Стабильная негигроскопичная кристаллическая форма n-[n-n-(4-(пиперидин-4-ил)бутаноил)-n-этилглицилового] соединения
Номер патента: 1362
Опубликовано: 26.02.2001
Авторы: Ванасс Бенуа Дж., Менсел Джеймс Дж., Кубиак Грегори Г., Следески Адам В., Пауэрс Мэттью Р., Хшан Зофия Дж., Лиу Роберт С., Родригез Вальтер, Виндиш Винсент, Толедо-Веласкез Дэвид, Вемури Нарасимха М., Гардетто Энтони Дж., Шербайн Джеймс П., Вудвард Рик Г., Салазар Дайан К.



















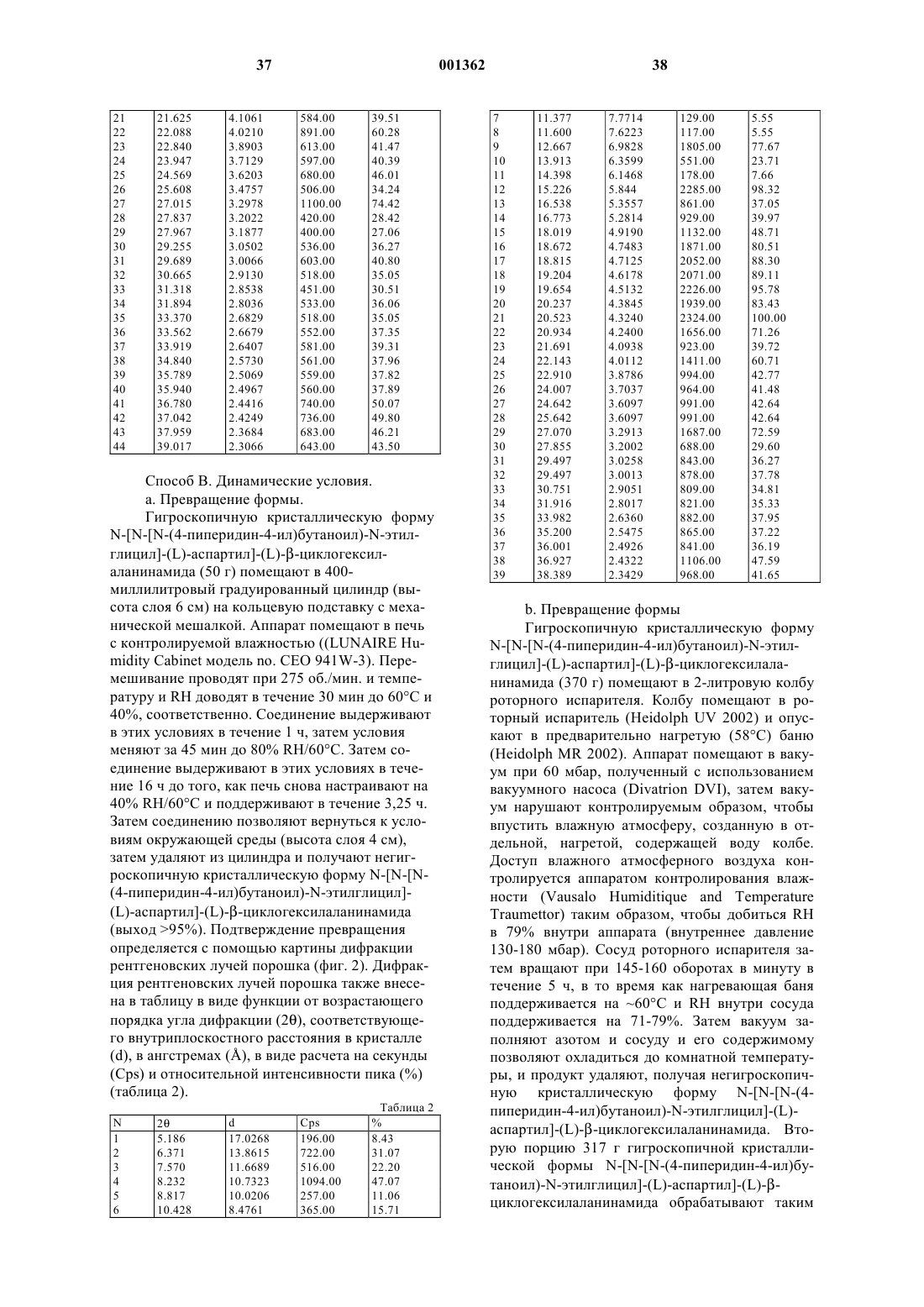
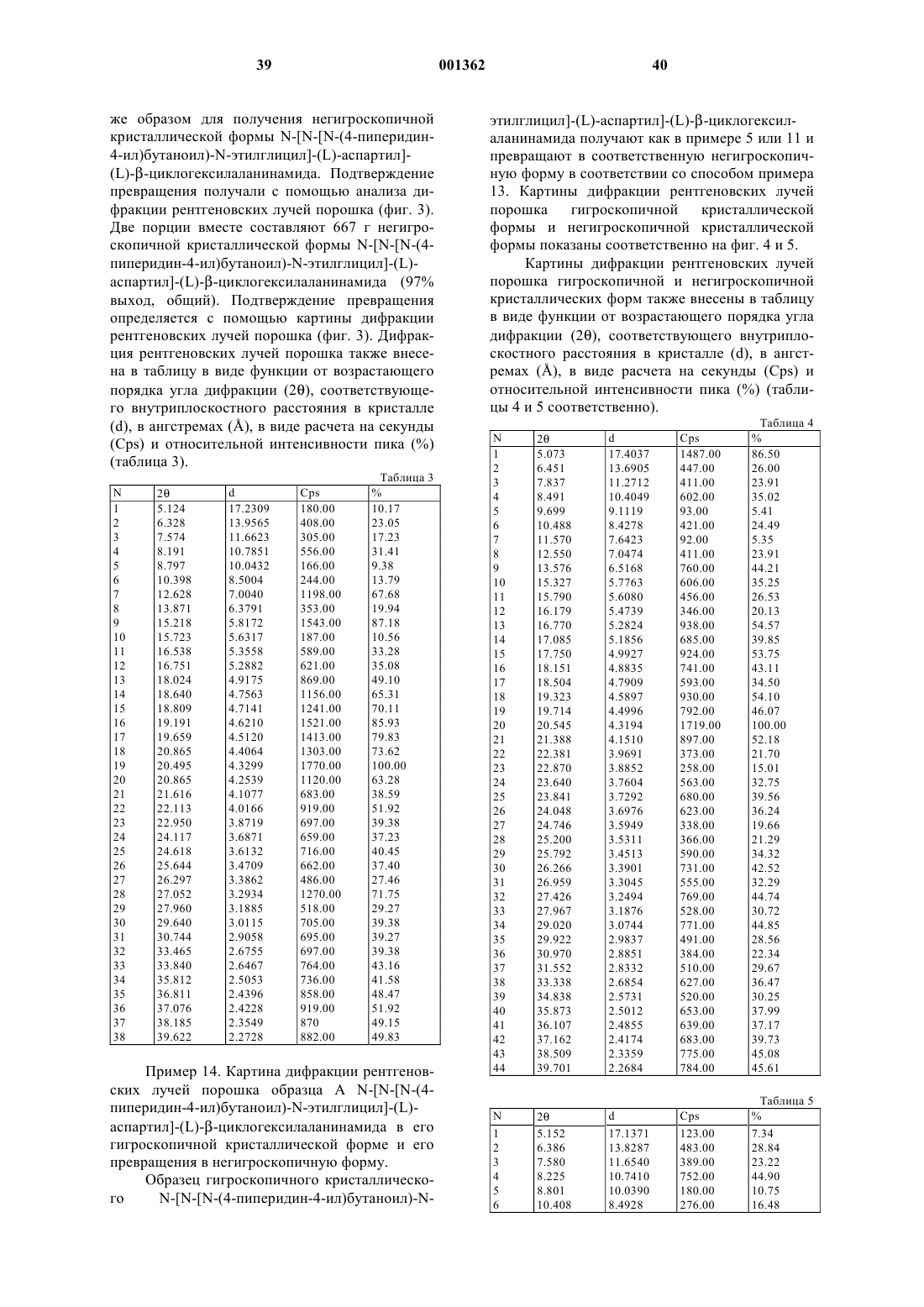





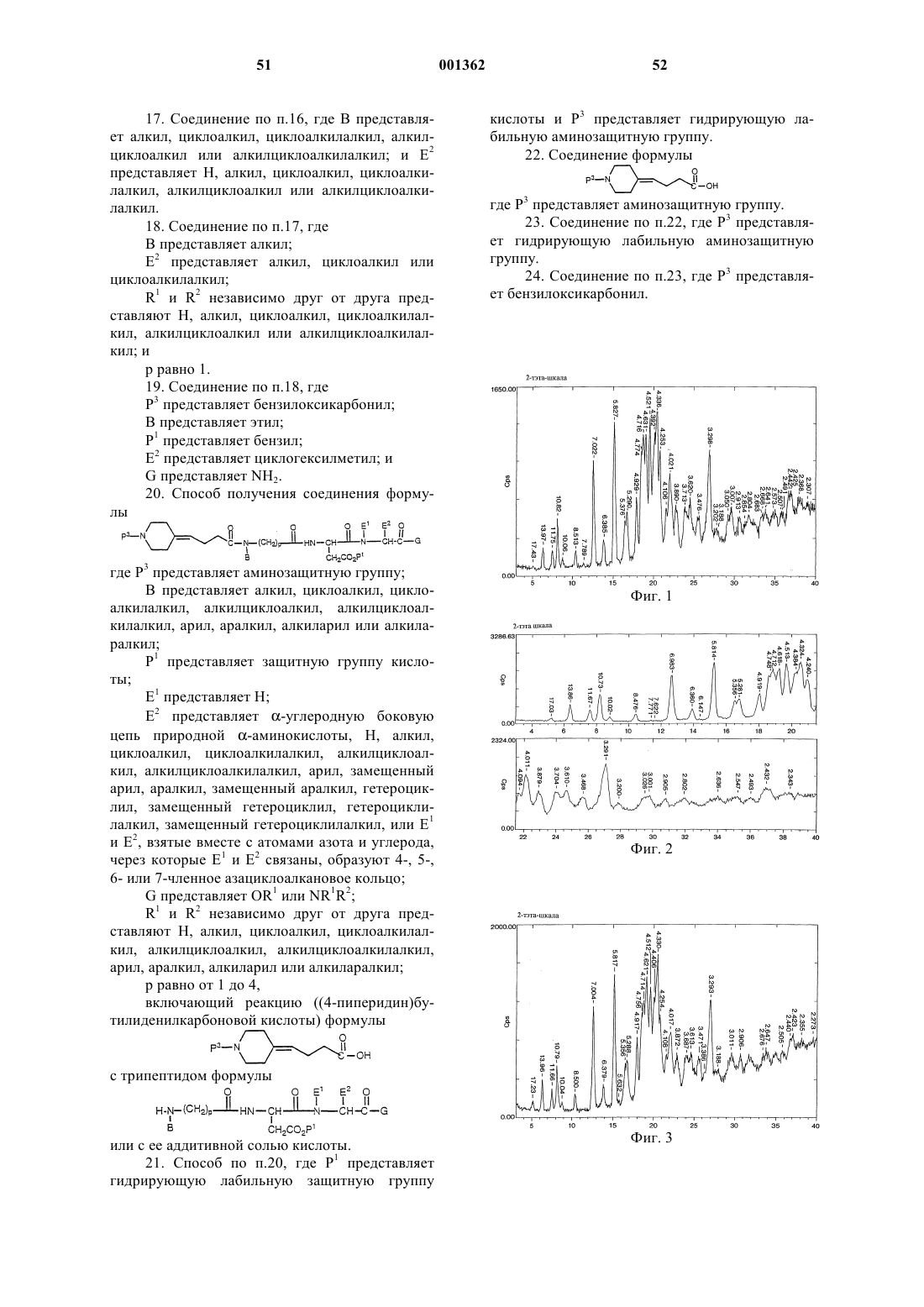
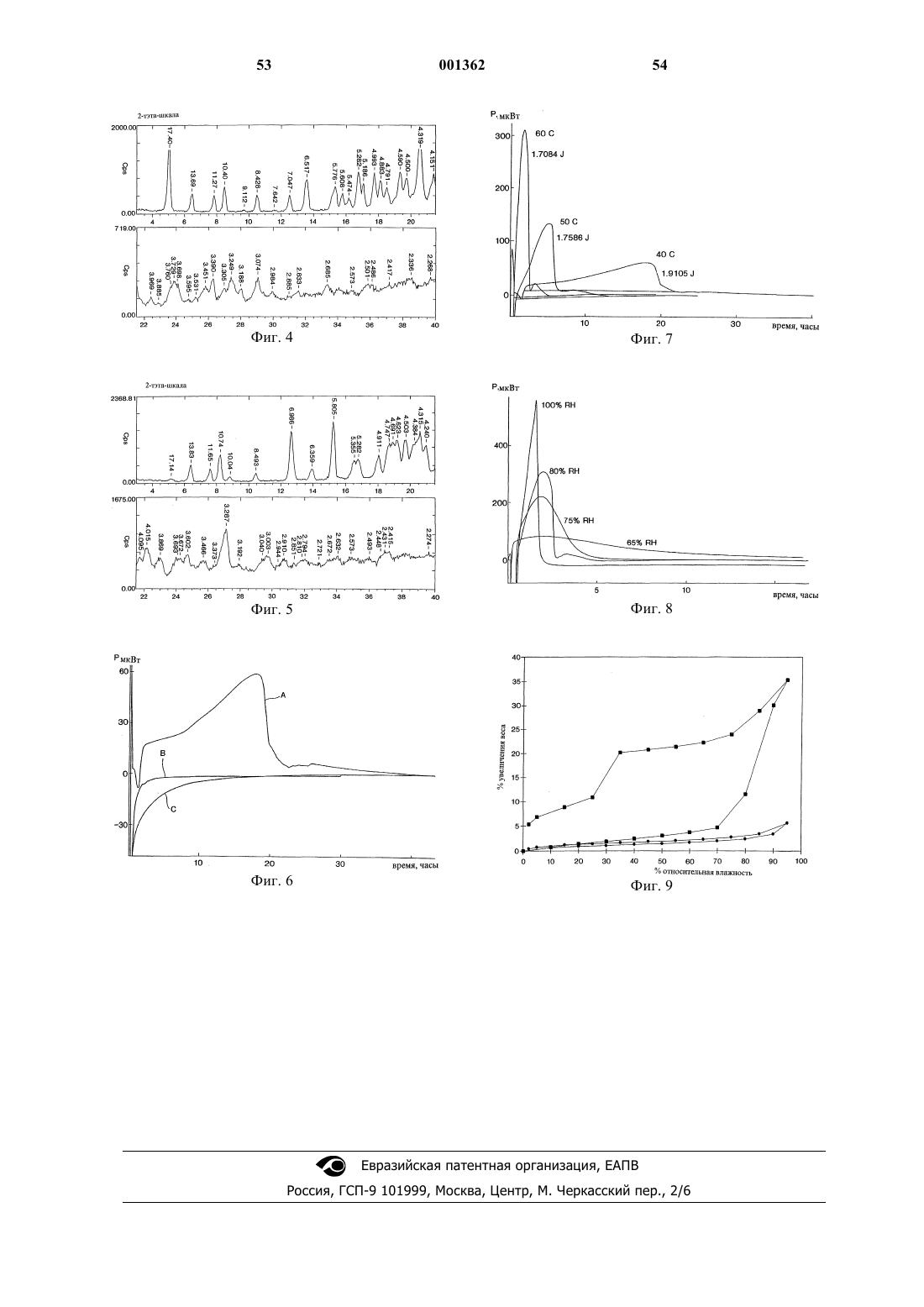
Формула / Реферат
1. Соединение, представляющее N-(N-трет-бутоксикарбонил-N-этилглицил)-(L)-аспарагиновой кислоты b-бензиловый сложный эфир.
2. Соединение, представляющее N-[N-[N-[4-[N-бензилоксикарбонилпиперидин-4-ил]бутаноил]-N-этилглицил]-(L)-аспартил] b-бензиловый сложный эфир]-(L)-b-циклогексилаланинамид.
3. Соединение, представляющее 4-(4-пиперидин)бутилиденилкарбоновую кислоту.
4. Соединение, представляющее N-[N-[N-[3-[N-бензилоксикарбонил-4-пиперидин]пропилиденилкарбонил]-N-этилглицил]-(L)-аспартил] b-бензиловый сложный эфир]-(L)-b-циклогексилаланинамид.
5. Соединение, представляющее N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-b-циклогексилаланинамид или его фармацевтически приемлемую соль.
6. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель.
7. Способ получения негигроскопичного кристаллического N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-b-циклогексилаланинамида, включающий воздействие на гигроскопичный кристаллический N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-b-циклогексилаланинамид относительной влажностью от около 40 до 100% и температурой от около 20 до около 80шС.
8. Способ по п.7, где относительная влажность составляет от около 65% до около 80%.
9. Способ по п.7, температура составляет от около 40 до около 80шС.
10. Способ по п.7, где воздействие осуществляют в статических условиях.
11. Способ по п.7, где воздействие осуществляют в динамических условиях.
12. Способ получения соли соединения формулы

где В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
Е1 представляет Н;
Е2 представляет a-углеродную боковую цепь природной a-аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е1 и Е2, взятые вместе с атомами азота и углерода, через которые Е1 и Е2 связаны, образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;
G представляет OR1 или NR1R2;
R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
р представляет целое число от 1 до 4;
Р1 представляет группу, защищающую кислоту;
включающий реакцию соединений формулы

где Р2 представляет лабильную аминозащитную группу, с образованием первого промежуточного соединения формулы

с последующей реакцией полученного первого промежуточного продукта с соединением формулы

с образованием второго промежуточного соединения формулы

с последующим удалением Р2 защитной группы из второго промежуточного соединения трифторуксусной кислотой, получая соединение в виде соли.
13. Способ по п.12, где Р1 представляет гидрирующую лабильную защитную группу кислоты.
14. Соединение формулы

где Р3 представляет аминозащитную группу;
В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
Р1 представляет защитную группу кислоты,
Е1 представляет Н;
Е2 представляет a-углеродную боковую цепь природной a-аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е1 и Е2 вместе с атомами азота и углерода, через которые Е1 и Е2 связаны, образуют 4-, 5-, 6- или 7- членное азациклоалкановое кольцо;
G представляет OR1 или NR1R2;
R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
р равно от 1 до 4,
15. Соединение по п.14, где Р1 представляет гидрирующую лабильную защитную группу кислоты, а Р3 представляет гидрирующую лабильную аминозащитную группу.
16. Соединение по п.15, где
В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
Е1 представляет Н;
Е2 представляет Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил или замещенный аралкил;
L представляет OR1 или NR1R2;
R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил;
р равно 1 или 2.
17. Соединение по п.16, где В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил; и Е2 представляет Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил.
18. Соединение по п.17, где
B представляет алкил;
Е2 представляет алкил, циклоалкил или циклоалкилалкил;
R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил; и
р равно 1.
19. Соединение по п.18, где
Р3 представляет бензилоксикарбонил;
B представляет этил;
Р1 представляет бензил;
Е2 представляет циклогексилметил; и
G представляет NН2.
20. Способ получения соединения формулы

где Р3 представляет аминозащитную группу;
В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил
Р1 представляет защитную группу кислоты; Е1 представляет Н; Е2 представляет a-углеродную боковую цепь природной a-аминокислоты, Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е1 и Е2, взятые вместе с атомами азoта и углерода, через которые Е1 и Е2 связаны, образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;
G представляет OR1 или NR1R2;
R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил шыш алкиларалкил;
р равно от 1 до 4,
включающий реакцию ((4-пиперидин)бутилиденилкарбоновой кислоты) формулы

с трипептидом формулы

или с ее аддитивной солью кислоты.
21. Способ по п.20, где Р1 представляет гидрирующую лабильную защитную группу кислоты и Р3 представляет гидрирующую лабильную аминозащитную группу.
22. Соединение формулы

где Р3 представляет аминозащитную группу.
23. Соединение по п.22, где Р3 представляет гидрирующую лабильную аминозащитную группу.
24. Соединение по п.23, где Р3 представляет бензилоксикарбонил.
Текст
1 1. Область изобретения Изобретение относится к негигроскопичной стабильной форме кристаллической формы Это соединение обладает противотромботической активностью, включая ингибирование агрегации тромбоцитов и образования тромбов у млекопитающих, и пригодно для предупреждения и лечения тромбоза, связанного с такими заболеваниями, как инфаркт миокарда, инсульт,артериальные периферические заболевания и диссеминированная внутрисосудистая коагуляция. Кроме того, изобретение относится к процессам получения кристаллической формы N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексил-аланинамида, его фармацевтической композиции и промежуточных продуктов. Гемостаз, биохимия свертывания крови,представляет комплексное явление, в котором нормальная цельная кровь и ткани тела спонтанно прекращают кровотечение из поврежденных кровеносных сосудов. Для эффективного гемостаза требуется сочетание активности сосуда, тромбоцита и плазменных факторов, так же,как и наличие контролирующего механизма для предупреждения чрезмерного тромбообразования. Дефекты, ошибки или неумеренность в любом из компонентов этого механизма могут привести к геморрагическим или тромботическим осложнениям. Адгезия тромбоцитов, распределение и агрегация на внеклеточном матриксе являются главными процессами в образовании тромба. Эти процессы медиируются группой родственных адгезивных гликопротеинов, а именно фибриногеном, фибронектином и фактором фон Виллебранда. Фибриноген является кофактором агрегации тромбоцитов, в то время как фибронектин поддерживает связывание тромбоцитов и реакцию распределения, и фактор фон Виллебранда важен для присоединения тромбоцитов к субэндотелиальным матриксам и для распределения на них. Участки связывания для фибриногена, фибронектина и фактора фон Виллебранда располагаются на протеиновом комплексе мембраны тромбоцита, известном как гликопротеин IIb/IIIa. Адгезивные гликопротеины, типа фибриногена, не связываются нормальными, находящимися в покое тромбоцитами. Однако, когда тромбоциты активированы агонистом, таким,как тромбин или аденозиндифосфат, тромбоцит 2 меняет свое состояние, возможно делая участок для связывания GPIIb/IIIa доступным для фибриногена. Соединение, лежащее в области настоящего изобретения, блокирует рецептор для фибриногена, и таким образом обладает вышеназванной антитромботической активностью. Сообщения о последних разработках Обнаружено, что в фибриногене, фибронектине и факторе фон Виллебранда для их взаимодействия с рецептором поверхности клетки необходимо присутствие Arg-Gly-Asp(RGD) (Ruoslahti E., Pierschbacher, Cell 1986, 44,517-18). Возможно, в функции фибриногена по присоединению тромбоцитов принимают участие две другие аминокислотные последовательности, а именно Gly-Pro-Arg последовательность и додекапептид, His-His-Leu-Gly-GlyAla-Lys-Gln-Ala-Gly-Asp-Val последовательность. Показано, что небольшие синтетические пептиды, содержащие RGD или додекапептид,связываются с тромбоцитарным GPIIb/IIIa рецептором и конкурентно ингибируют связывание фибриногена, фибронектина и фактора фон Виллибранда, а также ингибируют агрегацию активированных тромбоцитов. (Plow, et al., Proc.Gartner, et al., J. Biol. Chem. 1987. 260, 11, 89194). Индолиловые соединения, содержащие гуанидиноалканоил- и гуандиноалкеноиласпартиловые остатки, являются ингибиторами агрегации тромбоцитов, как сообщается у Tjoeng, etal., патент США 5 037 808 и 4 879 313. Патент США 4 992 463 (Tjoeng, et al.),выданный 12 февраля 1991, в целом раскрывает,что ряд арил и аралкил гуанидиноалкилпептидоподобных соединений проявляет активность как ингибитор агрегации тромбоцитов, и конкретно раскрывает моно- и диметоксифенильные пептидмиметические соединения и бифенилалкильное пептидмиметическое соединение. Патент США 4 857 508 (Adams, et al.),выданный 15 августа 1989 г, в целом раскрывает, что ряд гуанидиноалкилпептидных производных, содержащих концевые алкильные заместители, проявляет активность в качестве ингибитора агрегации тромбоцитов, и конкретно раскрывает ряд производных Ометилтирозина, бифенила и нафтила, содержащих концевую амидную функциональность.(1985), раскрывает, что ряд синтетических пептидов, включая arg-gly-asp-ser и gly-arg-gly-aspser способны ингибировать вызванную тромбином агрегацию тромбоцитов.Plow, E.F. et al., Procl. Natl. Acad. Sci. USA 79, 3711-3715 (1982), раскрывают, что тетрапептид глицил-L-пролил-L-аргинил-L-пролин ингибирует присоединение фибриногена к тромбоцитам человека.al.), выданный 28 июля 1987 г., раскрывает, что ряд пептидов, содержащих от шести до сорока аминокислот, которые содержат последовательность -arg-gly-asp, являются ингибиторами связывания тромбоцитов. Публикация Европейской Заявки 0 319 506, опубликованная 7 июня 1989 г., раскрывает,что ряд производных тетра-, пента и гексапептида, содержащих -arg-gly-asp-последовательность, являются ингибиторами агрегации тромбоцитов. В патенте США 5 023 233 сообщается, что аналоги циклического пептида, содержащие фрагмент Gly-Asp, являются антагонистом рецептора фибриногена. В рассматриваемых заявках США серийные номера 07/677 006, 07/534 385 и 07/460 777,поданных 28 марта 1991 г., 7 июня 1990 и 4 января 1990, соответственно, а также в патенте США 4 952 562 и в международной заявкеРСТ/США 90/05448, поданной 25 сентября 1990, все из которых представлены тем же заявителем, что и настоящее изобретение, сообщается, что пептиды и псевдопептиды, содержащие амино-, гуанидино, имидизалоильный и/или амидиноалканоильный и алкеноильный остатки, являются противотромботическими агентами. Пептиды и псевдопептиды, содержащие амино- и гуанидино алкил- и алкенил-бензоил,фенилалканоильные и фенилалкеноильные остатки являются противотромботическими агентами, как сообщается в рассматриваемой заявке США серийный номер 07/475 043, поданной 5 февраля 1990, и в международной заявкеРСТ/США 91/02471, поданной 11 апреля 1991,опубликованной в качестве международной публикацииWO 92/13117 29 октября 1992, и представленной тем же заявителем, что и настоящее изобретение. Алканоильные и замещенные алканоильные азациклоалкилформильные производные аспарагиновой кислоты являются ингибиторами агрегации тромбоцитов, как сообщается в патенте США 5053 392, поданном 1 декабря 1989 и заявленном тем же заявителем, что и настоящее заявление.N-замещенные производные азациклоалкилкарбонил циклической аминоациласпарагиновой кислоты являются противотромботическими агентами, как сообщается в патенте США 5 064 814, поданном 5 апреля 1990 теми же изобретателями, и заявлены тем же заявителем,что и настоящее изобретение. Производные азациклоалкилформилглицил аспарагиновой кислоты являются противотромботическими аген 001362 4 тами, как сообщается в патенте США 5 051 405, поданном 10 октября 1989, и заявленном тем же заявителем, что настоящее изобретение. Европейская патентная заявка 0 479 481,опубликованная 8 апреля 1992, раскрывает азациклоалкилалканоилглициласпартил аминокислоту как антагониста рецептора фибриногена. Европейская патентная заявка 0 478 362,опубликованная 1 апреля 1992, раскрывает азациклоалканоил пептидил -аланины в качестве антагонистов фибриногеновых рецепторов. Публикация РСТ патентной заявкиWO 95/10295 раскрывает азациклоалкилалканоильные пептиды и псевдопептиды формулы II: и, в частности, N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамид, ингибирующий агрегацию тромбоцитов и образование тромбина у млекопитающих и пригодный для предупреждения и лечения тромбоза. N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамид, полученный в соответствии с публикацией РСТ патентной заявкиWO 95/10295, является аморфным, гигроскопичным и физически нестабильным, так как он поглощает влагу. Публикация РСТ патентной заявкиWO 95/10295 не раскрывает негигроскопичную стабильную кристаллическую форму N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексил-аланинамида. Публикация РСТ патентной заявкиWO 95/10295 также раскрывает, что азациклоалкилалканоильные пептиды и псевдопептиды получают в целом с помощью стандартных методик твердофазного или жидкофазного синтеза пептидов с использованием исходных веществ и/или легко доступных промежуточных продуктов, полученных от компаний, поставляющих химические вещества, таких как Aldrich; Sigma;after 25 Years: The Design and Synthesis of Antagonists of Glucagon", Makromol. Chem. Macromol. Symp. 19, 31 (1988. Кроме того, РСТ патентная заявка публикацияWO 95/10295 раскрывает, что аморфные и гигроскопичные формыN-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида получают путем последовательно 5 го синтеза с С-концевой аминокислоты, как показано на схеме 1. Схема 1 РСТ патентная заявка публикацияWO 95/10295 не раскрывает образование тетраазациклоалкилалканоильных пептидов и псевдопептидов или, в частности, N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида из центрального ди(псевдопептида или пептида),где и N- и С-терминальные окончания центрального ди(псевдопептида или пептида) соединяются (сочетаются) с псевдоаминокислотами и/или аминокислотами с образованием тетраазациклоалкилалканоильных пептидов и псевдопептидов. Сущность изобретения Настоящее изобретение относится к негигроскопичным стабильным кристаллическим формам N-[N-[N-(4-пиперидин-4-ил)бутаноил)N-этилглицил]-(L)-аспартил]-(L)-циклогексил-аланинамида формулы I: Соединение обладает противотромботической активностью, включая ингибирование агрегации тромбоцитов и образования тромбов у млекопитающих, и пригодно в предупреждении и лечении тромбоза, связанного с такими заболеваниями, как инфаркт миокарда, инсульт, периферические артериальные заболевания и диссеминированное внутрисосудистое свертывание. Изобретение также относится к фармацевтической композиции негигроскопичной стабильной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида и ее промежуточных продуктов. Изобретение также относится к получению тетра-азациклоалкилалканоильного пептида или псевдопептида соединения формулы II: Е 1 представляет Н; Е 2 представляет -углеродную боковую цепь природной -аминокислоты, Н, алкил,циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил,или Е 1 и Е 2 взяты вместе с азотом и атомами углерода, через которые Е 1 и Е 2 связаны, и образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,арил, аралкил, алкиларил или алкиларалкил;n равно 0-6; и р равно 1-4,и, в частности, к получению негигроскопичной стабильной кристаллической формы N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексил-аланинамида. Краткое описание фигур Фиг. 1 представляет картину дифракции рентгеновских лучей порошка образца негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способом А. Фиг. 2 представляет картину дифракции рентгеновских лучей порошка образца негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способом В(а). Фиг. 3 представляет картину дифракции рентгеновских лучей порошка образца негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 13, способом B(b). Фиг. 4 представляет картину дифракции рентгеновских лучей порошка образца негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 14. Фиг. 5 представляет картину дифракции рентгеновских лучей порошка образца негигро 7 скопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида, полученного в примере 14. Фиг. 6 представляет график изотермальной микрокалориметрии интенсивности выхода как функцию от времени для трех различных экспериментов, которые выполнены, как описано в примере 15. Эксперименты определяли термальную активность различных кристаллических форм N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексил-аланинамида при воздействии паров различных растворителей. Кривая (А) на фиг. 6 показывает, что сильная экзотермическая реакция происходила, когда N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексил-аланинамид, полученный в соответствии с примерами 5 или 11,подвергался воздействию 80% RH (относительная влажность) (насыщенный раствор КСl) при 40 С в течение 30 мин, причем во время этого воздействия гигроскопичная форма соединения превращается в негигроскопичную форму соединения. Кривая (В) на фиг. 6 показывает отсутствие экзотермического превращения, происходящего, когда гигроскопичный N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексил-аланинамид, полученный в соответствии с примерами 5 и 11, подвергается воздействию паров метанола при 40 С(растворитель отличный от воды, в котором соединение растворимо), и таким образом метанол не обеспечивает подвижности в кристаллах этой формы для превращения в негигроскопичную форму. Кривая (С) на фиг. 6 показывает отсутствие экзотермического превращения, происходящего, когда негигроскопичный N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексил-аланинамид, полученный в соответствии с примером 13, подвергается воздействию 40 С/80% RH, и, таким образом, негигроскопичная форма соединения при таких условиях не претерпевает изменение формы, то есть она является стабильной формой. Фиг. 7 представляет график изотермальной микрокалориметрии интенсивности выхода как функцию от времени для трех различных экспериментов, которые выполнены, как описано в примере 15. Эксперимент определял термальную активность превращения гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида в свою не гигроскопичную форму, когда она подвергается воздействию 80% RH при 40 С, 50 С и 60 С. Фигура показывает, что превращение занимает приблизительно 24 ч при 40 С, 6,5 ч при 50 С и 3 ч при 60 С. 8 Фиг. 8 представляет график изотермальной микрокалориметрии интенсивности выхода как функцию от времени для трех различных экспериментов, которые выполнены, как описано в примере 15. Эксперимент определял термальную активность превращения гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида в свою негигроскопичную форму, когда она подвергается воздействию при 60 С 65% RH, 75% RH, 80% RH и 100% RH. Рельеф пиков фиг. 8 показывает, что наиболее высокая относительная влажность вызывает более быстрое превращение. Кроме того, кривые показывают, что негигроскопичная форма соединения появляется при 100% RH и 60 С без поглощения жидкости, которая происходит с гигроскопичной формой при комнатной температуре. Основываясь на этих результатах можно ожидать, что скорость превращения в негигроскопичную форму выше, чем скорость поглощения жидкости гигроскопичной формой при 60 С. Фиг. 9 представляет сравнение % увеличения веса относительно % RH для гигроскопичной (темный квадрат) и негигроскопичной (темный кружок) форм N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида при 25 С, при проведении эксперимента, как описано в примере 16. Фиг. 9 показывает, что гигроскопичная форма поглощает больше воды, чем негигроскопичная форма при повышении RH, и более резко выражено при относительной влажности более 60%. Кроме того, фиг. 9 показывает, что гигроскопичная форма соединения не возвращается к своему исходному % весу, тогда как негигроскопичная форма соединения возвращается к своему первоначальному % весу. Подробное описание изобретения При использовании выше и при описании изобретения следующие термины, если не указано другое, должны пониматься таким образом. Следующие сокращения, используемые здесь, включают: ВОС (трет-бутилоксикарбонил), CBZ (бензилоксикарбонил), Gly (глицин), Asp (аспарагиновая кислота), Obzl (бензилокси), TFA (трифторуксусная кислота), Cha (-циклогексилаланин), EtOAc (этилацетат), DMF (диметилформамид),DCC(дициклогексилкарбодиимид), НОВТ (гидроксибензотриазол),TBTU (2-1H-бензотриазол-1-ил-1,1,3,3-тетраметилуроний тетра-фторборат), DI (деионизированная вода), PNP (п-нитрофенол), PFP (пентафторфенол), DCU (дициклогексилмочевина),NMM (N-метилморфолин), МТВЕ (метил третбутиловый эфир), RH (относительная влажность), THF (тетрагидрофуран), PipBu (4 пиперидинмасляная кислота) и PipBuen (4 9 пиперидин) бутилиденилкарбоновая представляет соединение формулы"Фармацевтически приемлемая соль" означает солевую форму исходного соединения формулы I, которая относительно безвредна для пациента при использовании в терапевтических дозах таким образом, что полезные фармацевтические свойства исходного соединения формулы I не нарушаются от побочных эффектов,приписываемых противоположному иону этой солевой формы. Фармацевтически приемлемая соль также включает цвиттер-ион или промежуточную соль соединения формулы I."Алкил" означает насыщенную алифатическую углеводородную группу, которая может иметь прямую или разветвленную цепь, содержащую от около 1 до около 20 атомов углерода. Разветвленность означает, что низшая алкильная группа, такая как метил, этил или пропил,присоединяется к линейной алкильной цепи. Предпочтительно прямые или разветвленные алкильные группы представляют группы "низшего алкила", которые являются алкильными группами, имеющими от 1 до 10 атомов углерода. Наиболее предпочтительные низшие алкильные группы имеют от 1 до около 6 атомов углерода."Циклоалкил" означает карбоциклическую группу, имеющую одно или большее количество колец и от около 3 до около 10 атомов углерода. Предпочтительные циклоалкильные группы включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и декагидронафтил."Циклоалкилалкил" означает алкильную группу, замещенную циклоалкильной группой. Предпочтительные циклоалкильные группы включают циклопентилметил, циклогексилметил,циклогексилэтил,декагидронафт-1 илметил и декагидронафт-2-илметил."Алкилциклоалкил" означает циклоалкильную группу, замещенную алкильной группой. Например, алкилциклоалкильные группы включают 1-,2-,3- или 4-метил или этилциклогексил."Алкилциклоалкилалкил" означает алкильную группу, замещенную алкилциклоалкильной группой. Например, алкилциклоалкильные группы включают 1-,2-,3-или 4-метилили этилциклогексилметил или 1-, 2-, 3- или 4 метил или этилциклогексилэтил."Азациклоалкан" означает алифатическое кольцо, содержащее атом азота. Предпочтительные азациклоалканы включают пиролидин и пиперидин."-углеродная боковая цепь природной аминокислоты" означает остаток, замещенный-аминокислоты включают изопропил, метил и карбоксиметил для валина, аланина и аспарагиновой кислоты, соответственно. Термин "аминозащитная группа" означает легко удаляемую группу, которая известна в настоящем уровне техники как аминогруппа,защищающая от нежелательных реакций во время процесса синтеза, и которая может селективно удаляться. Хорошо известно, что защитные аминогруппы используются для защиты групп от нежелательных реакций во время процесса синтеза, и многие подобные защитные группы хорошо известны, например, по работе Т.Н. Greene and P.G.M. Wuts, Protective GroupsSons. New York (1991), включенной здесь в качестве ссылки. Предпочтительные защитные аминогруппы представляют ацил, включая формил, ацетил, хлорацетил, трихлорацетил, онитрофенилацетил,о-нитрофеноксиацетил,трифторацетил, ацетоацетил, 4-хлорбутирил,изобутирил, о-нитроциннамоил, пиколиноил,ацилизотиоцианат, аминокапроил, бензоил и им подобные, и ацилокси, включая метоксикарбонил,9-флуоренилметоксикарбонил,2,2,2 трифтороэтоксикарбонил,2 триметилсилилэтоксикарбонил, винилоксикарбонил, аллилоксикарбонил,t-бутилоксикарбонил(ВОС),1,1 диметилпропинилоксикарбонил,бензилоксикарбонил (CBZ), р-нитробензилоксикарбонил, 2,4-дихлорбензилоксикарбонил и им подобные. Термин "кислотолабильные аминозащитные группы" означает защитные аминогруппы,определенные выше, которые легко удаляются при обработке кислотой, оставаясь стабильными при обработке другими реагентами. Предпочтительная кислотолабильная защитная аминогруппа представляет трет-бутоксикарбонил(ВОС). Термин "гидрирующая лабильная (неустойчивая к восстановлению) аминозащитная группа" означает аминозащитную группу, определенную выше, которая легко удаляется при гидрогенации, оставаясь относительно стабильной при обработке другими реагентами. Предпочтительная неустойчивая к восстановлению аминозащитная группа представляет бензилоксикарбонил (CBZ). Термин "защитная группа кислоты" означает легко удаляемую группу, которая известна в настоящем уровне техники как группа, защи 11 щающая карбоновую кислоту (-СO2 Н) против нежелательных реакций во время процесса синтеза и селективно удаляемая. Использование защищающих карбоновую кислоту групп хорошо известно в настоящем уровне техники, а также известны и многие подобные группы,например, по работе Т.Н. Green and P.G.M.edition, John WileySons. New York (1991),включенной здесь в качестве ссылки. Примеры групп, защищающих карбоновую кислоту,включают сложные эфиры, такие как метоксиметил, метилтиометил, тетрагидропиранил, бензилоксиметил, замещенный и незамещенный фенацил, 2,2,2-трихлорэтил, трет-бутил, циннамил, замещенный и незамещенный бензил, триметилсилил и им подобные, и амиды и гидразиды, включающие N,N-диметил,7-нитроиндолил,гидразид, N-фенилгидразид и им подобные. Термин "гидрирующая лабильная защитная группа кислоты" означает такие из указанных выше кислотных защитных групп, которые легко удаляются при гидрогенации, оставаясь относительно стабильными при обработке другими реагентами. Предпочтительной гидрирующей лабильной защитной группой кислоты является бензил."Арил" означает фенильную или нафтильную группу."Замещенный арил" означает фенильную или нафтильную группу, замещенную одной или большим количеством арильных групп, которые могут быть такими же или отличными от нее, причем "заместитель арильной группы" включает алкил, алкенил, алкинил, арил, аралкил, гидрокси, алкокси, арилокси, аралкокси,гидроксиалкил, ацил, формил, карбокси, алкеноил, ароил, гало, нитро, тригалометил, циано,алкоксикарбонил, арилоксикарбонил, аралкоксикарбонил, ациламино, ароиламино, карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбамоил, аралкилкарбамоил, алкилсульфонил,алкилсульфинил, арилсульфонил, арилсульфинил, аралкилсульфонил, аралкилсульфинил или"Аралкил" означает алкильную группу,замещенную арильным радикалом. Предпочтительные аралкильные группы включают бензил,нафт-1-илметил, нафт-2-илметил и фенетил."Замещенный аралкил" означает аралкильную группу, замещенную в арильной части одной или большим количеством заместителей арильной группы."Гетероциклил" означает от около 4- до около 15-членную моноциклическую или полициклическую кольцевую систему, в которой один или большее количество атомов в кольце представляет элемент, отличный от углерода,например, азот, кислород и сера. Предпочтительные гетероциклические группы включают пиридил, пиримидил и пирролидил."Замещенный гетероциклилозначает гетероциклическую группу, замещенную одним или большим количеством заместителей арильной группы."Гигроскопичность" означает сорбцию,имея в виду приобретенное или уже имеющееся количество воды, необходимое для осуществления химических или физических свойств вещества (Eds. J. Swarbrick and J.C. Boylan, Encyclopedia of Pharmaceutical Technology, Vol. 10, p. 33). Предпочтительные воплощения Предпочтительные соединения, полученные в соответствии с настоящим изобретением,описаны формулой II, где Е 2 представляет Н,алкил, гидроксиметил, 1-гидроксиэтил, меркаптометил, 2-метилтиоэтил, карбоксиметил, 2 карбоксиэтил,4-аминобутил,3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замешенный аралкил,гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2, взятые вместе с атомами азота и углерода, через которые Е 1 и Е 2 связываются,образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо, при условии, что гетероциклилалкил отличен от индол-3-илметила. Более предпочтительные соединения, полученные в соответствии с настоящим изобретением, описаны формулой II, где Е 2 представляет Н, алкил, гидроксиметил, 1-гидроксиэтил,меркаптометил, 2-метилтиоэтил, карбоксиметил, 2-карбоксиэтил, 4-аминобутил, 3-гуанидинопропил, циклоалкил, циклоалкилалкил,алкилциклоалкил, алкилциклоалкилалкил, арил,замешенный арил, аралкил, замещенный аралкил, или Е 1 и Е 2, взятые вместе с атомами азота и углерода, через которые Е 1 и Е 2 связываются,образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо. Еще более предпочтительное соединение настоящего изобретения описано формулой II,где Е 2 представляет Н, алкил, гидроксиметил, 1 гидроксиэтил, меркаптометил, 2-метилтиоэтил,карбоксиметил, 2-карбоксиэтил, 4-аминобутил,3-гуанидинопропил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, или Е 1 и Е 2, взятые вместе с атомами азота и углерода, через которые Е 1 и Е 2 связываются,образуют 4-, 5-, 6- или 7-членное азациклоалкановое кольцо. Еще одно предпочтительное соединение по настоящему изобретению описано формулойJ представляет Н, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил или замещенный аралкил;n равно 2-6; и р равно 1-2. Более предпочтительное конкретное воплощение, полученное в соответствии с настоящим изобретением, описано формулой IIа,где В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил;n равно 3 или 4. Еще одно конкретное воплощение, полученное в соответствии с настоящем изобретением, описано формулой II, где В представляет алкил;n равно 3 или 4; и р равно 1. Еще одно конкретное воплощение, полученное в соответствии с настоящим изобретением, описано формулой IIа, которая представляетN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамид. Другое воплощение настоящего изобретения представляет образование стабильной негигроскопичной кристаллической формы N-[N[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. В соответствии с изобретением, эта форма соединения имеет перспективу в качестве стабильной готовой препаративной формы соединения. Стабильная негигроскопичная кри 001362 14 сталлическая форма N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида также имеет высокую точку плавления и показывает отсутствие тенденции к абсорбции воды. Стабильная форма также демонстрирует уникальную и неожиданную стабильность при воздействии влажности и температуры даже при их избытке при транспортировке,при производстве дозированных форм или при длительной перевозке или при хранении. Наличие этих свойств также удобно при производстве дозированных форм. Также в результате превращения в стабильную форму не теряется вещество или его чистота, и не происходит пагубного влияния на свойства частиц. Следует понимать, что настоящее изобретение охватывает все комбинации определенных здесь предпочтительных соединений, предпочтительных воплощений и конкретных воплощений. Соединения настоящего изобретения можно применять в форме свободного основания или кислоты, их цвиттер-ионной соли или в форме их фармацевтически приемлемой соли. Все формы входят в область настоящего изобретения. Когда соединение настоящего изобретения замещено основным остатком, образуется аддитивная соль кислоты, которая является простой и более удобной формой для использования; и на практике, используется солевая форма в количестве, соответствующем форме свободного основания. Кислота, которая может быть использована для получения аддитивной соли кислоты, предпочтительно включает такую кислоту, которая при соединении со свободным основанием образует такую фармацевтически приемлемую соль, анион которой не является токсичным для пациента в фармацевтических дозах этой соли, так что полезный ингибирующий эффект по агрегации тромбоцитов и образованию тромба, присущий свободному основанию,не нарушается побочными эффектами, вызываемыми анионом. Хотя фармацевтически приемлемые соли названного основного соединения являются предпочтительными, все соли присоединения кислоты пригодны в качестве источников для формы свободного основания, даже если конкретная соль, как таковая, желательна только в качестве промежуточного продукта,как, например, в случае, если соль образовалась только с целью очистки и идентификации, или если она используется в качестве промежуточного вещества при получении фармацевтически приемлемой соли путем ионообменного процесса. Фармацевтически приемлемые соли, входящие в область настоящего изобретения, являются производными следующих кислот: минеральные кислоты, такие как соляная кислота,серная кислота, фосфорная кислота и сульфаминовая кислота; органические кислоты, такие, 15 как уксусная кислота, лимонная кислота, молочная кислота, винная кислота, малоновая кислота, метансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, птолуолсульфоновая кислота, циклогексилсульфаминовая кислота и хинная кислота и т.д. Соответствующие аддитивные соли кислот включают следующие: галогеноводороды, например, хлоргидрат и бромгидрат, сульфат,фосфат, нитрат, сульфамат, ацетат, цитрат, лактат, тартрат, малонат, оксалат, салицилат, пропионат, сукцинат, фумарат, малеат, метиленбисгидроксинафтоаты, гентизаты, мезилаты,изетионаты и ди-п-толуолтартратметансульфонаты, этансульфонат, бензолсульфонат,п-толуолсульфонат, циклогексилсульфамат и соль хинной кислоты, соответственно. В соответствии с другими признаками изобретения аддитивные соли кислоты соединений данного изобретения получают путем реакции свободного основания с соответствующей кислотой при применении или при использовании известных способов. Например, аддитивные соли кислоты соединения данного изобретения получают или путем растворения свободного основания в водном или водно-спиртовом растворе или в других подходящих растворителях,содержащих соответствующие кислоты, и выделяя соль путем выпаривания раствора, или реакцией свободного основания и кислоты в органическом растворителе, причем в этом случае соль выделяется непосредственно или может быть получена путем концентрации раствора. Соединения данного изобретения могут быть заново выделены из солей при применении или адаптировании известных способов. Например, исходные соединения изобретения могут быть получены из своих кислых аддитивных солей путем обработки щелочью, например,водным раствором бикарбоната натрия или водным раствором аммиака. Когда соединение изобретения замещено остатком кислоты, могут быть получены основные соли, которые являются более удобной для использования формой; и на практике, использование солевой формы равняется использованию свободной кислой формы. Основания, которые могут быть использованы для получения аддитивных и основных солей включают предпочтительно такие основания, которые со свободной кислотой образуют фармацевтически приемлемые соли, катионы которых являются нетоксичными для организма животного при применении фармацевтических доз солей, так что полезный ингибиторный эффект по агрегации тромбоцитов и образованию тромба, присущий свободной кислоте, не уничтожается побочными эффектами, приписываемыми катиону. Фармацевтически приемлемые соли, включая,например, соли щелочных и щелочно-земельных металлов, входящие в область данного изо 001362 16 бретения, являются производными следующих оснований: гидрид натрия, гидроксид натрия,гидроксид калия, гидроксид кальция, гидроксид аммония, гидроксид лития, гидроксид магния,гидроксид цинка, аммиак, этилендиамин, Nметилглюкамин, лизин, аргинин, орнитин, холин, N,N'-дибензолэтилен-диамин, хлорпрокаин,диэтаноламин,прокаин,Nбензолфенэтиламин, диэтиламин, пиперазин,трис(гидроксиметил)-аминометан,гидроксид тетраметиламмония и им подобные. Соли металлов соединений настоящего изобретения могут быть получены при взаимодействии гидрида, гидроксида, карбоната или подобных реактивных соединений выбранного металла в водном или органическом растворителе со свободной кислой формой соединения. Применяемым водным растворителем может быть вода, или это может быть смесь воды и органического растворителя, предпочтительно спирта, такого, как метанол или этанол, кетона,такого, как ацетон, алифатического эфира, такого, как тетрагидрофуран, или сложного эфира,такого как этилацетат. Такие реакции обычно проводятся при температуре окружающей среды, но при желании они могут проводится с нагреванием. Соли амина соединений настоящего изобретения могут быть получены взаимодействием амина в водном или органическом растворителе со свободной кислой формой соединения. Подходящие водные растворители включают воду и смеси воды со спиртами, такими как метанол или этанол, эфирами, такими, как тетрагидрофуран, нитрилами, такими как ацетонитрил, или кетонами, такими как ацетон. Соли аминокислот могут быть получены таким же образом. Соединения данного изобретения могут быть выделены из солей путем применения или адаптирования известных способов. Например,исходные соединения изобретения могут быть выделены из их аддитивных основных солей при обработке кислотой, например, соляной кислотой. Полезные сами по себе в качестве активных соединений, соли соединений изобретения полезны также для очистки соединений, например, при использовании разницы в растворимости между солями и исходными соединениями,побочными продуктами и/или исходными веществами при использовании методик, хорошо известных квалифицированным специалистам. Соединения настоящего изобретения могут иметь центры асимметрии. Эти центры асимметрии могут независимо быть или в R,или в S конфигурации. Для квалифицированных специалистов также очевидно, что некоторые соединения формулы I могут иметь геометрические изомеры. Геометрические изомеры включают цис и транс формы соединений изобретения, имеющих алкенильные остатки. Настоящее 17 изобретение включает отдельные геометрические изомеры и стереоизомеры и их смеси. Такие изомеры могут быть выделены из их смеси при применении или адаптировании известных способов, например, при использовании хроматографических методик и методик перекристаллизации, или их получают раздельно из соответственных изомеров их промежуточных продуктов, например, при применении или адаптировании описанных здесь способов. Патентная заявка США 08/138 820 и 08/476 750, включенная здесь в качестве ссылки, описывает способ получения аморфного соединения формулы II, и, в частности, аморфного соединения формулы I. Новый способ по изобретению получения соединения формулы II, и в частности, кристаллического соединения формулы I по данному изобретению, описан в синтезе, показанном на схеме II, где В, Е 1, Е 2, G, R, m, n и р такие, как определено выше, и Р 1 представляет неустойчивую к гидрированию кислотную защитную группу, такую как бензил, Р 2 представляет лабильную (неустойчивую) аминозащитную группу, такую как трет-бутоксикарбонил (ВОС), и Р 3 представляет неустойчивую к гидрированию аминозащитную группу, такую как бензилоксикарбонил (CBZ). Схема II 18 быть оставлены или, если требуется, последовательно удалены известными способами для получения желаемых продуктов или промежуточных продуктов (смотри, например, Green, "Protective Groups in Organic Synthesis". Wiley, NewYork, 1981). Выборочная защита или ее снятие также могут быть необходимы или желательны для того, чтобы обеспечить превращение или удаление существующих заместителей или для того, чтобы обеспечить возможность протекания последовательных реакций с целью получения конечного желаемого продукта. Процесс, показанный на схеме II, является примером получения соединения формулы II,однако следует понимать, что соединение формулы I получают, используя соответствующие исходные вещества. При получении соединения формулы I в соответствии со схемой II В представляет этил, Е 1 представляет Н, Е 2 представляет циклогексилметил, G представляет NH2, R представляет Н, m представляет 3, n представляет 3, р представляет 1, Р 1 представляет бензил, Р 2 представляет ВОС и Р 3 представляетCBZ. В соответствии с изобретением альтернативный процесс получения соединения формулы I такой же, как показано на схеме II, за исключением того, что соединение формулы III,где Р 3 такой же, как определено выше, используется вместо соединения формулы IV:CBZ для получения промежуточного продукта формулы V: где В представляет этил, Е 1 представляет Н, Е 2 представляет циклогексилметил, G представляет NH2, р представляет 1, Р 1 представляет бензил и Р 3 представляет CBZ. На схеме II показан пятистадийный способ получения соединения изобретения, начинающийся с получения центрального дипептидного промежуточного продукта по изобретению формулы VI: В процессе получения соединений формулы II или их промежуточных продуктов желательно или необходимо предотвратить перекрестные реакции между химически активными заместителями, присутствующими на природных аминокислотах или псевдоаминокислотах. Заместители могут быть защищены стандартными блокирующими группами, которые могут где В, р, Р 2 и Р 1 такие же, как определено выше. В случае получения соединения формулы I центральное дипептидное промежуточное вещество в соответствии с изобретением представляет из себя BOC-N(Et)Gly-(L)-Asp(OBzl)-ОН. Центральное дипептидное промежуточное вещество 19 получают без защиты свободного остатка карбоновой кислоты. На стадии 2 схемы II реакция образования дипептида может быть осуществлена или в дихлорметане или в смеси этилацетата - с или безDMF в качестве сорастворителя - и органических оснований, таких, как NMM, и может проводиться при температуре от примерно комнатной и до примерно 40 С. Активация аминокислот или псевдоаминокислот следующей формулы: для реакции может быть произведена с использованием невыделенных активных сложных эфиров с п-нитрофенолом, пентафторфенолом иN-гидроксисукцинимидом посредством действия дициклогексилкарбодиимида. Время реакции колеблется от около 1 до около 20 ч в зависимости от того, какие аминокислоты или псевдоаминокислоты должны быть соединены, в зависимости от активирующего агента, растворителя и температуры. Центральный дипептидный продукт стадии I не обязательно должен выделяться. Реакционную смесь стадии I обычно промывают водой или разбавленной водной кислотой (например водной НСl), и затем непосредственно используют на стадии 2 без высушивания. Например, когда используется активный сложный эфир фенольного основания центральный дипептидный продукт экстрагируют из реакционной смеси в подщелоченную воду,затем повторно экстрагируют из подкисленного водного раствора обратно в органический растворитель; и раствор реагирует непосредственно как на стадии 2. Дипептидное промежуточное вещество формулы VI используется для получения трипептидного промежуточного вещества по изобретению формулы VII: где B, E, F, G, р и Р 1 такие же, как определено выше, и Р 2' представляет Р 2 или TFAH-. Когда Р 2' представляет TFAH-, символ обозначает диссоциацию TFA с образованием F3 ССО-2 Н+,где H+ (протонирует) обеспечивает положительный заряд концевому амину в соединении формулы VII, то есть образуя TFA соль формулы В случае получения соединения формулыI, трипептидное промежуточное вещество по изобретению представляетP2'-N(Et)Gly-LAsp(Obzl)-(L)-Cha-NH2. На стадии 2 реакция аминокислоты или псевдоаминокислоты для образования цен 001362 20 трального дипептида может быть произведена или в дихлорметане или в смеси этилацетата иDMF или THF, и при температуре около или выше комнатной. Активирование центрального дипептида следующей формулы: для реакции может быть произведено с использованием невыделенного активного сложного эфира пентафторфенола или N-гидроксисукцинимида посредством воздействия дициклогексилкарбодиимида. Активация также может быть осуществлена с использованием хлорформиата изопропила. Время реакции может варьироваться в зависимости от соединяемой аминокислоты или псевдоаминокислоты, активирующего агента, температуры и колеблется от около 1 до около 20 ч. Трипептидный продукт не обязательно должен выделяться. Если трипептидный продукт не выделяется, реакционную смесь промывают водным органическим основанием,таким как N-метилморфолин, и водным раствором кислоты, таким как водная НСl, и реагирует"как есть" через способ стадии 3 после промывания водой и без высушивания. На стадии 3 схемы II удаление защитных групп, таких как ВОС, из трипептидного продукта стадии 2 может быть осуществлено с использованием трифторуксусной кислоты в дихлорметане или с использованием смеси НВr в уксусной кислоте и этилацетата. Реакция может протекать при комнатной температуре и требует около 1 ч (НВr способ) и около 2 ч (TFA способ). Кислый солевой продукт трипептида выделяют фильтрацией в виде кристаллического твердого вещества непосредственно из реакционной смеси (НВг способ) или после частичного удаления растворителя путем дистилляции и добавления неполярного растворителя к остатку в сосуде. Дальнейший процесс по изобретению описан как единичный связанный процесс быстрого и простого получения TFAH-N(Et)Gly-(L)Asp(Obzl)-(L)-Cha-NH2 из BOC-N(Et)Gly-OH,который является реакцией в одном сосуде,включающей две первые стадии реакций по схеме II и обработку TFA.TFAH-N(Et)Gly-(L)-Asp(Obzl)-(L)-ChaNH2 получают в единственном числе так, как оно непосредственно кристаллизуется из концентрированного реакционного раствора. Процесс концентрации помогает избежать трех последующих отдельных реакций в схеме II и решает проблему осуществления простого, доступного по времени и по цене синтеза, который пригоден в условиях производства. Схема II показывает построение полипептида в обратном порядке, начиная с Nзащищенной аминокислоты и затем последовательного добавления к карбоксильному концу,что является противоположным общепринятому 21 порядку, когда полипептид конструируется путем последовательного амидирования аминных окончаний защищенной С-концевой аминокислоты. Этот обратный способ синтеза по изобретению требует защиты азота только первой аминокислоты, что дает возможность начинать с этой точки, двигаясь вперед по последовательности аминокислот, не имеющих защиты как на аминном, так и на кислотном окончании(исключая функциональные группы боковых цепей). Обратный метод синтеза также упрощает получение соединений формулы II, и, в частности, соединения формулы I, позволяя использовать поточную производственную технологию в противоположность способу периодической загрузки, который обычно используется в жидкофазной пептидной химии. Новый подход снижает стоимость производства,снимая требования к приобретению аминокислот, защищенных на концевых аминах. Специального оборудования, реагентов или аналитической методологии не требуется. Другой способ по изобретению состоит в образовании стабильного негигроскопичного кристаллического N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, репродуктивно пропущенного с помощью нового способа превращения в твердом состоянии из гигроскопичного кристаллического N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, полученного способом, описанным в схеме II и с помощью отмеченных альтернативных стадий реакции. Гигроскопичная кристаллическая формаN-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида физически нестабильна и превращается под воздействием влажности и температуры в высоко стабильную, негигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида. Общие условия в соответствии с изобретением превращения из гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида в высокостабильную негигроскопичную кристаллическую форму N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этиглицил]-(L)-аспартил]-(L)циклогексилаланинамида осуществляются при статических и динамических условиях. Статический процесс по изобретению описан как статическое превращение, так как он включает воздействие на гигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, помещенного в неподвижную емкость, такую как пробирка или лоток, в определенные условия температуры и 22 влажности в камере с контролируемыми условиями среды. Это "статическое" превращение производится при температуре и относительной влажности от около 20 С до около 80 С, более предпочтительно при от около 40 С до около 80 С и при от около 40% до около 100% RH,предпочтительно от около 65 до около 80% RH. Динамический процесс по изобретению описан как динамическое превращение, так как он включает воздействие на гигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида при инкубировании его при такой же температуре и влажности, как и при статическом превращении, но также и при использовании средств перемешивания, включающих обработку в барабане гигроскопичной кристаллической формы N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида в колбе роторного испарителя или в цилиндрическом сосуде (во влажной печи) с пропеллерной мешалкой. Следующие примеры иллюстрируют данное изобретение, не ограничивая его. Если не указано другого, данные массспектрального анализа получены способом бомбардировки быстрыми атомами низкого разрешения, производимым на VG 70SE с "расчетными" значениями (М+Н)+. Спектральные данные ядерно-магнитного резонанса (ЯМР) получены на Brucker ACF 300 в D2O. Флэшхроматография выполнена на силикагеле. Жидкостная хроматография высокого разрешения(ЖХВР) выполнена на колонках с обращенной фазой С-18 для размера частиц от 8 до 15 мк. Если не указано другого, картины рентгеновской дифракции порошка получены с использованием дифрактометра Siemens D5000 с Сu источником радиации (1,8 кВт, 45 кВ и 40 мА) для сканирования образцов. Образцы до измерения размалывали, чтобы исключить влияние размера частиц на интенсивность пика. Приблизительно 60 мг образца загружали в 1,5 х 1 см емкость для образца и сканировали в интервале 3-40 2 тэта (2) с размером шага 0,04 и полной экспозицией в 1 с на шаг. Пример 1. Получение BOC-N(Et)Gly-(L)Asp(OBzl)-ОН (стадия I схемы II). В трехгорлую однолитровую колбу с круглым дном помещают 51 г (0,25 моля) BOCN(Et)Gly-OH, 35 г (0,25 моля) PNP, 400 мгEtOAc и 100 мл DMF. Смесь перемешивают до образования раствора и охлаждают до 4-6 С. Раствор 51,5 г (0,25 моля) DCC в 125 мл EtOAc добавляют по каплям в течение 10 мин, одновременно поднимая температуру с около 5 С до около 8 С. После того, как весь DCC добавлен,удаляют охлаждающую баню и оставляют смесь перемешиваться на 1,5 ч, причем она нагревается до комнатной температуры (20-22 С). В это 23 время образуется твердый осадок DCU. Завершение образования PNP эфира определяется с помощью ЖХВР (по исчезновению BOCN(Et)Gly-OH). Реакционную смесь фильтруют и остаток DCU промывают 2-50 миллилитровыми порциями EtOAc и смыв добавляют к фильтрату. DCU отбрасывают. К перемешанному профильтрованному раствору добавляют 67 г (0,3 моля) H2N-(L)Asp(Obzl)-ОН в виде взвеси в 150 мл (138 г, 1,36 моля) NMM. Смесь нагревают до 38-40 С и поддерживают эту температуру в течение 41 ч,это тот момент, при котором аналитическая ЖХВР показывает полное использование BOCN(Et)Gly-OPNP. Реакционную смесь охлаждают до 25 С и непрореагировавший H2N-(L)Asp(Obzl)-ОН отфильтровывают. Раствор охлаждают и повторно фильтруют для получения дополнительных 1,2 г (21,7 г выделено; 11,2 г представляют 20% избыток и 10,5 г (0,047 моля) представляют непрореагировавшее вещество). Профильтрованный раствор экстрагируют в двухлитровой воронке Сквиба одной 500 миллилитровой порцией деионизированной воды, а затем двумя 250-миллилитровыми порциями. Объединенный водный раствор экстрагируют тремя 300-миллилитровыми порциями 1:1 MTBE/EtOAc для удаления остаточного PNP(аналитическая ЖХВР показывает только следовые остатки), затем охлаждают до 5 С и подкисляют от рН 8,9 до рН 1,79 добавлением по каплям 150 мл концентрированной НСl. Подкисленный водный раствор экстрагируют двумя 200-миллилитровыми порциями EtOAc. ЖХВР анализ жидкости не показывает остатка желаемого продукта. EtOAc экстракты объединяют,сушат над MgSO4, фильтруют и концентрируют на роторном испарителе при 35 С. Полученное в результате бледно-оранжевое масло выкачивают при 35 С для максимального удаления остаточного растворителя для получения 85,68 г(21,3 ммоля, выход 85,5%, непроверенного на остаточный растворитель). Характеристика: ЯМР (250 МГц): 7,3 м.д. (с), 5,1 м.д. (с), 3,3 м.д. (д.кв.), 3,0 (д.кв.), 1,4 м.д. (с), 1,1 (т); МС: М=408; М+1 набл=409; ЖХВР: 90,79 А% (3,87 А% п-нитрофенол,нескорректированный для е); Элементный анализ: C20H28N2O7: Н, N,Снайдено 57, 54, Срассчитано 58, 81. Пример 2. Получение BOC-N(Et)Gly-(L)Asp(OBzl)-(L)-Cha-NH2 (стадия 2 схемы II). Способ А. Изопропилхлорформиатный способ. Один эквивалентBOC-N(Et)Gly-(L)Asp(OBzl)-ОН растворяют в EtOAc, (6-8 объемов; 1:6,5 вес:объем) и поддерживают температуру между -15 и 0 С. NMM (1 эквивалент) добавляют во время поддерживания температуры от около -15 С до около 0 С. Изопропилхлор 001362 24 формиат (1-1,1 эквивалента) добавляют в защищенный дипептидный раствор при температуре от -15 до 0 С. Реакцию поддерживают при температуре от -15 до около 0 С. Раствор H2N-(L)Cha-NH2 (1 эквивалент) в THF (10 объемов; 1:10 вес/объем) добавляют к охлажденному дипептидному раствору, поддерживая температуру от около -15 до 0 С. Реакция проводится под текущем контролем (ЖХВР) образцов, полученных на 15 мин, 1 ч и 2 ч, чтобы оценить завершенность реакции. (Реакция закончена, когда количество наблюдаемого дипептида составляет меньше 10% площади при ЖХВР исследовании). ВОС-трипептидный продукт осаждают непосредственно из реакционного раствора и фильтруют из реакционной смеси, промываютEtOAc (2 х, 1 объем; вес:объем) и сушат в вакууме. Обычный выход составляет 60%, с очисткой 90 А%; обычно наблюдается 1 А% эпимерного диастереомера аспарагиновой кислоты.EtOAc повторно образующаяся взвесь дает конечный выход 60% BOC-N(Et)Gly-(L)Asp(OBzl)-(L)-Cha-NH2 и улучшает чистоту до 95 А% при снижении диастереомера до 0,05%. Конкретный пример изопропилхлорформиатного способа состоит в проведении основного процесса примера а с использованием 4,55 г (8,1 ммоля) BOC-N(Et)Gly-(L)-Asp(OBzl)-ОН,причем количество полученного BOC-N(Et)Gly(L)-Asp(OBzl)-(L)-Cha-NH2 составляет 3,26 гDCC (1 эквивалент) растворяют в EtOAc, (5 объемов; 1:5 вес:объем) при комнатной температуре и охлаждают до температуры от -15 до 0 С. Один эквивалент BOC-N(Et)Gly-(L)Asp(OBzl)-ОН растворяют в EtOAc (6 объемов; 1:6 вес:объем) и смешивают с 1 эквивалентомH2N-(L)-Cha-NH2, предварительно растворенном в DMF (10 объемов: 1: 10 вес:объем). Раствор дипептид/H2N-(L)-Cha-NH2 добавляют по каплям к раствору PFP и DCC, поддерживая температуру от -15 до 0 С. При проведении реакции поддерживается температура от 15 до 22 С в течение 5-16 ч при текущем контроле образцов (ЖХВР), полученных на 1, 2, 3, 4 и 16 ч для оценки завершенности реакции. (Реакция завершена, когда количество наблюдаемого дипептида менее 2% площади при ЖХВР исследовании). Реакционную смесь фильтруют и осадок на фильтре (DCU) промывают EtOAc (2 х 0,5 объема; вес/объем). Фильтрат обрабатывают водой (10 объемов; 1:10 вес:объем) и водный слой удаляют. Слой EtOAc промывают водой(1X, 5 объемов; 1:5 вес:объем). Слой EtOAc охлаждают до выпадения продукта в осадок, кото 25 рый фильтруют, промывают EtOAc (2 х 0,4 объемов; 1:0,4 вес:объем). Выделенные молярные выходы составляют 60% с обычной чистотой 90 А%, с 1-4 А% эпимерного диастереомера аспарагиновой кислоты. Повторное образование взвеси с EtOAc дает конечный выход 60% BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 и улучшение чистоты до 99 А% при снижении диастереоизомеров до 0,5%. Конкретный пример пентафторфенол-DCC комплексного способа состоит в проведении общего процесса способа В с использованием 10 г (24,5 ммоля) BOC-N(Et)Gly-(L)-Asp(OBzl)-ОН, причем получаютBOC-N(Et)Gly-(L)Asp(OBzl)-ОН растворяют в DMF (9-10 объемов; 1:10 вес:объем) и поддерживают температуру окружающей среды. К этому раствору добавляют H2N-(L)-Cha-NH2 (1 эквивалент) и гидроксибензотриазол (НОВТ, 1 эквивалент). Полученный в результате раствор охлаждают до от около 0 до 10 С и добавляют NMM (1-1,1 эквивалента). Связывающий реагент TBTU (1-1,1 эквивалента) растворяют в DMF (4-5 объемов; 1:5 вес:объем) и добавляют к защищенному дипептидному раствору при температуре от 0 до 10 С. Этот раствор перемешивают при около 10-25 С в течение 3 ч, пока ЖХВР анализ не покажет, что реакция завершена (менее 2% площади исходного материала). Реакционную смесь добавляют к перемешиваемой смеси 5%ного водного раствора хлорида натрия (около 4 объемов на объем реакционной смеси) и EtOAc(около 2 объемов на объем реакционной смеси). Фазы разделяют и водную фазу экстрагируют дополнительной порцией EtOAc (около 1,5 объемов на объем реакционной смеси). Органические фазы объединяют и последовательно промывают 0,5 Н водной лимонной кислотой (около 0,6-0,7 объемов на объем органической фазы),10%-ным водным раствором бикарбоната натрия (дважды, каждый около 0,6-0,7 объемов на объем органической фазы) и 25%-ным водным раствором хлорида натрия (около 0,3-0,4 объемов на объем органической фазы). Полученную в результате органическую фазу концентрируют до от около 1/4 до около 1/2 объема при пониженном давлении при температуре около 3050 С и к этому теплому раствору добавляют равное количество гептана. Смесь перемешивают и дают охладиться до около 0-20 С для осаждения желаемого трипептида. Это твердое вещество фильтруют, промывают смесью EtOAc и гептана и сушат. Обычный выход составляет 60% с обычной чистотой 95,7 А% и уровнем эпимерного диастереомера аспарагиновой кислоты 2 А%. 26 Конкретный пример HOBT/TBTU способа состоит в проведении общего процесса с использованием 10 г (24,5 ммоля) BOC-N(Et)Gly(L)-Asp(OBzl)-ОН, причем получают BOCN(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 (96,1A%. 1,77A% диастереомера у Asp), 67,7% теоретического выхода 9,3 г. Мас. спектр: Мрасч 560,7; М+1 набл.561. Т.п. 182,17 (ДСК). 1BOC-N(Et)Gly-(L)-Asp(Obzl)-(L)-Cha-NH2 растворяют в дихлорметане (1:12 вес/объем) и к этому раствору добавляют TFA при температуре окружающей среды. Затем все перемешивают, пока ЖХВР не покажет, что реакция завершена (3-5 ч). Раствор концентрируют до около 1/2 объема при 40-45 С. К этому теплому раствору добавляют МТВЕ (1:10 вес/объем по отношению к BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)Cha-NH2), причем поддерживается температура 40 С. Эту смесь медленно охлаждают до около 5 С и перемешивают в течение 1 ч, чтобы убедиться в полноте кристаллизации. Полученный раствор фильтруют и промывают охлажденной МТВЕ. Раствор сушат при пониженном давлении и анализируют на присутствие TFAN(Et)Gly-(L)-Asp(Obzl)-(L)-Cha-NH2(ЖХВР вес/вес анализ). Выход обычно бывает количественным,чистота 95 А%. Мас. спектр: Мрас. 460 (свободное основание); М+1 набл.461. Элементный анализ: C26H37N4O7F3: H, N, F,С 54,35, найдено 53,82. 1H ЯМР: ( относительно ТМС, Д 6 ДМСО) 0,9 м (2 Н); 1,15 т (6 Н); 1,5 м (1 Н); 1,5-1,8 м (6 Н); 2,65 д.д. (1 Н); 2,9 м (3 Н); 3,7 с (2 Н); 3,9 м (2 Н); 4,2 м (1 Н); 4,75 м (1 Н); 5,1 с (2 Н); 7,0 с (1 Н); 7,15 с (1 Н); 7,2 с (5 Н); 8,13 д (1 Н); 8,7-8,8 м (3 Н). 13 С ЯМР (стандартные сигналы,относительно ТМС, Д 6 ДМСО): 10,76; 25,49; 25,68; 25,96; 31,66; 33,07; 33,36; 36,25; 38,59; 41,88; 47,02; 49,40; 50,47; 65,71; 127,81-128,34; 135,82; 165,10; 169,34; 173,79. Конкретные примеры снятия защиты показаны в таблице А. Лаб. Пример Таблица А Реакционное весовое Выход и А% чистота количествоCBZ-PipBuN(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 (стадия 4 схемы II). Готовят суспензию эквимолярного количества TFAN(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2,CBZ-PipBu и TBTU в EtOAc, DMF и воде(100:8:4 объем/объем, 11:1 общего объем/вес по отношению к TFAN(Et)Gly-(L)-Asp(Obzl)(L)-Cha-NH2. Эту суспензию охлаждают до 010 С и добавляют около 3-4 эквивалентовNMM. Эту смесь оставляют нагреваться до комнатной температуры и перемешивают до тех пор, пока ЖХВР не покажет завершение реакции (1-3 ч; раствор получается в течение этого времени). Добавляют воду (2-3 Х начального количества добавленной воды) и фазам дают разделиться. Водную фазу сохраняют и органическую фазу промывают двумя порциями воды. Эти объединенные водные смывы экстрагируют обратно в EtOAc и объединенные органические фазы промывают 25%-ным водным раствором хлорида натрия. Органическую фазу концентрируют при пониженном давлении до 1/2 объема и добавляют МТВЕ (1/2 объем/объем по отношению к объему раствора). Эту смесь оставляют для кристаллизации (несколько часов) и твердые вещества собирают фильтрацией,промывая охлажденной смесью EtOAc и МТВЕ. Твердые вещества сушат при пониженном давлении. Содержание CBZ-PipBu-N(Et)Gly-(L)Asp(OBzl)-(L)-Cha-NH2 анализируют с помощью ЖХВР весового анализа. Выход обычно составляет 80%, чистота 95 А%. Конкретным примером является проведение основного процесса стадии 4 с 7,25 г[стандартные пики] 66,93 (оба углерода бензильной группы), 127,78-128,64 (оба фенильные кольца), 155,249; 170,00; 170,24; 171,69; 174,27; 174,21 (все карбонильные углероды). Пример 5. Получение гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил] 001362(Obzl)-(L)-Cha-NH2). Эту смесь нагревают до 40-50 С и перемешивают, пока ЖХВР не покажет завершение реакции (1-2 ч). Смесь охлаждают до комнатной температуры и фильтруют для удаления катализатора. Полученный раствор нагревают до 40-50 С и добавляют ацетон( равный объем по отношению к профильтрованному раствору), оставляя раствор для охлаждения до 30-40 С. Затравки N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида добавляют к смеси и на них кристаллизуют гигроскопичную форму N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, одновременно охлаждая до комнатной температуры (несколько часов). Твердые вещества собирают фильтрацией в токе азота, промывая ацетоном. Твердые вещества сушат при пониженном давлении и анализируют на содержание гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)цикло-гексилаланинамида (ЖХВР вес/вес анализ). Выход в целом составляет 85%, чистота 95 А%. Конкретный пример вышеприведенного получения состоит в проведении общего процесса стадии 5, когда 5 г CBZ-PipBu-N(Et)Gly(L)-Asp(Obzl)-(L)-Cha-NH2 дают 3,1 г гигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида в виде белого твердого вещества (99,6 А% чистоты),стехиометрический выход 89,4%. Другие соединения, получаемые в соответствии с вышеприведенными примерами 1-5, но с использованием соответствующих исходных веществ, включают следующие: 30 Смесь 40 кг N-бензилоксикарбонилокси сукцинимида и 26 кг (175 моля) 4-пиперидонНСlН 2 О в 38,8 кг воды и 88 кг TNF перемешивают при 155C до полного растворения (15 мин). NMM (28,8 кг) добавляют к взбалтываемой смеси (экзотермически), поддерживая температуру в 20 С или ниже. Всю порцию взбалтывают при 205 С в течение 2,5 ч, пока ЖХВР не покажет завершение реакции. Смесь разбавляют 115,2 кг МТВЕ и 38,8 кг воды и взбалтывают при 205 С в течение 5 мин. Взбалтывание прекращают, слоям дают разделиться, и водный (нижний) слой удаляют и отбрасывают. Органический слой промывают 2 х 129,6 кг воды (взбалтывают 5 мин, разделяют слои, удаляют/отбрасывают водный [нижний] слой). Органический слой промывают 5,2 кгNaCl в 46,8 кг воды (взбалтывают 5 мин, разделяют фазы, удаляют/отбрасывают водный [нижний] слой). Органический слой обрабатывают 11,5 кг MgSО 4 при взбалтывании в течение 1 ч,затем смесь фильтруют. Реактор промывают 8 кг МТВЕ (профильтрованный, соединенный с основным фильтратом; общее содержание воды в фильтрате 0,52%). Объем смеси уменьшают наполовину путем дистилляции при пониженном давлении при 30 С. Вакуум заполняют азотом и остаток охлаждают до 20 С (остаточное содержание воды в сосуде: 0,43%). Остаток разбавляют 57,6 кг МТВЕ, затем объем смеси снова уменьшают наполовину путем дистилляции в вакууме при 30 С. Вакуум заполняют азотом и смесь охлаждают до 20 С (остаточное содержание воды в сосуде: 0,25%). Это повторяют еще 5 раз. Конечный остаток в сосуде разбавляют 28,8 кг МТВЕ и перемешивают в течение 5 мин, затем определяют содержание воды и 4-N-СВZпиперидона (вода: 0,05%; вес/вес анализ 4-NСВZ-пиперидона: 22,66 вес.%, 35,36 кг, 155 молей, 88,6 стехиометр. выхода). Пример 7. Получение PipBu. Под чистым N2 и при взбалтывании получают раствор 53,5 кг 3-карбоксипропилтрифенилфосфоний бромида в 230,1 кг 1.2 диметоксиэтана. Калий-трет-бутоксид/TNF (20 вес.%, 141,8 кг раствора) добавляют в течение 35 мин, поддерживая температуру на 24-28 С. Смесь перемешивают при этой температуре в течение 0,5 ч, пока ЖХВР не покажет завершение реакции. Взбалтываемую смесь охлаждают до 102 С, затем к смеси добавляют 96,45 кг(титр: 1,15 отн. мол. экв.) 4-CBZ- пиперидона в МТВЕ в течение 40 мин, так, чтобы сохранить температуру партии на 122 С. Смесь взбалтывают при этой температуре в течение 10 мин,затем нагревают до 202 С и взбалтывают при этой температуре в течение 2 ч. К взбалтываемой смеси добавляют раствор 22,5 кг концентрированной водной НСl в 245,6 кг воды таким образом, чтобы поддержать температуру на 202 С; конечная рН - 0,5. Смесь экстрагируют, 31 вновь взбалтывают с 214,0 кг метил-третбутиловым эфиром. Взбалтывание прекращают,дают фазам разделиться и водный слой (нижний) удаляют и отбрасывают. Органическую фазу промывают 133,75 кг воды (взбалтывают 5 мин, разделяют, удаляют/отбрасывают водный[нижний] слой), затем 10,7 кг 50% NaOH в 126,8 кг воды (взбалтывают 10 мин, разделяют слои,удаляют/отбрасывают органический (верхний) слой). Водный слой экстрагируют 2 х 123,05 кгEtOAc (взбалтывают 5 мин, разделяют слои,удаляют/отбрасывают органические [верхние] слои). К взболтанному органическому слою добавляют 13,1 кг концентрированной водной НСl до рН около 1,2-3,5 (конечная: 2,82), затем смесь экстрагируют 123,05 кг EtOAc (взбалтывают 5 мин, разделяют слои, удаляют/отбрасывают водный [нижний] слой), EtOAc раствор промывают 133,75 кг воды (взбалтывают 5 мин,разделяют слои,удаляют/отбрасывают водный [нижний] слой), затем проводят анализ (вес/вес) на содержание CBZPipBuen (общий вес: 194,86 кг, 17,05% CBZPipBuen [33,22 кг, 108 молей] 87,9% стехиометр. выход). Раствор PipBuen в EtOAc сам по себе с 6,6 кг 5% Pd/C (50% воды по весу) загружают при взбалтывании в емкость из нержавеющей стали под давлением, затем смесь нагревают до 55 С 2 С. Формиат калия (38,2 кг), растворенный в 66,4 кг воды добавляют таким образом, чтобы температура реакционной смеси оставалась 552 С (30 мин). Смесь взбалтывают при 552 С в течение 2 ч, и за это время реакция завершается (ЖХВР). В реактор добавляют 6,6 кг целита и 33,2 кг воды, смесь взбалтывают, а затем фильтруют. Реактор промывают 33,2 кг воды (профильтрованной, добавленной к основному фильтрату). Фильтрат помещают в новый сосуд,охлаждают до 20-25 С, слоям дают разделиться и органический слой удаляют и отбрасывают. Водный слой подкисляют 52,1 кг концентрированной водной НСl до рН 2-3 (конечная: 2,82),затем экстрагируют 4 х 129,5 кг метиленхлорида (взбалтывают 5 минут, разделяют слои, удаляют/отбрасывают органический[нижний] слой). рН водной фазы доводят до 6,1 добавлением при взбалтывании 17,85 кг 50% воднойNaOH. Смесь фильтруют для получения 224 кг раствора, содержащего 17,6 кг (103 моля) 4-(3'карбоксипропил)пиперидина. Пример 8. Получение CBZ-PipBu. 224 кг раствора 4-(3'-карбоксипропил) пиперидина в водной NaOH соединяют с 55,3 кгTHF и смесь охлаждают при взбалтывании до 82 С. NММ (20,9 кг) добавляют, поддерживая температуру 10 С. После завершения добавления температуру доводят до 82 С, затем в течение 1 ч добавляют 25,7 кг 1(бензилоксикарбонил)сукцинимида, растворенного в 49,8 кг THF, поддерживая температуру 15 С. При 10-15 С реакция заканчивается че 001362 32 рез 3 ч (ЖХВР). Добавляют концентрированную водную НС 1 (29,9 кг) для доведения рН до 2,53,5 (конечная: 3,3), затем добавляют 61,4 кг МТВЕ и смесь взбалтывают в течение 5 мин. Взбалтывание прекращают, слоям дают разделиться и водный (нижней) слой отделяют (как ненужный). МТВЕ слой промывают тремя порциями воды по 83,1 кг (время взбалтывания 10 мин, затем 5 мин и 5 мин); водной фазе дают отделиться и каждый раз удаляют (как ненужную). Раствор 8,3 кг 50% раствора NaOH в 19,7 кг воды добавляют без взбалтывания, а затем после завершения добавления взбалтывают в течение 5 мин. Взбалтывание прекращают,фазам дают разделиться и органический (верхний) слой отделяют и отбрасывают. Водный слой возвращают в реактор и экстрагируют 2 х 38,4 кг метил-трет-бутиловым эфиром (взбалтывают 5 мин, слои разделяют, органический[верхний] слой удаляют/отбрасывают). Эту операцию повторяют с использованием 18,5 кг метил-трет-бутилового эфира. Водный слой, возвращенный в реактор, подкисляют до рН 2,5-3,5(конечная: 3,37) 9,9 кг концентрированной водной НСl. Смесь экстрагируют 76,4 кг метилтрет-бутиловым эфиром (взбалтывают 5 мин,разделяют слои, нижний [водный] слой удаляют/отбрасывают). Органический слой промывают (взбалтывают 5 мин) раствором 1,1 кг[нижний] удаляют/отбрасывают). Реактор помещают под пониженное давление и испаряющиеся растворители удаляют при 55 С до прекращения потока дистилляции. Добавляют толуол (32,4 кг) и смесь дистиллируют при атмосферном давлении до прекращения потока дистилляции, в то время как температура смеси поднимается до 90-95 С. Затем смесь охлаждают до 30-35 С, в реактор загружают 56,85 кг гептана (две фазы), смесь нагревают до 90-95 С(одна фаза), затем снова охлаждают до 38-42 С. Добавляют затравочные кристаллы CBZ-PipBu и продукт кристаллизуют из смеси в течение 1 ч. Твердое вещество собирают фильтрацией и промывают 19,35 кг 1:2 толуол/гептан, затем 33,4 гептана. Осадок на фильтре сушат под вакуумом при 40 С (до потери 0,13% при анализе высушивания) для получения 22,4 кг (72,96 моля, 42% стехиометр. выход из 4-пиперидона)CBZ-PipBu. Пример 9. Получение CBZ-PipBuen. К суспензии 82 г 3-карбоксипропилтрифенилфосфония бромид в 407 мл 1,2-диэтоксиэтана при 14 С добавляют в течение 25 мин 220 г 20% (по весу) калий-трет-бутоксида в тетрагидрофуране, поддерживая температуру реакционной смеси 24-28 С. Смесь перемешивают в течение 1 ч, охлаждают до 10 С, затем раствор 52,5 г 4-N-СВZ-пиперидона в 246 мл трет 33 бутилметилового эфира добавляют в течение 30 мин, сохраняя охлаждение. Когда добавление закончено, смесь перемешивают при 12 С в течение 10 мин, затем нагревают до 20 С и перемешивают еще 30 мин. Реакционную смесь обрабатывают 410 мл 1 Н водной НСl в течение 10 мин, разбавляют 328 миллилитрами третбутилметилового эфира и затем фазы разделяют. Органическую фазу промывают 205 мл воды, затем 210 мл 1 Н водной NaOH. Слой NaOH,содержащий продукт, собирают отдельно, промывают порциями этилацетата по 189 г, подкисляют до рН 3,48 концентрированной НСl,затем экстрагируют 189 мл этилацетата. Слой этилацетата отделяют, промывают 211 мл воды,затем сушат в течение 30 мин над 10 г МgSO4,фильтруют и концентрируют в вакууме. Маслянистый остаток (50,7 г) кристаллизуют из толуола/гептана с получением 29,46 г CBZPipBuen (50,9% выход;95 А% чистоты). Мас. спектр: Мрассчит. 303, М+1 набл. 304. 1CBZ-PipBuen (70 г, 0,23 моля) и DMF (230 мл) добавляют в 1 л колбу с наружным обогревом и перемешивают при охлаждении до 0 С,затем сразу добавляют TBTU (74,9 г, 0,23 моля). Температуру поддерживают на 0 С и начинают добавление DIPEA (61,9 г 0,61 моль). Через 45 мин TFA-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2(138,7 г, 0,24 моля) добавляют в DMF (230 мл). рН доводят до 7-8 добавлением DIPEA (45 мл) и смеси позволяют достигнуть комнатной температуры. Через 2 ч реакция заканчивается (ЖХВР анализ). Смесь гасят в воде (2,5 л) и экстрагируют EtOAc (1 л). Водную фазу экстрагируют обратно в EtOAc (0,3 л). Органические слои объединяют, промывают водной лимонной кислотой (5 % вес/вес, 2 х 1 л), промывают воднымNаНСО 3 (5% вес/вес, 2 х 2 л) и промывают водой (2 л). Слой EtOAc переносят в двухлитровую колбу и добавляют гептан (500 мл) при перемешивании для проведения кристаллизации. Твердое вещество собирают путем пропускания через воронку Бюхнера,промываютEtOAc/гептаном (2:1 объем/объем, 1 л) и сушат до постоянного веса с получением CBZPipBuen-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2(1 Н); 2,2 б.д. (4 Н); 2,35 м (1 Н); 2,4 м (2 Н); 2,74 д.д. (1 Н); 3,07 м (4 Н); 3,52 д (1 Н); 3,85 д (1H); 4,12 кв (1 Н); 4,49 кв.д. (1 Н); 4,68 д.т. (1 Н); 5,07 д (1 Н); 5,14 с (1 Н); 5,16 д (1H); 5,22 т (2 Н); 6,45 с (1 Н); 7,28 д (1H); 7,26 с (5 Н); 7,35 с (5 Н); 7,56 д (1H). 13 С ЯМР ( относительно ТМС, СDСl3): 14,15; 22,68; 24,95; 25,61; 26,03; 26,45; 28,20; 31,71; 32,89; 33, 80; 33, 89; 34,00; 35,63; 38,37; 44,79; 45,13; 45,65; 50,23; 51,34; 60,40: 66,87; 67,06; 76,50; 77,13; 77,77; 122,46; 126,88; 127,80128,69; 135,15; 155,19; 170,11; 170,20; 171,61; 173,76; 175,35. Пример 11. Получение гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида (альтернативно стадии 5 схемы II).(ДИ, 70 г). Смесь нагревают до 40-50 С и перемешивают, пока ЖХВР не покажет завершение реакции (5 ч). Смесь охлаждают до комнатной температуры и фильтруют для удаления катализатора. Полученный раствор нагревают до 4050 С и добавляют ацетон (объем, равный объему профильтрованного раствора), давая раствору охладиться до 35-40 С. Затравку N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида добавляют к смеси, и гигроскопичная форма N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида кристаллизуется оттуда во время охлаждения до комнатной температуры (несколько часов). Твердое вещество собирают при пропускании через воронку Бюхнера в токе азота, осадок промывают ацетоном и сушат до постоянного веса с получением N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида (84,3 г 0,16 моля, выход 84,8%, 95 А%). Пример 12. Каскадное получение 35 1 ч, в течение которого наблюдается небольшое выделение тепла (температура поднимается с 10 до 28 С) и осаждают DCU. Полученную таким образом суспензию фильтруют под вакуумом с использованием воронки Бюхнера, снабженной фильтровальной бумагой Whatman 1. Осадок на фильтре промывают дихлорметаном (2 х 25 мл). Фильтраты возвращают обратно в первоначальные 500-миллилитровые колбы и затем последовательно добавляют (L)Asp(OBzl) (22,3 г,0,1 моля), NNM (33,8 мл, 0,3 моля) и DMF (80 г,1,01 моля). После перемешивания в течение 2 ч при комнатной температуре, образование BOCN(Et)Gly-(L)-Asp(OBzl) закончено (проверка с ЖХВР). Реакционную смесь выливают в воронку для экстракции, содержащую ледяную воду(100 мл). Смесь подкисляют НСl (36%, 25 мл) до рН 1. Слои разделяют и слой дихлорметана промывают ледяной водой (100 мл), и фазы разделяют (водная фаза рН 3-4). Дихлорметановый слой возвращают обратно в 500-миллилитровую колбу, которая наполняется последовательноNH2-(L)-Cha-NH2 (17 г, 0,1 моля), N-гидроксисукцинимидом (11,5 г, 0,1 моля) и DCC (20,6 г,0,1 моля) одной порцией в виде твердого вещества. После перемешивания в течение 2 ч при комнатной температуре образование BOC-N(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 завершено (проверка ЖХВР) и DCU фильтруют под вакуумом с использованием воронки Бюхнера, снабженной фильтровальной бумагой Whatman 1. Осадок на фильтре промывают дихлорметаном (2 х 25 мл). Фильтрат переносят в экстракционную воронку и промывают деионизированной водой(200 мл), содержащей N-метилморфолин 915 мл, рН 8-9). Фазы разделяют и дихлорметановый слой снова промывают водой (ДИ, 2 х 150 мл). Дихлорметановую фазу промывают 150 мл 1 Н НСl (рН 1). Фазы разделяют и дихлорметановый слой промывают деионизированной водой (200 мл, рН 3). Дихлорметановый растворTFAHN(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 полностью заканчивается (проверка ЖХВР). Реакционную смесь дистиллируют под вакуумом для удаления дихлорметана и основного количества TFA, затем добавляют МТВЕ и кристаллы, чтобы вызвать кристаллизацию продукта. Смесь фильтруют под вакуумом, используя воронку Бюхнера, снабженную фильтровальной бумагой Whatman 1. Осадок промывают МТВЕ(2 х 25 мл) и сушат воздухом для полученияTFAHN(Et)Gly-(L)-Asp(OBzl)-(L)-Cha-NH2 (46,8 г, выход 81,5%) в виде белого твердого вещества (97 А% чистоты, 0,2 А% D-Asp диастереомера). Пример 13. Получение стабильной негигроскопичной формы N-[N-[N-(4-пиперидин-4 001362N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (7,45 кг) размалывают на молотковой мельнице. Полученное твердое вещество 7,35 кг помещают в сушильный лоток из нержавеющей стали и покрывают его перфорированной алюминиевой фольгой. Затем лоток герметично закрывают во влажной печи (LUNAIREHumidity Cabinet модель no CEO 941W-3); печь оставляют герметично закрытой во время всего процесса превращения, кроме моментов забора образцов для анализа. Печь доводят до 40%RH/60C и поддерживают на этом уровне в течение 1 ч. Влажность печи затем доводят до 80% RH/60C и поддерживают на этом уровне 12 ч. Образцы забирают через 18 ч при 80%RH/60C и проверяют с помощью анализа дифракции рентгеновских лучей порошком для оценки превращения в негигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида. Влажную печь снова герметично закрывают и доводят до 40%RH/60C и поддерживают в таком виде в течение 2 ч. Печь доводят до температуры окружающей среды, затем лоток вынимают из печи и получают негигроскопичную кристаллическую форму N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида(7,2 кг, выход 96,6%). Подтверждение превращения определяется с помощью картины дифракции рентгеновских лучей порошка (фиг. 1). Дифракция рентгеновских лучей порошка также внесена в таблицу в виде функции от возрастающего порядка угла дифракции (2), соответствующего внутриплоскостного расстояния в кристалле (d), в ангстремах, в виде расчета на секунды (Cps) и относительной интенсивности пика (%) (таблица 1).N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида (50 г) помещают в 400 миллилитровый градуированный цилиндр (высота слоя 6 см) на кольцевую подставку с механической мешалкой. Аппарат помещают в печь с контролируемой влажностью LUNAIRE Humidity Cabinet модель no. СЕО 941W-3). Перемешивание проводят при 275 об./мин. и температуру и RH доводят в течение 30 мин до 60 С и 40%, соответственно. Соединение выдерживают в этих условиях в течение 1 ч, затем условия меняют за 45 мин до 80% RH/60C. Затем соединение выдерживают в этих условиях в течение 16 ч до того, как печь снова настраивают на 40% RH/60C и поддерживают в течение 3,25 ч. Затем соединению позволяют вернуться к условиям окружающей среды (высота слоя 4 см),затем удаляют из цилиндра и получают негигроскопичную кристаллическую форму N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида(выход 95%). Подтверждение превращения определяется с помощью картины дифракции рентгеновских лучей порошка (фиг. 2). Дифракция рентгеновских лучей порошка также внесена в таблицу в виде функции от возрастающего порядка угла дифракции (2), соответствующего внутриплоскостного расстояния в кристалле(d), в ангстремах , в виде расчета на секунды(Cps) и относительной интенсивности пика (%)b. Превращение формы Гигроскопичную кристаллическую форму(Heidolph MR 2002). Аппарат помещают в вакуум при 60 мбар, полученный с использованием вакуумного насоса (Divatrion DVI), затем вакуум нарушают контролируемым образом, чтобы впустить влажную атмосферу, созданную в отдельной, нагретой, содержащей воду колбе. Доступ влажного атмосферного воздуха контролируется аппаратом контролирования влажности (Vausalo Humiditique and TemperatureTraumettor) таким образом, чтобы добиться RH в 79% внутри аппарата (внутреннее давление 130-180 мбар). Сосуд роторного испарителя затем вращают при 145-160 оборотах в минуту в течение 5 ч, в то время как нагревающая баня поддерживается на 60 С и RH внутри сосуда поддерживается на 71-79%. Затем вакуум заполняют азотом и сосуду и его содержимому позволяют охладиться до комнатной температуры, и продукт удаляют, получая негигроскопичную кристаллическую форму N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида. Вторую порцию 317 г гигроскопичной кристаллической формы N-[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида обрабатывают таким же образом для получения негигроскопичной кристаллической формы N-[N-[N-(4-пиперидин 4-ил)бутаноил)-N-этилглицил]-(L)-аспартил](L)циклогексилаланинамида. Подтверждение превращения получали с помощью анализа дифракции рентгеновских лучей порошка (фиг. 3). Две порции вместе составляют 667 г негигроскопичной кристаллической формы N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида (97% выход, общий). Подтверждение превращения определяется с помощью картины дифракции рентгеновских лучей порошка (фиг. 3). Дифракция рентгеновских лучей порошка также внесена в таблицу в виде функции от возрастающего порядка угла дифракции (2), соответствующего внутриплоскостного расстояния в кристалле(d), в ангстремах , в виде расчета на секунды(Cps) и относительной интенсивности пика (%) Пример 14. Картина дифракции рентгеновских лучей порошка образца A N-[N-[N-(4 пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)аспартил]-(L)циклогексилаланинамида в его гигроскопичной кристаллической форме и его превращения в негигроскопичную форму. Образец гигроскопичного кристаллическогоN-[N-[N-(4-пиперидин-4-ил)бутаноил)-N 40 этилглицил]-(L)-аспартил]-(L)циклогексилаланинамида получают как в примере 5 или 11 и превращают в соответственную негигроскопичную форму в соответствии со способом примера 13. Картины дифракции рентгеновских лучей порошка гигроскопичной кристаллической формы и негигроскопичной кристаллической формы показаны соответственно на фиг. 4 и 5. Картины дифракции рентгеновских лучей порошка гигроскопичной и негигроскопичной кристаллических форм также внесены в таблицу в виде функции от возрастающего порядка угла дифракции (2), соответствующего внутриплоскостного расстояния в кристалле (d), в ангстремах , в виде расчета на секунды (Cps) и относительной интенсивности пика (%) (таблицы 4 и 5 соответственно). Пример 15. Изотермальные микрокалориметрические эксперименты с гигроскопичными и негигроскопичными кристаллическими формами N-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Изотермальные микрокалориметрические эксперименты с гигроскопичными и негигроскопичными формами N-[N-[N-(4-пиперидин-4 ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида выполняются на Мониторе термальной активности (МТА) Thermometric. Превращение твердого состояния различных кристаллических форм изучают,подвергая их воздействию различной влажности или паров растворителя при различных температурах. Для получения различной влажности используются следующие солевые растворы: КСl (80% RH), NaCl (75% RH) и NaBr (66% RH). Приблизительно 100 миллиграммовые порции взвешивают в стеклянной ампуле МТА и микрогидростат, содержащий солевой раствор (с избытком твердого вещества) или органический растворитель, помещают внутрь ампулы. Ампулу запаивают, доводят до температуры эксперимента и опускают в ТАМ в положение измерения. Идентичную систему, содержащую вместо тестируемого объекта промытый морской песок, помещают для сравнения. Мощность вы 42 работки (мкВт) измеряется как функция времени (фиг.6-8). Пример 16. Изотермы поглощения влажности гигроскопичной и негигроскопичной форм N-[N-[N-(4-пиперидин-4-ил)бутаноил)-Nэтилглицил]-(L)-аспартил]-(L)циклогексилаланинамида. Изотермы поглощения влажности гигроскопичной и негигроскопичной форм N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамида получают на VTI MB 300G балансе влажности. Эксперименты проводят или путем воздействия на приблизительно 15 мг исследуемой кристаллической формы, повышая и снижая % RH, и следя за увеличением веса (на каждом этапе уравновешивания), как функцией % RH (фиг.9),или путем выдерживания кристаллической формы при постоянной влажности, следя за увеличением веса, как функцией времени. Соединение формулы II проявляет пригодную фармацевтическую активность и соответственно включается в фармацевтические композиции и может использоваться для лечение больных, страдающих от некоторых патологий. Настоящее изобретение также относится к способам лечения больных, страдающих от или подверженных состояниям, которые могут быть улучшены или предупреждены при применении ингибиторов агрегации тромбоцитов путем ингибирования связывания фибриногена для активации тромбоцитов, и других адгезивных гликопротеинов, включенных в агрегацию тромбоцитов и образование тромба. Более того, настоящее изобретение направлено на способ предупреждения или лечения тромбоза, связанного с определенными заболеваниями, такими как инфаркт миокарда, инсульт, периферические артериальные заболевания и диссеминированное внутрисосудистое свертывание у людей и других млекопитающих. Надо понимать, что приведенные здесь ссылки на лечение включают профилактическую терапию, а также и лечение основного заболевания. Настоящее изобретение также включает в себя фармацевтические композиции, в которые входят фармацевтически приемлемые количества по крайней мере одного соединения формулыI в сочетании с фармацевтически приемлемым носителем или эксципиентом. На практике соединения или композиции для лечения в соответствии с настоящим изобретением могут применяться любыми подходящими способами, например, местно, ингаляционно, парентерально, ректально или орально,но предпочтительным является оральное применение. Соединение формулы II может быть представлено в формах, позволяющих применять его наиболее подходящим путем, а также изобрете 43 ние относится к композициям, содержащим по крайней мере одно соединение по изобретению,которое пригодно для использования человеком или в ветеринарии. Эти композиции могут быть приготовлены в соответствии с общепринятыми способами, используя один или более фармацевтически приемлемых адъювантов или эксципиентов. Адъюванты включают, помимо прочих, разбавители, стерильную водную среду и различные нетоксичные органические растворители. Композиции могут быть представлены в форме таблеток, пилюль, капсул, гранул, порошков, водных растворов и суспензий, растворов для инъекций, эликсиров, сиропов и им подобных, и могут содержать один или большее количество агентов, выбранных из группы, состоящей из подслащивающих агентов, ароматизаторов, стабилизаторов или консервантов с целью получения фармацевтически приемлемых препаратов. Выбор наполнителя и количество активного вещества в наполнителе обычно определяется в соответствии с растворимостью и химическими свойствами продукта, в частности, в соответствии со способом применения и с веществами, принятыми в настоящее время в фармацевтической практике. Например, такие эксципиенты как лактоза, цитрат натрия, карбонат кальция, дикальций фосфат, и дизинтеграционные агенты, такие как крахмал, альгиновые кислоты и определенные комплексные силикагели, объединенные со смазывающими веществами, такими как стеарат магния, натрий лаурилсульфат и тальк, могут быть использованы для приготовления таблеток. Чтобы приготовить капсулы, полезно использовать лактозу и полиэтиленгликоли с высоким молекулярным весом. При использовании водных суспензий, они могут содержать эмульгирующие агенты или агенты, способствующие получению суспензии. Разбавители, такие как сахароза, этанол, полиэтиленгликоль, пропиленгликоль, глицерин и хлороформ или их сочетания, могут применяться наряду с другими веществами. Для парентерального применения используются эмульсии, суспензии и растворы соединений по изобретению в растительном масле,например, кунжутовом масле, арахисовом масле или оливковом масле, или водно-органических растворителях, таких как вода и пропиленгликоль, инъекционных органических сложных эфирах, таких как этилолеат, а также стерильных водных растворах фармацевтически приемлемых солей. Растворы солей продукта по изобретению также пригодны для применения при внутримышечных или подкожных инъекциях. Водные растворы, также содержащие растворы солей в чистой дистиллированной воде, могут быть использованы для внутривенного применения при условии, что их рН доведена до нужной величины, и что они правильно забуферены и сделаны изотоничными с помощью соответст 001362 44 вующего количества глюкозы или хлорида натрия, и что они стерилизованы нагреванием,радиацией и/или микрофильтрацией. Могут использоваться препараты для местного применения, гели (на основе воды или спирта), кремы или бальзамы, содержащие соединения по изобретению. Соединения по изобретению также могут включаться в гели или в матриксную основу для повязок (пластырей),которые позволяют осуществить контролируемое проникновение соединения через трансдермальный барьер. Твердые композиции для ректального применения включают суппозитории в соответствии с известными способами, приготовленные в соответствии с известными способами и содержащие, по крайней мере, одно соединение формулы II. Процентное содержание активных ингредиентов в композиции в соответствии с изобретением может варьироваться так, чтобы составлять пропорционально подходящую дозу. Очевидно, что несколько единичных дозированных форм могут применяться в одно и то же время. Применяемая доза может быть определена врачом или квалифицированным медицинским персоналом и зависит от желаемого терапевтического эффекта, пути введения и длительности лечения и от состояния пациента. Режим введения дозы является способом данного изобретения, при котором обеспечивается максимальный терапевтический ответ, пока не будет достигнуто улучшение, а затем применяется минимальная эффективная доза, приносящая облегчение. В общем, оральная доза может составлять от около 0,1 мг/кг до около 100 мг/кг, предпочтительно от около 0,1 мг/кг до 20 мг/кг, и наиболее предпочтительно от около 1 мг/кг до 20 мг/кг, и внутривенная доза составляет от около 0,1 мкг/кг до около 100 мкг/кг, предпочтительно от около 0,1 мг/кг до около 50 мг/кг. В каждом отдельном случае доза определяется в соответствии с особенностями конкретного пациента,такими как возраст, вес, общее состояние здоровья и другие характеристики, которые могут влиять на эффективность соединения по изобретению. Далее, соединение формулы II может применяться так часто, как это необходимо для достижения желаемого терапевтического эффекта. Некоторые пациенты быстро дают ответ на высокие или низкие дозы, и для них адекватным является поддержание более слабых доз. Для других пациентов может быть необходимым длительное лечение при 1-4 оральных дозах в день, предпочтительно один или два раза в день,в соответствии с физиологическими требованиями каждого конкретного пациента. В общем,активный продукт может назначаться орально от 1 до 4 раз в день. Конечно, для других пациентов может быть необходимо прописывать не более одной или двух доз в день. 45 Соединения формулы II проявляют отмеченную фармакологическую активность в соответствии с описанными в литературе анализами,результаты которых коррелируют с фармакологической активностью у человека и других млекопитающих. Следующие результаты фармакологических анализов in vivo и in vitro типичны для характеристики соединений формулы II. Следующие фармакологические анализы оценивают ингибиторную активность соединений формулы II по фибриноген-медиированной агрегации тромбоцитов, связыванию фибриногена со стимулированными тромбином тромбоцитами и по ингибированию ADP-вызванной агрегации тромбоцитов ex-vivo, причем результаты этих анализов коррелируют с ингибиторными свойствами соединения формулы II invivo. Анализ агрегации тромбоцитов основан на анализе, описанном в Blood 66(4), 946-952(1985). Анализ связывания фибриногена подобен описанному у Ruggeri Z.M. et al., Proc. Natl.(1985). Анализ ADP-индуцированного ингибирования агрегации тромбоцитов ex-vivo основан на работе Zucker, "Platelet Aggregation Measuredby the Photoelectric Method", Methods in Enzymology 169, 117-133 (1989). Анализ агрегации тромбоцитов Получение фиксированного препарата активированных тромбоцитов Тромбоциты выделяют из концентрата тромбоцитов человека,используя гельфильтрационную методику, как описано у Marguerie, G.A. et al., J. Biol. Chem. 254, 5357-5363(1979) и Ruggeri, Z.M. et al., J. Clin. Invest. 72, 112 (1983). Тромбоциты суспендируют при концентрации 2 х 108 клеток/мл в модифицированном свободном от кальция буфере Тирода, содержащем 127 мМ хлорида натрия, 2 мМ хлорида магния, 0,42 мМ Na2HPO4, 11,9 мМHEPES при рН 7,35 и 0,35% сывороточного альбумина человека. Эти промытые тромбоциты активируют -тромбином человека при конечной концентрации 2 единицы/мл, с последующей обработкой ингибитором тромбина 1-2581 при конечной концентрации 40 мкМ. К активированным тромбоцитам добавляют параформальдегид до конечной концентрации 0,50% и инкубируют их при комнатной температуре в течение 30 мин. Зафиксированные активированные тромбоциты затем собирают центрифугированием при 650 хg в течение 15 мин. Тромбоцитарные пластинки промывают четыре раза вышеуказанным буфером Тирода с 0,35% сывороточного альбумина человека и ресуспендируют в том же буфере до концентрации 2 х 108 клеток/мл. 46 Анализ агрегации тромбоцитов Фиксированный препарат активированных тромбоцитов инкубируют с выбранной дозой тестируемого соединения для ингибирования агрегации тромбоцитов в течение одной минуты, и инициируют агрегацию добавлением фибриногена человека до конечной концентрации 250 мкг/мл. Для регистрации агрегации тромбоцитов применяется аппарат для измерения уровня агрегации тромбоцитов Модель РАР-4. Степень ингибирования агрегации выражается в процентах от нормы агрегации, наблюдаемой при отсутствии ингибитора. Затем для каждого соединения высчитывается IС 50, то есть количество ингибитора, необходимое для снижения степени агрегации на 50% (смотри, например,Plow E.F. et al., Proc. Natl. Acad. Sci., USA 82,8057-8061 (1985. Анализ связывания фибриногена Тромбоциты отмывают от компонентов плазмы в градиенте плотности альбумина (методика Walsh P.N. et al., Br. J. Haematol. 281-296et al., J. Clin. Invest. 76, 1950-1958 (1985. Каждую экспериментальную смесь тромбоцитов в модифицированном буфере Тирода (Ruggeri,Z.M. et al., J. Clin. Invest. 72, 1-12 (1983 стимулируют -тромбином человека при 22-25 С в течение 10 мин (3, 125 х 1011 тромбоцитов на литр и тромбин при 0,1 NIH единиц/мл). Затем добавляют гирудин с 25-кратным избытком(единица/единица) за 5 мин до добавления 125Iмеченного фибриногена и тестируемого соединения. После этого добавления конечная концентрация тромбоцитов в смеси составляет 1 х 1011/л. После инкубирования в течение еще 30 мин при 22-25 С связанный и свободный лиганд выделяют центрифугированием 50 мкл смеси через 300 мкл 20% сахарозы при 12000 х g в течение 4 мин. Затем тромбоцитарные пластинки выделяют из остальной смеси для определения связанной с тромбоцитами радиоактивности. Неспецифическое связывание измеряют в смесях, содержащих избыток немеченного лиганда. При анализе кривых связывания с помощью анализа Scatchard, неспецифическое связывание выводят как соответствующий параметр из изотермы связывания с помощью компьютерной программы (Munson P. J. Methods Enzymol. 92, 542-576 (1983. Для определения концентрации каждого соединения, необходимого для ингибирования 50% фибриногена, связанного с тромбин-стимулированными тромбоцитами(IC50), каждое соединение тестируется в 6 и более концентрациях с фибриногеном, меченным 125I, при 0,176 мкмоля/л. IC50 получают путем нанесения на график связывания остаточного фибриногена против логарифма концентрации образца соединения. Ингибирование ADP-индуцированной агрегации тромбоцитов ex-vivo Протокол эксперимента Контрольный образец крови получают за 5-10 мин до применения тестируемого соединения нечистокровным собакам весом от 10 до 20 кг. Соединение вводят внутрижелудочно, через водяной желудочный зонд, или орально, в желатиновых капсулах. Затем получают образцы крови (5 мл) с 30-минутным интервалом в течение 3 ч и на 6, 12 и 24 ч после применения дозы. Каждый образец крови получают путем венопунктуры краниальной вены и собирают непосредственно в пластмассовый шприц, содержащий одну часть 3,8% тринатрий цитрата на девять частей крови. Агрегация тромбоцитов у собак ех vivo Образцы крови центрифугируют при 1000 об/мин для получения обогащенной тромбоцитами плазмы (ПОТ). После удаления ПОТ образцы центрифугируют еще 10 мин при 2000 об/мин для получения бедной тромбоцитами плазмы (БТП). Подсчет тромбоцитов в ПОТ производят с использованием счетчика Coulter(Coulter Electronics, Hialeah, FL). Если концентрация тромбоцитов в ПОТ больше 300 000 тромбоцитов/мкл, тогда ПОТ разбавляют БТП до получения 300 000 тромбоцитов/мкл. Аликвоты ПОТ (250 мкл) затем помещают в силиконовые стеклянные кюветы (7,25 х 55 мм,Bio/Data Corp., Horsham, PA). Затем к ПОТ добавляют эпинефрин (конечная концентрация 1 мкМ) и инкубируют в течение 1 мин при 37 С. Затем к ПОТ добавляют стимулятор агрегации Ингибирование фиксированной агрегации тромбоцитов (IC50 мкМ) Квалифицированные специалисты легко определят, что настоящее изобретение хорошо приспособлено к выполнению целей изобретения, и достигает преимуществ, присущих изобретению. Соединения, композиции и способы,описанные здесь, приведены для представления предпочтительных воплощений, или как пример, и не должны рассматриваться, как ограничение для настоящего изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, представляющее N-(Nтрет-бутоксикарбонил-N-этилглицил)-(L)-аспарагиновой кислоты -бензиловый сложный эфир. 2. Соединение, представляющее N-[N-[N[4-[N-бензилоксикарбонилпиперидин-4-ил]бута 48 тромбоцитов, ADP в конечной концентрации 10 мкМ. Агрегация тромбоцитов регистрируется спектрофотометрически с использованием светового трансмиссионного агрегометра (Bio/DataPlatelet Aggregation Profiler. Model PAP-4,Bio/Data Corp. Horsham, PA). Для анализа соединения скорость изменения (наклон) световой трансмиссии и максимум трансмиссии (максимум агрегации) определяются дважды. Данные агрегации тромбоцитов представлены в процентах повышения (среднеесреднеквадратичное отклонение) наклона или максимума агрегации по сравнению с данными, полученными с контрольным образцом ПОТ, который готовят из образцов крови, полученных до применения тестируемого соединения. Соединение формулы II проявляет отмеченную активность в вышеприведенных тестах и считается пригодным для предупреждения и лечения тромбоза, связанного с определенными болезненными состояниями. Противотромботическая активность в анализе, проведенном на собаках ех vivo, предсказывает такую же активность у человека (смотри, например, CatalfamoA, 27 (1989. Результаты анализа соединений формулы II вышеприведенными способами представлены ниже в таблице 6. В таблице также представлены результаты сравнительного анализа 4-4(пиперидил)бутаноилглициласпартилтриптофана, то есть соединения, раскрытого в публикации Европейской патентной заявки 0479 481. Таблица 6 Ингибирование ADP-вызванной агрегации тромбоцитов ex-vivo Дозаex-vivo после орального применения 1 ч 3 ч 6 ч 12 ч 24 ч 5 100 100 100 98 50 5 53 29 ноил]-N-этилглицил]-(L)-аспартил] -бензиловый сложный эфир]-(L)циклогексилаланинамид. 3. Соединение, представляющее 4-(4 пиперидин)бутилиденилкарбоновую кислоту. 4. Соединение, представляющее N-[N-[N[3-[N-бензилоксикарбонил-4-пиперидин]пропилиденилкарбонил]-N-этилглицил]-(L)-аспартил] -бензиловый сложный эфир]-(L)-циклогексилаланинамид. 5. Соединение, представляющее N-[N-[N(4-пиперидин-4-ил)бутаноил)-N-этилглицил](L)-аспартил]-(L)циклогексилаланинамид или его фармацевтически приемлемую соль. 6. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель. 49 7. Способ получения негигроскопичного кристаллического N-[N-[N-(4-пиперидин-4-ил) бутаноил)-N-этилглицил]-(L)-аспартил]-(L)-циклогексилаланинамида, включающий воздействие на гигроскопичный кристаллический N[N-[N-(4-пиперидин-4-ил)бутаноил)-N-этилглицил]-(L)-аспартил]-(L)циклогексилаланинамид относительной влажностью от около 40 до 100% и температурой от около 20 до около 80 С. 8. Способ по п.7, где относительная влажность составляет от около 65 до около 80%. 9. Способ по п.7, температура составляет от около 40 до около 80 С. 10. Способ по п.7, где воздействие осуществляют в статических условиях. 11. Способ по п.7, где воздействие осуществляют в динамических условиях. 12. Способ получения соли соединения формулы где В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Е 1 представляет Н; Е 2 представляет -углеродную боковую цепь природной -аминокислоты, Н, алкил,циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2, взятые вместе с атомами азота и углерода,через которые Е 1 и Е 2 связаны, образуют 4-, 5-,6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,арил, аралкил, алкиларил или алкиларалкил; р представляет целое число от 1 до 4; Р 1 представляет группу, защищающую кислоту; включающий реакцию соединений формулы где Р 2 представляет лабильную аминозащитную группу, с образованием первого промежуточного соединения формулы с последующей реакцией полученного первого промежуточного продукта с соединением формулы с образованием второго промежуточного соединения формулы с последующим удалением Р 2 защитной группы из второго промежуточного соединения трифторуксусной кислотой, получая соединение в виде соли. 13. Способ по п.12, где Р 1 представляет гидрирующую лабильную защитную группу кислоты. 14. Соединение формулы где Р 3 представляет аминозащитную группу; В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Р 1 представляет защитную группу кислоты; Е 1 представляет Н; Е 2 представляет -углеродную боковую цепь природной -аминокислоты, Н, алкил,циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2 вместе с атомами азота и углерода, через которые Е 1 и Е 2 связаны, образуют 4-, 5-, 6- или 7- членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,арил, аралкил, алкиларил или алкиларалкил; р равно от 1 до 4,15. Соединение по п.14, где Р 1 представляет гидрирующую лабильную защитную группу кислоты, а Р 3 представляет гидрирующую лабильную аминозащитную группу. 16. Соединение по п.15, где В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Е 1 представляет Н; Е 2 представляет Н, алкил, циклоалкил,циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил или замещенный аралкил;R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,арил, аралкил, алкиларил или алкиларалкил; р равно 1 или 2.R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил или алкилциклоалкилалкил; и р равно 1. 19. Соединение по п.18, где Р 3 представляет бензилоксикарбонил;G представляет NН 2. 20. Способ получения соединения формулы где Р 3 представляет аминозащитную группу; В представляет алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, аралкил, алкиларил или алкиларалкил; Р 1 представляет защитную группу кислоты; Е 1 представляет Н; Е 2 представляет -углеродную боковую цепь природной -аминокислоты, Н, алкил,циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил, арил, замещенный арил, аралкил, замещенный аралкил, гетероциклил, замещенный гетероциклил, гетероциклилалкил, замещенный гетероциклилалкил, или Е 1 и Е 2, взятые вместе с атомами азoта и углерода,через которые Е 1 и Е 2 связаны, образуют 4-, 5-,6- или 7-членное азациклоалкановое кольцо;R1 и R2 независимо друг от друга представляют Н, алкил, циклоалкил, циклоалкилалкил, алкилциклоалкил, алкилциклоалкилалкил,арил, аралкил, алкиларил или алкиларалкил; р равно от 1 до 4,включающий реакцию 4-пиперидин)бутилиденилкарбоновой кислоты) формулы 52 кислоты и Р 3 представляет гидрирующую лабильную аминозащитную группу. 22. Соединение формулы где Р 3 представляет аминозащитную группу. 23. Соединение по п.22, где Р 3 представляет гидрирующую лабильную аминозащитную группу. 24. Соединение по п.23, где Р 3 представляет бензилоксикарбонил. или с ее аддитивной солью кислоты. 21. Способ по п.20, где Р 1 представляет гидрирующую лабильную защитную группу
МПК / Метки
МПК: A61P 7/02, A61K 31/445, C07D 211/06
Метки: кристаллическая, форма, соединения, стабильная, n-[n-n-(4-(пиперидин-4-ил)бутаноил)-n-этилглицилового, негигроскопичная
Код ссылки
<a href="https://eas.patents.su/28-1362-stabilnaya-negigroskopichnaya-kristallicheskaya-forma-n-n-n-4-piperidin-4-ilbutanoil-n-etilglicilovogo-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Стабильная негигроскопичная кристаллическая форма n-[n-n-(4-(пиперидин-4-ил)бутаноил)-n-этилглицилового] соединения</a>
Альтернативная кристаллическая форма тазофелона

Номер патента: 258
Опубликовано: 25.02.1999
Авторы: Хансен Марвин М., Харкнесс Аллен Р., Рейтцель Сюзн М.
МПК: C07D 277/14, A61K 31/425
Метки: альтернативная, тазофелона, кристаллическая, форма
Формула / Реферат:
1. По существу чистая форма II (±)-5-{[3,5-бис( 1,1 -диметилэтил)-4 -гидроксифенил] метил }-4-тиазолидинона, имеющая следующие характеристики дифракции рентгеновских лучей, получаемые при использовании СиКос облучения с ?i=l,53056A:Межплоскостное Относительная расстояние d, А интенсивность Шо5,64 5,16 4,90 4,66 4,4910 38 37 57 412. Способ получения и выделения по существу чистой формы II...
Новая кристаллическая форма морфин-6-глюкуронида

Номер патента: 755
Опубликовано: 24.04.2000
Авторы: Францмайр Рудольф, Шнайдер Хервиг, Ровенски Франц, Кох Андреас
МПК: C07H 17/00
Метки: новая, морфин-6-глюкуронида, форма, кристаллическая
Формула / Реферат:
1. Кристаллический морфин-6-глюкуронид, обозначаемый как форма А, характеризующийся инфракрасным спектром со следующими основными пиками: 3400 1060 2920 1020 2880 985 2845 935 1610 870 1505 840 1470 790 1420 760 1400 660 ...
Кристаллическая кислая кальциевая соль [r-(r, r)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1h-пиррол-1гептановой кислоты (аторвастатин)

Номер патента: 474
Опубликовано: 26.08.1999
Авторы: Бриггс Кристофер, Вейд Роберт, Минохара Казуо, Харасава Кикуко, Ичикава Шигеру, Дженнингс Рекс Аллен, Накагава Шинсуке
МПК: A61K 31/40, C07D 207/34
Метки: аторвастатин, соль, кристаллическая, кислая, r-(r, кальциевая, кислоты, r)]-2-(4-фторфенил)-бета, дельта-дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1h-пиррол-1гептановой
Формула / Реферат:
1. Кристаллическая форма I аторвастатина или его гидрата, имеющая в форме порошка дифракцию рентгеновских лучей, содержащую в качестве основного пика следующую величину 2q , измеренную с использованием излучения СuKa : 21,6. 2. Кристаллическая форма I аторвастатина или его гидрата согласно п.1, характеризующаяся также следующими величинами 2q , измеренными с использованием излучения СuKa : 17,1, 19,5 и 23,7. 3. Кристаллическая форма I...
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Икрами Хуссейн М., Шен Вей Чанг
МПК: A61K 31/44, C07D 213/70, C07H 19/048...
Метки: получения, сохранения, способы, содержащего, группу, соединения, конъюгаты, промежуточные, пролонгированного, жирной, абсорбции, млекопитающего, повышения, кислоты, сульфгидрильную, тканях, крови, производного, конюгатов, способ
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
5-замещенные-3-(1,2,3,6-тетрагидропиридин-4-ил)- и 3-(пиперидин-4-ил)-1н-индолы и их фармацевтически приемлемые соли и сольваты, фармацевтическая композиция на их основе, способ активации рецепторов 5-нт1 и способ ингибирования нейронной белковой транссудации.

Номер патента: 1113
Опубликовано: 30.10.2000
Авторы: Кэлдор Стефен Уоррен, Кох Дэниел Джеймс, Дрессман Брюс Энтони, Шос Джон Мехнет, Дрост Джеймс Джозеф, Томпсон Деннис Чарльз, Ниссен Джеффри Скотт, Одиа Джеймс Эдмунд, Фриц Джеймс Эрвин, Рокко Винсент Патрик
МПК: C07D 401/04, A61K 31/4439
Метки: рецепторов, 3-(пиперидин-4-ил)-1н-индолы, основе, способ, транссудации, композиция, приемлемые, 5-замещенные-3-(1,2,3,6-тетрагидропиридин-4-ил, фармацевтическая, фармацевтически, активации, соли, нейронной, белковой, сольваты, ингибирования, 5-нт1
Формула / Реферат:
1. 5-Замещенные -3-(1,2,3,6-тетрагидропиридин-4-ил)-3-(пиперидин-4-ил)индолы общей формулы I в которой А-В представляет -CH-CH2- или -С=СН-; R представляет Н или C1-C6-алкил; R1 представляет Н или С1-С4-алкил; Х представляет -C(О)NR4R15, -NR5R6, -NR7SO2R8, -NHC(Q)NR10R11, -NHC(O)OR12 или NR13C(O)R14; где Q представляет О или S; R4 представляет гетероарил, замещенный гетероарил, гетероарил(C1-C4 алкил) или замещенный гетероарил(C1-C4...
Предыдущий патент: Схема умножения для устройства измерения мощности и способ измерения мгновенного значения мощности
Следующий патент: Кристаллический фармацевтический продукт
Случайный патент: Система защиты персонального устройства от несанкционированного доступа к нему