Способ получения производных бисфуранового спирта, соединения, соль, фармацевтическая композиция на ее основе, способ лечения или профилактики ретровирусных инфекций с ее помощью
Номер патента: 20088
Опубликовано: 29.08.2014
Авторы: Гутиерес Арнольд, Юй Ричард Хун Чуй, Полняшек Ричард П., Кроуфорд Кеннет Р., Дауди Эрик Д.







Формула / Реферат
1. Соединение, имеющее структурную формулу С

или его фармацевтически приемлемая соль.
2. Соединение, имеющее структурную формулу М

или его фармацевтически приемлемая соль.
3. Соединение, имеющее структурную формулу N

или его фармацевтически приемлемая соль.
Текст
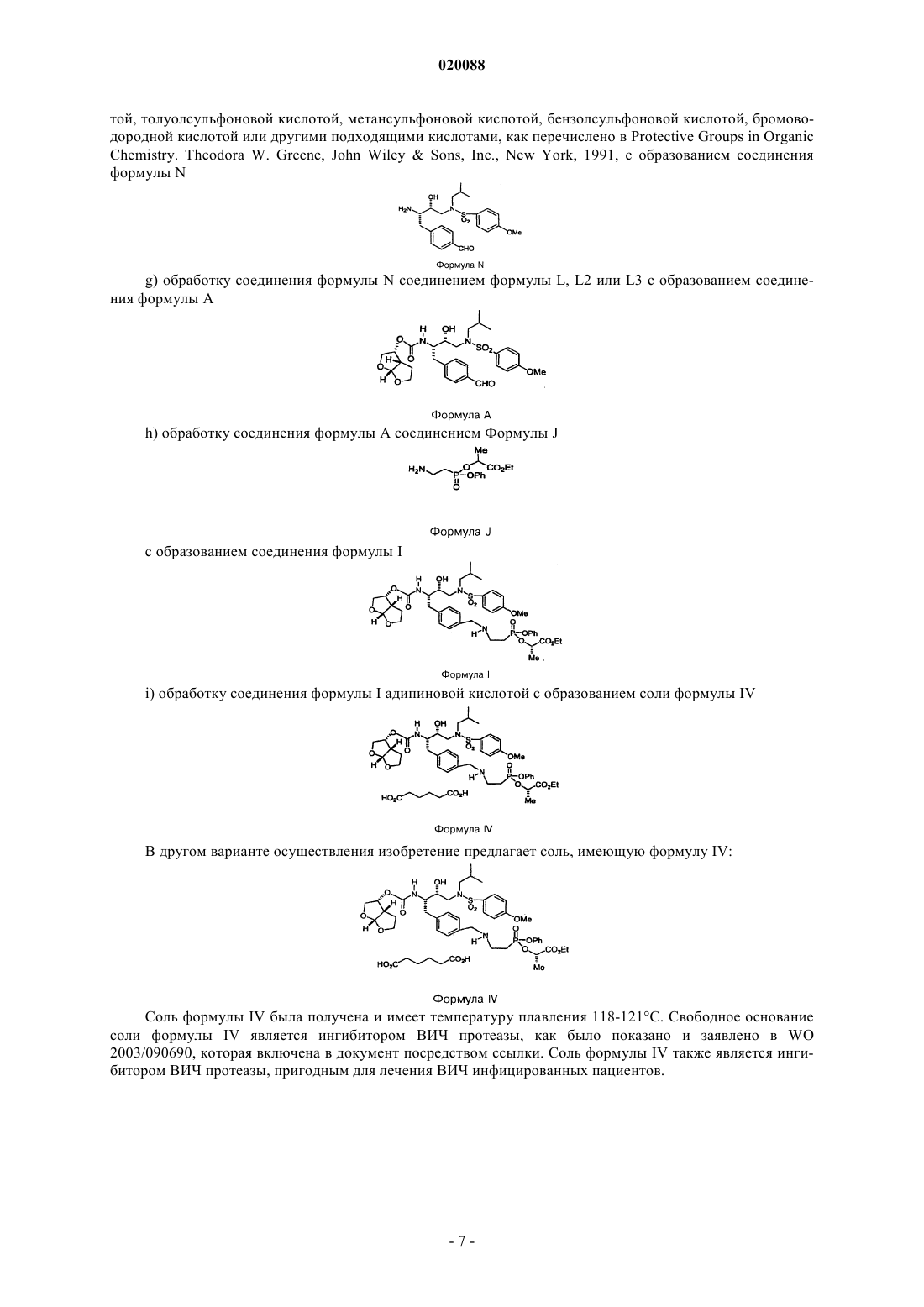
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БИСФУРАНОВОГО СПИРТА,СОЕДИНЕНИЯ, СОЛЬ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ЕЕ ОСНОВЕ,СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ РЕТРОВИРУСНЫХ ИНФЕКЦИЙ С ЕЕ ПОМОЩЬЮ Предложен способ синтеза бисфурановых промежуточных соединений, полезных для получения противовирусных ингибиторов протеазы ВИЧ. Кроуфорд Кеннет Р., Дауди Эрик Д.,Гутиерес Арнольд, Полняшек Ричард П., Юй Ричард Хун Чуй (US) Нилова М.И. (RU) Область техники, к которой относится изобретение Изобретение относится в основном к получению противовирусных соединений со свойствами против ВИЧ протеазы. Изобретение относится к способам получения эфиров карбаматсульфонамидаминофосфорной кислоты и их интермедиатов. Изобретение также отностися к новым интермедиатам, полученным данными способами. Эфиры карбаматсульфонамидаминофосфорной кислоты, полученные представленными способами, являются ингибиторами ВИЧ-протеазы, которые могут использоваться для лечения человеческого синдрома приобретенного иммунодефицита (СПИД). Сведения о предшествующем уровне техники СПИД является огромной проблемой общественного здоровья во всем мире. Хотя лекарства, направленные на вирусы ВИЧ, широко используются и уже показали свою эффективность, их применение ограничивается токсичностью и появлением устойчивых штаммов. Способы анализа, способные определять наличие, отсутствие или количество вирусов ВИЧ, используются на практике как для поиска ингибиторов, так и для диагностики наличия ВИЧ. Традиционный способ для получения ингибитора ВИЧ протеазы (ИП) общей формулы I является длительным с низким выходом около 1% и непостоянной воспроизводимостью, требует многочисленных стадий хроматографической очистки, и использует нежелательные реагенты, такие как озон, цианоборгидрид натрия и гидрид трибутилолова. Соединения формулы I являются ингибиторами ВИЧ протеазы, что было показано и заявлено в WO 2003/090690. Способы получения бисфурановых спиртов как интермедиатов, используемых при синтезе соединений формулы I, были описаны Pezechk (Pezechk, M. Et.al., Tetrahedron Letters, 1986, 27, 3715) и Ghosh(Ghosh, А.К. et. al., J. Med. Chem., 1994, 37, 2506; Ghosh, A.K. et. al., J. Med. Chem., 1996, 39, 3278; Ghosh,A.K. et. al., Tetrahedron Letters, 1995, 36, 505). На схеме 1 показан синтез бисфурановых спиртов по Традиционный способ включает много стадий и использует токсичные реагенты. По одному из этих способов (Ghosh, A.K. et. al., Tetrahedron Letters, 1995, 36, 505) разделение рацемической смеси достигалось обработкой иммобилизованным ферментом с последующим хроматографическим разделением. Реактивные карбонатные эфиры были получены из бисфуранового спирта (I) и дипиридилкарбонатаand Medicinal Chemistry Letters, 1996, 6, 2847). Эти реагенты вводились в реакцию с соответствующими нуклеофилами, но не всегда проявляли достаточную реактивность и эффективность. Существуют способы для получения хиральных галоспиртов из N-защищенных аминокислот (Albeck, A. et al., Tetrahedron, 1994, 50, 6333). Способы превращения таких хлорсодержащих спиртов в карбаматные производные сульфонамида известны (Malik, A. et al., WO 01/46120AI). Подобным образом,галогидрины также могут быть превращены в эпоксиды и затем в карбаматные производные сульфонамида (WO 03/090690). Получение карбаматных производных аминофосфорных кислот и их последующее превращение в моно- и диэфиры фосфорных кислот было описано в Yamauchi, K. et al., J. Org. Chem.,1984, 49, 1 158; Yamauchi, K. et al., J. Chem. Soc. Perkin Trans. 1,1986, 765. Диэфиры аминоэтилфосфорной кислоты могут быть получены способом, включающим ацилирование аминофосфорной кислоты ацилгалидом или бензилхлорформиатом (CBZCI), дающим соединения формулы VII Соединения формулы VII могут быть активированы и сконденсированы с фенолом, образуя соединения формулы VIII Соединения формулы VIII могут быть активированы и сконденсированы с вторым спиртом или фенолом, образуя IX Соединение формулы IX может быть деацилировано, образуя аминофосфонат формулы X Соединение формулы X может быть выделено в виде соли органической или неорганической кислоты. Способ Ghosh для бисфурановых спиртов (Ghosh, A.K. et al, J. Org. Chem., 1995, 36, 505) требует использование гидрида трибутилолова и озона. Соединение формулы I в виде свободного основания является некристаллическим и гигроскопичным с ограниченной стабильностью в протонных растворителях. Таким образом, существует потребность в разработке синтеза более стабильных форм ИП формулыI. Также существует потребность в разработке более эффективных способов синтеза ИП формулы I. Сущность изобретения Изобретение предлагает усовершенствованные способы получения производных бисфурановых спиртов, производных аминофосфонатов и способ получения диэфиров карбаматсульфонамидаминоэтилдиэтилфосфорной кислоты, применимых для лечения синдрома приобретенного иммунодефицита человека (СПИД). В одном из вариантов осуществления изобретение предлагает способ получения бисфуранового спирта формулы 0: включающий в себя реакцию между 2,3-дигидрофураном и гликолевым альдегидом или димером гликолевого альдегида в присутствии лантаноида или переходного металла в качестве катализатора с образованием бисфуранового спирта формулы 0. Сведения, подтверждающие возможность осуществления изобретения Если не указано иного, следующие термины и фразы, используемые в данном документе, подразумевают следующие значения. Когда используются торговые названия, заявители намерены независимо включать продукт торговой марки и активные фармацевтические ингредиенты продукта торговой марки."Защитная группа" означает фрагмент соединения, который маскирует или изменяет свойства функциональной группы или свойства соединения в целом. Химические защитные группы и стратегии защиты/снятия защиты хорошо известны специалистам. См. например Protective Groups in Organic Chemistry, Theodora W. Greene, John WileySons, Inc., New York, 1991. Защитные группы часто используются для маскировки реактивности определенных функциональных групп с целью способствования эффективности целевой химической реакции, например, создание и разрыв химических связей в регулярном и запланированном порядке. Защита функциональных групп в соединении кроме реактивности защищен-2 020088 ной функциональной группы, изменяет другие физические свойства, такие как полярность, липофильность (гидрофобность) и другие свойства, которые могут быть измерены обычными аналитическими способами. Химически защищенные интермедиа могут в свою очередь быть биологически активными или неактивными. Термин "хиральный" относится к молекулам, которые обладают свойством несовместимости со своим зеркальным изображением, в то время как "ахиральный" относится к молекулам, которые накладываются на свое зеркальное отображение. Термин "стереоизомеры" относится к соединениям, которые имеют одинаковое химическое строение, но отличаются расположением атомов или групп в пространстве."Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, и молекулы которых не являются зеркальными отображениями друг друга. Диастереомеры обладают разными физическими свойствами, такими как температура плавления, температура кипения, спектральными свойствами и реактивностью. Смеси диастереомеров могут быть разделены аналитическими способами высокого разрешения, такими как электрофорез и хроматография. Термин "энантиомеры" относится к двум стереоизомерам соединения, которые несовместимы с зеркальными отображениями друг друга. "Лантаноиды" относятся к следующим элементам и их ионам:"Переходные металлы" относятся к следующим элементам и их ионам: Sc, Ti, V, Cr, Mn, Fe, Co, Ni,Cu, Zn, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, Hf, Та, W, Re, Os, Ir, Pt, Au, Hg. Лиганды, входящие в металл-содержащий катализатор, могут быть хиральными, ахиральными или рацемическими. Схемы и примеры Общие аспекты этих иллюстративных способов описаны ниже и в примерах. Каждый из продуктов следующих способов необязательно отделяется, выделяется, и/или очищается перед его использованием в последующих процедурах. Реакции окисления и восстановления, как правило, проводятся при температурах, близких к комнатной (около 20 С), хотя при восстановлении металлическими гидридами температуру часто понижают до 0 - (-100)С, растворители, как правило, являются апротонными для восстановления, но могут быть либо протонными, либо апротонными для окисления. Время реакции подбирают для достижения желаемой конверсии. Реакции конденсации, как правило, проводят при температурах, близких к комнатной, хотя неравновесные, кинетически контролируемые конденсации часто также проводят при пониженных температурах (от 0 С до -100 С). Растворитель может быть либо протонным (обычно для равновесных реакций),либо апротонный (обычно для кинетически контролируемых реакций). Стандартные синтетические процедуры, такие как азеотропная отгонка побочных продуктов реакции и использование безводных условий реакции (например, среда инертного газа), являются обычными в данной области и будут применяться, когда эти возможно. Термины "обработанный", "которым обрабатывают", "обработка" и подобные при использовании в связи с химической синтетической операцией означают контакт, смешивание, реагирование, позволение реагировать, приведение в контакт и другие термины, обычные в этой области для обозначения того, что один или более одного химических компонентов обработаны таким образом, чтобы превратить его (их) в один или более одного других химических компонентов. Это означает, что "обработка соединения один соединением два" является синонимом выражениям "позволение соединению один реагировать с соединением два", "контактирование соединения один с соединением два", "реагирование соединения один с соединением два" и другим выражениям, корректно употребляемым в области органического синтеза для обозначения того, что соединение один было "обработано", "прореагировало", "оставлено реагировать" и т.п. с соединением два. Например, обработка обозначает корректный и разумный способ, при котором органическим химическим реагентам позволяется реагировать. Если не указано другого, подразумеваются нормальные концентрации (от 0,01 М до 10 М, обычно от 0,1 М до 1 М), температуры (от -100 до 250 С, обычно от -78 до 150 С, наиболее обычно от -78 до 100 С, еще более обычно от 0 до 100 С), реакционные сосуды (обычно стеклянные, пластиковые или металлические), растворители, давление, среды (обычно воздушная для реакций, не чувствительных к кислороду и воде, или азот или аргон для реакций, чувствительных к кислороду или воде) и т.п. При выборе условий и аппаратуры для данного процесса используются знания о подобных реакциях, известных в области органического синтеза. В частности, человек, обладающий обычными навыками органического синтеза, подберет условия и аппаратуру для вероятного успешного выполнения химических реакций, основываясь на знаниях в этой области. В каждой иллюстративной схеме может быть целесообразным разделение продкутов реакции друг от друга и/или от исходных материалов. Целевой продукт на каждой стадии или серии стадий выделяют и/или очищают (в дальнейшем выделяют) до желаемой степени гомогенности способами, известными в данной области. Обычно такое выделение включает многоступенчатую экстракцию, кристаллизацию из растворителя или смеси растворителей, перегонку, сублимацию или хроматографию. Хроматография может использовать любой набор способов, включающих, например, хроматографию на нормальной или обращенной фазе, гель-фильтрацию, ионобменную хроматографию, высоко, умеренно и низко эффективные жидкостные хроматографические способы и аппаратуру, аналитическую с небольшими загрузками, противоточную хроматографию псевдоподвижного слоя и препаративную толсто- и тонкослойную, а также способы тонкослойной и флэш-хроматографии на небольших загрузках. Другой класс способов разделения включает обработку смеси реагентом, селективно связывающимся с целевым продуктом или другим способом, делающим его выделяемым, но не связывающимся с исходными материалами, побочными продуктами реакции и т.п. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ион-обменные смолы и так далее. В качестве альтернативы, реагент может быть кислотой - в случае основных материалов, основанием - в случае кислых материалов, связывающим реагентом, таким как антитела, связывающимися белками, селективными хелаторами, такими как краун-эфиры, жидкостно-жидкостные ион-экстрагирующие реагенты и так далее. Отдельный стереоизомер, например, энантиомер, практически свободный от своего стереоизомера,может быть получен путем разделения рацемической смеси с использованием такого способа, как образование диастереомеров при действии оптически активных разделяющих реагентов (Stereochemistry ofCarbon Compounds. (1962) by E. L Eliel, McGraw Hill; Lochmuller, С. Н., (1975) J. Chromatogr., 113:(3) 283302). Рацемические смеси хиральных соединений данного изобретения могут быть разделены и изолированы любым подходящим способом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение дробной кристаллизацией или другими способами, (2) образование диастереомерных соединений с хиральным модифицирующими реагентами, разделение диастереомеров и превращение производных в чистые стереоизомеры и (3) прямое разделение достаточно чистых или обогащенных стереоизомеров в хиральных условиях. Иллюстративные варианты осуществления В одном варианте осуществления, изобретение предлагает соединение формулы С и его фармацевтически приемлемые соли: В другом варианте осуществления, изобретение предлагает способ получения соединения формулы М, включающий в себя: а) обработку соединения формулы Е таким амином, как 1-амино-2-метилпропан с образованием соединения формулы Fb) обработку соединения формулы F соединением формулы G с образованием соединения формулы С с) обработку соединения формулы С восстанавливающим агентом с образованием соединения формулы М Типичный восстанавливающий агент, который может быть использован для превращения нитриль-4 020088 ной группы в карбальдегидную группу может быть найден в Larock, Richard, С. "Comprehensive OrganicTransformations 2nd Ed. 1999 John Wiley and Sons publisher, pages 1271-1272. В другом варианте осуществления изобретение предлагает соединение В другом варианте осуществления изобретение предлагает способ получения соединения формулы М: включающий в себя обработку соединения формулы С восстанавливающим реагентом с образованием соединения формулы М В другом варианте осуществления изобретение предлагает способ получения соединения формулы М, где восстанавливающим реагентом служит диизобутилалюминийгидрид. В другом варианте осуществления изобретение предлагает способ получения соединения формулы 0, включающий в себя обработку бисфуранового спирта формулы 0 дисукцинимидилкарбонатом с образованием соединения формулы L1 В другом варианте осуществления изобретение предлагает способ получения соединения формулы 0, включающий в себя обработку бисфуранового спирта формулы 0 бис(п-нитрофенил)карбонатом или п-нитрофенолхлорформиатом с образованием соединения формулы L2 В другом варианте осуществления изобретение предлагает способ получения соединения формулы 0, включающий в себя обработку бисфуранового спирта формулы 0 дипиридилкарбонатом с образованием соединения формулы L3 В другом варианте осуществления изобретение предлагает соединение, имеющее формулу N, и его фармацевтически приемлемые соли В другом варианте осуществления изобретение предлагает соединение, имеющее формулу А, и его В другом варианте осуществления изобретение предлагает способ получения эфиров карбаматсульфонамидаминофосфорной кислоты, включающий в себя: а) прибавление дигидрофурана к димеру гликолевого альдегида в присутствии Yb, Pr, Cu, Eu или SC в качестве катализатора с образованием бисфуранового спирта формулы 0b) обработку продукта стадии (а) дисукцинимидилкарбонатом, бис(п-нитро)фенилкарботатом, пнитрофенолхлорформиатом или дипиридил карбонатом с образованием соединения формулы L1, формулы L2, или формулы L3 формулы L соответственно, с) обработку соединения формулы Е амином с образованием соединения формулы Fd) обработку соединения формулы F соединением формулы G с образованием соединения формулы С е) обработку соединения формулы С восстанавливающим реагентом с образованием соединения формулы Мf) снятие защитной группы в соединении формулы М трифторуксусной кислотой, соляной кисло-6 020088 той, толуолсульфоновой кислотой, метансульфоновой кислотой, бензолсульфоновой кислотой, бромоводородной кислотой или другими подходящими кислотами, как перечислено в Protective Groups in OrganicChemistry. Theodora W. Greene, John WileySons, Inc., New York, 1991, с образованием соединения формулы Ng) обработку соединения формулы N соединением формулы L, L2 или L3 с образованием соединения формулы Аh) обработку соединения формулы А соединением Формулы J с образованием соединения формулы Ii) обработку соединения формулы I адипиновой кислотой с образованием соли формулы IV В другом варианте осуществления изобретение предлагает соль, имеющую формулу IV: Соль формулы IV была получена и имеет температуру плавления 118-121 С. Свободное основание соли формулы IV является ингибитором ВИЧ протеазы, как было показано и заявлено в WO 2003/090690, которая включена в документ посредством ссылки. Соль формулы IV также является ингибитором ВИЧ протеазы, пригодным для лечения ВИЧ инфицированных пациентов. Таблица 1. Хиральные катализаторы при образовании бисфуранового спиртаDNA - данные не доступны ГХ анализ был выполнен при превращении бисфуранового спирта в трифторацетат действием трифторуксусного ангидрида в ДХМ. Таблица 2. Применение скандиевого (III) катализатора и хиральных лигандов для прямого получения (-)-1NA - не доступно ГХ анализ был выполнен при превращении бисфуранового спирта в трифторацетат действием трифторуксусного ангидрида в ДХМ. Таблица 3. Применение катализаторов и хиральных лигандов для прямого получения (-)-1 ГХ анализ был выполнен при превращении бисфуранового спирта в трифторацетат действием трифторуксусного ангидрида в ДХМ. Таблица 4. Применение колоночного способа для энантиомерного расщепления (+)-бисфуранового спирта Схема 1. Способ, используемый для получения бисфуранкарбонатов из бисфуранового спирта с использованием нового способа синтеза бисфурановых спиртов Схема 2. Аминирование хлоргидринов до ВОС(OBn)тирозина Схема 3. Реакция продкута, полученного по схеме 2, с бисфуранкарбонатом, полученный, как описано на схеме 1 Теперь изобретение будет проиллюстрировано следующими неограничивающими примерами. Схема 8. Кинетический гидролиз бисфуранацетата, вызываемый липазой Получение (3R,3aS,6aR) гексагидрофуро[2,3-b]фуран-3-ола (1). В реакционный сосуд загружают димер гликолевого альдегида (4,45 кг), катализатор Yb(fod)3 (0,29 кг) и дигидрофуран (20,5 кг). Содержимое перемешивают и нагревают до 50 С в течение приблизительно 5 ч. Реакционную смесь концентрируют до сырого масла, растворяют в насыщенном водном раствореNaHCO3 (60 кг) и промывают дихлорметаном (6 кг). К водному слою прибавляют дихлорметан (58 кг),KBr (0,89 кг), TEMPO (0,116 кг) и смесь охлаждают до 0 С. К этой смеси медленно добавляют гипохло- 15020088 рит натрия (NaOCl, приблизительно 11% Cl, 55 кг). По окончании реакции органическому и водному слоям позволяют разделиться. Водный слой промывают дихлорметаном (29 кг). Органические слои объединяют и промывают водой, 10% HCl с Kl и 10% тиосульфатом натрия. Органический слой высушивают над сульфатом натрия, отфильтровываюттвердое вещество, и фильтрат охлаждают до температуры ниже 0 С. Поддерживая температуру реакционной смеси ниже 0 С, добавляют раствор боргидрида натрия (0,36 кг) в этаноле (7,1 кг). По завершении реакции для ее подавления добавляют уксусную кислоту(1,4 кг) и воду (13,4 кг). Смесь концентрируют вакуумной перегонкой. К полученной сырой маслообразно-полутвердой смеси добавляют этилацетат (31 кг). Органический слой высушивают над сульфатом натрия, отфильтровывают твердое вещество и концентрируют путем вакуумной перегонки, выделяя -1(рацемат гексагидрофуро[2,3-b]фуран-3-ола) в виде масла. Ферментативное разрешение. Сырой маслообразный продукт обрабатывают диметиловым эфиром этиленгликоля (DME, 14,7 кг) и уксусным ангидридом (4,6 кг). Этот раствор многократно пропускают через колонку, набитую смесью Липазы PS-C"Amano I" (0,36 кг) и песка (6 кг). По окончании энантиомерного разрешения, раствор концентрируют путем вакуумной перегонки. Для растворения продукта добавляют воду (18 кг) и раствор промывают дихлорметаном (28 кг). Продукт, содержащий водный слой, концентрируют путем вакуумной перегонки. Полученное масло растворяют в этилацетате (16 кг) и сушат над сульфатом натрия. Дополнительное количество продукта может быть выделено повторной экстракцией дихлорметанового слоя водой несколько раз. Объединенные водные фракции концентрируют путем вакуумной перегонки. Полученное масло растворяют в этилацетате, сушат над сульфатом натрия и отфильтровывают твердое вещество. Объединенные этилацетатные фракции концентрируют путем вакуумной перегонки, получая продукт (3R, 3aS,6aR) гексагидрофуро[2,3-b]фуран-3-ол, (-)-(1) в виде масла (1,6 кг, 97% ее, выход 33%), загрязненный примерно 15 весовыми % соответствующего ацетата. Аналитические даные: 1 Н ЯМР (ДМСО-d6, 300 МГц)5,52 (дд, 1 Н), 4,25-4,15 (м, 1 Н), 3,85-3,75 (м, 2 Н), 3,7-3,6 (м, 1 Н), 3,3 (т, 1 Н), 2,75-2,65 (м, 1 Н),2,23- 2,13 (м, 1 Н), 1,75-1,6 (м, 1 Н). Получение (3R,3aS,6aR)гексагидрофуро[2,3-b]фуран-3-ил 4-нитрофенилкарбоната. В реакционный сосуд помещают бис(4-нитрофенил)карбонат (2,58 кг) и дихлорметан (33,4 кг). К этому раствору добавляют (3R, 3aS, 6aR) гексагидрофуро[2,3-b]фуран-3-ол, (-)-1 (1,2 кг, 98,5% ее, загрязненный приблизительно 36% ацетата), растворенный в дихлорметане (6,7 кг). Добавляют триэтиламин (1,6 кг), и полученная реакционная смесь перемешивается при 20-25 С. По окончании реакции содержимое промывают водой (16,8 кг). Слои разделяют и дихлорметановый слой концентрируют путем вакуумной перегонки. Продукт, содержащий масло, растворяют в этилацетате (21,2 кг) и последовательно промывают водой, водным карбонатом калия и насыщенным солевым раствором. Этилацетатную фракцию сушат над сульфатом натрия, отфильтровывают твердое вещество и фильтрат концентрируют путем вакуумной перегонки. Концентрированную смесь продуктов растворяют в этилацетате (9,3 кг) и нагревают до 45 С. Медленно добавляют гексан (6,7 кг) и конечную смесь медленно охлаждают до 0 С. Полученный мутный раствор отфильтровывают, получая 12 (конечный продукт реакции, показанной на стр. 35). Твердую лепешку промывают раствором этилацетата и гексана (1:1 по объему, 5,3 кг). Продукт высушивают до постоянного веса, получая 1,5 кг 12 (55%) в виде грязно-белого твердого вещества. Дополнительное количество продукта может быть получено концентрированием маточного раствора путем вакуумной перегонки и многократной перекристаллизацией. Аналитические данные: 1 Н ЯМР (CDCl3,300 МГц)8,3 (д, 2 Н), 7,4 (д, 2 Н), 5,8 (д, 1 Н), 5,3-5,2 (м, 1 Н), 4,2-4,1 (м, 1 Н), 4,1-3,9 (м, 3 Н), 3,25-3,1(м, 1 Н), 2,3-2,1 (м, 1 Н), 2,1-1,9 (м, 1 Н); Анал. ВЭЖХ = 98,5%. Способ для соединения формулы 12, гексагидрофуро[2,3-b]фуран-3-илового эфира (2S,3R)-1-(4 бензилоксибензил)-2-гидрокси-3-[изобутил-(4-метоксибензолсульфонил)амино]пропил-[3R,3aS,6aR]карбаминовой кислоты В колбу помещают соединение формулы 27 (1,3 кг), а затем соединение формулы 11 (0,65 кг) и этилацетат и перемешивают, затем добавляют триэтиламин (0,65 кг) и диметиламинопиридин (24 г) и перемешивают при комнатной температуре несколько часов. Реакционную смесь последовательно промывают водой (8 кг), насыщенным водным раствором NaHCO3 (8 л), разбавленным водным раствором HCl (8 л) и насыщенным солевым раствором (8 л). Реакционную смесь обрабатывают активированным углем(0,13 кг), перемешивают несколько часов, отфильтровывают через целит и промывают этилацетатом. Добавляют гептан (6 л), смесь перемешивают несколько часов и выделяют продукт фильтрованием, а затем промывают его смесью ЕЮ Ас/гептан 1:1. Продукт высушивают до постоянной массы, получая 1 кг соединения формулы 12 (70%) в виде не совсем белого твердого вещества, т.пл. 127,5 С, чистота по ВЭЖХ 98,4. 1 Н ЯМР (CDCI3) 7,7- 7,75 (д, 2 Н), 7,26-7,48 (м, 5 Н), 7,12-7,20 (д, 2 Н), 6,96-7,03 (д, 2 Н), 6,856,94 (д, 2 Н), 5,65 (д, 1 Н), 5.3 (уш.д, 1 Н), 5,01 (с, 2 Н), 4,96-5,06 (уш, 1 Н), 3,63-3,96 (м, 7 Н), 3,84 (с, 3 Н),2,62-3,20 (м, 7 Н), 1,8-1,95 (м, 1 Н), 1,40-1,69 (м, 2 Н), 0.95 (дд, 6 Н). Способ для соединения формулы 13, гексагидро-[3R,3aS,6aR]-фуро[2,3-b]фуран-3-илового эфира В колбу помещают соединение формулы 12(1 кг) и продувают колбу азотом. Добавляют влажный 10% вес. палладий на активированном угле (0,2 кг), колбу продувают азотом, добавляют этилацетат (10 л), и реакционную смесь нагревают до 50 С, затем реакционную смесь насыщают водородом в течение 2,5 ч до окончания реакции. Реакционную смесь продувают азотом, затем под азотом отфильтровывают через целит, затем промывают этилацетатом. Фильтрат концентрируют до 2,5 л и к теплому раствору добавляют гептан (7,5 л). Полученный мутный раствор охлаждают на ледяной бане, отфильтровывают,промывают н-гептаном и высушивают до постоянной массы, получая соединение формулы 13 в виде твердого вещества, 0,82 кг, т.пл.: два эндотерма 98,2 и 133,8 С, чистота по ВЭЖХ 97,4%. 1 Н ЯМР(CDCl3) 7,61-7,75 (д, 2 Н), 7,01-7,10 (м, 2 Н), 6,91-6,99 (д, 2 Н), 6,63-6,79 (д, 2 Н), 5,62 (д, 2 Н), 5,51 (уш.с,1 Н), 4,96-5,09 (д, 2 Н), 3,81 (с, 3 Н), 3,59-3,98 (м, 6 Н), 2,62-3,18 (м, 7 Н), 1,42-1,91 (м, 3 Н), 0,78-0,95 (дд,6 Н). Способ для соединения формулы 14, 4-(2S,3R)-2-([2R,3S]-гексагидрофуро[2,3-b]фуран-(3R)-3 илоксикарбониламино)-3-гидрокси-4-[N-изобутил-(N-4-метоксибензолсульфонил)амино]бутилфенилового эфира трифторметансульфоновой кислоты. Соединение формулы 13 (0,82 кг) и дихлорметан (8 кг) были помещены в колбу и немного нагреты до растворения соединения формулы 13. В отдельную колбу поместили N-фенилтрифлимид (0,61 кг) и дихлорметан (2,6 кг) и немного нагрели до образования раствора. Раствор трифлирующего реагента был добавлен к раствору, содержащему соединение формулы 13, затем был добавлен карбонат цезия (0,55 кг), и перемешивание продолжалось при комнатной температуре в течение нескольких часов до завершения реакции. Добавили воду (4 кг), разделили слои, водный слой был снова экстрагирован дихлорметаном, объединенные органические фракции высушили над безводным сульфатом натрия. Раствор был профильтрован и сконцентрирован до небольшого объема, затем последовательно разбавлен метил-третбутиловым эфиром (7 л) и гептаном (16 л) и перемешивался при комнатной температуре до образования твердого вещества, которое было собрано и высушено до постоянной массы, давая соединение формулы 14 в виде твердого вещества, 0,68 кг, т.пл. 133,7 С, 19F ЯМР (CDCl3)-73,5 мд, чистота по ВЭЖХ 97.2%. 1 Н ЯМР (CDCl3) 7,70-7,78 (д, 2 Н), 7,29-7,38 (д, 2 Н), 7,16-7,23 (д, 2 Н), 6,96-7,06 (д, 2 Н), 5,67 (д, 1 Н), 4,95-5,04(м, 2 Н), 3,87 (с, 3 Н), 3,64-4,01 (м, 7 Н), 2,78-3,21 (м, 7 Н), 1,51-1,90 (м, 3 Н), 0,87-0,97 (дд, 6 Н). Способ получения соединения формулы 15, [3R,3aS,6aR]-гексагидрофуро[2,3-b]фуран-3-илового эфира В колбу помещают соединение формулы 14 (0,15 кг), а затем Pd(OAc)2 (0,06 кг), dppp (1,3 бис(дифенилфосфино)пропан) (0,1 кг) и диметилформамид (1,9 кг) и последовательно несколько раз создают вакуум и продувают колбу азотом, затем нагревают под азотом в температурном интервале от 60 до 65 С и добавляют хлорид лития (3 г). Смесь нагревают до 65-70 С и насыщают монооксидом углерода в течение 30 мин. К раствору добавляют триэтиламин (86 г), а затем медленно добавляют триэтилсилан(0,05 кг). Реакцию выдерживают при 65-70 С под атмосферой СО до завершения процесса. Реакционную смесь охлаждают до комнатной температуры, разбавляют этилацетатом (1,8 кг) и промывают водой (4 кг). Этилацетат вновь экстрагируют водой (1 кг) и объединенные водные фракции вновь экстрагируют этилацетатом (0,5 кг). Объединенные этилацетатные экстракты несколько раз промывают водой и фильтруют этилацетат через целит, разбавляя ацетонитрилом (0,2 кг). Добавляют HF (48% в воде, 0,23 кг) и насыщенный NaHCO3 (3 кг), реакционную смесь разделяют и водный слой выбрасывают. Органический слой сушат над безводным сульфатом натрия, фильтруют, фильтрат нагревают до температуры 50-55 С, обрабатывают тримеркаптотриазином (23 г) в течение нескольких минут, добавляют активированный уголь (10 г), смесь нагревают до 50-55 С по крайней мере 30 мин, охлаждают до комнатной температуры и фильтруют через слой целита. Фильтрат промывают насыщенным NaHCO3 (0,7 кг), отделяют,сушат над безводным сульфатом натрия, концентрируют и очищают остаток колоночной хроматографией на силикагеле, элюируя смесью этилацетата и гептана. Фракции, содержащие целевой продукт формулы 15, собирают и концентрируют, получая не совсем белое твердое вещество, которое перекристаллизовывают, растворяя в диметиловом эфире этиленгликоля при повышенной температуре, медленно добавляя гептан, а затем охлаждая до комнатной температуры. Отфильтровывание твердого вещества,промывание его гептаном и высушивание до постоянной массы дает соединение формулы 15 в виде белого твердого вещества, 72%, 0,125 кг, т.пл. 140,2 С, чистота по ВЭЖХ 98,3%. 1 Н ЯМР (CDCl3) 9,98 (с,1 Н), 7,80-7,85 (д, 2 Н), 7,67-7,76 (д, 2 Н), 7,39-7,45 (д, 2 Н), 6,95-7,04 (д, 2 Н), 5,65 (д, 1 Н), 4,96-5,12 (м, 2 Н),3,85 (с, 3 Н), 3,64-4,02 (м, 7 Н), 2,75-3,21 (м, 7 Н), 1,72-1,89 (м, 1IH), 1,42-1,70 (м, 2 Н), 0,84-0,98 (дд, 6 Н). Способ для соединения формулы 16, 2-(N-бензилоксикарбамоил)аминоэтилфосфорной кислоты В колбу помещают деионизованную воду (9 кг) и при перемешивании в инертных условиях добавляют поциями гидроксид натрия (2,7 кг), поддерживая температуру ниже 35 С. Порциями в колбу помещают аминоэтилфосфорную кислоту (АЕР, 3 кг). В несколько порций добавляют бензилхлорформиат (5,6 кг), поддерживая температуру около 40 С. Смесь оставляют реагировать при комнатной температуре на несколько часов до завершения реакции. Смесь дважды экстрагируют этилацетатом (порции 16 кг). Водный слой подкисляют концентрированной HCl до рН 1,3 и оставляют на несколько часов. Твердое вещество отделяют и промывают ацетонитрилом (2,3 кг). Затем твердое вещество и метанол (9,6 кг) помещают в колбу и обрабатывают смолой Dowex (8,7 кг), предварительно промытой водой и метанолом. Смесь перемешивают при комнатной температуре 1 ч, фильтруют и промывают метанолом (3 кг). Фильтрат концентрируют до густого масла, разбавляют ацетонитрилом и неоднократно азеотропно отгоняют ацетонитрил до тех пор, пока остаточный метанол не будет удален. Раствор затем разбавляют ацетонитрилом, нагревают до растворения, фильтруют и оставляют постепенно остывать на ледяной бане. Твердое вещество отделяют, сушат до постоянной массы, получая соединение Формулы 16 (CBZ-AEP), 4,8 кг, 77%, т.пл. 107 С, 31 Р ЯМР (D2O) 26,6 мд. 1H ЯМР (D20) 7,2-7,36(уш.с, 5 Н), 4,95 (уш.с, 2 Н), 3,16-3,30 (м, 2 Н), 1,78-1,94 (м, 2 Н). Способ для соединения формулы 17, фенил-2-(N-бензилоксикарбамоил)аминоэтилфосфонатCBZ-AEP (2,5 кг) и ацетонитрил перемешивали при нагревании до 60-65 С. В отдельной колбе нагревали фенол (4,5 кг) и ацетонитрил (3,5 кг) до получения раствора, и этот раствор добавляли к смесиCBZ-AEP и перемешивали до образования раствора. К этому раствору добавляли взвесь диметиламинопиридина (DMAP, 1,4 кг) в ацетонитриле (3,1 кг). В отдельную колбу помещали ацетонитрил (0,8 кг) и дициклогексилкарбодиимид (3 кг). Этот ДЦГК раствор добавляли к теплому АЕР раствору. Сразу после окончания прибавления реакционную смесь кипятили несколько часов до завершения реакции. Реакционную смесь охладили до комнатной температуры, отфильтровали, фильтрат сконцентрировали и разбавили водой (20 л) и водным NaOH. Раствор дважды экстрагировали этилацетатом (13,5 л). Водную фазу подкислили до рН 1, добавляя 6 М HCl, получившееся твердое вещество собрали и снова суспендировали в воде (19 л), и снова собрали и высушили до постоянной массы, получая соединение формулы 17 в виде белого твердого вещества, 2,47 кг, т.пл. 124 С, чистота по ВЭЖХ 99,2%. 31 Р ЯМР (CDCl3) 29,8 мд(приблизительно 90%) и 28,6 мд (приблизительно 10%), относящиеся к ротамерам карбаматной функциональной группы. 1 Н ЯМР (CDCl3) 7,05-7,40 (м, 10 Н), 5,10 (уш.с, 2 Н), 3,41-3,59 (м, 2 Н), 2,01-2,20 (м,2 Н). Способ для соединения формулы 18, фенил, (этил (S)-2-пропионил)-2-(N-бензилоксикарбамоил) Соединение формулы 17 (4,8 кг) поместили в реактор вместе с толуолом (24 кг) и ДМФА (4 г). Смесь нагрели до 70 С. Медленно добавили SOCl2, поддерживая внутреннюю температуру содержимого 67-72 С, затем реакционную смесь перемешивали при 75 С до завершения реакции. Раствор охладили до 45 С и сконцентрировали в вакууме примерно до половинного объема. В отдельном реакторе приготовили и охладили до -1 С сухой раствор (3)-этиллактата (1,9 кг), толуола (15 кг) и пиридина (1,5 кг). Медленно добавили хлорирующий раствор, поддерживая внутреннюю температуру от -3 до 3 С, а затем полученный раствор нагрели до 20 С и перемешивали до окончания реакции. Реакционную смесь вылили в водный раствор 10% лимонной кислоты (10 кг), слои разделили и органический слой промыли 10% водным NaH2PO4 (10 кг). Органический слой высушили над безводным сульфатам натрия (5 кг), концентрировали и упарили из этилацетата (4 кг), получая вязкое масло, которое очищали пропусканием через слой силикагеля (9,2 кг), элюируя смесью этилацетата и гептана. Фракции, содержащие соединение формулы 18, объединили и сконцентрировали, получая масло. Растворитель заменили, дважды упарив с ацетонитрилом (2x3 кг), получая густую жидкость (4,7 кг,80%) с чистотой по ВЭЖХ 98%, являющуюся смесью двух диастереомеров (верно для бензилхлорида). Смесь изомеров разделяли на Chromasil силикагеле, элюируя смесью этилацетата и гептана. Целевой изомер формулы 20 показал следующие физические свойства: масло, 31 Р ЯМР (CDCl3) 26,1 (-90%) и 25.4 (-10%) принадлежащие ротамерам карбаматной функциональной группы; 1 Н NMR (CDCl3) 7,24-7,4(м, 8 Н), 7,14-7,21 (м, 2 Н), 5,65 (уш.с, 1 Н), 5,1 (с, 2 Н), 5,02-5,06 (м, 1 Н), 4,12-4,17 (кв, 2 Н), 3,52-3,70 (м,2 Н), 2,15-2,36 (м, 2 Н), 1,57 (д, 3 Н), 1,22 (т, 3 Н). Способ для соединения формулы 19, фенил, (этил (S)-2-пропионил)-2-аминоэтилфосфоната, соли уксусной кислоты В колбу помещают влажный 10% вес. палладий на активированном угле (0,28 кг), уксусную кислоту (0,15 л), соединение формулы 20 (0,56 кг) и этанол (5,6 л), и колбу продувают азотом примерно 30 мин. Реакционную смесь несколько часов насыщают водородом до поглощения исходного вещества. Реакционную смесь продувают азотом 60 мин, реакционную смесь фильтруют через целит и промывают этиловым спиртом (2 л). Фильтрат концентрируют при комнатной температуре до небольшого объема,разбавляют ацетонитрилом (5,6 л), концентрируют до половинного объема и обрабатывают активированным углем (0,3 кг), затем фильтруют через целит и промывают ацетонитрилом (2,5 л). Фильтрат упаривают при комнатной температуре, разбавляют ацетонитрилом и упаривают. Это повторяют несколько раз для удаления всего этанола и воды и наконец, раствор концентрируют до небольшого объема и хранят при 5 С. Упаривание аликвоты дает вещество в виде масла, 90%, 0,49 кг, 31 Р ЯМР (CDCl3) 25,2. Материал использовали в следующей стадии без дополнительной очистки. Способ для соединения формулы 21 (3R,3aS,6aR)-гексагодрофуро[2,3-b]фуран-3-иловый эфир 2[(2S,3R)-4-[4-метоксибензол) сульфонил)(2-метилпропил)амино]-3-гидроксибутил]-(фенокси) (2(2R) -пропионовой кислоты этиловый эфир)окси]фосфинил]этиламино]бензил]-карбаминовой кислоты,соль гександикарбоновой кислоты (1:1) В колбу помещают соединение формулы 15 (0,5 кг), ацетонитрил (1,6 л) и раствор соединения формулы 19 (0,46 кг) в ацетонитриле (1 л), а затем ацетонитрил (2,4 л). Смесь перемешивают при комнатной температуре несколько часов. Порциями добавляют NaBH(OAc)3 (0,27 кг) так, чтобы температура оставалась комнатной. Реакционную смесь перемешивают несколько часов до завершения реакции. Добавляют целит (0,24 кг), реакционную смесь фильтруют и промывают ацетонитрилом и изопропилацетатом. Фильтрат концентрируют до небольшого объема, разбавляют изопропилацетатом (12,5 л) и последовательно промывают три-четыре раза насыщенным NaHCO3 (7,5 л порция) и насыщенным солевым раствором (3,8 л), органический слой высушивают над сульфатом натрия, фильтруют, концентрируют до небольшого объема, разбавляют изопропилацетатом и азеотропно удаляют остаточную воду. Раствор разбавляют ацетонитрилом, подогревают и добавляют адипиновую кислоту (0,13 кг). Раствор постепенно охлаждают и собирают твердое вещество, которое промывают изопропилацетатом и получают соединение формулы 21 в виде твердого вещества, 0,69 кг, 79%, т.пл. 119 С, чистота по ВЭЖХ 95,3%. Спектральные данные согласуются с указанными в ссылке: 31 Р ЯМР (ацетон-d6) 27,6; 13 С ЯМР (ацетон-d6) мд 173,4; 170; 162,6; 155,0; 150,4; 137,9; 137,4; 130,7; 129,3; 129,2; 129,1; 127,6; 124,5; 120,4; 113,9; 108,9; 72,7; 72,6; 70,4; 70,4; 68,6; 60,7; 57,8; 55,6; 54,9; 52,8; 52,3; 45,1; 42,1; 34,9; 32,6; 26,5; 26,5; 25,4; 24,0; 19,2; 18,6; 13,1; 1 Н ЯМР (ацетон d-6) мд 7,80 (д, 2 Н), 7,38 (т, 2 Н), 7,29 (д, 2 Н), 7,28 (д, 2 Н), 7,26 (д, 2 Н), 7,21 (т,1 Н), 7,12 (д, 2 Н), 5,53 (д, 1 Н), 5,04 (дкв, 1 Н), 4,95 (ддд, 1 Н), 4,14 (кв, 2 Н), 3,92 (с, 3 Н), 3,89 (м, 1 Н), 3,88(уш), 2959, 1755, 1703, 1599, 1497, 1308, 1343, 1 152, 991 , 950. Способ для соединения формулы 21 b, (3R,3aS,6aR)-гексагодрофуро[2,3-b]фуран-3-иловый эфир 2[(2S,3R)-4-[4-метоксибензол)сульфонил)(2-метилпропил)амино]-3-гидроксибутил]-(фенокси)(2(2R)-пропионовой кислоты этиловый эфир)окси]фосфинил]этиламино]бензил]карбаминовой кислоты,соль бутандикарбоновой кислоты (1:1). Получали растворением при перемешивании 7,8 г свободного основания соединения формулы 29 в горячем изопропилацетате (приблизительно 200 мл), обработкой янтарной кислотой (1 эквивалент), и,после образования раствора, постепенным охлаждением до комнатной температуры, а затем охлаждением несколько минут на ледяной бане; продукт отделяли, промывали изопропилацетатом и высушивали до постоянной массы, получая соль янтарной кислоты соединения формулы 21b, 7,7 г, 86%, чистота по ВЭЖХ 98,6%, т.пл. 106,5 С. 13 С ЯМР (CDCl3) 129,8; 129,4; 129,2; 124,9; 120,3; 114,1; 109,0; 70,9; 72,7; 71,4; 70,33; 70,28; 69,34; 69,30; 61,3; 56,51; 56,47; 55,3; 54,95; 52,24; 52,22; 51,74; 51,72; 44,93; 42,42; 30,65; 24,84; 24,79; 26,48; 25,42; 19,7; 19,6; 19,24; 13,7. 1 Н ЯМР (CDCl3) 7,75-7,79 (д, 2 Н), 7,38-7,43 (д,2 Н), 7,33-7,36 (м, 2 Н), 7,24-7,29 (д, 2 Н), 7,15-7,0 (т, 1 Н), 6,98-7,05 (4 Н), 5,63 (д, 1 Н), 5,00-5,08 (м, 1 Н),5,84-4,92 (м, 1 Н), 4,09-4,18 (м, 3 Н), 3,93-3,98 (м, 1 Н), 3,91 (с, 3 Н), 3,79-3,92 (м, 4 Н), 3,66-3,74 (м, 1 Н),3,22-3,56 (м, 4 Н), 2,96-3,02 (м, 2 Н), 2,51-2,83 (м, 10 Н), 1,74-1,82 (м, 2 Н), 1,6 (д, 3 Н), 1,46-2,01 (3 Н), 1,21 (т,3 Н), 1,83 (д, 3 Н), 1,63 (д, 3 Н). Способ для соединения формулы 22 В колбу помещают 14,8 г дисукцинимидилкарбоната, CH2Cl2(25 мл), 5,0 г соединения формулы 10 в виде раствора в CH2Cl2 (20 мл) и пиридин (7,8 мл). Раствор нагревают до слабого кипения несколько часов до завершения реакции. Нагревание убирают и добавляют воду (35 мл), смесь перемешивают несколько минут и слои разделяют. Органическую фазу последовательно промывают водой (35 мл) и насыщенным солевым раствором (30 мл). Органическую фазу высушивают над сульфатом натрия, фильтруют и концентрируют. Остаток вновь растворяют в дихлорметане CH2Cl2 (13 мл) при нагревании и к теплому раствору добавляют гептан (10 мл). Смесь постепенно охлаждают примерно до 10 С, твердое вещество отфильтровывают, промывают гептаном и высушивают до постоянной массы, получая приблизительно 8,9 г, 87,5%. В колбу помещают неочищенное соединение формулы 22 (106 г), активированный уголь (23 г) и толуол (5,7 кг). После перемешивания в течение 2 ч смесь фильтруют через целит и фильтрат упаривают,получая 100 г (94,3% возврата) соединения формулы 22 в виде грязно-белого твердого вещества. В колбу помещают соединение формулы 22 (12 г) и ацетон (24 г) и нагревают до 52 С до образования раствора. При перемешивании к теплому раствору добавляют гептан (60 г). Смесь охлаждают за 2 ч примерно до 10 С, твердое вещество отделяют, промывают смесью ацетон:гептан 3:1 и высушивают до постоянной массы, получая соединение формулы 22, 11,4 г, 95% возврата, в виде белого твердого вещества. 1 Н ЯМР(CDCl3) 5,75 (д, 1 Н), 5,21-5,30 (дд, 1 Н), 3,90-4,16 (м, 4 Н), 3,07-3,18 (м, 1 Н), 2,85 (с, 4 Н), 2,10-2,22 (м, 1 Н),1,92-2,06 (м, 1 Н). Получение соединения формулы 24 В колбу помещают соединение формулы 24 (10 г), питьевую воду (7,5 г, 13,5 экв.) и изобутиламин(22,08 г, 9,8 экв.), густую смесь нагревают до приблизительно 60 С и перемешивают при этой температуре до завершения реакции. Реакционную смесь обрабатывают 100 мл питьевой воды за приблизительно 30 мин, поддерживая внутреннюю температуру выше 55 С. Смесь охлаждают до 5 С за 1,5 ч и выдерживают при этой температуре еще 30 мин. Взвесь фильтруют, промывают 20 мл питьевой воды и высушивают до постоянной массы, получая соединение формулы 23, 10,94 г; 98,4%, чистота по ВЭЖХ 97,9%. 1 Н ЯМР (CDCl3) 7,55-7,62 (д, 2 Н), 7,32-7,38 (д, 2 Н), 4,62-4,72 (уш.с, 1 Н), 3,78-3,90 (уш.м, 1 Н),3,42-3,50 (м, 1 Н), 3,08-3,16 (дд, 1 Н), 2,63-2,90 (м, 3 Н), 2,42 (д, 2 Н), 1,65-1,81 (м, 1 Н), 1,35 (с, 9 Н), 0, 93 (Д,6 Н). Получение соединения формулы 25 В колбу помещают соединение формулы 23 (10,5 г), дихлорметан (63 мл) и триэтиламин (3,1 г, 1,05 экв.), и в течение приблизительно 10 минут добавляют раствор 4-метоксифенилсульфонил хлорида (6,1 г,1,02 экв.) в дихлорметане (18 мл), поддерживая во время прибавления внутреннюю температуру менее 25 С. После завершения реакции (приблизительно 2 ч при комнатной температуре) добавляют 1 М водную HCl (5 мл), перемешивают 5 мин и разделяют слои. К органической фазе добавляют 1 М воднуюNaHCO3 (5 мл), смесь перемешивают 5 мин, слои разделяют и органическую фазу концентрируют до образования пены. Сырой продукт растворяют в 100 мл EtOH при 65 С, добавляют за период приблизительно 45 мин воду (120 мл), поддерживая внутреннюю температуру выше 57 С, и смесь постепенно охлаждают до 10 С примерно за 4,5 ч. Взвесь фильтруют и промывают 50 мл 30% водного EtOH, продукт высушивают до постоянной массы, получая 14,5 г, 98%, чистота по ВЭЖХ 99,86%. 1 Н ЯМР (CDCl3) 7,70-7,76 (д, 2 Н), 7,55-7,64 (д, 2 Н), 7,36-7,43 (д, 2 Н), 6,96-7,04 (д, 2 Н), 4,63-4,72 (уш.с, 1 Н), 3,88 (с, 3 Н),3,72-3,90 (м, 2 Н), 3,04-3,18 (м, 3 Н), 2,79-3,01 (м, 3 Н), 1,78-1,92 (м, 1 Н), 1,62 (уш.с, 1 Н), 1,35 (с, 9 Н), 0,850,95 (дд, 6 Н). Способ для соединение формулы 26 В колбу при инертных условиях загружают соединение формулы 25 (35 г) и толуол (525 мл) и охлаждают до -20 С. Постепенно добавляют 1,5 М раствор DIBAL-H в толуоле (154 мл, 1,5 М, 3,5 экв.),поддерживая температуру ниже -10 С. Реакцию перемешивают при этой температуре несколько часов до ее завершения. Постепенно добавляют метанол (9,3 мл, 3,5 экв.), а затем -ТГФ (88 мл), и смесь нагревают до 0 С. В течение 5 мин добавляют водную лимонную кислоту (220 мл 40 вес.% лимонной кислоты, 7 экв., разбавленной 130 мл воды), затем смесь нагревают до приблизительно 60 С примерно 1 ч. Смесь охлаждают до комнатной температуры, слои разделяют, и органический слой выливают в 175 мл 1 МHCl и 35 мл воды. Делительную воронку споласкивают 105 мл ТГФ. Получившуюся смесь перемешивают при комнатной температуре примерно 1 ч, разбавляют ТГФ (35 мл), разделяют слои, органическую фракцию объединяют с 35 мл 1 М NaHCO3 и перемешивают 30 мин. Слои разделяют, фильтруют через слой безводного сульфата магния (примерно 2 г) и промывают толуолом (35 мл). Раствор концентрируют и азеотропно отгоняют с толуолом три раза, чтобы уменьшить количество остаточного ТГФ. Конечный объем доводят примерно до 275 мл, и мутный раствор нагревают до приблизительно 65 С до образования раствора. Постепенно добавляют гептан (132 мл), и смесь постепенно за 4 ч охлаждают до комнатной температуры. Продукт отфильтровывают, промывают смесью толуол:гептан 2:1 и высушивают до постоянной массы, получая соединение формулы 26, 31 г, 88%, т.пл. 120,5 С, чистота по ВЭЖХ 99,6%. 1 Н ЯМР (CDCl3) 10,0 (с, 1 Н), 7,80-7,85 (м, 4 Н), 7,27-7,50 (д, 2 Н), 7,09-7,10 (д, 2 Н), 5,99-6,07 (уш.д, 1 Н),3,91 (с, 3 Н), 3,78-3,93 (м, 3 Н), 3,41-3,51 (дд, 1 Н), 3,24-3,34 (дд, 1 Н), 2,79-3,05 (м, 5 Н), 1,29 (с, 9 Н), 0,870,93 (дд, 6 Н). Способ для соединения формулы 15, [3R,3aS,6aR]-гексагидрофуро[2,3-b]фуран-3-илового эфира В колбу помещают соединение формулы 26 (2,0 г) и 20 мл ТГФ. К раствору по каплям добавляют метансульфоновую кислоту. Раствор нагревают до 40 С до завершения реакции снятия защитной группы. Раствор охлаждают до 20 С, и в реактор добавляют N-метилимидазол (2,39 г). Затем прибавляют соединение формулы 22 (1,52 г) и реакционную смесь нагревают до 50 С до завершения реакции. Добавляют этилацетат (150 мл) и раствор последовательно промывают 0,5 М водной лимонной кислотой (20 г),10% водным NaH2PO4 (20 г), насыщенным NaHCO3 (20 г) и 10% водн. NaH2PO4 (20 г). Органический слой высушивают над безводным сульфатом натрия (2 г), фильтруют и концентрируют до вязкого масла,которое очищают колоночной хроматографией на силикагеле, элюируя смесью этилацетата и гептана. Фракции, содержащие целевое соединение формулы 15, объединяют и концентрируют, получая белое твердое вещество, 95%, 2,13 г, чистота по ВЭЖХ 97%. Была сделана ссылка на определенный вариант осуществления изобретения, примеры которого иллюстрируются в описании, а также структурами и формулами. Хотя изобретение было описано на примере ограниченного числа вариантов осуществления, должно быть понятно, что изобретение не ограничивается этими вариантами осуществления. Наоборот, подразумевается, что изобретение покрывает все альтернативы, модификации и эквиваленты, которые могут быть включены в пределы действия настоящего изобретения, как определено в формуле изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, имеющее структурную формулу С или его фармацевтически приемлемая соль. 2. Соединение, имеющее структурную формулу М или его фармацевтически приемлемая соль. 3. Соединение, имеющее структурную формулу N или его фармацевтически приемлемая соль.
МПК / Метки
МПК: C07F 9/40, A61P 31/18, C07D 493/04, C07C 311/15, A61K 31/34
Метки: помощью, ретровирусных, производных, соединения, профилактики, фармацевтическая, лечения, спирта, соль, основе, способ, получения, бисфуранового, композиция, инфекций
Код ссылки
<a href="https://eas.patents.su/23-20088-sposob-polucheniya-proizvodnyh-bisfuranovogo-spirta-soedineniya-sol-farmacevticheskaya-kompoziciya-na-ee-osnove-sposob-lecheniya-ili-profilaktiki-retrovirusnyh-infekcijj-s-ee-pomos.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения производных бисфуранового спирта, соединения, соль, фармацевтическая композиция на ее основе, способ лечения или профилактики ретровирусных инфекций с ее помощью</a>
N-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин или его соль, фармацевтическая композиция, содержащая это соединение, применение этого соединения для лечения рака и способ лечения рака с помощью этого соединения

Номер патента: 16052
Опубликовано: 30.01.2012
Авторы: Эрнандес-Абад Педро Э., Меклер Харолд, Иди Деннис Л., Кьэрсгорд Ханс Й., Жичкин Павел Э., Гевель Ронан И., Истам Стивен А., Шоу Стивен Р., Поломски Роберт Э., Херр Джейсон Р., Боуланджер Уилльям А., Колиер Стивен Дж.
МПК: A61P 35/00
Метки: рака, способ, это, соединение, этого, содержащая, применение, фармацевтическая, композиция, n-α-(бензилоксикарбонил)-l-γ-глутамил-3-[[2-[[бис[бис(2-хлорэтил)амино]фосфонил]окси]этил]сульфонил]-l-аланил-2(r)-фенилглицин, лечения, соль, помощью, соединения
Формула / Реферат:
1. Соединение формулыили его соль.2. Фармацевтическая композиция, включающая соединение по п.1 или его соль.3. Применение соединения по п.1 или его соли для приготовления лекарственного средства, предназначенного для лечения рака.4. Способ лечения рака у млекопитающего, включающий введение млекопитающему соединения по п.1 или его...
Средство для профилактики и лечения ринитов различной этиологии, способ его получения, фармацевтическая композиция на его основе, способ профилактики и лечения

Номер патента: 9610
Опубликовано: 28.02.2008
Авторы: Кожемякин Леонид Андреевич, Кетлинская Ольга Сергеевна
МПК: A61K 33/40, A61K 38/08, A61K 33/30...
Метки: композиция, получения, лечения, этиологии, ринитов, фармацевтическая, основе, средство, профилактики, способ, различной
Формула / Реферат:
1. Применение бис-(g-L-глутамил)-L-цистинил-бис-глицината лития (тетралитиевой соли дисульфидглутатиона) со стабилизированной дисульфидной связью в качестве средства для профилактики и лечения ринитов различной этиологии. 2. Способ получения средства для профилактики и лечения ринитов различной этиологии, включающий окисление исходного соединения - восстановленного глутатиона (g-L-глутамил-L-цистеинилглицина) - перекисью водорода в присутствии...
5-({6-[2,4-бис-(трифторметил)фенил]пиридазин-3-ил}метил)-2-(2-фторфенил)-5н-имидазо[4,5-c]пиридин, способ его получения, способ лечения или профилактики инфекционного вирусного гепатита с с его помощью, композиция и лекарственное средство на его основе

Номер патента: 19452
Опубликовано: 31.03.2014
Авторы: Тсе Уинстон К., Зиа Вахид, Ольяй Ризэ, Дал Теренс К., О Дэвид А., Бонди Стивн С.
МПК: C07D 403/14, A61P 31/12, A61K 31/4353...
Метки: способ, получения, основе, гепатита, профилактики, вирусного, композиция, помощью, средство, лечения, инфекционного, 5-({6-[2,4-бис-(трифторметил)фенил]пиридазин-3-ил}метил)-2-(2-фторфенил)-5н-имидазо[4,5-c]пиридин, лекарственное
Формула / Реферат:
1. Способ получения пиридазинового соединения формулы (1)включающий обработку 5-[6-хлорпиридазин-3-илметил]-2-(2-фторфенил)-5Н-имидазо[4,5-с]пиридина 2,4-бис-(трифторметил)фенилбороновой кислотой в присутствии растворителя и выделение соединения (1), где указанный растворитель имеет структуру R1OR2O(R4O)aR3, где каждый из R1, R2, R3 и R4 независимо выбран из (C1-С6)алкила и а представляет собой целое число 0 или 1.2. Способ по п.1, в котором...
Пиперазинил-бензилиденил-лактамовые соединения в качестве агонистов и антагонистов 5-нт1а и 5-нт1d рецепторов, фармацевтическая композиция на их основе и способ лечения или профилактики нарушения илисостояния, связанных с недостаточной серотонергической нейротрансмиссией у млекопитающего

Номер патента: 2157
Опубликовано: 24.12.2001
Автор: Ховард Гэрри Ральф
МПК: C07D 233/96, A61K 31/4166, A61P 25/00...
Метки: нейротрансмиссией, связанных, млекопитающего, 5-нт1d, нарушения, рецепторов, серотонергической, основе, фармацевтическая, способ, соединения, агонистов, пиперазинил-бензилиденил-лактамовые, недостаточной, композиция, лечения, качестве, профилактики, илисостояния, 5-нт1а, антагонистов
Формула / Реферат:
1. Пиперазинил-бензилиденил лактамовые соединения формулы где R1 представляет пиперазин-1-ил или пиперазин-1-ил, замещенный в положении 4 (C1-C4) алкилом; R2 представляет водород; R представляет-(СН2)mВ, где m равно 0 или 1 и В представляет водород, фенил, необязательно замещенный одним или несколькими заместителями, независимо выбранными из галогена, (C1-C6)алкила или трифторметила; Х представляет водород или галоген; Y представляет...
Способ получения бисфуранового спирта

Номер патента: 16140
Опубликовано: 28.02.2012
Авторы: Гутиерес Арнольд, Юй Ричард Хун Чуй, Дауди Эрик Д., Кроуфорд Кеннет Р., Полняшек Ричард П.
МПК: C07C 327/42, C07D 493/04, C07F 9/02...
Метки: спирта, получения, способ, бисфуранового
Формула / Реферат:
1. Способ получения бисфуранового спирта, имеющего структурную формулу 0Формула 0включающий в себя проведение реакции 2,3-дигидрофурана и гликолевого альдегида или димера гликолевого альдегида в присутствии катализатора, содержащего лантаноид или переходный металл, с образованием бисфуранового спирта формулы 0.2. Способ по п.1, в котором в качестве катализаторов используют комплексы Yb, Pr, Cu, Eu и Sc с лигандами, выбранными из3. Способ по п.2,...
Предыдущий патент: Способ удаления примесей цинка, железа, кальция, меди и марганца из водных растворов кобальта и/или никеля
Следующий патент: Модуль зоны межэтажного перекрытия системы сплошного остекления балконов и лоджий и способ (варианты) его монтажа
Случайный патент: Способ, устройство и рамная конструкция для очистки поверхностей труб и трубная конструкция для распределения частиц