 мл). Объединенные органические слои промывали гидрокарбонатом натрия(200 мл насыщенного водного раствора) до тех пор, пока не получали рН 8, рассолом (100 мл), сушили(MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэшхроматографией (бензин:этилацетат, 1:3) с получением целевого продукта, представляющего собой смесь аномеров, в виде белого аморфного твердого вещества (/, 15/85); H (500 мГц, CDCl3) 2,02, 2,03,2,04, 2,05, 2,06, 2,07, 2,08, 2,10, 2,13, 2,19, 2,22, 2,24 (66 Н, 12s, 22 ОАс), 3,89-4,14 (13 Н, m, H-4a, H-4b,H-4c, H-4d, H-4e, H-4f, Н-5 а, H-5b, H-5c, H-5d, H-5e, H-5f, H-5g), 4,25-4,34, 4,39 (1 Н, dd, J 4,0 Гц, J 12,3 Гц), 4,52-4,56 (13 Н, m, Н-6a, Н-6 а', Н-6b, Н-6b', Н-6c, Н-6 с', H-6d, H-6d', Н-6e, Н-6 е', H-6f, H-6f, H-6d, H6g'), 4,75-4,79 (H, m, H-2b, Н-2 с, H-2d, Н-2 е, Н-2 е, H-2f), 4,90 (1 Н, dd, J1,2 3,7 Гц, J2,310,5 Гц, H-2g), 5,00 1,2,3,6-Тетра-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)Dглюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)-D-глюкопиранозу (100 мг, 0,05 ммоль) растворяли в безводном ДХМ (5 мл). К этой смеси добавляли раствор бромоводорода (33% в уксусной кислоте, 0,5 мл). Смесь оставляли перемешиваться в атмосфере аргона при КТ. Через 40 мин ТСХ (бензин:этилацетат, 1:3) показала образование продукта (Rf 0,7) с полным расходованием исходного вещества (Rf 0,3). Реакционную смесь распределяли между ДХМ (10 мл) и водой (10 мл), и водный слой повторно экстрагировали ДХМ (310 мл). Объединенные органические слои промывали раствором гидрокарбонатом натрия (150 мл насыщенного водного раствора) до тех- 15011299 пор, пока не получали рН 7, рассолом (20 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с получением целевого продукта (98 мг, 96%) в виде белой пены; []D22 +162,0 (с, 1,0 в CHCl3); H 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)Dглюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозилбромид (1,08 г, 0,5 ммоль) и иодид тетрабутиламмония (19 мг, 0,05 ммоль) растворяли в безводном ацетоне (50 мл). К этой смеси добавляли сухую тиомочевину (52 мг, 0,7 ммоль),и реакционную смесь затем нагревали до температуры дефлегмации в атмосфере аргона. Через 8 ч ТСХ(бензин:этилацетат, 1:4) показала образование минорного продукта (промежуточного соединения) (Rf 0,0) с полным расходованием исходного вещества (Rf 0,6). Реакцию концентрировали в вакууме и титровали ДХМ для удаления органических веществ из избытка тиомочевины. Фильтрат концентрировали в вакууме, и остаток очищали колоночной флэш-хроматографией (этилацетат/метанол, 9:1) с получением промежуточного соединения 2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилOглюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)O-глюкопиранозил)O-глюкопиранозил)O-глюкопиранозил-1-изотиоурония бромида (212 мг, 19%), который использовали далее без идентификации. Это промежуточное соединение (210 мг, 0,09 ммоль) и Na2S2O5(22 мг, 0,11 ммоль) добавляли при перемешивании к смеси ДХМ (10 мл) и воды (5 мл). Эту смесь нагревали до температуры дефлегмации в атмосфере аргона. Через 4,5 ч ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,2) с полным расходованием исходного вещества (Rf 0,0), после чего реакционную смесь охлаждали до KT и разделяли фазы. Водный слой повторно экстрагировали ДХМ (210 мл). Объединенные органические слои промывали рассолом (20 мл), сушили (MgSO4), фильтровали, и растворитель удаляли в вакууме с получением целевого продукта (185 мг, 90%) в виде белого аморфного твердого вещества; []D24 +128,1 (с, 1,0 в CHCl3); H (500 МГц, CHCl3), 2,00, 2,01, 2,02, 2,03, 2,04, 2,05,2,07, 2,08, 2,12, 2,17, 2,19, 2,21, 2,22, 2,23 (66 Н, 14s, 22 СОСН 3), 2,27 (1 Н, d, J1,SH 9,8 Гц, SH), 3,76 (1 Н,dat, J4,5 9,7 Гц, J 3,5 Гц, Н-5 а), 3,92-4,08 (12 Н, m, H-4a, H-4b, H-4c, H-4d, H-4e, H-4f, H-5b, H-5c, H-5d, H5 е, H-5f, H-5g), 4,17-4,36, 4,49-4,56 (12 Н, m, H-6b, H-6b', Н-6c, Н-6 с', H-6d, H-6d', H-6e, Н-6 е', H-6f, H-6f,H-6g, H-6g'), 4,39 (1H, dd, J5,6 3,6 Гц, J6,6. 12,2 Гц, Н-6 а), 4,48 (1H, dd, J5,6 3,2 Гц, J6,6.12,3 Гц, H-6a), 4,62(12H, m, -1b, H-1c, H-1d, H-1e, H-1f, H-1g, H-3b, H-3c, H-3d, H-3e, H-3f, H-3g). Пример 17. Получение SBLCys156-S-SePh Модификацию по одному сайту исследовали, используя модель - цистеин-содержащий белок, представляющий собой сериновую протеазу субтилизин (SBL.Cys156) из мутантного штамма Bacillus lentusS156C. SBLCys156 (10 мг) растворяли в дегазированном водном буферном растворе (1 мл, 70 мМ CHES,5 мМ MES, 2 мМ CaCl2, рН 9,5). PhSeBr (5 мг, 0,02 ммоль) растворяли в ацетонитриле (200 мкл), 150 мкл- 16011299 которого (40 экв.) добавляли к раствору белка и помещали на ротатор для вертикального перемешивания(end-over-end rotator). Через 30 мин с помощью анализа Элмана было показано отсутствие свободного тиола (G. L. Ellman, K. D. Courtney, V. Andres, R. M. Featherstone, Biochem. Pharmacol. 1961, 7, 88). Реакционную смесь помещали на ротатор для вертикального перемешивания еще на 30 мин, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO предел отсечения мембраны по молекулярному весу от англ. molecular weight cut off 12-14 кДа) против воды (14 л в течение 1 ч, 22 л в течение 30 мин) с получением SBLS156C-S-SePh; m/z (ЭР+) найдено 26864, вычислено 26870. Пример 18. Получение SSGCys344Cys432-(S-SePh)2 Модификации по многим сайтам исследовали, используя мутант термофильной -гликозидазы из штамма Sulfolobus solfataricus, содержащий два цистеиновых остатка (SSG-Cys344Cys432). SSGCys344Cys432 (1 мг) растворяли в водном буферном растворе (1 мл, 70 мМ CHES, 5 мМ MES, 2 мМCaCl2, рН 9,5). PhSeBr (2 мг, 0,02 ммоль) растворяли в ацетонитриле (200 мкл), 20 мкл которого (74 экв.) добавляли к раствору белка и помещали на ротатор для вертикального перемешивания. Через 1 ч реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали (70 мМ HEPES, 2 мМ CaCl2, рН 7,0) с получением SSGCys344Cys432-(S-SePh)2; m/z (ЭР+) найдено 57700, вычислено 57697. Пример 19. Типичное гликозилирование белков сахарными тиолами и реакция с другими тиоламиSBLCys156-S-SePh (1 мг) растворяли в водном буферном растворе (1 мл, 70 мМ CHES, 5 мМ MES,2 мМ CaCl2, рН 9,5). Сахарный тиол или другой тиол растворяли в воде и добавляли к раствору белка в указанных количествах (см. данные эквивалентов тиола, приведенные в таблице ниже), и эту смесь помещали на ротатор для вертикального перемешивания. Через 1 ч реакцию анализировали с помощью масс-спектрометрии. Таблица Конв. = превращение, определяемое с помощью ИЭР-МС 1 Активированный реакцией с фенилселенбромидом с получением соответствующего соединения белок-S-Se-Ph или белок-(S-Se-Ph)2 перед добавлением тиола. 2 Прореагировавший с PMSF (фенилметилсульфонилфторидом) перед гликозилированием для предотвращения распада белка вследствие протеолитической активности. Результаты, приведенные в вышеуказанной таблице, демонстрируют, что способ согласно настоящему изобретению обеспечивает высокопроцентную конверсию с получением желаемых продуктов с- 17011299 использованием только одного эквивалента тиолового соединения. Более того, эти результаты демонстрируют, что способ согласно изобретению может быть использован для гликозилирования белков по одному и по многим сайтам. Три сайта гликозилирования в SBL-Cys156 и SSGCys344Cys432 обнаружены в сильно отличающихся белковых структурах и в условиях с различными уровнями воздействия, что иллюстрирует широкую применимость способа согласно изобретению. Пример 20. Типичное гликозилирование белкаGlcGlcGlcGlcGlcGlcGlc-SH 1-Тио-2,3,6-три-О-ацетил-4-О-(2,3,6-три-О-ацетил-4-О-(2,3,6-три-О-ацетил-4-О-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-O-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозил)D-глюкопиранозу (15 мг, 0,007 ммоль) и метоксид натрия (2 мг, 0,007 ммоль) добавляли при перемешивании к раствору МеОН (2 мл). Через 2 ч ТСХ (бензин:EtOAc, 1:2) показала образование продукта (Rf 0,0) с полным расходованием исходного вещества (Rf 0,2). Реакционную смесь нейтрализовали добавлением ионообменной смолы Dowex-50, после чего реакционную смесь фильтровали и концентрировали в вакууме. Неочищенную 1-тиоD-мальтогептаозу выливали в воду (5 мл), 300 мкл которой (11 экв.) добавляли к раствору SBLCys156-S-SePh (1 мг) в 500 мкл водного буфера (70 мМ CHES, 5 мМ MES, 2 мМ CaCl2, рН 9,5). Полученный раствор помещали на ротатор для вертикального перемешивания. Через 1 ч реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМHEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали с получением GlcGlcGLcGlcGlcGlcGlcSBLCys156; m/z (ЭР+) найдено 27878, вычислено 27881. Пример 21. Ферментативные удлинения SBLCys156-S-GlcNAcA. GlcNAc-SBLCys156 (3 мг) растворяли в 1 мл водного буфера. Добавляли фенилметилсульфонилфторид (PMSF) (50 мкл из 100 мг/мл раствора в ацетонитриле; 500-кратный избыток). Реакционную смесь инкубировали при комнатной температуре в течение 30 мин и очищали на обессоливающей колонке Sephadex G-25 (PD-10). Чистоту дезактивированного белка определяли с помощью ИЭР-масс-спектрометрии (найдено: 27100, вычислено: 27104). Белковую фракцию лиофилизировали и повторно растворяли в 1,0 мл 0,1 М натрий-какодилатного буфера (рН 7,52). Добавляли MnCl24H2O (3,2 мг, 16 мкмоль) и уридиндифосфатгалактозу (UDP-галактоза, 2,3 мг, 3,4 мкмоль, Kyowa Hakko; 30-кратный избыток). Добавляли рекомбинантную бычью (-1,4-галактозилтрансферазу из Spodoptera frugiperda (EC 2.4.1.22, 100 мЕд, Calbiochem), и реакционную смесь инкубировали при комнатной температуре в течение 40 мин с получением Gal1,4GlcNAc-SBL-Cys156 (ИЭР-МС, найдено 27265, вычислено 27266). В. Добавляли GDP-фукозу (3 мг, Kyowa Hakku) и человеческую -1,3-фукозилтрансферазу из Spodoptera frugiperda (EC 2.4.1.65, 100 мЕд, Calbiochem), и реакционную смесь инкубировали в течение ночи при комнатной температуре с получением Льюисx-S-SBL-Cys156 (ИЭР-МС, найдено 27410, вычислено 27412). Этот пример демонстрирует, что гликозилированные белки, полученные согласно способу изобретения, могут быть дополнительно модифицированы реакцией с подходящими углеводмодифицирующими ферментами,например гликозилтрансферазами,такими как-1,4 галактозилтрансфераза, которая селективно образует связь Gal1,4GlcNAc. Пример 22. Фенилтиосульфонат натрия (NaPTS) Бензолсульфинат натрия (10 г, 61 ммоль) и серу (1,95 г, 61 ммоль) растворяли в безводном пиридине (60 мл) с получением желтого раствора. Реакционную смесь перемешивали в атмосфере аргона и через 1 ч получали белую суспензию. Реакционную смесь фильтровали и осадок промывали безводным диэтиловым эфиром. После перекристаллизации из безводного этанола был получен целевой продукт(10,5 г, 88%) в виде белого твердого кристаллического вещества; т.пл. 305-306 С [Лит. 287 С, Sato, R.; 2,3,4,6-Тетра-O-ацетилD-глюкопиранозилбромид (207 мг, 0,5 ммоль) растворяли в безводном ацетонитриле (5 мл). К этой смеси добавляли фенилтиосульфонат натрия (201 мг, 1 ммоль) и бромид тетрабутиламмония (16 мг, 0,05 ммоль). Полученную смесь перемешивали в атмосфере аргона при 70 С.(Rf 0,5) с полным расходованием исходного вещества (Rf 0,3). Раствор концентрировали в вакууме. Неочищенное твердое вещество распределяли между дихлорметаном (ДХМ, 20 мл) и водой (20 мл), и водный слой повторно экстрагировали ДХМ (220 мл). Объединенные органические экстракты промывали рассолом (20 мл), сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 1:1) с получением целевого продукта (225 мг, 88%) в виде белого твердого кристаллического вещества; т.пл. 129-130 С; []D25 +51,2 (с, 1,0 в CHCl3); max(KBr) 1754 (s, C=O), 1376 (s, С=С) см-1; H (400 МГц, C6D6) 1,68, 1,72, 1,73, 1,75 (43H, 4s, 4 х ОАс),3,09 (1 Н, ddd, J4,5 10,2 Гц, J5,6 2,4 Гц, J5,6' 4,2 Гц, Н-5), 3,83 (1 Н, dd, J5,6 2,4 Гц, J6,6. 12,7 Гц, Н-6), 4,08 (1 Н,dd, J5,6. 4,2 Гц, J6,6. 12,6 Гц, Н-6'), 5,17-5,23 (2 Н, m, Н-2, Н-4), 5,40 (1 Н, d, J 1,2 10,2 Гц, Н-1), 5,44 (1 Н, at, J 9,4 Гц, Н-3), 6,98-7,03 (3H, m, Ar-Н), 7,90-7,92 (2 Н, m, Ar-Н). Структуру продукта дополнительно подтверждали дифракцией рентгеновских лучей на монокристаллах. Пример 24. 2,3,4,6-Тетра-O-ацетилD-галактопиранозил-фенилтиосульфонат 2,3,4,6-Тетра-O-ацетилD-галактопиранозилбромид (2,0 г, 5 ммоль) растворяли в безводном ацетонитриле (80 мл). К этой смеси добавляли фенилтиосульфонат натрия (2,02 г, 10,3 ммоль) и бромид тетрабутиламмония (160 мг, 0,5 ммоль). Полученную смесь перемешивали в атмосфере аргона при 70 С. Через 5 ч ТСХ (бензин:этилацетат, 1:1) показала образование продукта (Rf 0,4) с полным расходованием исходного вещества (Rf 0,6). Раствор концентрировали в вакууме. Неочищенное масло распределяли между ДХМ (50 мл) и водой (50 мл), и водный слой повторно экстрагировали ДХМ (250 мл). Объединенные органические экстракты промывали рассолом (100 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 2:1) с получением целевого продукта (1,7 г, 65%, 2 стадии) в виде белого твердого кристаллического вещества; т.пл. 53-54C; []D27 +24,2 (с, 1,0 в CHCl3); H (400 мГц, CHCl3) 1,98, 2,03, 2,06, 2,11 (43H, 4s, 4 ОАс), 3,85 (1 Н, dd, J5,6 8,8 Гц, J6,6. 14,0 Гц, Н-6), 3,95-4,00 (2 Н, m, Н-5, Н-6), 5,11 (1 Н, dd, J2,3 9,7 Гц, J3,4 3,3 Гц, Н-3), 5,23 (1 Н, at, J 10,3 Гц, Н-2), 5,25 (1 Н, d, J1,2 10,2 Гц, Н-1), 5,43 (1 Н, dd, J3,4 3,6 Гц, J4,5 1,0 Гц, Н-4),7,54-7,68 (3H, m, Ar-Н), 7,93-7,97 (2 Н, m, Ar-Н). Пример 25. Этил-2,3,4,6-тетра-O-ацетил-1-дитиоD-глюкопиранозил-дисульфид Способ 1: 2,3,4,6-Тетра-O-ацетилD-глюкопиранозил-фенилтиосульфонат (100 мг, 0,2 ммоль) и триэтиламин (0,03 мл, 0,2 ммоль) растворяли в безводном ДХМ (10 мл) и перемешивали при комнатной температуре (КТ) в атмосфере аргона. Медленно по каплям добавляли раствор этантиола (0,016 мл, 0,2 ммоль) в безводном ДХМ (10 мл) через шприцевой насос в течение 30 мин. Через 40 мин ТСХ (бензин:этилацетат,1:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,3). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией(бензин:этилацетат, 1:1) с получением целевого продукта (70 мг, 82%) в виде белого твердого кристаллического вещества; т.пл. 95-96 С [Лит. 100-102 С, (Davis, В. G.; Ward, S. J.; Rendle, P. M. Chem. Commun. 2001, 189)]; []D22 -164,9 (c, 0,2 в CHCl3) [Лит. []D24 -178,0 (c, 1,0 в МеОН) (Davis, B. G.; Ward, S. J.;Rendle, P. M. Chem. Commun. 2001, 189)]; H (400 мГц, CHCl3) 1,30 (1H, t, J 7,4 Гц, CH3), 2,00, 2,02, 2,03,2,06 (43H, 4s, 4CH3), 2,79 (2H, dq, JCH3-H 7,5 Гц, JHH 2,7 Гц), 3,73 (1H, , J4,510,2 Гц, J5,6 2,5 Гц, J5,64,8 Гц, H-5), 4,14 (1H, dd, J5,6 2,4 Гц, J6,6. 12,4 Гц, Н-6), 4,22 (1 Н, dd, J5,6. 4,7 Гц, J6,6. 12,4 Гц, Н-6'), 4,52 (1 Н,d, J1,2 9,8 Гц, Н-1), 5,10 (1H, at, J 9,8 Гц, Н-4), 5,21-5,26 (2 Н, m, Н-2, Н-3). Способ 2: Фенил-2,3,4,6-тетра-O-ацетил-1-селененилсульфид-Dглюкопиранозид (75 мг, 0,15 ммоль) и триэтиламин (30 мкл, 0,15 ммоль) растворяли в свежеперегнанном ДХМ (10 мл). Этот раствор перемешивали при КТ в атмосфере аргона. Раствор этантиола (11 мкл, 0,15 ммоль) в безводном ДХМ (10 мл) добавляли по каплям в течение 2,5 ч. Через 3 ч ТСХ (бензин:EtOAc, 1:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,5). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:EtOAc, 5:3) с получением целевого продукта (50 мг, 82%) в виде белого твердого кристаллического вещества. Способ 1: 2,3,4,6-Тетра-O-ацетилD-галактопиранозил-фенилтиосульфонат (100 мг, 0,2 ммоль) и триэтиламин (0,03 мл, 0,2 ммоль) растворяли в безводном ДХМ (10 мл) и перемешивали при КТ в атмосфере аргона. В течение 30 мин медленно по каплям добавляли раствор этантиола (0,016 мл, 0,2 ммоль) в безводном ДХМ (10 мл) через шприцевой насос. Через 40 мин ТСХ (бензин:этилацетат, 1:1) показала образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,3). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат,1:1) с получением целевого продукта (78 мг, 91%) в виде белого твердого кристаллического вещества; т.пл. 65-66 С; []D25 -52,1 (с, 1,4 в CHCl3); max (KBr) 1746 (s, С=O) см-1; H (400 мГц, CHCl3) 1,30 (1 Н, t, J 7.4 Гц, СН 3), 1,95, 2,01, 2,02, 2,13 (43H, 4s, 4 СН 3), 2,79 (2 Н, dq, JCH3-H 7,2 Гц, JHH 1,7 Гц), 3,94 (1 Н,td, J4,5 0,9 Гц, J5,6 6,3 Гц, J5,6' 7,0 Гц, Н-5), 4,06 (1 Н, dd, J5,6 6,3 Гц, J6,6 11,3 Гц, Н-6), 4,12 (1 Н, dd, J5,6. 7,0 Гц,J6,6' 11,2 Гц, Н-6'), 4,51 (1 Н, d, J1,2 9,9 Гц, Н-1), 5,05 (1 Н, dd, J2,3 9,9 Гц, J3,4 3,6 Гц, Н-3), 5,35-5,40 (2 Н, m, Н 2, Н-4). Способ 2. Фенил-2,3,4,6-тетра-O-ацетил-1-селененилсульфид-Dгалактопиранозид(75 мг, 0,15 ммоль) и триэтиламин (30 мкл, 0,15 ммоль) растворяли в свежеперегнанном ДХМ (10 мл). Этот раствор перемешивали при КТ в атмосфере аргона. Раствор этантиола (11 мкл, 0,15 ммоль) в безводном ДХМ (10 мл) добавляли по каплям в течение 2,5 ч. Через 3 ч ТСХ (бензин:EtOAc, 1:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,5). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:EtOAc, 5:3) с получением целевого соединения (50 мг, 82%) в виде белого твердого кристаллического вещества. Пример 27. Этил-3,4,6-три-O-ацетил-2-ацетамидо-2-дезоксиD-глюкопиранозилдисульфид Фенил-3,4,6-три-O-ацетил-2-ацетамидо-2-дезокси-1-селененилсульфид-Dглюкопиранозид (100 мг, 0,19 ммоль) и триэтиламин (0,03 мл, 0,19 ммоль) растворяли в свежеперегнанном ДХМ (20 мл). Раствор перемешивали при КТ в атмосфере аргона. В течение 1 ч по каплям добавляли раствор этантиола(0,014 мл, 0,19 ммоль) в безводном ДХМ (10 мл). Через 3 ч ТСХ (EtOAc) показала образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,5). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (EtOAc) с получением целевого продукта (75 мг, 93%) в виде белого аморфного твердого вещества. []D25 -70,1 (с, 2,5 в CHCl3); H (400 мГц, CHCl3), 1,32 (3H, d, JCH,CH3 6,6 Гц, CHCH3), 1,96, 2,04, 2,05, 2,08 (12 Н, 4s, 4 СОСН 3), 2,82 (2 Н, q,J 7,4 Гц, СН 2), 3,75 (1 Н, ddd, J4,5 10,1 Гц, J5,6 2,5 Гц, J5,6' 4,7 Гц, Н-5), 4,12-4,25 (3H, m, Н-2, Н-6, Н-6'), 4,73 Метиловый эфир бис-L-цистеинил-L-серина (100 мг, 0,23 ммоль) растворяли в метаноле (5 мл). К этой смеси добавляли раствор уксусного ангидрида (0,09 мл, 0,92 ммоль) и пиридина (0,075 мл, 0,92 ммоль). Через 15 мин ТСХ (этилацетат:метанол, 5:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,1). Реакционную смесь концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат:метанол, 5:1) с получением целевого продукта (60 мг, 50%) в виде белого твердого кристаллического вещества; т.пл. 145-147 С; Метиловый эфир бис-N-ацетил-L-цистеинил-L-серина (1,92 г, 3,96 ммоль) растворяли в необезвоженном хлороформе (100 мл) и метаноле (10 мл) и перемешивали. К этой смеси при перемешивании добавляли раствор трибутилфосфина (1,1 мл, 4,36 ммоль). Через 2 ч ТСХ (этилацетат:метанол, 10:1) показала образование продукта (Rf 0,6) наряду с полным расходованием исходного вещества (Rf 0,3). Реакционную смесь концентрировали в вакууме. После перекристаллизации из смеси этилацетат/метанол был получен целевой продукт (1,77 г, 93%) в виде белого твердого кристаллического вещества; т.пл. 127128 С; []D25 -32,0 (с, 1,0 в МеОН); H (400 мГц, CHCl3) 1,89 (1 Н, at, J 8,9 Гц, SH), 2,06 (3H, s, COCH3),2,84-2,93 (1 Н, m, CysCHH), 2,97-3,04 (1 Н, m, CysCHH), 3,79 (3H, s, ОМе), 3,91 (1 Н, dd, JCH,H 11,4 Гц, JCH,H 3,1 Гц, SerCHH), 4,03 (1 Н, dd, JCH,H 11,7 Гц, JCH,H 4,2 Гц, SerCHH), 4,61-4,65 (1 Н, m, HSer), 4,71-4,76 2,3,4,6-Тетра-O-ацетилD-глюкопиранозил-фенилтиосульфонат (61 мг, 0,12 ммоль) растворяли в безводном ДХМ (5 мл) и перемешивали при КТ в атмосфере аргона. К этой смеси медленно по каплям в течение 4 ч добавляли метиловый эфир N-ацетил-L-цистеин-L-серина (32 мг, 0,12 ммоль) и триэтиламин(0,015 мл, 0,11 ммоль) в безводном ДХМ (10 мл) и безводном метаноле (0,5 мл) через шприцевой насос. Через 5 ч ТСХ (этилацетат:метанол, 10:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,3, (система элюентов для ТСХ - бензин:этилацетат,1:1. Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат:метанол, 10:1) с получением целевого продукта (75 мг, 99%) в виде белого твердого кристаллического вещества; т.пл. 126-128 С [Лит. 125-128 С (Davis, В. G; Ward, S. J.; Rendle, P. M. Chem. Commun. 2001, 189)]; []D25 -47,9 (c, 0,7 в CHCl3) [Лит. []D24 -178,0 (c, 1,0 в MeOH) (Davis, B. G.; Ward, S. J.; 2,3,4,6-Тетра-O-ацетилD-галактопиранозил-фенилтиосульфонат (50 мг, 0,1 ммоль) растворяли в безводном ДХМ (5 мл) и перемешивали при КТ в атмосфере аргона. В течение 2 ч медленно по каплям добавляли раствор метилового эфира N-ацетил-L-цистеин-L-серина (31 мг, 0,12 ммоль) и триэтиламина(0,015 мл, 0,11 ммоль) в безводном ДХМ (10 мл) и безводном метаноле (0,5 мл) через шприцевой насос. Через 2 ч ТСХ (этилацетат:метанол, 10:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,5, система элюентов для ТСХ - бензин:этилацетат, 1:1). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат:метанол, 10:1) с получением целевого продукта (59 мг, 95%) в виде белого аморфного твердого вещества; []D25 -48,8 (с, 0,25 в CHCl3); H (400 мГц, CHCl3) 1,99, 2,04, 2,05, 2,08, 2,18 (53H, 4s, 5 СН 3),2,80 (1 Н, bs, ОН), 2,99 (1 Н, dd, JCH,H 14,1 Гц, JCH,H 9,2 Гц, CysCHH), 3,32, 3,77 (3H, s, ОМе), 3,92 (1 Н, dd,JCH,H 11,7 Гц, JCH,H 3,0 Гц, SerCHH), 4,01 (1 Н, dd, JCH,H 11,7 Гц, JCH,H 3,7 Гц, SerCHH), 4,06-4,14 (2 Н, m, Н 5, Н-6), 4,20-4,26 (1 Н, m, Н-6'), 4,61-4,63 (1 Н, m, HSer), 4,65 (1 Н, d, J1,2 9,8 Гц, Н-1), 4,88-4,93 (1 Н, m,HCys), 5,11 (1 Н, dd, J2,3 9,8 Гц, J3,4 3,3 Гц, Н-3), 5,42-5,47 (2 Н, m, Н-2, Н-4), 6,68 (1 Н, d, JNH,H 7,8 Гц,NHAc), 7,28 (1 Н, d, 8,1 Гц, NHSer). Пример 32. 2,3,4,6-Тетра-O-бензилD-глюкопиранозилбромид 2,3,4,6-Тетра-O-бензил-D-глюкопиранозу (1,0 г, 1,9 ммоль) растворяли в безводном ДХМ (6 мл) и безводном ДМФА (0,4 мл) в атмосфере аргона. Полученный раствор перемешивали при 0 С. В течение 5 мин по каплям добавляли оксалилбромид (4 мл, 2M в ДХМ, 24 ммоль). Реакционную смесь перемешивали при КТ. Через 40 мин ТСХ (бензин:этилацетат, 2:1) показала образование основного продукта (Rf 0,7). Реакционную смесь охлаждали до 0 С и гасили ледяной водой (30 мл), добавляемой в течение 5 мин. Реакцию распределяли между ДХМ (20 мл) и водой. Водный слой повторно экстрагировали ДХМ (320 мл), объединенные органические слои промывали рассолом (40 мл), сушили (MgSO4), фильтровали и концентрировали в вакууме с получением целевого продукта (1,10 г, 95%) в виде неочищенного желтого масла; H (400 мГц, CHCl3), 3,57 (1 Н, dd, J1,2 3,5 Гц, J2,3 9,1 Гц, Н-2), 3,68 (1 Н, dd, J5,6 2,1 Гц, J5,6' 11,0 Гц,Н-6), 3,79-3,84 (2 Н, m, Н-4, Н-6'), 4,07 (1 Н, at, J 9,1 Гц, Н-3), 4,07-4,11 (1 Н, m, Н-5), 4,47-4,62 (3H, m,PhCH2), 4,74 (s, 2 Н, PhCH2), 4,84-4,89 (2 Н, m, PhCH2), 5,10 (1 Н, d, J 11,1 Гц, PhCH2), 6,46(1 Н, d, Н-1),7,15-7,41 (20 Н, m, Ar-Н). Пример 33. 2,3,4,6-Тетра-O-бензилD-глюкопиранозил-фенилтиосульфонате 2,3,4,6-Тетра-O-бензил-Dглюкопиранозоилбромид (3,55 г, 5,88 ммоль) и фенилтиосульфонат натрия (4,76 г, 24,3 ммоль) растворяли в безводном 1,4-диоксане (90 мл). Реакционную смесь нагревали до 70 С в атмосфере аргона. Через 20 ч ТСХ (бензин:этилацетат, 2:1) показала образование основного продукта (Rf 0,6) с полным расходованием исходного вещества (Rf 0,7). Реакционную смесь охлаждали до КТ и фильтровали, осадок промывали смесью бензин/этилацетат, и фильтрат концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 4:1) с получением 2,3,4,6 тетра-O-бензил-O-глюкопиранозил-фенилтиосульфоната (3,18 г, 78%) в виде белой вязкой смолы, представляющей собой смесь ,-соединений в соотношении : 3:1. После избирательной перекристаллизации из смеси этилацетат/бензин был получен чистый 2,3,4,6-тетра-О-бензилD-глюкопиранозилфенилтиосульфонат в виде белого твердого кристаллического вещества; т.пл. 106-108 С; []D22 +21,4 (с,0,35 в CHCl3); H (500 мГц, C6D6) 3,21 (1 Н, ddd, J4,5 9,7 Гц, J5,6 1,4 Гц, J5,6' 3,8 Гц, Н-5), 3,29 (1 Н, dd, J5,6 1-4 Гц, J6,6' 11,1 Гц, Н-6), 3,34 (1 Н, dd, J1,2 9,9 Гц, J2,3 8,7 Гц, Н-2), 3,49 (1 Н, dd, J5,6 3,8 Гц, J6,6. 11,1 Гц, Н-6'),3,51 (1 Н, at, J 9,4 Гц, Н-3), 3,60 (1 Н, at, J 9,4 Гц, Н-4), 4,15, 4,25 (2 Н, ABq, J 12,1 Гц, PhCH2), 4,52, 4,58 2,3,4,6-Тетра-O-ацетилD-глюкопиранозил-фенилтиосульфонат (100 мг, 0,14 ммоль) и триэтиламин (0,02 мл, 0,14 ммоль) растворяли в безводном ДХМ (10 мл) и перемешивали при КТ в атмосфере аргона. К этой смеси в течение 90 мин медленно по каплям добавляли этантиол (11 мкл, 0,14 ммоль) в безводном ДХМ (10 мл) через шприцевой насос. Через 90 мин ТСХ (бензин:этилацетат, 6:1) показала- 22011299 образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,2). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 7:1) с получением целевого продукта (83 мг, 95%) в виде прозрачного масла; []D22 -164,9 2,3,4,6-Тетра-O-бензилD-глюкопиранозил-фенилтиосульфонат (50 мг, 0,07 ммоль) растворяли в безводном ДХМ (5 мл) и перемешивали при КТ в атмосфере аргона. К этой смеси в течение 5 ч медленно по каплям добавляли метиловый эфир N-ацетил-L-цистеин-L-серина (19 мг, 0,07 ммоль) и триэтиламин(11 мкл, 0,08 ммоль) в безводном ДХМ (5 мл) и безводном метаноле (0,5 мл) через шприцевой насос. Через 5 ч ТСХ (этилацетат) показала образование основного продукта (Rf 0,6) наряду с полным расходованием исходного вещества (Rf 0,9). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат) с получением целевого продукта (48 мг, 82%) в виде белого твердого кристаллического вещества; т.пл. 96-97 С; []D22 +56,2 (с, 1 в CHCl3); H (400 мГц, CHCl3) 2,03 (3H, s,COCH3), 3,19 (1 Н, dd, JCH,H 14,0 Гц, JCH,H 8,3 Гц, CysCHH), 3,37 (1 Н, dd, JCH,H 14,3 Гц, JCH,H 6,0 Гц,CysCHH), 3,64 (1 Н, ddd, J4,5 9,6 Гц, J5,6 1,8 Гц, J5,6' 3,9 Гц, H-5), 3,72 (1 Н, at, J 9,2 Гц, Н-4), 3,77 (1 Н, at, J 8,8 Гц, Н-3), 3,82 (3H, s, ОМе), 3,84-3,90 (4 Н, m, SerCHH, Н-2, Н-6, Н-6'), 3,96 (1 Н, dd, JCH,H 11,7 Гц, JCH,H 3,3 Гц, SerCHH), 4,50 (1 Н, d, J1,2 9,6 Гц, Н-1), 4,51, 4,70 (2 Н, ABq, J 1,6 Гц, PhCH2), 4,55, 4,85 (2 Н, ABq, J 10,4 Гц, PhCH2), 4,59-4,62 (1 Н, m, HSer), 4,81, 4,87 (2 Н, ABq, J 10,6 Гц, PhCH2), 4,91, 4,97 (2 Н, ABq, J 11,0 Гц, PhCH2), 4,93-4,98 (1 Н, m, HCys), 6,88 (1 Н, bd, JNH,H 7,9 Гц, NHAc), 7,13-7,39 (20 Н, m, 20Ar-С), 7,48 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-D-глюкопиранозил)D-глюкопиранозилбромид (200 мг, 0,21 ммоль) растворяли в безводном ацетонитриле (10 мл). К этой смеси добавляли бензолтиосульфонат натрия (80 мг, 0,41 ммоль) и иодид тетрабутиламмония (10 мг, 0,02 ммоль). Полученную смесь перемешивали в атмосфере аргона при 70 С. Через 2 ч ТСХ (бензин:этилацетат, 1:2) показала образование УФ-активного продукта (Rf 0,5) с полным расходованием исходного вещества (Rf 0,5), после чего раствор оставляли охлаждаться до КТ и фильтровали,фильтрат концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 1:2) с получением целевого продукта (140 мг, 62%) в виде белого аморфного твердого вещества; []D22 +69,9 (с, 0,75 в CHCl3); H (500 мГц, CHCl3) 2,03, 2,04, 2,06, 2,08, 2,11, 2,15, 2,19, (30H, 10COCH3), 3,77-3,79 (1 Н, m, Н-5 а), 3,94-4,00 (4 Н, m, Н-4 а, Н-4 с, Н-5b, Н-5 с), 4,10 (1 Н, dd, J5,6 2,1 Гц, J6,6. 12,4 Гц, H-6b), 4,17-4,22 (3H, m, Н-6a, Н-6c, Н-6 а'), 4,29 (1 Н, dd, J5,6 3,3 Гц, J6,6. 12,6 Гц, H-6b'), 4,46 (1 Н,dd, J5,6 1,9 Гц, J 6,6' 12,4 Гц, Н-6 с'), 4,76 (1 Н, dd, J1,2 3,9 Гц, J2,3 10,4 Гц, Н-2 а), 4,89-4,94 (2 Н, m, Н-2b, Н- 23011299 2 с), 5,12 (1 Н, at, J 9,9 Гц, H-4b), 5,28 (1 Н, d, J1,2 3,8 Гц, Н-1 а), 5,34 (1 Н, d, J1,2 9,7 Гц, Н-1 с), 5,37 (1 Н, at, J 9.1 Гц, Н-3c), 5,41 (1 Н, at, J 10,1 Гц, Н-3b), 5,41-5,45 (2 Н, m, Н-1b, Н-3a), 7,62-7,65 (2 Н, m, Ar-Н), 7,71 (1 Н,m, Ar-Н), 8,00-8,02 (2 Н, m, Ar-Н). Пример 37. Этил-2,3,6-три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилOглюкопиранозил)D-глюкопиранозил)-1-дитиоD-глюкопиранозилдисульфид 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-O-глюкопиранозил)D-глюкопиранозил-фенилтиосульфонат (50 мг, 0,05 ммоль) растворяли в безводном ДХМ (10 мл) и перемешивали при КТ в атмосфере аргона. В течение 1 ч медленно по каплям добавляли раствор триэтиламина (7 мкл, 0,05 ммоль) и этантиола (3 мкл, 0,05 ммоль) и безводный ДХМ (10 мл) через шприцевой насос. Через 1 ч ТСХ (бензин:этилацетат, 1:2) показала образование основного продукта (Rf 0,6) наряду с полным расходованием исходного вещества (Rf 0,4). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 1:2) с получением целевого этилового продукта (43 мг, 93%) в виде прозрачного масла; []D24 +26,4 (с, 1,5 в CHCl3); H (500 МГц, CHCl3) 1,30 (1 Н, t, J 7,2 Гц, СН 3), 2,04, 2,05, 2,06, 2,07, 2,10, 2,14, 2,19, 2,20 (30H, 8s, 10 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-D-глюкопиранозил)D-глюкопиранозил-фенилтиосульфонат (89 мг, 0,08 ммоль) растворяли в безводном ДХМ (5 мл) и перемешивали при КТ в атмосфере аргона. В течение 3 ч медленно по каплям добавляли раствор триэтиламина (0,014 мл, 0,2 ммоль) и метилового эфира N-бутоксикарбонил-L-цистеинилL-серина (30 мг, 0,09 ммоль) в безводном ДХМ (10 мл) и безводном метаноле (1 мл) через шприцевой насос. Через 3 ч ТСХ (этилацетат) показала образование основного продукта (Rf 0,6) наряду с полным расходованием исходного вещества (Rf 0,7). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат) с получением целевого продукта (66 мг, 74%) в виде белого аморфного твердого вещества; []D24 +25,1 (с, 1,25 в CHCl3); H (500 мГц, CHCl3) 1,47 (9 Н, s, С(CH3)3),2,00, 2,01, 2,02, 2,03, 2,06, 2,09, 2,15, 2,18 (30H, 8s, 10COCH3), 2,75-2,87 (1 Н, m, CHHCys), 3,16-3,19 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-O-ацетил-4-O-(2,3,4,6-тетра-O-ацетилO-глюкопиранозил)-D-глюкопиранозил)D-глюкопиранозилтиол (500 мг, 0,53 ммоль) и фенилселенбромид (200 мг, 0,9 ммоль) растворяли в безводном ДХМ (20 мл). Через 5 мин ТСХ (бензин:этилацетат 1:2) показала образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,3). Реакцию гасили добавлением триэтиламина (5 мл) и затем концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат 1:2) с получением целевого продукта (527 мг, 91%) в виде желтоватого аморфного твердого вещества; []D25 -2,6 (с, 1,0 в CHCl3); H (400 мГц, CHCl3), 1,99,2,01, 2,02, 2,04, 2.06, 2,10, 2,14 (30H, 9s, 10 ОАс), 3,79 (1 Н, dat, J4,5 9,7 Гц, J3.4 Гц, Н-5 а), 3,92 (3H, m,H-4b, H-5b, H-5 с), 4,00 (1 Н, at, J 9,3 Гц, Н-4 а), 4,05 (1 Н, dd, J5,6 2,8Гц, J6,6. 12,8 Гц, Н-6c), 4,15 (1 Н, dd, J5,6 2,8 Гц, J6,6' 12,6 Гц, H-6b), 4,22 (1 Н, dd, J5,6 3,7 Гц, J6,6' 12,0 Гц, Н-6a), 4,25 (1 Н, dd, J5,6 3,3 Гц, J6,6' 12,0 Гц,Н-6 с'), 4,42-4,46 (2 Н, m, Н-6 а', Н-6b'), 4,66 (1 Н, d, J1,2 9,9 Гц, Н-1 а), 4,74 (1 Н, dd, J1,2 4,1 Гц, J2,3 10,4 Гц, H2b), 4,86 (1 Н, dd, J1,2 4,1 Гц, J2,3 10,5 Гц, Н-2 с), 5,06 (1 Н, at, J 9,6 Гц, Н-4 с), 5,07 (1 Н, at, J 9,8 Гц, Н-2 а), 5,27 бис-1 М-Бутоксикарбонил-L-цистеин (4,0 г, 9,1 ммоль), метиловый эфир L-треонина (2,42 г, 18,2 ммоль), DCC (3,75 г, 18,2 ммоль), HOBt (2,46 г, 18,2 ммоль) и DIPEA (2,5 мл, 18,2 ммоль) растворяли в свежеперегнанном ДХМ (150 мл). Через 18 ч ТСХ (этилацетат:метанол, 9:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,0). Реакционную смесь разбавляли водой (2100 мл), и фазы разделяли. Органическую фазу промывали рассолом (100 мл), сушили (MgSO4), фильтровали, и растворитель удаляли в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат:метанол, 9:1), и после перекристаллизации из смеси метанол/диэтиловый эфир получали целевой продукт (3,26 г, 60%) в виде белого твердого кристаллического вещества; т.пл. 145-147 С; []D25 +20,8 (с, 1,0 в CHCl3); H (400 мГц, CHCl3), 1,23 (3H, d, JCHCH3 6,6 Гц,CHCH3), 1,44 (9 Н, s, С(CH3)3), 3,11-3,12 (2 Н, m, CH2Cys), 3,26 (1 Н, bs, ОН), 3,75 (3H, s, ОМе), 4,32-4,36 Метиловый эфир бис-N-бутоксикарбонил-L-цистеинил-L-треонина (2,0 г, 3,3 ммоль) растворяли в необезвоженном хлороформе (100 мл) и метаноле (10 мл) и перемешивали. К этой смеси при перемешивании добавляли раствор трибутилфосфина (1,0 мл, 4,0 ммоль). Через 2 ч ТСХ (этилацетат:метанол, 9:1) показала образование продукта (Rf 0,8) наряду с полным расходованием исходного вещества (Rf 0,7). Реакционную массу концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией(этилацетат) с получением целевого продукта (2,0 г, 99%) в виде белой пены; []D25 -11,4 (с, 1,0 в CHCl3); Фенил-2,3,4,6-тетра-O-ацетил-1-селененилсульфид-Dглюкопиранозид (130 мг, 0,25 ммоль) и триэтиламин (0,02 мл, 0,18 ммоль) растворяли в свежеперегнанном ДХМ (10 мл). Полученный раствор перемешивали при КТ. К вышеуказанному раствору медленно добавляли раствор метилового эфира Nбутоксикарбонил-L-цистеин-L-треонина (30 мг, 0,089 ммоль) в безводном метаноле (4 мл). Через 10 мин ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,2) наряду с полным расходованием исходного вещества (Rf 0,5). Раствор концентрировали в вакууме. Остаток очищали колоночной флэшхроматографией (бензин:этилацетат, 1:2) с получением целевого продукта (32 мг, 51%) в виде белого аморфного твердого вещества; []D25 -81,2 (с, 0,25 в CHCl3); H (400 мГц, CHCl3) 1,28 (3H, d, JCHCH3 6,7 Гц,CHCH3), 1,51 (9 Н, s, С(CH3)3), 2,06, 2,08, 2,10, 2,14 (12 Н, 4s, 4 ОАс), 2,86 (1 Н, bs, ОН), 3,06 (1 Н, dd,JCHH 8,8 Гц, JCHCH 13,4 Гц, CHHCys), 3,31 (1 Н, dd, JCHH 4,2 Гц, JCHCH 13,1 Гц, CHHCys), 3,82 (3H, s,OCH3), 3,87-3,89 (1 Н, m, H-5), 4,32-4,38 (2 Н, m, H-6, Н-6'), 4,39 (1 Н, dd, JCHCH3 6,4 Гц, JCHH 2,5 Гц,СНОН), 4,60-4,65 (3H, m, Н-1, HThr, HCys), 5,20-5,32 (3H, m, Н-2, Н-3, Н-4), 5,42 (1 Н, d, JNHaH 8,0 Гц,NHCys), 7,12 (1 Н, d, JNHaH 8,9 Гц, NHThr). Пример 43. Метиловый эфир N-бутоксикарбонил-L-цистеин (2,3,4,6-тетра-O-ацетил-1-дитиоDгалактопиранозил-дисульфид)-L-треонина Фенил-2,3,4,6-тетра-O-ацетил-1-селененилсульфид-Dгалактопиранозид (140 мг, 0,27 ммоль) и триэтиламин (0,01 мл, 0,089 ммоль) растворяли в свежеперегнанном ДХМ (5 мл). Полученный раствор перемешивали при КТ. К вышеуказанному раствору медленно добавляли раствор метилового эфира Nбутоксикарбонил-L-цистеин-L-треонина (26 мг, 0,077 ммоль) в безводном ДХМ (5 мл) и безводном метаноле (4 мл). Через 10 мин ТСХ (бензин:этилацетат, 1:2) показала образование продукта (Rf 0,2) наряду с полным расходованием исходного вещества (Rf 0,6). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (бензин:этилацетат, 1:2) с получением целевого продукта(49 мг, 93%) в виде белого аморфного твердого вещества; []D25 -81,2 (с, 0,25 в CHCl3); H (400 мГц,CHCl3) 1,24 (3H, d, JCH,CH3 6,4 Гц, CH3), 1,46 (9 Н, s, С(СН 3)3), 2,01, 2,06, 2,08, 2,20 (12 Н, 4s, 4 ОАс),2,79 (1 Н, bd, JCH,OH 4,1 Гц, ОН), 2,99 (1 Н, dd, JH,CH2 8,8 Гц, JCH,H 13,9 Гц, CHHCys), 3,32-3,35 (1 Н, m,CHHCys), 3,76 (3H, s, ОСН 3), 4,04 (1 Н, at, J 6,2 Гц, Н-5), 4,10-4,16 (1 Н, m, Н-6), 4,19 (1 Н, dd, J5,6' 6,1 Гц,J6,6' 10,8 Гц, Н-6'), 4,36-4,46 (1 Н, m, СНОН), 4,56 (1 Н, dd, JHThr,CH 2,4 Гц, JH,NH 8,9 Гц, HThr), 4,57-4,64 (1 Н,m, HCys), 4,65 (1 Н, d, J1,2 9,0 Гц, Н-1), 5,13 (1 Н, dd, J2,3 9,8 Гц, J2,3 9,8 Гц, Н-3), 5,31 (1 Н, d, JHCys,NH 8,3 Гц, NHCys), 5,47 (1 Н, d, J3,4 3,2 Гц, Н-4), 5,52 (1 Н, at, J 9,6 Гц, Н-2), 6,91 (1 Н, d, JHThr,NH 9,0 Гц, NHThr). Целевой продукт получали (55 мг, 88%) в виде белого аморфного твердого вещества по способу,аналогичному способу, описанному в примере 43, используя в качестве исходного вещества фенил-3,4,6 три-O-ацетил-2-ацетамидо-2-дезокси-1-селененилсульфид-Dглюкопиранозид. []D25 -47,1 (с, 0,1 в Фенил-1-селененилсульфид-Dглюкопиранозид (70 мг, 0,2 ммоль) и триэтиламин (0,01 мл, 0,1 ммоль) растворяли в МеОН (8 мл). Полученный раствор перемешивали при КТ. К вышеуказанному раствору медленно добавляли раствор метилового эфира N-бутоксикарбонил-L-цистеин-L-треонина (22 мг,0,07 ммоль) в МеОН (5 мл). Через 10 мин ТСХ (EtOAc:МеОН, 9:1) показала образование основного продукта (Rf 0,4). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией(EtOAc:МеОН, 9:1) с получением целевого соединения (32 мг, 91%) в виде белого аморфного твердого вещества; []D25 -139,5 (с, 0,6 в МеОН); H (500 мГц, CD3OD) 1,19 (3H, d, JCH,CH3 6,2 Гц, СНСН 3), 1,49 (9 Н,s, С(СН 3)3), 2,93 (1 Н, dd, JCHH,CHH 13,5 Гц, JCH,H 9,5 Гц, CH2Cys), 3,32-3,46 (4 Н, m, Н-3, Н-4, Н-5, СНН),3,60-3,63 (1 Н, m, Н-2), 3,73-3,77 (1 Н, m, Н-6), 3,78 (3H, s, ОМе), 3,92-3,94 (1 Н, m, Н-6'), 4,31-4,36 (1 Н, m,СНСН 3), 4,39 (1 Н, d, J1,2 9,3 Гц, Н-1), 4,48 (1 Н, d, JH,CH 2,9 Гц, HThr), 4,69 (1 Н, dd, JH,CHH 9,0 Гц, JH,CHH 5.2 Гц, HCys). Пример 46. Метиловый эфир N-бутоксикарбонил-L-цистеинил-(S-2-ацетамино-2-дезокси-1Dглюкопиранозилдисульфид)-L-треонина Целевой продукт получали (32 мг, 91%) в виде белого аморфного твердого вещества по способу,аналогичному способу, описанному в Примере 45, используя в качестве исходного вещества фенил-2 ацетамидо-2-дезокси-1-селененилсульфидD-глюкопиранозид. []D25 +6,21 (с, 0,45 в МеОН); H (500 мГц, CD3OD) 1,19 (3H, d, JCHCH3 6,7 Гц, CHCH3), 1,49 (9 Н, s, С(СН 3)3), 1,99 (3H, s, COCH3), 2,97 (1 Н, dd,JCH,H 13,8 Гц, JCH,H 9,6 Гц, CHHCys), 3,31-3,33 (1 Н, m, СНН), 3,38-3,41 (1 Н, m, Н-5), 3,45 (1 Н, at, J 9,3 Гц,Н-4), 3,54 (1 Н, dd, J2,3 8,6 Гц, J3,4 9,8 Гц, Н-3), 3,76-3,77 (1 Н, m, Н-6), 3,78 (3H, s, ОМе), 3,79-4,01 (2 Н, m,Н-2, Н-6'), 4,33 (1 Н, dq, JCHCH3 6,3 Гц, JCH,H, 3,0 Гц, СНСН 3), 4,48 (1H, d, JH,CH H 3,0 Гц, HThr), 4,59 (1 Н,d, J1,2 10,3 Гц, H-1), 4,63-4,67 (1 Н, m, Hcys). Пример 47. Фенил-1-селененилсульфидD-глюкопиранозид 1-ТиоD-глюкопиранозид (200 мг, 0,9 ммоль) и фенилселененилбромид (230 мг, 1,0 ммоль) добавляли к безводному 1,4-диоксану (5 мл), перемешивали в атмосфере аргона. Через 1 мин ТСХ (этилацетат) показала образование основного продукта (Rf 0,2). Реакцию гасили добавлением триэтиламина (2 мл). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилаце- 27011299 тат:метанол, 9:1) с получением целевого продукта (165 мг, 57%) в виде желтоватого аморфного твердого вещества; []D22 +56,2 (с, 1 в CHCl3); H (400 мГц, MeOD) 3,31-3,33 (2 Н, m, Н-3, Н-5), 3,39-3,45 (2 Н, m, Н 2, Н-4), 3,62 (1 Н, dd, J5,6 5,3 Гц, J6,6' 12,1 Гц, Н-6), 3,83 (1 Н, dd, J5,6' 1,9 Гц, J6,6' 12,2 Гц, Н-6), 4,47 (1 Н, d, J1,2 9,4 Гц, Н-1), 7,27-7,34 (3H, m, Ar-Н), 7,75-7,78 (2 Н, m, Ar-Н). Пример 48. Фенил-1-селененилсульфидD-галактопиранозид Целевое соединение получали (193 мг, 20%) в виде желтоватого аморфного твердого вещества по способу, аналогичному способу, описанному в примере 47, используя в качестве исходного вещества 1 тиоD-галактопиранозид. []D25 -111,4 (с, 1 в МеОН); H (400 мГц, CD3OD) 3,52 (1 Н, dd, J2,3 9,4 Гц, J3,4 3,3 Гц, Н-3), 3,56 (1 Н, at, J4,5 0,9 Гц, J 6,5 Гц, Н-5), 3,67-3,69 (2 Н, d, J 6,0 Гц, Н-6, Н-6'), 3,74 (1 Н, at, J 9,3 Гц, Н-2), 3,91 (1 Н, dd, J3,4 3,2 Гц, J4,5 0,7 Гц, Н-4), 4,45 (1 Н, d, J1,2 9,7 Гц, Н-1), 7,27-7,30 (3H, m, Ar-Н),7,76-7,79 (2 Н, m, Ar-Н). Пример 49. Фенил-2,3,4,6-тетра-O-ацетил-1-селененилсульфидD-глюкопиранозид 1-Тио-2,3,4,6-тетра-O-ацетилD-глюкопиранозу (200 мг, 0,6 ммоль) и PhSeBr (150 мг, 0,6 ммоль) добавляли к свежеперегнанному ДХМ (5 мл) и перемешивали в атмосфере аргона при КТ. Через 5 мин ТСХ (бензин:EtOAc, 1:1) показала образование основного продукта (Rf 0,5) наряду с полным расходованием исходного вещества (Rf 0,4). Реакционную смесь гасили добавлением триэтиламина (2 мл) и перемешивали в течение 5 мин. Остаток распределяли между ДХМ (5 мл) и водой (10 мл), и водную фазу повторно экстрагировали ДХМ (35 мл). Объединенные органические экстракты промывали рассолом(10 мл), сушили над MgSO4, фильтровали, и растворитель удаляли в вакууме. Полученный остаток очищали колоночной флэш-хроматографией (бензин:EtOAc, 2:1) с получением целевого продукта (260 мг,93%) в виде желтого твердого кристаллического вещества, т.пл. 111-112 С; []D25 -250,1 (с, 1,0 в CHCl3); Целевое соединение получали (402 мг, 95%) в виде желтого твердого кристаллического вещества,используя способ, аналогичный способу, описанному в примере 49, с использованием в качестве исходного вещества 1-тио-2,3,4,6-тетра-O-ацетилD-галактопиранозы. Т.пл. 123-125 С; []D25 -172,4 (с, 1,0 в Целевое соединение получали (300 мг, 66%) в виде белого твердого кристаллического вещества,используя способ, аналогичный способу, описанному в примере 49, с использованием в качестве исходного вещества 1-тио-3,4,6-три-O-ацетил-2-ацетамидо-2-дезоксиD-глюкопиранозы. Т.пл. 177-179 С; 1-Тио-2-ацетиламино-2-дезоксиD-глюкопиранозид (230 мг, 0,98 ммоль) и фенилселененилбромид (250 мг, 1,08 ммоль) добавляли к безводному 1,4-диоксану (5 мл) и безводному метанолу (3 мл), перемешивали в атмосфере аргона. Через 1 мин ТСХ (этилацетат:метанол, 9:1) показала образование основного продукта (Rf 0,4). Реакционную смесь гасили добавлением триэтиламина (5 мл). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (этилацетат:метанол, 9:1) с получением целевого продукта (270 мг, 70%) в виде белого аморфного твердого вещества; []D22 -174,0 Фенил-1-селененилсульфидD-глюкопиранозид (140 мг, 0,4 ммоль) растворяли в МеОН (10 мл) и перемешивали при КТ. К этой смеси в течение 1 ч по каплям добавляли раствор этантиола (10 мкл, 0,1 ммоль) и триэтиламина (60 мкл, 0,4 ммоль) в МеОН (5 мл). Через 1 ч ТСХ (EtOAc:МеОН, 9:1) показала образование основного продукта (Rf 0,4) наряду с полным расходованием исходного вещества (Rf 0,5). Раствор концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией(EtOAc:MeOH, 5:1) с получением целевого продукта (30 мг, 90%) в виде белого аморфного твердого вещества; []D22 -65,3 (с, 0,4 в CHCl3); H (500 мГц, CD3OD), 1,33 (3H, t, J 7,4 Гц, СН 3), 2,86 (2 Н, q, J 7,4 Гц,СН 2), 3,30-3,34 (2 Н, m, Н-4, Н-5), 3,41 (1 Н, at, J 9,0 Гц, Н-3), 3,49 (1 Н, at, J Гц, Н-2), 3,67 (1 Н, dd, J5,6 5,3 Гц, J6,6' 12,0 Гц, Н-6), 3,88 (1 Н, dd, J5,6. 2,1 Гц, J6,6' 12,0 Гц, Н-6'), 4,35 (1 Н, d, J1,2 9,1 Гц, Н-1). Пример 54. Этил-2-ацетиламидо-2-дезокси-1-дисульфидD-глюкопиранозид Фенил-2-ацетамидо-2-дезокси-1-селененилсульфидD-глюкопиранозид (140 мг, 0,4 ммоль) растворяли в МеОН (10 мл) и перемешивали при КТ. К этой смеси в течение 1 ч по каплям добавляли раствор этантиола (10 мкл, 0,13 ммоль) и триэтиламина (55 мкл, 0,4 ммоль) в МеОН (5 мл). Через 1 ч ТСХ(EtOAc:МеОН, 9:1) показала образование основного продукта (Rf 0,2). Раствор концентрировали в вакууме. Полученный остаток очищали колоночной флэш-хроматографией (EtOAc:МеОН, 9:1) с получением целевого продукта (38 мг, 99%) в виде белого аморфного твердого вещества; []D25 -7,9 (с, 1,0 вCHCl3); H (400 мГц, CD3OD) 1,30 (3H, t, J 7,3 Гц, СН 3), 2,01 (3H, s, ОАс), 2,83-2,86 (2 Н, m, СН 2), 3,313,39 (2 Н, m, Н-4, Н-5), 3,51-3,56 (1 Н, m, Н-3), 3,68-3,72 (1 Н, m, Н-6), 3,84-3,91 (2 Н, m, Н-2, Н-6'), 4,57 (1 Н,d, J1,2 10,3 Гц, Н-1). Пример 55. Способы гликозилирования белков с использованием тиосульфонатных реагентовA. Мутантный белок SBLS156C (24 мг, 0,89 мкмоль) растворяли в водном буферном растворе (2,4 мл, 70 мМ HEPES, 2 мМ CaCl2, рН 6,9). 2,3,4,6-Тетра-О-ацетилD-глюкопиранозил-фенилтиосульфонат(50 мг, 0,1 ммоль) растворяли в смеси вода/ацетонитрил (1,6 мл, 9/7, об./об.). Часть раствора сахара (50 мкл) добавляли к раствору белка и помещали на ротатор для вертикального перемешивания. Через 25 мин с помощью анализа Элмана было показано отсутствие свободного тиола (Ellman, G. L. Arch. Biochem. Biophys. 1959, 82, 70), после чего добавляли другую часть раствора сахара (50 мкл). Реакционную смесь помещали на ротатор для вертикального перемешивания еще на 5 мин, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против 10 мМ MES, 1 мМ CaCl2, рН 5,8 (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найде- 29011299 но 27072, вычислено 27078.B. Мутантный белок SBLS156C (24 мг, 0,89 мкмоль) растворяли в водном буферном растворе (2,4 мл, 70 мМ HEPES, 2 мМ CaCl2, рН 6,9). 2,3,4,6-тетра-О-ацетилD-галактопиранозилфенилтиосульфонат (50 мг, 0,1 ммоль) растворяли в смеси вода/ацетонитрил (1,0 мл, соотношение 1/1). К раствору белка добавляли раствор сахара (50 мкл) и помещали на ротатор для вертикального перемешивания. Через 25 мин с помощью анализа Элмана было показано отсутствие свободного тиола, после чего добавляли другую часть раствора сахара (50 мкл). Реакционную смесь помещали на ротатор для вертикального перемешивания еще на 5 мин, после чего реакционную смесь наносили на колонку PD10Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против 10 мМ MES, 1 мМ CaCl2, рН 5,8 (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 27072, вычислено 27078.C. Мутантный белок SBLS156C (10 мг, 0,37 мкмоль) растворяли в дегазированном водном буферном растворе (1 мл, 70 мМ CHES, 5 мМ MES, 2 мМ CaCl2, рН 9,5). 2,3,6-Три-O-ацетил-4-O-(2,3,6-три-Oацетил-4-O-(2,3,4,6-тетра-O-ацетилD-глюкопиранозил)O-глюкопиранозил)O-глюкопиранозилфенилтиосульфонат (30 мг, 0,03 ммоль) растворяли в ацетонитриле (150 мкл). К раствору белка добавляли раствор сахара (75 мкл) и помещали на ротатор для вертикального перемешивания. Через 30 мин с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против 10 мМ MES, 1 мМ CaCl2, рН 5,8 (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 27654, вычислено 27653.D. БСА (10 мг, 0,14 мкмоль) растворяли в водном буферном растворе (1 мл, 50 мМ Трис, рН 7,7). 2,3,4,6-Тетра-O-ацетилD-глюкопиранозил-фенилтиосульфонат (10 мг, 0,02 ммоль) растворяли в смеси вода/ацетонитрил (1,0 мл, соотношение 8/2). К раствору белка добавляли раствор сахара (150 мкл) и помещали на ротатор для вертикального перемешивания. Через 30 мин с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против чистой воды (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 66798, вычислено 66794.E. БСА (10 мг, 0,14 мкмоль) растворяли в водном буферном растворе (1 мл, 50 мМ Трис, рН 7,7). 2,3,4,6-Тетра-O-ацетилD-галактопиранозил-фенилтиосульфонат (25 мг, 0,05 ммоль) растворяли в ацетонитриле (0,5 мл). К раствору белка добавляли раствор сахара (75 мкл) и помещали на ротатор для вертикального перемешивания. Через 30 мин с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против чистой воды (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 66792, вычислено 66794. Пример 56. Способы гликозилирования белков с использованием селененилсульфидных реагентовA. Мутантный белок SBLS156C (5 мг) растворяли в дегазированном водном буферном растворе (1 мл, 70 мМ CHES, 5 мМ MES, 2 мМ CaCl2, рН 9,5). Фенил-2,3,4,6-тетра-О-ацетилD-селененилсульфидглюкопиранозид (10 мг, 0,02 ммоль) растворяли в ацетонитриле (500 мкл). К раствору белка добавляли раствор сахара (500 мкл) и помещали на ротатор для вертикального перемешивания. Через 1 ч с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против воды (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 27074, вычислено 27077.B. БСА (5 мг) растворяли в дегазированном водном буферном растворе (1 мл, 70 мМ CHES, 5 мМMES, 2 мМ CaCl2, рН 9,5). Фенил-2,3,4,6-тетра-O-ацетилD-селененилсульфид-глюкопиранозид (10 мг,0,02 ммоль) растворяли в ацетонитриле (800 мкл). К раствору белка добавляли раствор сахара (800 мкл) и помещали на ротатор для вертикального перемешивания. Через 1 ч с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фракцию собирали и диализовали (MWCO 12-14 кДа) против воды (14 л в течение 1 ч, 22 л в течение 30 мин), с получением гликозилированного продукта; m/z (ЭР) найдено 66792, вычислено 66794.C. Мутантный белок SBLS156C (5 мг) растворяли в дегазированном водном буферном растворе (1 мл, 70 мМ CHES, 5 мМ MES, 2 мМ CaCl2, рН 9,5). Фенил-2,3,4,6-тетра-О-ацетилD-селененилсульфидгалактопиранозид (10 мг, 0,02 ммоль) растворяли в ацетонитриле (500 мкл). К раствору белка добавляли раствор сахара (500 мкл) и помещали на ротатор для вертикального перемешивания. Через 1 ч с помощью анализа Элмана было показано отсутствие свободного тиола, после чего реакционную смесь наносили на колонку PD10 Sephadex G25 и элюировали 70 мМ HEPES, 2 мМ CaCl2, рН 7,0. Белковую фрак- 30
МПК / Метки
МПК: C12N 9/44, C07H 5/10, C12N 9/56
Метки: образования, реагенты, применяемые, способах, дисульфидных, связей, способы, гликозилирования, белков, этих
Код ссылки
<a href="https://eas.patents.su/30-11299-sposoby-obrazovaniya-disulfidnyh-svyazejj-i-glikozilirovaniya-belkov-i-reagenty-primenyaemye-v-etih-sposobah.html" rel="bookmark" title="База патентов Евразийского Союза">Способы образования дисульфидных связей и гликозилирования белков и реагенты, применяемые в этих способах</a>
Способы детекции амилоидогенных белков

Номер патента: 5582
Опубликовано: 28.04.2005
Автор: Кришнамурти Гириджа
МПК: G01N 33/68
Метки: амилоидогенных, белков, способы, детекции
Формула / Реферат:
1. Способ определения агрегационной конформации амилоидного полипептида, предусматривающий сравнение (a) спектроскопического свойства амилоидоспецифического спектроскопического зонда, образовавшего комплекс с амилоидным полипептидом, имеющим неизвестную агрегационную конформацию, и (b) предварительно определенного спектроскопического свойства амилоидоспецифического спектроскопического зонда, образовавшего комплекс с амилоидным полипептидом...
Способы инактивации клеток-мишеней, агенты и композиции, вызывающие цитолиз, и соединения, используемые для получения этих агентов

Номер патента: 2688
Опубликовано: 29.08.2002
Авторы: Дохлстен Микаэль, Абрахмсен Ларс, Форсберг Геран, Ландо Петер, Калланд Терье, Согорд Мортен
МПК: A61K 47/48, A61K 39/385, A61K 39/085...
Метки: используемые, агентов, инактивации, вызывающие, способы, получения, цитолиз, композиции, соединения, этих, клеток-мишеней, агенты
Формула / Реферат:
1. Способ инактивации клеток-мишеней в присутствии Т-клеток, включающий контактирование клеток мишеней и Т-клеток с суперантигеном в присутствии иммуномодулятора, когда, по меньшей мере, один из элементов - суперантиген или иммуномодулятор - конъюгирован с нацеливающим компонентом, причем нацеливающий компонент выбран из группы, состоящей из антитела, антигенсвязывающего фрагмента антитела, Fab-фрагмента антитела, Fаb2-фрагмента антитела или...
Бензконденсированные гетероариламидные производные тиенопиридинов, применяемые в качестве терапевтических агентов, фармацевтические композиции, включающие их, и способы их применения

Номер патента: 8255
Опубликовано: 27.04.2007
Авторы: Кания Роберт Стивен, Лоу Цзихун, Роминес Уилльям Генри III, Хэ Минин, Криппс Стефан Джеймс, Коллинс Майкл Рэймонд, Дил Джудит Гейл, Палмер Синтия Луиза, Чжоу Жу
МПК: C07D 495/04, A61P 35/00, A61K 31/4365...
Метки: гетероариламидные, бензконденсированные, терапевтических, их, применения, качестве, тиенопиридинов, фармацевтические, производные, применяемые, способы, композиции, агентов, включающие
Формула / Реферат:
1. Соединение, представленное формулой I где Y является -NH-, -O-, -S- или -СН2-; Z является -O-, -S- или -N-; R14 является C1-C6алкилом, C1-C6алкиламино, C1-C6алкилгидрокси, C3-C10циклоалкилом, С3-C10циклоалкиламино, C1-C6алкилC3-C10циклоалкилом или метилуреидогруппой; R15 и R17 независимо являются Н, галогеном или C1-C6алкилом, незамещенным или замещенным одним или более R5; R16 является Н или C1-C6алкилом, когда Z является N, и R16...
Новые гетероциклические соединения, применяемые для лечения нарушений аллергической или воспалительной природы: способы синтеза и содержащие их фармацевтические составы

Номер патента: 10634
Опубликовано: 30.10.2008
Авторы: Лакдавала Афтаб Давудбхай, Караунакаран Уша, Гопалан Баласубраманиан, Гхарат Лаксмикант Атмарам
МПК: A61P 29/00, C07D 307/91, A61K 31/4427...
Метки: гетероциклические, природы, применяемые, фармацевтические, нарушений, новые, воспалительной, содержащие, составы, способы, аллергической, синтеза, лечения, соединения
Формула / Реферат:
1. Соединение общей формулы (1) где в каждом отдельном случае R1 представляет собой водород или C1-C8алкил, возможно замещенный одним или более галогеном; R2 и R3 представляют собой водород; R4 представляет собой -NR5R6; где R5 и R6 могут быть одинаковыми или различными и независимо выбраны из группы, включающей: (i) водород; (ii) C1-C8алкил, возможно замещенный -C(O)ORa; (iii) -C(O)-Rb; (iv) -C(O)ORc; (v) -C(O)NRdRd; (vi) -S(O)qRa; (vii)...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Корнберг Брайэн Эдвард, Рафферти Майкл Фрэнсис, Никам Шам
МПК: C07D 241/44
Метки: фармацевтические, обладающие, лечения, глютамата, помощи, этих, отношении, удара, производные, рецептора, способы, фармацевтически, антагонистической, страдающих, заболеваний, приемлемые, композиции, активностью, соли, противосудорожной, хиноксалин-2,3-диона, пациентов, соединений
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Предыдущий патент: Применение бактерицидных пиримидинов для профилактики передачи вич половым путём и фармацевтические композиции на их основе
Следующий патент: Производные триазолопиримидина в качестве ингибиторов киназы гликогенсинтазы-3
Случайный патент: Кристаллизатор для непрерывной разливки блюмов, слябов или сортовых заготовок