Антагонист рецептора cgrp
Номер патента: 22815
Опубликовано: 31.03.2016
Авторы: Макор Джон Е., Дубовчик Джин М., Чатурведула Прасад В.


















Формула / Реферат
1. Соединение (R)-N-(3-(7-метил-1Н-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамид или его фармацевтически приемлемая соль

2. Фармацевтическая композиция для лечения состояния, связанного с аберрантными уровнями CGRP или сигнального CGRP рецептора, содержащая фармацевтически эффективное количество (R)-N-(3-(7-метил-1H-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамида в сочетании с фармацевтически приемлемым адъювантом, носителем или растворителем.
3. Способ лечения состояния, связанного с аберрантными уровнями CGRP или сигнального CGRP рецептора, включающий введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту.
4. Способ по п.3, отличающийся тем, что состояние представляет собой мигрень.
5. Способ по п.3, отличающийся тем, что состояние представляет собой нейропатическую боль.
6. Способ лечения пролиферативного заболевания, включающий в себя введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту.
7. Способ по п.6, отличающийся тем, что пролиферативное заболевание представляет собой рак молочной железы или глиому.
Текст
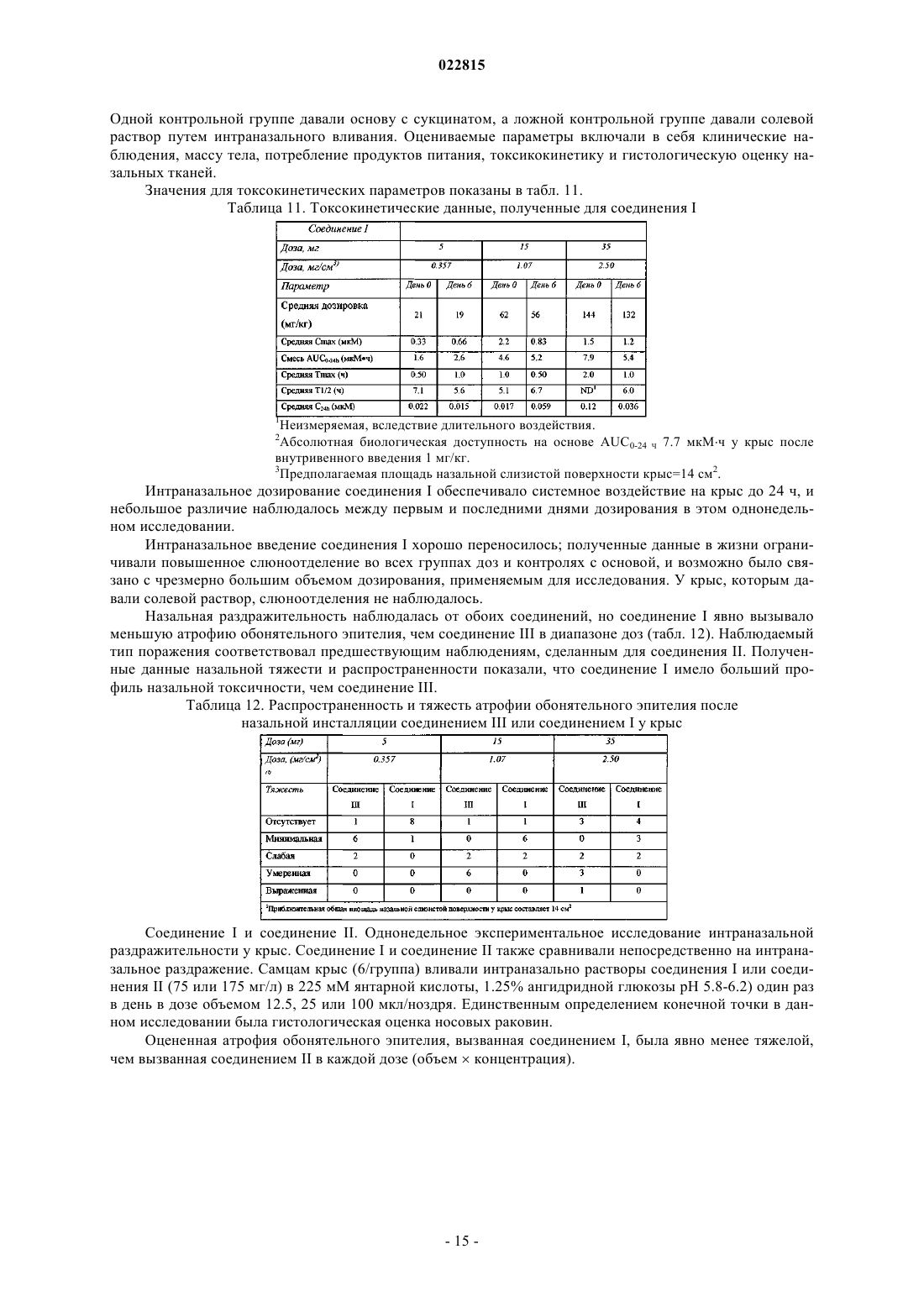
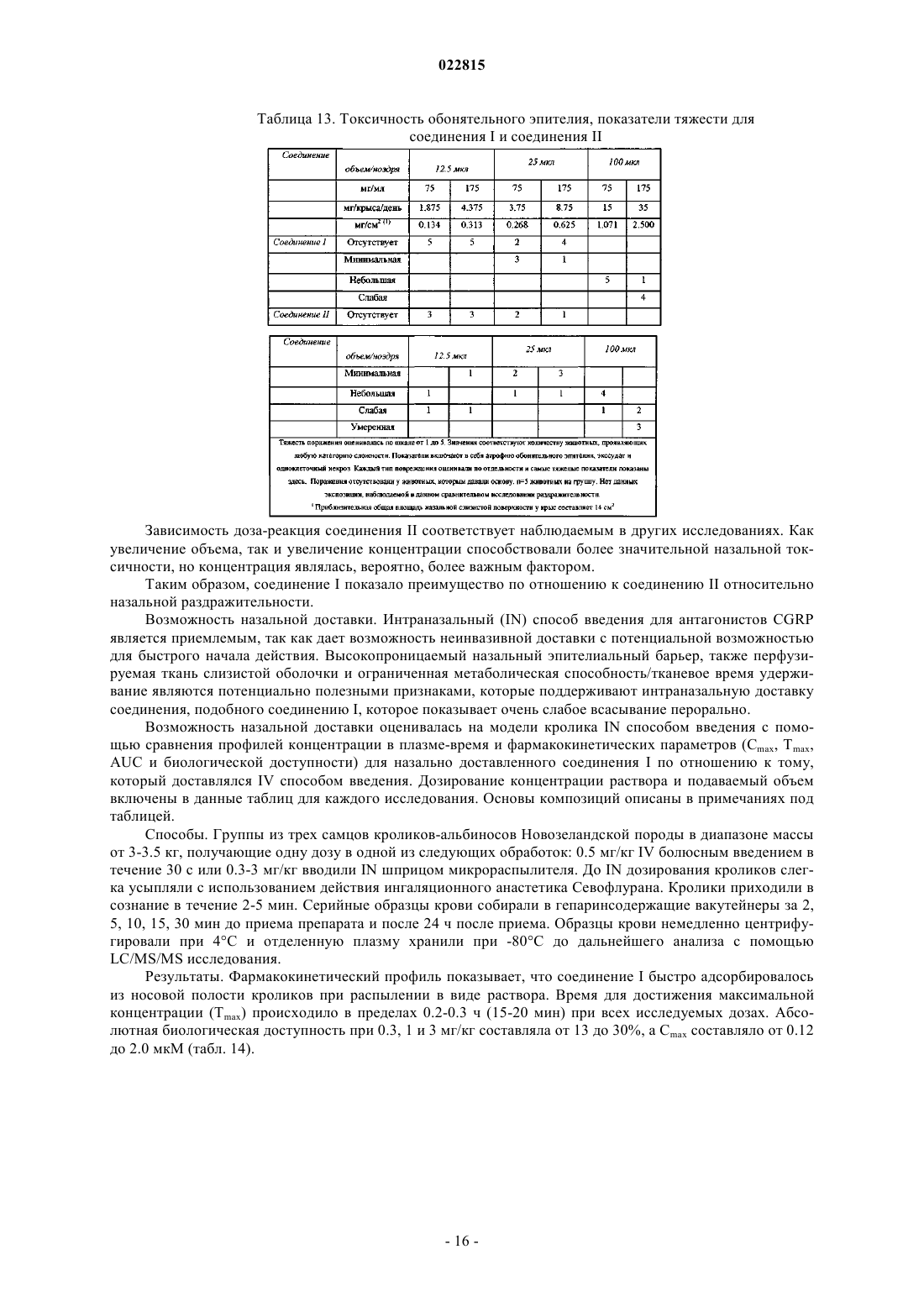
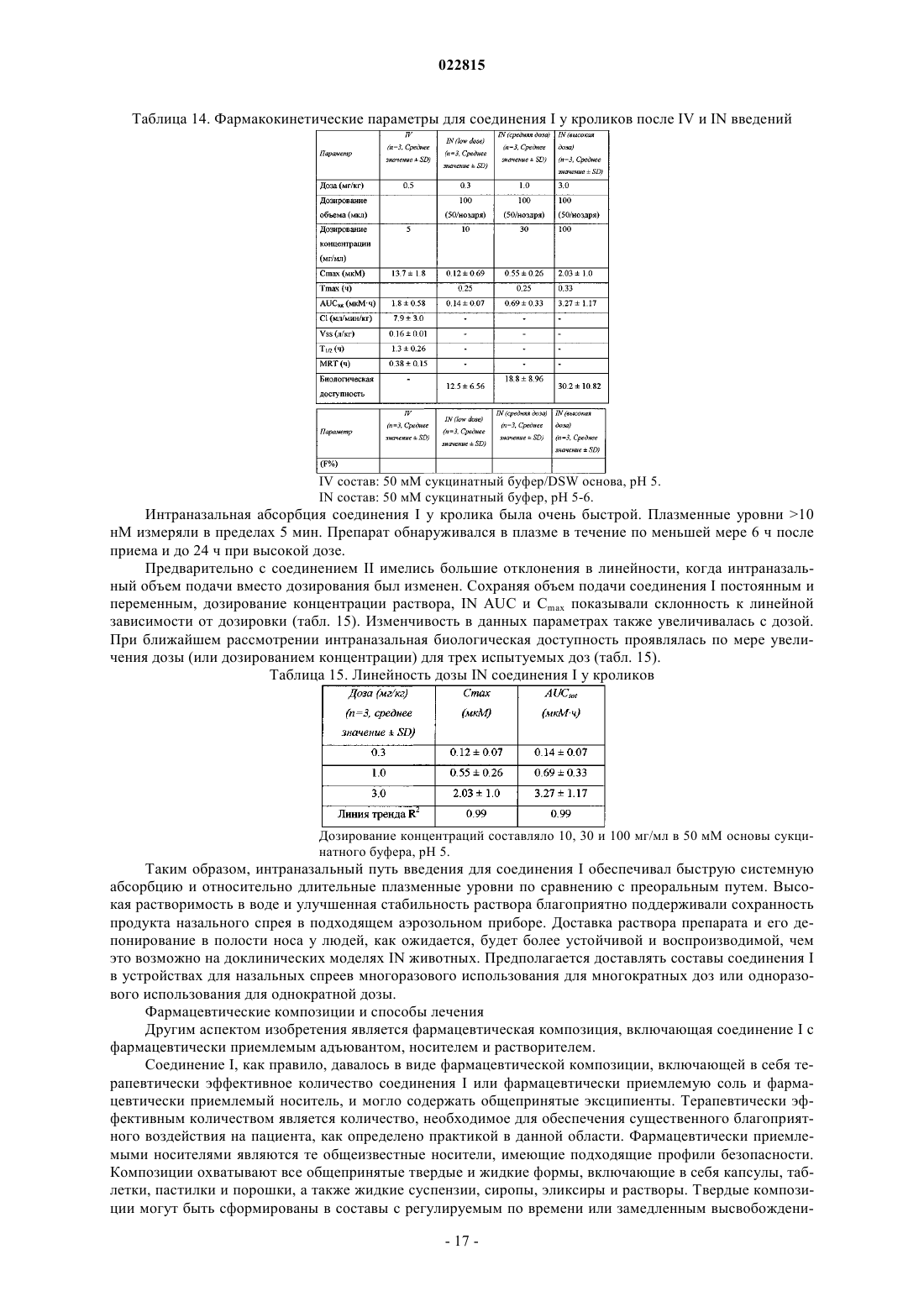
Изобретение, в целом, относится к соединению формулы I, (R)-N-(3-(7-метил-1 Н-индазол-5-ил)-1(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2 дигидрохинолин-3-ил)пиперидин-1-карбоксамиду, включающему в себя фармацевтически приемлемые соли, которое представляет собой антагонист CGRP-рецептора. Изобретение также относится к фармацевтическим композициям и способам для применения соединения в лечении расстройств, связанных с CGRP, включающих в себя мигренозные головные боли,нейрогенную вазодилатацию, нейрогенное воспаление, термическое повреждение, циркуляторный шок, гиперемию, связанную с менопаузой, воспалительные заболевания дыхательных путей, такие как астма, хроническое обструктивное заболевание легких (ХОЗЛ), и раковая опухоль. Перекрестная ссылка на родственные заявки Данная заявка на изобретение испрашивает приоритет по предварительной заявке США 61/319015, поданной 30 марта 2010 г. Уровень техники Изобретение, в целом, относится к соединению (R)-N-(3-(7-метил-1H-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1 карбоксамид (соединение I или соединение формулы I), включая его фармацевтически приемлемые соли,которое представляет собой антагонист CGRP-рецептора. Изобретение также относится к фармацевтическим композициям и способам применения соединения для лечения расстройств, связанных с CGRP,включающих в себя мигренозную головную боль, нейрогенную вазодилатацию, нейрогенное воспаление,термическое повреждение, циркуляторный шок, гиперемию, связанную с менопаузой, воспалительные заболевания дыхательных путей, таких как астма, хроническое обструктивное заболевание легких(ХОЗЛ), и раковую опухоль. Пептид, связанный с геном кальцитонина (CGRP), представляет собой естественный 37 аминокислотый пептид, впервые выявленный в 1983 г. (Amara S.G. и др. Science 1982, 298, 240-244). Экспрессируются две формы пептида (CGRP и CGRP), которые различаются первой и третьей аминокислотами у крыс и людей соответственно. Пептид имеет широкое распространение как в периферической (ПНС), так и центральной нервной системе (ЦНС), преимущественно локализован в сенсорных афферентных и центральных нейронах и отображает количество биологических эффектов, включающих вазодилатацию. При освобождении из клетки CGRP связывается со специфическими G белок-сопряженными рецепторами, расположенными на клеточной поверхности, и оказывает свое биологическое действие преимущественно путем активации внутриклеточной аденилатциклазы (Poyner D.R. и др. Br J Pharmacol 1992,105, 441-7; Van Valen F. и др. Neurosci Lett 1990, 119, 195-8). Два класса CGRP рецепторов, CGRP1 иCGRP2, были предложены на основе антагонистических свойств фрагмента пептида CGRP(8-37) и способности линейных аналогов CGRP к активизации CGRP2 рецепторов (Juaneda С. и др. TiPS 2000, 21,432-438). Однако существует недостаточно молекулярных данных в отношении GCRP2 рецепторов(Brain S.D. и др. TiPS 2002, 23, 51-53). CGRP1 рецептор включает три компонента: (i) 7 трансмембранных рецепторов, подобных рецепторам кальцитонина (CRLR); (ii) один сигнальный трансмембранный белок 1 типа, модифицирующий активность рецептора (RAMP1); и (iii) внутриклеточный белок-компонент рецептора (RCP) (Evans В.N. и др. J Biol Chem. 2000, 275, 31438-43). RAMP1 необходим для транспортировки CRLR к клеточной мембране и для связывания лигандов с CGRP-рецептором (McLatchie L.М. и др.Nature 1998, 393, 333-339). RCP необходим для сигнальной трансдукции (Evans В.N. и др. J Biol Chem. 2000, 275, 31438-43). Известны видоспецифичные различия связывания низкомолекулярных антагонистов с CGRP-рецептором обычно с более высокой аффинностью, наблюдаемой при антагонизме в отношении рецепторов человека, чем в отношении других видов (Brain S.D. и др. TiPS 2002, 23, 51-53). Аминокислотная последовательность RAMP1 определяет типы селективности, в частности аминокислотный остаток Trp74, отвечает за фенотип рецептора человека (Mallee и др. J Biol Chem. 2002, 277, 14294-8). Предполагается, что ингибиторы CGRP на рецепторном уровне применимы при патофизиологических состояниях, при которых наблюдается чрезмерная активация CGRP рецептора. Некоторые из них включают нейрогенную вазодилатацию, нейрогенное воспаление, мигрень, кластерную головную боль и другие головные боли, термическую травму, циркуляторный шок, менопаузальные приливы и астму. Активация CGRP рецептора была вовлечена в патогенез мигрени (Edvinsson L. CNS Drugs 2001; 15(10): 745-53; Williamson D.J. Microsc. Res. Tech. 2001, 53, 167-178.; Grant A.D. Brit. J. Pharmacol. 2002. 135, 356362). Уровень CGRP в сыворотке повышается во время мигрени (Goadsby P.J. и др. Ann Neurol 1990; 28: 183-7), а лечение лекарствами против мигрени возвращает уровень CGRP к нормальному, совпадающему с облегчением головной боли (Gallai V. и др. Cephalalgia 1995; 15: 384-90). Лицо, страдающее мигренозными головными болями, показывает повышенные основные уровни CGRP по сравнению с контролями(Ashina M. и др. Pain 2000, 86(1-2): 133-8.2000). Внутривенное вливание CGRP продуцирует продолжительную головную боль в мигренозную (Lassen L.H. и др. Cephalalgia 2002 Feb; 22(1): 54-61). Доклинические исследования на собаке и крысе сообщают, что системная блокада CGRP с пептидом антагонистомCGRP(8-37) не изменяет состояние покоя системной гемодинамики, ни регионального кровотока (ShenY-T. и др. J Pharmacol Exp Ther 2001, 298, 551-8). Таким образом, антагонисты CGRP-рецептора могут представлять собой новое для лечения мигрени, что позволяет избегать сердечно-сосудистой недостаточности в результате активной вазоконстрикции, связанной с неселективными 5-HT1B/1D агонистами,триптанами (например, суматриптан). Антагонисты CGRP показали эффективность при клинических испытаниях на человеке (см. DavisA.M.; Neurology 2008, 70: 1304. Epub 2007 Oct 3). Изобретение обеспечивает техническое преимущество, например, соединение является новым и ингибирует CGRP. Кроме того, соединение обеспечивает преимущества для фармацевтического применения, например, относительно некоторых своих механизмов действия, связывания, эффективности ингибирования, дискриминации мишеней, растворимости, профиля безопасности или биологической доступности. Антагонисты CGRP рецептора были описаны в публикации РСТ, включая WO 2003/104236. Подробное описание изобретения Изобретение включает (R)-N-(3-(7-метил-1H-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамид (соединение I или соединение формулы I) и фармацевтические композиции и способы для модуляции CGRP и лечения пациентов с заболеваниями, связанными с аберрантным уровнем CGRP или сигнального CGRP рецептора Изобретение включает в себя все фармацевтически приемлемые формы солей соединений. Фармацевтически приемлемые соли представляют собой соли, с помощью которых противоионы не вносят существенный вклад в физиологическую активность или токсичность соединений и, по существу, выполняют функцию фармакологических эквивалентов. Данные соли могут быть получены согласно общепринятым органическим способам с использованием коммерчески доступных реагентов. Некоторые формы анионных солей включают в себя ацетат, ацистрат, безилат, бромид, хлорид, цитрат, фумарат, глюкоуронат, гидробромид, гидрохлорид, йодгидрат, йодид, лактат, малеат, мезилат, нитрат, памоат, фосфат, сукцинат, сульфат, тартрат, тозилат и ксинофоат. Некоторые формы катионных солей включают в себя аммоний, алюминий, бензатин, висмут, кальций, холин, диэтиламин, диэтаноламин, литий, магний, меглюмин, 4-фенилциклогексиламин, пиперазин, калиевые, натриевые, трометамин и цинк. Изобретение предназначено для включения в себя всех изотопов атомов, возникающих в представленных соединениях. Изотопы включают в себя те атомы, имеющие одинаковый атомный номер, но разное массовое число. В качестве общего примера и не ограничиваясь, изотопы водорода включают в себя дейтерий и тритий. Изотопы углерода включают в себя 13 С и 14 С. Изотопно-меченные соединения настоящего изобретения, как правило, могут быть получены с помощью общепринятых способов, известных специалистам в данной области техники, или с помощью способов, аналогичных описанным в данной заявке, при использовании соответствующего изотопно-меченного реагента вместо иного используемого немеченого реагента. Такие соединения могут иметь различные возможные применения, например, в качестве стандартов или реагентов при определении биологической активности. Что касается стабильных изотопов, такие соединения могут иметь потенциал для благоприятного изменения биологических, фармакологических и фармакокинетических свойств. Синтетические способы Сокращения, как правило, заменяют условные обозначения, используемые в данной области техники. Химические сокращения, используемые в описании и примерах, определяются следующим образом:"NaHMDS" для натрия бис-(триметилсилил)амида; "DMF" для N,N-диметилформамида; "МеОН" для метанола; "NBS" для N-бромсукцинимида; "Ar" для арила; "TFA" для трифторуксусной кислоты; "LAH" для алюмогидрида лития; "ВОС", "DMSO" для диметилсульфоксида; "ч" для часов; "rt" для комнатной температуры или времени удержания (определяется контекстом); "мин" для минут; "EtOAc" для этилацетата; "THF" для тетрагидрофурана; "ЭДТА" для этилендиаминтетрауксусной кислоты; "Et2O" для диэтилового эфира, "DMAP" для 4-диметиламинопиридина, "DCE" для 1,2-дихлорэтана; "ACN" для ацетонитрила; "DME" для диметоксиэтана; "HOBt" для 1-гидроксибензотриазола гидрата; "DIEA" для диизопропилэтиламина, "Nf для CF3(CF2)3SO2- и "TMOF" для триметоксиметана. Сокращения, используемые в данной заявке, определяются следующим образом: "1" для одного раза, "2" для двух раз, "3" для трех раз подряд, "С" для градусов Цельсия, "eq" для эквивалента или эквивалентов, "г" для грамм или граммов, "мг" для миллиграмма или миллиграммов, "л" для литра или литров, "мл" для миллилитра или миллилитров, "мкл" для микролитра или микролитров, "N" для нормы,"М" для молярного, "'ммоль" для миллимоля или миллимолей, "мин" для минуты или минут, "ч" для часа или часов, "rt" для комнатной температуры, "RT" для времени удержания, "atm" для атмосферы, "psi" для фунтов на квадратный дюйм, "cone." для концентрата, "sat" или "sat'd " для насыщенного, "MW" для молекулярной массы, "mp" для температуры плавления, "ее" для энантиомерного избытка, "MS" или "MassSpec" для масс-спектрометрии, "ESI" для масс-спектроскопии с электрораспылительной ионизацией,"HR" для высокого разрешения, "HRMS" для масс-спектрометрии с высоким разрешением, "LCMS" для жидкостной хроматографии - масс-спектрометрии, "HPLC" для жидкостной хроматографии высокого давления, "RP HPLC" для обращенно-фазовой HPLC, 'TLC" или "tlc" для тонкослойной хроматографии,"NMR" для спектроскопии ядерного магнитного резонанса, 1 Н" для протона, для дельты, "s" для синглета, "d" для дуплета, "t" для триплета, "q" для квартета, "m" для мультиплета, "br" для общего, "Hz" для герц, а "а", "р", "R", "S", "Е" и "Z" представляют собой стереохимические обозначения, известные специалистам в данной области техники. Соединение I может быть приготовлено согласно схеме 1 Данный синтез представляет собой 14 химических этапов и сильно конвергентное соединение трех главных фрагментов в течение последних трех этапов. В связи с этим синтез начинается с приготовления главных фрагментов А (схема 2) и В Синтез фрагмента А начинается с реакции Хорнера-Эммонса N-Boc-4-пиперидона с илидом, полученным от триметилфосфоноацетата, с получением трет-бутил 4-(2-метокси-2-оксоэтилиден)пиперидин 1-карбоксилата в превосходном количестве произведенного продукта (схема 2). Каталитическое гидрирование, опосредованное палладиевым катализатором на углеродном носителе, снижает ненасыщенные двойные связи. Обработка трет-бутил 4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата с LDA образовывает еноляты, которые в результате захвата 2-нитробензальдегида обеспечивают нитроэтанол. Снижение нитрогруппы железом в уксусной кислоте с последующей обработкой хлороводородом в диоксане завершает синтез фрагмента А. Схема 2 Синтез индазолсодержащей аминокислоты В начинается с йодирования 2,6-диметиланилина под действием монохлорида йода (схема 3). Данный промежуточный продукт был временно отделен. Сложный метиловый эфир N-CBZ-L-серина подвергался реакции метансульфонилирования/элиминации, проходящей в одном реактивном сосуде для получения сложного метилового эфира N-CBZ-дегидроаланина. При наличии йодида и дегироаланина они эффективно связываются с использованием палладия (II) ацетата в реакции сочетания Хека для получения продукта с выходом 65%. На этой стадии хиральный центр получают с помощью каталитического ассиметричного гидрирования с использованием (-)-1,2-бис 2R,5R)-2,5-диэтилфосфолано)бензен(циклооктадиен)родий(I) тетрафторбората и водорода (60 psi) для получения хиральной аминокислоты в 96% ее. Кольцо индоназола затем формируется под действием изоамилнитрита. Полученный инданозол является высококристаллическим. Одна рекристаллизация из ацетона/гексана дает инданозолсодержащую аминокислоту отличной чистоты и с улучшением 99,8% ее. Удаление CBZ защитной группы в условиях гидрогенизации завершает приготовление фрагмента В. Индазолсодержащая аминокислота В может быть также приготовлена с применением ферментативного расщепления рацемической аминокислоты или кетокислоты (Hanson, Ronald L.; Davis, Brian L.; Goldberg,Steven L.; Johnston, Robert M.; Parker, William L.; Tully, Thomas P.; Montana, Michael A.; Patel, Ramesh N. Фрагменты А и В эффективно связываются с использованием N,N'-дисукцинимидилкарбоната для введения фрагмента мочевины с выходом 78% (схема 4). Омыление сложного метилового эфира с гидроксидом лития дает почти количественный выход карбоновой кислоты. TBTU опосредованно соединяет кислоту с 1-(1-метилпиперидин-4-ил)пиперазином, завершая синтез соединения I. Флэш-хроматография обеспечивает продукт в виде аморфного порошка, который может быть кристаллизирован из ацетона для получения соединения I в виде мелкого белого кристаллического порошка. Схема 4 трет-Бутил 4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилат Гидрид натрия в минеральном масле (60%, 7.92 г, 198.02 ммоль) промывали с гексаном, затем суспендировали в диметилформамиде (220 мл). Смесь охлаждали до 0 С. Триметилфосфоноацетата (29.0 мл, 189.82 ммоль) добавляли по каплям при перемешивании реакционной смеси. После 20 мин при 0 С раствор N-трет-бутоксикарбонил-4-пиперидона (30.41 г, 152.62 ммоль) в диметилформамиде (80 мл) добавляли к смеси по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и затем разбавляли диэтиловым эфиром (650 мл). Смесь промывали один раз водой, а водный слой экстрагировали один раз с диэтиловым эфиром. Объединенные органические слои промывали 4 раза водой, а водную фазу удаляли. Органическую фазу промывали солевым раствором и сушили над сульфатом магния, фильтровали и концентрировали досуха Титульное соединение получали в виде белого твердого вещества с выходом 92%. 1H-NMR (300 MHz,CDCl3)5.68 (s, 1 Н), 3.66 (s, 3 Н), 3.40-3.51 (m, 4 Н), 2.90 (t,J= 5.49 Hz, 2H), 2.25 (t, J= 5.49 Hz, 2H), 1.44 (s, 9H). трет-Бутил 4-(2-метокси-2-оксоэтип)пиперидин-1-карбоксилат Раствор трет-бутил 4-(2-метокси-2-оксоэтилиден)пиперидин-1-карбоксилата (35.71 г, 140 ммоль) в смеси 1:1 этилацетата/метанола (220 мл) осторожно обрабатывали с 50% увлажненным 10%-ным палладиевым катализатором на угле (3.3 г). Реакционный сосуд был наполнен 55 psi газообразного водорода и смесь встряхивали в аппарате Парра при комнатной температуре в течение 16 ч. Затем реакционную смесь фильтровали для удаления катализатора и концентрированного фильтрата в вакууме. Титульное соединение получали в виде прозрачного бесцветного масла с выходом 97%. 1H-NMR (300 MHz, CDCl3)4.04 (d, J= 10.25 Hz, 2H), 3.64 (s, 3H), 2.68 (t, J= 12.44 Hz, 2H), 2.21 (d, J= 6.95 Hz, 2H), 1.98-1.77 (m, 1H),1.64 (d, J= 13.54 Hz, 2H), 1.41 (s, 9H), 1.25-0.99 (m, 2H).N,N-диизопропиламин (4.40 мл, 31.3 ммоль) растворяли в тетрагидрофуране (50 мл). Смесь охлаждали до -78 С. Бутиллитий (2.5 М в гексане, 12.4 мл, 31 ммоль) добавляли по каплям к перемешиваемому раствору. После перемешивания при -78 С в течение 30 мин раствор трет-бутил 4-(2-метокси-2 оксоэтил)пиперидин-1-карбоксилата (6.65 г, 25.8 ммоль) в тетрагидрофуране (15 мл) добавляли по каплям в смесь. Перемешивание продолжали при -78 С в течение 1 ч. Раствор 2-нитробензальдегида (3.90 г,25.8 ммоль) в тетрагидрофуране (20 мл) был затем добавлен к смеси по каплям и перемешивание продолжали при -78 С в течение еще 2.5 ч. Реакционную смесь резко охлаждали холодным водным хлоридом аммония и затем разводили водой. Смесь экстрагировали два раза этилацетатом, а водную фазу удаляли. Вещество сушили (сульфат магния) фильтрованием и концентрировали досуха. Хроматографией на силикагеле получали желаемый продукт с выходом 94% в виде светло-желтой пены. MS m/e (MC4H8+H)+ = 353.1. трет-Бутиловый эфир 4-(4-гидрокси-2-оксо-1,2,3,4-тетрагидрохинолин-3-ил)пиперидин-1-карбоновой кислоты В трехголовой колбе, оснащенной выпускным отверстием для азота, термометром и механической мешалкой, трет-бутиловый эфир 4-[2-гидрокси-1-метоксикарбонил-2-(2-нитрофенил)этил]пиперидин-1 карбоновой кислоты (9.93 г, 24.3 ммоль) разводили в уксусной кислоте (1.75 моль, 100 мл). Железный порошок (8.90 г, 159 ммоль) добавляли в колбу с перемешиванием. Перемешиваемую смесь медленно нагревали до 80 С в течение 30 мин, а затем охлаждали до комнатной температуры. Затем смесь разводили этилацетатом и фильтровали через прокладку из целита. Твердые вещества промывали 20% метанолом/этилацетатом, а затем метанолом. Фильтрат концентрировали и остаток разделяли между этилацетатом и водным раствором бикарбоната натрия. Слои разделяли. Полученные водные фазы экстрагировали два раза этилацетатом. Органические слои объединяли. Смесь промывали два раза водой, а водную фазу удаляли. Вещество сушили (сульфат магния) фильтрованием и концентрировали досуха. Хроматографией на силикагеле получали титульное соединение в виде светло-желтой пены с выходом 77%. MS 3-(Пиперидин-4-ил)хинолин-2(1 Н) гидрохлорид Перемешиваемый раствор трет-бутилового эфира 4-(4-гидрокси-2-оксо-1,2,3,4-тетрагидрохинолин 3-ил)пиперидин-1-карбоновой кислоты (5.60 г, 16.2 ммоль) в этилацетате (70 мл) обрабатывали HCl в диоксане (4 N, 40 ммоль, 10 мл). Смесь перемешивали при комнатной температуре в течение 45 мин. Дополнительно HCl в диоксане (4 N, 120 ммоль, 30 мл) затем добавляли и перемешивание продолжали при комнатной температуре в течение 16 ч. Полученное твердое вещество собирали с помощью фильтрации и промывали этилацетатом. Затем суспендировали в 5% водном растворе изопропанола (100 мл) и смесь нагревали с обратным холодильником и перемешивали в течение 20 мин. Смесь охлаждали до комнатной температуры в течение 16 ч. Твердое вещество собирали с помощью фильтрации, промывали изопропанолом и сушили под высоким вакуумом. Титульное соединение было получено в виде белого твердого вещества с выходом 75%. 1H-NMR (DMSO-d6)11.85 (s, 1 Н), 9.02 (bs, 1 Н), 8.88 (bs, 1 Н), 7.70 (t, J= 3.81 4-Иод-2,6-диметилбензенамина гидрохлорид К суспензии бикарбоната натрия (126 г, 1.5 моль) и 2,6-диметилаланина (61.5 мл, 500 ммоль) в метаноле (700 мл) добавляли монохлорид иода (1.0 М в дихлорметане, 550 мл, 550 ммоль) при комнатной температуре в течение 1 ч. После завершения добавления перемешивание продолжали в течение 3 ч. Реакционную смесь фильтровали для удаления избытка бикарбоната натрия, а растворитель удаляли под вакуумом. Остаток повторно растворяли в диэтиловом эфире (1.5 л) и обрабатывали соляной кислотой (2 М в эфире, 375 мл, 750 ммоль). Полученную суспензию хранили в морозильном аппарате (-15 С) всю ночь. Твердое вещество фильтровали и промывали диэтиловым эфиром до тех пор, пока оно не становилось бесцветным, для получения 126.5 г (89%) в виде серо-зеленого порошка. 1H-NMR (DMSO-d6)2.33 Метил 2-(бензилоксикарбонил)акрилат В высушенную пламенем круглодонную трехгорловую колбу, снабженную механической мешалкой, добавляли (S)-метил 2-(бензилоксикарбонил)-3-гидроксипропаноат (129 г, 509 ммоль), безводный дихлорметан (2 л) и метансульфонилхлорид (49,3 мл, 636 ммоль). Смесь охлаждали до -15 С и обрабатывали триэтиламином (213 мл, 1527 ммоль) по каплям в целях обеспечения температуры реакционной смеси, не превышающей 0 С. Дополнительный первый эквивалент триэтиламина был экзотермический. После добавления триэтиламина смесь перемешивали при 0 С в течение 30 мин. Охлаждающую баню убирали и смесь перемешивали при комнатной температуре в течение 1,5 ч. Реакционную смесь резко охлаждали добавлением метанола (21 мл). Смесь промывали 0,5% водным раствором бисульфата калия до тех пор, пока обогащение будет рН 5, затем насыщенным раствором бикарбоната натрия и солевым раствором сушили над сульфатом натрия и концентрировали. Флэш-хроматография (силикагель, 1:9 этилацетат/гексан) дает 111 г (92%) в виде вязкого бесцветного масла, которое кристаллизуется в неподвижности. 1H-NMR (DMSO-d6)3.71 (s, 3 Н), 5.10 (s, 2 Н), 5.60 (s, 1 Н), 5.76 (s, 1 Н), 7.39-7.35 (m, 5 Н), 8.96(Z)-Метил 3-(4-амино-3,5-диметилфенил)-2-(бензилоксикарбонил)акрилат 2-л Круглодонная колба была наполнена 4-иод-2,6-диметилбензенамин хлористо-водородной солью(55 г, 194 ммоль), метил 2-(бензилоксикарбонил)акрилатом (59.2 г, 252 ммоль), хлоридом тетрабутиламмония (59.2 г, 213 ммоль), палладия (II) ацетатом (4.34 г, 19.4 ммоль) и тетрагидрофураном (1.2 л, дегазированный потоком азота в течение 30 мин). Смесь перемешивали так, чтобы была образована суспензия, а затем дегазировали потоком азота в течение 30 мин. Триэтиламин (110 мл, 789 ммоль) добавляли и полученную смесь нагревали с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали через прокладку из целита, промывали тетрагидрофураном (2100 мл) и концентрировали. Остаток растворяли в дихлорметане, промывали водой (3) и солевым раствором (2), сушили над сульфатом натрия и концентрировали. Флэш-хроматография (силикагель, применяя 1:9 этилацетат/дихлорметан) дает желтовато-коричневое твердое вещество. Твердое вещество рекристаллизовали с нагретым метанолом (210 мл) и водой (100 мл). Смесь выдерживали при комнатной температуре всю ночь, затем при 0 С в течение 2 ч и в конце при -15 С в течение 2 ч. Полученное твердое вещество фильтровали, промывали очень холодным 1:1 метанолом/водой и сушили под высоким вакуумом для получения 44.7 г (65%) в виде светло-коричневого твердого вещества, которое представляет собой смесь Z/E изомеров (73:27). 1H-NMR (DMSO-d6)2.05 (s, 6 Н), 3.61 (s, 0.8 Н), 3.68 (s,2.2 Н), 5.00 (s, 0.54 Н), 5.13 (s, 1.46 Н), 5.24 (s, 2 Н), 7.40-7.21 (m, 8 Н), 8.51 (s, 0.27 Н), 8.79 (s, 0.73 Н); 13CNMR (DMSO-d6)17.8, 51.7, 65.3, 119.4, 120.0, 120.3, 127.3, 127.7, 128.3, 130.9, 135.8, 137.2, 146.9, 154.7,166.0.(R)-Метил 3-(4-амино-3,5-диметилфенил)-2-(бензилоксикарбонил)пропаноат Высушенная пламенем 2-л колба Парра для гидрогенизации была наполнена (Z)-метил 3-(4-амино 3,5-диметилфенил)-2-(бензилоксикарбонил)акрилатом (84.5 г, 239 ммоль), дихлорметаном (300 мл) и метанолом (300 мл). Колбу перемешивали путем вращения так, чтобы образовалась светло-коричневая суспензия. Смесь дегазировали, применяя поток азота в течение 30 мин. К этому быстро добавляли (-)1,2-бис-2R,5R)-2,5-диэтилфосфолано)бензен(циклооктадиен)родия (I) тетрафторборат ([(2R,5R)-EtDuPhosRh]BF4) (2.11 г, 3.20 ммоль). Колбу сразу прикрепляли к гидрогенезатору Парра после 5 циклов водорода (60 psi) и вакуума колба была под давлением до 65 psi и суспензию перемешивали при комнатной температуре в течение 16 ч. Реакционная смесь становилась однородной. Реакционную смесь концентрировали, а полученный остаток очищали флэш-хроматографией (силикагель, 1:9 этилацетат/дихлорметан) до получения 82.9 г (98%). 1H-NMR (DMSO-d6)2.04 (s, 6 Н), 2.65 (dd, J= 13.4, 9.8 Hz, 1H),2.82 (dd, J= 13.7, 5.2 Hz, 1H), 3.62 (s, 3H), 4.15-4.10 (m, 1H), 4.41 (s, 2H), 5.00 (s, 2H), 6.68 (s, 2H), 7.37-7.28(R)-Метил 3-(4-амино-3,5-диметилфенил)-2-(бензилоксикарбонил)пропаноат (50.0 г, 140 ммоль) взвешивали в высушенной пламенем круглодонной трехгорловой колбе с последующим добавлением толуола (2.4 л) и ледяной уксусной кислоты (120 мл, 2.1 моль). Смесь механически перемешивали для получения прозрачного раствора, а затем добавляли ацетат калия (103 г, 1.05 моль). В полученную белую суспензию изоамилнитрит (20.7 мл, 154 ммоль) добавляли по каплям при комнатной температуре и полученную смесь перемешивали при комнатной температуре в течение 16 ч. Добавляли насыщенный бикарбонат натрия(1 л) с последующим осторожным добавлением твердого бикарбоната натрия для нейтрализации уксусной кислоты. Смесь экстрагировали со смесью дихлорметана (2 л) и солевого раствора (1.5 л). После разделения водный слой экстрагировали с дихлорметаном (500 мл). Объединенные органические слои сушили над безводным сульфатом натрия и фильтровали. Растворители удаляли для получения твердого вещества желтовато-коричневого цвета, которое промывали с гексаном (2 л) и толуолом (150 мл). Твердое вещество рекристаллизировали из горячего ацетона (260 мл) и гексана (700 мл). Слегка мутной смеси медленно давали остыть до комнатной температуры, затем до 0 С в течение 1.5 ч и в конце до -15 С в течение 1.5 ч. Полученное твердое вещество фильтровали и промывали очень холодным ацетоном/гексаном (1:1, 200 мл) для получения 39.1 г(76% выход). Аналитический HPLC показал 98% УФ-чистоты. Энантиомерный избыток (ее) был определен как 99.8% (условия: колонка Chiralpak AD, 4.6250 мм, 10 мкм; А = этанол, В = 0.05% диэтиламин/гептан; 85%В 1.0 мл/мин. В течение 55 мин. Время удержания для R составляло 44.6 мин, а для S составляло 28.8 мин). 1H-NMR (DMSO-d6)2.48 (s, 3 Н), 2.93 (dd, J= 13.4, 10.7 Hz, 1H), 3.10 (dd, J= 13.7, 4.9 Hz, 1H), 3.63 (s,3H), 4.32-4.27 (m, 1H), 4.97 (s, 2H), 7.03 (s, 1H), 7.24-7.22 (m, 2H), 7.29-7.27 (m, 3H), 7.41 (s, 1H), 7.83 (d, J= 8.2(R)-Метил 2-амино-3-(7-метил-1 Н-индазол-5-ил)пропаноат Сосуд гидрогенизации Парра был наполнен (R)-метил 2-(бензилоксикарбонил)-3-(7-метил-1 Н-индазол-5-ил)пропаноатом (11.0 г, 29.9 ммоль) и метанолом (75 мл). Суспензию очищали азотом и обрабатывали палладием (10%-ным на угле, 700 мг). Колбу встряхивали в водороде (15 psi) на протяжении всей ночи. Полученную смесь фильтровали через прокладку из целита для удаления катализатора. Концентрация элюента давала 7.7 г (quant.) в виде масла, которое применяли без дополнительной очистки. 1HNMR (CD3OD)2.54 (s, 3 Н), 2.98 (dd, J= 135, 7.0 Hz, 1H), 3.09 (dd, J= 13.5, 5.9 Hz, 1H), 3 68 (s, 3H), 3.75(R)-метил 3-(7-метил-1 Н-индазол-5-ил)-2-(4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамидо)пропаноат К раствору (R)-метил 2-амино-3-(7-метил-1 Н-индазол-5-ил)пропаноат гидрохлорида (7.26 г, 27.0 ммоль) в диметилформамиде (50 мл) при комнатной температуре был добавлен N,N-дисукцинимидилкарбонат (7.60 г, 29.7 ммоль), а затем триэтиламин (11.29 мл, 81 ммоль). Полученную смесь перемешивали в течение 30 мин и обрабатывали 3-(пиперидин-4-ил)хинолин-2(1 Н)-он (6.77 г, 29.9 ммоль) в частях. Реакционную смесь перемешивали в течение 24 ч. Смесь концентрировали, растворяли в этилацетате и промывали последовательно водой, солевым раствором и 0.5 N HCl (2). Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали. Полученный остаток очищали с помощью флэш-хроматографии (силикагель, 20:1 этилацетат/метанол) для получения 11.9 г (78%). 1H-NMR (CD3OD)13.0 (s, 1H), 11.8 (s, 1H), 7.98 (s, 1H), 7.63 (d,J= 7.6 Hz, 1H), 7.57 (s, 1H), 7.45-7.41 (m, 2H), 7.27 (d, J= 8.2 Hz, 1H), 7.16 (t, J= 7.9 Hz, 1H), 7.03 (s, 1H), 6.85 (d,J= 7.9 Hz, 1H), 4.31-4.26 (m, 1H), 4.10-4.08 (m, 2H), 3.60 (s, 3H), 3.07-3.01 (m, 2H), 2.93-2.88 (m, 1H), 2.77-2.67(R)-3-(7-Метил-1 Н-индазол-5-ил)-2-(4-(2-оксо-1,2-дигидрохинолин-3-ип)пиперидин-1-карбоксамидо)пропановая кислота Раствор (R)-метил 3-(7-метил-1H-индазол-5-ил)-2-(4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин 1-карбоксамидо)пропаноата (5.50 г, 11.3 ммоль) в тетрагидрофуране (50 мл) и метаноле (10 мл) охлаждали до 0 С. К этому добавляли холодный (0 С) раствор моногидрата гидрооксида лития (0.95 г, 22.6 ммоль) в воде (20 мл) по каплям в течение 15 мин. Реакционную смесь перемешивали при комнатной температуре в течение дополнительных 3 ч. Смесь концентрировали для удаления органических растворителей. Полученный остаток растворяли в минимальном количестве воды, охлаждали до 0 С и обрабатывали холодным (0 С) 1 N HCl до достижения рН 2. Полученное твердое вещество собирали фильтрованием, промывали холодной водой и эфиром, а затем сушили всю ночь под высоким вакуумом, получая 5.0 г (94%) в виде белого твердого вещества. 1H-NMR (DMSO-d6)13.05 (bs, 1 Н), 11.77 (s, 1 Н), 7.98 (s,1 Н), 7.62 (d, J= 8.0 Hz, 1H), 7.55 (s, 1H), 7.44 (d, J= 8.2 Hz, 1H), 7.42 (s, 1H), 7.27 (d, J= 8.2 Hz, 1H), 7.16 (t,J= 7.6 Hz, 1H), 7.05 (s, 1H), 6.65 (d, J= 7.9 Hz, 1H), 4.27-4.22 (m, 1H), 4.10-4.07 (m, 2H), 3.12-3.07 (m, 1H),3.03-2.99 (m, 1H), 2.93-2.88 (m, 1H), 2.77-2.66 (m, 2H), 2.47 (s, 3H), 1.77-1.74 (m, 2H), 1.34-1.27 (m, 2H). Масс-спектр: 474.30 (МН)+.(R)-3-(7-метил-1H-индазол-5-ил)-2-(4-(2-оксо-1,2-дигидрохинолин-3 ил)пиперидин-1-карбоксамидо)пропановой кислотой (2.9 г, 6.11 ммоль), триэтиламином (3.00 мл, 21.5 ммоль), 1-(1-метилпиперидин-4-ил)пиперазином (1.23 г, 6.72 ммоль) и диметилформамидом (10 мл). Полученный раствор обрабатывали 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборатом(2.26 г, 7.03 ммоль) в частях. Реакционную смесь перемешивали при комнатной температуре всю ночь. Смесь концентрировали под вакуумом для удаления диметилформамида. Неочищенный продукт растворяли в 7% метаноле в дихлорметане и очищали флэш-хроматографией с использованием 7% метанола в дихлорметане, содержащем 2% водного раствора гидроксида алюминия в качестве элюента. Чистые фракции собирали и растворитель удаляли под вакуумом. Целевой продукт кристаллизировали из горячего ацетона для получения соединения I с выходом 77%. Аналитический HPLC показал 99.0% УФчистоты при 230 нм. Энантиометрический избыток (ее) был определен как 99.9% (условия: колонка Биологические способы и другие характеристики Схема 5. Соединение I и препараты сравнения соединения II и III Растворимость в воде. Твердое вещество в виде свободного основания смешивали с карбонатным и этаноламиновым буферами. рН третьего образца устанавливали с использованием HCl. Твердые вещества были смешаны с носителями с использованием мешалки, которая была помещена в инкубатор, установленный на 25 С. После уравновешивания образцы супернатанта удаляли, разбавляли по необходимости и анализировали с помощью HPLC. Растворимость в водной среде, рН зависимость. Кристаллическое соединение I преобразовывалось в гелеобразную фазу при измерении рН профиля растворимости. Отсутствие кристаллической фазы в равновесии с водой не дает возможности уверенно оценить диапазона рН, в котором соединение I может быть получено в виде термодинамически стабильного раствора. Действия для получения кристаллической формы свободного основания соединения I, которое может быть уравновешано в воде, непрерывные. Данные, собранные на гелеобразной фазе, перечислены в табл. 1. Таблица 1. Растворимость соединения I в водной среде при 25 С Стабильное состояние раствора. Стабильное состояние раствора соединения I было оценено в зависимости от температуры, рН, светового излучения высокой эффективности (HIL), концентрации буфера и концентрации препарата. Применяемые экспериментальные матрицы показаны в табл. 2. Таблица 2. Стабильное состояние матричного раствора Сукцинатный буфер применяли для всех растворов. Для образцов, подвергающихся воздействию света, использовали камеру для испытания фотостабильности согласно руководству ICH (экспозиция 1.2 миллион люкс в час для видимого света и 200 Ватт ч/м 2 для УФ). Анализ осуществляли на 4, 8 и 12 недели. Десять продуктов распада были обнаружены в ходе исследования стабильности раствора. Процент-9 022815 ное содержание каждого продукта распада обнаружено в образцах после 12 недель хранения, приведены в табл. 3. Необходимо отметить, что в начальный момент времени продукты распада В и G были единственными представленными продуктами распада и их начальная концентрация составляла 0.17 и 0.06% соответственно. Масс-спектроскопическую оценку осуществляли на этих продуктах распада, присутствующих в достаточном количестве для возможности измерения массы. Результаты приведены в табл. 4. Приведены два пика под продуктом распада В, потому что было обнаружено, что два продукта распада недостаточно хорошо разделяются текущим способом HCLP. Продукты распада А и С соответствуют продуктам гидролиза. В наиболее благоприятных условиях исследования условия I в табл. 2 соединение I показало только 0,3% разрушения после 12 недель при 40 С. Это означает, что приведен водосодержащий состав, который соответствует руководству ICH, по меньшей мере, в химической стабильности на год. Таблица 3. Площадь процентного содержания продуктов распада, увиденная на двенадцатую неделю, и их время удержания (мин) Таблица 4. Молекулярная масса продукта распада Стабильность раствора: температурное воздействие. Температурное воздействие кратко изложено в табл. 5. Повышение в скорости распада с температурой в значительной степени связано с усилением в гидролизе. При высокой температуре распад продолжается непрерывно до продукта С, но при низкой температуре распад прекращается на продукте А. Таблица 5. Стабильность раствора и температура Стабильность раствора: воздействие света. Воздействие света на стабильность кратко изложено в табл. 6. Семь продуктов распада были отмечены для образцов под воздействием света, трое из которых не присутствовали в образцах, хранились в любом другом состоянии. Гидролиз (продукт распада А) усиливался под воздействием света. Таблица 6. Стабильность раствора и освещение Стабильность раствора: влияние рН. Влияние рН на скорость распада кратко изложено в табл. 7. Усиление в скорости распада изменялось обратно пропорционально рН и было преобладание усиления в гидролизе. При рН 4 распад переходил к гидролизу продукта С, но при рН 5 и 6 распад останавливался на гидролизе продукта А. Таблица 7. Стабильность раствора и рН Стабильность раствора: воздействие концентрации буфера. Высокие концентрации сукцинатного буфера, как правило, вызывали высокую скорость гидролиза продукта распада А (табл. 8). Таблица 8. Стабильность раствора и Концентрация сукцинатного буфера Стабильность раствора: воздействие концентрации препарата. Высокая загрузка препарата замедляла скорость гидролиза и ограничивала гидролиз для образования продукта распада А (табл. 9). Продукт распада G, который определялся только в образцах высокой концентрации, незаметно увеличивался в концентрации в процессе исследования и возможно только был примесью. Продукт распада J можно только было обнаружить в конечной точке времени. Таблица 9. Стабильность раствора и концентрация соединения I Конкуренция за [125I]CGRP связывание Анализ связывания. Гомогенат мембран клеток SK-N-MC выступал в качестве источника рецепторов. Клетки нейробластомы человека SK-N-MC применяли для исследования in vitro, потому что они экспрессируют эндогенно CGRP рецептор с индентичной последовательностью к клонированному человеческому CGRP рецептору (Aiyar и др., 2001). Клетки выращивали при 37 С в 5% CO2 среде, состоящей из MEM с солями Эрла и L-глутамина, дополненной 10% эмбриональной бычьей сывороткой для достижения слияния. Клетки собирали путем двойного промывания фосфатно-солевым буферным раствором и инкубировали в течение 5-10 мин при 4 С в гипотоническом лизирующем буфере, состоящем из 10 мМ трис (рН 7.4) и 5 мМ ЭДТА. Клетки собирали и переносили в пропиленовые трубки и гомогенизировали,применяя политрон. Гомогенаты центрифугировали при 32.000g в течение 30 мин. Осадок в пробирке ресуспендировали в холодном гипотоническом лизирующем буфере с 0.1% смеси ингибиторов протеазы млекопитающих и исследовали на концентрацию белка. Гомогенат мембран затем аликвотировали и хранили при -80 С до дня исследования. Способность соединения I к конкуренции за эндогенный человеческий пептид альфа CGRP с радиоактивной меткой ([125I]CGRP Amersham Biosciences) измеряли с использованием радиолиганда конкурентным анализом. Соединение I сначала растворяли и осуществляли последовательные разведения с использованием 100% DMSO. Далее соединение разбавляли 25 раз в аналитическом буфере (50 мМ трисCl рН 7.5, 5 мМ MgCl2, 0.005% Тритон Х-100) и переносили (50 мкл) в 96-луночные аналитические планшеты. [125I]-CGRP разводили до 600 рМ в аналитическом буфере и объем 50 мкл добавляли в каждую лунку (конечной концентрацией 15 рМ в анализе). Осадок мембран SK-N-MC размораживали, разбавляли в аналитическом буфере со свежей 0.1% смесью ингибиторов протеаз млекопитающих и гомогенизи- 11022815 ровали, как описано выше. От пяти до десяти мкг белка на лунку затем добавляли в объеме 100 мкл. Аналитические планшеты затем инкубировали при комнатной температуре (25 С) в течение 2 ч. Анализ прекращали добавлением избытка холодного промывочного буфера (20 мМ трис-Cl рН 7.5, 0.1% BSA) непосредственно с последующей фильтрацией через фильтры из стекловолокна, предварительно пропитанные 0.5% PEL. Неспецифическое связывание определяли с 1 мкМ -CGRP. Радиоактивность связанного белка измеряли с использованием сцинтилляционного гамма-счетчика. IC50 определяется как концентрация соединения, необходимая для ингибирования 50% радиолигандного связывания. Результаты. Соединение I отображает зависимость от концентрации ингибирования [125I]CGRP связывания с CGRP рецептором, эндогенно экспрессирующимся в мембранах клеток SK-N-MC. Среднее Ki 22.7 1.6 рМ. Взаимодействие с CGRP пептидом Способы. Природу взаимодействия между эндогенным CGRP пептидом и соединением I подробно изучали с применением экспериментов насыщения соединения. Вкратце, связывание [125I]CGRP с приготовленной мембраной клеток SK-N-MC измеряли с увеличением концентрации [125I]CGRP в отсутствие(условие контроля) или в присутствии (условие теста) одной из двух концентраций (30 и 100 рМ) соединения I. Данные насыщения анализировали с гиперболическим уравнением, используя программу Келли(Biosoft, Cambridge, UK) для оценки константы диссоциации (Kd) и максимального количества сайтов связывания (Bmax). Влияние добавления соединения I на параметры связывания (Kd, Bmax) [125I]CGRP измеряли и сравнивали. Результаты. Соединение I зависимым от концентрации образом увеличивает константу диссоциации Kd связывания [125I]CGRP (снижает аффинность) без значительного изменения максимального числа связывающих сайтов при связывании [125I]CGRP (Bmax). Это указывает на конкурентный механизм ингибирования соединением I связывания [125I]CGRP на рецепторе человека (табл. 10). Таблица 10. Kd, Bmax [125I]CGRP к приготовленной клеточной мембране SK-N-MC в присутствии или отсутствие соединения I Клеточный функциональный анализ - анализ циклической АМФ Способы. Рецепторный комплекс CGRP связан с белками G класса Gs. Связывание CGRP с этим комплексом приводит к образованию циклической АМФ (аденозин 3'5'-цикломонофосфат) через Gsзависимую активацию аденилатциклазы. Функциональный антагонизм соединения I определяется с помощью измерения его способности к ингибированию CGRP-стимулированного образования циклической АМФ в прикрепленных целых клетках SK-N-MC. Клетки SK-N-MC инкубировали при комнатной температуре только с 0.3 нМ CGRP в течение 30 мин или предварительно инкубировали с различными концентрациями соединения I в течение 15 мин перед добавлением 0.3 нМ CGRP, а затем продолжали инкубировать в течение 30 мин. Циклическую АМФ получали экстрагированием, используя лизирующий реагент, а ее концентрацию определяли с помощью радиоиммуноанализа, применяя набор реактивов RPA559 cAMP SPA Direct Screening Assay Kit (Amersham Pharmacia Biotech). Значения IC50 определяют как концентрацию соединения, необходимую для ингибирования 50% 0.3 нМ CGRP-стимулированного образования цАМФ. Ymax определяется как максимальный процент ингибирования 50% 0.3 нМ CGRP-стимулированного образования цАМФ. Результаты. Соединение I отображает зависимое от концентрации ингибирование CGRPстимулированного образования цАМФ в прикрепленных целых клетках SK-N-MC, с IC50 38.64.2 рМ иYmax 95.4 (1.3)%. Наблюдаемое максимальное (са. 100%) ингибирование указывает на полный антагонизм к CGRP рецептору. Анализ Шильда Способы. Анализ Шильда используется для характеристики природы антагонизма соединения I. Дозозависимый эффект CGRP-стимулированного образования цАМФ возникает либо только с CGRP,либо с CGRP в присутствии различных концентраций соединения I. При этом дозозависимую стимуляцию цАМФ с помощью CGRP исследовали с использованием или без пяти различных концентраций соединения I. Концентрация соединения I наносится на ось X против соотношения доза минус 1 на оси Y(соотношение дозы определяется как ЕС 50 CGRP в присутствии соединения I, деленное на ЕС 50 толькоCGRP). Линейную регрессию затем осуществляли логарифмическим преобразованием обеих осей X и Y. Наклон, который не значительно отличался от единицы (1), указывает на конкурентный антагонизм. Kb представляет собой константу диссоциации антагониста. Результаты. Анализ Шильда на соединение I показывал средний уклон 1.020.04 и среднюю величину константы диссоциации антагониста Kb 21.59.4 рМ. Параллельный сдвиг направо от влияния концентрации CGRP в присутствии возрастающей концентрации соединения I указывал на конкурентный антагонизм соединением I CGRP-стимулированного образования цАМФ. Наклон 1.02 от графика Шильда дополнительно поддерживал конкурентное взаимодействие между соединением I и функциейCGRP. Kb 21.5 рМ находился в соответствии со связыванием Ki (22.7 рМ). Обратимое CGRP-индуцированное расширение внутричерепной артерии человека в условиях Exvivo Для обеспечения в условиях ex vivo системы измерения, которая имитирует клиническое состояние(мигрень-связанное высвобождение CGRP предшествует началу лечения), сосуды сначала расширяли с помощью CGRP, а затем расширение реверсировали соединением I. В данном протоколе реверсированияCGRP-индуцированное расширение артерии реверсировало после обработки антагонистом (применяя единичную дозу агониста и несколько доз антагониста). Вкратце, кольца артерий с проволочным держателем сжимаются под действием иона калия (для имитации эндогенного тона), полностью расширяются под действием CGRP и это расширение реверсировало с повышением концентраций антагониста CGPR соединения I. Соединение I после обработки является эффективным при обратимо созданном CGRPиндуцированном расширении внутричерепной артерии человека в условиях ex vivo. Образцы тканей. Вскрытие образцов артерий человека получают от закупок тканей поставщиками. Все сосуды переносят в очень холодный HEPES буфер (состав в мМ: NaCl 130, KCl 4, KH2PO4 1.2,MgSO4 1.2, CaCl2 1.8, глюкоза 6, NaHCO3 4, HEPES 10, ЭДТА 0.025). После получения сосуды помещали в холодный буфер Кребса (состав в мМ: NaCl 118.4, KCl 4.7, KH2PO4 1.2, MgSO4 1.2, CaCl2 1.8, глюкоза 10.1, NaHCO3 25) насыщенный карбогеном (5% СО 2 и 95% кислорода). Способы. Сосуды очищали от соединительной ткани и нарезали цилиндрическими сегментами длиной 4-5 мм. Затем сосуды помещали в инкубаторы тканевых культур между двумя крючками из нержавеющей стали; один из которых зафиксирован, а другой из которых соединен с датчиком смещения действия. Напряженность сосуда непрерывно регистрировалась с использованием системы приема данных(Powerlab, AD Instruments, Mountain View, CA). Инкубаторы тканевых культур, содержащие буфер Кребса и установленные сосуды, находились под контролем температуры (37 С) и рН (7.4) и непрерывно газировались карбогеном. Сегменты артерий допускали уравновешивания в течение 30-45 мин до тех пор,пока достигался стабильный тон покоя (от 0.25 до 0.5 g). До исследования сосуды заливали (условно) с 100 мМ KCl, а затем промывали. Для тестирования противорасширяющего действия соединения I сосуды сначала заключали с 10 мМ хлоридом калия (KCl) для имитирования эндогенного тона, затем полностью расширяли с 1 нМhCGRP и в конце реверсировали расширение путем кумулятивного добавления возрастающих концентраций соединения I половины единиц логарифма (с учетом рассчета EC50). При каждой концентрации действия препаратов выражаются в % реверсирования CGRP-индуцированного расширения в каждом сосуде. Анализ полученных данных осуществляется для каждого сосуда отдельно, подбирают данные об ответной реакции на концентрацию для четырех параметров логистической функции с помощью анализа нелинейной регрессии для оценки значения EC50. Результаты. Соединение I показывало эффективное и полное реверсирование CGRP-индуцированного расширения внутричерепной артерии человека в условиях ех vivo с ЕС 50 = 88050 рМ. Анализ Шильда. Концентрационная кривая ингибирования CGRP внутричерепной артерии человека в условиях ex vivo Для оценки функционального антагонизма против ряда концентраций CGRP соединение I предварительно инкубировали с отдельными кольцами артерий в инкубаторе тканевых культур, а затем создавали концентрационную кривую CGRP, вызывая полное расширение (с применением нескольких доз агонистов и нескольких доз антагонистов). Более высокие концентрации антагониста образовывали сдвиг вправо в концентрационной кривой CGRP, требующие большие концентрации агониста для преодоления присутствия антагониста и достижения полного расширения. Вкратце, кольца артерий с проволочным держателем предварительно инкубировали с антагонистом, затем заключали с KCl (для имитации эндогенного тона) и далее путем добавления возрастающих концентраций CGRP достигали полного расслабления. Предварительная обработка соединением I являлась эффективной в ингибировании CGRPиндуцированного расширения внутричерепной артерии человека в условиях ex vivo и показывала параллельные сдвиги вправо на концентрационной кривой CGRP. Способы. Каждое артериальное кольцо с установленной проволокой предварительно инкубировалось в течение 30 мин с одной концентрацией (0.1-30 нМ) антагониста соединения I, затем заключалось с 10 мМ KCl (для имитирования эндогенного тона), далее путем добавления возрастающих концентрацийCGRP достигали полного расслабления. Заключение с KCl позволяло стабилизировать так, что общее время предварительной обработки антагонистом составляло приблизительно 45 мин до примененияCGRP. Результаты. Соединение I приводило к параллельному сдвигу вправо концентрационной кривойCGRP внутричерепной артерии человека в условиях ех vivo. Анализ Шильда показывал Kb 91 рМ. Данные результаты выгодно отличались от анализов связывания (Ki = 22.7 рМ) и функционального (Kb = 21.5 рМ) в условиях in vitro. Эффективность соединения I в условиях in vivo в лицевом кровообращении обезьянки Для оценки эффективности новых антагонистов CGRP рецептора в условиях in vivo обезьянки получали серию четырех внутривенных инъекций hCGRP (при 45-мин интервалах). Первая выступала в качестве основного контроля и сопровождалась подкожной доставкой исследуемого препарата. В последующих трех CGRP пробах проводили оценку функционального антагонизма CGRP в условиях in vivo. В настоящем исследовании соединение I показало сильный, длительный CGRP антагонизм. Способы. Обезьянкам давали наркоз и лицевой кровоток увеличивали путем внутривенного (IV) введения hCGRP при 45-мин интервалах (-30, 15, 60 и 105 мин). Влияние исследуемого соединения,доставленного в 0 мин, на hCGRP-индуцированные изменения в лицевом кровотоке измеряли с помощью лазерной допплер-флоуметрии. Эффективные соединения подавляли hCGRP-индуцированное увеличение в лицевом кровотоке, наблюдаемое на 15, 60 и 105 мин (по сравнению с основными данными влияния hCGRP, наблюдаемыми на -30 мин). Объекты. Взрослые особи мужского и женского пола обыкновенных игрунок (Callithrix jacchus) весом 350-650 г выступали в качестве объектов исследования. Анестезия и приготовление. Животных анестезировали путем ингаляции изофлураном в индукторной камере (4-5% быстрой индукции, поддерживается с 1-2.5% Solomon и др., 1999). Анестезия поддерживалась с помощью непрерывного снабжения воздуха:кислорода (50:50) и изофлурана посредством интубации и вентиляции (с контролированием газа крови). Температура тела поддерживалась 380.5 С с помощью помещения на автоматизированной температурорегулирующей поверхности с ректальным зондом. Небольшой участок шерсти (прибл. 1.5 см 2) удаляли с одной или обеих сторон лица с применением крема для удаления волос и/или бритья. Операционную область клипировали и обрабатывали с бетадином. Капельницу размещали в подкожной вене для введения исследуемых соединений и агонистаhCGRP рецептора CGRP. Кроме того, данная капельница обеспечивала забор образцов крови (максимально 2.5 мл, 10%) для контролирования газа крови и анализа уровня соединения в плазме. 5% Раствор декстрозы вводили внутривенно для поддержания уровня сахара в крови. Глубину анестезии контролировали путем измерения кровяного давления и частоты ударов сердца с использованием неинвазивного способа нарукавной манжетой и пульсоксиметра соответственно. Внутривенно 5-10 мг/кг гуанетидина,дополненного внутривенно 5 мг/кг по мере необходимости, давали для стабилизации максимальных потоков в лицевом кровообращении, которые в противном случае показывали постепенное сокращение после повторной стимуляции (Escott и др., 1995). Микроциркуляторный кровоток контролировался путем закрепления самоприлипающего лазерного допплеровского потока зонда на коже лица. Зонд регистрировал количество эритроцитов, пересекающих пути двух лазерных лучей, умноженное на их скорость(сообщалось как изменение в потоке). Доставка лекарственного средства. Исследуемые соединения вводили SC (0.1-0.6 мл/кг) в заднюю часть шеи. Агонист CGRP-рецептора, hCGRP, доставлялся внутривенно (1 мл/кг) в дозе 10 мкг/кг. Протокол исследования. Для оценки эффективности и продолжительности действия в условиях invivo контролировали усиление в лицевом кровотоке, индуцированное введением hCGRP (10 мкг/кг IV) 30 мин до (-0.5 ч) доставки лекарственного средства. Соединение I затем вводили в нулевой момент времени (0 мин) и повторяли доставку hCGRP с 45-мин интервалами в течение 2 ч (данные собирали на 0.25, 1 и 1.75 ч после приема). Соединение I дозировалось на 0.003, 0.01 и 0.03 мг/кг, SC. Образцы плазмы получали непосредственно перед каждым введением hCGRP. После испытания животных возвращали в транспортные клетки, которые находились на температурорегулирующейся поверхности, чтобы держать животных в тепле, пока полностью проснутся, и амбулаторно. Животные могли быть исследованы заново после 14-21 дней отдыха и периода отмывки после предыдущего периода исследования. Результаты. Соединение I (0.003-0.03 мг/кг, SC) показало дозозависимое ингибирование CGRPиндуцированного усиления в лицевом кровотоке обезьян. Сильное (53-80%) ингибирование наблюдалось при 0.03 мг/кг на 0.25, 1 и 1.75 ч после приема. Значительное (35-40%) ингибирование видно на протяжении всего времени испытаний после приема 0.01 мг/кг. При 0.003 мг/кг умеренное (20%), но значительное ингибирование наблюдалось на 0.25 ч при отсутствии эффекта после времени испытания. Сравнивая эффективность против экспозиции, уровни плазмы 8 нМ соответствовали значительной эффективности в условиях in vivo, а уровни 25 нМ с максимальной эффективностью для соединения. Исследования интраназальной раздражительности у крыс Соединение I и соединение III: однонедельное сравнительное интраназальное раздражение Исследование на крысах. Данное исследование выполняли для сравнения возможности назальной раздражительности соединением I по отношению к соединению III при введении интраназально самцам крыс в течение 1 недели. Самцам крыс (10/группа) вливали интраназально растворы соединения I или соединения III (25, 75 или 175 мг/л в 225 мМ янтарной кислоты, 0.02% бензалкония хлорида, 1.25% ангидридной глюкозы, рН 5.8-6.2) один раз в день в дозе объемом 100 мкл/ноздря. При использовании данных дозированных парадигм фиксированные дозы 5, 15 или 35 мг исследуемого препарата доставляли ежедневно. В результате дозы, нормированные на массу тела, снижались по мере того, как крысы росли. Одной контрольной группе давали основу с сукцинатом, а ложной контрольной группе давали солевой раствор путем интраназального вливания. Оцениваемые параметры включали в себя клинические наблюдения, массу тела, потребление продуктов питания, токсикокинетику и гистологическую оценку назальных тканей. Значения для токсокинетических параметров показаны в табл. 11. Таблица 11. Токсокинетические данные, полученные для соединения I Неизмеряемая, вследствие длительного воздействия. Абсолютная биологическая доступность на основе AUC0-24 Ч 7.7 мкМч у крыс после внутривенного введения 1 мг/кг. 3 Предполагаемая площадь назальной слизистой поверхности крыс=14 см 2. 2 Интраназальное дозирование соединения I обеспечивало системное воздействие на крыс до 24 ч, и небольшое различие наблюдалось между первым и последними днями дозирования в этом однонедельном исследовании. Интраназальное введение соединения I хорошо переносилось; полученные данные в жизни ограничивали повышенное слюноотделение во всех группах доз и контролях с основой, и возможно было связано с чрезмерно большим объемом дозирования, применяемым для исследования. У крыс, которым давали солевой раствор, слюноотделения не наблюдалось. Назальная раздражительность наблюдалась от обоих соединений, но соединение I явно вызывало меньшую атрофию обонятельного эпителия, чем соединение III в диапазоне доз (табл. 12). Наблюдаемый тип поражения соответствовал предшествующим наблюдениям, сделанным для соединения II. Полученные данные назальной тяжести и распространенности показали, что соединение I имело больший профиль назальной токсичности, чем соединение III. Таблица 12. Распространенность и тяжесть атрофии обонятельного эпителия после назальной инсталляции соединением III или соединением I у крыс Соединение I и соединение II. Однонедельное экспериментальное исследование интраназальной раздражительности у крыс. Соединение I и соединение II также сравнивали непосредственно на интраназальное раздражение. Самцам крыс (6/группа) вливали интраназально растворы соединения I или соединения II (75 или 175 мг/л) в 225 мМ янтарной кислоты, 1.25% ангидридной глюкозы рН 5.8-6.2) один раз в день в дозе объемом 12.5, 25 или 100 мкл/ноздря. Единственным определением конечной точки в данном исследовании была гистологическая оценка носовых раковин. Оцененная атрофия обонятельного эпителия, вызванная соединением I, была явно менее тяжелой,чем вызванная соединением II в каждой дозе (объемконцентрация). Таблица 13. Токсичность обонятельного эпителия, показатели тяжести для соединения I и соединения II Зависимость доза-реакция соединения II соответствует наблюдаемым в других исследованиях. Как увеличение объема, так и увеличение концентрации способствовали более значительной назальной токсичности, но концентрация являлась, вероятно, более важным фактором. Таким образом, соединение I показало преимущество по отношению к соединению II относительно назальной раздражительности. Возможность назальной доставки. Интраназальный (IN) способ введения для антагонистов CGRP является приемлемым, так как дает возможность неинвазивной доставки с потенциальной возможностью для быстрого начала действия. Высокопроницаемый назальный эпителиальный барьер, также перфузируемая ткань слизистой оболочки и ограниченная метаболическая способность/тканевое время удерживание являются потенциально полезными признаками, которые поддерживают интраназальную доставку соединения, подобного соединению I, которое показывает очень слабое всасывание перорально. Возможность назальной доставки оценивалась на модели кролика IN способом введения с помощью сравнения профилей концентрации в плазме-время и фармакокинетических параметров (Cmax, Tmax,AUC и биологической доступности) для назально доставленного соединения I по отношению к тому,который доставлялся IV способом введения. Дозирование концентрации раствора и подаваемый объем включены в данные таблиц для каждого исследования. Основы композиций описаны в примечаниях под таблицей. Способы. Группы из трех самцов кроликов-альбиносов Новозеландской породы в диапазоне массы от 3-3.5 кг, получающие одну дозу в одной из следующих обработок: 0.5 мг/кг IV болюсным введением в течение 30 с или 0.3-3 мг/кг вводили IN шприцом микрораспылителя. До IN дозирования кроликов слегка усыпляли с использованием действия ингаляционного анастетика Севофлурана. Кролики приходили в сознание в течение 2-5 мин. Серийные образцы крови собирали в гепаринсодержащие вакутейнеры за 2,5, 10, 15, 30 мин до приема препарата и после 24 ч после приема. Образцы крови немедленно центрифугировали при 4 С и отделенную плазму хранили при -80 С до дальнейшего анализа с помощьюLC/MS/MS исследования. Результаты. Фармакокинетический профиль показывает, что соединение I быстро адсорбировалось из носовой полости кроликов при распылении в виде раствора. Время для достижения максимальной концентрации (Tmax) происходило в пределах 0.2-0.3 ч (15-20 мин) при всех исследуемых дозах. Абсолютная биологическая доступность при 0.3, 1 и 3 мг/кг составляла от 13 до 30%, а Cmax составляло от 0.12 до 2.0 мкМ (табл. 14). Таблица 14. Фармакокинетические параметры для соединения I у кроликов после IV и IN введений Интраназальная абсорбция соединения I у кролика была очень быстрой. Плазменные уровни 10 нМ измеряли в пределах 5 мин. Препарат обнаруживался в плазме в течение по меньшей мере 6 ч после приема и до 24 ч при высокой дозе. Предварительно с соединением II имелись большие отклонения в линейности, когда интраназальный объем подачи вместо дозирования был изменен. Сохраняя объем подачи соединения I постоянным и переменным, дозирование концентрации раствора, IN AUC и Cmax показывали склонность к линейной зависимости от дозировки (табл. 15). Изменчивость в данных параметрах также увеличивалась с дозой. При ближайшем рассмотрении интраназальная биологическая доступность проявлялась по мере увеличения дозы (или дозированием концентрации) для трех испытуемых доз (табл. 15). Таблица 15. Линейность дозы IN соединения I у кроликов Дозирование концентраций составляло 10, 30 и 100 мг/мл в 50 мМ основы сукцинатного буфера, рН 5. Таким образом, интраназальный путь введения для соединения I обеспечивал быструю системную абсорбцию и относительно длительные плазменные уровни по сравнению с преоральным путем. Высокая растворимость в воде и улучшенная стабильность раствора благоприятно поддерживали сохранность продукта назального спрея в подходящем аэрозольном приборе. Доставка раствора препарата и его депонирование в полости носа у людей, как ожидается, будет более устойчивой и воспроизводимой, чем это возможно на доклинических моделях IN животных. Предполагается доставлять составы соединения I в устройствах для назальных спреев многоразового использования для многократных доз или одноразового использования для однократной дозы. Фармацевтические композиции и способы лечения Другим аспектом изобретения является фармацевтическая композиция, включающая соединение I с фармацевтически приемлемым адъювантом, носителем и растворителем. Соединение I, как правило, давалось в виде фармацевтической композиции, включающей в себя терапевтически эффективное количество соединения I или фармацевтически приемлемую соль и фармацевтически приемлемый носитель, и могло содержать общепринятые эксципиенты. Терапевтически эффективным количеством является количество, необходимое для обеспечения существенного благоприятного воздействия на пациента, как определено практикой в данной области. Фармацевтически приемлемыми носителями являются те общеизвестные носители, имеющие подходящие профили безопасности. Композиции охватывают все общепринятые твердые и жидкие формы, включающие в себя капсулы, таблетки, пастилки и порошки, а также жидкие суспензии, сиропы, эликсиры и растворы. Твердые композиции могут быть сформированы в составы с регулируемым по времени или замедленным высвобождени- 17022815 ем. Композиции сделаны с применением общепринятых способов технологий изготовления и общепринятых эксципиентов (таких как связывающие и увлажняющие средства) и основы (такие как вода и спирты). Твердые композиции, обычно, формируют в единицах дозирования, обеспечивающих от приблизительно 1 до приблизительно 1000 мг активного вещества на дозу. Некоторыми примерами твердых единиц дозирования являются 0.1, 1, 10, 100, 500 и 1000 мг. Жидкие композиции, обычно, в диапазоне единиц дозирования 1-100 мг/мл. Некоторыми примерами жидких единиц дозирования являются 0.1, 1, 10,25, 50 и 100 мг/мл. Изобретение охватывает все общепринятые модели введения, включающие в себя преоральный, парентальный, интраназальный, подъязычный и трансдермальный способы. Как правило, суточная доза составляет 0.01-100 мг/кг массы тела в день. Обычно, соединение более нуждается в преоральном и менее парентальном введении. Особый режим дозирования, однако, должен определяться врачом при использовании тщательного медицинского обследования. Другим аспектом изобретения является интраназальное введение. Предполагается, что ингибиторы CGRP на рецепторном уровне применимы при патофизиологических состояниях, при которых наблюдается чрезмерная активация CGRP рецептора. Некоторые из них включают в себя нейрогенную вазодилатацию, нейрогенное воспаление, мигрень, кластерную головную боль и другие головные боли, термическое повреждение, циркуляторный шок, менопаузальные приливы и астму. Активация CGRP рецептора была вовлечена в патогенез мигренозной головной боли (EdvinssonJ. Pharmacol. 2002, 135, 356-362). Уровень CGRP в сыворотке поднимается во время мигрени (GoadsbyP.J. и др. Ann. Neurol. 1990, 28, 183-7), а лечение препаратами против мигрени возвращают уровеньCGRP к норме, совпадающей с облегчением головной боли (Gallai V. и др. Cephalalgia 1995, 15, 384-90). Лицо, страдающее мигренозными головными болями, показывало повышенные основные уровни CGRP по сравнению с контролем (Ashina М. и др. Pain 2000, 86(1-2), 133-8). Внутривенное вливание CGRP вызывало продолжительные головные боли у лиц, страдающих мигренозными головными болями (LassenL.H. и др. Cephalalgia. 2002, 22(1), 54-61). Доклинические исследования на собаке и крысе сообщали, что системная блокада CGRP с пептидом антагонистом CGRP(8-37) не изменяла покоя системной гемодинамики ровно, как и регионального кровотока (Shen Y-T. и др. J. Pharmacol. Exp. Ther. 2001, 298, 551-8). Таким образом, антагонисты CGRP-рецепторов могут представлять собой новое лечение мигрени, чтобы избежать сердечно-сосудистой недостаточности в результате активной вазоконстрикции, связанной с неселективными 5-HT1B/1D агонистами, триптанами (например, суматриптан). Другим аспектом изобретения является способ ингибирования CGRP рецептора, включающий в себя контактирование CGRP рецептора с соединением формулы I или его фармацевтически приемлемой солью. Другим аспектом изобретения является способ лечения состояния, связанного с аберрантными уровнями CGRP или сигнальных CGRP рецепторов, включающий в себя введение терапевтически эффективного количества соединения формулы I пациенту. Другим аспектом изобретения является применение соединения формулы I в производстве лекарственного средства для лечения состояний, связанных с аберрантными уровнями CGRP или сигнальныхCGRP рецепторов. Другим аспектом изобретения является способ лечения мигрени или головной боли. Другим аспектом изобретения является способ лечения нейропатической боли. Другой аспект изобретения относится к способу лечения воспаления (в частности, нейрогенного воспаления), боли, термического повреждения, циркуляторного шока, диабета, синдрома Рейно, периферической артериальной недостаточности, субархаидального/краниального кровоизлияния, опухолевого роста, гиперемии, связанной с менопаузой, и других состояний, лечение которых может быть эффективным с помощью антагонизма CGRP рецептора путем введения фармацевтической композиции, включающей в себя соединение формулы I, как указано в данной заявке. Другой аспект изобретения относится к способам, выбранным из группы, состоящей из (а) иммунной регуляции в слизистой кишечника, (b) защитного воздействия против сердечного анафилактического повреждения, (с) стимулирования или предотвращения интерлейкин-1b(IL-1b)-стимуляции резорбции кости, (d) модуляция экспрессии NK1 рецептора в спинальных нейронах и (е) воспалительного заболевания дыхательных путей и хронического обструктивного заболевания легких, включающих в себя астму.Szolcsanyi, Janos. Department of Pharmacology and Pharmacotherapy, Faculty of Medicine, University of Pecs,Pecs, Hung. Current Medicinal Chemistry: Anti-InflammatoryAnti-Allergy Agents (2003), 2(2), 191-218). Другой аспект изобретения относится к способу лечения раковой опухоли и пролиферативных заболеваний и состояний. Антагонисты CGRP были также предложены, чтобы показать пользу в лечении злокачественных заболеваний, в частности против глиомы и рака молочной железы, которые метастазируют в мозг. Антагонисты CGRP могут быть особенно полезны против гипоксических опухолей и в предотвращении метастатической имплантации (см. публикацию заявки РСТ WO 2010006168). Другой аспект данного изобретения относится к способу лечения с применением сочетаний соединений формулы I с одним или несколькими агентами, выбранными из группы, состоящей из СОХ-2 ингибиторов, NSAIDS, аспирина, ацетоминофена, триптанов, эрготамина и кофеина, для лечения мигрени. Мигрень, головная боль и связанные с ними термины как их понимают врачи. Мигрень включает все классы мигреней, включающие в себя общую, классическую, кластерную, пронзающую, гемиплегическую, офтальноплегическую и офтальмическую. Терапевтически эффективное означает, что имеется значимая польза пациенту в понимании врачей. Пациент означает лицо, которое может извлекать пользу от лечения, что устанавливают врачи. Специалисту в данной области техники очевидно, что настоящее изобретение не ограничивается вышеизложенными наглядными примерами и что оно может быть воплощено в других конкретных формах без отклонения от его существенных признаков. Таким образом, желательно, чтобы эти примеры рассматривались во всех отношениях как иллюстрирующие, а не ограничивающие, ссылка сделана на прилагаемую формулу изобретения, а не на представленные выше примеры, и все изменения, которые входят в понятие и пределы эквивалентности формулы изобретения, следовательно, охватываются настоящей заявкой. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение (R)-N-(3-(7-метил-1 Н-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамид или его фармацевтически приемлемая соль 2. Фармацевтическая композиция для лечения состояния, связанного с аберрантными уровнямиCGRP или сигнального CGRP рецептора, содержащая фармацевтически эффективное количество (R)-N(3-(7-метил-1H-индазол-5-ил)-1-(4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-1-оксопропан-2-ил)-4-(2-оксо-1,2-дигидрохинолин-3-ил)пиперидин-1-карбоксамида в сочетании с фармацевтически приемлемым адъювантом, носителем или растворителем. 3. Способ лечения состояния, связанного с аберрантными уровнями CGRP или сигнального CGRP рецептора, включающий введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту. 4. Способ по п.3, отличающийся тем, что состояние представляет собой мигрень. 5. Способ по п.3, отличающийся тем, что состояние представляет собой нейропатическую боль. 6. Способ лечения пролиферативного заболевания, включающий в себя введение терапевтически эффективного количества соединения по п.1 или его фармацевтически приемлемой соли пациенту. 7. Способ по п.6, отличающийся тем, что пролиферативное заболевание представляет собой рак молочной железы или глиому.
МПК / Метки
МПК: A61P 29/00, C07D 401/14, A61K 31/496
Метки: антагонист, рецептора
Код ссылки
<a href="https://eas.patents.su/21-22815-antagonist-receptora-cgrp.html" rel="bookmark" title="База патентов Евразийского Союза">Антагонист рецептора cgrp</a>
Антагонист рецептора il-8

Номер патента: 15520
Опубликовано: 31.08.2011
Авторы: Буш-Петерсен Якоб, Брук Кристофер С., Гудмэн Ричард М., Уэбб Эдвард К.
МПК: A01N 43/00, A61K 31/33
Метки: антагонист, рецептора
Формула / Реферат:
1. Соединение, которое является солью п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной.2. Фармацевтическая композиция, содержащая соль п-толуолсульфокислоты с N-[4-хлор-2-гидрокси-3-(пиперазин-1-сульфонил)фенил]-N'-(2-хлор-3-фторфенил)мочевиной и фармацевтически приемлемый носитель или разбавитель.3. Применение соли п-толуолсульфокислоты с...
Антагонисты рецептора cgrp

Номер патента: 20409
Опубликовано: 30.10.2014
Авторы: Луо Гуанглин, Дубовчик Джин М., Мэйкор Джон Е.
МПК: A61K 31/473, A61K 31/4745, A61K 31/4704...
Метки: антагонисты, рецептора
Формула / Реферат:
1. Соединение формулы Iгде R1 представляет собой водород;R2 представляет собойили пиперидинил, замещенныйR3 представляет собой водород;R4 представляет собой водород;R5 представляет собой водород или гидроксигруппу;R6 представляет собой водород;R7 представляет собой водород;R8 представляет собой водород;R9 представляет собой водород или гидроксигруппу;R10 представляет собой водород;R11 представляет собой водород, гидрокси-, азидо-, амино-,...
Антагонист витронектинового рецептора

Номер патента: 3254
Опубликовано: 27.02.2003
Авторы: Мэнли Питер Дж., Миллер Вилльям Х.
МПК: A61P 9/10, A61K 31/4375
Метки: антагонист, витронектинового, рецептора
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль. 2. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель. 3. Фармацевтическая композиция, включающая соединение по п.1, противоопухолевый агент и фармацевтически приемлемый носитель. 4. Фармацевтическая композиция по п.3, где противоопухолевый агент представляет собой топотекан. 5. Фармацевтическая композиция по п.3, где...
Антагонист минералокортикоидного рецептора и способы его применения

Номер патента: 19179
Опубликовано: 30.01.2014
Авторы: Джадхав Прабхакар Кондаджи, Коутс Дэвид Эндрю, Гавардинас Константинос
МПК: A61P 13/12, A61K 31/5383, A61K 31/4184...
Метки: антагонист, применения, рецептора, способы, минералокортикоидного
Формула / Реферат:
1. Соединение, представляющее собой 5-((E)-(3-фтордибензо[b,e]оксепин-11(6Н)-илиден)метил)-1-((7R,8aR)-гексагидро-1Н-пирроло[2,1-c][1,4]оксазин-7-ил)-1Н-бензо[d]имидазол-2(3H)-он, или его фармацевтически приемлемая соль.2. Соединение по п.1, представляющее собой 5-((E)-(3-фтордибензо[b,е]оксепин-11(6Н)-илиден)метил)-1-((7R,8aR)-гексагидро-1Н-пирроло[2,1-c][1,4]оксазин-7-ил)-1Н-бензо[d]имидазол-2(3H)-он.3. Соединение по п.2 в кристаллической...
Антагонист хемокинового рецептора и циклоспорин в комбинированной терапии

Номер патента: 3181
Опубликовано: 27.02.2003
Авторы: Нельсон Питер Дж., Праудфут Аманда, Веллс Тимоти Н.С., Гроне Херманн-Йозеф
МПК: A61K 38/19, A61K 38/13
Метки: комбинированной, хемокинового, антагонист, циклоспорин, терапии, рецептора
Формула / Реферат:
1. Применение антагониста хемокинового рецептора в комбинации с циклоспорином с целью получения фармацевтической композиции для лечения или профилактики отторжения трансплантированных органов, тканей или клеток. 2. Применение по п.1, отличающееся тем, что антагонист хемокинового рецептора и циклоспорин используются одновременно, раздельно или последовательно. 3. Применение по любому из предшествующих пунктов, отличающееся тем, что антагонистом...
Предыдущий патент: Датчик системы контроля занятости пути
Следующий патент: Ацилированные аналоги глюкагона
Случайный патент: Полупроводниковая полимерная композиция