Производные пиримидин-2,4-диона в качестве антагонистов рецептора гонадотропин-высвобождающего гормона
Номер патента: 10370
Опубликовано: 29.08.2008
Авторы: Чен Чен, У Дунпэй, Уэйд Уоррен, Дуайт Уэсли Дж., Го Чжицян, Туччи Фабио К., Чень Юншэн, Хуанг Чарльз К.






Формула / Реферат
Соединение, имеющее следующую структуру:

или его стереоизомер или фармацевтически приемлемая соль, где
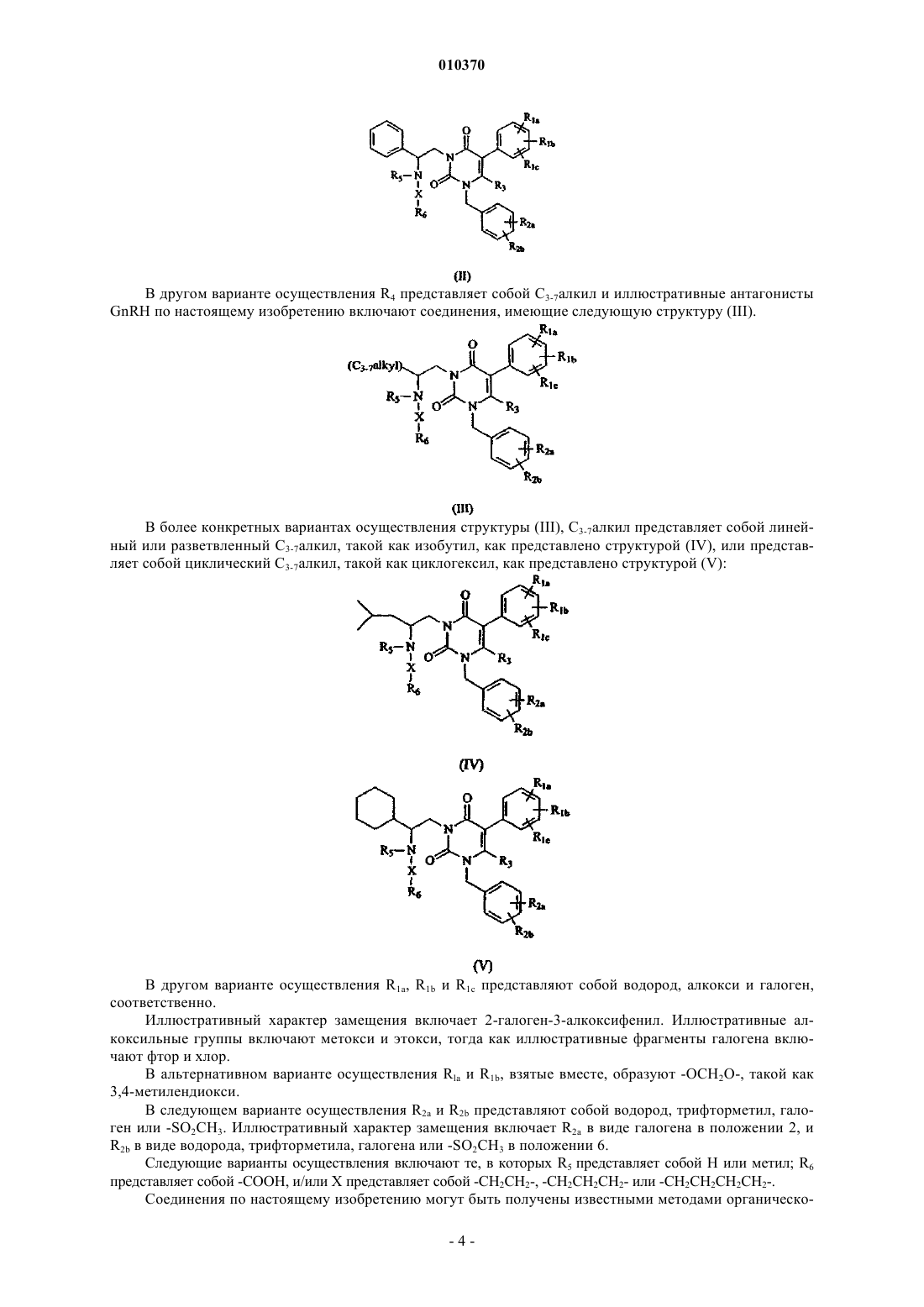
R1a, R1b, R1c являются одинаковыми или разными и независимо представляют собой водород, галоген, С1-4алкил, гидрокси или алкокси, или R1a и R1b, взятые вместе, образуют -OCH2O- или -ОСН2СН2-;
R2a и R2b являются одинаковыми или разными и независимо представляют собой водород, галоген, трифторметил, циано или -SO2CH3;
R3 представляет собой водород или метил;
R4 представляет собой фенил или C3-7алкил;
R5 представляет собой водород или С1-4алкил;
R6 представляет собой -СООН или тетразолил и
X представляет собой C1-6алкандиил, необязательно замещенный 1-3 C1-6алкильными группами.
2. Соединение по п.1, где R1a представляет собой галоген.
3. Соединение по п.1, где R1b представляет собой алкокси.
4. Соединение по п.3, где R1b представляет собой метокси.
5. Соединение по п.1, где R1c представляет собой галоген.
6. Соединение по п.5, где R1c представляет собой фтор или хлор.
7. Соединение по п.1, где R2a представляет собой галоген.
8. Соединение по п.1, где R2b представляет собой водород, галоген или -SO2CH3.
9. Соединение по п.1, где R3 представляет собой водород.
10. Соединение по п.1, где R3 представляет собой метил.
11. Соединение по п.1, где R4 представляет собой фенил.
12. Соединение по п.1, где R4 представляет собой C3-7алкил.
13. Соединение по п.12, где C3-7алкил представляет собой циклопентил или циклогексил.
14. Соединение по п.1, где R5 представляет собой Н или метил.
15. Соединение по п.1 где R6 представляет собой -СООН.
16. Соединение по п.1 где R6 представляет собой тетразолил.
17. Соединение по п.16 где тетразолил представляет собой тетразол-5-ил.
18. Соединение по п.1, где X представляет собой линейный C1-6алкандиил.
19. Соединение по п.18, где линейный Cl-6алкандиил представляет собой -СН2СН2СН2-.
20. Соединение по п.19, где R4 представляет собой фенил.
21. Соединение по п.20, где R1a и R2a представляют собой галоген.
22. Соединение по п.21, где R3 представляет собой метил.
23. Соединение по п.21, где R3 представляет собой водород.
24. Соединение по п.1, где X представляет собой разветвленный C1-6алкандиил.
25. Соединение по п.1, где соединение представляет собой
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(3-изопропилфенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-(циклогексил)этил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1Н,3H)дион или
3-[2(R)-{2-[1-(5-тетразоил)пропил]амино}-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6-метилсульфонилбензил]-6-метилпиримидин-2,4(1Н,3H)дион.
26. Соединение по п.1, где соединение представляет собой
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-хлор-3-метилфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6-(метилсульфонил)бензил]пиримидин-2,4(1Н,3H)дион,
3-[2(R)-{гидроксикарбонилпропиламино}-2-фенилэтил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1Н,3H)дион или
3-[2(R)-{гидроксикарбонилпропиламино}-2-(изобутил)этил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1Н,3H)дион.
27. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель или разбавитель.
28. Способ антагонизации гонадотропин-высвобождающего гормона в организме нуждающегося в этом субъекта, включающий введение субъекту эффективного количества соединения по п.1.
29. Способ лечения состояния, связанного с половым гормоном, у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции по п.27.
30. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой рак, доброкачественную гипертрофию предстательной железы или миому матки.
31. Способ по п.30, где рак представляет собой рак предстательной железы, рак матки, рак молочной железы или гипофизарную гонадотропную аденому.
32. Способ по п.31, где рак представляет собой рак предстательной железы.
33. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой эндометриоз, поликистозное заболевание яичников, фиброзные опухоли матки или преждевременное половое созревание.
34. Способ по п.33, где состояние, связанное с половым гормоном, представляет собой эндометриоз.
35. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой фиброзные опухоли матки.
36. Способ лечения бесплодия у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции по п.27.
37. Способ лечения красной волчанки, синдрома раздраженного кишечника, предменструального синдрома, волосатости, низкого роста или нарушений сна у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции по п.27.
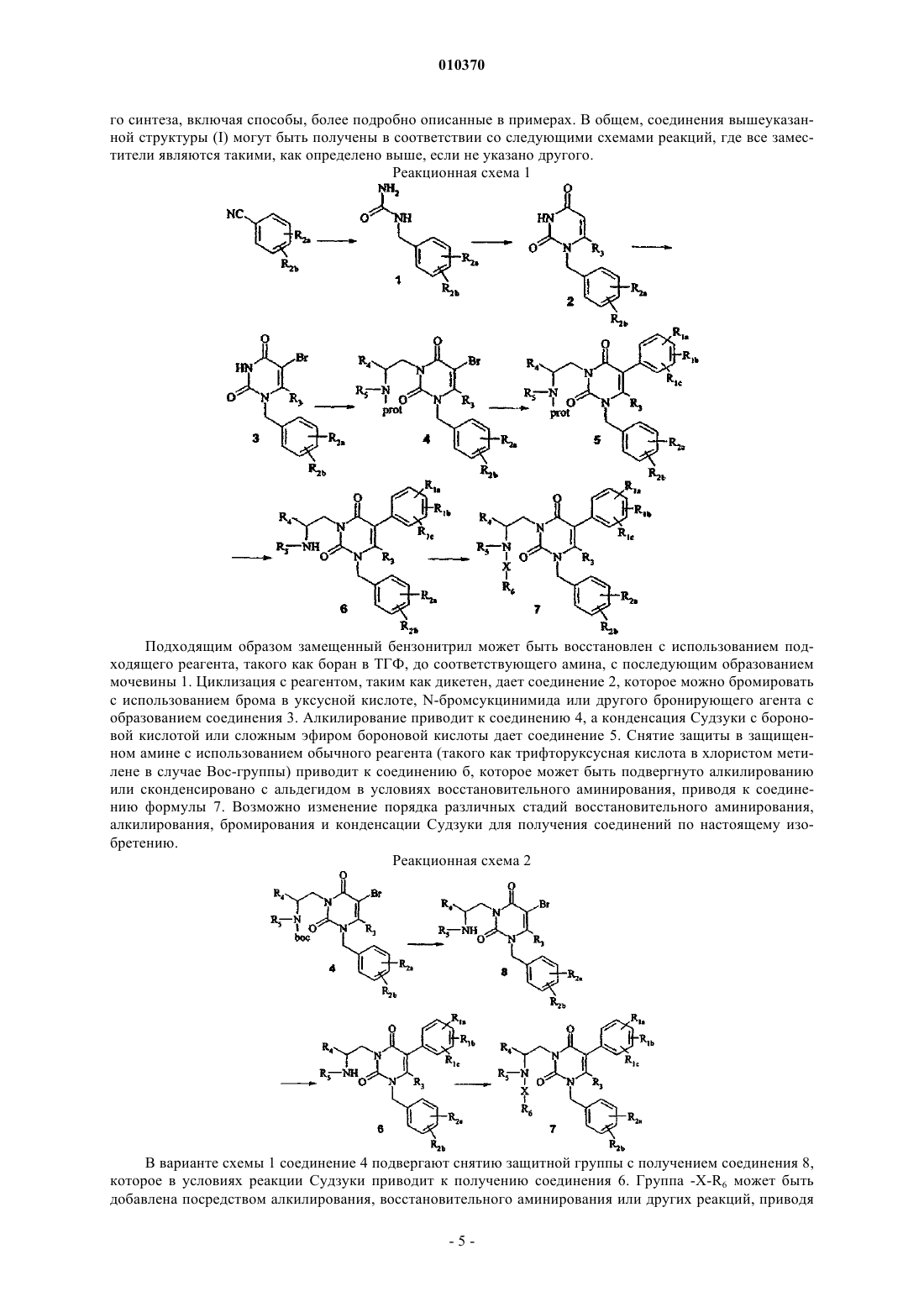
Текст
010370 Заявление о государственных интересах Частичная финансовая поддержка описанного исследования обеспечена правительством США,гранты 1-R43-HD38625 и 2R44-HD38625-02, предоставленные Национальным Институтом Здоровья. Правительство США может обладать определенными правами на данное изобретение. Предпосылки создания изобретения Область изобретения Данное изобретение относится, в общем, к антагонистам рецептора гонадотропинвысвобождающего гормона (GnRH) и к способам лечения нарушений путем введения таких антагонистов нуждающимся в этом теплокровным животным. Описание области техники Гонадотропин-высвобождающий гормон (гонадолиберин, GnRH), также известный как гормон, высвобождающий лютеинизирующий гормон (LHRH), представляет собой декапептид (pGlu-His-Trp-SerTyr-Gly-Leu-Arg-Pro-Gly-NH2), играющий важную роль в размножении человека. GnRH высвобождается из гипоталамуса и воздействует на гипофиз, стимулируя биосинтез и высвобождение лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH). LH, высвобождающийся из гипофиза, является ответственным за регулирование продуцирования гонадных стероидов как у самцов, так и у самок,тогда как FSH регулирует сперматогенез у самцов и развитие фолликулов у самок. Вследствие их биологической важности синтетические антагонисты и агонисты GnRH находились в центре пристального внимания, особенно в контексте рака предстательной железы, рака молочной железы, эндометриоза, лейомиомы матки (фиброзные опухоли), рака яичников, гиперплазии предстательной железы, вспомогательной репродуктивной терапии и преждевременного полового созревания (The Lancet 358: 1793-1803, 2001; Mol. Cell. Endo. 166: 9-14, 2000). Например, пептидные агонисты GnRH, такие как лейпрорелин (pGlu-His-Trp-Ser-Tyr-d-Leu-Leu-Arg-Pro-NHEt), использовали для лечения таких состояний. Оказалось, что такие агонисты функционируют за счет связывания с рецептором GnRH в гипофизарных гонадотропинах, индуцируя таким образом синтез и высвобождение гонадотропинов. Постоянное введение агонистов GnRH исчерпывает запасы гонадотропинов и впоследствии осуществляет понижающую регуляцию рецептора, приводя к подавлению стероидных гормонов после определенного промежутка времени (например, порядка 2-3 недель после начала постоянного введения). Напротив, предполагается, что антагонисты GnRH подавляют начало действия гонадотропинов, и соответственно привлекают наибольшее внимание в течение последних двух десятилетий. До настоящего времени некоторыми основными препятствиями для клинического применения таких антагонистов оказывалась их относительно низкая биодоступность и неблагоприятные побочные действия, вызываемые высвобождением гистамина. Однако сообщалось о нескольких пептидных антагонистах, обладающих свойствами низкого высвобождения гистамина, хотя их все еще нужно доставлять в организм с помощью путей продолжительной доставки (таких как подкожные инъекции или внутриназальные спреи) вследствие ограниченной биодоступности. Принимая во внимание ограничения, связанные с пептидными антагонистами GnRH, был предложен ряд непептидных соединений. Например, Cho et al. (J. Med. Chem. 41: 4190-4195, 1998) описали тиено[2,3-b]пиридин-4-оны для применения в качестве антагонистов рецепторов GnRH; в патентах США 5780437 и 5849764 предложены замещенные индолы в качестве антагонистов рецепторов GnRH(как в опубликованных заявках РСТ WO 97/21704, 98/55479, 98/55470, 98/55116, 98/55119, 97/21707,97/21703 и 97/21435); в опубликованной заявке РСТ WO 96/38438 описаны трициклические диазепины в качестве антагонистов рецептора GnRH; в опубликованных заявках РСТ WO 97/14682, 97/14697 и 99/09033 описаны производные хинолина и тиенопиридина в качестве антагонистов GnRH; в опубликованных заявках РСТ WO 97/44037, 97/44041, 97/44321 и 97/44339 предложены замещенные хинолин-2 оны в качестве антагонистов рецептора GnRH; и в опубликованной заявке РСТ WO 99/33831 описаны некоторые фенил-замещенные конденсированные азотсодержащие бициклические соединения в качестве антагонистов рецептора GnRH. В недавно опубликованных заявках РСТ WO 02/066459 и WO 02/11732 описано применение производных индола и новых бициклических и трициклических производных пирролидина в качестве антагонистов GnRH, соответственно. Другие недавно опубликованные заявки РСТ,в которых описаны соединения и их применение в качестве антагонистов GnRH, включают WO 00/69859, WO 01/29044, WO 01/55119, WO 03/013528, WO 03/011870, WO 03/011841, WO 03/011839 иWO 03/011293. Несмотря на то, что в данной области были проведены важные исследования, все еще сохраняется необходимость в эффективных антагонистах рецептора GnRH, имеющих небольшие молекулы. Также существует необходимость в фармацевтических композициях, содержащих такие антагонисты рецепторов GnRH, а также способах, относящихся к их применению для лечения, например, состояний, связанных с половым гормоном. Настоящее изобретение удовлетворяет таким требованиям и обеспечивает другие относящиеся к данному вопросу преимущества. Краткое изложение сущности изобретения Кратко, в общем, данное изобретение относится к антагонистам рецептора гонадотропин-1 010370 высвобождающего гормона (GnRH), а также к способам их получения и применения, и к содержащим их фармацевтическим композициям. Более конкретно, антагонистами рецептора GnRH по данному изобретению являются соединения, имеющие следующую общую структуру (I): включая их стереоизомеры и фармацевтически приемлемые соли, где R1a, R1b, R1c, R2a, R2b, R3, R4, R5, R6 иX являются такими, как определено ниже. Антагонисты рецептора GnRH по данному изобретению имеют применение для широкого ряда терапевтических назначений и могут использоваться для лечения множества состояний, связанных с половым гормоном, как у мужчин, так и у женщин, а также у млекопитающих в общем (также упоминаемых в данном изобретении как субъект). Например, такие состояния включают эндометриоз, фиброзные опухоли матки, поликистозное заболевание яичников, гирсутизм (волосатость), преждевременное половое созревание, гонадную стероид-зависимую неоплазию, такую как рак предстательной железы, молочной железы и яичников, гонадотропно-гипофизарные аденомы, апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественную гипертрофию предстательной железы, для контрацепции и при бесплодии (например, вспомогательная репродуктивная терапия, такая как оплодотворение in vitro). Соединения по данному изобретению также можно использовать в качестве вспомогательной терапии дефицита гормона роста и низкорослости, и для лечения системной красной волчанки. Соединения также можно использовать в комбинации с андрогенами, эстрогенами, прогестеронами и антиэстрогенами и антипрогестеронами для лечения эндометриоза, фиброзных опухолей и в контрацепции, а также в комбинации с ингибитором ангиотензин-превращающего фермента, антагонистов рецептора ангиотензина II или ингибитором ренина для лечения фиброзных опухолей матки. Кроме того соединения можно использовать в комбинации с бисфосфонатами и другими агентами для лечения и/или профилактики нарушений кальциевого и фосфатного баланса и костного метаболизма и в комбинации с эстрогенами, прогестеронами и/или андрогенами для профилактики или лечения потери костной массы или гипогонадальных симптомов, такие как приливы во время терапии антагонистом GnRH. Соединения по настоящему изобретению, помимо их антагонистической активности в отношении рецептора GnRH, обладают пониженным взаимодействием с основными метаболическими ферментами печени, а именно ферментами Цитохром Р 450. Данное семейство ферментов, которое включает подтипыCYP2D6 и CYP3A4, отвечает за метаболизм лекарственных средств и токсинов, приводя к их выведению из организма. Ингибирование данных ферментов может привести к состояниям, угрожающим жизни, при которых фермент не способен выполнять свою функцию. Способы данного изобретения включают введение эффективного количества антагониста рецептораGnRH, предпочтительно в виде фармацевтической композиции, нуждающемуся в этом млекопитающему. Таким образом, в следующем варианте осуществления, описаны фармацевтические композиции, содержащие один или несколько антагонистов рецептора GnRH по данному изобретению в комбинации с фармацевтически приемлемым носителем и/или разбавителем. Эти и другие аспекты изобретения станут очевидными со ссылкой на следующее подробное описание. Наконец, в данном описании указаны различные ссылки, в которых описана более подробно определенная информация, касающаяся предшествующего уровня техники, способы, соединения и/или композиции, каждая из которых включена посредством ссылки в данное описание во всей своей полноте. Подробное описание изобретения Как отмечено выше, в общем, настоящее изобретение относится к соединениям, полезным в качестве антагонистов рецептора гонадотропин-высвобождающего гормона (GnRH). Соединения данного изобретения имеют следующую структуру (I): или представляют собой ее стереоизомер или фармацевтически приемлемую соль, гдеR1a, R1b и R1c являются одинаковыми или разными и независимо представляют собой водород, галоген, C1-4 алкил, гидрокси или алкокси, или R1a и R1b, взятые вместе, образуют -OCH2O- или -ОСН 2 СН 2-;R2a и R2b являются одинаковыми или разными и независимо представляют собой водород, галоген,трифторметил, циано или -SO2CH3;R3 представляет собой водород или метил;R4 представляет собой фенил или C3-7 алкил;R5 представляет собой водород или C1-4 алкил;X представляет собой C1-6 алкандиил, необязательно замещенный 1-3 C1-6 алкильными группами. Как использовано в данном описании, вышеуказанные термины имеют следующие значения. Термин C1-6 алкил означает линейный или разветвленный, нециклический или циклический, ненасыщенный или насыщенный алифатический углеводород, содержащий от 1 до 6 атомов углерода. Иллюстративные насыщенные линейные алкилы включают метил, этил, н-пропил, н-бутил, н-пентил, н-гексил и тому подобные, тогда как насыщенные разветвленные алкилы включают изопропил, втор-бутил, изобутил, трет-бутил, изопентил и тому подобные. Иллюстративные насыщенные циклические алкилы включают циклопропил, циклобутил, циклопентил, циклогексил и тому подобные; в то время как ненасыщенные циклические алкилы включают циклопентенил и циклогексенил и тому подобные. Ненасыщенные алкилы содержат по крайней мере одну двойную или тройную связь между смежными атомами углерода (упоминаемые как алкенил или алкинил, соответственно). Иллюстративные линейные и разветвленные алкенилы включают этиленил, пропиленил, 1-бутенил, 2-бутенил, изобутиленил, 1 пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил и тому подобные,тогда как иллюстративные линейные и разветвленные алкилинилы включают ацетиленил, пропинил, 1 бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-бутинил и тому подобные. Термин С 1-4 алкил означает линейный или разветвленный, нециклический или циклический углеводород, содержащий от 1 до 4 атомов углерода. Иллюстративные линейные алкилы включают метил,этил, н-пропил, н-бутил и тому подобные, разветвленные алкилы включают изопропил, втор-бутил, изобутил, трет-бутил и тому подобные; тогда как циклические алкилы включают циклопропил и тому подобные. Термин C3-7 алкил означает линейный или разветвленный, нециклический или циклический углеводород, содержащий от 3 до 7 атомов углерода. Иллюстративные линейные алкилы включают нпропил, н-бутил, н-гексил и тому подобные; тогда как разветвленные алкилы включают изопропил, вторбутил, изобутил, трет-бутил, изопентил и тому подобные. Иллюстративные циклические алкилы включают циклопропил, циклопентил, циклогексил и тому подобные. Термин C1-6 алкандиил означает двухвалентный C1-6 алкил, у которого два атома водорода удалены от одного и того же атома углерода или от различных атомов углерода, такие, например, как -СН 2-, СН 2 СН 2-, -СН 2 СН 2 СН 2-, -СН(СН 3)СН 2 СН 2-, -СН 2 С(СН 3)2 СН 2- и тому подобные. Термин галоген означает фтор, хлор, бром или иод, обычно фтор и хлор. Термин гидрокси означает -ОН. Термин алкокси означает -О-(C1-6 алкил). Термин циано означает CN. Термин изостера кислоты означает фрагмент, который проявляет свойства, аналогичные свойствам карбоновой кислоты и имеет pKa менее 8 предпочтительно менее 7. Иллюстративные изостеры кислот включают тетразол, 3H-[1,3,4]оксадиазол-2-он, [1,2,4]оксадиазол-3-он, 1,2-дигидро-[1,2,4]триазол-3-он, 2 Н[1,2,4]оксадиазол-5-он, триазол, замещенный сульфонильной или сульфоксидной группой, имидазол, замещенный сульфонильной или сульфоксидной группой, [1,2,4]оксадиазолидин-3,5-дион, [1,2,4]тиадиазолидин 3,5-дион, имидазолидин-2,4-дион, имидазолидин-2,4,5-трион, пирролидин-2,5-дион и пирролидин-2,3,5 трион. Изостеры кислот также включают -C(=O)NHSO2NRaRb, -C(=O)NHSO2Rb, -C(=O)NHC(=O)NRaRb иRa представляет собой водород или C1-4 алкил, и Rb представляет собой C1-4 алкил. В одном варианте осуществления R4 представляет собой фенил, и иллюстративные антагонистыGnRH включают соединения, имеющие следующую структуру (II). В другом варианте осуществления R4 представляет собой C3-7 алкил и иллюстративные антагонистыGnRH по настоящему изобретению включают соединения, имеющие следующую структуру (III). В более конкретных вариантах осуществления структуры (III), C3-7 алкил представляет собой линейный или разветвленный С 3-7 алкил, такой как изобутил, как представлено структурой (IV), или представляет собой циклический С 3-7 алкил, такой как циклогексил, как представлено структурой (V): В другом варианте осуществления R1a, R1b и R1c представляют собой водород, алкокси и галоген,соответственно. Иллюстративный характер замещения включает 2-галоген-3-алкоксифенил. Иллюстративные алкоксильные группы включают метокси и этокси, тогда как иллюстративные фрагменты галогена включают фтор и хлор. В альтернативном варианте осуществления Rla и R1b, взятые вместе, образуют -OCH2O-, такой как 3,4-метилендиокси. В следующем варианте осуществления R2a и R2b представляют собой водород, трифторметил, галоген или -SO2CH3. Иллюстративный характер замещения включает R2a в виде галогена в положении 2, иR2b в виде водорода, трифторметила, галогена или -SO2CH3 в положении 6. Следующие варианты осуществления включают те, в которых R5 представляет собой Н или метил; R6 представляет собой -СООН, и/или X представляет собой -СН 2 СН 2-, -СН 2 СН 2 СН 2- или -CH2CH2CH2CH2-. Соединения по настоящему изобретению могут быть получены известными методами органическо-4 010370 го синтеза, включая способы, более подробно описанные в примерах. В общем, соединения вышеуказанной структуры (I) могут быть получены в соответствии со следующими схемами реакций, где все заместители являются такими, как определено выше, если не указано другого. Реакционная схема 1 Подходящим образом замещенный бензонитрил может быть восстановлен с использованием подходящего реагента, такого как боран в ТГФ, до соответствующего амина, с последующим образованием мочевины 1. Циклизация с реагентом, таким как дикетен, дает соединение 2, которое можно бромировать с использованием брома в уксусной кислоте, N-бромсукцинимида или другого бронирующего агента с образованием соединения 3. Алкилирование приводит к соединению 4, а конденсация Судзуки с бороновой кислотой или сложным эфиром бороновой кислоты дает соединение 5. Снятие защиты в защищенном амине с использованием обычного реагента (такого как трифторуксусная кислота в хлористом метилене в случае Вос-группы) приводит к соединению б, которое может быть подвергнуто алкилированию или сконденсировано с альдегидом в условиях восстановительного аминирования, приводя к соединению формулы 7. Возможно изменение порядка различных стадий восстановительного аминирования,алкилирования, бромирования и конденсации Судзуки для получения соединений по настоящему изобретению. Реакционная схема 2 В варианте схемы 1 соединение 4 подвергают снятию защитной группы с получением соединения 8,которое в условиях реакции Судзуки приводит к получению соединения 6. Группа -Х-R6 может быть добавлена посредством алкилирования, восстановительного аминирования или других реакций, приводя Сложный эфир замещенной фенилуксусной кислоты 9 (полученный из соответствующей кислоты или полученный коммерчески) и такой реагент, как диметилацеталь диметилформамида, конденсируют с получением соединения 10. Циклизация с мочевиной приводит к соединению формулы 11. Алкилирование с использованием, например, замещенного бромистого бензила приводит к соединению 12, которое может быть подвергнуто алкилированию с подходящим алкилгалогенидом, реакции конденсации Мицунобу с подходящим спиртом или взаимодействию с мезилатом или сульфонатом, приводя к соединению 5. Соединения по настоящему изобретению в общем можно использовать в виде свободной кислоты или свободного основания. Альтернативно, соединения по данному изобретению можно использовать в виде кислотных или основных аддитивных солей. Кислотно-аддитивные соли соединений со свободной аминогруппой по настоящему изобретению могут быть получены способами, хорошо известными в данной области, и могут быть образованы органическими и неорганическими кислотами. Подходящие органические кислоты включают малеиновую, фумаровую, бензойную, аскорбиновую, янтарную, метансульфоновую, уксусную, трифторуксусную, щавелевую, пропионовую, винную, салициловую, лимонную, глюконовую, молочную, миндальную, коричную, аспарагиновую, стеариновую, пальмитиновую,гликолевую, глутаминовую и бензолсульфоновую кислоты. Подходящие неорганические кислоты включают хлористо-водородную, бромисто-водородную, серную, фосфорную и азотную кислоты. Основноаддитивные соли включают соли, которые образованы карбоксилатным анионом, и включают соли, образованные органическими и неорганическими катионами, например, выбранными из щелочных и щелочно-земельных металлов (например, литий, натрий, калий, магний, барий и кальций). А также ионом аммония и его замещенными производными (например, дибензиламмоний, бензиламмоний, 2 гидроксиэтиламмоний и тому подобные). Таким образом, подразумевается, что термин фармацевтически приемлемая соль структуры (I) охватывает любые и все приемлемые солевые формы. Кроме того, пролекарства также включены в контекст данного изобретения. Пролекарства представляют собой любые ковалентно связанные носители, которые высвобождают соединение структуры(I) in vivo, когда такое пролекарство вводят пациенту. Пролекарства обычно получают модификацией функциональных групп таким образом, что модифицированная группа расщепляется или при обычной обработке или in vivo, давая исходное соединение. Пролекарства включают, например, соединения по данному изобретению, где гидроксильная, амино или сульфгидрильная группы связаны с любой группой, которая при введении пациенту отщепляется с образованием гидроксильной, амино или сульфгидрильной группы. Таким образом, иллюстративные примеры пролекарств включают (но не ограничиваются указанным) ацетатные, формиатные и бензоатные производные спиртовых и аминовых функциональных групп соединений структуры (I). Кроме того,в случае карбоновых кислот (-СООН), можно использовать сложные эфиры, такие как метиловые сложные эфиры, этиловые сложные эфиры и тому подобные. Что касается стереоизомеров, соединения структуры (I) могут иметь хиральные центры и могут существовать в виде рацематов, рацемических смесей и индивидуальных энантиомеров или диастереомеров. Все такие изомерные формы входят в объем данного изобретения. Кроме того, некоторые кристаллические формы соединений структуры (I) могут существовать в виде полиморфов, которые включены в объем настоящего изобретения. Дополнительно, некоторые соединения структуры (I) также могут образовывать сольваты с водой или другими органическими растворителями. Аналогичным образом такие сольваты включены в объем данного изобретения. Эффективность соединения в качестве антагониста рецептора GnRH может быть определена с использованием различных методов анализа. Методы анализа, хорошо известные в данной области, вклю-6 010370 чают применение культивированных гипофизарных клеток для измерения активности GnRH (Vale et al.,Endocrinology 91: 562-572, 1972) и измерение связывания радиоактивно меченного лиганда с мембранами гипофиза крыс (Perrin et al., Mol. Pharmacol. 23: 44-51, 1983) или с мембранами клеток, экспрессирующих клонированные рецепторы, как описано ниже. Другие методы анализа включают (но не ограничиваются указанным) измерение влияния антагонистов рецептора GnRH на ингибирование стимулированногоGnRH потока кальция, модулирование гидролиза фосфоинозита и циркулирующие концентрации гонадотропинов у кастрированных животных. Описание данных методов, синтез радиоактивно меченых лигандов, применение радиоактивно меченного лиганда в радиоиммуноанализе и измерение эффективности соединения в качестве антагониста рецептора GnRH приведены далее. Ингибирование высвобождения LH, стимулированного GnRH Подходящие антагонисты GnRH способны ингибировать специфическое связывание GnRH с его рецептором и антагонизировать активность, связанную с GnRH. Например, ингибирование стимулированного GnRH высвобождения LH у молодых крыс может быть измерено в соответствии со способомVilchez-Martinez (Endocrinology 96: 1130-1134, 1975). Кратко, самцам крыс Spraque-Dawley возрастом двадцать пять дней вводили антагонист GnRH в физиологическом растворе или в виде другой подходящей композиции перорально с помощью зонда, в виде подкожной инъекции или внутривенной инъекции. После этого с помощью подкожной инъекции вводили 200 нг GnRH в 0,2 мл физиологического раствора. Через тридцать минут после последней инъекции животных обезглавливали и собирали кровь из туловища. После центрифугирования отделенную плазму крови хранили при -20 С до определения концентрации LH и/или FSH методом радиоиммуноанализа (см. ниже). Анализ антагонистов GnRH в культуре клеток переднего гипофиза крыс Передние доли гипофиза получали от самок крыс Sprague-Dawley возрастом 7 недель, и собранный гипофиз расщепляли коллагеназой в дисперсионной колбе в течение 1,5 ч при 37 С. После расщепления коллагеназой гипофиз дополнительно обрабатывали нейраминидазой в течение 9 мин при 37 С. Обработанную ткань затем промывали средой 0,1% БСА/McCoy's 5 А и промытые клетки суспендировали в среде 3% FBS/0,1 BSA/McCoy's 5 А и помещали в 96-луночные планшеты для культур тканей при плотности клеток 40000 клеток на лунку в 200 мкл среды. Клетки затем инкубировали при 37 С в течение 3 дней. Для анализа антагониста GnRH инкубированные клетки первоначально промывали один раз средой 0,1% БСА/McCoy's 5 А с последующим добавлением тестируемого образца плюс 1 нМ GnRH в 200 мкл среды 0,1% БСА/McCoy's 5 А в трехкратном повторе. Каждый образец анализировали при 5 уровнях дозировки для получения доза-зависимой кривой для определения эффективности ингибирования высвобожденияLH и/или FSH, стимулированного GnRH. После инкубирования в течение 4 ч при 37 С среду собирали и определяли уровни LH и/или FSH, секретированные в среду, методом радиоиммуноанализа (RIA). Анализы мембранного связывания 1 Клетки, стабильно или временно трансфицированные векторами экспрессии рецептора GnRH, собирали, суспендировали в 5%-ном растворе сахарозы и гомогенизировали с использованием политронного гомогенизатора (215 с). Ядра удаляли центрифугированием (3000g в течение 5 мин), и супернатант центрифугировали (20000g в течение 30 мин, 4 С), собирая мембранную фракцию. Конечный препарат мембран повторно суспендировали в буфере для связывания (10 мМ Hepes (pH 7,5), 150 мМ NaCl,и 0,1% БСА) и хранили при -70 С. Реакции связывания проводили на сборном 96-луночном планшетеMillipore Multiscreen для фильтрования с мембранами GF/C, покрытыми полиэтиленимином. Реакцию инициировали добавлением мембран (40 мкг белка в 130 мкл буфера для связывания) к 50 мкл [125I]меченного пептида GnRH (100000 импульсов в минуту) и 20 мкл конкурента при различных концентрациях. Реакцию заканчивали через 90 мин, прилагая вакуум и промывая (2 Х) забуференным фосфатом физиологическим раствором. Связанную радиоактивность измеряли с использованием сцинтилляционного 96-луночного счетчика (Packard Topcount) или удаляя фильтры с планшета и непосредственно подсчитывая гамма-излучение. Величины Ki рассчитывали из данных конкурентного связывания с использованием нелинейной регрессии методом наименьших квадратов с использованием пакета программного обеспечения Prism (GraphPad Software). Анализы мембранного связывания 2 Для проведения дополнительного анализа мембранного связывания стабильно трансфицированные клетки НЕК 293 собирали, постукивая колбы с культурами тканей о зафиксированную поверхность и центрифугируя при 1000g в течение 5 мин. Осадок клеток повторно суспендировали в 5%-ном растворе сахарозы и гомогенизировали с помощью политронного гомогенизатора с использованием двух стадий гомогенизации в течение 15 с. Гомогенаты клеток затем центрифугировали в течение 5 минут при 3000g для удаления ядер и после чего супернатант центрифугировали в течение 30 мин при 44000g,собирая мембранную фракцию. Осадок мембран повторно суспендировали в буфере для связыванияGnRH (10 мМ HEPES, рН 7,5, 150 мМ NaCl и 0,1% БСА) и аликвоты немедленно подвергали мгновенному замораживанию в жидком азоте и хранили при -80 С. Содержание белка в суспензии мембран определяли с использованием набора для анализа белка (Bio-Rad, Hercules, СА). Анализы конкурентного радиолигандного связывания с препаратами мембран проводили в 96-7 010370 луночных планшетах для фильтрования Millipore с использованием мембранных фильтров GF/C, которые предварительно были покрыты 200 мкл 0,1%-ного раствора полиэтиленимина (Sigma, St. Louis. МО). Перед применением планшеты промывали 3 Х забуференным фосфатом физиологическим раствором. Фракцию мембран в буфере для связывания GnRH (130 мкл, содержащие 25 мкг белка для рецепторов человека или макаки или 12 мкг для рецепторов крысы) добавляли в лунки вместе с 20 мкл конкурирующего лиганда при различных концентрациях. Реакцию связывания инициировали добавлением радиоактивно меченого лиганда (0,1 нМ в 50 мкл буфера для связывания GnRH). Реакцию оставляли протекать в течение 90 мин на платформе встряхивающего устройства при комнатной температуре и затем заканчивали реакцию, помещая планшет для анализа на вакуумный коллектор Millipore (Millipore, Bedford, MA), удаляя растворитель и промывая лунки дважды 200 мкл охлажденного на льду забуференного фосфатом физиологического раствора (PBS). Фильтры из лунок удаляли и считывали с использованием счетчика гамма-излучения. Значения Ki рассчитывали из каждой кривой конкурентного связывания с использованием нелинейной регрессии методом наименьших квадратов и корректировали для концентрации радиоактивно меченого лиганда с использованием уравнения Ченга-Прусова (Prism, GraphPadSoftware, San Diego, СА), предполагая аффинность радиоактивно меченого лиганда равной 0,5 нм. Средние значения Ki рассчитывали из антилогарифма среднего значения их величин pKi для каждой пары рецепторов. Анализ мембранного связывания 3 Стабильно трансфицированные рецептором GNRH клетки RBL выращивали до слияния. Среду удаляли и монослой клеток промывали один раз с использованием DPBS. Раствор 0,5 мМ EDTA/PBS (не содержащий Са, Mg) добавляли к планшету, который затем инкубировали при 37 С в течение 10 мин. Затем клетки удаляли путем осторожного постукивания колб. Клетки собирали и осаждали ценрифугированием при 800g в течение 10 мин при 4 С. Осадок клеток затем повторно суспендировали в буфереEGTA, рН = 7,4 с NaOH]. Затем проводили клеточный лизис с использованием ячейки под давлением и прилагая N2 при давлении 900 фт/кв.дюйм в течение 30 мин при 4 С. Неразрушенные клетки и больший дебрис удаляли центрифугированием при 1200 g в течение 10 мин при 4 С. Супернатант клеточных мембран затем центрифугировали при 45000g и полученный осадок мембран затем повторно суспендировали в буфере для анализа и гомогенизировали на льду с использованием гомогенизатора тканей. Концентрации белка определяли с использованием набора реагентов Кумассин плюс белок (Coomassie Plus Protein Reagent kit (Pierce, Rockford, IL, используя в качестве стандарта бычий сывороточный альбумин. Осадок делили на аликвоты и хранили при -80 С до использования. С помощью анализов титрования с использованием диапазона концентраций белка определяли оптимальную концентрацию белка как равную 15 мкг на лунку для конечной концентрации. Фильтровальные планшеты UniFilter GF/C (Perkin Elmer, Boston MA) предварительно обрабатывали раствором 0,5% полиэтиленимина в дистиллированной воде в течение 30 мин. Фильтры предварительно споласкивали 200 мкл на лунку PBS, 1% БСА (Фракция V) и 0,01% Tween-20, рН = 7,4), используя устройства для сбора клеток (UniFilter-96 Filtermate; Packard). Мембраны собирали путем быстрого фильтрования в вакууме и промывали 3 раза 250 мкл охлажденного на льду буфера (PBS, 0,01% Tween-20, рН= 7,4). Планшеты сушили на воздухе, добавляли 50 мкл сцинтилляционной жидкости (Microscint 20;Packard) и контролировали радиоактивность планшета с использованием счетчика TopCount NXT (Packard Instruments, IL). Эксперименты связывания проводили в буфере, содержащем 10 мМ HEPES, 150 мМ NaCl и 0,1% БСА, рН=7,5. Мембраны инкубировали с 50 мкл [125I] His5, D-Tyr6 GnRH (конечная концентрация 0,2 нм) и 50 мкл конкурентных лигандов, имеющих небольшие молекулы, при концентрациях, колеблющихся в диапазоне от 30 пМ до 10 мкМ для общего объема в каждой лунке, составляющего 200 мкл. Инкубацию проводили в течение 2 ч при комнатной температуре. Реакцию заканчивали быстрым фильтрованием через фильтры GF/C, как описано ранее. Подгонку кривой проводили с использованием программного обеспечения Excel Fit (IDBS, Emeryville, CA). Значения Ki рассчитывали способом Ченга и Прусова(Cheng и Prusoff, 1973) с использованием величины Kd, равной 0,7 нм, для радиоактивно меченого лиганда, которая предварительно была определена в экспериментах насыщения связывания. Измерения потока Са Для определения ингибирования стимулированного GnRH потока кальция в клетках, эспрессирующих рецептор GnRH человека, в 96-луночный планшет высевали клетки RBL, стабильно трансфицированные рецептором GnRH человека, при плотности 50000 клеток/лунку и оставляли присоединяться в течение ночи. Клетки нагружали в течение 1 ч при 37 С в следующей среде: DMEM (модифицированная по способу Дульбекко среда Игла) с 20 мМ HEPES, 10% FBS, 2 мкМ Fluo=4, 0,02% плуроновой кислоты и 2,5 мМ пробенецида. Клетки промывали 4 раза промывочным буфером (сбалансированная по Хенку соль, 20 мМ HEPES, 2,5 мМ пробенецида) после нагрузки, оставляя 150 мкл в лунке после последнего промывания. GnRH разводили в 0,1% БСА, содержащем буфер FLIPR (сбалансированная по Хенку соль, 20 мМHEPES) до концентрации 20 нм и распределяли в лунки 96-луночного планшета (низкое связывание бел-8 010370 ка). Различные концентрации антагониста получали в 0,1% БСА/FLIPR буфере в третьем 96-луночном планшете. Измерение флуоресценции вследствие стимулированного GnRH (50 мкл 20 нМ, или 4 нМ конечной концентрации) потока Са проводили в соответствии с инструкциями производителя на системеFLIPR (Molecular Devices, система FLIPR384, Sunnyvale, CA) после инкубирования в течение 1 мин с 50 мкл антагониста в различной концентрации. Анализ гидролиза фосфоинозита Использованный способ представляет собой модификацию опубликованных протоколов (W. Zhouet al; J. Biol. Chem. 270 (32), pp 18853-18857, 1995). Кратко, клетки RBL, стабильно трансфицированные рецепторами GnRH человека, высевали в 24-луночные планшеты при плотности 200000 клеток/лунку в течение 24 ч. Клетки промывали однократно не содержащей инозита средой, содержащей 10% диализованного FBS и затем метили 1 мкКи/мл [myo-3 Н]-инозита. Через 20-24 часа клетки промывали буфером(140 нМ NaCl, 4 мМ KCl, 20 мМ Hepes, 8,3 мМ глюкозы, 1 мМ MgCl2, 1 мМ CaCl2 и 0,1% БСА) и обрабатывали природным пептидом GnRH в том же буфере в присутствии различных концентраций антагониста или без него и 10 мМ LiCl в течение 1 ч при 37 С. Клетки экстрагировали 10 мМ муравьиной кислотой при 4 С в течение 30 мин и загружали на колонку Dowex AG1-X8, промывали и элюировали 1 М формиатом аммония и 0,1 М муравьиной кислотой. Элюат считывали в сцинтилляционном счетчике. Для данных анализа гидролиза PI строили график с использованием нелинейной регрессии методом наименьших квадратов с помощью программы Prism (Graphpad, GraphPad Software, San Diego, CA), из которого также рассчитывали соотношение доз. Генерировали линейный график Шилда из соотношений доз,полученных в четырех независимых экспериментах путем линейной регрессий и Х-пересечение использовали для определения аффинности антагониста. Исследование кастрированных животных Исследования кастрированных животных обеспечивают чувствительный in vivo анализ влияния антагониста GnRH (Andrology 25: 141-147, 1993). Рецепторы GnRH в гипофизе опосредуют стимулированное GnRH высвобождение LH в круг кровообращения. Кастрация приводит к повышенным уровням циркулирующего LH вследствие восстановления негативной обратной реакции гонадных стероидов, приводя к повышению стимулированного GnRH высвобождения LH. Следовательно, измерение подавления уровней циркуляции LH у кастрированных макак можно использовать в качестве чувствительного измерения антагонизма GnRH in vivo. Таким образом, самцам макак проводили хирургическую кастрацию и предоставляли четырехнедельный период для выздоровления, где в данной временной точке присутствуют повышенные уровни LH. Затем животным вводили тестируемое соединение в виде пероральной или внутривенной дозы, и серийные образцы крови отбирали для измерения уровня LH. КонцентрацииLH в сыворотке крови данных животных могут быть определены иммуноанализом или методами биоанализа. (Endocrinology 107: 902-907, 1980). Получение радиоактивно меченого GnRH лиганда Аналог GnRH метили хлорамин-Т способом. К 10 мкг пептида в 20 мкл 0,5 М натрийфосфатного буфера, рН 7,6, добавляли 1 мКи Na125I, с последующим добавлением 22,5 мкг хлорамина-Т в 15 мкл 0,05 М натрийфосфатного буфера и смесь перемешивали вихревой мешалкой в течение 20 секунд. Реакцию останавливали добавлением 60 мкг метабисульфата натрия в 30 мкл 0,05 М натрийфосфатного буфера и свободный иод удаляли, пропуская реакционную смесь через катридж С-8 Sep-Pak (Millipore Corp., Milord, MA). Пептид элюируют небольшим объемом смеси 80% ацетонитрил/вода. Выделенный радиоактивно меченый пептид дополнительно очищают ВЭЖХ с обращенной фазой на аналитической колонкеVydac C-18 (The Separations Group, Hesperia, CA) на градиентной ВЭЖХ системе Beckman 334 с использованием градиента ацетонитрил в 0,1% ТФУК. Очищенный радиоактивный пептид хранят в 0,1% БСА/20% ацетонитрил/0,1% ТФУК при -80 С, и его можно использовать в течение 4 недель. Радиоиммуноанализ (RIA) LH и FSH Для определения уровней LH каждый образец среды анализировали в двухкратном повторе, и все разведения проводили с использованием буфера для радиоиммуноанализа (0,01 М натрийфосфатный буфер /0,15 NaCl/1% БСА/0,01% NaN3, рН 7,5) и набор для анализа получен от Государственной программы Гормон и гипофиз (Nation Hormone и Pituitary Program), поддержанной NIDDK. В полиэтиленовую тест-пробирку 1275 мм добавляли 100 мкл образца среды, разведенной в соотношении 1:5 стандартомrLH в буфере для радиоиммуноанализа, и 100 мкл [125I]-меченного rLH (30000 импульсов в минуту) плюс 100 мкл кроличьего антитела против rLH, разведенного в соотношении 1:187500, и 100 мкл буфера для радиоиммуноанализа. Смесь инкубировали при комнатной температуре в течение ночи. На следующий день добавляли 100 мкл козьего противокроличьего IgG, разведенного в соотношении 1:20, и 100 мкл обычной сыворотки крови кролика, разведенной 1:1000, и смесь инкубировали дополнительно в течение 3 ч при комнатной температуре. Инкубированные пробирки затем центрифугировали при 3000 об/мин в течение 30 мин и супернатант удаляли отсасыванием. Оставшийся в пробирках осадок считывали с использованием счетчика гамма-излучения. Радиоиммуноанализ FSH проводили аналогичным образом, как и анализ для LH, заменяя антитело в отношении LH на антитело в отношении FSH, разведенное в соотношении 1:30000, и меченый rLH на меченый rFSH.-9 010370 Активность антагонистов рецептора GnRH Активность антагонистов рецептора GnRH обычно рассчитывают из значений IC50 как концентрации соединения, необходимой для замещения 50% радиоактивно меченого лиганда из рецептора GnRH и представляют как величину "Ki", рассчитанную по следующему уравнению: где L = радиоактивно меченый лиганд и KD = афинность меченого лиганда по отношению к рецептору (Cheng and Prusoff, Biochem. Pharmacol. 22: 3099, 1973). Антагонисты рецептора GnRH по данному изобретению имеют величину Ki, составляющую 100 мкМ или менее. В предпочтительном варианте осуществления данного изобретения антагонисты рецептора GnRH по данному изобретению имеют величину Ki менее 10 мкМ, и более предпочтительно менее 1 мкМ, и еще более предпочтительно менее чем 0,1 мкМ (т.е. 100 нМ). В этой связи все соединения, конкретно описанные в примерах, имеют величину Ki менее 100 нМ в одном или нескольких анализах мембранного связывания с первого по третий,описанных выше. Способность антагонистов GnRH ингибировать основные ферменты, метаболизирующие лекарственные средства в печени человека, а именно, CYP2D6 и CYP3A4, может быть оценена in vitro с помощью флуориметрического способа, основанного на планшетах для микротитрования, описанного CrespiKm (то есть концентрации субстрата, которая дает половину максимальной скорости) использовали в качестве субстратов-маркеров для CYP2D6 и CYP3A4, соответственно. Кратко, рекомбинантныеCYP2D6 или CYP3A4 инкубировали с субстратом-маркером и генерирующей NADPH системой (состоящей из 1 мМ NADP+, 46 мМ глюкоза-6-фосфата и 3 единиц/мл глюкоза-6-фосфат-дегидрогеназы) при 37 С, в отсутствии или в присутствии 0,03, 0,09, 0,27, 0,82, 2,5, 7,4, 22, 67 и 200 мкМ образца антагониста GnRH. Реакции останавливали добавлением равного объема ацетонитрила. Выпавший в осадок белок удаляли центрифугированием и прозрачную надосадочную жидкость анализировали с использованием флуориметра для планшетов для микротитрования. Антагонисты GnRH по настоящему изобретению предпочтительно имеют величины Ki, превышающие 250 нМ, более предпочтительно, превышающие 1 мкМ, и наиболее предпочтительно, превышающие 5 мкМ. Как отмечено выше, антагонисты рецептора GnRH по данному изобретению имеют применение для широкого ряда терапевтических назначений и могут использоваться для лечения множества состояний,связанных с половыми гормонами, как у мужчин, так и у женщин, а также у млекопитающих в общем. Например, такие состояния включают эндометриоз, фиброзные опухоли матки, поликистозные заболевания яичников, гирсутизм, преждевременное половое созревание, неоплазию, зависимую от половых гормонов, такую как рак предстательной железы, молочной железы и яичников, или гипофизарную гонадотропную аденому, апноэ во сне, синдром раздраженного кишечника, предменструальный синдром, доброкачественную гиперплазию предстательной железы, для контрацепции и при бесплодии (например,вспомогательная репродуктивная терапия, такая как оплодотворение in vitro). Соединения по данному изобретению также могут использоваться в качестве дополнительного средства при лечении дефицита гормона роста и низкого роста и для лечения системной красной волчанки. Кроме того, соединения могут использоваться в сочетании с андрогенами, эстрогенами, прогестеронами и антиэстрогенами и антипрогестогенами для лечения эндометриоза, фиброзных опухолей и для контрацепции, а также в сочетании с ингибитором ангиотензин-превращающего фермента, антагонистом рецептора ангиотензин-11 или ингибитором ренина для лечения фиброзных опухолей матки. Соединения также могут использоваться в сочетании с бисфосфонатами и другими агентами для лечения и/или профилактики нарушений кальциевого, фосфатного и костного метаболизма, и в сочетании с эстрогенами,прогестеронами и/или андрогенами для профилактики или лечения потери костной массы или гипогонадальных симптомов, такие как приливы во время терапии с использованием антагониста GnRH. В другом варианте осуществления изобретения описаны фармацевтические композиции, содержащие один или несколько антагонистов рецептора GnRH. В целях введения соединения по настоящему изобретению могут быть введены в состав фармацевтических композиций. Фармацевтические композиции по настоящему изобретению включают антагонист рецептора GnRH по настоящему изобретению и фармацевтически приемлемый носитель и/или разбавитель. Антагонист рецептора GnRH присутствует в композиции в количестве, которое является эффективным для лечения конкретного заболевания, то есть в количестве, достаточном для достижения антагонистической активности в отношении рецептора GnRH и предпочтительно при приемлемой для пациента токсичности. Обычно фармацевтические композиции по настоящему изобретению могут включать антагонист рецептора GnRH в количестве примерно от 0,1 мг до 250 мг на дозированную форму в зависимости от пути введения и более обычно от 1 мг до 60 мг. Подходящие концентрации и дозы легко могут быть определены специалистом в данной области. Фармацевтически приемлемые носитель и/или разбавители знакомы специалистам в данной облас- 10010370 ти. Для композиций, полученных в виде жидких растворов, приемлемые носители и/или разбавители включают физиологический раствор и стерильную воду и необязательно могут включать антиоксиданты,буферы, бактериостатические агенты и другие обычные добавки. Композиции также могут быть получены в виде пилюль, капсул, гранул или таблеток, которые содержат, в дополнение к антагонисту рецептора GnRH, разбавители, диспергирующие и поверхностно-активные агенты, связующие вещества и лубриканты. Специалист в данной области может кроме того получить препарат антагониста рецептораGnRH подходящим образом и в соответствии с общепринятой практикой, как, например, описано в Remington's Pharmaceutical Sciences, Gennaro, Ed., Mack Publishing Co., Easton, PA 1990. В другом варианте осуществления настоящее изобретение относится к способу лечения состояний,связанных с половыми гормонами, как обсуждалось выше. Такие способы включают введение соединения по настоящему изобретению теплокровному животному в количестве, достаточном для лечения состояния. В данном контексте термин лечение включает профилактическое введение. Такие способы включают системное введение антагониста рецептора GnRH по данному изобретению, предпочтительно в виде фармацевтической композиции, как обсуждалось выше. Как использовано в данном описании,системное введение включает пероральные и парентеральные способы введения. Для перорального введения подходящие фармацевтические композиции антагонистов рецептора GnRH включают порошки,гранулы, пилюли, таблетки и капсулы, а также жидкости, сиропы, суспензии и эмульсии. Данные композиции могут также включать ароматизаторы, консерванты, суспендирующие, загущающие и эмульгирующие агенты, а также другие фармацевтически приемлемые добавки. Для парентерального введения соединения по настоящему изобретению могут быть введены в состав водных растворов для инъекций,которые могут содержать, в дополнение к антагонисту рецептора GnRH, буферы, антиоксиданты, бактериостатические агенты и другие добавки, обычно используемые в таких растворах. Следующий пример приведен в целях иллюстрации, а не ограничения. Подводя итог вышесказанному, антагонисты рецепторов GnRH по данному изобретению могут быть проанализированы с использованием общих способов, обсуждавшихся выше, тогда как в следующих примерах описан синтез иллюстративных соединений по данному изобретению Примеры А. ВЭЖХ методы анализа образцов. Время удерживания, tR, в минутах Способ 1 - Хроматография сверхкритической жидкостью масс-спектрометрия (SFC-MC) Колонка: 4,6150 мМ Deltabond Cyano 5 мкМ от Thermo-Hypersil-Keystone Подвижная фаза: диоксид углерода марки SFC и метанол наилучшего качества с добавлением модификатора 1 мМ диэтилмалоната динатрия Температура: 50 С Давление:120 бар Скорость потока: 4,8 мл/мин Градиент: от 5 до 55% метанола в течение 1,7 мин и выдерживание при 55% в течение 0,8 мин, затем возврат к 5% в течение 0,1 мин для общего времени проведения эксперимента в течение 2,6 мин Способ 2 (ВЭЖХ-масс-спектрометрия) Колонка: Waters ODS-AQ, 2,050 мм Подвижная фаза: А = вода с 0,05% трифторуксусной кислоты; В = ацетонитрил с 0,05% трифторуксусной кислоты Градиент: от 95% А/5% В до 5% А/95% В в течение 13,25 мин и выдерживание при 5% А/95% В в течение 2 мин, затем возврат до 95% А/5% В в течение 0,25 мин Скорость потока: 1 мл/мин УФ длина волны: 220 и 254 нм Способ 3 (ВЭЖХ-масс-спектрометрия) Колонка: ВНК LabODS-O/B, 4,650 мм, 5 мкМ Подвижная фаза: А = вода с 0,05% трифторуксусной кислоты; В = ацетонитрил с 0,05% трифторуксусной кислоты Градиент: 95% А/5% В в течение 0,5 минуты, затем до 90% А/10% В в течение 0,05 минуты, от 90% А/10% В до 5% А/95% В в течение 18,94 минуты, затем до 1% А/99% В в течение 0,05 мин и выдерживание при 1% А/99% В в течение 2,16 мин, затем возврат до 95% А/5% В в течение 0,50 мин Скорость потока: 2,5 мл/мин. УФ длина волны: 220 и 254 нм Способ 4 (ВЭЖХ-масс-спектрометрия) Колонка: Waters ODS-AQ, 2,050 мм Подвижная фаза: А = вода с 0,05% трифторуксусной кислоты; В = ацетонитрил с 0,05% трифторуксусной кислоты Градиент: от 95% А/5% В до 10% А/90% В в течение 2,25 мин и выдерживание при 10% А/90% В в течение 1,0 мин, затем возврат до 95% А/5% В в течение 0,1 мин- 11010370 Скорость потока: 1 мл/мин УФ длина волны: 220 и 254 нм Способ 5 (ВЭЖХ) Колонка: Agilent, ZorbaxSB-C18, 5 мкм, 4,6250 мм Подвижная фаза: А = вода с 0,05% трифторуксусной кислоты; В = ацетонитрил с 0,05% трифторуксусной кислоты Градиент: от 95% А/5% В до 5% А/95% В в течение 50 мин, затем от 5% А/95% В до 1% А/99% В в течение 0,1 мин, затем выдерживание при 1% А/99% в течение 0,8 мин и обратно до 95% А/5% В в течение 0,2 мин, выдерживание такого градиента в течение 4 мин Скорость потока 2,0 мл/мин УФ длина волны: 220 и 254 нм Способ 6 (ВЭЖХ-масс-спектрометрия) Колонка: Phenomenex Synergi 4 мкм Max-RP 80 А, 50,02,0 мм Подвижная фаза: А = вода с 0,025% трифторуксусной кислоты; В = ацетонитрил с 0,025% трифторуксусной кислоты Градиент: 95% А/5% В 0,25 мин, затем от 95% А/5% В до 95% В/5% А в течение 13 мин, поддержание от 95% А/5% В до 95% В/5% А в течение 2 мин, затем обратно до 95% А/5% В в 0,25 мин Скорость потока: 1 мл/мин УФ длина волны: 220 нм и 254 нм Пример 1. 3-[2(R)-Гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион Стадия 1 А. Получение 2-фтор-6-(трифторметил)бензиламина 1 а К 2-фтор-6-(трифторметил)бензонитрилу (45 г, 0,238 ммоль) в 60 мл ТГФ медленно добавляли при 60 С 1 М ВН 3:ТГФ и полученный раствор нагревали при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали до температуры окружающей среды. Медленно добавляли метанол(420 мл) и хорошо перемешивали. Растворители затем упаривали и остаток распределяли между EtOAc и водой. Органический слой сушили над Na2SO4. Упаривание давало 1 а в виде желтого масла (46 г, 0,238 ммоль). Масс-спектр (CI) m/z 194,0 (МН+). Стадия 1 В. Получение [2-фтор-6-(трифторметил)бензил]мочевины 1b К 2-фтор-6-(трифторметил)бензиламину 1 а (51,5 г, 0,267 ммоль) в колбе добавляли мочевину (64 г,1,07 ммоль), HCl (конц., 30,9 ммоль, 0,374 ммоль) и воду (111 мл). Смесь нагревали при кипении с обратным холодильником в течение 6 ч. Смесь охлаждали до температуры окружающей среды, дополнительно охлаждали льдом и фильтровали, получая желтое твердое вещество. Перекристаллизацией из 400 мл EtOAc получали 1b в виде белого твердого вещества (46,2 г, 0,196 ммоль). Масс-спектр (CI) m/z 237,0NaI (43,9 г, 293 ммоль) добавляли к N-[2-фтор-6-(трифторметил)бензил]мочевине 1b (46,2 г, 19,6 ммоль) в 365 мл ацетонитрила. Полученную смесь охлаждали на ледяной бане. Медленно через капельную воронку добавляли дикетен (22,5 мл, 293 ммоль) с последующим добавлением TMSCI (37,2 мл, 293 ммоль) таким же образом. Полученную желтую суспензию оставляли медленно нагреваться до комнатной температуры и перемешивали в течение 20 ч. ЖХ-МС показал исчезновение исходного вещества. К желтой смеси добавляли 525 мл воды и перемешивали в течение ночи. После перемешивания в течение дополнительных 20 ч осадок отфильтровывали на воронке Бюхнера и желтое твердое вещество промывали водой и EtOAc, получая 1 с в виде белого твердого вещества (48,5 г, 16 ммоль). 1 Н ЯМР (CDCl3)2,15 (с, 3H), 5,37 (с, 2 Н), 5,60 (с, 1 Н), 7,23-7,56 (м, 3H), 9,02 (с, 1 Н); Масс-спектр (CI) m/z 303,0(MH+). Стадия 1D. Получение 5-бром-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин 2,4(1 Н,3H)диона 1d Бром (16,5 мл, 0,32 ммоль) добавляли к 1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин 2,4(1 Н,3H)диону 1 с(48,5 г, 0,16 моль) в 145 мл уксусной кислоты. Полученная смесь стала прозрачной,затем в течение часа образовывался осадок. После перемешивания в течение 2 ч, желтое твердое вещество отфильтровывали и промывали холодным EtOAc до тех пор, пока получали почти белое твердое вещество. Фильтрат промывали насыщенным раствором NaHCO3 и сушили над Na2SO4. После упаривания получали желтое твердое вещество, которое промывали EtOAC, получая светло-желтое твердое вещест- 12010370 во. Два твердых вещества объединяли, получая всего 59,4 г 1d (0,156 моль). 1 Н ЯМР (CDCl3)2,4 (с, 3H),5,48 (с, 2 Н), 7,25-7, 58 (м, 3H), 8,61 (с, 1 Н); Масс-спектр (CI) m/z 380,9 (МН+). 5-Бром-1-[2,6-дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)-дион 1d.1 получали с использованием того же способа. Стадия 1E. Получение 5-бром-1-[2-фтор-6-(трифторметил)бензил]-6-метил-3-[2(R)-третбутоксикарбониламино-2-фенилэтил]пиримидин-2,4(1 Н,3H)диона 1 е К 5-бром-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)диону 1d (15 г, 39,4 ммоль) в 225 мл ТГФ добавляли N-t-Boc-D-фенилглицинол (11,7 г, 49,2 ммоль) и трифенилфосфин (15,5 г, 59,1 ммоль), с последующим добавлением ди-трет-бутилазодикарбоксилата (13,6 г, 59,1 ммоль). Полученный желтый раствор перемешивали в течение ночи. Летучие компоненты упаривали и остаток очищали на силикагеле с использованием смеси EtOAc/гексан в соотношении 3:7, получая 1 е в виде белого твердого вещества (23,6 г, 39,4 ммоль). Масс-спектр (CI) m/z 500,0 (МН+-Вос). Стадия 1F. Получение 3-[2(R)-амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)диона 1f К 5-бром-1-[2-фтор-6-(трифторметил)бензил]-6-метил-3-[2(R)-трет-бутоксикарбониламино-2 фенилэтил]пиримидин-2,4(1 Н,3H)диону 1 е (15 г, 25 ммоль) в смеси H2O/диоксан в соотношении 30 мл/90 мл в ампуле для проведения экспериментов под давлением добавляли 2-фтор-3-метоксифенилбороновую кислоту (4,25 г, 25 ммоль) и карбонат натрия (15,75 г, 150 ммоль). Через смесь барботировали газообразный N2 в течение 10 мин. Добавляли к смеси тетракис(трифенилфосфин)палладий (2,9 г, 2,5 ммоль), ампулу герметично закупоривали и полученную смесь нагревали при перемешивании при 90 С в течение ночи. После охлаждения до температуры окружающей среды осадок удаляли фильтрованием. Летучие компоненты удаляли упариванием и остаток распределяли между EtOAc/насыщенным раствором NaHCO3. Органический растворитель упаривали и остаток хроматографировали с использованием смесиEtOAc/гексан в соотношении 2:3, получая 13,4 г (20,8 ммоль, 83%) желтого твердого вещества. Данное желтое твердое вещество (6,9 г, 10,7 ммоль) растворяли в смеси CH2Cl2/ТФУК в соотношении 20 мл/20 мл. Полученный желтый раствор перемешивали при комнаткой температуре в течение 2 ч. Летучие компоненты упаривали, и остаток распределяли между EtOAc/насыщенным раствором NaHCO3. Органическую фазу сушили над Na2SO4. Упаривание давало 1f в виде желтого масла (4,3 г, 7,9 ммоль,74%). 1 Н ЯМР (CDCl3)2,03 (о, 3H), 3,72-4,59 (м, 6 Н), 5,32-5,61 (м, 2 Н), 6,74-7,56 (м, 11 Н); Масс-спектр(CI) m/z 546,0 (МН+). 3-[2(R)-амико-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2,6-дифторбензил]-6-метилпиримидин 2,4(1 Н,3H)дион 1f.1 получали с использованием того же способа, который описан в данном примере. Стадия 1G. Получение 3-[2(R)-этоксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н, 3H)диона 1g К соединению 3-[2(R)-амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4 (1 Н,3H)диону 1f (5 г, 9,4 ммоль) в 100 мл ацетонитрила добавляли этил-4-бромбутират (4 мл, 28,2 ммоль) и основание Хунига (1,6 мл, 9,4 ммоль). После нагревания при кипении с обратным холодильником при 95 С в течение ночи, реакционную смесь охлаждали до температуры окружающей среды и летучие компоненты удаляли. Остаток хроматографировали с использованием смеси EtOAc/гексан/Et3N в соотношении 10:10:1, получая 1g в виде желтого масла (3,0 г, 4,65 ммоль). Масс-спектр (CI) m/z 646,2 (МН+). Стадия 1 Н. Получение 3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)диона 1-1 Соединение 3-[2(R)-этоксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2 фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)-дион 1g (2,6 г, 4,0 ммоль) растворяли в смеси ТГФ/вода в соотношении 30 мл/30 мл. Добавляли твердый NaOH (1/6 г, 40 ммоль) и полученную смесь нагревали при 50 С в течение ночи. Смесь охлаждали до температуры окружающей среды и летучие компоненты упаривали. К водному раствору добавляли лимонную кислоту до рН=3. ЭкстракцияEtOAc с последующим упариванием растворителя давала 1,96 г белого геля. Гель пропускали через колонку с макропористой сильной катионообменной смолой Dowex MSC-1 для преобразования в натриевую соль. Лиофилизация давала белое твердое вещество 1-1 в виде натриевой соли (1,58 г, 2,47 ммоль). 1 Н ЯМР (CD3OD)1,69-1,77 (м, 2 Н), 2,09 (с, 3H), 2,09-2,19 (т, J = 7,35 Гц, 2 Н), 2,49-2,53 (т, J = 7,35 Гц,2 Н), 3,88 (с, 3H), 4,15-4,32 (м, 3H), 5,36-5,52 (м, 2 Н), 6,60-7,63 (м, 11 Н). ВЭЖХ-масс-спектр (CI) m/z 632,2(МН+), tR = 26,45 (способ 5). Следующие соединения синтезировали в соответствии с вышеуказанным способом,К соединению 1-1 (0,045 ммоль) в 1 мл МеОН, добавляли формальдегид (0,0475 ммоль) с последующим добавлением 8 М BH3:пиридина (0,0475 ммоль). После встряхивания в течение ночи соединение 1-4 очищали препаративной ЖХ-МС. ВЭЖХ-Масс-спектр (CI) m/z 646,5 (МН+), tR = 2,231, (способ 4) Следующие соединения синтезировали в соответствии с вышеуказанным способом. Стадия 2 А. Получение 5-бром-1-[2-фтор-6-(трифторметил)бензил]-6-метил-3-[2(R)-амино-2 фенилэтил]пиримидин-2,4(1 Н,3H)дион 2 а 5-Бром-1-[2-фтор-6-(трифторметил)бензил]-6-метил-3-[2(R)-трет-бутоксикарбониламино-2-фенилэтил]пиримидин-2,4(1 Н,3H)дион 1 е растворяли в смеси 20 мл/20 мл CH2Cl2/ТФУК. Полученный желтый раствор перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты упаривали и остаток распределяли между EtOAc/нас.NaHCO3. Органическую фазу сушили над Na2SO4. Упариванием получали 2 а в виде желтого масла. Стадия 2 В. Получение 5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]-6-метил-3-[2(R)-амино 2-фенилэтил]пиримидин-2,4(1 Н,3H)диона 2b. К соединению 2 а (40 мг, 0,08 ммоль) в смеси H2O/диоксан в соотношении 0,25 мл/0,75 мл в ампуле объемом 4 мл добавляли 2-хлорфенилбороновую кислоту (0,12 ммоль) и карбонат натрия (51 мг, 48 ммоль, 6 эквив.). Через раствор в течение 1 мин пропускали газообразный азот и добавляли тетракис(трифенилфосфин) палладий (9,24 мг, 0,008 ммоль). Полученную смесь герметично закрывали и нагревали при 90 С в течение ночи. После охлаждения до температуры окружающей среды осадок удаляли фильтрованием и очищали препаративной ЖХ-МС, получая 2b. Стадия 2 С. 3-(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион 2-1 К соединению 2b (0,03 ммоль) в 1 мл МеОН добавляли янтарный полуальдегид (0,03 ммоль) с последующим добавлением 8 М BH3:пиридина (0,03 ммоль). После встряхивания в течение ночи, соединение 2-1 очищали препаративной ЖХ-МС. Масс-спектр (CI) m/z 618,2 (МН+); tR = 1,005 (способ 1) Следующие соединения синтезировали в соответствии с вышеуказанным способом. Стадия 3 А. 3-[2(R)-3-цианопропиламино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион 3 а Соединение 1f (110 мг, 0,2 ммоль) растворяли в ацетонитриле (5 мл) и добавляли диизопропилэтиламин (52 мг, 0,4 ммоль) с последующим добавлением 4-бромбутиронитрила (90 мг, 0,6 ммоль). Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 ч. Летучие компоненты упаривали и остаток очищали флэш-хроматографией (диоксид кремния, 5% МеОН/CH2Cl2), получая соединение 3 а (115 мг, 94%). Масс-спектр (CI) m/z 613,3 (МН+). Стадия 3 В. 3-[2(R)-2-[5-тетразоилпропил]амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2 фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н, 3H)дион 3-1 К раствору 3 а (38 г, 0,06 ммоль) в толуоле (5 мл) добавляли азид трибутилолова (42 мг, 0,12 ммоль) и реакционную смесь нагревали при 100 С в течение 14 ч. Смесь охлаждали, распределяли между EtOAc и 1 н NaOH и органический слой промывали 1 н HCl и насыщенным раствором соли. Органический слой сушили (сульфат натрия), упаривали и остаток очищали флэш-хроматографией (диоксид кремния, 7% МеОН/CH2Cl2), получая соединение 3-1 (10 мг, 25%). ВЭЖХ-Масс-спектр (CI) m/z 656,2 (MH+), tR = 2,128 мин (способ 4) Пример 4. 3-[2(R)-гидроксикарбонилпропиламино-2-циклогексилэтил]-5-(2-фтор-3-метоксифенил-1-[2,6 дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)дион Стадия 4 А. Получение трет-бутил-1-циклогексил-2-гидроксиэтилкарбамата 4 а Раствор N-(трет-бутилоксикарбонил)циклогексилглицина (2,0 г, 7,77 ммоль) в безводном ТГФ (10 мл) охлаждали до 0 С. Медленно добавляли раствор борана (1 М в ТГФ, 15,5 мл, 15,5 ммоль) и реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Реакцию гасили МеОН(5 мл), летучие компоненты упаривали и остаток распределяли между водой и EtOAc. Органический слой промывали насыщенным раствором NaHCO3 в воде, насыщенным раствором соли, сушили (сульфат натрия) и упаривали, получая трет-бутил-1-циклогексил-2-гидроксиэтилкарбамат 4 а (1,26 г, 66,7%). Масс-спектр (CI) m/z 144,2 (МН+-Вос). Стадия 4 В. Получение 5-бром-3-[2(R)-трет-бутоксикарбониламино-2-циклогексилэтил]-1-[2,6 дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 4b Раствор трет-бутил 1-циклогексил-2-гидроксиэтилкарбамата 4 а (638 мг, 2,62 ммоль) в ТГФ (10 мл) обрабатывали 5-бром-1-(2,6-дифторбензил)-6-метилпиримидин-2,4(1 Н,3H)-дионом 1d.1 (869 мг, 2,62 ммоль) и трифенилфосфином (1,03 г, 3,93 ммоль) при температуре окружающей среды, затем вводили ди-трет-бутилазодикарбоксилат (906 мг, 3,93 ммоль). Реакционную смесь перемешивали при температуре окружающей среды в течение 16 ч и летучие компоненты упаривали. Остаток распределяли между насыщенным раствором NaHCO3 в H2O и EtOAc. Органический слой сушили (сульфат натрия), упаривали и очищали флэш-хроматографией (диоксид кремния, 25% EtOAc/гексаны), получая соединение 4b(1,39 г, 95,4%). Масс-спектр (CI) m/z 456,1, 458,1 (МН+-Вос). Стадия 4 С. Получение 3-[2(R)-трет-бутоксикарбониламино-2-циклогексилэтил]-5-(2-фтор-3 метоксифенил)-1-[2,6-дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 4c К 5-бром-3-[2(R)-трет-бутоксикарбониламино-2-циклогексилэтил]-1-[2,6-дифторбензил]-6-метилпиримидин-2,4 (1 Н, 3H)-диону 4b (1,0 г, 1,79 ммоль) в смеси бензол/EtOH/диметиловый эфир этиленгликоля (20/2/22 мл) добавляли 2-фтор-3-метоксифенилбороновую кислоту (382 мг, 2,24 ммоль) и насыщенный раствор Ва(ОН)2 в воде (0,5 М, 15 мл). Реакционную смесь деоксигенировали с использованием N2 в течение 10 мин, добавляли тетракис(трифенилфосфин)палладий(0) (208 мг, 0,18 ммоль) и реакционную- 16010370 смесь нагревали при 80 С в течение ночи в атмосфере N2. Реакционную смесь распределяли между насыщенным раствором соли и EtOAc. Органический слой сушили (сульфат натрия), упаривали и очищали флэш-хроматографией (диоксид кремния, 30% EtOAc/гексанах), получая соединение 4 с (348 мг, 32,3%). Масс-спектр (CI) m/z 502,2 (МН+-Вос). Стадия 4D: получение 3-[2(R)-амино-2-циклогексилэтил]-5-(2-фтор-3-метоксифенил)-1-[2,6 дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 4d К соединению 4 с (300 мг, 0,5 ммоль) в дихлорметане (2 мл) добавляли ТФУК (2 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Летучие компоненты упаривали и остаток распределяли между насыщенным раствором NaHCO3 в воде и EtOAc. Органический слой сушили (сульфат натрия), упаривали и очищали ВЭЖХ с обращенной фазой (колонка С-18, 15-75%ACN/вода), получая соединение 4d. Масс-спектр (CI) m/z 502,2 (МН+). Стадия 4 Е: получение 3-[2(R)-гидроксикарбонилпропиламино-2-циклогексилэтил]-5-(2-фтор-3 метоксифенил)-1-[2,6-дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 4-1 Раствор соединения 4d (10 мг, 0,02 ммоль) в метаноле (2 мл) добавляли янтарный полуальдегид (15 мг, 15% водный раствор), с последующим добавлением борана/пиридина (8 М, 3 мкл). Реакционную смесь перемешивали при температуре окружающей среды в течение 1 ч. Летучие компоненты упаривали и остаток очищали непосредственно на препаративной пластине ТСХ, элюируя смесью 7% МсОН/CH2Cl2, получая соединение 4-1 (5 мг). Масс-спектр (CI) m/z 588,3 (МН+). 3-[2(R)-гидроксикарбонилпропиламино-2-циклогексилэтил]-5-(2-фтор-3-метоксифенил)-1-[2 фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион 4-2 синтезировали с использованием того же способа и промежуточного соединения 1d. Следующие соединения синтезировали в соответствии с вышеуказанным способом. Стадия 5 А. Получение 5-бром-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 5 а Суспензию 5-бромурацила (31,0 г) в 300 мл дихлорэтана обрабатывают N,Oбис(триметилсилил)ацетамидом (80 мл). Реакционную смесь нагревали в атмосфере азота. Раствор охлаждали до температуры окружающей среды, добавляли 2-фтор-6-(трифторметил)бензилбромид (50 г) и реакционную смесь нагревали в течение ночи в атмосфере азота. Реакционную смесь охлаждали, гасили МеОН и распределяли между дихлорметаном и водой. Органический слой промывали насыщенным раствором соли, сушили (сульфат натрия) и упаривали, получая твердое вещество. Неочищенный продукт растирали с простым эфиром,фильтровали и промывали три раза простым эфиром, получая 40,7 г 5-бром-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион 5 а. Масс-спектр (CI) m/z 366,0, 368,0 (МН+). Стадия 5 В. Получение 3-[2(R)-амино-2-фенилэтил]-5-бром-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н, 3H)диона 5b Раствор 5-бром-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)-диона 5 а (19,2 г, 52,3 ммоль) в ТГФ (180 мл) обрабатывали N-(трет-бутилоксикарбонил)-Dфенилглицинолом (13,6 г, 57,5 ммоль) и трифенилфосфином (20,6 г, 78,5 ммоль) при комнатной температуре, затем вводили несколькими порциями ди-трет-бутилазодикарбоксилат (18,0 г, 78,5 ммоль) в течение 5 мин. Смесь перемешивали при комнатной температуре в течение 16 ч, дополнительно добавляли ТГФ (90 мл) и смесь нагревали до 50 С. Добавляли концентрированную HCl (34,6 мл, 418 ммоль) и реакционную смесь перемешивали при 50 С в течение 40 часов. После разбавления этилацетатом (100 мл) твердое вещество отфильтровывали,промывали дополнительно этилацетатом (100 мл) и сушили, получая соединение 5b (26,9 г, 98%) в виде белого порошка. Масс-спектр (CI) m/z 485,0, 487,0 (МН+). Стадия 5 С. Получение 3-[2(R)-амино-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 5 с К соединению 5b (10,45 г, 20 ммоль) в смеси диоксан/вода (180/20 мл) добавляли 2 хлорфенилбороновую кислоту (6,26 г, 40 ммоль) и Na2CO3 (12,72 г, 120 ммоль). Из смеси удаляли кислород током N2 в течение 15 мин, добавляли тетракис(трифенилфосфин)палладий(0) (2,31 г, 2 ммоль) и реакционную смесь нагревали при 90 С в течение 16 ч. Реакционную смесь распределяли между EtOAc иH2O. Органический слой промывали насыщенным раствором соли, сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны/триэтиламин, начиная с соотношения 500/500/6 и до соотношения 800/200/7, получая соединение 5 с (7,26 г, 70%) в виде белого вспененного вещества. Масс-спектр (CI) m/z 518,0, 520,1 (МН+). Стадия 5D. Получение 3-[2(R)-этоксикарбонилпропиламино-2-фенилэтил]-5-(2-хлорфенил)-1-[2 фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 5d Смесь соединения 5 с (4,1 г, 7,93 ммоль), этил-4-бромбутирата (3,6 мл, 23,79 ммоль) и K2CO3 (2,2 г,15,86 ммоль) в MeCN (80 мл) нагревали при кипении с обратным холодильником в течение 16 ч. MeCN удаляли и остаток распределяли между EtOAc и H2O. Органический слой промывали насыщенным раствором соли, сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны/триэтиламин в соотношении 400/600/7, получая соединение 5dNaOH (1,588 г, 39,7 ммоль). Смесь перемешивали при 50 С в течение 16 ч. ТГФ удаляли в вакууме, водный раствор промывали простым эфиром и охлаждали до 0 С. Нейтрализация 10%-ным водным раствором лимонной кислоты (26,0 мл, 40,6 ммоль) приводила к образованию осадка, который промывали H2O и сушили, получая соединение 5-1 (1,88 г, 82%). ВЭЖХ-масс-спектр (CI) m/z 604,1, 606,1 (МН+), tR = 2,511 (способ 4), tR = 26,98(способ 5). Следующие соединения синтезировали в соответствии с вышеуказанным способом. Стадия 6 А. Получение соединения метил-(2-хлорфенил)ацетата 6 а К 2-хлорфенилуксусной кислоте (1,04 г, 6 ммоль) в МеОН (25 мл) добавляли серную кислоту (6 капель) и раствор нагревали при кипении с обратным холодильником в течение 16 ч. После концентрирования остаток помещали в этилацетат и промывали насыщенным водным раствором NaHCO3, H2O и насыщенным раствором соли. Органический слой сушили над Na2SO4 и концентрировали, получая метил(2-хлорфенил)ацетат 6 а (1,08 г, 97,5%) в виде желтоватого масла. ГХ-Масс-спектр (EI) m/z 184,186 (М+). Стадия 6 В. Получение метил-2-(2-хлорфенил)-3-(диметиламино)акрилата 6b Раствор метил-(2-хлорфенил)ацетата 6 а (1,08 г, 5,8 5 ммоль) в ДМФ-ДМА (10 мл, 70,8 ммоль) нагревали при кипении с обратным холодильником в течение 16 ч. После упаривания остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны в соотношении от 1/3 до 1/2, получая первоначально не вступивший в реакцию метил-(2-хлорфенил)ацетат 6 а (0,67 г, 62%), а затем метил-2-(2-хлорфенил)-3-(диметиламино)акрилат 6b (0,38 г, 27%; 71% из расчета на выделенное исходное вещество) в виде бесцветного сиропа. Масс-спектр (CI) m/z 240,2, 242,2 (МН+). Стадия 6 С. Получение 5-(2-хлорфенил)пиримидин-2,4(1 Н,3H)диона 6 с К смеси метил-2-(2-хлорфенил)-3-(диметиламино)акрилата 6b (0,26 г, 1,08 ммоль), мочевины (0,2 г,3,26 ммоль) и Nal (0,49 г, 3,26 ммоль) в ацетонитриле (5 мл) добавляли TMSCI (0,41 мл, 3,26 ммоль). Полученную смесь нагревали при кипении с обратным холодильником в течение 16 ч, охлаждали до ком- 19010370 натной температуры и добавляли 1,0 М NaOH (8 мл). Полученный раствор перемешивали в течение 20 ч и ацетонитрил удаляли в вакууме. Водный раствор промывали простым эфиром, охлаждали на ледяной бане и нейтрализовали 1 н HCl (8 мл). осадок отфильтровывали, промывали дополнительно H2O и сушили, получая 5-(2-хлорфенил)пиримидин-2,4(1 Н,3H)дион 6 с (0,16 г, 66%) в виде белого твердого вещества. Масс-спектр (CI) m/z 222,9, 224,9 (МН+). Стадия 6D. Получение 5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 6d К суспензии 5-(2-хлорфенил)пиримидин-2,4(1 Н,3H)-диона 6 с (0,16 г, 0,72 ммоль) в ацетонитриле (5 мл) добавляли бис(триметилсилил)ацетамид (0,36 мл, 1,44 ммоль), и полученный раствор нагревали при кипении с обратным холодильником в течение 1,5 ч. После охлаждения до комнатной температуры добавляли 2-фтор-3-трифторметилбензилбромид (0,22 г, 0,86 ммоль) и нагревание продолжали в течение 16 ч. Реакцию гасили добавлением МеОН (5 мл) и перемешивали в течение 2 ч. После концентрирования остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны в соотношении 1/1, получая 5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3 Н)-дион 6d (0,25 г, 87%) в виде белого твердого вещества. Масс-спектр (CI) m/z 398,9, 400,9 (МН+). Стадия 6 Е. Получение 3-[2(R)-трет-бутоксикарбониламино-2-фенилэтил]-5-(2-хлорфенил)-1-[2 фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 6e Смесь 5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)-диона 6d (125 мг,0,32 ммоль), K2CO3 (130 мг, 0,96 ммоль) и N-(трет-бутилоксикарбонил)-Dфенилглицинолмезилата (0,2 г, 0,64 ммоль) в ДМФ (3 мл) нагревали при 75 С в течение 16 ч. Реакционную смесь разбавляли этилацетатом, промывали H2O и насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны в соотношении 2/3, получая соединение 6 е (144 мг, 74%). Масс-спектр (CI) m/z 518,0, 520,0 (МН+-Boc). Стадия 6F. Получение 3-[2(R)-амино-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4 (1 Н,3H)диона 6f К раствору соединения 6e (0,144 г, 0,23 ммоль) в ДХМ (1 мл) добавляли ТФУК (0,5 мл, 6,5 ммоль) и смесь перемешивали при комнатной температуре в течение 1,5 ч. После концентрирования остаток помещали в ДХМ и добавляли насыщенный водный раствор NaHCO3. Водный слой экстрагировали ДХМ. Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение 6f(0,12 г). Масс-спектр (CI) m/z 518,0, 520,1 (МН+). Стадия 6G. Получение 3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлорфенил)-1[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 6-1 Раствор соединения 6f (0,1 г, 0,19 ммоль) и янтарного полуальдегида (15 вес.% раствор в воде; 0,13 мл, 0,21 ммоль) в MeCN перемешивали при комнатной температуре в течение 5 мин. Добавляли комплекс боран-пиридин (8 М; 72 мкл) и перемешивали в течение 16 ч. После концентрирования остаток очищали первоначально на препаративной пластине ТСХ, а затем препаративной ВЭЖХ, получая соединение 6-1. ВЭЖХ-Масс-спектр (CI) m/z 604,1, 606,1 (МН+), tR = 26,98 (способ 5), tR = 2,511 (способ 4). Пример 7. 3-[2(R)-Гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион Стадия 7 А. Получение 2-хлор-3-метоксибензальдегида 7 а К суспензии 3-гидроксибензальдегида (20,12 г, 160 ммоль) в НОАс (40 мл) осторожно добавляли при перемешивании tBuOCl (20 мл, 176 ммоль). Реакционная смесь превращается в прозрачный раствор,и реакция является сильно экзотермичной. Реакционную смесь оставляли охлаждаться и перемешивали в течение 16 ч, что приводило к образованию белого осадка. Твердое вещество отфильтровывали, промывали H2O и сушили, получая 2-хлор-3-гидроксибензальдегид (13,77 г, 55%), ЖХ-масс-спектр (EI) m/z 156, 158 (М+). К раствору 2-хлор-3-гидроксибензальдегида (4,55 г, 29 ммоль) в ДМФ (30 мл) добавляли K2CO3 (4,8 г, 34,9 ммоль) с последующим добавлением MeI (2,7 мл, 43,6 ммоль) и смесь перемешивали при комнатной температуре в течение 16 ч. После концентрирования в вакууме остаток помещали в этилацетат,промывали H2O, насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Очистка колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны в соотношении 1/5,получая 2-хлор-3-метоксибензальдегид 7 а (4,68 г, 94%) в виде бесцветного масла, которое затвердевало при хранении. ГХ-масс-спектр (EI) m/z 17 0, 172 (М+).(4,65 г,27,3 ммоль) и метил(метилтио)метилсульфоксида (4,3 мл, 43,9 ммоль) в ТГФ (25 мл) добавляли 40%-ный метанольный раствор Тритона В (6,2 мл, 13,6 ммоль) и полученный раствор нагревали при кипении с обратным холодильником в течение 16 ч. Затем ТГФ удаляли, остаток помещали в этилацетат, промывали 1 н HCl, H2O и насыщенным раствором соли, затем сушили над Na2SO4 и концентрировали. После очистки колоночной хроматографией на силикагеле с использованием дихлорметана получали 2-хлор-1-метокси-3-[2(метилсульфанил)-2-(метилсульфинил)винил]бензол 7b (3,61 г, 48%) в виде желтого масла. ГХ-массспектр (EI) m/z 225 (М+-Cl-16), 210 (М+-Cl-OMe). Стадия 7 С. Получение этил(2-хлор-3-метоксифенил)ацетата 7 с К раствору 2-хлор-1-метокси-3-[2-(метилсульфанил)-2-(метилсульфинил)винил]бензола 7b (3,58 г,12,9 ммоль) в этаноле (20 мл) добавляли 5 М раствора HCl в этаноле (5,2 мл) и полученный раствор нагревали при кипении с обратным холодильником в течение 3 ч. После упаривания остаток очищали колоночной хроматографией на силикагеле, элюируя дихлорметаном, получая этил (2-хлор-3 метоксифенил)ацетат 7 с (2,78 г, 94%) в виде желтого масла. ГХ-масс-спектр (EI) m/z 228, 230 (М+). Стадия 7D. Получение этил-2-(2-хлор-3-метоксифенил)-3-(диметиламино)акрилата 7d Раствор этил(2-хлор-3-метоксифенил)ацетата 7 с (2,78 г, 12 ммоль) в ДМФ-ДМА (16 мл, 120 ммоль) нагревали при кипении с обратным холодильником в течение 16 ч. После упаривания остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны в соотношении от 1/2 до 1/1, получая первоначально не вступивший в реакцию этил(2-хлор-3-метоксифенил) ацетат 7 с (1,8 г,65%), а затем этил-2-(2-хлор-3-метоксифенил)-3-(диметиламино)акрилат 7d (1,1 г, 32%; 90% из расчета на выделенное исходное вещество) в виде желтого сиропа. Масс-спектр (CI) m/z 284,0, 286,0 (МН+). Стадия 7 Е. Получение 5-(2-хлор-3-метоксифенил)пиримидин-2,4(1 Н,3H)диона 7 е К смеси этил-2-(2-хлор-3-метоксифенил)-3-(диметиламино)акрилата 7d (1,7 г, 6 ммоль), мочевины(1,08 г, 18 ммоль) и NaI (2,7 г, 18 ммоль) в ацетонитриле (20 мл) добавляли TMSCI (2,3 мл, 18 ммоль). Полученную смесь нагревали при кипении с обратным холодильником в течение 16 ч, охлаждали до комнатной температуры и добавляли 1,0 М NaOH (30 мл). Полученный раствор перемешивали в течение 20 ч и ацетонитрил удаляли в вакууме. Водный раствор промывали простым эфиром, охлаждали на ледяной бане и нейтрализовали 1 н HCl (30 мл). Осадок отфильтровывали, промывали дополнительным количество H2O и сушили, получая 5-(2-хлор-3-метоксифенил)пиримидин-2,4(1 Н,3H)-дион 7 е (1,24 г,82%) в виде бледно-желтого твердого вещества. Масс-спектр (CI) m/z 253,1, 255,1 (МН+). Стадия 7F. Получение 5-(2-хлор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин 2,4(1 Н,3H)-диона 7f К суспензии 5-(2-хлор-3-метоксифенил)пиримидин-2,4(1 Н,3H)-диона 7 е (2,2 г, 8,7 ммоль) в ацетонитриле (25 мл) добавляли бис(триметилсилил)ацетамид (4,3 мл, 17,4 ммоль), и полученный раствор нагревали при кипении с обратным холодильником в течение 1,5 ч. Смесь охлаждали до комнатной температуры, добавляли 2-фтор-3-трифторметилбензилбромид (2,7 г, 10,5 ммоль) и нагревание при кипении с обратным холодильником продолжали в течение 16 ч. Реакцию гасили добавлением МеОН (25 мл) и перемешивали в течение 2 ч. После концентрирования остаток очищали колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны в соотношении 1/1, получая 5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)-дион 7f (3,3 г, 88%) в виде белого твердого вещества. Масс-спектр (CI) m/z 429,0, 431,0 (МН+). Стадия 7G. Получение 3-[2(R)-(трет-бутоксикарбониламино)-2-фенилэтил]-5-(2-хлор-3 метоксифенил-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 7g Смесь 5-(2-хлор-3-метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)-диона 7f (75 мг, 0,175 ммоль), K2CO3 (72 мг, 0,525 ммоль) и N-(трет-бутилоксикарбонил)-D-фенилглицинолмезилата (0,11 г, 0,35 ммоль) в ДМФ (2 мл) нагревали при 75 С в течение 16 ч. Реакционную смесь разбавляли этилацетатом, промывали H2O и насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны в соотношении 2/3, получая соединение 7 д (82 мг, 72%) в виде белого твердого вещества. Масс-спектр (CI) m/z 548,0, 550,0 (МН+-Вос). Стадия 7 Н. Получение 3-[2(R)-амино-2-фенилэтил]-(2-хлор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 7h Соединение 7g (2,7 г, 4,2 ммоль) растворяли в дихлорметане (10 мл), добавляли ТФУК (14 мл, 175 ммоль), и смесь перемешивали при комнатной температуре в течение 4,5 ч. После концентрирования остаток помещали в ДХМ и добавляли насыщенный водный раствор NaHCO3. Водный слой экстрагировали ДХМ. Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение 7h (2,2 г, 96%). Масс-спектр (CI) m/z 548,0, 550,0 (МН+). 3-[2(R)-амино-2-фенилэтил]-5-(2-хлор-3-метоксифенил)-1-[2,6-дифторбензил]пиримидин 2,4(1 Н,3H)-дион 7h.1 получали заменой на подходящее исходное вещество и с использованием описанного выше способа.- 21010370 Стадия 7I. Получение 3-[2(R)-этоксикарбонилпропиламино-2-фенилэтил]-5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 7i К раствору соединения 7h (2,0 г, 3,65 ммоль) в ДМФ (8 мл) добавляли Na2CO3 (0,47 г, 4,38 ммоль) с последующим добавлением зтил-4-бромбутирата (0,83 мл, 5,48 ммоль). Смесь нагревали при 95 С в течение 1,5 ч, охлаждали до комнатной температуры и распределяли между этилацетатом и H2O. Органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат/гексаны/триэтиламин в соотношении 500/500/5, получая соединение 7i (1,29 г) в виде белого твердого вещества. Масс-спектр(CI) m/z 662,2, 664,2 (МН+) Стадия 7J. Получение 3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)-диона 7-1 К соединению 7i (0,7 г, 1,06 ммоль) добавляли ТГФ (6 мл) и H2O (6 мл) с последующим добавлением NaOH (0,17 г, 4,24 ммоль). Смесь перемешивали при 50 С в течение 16 ч. ТГФ удаляли в вакууме. Водный раствор промывали простым эфиром и охлаждали до 0 С. Нейтрализация 5%-ной водной лимонной кислотой (6,0 мл, 4,7 ммоль) приводила к образованию осадка, который собирали и дополнительно очищали колоночной хроматографией на силикагеле с использованием смеси МеОН/ДХМ/триэтиламин в соотношении 8/100/2, получая соединение 7-1 (0,56 г, 84%) в виде белого твердого вещества. ВЭЖХ-масс-спектр (CI) m/z 634,2, 636,2 (МН+), tR = 24,925, (способ 5) Пример 8. 3-[2(R)-Гидроксикарбонилпропиламино-2-(изобутил)этил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор 6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион Стадия 8 А. Получение 3-[2(R)-трет-бутоксикарбониламино-2-(изобутил)этил]-5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4 (1 Н,3H)диона 8 а К раствору N-(трет-бутилоксикарбонил)-Dлейцинола (1,21 г, 5,57 ммоль) в пиридине (6 мл) добавляли тозилхлорид (1,6 г, 8,35 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч, разбавляли EtOAc, и промывали последовательно 1 н HCl, H2O, насыщенным водным раствором NaHCO3 и насыщенным раствором соли. Органический слой сушили над Na2SO4, концентрировали и очищали колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны в соотношении 1/3,получая[3-метил-1-(4 метилфенил)сульфонил]окси]метил]бутил]-1,1-диметилэтилкарбаминового эфира (97 мг, 0,26 ммоль) в ДМФ (2 мл) нагревали при 95 С в течение 16 ч. Реакционную смесь разбавляли этилацетатом, промывали H2O и насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны в соотношении 1/1,получая выделенный(МН+-Вос). Стадия 8 В. Получение 3-[2(R)-амино-2-(изобутил)этил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 8b К раствору соединения 8 а (30 мг, 0,048 ммоль) в ДХМ (1 мл) добавляли ТФУК (0,1 мл, 1,3 ммоль) и перемешивали при комнатной температуре в течение 1,5 ч. После концентрирования остаток помещали в ДХМ и добавляли насыщенный водный раствор NaHCO3. Водный слой экстрагировали ДХМ. Объединенные органические экстракты сушили над Na2SO4 и концентрировали, получая соединение 8b. Массспектр (CI) m/z 528,0, 530,0 (МН+). Стадия 8 С. Получение 3-[2(R)-этоксикарбонилпропиламино-2-(изобутил)этил]-5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)диона 8 с К раствору соединения 8b (25 мг, 0,048 ммоль) в ДМФ (1 мл) добавляли K2CO3 (21 мг, 0,15 ммоль) с последующим добавлением этил-4-бромбутирата (0,015 мл, 0,1 ммоль). Смесь нагревали при 95 С в течение 16 ч, охлаждали до комнатной температуры и распределяли между этилацетатом и H2O. Органический слой промывали насыщенным раствором соли, сушили над Na2SO4 и концентрировали. Остаток очищали колоночной хроматографией на силикагеле с использованием смеси этилацетат/гексаны/триэтиламин в соотношении 500/500/5, получая соединение 8 с. Масс-спектр (CI) m/z 642,2,- 22010370 644,2 (МН+). Стадия 8D. Получение 3-[2(R)-гидроксикарбанилпропиламино-2-(изобутил)этил]-5-(2-хлор-3 метоксифенил)-1-[2-фтор-6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион 8-1 К соединению 8 с (10 мг, 0,016 ммоль) добавляли ТГФ (0,3 мл) и H2O (0,3 мл) с последующим добавлением NaOH (6,4 мг, 0,16 ммоль). Смесь перемешивали при 50 С в течение 16 ч и очищали препаративной ВЭЖХ, получая соединение 8-1. Масс-спектр (CI) m/z 614,1, 616,1 (МН+), tR = 6,550 мин (способ 6) Пример 9. 3-[2(R)-2-[1-(5-Тетразоил)пропил]амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2,6-дифторбензил]пиримидин-2,4(1 Н,3H)дион Стадия 9 А. Получение 3-[2(R)-2-[3-цианопропил]амино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2,6-дифторбензил]пиримидин-2,4(1 Н,3H)диона 9 а К раствору 7h.1 (2,59 г, 5 ммоль) в CH3CN (25 мл) добавляли диизопропилэтиламин (2,61 мл, 15 ммоль) с последующим доставлением 4-бромбутиронитрила (2,22 г, 15 ммоль), Реакционную смесь нагревали при кипении с обратным холодильником в течение 16 ч. Летучие компоненты упаривали и остаток очищали флэш-хроматографией (диоксид кремния, 4% МеОН/CH2Cl2), получая соединение 9 а (2,62 г, 95,5%). Масс-спектр (CI) m/z 549,1 (МН+). Стадия 9 В. Получение 3-[2(R)-2-[5-тетразоилпропил]амино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2,6-дифторбензил]пиримидин-2,4(1 Н,3H)диона 9-1 К раствору 9 а (274 мг, 0,5 ммоль) в ДМФ (5 мл) добавляли азид натрия (97 мг, 1,5 ммоль) и хлорид аммония (120 мг, 2,25 ммоль). Реакционную смесь нагревали при 110 С в течение 12 ч. Смесь охлаждали, распределяли между EtOAc и насыщенным раствором NaHCO3 в воде, промывали насыщенным раствором соли, сушили (сульфат натрия) и упаривали. Остаток очищали флэш-хроматографией (диоксид кремния, 6% МеОН/CH2Cl2), получая соединение 9-1 (52 мг, 17,6%). ВЭЖХ-Масс-спектр (CI) m/z 592,3 Стадия 10 А. Получение 3-[2(R)-трет-бутоксикарбониламино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2,6-дифторбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 10 а К раствору соединения 1f.1 (28 г, 56 ммоль) в дихлорметане (200 мл) добавляли по каплям с помощью капельной воронки раствор ди-трет-бутилдикарбоната (12 г, 56 ммоль) в дихлорметане (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали в вакууме, получая целевой продукт 10 а в виде светло-желтого твердого вещества (33 г, 56 ммоль, 100%). ВЭЖХ-Масс-спектр (CI) m/z = 496,1 (M+H+-Boc), tR = 3,052 (способ 4). Стадия 10 В. Получение 3-[2(R)-трет-бутоксикарбониламино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-1-[2-фтор-6-метилтиобензил]-6-метилпиримидин-2,4 (1 Н,3H)диона 10b К раствору соединения 10 а (33 г, 56 ммоль) в сухом ДМСО (100 мл) добавляли тиометоксид натрия(4,0 г, 56 ммоль) в атмосфере азота. Реакционную смесь нагревали при 100 С в атмосфере азота в течение 1 ч. Добавляли дополнительно 0,28 эквивалента тиометоксида натрия (1,1 г, 16 ммоль) и реакционную смесь нагревали при 100 С в атмосфере азота в течение 1 ч. Реакционную смесь охлаждали и распределяли между диэтиловым эфиром и водой. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии на силикагеле,элюируя 50%-ным этилацетатом в гексане, получая соединение 10b в виде бледно-желтого твердого ве- 23010370 щества (27 г, 44 ммоль, 78%). ВЭЖХ-Масс-спектр (CI) m/z = 524,1 (М+Н+-Вос), tR = 3,134 (способ 4). 1 Н ЯМР (CDCl3): 1,38 (с, 9 Н), 2,07 (с, 3H), 2,51 (с, 3H), 3,90 (с, 3H), 4,07-4,13 (м, 1 Н), 4,29-4,39 (м, 1 Н), 5,305,53 (м, 2 Н), 5,79-5,85 (м, 1 Н), 6,80-6,91 (м, 2 Н), 6,70 (дд, 1 Н), 7,06-7,15 (м, 2 Н), 7,22-7,41(м, 6 Н). Стадия 10 С. Получение 3-[2(R)-трет-бутоксикарбониламино-2-фенилэтил]-5-(2-фтор-3 метоксифенил)-[2-фтор-6-метилсульфонилбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 10 с К раствору соединения 10b (27 г, 44 ммоль) в безводном дихлорметане (400 мл) добавляли 3 хлорпероксибензойную кислоту (mCPBA, 30 г, 180 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между дихлорметаном и водой. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили сульфатом натрия, фильтровали и концентрировали. Неочищенный продукт очищали с помощью флэш-хроматографии на силикагеле, элюируя 50%-ным этилацетатом в гексане, получая соединение 10c в виде бледно-желтого твердого вещества (15 г, 24 ммолъ, 53%). ВЭЖХ-Масс-спектр(CI) m/z = 556,0 (М+Н+-Boc), tR =2,941 (способ 4). 1 Н ЯМР (CDCl3): 1,38 (с, 9 Н), 2,27 (ушир.с, 3H), 3,48 (с, 3H),3,92 (с, 3H), 4,01-4,15 (м, 1 Н), 4,24-4,40 (м, 1 Н), 4,95-5,05 (м, 1 Н), 5,58-5,68 (м, 2 Н), 6,85-6,91 (дд, 1 Н), 7,02 (дд,1 Н), 7,14 (д, J = 7,6 Гц, 1 Н), 7,19-7,55 (м, 7 Н), 7,97 (д, J = 7,6 Гц, 1 Н). Стадия 10D. Получение 3-[2(R)-амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6 метилсульфонилбензил]-6-метилпиримидин-2,4(1 Н,3H)диона 10-1 К раствору соединения 10 с (10 г, 15 ммолъ) в безводном дихлорметане (60 мл) добавляли трифторуксусную кислоту (ТФУК, 16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь концентрировали и распределяли между этилацетатом и разбавленным водным раствором NaOH. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли, сушили сульфатом натрия, фильтровали и концентрировали, получая 10-1 в виде золотисто-желтого твердого вещества (8,0 г, 14 ммоль, 94%). ВЭЖХ-Масс-спектр (CI)(10,6 мл, 80 ммоль), и оксид дибутилолова (2,48 г, 4 ммоль) суспендировали в 40 мл толуола и 40 мл диоксана и нагревали при кипении с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли 100 мл гексана. Твердый осадок собирали, промывали гексаном (230 мл) и сушили на воздухе. Твердое вещество суспендировали в 100 мл 5%-ного раствора карбоната натрия, добавляли достаточное количество этилацетата для растворения большей части твердого вещества и смесь перемешивали в течение 1 ч. Слои разделяли, водный слой промывали этилацетатом (2100 мл), и органические слой опять экстрагировали 5%-ным раствором карбоната натрия (150 мл). Водные слои объединяли, подкисляли до рН 7 концентрированной соляной кислотой, фильтровали через целит и подкисляли до рН 3. Твердое вещество собирали, промывали водой (250 мл) и ацетоном- 24010370 весом 25 мг (70 мкмоль) и п-толуолсульфоновую кислоту (20 мг, 100 мкмоль) суспендировали в 1 мл воды и нагревали при 80 С в течение 18 ч. Раствор охлаждали и добавляли к соединению 10-1 (29 мг, 50 мкмоль), растворенному в 1 мл этанола и 17 мкл триэтиламина (100 мкмоль). Затем добавляли комплекс боранпиридин (24 мкл, 240 мкмоль) и смесь перемешивали в течение 0,25 ч до прекращения выделения газа. Летучие компоненты удаляли и остаток помещали в 2 мл этилацетата и промывали водой (10,5 мл). Этилацетатный слой упаривали и очищали препаративной ЖХ/МС, получая 11-1 (5 мг, 12% выход). Следующие соединения были синтезированы в соответствии с вышеуказанным способом. Следует понимать, что хотя конкретные варианты осуществления данного изобретения были описаны здесь в целях иллюстрации, могут быть проведены различные модификации, не отступая от сути и объема изобретения. Соответственно, изобретение ограничено только прилагаемой формулой изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ Соединение, имеющее следующую структуру: или его стереоизомер или фармацевтически приемлемая соль, гдеR1a, R1b, R1c являются одинаковыми или разными и независимо представляют собой водород, галоген, С 1-4 алкил, гидрокси или алкокси, или R1a и R1b, взятые вместе, образуют -OCH2O- или -ОСН 2 СН 2-;R2a и R2b являются одинаковыми или разными и независимо представляют собой водород, галоген,трифторметил, циано или -SO2CH3;R3 представляет собой водород или метил;R4 представляет собой фенил или C3-7 алкил;R5 представляет собой водород или С 1-4 алкил;X представляет собой C1-6 алкандиил, необязательно замещенный 1-3 C1-6 алкильными группами. 2. Соединение по п.1, где R1a представляет собой галоген. 3. Соединение по п.1, где R1b представляет собой алкокси. 4. Соединение по п.3, где R1b представляет собой метокси. 5. Соединение по п.1, где R1c представляет собой галоген. 6. Соединение по п.5, где R1c представляет собой фтор или хлор. 7. Соединение по п.1, где R2a представляет собой галоген. 8. Соединение по п.1, где R2b представляет собой водород, галоген или -SO2CH3. 9. Соединение по п.1, где R3 представляет собой водород. 10. Соединение по п.1, где R3 представляет собой метил. 11. Соединение по п.1, где R4 представляет собой фенил.- 25010370 12. Соединение по п.1, где R4 представляет собой C3-7 алкил. 13. Соединение по п.12, где C3-7 алкил представляет собой циклопентил или циклогексил. 14. Соединение по п.1, где R5 представляет собой Н или метил. 15. Соединение по п.1 где R6 представляет собой -СООН. 16. Соединение по п.1 где R6 представляет собой тетразолил. 17. Соединение по п.16 где тетразолил представляет собой тетразол-5-ил. 18. Соединение по п.1, где X представляет собой линейный C1-6 алкандиил. 19. Соединение по п.18, где линейный Cl-6 алкандиил представляет собой -СН 2 СН 2 СН 2-. 20. Соединение по п.19, где R4 представляет собой фенил. 21. Соединение по п.20, где R1a и R2a представляют собой галоген. 22. Соединение по п.21, где R3 представляет собой метил. 23. Соединение по п.21, где R3 представляет собой водород. 24. Соединение по п.1, где X представляет собой разветвленный C1-6 алкандиил. 25. Соединение по п.1, где соединение представляет собой 3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(3-изопропилфенил)-1-[2-фтор-6(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-(циклогексил)этил]-5-(2-фтор-3-метоксифенил)-1-[2 фтор-6-(трифторметил)бензил]-6-метилпиримидин-2,4(1 Н,3H)дион или 3-[2(R)-2-[1-(5-тетразоил)пропил]амино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6 метилсульфонилбензил]-6-метилпиримидин-2,4(1 Н,3H)дион. 26. Соединение по п.1, где соединение представляет собой 3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-фтор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлор-3-метилфенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлорфенил)-1-[2-фтор-6-(метилсульфонил)бензил]пиримидин-2,4(1 Н,3H)дион,3-[2(R)-гидроксикарбонилпропиламино-2-фенилэтил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор-6(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион или 3-[2(R)-гидроксикарбонилпропиламино-2-(изобутил)этил]-5-(2-хлор-3-метоксифенил)-1-[2-фтор 6-(трифторметил)бензил]пиримидин-2,4(1 Н,3H)дион. 27. Фармацевтическая композиция, содержащая соединение по п.1 и фармацевтически приемлемый носитель или разбавитель. 28. Способ антагонизации гонадотропин-высвобождающего гормона в организме нуждающегося в этом субъекта, включающий введение субъекту эффективного количества соединения по п.1. 29. Способ лечения состояния, связанного с половым гормоном, у нуждающегося в этом субъекта,включающий введение субъекту эффективного количества фармацевтической композиции по п.27. 30. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой рак, доброкачественную гипертрофию предстательной железы или миому матки. 31. Способ по п.30, где рак представляет собой рак предстательной железы, рак матки, рак молочной железы или гипофизарную гонадотропную аденому. 32. Способ по п.31, где рак представляет собой рак предстательной железы. 33. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой эндометриоз, поликистозное заболевание яичников, фиброзные опухоли матки или преждевременное половое созревание. 34. Способ по п.33, где состояние, связанное с половым гормоном, представляет собой эндометриоз. 35. Способ по п.29, где состояние, связанное с половым гормоном, представляет собой фиброзные опухоли матки. 36. Способ лечения бесплодия у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции по п.27. 37. Способ лечения красной волчанки, синдрома раздраженного кишечника, предменструального синдрома, волосатости, низкого роста или нарушений сна у нуждающегося в этом субъекта, включающий введение субъекту эффективного количества фармацевтической композиции по п.27. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2/6
МПК / Метки
МПК: C07D 239/54, C07D 405/04, A61K 31/513, A61P 5/02, C07D 401/12
Метки: гонадотропин-высвобождающего, рецептора, гормона, качестве, производные, антагонистов, пиримидин-2,4-диона
Код ссылки
<a href="https://eas.patents.su/27-10370-proizvodnye-pirimidin-24-diona-v-kachestve-antagonistov-receptora-gonadotropin-vysvobozhdayushhego-gormona.html" rel="bookmark" title="База патентов Евразийского Союза">Производные пиримидин-2,4-диона в качестве антагонистов рецептора гонадотропин-высвобождающего гормона</a>
Антагонисты гормона, высвобождающего гонадотропин.

Номер патента: 992
Опубликовано: 28.08.2000
Авторы: Джиротра Нариндар Н., Лин Питер, Чу Лин, Виврэтт Мэтью Дж., Фишер Майкл Х., Эштон Уоллес Т., Гаулет Марк
МПК: C07D 401/12, A61P 13/08, A61K 31/4045...
Метки: гонадотропин, высвобождающего, антагонисты, гормона
Формула / Реферат:
1. Соединение формулы где А представляет C1-C6алкил, замещенный C1-C6алкил, С3-С7 циклоалкил, замещенный С3-С7циклоалкил, С3-С6алкенил, замещенный С3-С6алкенил, С3-С6алкинил, замещенный С3-С6 алкинил, C1-C6 алкокси или (C0-C5алкил)-S(О)n-(С0-С5алкил), (C0-C5алкил)-O-(C0-C5алкил), (C0-C5алкил)-NR18-(C0-C5алкил), где R18 и (C0-C5 алкил) могут быть соединены, образуя кольцо, или одинарную связь; R0 представляет водород, C1-C6алкил,...
Азабициклические производные в качестве антагонистов мускаринового рецептора

Номер патента: 9942
Опубликовано: 28.04.2008
Авторы: Дхармараджан Санкаранарайанан, Кхугх Анита, Кумар Нареш, Салман Мохаммад, Каур Кирандип, Мехта Анита, Сарма Пакала Кумара Савитхру
МПК: A61K 31/403, A61P 11/00, A61P 1/00...
Метки: рецептора, мускаринового, качестве, производные, антагонистов, азабициклические
Формула / Реферат:
1. Азабициклические производные в качестве антагонистов мускаринового рецептора, выбранные из следующих соединений: (2R,2S)(1a,5a,6a)-N-{[4-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-бутил]-3-азабицикло[3.1.0]гекс-6-ил-метил}-2-гидрокси-2-циклопентил-2-фенилацетамид (соединение 1), (2R)(1a,5a,6a)-N-(3-бензил-3-азабицикло[3.1.0]гекс-6-ил-метил)-2-гидрокси-2-циклопент-1-енил-2-фенилацетамид (соединение 2),...
Производные (тио) карбамоилциклогексана в качестве антагонистов d3/d2 рецептора

Номер патента: 9022
Опубликовано: 26.10.2007
Авторы: Ласи Юдит, Саги Каталин, Дьертьян Иштван, Кишш Бела, Галамбош Янош, Ваго Иштван, Ногради Каталин, Агаине Чонгор Эва, Ласловски Иштван
МПК: A61P 25/00, A61K 31/495, C07D 243/08...
Метки: антагонистов, карбамоилциклогексана, рецептора, тио, качестве, производные
Формула / Реферат:
1. Соединение формулы (I) где R1 и R2 независимо представляют собой заместитель, выбранный из водорода, C1-6алкила, С2-7алкенила, арила, циклоалкила, ароила, или R1 и R2 могут образовывать гетероциклическое кольцо со смежным атомом азота; X представляет собой атом кислорода или серы; n является целым числом от 1 до 2, и/или его геометрические изомеры, и/или стереоизомеры, и/или диастереомеры, и/или соли, и/или гидраты, и/или сольваты. 2....
Производные циклопентена в качестве антагонистов рецептора мотилина

Номер патента: 3252
Опубликовано: 27.02.2003
Авторы: Ксианг Мин, Биверз Мэри Пэт, Мур Джон Б.Мл., Чен Роберт Х.
МПК: C07C 233/41, A61K 31/5375, A61P 1/00...
Метки: мотилина, производные, циклопентена, антагонистов, качестве, рецептора
Формула / Реферат:
1. Соединение формулы I в которой R1 представляет H, C1-5-алкил, замещенный C1-5-алкил (где заместителями являются один или несколько галогенов), амино-C1-5-алкил, C1-5-алкиламино-C1-5-алкил, ди-C1-5-алкиламино-C1-5-алкил, RaRbN-C1-5-алкил (где Ra и Rb независимо выбраны из H и C1-5-алкила или вместе образуют морфолин, пиперазин, пиперидин или N-замещенный пиперидин, где N-заместитель представляет C1-5-алкил или фенил-C1-5-алкил),...
Замещенные производные азабициклогексана в качестве антагонистов мускаринового рецептора

Номер патента: 9059
Опубликовано: 26.10.2007
Авторы: Гупта Джанг Бахадур, Силамкоти Арундутт Висванатхам, Мехта Анита
МПК: A61P 11/00, A61P 1/00, A61K 31/403...
Метки: антагонистов, замещенные, мускаринового, качестве, производные, азабициклогексана, рецептора
Формула / Реферат:
1. Соединения структурной формулы I и их фармацевтически приемлемые соли, фармацевтически приемлемые сольваты, эфиры, энантиомеры, диастереомеры, N-оксиды, полиморфные формы, где Ar представляет собой арильное или гетероарильное кольцо, имеющее 1-2 гетероатома, выбранных из группы, состоящей из атомов кислорода, серы и азота, арильное или гетероарильное кольца могут быть незамещенными или замещенными от одного до трех заместителями, независимо...
Предыдущий патент: Замещённые хинолиновые соединения
Следующий патент: Тканевые протективные цитокины для защиты, восстановления и стимуляции реактивных клеток, тканей и органов
Случайный патент: Способ оценки результатов обследования скважины