Новые гетероциклические соединения, применяемые для лечения нарушений аллергической или воспалительной природы: способы синтеза и содержащие их фармацевтические составы
Номер патента: 10634
Опубликовано: 30.10.2008
Авторы: Гхарат Лаксмикант Атмарам, Гопалан Баласубраманиан, Караунакаран Уша, Лакдавала Афтаб Давудбхай







Формула / Реферат
1. Соединение общей формулы (1)

где в каждом отдельном случае R1 представляет собой водород или C1-C8алкил, возможно замещенный одним или более галогеном;
R2 и R3 представляют собой водород;
R4 представляет собой -NR5R6; где R5 и R6 могут быть одинаковыми или различными и независимо выбраны из группы, включающей: (i) водород; (ii) C1-C8алкил, возможно замещенный -C(O)ORa; (iii) -C(O)-Rb; (iv) -C(O)ORc; (v) -C(O)NRdRd; (vi) -S(O)qRa; (vii) -S(O)q-NRaRa; (viii) -C(=S)-NRaRa и (ix) защитные группы, выбираемые из группы, включающей карбобензилокси и трет-бутилоксикарбонил, при условии, что R4 не является NH2;
Ar выбран из группы, включающей: (i) фенил, возможно замещенный C1-C8алкокси; (ii) (С6арил)(С1-С8алкил); (iii) пиридил или пиридил-N-оксид, возможно замещенный галогеном;
в каждом отдельном случае Ra независимо представляет собой водород или С1-С8алкил;
Rb представляет собой: (i) водород; (ii) C1-C8алкил, возможно замещенный галогеном; (iii) С3-С5циклоалкил; (iv) C5гетероарил; (v) C6гетероциклил, который содержит от 1 до 2 гетероатомов, выбираемых из кислорода и азота, и может быть замещен C1-C8алкилом или гидрокси; или (vi) -C(O)O-Ra;
Rc представляет собой водород, C1-C8алкил, возможно замещенный галогеном, фенил или (С3-С5циклоалкил)(C1-C8алкил);
в каждом отдельном случае Rd независимо представляет собой водород, C1-C8алкил, возможно замещенный -C(O)O-Ra, или С3-С5циклоалкил;
X выбран из группы, включающей О, S и NCH3;
Y представляет собой -C(O)NH;
Р представляет собой О;
m представляет собой 0;
n представляет собой 1;
q представляет собой 2;
а также его фармацевтически приемлемые соли и N-оксиды.
2. Соединение по п.1, в котором Ar представляет собой фенил, возможно замещенный C1-C8алкокси, выбранным из 4-пиридила, 3-пиридила, 2-пиридила, 4-пиридил-N-оксида, 3-пиридил-N-оксида и 2-пиридил-N-оксида, возможно замещенного галогеном.
3. Соединение по п.1, в котором R1 представляет собой C1-C8алкил, возможно замещенный одним или более галогеном, например метил или -CHF2.
4. Соединение по пп.1-3, в котором Ar представляет собой 4-пиридил, 4-пиридил-N-оксид или 3-пиридил, возможно замещенный одним или более галогеном.
5. Соединение по п.4, в котором Ar замещен одним или более галогеном, таким как хлор.
6. Соединение по п.5, в котором Ar выбран из группы

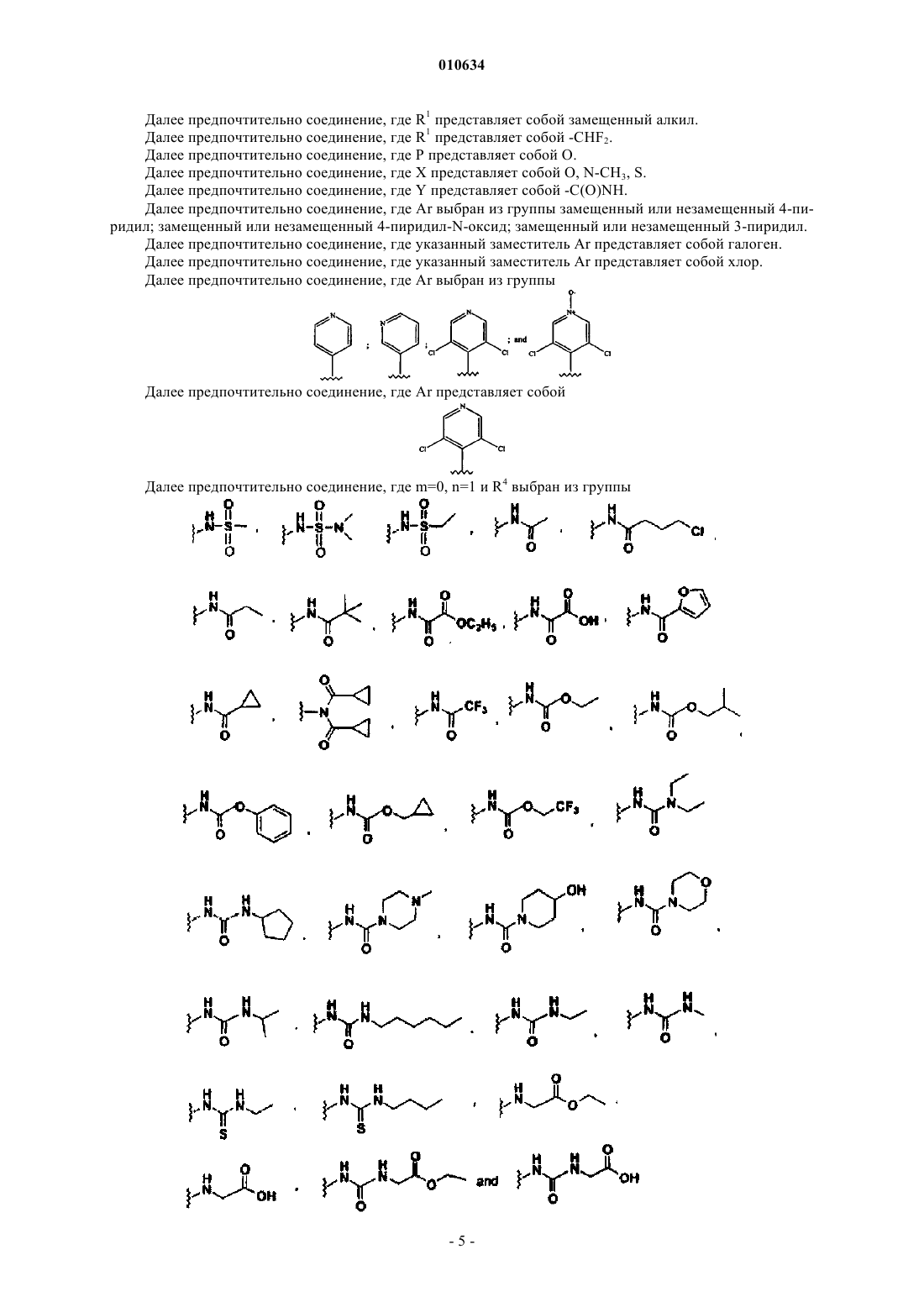
7. Соединение по пп.1-6, в котором m=0, n=1, a R4 выбран из группы

8. Соединение по п.7, в котором m=0, n=1, a R4 выбран из группы

9. Соединение по п.1, выбранное из следующей группы:
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(N,N-диметилсульфонамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(этансульфонамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(3-хлорпропилкарбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-t-бутилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этоксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
натриевая соль N-(3,5-дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(фур-2-ил-карбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(циклопропилкарбониламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(N,N-дициклопропилкарбониламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-трифторацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этоксикарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-изобутилоксикарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-феноксикарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-циклопропилметоксикарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-трифторметилметоксикарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-N,N-диэтиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-циклопентиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(N-метилпиперазин-4-ил-карбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
гидрохлорид N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(N-метилпиперазин-4-илкарбоксамид)дибензо
[b,d]фуран-1-карбоксамида;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(4-гидроксипиперидин-1-ил-карбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(морфол-4-ил-карбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-изопропиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемые соли;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-n-гексиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-метиламинкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамида;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-этансульфонамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-N,N-диметиламинсульфонамиддибензю[b,d]фуран-1-карбоксамид и его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-(1-хлопропилкарбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-циклопропилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-этоксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
двунатриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида;
N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-фур-2-ил-карбоксамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N1-фенил-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N1-(4-метоксифенил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N1-бензил-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(этиламинтиокарбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(п-бутиламинтиокарбоксамид)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N1-(пирид-3-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид-N-оксид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамид-N-оксид или его фармацевтически приемлемая соль;
N-(пирид-4-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(2-этокси-2-оксоэтиламинкарбониламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(2-гидрокси-2-оксоэтиламинкарбониламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(2-этокси-2-оксоэтиламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(2-гидрокси-2-оксоэтиламин)дибензо[b,d]фуран-1-карбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-ацетамид-9H-4-карбазолкарбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-метансульфонамид-9H-4-карбазолкарбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-этансульфонамид-9H-4-карбазолкарбоксамид или его фармацевтически приемлемая соль;
N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-пропионамид-9H-4-карбазолкарбоксамид или его фармацевтически приемлемая соль;
двунатриевая соль 1N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамида;
N-(3,5-дихлорпирид-4-ил)-1-метокси-6-ацетамиддибензо[b,d]тиофен-4-карбоксамид или его фармацевтически приемлемая соль;
натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамида;
натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-фур-2-ил-карбоксамиддибензо[b,d]фуран-1-карбоксамида.
10. Соединение по п.1, где указанное соединение представляет собой N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамидодибензо[b,d]фуран-1-карбоксамид или его фармацевтические приемлемую соль.
11. Соединение по п.10, где указанное соединение представляет собой натриевую соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамидодибензо[b,d]фуран-1-карбоксамида.
12. Соединение по п.10, где указанное соединение представляет собой мононатриевую соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамидодибензо[b,d]фуран-1-карбоксамида.
13. Фармацевтический состав, содержащий эффективное количество соединения по пп.1-12, при условии, что он не включает нижеописанную структуру:

и фармацевтически приемлемый носитель.
14. Способ получения соединений общей формулы (1)

по любому из пп.1-12, включающий следующую последовательность операций:
а) приведение во взаимодействие соединения общей формулы (10) с соединением общей формулы (11) в присутствии основания

где Z представляет собой галоген, W представляет собой галоген; в результате образуется полупродукт общей формулы (12);
b) преобразование полупродукта общей формулы (12) с применением реагента, выбранного из группы ацетат палладия в ДМФ или ледяной уксусной кислоте, никелевый катализатор в пиридине или ДМФ или тетракистрифенифосфин палладия в ДМФ, в трициклический полупродукт общей формулы (13)

с) окисление трициклического полупродукта общей формулы (13) до полупродукта общей формулы (14) через взаимодействие с хлоритом натрия или перманганатом калия

d) преобразование полупродукта общей формулы (14) в полупродукт общей формулы (15), где Y представляет собой -CONR7, через взаимодействие активированного полупродукта карбоновой кислоты, выбранного из группы кислый галид, смешанный ангидрид или активный сложный эфир общей формулы (14), с возможно замещенным арил- или гетороариламином (ArNHR7) в щелочной среде;

е) восстановление полупродукта общей формулы (15) до полупродукта общей формулы (16)

f) преобразование полупродукта общей формулы (16) в желательное соединение общей формулы (1), где Y представляет собой -CONR7, R4 представляет собой -NR5R6

g) возможно преобразование соединений общей формулы (1) в соответствующие соли и/или N-оксиды.
15. Способ получения соединений общей формулы (1)

по любому из пп.1-12, включающий следующую последовательность операций:
а) приведение во взаимодействие соединения общей формулы (17) с соединением общей формулы (18) в присутствии основания

где Z представляет собой галоген, W представляет собой галоген, FG выбран из группы СНО, COCH3, CN, COORa, в щелочной среде; в результате образуется полупродукт общей формулы (19)

b) преобразование полупродукта общей формулы (19) с применением реагента, выбранного из группы ацетат палладия в ДМФ или ледяной уксусной кислоте, никелевый катализатор в пиридине или ДМФ или тетракистрифенифосфин палладия в ДМФ, в трициклический полупродукт общей формулы (20)

с) окисление трициклических полупродуктов общей формулы (20), если FG представляет собой СНО или СОСН3, или гидролиз, если FG представляет собой CN или COORa, в полупродукт общей формулы (14)

d) преобразоваэшх полупродукта общей формулы (14) в полупродукт общей формулы (15), где Y представляет собой -CONR7, через взаимодействие активированного полупродукта карбоновой кислоты, выбранного из группы кислый галид, смешанный ангидрид или активный сложный эфир общей формулы (14), с возможно замещенным арил- или гетороариламином (ArNHR7) в щелочной среде

е) восстановление полупродукта общей формулы (15) в полупродукт общей формулы (16)

f) преобразование полупродукта общей формулы (16) в желательное соединение общей формулы (1), где Y представляет собой -CONR7, R4 представляет собой -NR5R6

g) возможно преобразование соединений общей формулы (1) в соответствующие соли и/или N-оксиды.
16. Способ получения соединений общей формулы (1)

по любому из пп.1-12, включающий следующую последовательность операций:
а) приведение во взаимодействие соединения общей формулы (21) с соединением общей формулы (22) в присутствии основания

где Z представляет собой галоген, в результате образуется полупродукт общей формулы (23)

b) восстановление полупродукта общей формулы (23) до полупродукта общей формулы (24)

с) цитирование полупродукта формулы (24) с образованием в результате трициклического полупродукта общей формулы (25)

d) преобразование ацетильной группы трициклического полупродукта формулы (25) в ацетамидную группу с образованием в результате интермедиата формулы (26)

е) формилирование интермедиата формулы (26) с образованием в результате интермедиата формулы (27)

f) окисление интермедиата формулы (27) с образованием интермедиата формулы (28)

g) активирование карбоксильной группы интермедиата формулы (28) и взаимодействие активированной карбоновой кислоты с возможно замещенным арилом или гетероариламином (ArNHR7) с образованием соединения формулы (1), при этом R4 представляет собой -NHC(O)CH3;
h) возможное превращение группы -NHC(O)CH3 в группу формулы -NR5R6 и
i) возможное превращение соединения, полученного на стадии (g) или (h), в соответствующую соль и/или N-оксид.
17. Способ лечения воспалительных заболеваний и иммунных нарушений, или заболеваний центральной нервной системы, или инсулинустойчивых форм диабета, или уменьшения воспаления в пораженном органе или ткани у субъектов, нуждающихся в данной терапии, заключающийся во введении в организм названного субъекта или доставке в названный орган или ткани терапевтически эффективного количества соединения по пп.1-12.
18. Способ лечения по п.17, где упомянутые воспалительные заболевания и иммунные нарушения выбраны из группы воспалительные заболевания, расстройства и состояния, которые характеризуются или ассоциированы с нежелательной воспалительной иммунной реакцией, и все болезни и состояния, которые вызваны избыточной секрецией TNF-a и PDE4 или ассоциированы с ней.
19. Способ лечения по п.17, где упомянутые воспалительные заболевания и иммунные нарушения выбраны из группы астма, бронхиальная астма, хроническое обструктивное легочное заболевание, аллергический ринит, эозинофильная гранулема, нефрит, ревматоидный артрит, кистозный фиброз, хронический бронхит, рассеянный склероз, болезнь Крона, псориаз, крапивная лихорадка, весенний конъюнктивит у взрослых, синдром респираторного заболевания, ревматоидный спондилит, остеартрит, подагрический артрит, воспаление сосудистой оболочки глазного яблока (увеит), аллергический конъюнктивит, воспалительные заболевания кишечника, язвенный колит, экзема, атопический дерматит и хроническое воспаление, или названное воспалительное состояние представляет собой аллергическое воспалительное состояние, или воспалительное состояние выбрано из группы бронхиальная астма, нефрит и аллергический ринит.
20. Способ по п.17, где указанные воспалительные состояния и иммунные нарушения выбраны из группы воспалительные состояния и иммунные расстройства легких, суставов, глаз, кишечника, кожи и сердца.
21. Способ по п.17, где указанные заболевания центральной нервной системы выбраны из группы депрессия, амнезия, слабоумие, болезнь Альцгеймера, сердечная недостаточность, шок и цереброваскулярное заболевание.
22. Способ лечения астмы у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения по одному из пп.1-12.
23. Способ лечения хронического обструктивного заболевания легких у субъекта, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества соединения по одному из пп.1-12.
Текст
010634 Область техники Настоящая заявка испрашивает приоритет на основании индийской предварительной патентной заявки 363/MUM/2003, поданной 11 апреля 2003 года, и предварительной патентной заявки США 60/519967, поданной 13 ноября 2003 года. Обе приоритетные заявки включены в настоящую заявку во всей полноте через ссылки. Настоящее изобретение относится к новым гетероциклическим соединениям, их аналогам, таутомерам, региоизомерам, стереомерам, энантиомерам, диастереомерам, полиморфным формам, фармацевтически приемлемым солям и содержащим их фармацевтическим составам. Настоящее изобретение относится, в частности, к новым ингибиторам фосфодиэстеразы типа 4 (PDE4) формулы (1), их аналогам,таутомерам, региоизомерам, стереомерам, энантиомерам, диастереомерам, полиморфным формам, фармацевтически приемлемым солям и содержащим их фармацевтическим составам. Таким образом, изобретение относится к соединениям формулы (1) где R1 в каждом отдельном случае представляет собой водород или C1-C8 алкил, возможно замещенный одним или более галогеном;R4 представляет собой -NR5R6, где R5 и R6 могут быть одинаковыми или различными и независимо выбраны из группы, включающей: (i) водород или (ii) C1-C8 алкил, возможно замещенный -C(O)ORa;Ar предпочтительно выбран из группы возможно замещенный фенил, возможно замещенный бензил, возможно замещенный пиримидин, возможно замещенный пиридил, выбранный из 4-пиридил, 3-пиридил и 2-пиридил, или возможно замещенный пиридил-N-оксид, выбранный из 4-пиридил-N-оксид, 3 пиридил-N-оксид и 2-пиридил-N-оксид, возможно замещенных галогеном, таким как хлор; в каждом отдельном случае Ra независимо представляет собой водород или C1-C8 алкил;Rb представляет собой: (i) водород; (ii) C1-C8 алкил, возможно замещенный галогеном; (iii) C3-C5 циклоалкил; (iv) C5 гетероарил, (v) C6 гетероциклил, который содержит от 1 до 2 гетероатомов, выбираемых из кислорода и азота, и может быть замещен C1-C8 алкилом или гидрокси; или (vi) -C(O)O-Ra;Rc представляет собой водород, C1-C8 алкил, возможно замещенный галогеном, фенил или (С 3-С 5 циклоалкил)(С 1-С 8 алкил); в каждом отдельном случае Rd независимо представляет собой водород, C1-C8 алкил, возможно замещенный -C(O)O-Ra, или C3-C5 циклоалкил;X выбран из группы О, S и NCH3;q представляет собой 2; а также их фармацевтически приемлемым солям и N-оксидам. Настоящее изобретение относится также к способу получения вышеуказанных новых гетероциклических соединений формулы (1). Соединения общей формулы (1), в частности, снижают или ингибируют выработку TNF-, поскольку являются ингибиторами PDE4, вследствие чего полезны при лечении различных аллергических и воспалительных нарушений, включая астму, хронический бронхит, атопический дерматит, крапивную лихорадку,аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильную гранулему,псориаз, ревматоидный артрит, септический шок, язвенный колит, болезнь Крона, реперфузное поражение миокарда и реперфузное поражение мозга, хронический гломерулонефрит, эндотоксический шок и синдром дыхательной недостаточности у взрослых. Соединения согласно настоящему изобретению, в частности, полезны при лечении астмы или хронического обструктивного легочного заболевания (COPD). Уровень техники Воспаление дыхательных путей возникает при некоторых серьезных легочных заболеваниях, таких-1 010634 как астма и хроническое обструктивное легочное заболевание (COPD). Среди причин, приводящих к обструкции дыхательных путей, - отек стенок дыхательных путей, инфильтрация клеток воспалительного очага в легкое, выработка различных медиаторов воспаления и повышенное отделение слизи. Дыхательные пути больных астмой инфильтрированы воспалительными лейкоцитами, среди которых преобладают эозинофилы. Сила астматических реакций прямо зависит от количества присутствующих в легких эозинофилов. Обнаружено, что в легких пациентов, больных астмой, значительно повышается количество эозинофилов; в то время как в легких здорового человека их совсем немного. Эозинофилы способствуют лизису или активации клеток и разрушают ткани. Активированные эозинофилы вырабатывают воспалительные цитокины, такие как IL-1, IL-3, TNF- и медиаторы воспаления, такие как PAF, LTD4 и соответствующие соединения кислорода, которые могут вызывать отеки и бронхостеноз. Известно, что в патогенезе ряда аутоиммунных и воспалительных заболеваний участвует фактор некроза опухоли (TNF-). Следовательно, путем воздействия на выработку сигнальных цитокинов или пути биосинтеза, связанные с этими белками, можно обеспечить дополнительный терапевтический эффект при лечении таких болезненных состояний. Убедительно показано, что выработка TNF- в провоспалительных клетках снижается при повышении внутриклеточной концентрации циклического аденозин 3'5'-монофосфата (сАМР). Этот вторичный мессенджер (посредник в передаче сигналов) регулируется ферментами семейства фосфодиэстеразы (PDE). Ферменты фосфодиэстеразы играют существенную роль в работе клеточных сигнальных механизмов через гидролиз сАМР и cGP в неактивные 5'-формы. Таким образом, ингибирование ферментов PDE проявляется в снижении уровня сАМР и/или cGP и изменяет внутриклеточную реакцию (внутриклеточный ответ) на внеклеточные сигналы посредством влияния на процессы, в которых медиаторами являются циклические нуклеотиды. Поскольку считается, что эозинофилы представляют собой основную провоспалительную мишень при астме, обнаружение экспрессии семейства генов PDE4 в эозинофилах привела к выбору PDE4 в качестве потенциальной терапевтической мишени при лечении астмы (Rogers, D.F., Giembycz, M.A., Trends Pharmacol. Sci., 19, 160-164 (1998); Barnes, P.J., Trends Pharmacol.Sci., 19, 415-423 (1998), включено во всей полноте через ссылку). Фосфодиэстеразы циклических нуклеотидов млекопитающих (PDEs) подразделяются на десять семейств по последовательностям аминокислот и/или последовательности ДНК, специфике субстрата и чувствительности к воздействию фармакологических агентов (Soderling, S.H., Bayuga, S.J., and Beavo, J.A., Proc.Natl. Acad. Sci., USA, 96, 7071-7076 (1999); Fujishige, K., Kotera, J., Michibata, H., Yuasa, K., Takebayashi, Si,Okamura, K. and Omori, K., J. Biol. Chem., 274, 18438-18445 (1999), включено во всей полноте через ссылку). Многие типы клеток экспрессируют более чем один тип PDE, причем распределение изоферментов от клетки к клетке варьирует значительно. Поэтому разработка высокоселективных ингибиторов изоферментов PDE предоставляет уникальную возможность для выборочного воздействия на различные патофизиологические процессы. Фосфодиэстераза типа 4 (PDE4) представляет собой фермент, регулирующий активность клеток,приводящую к воспалению легких. PDE4, сАМР-специфичный и Са+2-независимый фермент, является ключевым изоферментом в процессе гидролиза сАМР в мастоцитах, базофилах, эозинофилах, моноцитах и лимфоцитах. Обнаружение связи между увеличением количества сАМР в клетках очага воспаления и расслаблением гладкой мускулатуры дыхательных путей и ингибированием выработки медиатора привело к возникновению общей заинтересованности в разработке ингибиторов PDE4 (см. статью Trophy, T.J.,Am. J. Respir. Crit. Care Med., 157, 351-370 (1998), которая включена во всей полноте через ссылку). Считается, что развитие или осложнение некоторых нежелательных физиологических состояний, а именно ряда заболеваний, включая остеоартрит и другие артритные состояния, септический шок, синдром респираторного заболевания и болезни, связанные с резорбцией кости, является следствием избыточной или нерегулируемой продукции TNF-; поскольку TNF- также играет определенную роль в возникновении и прогрессировании аутоиммунных заболеваний, ингибиторы PDE4 могут найти применение в качестве терапевтических агентов при ревматоидном артрите, рассеянном склерозе и болезни Крона (Nature Medicine,1, 211-214 (1995) and ibid., 244-248, включено во всей полноте через ссылку). Повышенный интерес к препаратам, способным к селективному ингибированию PDE4, возник по нескольким причинам. Распределение PDE4 в тканях позволяет предположить, что на патологии, связанные с центральной нервной и иммунной системой, можно воздействовать с помощью селективных ингибиторов PDE4. Кроме того, повышение внутриклеточной концентрации сАМР, очевидного биохимического последствия ингибирования PDE4, хорошо известно для иммунокомпетентных клеток, где оно служит сигналом деактивации. Недавно семейство PDE4 расширилось за счет появления четырех подтипов - от PDE4A до PDE4D,каждый из них кодируется отдельным геном (British Journal of Pharmacology; 1999; v.128; p.1393-1398,включено во всей полноте через ссылку). Показано, что повышение уровня сАМР в таких клетках проявляется в подавлении клеточной активации, которая, в свою очередь, ингибирует продукцию и выделение провоспалительных цитокинов, таких как TNF-. Поскольку считают, что эозинофилы представляют собой важнейшую провоспалитель-2 010634 ную мишень при астме, идентификация экспрессии семейства генов PDE4 в эозинофилах выявила PDE4 как потенциальную терапевтическую мишень при лечении астмы. Применение некоторых ингибиторов PDE4, к сожалению, ограничено из-за ряда нежелательных,побочных эффектов, таких как тошнота и рвота (из-за воздействия PDE4 на центральную нервную систему) и повышение желудочной кислотной секреции (из-за воздействия PDE4 на париетальные клетки кишечника) (Barnette, M.S., Grous, M., Cieslinsky, L.B., Burman, M., Christensen, S.B., Trophy, T.J., J. Pharmacol.Exp. Ther., 273, 1396-1402 (1995), включено во всей полноте через ссылку). Один из первых препаратовингибиторов PDE4, Rolipram, был снят с клинических испытаний из-за возникновения ряда неприемлемых тяжелых побочных эффектов (Zeller E. et al., Pharmacopsychiatry 17, 188-190 (1984), включено во всей полноте через ссылку). Причина возникновения тяжелых побочных эффектов при использовании некоторых ингибиторовPDE4 в ходе клинических испытаний на человеческом организме выяснилась недавно. Существует два сайта связывания PDE4 млекопитающих, в которых молекулы ингибитора могут образовывать связь. Кроме того, PDE4 существует в двух различных формах, представляющих разные конформации. Их определяют как сайт связывания PDE4H высокого сродства к Rolipram и сайт связыванияPDE4L низкого сродства к Rolipram (Jacobitz, S., McLaughlin, M.M., Livi, G.P., Burman, M., Trophy, T.J.,Mol. Pharmacol., 50, 891-899 (1996), включено во всей полноте через ссылку). Показано, что некоторые нежелательные побочные эффекты (тошнота и желудочная кислотная секреция) связаны с PDE4H-ингибированием, в то время как некоторый благоприятный эффект ассоциировали с PDE4L ингибированием. Было обнаружено, что рекомбинантная PDE4 человека существует в четырех изоформах А, В, С и D(Muller, Т., Engels, P., Fozard, J.R., Trends Pharmacol. Sci., 17, 294-298 (1996), включено во всей полноте через ссылку). Соответственно, обнаружено, что соединения, проявляющие большую селективность в отношении изофермента PDE4D, вызывают меньше побочных эффектов, чем Rolipram (Hughes, В. et al.,Br. J. Pharmacol. 1996, 118, 1183-1191, включено во всей полноте через ссылку). Поэтому селективные ингибиторы изоферментов PDE4 могут иметь терапевтический эффект при лечении воспалительных заболеваний, таких как астма и другие респираторные заболевания. Несмотря на то, что по всему миру работает несколько групп исследователей, занимающихся поиском высоко селективных ингибиторов изоферментов PDE4, до сих пор им удалось добиться лишь ограниченного успеха. Ингибирующее воздействие на PDE4 проявляют многие соединения.Bay-19-8004 от Bayer формулы Е находятся на продвинутых этапах клинических испытаний на человеческом организме. Другие соединения, проявившие выраженное PDE4 ингибирующее действие, включаютCDP-840 от Celltech формулы В, D-4418 от Schering Plough формулы С, 5 СР-220,629 от Pfizer формулы F,PD-168787 от Parke Davis формулы G и Filaminast от Wyeth формулы Н. Однако недавно из-за проблем,связанных с эффективностью и побочными эффектами, Ariflo, CDP-840 и Bay-19-8004 в качестве средств лечения астмы были сняты с клинических испытаний. Другие соединения С и F в настоящее время проходят первую фазу клинических испытаний. В ходе исследования, целью которого являлось исследование новых антиастматических соединений, обладающих потенциальной PDE4 ингибирующей активностью, мы зарегистрировали заявку на патент WTO под 922/MUM/2002 от 23 октября 2002 года и заявкуPCT/IB03/04442 от 4 октября 2003 года, которые включены здесь во всей полноте через ссылки, на новые серии трициклических со-3 010634 единений, полезных при лечении нарушений воспалительной или аллергической природы. Краткое описание изобретения Соответственно, настоящее изобретение предлагает новые гетероциклические соединения общей формулы где в каждом отдельном случае R1 представляет собой водород или C1-C8 алкил, возможно замещенный одним или более галогеном;R4 представляет собой -NR5R6, где R5 и R6 могут быть одинаковыми или различными и независимо выбраны из группы, включающей: (i) водород; (ii) C1-C8 алкил, возможно замещенный -C(O)ORa; (iii) -C(O)-Rb;Ar предпочтительно выбран из группы возможно замещенный фенил, возможно замещенный бензил, возможно замещенный пиримидин, возможно замещенный пиридил, выбранный из 4-пиридил, 3-пиридил и 2-пиридил, или возможно замещенный пиридил-N-оксид, выбранный из 4-пиридил-N-оксид, 3 пиридил-N-оксид и 2-пиридил-N-оксид, возможно замещенных галогеном, таким как хлор; в каждом отдельном случае Ra независимо представляет собой водород или C1-C8 алкил;Rb представляет собой: (i) водород; (ii) C1-C8 алкил, возможно замещенный галогеном; (iii) С 3-С 5 циклоалкил; (iv) C5 гетероарил; (v) C6 гетероциклил, который содержит от 1 до 2 гетероатомов, выбираемых из кислорода и азота, и может быть замещен C1-C8 алкилом или гидрокси; или (vi) -C(O)O-Ra;Rc представляет собой водород, C1-C8 алкил, возможно замещенный галогеном, фенил или (С 3-С 5 циклоалкил)(С 1-С 8 алкил); в каждом отдельном случае Rd независимо представляет собой водород, C1-C8 алкил, возможно замещенный -C(O)O-Ra, или С 3-С 5 циклоалкил;X выбран из группы О, S и NCH3;q представляет собой 2; а также их фармацевтически приемлемые соли и N-оксиды. Настоящее изобретение относится также к способу получения вышеуказанных новых гетероциклических соединений формулы (1). Соединения общей формулы (1), в частности, снижают или ингибируют выработку TNF-, поскольку являются ингибиторами PDE4, вследствие чего могут применяться для лечения заболевания и состояний, которые связаны или ассоциированы с избыточной нежелательной секрецией TNF- или PDE4. К таким заболеваниям относятся различные аллергические и воспалительные заболевания и иммунные нарушения, а также заболевания центральной нервной системы и инсулинустойчивые формы диабета. Воспалительные заболевания и имунные нарушения предпочтительно выбраны из группы, включающей астму, хронический бронхит, атопический дерматит, крапивную лихорадку, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, эозинофильную гранулему, псориаз, ревматоидный артрит, септический шок, язвенный колит, болезнь Крона, реперфузное поражение миокарда и реперфузное поражение мозга, хронический гломерулонефрит, эндотоксический шок и синдром дыхательной недостаточности у взрослых. Соединения согласно настоящему изобретению, в частности, полезны при лечении астмы или хронического обструктивного легочного заболевания (COPD). Воспалительные состояния и иммунные нарушения предпочтительно выбраны из группы воспалительных состояний и иммунных расстройств легких, суставов, глаз, кишечника, кожи и сердца. Заболевания центральной нервной системы согласно настоящему изобретению предпочтительно выбраны из группы депрессия, амнезия, слабоумие, болезнь Альцгеймера, сердечная недостаточность, шок и цереброваскулярное заболевание. Далее предпочтительно соединение, где R1 представляет собой незамещенный алкил. Далее предпочтительно соединение, где R1 представляет собой метил.-4 010634 Далее предпочтительно соединение, где R1 представляет собой замещенный алкил. Далее предпочтительно соединение, где R1 представляет собой -CHF2. Далее предпочтительно соединение, где Р представляет собой О. Далее предпочтительно соединение, где X представляет собой О, N-CH3, S. Далее предпочтительно соединение, где Y представляет собой -C(O)NH. Далее предпочтительно соединение, где Ar выбран из группы замещенный или незамещенный 4-пиридил; замещенный или незамещенный 4-пиридил-N-оксид; замещенный или незамещенный 3-пиридил. Далее предпочтительно соединение, где указанный заместитель Ar представляет собой галоген. Далее предпочтительно соединение, где указанный заместитель Ar представляет собой хлор. Далее предпочтительно соединение, где Ar выбран из группы Настоящее изобретение особо исключает следующее соединение: Подробное описание изобретения Термин "алкил" относится к углеводородным радикалам, содержащим прямую или разветвленную углеводородную цепь, состоящую исключительно из атомов углерода и водорода, полностью насыщенным, содержащим от 1 до 8 атомов углерода, связанным с остальной молекулой простой связью. Примеры: метил, этил, н-пропил, 1-метилэтил (изопропил), н-бутил, н-пентил, 1,1-диметилэтил (t-бутил) и т.п. Термин "алкенил" относится к алифатическим углеводородным группам, содержащим двойную углерод-углеродную связь; углеродная цепь может быть прямой или разветвленной или иметь ответвление,содержащее от 2 до 10 атомов углерода. Примеры: этенил, 1-пропенил, 2-пропенил (аллил), изопропенил,2-метил-1-пропенил, 1-бутенил, 2-бутенил и т.п. Термин "алкинил" относится к углеводородным радикалам с прямой или разветвленной цепью, содержащим по меньшей одну тройную углерод-углеродную связь, с количеством атомов углерода от 2 до 12 (радикалы с количеством атомов углерода от 2 до 10 в данном случае предпочтительны). Примеры: этинил, пропинил, бутинил и т.п. Термин "алкоксил" обозначает описанную выше алкильную группу, присоединенную к остальной молекуле через кислородную связь. Репрезентативные примеры подобных групп: -OCH3, -ОС 2 Н 5 и т.п. Термин "алкилкарбонил" обозначает описанную выше алкильную группу, присоединенную к остальной молекуле через карбонильную связь. Репрезентативные примеры подобных групп: -С(O)CH3,-C(O)C2H5 и т.п. Термин "алкоксикарбонил" обозначает описанную выше алкоксильную группу, присоединенную к остальной молекуле через карбонильную связь. Репрезентативные примеры подобных групп: -C(O)-OCH3,-C(O)-OC2H5 и т.п. Термин "алкилкарбонилокси" обозначает описанную выше алкилкарбонильную группу, присоединенную к остальной молекуле через кислородную связь. Репрезентативные примеры подобных групп:-O-С(O)CH3, -O-С(O)C2H5 и т.п. Термин "алкиламино" обозначает описанную выше алкильную группу, присоединенную к остальной молекуле через аминную связь. Репрезентативные примеры подобных групп: -NH2CH3, -NH(CH3)2,-N(CH3)3 и т.п. Термин "циклоалкил" обозначает неароматическую моно или полициклическую кольцевую систему, содержащую приблизительно от 3 до 12 атомов углерода, например циклопропил, циклобутил, циклопентил, циклогексил; примеры мультициклических циклоалкильных групп включают пергидронафтил-, адамантил- и норборниловые группы, циклические группы, содержащие мостик, или спиробициклические группы, например спиро(4,4)-нон-2-ил. Термин "циклоалкилалкил" относится к циклическим радикалам, содержащим кольца, в состав которых входит приблизительно от 3 до 8 атомов углерода, прямо связанных с алкильной группой, в свою очередь, связанной с основной структурой через любой атом углерода алкильной группы, с образованием в результате стабильной структуры, такой как циклопропилметил, циклобутилэтил, циклопентилэтил и т.п. Термин "циклоалкенил" относится к циклическим радикалам, содержащим кольца, в состав которых входит приблизительно от 3 до 8 атомов углерода по меньшей мере с одной двойной углеродуглеродной связью, такие как циклопропенил, циклобутенил, циклопентенил и т.п. Термин "арил" относится к ароматическим радикалам, в состав которых входит приблизительно от 6 до 14 атомов углерода, таким как фенил, нафтил, тетрагидронафтил, инданил, бифенил и т.п. Термин "арилалкил" относится к описанной выше арильной группе, прямо связанной с алкильной-6 010634 группой, описанной выше. Примеры: -СН 2 С 6 Н 5, -C2H5C6H5 и т.п. Термин "гетероциклическое кольцо" относится к стабильному 3-15-членному кольцевому радикалу,состоящему из атомов углерода и от 1 до 5 гетероатомов, выбранных из группы азот, фосфор, кислород или сера. В рамках задач настоящего изобретения гетероциклический кольцевой радикал может представлять собой моноциклическую, бициклическую или трициклическую кольцевую систему, которая может включать конденсированную, содержащую мостик или спиралевидную кольцевую систему, а входящие в гетероциклический кольцевой радикал атомы азота, фосфора, углерода, кислорода или серы возможно окислены до различных степеней окисления. Кроме того, атом азота может быть кватернизирован; а кольцевой радикал может быть частично или полностью насыщенным (например, гетероароматическим или гетероариловым ароматическим). Примеры подобных гетероциклических кольцевых радикалов включают, без ограничения, азетинидил, акринидил, бензодиоксолил, бензодиоксанил, бензофуран, карбазолил, циннолинил, диоксоланил, индолизинил, нафтиридинил, пергидроазепинил, феназинил,фенотиазинил, феноксазинил, фталазинил, пиридил, птеридинил, пуринил, хиназолинил, хинооксалинил,хинолинил, изохинолинил, тетразоил, имидазолил, тетрагидроизоинолил, пиперидинил, пиперазинил, 2 оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксоазепинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолинил, оксазолидинил, триазолил, инданил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолинил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, изоиндолил, индолинил, изоиндолинил,октагидроиндолил, октагидроизоиндолил, хинолил, изохинолил, декагидроизохинолил, бензимидазолил,тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофуртил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиаморфолинил сульфоксид, тиаморфолинил сульфон, диоксафосфоланил, оксадиазолил, хроманил, изохроманил и т.п. Термин "гетероарил" относится к описанным выше гетероциклическим кольцевым радикалам. Гетероариловый кольцевой радикал может быть связан с основной структурой через любой из гетероатомов или через атом углерода, в результате чего образуется стабильная структура. Гетероциклический кольцевой радикал может быть связан с основной структурой через любой из гетероатомов или через атом углерода, в результате чего образуется стабильная структура. Термин "гетероарилалкил" относится к описанному выше гетероариловому кольцевому радикалу,прямо связанному с алкильной группой. Гетероарилалкильный радикал может быть связан с основной структурой через любой из атомов углерода алкильной группы, в результате чего образуется стабильная структура. Термин "гетероциклил" относится к описанному выше гетероциклическому кольцевому радикалу. Гетероциклический кольцевой радикал может быть связан с основной структурой через любой из гетероатомов или атомов углерода, в результате чего образуется стабильная структура. Термин "гетероциклилалкил" относится к описанному выше гетероциклическому кольцевому радикалу, прямо связанному с алкильной группой. Гетероциклилалкильный радикал может быть связан с основной структурой через атом углерода алкильной группы, в результате чего образуется стабильная структура. Термин "циклическое кольцо" относится к циклической группе, содержащей 3-10 атомов углерода. Термин "защитная группа" относится к карбобензилокси (CBZ) или трет-бутилоксикарбонилу(ВОС) и т.п. Термин "галоген" относится к фтор-, хлор-, бром- и иод-радикалам. Частью настоящего изобретения являются фармацевтически приемлемые соли неорганических оснований, таких как Li, Na, K, Са, Mg, Fe, Cu, Zn, Mn; органических оснований, таких как N,N'-диацетилэтилендиамин, глюкамин, триэтиламин, холин, гидроксид, дициклогесиламин, метформин, бензиламин,триалкиламин, тиамин и т.п.; хиральные основания, такие как алкилфениламин, глицинол, фенилглицинол и т.п., соли природных аминокислот, таких как глицин, аланин, валин, лейцин, изолейцин, норлейцин,тирозин, цистин, цистеин, метионин, пролин, гидроксипролин, гистидин, омитин, лизин, аргинин, серин и т.п.; четвертичные аммонийные соли соединений по настоящему изобретению и алкилгалидов, алкилсульфаты, такие как MeI, (Me)2SO4 и т.п.; неприродные аминокислоты, такие как D-изомеры или замещенные аминокислоты; гуанидин, замещенный гуанидин, в котором заместители выбраны из группы нитро, амино, алкил, алкенил, алкинил, аммоний или замещенные соли аммония и соли алюминия. Там,где это возможно, соли могут представлять собой кислые соли, например сульфаты, нитраты, фосфаты,перхлораты, бораты, гидрогалиды, ацетаты, тартраты, малеаты, цитраты, сукцинаты, пальмоаты, метансульфонаты, бензоаты, салицилаты, бензосульфонаты, аскорбаты, глицерофосфаты, кетоглутараты и т.п. Фармацевтически приемлемые сольваты могут представлять собой гидраты или содержать молекулы других растворителей, например спиртов. Еще одним объектом изобретения является способ лечения воспалительных заболеваний и состояний, которые характеризуются или ассоциированы с нежелательными иммунными воспалительными реакциями, а также всех заболеваний и состояний, вызванных избыточной секрецией TNF- и PDE4 или связанных с ней, включающий введение в организм субъекта терапевтически эффективного количества соединения по формуле (1).-7 010634 Еще одним объектом настоящего изобретения является способ лечения воспалительных состояний и иммунных нарушений у субъекта, нуждающегося в лечении, включающий введение в организм названного субъекта терапевтически эффективного количества соединения по формуле (1). Воспалительные заболевания и иммунные нарушения предпочтительно выбирают из группы астма,бронхиальная астма, хроническое обструктивное легочное заболевание, аллергический ринит, эозинофильная гранулема, нефрит, ревматоидный артрит, кистозный фиброз, хронический бронхит, рассеянный склероз, болезнь Крона, псориаз, крапивная лихорадка, весенний (vernal) конъюнктивит у взрослых, синдром респираторного заболевания, ревматоидный спондилит, остеоартрит, подагрический артрит, воспаление сосудистой оболочки глазного яблока, аллергический конъюнктивит, воспалительные заболевания кишечника, язвенный колит, экзема, атопический дерматит и хроническое воспаление. Более предпочтительными являются аллергические воспалительные состояния. Более предпочтительными являются воспалительные состояния и иммунные нарушения, выбранные из группы воспалительные состояния и иммунные расстройства легких, суставов, глаз, кишечника,кожи и сердца. Более предпочтительными являются воспалительные состояния, выбраные из группы бронхиальная астма, нефрит и аллергический ринит. Еще одним объектом настоящего изобретения является способ уменьшения воспаления в поврежденном органе или ткани, включающий доставку к названному органу или ткани терапевтически эффективного количества соединения, представленного соединением по формуле (1). Еще одним объектом настоящего изобретения является способ лечения заболеваний центральной нервной системы у субъекта, нуждающегося в лечении, включающий введение в организм названного субъекта терапевтически эффективного количества соединения по формуле (1). Предпочтительными являются заболевания центральной нервной системы, которые выбирают из группы депрессия, амнезия, слабоумие, болезнь Альцгеймера, сердечная недостаточность, шок и цереброваскулярное заболевание. Еще одним объектом изобретения является способ лечения инсулинрезистентного диабета у субъекта, нуждающегося в лечении, включающий введение в организм названного субъекта терапевтически эффективного количества соединения по формуле (1). Термин "лечение" состояния включает:(1) предотвращение или замедление проявления клинических симптомов состояния, нарушения или заболевания, развивающихся в организме млекопитающего, возможно находящегося или предрасположенного к состоянию, нарушению или заболеванию, в стадии, когда еще не проявились клинические или субклинические симптомы состояния, нарушения или заболевания;(2) облегчение состояния, нарушения или заболевания, т.е. предотвращение или замедление развития заболевания или по меньшей мере одного из его клинических или субклинических симптомов; или(3) облегчение течения заболевания, т.е. появление регрессии состояния, нарушения или заболевания или по меньшей мере одного из его клинических и субклинических симптомов. Польза для субъекта, получающего лечение, либо оценивается статистически как значительная,или, по меньшей мере, признается таковой пациентом или лечащим врачом. Термин "терапевтически эффективное количество" обозначает достаточное количество соединения,которое, будучи введенным в организм млекопитающего для лечения состояния, нарушения или заболевания, поддерживает данное лечение. "Терапевтически эффективное количество" варьирует в зависимости от соединения, заболевания и его тяжести, а также возраста, веса, физического состояния и реакции организма млекопитающего, получающего лечение. Четырьмя классическими симптомами острого воспаления являются покраснение, повышенная температура, отек и боль в пораженной области, а также нарушение функционирования пораженного органа. Симптомы и признаки воспаления, связанного с определенными состояниями, включают: ревматоидный артрит - боль, отечность, повышенная температура и чувствительность в пораженных суставах; общая или утренняя ригидность; инсулинзависимый диабет - mellitus-insulitis; это заболевание может приводить к множеству осложнений с воспалительной составляющей, включая ретинопатию, невропатию, нефропатию; заболевание коронарной артерии, периферическое васкулярное заболевание и цереброваскулярное заболевание; аутоиммунный тироидит - слабость, запор, одышка, отечность лица, рук и ступней, периферический отек, брадикардия; рассеянный склероз - спастичность, нечеткое зрение, головокружение, слабость в конечностях, парестезия; увеальный ретинит - ухудшение ночного зрения, потеря периферического зрения; красная волчанка - боль в суставах, сыпь, светочувствительность, лихорадка, боль в мышцах, отечность рук и ступней, расстройства мочевыделения (гематурия, цилиндурия, протеинурия), гломерулонефрит, когнитивное расстройство, тромбоз сосудов, перикардит; склеродерма - болезнь Рэйнода (Raynaud); припухлость кистей рук, верхних и нижних конечностей-8 010634 и лица; утолщение кожи; боль, отеки и припухлость пальцев и коленей, желудочно-кишечное расстройство, рестриктивный легочный процесс; перикардит; почечная недостаточность; другие артритные состояния с воспалительной составляющей, например ревматоидный спондилит,остеоартрит, септический артрит и полиартрит - лихорадка, боль, отечность, чувствительность; другие воспалительные заболевания мозга, такие как менингит, болезнь Альцгеймера, ассоциированный со СПИД синдром деменция энцефалит-фотофобия, когнитивное расстройство, потеря памяти; другие воспалительные заболевания глаз, например ретинит - снижение остроты зрения; воспалительные кожные нарушения, например экзема, другие дерматиты (например, атонический,контактный), псориаз, ожоги от ультрафиолетового излучения (солнечные лучи и другие источники УФизлучения) - эритема, боль, шелушение, отек, чувствительность; воспалительные заболевания кишечника, такие как болезнь Крона, язвенный колит - боль, диарея,запор, аноректальное кровотечение, лихорадка, артрит, астма - одышка, свистящее дыхание; другие аллергические расстройства, такие как аллергический ринит - чихание, зуд, насморк; состояния, связанные с острой травмой, такие как повреждения мозга вследствие ушиба - сенсорные нарушения, моторные нарушения, потеря сознания; повреждение сердечной ткани вследствие миокардиальной ишемии - боль, одышка; поражения легких, подобные возникающим при синдроме респираторного заболевания у взрослых одышка, гипервентиляция, снижение оксигенации, легочные инфильтраты; воспаление, сопровождающее инфекцию, такую как сепсис, септический шок, синдром токсического шока - лихорадка, респираторная недостаточность, тахикардия, гипотензия, лейкоцитоз; другие воспалительные состояния в отдельных органах или тканях, такие как нефрит (например,гломерулонефрит) - олигурия, нарушение мочевыделения; воспаление аппендикса - лихорадка, боль, чувствительность, лейкоцитоз; подагра - боль, чувствительность, припухлость и покраснение вокруг пораженного сустава, повышение количества сыворотки и/или мочевой кислоты в моче; воспаление желчного пузыря - боль и чувствительность в области живота, тошнота, лихорадка, лейкоцитоз; хроническое обструктивное легочное заболевание - одышка, свистящее дыхание; застойная сердечная недостаточность - одышка, влажные хрипы, периферический отек; диабет II типаприводит, в конце концов, к осложнениям во внутренних органах, включая возникновение кардиоваскулярных, глазных, почечных и периферических васкулярных заболеваний; легочный фиброз - гипервентиляция, одышка, снижение оксигенации; васкулярные заболевания, такие как атеросклероз и restenosis - боль, утрата чувствительности, ослабление пульса, нарушение функции и аллоиммунитет, приводящий к отторжению трансплантанта боль, чувствительность, лихорадка. Субклинические симптомы включают без ограничения диагностические маркеры воспаления, появление которых может предшествовать манифестации клинических симптомов. Один класс субклинических симптомов представляют иммунологические симптомы, такие как инвазия или аккумуляция в органе или ткани провоспалительных лимфоидных клеток, или местное или периферическое присутствие активированных провоспалительных лимфоидных клеток, распознающих (специфичных к) патоген или антиген, характерный для органа или ткани. Активацию клеток можно измерить способами, известными специалистам в данной области."Доставка" терапевтически эффективного количества активного ингредиента в конкретную часть организма пациента означает создание терапевтически эффективной концентрации активного ингредиента в крови пациента указанной части организма. Этого можно достичь, например, путем местного или системного введения активного ингредиента в организм пациента."Субъект" или "пациент" - термины, относящиеся к млекопитающим животным, предпочтительно к человеку. Отдельные репрезентативные соединения по настоящему изобретению представлены ниже, однако,они не ограничивают настоящего изобретения. 1. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамид. 2. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-(N,N-диметилсульфонамид)дибензо[b,d]фуран-1-карбоксамид. 3. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-(этансульфонамид)дибензо[b,d]фуран-1-карбоксамид. 4. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид. 5. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-(3-хлорпропилкарбоксамид)дибензо[b,d]фуран-1-карбоксамид. 6. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-этилкарбоксамиддибензо[b,d]фуран-1-карбоксамид. 7. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-t-бутилкабоксамиддибензо[b,d]фуран-1-карбоксамид. 8. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-этоксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид. 9. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид. 10. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо- 10010634 56. N-(3,5-Дихлорпирид-4-ил)-1-метокси-9-метил-6-этансульфонамид-9H-4-карбазолкарбоксамид. 57. N-(3,5-Дихлорпирид-4-ил)-1-метокси-9-метил-6-пропионамид-9H-4-карбазолкарбоксамид. 58. Двунатриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d] фуран-1-карбоксамида. 59. N-(3,5-Дихлорпирид-4-ил)-1-метокси-6-ацетамиддибензо[b,d]тиофен-4-карбоксамид. 60. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-ацетамиддибензо[b,d]фуран-1 карбоксамида. 61. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-фур-2-ил-карбоксамиддибензо[b,d] фуран-4-карбоксамида,а также фармацевтически приемлемые соли вышеупомянутых соединений. Соединения по настоящему изобретению можно приготовить следующими способами. Используемые в нижеприведенных формулах символы Р, Ar, X, Y, R1, R2, R3 и R4 представляют группы из описания к формуле (1), если не указано иное. Настоящее изобретение описывает процесс приготовления соединений общей формулы (1). По одному примеру реализации желательные соединения формулы (1), в которых Y представляет собой CONR7; R4 представляет собой -NR5R6; Р, Ar, X, Y, R1, R2, R3, R4, R5, R6, R7, m и n принимают значения, указанные в общем описании изобретения, можно синтезировать из общего полупродукта формулы (16). Общий полупродукт формулы (16) можно синтезировать по общей методике, описанной в схеме синтеза I. Схема синтеза I Согласно вышеописанной схеме синтеза соединение общей формулы (10), в котором Z представляет собой галоген, предпочтительно фтор, вступает во взаимодействие с соединением общей формулы(11), где W представляет собой галоген, предпочтительно бром или иод, в щелочной среде (калиевые соли в DMF или DMSO, NaH в DMF или DMSO и т.п.), в результате образуется полупродукт общей формулы (12). Полупродукт общей формулы (12) образует дополнительный цикл в ходе реакции с участием соединения металла или при образовании связи в присутствии металлического катализатора (ацетат палладия в ДМФ или ледяной уксусной кислоте, никелевый катализатор в пиридине или ДМФ, тетракистрифенилфосфин палладия в ДМФ и т.п.), предпочтительно ацетат палладия в ДМФ; при этом образуется трициклический полупродукт (13). Трициклический полупродукт общей формулы (13) далее окисляют до образования полупродукта общей формулы (14) при помощи стандартных способов (хлорит натрия или перманганат калия и т.п.), описанных в литературе. Полупродукт общей формулы (14) далее преобразуют в полупродукт общей формулы (15), где Y представляет собой -CONR7, приведением во взаимодействие соответствующим образом активированного полупродукта карбоновой кислоты (кислого галида, или смешанного ангидрида, или активного сложного эфира) общей формулы (14) с возможно замещенным арил- или гетороариламином (ArNHR7) в щелочной среде (NaH в ДМФ, диизопропиламино, или триэтиламин, или пиридин в THF и т.п.), способ описан в литературе. Полупродукт общей формулы (15) далее восстанавливают стандартными способами (никель Ранея/гидразин, железо/хлорид аммония, гидрогенизация в присутствии Pd/C и т.п.), описанными в литературе, до полупродукта общей формулы (16).- 11010634 Полупродукт общей формулы (16) преобразуют в желательное соединение общей формулы (1), где Полученные желательные соединения общей формулы (1) далее преобразуют в соответствующие соли и/или N-оксиды и, по желанию, преобразуют полученные соли соединений формулы (1) в свободные формы. По другому примеру реализации желательные соединения формулы (1), где Y представляет собой -CONR7; 4R представляет собой -NR5R6; Р, Ar, X, Y, R1, R2, R3, R4, R5, R6, R7, m и n принимают значения, указанные в общем описании изобретения, можно синтезировать из общего полупродукта формулы (16). Общий полупродукт формулы (16) можно синтезировать способом, описанным в схеме синтеза II. Схема синтеза II Согласно вышеописанной схеме соединение общей формулы (17), где Z представляет собой галоген, предпочтительно фтор; W также представляет собой галоген, предпочтительно бром или иод; вступает во взаимодействие с соединением общей формулы (18), где FG представляет собой СНО, СОСН 3,CN или -COORa, в щелочной среде (калиевые соли в ДМФ или ДМСО, NaH в ДМФ или ДМСО и т.п.); в результате образуется полупродукт общей формулы (19). Полупродукт общей формулы (19) образует дополнительный цикл в ходе реакции с участием соединения металла или при образовании связи в присутствии металлического катализатора (хлорид никеля, ацетат палладия и т.п.), предпочтительно ацетат палладия; при этом образуется трициклический полупродукт (20). Трициклический полупродукт общей формулы (20) далее окисляют при помощи KMnO4, NaOCl2 и т.п., если FG представляет собой СНО илиCOCH3; или подвергают гидролизу в присутствии NaOH или H2SO4, если FG представляет собой CN или-COORa, до полупродукта общей формулы (14); способ описан в литературе. Полупродукт общей формулы (14) далее преобразуют в полупродукт общей формулы (15), где Y представляет собой -CONR7,через взаимодействие соответствующим образом активированного полупродукта карбоновой кислоты(кислого галида, или смешанного ангидрида, или активного сложного эфира) общей формулы (14) с возможно замещенным арил- или гетороариламином (ArNHR7) в соответствующей щелочной среде, например NaH в ДМФ, или триэтиламин, или т.п., способ описан в литературе. Полупродукт общей формулы(15) далее восстанавливают стандартными способами (хлорид палладия или никель Ранея) до полупродукта общей формулы (16). Полупродукт общей формулы (16) преобразуют в желательное соединение общей формулы (1), где Y представляет собой -CONR7; R4 представляет собой -NR5R6; P, Ar, X, Y, R1, R2, R3, R4, R5, R6, R7, m и n принимают значения, указанные в общем описании изобретения, общепринятыми способами, описанными в литературе.- 12010634 Полученные желательные соединения общей формулы (1) далее преобразуют в соответствующие соли и/или N-оксиды через взаимодействие с m-хлорпербензойной кислотой или Н 2 О 2 и т.п.; далее, по желанию, полученные соли соединений формулы (1) преобразуют в свободные формы. По еще одному примеру реализации желательные соединения формулы (1), где Y представляет собой-CONR7; R4 представляет собой -NHCOCH3; n=1; Р, Ar, X, Y, R1, R2, R3, R4, R5, R6, R7 и m принимают значения, указанные в общем описании изобретения, можно синтезировать по схеме III. Далее, R4 там, где Согласно вышеупомянутой схеме соединение формулы (21), где Z представляет собой галоген,предпочтительно фтор, реагирует с соединением общей формулы (22) в щелочной среде (калиевые соли в ДМФ или ДМСО, NaH в ДМФ или ДМСО и т.п.); в результате образуется полупродукт общей формулы (23). Далее полупродукт общей формулы (23) восстанавливают до полупродукта общей формулы (24) при помощи стандартных восстановителей, таких как никель Ранея/гидразин или палладий на углероде в атмосфере водорода. Полупродукт общей формулы (24) преобразуют стандартными способами (оксид меди в 0,1N серной кислоте, медь в ДМСО) в трициклический полупродукт общей формулы (25) реакцией азосочетания. Ацетильную группу трициклического полупродукта общей формулы (25) далее преобразуют в ацетамидную группу через перегруппировку Бекмана; в результате образуется полупродукт общей формулы (26). Полупродукт общей формулы (26) формилируют стандартными способами, например приведением во взаимодействие с дихлорметилметиловым эфиром с хлоридом олова(IV) и т.п.; в результате образуется полупродукт общей формулы (27). Далее, полупродукт общей формулы (27) окисляют при помощи KMnO4 или NaOCl2 до полупродукта общей формулы (28) стандартными способами,описанными в литературе. Полупродукт общей формулы (28) преобразуют в желательное соединение общей формулы (1), где Y представляет собой -CONR7; R4 представляет собой -NHCOCH3; n=1; Р, Ar, X,Y, R1, R2, R3, R4, R5, R6, R7 и m принимают значения, указанные в общем описании, через взаимодействие с соответствующим образом активированным полупродуктом карбоновой кислоты (кислого галида, или смешанного ангидрида, или активного сложного эфира) общей формулы (28) с возможно замещенным арил- или гетороариламином (ArNHR7); способ описан в литературе. Далее, R4 там, где R4 представляет собой -NHCOCH3, можно преобразовать в -NR5R6 способами, описанными в литературе. Полученные желательные соединения общей формулы (1) далее преобразуют в соответствующие соли и/или N-оксиды; далее, по желанию, полученные соли соединений формулы (1) преобразуют в свободные формы. Способы получения N-оксидов хорошо известны специалистам, работающим в данной области, например через взаимодействие с m-перхлорбензойной кислотой в дихлорметане при комнатной температуре. Рядовому специалисту известны условия проведения данной реакции. Соединения согласно настоящему изобретению выделяют и очищают общепринятыми способами,например отгонкой растворителя в вакууме, рекристаллизацией осадка из соответствующего растворителя, или одним из традиционных методов очистки, например колоночной хроматографией на соответствующем носителе. Соли получают растворением свободного соединения в соответствующем растворителе, например в хлорированном углеводороде: метиленхлориде или хлороформе, или в низкомолекулярном алифатическом спирте (этанол, изопропанол), который содержит желательную кислоту или основание, или же к- 13010634 спирту потом добавляют желательную кислоту или основание. Соли получают фильтрацией, переосаждением, осаждением из жидкости, в которой они не растворимы, или испарением растворителя. Полученные соли можно преобразовать в свободные соединения реакцией с кислотой или основанием; их, в свою очередь, можно преобразовать в соли. В общем, эфирные растворители, применяемые в вышеописанном способе приготовления соединений по формуле (1), выбраны из следующих: диэтиловый эфир, 1,2-диметоксиэтан, тетрагидрофуран,диизопропиловый эфир, 1,4-диоксан и т.п. Применяемый хлорированный растворитель можно выбирать из следующих: дихлорметан, 1,2-дихлорэтан, хлороформ, четыреххлористый углерод и т.п. Применяемые ароматические растворители можно выбирать из бензола и толуола. Спиртовые растворители выбирают из следующих: метанол, этанол,n-пропанол, изопропанол, трет-бутанол и т.п. Апротонные растворители выбирают из N,N-диметилформамида, диметилсульфоксида и т.п. В общем, соединения, приготовленные по описанным выше методикам, получают в чистом виде благодаря применению хорошо известных способов очистки, таких как кристаллизация с применением таких растворителей, как пентан, диэтиловый эфир, изопропиловый эфир, хлороформ, дихлорметан,этилацетат, ацетон, метанол, этанол, изопропанол, вода или их сочетания; или колоночная хроматография на алюмо- или силикагеле с применением в качестве элюента таких растворителей, как гексан, петролейный эфир, хлороформ, этилацетат, ацетон, метанол или их сочетания. Различные полиморфные модификации соединений общей формулы (1), составляющие часть настоящего изобретения, можно приготовить кристаллизацией соединения формулы (1) в различных условиях, например с применением различных традиционных растворителей или их смесей для рекристаллизации; кристаллизацией при различных температурой, с различными режимами охлаждения - от очень быстрого до очень медленного режима кристаллизации. Полиморфные модификации можно получать нагреванием или плавлением соединения с последующим постепенным или быстрым охлаждением. Наличие полиморфных модификаций определяют ЯМР-спектроскопией образца твердого вещества, ИКспектроскопией, дифференциальной сканирующей калориметрией, исследованием дифракции рентгеновских лучей на порошке или подобными способами. Настоящее изобретение предлагает новые гетероциклические соединения, их аналоги, таутомеры,региоизомеры, стереоизомеры, энантиомеры, диастереомеры, полиморфные модификации, их фармацевтически приемлемые соли, соответствующие N-оксиды и их фармацевтически приемлемые соли. Настоящее изобретение предлагает также фармацевтически приемлемые составы, содержащие вышеописанные соединения общей формулы (1), их производные, аналоги, таутомерные формы, стереоизомеры, полиморфные модификации, энантиомеры, диастереомеры, их фармацевтически приемлемые соли или их фармацевтически приемлемые сольваты в сочетании с традиционными носителями, используемыми в фармакологии, разбавителями и т.п. Фармацевтические составы по настоящему изобретению можно применять для лечения нарушений аллергической природы. Желательно, чтобы некоторые вышеописанные соединения общей формулы (1) по настоящему изобретению содержали 1 или более асимметрично замещенных атомов углерода. Присутствие 1 или более асимметричных центров в соединениях общей формулы (1) порождает большое количество стереоизомеров, и в каждом отдельном случае изобретение распространяется на все стереоизомеры, включая энантиомеры и диастереомеры, а также их смеси, включая рацемические смеси. Изобретение может включать геометрические изомеры Е и Z там, где это возможно в соединениях общей формулы (1), это относится как к отдельному изомеру, так и к смеси обоих изомеров. Фармацевтические составы можно готовить в традиционно применяемых формах, таких как таблетки, капсулы, порошки, сиропы, растворы, суспензии и т.п., они могут также содержать ароматизаторы, подсластители и т.п. в соответствующих твердых или жидких носителях или растворителях или же в соответствующей стерильной среде для приготовления растворов и суспензий для инъекций. Активные соединения формулы (1) должны присутствовать в таких фармацевтических составах в количестве, обеспечивающем введение необходимой дозы в вышеуказанном диапазоне. Таким образом, для перорального введения соединения формулы (1) можно сочетать с соответствующим твердым или жидким носителем или разбавителем, изготавливая капсулы, таблетки, порошки, сиропы, растворы, суспензии и т.п. Фармацевтические составы могут, по желанию, содержать дополнительные компоненты, такие как ароматизаторы, подсластители, наполнители и т.п. Для парентерального введения соединения формулы (1) можно сочетать со стерильными водными или органическими средами, изготавливая растворы и суспензии для инъекций. Например, растворы в кунжутном или арахисовом масле, водном пропиленгликоле и т.п. можно применять наряду с водными растворами водорастворимых фармацевтически приемлемых кислых или основных солей соединений формулы (1). Приготовленные таким образом растворы для инъекций можно вводить в организм внутривенно, внутрибрюшинно, подкожно, внутримышечно; для человека предпочтительно внутримышечное введение. Соединения можно вводить путем ингаляции в случаях, когда необходимо введение препарата в дыхательные пути. Приготовление лекарственных составов, содержащих данные соединения, имеет особое значение в случае препаратов для респираторной ингаляции там, где соединение формулы (1) вводят- 14010634 в организм в виде аэрозоля под давлением. Предпочтительно тонкое измельчение частиц соединения формулы (1) после гомогенизации, например, в лактозе, глюкозе, высших жирных кислотах, натриевой соли диоктилсульфоянтарной кислоты или, наиболее предпочтительно, в карбоксиметилцеллюлозе, чтобы большинство микрочастиц размером не превышали 5 мкм. Состав для ингаляций, аэрозоль можно смешивать с газом или жидкостью для распыления активной субстанции. Возможно применение ингалятора, пульверизатора или распылителя. Подобные устройства известны. См., например, Newman et al.,Thorax, 1985, 40: 61-675; Berenberg, M., J. Asthma USA, 1985, 22: 87-92; включено в данное изобретение во всей полноте через ссылки. Возможно также применение распылителя Bird'a. См. также патенты США 6402733; 6273086 и 6228346, включенные в данное изобретение во всей полноте через ссылки. Соединение структуры (I) предпочтительно применять для ингаляции в виде сухого порошка с тонкоизмельченными частицами. Дозирование соединений по настоящему изобретению возможно также при помощи дозирующего ингалятора по способу, описанному в патенте США 6131566, включенному в данное изобретение во всей полноте через ссылки. Кроме соединений по формуле (1) фармацевтические соединения по настоящему изобретению можно применять или вводить в организм одновременно с одним или несколькими препаратами, выбранными из других клинически полезных терапевтических агентов. Подробное описание изобретения дано в описанных ниже примерах, представленных исключительно в качестве иллюстраций, ни в коей мере не ограничивающих изобретения, сведенных в табл. 1, представленную ниже. Таблица 1 Общая структурная формула (1 А) При синтезе соединений, представляющих собой репрезентативные примеры по настоящему изобретению, применялись следующие полупродукты. Полупродукт 1. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-аминодибензо[b,d]фуран-1-карбоксамид. Этап 1. 2-Бромизованилин. Изованилин (5 мг, 0,033 моль) растворили в ледяной уксусной кислоте (30 мл). К вышеназванному раствору добавили безводный ацетат натрия (5,4 мг), затем порошок железа (0,15 мг). Систему тщательно продули азотом. К вышеназванной суспензии в течение 15 мин добавляли при перемешивании раствор брома (5,79 мг, 0,0362 моль) в ледяной уксусной кислоте (10 мл) при комнатной температуре. Затем реакционную смесь перемешивали при комнатной температуре в течение 45 мин. Реакционную смесь вылили в 2% водный раствор бисульфита натрия (200 мл) и перемешивали в течение 10 мин. Отфильтрованный осадок промыли водой (100 мл), высушили и получили в результате 3,5 мг 2-бромизованилина в виде белого порошка; т.пл.: 200-202 С.IR (KBr): 3233, 2990, 2891, 2844, 1669, 1593, 1564, 1494, 1463, 1286, 1238, 1205, 1019, 987, 805, 786 см-1. 1 Н NMR (300 МГц, CDCl3):3,99 (s, 3H), 6,13 (s, 1H), 6,89 (d, 1H, J=8,4 Гц), 7,55 (d, 1H, J=8,4 Гц),10,23 (s, 1H). Этап 2. 2-Бром-3-(р-нитрофенокси)-4-метоксибензальдегид. К суспензии фторида калия (1,89 мг, 0,0326 моль) в безводном ДМСО (10 мл) добавили раствор 2 бромизованилина (5,0 мг, 0,0217 моль) в ДМСО (10 мл). Раствор 4-фторнитробензола (5,0 мг, 0,0260 моль) в ДМСО (5 мл) добавили к вышеназванной суспензии и реакционную смесь перемешивали при 140 С в течение 4 ч. Реакционную смесь остудили до комнатной температуры и вылили содержимое в воду (150 мл) и проэкстрагировали этилацетатом (50 мл 3). Органические экстракты объединили и промыли 1N гидроксидом натрия (25 мл 2), водой и солевым раствором, затем высушили над безводным сульфатом натрия. Обезвоженный органический слой сконцентрировали под вакуумом и очистили осадок колоночной- 20010634 хроматографией с использованием в качестве элюента 20% смеси этилацетат-петролейный эфир; в результате получили 2-бром-3-(р-нитрофенокси)-4-метоксибензальдегид в виде твердого вещества бледножелтого цвета (5,0 мг); т.пл.: 132-140 С.IR (KBr): 3084, 2874, 1689, 1584, 1506, 1486, 1348, 1285, 1253, 1234, 1114, 1025, 848, 815, 747 см-1. 1 Н NMR (300 МГц, CDCl3):3,86 (s, 3H), 6,89 (d, 2H, J=7,2 Гц), 7,07 (d, 1H, J=9,0 Гц), 7,92 (d, 1H,J=8,4 Гц), 8,17 (d, 2H, J=9,0 Гц), 10,24 (s, 1H). Этап 3. 4-Метокси-8-нитро-1-формилдибензо[b,d]фуран. 2-Бром-3-(р-нитрофенокси)-4-метоксибензальдегид (3,5 мг, 0,0087 моль), безводный карбонат натрия (1,125 мг, 0,0106 моль) и ацетат палладия(II) (0,19 мг, 0,0008 моль) в диметилацетамиде (15 мл) нагрели и перемешивали в атмосфере азота при 170 С в течение 2 ч. Воду (90 мл) добавили к охлажденной реакционной смеси. Осажденное твердое вещество отфильтровали и промыли 5% соляной кислотой, затем водой. В результате получили продукт в виде твердого вещества желтого цвета (3,4 мг). Этап 4. 4-Метокси-8-нитродибензо[b,d]фуран-1-карбоновая кислота. 4-Метокси-8-нитро-1-формилдибензо[b,d]фуран (1,1 мг, 0,0034 моль) в ацетоне (5 мл) нагревали до 60-70 С в течение 10 мин. К вышеназванной суспензии в течение 10 мин добавляли по каплям горячий раствор перманганата калия (1,07 мг, 0,0068 моль) в смеси вода:ацетон (1:3) (15 мл). Реакционную смесь нагревали до 60-70 С в течение 10 мин, охладили до комнатной температуры, отфильтровали. Осадок промыли ацетоном и проэкстрагировали фильтрат 10% раствором гидроксида натрия. После подкисления, последующего фильтрования и промывания осадка получили 4-метокси-8-нитродибензо[b,d]фуран 1-карбоновую кислоту (0,6 мг) в виде твердого вещества белого цвета; т.пл.: 178 С (dec).H NMR (300 МГц, DMSO):4,08 (s, 3H), 7,36 (d, 1H, J=8,4 Гц), 7,8 (d, 1H, J=9,0 Гц), 8,07 (d, 1H,J=8,4 Гц), 8,44 (dd, 1H, J=9,0 Гц, 2,7 Гц), 9,79 (d, 1H, J=2,4 Гц). Этап 5 а. 4-Метокси-8-нитродибензо[b,d]фуран-1-карбоновой кислоты хлорангидрид. Суспензию 4-метокси-8-нитродибензо[b,d]фуран-1-карбоновой кислоты (150 мг, 0,52 ммоль) (по этапу 4) в смеси бензола (2 мл) и свежеперегнанного тионилхлорида (2 мл) нагревали с обратным холодильником в течение 4 ч. Избыток тионилхлорида отогнали под вакуумом, в результате получили соответствующий хлорангидрид, вступивший в реакцию на следующем этапе. Этап 5b. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид. К предварительно промытой суспензии гидрида натрия (52 мг, 2,5 экв., 1,3 ммоль, 60% масляная дисперсия) в ДМФ (2 мл) добавили по каплям раствор 4-амино-3,5-дихлопиридина (93 мг, 0,52 ммоль) в ДМФ (2 мл) при температуре -10 С. Предварительно охлажденный раствор вышеназванного хлорангидрида (0,52 ммоль) (по этапу 5 а) в ТГФ (2 мл) добавили в один прием к реакционной смеси и перемешивали содержимое при -10 С в течение 30 мин. Реакцию погасили добавлением солевого раствора, разбавили водой и отфильтровали, в результате образовалось неочищенное твердое вещество; после промывания этанолом получили N-(3,5-дихлорпирид-4-ил)-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид в виде белого твердого вещества (80 мг); т.пл.: 315-317 С.(800 мг, 1,85 ммоль) в метаноле позволили свободно стечь в вышеназванную реакционную смесь при нагревании с обратным холодильником. Реакционную смесь нагревали с обратным холодильником в течение 3 ч и отфильтровали в горячем виде. После испарения метанола осадок промыли водой и использовали его без дополнительной очистки при синтезе соединений по следующим примерам. Полупродукт 2. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-аминодибензо[b,d]фуран-1-карбоксамид. Этап 1. 4-Циклопентокси-3-гидроксибензальдегид. Суспензию 3,4-дигидроксибензальдегида (5,0 мг, 0,0362 моль), безводного карбоната калия (6,0 мг,0,0434 моль) и циклопентил бромида (6,5 мг, 0,0434 моль) в безводном ДМФ (50 мл) нагревали в течение 24 ч при температуре 80 С. Затем реакционную смесь охладили и разбавили водой (500 мл), подкислили 1N HCl и проэкстрагировали этилацетатом (3100 мл). Этилацетатный экстракт промыли 5% бикарбона- 21010634 том натрия, затем солевым раствором и высушили над безводным сульфатом натрия. После концентрирования из безводного экстракта выделился осадок, который очистили колоночной хроматографией на силикагеле с использованием в качестве элюента 10% этилацетат в петролейном эфире; в результате образовалось 5,0 мг названного продукта в виде белого твердого вещества; т.пл.: 87-89 С.H NMR (300 МГц, CDCl3):1,65-2,04 (m, 8H), 4,93 (m, 1H), 5,83 (s, 1H), 6,94 (d, 1H), 7,38-7,43 (m,2H), 9,82 (s, 1H). Этап 2. 2-Бром-4-циклопентокси-3-гидроксибензальдегид. 4-Циклопентокси-3-гидроксибензальдегид (1,0 мг, 4,84 ммоль) растворили в ледяной уксусной кислоте (20 мл). Добавили к названному раствору безводный ацетат натрия (0,8 мг, 9,7 ммоль), затем порошок железа (0,22 мг). Систему тщательно продули азотом. К названной суспензии при перемешивании за 15 мин добавили раствор брома (0,854 мг, 5,32 ммоль) в ледяной уксусной кислоте (10 мл) при 15 С. Реакционную смесь перемешивали при 15 С в течение 45 мин. Реакционную смесь вылили в 2% водный раствор бисульфита натрия (100 мл) и перемешивали в течение 15 мин. Осадок отфильтровали, промыли водой (100 мл) и высушили; в результате получили 800 мг 2-бром-4-циклопентокси-3-гидроксибензальдегида в виде белого порошка; т.пл.: 107-109 С. 1 Н NMR (300 МГц, CDCl3):1,66-2,03 (m, 8H), 4,92 (m, 1H), 6,15 (s, 1H), 6,90 (d, 1H), 7,54 (d, 1H),10,25 (s, 1H). Этап 3. 2-Бром-4-циклопентокси-3-(р-нитрофенокси)бензальдегид. К суспензии фторида калия (125 мг, 2,104 ммоль) в безводном ДМСО (2,5 мл) добавили при перемешивании раствор 2-бром-4-циклопентокси-3-гидроксибензальдегид (500 мг, 1,754 ммоль) в ДМСО (2,5 мл). К названной суспензии добавили раствор 4-фторнитробензола (500 мг, 2,631 ммоль) в ДМСО (2,5 мл). Реакционную смесь перемешивали при 140 С в течение 6 ч, затем охладили до комнатной температуры и вылили содержимое в воду (100 мл) и проэкстрагировали этилацетатом (50 мл 3). Органические экстракты объединили и промыли 1N гидроксидом натрия (25 мл 2), водой и солевым раствором, высушили над безводным сульфатом натрия. Безводный органический слой сконцентрировали под вакуумом; в результате получили 2-бром-3-(р-нитрофенокси)-4-метоксибензальдегид в виде бледно-желтого твердого вещества (500 мг); т.пл.: 115-117 С. 1H NMR (300 МГц, CDCl3):1,18-1,23 (m, 2H), 1,39-1,53 (m, 4H), 1,73-1,81 (m, 2H), 5,01 (m, 1H),7,09 (dd, 2H), 7,43 (d, 1H), 7,87 (d, 1H), 8,24 (dd, 2H), 10,13 (s, 1H). Этап 4. 4-Циклопентокси-8-нитро-1-формилдибензо[b,d]фуран. Полупродукт 2-бром-4-циклопентокси-3-(р-нитрофенокси)бензальдегид (500 мг, 1,09 ммоль), безводный карбонат натрия (150 мг, 1,325 ммоль) и ацетат палладия(II) (25 мг, 0,096 ммоль) в диметилформамиде (10 мл) нагрели и перемшивали в атмосфере азота при 130 С в течение 7 ч. К охлажденной реакционной смеси добавили воду (90 мл) и проэкстрагировали этилацетатом (225 мл). Объединенные органические экстракты промыли 5% соляной кислотой, затем водой и высушили над безводным сульфатом натрия; в результате получили продукт в виде желтого твердого вещества (200 мг); т.пл.: 230240 С. 1 Н NMR (300 МГц, DMSO):1,70 (m, 2 Н), 1,77-1,92 (m, 4 Н), 2,09 (m, 2 Н), 5,25 (m, 1 Н), 7,53 (d, 1H),8,05 (d, 1H), 8,14 (d, 1H), 8,51 (d, 1H), 9,80 (s, 1H), 10,14 (s, 1H),Этап 5. 4-Гидрокси-8-нитро-1-формилдибензо[b,d]фуран. 4-Циклопентокси-8-нитро-1-формилдибензо[b,d]фуран (200 мг, 0,530 моль) в течение 7-8 ч нагревали в HBr (47% в уксусной кислоте) (5 мл) в ледяной уксусной кислоте (10 мл) при 50 С. Реакционную массу вылили в ледяную воду (200 мл) и проэкстрагировали этилацетатом (350 мл). Объединенные органические экстракты промыли насыщенным раствором бикарбоната натрия, затем водой и высушили над безводным сульфатом натрия. После удаления органического растворителя иод вакуумом образовался неочищенный продукт в виде белого твердого вещества (150 мг). Неочищенное твердое вещество использовали далее в синтезе без дополнительной очистки; т.пл.: 270 С. 1(s, 1H), 11,92 (s, 1H). Этап 6. 4-Дифторметокси-8-нитро-1-формилдибензо[b,d]фуран. Суспензию 4-гидрокси-8-нитро-1-формилдибензо[b,d]фурана (150 мг, 0,485 ммоль) и безводного карбоната калия (200 мг, 1,455 ммоль) в безводном ДМФ (5,0 мл) перемешивали в течение 10 мин при 80 С. Газообразный хлордифторметан пропускали через реакционную смесь в течение 45 мин. Реакционную смесь охладили, разбавили водой (50 мл) и проэкстрагировали этилацетатом (325 мл). Объединенный органический экстракт промыли водой и высушили над безводным сульфатом натрия. Удалили органический растворитель под вакуумом; в результате получили продукт в виде твердого вещества белого цвета (150 мг); т.пл.: 245-248 С. Этап 7. 4-Дифторметокси-8-нитродибензо[b,d]фуран-1-карбоновая кислота. 4-Дифторметокси-8-нитро-1-формилдибензо[b,d]фуран (150 мг, 0,48 ммоль) в ацетоне (20 мл) и воде (5 мл) нагревали в течение 10 мин до 60-70 С. К вышеназванному раствору добавили за 10 мин по- 22010634 каплям раствор перманганата калия (150 мг, 0,973 ммоль) в воде (5 мл). Реакционную смесь нагревали до 60-70 С в течение 30 мин, затем профильтровали в горячем виде через целитную подложку. Фильтрат подкислили, после чего произошло выпадение осадка, который отфильтровали и промыли водой, в результате получили 4-дифторметокси-8-нитродибензо[b,d]фуран-1-карбоновую кислоту (100 мг) в виде белого твердого вещества; т.пл.: 270 С. 1 Н NMR (300 МГц, DMSO):7,61 (t, 1H, J=72 Гц), 7,60 (d, 1H), 8,07 (d, 1H), 8,13 (d, 1H), 8,52 (d,1H), 9,77 (s, 1H), 13,80 (s, 1H). Этап 8. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-нитродибензо[b,d]фуран-1-карбоксамид. Раствор 4-дифторметокси-8-нитродибензо[b,d]фуран-1-карбоновой кислоты (100 мг, 0,30 ммоль) в смеси бензола (2 мл) и свежеперегнанного тионилхлорида (2 мл) нагревали до температуры рефлюкса в течение 4 ч. Избыток тионилхлорида удалили под вакуумом, в результате получили соответствующий тионилхлорид. К предварительно промытой суспензии гидрида натрия (25 мг, 60% масляная дисперсия) в ДМФ (3 мл) добавили по каплям раствор 4-амино-3,5-дихлорпиридина (53 мг, 0,30 ммоль) в ДМФ (2 мл) при -10 С. К реакционной смеси добавили предварительно охлажденный раствор вышеназванного хлорангидрида(0,30 ммоль) в ТГФ (5 мл), после чего реакционную массу перемешивали при -10 С в течение 30 мин. Реакцию погасили солевым раствором, разбавили водой и отфильтровали, образовавшийся неочищенный осадок, который очистили колоночной хроматографией на силикагеле с использованием в качестве элюента 10% ацетона в хлороформе; в результате получили 100 мг n-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-нитродибензо[b,d]фуран-1-карбоксамида в виде твердого вещества белого цвета; т.пл.: 270 С.(100 мг), метанола (10 мл) и 10% Pd/C (10 мг) гидрогенизировали при 60 psi в течение 12 ч. Реакционную смесь профильтровали через целитный слой, под разрежением удалили растворитель метанол; в результате получили N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-аминодибензо[b,d]фуран-1-карбоксамид в виде белого твердого вещества; т.пл.: 270 С. Этап 1. Метил-3-(2-бром-4-нитроанилин)-4-метоксибензоат. 3-Амино-4-метоксиметилбензоат (3,5 мг, 0,0193 моль) (технический) растворили в ДМФ (20 мл) и добавили к суспензии гидрида натрия (60% дисперсия) (1,54 мг, 0,0386 моль) в ДМФ (20 мл). Реакционную смесь перемешивали в атмосфере азота в течение 30 мин при комнатной температуре, затем при 0 С за 10 мин добавили раствор 3-бром-4-фторнитробензола (5,05 мг, 0,0231 моль) в ДМФ (20 мл). Реакционную массу перемешивали при комнатной температуре в течение 18 ч. Реакцию погасили солевым раствором и разбавили смесь ледяной водой (500 мл); образовавшийся осадок отфильтровали, высушили и получили неочищенный продукт. Продукт очистили колоночной хроматографией с использованием в качестве элюента 40% этилацетата в петролейном эфире; в результате получили чистый продукт в виде желтого твердого вещества.(2,96 мг, 0,0275 моль) и ацетат палладия(II) (1 мг, 0,0046 моль) в диметилформамиде (60 мл) нагревали и перемешивали в атмосфере азота при 140 С в течение 18 ч. Реакционную смесь охладили и профильтровали сквозь целитный слой. К охлажденной реакционной смеси добавили воду (90 мл). Образовавшийся осадок подкислили, отфильтровали и промыли водой. В результате получили продукт в виде твердого вещества желтого цвета (3,4 мг).H NMR (300 МГц, DMSO):3,99 (s, 3H), 4,11 (s, 3H), 7,24 (d, 1H, J=8,7 Гц), 7,67 (d, 1H, J=9,0 Гц),8,01 (d, 1H, J=9,0 Гц), 8,34 (dd, 1H, J=8,7 и 2,4 Гц), 9,95 (d, 1H, J=2,4 Гц), 12,57 (s, 1H). Этап 3. Метил-1-метокси-9-метил-6-нитро-9H-4-карбазолкарбоксилат. К суспензии гидрида натрия (104 мг, 0,0052 моль) добавили за 10 мин метил-1-метокси-6-нитро-9H4-карбазолкарбоксилат (1 мг, 0,0026 моль), растворенный в ДМФ (10 мл) при комнатной температуре. Раствор метилиодида (549 мг, 0,0039 моль) в ДМФ (10 мл) добавили к реакционной смеси и перемешивали ее еще в течение 1 ч. Затем реакцию погасили добавлением солевого раствора и разбавили реакционную смесь водой. Образовавшийся осадок подкислили, затем отфильтровали; в результате получили твердое вещество желтого цвета.H NMR (300 МГц, DMSO):3,97 (s, 3H), 4,05 (s, 3H), 4,19 (s, 3H), 7,22 (d, 1H, J=8,7 Гц), 7,75 (d, 1H,J=9,0 Гц), 7,95 (d, 1H, J=9,0 Гц), 8,34 (dd, 1H, J=8,7 и 2,4 Гц), 9,90 (d, 1H, J=2,4 Гц). Этап 4. 1-Метокси-9-метил-6-нитро-9H-4-карбазолкарбоновая кислота. Приготовили суспензию метил-1 метокси-9-метил-6-нитро-9H-4-карбазолкарбоксилата (500 мг) в метаноле (15 мл) и добавили 1 М NaOH (10 мл). Реакционную смесь нагревали с обратным холодильником в течение 18 ч. После испарения метанола соединение разбавили водой, затем добавили HCl. Осадок отфильтровали, в результате получили твердое вещество коричневого цвета. 1 Н NMR (300 МГц, DMSO):4,06 (s, 3H), 4,23 (s, 3H), 7,24 (d, 1 Н, J=8,7 Гц), 7,82 (d, 1H, J=9,0 Гц),7,95 (d, 1H, J=9,0 Гц), 8,36 (dd, 1H, J=9,0 и 2,4 Гц), 10,03 (d, 1H, J=2,4 Гц), 13,01 (brs, 1H). Этап 4 а. 1-Метокси-9-метил-6-нитро-9H-4-карбазолкарбоновой кислоты хлорангидрид. Приготовили суспензию 1-метокси-9-метил-6-нитро-9H-4-карбазолкарбоновой кислоты (300 мг) в свежеперегнанном тионилхлориде (2 мл) нагревали до температуры рефлюкса в течение 4 ч. Избыток тионилхлорида удалили под вакуумом и получили соответствующий хлорангидрид, который непосредственно использовали на следующем этапе синтеза. Этап 5. N-(3,5-Дихлорпирид-4-ил)-1-метокси-9-метил-6-нитро-9H-4-карбазолкарбоксамид. К предварительно промытой суспензии гидрида натрия (100 мг, 2,5 экв., 0,0025, 60% масляная дисперсия) в ДМФ (3 мл) добавили по каплям раствор 4-амино-3,5-дихлорпиридина (244 мг, 0,0015 моль) в ДМФ (3 мл) при температуре -10 С. К реакционной смеси добавили в один прием предварительно охлажденный раствор вышеназванного хлорангидрида) в ТГФ (5 мл) при температуре -50 С и перемешивали содержимое при -50 С в течение 1 ч. Реакцию погасили добавлением солевого раствора, разбавили водой и отфильтровали; в результате получили N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-нитро-9H-4-карбазолкарбоксамид в виде желтого твердого вещества (250 мг); т.пл.:250 С.(dd, 1H, J=9,0 и 2,4 Гц), 8,84 (s, 2H), 10,03 (d, 1H, J=2,4 Гц), 10,89 (s, 1H). Этап 6. N-(3,5-Дихлорпирид-4-ил)-1-метокси-9-метил-6-амино-9H-4-карбазолкарбоксамид. Приготовили суспензию N-(3,5-дихлорпирид-4-ил)-1-метокси-9-метил-6-нитро-9H-4-карбазолкарбоксамида (250 мг) в ДМФ (20 мл) и метаноле (10 мл), добавили к суспензии никель Ранея (25 мг, 10% вес./вес.) и проводили восстановление при повышенном давлении (60 psi) в течение 18 ч при комнатной температуре. Реакционную смесь профильтровали через целит, и после испарения ДМФ образовалось твердое вещество зеленого цвета, которое промыли водой, и получили в результате N-(3,5-дихлорпирид 4-ил)-1-метокси-9-метил-6-амино-9H-4-карбазолкарбоксамид. Проверено реакцией с нингидрином. Соединение без дополнительной очистки использовали при синтезе соединения по примерам 54, 55,56 и 57. Репрезентативные соединения по настоящему изобретению, никак не ограничивающие настоящего изобретения, представлены в табл. 1 общей структурной формулой (1 А). Пример 1. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамид.(полупродукт 1) обработали метансульфонилхлоридом (24 мг, 0,299 ммоль) в ТГФ (10 мл), содержащим пиридин (23 мг, 0,299 ммоль), при 0 С и выдерживали до нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После испарения ТГФ осадок промыли насыщенным раствором бикарбоната натрия и водой. Полученное твердое вещество очистили колоночной хроматографией на силикагеле, используя в качестве элюента 30% смесь ацетона и хлороформа; в результате получили 30 мг 1N-(3,5-дихлорпирид-4-ил)-4-метокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества; т.пл.: 315 С.- 24010634 Соединения по примерам 2 и 3 синтезировали в условиях, аналогичных условиям по примеру 1, за исключением применения соответствующего замещенного сульфонилхлорида вместо метансульфонилхлорида. Пример 2. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-(N,N-диметилсульфонамид)дибензо[b,d]фуран-1 карбоксамид.(23 мг, 0,299 ммоль), при 0 С и выдержали до нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После испарения ТГФ осадок промыли насыщенным раствором бикарбоната натрия и водой. Полученное твердое вещество очистили колоночной хроматографией на силикагеле, используя в качестве элюента 30% смесь ацетона и хлороформа; в результате получили 25 мг N-(3,5-дихлорпирид-4-ил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1 карбоксамида в виде белого твердого вещества; т.пл.: 252 С.IR (KBr): 3271, 2961, 2925, 2852, 1660, 1607, 1542, 1499, 1468, 1392, 1285, 1261, 1101, 1021, 805 см-1. 1 Н NMR (300 МГц, DMSO):2,01 (s, 3H), 4,07 (s, 3H), 7,32 (d, 1H, J=8,4 Гц), 7,65 (d, 1H, J=8,4 Гц),7,93 (m, 2H), 8,41 (s, 1H), 8,76 (s, 2H), 10,06 (s, 1H), 10,76 (s, 1H). Соединения по примерам 5, 6, 7, 8, 11, 12 и 13 синтезировали в условиях, аналогичных условиям по примеру 4, за исключением применения соответствующего замещенного хлорангидрида вместо ацетилхлорида. Пример 5. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-(3-хлорпропилкарбоксамид)дибензо[b,d]фуран-1 карбоксамид.(s, 1H). Пример 9. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран 1-карбоксамид. Соединение синтезировали путем гидролиза N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этоксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида (пример 8) с использованием гидроксида калия(3 экв.) в метаноле; т.пл.: 250 С. Полученное соединение непосредственно использовали для приготовления вещества по примеру 10 без дополнительной идентификации. Пример 10. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида. Соединение синтезировали из N-(3,5-дихлорпирид-4-ил)-4-метокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида (пример 9) с использованием 1% метанольного гидроксида натрия (1,0 экв.). 1(полупродукт 1) обработали трифторуксусным ангидридом (57 мг, 0,27 ммоль) в дихлорметане (5 мл),содержащим пиридин (23 мг, 0,299 ммоль), при комнатной температуре. Реакционную смесь перемешивали в течение 20 ч при комнатной температуре. После испарения дихлорметана осадок растирали с холодной водой; в результате образовалось твердое вещество белого цвета. Осадок отфильтровали и очистили колоночной хроматографией на силикагеле, используя в качестве элюента 6% смесь ацетона и хлороформа; получили 30 мг N-(3,5-дихлорпирид-4-ил)-4-метокси-8-трифторацетамиддибензо[b,d]фуран-1 карбоксамида в виде белого твердого вещества; т.пл.: 300 С.(полупродукт 1) обработали этилхлорформатом (40 мг, 0,374 ммоль) в ТГФ (10 мл), содержащим пиридин (29 мг, 0,374 ммоль), при 0 С и выдержали до нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После испарения ТГФ осадок промыли водой. Полученное твердое вещество очистили колоночной хроматографией на силикагеле, используя в качестве элюента 10% смесь ацетона и хлороформа; в результате получили 40 мг N-(3,5-дихлорпирид-4-ил)-4-метокси-8-этоксикарбоксамиддибензо[b,d]фуран-1-карбоксамида в виде твердого вещества белого цвета; т.пл.: 274 С.(d, 2H), 7,88 (d, 1H, J=8,1 Гц), 8,45 (s, 1H), 8,76 (s, 2H), 9,62 (s, 1H), 10,76 (s, 1H). Соединения по примерам 16 и 17 синтезировали при условиях, аналогичным условиям реакции по примеру 15, за исключением использования соответствующего замещенного хлорформата вместо этилхлорформата. Пример 16. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-изобутилоксикарбоксамиддибензо[b,d]фуран-1 карбоксамид. 1 Н NMR (300 МГц, DMSO-d6):0,91 (d, J=6,6 Гц, 6H), 1,84-1,96 (m, 1H), 3,84 (d, J=6,9 Гц, 2H), 4,08IR (KBr): 3358, 2918, 1777, 1750, 1610, 1560, 1493, 1391, 1284, 1235, 1192, 1003, 803, 619 см-1. Пример 18. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-циклопропилэтоксикарбоксамиддибензо[b,d]фуран-1-карбоксамид. Раствор циклопропилметанола (70 мг, 0,273 ммоль) в ТГФ (3 мл) охладили до -30 С. К этому раствору добавили триэтиламин (37 мг, 0,374 ммоль) и перемешивали в течение 10 мин. К названному раствору добавили раствор трифосгена (73 мг, 0,249 ммоль) в ТГФ (3 мл) при -30 С и перемешивали в течение 30 мин при комнатной температуре. Далее этот раствор добавили к суспензии N-(3,5-дихлорпирид-4- 26010634 ил)-4-метокси-8-аминодибензо[b,d]фуран-1-карбоксамида (100 мг, 0,249 ммоль) (полупродукт 1) и триэтиламина в ТГФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. После испарения ТГФ осадок промыли водой. Полученное твердое вещество очистили колоночной хроматографией на силикагеле, используя в качестве элюента 10% смесь ацетона и хлороформа; в результате получили 15 мг N-(3,5-дихлорпирид-4-ил)-4-метокси-8-циклопропилметоксикарбоксамиддибензо[b,d] фуран-1-карбоксамида в виде твердого вещества белого цвета; т.пл.: 272 С. 1 Н NMR (300 МГц, DMSO-d6):0,29 (q, 2 Н, J=6,0 Гц), 0,52 (q, 2H, J=6,3 Гц), 1,14 (m, 1H), 3,88 (d,2H, J=7,2 Гц), 4,06 (s, 3H), 7,32 (d, 1H, J=8,1 Гц), 7,60 (d, 1H, J=8,4 Гц), 7,66 (d, 1H, J=8,7 Гц), 7,90 (ds, 1H,J=8,1 Гц), 8,48 (d, 1H, J=2,4 Гц), 8,76 (s, 2H), 9,69 (s, 1H), 10,75 (s, 1H).IR (KBr): 2960, 2735, 1672, 1596, 1473, 1461, 1323, 1271, 1180, 1113, 1089, 1001, 945, 817, 767 см-1. Пример 19. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-трифторметилметоксикарбоксамиддибензо[b,d] фуран-1-карбоксамид. Соединение синтезировали при условиях, аналогичным условиям реакции по примеру 17, за исключением использования 2,2,2-трифторэтанола вместо циклопропилметанола; т.пл.: 250 С. 1 Н NMR (300 МГц, DMSO-d6):4,07 (s, 3H), 4,77 (q, 2H, J=9,0 Гц), 7,35 (d, 1H, J=8,4 Гц), 7,63 (d, 1H,J=9,3 Гц), 7,72 (d, 1H, J=9,0 Гц), 7,93 (d, 1H, J=8,4 Гц), 8,52 (s, 1H), 8,76 (s, 2H), 10,18 (brs, 1H), 10,76 (s, 1H).(полупродукт 1) обработали фенилхлорформатом (190 мг, 1,09 ммоль) в ТГФ (15 мл), содержащим пиридин (0,5 мл), при 0 С и выдержали до нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 12 ч. После испарения ТГФ осадок промыли водой и горячим этанолом; в результате получили 400 мг N-(3,5-дихлорпирид-4-ил)-4-метокси-8-феноксикарбоксамиддибензо[b,d]фуран-1-карбоксамида в виде твердого вещества белого цвета, которое использовали в следующем этапе синтеза. Этап 2. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-N,N-диэтиламинокарбоксамиддибензо[b,d]фуран-1 карбоксамид. К N-(3,5-дихлорпирид-4-ил)-4-метокси-8-феноксикарбоксамиддибензо[b,d]фуран-1-карбоксамиду (по этапу 1) (100 мг, 0,19 ммоль), растворенному в ДМСО (2,0 мл), медленно добавили раствор N,N-диэтиламина (20 мг, 0, 28 ммоль) в ДМСО (1 мл). Реакционную смесь перемешивали в течение 5 ч при 50 С, затем охладили до комнатной температуры и разбавили ледяной водой (25 мл). Выделившийся осадок отфильтровали, высушили и очистили колоночной хроматографией на силикагеле с использованием в качестве элюента 10% метанола в хлороформе; в результате получили 45 мг N-(3,5-дихлорпирид 4-ил)-4-метокси-8-N,N-диэтиламинокарбоксамиддибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества; т.пл.: 250 С. 1 Н NMR (300 МГц, DMSO-d6):1,08 (t, 6 Н, J=7,2 Гц), 3,25 (q, 4H, J=5,1 Гц), 4,06 (s, 3H), 7,31 (d, 1H,J=8,4 Гц), 7,59 (s, 2H), 7,90 (d, 1H, J=8,4 Гц), 8,32 (s, 1H), 8,75 (s, 2H), 10,74 (s, 1H).IR (KBr): 3357, 2932, 1673, 1631, 1552, 1474, 1396, 1285, 1198, 1101, 952, 805, 670 см-1. Соединения по примерам 21, 22, 24, 25, 26, 27, 28 и 29 синтезировали в условиях, аналогичным условиям реакции по этапу 2 примера 21, за исключением использования соответствующего первичного или вторичного амина вместо N,N-диэтиламина. Пример 21. N-(3,5-Дихлорпирид-4-ил)-4-метокси-8-циклопентиламинокарбоксамиддибензо[b,d]фуран 1-карбоксамид. 1 Н NMR (300 МГц, DMSO):1,355 (m, 2H), 1,57 (m, 4H), 1,81 (m, 2H) 3,94 (m, 1H), 4,07 (s, 3H), 6,03[b,d]фуран-1-карбоксамида гидрохлорид. Соединение синтезировали из N-(3,5-дихлорпирид-4-ил)-4-метокси-8-(N-метилпиперазин-4-ил-карбоксамид)дибензо[b,d]фуран-1-карбоксамида реакцией с метанольным HCl; т.пл.: 250 С. 1 Н NMR (300 МГц, DMSO-d6):2,77 (d, 3H, J=4,2 Гц), 3,02 (m, 2H), 3,24-3,43 (brm, 4H), 4,08 (s, 3H),4,23 (m, 2H), 7,34 (d, 1H, J=8,7 Гц), 7,63 (s, 2H), 7,89 (d, 1H, J=8,4 Гц), 8,37 (s, 1H), 9,03 (s, 1H), 10,63 (s,2H), 11,23 (s, 1H).(полупродукт 2) обработали метансульфонилхлоридом (22 мг, 0,194 ммоль) в ТГФ (10 мл), содержащим пиридин (0,5 мл), при 0 С и выдержали до нагревания до комнатной температуры. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. После испарения ТГФ осадок промыли насыщенным раствором бикарбоната натрия, водой. Образовавшийся твердый осадок очистили колоночной хроматографией на силикагеле с использованием в качестве элюента 12% смеси ацетон-хлороформ; в результате получили 37 мг N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d] фуран-1-карбоксамида в виде белого твердого вещества; т.пл.: 250 С. 1IR (KBr): 3323, 2926, 1698, 1636, 1489, 1396, 1283, 1266, 1142, 1040, 812, 621 см-1. Пример 31. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамида. Соединение синтезировали из N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-метансульфонамиддибензо[b,d]фуран-1-карбоксамида (пример 30) реакцией с 1% метанольным гидроксидом натрия (1,0 экв.). 1IR (KBr): 2920, 1651, 1524, 1463, 1391, 1278, 1194, 1105, 1005, 882, 815 см-1. Соединения по примерам 32 и 33 синтезировали в условиях, аналогичных условиям реакции по примеру 30, за исключением использования соответствующего замещенного сульфонилхлорида вместо метансульфонилхлорида. Пример 32. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-этансульфонамиддибензо[b,d]фуран-1-карбоксамид. 1 Н NMR (300 МГц, DMSO-d6):1,19 (t, 3H, J=7,2 Гц), 3,03 (q, 2H, J=7,2 Гц), 7,49 (dd, 1H, J=8,7 Гц и(0,5 мл), при 0 С и выдержали до нагревания до комнатной температуры. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. После испарения ТГФ осадок промыли насыщенным раствором бикарбоната натрия, 5% HCl, водой. Образовавшийся твердый осадок очистили колоночной хроматографией на силикагеле с использованием в качестве элюента 10% смеси ацетон-хлороформ; в результате получили 25 мг N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-ацетамиддибензо[b,d]фуран-1 карбоксамида в виде белого твердого вещества; т.пл.: 234 С. 1 Н NMR (300 МГц, DMSO-d6):2,15 (s, 3H), 7,4 (d, 1H, J=8,4 Гц), 7,53 (t, 1H, J=73 Гц), 7,56 (d, 1H,J=8,4 Гц), 7,77-7,86 (m, 3H), 8,36 (d, 1H, J=2,4 Гц), 8,81 (s, 2H), 10,25 (s, 1H).IR (KBr): 3344, 2924, 1727, 1712, 1686, 1555, 1390, 1367, 1288, 1269, 1116, 1047, 822, 584 см-1. Соединения по примерам 35, 36, 37 и 40 синтезировали в условиях, аналогичных условиям реакции по примеру 34, за исключением использования соответствующего замещенного хлорангидрида вместо ацетилхлорида. Пример 35. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-(1-хлорпропилкарбоксамид)дибензо[b,d] фуран-1-карбоксамид. 1IR (KBr): 3206, 3107, 2987, 1759, 1702, 1669, 1548, 1499, 1384, 1296, 1279, 1222, 1191, 1118, 1055, 810 см-1. Пример 38. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-гидроксикарбонилкарбоксамиддибензо[b,d] фуран-1-карбоксамид. Соединение синтезировали путем гидролиза N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-этоксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамид (пример 37) в присутствии гидроксида калия (3 экв.) в метаноле. 1H NMR (300 МГц, DMSO-d6):7,58 (t, 1H, J=12 Гц), 7,62 (d, 1H, J=8,4 Гц), 7,86 (d, 1H, J=8,7 Гц),7,91-7,96 (m, 2H), 8,76 (d, 1H, J=1,8 Гц), 8,81 (s, 2H), 10,97 (s, 1H), 11,04 (s, 1H). Пример 39. Натриевая соль N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида. Соединение синтезировали из N-(3,5-дихлорпирид-4-ил)-4-дифторметокси-8-гидроксикарбонилкарбоксамиддибензо[b,d]фуран-1-карбоксамида (пример 38) реакцией с этанольным этоксидом натрия (2,0 экв.). 1 Н NMR (300 МГц, DMSO-d6):7,30 (d, 1H, J=8,7 Гц), 7,42 (t, 1H, J=72 Гц), 7,65 (d, 1H, J=8,7 Гц),7,98 (d, 1H, J=8,7 Гц), 8,06 (dd, 1H, J=9,0 и 1,8 Гц), 8,21 (s, 2H), 9,08 (d, 1H, J=2,4 Гц), 10,08 (s, 1H). Пример 40. N-(3,5-Дихлорпирид-4-ил)-4-дифторметокси-8-фур-2-ил-карбоксамиддибензо[b,d]фуран 1-карбоксамид. 1IR (KBr): 3288, 3031, 1651, 1586, 1556, 1518, 1498, 1386, 1281, 1193, 1117, 1046, 809, 750 см-1. Пример 41. N-Фенил-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид. Этап 1. 1H-Фенил-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид. Реакцию взаимодействия 4-метокси-8-нитродибензо[b,d]фуран-1-карбоксилатхлорида (200 мг,- 29010634 0,696 ммоль) (по этапу 5 а синтеза полупродукта 1) с анилином (2 экв.) в ТГФ (10 мл) в присутствии диизопропилэтиламина (3 экв.) проводили при комнатной температуре в течение 16 ч. Желтую суспензию отфильтровали и полученный твердый осадок промыли 5% HCl и водой; в результате получили 100 мгN1-фенил-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества. 1 Н NMR (300 МГц, DMSO-d6):4,15 (s, 3H), 7,18 (t, 1H), 7,43 (m, 3H), 7,80 (d, 2H), 7,95 (d, 1H), 8,01N1-Фенил-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид (100 мг) (по этапу 1) восстанавливали при помощи никеля Ранея (100 мг) в метаноле (40 мл) и ДМФ (10 мл) в присутствии гидразингидрата (5 мл) в условиях мягкого рефлюкса в течение 1 ч. Реакционную смесь профильтровали сквозь целит и сконцентрировали фильтрат под вакуумом. Осадок растирали с водой, в результате образовалось твердое вещество, которое отфильтровали и высушили; в результате получили продукт реакции в виде белого твердого вещества (90 мг). 1H NMR (300 МГц, DMSO-d6):4,15 (s, 3H), 5,03 (brs, 2H), 6,80 (d, 1H), 7,08 (t, 1H), 7,11 (d, 1H),7,40 (m, 4H), 7,60 (d, 1H), 7,93 (d, 2H), 10,43 (s, 1H). Этап 3. 1H-Фенил-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид. 1H-Фенил-4-метокси-8-аминодибензо[b,d]фуран-1-карбоксамид (69 мг, 0,207 ммоль) (этап 2) обработали ацетилхлоридом (1,1 экв.) в ТГФ (10 мл), содержащим пиридин (1,1 экв.), при 0 С и оставили для нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. После испарения ТГФ осадок промыли этанолом; в результате получили 40 мг 1H-фенил-4 метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества; т.пл.: 252 С.(m, 2H), 7,82 (d, 2H), 7,91 (dd, 1H, J=9,0 и 2,7 Гц), 8,37 (d, 1H, J=2,4 Гц), 10,10 (s, 1H), 10,50 (s, 1H). Пример 42. N1-(4-Метоксифенил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид. Этап 1. N1-(4-Метоксифенил)-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид. Реакцию взаимодействия хлорангидрида 4-метокси-8-нитро дибензо[b,d]фуран-1-карбоновой кислоты (200 мг, 0, 696 ммоль) (по этапу 5 а синтеза полупродукта 1) с 4-метокси анилином (2 экв.) в ТГФ(10 мл) в присутствии диизопропилэтиламина (3 экв.) проводили при комнатной температуре в течение 16 ч. Желтую суспензию отфильтровали, и полученный твердый осадок промыли 5% HCl и водой; в результате получили 153 мг N1-(4-метоксифенил)-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества. 1N1-(4-Метоксифенил)-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид (140 мг) (по этапу 1) восстанавливали при помощи никеля Ранея (100 мг) в метаноле (40 мл) и ДМФ (10 мл) в присутствии гидразингидрата (1 мл) в условиях мягкого рефлюкса в течение 1 ч. Реакционную смесь профильтровали сквозь целит,и сконцентрировали фильтрат под вакуумом. Осадок растирали с водой, в результате образовалось твердое вещество, которое отфильтровали и высушили, в результате получили продукт в виде белого твердого вещества (90 мг). 1(этап 2) обработали ацетилхлоридом (1,1 экв.) в ТГФ (10 мл), содержащим пиридин (1,1 экв.), при 0 С и оставили для нагревания до комнатной температуры. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. После испарения ТГФ осадок промыли этанолом; в результате получили 40 мгN1-(4-метоксифенил)-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества; т.пл.: 285 С. 1IR (KBr): 3256, 2938, 2839, 1645, 1599, 1531, 1514, 1469, 1412, 1291, 1195, 1178, 1099, 1023, 825, 812 см-1. Пример 43. 1H-Бензил-4-метокси-8-ацетамиддибензо[b,d]фуран-1-карбоксамид. Этап 1. N-Бензил-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамид. Реакцию взаимодействия хлорангидрида 4-метокси-8-нитродибензо[b,d]фуран-1-карбоновой кислоты (200 мг, 0,696 ммоль) (по этапу 5 а синтеза полупродукта 1) с бензиламином (2 экв.) в ТГФ (10 мл) в присутствии диизопропилэтиламина (3 экв.) проводили при комнатной температуре в течение 16 ч. Суспензию отфильтровали и полученный твердый осадок промыли 5% HCl и водой; в результате получили 170 мг N1-бензил-4-метокси-8-нитродибензо[b,d]фуран-1-карбоксамида в виде белого твердого вещества. 1 Н NMR (300 МГц, DMSO-d6):4,08 (s, 3H), 4,62 (d, 2 Н), 7,15 (d, 1 Н), 7,18-7,23 (m, 5H), 7,84 (d, 1H),- 30
МПК / Метки
МПК: A61P 29/00, C07D 401/12, C07D 405/14, C07D 409/12, C07D 405/12, A61K 31/4427, C07D 307/91
Метки: гетероциклические, применяемые, лечения, составы, природы, воспалительной, соединения, способы, нарушений, новые, содержащие, аллергической, фармацевтические, синтеза
Код ссылки
<a href="https://eas.patents.su/30-10634-novye-geterociklicheskie-soedineniya-primenyaemye-dlya-lecheniya-narushenijj-allergicheskojj-ili-vospalitelnojj-prirody-sposoby-sinteza-i-soderzhashhie-ih-farmacevticheskie-sostavy.html" rel="bookmark" title="База патентов Евразийского Союза">Новые гетероциклические соединения, применяемые для лечения нарушений аллергической или воспалительной природы: способы синтеза и содержащие их фармацевтические составы</a>
Трициклические соединения для лечения воспалительных и аллергических нарушений, способы их приготовления и содержащие их фармацевтические составы

Номер патента: 10408
Опубликовано: 29.08.2008
Авторы: Лакдавала Афтаб Давудбхай, Анупинди Рагху Рам, Баласубраманиан Гопалан, Гхарат Лаксмикант Атмарам
МПК: A61K 31/403, A61K 31/381, A61K 31/34...
Метки: воспалительных, аллергических, лечения, приготовления, соединения, составы, содержащие, способы, фармацевтические, нарушений, трициклические
Формула / Реферат:
1. Соединение общей формулы (1) где R1 представляет собой водород, С1-С8 алкил, С3-С12 циклоалкил, (С3-С12 циклоалкил)(С1-С8 алкил), (С6-С14 арил)(С1-С8 алкил) или защитную группу, где С1-С8 алкил, С3-С12 циклоалкил, (С3-С12 циклоалкил)(С1-С8 алкил) и (C6-C14 арил)(С1-С8 алкил) возможно имеют от 1 до 3 заместителей, выбранных из группы, включающей С1-С8 алкил, галоген, -ORx, оксо- и -COORx; R2 представляет собой водород, С1-С8 алкил, С3-С15...
Новые соединения 1,2-дитиолана, способ их получения и содержащие их фармацевтические составы

Номер патента: 3424
Опубликовано: 24.04.2003
Авторы: Локар Бриан, Гиллонно Клод, Шартон Ив, Лестаж Пьер, Гольдстейн Соло
МПК: A61K 31/5377, C07D 413/12
Метки: соединения, составы, фармацевтические, получения, 1,2-дитиолана, способ, содержащие, новые
Формула / Реферат:
1. Соединения формулы (I) где Ra представляет линейную или разветвленную (C1-C8)алкиленовую группу, Rb представляет простую связь или линейную или разветвленную (C1-C6)алкиленовую группу, Z представляет тиокарбаматную группу где атом кислорода связан с Ra группой и R1 представляет атом водорода, или тиоамидную группу где тиокарбонильная группа связана с Ra группой, и R1 является таким, как определено выше, T представляет группу где...
Гетероциклические соединения, способы их получения и фармацевтические композиции, содержащие эти соединения, и их использование в медицине

Номер патента: 8303
Опубликовано: 27.04.2007
Авторы: Джаин Мукул Р., Пател Гаутам Д., Лохрай Видья Бхушан, Лохрай Брадж Бхушан, Пингали Харикишоре
МПК: A61K 31/34, A61K 31/42, A61K 31/415...
Метки: гетероциклические, содержащие, фармацевтические, способы, получения, медицине, эти, композиции, соединения, использование
Формула / Реферат:
1. Соединение формулы (I), где G представляет собой группы А, В, С, D, Е или F, описанные ниже: где R1 представляет собой водород, ацильную, галогенацильную группу; R2 представляет собой (C1-С3)алкил; X1, X2, Х3, Х4 могут быть одинаковыми или различными и представляют собой водород или галоген; и если G представляет собой гетероцикл "D", то по меньшей мере одна из групп, определенных как X1, X2, Х3, Х4, не является водородом; R3 выбран из...
Циклоалкильные соединения, лактамы, лактоны и родственные соединения, содержащие их фармацевтические композиции и способы ингибирования высвобождения и/или синтеза β-амилоидного пептида с помощьюуказанных соединений

Номер патента: 2100
Опубликовано: 24.12.2001
Авторы: Джон Варгес, Дрост Джеймс Дж., Фридман Стефен, Дрессман Брюс А., Торсетт Юджин Д., Портер Варрен Дж., Ву Джинг, Танг Джей С., Плейсс Майкл А., Скотт Уильям Леонард, Бриттон Томас К., Кви Синтия Л., Латимер Ли Х., Одия Джеймс Е., Мабри Томас Э., Ниссен Джеффри С., Генри Стивен С., Макданиел Стейси Л., Стаки Расселл Д., Нейц Джеффри, Рил Джон К.
МПК: A61P 25/28, A61K 31/55, C07D 243/10...
Метки: лактамы, способы, помощьюуказанных, высвобождения, циклоалкильные, соединения, содержащие, соединений, синтеза, лактоны, beta;-амилоидного, композиции, пептида, фармацевтические, ингибирования, родственные
Формула / Реферат:
1. Способ ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, который заключается в том, что в такую клетку вводят соединение или смесь соединений в количестве, эффективном для ингибирования высвобождения и/или синтеза b -амилоидного пептида в клетке, и указанные соединения имеют формулу I в которой R1 выбирают из группы, включающей C1-С10алкил, необязательно замещенный 1-3 заместителями, независимо выбранными из...
Новые соединения, фармацевтические композиции, содержащие их, и способы их использования

Номер патента: 10484
Опубликовано: 30.10.2008
Авторы: Медгалчи Сьюзен М., Таунсенд Крэйг А., Кухаджа Фрэнсис П., Макфэдден Джилл М., Тхупари Джаган Н.
МПК: C07D 307/56, A61K 31/34
Метки: фармацевтические, их, способы, соединения, новые, использования, содержащие, композиции
Формула / Реферат:
1. Соединения формулы I в которой R1 означает C1-С20алкил или группу =СН2; R2 означает C1-С20алкил; X1 означает NHR4, где R4 представляет собой C1-С20алкил, С3алкенил и необязательно содержит карбоксильную группу, спиртовую группу, или когда R1 означает группу =СН2, и R2 означает С8алкил, X1 означает группу -NHCH2COOMe или группу -NHCH2COOtrBu. 2. Соединения по п.1, в которых R1 представляет C1-С10алкил или группу =СН2. 3. Соединения по п.2, в...
Предыдущий патент: Новые бензальдоксимы, способ получения бензальдоксимов и его применение
Следующий патент: Цеолит, его применение в процессе конверсии углеводородов
Случайный патент: Способ выщелачивания металлсодержащего сульфидного концентрата