3-замещенные 6-(пиридинилметокси)пирролопиридиновые соединения
Номер патента: 21781
Опубликовано: 31.08.2015
Авторы: Ламас-Петейра Карлос, Ричардс Саймон Джеймс, Сапмаз Селма, Уолтер Магнус Вильгельм



















Формула / Реферат
1. Соединение следующей формулы

или его фармацевтически приемлемая соль, где
R1 представляет собой 2-пиридинил, 3-пиридинил, 4-пиридинил, 5-фтор-2-пиридинил, 5-метил-2-пиридинил, 5-метокси-2-пиридинил, 3-метил-2-пиридинил или 6-метил-2-пиридинил;
R2 представляет собой метил, этил или циклопропил;
R3 представляет собой метил, этил, н-пропил, изопропил, 2-фторэтил, 2-метоксиэтил или циклобутил и
R4 представляет собой водород, фтор, хлор или метил.
2. Соединение по п.1, или его фармацевтически приемлемая соль, отличающееся тем, что R1 представляет собой 2-пиридинил.
3. Соединение по любому из пп.1-2, или его фармацевтически приемлемая соль, отличающееся тем, что R2 представляет собой метил.
4. Соединение по любому из пп.1-3, или его фармацевтически приемлемая соль, отличающееся тем, что R3 представляет собой этил.
5. Соединение по любому из пп.1-4, или его фармацевтически приемлемая соль, отличающееся тем, что R4 представляет собой водород.
6. Соединение по п.1, которое представляет собой этил-4-[1-метил-6-(пиридин-2-илметокси)-1Н-пирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилат или его фармацевтически приемлемую соль.
7. Фармацевтическая композиция для лечения болезни Паркинсона, содержащая соединение по любому из пп.1-6 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель.
8. Способ лечения болезни Паркинсона, включающий введение пациенту, который в этом нуждается, эффективного количества соединения по любому из пп.1-6 или его фармацевтически приемлемой соли.
9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в терапии заболеваний, связанных с активностью mGluR5.
10. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения болезни Паркинсона.
11. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для лечения болезни Паркинсона.
Текст
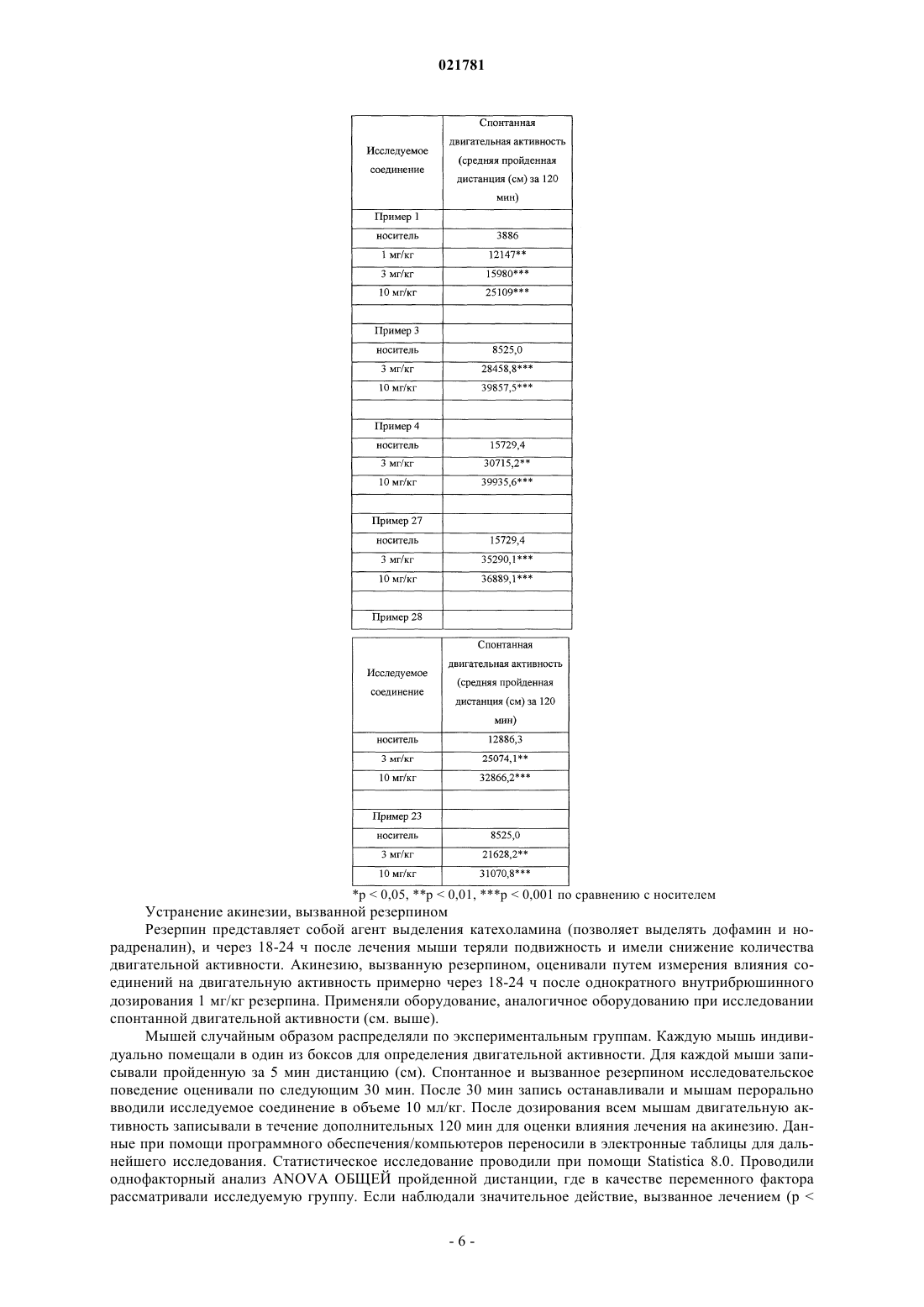
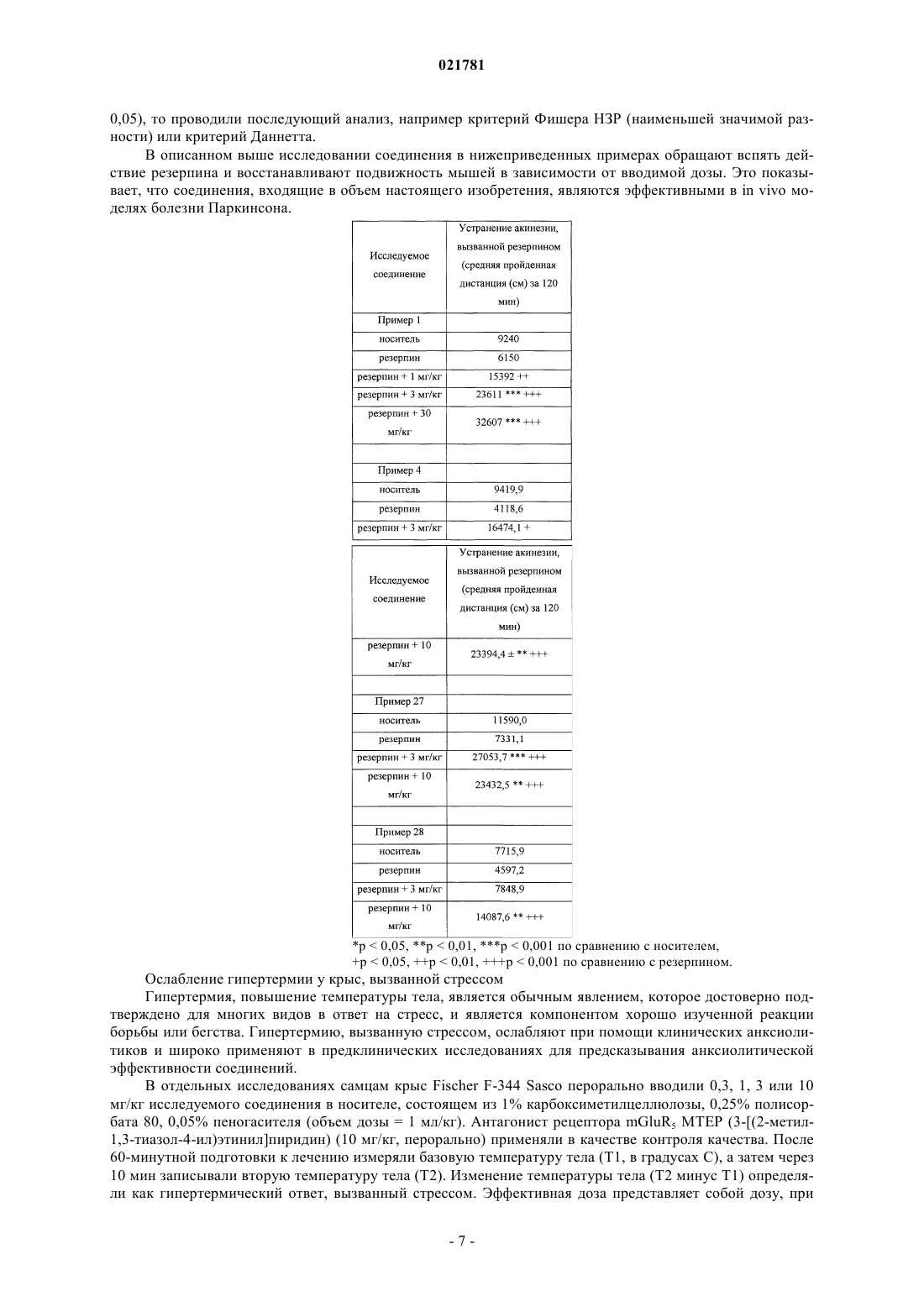
В настоящем изобретении предложены некоторые 3-замещенные 6(пиридинилметокси)пирролопиридиновые соединения, в частности соединения формулы I, и их фармацевтические композиции. Также в настоящем изобретении предложены способы применения соединения формулы I для лечения болезни Паркинсона.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) В настоящем изобретении предложены некоторые 3-замещенные 6-(пиридинилметокси)пирролопиридиновые соединения, в частности некоторые 3-оксопиперазинил-6-(пиридинилметокси)пирролопиридиновые соединения, их фармацевтические композиции, способы их применения и способы их получения.L-Глутамат является главным возбуждающим нейромедиатором в центральной нервной системе и относится к возбуждающим аминокислотам. Глутаматные рецепторы состоят из двух основных подтипов: лиганд-управляемые ионотропные рецепторы, сопряженные с ионными каналами, и G-белоксопряженные метаботропные рецепторы с семью трансмембранными доменами (mGluRs). Метаботропное семейство включает восемь представителей и подразделяется на три группы на основе сходства последовательностей, передачи сигнала и фармакологии. Рецепторы группы I (mGluR1 и mGluR5, а также их сплайс-варианты) при связывании активируют гидролиз инозитолфосфата и выработку внутриклеточных кальциевых сигналов. Рецепторы группы II (mGluR2 и mGluR3) и рецепторы группы III (mGluR4, mGluR6,mGluR7 и mGluR8) при связывании подавляют аденилатциклазу и регулируют содержание циклического АМФ (аденозинмонофосфата) путем непрямого ингибирования активности аденилилциклазы. Указанные подтипы рецепторов mGlu обладают уникальными характеристиками экспрессии в центральной нервной системе, на которую можно направленно воздействовать с применением новых и селективных агентов(см., например, Slassi A. et al., Current Topics in Medicinal Chemistry (2005), 5, 897-911, где описаны антагонисты mGluR5, подходящие для применения в качестве противопаркинсонического средства на животной модели болезни Паркинсона). Кроме того, как полагают, антагонисты mGluR5 подходят для применения в моделях беспокойства, синдрома ломкой Х-хромосомы, наркотической зависимости и абстиненции, в том числе алкоголизма, а также в моделях воспалительной и нейропатической боли. В публикации заявки на патент США 2009/0197881 предложены некоторые азаиндольные производные соединений в качестве антагонистов рецептора простагландина DP, а также соединения, подходящие для применения для лечения аллергических заболеваний, включая астму. Соединения согласно настоящему изобретению являются селективными антагонистами метаботропных рецепторов группы I, в частности рецептора mGluR5 (mGluR5), особенно в отношении селективности по сравнению с mGluR2, mGluR3 и mGluR4. Неожиданным образом, в группе I метаботропных рецепторов соединения согласно настоящему изобретению являются селективными в отношении mGluR5 по сравнению с mGluR1. Полагают, что соединения согласно настоящему изобретению подходят для применения для лечения состояний, связанных с рецепторами mGluR5, таких как болезнь Паркинсона,боль, зависимость от психоактивных веществ и абстиненция, беспокойство, в том числе генерализованное тревожное расстройство, депрессия, в том числе большое депрессивное расстройство, а также беспокойство с сопутствующей депрессией (смешанное расстройство депрессии и беспокойства), в том числе генерализованное тревожное расстройство с сопутствующим большим депрессивным расстройством. В настоящем изобретении предложены новые соединения, которые являются антагонистамиmGluR5 и, следовательно, как полагают, подходят для применения для лечения расстройств, обсуждаемых выше. Такие новые соединения могли бы удовлетворить потребность в безопасных и эффективных способах лечения состояний, связанных с описанными выше рецепторами, не вызывающих побочных эффектов. В настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль где R1 представляет собой пиридинил, возможно замещенный одной группой, выбранной из фтора,метила или метокси;R4 представляет собой водород, фтор, хлор или метил. Кроме того, в настоящем изобретении предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или наполнитель. В одном из вариантов реализации композиция дополнительно содержит один или более других терапевтических агентов. Кроме того, в настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль для применения в терапии. Кроме того, в настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль для применения для лечения болезни Паркинсона. Кроме того, в настоящем изобретении предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения болезни Паркинсона. Кроме того, в настоящем изобретении предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения депрессии. Кроме того, в настоящем изобретении предложен способ лечения болезни Паркинсона, включающий введение пациенту, нуждающемуся в лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Термин "C1-С 3 алкил" относится к прямой или разветвленной алкильной цепи, содержащей от одного до трех атомов углерода, и включает метил, этил, п-пропил и изопропил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляют собой соединение, гдеR4 представляет собой водород, фтор, хлор или метил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R1 представляет собой 2-пиридинил, 3-пиридинил, 4-пиридинил, 5-фтор-2-пиридинил, 5 метил-2-пиридинил, 5-метокси-2-пиридинил, 3-метил-2-пиридинил или 6-метил-2-пиридинил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R2 представляет собой метил, этил или циклопропил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R3 представляет собой метил, этил, п-пропил, изопропил, 2-фторэтил, 2-метоксиэтил или циклобутил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R4 представляет собой водород, фтор, хлор или метил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где представляет собой 2-пиридинил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R2 представляет собой метил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R3 представляет собой этил. Конкретное соединение формулы I или его фармацевтически приемлемая соль представляет собой соединение, где R4 представляет собой водород. Конкретное соединение формулы I представляет собой этил-4-[1-метил-6-(пиридин-2-илметокси)1 Н-пирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилат или его фармацевтически приемлемую соль. Один из вариантов реализации согласно настоящему изобретению включает способ получения соединения формулы I или его фармацевтически приемлемой соли, включающий А) взаимодействие соединения формулы II, где Х 1 представляет собой галогеновую группу В) ацилирование соединения формулы III где в случае, когда требуется фармацевтически приемлемая соль соединения формулы I, ее получают путем взаимодействия основного соединения формулы I с физиологически приемлемой кислотой или при помощи других традиционных способов. Следует понимать, что соединения согласно настоящему изобретению могут существовать в виде стереоизомеров. Хотя в рамках настоящего изобретения рассматриваются все энантиомеры, диастереомеры и их смеси, предпочтительными вариантами реализации являются индивидуальные диастереомеры,а более предпочтительными вариантами реализации являются индивидуальные энантиомеры. Следует понимать, что соединения согласно настоящему изобретению могут существовать в таутомерных формах. Если существуют таутомерные формы, в настоящем изобретении рассматривается каждая форма и их смеси. Термин "фармацевтически приемлемая соль" включает соль присоединения кислоты, которая существует в сопряжении с основной частью соединения формулы I. Такие соли включают фармацевтически приемлемые соли (см. перечисленные в Handbook of Pharmaceutical Salts: Properties, Selection and Use,P.H. Stahl and С.G. Wermuth (Eds.), Wiley-VCH, New York, 2002, которые известны специалистам в данной области техники). В дополнение к фармацевтически приемлемым солям в объем изобретения включены и другие соли. Они могут служить в качестве промежуточных соединений при очистке соединений или при получении других фармацевтически приемлемых солей или могут подходить для применения для идентификации, исследования или очистки. Полагают, что соединение согласно настоящему изобретению подходит для всех случаев, когда показано применение антагониста рецептора mGluR5. В одном из вариантов реализации соединение согласно настоящему изобретению предположительно будет подходить для применения для лечения болезни Паркинсона или расстройств, связанных с болезнью Паркинсона. В частности, соединение согласно настоящему изобретению предположительно подходит для применения для лечения дискинезии, в том числе дискинезии, вызванной леводопа (L-dopa), применяемой для лечения болезни Паркинсона(PD-LID). Согласно настоящему описанию термин "пациент" относится к теплокровному животному, такому как млекопитающее, и включает человека. Человек является предпочтительным пациентом. Специалист в данной области техники может оказывать влияние на болезнь Паркинсона путем лечения пациента с выраженными симптомами при помощи эффективного количества соединения формулы I. Таким образом, термины "лечение" и "лечить" относятся ко всем действиям, которые способны замедлять, прерывать, блокировать, контролировать или останавливать развитие существующего расстройства и/или его симптомов, но не обязательно обозначают полное устранение всех симптомов. Специалист в данной области техники может оказывать влияние на болезнь Паркинсона путем лечения пациента, находящегося в зоне риска будущего развития симптомов, при помощи эффективного количества соединения формулы I, что включает профилактическое лечение. Согласно настоящему описанию термин "эффективное количество" соединения формулы I относится к количеству, т.е. дозировке, которое является эффективным для лечения расстройства, такого как указанная в настоящем описании болезнь Паркинсона. Лечащий врач, являющийся специалистом в данной области техники, способен легко определять эффективное количество при помощи стандартных способов и изучения результатов, полученных в аналогичных условиях. При определении эффективного количества, т.е. дозы соединения формулы I, лечащий врач учитывает ряд факторов, включая, но, не ограничиваясь ими, вводимое соединение формулы I; если применяются, другие совместно вводимые агенты; вид млекопитающего; его размер, возраст и общее состояние здоровья; степень поражения или тяжести расстройства, такого как болезнь Паркинсона; ответную реакцию у данного пациента; способ введения; характеристики биодоступности вводимого лекарственного средства; выбранный режим дозирования; применение других сопутствующих лекарственных средств и другие соответствующие условия. Соединение формулы I можно применять в комбинации с другими лекарственными средствами, ко-3 021781 торые применяют для лечения/профилактики/подавления или облегчения заболеваний или состояний, в том числе болезни Паркинсона, для которых соединения формулы I являются подходящими для применения. Указанные другие лекарственные средства можно вводить обычно применяемым способом и в обычно применяемом количестве одновременно или последовательно с соединением формулы I. В случае одновременного применения соединения формулы I с одним или более другими лекарственными средствами предпочтительной является фармацевтическая лекарственная форма, содержащая указанные лекарственные средства в дополнение к соединению формулы I. Соответственно, фармацевтические композиции согласно настоящему изобретению включают фармацевтические композиции, которые также содержат один или более других активных ингредиентов в дополнение к соединению формулы I. Примеры других активных ингредиентов, эффективных при лечении болезни Паркинсона, которые можно комбинировать с соединением формулы I и вводить или отдельно, или в той же лекарственной форме,включают, но не ограничиваются ими:(c) ингибиторы монаминоксидазы Б (МАО-Б), такие как селегилин и разагилин;(d) ингибиторы катехин-О-метилтрансферазы (КОМТ), такие как толкапон и энтакапон;(f) лекарственные средства, блокирующие глутаматные рецепторы (NMDA (N-метил-D-аспартат,такие как амантадин;(g) антагонисты аденозина А 2 а рецептора, такие как истрадефиллин и преладенант;(h) антагонисты 5-НТ 1 а, такие как пиклозотан и пардопрунокс; или(i) альфа-2-антагонисты, такие как атипамезол и фипамезол. Соединения согласно настоящему изобретению можно вводить отдельно или в виде фармацевтической композиции, т.е. в сочетании с фармацевтически приемлемыми носителями или наполнителями,пропорции и свойства которых определяются растворимостью и химическими свойствами, в том числе стабильностью, выбранного соединения, выбранного способа введения и стандартной фармацевтической практикой. Соединения согласно настоящему изобретению, обладающие самостоятельно эффективностью, можно получать и вводить в виде их фармацевтически приемлемых солей для удобства кристаллизации, повышенной растворимости и т.п. Таким образом, в настоящем изобретении предложены фармацевтические композиции, содержащие соединение формулы I и фармацевтически приемлемый носитель, разбавитель или наполнитель. Специалист в области приготовления композиций способен легко выбрать надлежащую форму и способ введения в зависимости от конкретных характеристик выбранного соединения, расстройства или состояний, подлежащих лечению, стадии расстройства или состояния, а также других соответствующих условий (см., например, Remington: The Science and Practice of Pharmacy, D.B. Troy, Editor, 21st Edition.,Lippincott, WilliamsWilkins, 2006). Функциональные исследования рецепторов mGluR5 и mGluR1 человека in vitro Активация рецепторов, связанных с G-белком (GPCRs), которые связаны с Gq-белками, приводит к изменению внутриклеточной концентрации кальция. Данный функциональный ответ можно измерить при помощи кинетического анализа с применением кальций-чувствительных красителей и спектрофотометра для чтения планшетов для визуализации флуоресценции с применением стандартного способа,известного как FLIPR (MDS Analytical Technologies, Sunnyvale, CA). Стабильный состав клеточной линии и способы исследования брали из Kingston А.Е. et al. (1995)Neuropharmacology 34: 887-894. Вкратце, клональные клеточные линии, экспрессирующие рекомбинантные рецепторы mGlu5a и mGlu1 человека, трансфицировали в AV-12 клетки (American Type CultureCollection, Manassas, VA), содержащие крысиный глутаматный транспортер ЕААТ 1. Клетки выращивали в среде Игла, модифицированной по способу Дульбекко (ДМЕМ; Invitrogen, Carlsbad, СА), с добавлением 5% фетальной бычьей сыворотки, 1 мМ L-глутамина, 1 мМ пирувата натрия, 10 мМ ГЭПЭС (4-(2 гидроксиэтил)-1-пиперазинэтансульфоновая кислота), 0,75 мг/мл генетицина и 0,3 мг/мл гигромицина Б при 37 С в инкубаторе с 95% относительной влажностью и 5% CO2. Конфлюэнтные культуры пассажировали раз в две недели. Для функциональных исследований клетки высеивали в питательной среде с недостаточным количеством антибиотиков с плотностью 65 тыс. на лунку в 96-луночные, черные с прозрачным дном, покрытые поли-D-лизином микропланшеты и инкубировали в течение 18-20 ч до начала эксперимента. После удаления среды клетки вводили 8 мкМ Fluo-3 (Invitrogen) в буфере для исследований, состоящем из сбалансированного солевого раствора Хенкса (Invitrogen), с добавлением 20 мМ ГЭПЭС в течение 1,5 ч при 25 С. Соединения серийно разбавляли в ДМСО (диметилсульфоксид), а затем однократно разбавляли в буфере для исследований; конечная концентрация ДМСО в исследовании составляла 0,625%. Для оценки количества агониста, необходимого для индуцирования ЕС 90 ответа, перед каждым экспериментом проводили исследование на FLIPR с однократным добавлением с получением 11-точечной кривой зависимо-4 021781 сти доза-эффект для агониста глутамата. Действие антагонистов соединений количественно оценивали при помощи FLIPR прибора по 10-точечным кривым доза-эффект путем сравнения пиков флуоресцентных ответов на агониста глутамата в присутствии и в отсутствие соединения. В частности, действие соединения измеряли как разницу между высотой максимального и минимального пиков в относительных единицах флуоресценции с поправкой на базальную флуоресценцию, измеренную в отсутствие глутамата. Все данные рассчитывали в виде относительных значений IC50 с применением программного обеспечения для построения четырехпараметрических логистических кривых (ActivityBase v5.3.1.22). В описанном выше исследовании соединения, приведенные в качестве примера, демонстрировали значения IC50 менее 750 нМ для рецептора mGluR5 человека. В частности, соединение из примера 1 обладает IC50, равным 184 нМ, измеренным для рецептора mGluR5 человека. Это показывает, что соединения, входящие в объем настоящего изобретения, являются сильными антагонистами рецептора mGluR5. Некоторые соединения, приведенные в качестве примера, оценивали для рецептора mGluR1. В описанном выше исследовании соединения из примеров 1-6, 8, 11, 20 и 22-28 демонстрировали значения IC50 более 6000 нМ, для рецептора mGluR1 человека. В частности, соединение из примера 1 обладает IC50 более 12,500 нМ, измеренным для рецептора mGluR1 человека. Это показывает, что соединения, входящие в объем настоящего изобретения, являются селективными антагонистами рецептора mGluR5 по сравнению с рецептором mGluR1. Антипаркинсоническое действие соединений согласно настоящему изобретению можно определять при помощи способов, хорошо известных в данной области техники, таких как животные модели двигательной активности. Например, соединения согласно настоящему изобретению демонстрировали влияние на спонтанную (приобретенную) двигательную активность и на акинезию, вызванную резерпином, у С 57/черных 6J самцов мышей. Спонтанная (приобретенная) двигательная активность Двигательную активность измеряли при помощи автоматизированной системы отслеживания движения мышей. Мышей помещали в камеры и позволяли привыкнуть к камерам в течение 30 мин. В течение данного времени они демонстрировали снижение передвижений с течением времени. После введения соединения согласно настоящему изобретению движения животных вернулись на уровни, соответствующие первоначальным. В частности, боксы для определения двигательной активности [404040 см с гладкой поверхностью] размещали группами по 4 бокса в инфракрасные камеры и проводили исследование без доступа света. Двигательную активность записывали и измеряли при помощи инфракрасного видеослежения. Двигательную активность записывали между 8:30 и 17:00 ч. В некоторых случаях двигательную активность измеряли при помощи открытых областей, которые используют прерывание инфракрасных лучей в качестве оценки движения. Мышей случайным образом распределяли по экспериментальным группам. Каждую мышь индивидуально помещали в один из боксов для определения двигательной активности. Для каждой мыши записывали пройденную за 5 мин дистанцию (см). Исследовательское поведение оценивали по следующим 30 мин. После 30 мин запись останавливали имышам перорально вводили исследуемое соединение или носитель в объеме 10 мл/кг. После дозирования всем мышам двигательную активность записывали в течение дополнительных 120 мин для оценки влияния лечения на приобретенную двигательную активность. Данные при помощи программного обеспечения/компьютеров переносили в электронные таблицы для дальнейшего исследования. Статистическое исследование проводили при помощи Statistica 8.0. Проводили однофакторный анализ ANOVA ОБЩЕЙ пройденной дистанции, где в качестве переменного фактора рассматривали исследуемую группу. Если наблюдали значительное действие, вызванное лечением (р 0,05), то проводили последующий анализ, например критерий Фишера НЗР (наименьшей значимой разности) или критерий Даннетта. В описанном выше исследовании соединения в нижеприведенных примерах облегчают подвижность мышей в зависимости от вводимой дозы. Это показывает, что соединения, входящие в объем настоящего изобретения, являются эффективными в in vivo моделях болезни Паркинсона. Устранение акинезии, вызванной резерпином Резерпин представляет собой агент выделения катехоламина (позволяет выделять дофамин и норадреналин), и через 18-24 ч после лечения мыши теряли подвижность и имели снижение количества двигательной активности. Акинезию, вызванную резерпином, оценивали путем измерения влияния соединений на двигательную активность примерно через 18-24 ч после однократного внутрибрюшинного дозирования 1 мг/кг резерпина. Применяли оборудование, аналогичное оборудованию при исследовании спонтанной двигательной активности (см. выше). Мышей случайным образом распределяли по экспериментальным группам. Каждую мышь индивидуально помещали в один из боксов для определения двигательной активности. Для каждой мыши записывали пройденную за 5 мин дистанцию (см). Спонтанное и вызванное резерпином исследовательское поведение оценивали по следующим 30 мин. После 30 мин запись останавливали и мышам перорально вводили исследуемое соединение в объеме 10 мл/кг. После дозирования всем мышам двигательную активность записывали в течение дополнительных 120 мин для оценки влияния лечения на акинезию. Данные при помощи программного обеспечения/компьютеров переносили в электронные таблицы для дальнейшего исследования. Статистическое исследование проводили при помощи Statistica 8.0. Проводили однофакторный анализ ANOVA ОБЩЕЙ пройденной дистанции, где в качестве переменного фактора рассматривали исследуемую группу. Если наблюдали значительное действие, вызванное лечением (р 0,05), то проводили последующий анализ, например критерий Фишера НЗР (наименьшей значимой разности) или критерий Даннетта. В описанном выше исследовании соединения в нижеприведенных примерах обращают вспять действие резерпина и восстанавливают подвижность мышей в зависимости от вводимой дозы. Это показывает, что соединения, входящие в объем настоящего изобретения, являются эффективными в in vivo моделях болезни Паркинсона. Ослабление гипертермии у крыс, вызванной стрессом Гипертермия, повышение температуры тела, является обычным явлением, которое достоверно подтверждено для многих видов в ответ на стресс, и является компонентом хорошо изученной реакции борьбы или бегства. Гипертермию, вызванную стрессом, ослабляют при помощи клинических анксиолитиков и широко применяют в предклинических исследованиях для предсказывания анксиолитической эффективности соединений. В отдельных исследованиях самцам крыс Fischer F-344 Sasco перорально вводили 0,3, 1, 3 или 10 мг/кг исследуемого соединения в носителе, состоящем из 1% карбоксиметилцеллюлозы, 0,25% полисорбата 80, 0,05% пеногасителя (объем дозы = 1 мл/кг). Антагонист рецептора mGluR5 MTEP (3-[(2-метил 1,3-тиазол-4-ил)этинил]пиридин) (10 мг/кг, перорально) применяли в качестве контроля качества. После 60-минутной подготовки к лечению измеряли базовую температуру тела (Т 1, в градусах С), а затем через 10 мин записывали вторую температуру тела (Т 2). Изменение температуры тела (Т 2 минус Т 1) определяли как гипертермический ответ, вызванный стрессом. Эффективная доза представляет собой дозу, при которой соединение вызывает 35% снижение гипертермии, вызываемой стрессом, по отношению к ответу при введении носителя и определяется как Т 35 доза. В описанном выше исследовании соединение из примера 1 приводит к снижению гипертермии, вызываемой стрессом, с Т 35 дозой = 0,55 мг/кг. Соединение из примера 3 приводит к снижению гипертермии, вызываемой стрессом, с Т 35 дозой = 0,93 мг/кг. Это показывает, что соединения, входящие в объем настоящего изобретения, являются эффективными в in vivo модели беспокойства. Тест принудительного плавания мышей Самцов мыши NIH-Swiss (20-25 г, Harlan Sprague-Dawley, Indianapolis, IN) применяли в способе,отличающемся от способа, представленного в Porsolt RD, Le Pichon M, Jalfre M Depression: new animalmodel sensitive to antidepressant treatments. Nature. 1977 Apr 21; 266(5604):730-2. Мышей помещали в прозрачные пластиковые цилиндры (диаметр 10 см; высота 25 см), заполненные до уровня 6 см 22-25 С водой на 6 мин. Продолжительность неподвижности записывали в течение последних 4 мин из шестиминутного теста. Мышь считали неподвижной, если она плавала неподвижно или совершала только движения, необходимые для того, чтобы держать голову над водой. Данные анализировали при помощи последующего анализа по критерию Даннетта при уровне значимости 0,05. Измеряли количество времени,проведенное в неподвижности. Среднее + СОС (стандартная ошибка среднего) подвергали анализуANOVA, а затем критерию Даннетта с р 0,05, установленным в качестве вероятности ошибки для статистической значимости. ED60 значения экстраполировали из линейной части кривой дозовой зависимости и представляли предполагаемую дозу, снижающую спонтанную неподвижность (100% при 0 мг/кг соединения) на 60%. Максимальное снижение времени неподвижности рассчитывали при помощи наибольшего снижения в потере подвижности, полученного при любой дозе соединения по формуле: (100 потеря подвижности с соединением/неподвижность при введении носителя)%. В описанном выше исследовании соединение из примера 1 приводит к снижению неподвижности принудительного плавания с ED60 дозой = 8,6 мг/кг и максимальному снижению неподвижности 50,6%. Это показывает, что соединения, входящие в объем настоящего изобретения, являются подходящими для применения в in vivo модели депрессии. Соединения формулы I можно получать при помощи способов, известных в химической области получения структурно аналогичных соединений, или при помощи нового способа, представленного в настоящем описании. В настоящем изобретении также предложены способ получения соединения формулы I или его фармацевтически приемлемой соли и новые промежуточные соединения для получения соединения формулы I, представленные в следующих способах, в которых заместители, такие как R1, R2,R3 и R4, являются такими, как описано выше, если не указано иное. В целом, соединение формулы I можно получать из соединения формулы II, где X1 представляет собой подходящую связывающую группу (схема 1). В одном из способов соединение формулы II, где X1 представляет собой галогеновую группу, такую как бромо, взаимодействует с R3-3-оксопиперазин-1 карбоксилатом и подходящим металлическим катализатором, таким как иодид меди (I), в присутствии основания, такого как фосфат калия, и аминного лиганда, такого как N,N'-диметилэтилендиамин, в подходящем растворителе с получением соединения формулы I. Подходящие растворители включают 1,4 диоксан и диметилформамид. Альтернативно, соединение формулы I можно получать из соединения формулы II через соединение формулы III (схема 1). В частности, соединение формулы II, где X1 представляет собой галогеновую группу, такую как бромо, взаимодействует с 1-Pg-3-оксопиперазином, где Pg представляет собой подходящую защитную группу для аминогруппы, такую как трет-бутилоксикарбонил, и подходящим металлическим катализатором, таким как иодид меди (I), в присутствии основания, такого как фосфат калия, и аминного лиганда, такого как N,N'-диметилэтилендиамин, в подходящем растворителе с получением соединения формулы IV, где Pg представляет собой трет-бутилоксикарбонил. Подходящие растворители включают 1,4-диоксан и диметилформамид. Соединение формулы IV взаимодействует с подходящим агентом снятия защиты, таким как соляная кислота или трифторуксусная кислота, в растворителе с получением соединения формулы III. Подходящие растворители включают дихлорметан и этилацетат. Соединение формулы III ацилируют в растворителе с R3-карбонохлоридатом в присутствии основания, такого как триэтиламин, с получением соединения формулы I. Подходящие растворители включают дихлорметан. Согласно схеме 2 соединение формулы II можно получать из соединения формулы V, где Х 2 представляет собой подходящую уходящую группу, а X1 является таким, как определено выше. В частности,соединение формулы V, где Х 2 представляет собой галогеновую группу, такую как фторо, в растворителе взаимодействует с R1CH2OH в присутствии подходящего основания с получением соединения формулыII. Подходящие основания включают гидрид натрия и трет-бутоксид калия. Подходящие растворители включают диметилсульфоксид и диметилформамид. В способе, альтернативном схеме 1, соединение формулы IV можно получать из соединения формулы V (схема 2). В частности, соединение формулы V, где Х 1 и Х 2 являются такими, как определено выше, взаимодействует с l-Pg-3-оксопиперазином, где Pg представляет собой подходящую защитную группу для аминогруппы, такую как трет-бутилоксикарбонил, и подходящим металлическим катализатором, таким как иодид меди (I), в присутствии основания, такого как фосфат калия, и аминного лиганда,такого как N,N'-диметилэтилендиамин, в подходящем растворителе с получением соединения формулыVI, где Pg представляет собой трет-бутилоксикарбонил. Подходящие растворители включают 1,4 диоксан и диметилформамид. Соединение формулы VI, где Х 2 представляет собой галогеновую группу,такую как бромо, в подходящем растворителе взаимодействует с R1CH2OH в присутствии подходящего основания с получением соединения формулы IV. Подходящие основания включают гидрид натрия и трет-бутоксид калия. Подходящие растворители включают диметилсульфоксид и диметилформамид. Соединение формулы V, где X1 и Х 2 являются такими, как определено выше, можно получать, как описано в способах приготовления, или при помощи способов, известных в химической области получения структурно аналогичных соединений. Схема 2 В следующих иллюстративных примерах получения и примерах применяются следующие значения и сокращения: ДМСО, диметилсульфоксид (пердейтерированный [-d6], если для ЯМР (ядерный магнитный резонанс; ДСК, дифференциальная сканирующая калориметрия; МС, масс-спектр; EtOAc, этилацетат; ТГФ, тетрагидрофуран; мин, минуты; ч, часы; ВЭЖХ, высокоэффективная жидкостная хроматография; ЖХМС, ВЭЖХ-масс-спектрометрия; ГХ, газовая хроматография; ДМФ, диметилформамид; Et2O,диэтиловый эфир; ДХМ, дихлорметан; МеОН, метанол; МТБЭ, метил t-бутиловый эфир; SCX-2, катионообменная смола; Тпл, температура плавления; ЯМР, ядерная магнитно-резонансная спектроскопия или спектр; СФХ, сверхкритическая флюидная хроматография; ДМЭА, диметилэтиламин; и CHCl3, хлороформ. Реагенты получали из различных коммерческих источников. Как правило, растворители удаляли при пониженном давлении (выпаривали). В некоторых способах указанные выходы указывали для сырых продуктов, которые выделяли путем выпаривания или фильтрования и применяли непосредственно без дальнейшей очистки. Пример получения 1 Синтез 6-фтор-1-метил-1 Н-пирроло[2,3-b]пиридина К раствору 6-фтор-1 Н-пирроло[2,3-b]пиридина (250 г, 1,84 моль) в диметилформамиде (2,50 л) добавляли карбонат калия (507,6 г; 3,67 моль), а затем метилиодид (171,6 мл, 2,75 моль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в воду(3000 мл) и экстрагировали Et2O (31500 мл). Органические экстракты объединяли и промывали водой(41000 мл), а затем солевым раствором, и сушили над Na2SO4. Растворитель выпаривали с получением светло-коричневого масла, которое при хранении образовывало прозрачные бесцветные кристаллы с небольшим количеством подвижной жидкости поверх кристаллов. Жидкость декантировали и удаляли с получением продукта в виде кристаллического твердого вещества (257,3 г, 1,71 моль). 1H-ЯМР (400 МГц,CDCl3):7,93 (t, 1 Н), 7,11 (d, 1H), 6,69 (d, 1 Н), 6,46 (d, 1H), 3,83 (s, 3 Н). Пример получения 2 Синтез 3-бром-6-фтор-1-метилпирроло[2,3-b]пиридина К раствору 6-фтор-1-метил-1 Н-пирроло[2,3-b]пиридина (257,3 г, 1,71 моль) в ДХМ (3,86 л) добавляли N-бромсукцинимид (320,3 г; 1,80 моль) пятью порциями в течение 30 мин. Смесь перемешивали без нагревания или охлаждения в течение ночи, а затем фильтровали и концентрировали до приблизительно 1 л. Смесь очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 30%EtOAc в изогексане. Соответствующие фракции выпаривали с получением продукта в виде грязнобелого твердого вещества (391,3 г, 1,7 моль). 1H-ЯМР (400 МГц, CDCl3):7,91-7,87 (m, 1H), 7,15 (s, 1H),6,77 (d, 1H), 3,81 (s, 3H). Пример получения 3 Синтез 3-бром-1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридина Трет-бутоксид калия (137,4 г; 1,19 моль) и ДМФ (1,44 л) загружали в колбу и в течение 15 мин добавляли раствор 2-пиридилметанола (145,8 г, 1,34 моль) в ДМФ (306 мл). При необходимости колбу охлаждали для поддержания комнатной температуры. Смесь перемешивали при комнатной температуре в течение 40 мин. В течение 15 мин добавляли раствор 3-бром-6-фтор-1-метилпирроло[2,3-b]пиридина(170 г, 742,2 ммоль) в ДМФ (306 мл), поддерживая температуру между 20 и 25 С. Смесь перемешивали в течение 2 ч. К смеси медленно добавляли воду (1,7 л), при необходимости охлаждая, а затем экстрагировали EtOAc (41,0 л). Объединенные экстракты промывали водой (41,0 л), а затем солевым раствором, и сушили над сульфатом натрия, фильтровали и концентрировали с получением продукта в виде желтого твердого вещества (235,1 г, 0,74 моль). МС (m/z): 318,0/320,0. Пример получения 4 Синтез 3-бром-1-метил-6-(пиридин-4-илметокси)-1 Н-пирроло[2,3-b]пиридина К раствору 3-бром-6-фтор-1-метил-1H-пирроло[2,3-b]пиридина (0,4 г, 1,74 ммоль) и пиридин-4 илметанола (0,21 г, 1,93 ммоль) в ДМФ (5,0 мл) порциями добавляли гидрид натрия (0,05 г, 2,11 ммоль) при комнатной температуре и перемешивали полученную реакционную смесь в течение 1 ч. Реакцию гасили холодным солевым раствором и экстрагировали EtOAc (4100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали путем кристаллизации из смеси Et2O/пентан с получением титульного соединения (0,300 г, 0,942 ммоль) в виде оранжево-красного твердого вещества. МС (m/z): 318, 320 (М+1). 1H-ЯМР (400 МГц, ДМСО-d6):3,73 (s,3H), 5,48 (s, 2H), 6,76 (d, 1H), 7,47 (d, 2H), 7,50 (s, 1H), 7,78 (d, 1H), 8,56 (d, 2H). Следующие соединения получали, по существу, при помощи способа из примера получения 4. К раствору 2-пиперазинона (5,0 г, 50,0 ммоль) и триэтиламина (11,09 г, 110,0 ммоль) в ДХМ (15 мл) при комнатной температуре добавляли этилхлорформиат (5,9 г, 55,0 ммоль) и перемешивали реакционную смесь в течение 2 ч. Реакцию гасили водой (100 мл) и экстрагировали ДХМ (3100 мл). Объединенные органические слои сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме. Остаток растирали с Et2O с получением титульного соединения (5,0 г, 29,05 ммоль) в виде бледно-желтого твердого вещества. 1 Н-ЯМР (400 МГц, ДМСО-d6):1,19 (t, 3 Н), 3,16-3,19 (m, 2 Н),3,48-3,51 (m, 2 Н), 3,85 (s, 2H), 4,05 (q, 2H), 8,06 (s, 1H). Следующие соединения получали, по существу, при помощи способа из примера получения 13. К раствору 6-фтор-1 Н-пирроло[2,3-b]пиридина (6,2 г, 45,55 ммоль) в сухом ДХМ (250 мл) добавляли циклопропилборную кислоту (7,82 г, 91,09 ммоль), а затем ацетат меди (8,36 г, 45,55 ммоль), карбонат натрия (9,65 г, 91,09 ммоль) и 2,2'-бипиридин (7,11 г, 45,55 ммоль). Полученную смесь перемешивали и нагревали при 50 С в течение 15 ч. Смесь охлаждали до комнатной температуры и добавляли ацетат меди (4,18 г, 22,77 ммоль) и карбонат натрия (2,41 г, 22,77 ммоль), а затем циклопропилборную кислоту(1,96 г, 22,77 ммоль). Смесь перемешивали и нагревали при 50 С в течение дополнительных 15 ч, а затем добавляли ацетат меди (1,5 г, 8,25 ммоль) и циклопропилборную кислоту (1,49 г, 17,34 ммоль). Смесь перемешивали при комнатной температуре в течение 4 дней, а затем вливали в насыщ. вод. NH4Cl, разбавляли водой и экстрагировали ДХМ. Органические слои объединяли, промывали солевым раствором,сушили (сульфат магния) и концентрировали в вакууме с получением зеленого масла, которое очищали при помощи колоночной хроматографии на силикагеле, элюировали ДХМ с получением титульного соединения (2,03 г, 11,52 ммоль). МС (m/z): 177 (М+1). Непрореагировавший 6-фтор-1 Н-пирроло[2,3-b]пиридин также выделяли (3,012 г, 22,1 ммоль). МС (m/z): 137 (М+1). Пример получения 18 Синтез 3-бром-1-циклопропил-6-фтор-1H-пирроло[2,3-b]пиридина К раствору 1-циклопропил-6-фтор-1 Н-пирроло[2,3-b]пиридина (2,03 г, 11,52 ммоль) в ДМФ (38 мл) добавляли гидроксид натрия (0,506 г, 12,67 ммоль), а затем порциями в течение 5 мин добавляли N- 12021781 бромсукцинимид (2,26 г, 12,67 ммоль), что приводило к экзотермической реакции (28 С). Смесь перемешивали при комнатной температуре в течение 15 мин, а затем добавляли гидроксид натрия (46,1 мг, 1,15 ммоль) и N-бромсукцинимид (0,205 г, 1,15 ммоль). Перемешивание продолжали при комнатной температуре в течение 30 мин. Затем добавляли гидроксид натрия (46,1 мг, 1,15 ммоль) и N-бромсукцинимид(0,205 г, 1,15 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, а затем вливали в солевой раствор (прибл. 500 мл) и экстрагировали CHCl3 (прибл. 2300 мл). Органические слои объединяли и сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла, которое очищали при помощи колоночной хроматографии на силикагеле,элюировали от 0 до 100% ДХМ в изогексане с получением титульного соединения в виде белого порошка (2,32 г, 9,10 ммоль). МС (m/z): 255/257 (М+1). Пример получения 19 Синтез 3-бром-1-циклопропил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридина К раствору 3-бром-1-циклопропил-6-фтор-1 Н-пирроло[2,3-b]пиридина (0,249 г, 0,98 ммоль) в диметилсульфоксиде (5 мл) добавляли 2-пиридинметанол (188 мкл, 1,95 ммоль), а затем порциями добавляли гидрид натрия (97,6 мг, 2,44 ммоль). Смесь перемешивали при комнатной температуре в течение 5 мин, а затем вливали в солевой раствор и экстрагировали EtOAc. Органические слои объединяли, сушили(сульфат магния) и концентрировали в вакууме с получением коричневого масла. Смесь растворяли в метаноле и вливали в SCX-2 ионообменную колонку. Смесь промывали 3 объемами колонки МеОН, а затем продукт собирали после промывки 7 М раствором аммиака в метаноле с применением одного объема колонки. Затем раствор концентрировали в вакууме с получением титульного соединения в виде желтого масла (0,263 г, 0,76 ммоль). МС (m/z): 344/346 (М+1). Пример получения 20 Синтез трет-бутил-4-[1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилата Смесь 3-бром-1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридина (0,226 г, 0,71 ммоль),трет-бутил-3-оксопиперазин-1-карбоксилата (0,20 г, 1 ммоль), иодида меди (I) (0,027 г, 0,142 ммоль) и фосфата калия (0,212 г, 1 ммоль) продували в атмосфере азота в реакционной трубке. Добавляли 1,4 диоксан (3 мл) и N,N'-диметилэтилендиамин (0,031 мл, 0,288 ммоль), трубку плотно закрывали и реакционную смесь нагревали при 100 С в течение 25 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в воду и экстрагировали EtOAc. Органическую фазу сушили над сульфатом натрия,фильтровали и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле и элюировали смесью гексан/EtOAc (1:1), а затем чистым EtOAc с получением титульного соединения (0,275 г, 0,629 ммоль) в виде бледно-желтого твердого вещества. МС (m/z): 438 (М+1). Пример получения 21 Синтез гидрохлорида 1-[1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]пиперазин-2-она К раствору трет-бутил-4-[1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]-3 оксопиперазин-1-карбоксилата (0,278 г, 0,653 ммоль) в ДХМ (10 мл) добавляли насыщенный раствор соляной кислоты в EtOAc (6 мл) при комнатной температуре и перемешивали реакционную смесь при комнатной температуре в течение 67 ч. Растворитель осторожно декантировали и остаток растирали со смесью Et2O/MeOH. Растворитель снова декантировали и остаток опять растирали со смесьюEt2O/MeOH. Полученный остаток сушили в вакууме в течение 3 ч с получением титульного соединения К раствору циклобутанола (5,0 г, 69,4 ммоль) и пиридина (5,4 г, 69,4 ммоль) в ДХМ (30 мл) порциями добавляли трифосген (10,2 г, 34,7 ммоль) при 0 С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 3 ч. Реакцию гасили 10% водным раствором серной кислоты(100 мл) и экстрагировали ДХМ (5100 мл). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме с получением титульного соединения (4,1 г, 30,47 ммоль) в виде бесцветного вязкого масла, загрязненного исходным спиртом. Продукт применяли на следующей стадии без дополнительной очистки. 1H-ЯМР (400 МГц, CDCl3):1,57-1,64 (m, 2H), 2,06-2,16 К раствору 6-фтор-1 Н-пирроло[2,3-b]пиридина (15,00 г, 110,19 ммоль) в ДМФ (100 мл) в атмосфере азота при перемешивании добавляли карбонат калия (22,84 г, 165,3 ммоль), а затем этилбромид (12,36 мл, 165,3 ммоль). Реакцию нагревали до 70 С в течение 4 ч. Затем добавляли этилбромид (3,00 мл, 27,6 ммоль) и выдерживали реакцию при 70 С в течение ночи. После охлаждения добавляли карбонат калия(8,00 г, 57,9 ммоль) и этилбромид (3,00 мл, 27,6 ммоль) и реакцию нагревали при 70 С в течение 4 ч. Реакцию охлаждали, выливали в солевой раствор (прибл. 500 мл) и экстрагировали продукт CHCl3 (прибл. 2300 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 70% ДХМ в гексане с получением титульного соединения в виде светло-желтого масла (16,38 г, 99,77 ммоль). МС (m/z): 165 (М+1). Пример получения 24 Синтез 3-бром-1-этил-6-фтор-1 Н-пирроло[2,3-b]пиридина К раствору 1-этил-6-фтор-1H-пирроло[2,3-b]пиридина (16,38 г, 99,77 ммоль) в ДМФ (300 мл) охлаждали до 15 С при перемешивании. К смеси добавляли гидроксид натрия (4,39 г, 109,7 ммоль), а затем порциями в течение 5 мин добавляли N-бромсукцинимид (19,53 г, 109,7 ммоль), а затем реакцию оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь выливали в солевой раствор (прибл. 500 мл) и продукт экстрагировали CHCl3 (прибл. 2300 мл). Объединенную органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 60% ДХМ в гексане с получением титульного соединения в виде светло-желтого масла (23,4 г, 96,4 ммоль). МС (m/z): 243/245 (М+1). Пример получения 25 Синтез трет-бутил-4-(1-этил-6-фтор-1 Н-пирроло[2,3-b]пиридин-3-ил)-3-оксопиперазин-1-карбоксилата диоксан (250 мл) при перемешивании объединяли в атмосфере азота и нагревали с обратным холодильником в течение ночи. Реакцию охлаждали, выливали в солевой раствор (прибл. 500 мл) и продукт экстрагировали CHCl3 (прибл. 2300 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением желтого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 90% EtOAc в гексане с получением титульного соединения в виде оранжевого масла (20,039 г, 55,29 ммоль). МС (m/z): 363 (М+1). Пример получения 26 Синтез трет-бутил-4-[1-этил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилата К раствору трет-бутил-4-(1-этил-6-фтор-1H-пирроло[2,3-b]пиридин-3-ил)-3-оксопиперазин-1-карбоксилата (5,00 г, 13,80 ммоль) и 2-пиридинметанола (1,60 мл, 16,56 ммоль) в диметилсульфоксиде (50 мл) в атмосфере азота при перемешивании порциями добавляли 60% гидрид натрия (0,662 г, 16,56 ммоль). Реакцию перемешивали при комнатной температуре в течение 1 ч, а затем нагревали при 135 С в течение ночи. Реакцию охлаждали до комнатной температуры и добавляли 60% гидрид натрия (0,662 г, 16,56 ммоль) и реакцию перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в солевой раствор (прибл. 500 мл) и продукт экстрагировали CHCl3 (прибл. 2300 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 80% EtOAc в ДХМ с получением титульного соединения в виде оранжевой пены(4,15 мл, 54,84 ммоль) в течение 2 мин и реакцию перемешивали в течение 90 мин. Реакционную смесь разбавляли МеОН и вливали в SCX-2 ионообменную колонку. Смесь промывали МеОН одним объемом колонки, а затем продукт собирали после промывки 7 М раствором аммиака в метаноле с применением одного объема колонки. Затем раствор концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 40% МеОН в EtOAc с получением титульного соединения в виде оранжевого масла (2,989 г, 8,51 ммоль). МС К раствору 5-фтор-1H-пирроло[2,3-b]пиридина (5,00 г, 36,73 ммоль) в Et2O (120 мл) в атмосфере азота при перемешивании порциями в течение 5 мин добавляли 3-хлорпероксибензойную кислоту (11,09 г, 64,28 ммоль) и перемешивали реакцию в течение 3 ч. Затем реакцию охлаждали до 5 С, отфильтровывали и твердое вещество промывали Et2O (прибл. 100 мл). Продукт сушили в вакууме с получением титульного соединения в виде бледно-зеленого кристаллического твердого вещества (4,317 г, 28,38 ммоль). МС (m/z): 153 (М+1). Следующие соединения получали, по существу, при помощи способа из примера получения 28. К раствору 7-оксида 5-фтор-1 Н-пирроло[2,3-b]пиридина (4,317 г, 28,38 ммоль) в ТГФ (150 мл) при перемешивании добавляли гексаметилдисилазан (6,54 мл, 31,22 ммоль). Реакционную смесь охлаждали до 5 С и по каплям добавляли метилхлорформиат (5,49 мл, 70,94 ммоль). После перемешивания при 5 С в течение 3 ч по каплям добавляли 2 М гидроксид натрия (80 мл, 0,16 моль), поддерживая температуру ниже 10 С. Через 2 ч добавляли 2 М раствор соляной кислоты до рН 7. Реакционную смесь выливали в солевой раствор (прибл. 500 мл) и продукт экстрагировали CHCl3 (прибл. 4300 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением светло-коричневого твердого вещества (4,15 г, 24,33 ммоль). МС (m/z): 171/173 (М+1). Следующие соединения получали, по существу, при помощи способа из примера получения 31. К раствору 6-хлор-5-фтор-1 Н-пирроло[2,3-b]пиридина (4,15 г, 24,33 ммоль) в ДМФ (50 мл) в атмосфере азота добавляли карбонат калия (6,73 г, 48,66 ммоль), а затем метилиодид (2,27 мл, 36,49 ммоль) и реакционную смесь нагревали до 70 С в течение 2 ч. Реакционную смесь охлаждали, выливали в солевой раствор (прибл. 50 мл) и продукт экстрагировали CHCl3 (прибл. 230 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого твердого вещества. Продукт очищали при помощи колоночной хроматографии на силикагеле,элюировали от 0 до 50% ДХМ в гексане с получением титульного соединения в виде белого твердого вещества (1,274 г, 6,90 ммоль). МС (m/z): 185/187 (М+1). Следующие соединения получали, по существу, при помощи способа из примера получения 34. К раствору 6-хлор-5-фтор-1-метил-1 Н-пирроло[2,3-b]пиридина (1,276 г, 6,91 ммоль) и 2-пиридинметанола (0,800 мл, 8,29 ммоль) в диметилсульфоксиде (10 мл) в атмосфере азота порциями при перемешивании добавляли 60% гидрид натрия (0,332 г, 8,29 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Затем реакционную смесь нагревали до 80 С в течение 1 ч, охлаждали до комнатной температуры и добавляли 60% гидрид натрия (0,090 г, 2,32 ммоль). Через 30 мин перемешивания при комнатной температуре реакционную смесь дополнительно нагревали при 80 С в течение 30 мин. Реакционную смесь охлаждали, выливали в солевой раствор (прибл. 50 мл) и продукт экстрагировали CHCl3 (прибл. 230 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 80% EtOAc в гексане с получением титульного соединения в виде светло-зеленого масла (1,445 г, 5,62 ммоль). МС (m/z): 258 (М+1). Следующие соединения получали, по существу, при помощи способа из примера получения 37. К раствору 5-фтор-1-метил-6-(пиридин-2-илметокси)-1H-пирроло[2,3-b]пиридина (1,00 г, 3,89 ммоль) в ДМФ (30 мл), охлажденному до 15 С, в атмосфере азота при перемешивании добавляли гидроксид натрия(0,171 г, 4,28 ммоль), а затем порциями в течение 5 мин добавляли N-бромсукцинимид (0,761 г, 4,28 ммоль). Через 15 мин реакционную смесь выливали в солевой раствор (прибл. 50 мл) и продукт экстрагировали CHCl3(прибл. 230 мл). Объединенные органические экстракты сушили над сульфатом магния, фильтровали и концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 100% EtOAc в гексане с получением титульного соединения в виде светло-желтого твердого вещества (1,195 г, 3,55 ммоль). МС (m/z): 336/338 (М+1). Следующие соединения получали, по существу, при помощи способа из примера получения 40. Этил-3-оксопиперазин-1-карбоксилат (161,4 г, 937,2 ммоль), иодид меди (I) (27,65 г, 145,20 ммоль) и фосфат калия (трехосновный, n-гидрат) (205,1 г, 937,2 ммоль) загружали в стеклянный реактор при комнатной температуре в атмосфере азота. Добавляли раствор 3-бром-1-метил-6-(пиридин-2-илметокси)1 Н-пирроло[2,3-b]пиридина (210 г, 660,0 ммоль) в 1,4-диоксане (2,73 л), а затем N,N'-диметилэтан-1,2 диамин (24,34 г; 270,6 ммоль). Реакционную смесь нагревали до 100 С и перемешивали в течение 20 ч. Затем добавляли иодид меди (I) (10,06 г, 52,80 ммоль) и N,N'-диметилэтан-1,2-диамин (10,95 г, 105,6 ммоль), а затем реакционную смесь перемешивали в течение 23 ч. Реакционную смесь объединяли с меньшей партией, полученной аналогично 23,68 г 3-бром-1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридина. Смесь охлаждали до комнатной температуры, а затем выливали в воду (4,2 л) и экстрагировали EtOAc (31,7 л). Органические экстракты объединяли и промывали 3% мас./мас., водным аммиаком (3400 мл), а затем водой (22 л) и солевым раствором (600 мл) и сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле, элюировали от 50 до 100% EtOAc в изогексане. Соответствующие фракции объединяли, выпаривали и перекристаллизовывали из этанола (525 мл). Твердое вещество сушили с получением титульного соединения (125,6 г, 0,3 моль). МС (m/z): 410,1 (М+1). 1H-ЯМР (400 МГц, CDCl3):1,31(t, 3 Н), 3,72 (s, 3 Н), 3,80-3,78 (t, 2H), 3,86 (t, 2H), 4,22 (q, 2H), 4,34 (s, 2H), 5,59 (s, 2H), 6,71 (d, 1H), 7,01 (s,1H), 7,20 (dd, 1H), 7,49 (d, 1H), 7,70-7,66 (m, 2H), 8,60 (d, 1H). ДСК (старт) Тпл=143,42 С. Дополнительно продукт получали путем промывки хроматографической колонки большим количеством EtOAc и выпариванием соответствующих фракций. При помощи рекристаллизации остатка из этанола (100 мл) получили дополнительное количество титульного соединения (26,29 г, 64,26 ммоль). Альтернативный синтез этил-4-[1-метил-6-(пиридин-2-илметокси)-1H-пирроло[2,3-b]пиридин-3 ил]-3-оксопиперазин-1-карбоксилата К раствору гидрохлорида 1-[1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]пиперазин-2-она (0,10 г, 0,267 ммоль) и триэтиламина (160 мкл, 1,148 ммоль) в ДХМ (1,5 мл) по каплям добавляли этилхлорформиат (35 мкл, 0,366 ммоль) при комнатной температуре и реакционную смесь перемешивали в течение 1,5 ч. Реакционную смесь выливали в воду и экстрагировали ДХМ. После разделения органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали на силикагеле (10 г Isolute картридж), элюировали EtOAc с получением очень плотного бледно-желтого масла, которое затвердевало при добавлении Et2O. Продукт растирали в Et2O, отфильтровывали и дважды промывали Et2O с получением титульного соединения (0,066 г, 0,161 ммоль) в виде бесцветного твердого вещества. МС (m/z) 410 (М+1). Пример 2 Синтез метил-4-[1-метил-6-(пиридин-2-илметокси)-1H-пирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилата(0,467 г, 2,20 ммоль). Смесь дегазировали азотом в течение 15 мин, а затем добавляли иодид меди (I)(0,060 г, 0,31 ммоль) и N,N'-диметилэтилендиамин (0,055 г, 0,63 ммоль). Реакционный сосуд закрывали и нагревали при 100 С в течение 16 ч. Реакцию охлаждали до комнатной температуры, гасили водой (50 мл) и экстрагировали EtOAc (3100 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии на нейтральной окиси алюминия, элюировали 1% МеОН в ДХМ. Полученный продукт растирали со смесью Et2O/пентан (1:1) с получением титульного соединения (0,5 г, 1,26 ммоль) в виде грязно-белого твердого вещества. МС (m/z): 396 (М+1). 1H-ЯМР (400 МГц, ДМСО-d6):3,66 (s,- 18021781 3 Н), 3,68 (s, 3 Н), 3,75 (m, 4 Н), 4,14 (s, 2 Н), 5,49 (s, 2 Н), 6,66 (d, 1H), 7,30-7,33 (m, 2 Н), 7,50 (d, 1H), 7,777,82 (m, 2H), 8,56 (d, 1H). Следующие соединения получали, по существу, при помощи способа из примера 2.(0,179 г, 841 мкмоль) в ДМФ (10 мл) нагревали при 120 С в течение 15 ч. Смесь охлаждали до комнатной температуры, добавляли иодид меди (I) (0,145 г, 765 мкмоль), фосфат калия (трехосновный, n-гидрат)(0,536 г, 2,52 ммоль) и N,N'-диметилэтан-1,2-диамин (165 мкл, 1,53 ммоль), а затем этил-3-оксопиперазин-1-карбоксилат (0,132 г, 0,765 ммоль). После дополнительного нагревания в течение 24 ч в атмосфере азота при 100 С смесь концентрировали в вакууме, разбавляли МеОН и вливали в SCX-2 ионообменную колонку. Смесь промывали МеОН 3 объемами колонки, а затем продукт собирали после промывки 7 М раствором аммиака в метаноле с применением одного объема колонки. Раствор концентрировали в вакууме и очищали при помощи колоночной хроматографии на силикагеле, элюировали от 0 до 100% EtOAc в изогексане с получением титульного соединения в виде желтого масла (40 мг, 0,09 ммоль). МС (m/z): 436 (М+1). 1 Н-ЯМР (400 МГц, CDCl3): 0,89-0,99 (m, 4H), 1,31 (t, 3H), 3,31-3,37 (m, 1H), 3,76- 21021781 К раствору гидрохлорида 1-[1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин-3-ил]пиперазин-2-она (0,15 г, 0,40 ммоль) и триэтиламина (0,121 г, 1,20 ммоль) в ДХМ (15 мл) при комнатной температуре по каплям добавляли 2-фторэтилхлорформиат (0,076 г, 0,602 ммоль) и реакционную смесь перемешивали в течение 3 ч. Реакцию гасили насыщенным раствором бикарбоната натрия (20 мл) при 0 С и экстрагировали ДХМ (350 мл). Объединенные органические слои сушили над сульфатом натрия,фильтровали и концентрировали в вакууме. Остаток очищали на нейтральной окиси алюминия, элюировали 1% МеОН в ДХМ с получением продукта, который растирали в Et2O, с получением титульного соединения (0,07 г, 0,164 ммоль) в виде беловатого твердого вещества. МС (m/z): 428 (М+1). 1H-ЯМР (400 МГц, ДМСО-d6):3,68 (s, 3 Н), 3,77 (m, 4H), 4,17 (bs, 2H), 4,28 (t, 1H), 4,35 (t, 1H), 4,58 (t, 1H), 4,71 (t,1H), 5,49 (s, 2H), 6,66 (d, 1H), 7,30-7,34 (m, 2H), 7,50 (d, 1H), 7,77-7,83 (m, 2H), 8,56 (d, 1H). Следующие соединения получали, по существу, при помощи способа из примера 20.(0,200 г, 0,569 ммоль) в ДХМ (10 мл) при перемешивании добавляли триэтиламин (0,095 мл, 0,683 ммоль), а затем метилхлорформиат (0,052 мл, 0,683 ммоль). Затем реакционную смесь перемешивали в течение ночи. Реакционную смесь разбавляли МеОН и вливали в SCX-2 ионообменную колонку. Смесь промывали МеОН одним объемом колонки, а затем продукт собирали после промывки 7 М раствором аммиака в метаноле с применением одного объема колонки. Затем раствор концентрировали в вакууме с получением титульного соединения в виде оранжевого масла (0,1643 г, 0,401 ммоль). МС (m/z): 410(m, 2H), 8,60 (d, 1H). Следующие соединения получали, по существу, при помощи способа из примера 23. 3-Бром-5-фтор-1-метил-6-(пиридин-2-илметокси)-1 Н-пирроло[2,3-b]пиридин (0,233 г, 0,693 ммоль),этиловый эфир 3-оксопиперазин-1-карбоновой кислоты (0,131 г, 0,762 ммоль), N,N'-диметилэтан-1,2 диамин (0,031 мл, 0,284 ммоль), иодид меди (I) (0,029 г, 0,152 ммоль), фосфат калия (трехосновный, nгидрат) (0,162 г, 0,762 ммоль) и 1,4-диоксан (15 мл) совместно при перемешивании нагревали до кипения в атмосфере азота в течение ночи. Затем добавляли иодид меди (I) (0,120 г, 0,630 ммоль) и N,N'диметилэтан-1,2-диамин (0,120 мл, 1,099 ммоль) и выдерживали реакционную смесь при 105 С в течение дополнительных 5 ч. Реакционную смесь охлаждали, выливали в солевой раствор (прибл. 50 мл) и продукт экстрагировали CHCl3 (прибл. 330 мл). Объединенные органические экстракты разбавляли МеОН и вливали в SCX-2 ионообменную колонку. Смесь промывали МеОН одним объемом колонки, а затем продукт собирали после промывки 7 М раствором аммиака в метаноле с применением одного объема колонки. Затем раствор концентрировали в вакууме с получением коричневого масла. Продукт очищали при помощи сверхкритической флюидной хроматографии (RT (время удерживания) = 4,7 мин (УФ); СФХ колонка: бензолсульфонамид 21,2 мм 500 мм 5 мкм; градиент СО 2: 15-30% МеОН мас./0,2% ДМЭА в 5,5 мин, а затем повышали до 50% МеОН и удерживали в течение 3,5 мин; температура колонки: 40 С; скорость потока: 50,0 мл/мин) с получением оранжевого твердого вещества. Затем продукт очищали при помощи ВЭЖХ хроматографии (RT = 4,67 мин (УФ); ЖХ колонка: Waters Xbridge C18 100 мм 30 мм 5 мкм; Н 2 О мас./0,2% NH4HCO3; градиент: 9-100% ACN мас./0,2% NH4HCO3 в 6,0 мин, затем удерживали при 100% в течение 3,0 мин; температура колонки: 50 С; скорость потока: 3,0 мл/мин) с получением титульного соединения в виде белого твердого вещества (0,0621 г, 0,145 ммоль). МС (m/z): 428(q, 2H), 4,34 (br, 2 Н), 5,66 (s, 2 Н), 7,03 (s, 1H), 7,21 (t, 1H), 7,49 (d, 1H), 7,51-7,55 (m, 1H), 7,70 (td, 1H),8,60 (d, 1H). Следующие соединения получали, по существу, при помощи способа из примера 26. или его фармацевтически приемлемая соль, гдеR4 представляет собой водород, фтор, хлор или метил. 2. Соединение по п.1, или его фармацевтически приемлемая соль, отличающееся тем, что R1 представляет собой 2-пиридинил. 3. Соединение по любому из пп.1-2, или его фармацевтически приемлемая соль, отличающееся тем,что R2 представляет собой метил. 4. Соединение по любому из пп.1-3, или его фармацевтически приемлемая соль, отличающееся тем,что R3 представляет собой этил. 5. Соединение по любому из пп.1-4, или его фармацевтически приемлемая соль, отличающееся тем,что R4 представляет собой водород. 6. Соединение по п.1, которое представляет собой этил-4-[1-метил-6-(пиридин-2-илметокси)-1 Нпирроло[2,3-b]пиридин-3-ил]-3-оксопиперазин-1-карбоксилат или его фармацевтически приемлемую соль. 7. Фармацевтическая композиция для лечения болезни Паркинсона, содержащая соединение по любому из пп.1-6 или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель,разбавитель или наполнитель. 8. Способ лечения болезни Паркинсона, включающий введение пациенту, который в этом нуждается, эффективного количества соединения по любому из пп.1-6 или его фармацевтически приемлемой соли. 9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли в терапии заболеваний, связанных с активностью mGluR5. 10. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для получения лекарственного средства для лечения болезни Паркинсона. 11. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли для лечения болезни Паркинсона. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: A61K 31/437, C07D 471/04, A61P 25/16
Метки: соединения, 3-замещенные, 6-(пиридинилметокси)пирролопиридиновые
Код ссылки
<a href="https://eas.patents.su/25-21781-3-zameshhennye-6-piridinilmetoksipirrolopiridinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">3-замещенные 6-(пиридинилметокси)пирролопиридиновые соединения</a>
Четырехциклические конденсированные соединения с гетероатомами, замещенные арилом, промежуточные соединения, способы получения, композиции и методы лечения

Номер патента: 1649
Опубликовано: 25.06.2001
Автор: Гриз Тимоти А.
МПК: A61K 31/35, C07D 471/00, A61P 5/30...
Метки: получения, четырехциклические, лечения, методы, замещенные, гетероатомами, промежуточные, конденсированные, соединения, композиции, способы, арилом
Формула / Реферат:
1. Соединение формулы I или II где Y обозначает -О-, -S-, -СН2-, -СН2СН2-, -СН=СН- или -NR4-; В обозначает -СН2- или -СО-; R1 и R2 обозначают каждый независимо -Н, -ОН, -O(C1-C4алкил), -ОСОС6Н5, -ОСО(С1-С6алкил), -ОSО2(С4-С6алкил), OSO2СF3, Сl или F; n равно 1 или 2; W обозначает -СН2- или >С=O; R3 обозначает 1-пиперидинил, 2-оксо-1-пиперидинил, 1-пирролидинил, метил-1-пирролидинил, диметил-1-пирролидинил, 2-оксо-1-пирролидинил,...
Замещенные трициклические соединения.

Номер патента: 2347
Опубликовано: 25.04.2002
Авторы: Хайт Гари Алан, Бах Николас Джеймс, Морин Джон Майкл, Василефф Роберт Теодор, Харн Нэнси Кей, Сойер Джейсон Скотт, Харпер Ричард Вальтц, Бастиан Жоли Анн, Макджилл Джон Макнейлл, Шевитц Ричард Вальтер, Лин Хо-Шен, Михелик Эдвард Дэвид, Ричетт Майкл Энрико, Лончарик Ричард Джеймс, Филлипс Майкл Лерой, Кинник Майкл Дин, Андерсон Бенджамин Алан, Солл Дэниел Джон
МПК: C07C 229/48, A61K 31/403, A61P 29/00...
Метки: трициклические, замещенные, соединения
Формула / Реферат:
1. Соединение формулы (I) где Z представляет собой циклогексенил или фенил; R20 представляет собой бензил, необязательно замещенный одним или двумя заместителями, выбранными из группы: галоген, низший алкил, фенокси, бензил, -ОСF3 или пиридилметил, циклогексилметил, циклопентилметил, нафтилметил; R21 представляет собой СF3фенил; R1 представляет собой -NHNH2, -NH2 или -CONH2; R2' выбран из группы, включающей -ОН и -O(CH2)tR5', где R5' обозначает...
Замещенные 2-бензиламино-2-фенилацетамидные соединения

Номер патента: 3097
Опубликовано: 26.12.2002
Авторы: Варази Марио, Певарелло Паоло, Сальвати Патричия, Пост Клаес
МПК: C07C 237/20, A61K 31/16, A61P 25/00...
Метки: соединения, 2-бензиламино-2-фенилацетамидные, замещенные
Формула / Реферат:
1. Соединение, представляющее замещенный 2-бензиламино-2-фенилацетамид формулы (I) в которой n равно нулю, 1, 2 или 3; X представляет -О-, -S- -СН2- или -NH-; каждый из R, R1, R2 и R3 независимо представляет водород, C1-С6алкил, галоген, гидрокси, C1-С6алкокси или трифторметил; каждый из R4 и R5 независимо представляет водород, C1-С6алкил, или C3-С7циклоалкил; или его фармацевтически приемлемую соль. 2. Соединение по п.1, в котором n равно 1...
Замещенные трициклические соединения.

Номер патента: 3129
Опубликовано: 27.02.2003
Авторы: Мартинелли Майкл Джон, Бастиан Жоли Анн, Ванг Кьюпинг, Бах Николас Джеймс, Смит Эдвард С.Р., Солл Дэниел Джон, Сойер Джейсон Скотт, Кинник Майкл Дин, Вильсон Томас Майкл, Михелик Эдвард Дэвид, Бейт Дуглас Вейд, Суарес Тулио
МПК: A61K 31/40, C07D 209/88
Метки: соединения, трициклические, замещенные
Формула / Реферат:
1. Соединение, выбранное из группы, включающей (R,S)-(9-бензил-4-карбамоил-1-оксо-3-тиа-1,2,3,4-тетрагидрокарбазол-5-ил)оксиуксусную кислоту; (R,S)-(9-бензил-4-карбамоил-3-тиа-1,2,3,4-тетрагидрокарбазол-5-ил)оксиуксусную кислоту; [N-бензил-1-карбамоил-1-аза-1,2,3,4-тетрагидрокарбазол-8-ил]оксиуксусную кислоту; 4-метокси-6-метоксикарбонил-10-фенилметил-6,7,8,9-тетрагидропиридо[1,2-a]индол;...
Замещенные соединения бифенила, способ их получения и фармацевтические композиции содержащие их

Номер патента: 5766
Опубликовано: 30.06.2005
Авторы: Ренар Пьер, Гийом Жеральд, Делагранж Филипп, Йо Саид, Вио Мари-Клод, Бенжан Каролин, Лесьё Даниэль, Декамп-Франсуа Кароль, Да Коста Эрве
МПК: A61K 31/165, C07C 233/18, C07D 307/81...
Метки: бифенила, содержащие, способ, получения, фармацевтические, замещенные, композиции, соединения
Формула / Реферат:
1 Соединения формулы (I) в которой B обозначает атом водорода, COOR-группу или CH2OR-группу (в которых R обозначает атом водорода или линейную или разветвленную (C1-C6)алкильную группу), G1 обозначает -X'-(CH2)n-X-(CH2)m-X"-цепь, в которой X обозначает CH2-группу, X' и X" каждый обозначает атом кислорода, n и m, которые могут быть одинаковыми или разными, каждый представляет 0, 1, 2 или 3, Cy обозначает группу формулы (II) в которой D...
Предыдущий патент: Комплексная система очистки биогаза с целью удаления воды, силоксанов, серы, кислорода, хлоридов и летучих органических соединений
Следующий патент: Цементно-стружечная плита
Случайный патент: Смесь ферментов, выделенных из бактерий clostridium histolyticum